Embed Size (px)
Citation preview

Epigenetic Deregulation of Genomic Imprinting inHuman Disorders and Following AssistedReproduction
Philippe Arnaud* and Robert Feil
INTRODUCTIONMammals are diploid organisms andfor each autosomal chromosome (22autosomes in humans, 19 in mice),one copy is inherited from themother, and one from the father. Un-til the 1980s, it was thought that thetwo parental copies of autosomalchromosomes were functionallyequivalent. However, this assump-tion was challenged by elegant em-bryological studies in the mouse thatled to the discovery of genomic im-printing (McGrath and Solter, 1984;Barton et al., 1984), and by the sub-sequent discovery of human dis-eases for which their patterns of in-
heritance could not be explained byMendelian genetics (Reik, 1989).
The mouse studies that indicatedthe nonequivalence of maternal andpaternal genomes involved trans-plantation of pronuclei directly fol-lowing fertilization of the egg by thesperm. In this way, zygotes wereproduced that contained either twosets of maternal chromosomes(called gynogenotes), or two sets ofpaternal chromosomes (called an-drogenotes). When transplantedback into recipient female mice, suchmonoparental embryos were foundto arrest during embryonic develop-ment. Intriguingly, different and
rather opposite developmental ab-normalities were observed in the gy-nogenotes as compared to the an-drogenotes. Gynogenotes developedto only about 10 days of gestation,with an apparently normal butsmall embryo. In contrast, theirextraembryonic tissues were se-verely deficient. The phenotype ofandrogenotes was rather oppo-site: whereas the extraembryonicmembranes were largely normal,development of the embryo itselfwas very poor and rarely pro-gressed beyond the four- to six-somite stage (Barton et al., 1984;McGrath and Solter, 1984). Theseexperiments established that dip-loidy alone is not sufficient for em-bryonic development, but thatboth a maternal and a paternal ge-nome are required, because theyplay different developmental roles.
This conclusion was reinforced bygenetic studies on mouse embryosthat were uniparentally disomic(UPD) for individual chromosomes,or for chromosomal regions (i.e.,mice that had inherited two maternalcopies and no paternal copy, or viceversa, of a chromosome or portion ofchromosome). UPD mice were gen-erated for many chromosomal re-gions, together covering almost theentire mouse genome (Cattanachand Kirk, 1985) (Medical ResearchCouncil [MRC] Mammalian Genetics
Imprinted genes play important roles in the regulation of growth anddevelopment, and several have been shown to influence behavior. Theirallele-specific expression depends on inheritance from either the motheror the father, and is regulated by “imprinting control regions” (ICRs). ICRsare controlled by DNA methylation, which is present on one of the twoparental alleles only. These allelic methylation marks are established ineither the female or the male germline, following the erasure ofpreexisting DNA methylation in the primordial germ cells. Afterfertilization, the allelic DNA methylation at ICRs is maintained in allsomatic cells of the developing embryo. This epigenetic “life cycle” ofimprinting (germline erasure, germline establishment, and somaticmaintenance) can be disrupted in several human diseases, includingBeckwith-Wiedemann syndrome (BWS), Prader-Willi syndrome (PWS),Angelman syndrome and Hydatidiform mole. In the neurodevelopmentalRett syndrome, the way the ICR mediates imprinted expression is perturbed.Recent studies indicate that assisted reproduction technologies (ART) cansometimes affect the epigenetic cycle of imprinting as well, and that thisgives rise to imprinting disease syndromes. This finding warrants carefulmonitoring of the epigenetic effects, and absolute risks, of currently used andnovel reproduction technologies. Birth Defects Research (Part C) 75:81–97, 2005. © 2005 Wiley-Liss, Inc.
Philippe Arnaud and Robert Feil are from the Institute of Molecular Genetics, Centre National de la Recherche Scientifique (CNRS)-Universite de Montpellier II, Montpellier, France.
Grant sponsor: Association pour la Recherche Contre le Cancer (ARC); Grant sponsor: Marie Curie European Reintegration Grant; Grantnumber: MERG-CT-2004-510972.
*Correspondence to: Philippe Arnaud and Robert Feil, Institute of Molecular Genetics (IGMM) UMR5535 CNRS Universite de MontpellierII, 1919 Route de Mende, 34293 Montpellier Cedex 05, France. E-mail: [email protected]; [email protected]
Published online in Wiley InterScience (www.interscience.wiley.com). DOI: 10.1002/bdrc.20039
REVIE
WBirth Defects Research (Part C) 75:81–97 (2005)
© 2005 Wiley-Liss, Inc.

Unit, Harwell; http://www.mgu.har.mrc.ac.uk/research/imprinting/).Significantly, uniparental disomy forindividual chromosomes caused abroad range of phenotypic abnor-malities, including abnormal growthand embryonic lethality. In addition,as observed at the whole genomelevel in the pronuclear transplanta-tion experiments, it was found thatthe maternal and paternal copies ofindividual chromosomal regionshave frequently opposite roles in de-velopment, and after birth (http://www.mgu.har.mrc.ac.uk/research/imprinting/).
Combined, the studies on UPDand monoparental embryos pro-vided clear evidence for the exis-tence of genes whose expressiondepends on whether they are inher-ited from the mother or the father.Such differentially expressed genesare referred to as “imprintedgenes.” The first ones to be identi-fied in the mouse were the insulin-like growth factor 2 (Igf2) gene andits neighboring gene H19 on chro-mosome 7, and the Igf2-receptor(Igf2r) gene on chromosome 17(Barlow et al., 1991; Bartolomei etal., 1991; DeChiara et al., 1991).About 80 imprinted genes havesince been discovered in humanand mouse. About half of these areexpressed from the maternal alleleexclusively, and the other half fromthe paternal allele only. Up-to-daterepertories of imprinted genes andtheir diverse functions are pre-sented elsewhere (see Websites ofInterest). Briefly, many imprintedgenes are involved in the regulationof cellular proliferation and growthin the placenta and the embryo.Other groups of imprinted genesplay key roles in neurological pro-cesses and in behavior (Reik andWalter, 2001a; Tycko and Morison,2002). Not surprisingly, therefore,deregulation of imprinted genesgives rise to aberrant development,and is causally involved in differentgrowth and behavioral syndromesin humans (see below).
The majority of imprinted genesare organized in gene clusters thatare up to several megabases insize, and these imprinted domainsare structurally conserved between
human and mouse. Interestingly,most of the imprinted domainscomprise both maternally and pa-ternally expressed genes, as well asgenes that are not imprinted(Paulsen et al., 2000; Reik andWalter, 2001a). The allelic repres-sion along imprinted gene domainsis brought about by specific cis-act-ing control elements. Recently, theuse of genomewide screens to iden-tify imprinted genes (reviewed inSmith et al., 2004) revealed thatsome imprinted genes are locatedapparently on their own, and arenot part of a cluster. For these “sin-gletons,” the cis-acting control ele-ment is thought to be located in thevicinity of the gene, constituting a“microimprinting domain” (Evanset al., 2001).
Studies on species other than hu-man and mouse indicate that im-printing is broadly conservedamong placental mammals, includ-ing primates, rodents, ruminants,and marsupials (Feil et al., 1998;Young et al., 2003). Imprinting hasnot been detected in monotreme oroviparous mammals, as in nonmam-malian vertebrates (Killian et al.,2000; O’Neill et al., 2000; Yokomineet al., 2001). Interestingly, genomicimprinting evolved independently inangiosperm plants, as suggested bythe presence of parental-specificgene expression in the endosperm(the plant “equivalent” of the pla-centa) in Arabidopsis (Messing andGrossniklaus, 1999).
In this review, we focus on thenature of “imprinting control re-gions” (ICRs), the cis-acting ele-ments that control imprinting. Weaddress how and when these keyelements become marked by epi-genetic modifications, and discussthe developmental “life cycle” ofsuch imprints. To make the linkwith human pathologies, in the sec-ond part of this article we provideexamples of how imprints can be-come altered at different develop-mental stages, leading to variousimprinting-related diseases in hu-mans. Additionally, we review re-cent publications that suggest thepossibility that the cycle of imprint-ing can become disrupted as a con-sequence of assisted reproductionprocedures as well.
DIFFERENTIALLYMETHYLATED REGIONSCONTROL IMPRINTING
A remarkable feature of imprintedgenes is that both an active and aninactive allele coexist in the samecell. To achieve such allele-specificexpression, imprinted loci are reg-ulated by epigenetic modificationsthat differentially mark the parentalalleles as either active or re-pressed. These epigenetic modifi-cations are found at key control el-ements, which mediate theimprinted gene expression. An es-sential component of this epige-netic mark is differential DNA meth-ylation (Li et al., 1993). However,DNA methylation is not the onlycomponent of the imprint. In so-matic cells, the parental alleles ofimprinted genes are also differen-tially marked by modifications onthe histone proteins of the nucleo-somes (Hu et al., 2000; Gregory etal., 2001; Xin et al., 2001; Fournierat al., 2002; Umlauf et al., 2004).These covalent modifications of thehistone tails, by methylation andacetylation, are associated with dif-ferences in chromatin conformationbetween the two parental alleles(Feil et al., 1997; Feil and Khosla,1999; Khosla et al., 1999).
So far, at almost all imprinted lociexamined, differentially methyl-ated regions (DMRs) have beenidentified. These are elements of upto several kilobases in size in whichthe DNA methylation status differsbetween the parental alleles. SomeDMRs are “germline DMRs,” inwhich the allelic DNA methylationoriginates from either the male orthe female gamete, and is subse-quently maintained throughout de-velopment. In contrast, at otherDMRs, the parental-allele-specificmethylation is established duringembryonic development. TheseDMRs are sometimes referred to as“somatic DMRs” (Constancia et al.,1998). By gene targeting experi-ments in the mouse, ICRs havebeen defined for several imprintedloci (Thorvaldsen et al., 1998; Bie-linska et al., 2000; Wutz et al.,2001; Fitzpatrick at al., 2002; Yoonet al., 2002; Lin et al., 2003; Wil-liamson et al., 2004). Importantly,
82 ARNAUD AND FEIL
Birth Defects Research (Part C) 75:81–97, (2005)

all these proven ICRs coincide withgermline DMRs. Given their consis-tently found involvement in thecontrol of imprinting, other, not-yet-functionally-explored, germ-line DMRs are considered to be pu-tative ICRs as well. Thus, ICRs areessential sequence elements of fewkilobases in size that bear differen-tial histone modifications andgermline inherited DNA methyl-ation, and that control imprintedgene expression. Whereas someICRs regulate the imprinting of in-dividual genes only, others werefound to be essential for entire clus-ters of imprinted genes coveringhundreds to thousands of kilobases(Bielinska et al., 2000; Fitzpatricket al., 2002; Williamson et al.,2004).
THE LIFE CYCLE OF THEIMPRINT
Following their acquisition in eitherthe male or the female germ line,the allelic methylation marks at
ICRs are maintained in the zygote,and are propagated throughout de-velopment in all somatic tissues(Fig. 1). Nevertheless, imprintsneed to be reset between subse-quent generations, to ensure thatnovel imprints can be properly re-established according to the sex(testis or ovary) of the newly-formed germline. This crucial stepof imprinting is limited in space andin time. The initiation of the imprintis confined to the developing germline lineages, first by erasure of theexisting imprint at the ICR or “im-printing erasure,” followed by ac-quisition of a new set of imprintsaccording to the sex of the germline or “imprinting acquisition.” Webriefly discuss these two steps inthe imprinting process below.
Imprinting Erasure
Erasure of imprinting occurs in pri-mordial germ cells, at an earlystage during differentiation of thefemale and male germlines. In mice
and humans, prior to their implan-tation, blastocysts consist of twomain cell types, the inner cell massand trophectoderm cells. The epi-blast cells within the inner cell massare pluripotent, and these cells giverise to all the somatic cells of thedeveloping embryo, as well as togerm cells. In particular, germ cellsare recruited from the proximal epi-blast at around 7 days postcoitus(dpc) in the mouse. These primor-dial germ cells (PGCs) then migratethrough the hindgut and mesen-tery, and eventually colonize thedifferentiating genital ridges ataround 10.5 dpc, to give rise to thegametes. At 13.5 dpc, female germcells enter meiosis, whereas malegerm cells undergo mitotic arrest.During this process, germ cells ofboth sexes undergo unique and ex-tensive epigenetic reprogramming.In addition to resetting imprints,this global reprogramming is be-lieved to erase aberrant epigeneticmodifications in the germ cells, soas to prevent passing on epigenetic
Figure 1. The life cycle of imprints. This depicted cycle is based on epigenetic modification by DNA methylation (black lollipop), aconsistent hallmark of imprints (see The Life Cycle of the Imprint section). As examples, two ICRs are depicted (as red rectangles) ona hypothetical chromosome: ICR1 has paternally-derived DNA methylation and ICR2 has maternal methylation. The maternallyinherited chromosome is shown in red, the paternal chromosome in hatched blue. The methylation marks at ICR1 and ICR2 areestablished in the male and the female germline, respectively. After fertilization, these opposite imprints are somatically maintainedthroughout development. Their recognition (the “reading”) in the embryo induces stable monoallelic expression. In the developinggermline, the methylation imprints are erased in early germ cells. They are reestablished in later-stage germ cells according to the sexof embryo, to be transmitted to the next generation.
IMPRINTING IN HUMAN DISEASE 83
Birth Defects Research (Part C) 75:81–97, (2005)

“mutations” to the next generation.It was shown recently by immuno-histochemistry that the reprogram-ming initiates at around 8 dpc, andleads to a genomewide, and possi-bly passive, decrease in DNA meth-ylation and of another repressivemodification, methylation at lysine9 of histone H3 (H3-K9) (Seki et al.,2005). Apparently, the processdoes not affect the entire genome,since at 9.5–10.5 dpc, ICRs as theother DMRs, remain unaffected(Hajkova et al., 2002; Sato et al.,2003). It is only at later stages, fol-lowing entry of the PGCs in the maleor the female genital ridges, thatthere is a rapid and possibly activeerasure of DNA methylation at ICRs(Hajkova et al., 2002). This obser-vation is reinforced by elegant nu-clear transfer experiments, inwhich single 11.5 to 13.5 dpc germcells were used to produce clonedembryos (Kato et al., 1999; Lee etal., 2002). The imprinting status inthese embryos suggests that era-sure of functional imprints pro-ceeds from 10.5 dpc, and is com-pleted at 13.5 dpc. This timing isprecisely controlled for each im-printed gene and correlates witherasure of DNA methylation at ICRs(Hajkova et al., 2002; Lee et al.,2002). Thus, different ICRs are de-methylated differently, althoughthe precise underlying mechanismsare unknown. A recent studyshowed that the DNA deaminaseAID can deaminate 5-methylcy-tosine (5meC), and is expressed inPGCs (Morgan et al., 2004). Thisgave rise to the hypothesis that atwo-step process could be involvedin DNA demethylation. Deamina-tion of 5meC by AID could be re-paired by a mismatch glycosylase,and could thus trigger nucleotideexcision repair, replacing 5meC bycytosine. Although attractive, itneeds to be explored yet whethersuch a two-step scenario could beinvolved in demethylation of ICRsin developing germ cells.
Imprinting Acquisition
Acquisition of new imprints occursat late stages of germ cell develop-ment. Interestingly, most of theknown ICRs have oocyte-derived
methylation (Reik and Walter,2001b). Only three imprinted do-mains were found to have an ICRthat is marked by paternally-de-rived DNA methylation (Olek andWalter, 1997; Takada et al., 2002;Yoon et al., 2002). The reason forthis apparent asymmetry betweenthe germlines is unknown, butcould be related to the process ofactive demethylation that affectsthe sperm genome in the fertilizedegg in different (but not all) mam-malian species (Oswald et al.,2000; Reik and Walter, 2001b).
The timing of acquisition is mark-edly different between the maleand the female germlines in mouse.For the male germline, analysis ofthe H19 DMR (at the Igf2-H19 do-main; see Fig. 2) revealed that ac-quisition of DNA methylation startsbefore birth in mitotically arrestedprospermatogonia (between 13.5and 18.5 dpc), and is completedpostnatally by the pachytene stageof meiosis (Davis et al., 1999; Uedaet al., 2000). A similar temporalregulation of imprint establishmentwas observed at the two otherknown ICRs with paternal methyl-ation (Li et al., 2004). By sperm in-jection, it was confirmed that aftermeiosis, in haploid germ cells, pa-ternal imprints are functionally fullyin place (Kimura et al., 1998 Sha-manski et al., 1999). In the femalegermline, in contrast, methylationacquisition at ICRs occurs afterbirth only, during the maturation ofthe oocytes (10–25 days after birthin the mouse). Maternal methyl-ation marks are acquired asynchro-nously at different loci but are, in allcases, completed by the meta-phase II stage (Lucifero et al.,2002, 2004). Nuclear experimentshave shown that, similarly to theDNA methylation imprints, thecompetence for imprinting is ac-quired during the postnatal growthof the oocyte (Obata et al., 1998;Obata and Kono, 2002).
Little is known about the timing ofimprint establishment in humans.The ICR regulating the IGF2-H19locus (Fig. 2) is unmethylated inmale germ cells at fetal stages. Ac-quisition of methylation appears toinitiate in adult spermatogoniaonly, before meiotic division, and
this methylation mark is main-tained during subsequent spermcell development (Kerjean et al.,2000). With regard to the femalegermline in humans, two differentstudies focused on the SNRPN ICR,which controls the Prader-Will syn-drome (PWS) and Angelman syn-drome (AS) domain on chromo-some 15q11–13 (see Fig. 3), but ledto rather different conclusions. Inone study, this ICR was found to beunmethylated in fully-grown hu-man oocytes. This suggested thatthe methylation mark is acquiredonly upon, or after, fertilization inhumans (El-Maarri et al., 2001).This rather unexpected result wasrecently challenged in a secondstudy that applied the same techni-cal approach, and in which theSNRPN ICR was found to be fullymethylated in mature human oo-cytes (Geuns et al., 2003), as in themouse (Lucifero et al., 2004). Fu-ture work on the SNRPN, and onother maternally methylated ICRs,may shed light on the precise tim-ing of imprints establishment in thehuman oocytes, but for obviousethical reasons such studies are noteasily performed. Whatever its pre-cise timing in humans, after estab-lishment of methylation imprints,these are stably maintained in thedeveloping embryo, till birth andinto adulthood (see below).
Factors Mediating ImprintEstablishment
The methylation at ICRs is estab-lished in the male or the femalegermline only. This suggests thatthe protein factors involved in thisprocess are specific to the germlinelineage (Tucker et al., 1996). Someof these key factors were identifiedrecently, and one of these is theDNMT3L protein. DNMT3L is a pro-tein belonging to the DNMT3 denovo methyltransferase family, al-though lacking a functional methyl-transferase domain (Aapola et al.,2000, 2002). Mouse embryos pro-duced from Dnmt3l–/– femalesshowed a complete absence of ma-ternal methylation at ICRs, andhence, had aberrant imprintedgene expression and failed to de-velop beyond 10.5 dpc (Bourc’his et
84 ARNAUD AND FEIL
Birth Defects Research (Part C) 75:81–97, (2005)

al., 2001; Hata et al., 2002). Bio-chemical studies showed that incultured cells, DNMT3L can interactwith the de novo methyltransferaseDNMT3A, and thereby enhances itsDNA methylating activity (Chedinet al., 2002). That DNMT3A is alsoessential for the acquisition of ma-ternal imprint was shown subse-quently (Hata et al., 2002). Thus,DNMT3L is postulated to be a regu-lator of maternal imprint establish-ment, through the interaction withthe essential de novo methyltrans-ferase DNMT3A.
Regarding the male germline,conditional mutant males forDnmt3a showed severely impairedspermatogenesis, and lack of DNAmethylation at two (including H19)of the three paternally-methylatedICRs (Kaneda et al., 2004). The in-volvement of DNMT3L in imprintedmethylation seems to be less criti-cal than in the female germline(Bourc’his and Bestor, 2004;
Kaneda et al., 2004). Rather, re-cent studies show that DNMT3l isresponsible for de novo methyl-ation of dispersed repeated se-quences in spermatogonial stemcells. Inactivation of Dnmt3l in-duces transcriptional reactivationof these retrotransposons as well asto meiotic failure in spermatocytes,leading to arrest of spermatogene-sis (Bourc’his and Bestor, 2004;Webster et al., 2005).
Although these studies have pin-pointed proteins that are essentialfor methylation at ICRs, it is unclearwhy these CpG dinucleotide (CpG)-rich stretches of DNA attract DNAmethylation, and why this happensin one germline only. The normalpattern for such CpG-rich se-quences (so called CpG islands) isthat they are protected againstDNA methylation and stay unmeth-ylated (Jaenisch and Bird, 2003).Given this unusual behavior, it isthought that the capacity of im-
printed CpG islands to attract denovo methylation in the female orthe male germline is mediated byother sequence elements in the vi-cinity. It was postulated that thesecould be stretches of specific repeatsequences, as such repeats werefound to flank several ICRs (Neu-mann et al., 1995). So far, for oneimprinted mouse gene (Rasgrf1) itwas convincingly shown that thedifferential methylation at its ICR isregulated by a nearby repeat se-quence (Yoon et al., 2002). Similartargeted deletion experiments onrepeat elements close to other ICRsdid not, however, alter their im-printing status (Sunahara et al.,2000; Reed et al., 2001; Lewis etal., 2004). Possibly, at these im-printed loci, other sequence ele-ments in the vicinity are importantfor imprint establishment or main-tenance. Whatever the preciserole(s) of repeats in imprinting, it isthought that they could also be in-
Figure 2. The BWS chromosomal region on 11p15.5. The chromosomal region affected in BWS contains two imprinted domains: theKCNQ1 domain, and the neighboring IGF2-H19 domain. These two domains are regulated independently and both have their own ICR(red rectangle). For simplicity, only the essential genes are depicted (but see Delaval and Feil, 2004, Reik et al., 2004). The KCNQ1domain is controlled by the KCNQ1 imprinting control region. This ICR is methylated on the maternal chromosome (filled lollipops), andproduces a noncoding RNA (KCNQ1OT1, also called LIT1) from the paternal chromosome. This noncoding RNA could be involved in thepaternal gene repression along the domain. The neighboring imprinted IGF2-H19 domain is controlled by an ICR located upstream ofthe H19 gene (H19 ICR). This ICR has paternal DNA methylation, and is bound by CTCF proteins on the unmethylated maternal allele.The latter creates an insulating chromatin structure, which insulates IGF2 from its enhancers (blue ovals) located downstream of H19.As a consequence, IGF2 is repressed on the maternal allele. On the paternal chromosome, the H19 ICR methylation spreads into thenearby H19 promoter, and thus leads to paternal repression of H19.
IMPRINTING IN HUMAN DISEASE 85
Birth Defects Research (Part C) 75:81–97, (2005)

volved in how imprints are “read” insomatic cells (Hikichi et al., 2003),or, through their CpG-rich content,could play an evolutionary role inmaintaining a CpG content thresh-old at differentially methylated re-gions (Arnaud et al., 2003).
Concerning the acquisition of dif-ferential methylation in the germ-lines, there is the intriguing possi-bility that other epigeneticmodifications are established at anearlier point in time, as a mark toindicate which DNA sequencesneed to become methylated (Jae-nisch and Bird, 2003). For instance,it was observed that, in male germcells, after methylation erasure,methylation is first reacquired atthe H19 ICR on the paternally-in-herited allele, whereas methylationacquisition on the maternally-in-herited allele is a much later eventand continues even after birth(Davis et al., 2000). A similar ob-servation was made in the femalegermline for the Snrpn ICR. This
ICR first acquires methylation onthe maternally-inherited allele, ingrowing oocytes, whereas gain ofmethylation on the paternally-in-herited allele is detected much later(Lucifero et al., 2004). Thus, inboth the male and the femalegermline, other epigenetic modifi-cations could allow parental allelesto “remember” their origin in theabsence of DNA methylation. Thenature of such modifications is un-known at present. However, giventheir involvement in somatic cells(Gregory et al., 2001, Fournier etal., 2002), covalent histone modifi-cations could be involved (Delavaland Feil., 2004). Alternatively, andnonexclusively, such an earlymarking process prior to the estab-lishment of DNA methylation couldbe somehow linked to the timing ofDNA replication (Simon et al.,1999; Kerjean et al., 2000). Thechallenge for future research will beto show whether ICRs are, indeed,marked by epigenetic modifications
prior to the acquisition of DNAmethylation. Another mechanism isthat ICRs are specifically protectedagainst methylation in one germ-line, but not in the other. This pro-tection could be via binding of non-histone proteins. For instance, inone study the zinc finger proteinCTCF (CCCTC binding factor) wasshown to protect the H19 ICRagainst becoming methylated inthe female germline (Fedoriw et al.,2004). However, no such effectswere observed in another study ex-ploring the role of CTCF in imprintestablishment (Schoenherr et al.,2003).
The Maintenance ofImprints in Somatic Cells
After their establishment in thegermline, methylation imprints aremaintained from the zygote to theembryo, and throughout adult-hood. Remarkably, the differentialmethylation at ICRs is maintained
Figure 3. The PWS and AS domain on 15q11–13. This large imprinted domain is controlled by a bipartite ICR (red rectangles) whichconsists of a small region frequently deleted in AS (AS region), and a minimal region, which is frequently affected in PWS (PWS region).The latter region has differential DNA methylation (filled lollipops), and is also referred to as the SNRPN ICR. The AS region upstreamof the SNRPN ICR is essential for the acquisition of its differential DNA methylation. For simplicity, only the key genes of the domain aredepicted. The paternally-expressed genes (green arrows) in the domain are thought to be activated by the SNRPN ICR on the paternalchromosome. The paternally-expressed UBE3A antisense RNA (UBE3Aas) is thought to repress the UBE3A and ATC10 genes on thepaternal chromosome. This gives rise to maternal allele-specific expression of UBE3A and ATPC10.
86 ARNAUD AND FEIL
Birth Defects Research (Part C) 75:81–97, (2005)

during preimplantation develop-ment, in spite of genomewidechanges in DNA methylation thatoccur at these early stages (Reikand Walter, 2001a). For instance, ithas been observed that, in themouse, after fertilization of theegg, the paternal genome under-goes active demethylation leadingto the removal of almost all its DNAmethylation. The maternal chromo-somes, on the other hand, undergoa passive demethylation during thefirst cell divisions of development.Following implantation, there is awave of de novo methylation, lead-ing to the establishment of newmethylation at many chromosomalregions (Santos and Dean, 2004;and references therein). ICRs arefully resistant to these dramaticglobal changes in DNA methylation.Hence, the methylated parental al-lele at ICRs is protected against de-methylation, whereas the unmeth-ylated allele is protected againstthe acquisition of DNA methylation.
It is poorly understood which pro-tein factors are involved in the so-matic maintenance of the differen-tial methylation at ICRs. Clearly,the maintenance methyltrans-ferase Dnmt1 and its early-embry-onic isoforms (such as Dnmt1o) areessential for the propagation of themethylated allele from one somaticcell generation to the next (Li et al.,1993; Howell et al., 2001). Atpostimplantation stages, the un-methylated allele at ICRs could be-come protected against the de novomethylation machinery by nonhis-tone proteins (Birger et al., 1999).This has been reported in detail forCTCF, which binds to the unmethyl-ated allele of the H19 ICR (Pant etal., 2003; Schoenherr et al., 2003;Szabo et al., 2004).
Covalent histone modificationsare involved in the somatic mainte-nance of imprints at ICRs as well(Fournier et al., 2002). At all ICRsanalyzed, specific histone modifica-tions were found to mark the chro-matin that is associated with themethylated allele. These repressivemodifications include lysine-9 meth-ylation on histone H3, and could berequired for the somatic mainte-nance of the DNA methylation. Onthe opposite parental allele, where
the DNA is not methylated, differenthistone modifications mark the chro-matin. This “ensemble” of modifica-tions includes histone acetylation,and could be involved in keeping theDNA unmethylated (Delaval and Feil,2004).
“Reading” of the Imprint:Chromatin Boundaries andNoncoding RNA
Their opposite configuration on thepaternal versus the maternal alleleensures that ICRs exert their actionin an allele-specific manner in so-matic tissues, and thus mediate pa-rental allele-specific gene expres-sion (Reik and Walter 2001a;Delaval and Feil, 2004). Despite thefact that differential methylation atICRs is maintained in all the so-matic lineages throughout develop-ment, at some imprinted loci theallelic expression is limited to spe-cific tissues, or to certain develop-mental stages (Liu et al., 2000). Atsome imprinted loci, such a re-stricted pattern of imprinted ex-pression correlates with tissue-spe-cific differential DNA methylation insecondary DMRs (Feil et al., 1994;Constancia et al., 2000; Weber etal., 2003a). Differential “reading”of imprints could also explain thedifference in allelic gene expressionbetween human and mouse atsome imprinted loci (Smrzka et al.,1995; Arnaud et al., 2003).
How ICRs give rise to allele-spe-cific gene expression differs betweenimprinted loci. In fact, multiple read-ing mechanisms were discoveredduring the last years. The simplestscenario is found at loci in which theICR constitutes the promoter of agene. DNA methylation and chroma-tin compaction at the ICR repressespromoter activity on one of the twoparental alleles, whereas chromatinon the opposite allele is permissive togene transcription. The imprintedZac1 and U2af1-rs1 loci in the mouseare likely to be controlled throughsuch a direct mechanism (Feil et al.,1997; Arima et al., 2000; Fournier etal., 2002; Smith et al., 2002). Atother imprinted domains, more com-plex reading mechanisms were de-tected (Figs. 2 and 3) and, indeed,
some ICRs regulate gene expressionalong thousands of kilobases.
The regulation of large imprinteddomains is thought to involve a se-quence of epigenetic events that isinitiated by the ICR, and that even-tually affects gene expression alongthe entire domain. For such “indi-rect” mechanisms, two main para-digms have emerged: regulation bychromatin boundaries and regula-tion by noncoding RNAs. These twomodels of imprinted gene regulationare summarized below. Comprehen-sive descriptions can be found in re-cent review texts published else-where (Verona et al., 2003; Reik etal., 2004; Weber et al., 2005). Themodel that ICRs can act as chromatinboundaries comes from functionalstudies on the Igf2/H19 locus and isdescribed in Figure 2.
The involvement of noncoding RNAin genomic imprinting became ap-parent from the Igf2r domain onmouse chromosome 17. Here, theoocyte-derived methylation at theICR covers the promoter of a non-coding transcript, called Air. Air RNAis transcribed from the unmethyl-ated paternal allele only. Via a stillunclear mechanism, this paternalRNA, or its transcription, repressesthe Igf2r gene on the paternal al-lele, and in the placenta, two flank-ing genes are transcriptionally re-pressed as well (Wutz et al., 2001;Sleutels et al., 2002). NoncodingRNAs have been identified withinother imprinted domains (Figs. 2and 3), and at some, they are pro-duced from the ICR at the unmeth-ylated allele only, similar to theIgf2r locus. Although still poorly un-derstood, the putative involvementof noncoding RNA at imprinted do-mains shows similarities to X-chro-mosome inactivation in human andmouse. Future work should unravelto what extent these two main epi-genetic phenomena are mechanis-tically linked (Ogawa and Lee,2002; Delaval and Feil, 2004).
IMPRINTING AND HUMANDISEASE
In some human diseases, the pheno-typic manifestations depend on in-heritance from mother or fromfather. These complex pathologies
IMPRINTING IN HUMAN DISEASE 87
Birth Defects Research (Part C) 75:81–97, (2005)
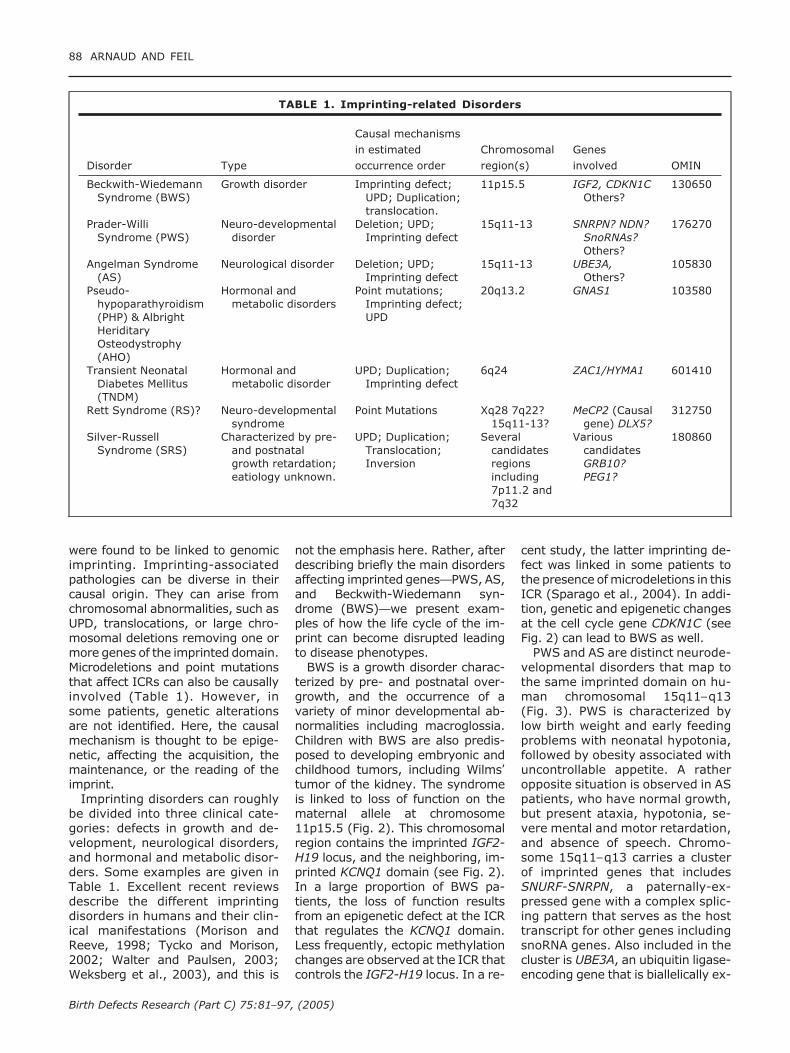
were found to be linked to genomicimprinting. Imprinting-associatedpathologies can be diverse in theircausal origin. They can arise fromchromosomal abnormalities, such asUPD, translocations, or large chro-mosomal deletions removing one ormore genes of the imprinted domain.Microdeletions and point mutationsthat affect ICRs can also be causallyinvolved (Table 1). However, insome patients, genetic alterationsare not identified. Here, the causalmechanism is thought to be epige-netic, affecting the acquisition, themaintenance, or the reading of theimprint.
Imprinting disorders can roughlybe divided into three clinical cate-gories: defects in growth and de-velopment, neurological disorders,and hormonal and metabolic disor-ders. Some examples are given inTable 1. Excellent recent reviewsdescribe the different imprintingdisorders in humans and their clin-ical manifestations (Morison andReeve, 1998; Tycko and Morison,2002; Walter and Paulsen, 2003;Weksberg et al., 2003), and this is
not the emphasis here. Rather, afterdescribing briefly the main disordersaffecting imprinted genes—PWS, AS,and Beckwith-Wiedemann syn-drome (BWS)—we present exam-ples of how the life cycle of the im-print can become disrupted leadingto disease phenotypes.
BWS is a growth disorder charac-terized by pre- and postnatal over-growth, and the occurrence of avariety of minor developmental ab-normalities including macroglossia.Children with BWS are also predis-posed to developing embryonic andchildhood tumors, including Wilms’tumor of the kidney. The syndromeis linked to loss of function on thematernal allele at chromosome11p15.5 (Fig. 2). This chromosomalregion contains the imprinted IGF2-H19 locus, and the neighboring, im-printed KCNQ1 domain (see Fig. 2).In a large proportion of BWS pa-tients, the loss of function resultsfrom an epigenetic defect at the ICRthat regulates the KCNQ1 domain.Less frequently, ectopic methylationchanges are observed at the ICR thatcontrols the IGF2-H19 locus. In a re-
cent study, the latter imprinting de-fect was linked in some patients tothe presence of microdeletions in thisICR (Sparago et al., 2004). In addi-tion, genetic and epigenetic changesat the cell cycle gene CDKN1C (seeFig. 2) can lead to BWS as well.
PWS and AS are distinct neurode-velopmental disorders that map tothe same imprinted domain on hu-man chromosomal 15q11–q13(Fig. 3). PWS is characterized bylow birth weight and early feedingproblems with neonatal hypotonia,followed by obesity associated withuncontrollable appetite. A ratheropposite situation is observed in ASpatients, who have normal growth,but present ataxia, hypotonia, se-vere mental and motor retardation,and absence of speech. Chromo-some 15q11–q13 carries a clusterof imprinted genes that includesSNURF-SNRPN, a paternally-ex-pressed gene with a complex splic-ing pattern that serves as the hosttranscript for other genes includingsnoRNA genes. Also included in thecluster is UBE3A, an ubiquitin ligase-encoding gene that is biallelically ex-
TABLE 1. Imprinting-related Disorders
Disorder Type
Causal mechanismsin estimatedoccurrence order
Chromosomalregion(s)
Genesinvolved OMIN
Beckwith-WiedemannSyndrome (BWS)
Growth disorder Imprinting defect;UPD; Duplication;translocation.
11p15.5 IGF2, CDKN1COthers?
130650
Prader-WilliSyndrome (PWS)
Neuro-developmentaldisorder
Deletion; UPD;Imprinting defect
15q11-13 SNRPN? NDN?SnoRNAs?Others?
176270
Angelman Syndrome(AS)
Neurological disorder Deletion; UPD;Imprinting defect
15q11-13 UBE3A,Others?
105830
Pseudo-hypoparathyroidism(PHP) & AlbrightHeriditaryOsteodystrophy(AHO)
Hormonal andmetabolic disorders
Point mutations;Imprinting defect;UPD
20q13.2 GNAS1 103580
Transient NeonatalDiabetes Mellitus(TNDM)
Hormonal andmetabolic disorder
UPD; Duplication;Imprinting defect
6q24 ZAC1/HYMA1 601410
Rett Syndrome (RS)? Neuro-developmentalsyndrome
Point Mutations Xq28 7q22?15q11-13?
MeCP2 (Causalgene) DLX5?
312750
Silver-RussellSyndrome (SRS)
Characterized by pre-and postnatalgrowth retardation;eatiology unknown.
UPD; Duplication;Translocation;Inversion
Severalcandidatesregionsincluding7p11.2 and7q32
VariouscandidatesGRB10?PEG1?
180860
88 ARNAUD AND FEIL
Birth Defects Research (Part C) 75:81–97, (2005)

pressed in most tissues except inbrain, where the paternal allele is si-lenced. The regulation of this com-plex imprinting cluster remains enig-matic, but is believed to depend on abipartite ICR controlling the epige-netic status of the entire domain. Thebipartite control element comprisesthe maternally methylated 5� portionof the SNRPN gene (Geuns et al.,2003), and a short region 35-kb up-stream of SNRPN that does not carrydifferential DNA methylation (Buitinget al., 1999) (Fig. 3). Most PWS pa-tients have either a sporadically-ac-quired deletion of 15q11–q13 on thepaternal allele, or present a maternalduplication of chromosome 15. Inonly a small percentage of patients,the disease is caused by an epige-netic defect (Buiting et al., 2003).AS, on the other hand, is caused bypaternal duplication, or by deletion ofthe maternal allele within the regioncomprising UBE3A, a gene which iscausally involved in the disease. Epi-genetic defects are found in less than5% of AS patients (Paoloni-Giaco-bino and Chaillet, 2004).
Failure to Erase ImprintsCan Lead to Disease
From mouse studies, it has becomeapparent that epigenetic marks atretroviral and certain repeated ele-ments are not always completelyerased from one generation to thenext (Morgan et al., 1999; Kearns etal., 2000; Lane et al., 2003). In thecase of such an incomplete erasurein the germline occurring at im-printed loci, this may give rise to dif-ferent phenotypes according to thegrandparental origin of the chromo-some transmitted to the progeny. Anallele for a given locus, inherited forexample from the mother, may be-have differently depending onwhether it originated from the ma-ternal grandfather, or from the ma-ternal grandmother. Interestingly, agrandparental effect was observed ina subset of PWS patients. In thesePWS patients, there is a maternalimprint at 15q11–q13 on both thematernally- and the paternally-in-herited allele, in absence of any se-quence abnormalities at the bipartitecontrol region. Detailed molecularstudies suggested that the aberrant
imprinting in these PWS patients re-sulted from a failure to erase thegrandmaternal imprint in the pater-nal germline (Buiting et al., 2003).
Failure in Acquisition inBiparental HydatidiformMoles
The abnormal “hydatidiform mole”pregnancy belongs to the group ofgestational trophoblastic diseases (Liet al., 2002; Fisher and Hodges,2003). Most hydatidiform moles cor-respond to conceptuses that carrytwo paternal genomes, and no ma-ternal genome. These moles are re-ferred to as “complete moles” and,as in androgenetic mouse embryos,exhibit trophoblastic hyperplasia andgrossly abnormal embryonic devel-opment (Devriendt, 2005). In addi-tion, trophoblastic tumors are fre-quently associated with hydatidiformmoles (Devriendt, 2005). Most hy-datidiform moles are sporadic, andthey occur about once every 1000pregnancies in Caucasians (Grimes,1984). Their exact etiology isunknown, but clearly abnormalgenomic imprinting underlies the pa-thology, since there is an imbalancebetween the maternal and the pater-nal genome. In a less-common classof hydatidiform moles, there are twopaternal and one maternal genomes.In contrast to complete moles, these“partial moles” undergo develop-ment of the embryo, at least until theearly fetal stages.
Consistent with the view that im-printing is causally involved (Dev-riendt, 2005), complete hydatidiformmoles have a paternal epigenotype onboth alleles at all imprinted loci ana-lyzed (Mowery-Rushton et al., 1996;Arima et al., 2000; Kamiya et al.,2000; Judson et al., 2002). In addi-tion, imprinted genes that are nor-mally transcribed from the maternal-ly-inherited allele do not show anyexpression at all in complete moles(Chilosi et al., 1998; Castrillon et al.,2001; Saxena et al., 2003).
Exceptionally, some hydatidiformmoles have a biparental genomiccontribution with both a maternaland a paternal genome. Such “bipa-rental moles” can be familial in ori-gin. Given their aberrant develop-ment, a major imprinting defect is
likely to be involved in these molesas well. Aberrant imprinting on thepaternally-inherited genome seemsunlikely in this specific disorder,since it was reported that biparentalmoles can occur in the same womanwith different partners (Fisher et al.,2000). It seems more likely from fa-milial studies that in biparentalmoles, there is a genetic defect thatprevents the establishment of germ-line maternal imprints. Indeed, in arecent study on a family with bipa-rental moles (Judson et al., 2002),several ICRs that normally have ma-ternal DNA methylation (includingthe SNRPN and the KCNQ1 ICR)were completely lacking methylationon both the parental alleles. The H19ICR, which is normally methylatedon the paternal allele, displayed anunaltered pattern of DNA methyl-ation. These observations are con-sistent with the idea that biparentalmoles are caused by a (recessive)maternal mutation that leads to fail-ure to establish maternal imprints.Molecular studies on the family indi-cated that DNMT3L and other DNMTgenes were not affected in the bipa-rental moles (Hayward et al., 2003),in addition, a mutation in the 19q13locus (see below) has been excludedin this specific family (Judson et al.,2002). It remains unknown whichfactors are involved in the aberrantmaternal imprints.
Linkage analyses performed onother families with biparental molesassigned one causal locus to chro-mosome 19q13.4, a region that isrich in genes encoding zinc fingerproteins (Moglabey et al., 1999;Hodges et al., 2003). However, ad-ditional genetic loci could be causallyinvolved in biparental moles, or themutation on 19q13 is not alwayspenetrant. The latter assumption issupported by the observation that insome families there is reduction inDNA methylation at ICRs (SNRPN,KCNQ1, and others), but the loss ofDNA methylation is not complete inall conceptuses analyzed. Further-more, some biparental moles showan H19 ICR that has ectopic methyl-ation on the maternal allele (El-Maarri et al., 2003). Possibly, bipa-rental moles linked to a defective19q13.4 locus are due to an error,rather than a failure, in establishing
IMPRINTING IN HUMAN DISEASE 89
Birth Defects Research (Part C) 75:81–97, (2005)

maternal imprints during oogenesis.Alternatively, there may be a prob-lem with their postzygotic mainte-nance (El-Maarri et al., 2003, 2005).Consistent with the idea that causalmutations do not always manifestthemselves (partial penetrance?),some women that carried the disor-der nevertheless had a normal preg-nancy (Helwani et al., 1999).
Given the broad deregulation ofmaternal imprints, it is difficult topinpoint specific imprinted genesthat are involved in hydatidiformmole. However, some imprintedgenes seem to be of critical impor-tance. CDKN1C, coding for a nega-tive regulator of the cell cycle, ismaternally expressed in the ex-traembryonic tissues. As expected,in partial and complete moles thereis hardly any expression of this genein the extraembryonic tissues (Fisheret al., 2002). However, in a singlecase of hydatidiform mole, indistin-guishable from others, a positive im-munostaining for CDKN1C has beenreported, explained by the selectiveretention of the maternal chromo-some 11, in addition to two paternalcopies. This argues against an exclu-sive role for this gene, or other ma-ternally-expressed genes such asH19 or IPL present on chromosome11, in the pathogenesis of this defect(Fisher et al., 2004). Another poten-tial candidate is the paternally-ex-pressed IGF2 gene, which has beenshown to be crucial for placental de-velopment in mice (Constancia et al.,2002). However, the discrepancy inthe methylation pattern of the ICRthat regulates its imprinted expres-sion (H19 ICR) between most of thecomplete moles (biallelically methyl-ated) and some phenotypically indis-tinguishable biparental moles (cor-rect paternal methylation; Judson etal., 2002) also argues against an ex-clusive role for this gene.
In Vivo Failure inMaintenance: DiscordantImprinting Disorder inTwins and Somatic MosaicImprinting
The preimplantation period of de-velopment is particularly critical forthe maintenance of imprinting. This
follows from the observation that inmonozygotic twin pairs, frequentlyonly one manifests BWS. In 10 suchdiscordant pairs of twins, all of theaffected twins of the pairs showedloss of methylation at the KCNQ1ICR. This correlated with aberrantimprinted expression along the im-printed domain. In fact, monozy-gotic twin births have an increasedrisk of BWS. Hence, the incidence ofmonozygotic twins among BWS pa-tients is considerably higher (up to20 times), compared to that of thegeneral population (Weksberg etal., 2002). This suggests that thereis a critical time period during earlydevelopment during which distur-bance in imprinting is associatedwith an increased likelihood ofmonozygotic twinning.
It is not known why monozygotictwinning and loss of imprinting aresometimes linked, and whetherthere are predisposing genetic fac-tors. Although these phenomena cancoexist in male twins as well (Leo-nard et al., 1996), the vast majorityof monozygotic twin pairs in whichone has BWS, are female (Bestor,2003). This striking discrepancyraises the question whether duringearly female development (as com-pared to male development), there isa higher probability to have dis-rupted maintenance of imprinting.Future research may unravelwhether this is linked to the earlyembryonic distribution, or the actionof DNA methyltransferases, or to amore indirect effect (Bestor, 2003).
Failure to maintain imprints dur-ing early development can lead tomosaic defects in individual babiesas well. Recently, a child with PWSwas shown to have a mosaic im-printing defect (Wey et al., 2005).This patient had an apparently nor-mal karyotype, with both a mater-nal and a paternal chromosome 15,but displayed the typical features ofPWS. Molecular analyses revealedthat only about one-half of its bloodcells had aberrant imprinting (DNAmethylation) at the SNRPN ICR. Al-though brain tissues were not stud-ied, this observation suggests thatloss of imprinted DNA methylationin a subset of cells can be sufficientto promote PWS.
In Vitro Failure inMaintenance: PerturbedImprinting followingAssisted Reproduction
Disruption of imprinting through fail-ure of maintenance can result fromculture of early embryos as well. Al-though such culturing effects havebeen mostly described in animalmodels, they could potentially be rel-evant to assisted reproductive tech-nologies (ART) in humans. Pheno-typic effects of in vitro culture werefirst observed in mouse, and aremost pronounced in ruminant spe-cies (reviewed in Khosla et al.,2001a). In sheep and cattle, cultureof preimplantation embryos fre-quently gives rise to overgrowth andto diverse developmental abnormal-ities during fetal and postnatal devel-opment. These aberrant phenotypesare collectively referred to as thelarge offspring syndrome (Young etal., 1998), which is reminiscent ofBWS in human. It is thought that im-printed genes could, at least in part,be causally involved in the syn-drome. In sheep, in vitro culture untilthe blastocyst stage, followed bytransfer into recipient females, givesrise to fetuses that have strongly re-duced methylation at the ICR con-trolling the imprinted IGF2R gene.This correlates with a loss of IGF2Rexpression (Young et al., 2001),which is likely involved in the fetalovergrowth (Wutz et al., 2001). In-terestingly, somatic cell nucleartransfer (“cloning”) in sheep alsoleads to loss of methylation at theimprinted IGF2R gene, possibly be-cause this procedure also involvesextensive in vitro culture of embryos(Young et al., 2003).
In addition, in mouse, culture ofembryos may affect postimplanta-tion developmental potential, andcan induce behavioral alterations aswell (Bowman and McLaren 1970;Doherty et al., 2000; Khosla et al.,2001b; Fernandez-Gonzalez et al.,2004). For instance, altered expres-sion of imprinted genes (H19, Igf2,and the growth-factor receptor bind-ing protein Grb10) was observed infetuses that derived from blastocyststhat had been cultured in mediumsupplemented with fetal calf serum(Khosla et al., 2001b). In these fe-
90 ARNAUD AND FEIL
Birth Defects Research (Part C) 75:81–97, (2005)

tuses, decreased expression of H19correlated with an increase in meth-ylation at its ICR. However, the cul-ture medium used did not affect allimprinted genes. In addition, nonim-printed genes were also found to beaffected, showing that the perturba-tion is not specific to imprintedgenes. For instance, Grb7, a growth-factor receptor binding protein that isnot imprinted, displayed a substan-tial reduction in expression. In an-other study, in a different culturemedium, a subset of cultured blasto-cysts displayed biallelic expression ofH19. This correlated with loss ofmethylation on the paternal allele(Doherty et al., 2000). The biallelicH19 expression persisted in the ex-traembryonic tissues, but becamesomehow corrected in the develop-ing embryo itself (Mann et al., 2004).Interestingly, some placentae dis-played biallelic expression of otherimprinted genes as well (Mann et al.,2004). Some of these, however,seemed unaffected in their expres-sion in the cultured blastocyststhemselves. This suggests that per-turbations in preimplantation em-bryos can manifest themselvesmuch later in development, long af-ter the embryos are removed fromthe culture medium.
The above studies establish that inseveral mammalian species, cultureof early embryos may have pheno-typic consequences later in develop-ment and that this could, in part, bedue to a perturbation in the somaticmaintenance of imprinting. Relativeto the situation in humans, thisraises the question as to whetherepigenetic alterations could arise asa consequence of ART as well (seeGosden et al., 2003; Maher et al.,2003a; Paoloni-Giacobino and Chail-let, 2004). Assisted reproductionprocedures involve manipulation andin vitro culturing of gametes and pre-implantation embryos. Potentially,these procedures could affect main-tenance of genomic imprinting, sim-ilarly as in other mammalian species(Feil, 2001).
Since 1978, when the first in vitrofertilization (IVF) baby was born,assisted reproduction has been in-creasingly used for the treatment ofinfertility. Intracytoplasmic sperminjection (ICSI) was introduced in
1992, for the treatment of someforms of male infertility. At present,ARTs account for no less than 1–3%of births in developed countries.Generally, these techniques are re-garded as safe, since the incidenceof abnormalities at birth is reassur-ingly low and children develop nor-mally. However, in recent years,several reports have suggested apossible link between assisted re-production and an increased risk ofbirth defects. In 42,463 infantsconceived by assisted reproduc-tion, a recent study reported a 2.6-fold increased risk for low and verylow birth weight for singleton preg-nancy babies (Schieve et al.,2002). In another large study, therisk of major birth defects arisingfrom assisted reproduction was re-ported to be as high as 9% for IVF,and 8.6% for ICSI, compared to4.2% following natural conception(Hansen et al., 2002). In both ofthese large studies, causes leadingto such abnormalities were notidentified. Generally, it is difficult todetermine whether birth defects af-ter assisted reproduction reflect thesuboptimal fertility of the parents,or whether they result directly fromthe procedures used.
A possible link between ART andcertain imprinting congenital disor-ders was suggested by severalgroups. Cox et al. (2002) andOrstavik et al. (2003) describedthree children with AS conceived byICSI. In all three cases, AS wascaused by a loss of methylation atthe SNRPN ICR (Fig. 3). In the gen-eral population, the occurrence ofAS is about 1 in 15,000, and in only5% of patients the syndrome iscaused by epigenetic changes.Given this extremely low frequencyof “epigenetic AS,” the findings ofthe two groups suggest a link be-tween ART and epigenetic defectsleading to aberrant imprinting.
Associations between assistedreproduction and a second imprint-ing disorder, BWS, have been doc-umented as well in several coun-tries. These studies showed thatthe link with imprinting disorders isnot restricted to ICSI procedures.In the United States, DeBaun et al.(2003) reported seven cases ofBWS, five of whom were conceived
by ICSI and one by IVF. It was un-known if the remaining child wasconceived by ICSI or IVF. The sameclinical group estimated that theconception by assisted reproduc-tion was 4.6% (3/65) among BWSchildren, which is significantlyhigher than in the normal popula-tion. In a British BWS registry, 6 of149 (4%) children were conceivedby assisted reproduction (three byIVF, three by ICSI) (Maher et al.,2003b). In a French registry, thesame percentage was reported(Gicquel et al., 2003). Combinedmolecular analyses establishedthat no fewer than 13 of the 14 BWSchildren for whom analysis waspossible, had an epigenetic imprint-ing defect. Loss of methylation atthe KCNQ1 ICR was observed in all13. For one of the BWS childrenthere was, in addition, ectopicmethylation at the H19 ICR. Thesetwo epimutations are known to oc-cur in �50% and �2% of overallpatients with BWS, respectively(Weksberg et al., 2003). The highoccurrence of epimutation at theKCNQ1 ICR in the 14 patients ana-lyzed here (93%) suggests that as-sisted reproduction procedureslead to the epigenetic alterations.
From the combined studies, it ap-pears that there is a four-fold higherrisk of BWS in babies conceived byART. In an earlier report, one case ofBWS was found in 73 children con-ceived by assisted reproduction (Ol-ivennes et al., 2001). Since BWS isestimated to occur in 1.3 per100,000 births (minimal estimation)(Maher et al., 2003b; and referencestherein), this report also suggests alink between assisted reproductionand an increased risk of BWS. How-ever, in all these studies, the numberof cases analyzed remains small andstudies on larger cohorts are war-ranted. Two such large studies werepublished recently, providing dis-crepant conclusions. A case-controlstudy was conducted in Australia,where a single clinical genetics ser-vice laboratory provided moleculartests for BWS. Among 1,316,500births that were monitored between1983 and 2003, 37 BWS cases weredetected. Assisted reproduction wasthe method of conception for four ofthem (one by ICSI, three by IVF).
IMPRINTING IN HUMAN DISEASE 91
Birth Defects Research (Part C) 75:81–97, (2005)

Molecular analysis performed onthree of these four BWS childrenshowed loss of methylation at theKCNQ1 ICR. During the period ofstudy, 14,894 babies were born as aresult of an ART procedure. Based onthese figures, the risk of BWS wasreported to be nine times higher fol-lowing assisted reproduction, com-pared to the general population (Hal-liday et al., 2004). Another largestudy assessed the incidence of im-printing disease in Danish singletonbabies born between 1995 and2001, including malignancies andneurodevelopmental diseases thatcould be linked to imprinting (Lide-gaard et al., 2005). During the sev-en-year study period, 442,349 ba-bies were conceived naturally, and6052 by assisted reproduction (28%of whom were by ICSI). In this largestudy, no increased risk of imprintingdisease syndromes, childhood can-cer, or neurodevelopmental distur-bance was apparent following ART. Itis noteworthy that no case of BWS isreported in this study. Thus, theDanish register data do not supportan increased risk of imprinting dis-ease after assisted reproduction.
Regarding ART, it is unclear atwhich stage(s) of the cycle of im-printing, the epigenetic alter-ations could arise. It seems un-likely that ARTs interfere withpaternal imprints in sperm, sincethese are already established atthe spermatid phase of spermato-genesis. Whether freezing of ma-ture sperm, or cryopreservativesused for freezing oocytes, coulddisturb imprints is not docu-mented. Concerns have been ex-pressed for ICSI in that it bypassesselection mechanisms that operatein natural conception, and that theprocedures used could have delete-rious effects. However, round sper-matid injections in the mouse indi-cate that imprinting is notperturbed by ICSI itself (Shaman-ski et al., 1999). In addition, stud-ies on BWS (Bonduelle et al., 2002)indicate that ICSI is not more riskythan conventional IVF.
It has been argued that the sper-matozoa used for ICSI originatefrom men with abnormal semen pa-rameters, and that this could affectimprinting. That indeed this could
be the case is suggested by a recentstudy showing that there is some-times reduced methylation at theH19 ICR in spermatozoa from oligo-zoospermic men compared to nor-mozoospermic men (Marques etal., 2004). Nevertheless, this ob-servation alone cannot account forthe majority of cases, in which theepigenetic alterations affect thematernal imprint (loss of maternalICR methylation in BWS, and inAS). The oocytes used for assistedreproduction are usually obtainedfrom women following hormonalhyperstimulation. Such hormonaltreatments could affect the estab-lishment of the complete array ofmaternal imprints. Hormonal hy-perstimulation could, for instance,cause the premature release of im-mature oocytes that have not yetcompleted the establishment of im-prints at all the regions. Similartreatments in mice show that therecan be abnormal overall levels ofDNA methylation as a consequenceof superovulation (Shi and Haaf,2002). Interestingly, in a study on12 BWS patients conceived with dif-ferent assisted reproduction proce-dures, the only identified commonfactor was that all 12 mothers hadundergone ovarian stimulation(Chang et al., 2005). This apparentrisk warrants care and strict moni-toring, particularly relative to theemerging technology of in vitro oo-cyte maturation (IVM). In IVM, oo-cytes are cultured from the germi-nal vesicle to the metaphase IIstage, and thus could affect genesthat acquire their imprint late in oo-cyte development. In one recentstudy, culture of mouse oocyteswas indeed shown to affect methyl-ation imprints at H19, Igf2r, andPeg (Kerjean et al., 2003).
In conclusion, several recent stud-ies suggest a link between assistedreproduction and increased frequen-cies of imprinting diseases. How-ever, it should be noted that theabsolute risk these procedures rep-resent is extremely low. AS causedby epigenetic alterations is observedat a frequency of 1 in 300,000 only.Therefore, even with a several-foldincrease following assisted repro-duction, it remains highly unlikelythat babies will develop the disease.
This important consideration also fol-lows from a study in which there wasno evidence of abnormal methyl-ation at the PWS/AS domain in 92children conceived by ICSI (Manninget al., 2000). Nonetheless, epimuta-tions found at the SNRPN and KCNQ1ICRs may reflect wider epigeneticchanges across the genome, includ-ing at less-well-characterized re-gions. Such epigenetic changes else-where in the genome may induceother health problems, such as child-hood cancer or neurodevelopmentalcomplications, later in life. For in-stance, an association between as-sisted reproduction procedures andretinoblastoma was reported in aDutch cohort of children (Moll et al.,2003), emphasizing the importanceof carefully monitoring the long-termconsequences for health. It is atpresent too early to concludewhether there may also be conse-quences to the next generation. Toprovide meaningful information tocouples seeking assisted reproduc-tion, it should be important to per-form large multination studies to as-sess the absolute risks of thedifferent pathologies, including im-printing disorders. For this, long-term follow up of physical and neu-robehavioral development, as well asof cancer incidence, is necessary.Such studies would have to be com-pleted with molecular analysis of thegametes and embryos used in as-sisted reproduction. Combined, suchstudies may shed light on whetherepigenetic alterations at imprintedand other genes are somehow a con-sequence of the compromised fertil-ity itself (including increased age ofparents, stress factors, etc.), or areindeed caused by the assisted repro-duction procedures.
Failure in the Reading:Loss of Imprinting andRett Syndrome
The epigenetic features at ICRs areread in differing ways to ensureproper parental allele-specific ex-pression. Conceptually, deficien-cies in factors that are involved inthe reading of ICRs could impair im-printed expression, and could thuslead to disease. Such a scenariowas recently shown to occur as a
92 ARNAUD AND FEIL
Birth Defects Research (Part C) 75:81–97, (2005)

consequence of the reduced expres-sion of MeCP2 in Rett syndrome. Rettsyndrome is a severe and progres-sive neurodevelopmental disorderlinked to the X-chromosome, whichoccurs almost exclusively in females.Approximately 80% of cases arecaused by mutations in the gene en-coding methyl CpG binding protein 2(MeCP2). MeCP2 is a nuclear proteinthat binds specifically to methylatedDNA, and is though to act as a tran-scriptional silencer by recruiting his-tone deacetylase complexes. Previ-ous studies in the mouse have shownthat MeCP2 binds to the methylatedallele of ICRs in the mouse (Gregoryet al., 2001; Drewell et al., 2002;Fournier et al., 2002). This sug-gested the possibility that this meth-ylated DNA binding protein could beinvolved in the reading of imprints.Another indication was that the neu-rodevelopmental imprinting disorderAS has several clinical features incommon with Rett syndrome. In AS,the UBE3A gene is repressed on boththe parental alleles. This raised thequestion as to whether this im-printed gene also loses its expressionin Rett syndrome. Indeed, two stud-ies (Samaco et al., 2005; Makedon-ski et al., 2005) have shown that inthe brains of Rett syndrome patients,UBE3A mRNA and protein weregreatly reduced. The imprinted ex-pression of UBE3A is controlled bythe bipartite ICR at the SNRPN gene,a region to which MeCP2 was shownto bind in vivo (Makedonski et al.,2005). MeCP2 deficiency induced analteration of the histone modificationpattern at this ICR, but did not affectits maternal DNA methylation. Byperforming detailed studies onMeCP2-deficient mice, the authorsshowed that the histone changes atthe ICR correlated with biallelic ex-pression of a noncoding RNA (andantisense to the UBE3A RNA), andthey proposed that this is responsi-ble for the loss of UBE3A expression(Makedonski et al., 2005).
Another recent study links MeCP2deficiency in Rett syndrome to lossof imprinting at the maternally ex-pressed DLX5 gene on chromo-some 7. The basis of this work wasthe finding that DLX5 displayed lossof imprinting in lymphoblast celllines derived from Rett patients
(Horike et al., 2005). Furthermore,higher-order organization of chro-matin into loops has been shown tobe involved in bringing about im-printed gene expression (Weber etal., 2003b; Murrell et al., 2004;Kato and Sasaki, 2005). Interest-ingly, by using MeCP2-deficientmice and by applying chromatin im-munoprecipitation techniques,MeCP2 was found to be involved inthe formation of a silent chromatinloop that includes the DLX5 im-printed gene. Absence of the pro-tein interferes with this silencingprocess and leads to loss of imprint-ing of DLX5. The DLX5 protein me-diates the production of GABAergicneurons in the brain. Its altered ex-pression may thus be a contributingfactor in the altered brain functionsobserved in Rett syndrome.
OUTLOOK
This review on genomic imprintingand its perturbation in human dis-ease describes that imprinted geneexpression is mediated by differen-tial epigenetic organization of keyregulatory elements or ICRs. Theepigenetic marks at these ICRs areestablished in one of the two germ-lines only, and after fertilization, theyare somatically maintained through-out development. In the newly-formed germ cells of the developingembryo, however, the epigeneticmodifications are removed to allowsubsequent establishment of newimprints. These three sequentialevents—establishment, mainte-nance, and erasure—constitute thelife cycle of imprinting (Fig. 1). Ithas become clear from studies dur-ing the last years that, apart fromgenetic mutations, imprinting-re-lated syndromes in humans can becaused by epigenetic changes aswell. We have presented severalexamples to show that such epimu-tations can arise at the establish-ment stage, or can affect the so-matic maintenance of imprinting,or may be explained by nonerasurein the germline.
During the last years, there hasbeen a growing interest in whetherassisted reproduction could affectthe epigenetic regulation of imprint-ing, in particular after it was found
that embryo culture and manipula-tion can affect phenotype and im-printed gene expression in mouseand ruminant species. In this review,we have presented studies that indi-cate that IVF and ICSI procedures inhumans may affect the epigeneticregulation of imprinted gene loci,giving rise to imprinting disorders.Although these studies clearly estab-lish a link between assisted repro-duction and imprinting syndromes, itshould be noted that in IVF and ICSI,the frequency of these disorders isstill extremely low in spite of an ap-parent several-fold increase. Otherrisks, such as low birth weight andpoor postnatal development, are sig-nificantly higher. For information andcounseling to prospective parents, itshould be important to establish theabsolute risks, in terms of imprint-ing, that are represented by differentreproduction procedures. Such fig-ures should be obtained from largestudies and may reveal that someapproaches affect imprinted generegulation more than others. For in-stance, does the extent of embryoculture (which in some clinics is per-formed until the blastocyst stage) af-fect the risk of acquiring imprintingdisorders? Possibly the main ques-tion for future research concernsnew technologies, such as in vitromaturation of oocytes, for which littleis known from laboratory animals.Since maternal imprints are acquiredlate in oogenesis, this approachcould interfere with imprint estab-lishment, and may thus increase therisk of imprinting disorders.
WEBSITES OF INTEREST
For up-to-date information on im-printed genes and their functions:Mammalian Genetics Unit, MRC,Harwell, Oxfordshire, UK, Mouse Im-printing; http://www.mgu.har.mrc.ac.uk/research/imprinting/
Cancer Genetics Laboratory,Department of Biochemistry, Uni-versity of Otago, Dunedin, NewZealand, Imprinted Gene Cata-logue, http://cancer.otago.ac.nz/IGC/Web/home.html
Laboratory for Genome Explora-tion Research Group, RIKENGenomic Sciences Center (GSC),RIKEN Yokohama Institute, Yoko-
IMPRINTING IN HUMAN DISEASE 93
Birth Defects Research (Part C) 75:81–97, (2005)

hama, Japan, Candidate ImprintedTranscript from Gene Expression(CITE); http://fantom2.gsc.riken.go.jp/imprinting/
ACKNOWLEDGEMENTWe thank Katia Delaval, AlexandreWagschal, and Thierry Forne for ad-vice and critical reading of themanuscript.
REFERENCES
Aapola U, Kawasaki K, Scott HS, et al.2000. Isolation and initial character-ization of a novel zinc finger gene,DNMT3L, on 21q22.3, related to thecytosine-5-methyltransferase 3 genefamily. Genomics 65:293–298.
Aapola U, Liiv I, Peterson P. 2002. Im-printing regulator DNMT3L is a tran-scriptional repressor associated withhistonedeacetylase activity.Nucleic Ac-ids Res 30:3602–3608.
Arima T, Drewell RA, Oshimura M, et al.2000. A novel imprinted gene, HYMAI,is located within an imprinted domainon human chromosome 6 containingZAC. Genomics 67:248–255.
Arnaud P, Monk D, Hitchins M, et al. 2003.Conserved methylation imprints in thehuman and mouse GRB10 genes withdivergent allelic expression suggestsdifferential reading of the same mark.Hum Mol Genet 12:1005–1019.
Barlow DP, Stoger R, Herrmann BG, etal. 1991. The mouse insulin-like growthfactor type-2 receptor is imprinted andclosely linked to the Tme locus. Na-ture 349:84–87.
Bartolomei MS, Zemel S, Tilghman SM.1991. Parental imprinting of the mouseH19 gene. Nature 351:153–155.
Barton SC, Surani MA, Norris ML. 1984.Role of paternal and maternal genomesin mouse development. Nature 311:374–6.
Bestor TH. 2003. Imprinting errors anddevelopmental asymmetry. PhilosTransR Soc Lond B Biol Sci 358:1411–1415.
Bielinska B, Blaydes SM, Buiting K, et al.2000. De novo deletions of SNRPN exon1 in early human and mouse embryosresult in a paternal to maternal im-print switch. Nat Genet 25:74–78.
Birger Y, Shemer R, Perk J, et al. 1999.The imprinting box of the mouse Igf2rgene. Nature 397:84–88.
Bonduelle M, Liebaers I, Deketelaere V, etal. 2002. Neonatal data on a cohort of2889 infantsbornafter ICSI(1991–1999)andof2995 infantsbornafter IVF (1983–1999). Hum Reprod 17:671–694.
Bourc’his D, Xu GL, Lin CS et al. 2001.Dnmt3L and the establishment of ma-ternal genomic imprints. Science 294:2536–2539.
Bourc’his D, Bestor TH. 2004. Meioticcatastrophe and retrotransposon re-activation in male germ cells lackingDnmt3L. Nature 431:96–99.
Bowman P, McLaren A. 1970. Viabilityand growth of mouse embryos after invitro culture and fusion. J Embryol ExpMorphol 23:693–704.
Buiting K, Lich C, Cottrell S, et al. 1999.A 5-kb imprinting center deletion in afamily with Angelman syndrome re-duces the shortest region of deletionoverlap to 880 bp. Hum Genet 105:665–666.
Buiting K, Gross S, Lich C, et al. 2003.Epimutations in Prader-Willi and An-gelman syndromes: a molecular studyof 136 patients with an imprinting de-fect. Am J Hum Genet 72:571–577.
Castrillon DH, Sun D, Weremowicz S, etal. 2001. Discrimination of completehydatidiform mole from its mimics byimmunohistochemistryof thepaternallyimprinted gene product p57KIP2. Am JSurg Pathol 25:1225–1230.
Cattanach BM, Kirk M. 1985. Differentialactivity of maternally and paternallyderived chromosome regions in mice.Nature 315:496–498.
Chang AS, Moley KH, Wangler M, et al.2005. Association between Beckwith-Wiedemann syndrome and assisted re-productive technology: a case seriesof 19 patients. Fertil Steril 83:349–354.
Chedin F, Lieber MR, Hsieh CL. 2002.The DNA methyltransferase-like pro-tein DNMT3L stimulates de novo meth-ylation by Dnmt3a. Proc Natl Acad SciUSA 99:16916–16921.
Chilosi M, Piazzola E, Lestani M, et al.1998. Differential expression ofp57kip2, a maternally imprinted cdkinhibitor, in normal human placentaand gestational trophoblastic disease.Lab Invest 78:269–276.
Constancia M, Pickard B, Kelsey G, et al.1998. Imprintingmechanisms.GenomeRes 8:881–900.
Constancia M, Dean W, Lopes S, et al.2000. Deletion of a silencer element inIgf2 results in loss of imprinting inde-pendent ofH19.NatGenet 26:203–206.
Constancia M, Hemberger M, Hughes J,et al. 2002. Placental-specific IGF-II isa major modulator of placental and fe-tal growth. Nature 417:945–948.
Cox GF, Burger J, Lip V, et al. 2002. In-tracytoplasmic sperm injection may in-crease the risk of imprinting defects.Am J Hum Genet 71:162–164.
Davis TL, Trasler JM, Moss SB, et al. 1999.Acquisition of the H19 methylation im-print occurs differentially on the pa-rental alleles during spermatogenesis.Genomics 58:18–28.
Davis TL, Yang GJ, McCarrey JR, et al.2000. The H19 methylation imprint iserased and re-established differentiallyon theparental alleles duringmalegermcell development. Hum Mol Genet9:2885–2894.
DeBaun MR, Niemitz EL, Feinberg AP.2003. Association of in vitro fertiliza-tion with Beckwith-Wiedemann syn-dromeandepigenetic alterationsof LIT1and H19. Am J Hum Genet 72:156–160.
DeChiara TM, Robertson EJ, EfstratiadisA. 1991. Parental imprinting of themouse insulin like growth factor II gene.Cell 64:849–859.
Delaval K, Feil R. 2004. Epigenetic reg-ulation of mammalian genomic imprint-ing. Curr Opin Genet Dev 14:188–195.
Devriendt K. 2005. Hydatidiform moleand triploidy: the role of genomic im-printing in placental development. HumReprod Update 11:137–142.
Doherty AS, Mann MR, Tremblay KD, etal. 2000. Differential effects of cultureon imprinted H19 expression in the pre-implantation mouse embryo. Biol Re-prod 62:1526–1535.
Drewell RA, Goddard CJ, Thomas JO, etal. 2002.Methylation-dependent silenc-ing at the H19 imprinting control re-gion by MeCP2. Nucleic Acids Res 30:1139–1144.
El-Maarri O, Buiting K, Peery EG, et al.2001. Maternal methylation imprintson human chromosome 15 are estab-lished during or after fertilization. NatGenet 27:341–344.
El-Maarri O, Seoud M, Coullin P, et al.2003. Maternal alleles acquiring pa-ternal methylation patterns in biparen-tal complete hydatidiform moles. HumMol Genet 12:1405–1413.
El-Maarri O, Seoud M, Riviere JB, et al.2005. Patients with familial biparentalhydatidiform moles have normal meth-ylation at imprinted genes. Eur J HumGenet 3:486–490.
Evans HK, Wylie AA, Murphy SK et al.2001 The neuronatin gene resides in a“micro-imprinted” domain on humanchromosome 20q11.2. Genomics 77:99–104.
Fedoriw AM, Stein P, Svoboda P, et al.2004. Transgenic RNAi reveals essen-tial function for CTCF in H19 gene im-printing. Science 303:238–240.
Feil R, Walter J, Allen ND, et al. 1994.Developmental control of allelic meth-ylation in the imprinted mouse Igf2 andH19 genes. Development 120:2933–2943.
Feil R, Boyano MD, Allen ND, et al. 1997.Parental chromosome-specific chroma-tin conformation in the imprintedU2af1-rs1 gene in the mouse. J Biol Chem272:20893–20900.
Feil R, Khosla S, Cappai P et al. 1998.Genomic imprinting in ruminants: allele-specific gene expression in parthenoge-netic sheep.MammGenome9:831–834.
Feil R, Khosla S. 1999.Genomic imprint-ing in mammals: an interplay betweenchromatin and DNA methylation?Trends Genet 15:431–435.
Feil R. 2001. Early-embryonic culture andmanipulation could affect genomic im-printing. Trends Mol Med 7:245–246.
Fernandez-Gonzalez R, Moreira P, Bil-bao A, et al. 2004. Long-term effect ofin vitro culture of mouse embryos withserum on mRNA expression of imprint-ing genes, development, and behav-ior. Proc Natl Acad Sci USA 101:5880–5885.
94 ARNAUD AND FEIL
Birth Defects Research (Part C) 75:81–97, (2005)

Fisher RA, Khatoon R, Paradinas FJ, etal. 2000 Repetitive complete hydatid-iform mole can be biparental in originand either male or female. Hum Re-prod 15:594–598.
Fisher RA, Hodges MD, Rees HC, et al.2002. The maternally transcribed genep57(KIP2) (CDNK1C) is abnormally ex-pressed in both androgenetic and bi-parental complete hydatidiform moles.Hum Mol Genet 11:3267–3272.
Fisher RA, Hodges MD. 2003. Genomicimprinting in gestational trophoblasticdisease—a review. Placenta 24(SupplA):S111–S118.
Fisher RA, Nucci MR, Thaker HM, et al.2004. Complete hydatidiform mole re-taining a chromosome 11 of maternalorigin: molecular genetic analysis of acase. Mod Pathol 17:1155–1160.
Fitzpatrick GV, Soloway PD, Higgins MJ.2002. Regional loss of imprinting andgrowth deficiency in mice with a tar-geted deletion of KvDMR1. Nat Genet32:426–431.
Fournier C, Goto Y, Ballestar E, et al.2002. Allele-specific histone lysinemethylation marks regulatory regionsat imprinted mouse genes. EMBO J 21:6560–6570.
Geuns E, De Rycke M, Van SteirteghemA, et al. 2003. Methylation imprints ofthe imprint control regionof theSNRPN-gene in human gametes and preim-plantation embryos. Hum Mol Genet12:2873–2879.
Gicquel C, Gaston V, Mandelbaum J, etal. 2003. In vitro fertilization may in-crease the risk of Beckwith-Wiedemannsyndrome related to the abnormal im-printing of the KCN1OT gene. Am JHum Genet 72:1338–1341.
Gosden R, Trasler J, Lucifero D, Faddy M.2003. Rare congenital disorders, im-printed genes, and assisted reproduc-tive technology. Lancet361:1975–1977.
Gregory RI, Randall TE, Johnson CA, etal. 2001. DNA methylation is linked todeacetylation of histone H3, but notH4, on the imprinted genes Snrpn andU2af1-rs1. Mol Cell Biol 21:5426–5436.
Grimes DA. 1984. Epidemiology of ges-tational trophoblastic disease. Am J Ob-stet Gynecol 150:309–318.
Hajkova P, Erhardt S, Lane N, et al. 2002.Epigenetic reprogramming in mouseprimordial germ cells. Mech Dev 117:15–23.
Halliday J, Oke K, Breheny S, et al. 2004.Beckwith-Wiedemann syndrome andIVF: a case-control study. Am J HumGenet 75:526–528.
Hansen M, Kurinczuk JJ, et al. 2002. Therisk of major birth defects after intracy-toplasmic sperm injection and in vitrofertilization. N Engl J Med 346:725–730.
Hata K, Okano M, Lei H, et al. 2002.Dnmt3L cooperates with the Dnmt3family of de novo DNA methyltrans-ferases to establish maternal imprintsin mice. Development 129:1983–1993.
Hayward BE, De Vos M, Judson H, et al.2003. Lack of involvement of knownDNA methyltransferases in familial hy-
datidiform mole implies the involve-ment of other factors in establishmentof imprinting in the human femalegermline. BMC Genet 4:2.
Helwani MN, Seoud M, Zahed L, et al.1999. A familial case of recurrent hy-datidiform molar pregnancies with bi-parental genomic contribution. HumGenet 105:112–115.
Hikichi T, Kohda T, Kaneko-Ishino T, etal. 2003. Imprinting regulation of themurine Meg1/Grb10 and human GRB10genes; roles of brain-specific promot-ers and mouse-specific CTCF-bindingsites. Nucleic Acids Res 31:1398–1406.
Hodges MD, Rees HC, Seckl MJ, et al.2003. Genetic refinement and physi-cal mapping of a biparental completehydatidiform mole locus on chromo-some 19q13.4. J Med Genet 40:e95.
Horike S, Cai S, Miyano M, et al. 2005.Loss of silent-chromatin looping andimpaired imprinting of DLX5 in Rettsyndrome. Nat Genet 37:31–40.
Howell CY, Bestor TH, Ding F, Latham KE,etal.2001.Genomic imprintingdisruptedby a maternal effect mutation in theDnmt1 gene. Cell 104:829–838.
Hu JF, Pham J, Dey I, et al. 2000. Allele-specific histone acetylation accompa-nies genomic imprinting of the insulin-like growth factor II receptor gene.Endocrinology 141:4428–4435.
Jaenisch R, Bird A. 2003. Epigenetic reg-ulation of gene expression: how thegenome integrates intrinsic and envi-ronmental signals. Nat Genet33(Suppl):245–254.
Judson H, Hayward BE, Sheridan E, etal. 2002. A global disorder of imprint-ing in the human female germ line.Nature 416:539–542.
Kamiya M, Judson H, Okazaki Y, et al.2000. The cell cycle control gene ZAC/PLAGL1 is imprinted—a strong candi-date gene for transient neonataldiabetes. Hum Mol Genet 9:453–460.
Kaneda M, Okano M, Hata K, et al. 2004.Essential role for de novo DNA meth-yltransferase Dnmt3a in paternal andmaternal imprinting. Nature 429:900–903.
Kato Y, Rideout WM 3rd, Hilton K, et al.1999. Developmental potential ofmouse primordial germ cells. Devel-opment 126:1823–1832.
Kato Y, Sasaki H. 2005. Imprinting andlooping: epigenetic marks control in-teractions between regulatory ele-ments. Bioessays 27:1–4.
Kearns M, Preis J, McDonald M, et al.2000. Complex patterns of inheritanceof an imprinted murine transgene sug-gest incomplete germline erasure. Nu-cleic Acids Res 28:3301–3309.
Kerjean A, Dupont JM, Vasseur C, et al.2000. Establishment of the paternalmethylation imprint of the human H19and MEST/PEG1 genes during spermat-ogenesis.HumMolGenet9:2183–2187.
Kerjean A, Couvert P, Heams T, et al.2003. In vitro follicular growth affectsoocyte imprinting establishment inmice. Eur J Hum Genet 11:493–496.
Khosla S, Aitchison A, Gregory R, et al.1999. Parental allele-specific chroma-tin configuration in a boundary-imprint-ing-control element upstream of themouseH19gene.MolCell Biol 19:2556–2566.
Khosla S, Dean W, Reik W, et al. 2001a.Culture of preimplantationembryosandits long-term effects on gene expres-sion and phenotype. Hum Reprod Up-date 7:419–427.
Khosla S, Dean W, Brown D, et al. 2001b.Culture of preimplantation mouse em-bryos affects fetal development andthe expression of imprinted genes. BiolReprod 64:918–926.
Killian JK, Byrd JC, Jirtle JV, et al. 2000.M6P/IGF2R imprinting evolution inmammals. Mol Cell 5:707–716.
Kimura Y, Tateno H, Handel MA, et al.1998. Factors affecting meiotic and de-velopmental competence of primaryspermatocytenuclei injected intomouseoocytes. Biol Reprod 59:871–877.
Lane N, Dean W, Erhardt S, et al. 2003.Resistance of IAPs to methylation re-programming may provide a mecha-nism for epigenetic inheritance in themouse. Genesis 35:88–93.
Lee J, Inoue K, Ono R, et al. 2002. Eras-ing genomic imprinting memory inmouse clone embryos produced fromday 11.5 primordial germ cells. Devel-opment 129:1807–1817.
Leonard NJ, Bernier FP, Rudd N, et al.1996. Two pairs of male monozygotictwins discordant for Wiedemann Beck-with syndrome. Am J Med Genet 61:253–257.
Lewis A, Mitsuya K, Constancia M, et al.2004. Tandem repeat hypothesis in im-printing: deletion of a conserved di-rect repeat element upstream of H19has no effect on imprinting in the Igf2–H19region.MolCell Biol 24:5650–5656.
Li E, Beard C, Jaenisch R. 1993. Role forDNA methylation in genomic imprint-ing. Nature 366:362–365.
Li HW, Tsao SW, Cheung AN. 2002. Cur-rent understandings of the moleculargenetics of gestational trophoblastic dis-eases. Placenta 23:20–31.
Li JY, Lees-Murdock DJ, Xu GL, et al.2004. Timing of establishment of pa-ternal methylation imprints in themouse. Genomics 84:952–960.
Lidegaard O, Pinborg A, Andersen AN.2005. Imprinting diseases and IVF:Danish National IVF cohort study. HumReprod 20:950–954.
LinSP, YoungsonN, TakadaS, et al. 2003.Asymmetric regulation of imprinting onthe maternal and paternal chromo-somes at the Dlk1-Gtl2 imprinted clus-ter on mouse chromosome 12. NatGenet 35:97–102.
Liu J, Yu S, Litman D, Chen W, et al.2000. Identification of a methylationimprint mark within the mouse Gnaslocus. Mol Cell Biol 20:5808–5817.
Lucifero D, Mertineit C, Clarke HJ, et al.2002. Methylation dynamics of im-printed genes in mouse germ cells.Genomics 79:530–538.
IMPRINTING IN HUMAN DISEASE 95
Birth Defects Research (Part C) 75:81–97, (2005)

Lucifero D, Mann MR, Bartolomei MS, etal. 2004. Gene-specific timing and epi-genetic memory in oocyte imprinting.Hum Mol Genet 13:839–849.
Maher ER, Afnan M, Barratt CL. 2003a.Epigenetic risks related to assisted re-productive technologies: epigenetics,imprinting, ART and icebergs? Hum Re-prod 18:2508–2511.
Maher ER, Brueton LA, Bowdin SC, et al.2003bBeckwith-Wiedemann syndromeand assisted reproduction technology(ART). J Med Genet 40:62–64.
Makedonski K, Abuhatzira L, KaufmanY, et al. 2005. MeCP2 deficiency in Rettsyndrome causes epigenetic aberra-tions at the PWS/AS imprinting centerthat affect UBE3A expression. Hum MolGenet 14:1049–1058.
Mann MR, Lee SS, Doherty AS, et al.2004. Selective loss of imprinting inthe placenta following preimplantationdevelopment in culture. Development131:3727–3735.
Manning M, Lissens W, Bonduelle M, etal. 2000. Study of DNA-methylationpatterns at chromosome 15q11–q13in children born after ICSI reveals noimprinting defects. Mol Hum Reprod6:1049–1053.
MarquesCJ,CarvalhoF,SousaM,etal.2004.Genomic imprinting indisruptivespermat-ogenesis. Lancet 363:1700–1702.
McGrath J, Solter D. 1984. Completionof mouse embryogenesis requires boththe maternal and paternal genomes.Cell 37:179–183.
Messing J,GrossniklausU. 1999.Genomicimprinting in plants. Results Probl CellDiffer 25:23–40.
Moglabey YB, Kircheisen R, Seoud M, etal. 1999. Genetic mapping of a mater-nal locus responsible for familial hyda-tidiform moles. Hum Mol Genet 8:667–671.
Moll AC, Imhof SM, Cruysberg JR, et al.2003. Incidence of retinoblastoma inchildren born after in-vitro fertilization.Lancet 361:309–310.
Morgan HD, Sutherland HG, Martin DI,et al. 1999. Epigenetic inheritance attheagouti locus in themouse.NatGenet23:314–318.
Morgan HD, Dean W, Coker HA, et al.2004. Activation-induced cytidinedeaminase deaminates 5-methylcy-tosine in DNA and is expressed in plu-ripotent tissues: implications forepigenetic reprogramming. J Biol Chem279:52353–52360.
Morison IM, Reeve AE. 1998. A catalogueof imprinted genes and parent-of-ori-gin effects in humans and animals. HumMol Genet 7:1599–1609.
Mowery-Rushton PA, Driscoll DJ, NichollsRD, et al. 1996. DNA methylation pat-terns in human tissues of uniparentaloriginusingazinc-fingergene (ZNF127)from the Angelman/Prader-Willi region.Am J Med Genet 61:140–146.
Murrell A, Heeson S, Reik W. 2004. In-teraction between differentially meth-ylated regions partitions the imprintedgenes Igf2 and H19 into parent-spe-
cific chromatin loops. Nat Genet 36:889–893.
Neumann B, Kubicka P, Barlow DP. 1995.Characteristics of imprinted genes. NatGenet 9:12–13.
Obata Y, Kaneko-Ishino T, Koide T, etal. 1998. Disruption of primary imprint-ing during oocyte growth leads to themodified expression of imprinted genesduring embryogenesis. Development125:1553–1560.
Obata Y, Kono T. 2002. Maternal primaryimprinting is established at a specifictime for each gene throughout oocytegrowth. J Biol Chem 277:5285–5289.
Ogawa Y, Lee JT. 2002. Antisense reg-ulation in X inactivation and autoso-mal imprinting. Cytogenet Genome Res99:59–65.
Olek A, Walter J. 1997. The pre-implan-tation ontogeny of the H19 methyl-ation imprint. Nat Genet 17:275–276.
Olivennes F, Mannaerts B, Struijs M, etal. 2001. Perinatal outcome of preg-nancy after GnRH antagonist (ganire-lix) treatment during ovarianstimulation for conventional IVFor ICSI:a preliminary report. Hum Reprod 16:1588–1591.
O’Neill MJ, Ingram RS, Vrana PB, et al.2000. Allelic expression of IGF2 in mar-supials and birds. Dev Genes Evol 210:18–20.
Orstavik KH, Eiklid K, van der Hagen CB,et al. 2003.Another case of imprintingdefect in agirlwithAngelman syndromewho was conceived by intracytoplas-mic semen injection. Am J Hum Genet72:218–219.
Oswald J, Engemann S, Lane N, et al.2000. Active demethylation of the pa-ternal genome in the mouse zygote.Curr Biol 10:475–478.
Pant V, Mariano P, Kanduri C, et al. 2003.The nucleotides responsible for the di-rect physical contact between the chro-matin insulator protein CTCF and theH19 imprinting control region mani-fest parent of origin-specific long-dis-tance insulation and methylation-freedomains. Genes Dev 17:586–590.
Paoloni-Giacobino A, Chaillet JR.2004.Genomic imprinting and assistedreproduction. Reprod Health 1:6.
Paulsen M, El-Maarri O, Engemann S, etal. 2000. Sequence conservation andvariability of imprinting in the Beckwith-Wiedemann syndrome gene cluster inhuman and mouse. Hum Mol Genet9:1829–1841.
Reed MR, Riggs AD, Mann JR. 2001. De-letion of a direct repeat element hasno effect on Igf2 and H19 imprinting.Mamm Genome 12:873–876.
Reik W. 1989. Genomic imprinting andgenetic disorders inman. TrendsGenet.5:331–336.
Reik W, Walter J. 2001a. Genomic im-printing: parental influence on the ge-nome. Nat Rev Genet 2:21–32.
Reik W, Walter J. 2001b. Evolution ofimprinting mechanisms: the battle ofthe sexes begins in the zygote. NatGenet 27:255–256.
Reik W, Murrell A, Lewis A, et al. 2004.Chromosome loops, insulators, and hi-stone methylation: new insights intoregulation of imprinting in clusters. ColdSpring Harbor Symposia on Quantita-tive Biology, 59. Cold Spring Harbor,NY: Cold Spring Harbor LaboratoryPress.
Samaco RC, Hogart A, LaSalle JM. 2005.Epigenetic overlap in autism-spectrumneurodevelopmental disorders: MECP2deficiency causes reduced expressionof UBE3A and GABRB3. Hum Mol Genet14:483–492.
Santos F, Dean W. 2004. Epigenetic re-programmingduringearly developmentin mammals. Reproduction 127:643–651.
Sato S, Yoshimizu T, Sato E, et al. 2003.Erasure of methylation imprinting ofIgf2r during mouse primordial germ-cell development. Mol Reprod Dev 65:41–50.
Saxena A, Frank D, Panichkul P, et al.2003. Theproduct of the imprintedgeneIPL marks human villous cytotropho-blast and is lost in complete hydatidi-form mole. Placenta 24:835–842.
Schieve LA, Meikle SF, Ferre C, et al.2002. Low and very low birth weightin infants conceived with use of as-sisted reproductive technology. N EnglJ Med 346:731–737.
Schoenherr CJ, Levorse JM, Tilghman SM.2003.CTCFmaintainsdifferentialmeth-ylation at the Igf2/H19 locus. Nat Genet33:66–69.
Seki Y, Hayashi K, Itoh K, et al. 2005.Extensive and orderly reprogrammingof genome-wide chromatin modifica-tions associated with specification andearly development of germcells inmice.Dev Biol 278:440–458.
Shamanski FL, Kimura Y, Lavoir MC, etal. 1999. Status of genomic imprint-ing in mouse spermatids. Hum Reprod14:1050–1056.
Shi W, Haaf T. 2002. Aberrant methyl-ation patterns at the two-cell stage asan indicator of early developmental fail-ure. Mol Reprod Dev 63:329–334.
Simon I, Tenzen T, Reubinoff BE, et al.1999. Asynchronous replication of im-printed genes is established in the ga-metes and maintained duringdevelopment. Nature 401:929–932.
Sleutels F, Zwart R, Barlow DP. 2002.The non-coding Air RNA is required forsilencing autosomal imprinted genes.Nature 415:810–813.
Smith RJ, Arnaud P, Konfortova G, et al.2002. The mouse Zac1 locus: basis forimprinting and comparison with humanZAC. Gene 292:101–112.
Smith RJ, Arnaud P, Kelsey G. 2004. Iden-tification and properties of imprintedgenes and their control elements. Cy-togenet Genome Res 105:335–345.
Smrzka OW, Fae I, Stoger R, et al. 1995.Conservation of a maternal-specificmethylation signal at the human IGF2rlocus. Hum Mol Genet 4:1945–1952.
Sparago A, Cerrato F, Vernucci M, et al.2004. Microdeletions in the human H19
96 ARNAUD AND FEIL
Birth Defects Research (Part C) 75:81–97, (2005)

DMR result in loss of IGF2 imprintingand Beckwith-Wiedemann syndrome.Nat Genet 36:958–960.
Sunahara S, Nakamura K, Nakao K, etal. 2000. The oocyte-specific methyl-ated region of the U2afbp-rs/U2af1-rs1 gene is dispensable for its imprintedmethylation. Biochem Biophys ResCommun 268:590–595.
Szabo PE, Tang SH, Silva FJ et al. 2004.Role of CTCF binding sites in the Igf2/H19 imprinting control region. Mol CellBiol 24:4791–4800.
Takada S, Paulsen M, Tevendale M, etal. 2002. Epigenetic analysis of theDlk1-Gtl2 imprinted domain on mousechromosome 12: implications for im-printing control from comparison withIgf2–H19. Hum Mol Genet 11:77–86.
Thorvaldsen JL, Duran KL, Bartolomei MS.1998. Deletion of the H19 differentiallymethylated domain results in loss ofimprinted expression of H19 and Igf2.Genes Dev 12:3693–3702.
Tucker KL, Beard C, Dausmann J, et al.1996. Germ-line passage is requiredfor establishment of methylation andexpression patterns of imprinted butnot of nonimprinted genes. Genes Dev10:1008–1020.
Tycko B, Morison IM. 2002. Physiologi-cal functions of imprinted genes. J CellPhysiol 192:245–258.
Ueda T, Abe K, Miura A, et al. 2000. Thepaternal methylation imprint of themouse H19 locus is acquired in thegonocyte stage during foetal testis de-velopment. Genes Cells 5:649–659.
Umlauf D, Goto Y, Cao R, et al. 2004.Imprinting along the Kcnq1 domain onmouse chromosome 7 involves repres-sive histone methylation and recruit-
ment of Polycomb group complexes.Nat Genet 36:1296–1300.
Verona RI, Mann MR, Bartolomei MS.2003. Genomic imprinting: intricaciesof epigenetic regulation in clusters.Annu Rev Cell Dev Biol 19:237–259.
Walter J, Paulsen M. 2003. Imprintingand disease. Semin Cell Dev Biol 14:101–110.
Weber M, Milligan L, Delalbre A et al.2003a. Extensive tissue-specific vari-ation of allelic methylation in the Igf2gene during mouse fetal development:relation to expression and imprinting.Mech Dev 101:133–141.
Weber M, Hagege H, Murrell A et al.2003b. Genomic imprinting controlsmatrix attachment regions in the Igf2gene. Mol Cell Biol 23:8953–8959.
Weber M, Hagege H, Aptel N, et al. 2005.Epigenetic regulationofmammalian im-printed genes: from primary to func-tional imprints. In: Epigenetics andchromatin. Progress in Molecular andSubcellular Biology. pp. 207–236. P.Jeanteur,ed.BerlinHeidelberg:Springer-Verlag.
Webster KE, O’Bryan MK, Fletcher S, etal. 2005. Meiotic and epigenetic de-fects in Dnmt3L-knockout mouse sper-matogenesis. Proc Natl Acad Sci USA102:4068–4073.
Weksberg R, Shuman C, Caluseriu O, etal. 2002. Discordant KCNQ1OT1 im-printing in sets of monozygotic twinsdiscordant for Beckwith-Wiedemannsyndrome. Hum Mol Genet 11:1317–1325.
Weksberg R, Smith AC, Squire J, et al.2003. Beckwith-Wiedemann syndromedemonstrates a role for epigenetic con-trol of normal development. Hum MolGenet 12(Spec No 1):R61–R68.
Wey E, Bartholdi D, Riegel M, et al. 2005.Mosaic imprinting defect in a patientwith an almost typical expression ofthe Prader-Willi syndrome. Eur J HumGenet 13:273–277.
Williamson CM, Ball ST, Nottingham WT,et al. 2004. A cis-acting control regionis required exclusively for the tissue-specific imprinting of Gnas. Nat Genet36:894–899.
Wutz A, Theussl HC, Dausman J, et al.2001. Non-imprinted Igf2r expressiondecreases growth and rescues the Tmemutation in mice. Development 128:1881–1887.
Xin Z, Allis CD, Wagstaff J. 2001. Parent-specific complementary patterns of hi-stone H3 lysine 9 and H3 lysine 4methylation at the Prader-Willi syn-drome imprinting center. Am J HumGenet 69:1389–1394.
Yokomine T, Kuroiwa A, Tanaka K, et al.2001. Sequence polymorphisms, allelicexpression status and chromosome lo-cations of the chicken IGF2 and MPR1genes. Cytogenet Cell Genet 93:109–113.
Yoon BJ, Herman H, Sikora A, et al. 2002.Regulation of DNA methylation of Ras-grf1. Nat Genet 30:92–96.
Young LE, Sinclair KD, Wilmut I. 1998.Large offspring syndrome in cattle andsheep. Rev Reprod 3:155–163.
Young LE, Fernandes K, McEvoy TG, etal. 2001. Epigenetic change in IGF2Ris associated with fetal overgrowth af-ter sheep embryo culture. Nat Genet27:153–154.
Young LE, Schnieke AE, McCreath KJ, etal. 2003.Conservationof IGF2–H19andIGF2R imprinting in sheep: effects ofsomatic cell nuclear transfer. Mech Dev120:1433–1442.
IMPRINTING IN HUMAN DISEASE 97
Birth Defects Research (Part C) 75:81–97, (2005)





























