Embed Size (px)
Citation preview

INSTITUTO POLITECNICO NACIONALESCUELA SUPERIOR DE INGENIERÍA QUÍMICA E
INDUSTRIAS EXTRACTIVASSECCIÓN DE ESTUDIOS DE POSGRADO E INVESTIGACIÓN
ESTUDIO EXPERIMENTAL Y MODELAMIENTO CINÉTICODE LA HIDRODESULFURACIÓN DE DIBENZOTIOFENO
EN FASE LÍQUIDA
T E S I S
QUE PARA OBTENER EL GRADO DE MAESTRO EN CIENCIAS CON ESPECIALIDAD EN INGENIERÍA QUÍMICA
P R E S E N T A
ELVA ARZATE BARBOSA
DIRECTOR: DR. Ir. JOSÉ ANTONIO MUÑOZ ARROYOCODIRECTOR: DR. MIGUEL ANGEL VALENZUELA ZAPATA
México, D. F. 2006






AGRADECIMIENTOS
A mi esposo Rogelio Márquez Nuño quien ha sido la alegría de mi vida, por todo el apoyo y amor,
sin el cual me hubiera sido muy difícil terminar este trabajo.
A mis preciosos hijos Diego y Alonso quienes han sido la chispa que enciende mi existencia.
A mi apreciada mamá Elba, que siempre me ha apoyado incondicionalmente.
Quiero agradecer de manera muy especial al Dr. Ir. José Antonio Muñoz Arroyo por la experiencia
profesional y sus consejos, que de manera personal ayudaron a incrementar mi formación
profesional y entusiasmo por seguir en el área experimental, lo cual considero fascinante.
Al M. en C. Javier Esteban Rodríguez Rodríguez. por la confianza depositada en mi persona para
adentrarme en esta fabulosa área experimental.
Al Dr. Ciro Humberto Ortiz Estrada por su amistad y sus consejos desinteresados en la realización
de este trabajo.
Al Dr. Ignacio Grossmann por sus valiosos consejos sobre técnicas de optimización.
Quiero agradecer al Instituto Mexicano del Petróleo, por haberme dado la oportunidad de colaborar
con un grupo de personas altamente capacitadas que participaron con su entusiasmo y experiencia
para la realización de esta tesis. A Evangelina Camacho Frías y Yolanda Figueroa por su amistad y
apoyo y por allanarme el camino del área analítica, así como por desarrollar técnicas y
metodologías, que hasta el momento no han sido publicadas y que contribuyeron en mucho al
desarrollo de este trabajo. A los excelentes profesionistas y técnicos del área de plantas piloto
(Ing. Virgilio Ramírez Hernández, Ing. Juan José Cuarenta García, M. en C. Armando Morales
Sánchez, Ing. Lázaro Moisés Garcia Moreno, Tec. Alberto Salvador Abrego Novo. Tec. Alfonso
Chávez Flamenco, Tec. Luis Roberto Bones Hernández) que intercambiaron su experiencia para
construir la planta piloto, así como al Ing. José David Ortega Jiménez y al Tec. Luis Manuel
Hernández Santander por sus conocimientos aportados a las técnicas de análisis de gases.
A la mí querida escuela la ESIQIE del Instituto Politécnico Nacional, por haberme brindado la
formación profesional de la cual estoy orgullosa y disfruto a cada minuto en mi trabajo.
A TODOS USTEDES MUCHAS GRACIAS.


I
CONTENIDO
Contenido I
Índice de Figuras III
Índice de Tablas V
NOTACIÓN VI
RESUMEN 1
ABSTRACT 3
1. INTRODUCCIÓN 5
2. ANTECEDENTES 11
2.1 Modelos cinéticos en la hidrodesulfurización de dibenzotiofeno 11
2.2 Trayectorias y mecanismos de reacción para la hidrodesulfuración 19
2.3 Esquemas de hidroprocesamiento 23
3. PROCEDIMIENTOS EXPERIMENTALES 31
3.1 Consideraciones para la construcción de la planta piloto de hidrotratamiento
31
3.1.1 Descripción de la planta piloto de hidrotratamiento 31
3.1.2 Sección de preparación de la carga 33
3.1.3 Sección de reacción 33
3.1.4 Sección de separación 36
3.1.5 Equipos periféricos 39
3.1.5.1 Gasómetro 39
3.1.5.2 Bomba de carga 39
3.1.5.3 Medidor de flujo de gas 39
3.2 Catalizador de HDS 39
3.3 Selección del tipo de disolvente 40
3.4 Métodos analíticos 41
3.4.1 Cromatografía de líquidos de alta eficiencia 42
3.4.2 Destilación simulada 46
3.4.2.1 Procedimiento analítico 47
3.4.3 Análisis de las corriente gaseosas 56
4. ANÁLISIS DE LOS DATOS EXPERIMENTALES 61
4.1 Metodología de análisis de las corrientes de la planta piloto 61
4.2 Análisis de las corrientes a la salida de la planta piloto 64
4.3 Análisis estadístico de los resultados 73

CONTENIDO
II
Continuación…
4.4 Análisis cuantitativo del comportamiento de la velocidad de reacción
74
5. MODELAMIENTO DE LOS DATOS EXPERIMENTALES 79
5.1 Modelamiento cinético de la Hidrodesulfuración de dibenzotiofeno 79
5.2 Condiciones experimentales 83
5.3 Procedimiento de estimación de parámetros 84
5.4 Resultados del modelado cinético 91
6. CONCLUSIONES 103
RECOMENDACIONES 105
REFERENCIAS 107
APÉNDICES
A Operación de la planta piloto A.1
A.1 Procedimiento de activación del catalizador A.1
A.2 Procedimiento de arranque de la planta piloto A.3
A.3 Estabilidad, medición y toma de muestras A.4
B CALIBRACIÓN DE LOS EQUIPOS DE MEDICIÓN B.1
B.1 Controladores y medidores de flujo másico B.1
B.1.1 Controladores para el flujo de carga líquida B.1
B.1.2 Controladores para el flujo de hidrógeno B.3
B.1.3 Medidor de flujo de gas amargo B.3
C CALIBRACIÓN DEL CROMATÓGRAFO DE GASES C.1
D ANÁLISIS ESTADÍSTICO DE LOS DATOS EXPERIMENTALES D.1
D.1 Análisis univariable D.1
D.2 Análisis multivariable D.3

III
INDICE DE FIGURAS
Figura Descripción
Página
2.1 Trayectoria de reacción para la hidrodesulfuración de dibenzotiofeno [Vanrysselberghe V. & Froment G. F., 1996]
15
2. 2 Trayectorias de reacción para la desulfuración de Dibenzotiofeno en CoMo o NiMo/Al2O3 [Bataille F., et al., 2000]
19
2. 3 Trayectorias de reacción para la desulfuración de 4,6-dimetildibenzotiofeno en CoMo o NiMo/Al2O3 [Mijoin J. et al., 2001]
22
2.4. Mecanismo de ruptura del enlace C-S en la hidrogenólisis sobre MoS2 23 2.5. Modelo catalítico CoMoS: S; Co (Ni); Mo [Bataille F. et al., 2000] 23 2.6 Esquema de una unidad de desulfuración de gasóleo [Whitehurst D. D. et al., 1998] 25 3.1 Diagrama simplificado del proceso de hidrotratamiento de destilados intermedios 32 3.2 Reactor Robinson Mahoney 35 3.3 Distribución del flujo líquido dentro del reactor 35 3.4 Diseño de las propelas y canasta catalítica del reactor Robinson Mahoney 36 3.5 Esquema del separador FA-02 de alta presión y temperatura 37 3.6 Sistema de separación de baja presión de gas y líquido flasheados 38 3.7 Vista general de la planta piloto 38 3.8 Tiempo de retención en función del porcentaje de ACN en la fase móvil para BPH,
DBT y CHB disueltos en la fase móvil utilizada 44
3.9 Cromatograma de los estándares de BPH, DBT y CHB disueltos en ACN-agua 65:35 (V/v) a las condiciones cromatográficas descritas en el texto.
45
3.10 Curva de calibración para la determinación de la composición del BPH, CHB y DBT 45 3.11 Curva de calibración de los puntos de ebullición contra los tiempos de retención de la
mezcla de n-parafinas de C5 a C40 50
3.12 Cromatograma del aceite mineral 85 NF sin reaccionar 53 3.13 Cromatograma obtenido del aceite mineral hidrotratado a 643 K 53 3.14 Cromatograma para la carga a hidrodesulfuración (aceite mineral más 2.09 %peso
de dibenzotiofeno) 53
3.15 Cromatograma obtenido por destilación simulada para el producto de hidrodesulfuración tratado a 643 K.
54
3.16 Programa de temperatura utilizado en la determinación de la composición de las corrientes gaseosas
57
3.17 Diagrama del arreglo de válvulas para el cromatógrafo 58 4.1 Diagrama esquemático de la planta piloto 62 4.2 Conversión de DBT y rendimiento de aromáticos para diferentes LHSV 67 4.3 Comparación de datos de planta piloto con resultados de Vanrysselberghe V. &
Froment G.F., 1996. 68
4.4 Comportamiento del DBT y los productos de la HDS con la temperatura, a un LHSV de 2, ∗ DBT, ∆ BPH, ♦ CHB, ■ DCH
70
4.5 Efecto del exceso de hidrógeno sobre la conversión a las condiciones experimentales de T = 553 K, P = 7.85 MPa, LHSV = 2.
72
4.6 Selectividad para la hidrogenación del ciclohexilbenceno y bifenilo en función de la relación H2/HC a las condiciones experimentales de T = 553 K, P = 7.85 MPa, LHSV = 2
73
4.7 Comportamiento de la velocidad de reacción del dibenzotiofeno como función del espacio-velocidad molar
75
4.8 Comportamiento de la velocidad de reacción del BPH con respecto a la concentración del DBT y BPH @ T= 553 K, P = 7.85 MPa y H2/Hc = 3.0
76
4.9 Comportamiento de la velocidad de reacción del CHB con respecto a la concentración del CHB, DBT y BPH @ T = 553 K, P = 7.85 MPa y H2/Hc = 3.0.
77
4.10. Comportamiento de la velocidad de reacción del DCH con respecto a la concentración del CHB, DBT y BPH @ T = 553 K, P = 7.85 MPa y H2/Hc = 3.0.
78

INDICE DE FIGURAS
IV
Continuación… 5.1 Determinación del equilibrio de fases de la mezcla de reacción presente en el reator
Robinson Mahoney 82
5.2 Comportamiento de las velocidades de reacción experimentales con las concentraciones para el DBT, BPH, CHB, DCH
85
5.3 Metodología de la estimación de los parámetros cinéticos y de adsorción para el proceso de hidrodesulfuración del DBT
90
5.4 Comportamiento de las constantes cinéticas y de adsorción reportadas por Vanrysselberghe V. & Froment G.F., 1996
94
5.5.a Diagrama de paridad de la velocidad de reacción calculada vs experimental del DBT obtenida por el método de Gams
98
5.5.b Diagrama de paridad de la velocidad de reacción calculada vs experimental del BPH obtenida por el método de Gams
98
5.5.c Diagrama de paridad de la velocidad de reacción calculada vs experimental del CHB obtenida por el método de Gams
99
5.5.d Diagrama de paridad de la velocidad de reacción calculada vs experimental del DCH obtenida por el método de Gams
99
5.6.a Gráfica de paridad para el flujo molar calculado vs experimental a T= 553K y 7.85 MPa obtenido por el método de Rosenbrock
100
5.6.b Gráfica de paridad para el flujo molar calculado vs experimental a T= 593K y 7.85 MPa obtenido por el método de Rosenbrock
101
5.6.c Gráfica de paridad para el flujo molar calculado vs experimental a T= 623K y 7.85 MPa obtenido por el método de Rosenbrock
101
5.6.d Gráfica de paridad para el flujo molar calculado vs experimental a T= 643K y 7.85 MPa obtenido por el método de Rosenbrock
102
B.1 Calibración de la bomba GA-01 Milton Roy en volumen B.2 B.2 Calibración de la bomba Minipump B.3 B.3 Estabilidad para la medición de flujo de gas amargo B.4 C.1 Curva de calibración de la concentración de H2S con las áreas C.2 D.1.a Curvas de contorno para la conversión del DBT en función de la temperatura y el
LHSV D.3
D.1.b Curvas de contorno para la conversión del DBT en función de la temperatura y la relación H2/HC
D.3
D.2 Curvas de contorno para el rendimiento de bifenilo en función de la temperatura y el LHSV
D.4
D.3 Curva de contorno para el rendimiento de bifenilo en función de la temperatura y el H2/Hc
D.4
D.4 Curvas de contorno para el rendimiento de Ciclohexilbenceno en función de la temperatura y el LHSV.
D.5
D.5 Curvas de contorno para el rendimiento de Ciclohexilbenceno en función de la temperatura y la relación H2/Hc
D.6
D.6 Curvas de contorno para el diciclohexilo en función de la temperatura y el LHSV D.6 D.7 Curvas de contorno para el diciclohexilo en función de la temperatura y la relación
H2/Hc D.7

V
INDICE DE TABLAS
No. Tabla
Descripción de la Tabla
Página
1.1 Especificaciones del contenido de azufre para el Diesel en México 6 2.1 Orden de reactividad de los compuestos azufrados en la Hidrodesulfuración
[Broderick D. H., 1980] 13
2.2 Velocidades de reacción de pseudo primer orden para diferentes compuestos azufrados [Broderick D. H., 1980; Xiaoliang Ma et al., 1994; Whitehurst D. D. et al., 1998]
13
2.3 Parámetros en las ecuaciones de velocidad para la Hidrogenólisis e Hidrogenación [Broderick D. H. and Gates B. C., 1981]
16
2.4 Parámetros en las ecuaciones de velocidad [Vanrysselberghe V. & Froment G. F., 1996] 18 2.5 Condiciones de operación típicas para diferentes fracciones del petróleo [Whitehurst D. D.
et al., 1998] 24
2.6 Hidrodesulfuración de compuestos modelo reportada en literatura 27 3.1 Características del solvente (aceite mineral) 34 3.2 Características del reactor Robinson Mahoney 36 3.3 Características de los equipos periféricos 39 3.4 Comparación de los catalizadores IMPDSD-14 y el utilizado por Vanrysselberghe V. &
Froment G. F., 1996 40
3.5 Temperaturas de equilibrio para diferentes solventes 41 3.6 Condiciones de operación para obtención de productos de hidrodesulfuración 47 3.7 Estándar de una mezcla de n-Parafinas para la validación del método 49 3.8 Puntos de ebullición para muestra de referencia SIMDIS D 2887 Lote No.40400 Parte No.
25650.150 49
3.9 Tiempos de retención, puntos de ebullición y peso molecular del DBT y productos de HDS
50
3.10 Intervalos de tiempo de retención correspondientes al número de átomos de carbono, de acuerdo a la curva de calibración
51
3.11 Resultados de los productos de reacción reportados por el cromatógrafo de gases con la técnica de destilación simulada
52
3.12 Composición en por ciento peso de los hidrocarburos del aceite mineral, del DBT y los productos de la HDS a 553 K y LHSV de 5.02 h-1
54
3.13 Concentración en peso para las muestras analizadas por destilación simulada 55 3.14 Comparación de la determinación del azufre total en las muestras analizadas. Por los
métodos IMP-QP03 y el de destilación simulada 56
3.15 Características del equipo utilizado para la cromatografía de gases 57 3.16 Funcionamiento de las válvulas del cromatógrafo 58 3.17 Gases utilizados en la planta piloto 58 3.18 Características de los gases para cromatografía realizada en planta piloto. 59 3.19 Composición de la mezcla de hidrocarburos 59 4.1 Hidrotratamiento de dibenzotiofeno: a diferentes LHSV, P=7.84 MPa y T=553 K , balance
de materia y % de error en el balance de átomos entre la carga y productos 65
4.2 Composición de la carga y el producto líquido en los experimentos de hidrotratamiento de DBT disuelto en aceite mineral a T=553 K y diferentes LHSV
66
4.3 Hidrotratamiento de dibenzotiofeno: condiciones de operación, balance de materia y % de error en el balance de átomos entre la carga y productos
68
4.4 Hidrodesulfuración de dibenzotiofeno: condiciones de operación, balance de materia y % de error en el balance de átomos entre la carga y productos
69
4.5 Composición de la carga y el producto líquido en los experimentos de hidrotratamiento de DBT disuelto en aceite mineral
70
4.6 Composición de la carga y el producto líquido en los experimentos de hidrotratamiento de DBT disuelto en aceite mineral
71

INDICE DE TABLAS
VI
Continuación… 4.7 Composición del gas amargo y del flash gas en los experimentos de Hidrodesulfuración
de DBT disuelto en aceite mineral 71
4.8 Composición del gas amargo y del flash gas en los experimentos de hidrotratamiento de DBT disuelto en aceite mineral
72
4.9 Velocidades de reacción de los productos 75 5.1 Agrupamiento de compuestos para el modelamiento 83 5.2 Condiciones de reacción consideradas para la estimación de parámetros 83 5.3 Comparación de los parámetros cinéticos y de adsorción con el modelo de Broderick D.H.
& Gates B.C., 1981 y el calculado por excel 93
5.4 Resultados de la optimización de parámetros cinéticos y de adsorción considerando el modelo de Vanrysselberghe V. & Froment G.F., 1996
95
5.5 Resultados de la optimización de parámetros cinéticos y de adsorción considerando el efecto del disolvente en el modelo de Vanrysselberghe V. & Froment G.F., 1996
97
B.1 Calibración de la bomba Milton Roy B.2 C.1 Parámetros base para determinar la exactitud de la concentración por componente C.2 C.2 Repetibilidad del tiempo de retención. C.3 C.3 Desviación de las áreas con respecto a la media. C.3 D.1 Análisis Estadístico para la significancia de los productos en función de la relación H2/HC. D.1 D.2 Modelo Estadístico para los productos de reacción en función de la relación H2/Hc a
P = 7.85 MPa (80Kg/cm2) y LHSV = 2.0 D.1
D.3 Análisis Estadístico para la significancia de los productos en función de la relación H2/HC D.1 D.4 Modelo Estadístico para los productos de reacción en función de la relación LHSV a
P = 7.85 MPa (80Kg/cm2) y H2/Hc = 1.33 D.2
D.5 Modelo lineal multivariable en función de la temperatura, LHSV y H2/HC D.2 D.6 Modelos para representar los productos de reacción en función de la Temperatura, LHSV
y H2/Hc D.7

VII
NOTACION Símbolo Parámetro
Unidades
A# Constante en la ecuación de Arrenius/ ecuación de Van´t Hoff m3 Kgmol-1 Balc Relación porcentual de los flujos molares a la salida y entrada de átomos
de carbono
BalH Relación porcentual de los flujos molares a la salida y entrada de átomos de Hidrógeno
BalS Relación porcentual de los flujos molares a la salida y entrada de átomos de azufre
CA Concentración molar de la especie química A para el modelo de Kittrel, J.R. 1996
CB Concentración molar de la especie química A para el modelo de Kittrel CAM.S Concentración de aceite mineral en un sitio activo Kgmol/m3 CDBT.S Concentración de dibenzotiofeno en un sitio activo σ ó τ Kgmol/m3 CCHB.S Concentración de ciclohexilbenceno en un sitio activo σ ó τ Kgmol/m3 CDCH.S Concentración de diciclohexilo en un sitio activo σ ó τ Kgmol/m3 CBPH Concentración de bifenilo en un sitio activo σ ó τ Kgmol/m3 CCHB Concentración de ciclohexilbenceno en un sitio activo σ ó τ Kgmol/m3 CDBT Concentración de dibenzotiofeno a la salida del reactor Kgmol/m3
2HC Concentración de hidrógeno a la salida del reactor Kgmol/m3
SH2C Concentración de ácido sulfhídrico a la salida del reactor Kgmol/m3
Ci Número de átomos de carbono
kC Concentración calculada de la mezcla en cada k repetición Kgmol/m3
hC Concentración calculada de la mezcla en cada h experimento Kgmol/m3
Ci Concentración del líquido del componente i Kgmol/m3 CV Concentración de sitios activos vacíos en el catalizador Kgmol/m3 Ea* Energía de Activación Aparente J mol-1 cj Número de átomos de carbono del hidrocarburo i CT Concentración total de sitios activos en el catalizador Kgmol/m3 H Número de átomos de hidrógeno Fio Flujo Molar del componente i a la entrada de la planta mol hr-1
F°DBT Flujo molar del DBT a la entrada del reactor mol hr-1
F 2Ho Flujo molar de H2 a la entrada del reactor mol hr-1
F cH
o Flujo molar de hidrocarburo a la entrada del reactor mol hr-1
Fj
o Flujo molar del componente j a la entrada del reactor mol hr-1
Fg Flujo molar Total del gas a la salida de la planta mol hr-1 Fg
i Flujo molar del componente i en el gas a la salida de la planta mol hr-1
FgH2
Flujo molar de H2 en la fase gaseosa a la salida de la planta mol hr-1
FI Flujo total molar del líquido a la salida del reactor mol hr-1 IiF Flujo molar del componente i en el líquido a la salida de la planta mol hr-1
FlH2
Flujo molar de H2 en la fase líquida a la salida de la planta mol hr-1
FI2 Flujo molar del producto líquido a la salida de la planta mol hr-1
l2iF
Flujo molar del componente i en el líquido a la salida de la planta mol hr-1

NOTACIÓN
VIII
Símbolo Parámetro
Unidades
FI1 Flujo molar del gas flasheado a la salida de la planta moll hr-1
1IiF Flujo molar del componente i en el gas flasheado a la salida de la planta mol hr-1
I2kh,F Flujo molar del líquido calculado por el modelo para las k´s respuestas en
los h´s experimentos mol hr-1
F°h,k Flujo molar a la entrada del reactor para la observación i en la respuesta j mol hr-1
(∆H) Calor de adsorción J Kgmol-1 hj Número de átomos de H2 en el componente i K Constante de velocidad aparente (Modelo de Broderick D. H. & Gates B.
C., 1981) m6/Kgmol.Kg de cat.s)
KA Constante de adsorción de la especie química A para el modelo de Kittrell KB Constante de adsorción de la especie química B para el modelo de Kittrell
'Hk Constante cinética para la hidrogenólisis del modelo Broderick D. H. &
Gates B. C., 1981 Kgmol/(Kgcat h)
KAM Constante de adsorción del aceite mineral para el modelo propuesto en este trabajo
m3/Kgmol
KDBT,H Constante de adsorción del DBT para la hidrogenólisis del modelo de Broderick D. H. & Gates B. C., 1981
m3/Kgmol
H,H2K Constante de adsorción del H2 para la hidrogenólisis del modelo de
Broderick D. H. & Gates B. C., 1981 m3/Kgmol
HS,H2K Constante de adsorción del H2S para la hidrogenólisis del modelo de
Broderick D. H. & Gates B. C., 1981 m3/Kgmol
k’ Constante cinética para la hidrogenación del modelo Broderick D. H. & Gates B. C., 1981
Kgmol/(Kgcat h)
KDBT,HG Constante de adsorción del DBT para la hidrogenación del modelo de Broderick D. H. & Gates B. C., 1981
m3/Kgmol
HG,H2K Constante de adsorción del H2 para la hidrogenación del modelo de
Broderick D. H. & Gates B. C., 1981 m3/Kgmol
kDBT,σ Constante cinética del DBT para la hidrogenólisis del modelo Vanrysselberghe V. & Froment G. F., 1996
Kgmol/(Kgcat h)
KDBT,σ Constante de adsorción del DBT para la hidrogenólisis del modelo de Vanrysselberghe V. & Froment G. F., 1996
m3/Kgmol
σH,K Constante de adsorción del H2 para la hidrogenólisis del modelo de Vanrysselberghe V. & Froment G. F., 1996
m3/Kgmol
σS,H2K Constante de adsorción del H2S para la hidrogenólisis del modelo de
Vanrysselberghe V. & Froment G. F., 1996 M3/Kgmol
kDBT, τ Constante cinética del DBT para la hidrogenación del modelo Vanrysselberghe V. & Froment G. F., 1996
Kgmol/(Kgcat h)
KDBT, τ Constante de adsorción del DBT para la hidrogenación del modelo de Vanrysselberghe V. & Froment G. F., 1996
M3/Kgmol
KH, τ Constante de adsorción del H2 para la hidrogenación del modelo de Vanrysselberghe V. & Froment G. F., 1996
M3/Kgmol
KBPH, τ Constante de adsorción del BPH para la hidrogenación del modelo de Vanrysselberghe V. & Froment G. F., 1996
M3/Kgmol
kBPH, τ Constante cinética del BPH para la hidrogenación del modelo Vanrysselberghe V. & Froment G. F., 1996
Kgmol/(Kgcat h)
kCHB, τ Constante cinética del CHB para la hidrogenación del modelo Vanrysselberghe V. & Froment G. F., 1996
Kgmol/(Kgcat h)
KCHB, τ Constante de adsorción del CHB para la hidrogenación del modelo de Vanrysselberghe V. & Froment G. F., 1996
M3/Kgmol
ki,s Coeficiente de velocidad del componente i en el sitio s Kgmol/(Kgcat h) Ki,s Coeficiente de adsorción del componente i en el sitio s M3/Kgmol LHSV Espacio velocidad del líquido hr-1

NOTACIÓN
IX
Símbolo Parámetro
Unidades
Ncom Número de componente N Número de experimentos ns Número de replicas experimentales PN Presión Normal MPa abs P Presión Total del sistema MPa Pa Presión Actual Mpa abs Ph Presión del sistema absoluta para cada h experimento MPa Ri Velocidad experimental del componente i Kmol/(Kgcat h) RDCH Velocidad total de aparición del diciclohexilo Kmol/(Kgcat h) RCHB Velocidad total de aparición del Ciclohexilbenceno Kmol/(Kgcat h) RDBT Velocidad total de desaparición del Dibenzotiofeno Kmol/(Kgcat h)
SH2R Velocidad total de aparición del ácido sulfhídrico Kmol/(Kgcat h)
2HR Velocidad total de desaparición del hidrógeno Kmol/(Kgcat h)
RBPH Velocidad total de aparición del Bifenilo Kmol/(Kgcat h)
kh,R Velocidad de reacción calculada en la k respuesta del h experimento Kmol/(Kgcat h)
R Velocidad de reacción del modelo de Kittrell, J.R. 1996 Kmol/(Kgcat h) rBPH, τ Velocidad de hidrogenación del bifenilo en Ciclohexilbenceno Kmol/(Kgcat h) rCHB, τ Velocidad de hidrogenación del ciclohexilbenceno en diciclohexilo Kmol/(Kgcat h) rDBT, τ Velocidad de hidrogenación del dibenzotiofeno en tetra y/o
hexahidrodibenziofeno Kmol/(Kgcat h)
isisHidrogenólr Velocidad de reacción en la ruta de hidrogenólisis Kmol/(Kgcat h)
iónHidrogenacr Velocidad de reaación en la ruta de hidrogenación Kmol/(Kgcat h)
Th Temperatura absoluta del sistema para cada h experimento K rDBT,σ Velocidad de hidrogenólisis del dibenzotiofeno en Bifenilo Kmol/(Kgcat h) VN Volumen Normal Lt Vi Volumen Indicado Lt T Temperatura Absoluta del sistema K TN Temperatura Normal K Ti Temperatura Indicada K S(θ ) Función objetivo propuesta por Martens G.G., 2000 S Sitio activo del catalizador si Número de átomos de azufre en el componente i W Cantidad de catalizador gr Wh Cantidad de catalizador para cada h experimento gr xi Conversión o Rendimiento del componente i presente en la reacción
I1ix Composición molar del componente i en el gas falseado I2i2x Composición molar del componente i en el producto líquido giy Composición molar del componente i en la fase gaseosa a la salida de la
planta
Símbolos griegos
kσ Función objetivo definida por Vanrysselberghe V. & Froment G.F., 1996 υ Número de respuestas γ Relación molar hidrógeno/hidrocarburo en la alimentación σ Sitio de Hidrogenólisis τ Sitio de Hidrogenación σh,k Factores de pesos para los experimentos k, y las repeticiones h

NOTACIÓN
X
Superíndices comp Componente de referencia
nob Número de observaciones
nresp Número de respuestas
L Referente a la fase líquida
G Referente a la fase vapor
° Referente a las condiciones de entrada o iniciales
a Orden de reacción de la especie química A en el modelo de Kittrell, J.R. 1996
b Orden de reacción de la especie química B en el modelo de Kittrell, J.R. 1996
Subíndices h Número de experimentos
k Número de repeticiones
Compuestos AM Aceite Mineral
BHDMDBT Bihidrodimetildibenzotiofeno
BPH Bifenilo
CHB Ciclohexilbenceno
DCH Diciclohexilo
DBT Dibenzotiofeno
DMBPH Dimetilbifenilo
DMCHB Dimetilcicclohexilbenceno
DMBPH Dimetilbifenilo
DMDBT Dimetildibenzotiofeno
HHDBT Hexahidrodibenzotiofeno HHDMDBT Hexahidrodimetildibenzotiofeno
MDBT Metildibenzotiofeno
PHCH Pentahidrociclohexilo
PHCHD Pentahidrodibenzotiofeno
THDBT Tetrahidrodibenzotiofeno THDMDBT Tetrahidrodimetildibenzotiofeno

RESUMEN
1
RESUMEN
Debido a la importancia que tiene el hecho de obtener productos combustibles con bajas
concentraciones de contaminantes y ajustarse así, a las normatividad que existe para estos
productos, se han realizado y desarrollado a lo largo de varios años por varios autores estudios
cinéticos para determinar el comportamiento que tiene los compuestos refractarios en cargas de
destilados intermedios. Estos estudios cinéticos consideran en la mayoría de los casos tomar como
base compuestos modelo para representar el comportamiento que tienen los compuestos
refractarios con la variación en las condiciones de operación. Sin embargo, en la mayoría de los
casos, estos autores no han considerado una experimentación abarcando las condiciones de
operación semejantes a las utilizadas en plantas industriales de hidrotratamiento. La otra
característica adversa en estos estudios, es que los modelos cinéticos presentados en la literatura
no consideran el efecto del disolvente en el desarrollo de las expresiones de velocidad de reacción.
En base a los comentarios anteriores el desarrollo del trabajo experimental se planeo utilizando el
DBT como compuesto modelo y representativo de los compuestos refractarios contenidos en los
destilados intermedios. En este trabajo se describe la construcción y validación de la planta piloto
utilizada para obtener la información experimental que permite el desarrollo de un modelo cinético.
La planta piloto esta constituida de un reactor sin gradientes Robinson Mahoney con capacidad de
un litro trabajando a condiciones de operación similares a las industriales. La dimensión de los
equipos de separación esta calculado en base a la capacidad del reactor, lo cual permite una
separación eficiente de los productos de reacción. El sistema de control de la planta, el tamaño del
reactor y el arreglo de líneas localizadas en la sección de separación a baja presión, son factores
determinantes para alcanzar el estado estacionario de la planta piloto permitiendo de esta manera
minimizar el tiempo de realización de cada experimento. Dentro de la infraestructura de medición
considerada, se incluye el análisis por cromatografía de gases en línea de las corrientes de gas
flasheado y gas amargo, permitiendo una reducción notable en el error para el balance de materia
y/o de átomos entre las corrientes.
El análisis cuantitativo por componente dio origen al desarrollo de dos técnicas de laboratorio. La
primera permitió determinar la composición de los productos de reacción por medio de
Cromatografía de alta eficiencia (CLAE) mientras que con la segunda se obtiene la composición del
disolvente en base al número de átomos de carbono a través de la destilación simulada (ASTM-
D2887). Esta determinación permitió realizar el cálculo del balance de masa y de átomos para las
corrientes de entrada y salida de la planta piloto, de manera satisfactoria.

RESUMEN
2
Las diferencias entre los resultados obtenidos en la planta piloto con aquellos reportados en la
literatura [Vanrysselberghe V. & Froment G.F., 1996] permite validar de manera satisfactoria la
operación de la planta en un intervalo amplio de condiciones de operación (553 -643 K, 80 MPa y
2 - 8 h-1).
La hidrodesulfuración del DBT se llevo a cabo en el intervalo de 553 -643 K, 80 MPa, 2 - 3
mol/mol para temperatura, presión y relación H2/Hc respectivamente. En este intervalo de
condiciones de operación se estableció el comportamiento de los productos de reacción (BPH,
CHB y DCH) encontrándose a partir de un análisis estadístico el efecto que tienen las variables
independientes (temperatura, presión, LHSV) sobre las variables respuesta (Xi, i = DBT, BPH,
CHB, DCH). Sin embargo la relación H2/HC en el rango de condiciones de operación industrial no
tiene un efecto significativo en el proceso de hidrodesulfuración dir|ecta (hidrogenólisis), no así
para la reacción de hidrogenación, donde la dependencia de esta variable es muy importante.
El análisis del comportamiento de la rapidez de reacción con respecto a la concentración de las
especies involucradas en la hidrodesulfuración, permite confirmar que las expresiones de velocidad
siguen un comportamiento de tipo Langmuir-Hinshelwood-Hougen-Watson (LHHW) como también
ya ha sido establecido por otros investigadores [Girgis M. J. & Gates B.C., 1991, y
Vanrysselberghe V. & Froment G.F., 1996].
La estimación de parámetros se realizó considerando este tipo de modelo (LHHW) y los
mecanismos y expresiones de velocidad de reacción publicados por Girgis M. J. & Gates B.C.,
1991, y Vanrysselberghe V. & Froment G.F., 1996 considerando la modificación por la adsorción
competitiva del disolvente en la mezcla de reacción. Para la obtención de los parámetros tanto
cinéticos como de adsorción se utilizaron tres métodos de optimización: Solver de Excel, Conopt de
GAMS y Rosenbrock. La comparación de los resultados obtenidos con estos métodos indica que
con el método de Rosenbrock la estimación de parámetros tiene una mejor aproximación a los
resultados experimentales. Finalmente utilizando la ley de Arrhenius y de Van´t Hoff se
establecieron tanto la energía de activación (Ea) como el calor de adsorción (-∆H) y los
parámetros constantes respectivos.

ABSTRACT
3
ABSTRACT
Due to the importance of developing ultra low sulphur fuel oil products, several research groups
have been working on methods concerning to understand the kinetic behavior of the refractory
compounds in middle distillates as feedstocks. These stidies are based on model compounds at
different operation conditions. However the most of these researchs, have not considered the
reaction conditions similar to those of industrial hydrotreating process. Another adverse
characteristic in these studies is that the kinetic models do not consider the solvent effect has in the
reaction rate expressions.
Based on the experimental work that has already been developed, this study uses DBT as a model
compound to represent the refractory compounds. The construction and the validation of the
experimental Set up has been described in detail, as well as the procedure to obtain the
experimental data that allow the development of a reliable kinetic model.
The experimental Setup was built with one liter, non gradient Robinson Mahoney reactor working at
operating conditions similar to those of industrial plants. The equipment upstream and downstream
was calculated considering the reactor dimensions which allows an efficient mixing and heating of
the hydrogen and liquid feedstock, as well as the separation of reactor effluent. The Setup control
system, the size of the reactor and the arrange of the lines located at the low pressure separation
play an important role to reach the steady state. Therefore the time on stream can be minimized for
each experiment. The measurement of the dependent variables includes the on-line gas
chromatography to monitor the flash and sour gas streams, allowing a remarkable error reduction in
the mass and/or atom balances.
The determination of the compound composition originated two new laboratory techniques. The first
technique is used to determine the reaction products composition by High Pressure Liquid
Chromatography (HPLC) whereas with the second it was possible determine the solvent
composition based on the carbon atoms number using simulated distillation (ASTM-D2887). Thus,
the mass and the atom balances for the Setup experiments were calculated in a satisfactory way.
The experimental results compared with those reported in the literature [Vanrysselberghe V. &
Froment G.F., 1996] have shown minimal deviations. Hence the validation of the Setup in a wide
range of operating conditions was possible (553 - 643 K, 80 MPa and 3 - 8 h-1).
The hydrodesulfurization of DBT was performed in the range of 553-643K, 80 MPa, 2-3 mol/mol for
temperature, pressure and molar ratio H2/Hc respectively. Thus, it was possible to follow the
behavior of the reaction products (BPH, CHB, DCH and H2S). In other hand the effect of

ABSTRACT
4
independent variables (temperature, pressure, LHSV) on the dependent variables (Xi, i = DBT,
BPH, CHB, DCH) was determined through a statistical analysis.
The analysis of the reaction scheme and the experimental data confirms the importance of the
hydrogenolysis compare with the hydrogenation reaction using a CoMo/AlO3 catalyst.
The analysis of the reaction rate versus concentration of the components involved in the
hydrodesulfurization process has confirmed that the reaction rate expressions follow the Langmuir-
Hinshelwood-Hougen-Watson (LHHW) mechanism.
The parameter estimation was performed considering LHHW reaction rate expressions as it was
published by Girgis M. J. & Gates B.C., 1991, and Vanrysselberghe V. & Froment G.F., 1996 and
also including the modification of the competitive adsorption of the solvent in the reaction mixture.
The optimization methods of: Excel (solver), GAMS (Conopt) and the method of Rosenbrock were
used to obtain the kinetic and adsorption parameters. A comparison of the parameters obtained
from these methods indicates a much better approach between the experimental and the calculated
data using the Rosenbrock method. Finally, the activation energy (Ea) and adsorption heat (-∆H)
as well as the constant parameters were calculated using the Arrhenius and Van´t Hoff expressions.

INTRODUCCION
5
1. INTRODUCCION El principal objetivo en una refinería moderna es la producción de combustibles con un alto valor
comercial a partir de petróleo crudo con composición variable. Diversos procesos catalíticos son
utilizados para cumplir con esta tarea, por citar algunos, la Reformación Catalítica de Naftas, la
Desintegración Catalítica de gasóleos FCC, el Hidrotratamiento de destilados medios e
Hidroisomerización de fracciones C6/C7. Sin embargo las regulaciones del medio ambiente con
respecto a la calidad de los combustibles para el transporte así como también las emisiones de las
mismas refinerías son los principales factores que han movido a los productores a la actualización
de las tecnologías existentes y al continuo desarrollo de tecnologías avanzadas [Babich I. V. &
Moulijn J. A., 2003].
La meta principal de las agencias de protección al medio ambiente en Europa y USA [Directive of
the European Parlament and of the Councyl, 2000; US EPA Clean Air Act Tier 2, 1999] es reducir
el contenido de azufre para los combustibles de transporte así como también limitar las emisiones
de CO2 a la atmósfera. Los principales combustibles de transporte producidos en una refinería son
la gasolina y el diesel por lo cual los procesos de hidrotratamiento están asociados a la producción
de estos. Por ejemplo el azufre presente en estos combustibles conduce a la contaminación del
aire con SOx generado por los motores combustión interna. De esta manera el nivel de azufre en
los combustibles debe disminuirse para eliminar los efectos negativos tanto en la salud como al
medio ambiente. Una nueva regulación en el rango entre 10-50 wppm de azufre para gasolina y
diesel aparecerá en el 2005 tanto en los Estados Unidos como en la Unión Europea [Hattiangadi U.
et al., 2000; Miller RB. et al, 2001]. En Alemania la legislación es más rigurosa, hasta 10 wppm ha
limitado las emisiones desde noviembre del 2001 [Larivé J.F., 2000]. De estos datos se puede
inferir que en un futuro inmediato no habrá tolerancia en las emisiones de azufre a nivel mundial y
de la que no escapa México, nuestro país.
Las especificaciones internacionales establecidas por la Environmental Protection Agency (EPA)
son por lo tanto cada vez más estrictas con relación a combustibles limpios, es por eso que uno de
los objetivos de las refinerías del Sistema de PEMEX-Refinación es obtener diesel con un
contenido máximo de azufre de 15 ppm, a partir del procesamiento de mezcla de crudos con un
contenido de Maya cada vez mayor integrando además a las cargas de las plantas
hidrodesulfuradoras de destilados intermedios, fracciones de Aceite Cíclico Ligero (ACL) y algunas
otras provenientes de procesos de coquización).
La última información disponible sobre la especificación del diesel como combustible se da en la
siguiente Tabla 1.1:

Capítulo 1
6
Tabla 1.1 Especificaciones del contenido de azufre para el Diesel en México.
Variable Año Todo el país
Azufre, ppm 2003 500
2006 300 máx. – 150 mín
2008 30 máx. – 15 mín
Por lo anterior, es de suma importancia cambiar la política existente en los procedimientos
experimentales y adaptar los intervalos de aplicabilidad de las metodologías analíticas existentes,
que están involucradas en los procesos de refinación para poder cumplir con las estrictas normas
ambientales que en un futuro cercano entrarán en vigor alrededor del mundo, y así poder competir
de manera eficiente en las posibles tecnologías que emanen de los desarrollos presentes.
Los procesos convencionales de hidrotratamiento están limitados para poder alcanzar el nivel de
cero para la composición de azufre en los combustibles, por lo cual es necesario el desarrollo de
nuevas tecnologías de hidrotratamiento para cumplir con las especificaciones. El diseño de estos
nuevos procesos involucra una extensiva experimentación en plantas pilotos para después realizar
una extrapolación a escala industrial basados en reglas de naturaleza heurística, las cuales son
parte del arte desarrollado en las diferentes industrias del petróleo [Froment G. F., 1999].
Los procesos de hidrotratamiento son muy importantes debido a que los derivados del petróleo
deben ser refinados para disminuir las emisiones contaminantes de óxidos de nitrógeno y azufre
las cuales contribuyen a la lluvia ácida en las grandes ciudades. Más aún muchos catalizadores los
cuales son utilizados en el procesamiento de las fracciones del petróleo no pueden tolerar el azufre
y/o metales, como consecuencia una gran parte de las corrientes de los derivados petrolíferos en
una refinería deben ser hidrotratados para eliminar los contaminantes. Es así, como el
hidrotratamiento llega a ser la más importante aplicación industrial de la catálisis sobre la base de
material procesado por año. Basados en la cantidad de catalizador vendido por año, el catalizador
de hidrotratamiento toma el tercer lugar en los grandes negocios de catalizadores, después de los
catalizadores del proceso de desintegración catalítica FCC y de los utilizados para la purificación
de gases de escape [Prada R., 2004, Santes V. et al., 2005].
Si bien los catalizadores de hidrotratamiento están constituidos por Molibdeno, Cobalto o Níquel
soportado en γ-Al2O3, el uso de promotores incrementa su actividad catalítica y por consiguiente los
catalizadores de Co-Mo/Al2O3 y Ni-Mo/Al2O3 presentan una más alta actividad catalítica que
Mo/Al2O3 [Koltai T. et al, 2002].

INTRODUCCION
7
Un aspecto importante en los procesos de hidrotratamiento dándole una atención precisa, es la
desulfuración profunda de destilados intermedios conteniendo especies de azufre orgánico difíciles
de desulfurar. La eliminación de estos compuestos puede darse a través de la comprensión de los
mecanismos involucrados en las reacciones de hidrodesulfuración, facilitando con esto el
desarrollo de nuevos catalizadores y esquemas de proceso.
Los gasóleos en general contienen cientos de compuestos, incluyendo compuestos de azufre,
nitrógeno y oxigeno en cantidades mensurables y su composición varia con el origen y tipo de
petróleo, por lo que las condiciones de reacción tienen que ser adaptadas para la eliminación de
estos heteroátomos (S, N, O) y de esta manera satisfacer la rigurosa especificación del producto.
Diferentes autores [Whitehurst D. D. et al., 1998; Girgis M. J. and Gates B. C., 1991; Shafi R. and
Hutchings J. G., 2000] han resumido los principales aspectos involucrando la desulfuración
profunda del gasóleo. Estos incluyen, la identificación y reactividad de las especies de azufre
contenida en la alimentación, las trayectorias y mecanismos de reacción, así como también la
actividad y selectividad de los catalizadores convencionales. También existen problemas asociados
con estos procesos que deben ser considerados. Así, en el proceso práctico de la
hidrodesulfuración de gasóleos tanto las especies aromáticas como las de azufre, compiten por los
sitios activos del catalizador. Más aún el H2S y algunos otros hidrocarburos producidos en la
primera sección del reactor, inhiben la hidrodesulfuración de las especies de azufre menos
reactivas.
La reactividad hacia la hidrodesulfuración de varias especies de azufre (incluyendo los refractarios)
en las diferentes etapas de sus respectivas trayectorias de reacción, necesitan ser descritas desde
el punto de vista de sus estructura molecular y/o electrónica a través de la química cuántica.
Bajo el esquema presentado anteriormente y a manera de encontrar la demanda de gasóleo con
bajo contenido de azufre, es necesario establecer nuevas rutas mecanísticas que mejoren la
reactividad, reduzcan la inhibición y permitan el diseño de nuevos catalizadores.
Sin embargo, las rigurosas regulaciones gubernamentales respecto a la especificación de los
combustibles y el procesamiento en las refinerías de crudo cada día más pesado ha traído consigo
el desarrollo de nuevos procesos para la desulfurización de los destilados intermedios. La
operación y el estudio del comportamiento de estos actualmente se realizan por medio de
simuladores basados en modelos cinéticos rigurosos.

Capítulo 1
8
La cinética de reacción es uno de los elementos principales en el modelamiento y simulación de un
reactor [Froment G. F. et al, 1994], sin embargo el desarrollo de tales modelos es una tarea
abrumadora, lo cual es consecuencia de la complejidad de la alimentación y sus transformaciones,
así como también del trabajo experimental involucrado.
En la actualidad en la literatura existe una gran cantidad de modelos cinéticos relacionados con la
conversión de compuestos modelo y/o mezclas complejas de compuestos de azufre, nitrógeno y
oxígeno [Vanrysselberghe V. et al, 1998; Vanrysselberghe V. & Froment G. F., 1998;
Vanrysselberghe V. & Froment G. F. 1996; Froment G. F. et al, 1994; Whitehurst D.D. et al, 1998;
Broderick D. H. and Gates B. C., 1981] los cuales han sido aplicados para seguir y representar el
comportamiento del proceso de hidrotratamiento.
Bajo este contexto, el modelamiento juega un papel importante en la desulfuración y aún más en el
diseño de un nuevo proceso. Un modelo confiable con cierto nivel de sofisticación puede contribuir
al desarrollo de nuevos catalizadores y/o a la normalización u optimización del proceso mismo. Es
aquí donde el modelo desarrollado puede reducir en gran manera el trabajo experimental, mejorar
la extrapolación, guiar la operación de la planta y servir como una base para su control.
Este trabajo de tesis se elaboró en el contexto del desarrollo de un modelo cinético para el proceso
de hidrodesulfuración, en el cual las condiciones de operación se aproximan a las del proceso
industrial de refinación. De esta manera se estable una visión de los avances recientes del
modelamiento de los datos experimentales obtenidos desde compuestos modelo y/o cargas reales,
de los esquemas y mecanismos de reacción utilizando catalizadores a base de Co y Ni, así como
también se hace una descripción de los reactores experimentales utilizados en la generación de la
información y finalmente se trata la influencia del tipo de disolvente utilizado para emular el
comportamiento de la carga industrial trabajando en un equipo experimental.
Debido a la necesidad de generar esta información se procedió a la construcción de una planta
piloto utilizando un reactor Robinson-Mahoney el cual elimina la resistencia al transporte de masa y
calor en las partículas catalíticas. La operación y cambio de las variables de operación de esta
planta piloto (temperatura, presión, LHSV, H2/HC) permitió hacer una descripción del
comportamiento que tiene el dibenzotiofeno (DBT) y los productos de la reacción de
hidrodesulfuración (BPH, CHB, DCH, H2S) utilizando un catalizador de CoMo/γ-Al2O3.
El efecto de las variables independientes sobre las variables respuesta se realiza a través de un
análisis estadístico uni - y- multivariable utilizando el software Design Experiment, 2003.

INTRODUCCION
9
Finalmente una vez establecida la reactividad y selectividad de los productos de la
hidrodesulfuración y considerando al reactor Robinson Mahoney como un reactor heterogéneo de
flujo mezclado fue posible determinar los parámetros de las expresiones de velocidad que
representan el esquema de reacción de la hidrodesulfuración de dibenzotiofeno.


ANTECEDENTES
11
2. ANTECEDENTES 2.1. MODELOS CINETICOS EN LA HIDRODESULFURACION DE DIBENZOTIOFENO
Diferentes autores han desarrollado modelos cinéticos fundamentales para la hidrodesulfuración de
compuestos de azufre poliaromáticos. La hidrodesulfuración de tiofeno y benzotiofeno ha sido
reportada por Van Parijs I. & Froment G. F., 1984, 1986 y para el último por Broderick D. H. &
Gates B. C., 1981, Edvinsson R. & Irandoust S., 1993 y Vanrysselberghe V. & Froment G. F., 1996.
Recientemente Vanrysselberghe V. et al., 1998 reportaron ecuaciones de velocidad de reacción
para la hidrodesulfuración de alquil-benzotiofenos. En todos los casos se considera la distinción
entre las reacciones de hidrogenólisis e hidrogenación y también se toma en cuenta la adsorción
competitiva de H2 y H2S, así como la de otras especies reactivas.
Se ha reportado recientemente en la literatura [Laredo S. C. G. et al., 2001] los efectos de
inhibición del nitrógeno básico y no básico sobre la hidrodesulfuración de dibenzotiofeno, así como
también el modelo cinético respectivo tomando en cuenta estos efectos; sin embargo, para otros
compuestos de azufre incluyendo los más refractarios no han sido reportados a la fecha.
Como ya ha sido previamente discutido los gasóleos son mezclas complejas de diferentes clases
de hidrocarburos contaminadas con pequeñas cantidades de heteroátomos que bajan la calidad
del producto y limitan el mercado de los gasóleos. La eliminación completa de estos compuestos
involucra considerar en detalle su composición. Actualmente es posible determinar detalladamente
la composición de todos los hidrocarburos, azufre, nitrógeno y oxigeno contenidos en el diesel por
medio de modernos procedimientos analíticos [Depauw G. & Froment G. F., 1996; Wiwel P. et al.,
2000].
El contenido de las diversas especies de azufre y su relativa reactividad en los procesos
convencionales de hidrodesulfuración para algunos combustibles ya se han reportado, [Froment G.
F. et al., 1994; Whitehurst D. D. et al., 1998] la combinación secuenciada de las técnicas de
cromatografía de líquidos de alta eficiencia (CLAE), cromatografía de gases ya sea con
espectrometría de masas (GC-MS) y/o detección de emisión atómica (GC-AED) los cuales utilizan
detectores específicos para compuestos de azufre y nitrógeno han permitido la identificación de la
mayoría de las especies de azufre individuales [Froment G. F. et al., 1994; Wiwel P. et al., 2000] y
algunas de nitrógeno, consecuentemente se puede dar un seguimiento a la cinética de
desaparición de ellos durante las reacciones de hidrotratamiento en los catalizadores de
CoMo/Al2O3.

Capítulo 2
12
Los primeros intentos para el modelamiento cinético del hidrotratamiento están referidos a la
hidrodesulfuración de compuestos tiofenicos, por ser los más refractarios [Van Parijs I. et al.,
1986(a); Van Parijs I. et al., 1986(b)]. Ecuaciones de velocidad para la hidrodesulfuración de
dibenzotiofeno se han reportado por diversos grupos de investigación [Girgis M. J. and Gates B. C.,
1991; Shafi R. and Hutchings J. G., 2000; Edvinsson R. & Irandoust S., 1993; Houalla M. et al.,
1978]. Si bien en estos trabajos se desarrollan las expresiones de velocidad, en algunos casos se
hacen simplificaciones que no consideran de manera completa la trayectoria de reacción, los
efectos directos de los subproductos en estas mismas, agrupan los productos de reacción y/o las
condiciones de operación están lejos de aquellas del proceso industrial. Más recientemente
trabajos relacionados con la hidrodesulfuración de alquil dibenzotiofenos, han establecido la
cinética de reacción de 4-metil - y - 4,6-dimetil dibenzotiofeno considerando expresiones de
velocidad desde la ley de potencias hasta aquellas de tipo Hougen-Watson [Broderick D. H. &
Gates B. C., 1981; Vanrysselberghe V. & Froment G. F., 1996; Vanrysselberghe V. et al., 1998;
Vanrysselberghe V. & Froment G. F., 1998; Froment G. F. et al., 1994].
Por otro lado en la literatura han aparecido recientemente estudios sobre la influencia de los
compuestos nitrogenados y aromáticos sobre la velocidad de reacción para la hidrodesulfuración
de dibenzotiofeno y alquildibenzotiofenos respectivamente [Laredo S. C. G. et al., 2001; Koitai T. et
al., 2002]. En estos trabajos se presenta una comparación del cambio en la reactividad de los
compuestos azufrados como resultado de la presencia del nitrógeno y aromáticos en la carga.
Como un intento para explicar las trayectorias de reacción para llevar a cabo la hidrodesulfuración
de dibenzotiofeno y alquildibenzotiofenos la literatura abierta presenta algunas consideraciones
mecanísticas relacionadas con la formación de los intermediarios en esta reacción, así como
también para una mejor comprensión del papel de los promotores en los catalizadores de
hidrotratamiento y en el efecto de los grupos metilo en la molécula de dibenzotiofeno [Bataille F. et
al., 2000; Mijoin J. et al., 2001].
En la búsqueda de nuevas rutas para alcanzar niveles muy bajos de azufre para los combustibles
del futuro, es muy importante conocer y comprender la naturaleza de los compuestos de azufre que
van a ser convertidos, así como también como estas transformaciones ocurren a través de la
interacción con las especies catalíticas en la superficie del catalizador, es decir cuales son las
trayectorias de reacción involucradas y sus limitaciones cinéticas y termodinámicas asociadas.

ANTECEDENTES
13
En este contexto la Tabla 2.1 muestra la reactividad a la hidrodesulfuración de los compuestos
tiofénicos presentes en los destilados medios y ligeros. De las constantes de velocidad mostradas
en esta Tabla se observa que la reactividad disminuye a medida que el impedimento estérico en la
molécula se incrementa.
Tabla 2.1 Orden de reactividad de los compuestos azufrados en la Hidrodesulfuración
[Broderick D. H., 1980]
Compuesto Constante de velocidad de reacción de Primer Orden
(m3s-1 Kg-1 de catalizador)
Tiofenos 1.389E-03 Benzotiofenos 8.056E-04 Benzonaftotiofenos 1.611E-04 Dibenzotiofenos 6.111E-05
Xiaoliang Ma et al., 1994 así como también Whitehurst D. D. et al., 1998 soportan este concepto,
agrupando los compuestos azufrados principalmente en 4 grupos. La Tabla 2.2 muestra las
velocidades de reacción de pseudo primer orden para estos grupos, donde la reactividad disminuye
nuevamente a medida que el impedimento estérico de las moléculas de azufre aumenta.
Tabla 2.2 Velocidades de reacción de pseudo primer orden para diferentes compuestos azufrados [Broderick D. H., 1980; Xiaoliang Ma et al., 1994; Whitehurst D. D. et al., 1998]
Grupo de compuesto azufrado Constante de velocidad de reacción de Pseudo Primer Orden (s-1)
La mayoría de Alquil Benzotiofenos, excepto C3-BT-4, C4-BT-7 y C7-BT-1
> 0.0017
C3-BT-4, C4-BT-7, C7-BT-1, DBT y Alquilados Homólogos del DBT sin sustituyentes en las posiciones 4 y 6.
0.00057 – 0.0017
Alquil DBT con un grupo alquilo ya sea en la posición 4 o 6 0.000217 – 0.00057
Alquil DBT con dos grupos alquilo en la posición 4 y 6 0.000083 – 0.00022
La hidrodesulfuración de destilados medios involucra la remoción de azufre contenido en forma de
benzotiofeno, dibenzotiofeno o alquildibenzotiofenos. Es bien conocido que sobre catalizadores
típicos sulfhidrados, el azufre es removido a través de dos trayectorias de reacción paralelas, una
conduciendo a productos tipo bifenilo y la otra a productos tipo tetrahidrodibenzotiofeno para
posteriormente llegar hasta ciclohexilbenceno. La primera reacción es conocida como
hidrogenólisis en la que el azufre se remueve de manera directa por rompimiento del enlace del
azufre y los átomos de carbono para formar el producto tipo bifenilo y el ácido sulfhídrico
(reacción de eliminación tipo E2). La segunda reacción se ha designado como hidrogenación

Capítulo 2
14
debido a que los anillos de la molécula conteniendo el azufre son primero hidrogenados para
posteriormente el azufre sea eliminado vía la hidrogenólisis descrita anteriormente. También
reacciones laterales ocurren durante el hidrotratamiento, la hidrodesintegración e hidrogenación de
compuestos insaturados, siendo estas responsables del incremento en el consumo de hidrógeno.
Como anteriormente se mencionó la hidrodesulfuración de compuestos tiofénicos se lleva a cabo a
través de dos trayectorias paralelas de reacción: La hidrogenólisis e hidrogenación [Broderick D. H.
& Gates B. C., 1981]. En base a estas trayectorias es posible determinar las expresiones de
velocidad representando el comportamiento de los principales productos de reacción en este
proceso.
Diversos investigadores en sus trabajos de hidrodesulfuración definen sus expresiones de
velocidad en términos del esquema de reacción presentado en la Figura 2.1. La formación y
selectividad de los productos de reacción depende de las condiciones de operación y del tipo de
catalizador utilizado, así como también de la concentración de H2S presente en la mezcla de
reacción. De esta manera con catalizadores de NiMo la selectividad hacía la hidrogenación se
incrementa [Bataille F. et al., 2000], manifestándose en una mayor formación de ciclohexilbenceno
(Figura 2.1). Este efecto también puede observarse con una relación alta H2S/H2 [Broderick D. H.
& Gates B. C., 1981; Vanrysselberghe V. & Froment G. F., 1996; Vanrysselberghe V. et al., 1998;
Vanrysselberghe V. & Froment G. F., 1998; Froment G. F. et al., 1994; Laredo S. C. G. et al., 2001;
Koltai T. et al., 2002; Bataille F.et al., 2000].
Utilizando catalizadores de Mo y CoMo la reacción de hidrogenólisis es predominante, de esta
manera una mayor concentración de bifenilo se puede observar en los productos de reacción con
respecto a los obtenidos en catalizadores NiMo.
Broderick D. H. & Gates B. C., 1981, realizaron la hidrodesulfuración de DBT en un reactor por
lotes, utilizando hexadecano como disolvente para un intervalo de temperatura entre 548-598K,
alrededor de 1.8 MPa de presión y un catalizador CoMo/γ-Al2O3. En base a los productos de
reacción establecieron un esquema de reacción muy similar al presentado en la Figura 2.1.
Principalmente bifenilo, ciclohexilbenceno y tetra- y – hexahidrodibenzotiofeno (THDBT, HHDBT)
se observaron como productos de reacción a las condiciones de operación antes mencionadas. A
estas condiciones de operación el diciclohexilo proveniente desde el ciclohexilbenceno no se
observo como producto de reacción.

ANTECEDENTES
15
S
SS
+ H2S
hidrogenólisishidrogenación
hidrogenación
hidrogenación
hidrogenólisis
DBT
THDBT HHDBT BPH
CHB
DCH
Figura 2.1 Trayectoria de reacción para la hidrodesulfuración de dibenzotiofeno
[Vanrysselberghe V. & Froment G. F., 1996]
Del análisis del esquema de reacción y el examen de diversas ecuaciones de velocidad vía los
datos de conversión diferencial, la cinética de las dos reacciones primarias se representa por las
siguientes dos expresiones de velocidad de reacción:
)CK1()CKCK1(
CCKKkr
2222
22
HH2
SHH,SHDBTDBT
HDBTH,HH,DBT'H
isishidrogenól +++= 2.1
)CK1(CCKKk
rDBTHG,DBT
HDBTHG,HHG,DBT´
iónhidrogenac22
+= 2.2
En estas expresiones se puede observar que la concentración del H2S solo tiene efecto en la
reacción de hidrogenólisis, por lo que un incremento en esta provoca la disminución de la rapidez
de reacción para la hidrodesulfuración de manera directa (hidrogenólisis). La Tabla 2.3 presenta los
parámetros cinéticos y de adsorción relativos a estas expresiones de velocidad de reacción.
El estudio cinético de todo el esquema de reacción de la Figura 2.1 involucrando expresiones de
velocidad de reacción para la hidrogenación e hidrogenólisis fue desarrollado por Vanrysselberghe

Capítulo 2
16
V. & Froment G. F., 1996. Un programa experimental utilizando dibenzotiofeno disuelto en una
mezcla de parafinas normales con composición conocida sobre un catalizador de CoMo/Al2O3 se
realizó en reactor de flujo mezclado (Robinson Mahoney) a una presión total entre 5.0 – 8.0 MPa y
temperaturas entre 513 y 573 K y una relación molar H2/HC entre 1.1 y 4.1.
Tabla 2.3 Parámetros en las Ecuaciones de Velocidad para la Hidrogenólisis e Hidrogenación [Broderick D. H. and Gates B. C., 1981]
Parámetro Cinético
Constantes cinéticas/energía de activación/calor de adsorción
Valor
K HDBT , A# 1.8E-01
∆H 1.9E07
K H,H2 A# 4.0E03
∆H -3.5E07
K HS,H2 A# 7.0E-01
∆H 2.2E07
KK HHDBT
k
2,
'Hk =
A# 7.87E05
Ea* -1.26E08
K HGDBT, A# 2.0
∆H 6.0E06
Kk HGH ,
'
2 A# 4.22E04
Ea** -1.16E08
Los productos de reacción de la hidrodesulfuración de dibenzotiofeno fueron, bifenilo,
ciclohexilbenceno, biciclohexilo y H2S. A estas condiciones tetra- y – hexahidrodibenzotiofeno no
fueron detectadas. Estos autores también proponen que las reacciones de hidrogenólisis e
hidrogenación se llevan a cabo en diferentes tipos de sitios (σ,τ). Por consiguiente fue necesario
derivar diversos mecanismos de reacción y sus correspondientes expresiones de velocidad tipo
Lagmuir-Hinshelwood-Hougen-Watson.
Los diversos mecanismos de reacción varían únicamente en la manera de cómo él hidrógeno se
adsorbe: atómicamente (A) o molecularmente (M); competitivamente en la hidrogenólisis (σc) e

ANTECEDENTES
17
hidrogenación (τc); no competitivamente sobre un tercer tipo de sitios activos (σnc, τnc), no
competitivamente sobre los sitios activos para la hidrogenación (στ) o hidrogenólisis (τσ)
[Vanrysselberghe V. & Froment G. F., 1996].
Para la adsorción atómica de hidrógeno se consideró la adición separada del primer átomo de
hidrógeno, el segundo átomo de hidrógeno y la adsorción simultánea de los dos átomos de
hidrógeno.
La adsorción con hidrógeno molecular también fue considerada (σER, τER). Después de la reacción
los átomos de DBT, THDBT o HHDBT permanecen en la superficie del catalizador, por lo que su
remoción ocurre vía reacción con H2 directamente desde la fase líquida o bien a través de un
mecanismo correspondiente con la hidrogenólisis sobre los sitios σ (Sσ, Sτ, Snc). La etapa
determinante de la velocidad de reacción puede darse ya sea por la adsorción de los reactivos o la
reacción en la superficie de las especies adsorbidas o la desorción de los productos de reacción
sobre ambos sitios activos σ y τ pueda ser. De esta manera la discriminación involucro 174
modelos de tipo Rival [Vanrysselberghe V. & Froment G. F., 1996]. Esta discriminación se realizó
considerando un examen estadístico así como también determinados criterios fisicoquímicos.
Las expresiones de velocidad involucrando las reacciones de hidrogenólisis e hidrogenación
determinadas a partir mecanismos de reacción bien definidos se muestran a continuación:
Hidrogenólisis
)( 3
,,,,
,,,,
222
2
1 CKCKCKCKCCKKkr
SHSHBPHBPHHHDBTDBT
HDBTDBTHDBT
DBT
σσσσ
σσσ
σ++++
= 2.3
Hidrogenación
)( 3
,,,
,,,,
2
2
1 CKCKCKCCKKkr
BPHBPHHHDBTDBT
HDBTDBTHDBT
DBT
τττ
τττ
τ+++
= 2.4
)( 3
,,,
,,,
2
2
1 CKCKCKCCKKkr
BPHBPHHHDBTDBT
HBPHBPHHBPH
BPH
τττ
ττττ
+++= 2.5

Capítulo 2
18
)( 3
,,,
,,,,
2
2
1 CKCKCKCCKKkr
BPHBPHHHDBTDBT
HCHBCHBHCHBCHB
τττ
ττττ
+++= 2.6
Nuevamente se puede observar en estas ecuaciones que solo el H2S tiene efecto en la expresión
de velocidad relativa a la reacción de hidrogenólisis. Por otro lado, también la adsorción de
dibenzotiofeno y los productos de reacción juegan un papel importante en estas expresiones. La
Tabla 2.4 muestra un resumen de los parámetros cinéticos y de adsorción correspondientes con
estas ecuaciones de velocidad.
Tabla 2.4 Parámetros en las Ecuaciones de Velocidad [Vanrysselberghe V. & Froment G. F., 1996]
Parámetro Cinético
Constantes
cinéticas/energía de activación/calor de
adsorción
Valor
K σDBT, 75.687
K σBPH, A# 17.904 ∆H -4.821E04 K σH, A# 3.080 ∆H -1.132E05 K σS,H2
A# 249.703 ∆H -1.057E05 k σDBT, A# 3.183E-02 Ea 1.228E05 KDBT,τ A# 6.877 ∆H -3.790E04 KBPH,τ A# 2.317 ∆H -3.790E04 KH,τ A# 9.138E-02 ∆H -1.4270E05 kDBT,τ A# 2.708E-02 Ea 1.862E05 kBPH,τ A# 5.992E-02 Ea 2.557E05 kCHB,τ KCHB,τ * 3.386E-01

ANTECEDENTES
19
La especiación de azufre en muestras de diesel utilizando las técnicas analíticas de cromatografía
de gases y un detector de emisión átomica (GC–AED) así como también con un detector
fotométrico de llama (FPD) muestra que la hidrodesulfuración de destilados intermedios involucra
la remoción de compuestos tiofénicos, de tipo benzotiofeno, dibenzotiofenos y alquil-
dibenzotiofenos como ha sido mostrado por Whitehurst D. D. et al., 1998 y Arzate B. E. et al.,
2001.
2.2. TRAYECTORIAS Y MECANISMOS DE REACCION PARA LA HIDRODESULFURACION La literatura abierta muestra que la descomposición de dibenzotiofeno y alquil-dibenzotiofenos son
utilizados como reactivos modelo para representar la hidrodesulfuración profunda de diesel
[Bataille F. et al., 2000; Mijoin J. et al., 2001]. La Figura 2.2 muestra el mecanismo de
hidrodesulfuración de dibenzotiofeno propuesto por Bataille F., et al., 2000.
SHHH
H
SHS
H HH
SHHH
H
SH
S
S
E2
+ H2S
+ H2S 2H2
H2
S
X
H2
DBT
H2
E2
H2
E2
E2
THDBT
HHDBT
BPH
CHB
Figura 2. 2 Trayectorias de reacción para la desulfuración de Dibenzotiofeno
en CoMo o NiMo/Al2O3 [Bataille F., et al., 2000]

Capítulo 2
20
En esta Figura se observa que el bifenilo y el tetrahidrodibenzotiofeno son los productos primarios
desde el dibenzotiofeno, además de que bajo condiciones de hidrodesulfuración el bifenilo no es
convertido fácilmente hacia ciclohexilbenceno, lo cual se contradice al mecanismo propuesto por
Vanrysselberghe V. & Froment G. F., 1996. En esta Figura puede ser también observado que para
la trayectoria de hidrogenación el rompimiento del segundo enlace no requiere que el segundo
anillo aromático sea totalmente hidrogenado produciendo de esta manera ciclohexilbenceno en
lugar de llegar hasta diciclohexilo.
La mayor contribución de ambas trayectorias a la desulfuración depende del tipo de catalizador a
utilizar, cuando se trata de catalizadores monometálicos (como Mo) la trayectoria dada por la
hidrogenación es la predominante, mientras que sobre catalizadores promovidos CoMo o NiMo la
hidrogenólisis es la principal trayectoria.
En la literatura abierta se ha reportado una diferente reactividad a las reacciones de hidrogenólisis
e hidrogenación entre las moléculas de dibenzotiofeno y 4,6 dimetildibenzotiofeno utilizando
catalizadores promovidos con cobalto [Bataille F. et al., 2000]. Sin embargo estos investigadores
también reportan que la actividad para la hidrodesulfuración de dibenzotiofeno y 4,6-
dimetildibenzotiofeno es similar considerando la trayectoria de hidrogenación cuando se utiliza solo
Mo como catalizador. Sin embargo el dibenzotiofeno es más reactivo que el 4,6-
dimetildibenzotiofeno por la trayectoria de hidrodesulfuración. La explicación de la baja reactividad
del 4,6-dimetildibenzotiofeno puede darse en términos del impedimento estérico del 4,6-dimetil
dibenzotiofeno debido a la presencia de grupos metilo en las posiciones 4 y 6.
En la Figura 2.3 se muestra de manera simplificada el mecanismo de hidrodesulfuración del 4,6
dimetildibenzotiofeno en catalizadores CoMo o NiMo/Al2O3. El inicio de la reacción involucra la
destrucción de uno de los anillos bencénicos, lo cual puede ser la etapa más difícil de reacción. Los
dihidroisomeros pueden ser formados por la adición 1,2 de dos átomos de hidrógeno al
dimetildibenzotiofeno. En esta Figura se muestran dos tipos de reacción que pueden ser
considerados para los dihidroisomeros a saber: a) ruptura del enlace C-S (hidrogenólisis) y b) la
hidrogenación hacia productos tetrahidrogenados.
La presencia de grupos metilo puede tener un efecto importante sobre la mayoría de las etapas de
reacción conduciendo a la hidrodesulfuración tanto de dibenzotiofeno como de 4,6
dimetildibenzotiofeno: 1) La adsorción de los reactivos o intermediarios (dihidrointermediarios), 2) la
adición de átomos de hidrógeno (formación de tetrahidro- y –hexahidrointermediarios) y 3) la
ruptura del enlace C-S vía la reacción de eliminación (E2), ver Figura 2.4. Estos efectos pueden
explicar las diferencias con respecto a las etapas controlantes de la velocidad de reacción para el
dibenzotiofeno y/o el 4,6-dimetildibenzotiofeno. Sin embargo, sobre catalizadores de Mo/Al2O3, la
ruptura C-S para ambos reactivos es la etapa controlante de la velocidad de reacción. Sobre

ANTECEDENTES
21
catalizadores promovidos (CoMo o NiMo) la ruptura del enlace C-S también es la etapa limitante de
la velocidad para la hidrogenólisis de 4,6 dimetildibenzotiofeno, mientras que para dibenzotiofeno
la etapa limitante de la velocidad esta dada por la formación del dihidrointermediario. Con respecto
a la trayectoria dada por la hidrogenación, sobre catalizadores de Mo la ruptura del enlace C-S
para ambos reactivos también es la etapa limitante de la velocidad, sin embargo en catalizadores
CoMo y/o NiMo/Al2O3, la formación de dihidrointermediarios en la transformación de dibenzotiofeno
es la etapa controlante de la velocidad. Para el 4,6 dimetildibenzotiofeno no esta definida la etapa
controlante de la reacción, se ha observado la formación de grandes cantidades de intermediarios
hidrogenados indicando que la ruptura C-S es un paso de reacción más lento.
Hasta ahora la discusión anterior se ha centrado a las diferentes etapas involucradas en la
hidrodesulfuración de moléculas tipo tiofeno, las cuales pertenecen a dos categorías: 1) la adición
de hidrógeno al anillo bencénico y 2) la ruptura por eliminación del enlace carbono- azufre (C-S).
Por consiguiente es de interés hacer algunos comentarios acerca de la naturaleza de los posibles
centros catalíticos responsables de estas dos categorías. Para lo cual los principales factores que
deben ser tomados en cuenta son: i) los centros activos deben tener el poder de activar alguno de
los anillos aromáticos de las moléculas sustrato con el fin de poder ser hidrogenadas, ii) estas
deben adsorberse y disociar la molécula de hidrógeno y iii) el catalizador debe de poder retener los
átomos de azufre resultado de la descomposición de las moléculas orgánicas [Bataille F. et al.,
2000], ver Figura 2.4.
A manera de cumplir con estos factores y antes de la adsorción de los reactivos, los sitios para las
dos etapas deben de tener al menos dos vacancias y un átomo vecino de azufre. Estos centros
catalíticos pueden ser obtenidos al remover los átomos de azufre de los bordes y esquinas de los
bloques de MoS2 con todas sus orillas alineadas por los átomos de azufre, presentando estos
bloques mayor estabilidad que aquellos terminados por átomos de molibdeno (Figura 2.5). La
adsorción de H2S en estos centros tendrá un efecto de inhibición tanto para la hidrogenación como
para la hidrogenólisis. Sin embargo, los átomos S2- remanentes pueden no tener la misma
basicidad si los iones de S2- removidos pertenecen al mismo o diferentes átomos de Molibdeno.
Por lo cual si el principal requerimiento para la etapa de eliminación E2 (ruptura del enlace C-S) es
la basicidad de los átomos S2- involucrados en el proceso de eliminación, la mayor sensibilidad de
la hidrogenólisis hacia H2S se puede explicar al suponer que el H2S (debido a sus propiedades
ácidas) se adsorbe preferencialmente sobre los sitios más básicos.

Capítulo 2
22
RR
3H2
S
R R
SR R
S
R R
H2
E2
2H2
RSHR
E2
SHR RS
R R
H2
RR
H2
E2b) a)
RR
S
R R
DMDBT DMDBT
BHDMDBT DMBPH
THDMDBT
SHR SHR RR
HHDMDBT
SHR R
RSHR
DMCHB
Figura 2. 3 Trayectorias de reacción para la desulfuración de 4,6-dimetildibenzotiofeno en CoMo o NiMo/Al2O3 [Mijoin J. et al., 2001]

ANTECEDENTES
23
SH H H
SMo Mo
S
S
MoMoS
SS
SMo Mo
SMo
SMo
SH
S
SMo MoS
S
MoMoS
S
HS
Figura 2.4. Mecanismo de ruptura del enlace C-S en la hidrogenólisis sobre MoS2
Figure 2.5. Modelo catalítico CoMoS: S; Co (Ni); Mo [Bataille F. et al., 2000]
Por otro lado el papel del promotor se dirige a disminuir la fuerza del enlace entre el molibdeno y
los átomos de azufre, resultado de la descomposición de las moléculas orgánicas. De la misma
manera se puede suponer que el promotor disminuye la fuerza del enlace metal-azufre en el
sulfuro mismo [Bataille F. et al., 2000] e incrementa la densidad electrónica sobre los átomos de
azufre, lo cual puede ser interpretado como un mejoramiento de la basicidad de los centros S2-. Por
consiguiente el principal efecto del promotor es el mejoramiento de la rapidez de reacción de la
hidrogenólisis. Esto es bastante fácil de comprender si se asume que el rompimiento del enlace C-
S ocurre a través de un mecanismo de eliminación (Figura 2.4). Este mecanismo involucra el
ataque de un átomo de hidrógeno (en posición β respecto al átomo de azufre) por un anión de
azufre actuando como un sitio básico, de esta manera, se sugiere que el promotor mejora la
basicidad de los iones de azufre favoreciendo la ruptura del enlace C-S.
2.3. ESQUEMA DE HIDROPROCESAMIENTO
Hoy en día los procesos de hidrotratamiento consisten de un reactor de lecho fijo con flujo
descendente como se muestra en la Figura 2.6 (tricked bed). De esta Figura puede ser observado
Capítulo 2
24
que el hidrógeno se utiliza en exceso y se recircula después de un agotamiento para eliminar el
H2S producido en el reactor. El control de esta última operación debe ser tal que sea posible
mantener un nivel bajo pero suficiente de H2S en la corriente de recirculación a manera de
mantener la actividad y estabilidad del catalizador, dado que el H2S es un fuerte inhibidor de las
reacciones de hidrodesulfuración de los compuestos tiofénicos.
Las capacidades instaladas para el hidrotratamiento de las diversas fracciones del petróleo utilizan
predominantemente reactores a presiones moderadas. La Tabla 2.5 muestra las condiciones
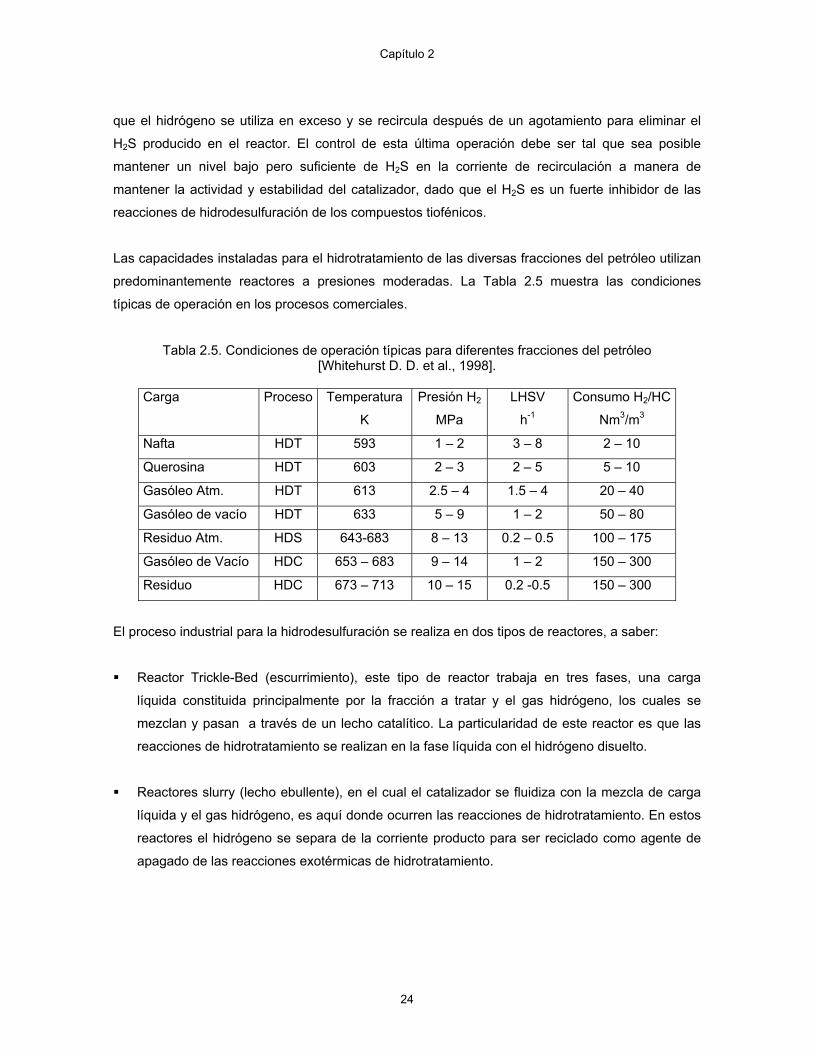
típicas de operación en los procesos comerciales.
Tabla 2.5. Condiciones de operación típicas para diferentes fracciones del petróleo [Whitehurst D. D. et al., 1998].
Carga Proceso Temperatura
K
Presión H2
MPa
LHSV
h-1
Consumo H2/HC
Nm3/m3
Nafta HDT 593 1 – 2 3 – 8 2 – 10
Querosina HDT 603 2 – 3 2 – 5 5 – 10
Gasóleo Atm. HDT 613 2.5 – 4 1.5 – 4 20 – 40
Gasóleo de vacío HDT 633 5 – 9 1 – 2 50 – 80
Residuo Atm. HDS 643-683 8 – 13 0.2 – 0.5 100 – 175
Gasóleo de Vacío HDC 653 – 683 9 – 14 1 – 2 150 – 300
Residuo HDC 673 – 713 10 – 15 0.2 -0.5 150 – 300
El proceso industrial para la hidrodesulfuración se realiza en dos tipos de reactores, a saber:
Reactor Trickle-Bed (escurrimiento), este tipo de reactor trabaja en tres fases, una carga
líquida constituida principalmente por la fracción a tratar y el gas hidrógeno, los cuales se
mezclan y pasan a través de un lecho catalítico. La particularidad de este reactor es que las
reacciones de hidrotratamiento se realizan en la fase líquida con el hidrógeno disuelto.
Reactores slurry (lecho ebullente), en el cual el catalizador se fluidiza con la mezcla de carga
líquida y el gas hidrógeno, es aquí donde ocurren las reacciones de hidrotratamiento. En estos
reactores el hidrógeno se separa de la corriente producto para ser reciclado como agente de
apagado de las reacciones exotérmicas de hidrotratamiento.

ANTECEDENTES
25
Figura 2.6 Esquema de una Unidad de Desulfuración de Gasóleo
[Whitehurst D. D. et al., 1998]
El estudio de procesos catalíticos multifásicos tales como el de hidrotratamiento del petróleo
requiere de reactores de laboratorio que aseguren un buen contacto entre las fases gas, líquida y
sólida a altas temperaturas y presiones. En el pasado se utilizaron reactores de lecho fijo y flujo
mezclado presentando pequeños gradientes de temperatura y concentración lo que impide el
desarrollo confiable de un modelo cinético. Sin embargo, actualmente se ha tenido un avance
significativo en el diseño y construcción de reactores experimentales que eliminan la resistencia al
transporte de masa y calor en la partícula catalítica [Mahoney J. A. at al., 1978].
Para sistemas catalíticos multifásicos, los estudios experimentales normalmente se desarrollan en
un reactor de lecho fijo escurrido. Este tipo de reactores presenta problemas en el patrón de flujo
que repercuten en el contacto del catalizador, provocando resistencias al transporte de masa
Gasóleo Pesado desulfurado
Carga Fresca
Hidrógeno reposición
Horno
Reactor
absorbedor
Separador
de
alta
presión
DEA limpia
DEA rica
Gas rico en Hidrógeno
Separador
de
baja
presión
Hidrógeno de recirculación
Gas combustible
Gasolina Ligera
Nafta Pesada
Gasóleo Ligero
Fraccionador
Carga Fresca
Hidrógeno reposición
Horno
Reactor
absorbedor
Separador
de
alta
presión
DEA limpia
DEA rica
Gas rico en Hidrógeno
Separador
de
baja
presión
Hidrógeno de recirculación
Gas combustible
Gasolina Ligera
Nafta Pesada
Gasóleo Ligero
Fraccionador

Capítulo 2
26
externa. Un contacto vigoroso del gas-líquido-catalizador frecuentemente elimina estas
resistencias. Los reactores de gradientes pequeños son mejores a los de lecho fijo en términos de
facilitar el muestreo, mantener la temperatura, transporte de masa y otras variables de operación.
Algunos reactores de gradiente pequeño están disponibles para estudios de reacción en fase gas
(reactor Berty), sin embargo ninguno de ellos se utiliza para estudios de sistemas multifásicos,
particularmente a una alta presión como la requerida en algunos procesos de refinación, como es
el caso del hidrotratamiento de destilados intermedios. Se han reportado estudios en reactores
multifásicos de gradiente pequeño trabajando solamente a presiones atmosféricas [Mahoney J. A.
et al., 1978]. Por otro lado se ha desarrollado una versión a alta presión para estudios de
reacciones de hidrotratamiento el cual contiene una canasta agitada dentro de un reactor
autoclave. Las aspas alrededor de la canasta aseguran un buen mezclado y contacto del líquido
así como también eliminan el vórtice de la interfase gas-líquido. Este tipo de reactor es conocido
como Robinson Mahoney [Koehloefl K., 2001], el cual se utilizó en la década pasada para
realizar estudios cinéticos de la hidrodesulfuración del dibenzotiofeno.
En los últimos años el diseño de reactores químicos para reacciones heterogéneas ha tenido un
auge intensivo. Aunque mucho de la investigación ha avanzado en gran parte por el desarrollo, la
capacidad y velocidad de cálculo de las computadoras, el problema todavía persiste debido a la
carencia de datos que son requeridos para la determinación de las expresiones de velocidad de
reacción involucradas en el proceso de hidrotratamiento. Estas expresiones se introducen en el
modelo del reactor y a su vez se usan en la simulación del proceso para hacer determinaciones en
tiempo real y obtener un panorama del comportamiento de este proceso, ya sea para resolver
problemas en la operación de la planta, cambios en el tipo de carga a procesar, así como también
para realizar un análisis de optimización de las condiciones de operación que estarán
determinadas en base principalmente a los productos de reacción obtenidos en el reactor. Cuando
alguno de estos factores cambia, se requiere determinar las nuevas propiedades termofísicas del
reactor, en este caso se utilizan correlaciones teóricas o empíricas que están especificadas en
función del cambio de presión, temperatura y composición. Sin embargo en el caso del cálculo de
las velocidades de reacción esto no es posible, se requiere de la evaluación de los parámetros
cinéticos del modelo que ha sido desarrollado particularmente para cada proceso. Estos
parámetros pueden ser estimados a través de la optimización de los datos experimentales.
En la literatura abierta se han publicado diversos datos experimentales y sus correspondientes
modelos cinéticos representado las trayectorias de reacción para el hidrotratamiento, sin embargo
estos se reportan a condiciones de operación muy por debajo de las utilizadas a nivel industrial
procesando los destilados intermedios en nuestro país [Vanrysselberghe V. & Froment G.F., 1996].
ANTECEDENTES
27
Adicionalmente a las especificaciones que pueda tener la experimentación en cuanto al diseño de
experimentos, tipos de equipo y sistemas de control especificados en cada sección de la planta
piloto, las variables físicas, como el tipo de disolvente y las sustancias base que serán utilizadas
como compuestos modelo, constituyen una complejidad de variables que deberán ser
consideradas para la construcción y operación de una planta piloto para el hidrotratamiento de
destilados intermedios.
Actualmente algunos investigadores [Vanrysselberghe V. et al., 1998; Vanrysselberghe V. &
Froment G. F., 1996; Edvinsson R. & Irandoust S., 1993; Tikhonov G. F. et al., 1986] han reportado
parámetros cinéticos determinados para la hidrodesulfuración de algunos compuestos azufrados a
condiciones de operación diferentes a las utilizadas a nivel industrial, más aún estos trabajos no
consideran todos los compuestos altamente reactivos de interés. En la Tabla 2.6 se presenta un
resumen de diversos trabajos para el hidrotratamiento de compuestos modelo incluyendo las
condiciones de operación, disolvente y tipo de reactor utilizado, así como también el número de
experimentos realizados.
Tabla 2.6. Hidrodesulfuración de compuestos modelo reportada en literatura.
VARIABLES COMPUESTOS MODELO
DBT(16) DBT,
DBT/BPH(14)
DBT(1) 4-
MDBT/DBT(3)
4,6-
DMDBT/DBT(3)
Temperatura, K 573 548-598 513 -573 533-593 533-590
Presión, MPa 10.2 18.0 5.0 - 8.0 8.0 6.0 – 8.0
Experimentos 6 54 98 12 24
Disolvente Hexano Hexano Parapur Parapur Parapur
Tipo de reactor HPPBFMR/AC* HPPBFMR/AC* RM** RM** RM** *Microreactor de cama empacada de alta presión/ Auto clave (High Pressure, Packed-Bed Flow Reactor/ AutoClave)
**Robinson Mahoney (RM) Como puede verse en la Tabla 2.6 en algunos casos el número de experimentos para un solo
compuesto modelo resulta ser muy grande, por lo cual es necesario tener claro los objetivos y el
tipo de resultado que se espera obtener en cada experimentación, ya que esto determinará el
número de experimentos y variables a estudiar, dando como resultado el tiempo de
experimentación necesario para determinar el comportamiento de cada variable. En la mayoría de
los casos el tiempo requerido para el análisis minucioso de cada una de las variables involucradas
en la experimentación es muy largo, por lo que la obtención de los parámetros cinéticos de
reacción será en todos los casos muy costosa.
Es así como surge la necesidad de obtener datos experimentales a condiciones de operación

Capítulo 2
28
similares a las utilizadas en el sistema de refinación en México. Bajo este contexto se decidió la
construcción de una planta piloto con la cual se generen datos confiables de hidrotratamiento de
destilados intermedios que puedan ser utilizados en el desarrollo y/o adaptación de los modelos
cinéticos.
En referencia a la información anterior se observa que existe un sin número de variables que
deben ser consideradas al realizar un estudio experimental, para poder contar con datos que
aproximen el modelo cinético de reacción al comportamiento que se tiene en el reactor industrial de
hidrotratamiento. Sin embargo de los estudios reportados en literatura abierta no existe información
experimental obtenida a condiciones de operación similares a las del proceso de hidrotratamiento
industrial. Como consecuencia en la presente tesis se pretende mostrar el comportamiento de la
hidrodesulfuración que tiene el Dibenzotiofeno, así como también el de la distribución de los
productos y subproductos de reacción obtenidos en la planta piloto a condiciones industriales
utilizando un catalizador a base de CoMo / Al2O3.
La cinética de reacción es y ha sido utilizada para el traslado seguro de datos de laboratorio y
planta piloto hacia el diseño y análisis de una unidad comercial [Berger R. J. et al., 2001]. Los
estudios cinéticos pueden ser llevados a cabo a través de diferentes juegos de condiciones
experimentales, dependiendo si los datos cinéticos se generan en presencia y/o ausencia de
resistencias a la transferencia de masa interna o externa. Por consiguiente los experimentos
pueden ser conducidos en tres niveles [Bos A. N. R. et al., 1997]:
I. Experimentos que se llevan a cabo usando catalizadores con tamaño de partícula catalítica
muy pequeña y bajo condiciones apropiadas para eliminar limitaciones tanto externas
como internas de transferencia de masa y calor , dando como resultado una cinética
intrínseca del catalizador.
II. Experimentos que se llevan a cabo con catalizadores de tamaño comercial, esto es se
incluyen los efectos de transferencia de masa interna, pero se eliminan las resistencias de
transferencia de masa externa y efectos de mezclado por lo que se puede desarrollar una
reacción de cinética aparente.
III. Experimentos que se llevan a cabo de tal manera que las resistencias difusionales internas
y/o las limitaciones a la transferencia de masa externa no se eliminan, dando como
resultado un modelo cinético extrínseco para una reacción particular.
Si bien los modelos cinéticos aparentes no se favorecen para estudios científicos y/o académicos,
estos han sido utilizados por los ingenieros de proceso para el escalamiento de los reactores

ANTECEDENTES
29
comerciales [Bos A. N. R. et al., 1997]. Esto es debido a que ayudan a predecir el comportamiento
del catalizador en la forma en la cual será utilizado en el reactor comercial.
Sin embargo tales modelos cinéticos no pueden ser extrapolados para diferentes formas de
catalizador. Por consiguiente es aconsejable realizar los estudios cinéticos cuando la forma del
catalizador ya ha sido decidida para la aplicación comercial.
Por otro lado a manera de comprender y/o desarrollar nuevos catalizadores, se puede realizar
trabajo experimental para la medición de la reactividad relativa de diversos compuestos modelo
presentes en las cargas reales así como también para la comprensión de las trayectorias
mecanísticas.
Muy pocos investigadores [Geneste P. et al., 1980; Krishnamyrthy S. et al., 1981; Daly F. P., 1978,
Le Page J. F. et al., 1987], han utilizado reactores por lotes tipo autoclave en estudios cinéticos
para las reacciones involucradas en el proceso de hidrotratamiento. En estos reactores el
catalizador puede utilizarse en forma de polvo fino, puede también ser introducido en una canasta
giratoria o estática localizada en el interior del autoclave.
Para los estudios cinéticos en las reacciones de hidrotratamiento se han utilizado ampliamente
reactores continuos de lecho fijo con catalizador en la forma de pellets o partículas fragmentadas
[Korsten H. & Hoffmann U., 1996; Miller J. T. & Hineman M. F., 1984; Tsamatsoulis D. &
Papayannakos N., 1998; Yui S. M. & Sanford E. C., 1989; Statterfield C. N. & Yang S. H., 1984;
Vradman L. et al., 1999]. Generalmente en este tipo de reactores el lecho catalítico se diluye con
partículas inertes de un tamaño apropiado con objeto de reducir la dispersión axial y canalizaciones
que pudiesen presentarse.
Si se trata de un reactor trifásico (trickled bed) el uso de este material favorece el humedecimiento
del catalizador. Estos reactores se utilizan bajo régimen diferencial o integral. En la operación del
primero, los gradientes de concentración y temperatura son pequeños y de esta manera el análisis
de los datos es simple. Para el segundo modo de operación existen altos gradientes de
concentración y temperatura y por consiguiente la interpretación de datos experimentales se hace
más difícil.
Los reactores con sistemas de recirculación interna aproximándose al comportamiento de un
reactor de flujo mezclado son los más útiles para obtener datos cinéticos para las reacciones de
hidroprocesamiento. Estos reactores generalmente se tipifican por los reactores tipo Berty,
Robinson Mahoney y Carberry. Debido al buen mezclado que presenta este tipo de reactores, la

Capítulo 2
30
concentración dentro del reactor como a la salida son similares. La información experimental
generada en un reactor sin gradientes proporciona ventajas para su análisis e interpretación y de
esta manera facilitan la determinación del modelo cinético.

PROCEDIMIENTOS EXPERIMENTALES
31
3. PROCEDIMIENTOS EXPERIMENTALES 3.1. CONSIDERACIONES PARA LA CONSTRUCCIÓN DE LA PLANTA PILOTO DE HIDROTRATAMIENTO En este capítulo se describirá las partes constitutivas y los parámetros operaciones que se tomaron
en cuenta para la construcción de la planta piloto, la cual se dividió en tres secciones a saber: a)
Sección de preparación de carga, b) Sección de reacción, c) Sección de separación.
3.1.1. Descripción de la planta piloto de hidrotratamiento
En el Figura 3.1 se muestra el diagrama de simplificado del proceso en planta piloto de
hidrotratamiento de destilados intermedios. El flujo de hidrógeno proveniente de límites de batería
pasa a través del controlador de flujo FIC-01, para posteriormente ser precalentado con una cinta
de calentamiento. La carga líquida se pesa en la báscula WI-21, y se precalienta por el mismo
medio que el hidrógeno. Esta corriente se mezcla con la de hidrógeno para ser introducida
posteriormente a la sección de reacción. La sección de reacción esta constituida principalmente de
un reactor tipo Robinson Mahoney con capacidad de 10-3 m3 (el cual tiene un sistema de control
para la presión, temperatura y la agitación) donde se efectúan las reacciones del hidrotratamiento.
La corriente de salida del reactor pasa a la sección de separación. En el separador FA-02 de alta
temperatura y alta presión se separa el líquido y el vapor que provienen del reactor el cual esta a
las mismas condiciones de reacción. El gas que sale por el domo de este separador se introduce a
un segundo separador FA-03 de alta presión y baja temperatura, en donde se atrapan las gotas de
líquido que se arrastran con el vapor. La fase gaseosa pasa a través de la válvula solenoide PCV-
46 la cual disminuye la presión del gas hasta aproximadamente 0.2 MPa, presión suficiente para
que este gas se introduzca al gasómetro FQI-61 con el objeto de ser medido. Una parte de este
gas pasa al cromatógrafo de gases (CG) donde se analiza y el gas sobrante se envía a la
atmósfera previo tratamiento cáustico en el tanque FA-06. El líquido que entra al separador FA-02
se acumula en la pierna de este, y por medio del sistema control se abre la válvula que esta
conectada en serie al separador dejando pasar el líquido acumulado a la sección de baja presión.
La sección de baja presión esta constituida de una bureta y una probeta conectadas en serie en
donde se separa el gas disuelto en la fase líquida. Por el fondo de la probeta FA-05 se obtiene el
líquido producto que se recolecta para su medición y posterior caracterización. El gas disuelto en el
domo de esta probeta se mide y una parte de este gas pasa por el compresor C-1 para elevar su
presión e introducirlo al cromatógrafo (CG) para determinar su composición.
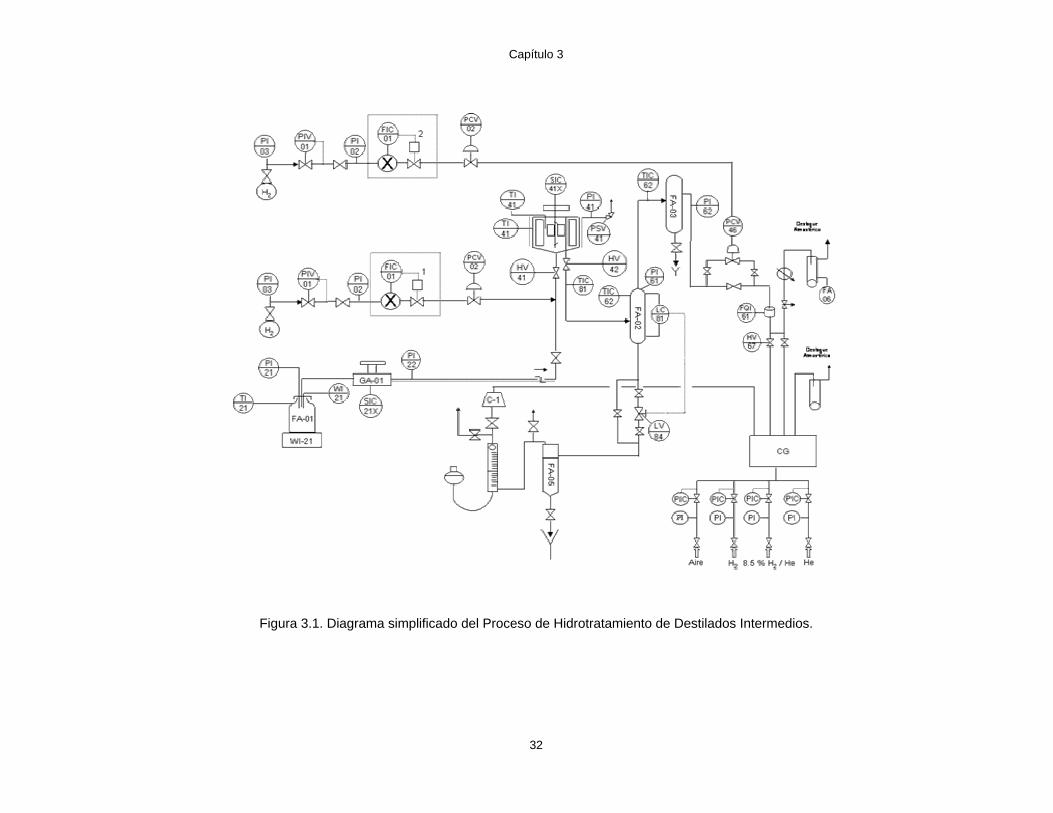
Capítulo 3
32
Figura 3.1. Diagrama simplificado del Proceso de Hidrotratamiento de Destilados Intermedios.

PROCEDIMIENTOS EXPERIMENTALES
33
3.1.2. Sección de preparación de carga
La construcción de la sección de carga de la planta piloto se llevo a cabo bajo las siguientes
consideraciones para las corrientes de entrada:
Hidrógeno de alta pureza a alta presión proveniente de los límites de batería
Carga líquida con características semejantes a un gasóleo ligero conteniendo compuestos
modelo.
Uso de estándares externos para el análisis analítico
La carga al reactor puede estar constituida de hidrógeno y un compuesto modelo disuelto en un
disolvente como el aceite mineral. La caracterización del disolvente (aceite mineral) se presenta en
la Tabla 3.1. En esta Tabla se reporta la curva de destilación, el contenido de aromáticos, nitrógeno
y azufre, así como el peso específico que contiene el aceite mineral que se eligió para la
experimentación. Este último es semejante al que tiene en los gasóleos ligeros en México (0.84
promedio). Actualmente esta sustancia se suministra por la compañía Parafinas y Aceites S.A. de
C.V.
La pureza del hidrógeno que se utiliza para la operación de la planta es de 99.8% como mínimo
suministrado por la compañía INFRA Air Products. Las características que debe cumplir el
disolvente para llevar a cabo la experimentación son que este debe estar libre de compuestos
aromáticos, nitrogenados y azufrados para no interferir en las reacciones de remoción de los
heteroátomos contenidos en la mezcla sintética preparada y de esta manera no repecutir
posteriormente en el análisis de los productos, los cuales se utilizarán en el modelamiento cinético.
Estos compuestos se calientan, mezclan y presionan hasta las condiciones a las cuales se llevará
a cabo la experimentación.
Un aspecto importante que mencionar en esta sección es la estabilidad del flujo a la entrada del
reactor, por lo que el recipiente (FA-01) de carga líquida se presiona con nitrógeno para mantener
constante el flujo del líquido a la entrada de la bomba y minimizar de esta manera las fluctuaciones
que se puedan presentar.
3.1.3. Sección de reacción La sección de reacción esta constituido por un reactor tipo Robinson Mahoney. La Figura 3.2

Capítulo 3
34
muestra un diagrama detallado de los internos del reactor. La carga al reactor se introduce por la
parte superior de este y por medio de un tubo buzo hasta la parte inferior del reactor.
Tabla 3.1 Características del solvente (aceite mineral)
Destilación ASTM D-1160 Temp. (K)
TIE 585
5 % 600
10% 611
30% 634
40% 641
50% 647
60% 651
80% 666
90% 675
95% 682
T.F.E. 686
Peso específico ASTM D-70 0.8467
Peso Molecular (espectometría) 334.01
Contenido de aromáticos < 0.1% peso
Contenido de azufre total < 0.3 ppm
Contenido de nitrógeno total < 2 ppm
La carga se agita por medio de las propelas (2, 3, 6) para pasar a través del catalizador, el cual
esta contenido en la canasta de catalizador (4). Las propelas están adheridas a la flecha (1) por
medio de tornillos (5) y giran por medio del agitador magnético del reactor. La flecha descansa en
la base del reactor (7) la cual tiene anillos especiales (8,9) para dejarla girar. El reactor tiene un
termopar (11) que sirve para monitorear la temperatura interna del reactor. Adicionalmente tiene
una chaqueta (no mostrada en el diagrama) que proporciona el calor necesario para operar el
reactor hasta una temperatura de 724 K. El sistema de control de este monitorea y controla la
temperatura interna, la temperatura de pared, la presión interna y la velocidad de la flecha. Para
asegurar que el líquido contenido dentro del reactor ocupe todo el espacio vacío, se coloco la
salida del reactor a un centímetro de la tapa de este. El reactor tiene un sistema de sellado (12)
para asegurar su hermeticidad.
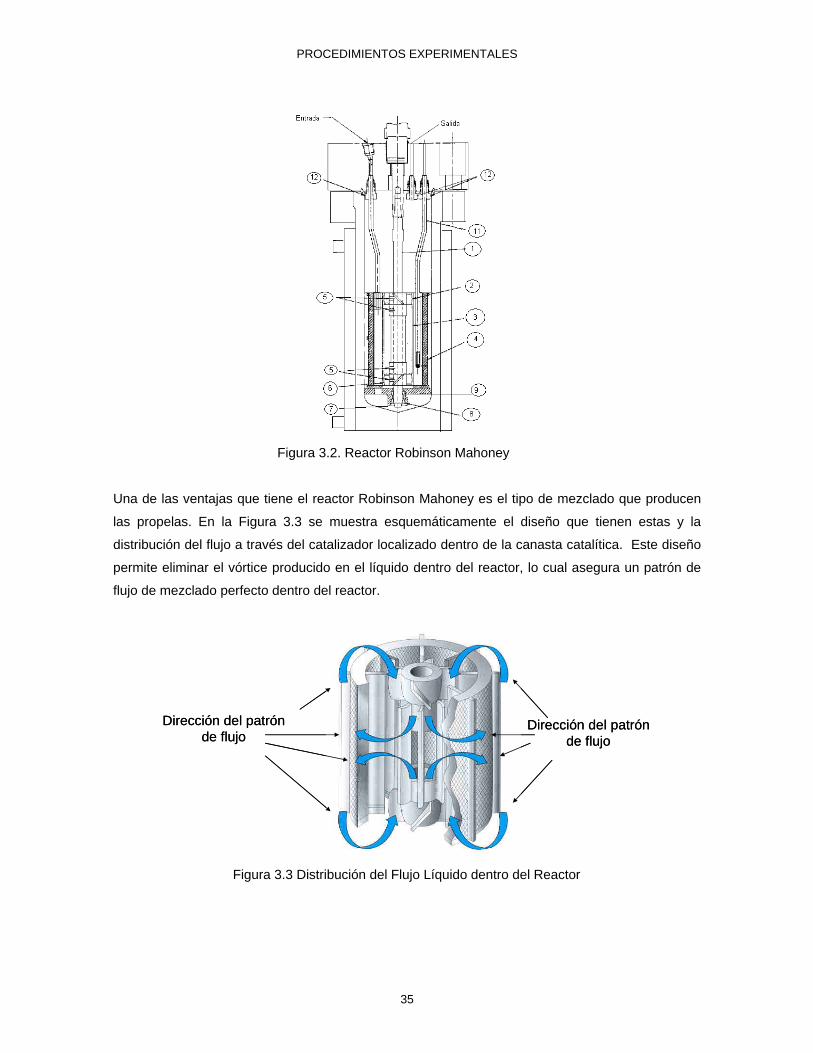
PROCEDIMIENTOS EXPERIMENTALES
35
Figura 3.2. Reactor Robinson Mahoney
Una de las ventajas que tiene el reactor Robinson Mahoney es el tipo de mezclado que producen
las propelas. En la Figura 3.3 se muestra esquemáticamente el diseño que tienen estas y la
distribución del flujo a través del catalizador localizado dentro de la canasta catalítica. Este diseño
permite eliminar el vórtice producido en el líquido dentro del reactor, lo cual asegura un patrón de
flujo de mezclado perfecto dentro del reactor.
Dirección del patrónde flujo
Dirección del patrónde flujo
Dirección del patrónde flujo
Dirección del patrónde flujo
Figura 3.3 Distribución del Flujo Líquido dentro del Reactor

Capítulo 3
36
En la Figura 3.4 se muestra una fotografía del diseño real que tienen las propelas, además se
puede observar el espacio destinado en la canasta catalítica para el catalizador.
Figura 3.4 Diseño de las Propelas y Canasta Catalítica del Reactor Robinson Mahoney
Las características que tiene el reactor se muestran en la Tabla siguiente:
Tabla 3.2 Características del Reactor Robinson Mahoney
Características del reactor Unidades Valor
Volumen m3 1x 10-3
Presión de diseño MPa 34.4
Temperatura de diseño K 727
Máxima velocidad de la agitación rpm 5000
Características d la canasta catalítica
Volumen de la canasta m3 1.31x 10-4
Tamaño de la malla 14X14 malla 0.020”
Calibre (0.51 mm)
3.1.4. Sección de Separación
El hidrotratamiento de la mezcla compleja de hidrocarburos y el compuesto modelo se lleva a cabo
en fase líquida, sin embargo el hidrógeno alimentado y la fase gas producida por las reacciones de
hidrodesintegración da lugar a un sistema multifásico, por consiguiente, el objetivo principal de esta

PROCEDIMIENTOS EXPERIMENTALES
37
sección es la de obtener el líquido y el vapor generado en el reactor a las mismas condiciones de
operación de este. Las fases líquida y vapor deben ser analizadas por separado, dado que la
composición de estas se utilizan en el modelamiento cinético del reactor. Un punto clave para
realizar esta operación, es el mantener el primer separador FA-02 a las mismas condiciones que el
reactor y realizar de esta manera la separación física-mecánica del líquido y vapor que entra al
separador. Este primer separador es de diseño especial como se muestra en la Figura 3.5. Tiene
una entrada tangencial parecida al comportamiento de un ciclón y un termopozo acoplado al
cuerpo de este. Este diseño tiene la ventaja de poseer una elevada eficiencia en la separación
líquido-vapor. El termopar esta acoplado a una cinta de calentamiento y por medio del sistema de
control de la planta se regula la temperatura del separador y de la línea que une a este con el
reactor. El segundo separador FA-03 es semejante al primero y tiene la función de separar el
líquido arrastrado en la fase gaseosa. El líquido obtenido en el separador FA-02 sufre una caída de
presión por medio de la válvula neumática LV-84 (intervalo de operación de 0 a 21 MPa, con el
objeto de separar el líquido y los gases disueltos en este gas flasheado) hasta la presión
atmosférica. Posteriormente esta corriente se dirige a la sección de separación de baja presión la
cual esta constituida por el conjunto bureta-probeta graduada el cual fue construido en el taller de
soplado de vidrio del Instituto Mexicano del Petróleo.
Figura 3.5 Esquema del separador FA-02 de alta presión y temperatura
En la Figura 3.6 se muestra este sistema, en el cual la bureta contiene un aceite mineral pesado
(350 USP) para realizar la medición del gas flasheado por dezplazamiento través del principio de
vasos comunicantes. Este aceite tiene la característica de no ser soluble con los gases que se
obtienen en esta sección.

Capítulo 3
38
Probeta -bureta
Recipiente paraEl aceite mineral pesadoProbeta -bureta
Recipiente paraEl aceite mineral pesado
Figura 3.6 Sistema de separación de baja presión de gas y líquido flasheados
Una vista general de la planta puede ser observada en la Figura 3.7, en donde se marca la
localización real de cada uno de los equipos.
Figura 3.7. Vista general de la planta piloto
Separadores Válvula
Cromatógrafo
Sección de Separación de bajapresión
Válvula neumáticaReactorBomba
Balanza electrónica
Recipiente decarga
Sistema decontrol
Separadores Válvula
Cromatógrafo
Sección de Separación de bajapresión
Válvula neumáticaReactorBomba
Balanza electrónica
Recipiente decarga
Sistema decontrol
Separadores Válvula
Cromatógrafo
Sección de Separación de bajapresión
Válvula neumáticaReactorBomba
Balanza electrónica
Recipiente decarga
Sistema decontrol
Separadores Válvula
Cromatógrafo
Sección de Separación de bajapresión
Válvula neumáticaReactorBomba
Balanza electrónica
Recipiente decarga
Sistema decontrol
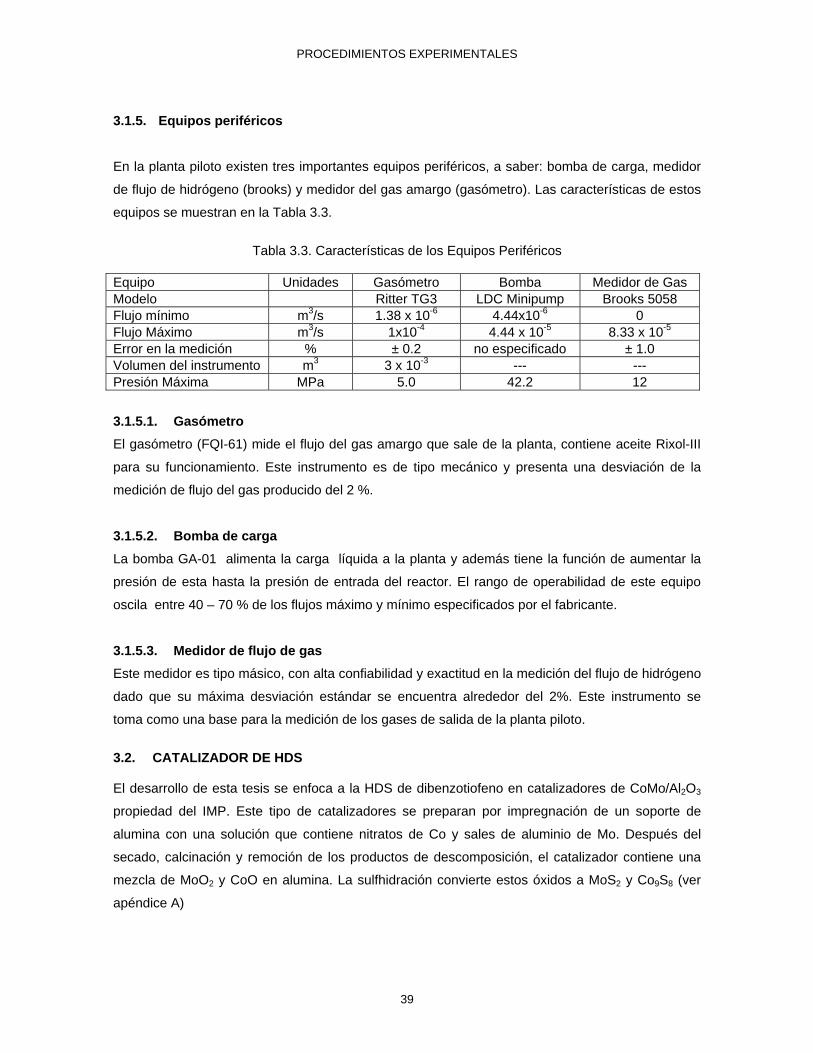
PROCEDIMIENTOS EXPERIMENTALES
39
3.1.5. Equipos periféricos
En la planta piloto existen tres importantes equipos periféricos, a saber: bomba de carga, medidor
de flujo de hidrógeno (brooks) y medidor del gas amargo (gasómetro). Las características de estos
equipos se muestran en la Tabla 3.3.
Tabla 3.3. Características de los Equipos Periféricos
Equipo Unidades Gasómetro Bomba Medidor de Gas Modelo Ritter TG3 LDC Minipump Brooks 5058 Flujo mínimo m3/s 1.38 x 10-6 4.44x10-6 0 Flujo Máximo m3/s 1x10-4 4.44 x 10-5 8.33 x 10-5 Error en la medición % ± 0.2 no especificado ± 1.0 Volumen del instrumento m3 3 x 10-3 --- --- Presión Máxima MPa 5.0 42.2 12 3.1.5.1. Gasómetro El gasómetro (FQI-61) mide el flujo del gas amargo que sale de la planta, contiene aceite Rixol-III
para su funcionamiento. Este instrumento es de tipo mecánico y presenta una desviación de la
medición de flujo del gas producido del 2 %.
3.1.5.2. Bomba de carga La bomba GA-01 alimenta la carga líquida a la planta y además tiene la función de aumentar la
presión de esta hasta la presión de entrada del reactor. El rango de operabilidad de este equipo
oscila entre 40 – 70 % de los flujos máximo y mínimo especificados por el fabricante.
3.1.5.3. Medidor de flujo de gas Este medidor es tipo másico, con alta confiabilidad y exactitud en la medición del flujo de hidrógeno
dado que su máxima desviación estándar se encuentra alrededor del 2%. Este instrumento se
toma como una base para la medición de los gases de salida de la planta piloto.
3.2. CATALIZADOR DE HDS El desarrollo de esta tesis se enfoca a la HDS de dibenzotiofeno en catalizadores de CoMo/Al2O3
propiedad del IMP. Este tipo de catalizadores se preparan por impregnación de un soporte de
alumina con una solución que contiene nitratos de Co y sales de aluminio de Mo. Después del
secado, calcinación y remoción de los productos de descomposición, el catalizador contiene una
mezcla de MoO2 y CoO en alumina. La sulfhidración convierte estos óxidos a MoS2 y Co9S8 (ver
apéndice A)
Capítulo 3
40
Tabla 3.4 Comparación de los catalizadores IMPDSD-14 y el utilizado por Vanrysselberghe V. & Froment G. F., 1996.
Composición Unidades IMP-DSD-14 Vanrysselberghe V. &
Froment G. F., 1996. CoO % peso 3.0 +/- 0.2 1 – 10 MoO3 % peso 12.5 +/- 0.5 5 – 30 Na2O % peso 0.06 máx Fósforo % peso 1.6 máx 0 – 10 SiO2 % peso 0 – 6 Área superficial M2/Kg 1.95E+05 2.64E+05 Volumen de poro M3/Kg 4.30E-05 5.20E-04
En la Tabla 3.4 se muestran las características del catalizador utilizado en la experimentación de la
HDS del DBT, también se presenta a manera de comparación el catalizador utilizado por
Varysselberghe V. & Froment G.F., 1996 en su trabajo de hidrodesulfuración del DBT. Como
podrá observarse, ambos catalizadores contienen CoMo en su composición, sin embargo, el
contenido de Mo y Co, tamaño de poro, así como el área superficial del catalizador reportado por
estos autores, es mayor al comparado con el catalizador seleccionado para la experimentación en
este trabajo. En el apéndice I, se muestra el procedimiento de activación y las reacciones
involucradas para convertir los óxidos u oxisulfuros metálicos de Co y Mo contenidos en el
catalizador a sulfuros metálicos, los cuales constituyen la especie catalíticamente activa.
3.3. SELECCIÓN DEL TIPO DE DISOLVENTE El tipo de disolvente juega un papel importante en la experimentación. La Tabla 3.5 muestra los
diferentes disolventes que pueden ser utilizados en la experimentación. Sin embargo, ninguno de
estos pudo ser considerado en la experimentación realizada en este trabajo de tesis, debido a que
el hexadecano resulta ser muy costoso y el Parapur (mezcla de parafinas normales) a las
condiciones de operación experimentales (alrededor de T=613 K, P=6.0 MPa) tiene una alta
relación vapor/hidrocarburo.
En la Tabla 3.5, se reporta la temperatura de equilibrio físico de diferentes disolventes a una
presión de 6.0 MPa, calculada a una vaporización del 75 % (para una relación 3 molar H2/Hc) que
es aproximadamente la que se maneja en los reactores industriales de hidrotratamiento. Puede
observarse que solamente aceites parafínicos puede ser utilizados para trabajar en el intervalo de
condiciones de operación que permita emular aquellas que se tienen en las plantas industriales.

PROCEDIMIENTOS EXPERIMENTALES
41
Tabla 3.5. Temperaturas de equilibrio para diferentes solventes.
Solvente Temperatura equilibrio
(K) Tetralina 442
Decalina 448
Hexadecano 598
Parapur 558
Mezcla parafinita 585
Aceite Mineral 779
Aceite Mineral 85 NF 743
Aceite Mineral 350 757
3.4. MÉTODOS ANALÍTICOS
Cualquier modelo predictivo, basado en un modelo cinético requiere que este sea desarrollado en
base a datos confiables para después este pueda ser usado como una herramienta segura de
análisis de un proceso. Para determinar si los resultados obtenidos en la planta piloto son
confiables, es necesario contar con la caracterización y cuantificación de los compuestos presentes
en las corrientes que se encuentran en la planta. A su vez estos se utilizan en los balances de
masa y de átomos de estas corrientes y de este modo se puede obtener el error global que se tiene
en los resultados de la planta. Por otro lado, en la experimentación de la hidrodesulfuración de
compuestos modelo se utiliza aceite mineral como disolvente, el cual está constituido por una
mezcla de parafinas libre de compuestos aromáticos, azufrados y nitrogenados. Para este tipo de
sustancias las técnicas que actualmente se tienen desarrolladas en el IMP, no aplican, debido a
que no tienen la capacidad de caracterizar y cuantificar por componente este tipo de mezclas. Por
lo que fue necesario desarrollar una metodología basada en la destilación simulada para
caracterizar por número de átomos de carbono las corrientes líquidas de la planta. Adicionalmente
se desarrolló la técnica por medio de cromatografía de líquidos de alta eficiencia (CLAE) para
determinar y cuantificar los compuestos de productos de la reacción de hidrodesulfuración de
dibenzotiofeno como son el bifenilo, el ciclohexilbenceno y el dibenzotiofeno no reaccionado.
Ambas técnicas se desarrollaron en el Laboratorio de Química del Petróleo del IMP y se describen
más adelante en este capítulo.

Capítulo 3
42
Por otro lado en la técnica para la determinación y cuantificación de los compuestos en las
corrientes en fase gaseosa, se utiliza un cromatógrafo de gases equipado con dos detectores: un
detector de conductividad térmica (TCD) en donde se determina el H2S e H2 y el detector de
ionización de llama (FID) donde se identifican los hidrocarburos. Esta técnica es muy conocida y es
la que se utiliza en el área de planta piloto, sin embargo fue necesario modificar esta técnica y
desarrollar una nueva metodología la cual incluye un programa con el movimiento de las válvulas
del cromatógrafo para poder analizar las dos corrientes gaseosas que salen de la planta (gas
amargo y gas flasheado) en línea y pasar estas a través de los dos detectores. En la aplicación de
esta técnica también incluye un compresor a la salida del cromatógrafo el cual ya ha sido descrito
en este capítulo.
3.4.1. Cromatografía de Líquidos de Alta Eficiencia
En el caso particular del producto de la hidrodesulfuración de compuestos modelo (DBT), se tiene
el problema de que las muestras a las que se les quiere determinar el contenido de estos
componentes tienen como matriz aceite mineral que es inmiscible con los disolventes polares. Sin
embargo la condición para poder cuantificar cualquier componente por CLAE es que éste sea
soluble en la fase móvil, por lo cual se necesitan extraer a los componentes de interés antes de su
cuantificación.
El hidrotratamiento de dibenzotiofeno (DBT) produce vía desulfuración directa (hidrogenólisis)
bifenilo (BPH) y por medio de la hidrogenación-hidrogenólisis el ciclohexilbenceno (CHB) y
dependiendo de las condiciones de operación el diciclohexilo (DCH) puede también ser producido.
Estos cuatro compuestos tienen diferente polaridad, por lo que es posible separarlos y
cuantificarlos por Cromatografía de Líquidos de Alta Eficiencia (CLAE) usando una columna de tipo
apolar y como fase móvil un disolvente polar (agua, metanol, acetonitrilo o mezcla de ellos). Para
cumplir con esta tarea se preparó una solución extractora a base de acetonitrilo para separar los
compuestos de interés. En la preparación de las disoluciones se utilizaron: aceite mineral 85 NF,
acetonitrilo Omnisolv grado cromatográfico (Alemania), agua Omnisolv grado cromatográfico
(Alemania), bifenilo Aldrich, 99.5 % de pureza (EUA), ciclohexilbenceno Aldrich, 98 % de pureza
(EUA), dibenzotiofeno Aldrich, más de 99 % de pureza (EUA).
Una observación importante esta relacionada con el nivel de absorción de energía ultravioleta del
DBT, BPH y CHB por lo que es fácil detectarlos usando un detector UV-Visible.
Desafortunadamente, no es el caso del DCH, que no presenta esta propiedad, por lo que no se

PROCEDIMIENTOS EXPERIMENTALES
43
puede detectar bajo estas condiciones. De esta manera el DCH no fue cuantificado por esta
técnica.
A continuación se hace una descripción de la separación y cuantificación de DBT, BPH y CHB en
muestras cuya matriz es aceite mineral.
Para la separación se usó un cromatógrafo de líquidos de alta eficiencia equipado con un sistema
de control de bombas Waters modelo 600 E, detector de arreglo de diodos Waters modelo 996
ajustado a 234 nm e inyector manual tipo Rhèodyne con bucle de inyección de 20 µL. Los datos
fueron procesados por medio del software Millenium 32 de Waters. La columna utilizada fue 3.9 x
150 mm, empacada con partículas de C8 Symmetry de Waters, con tamaño de partícula de 5 µm.
Como fase móvil se usó mezcla acetonitrilo – agua en una proporción 65:35 (v/v) y el flujo utilizado
fue de 1 mL/min. Cada análisis se realizó por triplicado.
Para las curvas de calibración, en un matraz aforado de 10 mL se pesaron alrededor de 0.045 g de
BPH, 0.035 g de CHB y 0.02 g de DBT, se añadió 1 mL de tolueno y se aforo con aceite mineral, a
manera a obtener una solución base madre (SM) con 4.5 mg/mL de BPH, 3.5 mg/mL de CHB y
2mg/mL de DBT. De esta SM se tomaron diferentes alícuotas (10 a 100 µL) y se aforaron a 10 mL
con mezcla acetonitrilo – tetrahidrofurano (ACN - THF) 90:10 (v/v). Cada solución preparada se
analizó por CLAE. Para las muestras, se tomaron 10 a 100 µL de la muestra a analizar y se
aforaron a 10 mL con mezcla ACN – THF 90:10 (v/v). La disolución final se analizó por CLAE.
El primer paso a seguir en la determinación analítica fue disolver un poco de cada uno de los
solutos a analizar en la fase móvil utilizada e inyectarlos en el cromatógrafo usando como fase
móvil diferentes mezclas de acetonitrilo y agua. Se inició utilizando 60 % de acetonitrilo tomando
en cuenta de que las muestras a analizar están en aceite mineral. En la Figura 3.8 se presentan
los resultados obtenidos. Aquí se observa que usando un porcentaje de acetonitrilo menor a 60 %,
se obtiene una buena separación, lo mismo que con una mezcla acetonitrilo – agua en una
proporción 65:35 (v/v), pero al aumentar el porcentaje de acetonitrilo arriba del 70 %, el BPH y el
DBT ya no se separan. Como resultado se decidió usar una fase móvil de acetonitrilo – agua en
proporción 65:35 (v/v). En la Figura 3.9 se presenta un cromatograma obtenido con esta fase
móvil.
Capítulo 3
44
0
2
4
6
8
10
55 60 65 70 75
% ACN
TR (min)
BPH CHB DBT
Figura 3.8. Tiempo de retención en función del porcentaje de ACN en la fase móvil para BPH, DBT y CHB disueltos en la fase móvil utilizada.
El siguiente paso consistió en extraer estos compuestos, ya que en las muestras a analizar están
en aceite mineral y éste al ser completamente inmiscible con la fase móvil, no puede inyectarse
directamente, ya que dañaría la columna, además de que no se obtendrían resultados confiables.
Se hicieron varias pruebas de solubilidad y se vio que estos compuestos son muy poco solubles en
aceite mineral (es necesario calentarlo para disolverlos), muy poco solubles en agua, solubles en
acetonitrilo y muy solubles en tetrahidrofurano (THF). Considerando que para el análisis, estos
compuestos deben estar disueltos en la fase móvil, se hicieron extracciones con acetonitrilo y
mezclas de éste con la menor cantidad posible de THF. Por otro lado, tomando en cuenta las
concentraciones estimadas en que estarían en las muestras, sería necesaria una fuerte dilución.
Así, se decidió tomar un volumen de 0 a 500 µL de muestra y extraer con 10 mL de disolvente. De
estas pruebas se concluyó que los mejores resultados se obtienen usando una mezcla ACN – THF
en una proporción 90:10 (v/v) y un volumen de muestra máximo de 100 µL.
Teniendo estas condiciones se preparó una solución base (madre) de aceite mineral conteniendo
el bifenilo, ciclohexilbenceno y dibenzotiofeno y a partir de esta solución se hicieron diferentes
diluciones tomando 10, 30, 50, 70 y 100 µL de solución y aforando a 10 mL con mezcla ACN –
THF en una proporción 90:10 (v/v). Para que se disolvieran todos los compuestos, antes de aforar
se tuvo que añadir tolueno al medio. Este disolvente no afecta a las condiciones de separación
encontradas.

PROCEDIMIENTOS EXPERIMENTALES
45
Figura 3.9. Cromatograma de los estándares de BPH, DBT y CHB disueltos en ACN-agua 65:35 (V/v) a las condiciones cromatográficas descritas en el texto.
En la Figura 3.10 se muestra la curva de calibración obtenida para cada compuesto.
0.0E+00
2.0E+06
4.0E+06
6.0E+06
8.0E+06
1.0E+07
- 10.0 20.0 30.0 40.0 50.0
Concentración [mg/ml]
Area[pA*s]
BPH
CHB
DBT
Figura 3.10. Curva de calibración para la determinación de la composición
del BPH, CHB y DBT

Capítulo 3
46
La concentración determinada por el análisis cromatográfico se expresa en masa/volumen, ya que
lo que se toma en cuenta durante el análisis es el volumen. La concentración en masa/masa se
calcula haciendo la conversión por medio de la densidad de la muestra problema, que se
considerará la misma que para el aceite mineral (0.857 g/mL) la cual fue determinada en el
laboratorio central del IMP por el método ASTM D-70.
3.4.2. Destilación Simulada
Durante el proceso de hidrodesulfuración (HDS) del dibenzotiofeno (DBT), se obtienen los
siguientes productos; bifenilo (BPH), ciclohexilbenceno (CHB) y diciclohexilo (DCH), todos ellos con
diferencia significativa en su punto de ebullición, que permite separarlos y cuantificarlos por
cromatografía de gases utilizando el método de destilación simulada ASTM D–2887.
Destilación simulada es el término utilizado para designar los resultados obtenidos por
cromatografía de gases equivalentes a los calculados por destilación física. Ambas técnicas sirven
para determinar la distribución en intervalos de puntos de ebullición para naftas, fracciones de
petróleo y crudos. El método ASTM D-2887 se emplea para fracciones y productos del petróleo con
puntos de ebullición entre 493 y 811 K (220°C y 538°C).
El método de destilación simulada utiliza la cromatografía de gases, pero no en la forma
convencional de lograr una separación óptima de los componentes de una mezcla. Por el contrario,
las condiciones cromatográficas se seleccionan de tal manera que la resolución y eficiencia de la
separación de los componentes de la mezcla este limitada.
Este método se fundamenta considerando que los hidrocarburos se separan (eluyen) en columnas
cromatográficas no polares, en un orden acorde con sus puntos de ebullición. Debido a la
regularidad de este orden de elusión, los tiempos de retención pueden ser correlacionados con la
temperatura de ebullición, cuya representación gráfica es lineal. La correlación empírica entre
tiempo de retención y punto de ebullición se establece mediante una mezcla de n-parafinas que
cubra el intervalo de ebullición deseado.
La respuesta acumulada del detector para cada intervalo de ebullición, es proporcional a la
cantidad destilada en ese intervalo y es equivalente al porcentaje de esa respuesta con respecto a
la respuesta total de la mezcla analizada.

PROCEDIMIENTOS EXPERIMENTALES
47
La baja resolución de los componentes de la mezcla hace que la especie que realmente eluye en
cualquier instante sea una mezcla de muchos componentes con similar punto de ebullición. Por lo
tanto, se considera que su respuesta al detector este representado por un componente promedio al
cual se le ha asignado un factor de respuesta igual a la unidad.
La solución base (matriz) que se utilizó para el proceso de hidrodesulfuración del dibenzotiofeno
fue aceite mineral ligero, que sufre una desintegración de moléculas durante el proceso de HDS.
Para poder dar seguimiento a este proceso, se realizaron experimentos con el aceite mineral ligero
sin el compuesto modelo (DBT) a las mismas condiciones de reacción que aquellas utilizadas en
el tratamiento del aceite mineral ligero mezclado con DBT.
Las muestras de productos de reacción se obtuvieron para un intervalo de condiciones de
operación, (ver Tabla 3.6) a las cuales es posible establecer un modelo cinético fundamental para
la hidrodesulfuración del DBT.
Tabla 3.6. Condiciones de operación para obtención de productos de hidrodesulfuración
Temperatura (K)* LHSV (h-1)*
553 5.02 y 7.26
593 2.00
623 2.00
643 2.00 * Todos los experimentos fueron llevados a cabo a presión de 7.85 MPa
A continuación se describirá la cuantificación de dibenzotiofeno (DBT), bifenilo (BPH),
ciclohexilbenceno (CHB) y diciclohexilo (DCH) en las muestras cuya matriz es aceite mineral
usando una modificación de la técnica de destilación simulada.
3.4.2.1. Procedimiento Analítico
En la preparación de las disoluciones se utilizó: disulfuro de carbono Aldrich grado
espectrofotometría al 99+% pureza (EUA). Para comprobar la identidad de los productos de HDS
se usaron: bifenilo Aldrich, 99.5 % de pureza (EUA), ciclohexilbenceno Aldrich, 98 % de pureza
(EUA), diciclohexilo Aldrich, 99 % de pureza (EUA), dibenzotiofeno Aldrich, más de 99 % de pureza
(EUA).

Capítulo 3
48
Para obtener la curva de calibración se utilizó un estándar de una mezcla de n-parafinas de C5 a
C40 con los puntos de ebullición que se muestran en la Tabla 3.7.
Para validar los resultados se utilizó una muestra de referencia, SIMDIS D 2887 Lote No.40400
Parte No. 25650.150.
Para la determinación de la concentración por destilación simulada del dibenzotiofeno y los
productos de la hidrodesulfuración, se utilizó:
• Un cromatógrafo de gases Hewlett Packard modelo HP 6890 equipado con detector de
ionización de llama programado a una temperatura de 623 K, puerto de inyección splitless
ajustado a una temperatura de 523 K y un inyector automático serie 7683.
• Una columna HP-1 (methyl Siloxane) con espesor de película de 2.65 µm, diámetro interno de
0.53 mm y longitud de 5 metros. El gas de arrastre utilizado fue helio. Los datos fueron
procesados por medio del software Chemstation y un programa de temperatura iniciando con
288 K en el horno, seguido por un incremento de 1.5 K por minuto hasta alcanzar 623 K.
Con el objeto de identificar el orden de elusión del DBT y los productos de la HDS, se inyectaron
soluciones de estos compuestos grado reactivo para determinar sus tiempos de retención.
En la Tabla 3.7 se puede observar los compuestos por número de átomos de carbono y sus
respectivos puntos de ebullición que contiene el estándar que se utilizó para construir la curva de
calibración de los hidrocarburos del aceite mineral, el cual se inyectó 1 µL de una disolución de la
mezcla de n- parafinas desde C5 hasta C40.
Antes de analizar las muestras experimentales se inyecta un blanco (disulfuro de carbono) para
verificar la estabilidad de la línea base y una disolución en disulfuro de carbono de una muestra de
referencia certificada a la que se le han determinado sus intervalos de puntos de ebullición por
destilación física, ver Tabla 3.8.
PROCEDIMIENTOS EXPERIMENTALES
49
Tabla 3.7. Estándar de una mezcla de n-Parafinas para la validación del método.
No. de
átomos de carbono
Punto de ebullición
(K)
No. de átomos de carbono
Punto de ebullición
(K)
No. de átomos de carbono
Punto de ebullición
(K)
C5 309 C13 508 C24 664
C6 342 C14 527 C26 685
C7 371 C15 544 C28 704
C8 399 C16 560 C30 722
C9 424 C17 575 C32 739
C10 447 C18 589 C34 754
C11 469 C20 617 C36 769
C12 489 C22 642 C38 782
C40 795
Tabla 3.8. Puntos de ebullición para muestra de referencia SIMDIS D 2887 Lote No.40400 Parte No. 25650.150.
Destilación Física Destilación Simulada
% peso Punto de Ebullición (K) Punto de Ebullición (K)
0.05 414.0 416.5
10 481.0 481.0
20 508.0 507.0
30 527.0 526.5
40 544.0 543.0
50 560.0 558.0
60 576.0 574.0
70 592.0 589.0
80 611.0 607.5
90 633.0 630.0
99.9 671.0 667.0
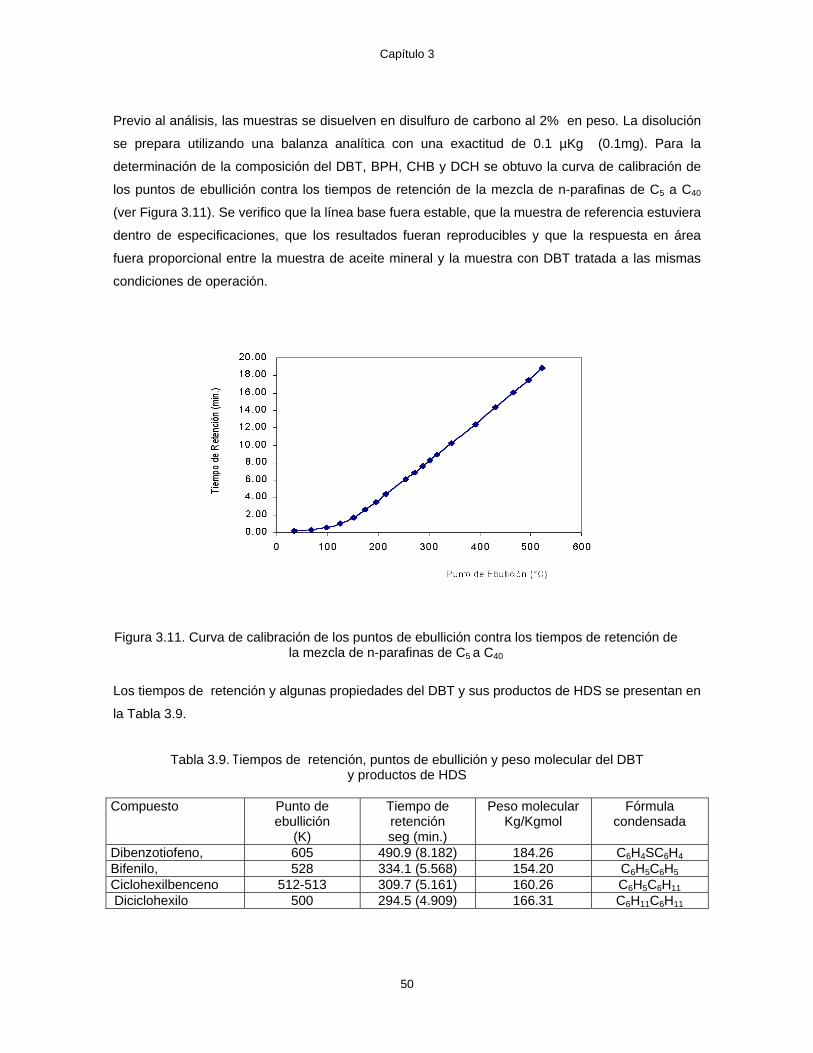
Capítulo 3
50
Previo al análisis, las muestras se disuelven en disulfuro de carbono al 2% en peso. La disolución
se prepara utilizando una balanza analítica con una exactitud de 0.1 µKg (0.1mg). Para la
determinación de la composición del DBT, BPH, CHB y DCH se obtuvo la curva de calibración de
los puntos de ebullición contra los tiempos de retención de la mezcla de n-parafinas de C5 a C40
(ver Figura 3.11). Se verifico que la línea base fuera estable, que la muestra de referencia estuviera
dentro de especificaciones, que los resultados fueran reproducibles y que la respuesta en área
fuera proporcional entre la muestra de aceite mineral y la muestra con DBT tratada a las mismas
condiciones de operación.
Figura 3.11. Curva de calibración de los puntos de ebullición contra los tiempos de retención de
la mezcla de n-parafinas de C5 a C40
Los tiempos de retención y algunas propiedades del DBT y sus productos de HDS se presentan en
la Tabla 3.9.
Tabla 3.9. Tiempos de retención, puntos de ebullición y peso molecular del DBT y productos de HDS
Compuesto Punto de
ebullición (K)
Tiempo de retención seg (min.)
Peso molecular Kg/Kgmol
Fórmula condensada
Dibenzotiofeno, 605 490.9 (8.182) 184.26 C6H4SC6H4 Bifenilo, 528 334.1 (5.568) 154.20 C6H5C6H5 Ciclohexilbenceno 512-513 309.7 (5.161) 160.26 C6H5C6H11 Diciclohexilo 500 294.5 (4.909) 166.31 C6H11C6H11

PROCEDIMIENTOS EXPERIMENTALES
51
Se inyectaron las disoluciones de las muestras de aceite mineral con y sin dibenzotiofeno
obtenidas a cada una de las condiciones de reacción y se integraron por intervalos de tiempos de
retención correspondientes al número de átomos de carbono, de acuerdo a la curva de calibración
anteriormente determinada (ver Tabla 3.10).
Tabla 3.10. Intervalos de tiempo de retención correspondientes al número de átomos de carbono, de acuerdo a la curva de calibración
Número de carbones Intervalo de Tiempo de Retención
Segundos Minutos
C7 y C8 27.8 a 96.2 0.463 a1.603
C9 96.2 a 144.4 1.603 a 2.407
C10 144.4 a 196.9 2.407 a 3.282
C11 196.9 a 249.4 3.282 a 4.157
C12 249.4 a 298.2 4.157 a 4.970
C13 298.2 a 319.9 4.970 a 5.332
C14 319.9 a 371.9 5.332 a 6.198
C15 371.9 a 435.7 6.198 a 7.262
C16 435.7 a 478.5 7.262 a 7.975
C17 478.5 a 519.5 7.975 a 8.658
C18 y C19 519.5 a 594.6 8.658 a 9.910
C20 + 594.6 > > 9.910
El tiempo de retención y el área respectiva de los picos contenidos en la curva de calibración se
muestra en la Tabla 3.11.
Las Figura 3.12 a 3.15 muestran el análisis cromatográfico obtenido por destilación simulada para
las muestras de aceite mineral sin reaccionar, el aceite mineral hidrotratado a 643 K, el aceite
mineral como carga (conteniendo dibenzotiofeno) y el producto de la hidrodesulfuración de
dibenzotiofeno. Los datos de la Figura 3.12 muestran el área de los picos con respecto al tiempo
de retención correspondiendo con moléculas en el rango de 10 – 36 átomos de carbono. El
hidrotratamiento del aceite mineral a una temperatura de 643 K produce moléculas de
hidrocarburos ligeros (ver Figura 3.13). El aceite mineral sufre una desintegración térmica de las
moléculas contenidas en las fracciones pesadas de este. Por medio de esta misma técnica el

Capítulo 3
52
dibenzotiofeno agregado al aceite mineral (carga) también puede determinarse cualitativamente y/o
cuantitativamente dado que el pico correspondiente a este compuesto eluye de manera separada
de los hidrocarburos que conforman el aceite mineral (Figura 3.14). La Figura 3.15 muestra los
productos de la hidrodesulfuración (DBT, BPH, CHB, y DCH) y la desintegración del aceite mineral
para una temperatura de 643 K. Los picos correspondientes a los productos de la
hidrodesulfuración pueden ser cualitativa- o -cuantitativamente determinados.
Tabla 3.11. Resultados de los productos de reacción reportados por
el cromatógrafo de gases con la técnica de destilación simulada.
No. Pico T inicial Area (pA*s) Segundos Minutos
1 1.56 0.026 2,934.60 2 27.78 0.463 448.07 3 55.38 0.923 409.28 4 96.18 1.603 342.59 5 144.40 2.407 266.27 6 196.92 3.282 218.15 7 249.42 4.157 256.61 8 298.20 4.970 1,243.89 9 319.92 5.332 3,523.68
10 371.88 6.198 2,925.51 11 435.72 7.262 6,448.76 12 478.50 7.975 11,256.75 13 519.36 8.658 23,905.33 14 594.60 9.910 278,965.88
En la determinación y la cuantificación de los compuestos parafínicos por número de átomos de
carbono, se observó que los productos principales de la reacción de hidrodesulfuración (DBT, BPH,
CHB y DCH) en las diferentes muestras eluyen al mismo tiempo que algunas parafinas que tienen
semejantes puntos de ebullición. Para poder realizar la cuantificación de cada una de las especies
contenidas en la mezcla, se consideró que el factor de respuesta de detección era igual a uno para
los productos de reacción. En este caso la integración se realizó por bloques, considerando los
intervalos de tiempo en los que cada familia eluye. Debe aclararse que, cada familia esta integrada
por moléculas con el mismo número de átomos de carbono.

PROCEDIMIENTOS EXPERIMENTALES
53
Figura 3.12. Cromatograma del aceite mineral 85 NF sin reaccionar
Figura 3.13. Cromatograma obtenido del aceite mineral hidrotratado a 643 K
Figura 3.14. Cromatograma para la carga a hidrodesulfuración (aceite mineral más 2.09 %peso
de dibenzotiofeno)

Capítulo 3
54
Figura 3.15. Cromatograma obtenido por destilación simulada para el producto de hidrodesulfuración tratado a 643 K.
La composición en por ciento en peso de cada componente del esquema de reacción se determinó
con la diferencia de áreas obtenida al analizar el aceite mineral sin reaccionar y el aceite mineral
con dibenzotiofeno hidrotratado a diferentes condiciones de operación (ver Tabla 3.12).
Tabla 3.12. Composición en por ciento peso de los hidrocarburos del aceite mineral, del DBT y los
productos de la HDS a 553 K y LHSV de 5.02 h-1
Tiempo de retención
(min)
No de átomos de carbono
Aceite mineral más DBT ( % peso)
Aceite mineral sin DBT
(% peso)
Diferencia (% peso)
0.923 C7 y C8 0.260 0.277 -.009 1.603 C9 0.104 0.102 0.002 2.407 C10 0.081 0.084 -.003 3.282 C11 0.066 0.083 -.017 4.157 C12 y DCH 0.078 0.094 -.017 4.970 C13 y DCH 0.377 0.062 0.314 5.332 C14 y BPH 1.067 0.224 0.843 6.198 C15 0.886 0.930 -.044 7.262 C16 1.953 1.987 -.034 7.975 C17 y Dibenzotiofeno 3.409 2.945 0.464 8.658 C18 y C19 7.239 7.218 0.022 9.910 C20 + 84.481 83.903 0.579

PROCEDIMIENTOS EXPERIMENTALES
55
El porcentaje en peso de los hidrocarburos contenidos en el aceite mineral y para cada uno de los
productos de la HDS del DBT de las muestras analizadas se presenta en la Tabla 3.13. Esta Tabla
presenta la composición de las muestras de aceite mineral conteniendo los productos de la
hidrodesulfuración a una temperatura de 553 K y dos diferentes LHSV (5.02 y 7.26 h-1).
Tabla 3.13. Concentración en peso para las muestras analizadas por destilación simulada
Temperatura No. de átomos de carbono
Carga ( % peso)
553 K* ( % peso)
553 K**(% peso)
593 K*
(% peso) 623 K*
(% peso) 643 K**
(% peso) C7 y C8 0.1717 0.260 0.306 0.275 0.513 0.920 C9 0.044 0.104 0.131 0.117 0.240 0.489 C10 0.023 0.081 0.111 0.100 0.253 0.560 C11 0.013 0.066 0.097 0.098 0.309 0.700 C12 0.021 0.078 0.094 0.104 0.182 0.521 Diciclohexilo 0.000 0.000 0.011 0.115 0.370 0.481 C13 0.025 0.062 0.062 0.069 0.113 0.279 Ciclohexilbenceno 0.000 0.314 0.247 0.763 0.721 0.753 C14 0.162 0.224 0.224 0.241 0.344 0.760 Bifenilo 0.000 0.843 0.778 0.646 0.529 0.537 C15 – C17 5.841 5.784 5.916 5.498 6.582 8.068 Dibenzotiofeno 1.925 0.464 0.665 0.319 0.084 0.033 C18 y C19 7.188 7.239 7.217 7.138 7.533 7.910 C20 + 84.587 84.481 84.139 84.517 82.227 77.988 Total 100.000 100.000 100.000 100.000 100.000 100.000
*muestra tratada a un LHSV de 5.02 h-1 **muestra tratada a un LHSV de 7.26 h-1
Finalmente en la Tabla 3.14 se presenta una comparación de la determinación del azufre total en
las muestras analizadas entre el método de prueba por espectroscopia de fluorescencia por
dispersión de energía de rayos “X” (IMP-QP03) y el método de destilación simulada. Por
experiencia acumulada se sabe, que la técnica IMP-QP03 tiene un mayor grado de confiabilidad
en la determinación de azufre total.
Los datos de esta Tabla muestran que la determinación de azufre total por el método IMP-QP03
resulta ser mayor en todos los casos considerados. Esta diferencia puede deberse a varios
factores que son:
La diferencia debida a la incertidumbre que presenta el método en el intervalo de temperatura
considerado, esto es, 89 ppm de azufre a la temperatura de 553 K (280°C) y de 16ppm a 643 K
(370°C).

Capítulo 3
56
El H2S disuelto en las muestras fue detectado por la primera técnica y no así por el método de
destilación simulada dado que estas se analizaron un tiempo después y el H2S disuelto en
estas se evaporó.
Tabla 3.14. Comparación de la determinación del azufre total en las muestras analizadas. Por los
métodos IMP-QP03 y el de destilación simulada
Método Temperatura (K)
% Peso 553 553 643
IMP-QP03 0.0999 0.1390 0.0103
SIMDIS 0.0806 0.1157 0.0057
Se observó (como podrá verse más adelante en el análisis de resultados) que el azufre molecular
juega un papel muy importante, debido a que esta molécula es muy sensible a cualquier cambio de
composición en el balance de átomos. Por lo que será necesario realizar la determinación del ácido
sulfhídrico disuelto en el producto líquido de forma inmediata por medio de la técnica de azufre total
IMP-QP03, una vez obtenida y sellada la muestra en la planta piloto.
3.4.3. Análisis de las corrientes gaseosas En la planta piloto de hidrotratamiento de destilados intermedios es necesario determinar la
composición por componente de las tres corrientes gaseosas involucradas a saber:
Hidrógeno de carga
Gas amargo producto
Gas flasheado
Las tres corrientes están constituidas de hidrocarburos ligeros que van desde C1 a C6, además del
hidrógeno y ácido sulfhídrico. La metodología más comúnmente utilizada para determinar la
composición de estas fases es por medio de cromatografía de gases. Para lo cual se utilizó la
técnica para análisis de gases de refinería y los métodos cromatográficos HDT(HC).M y
HDT(H2S).M implantado en el área de plantas piloto del IMP. Para realizar este análisis se utilizó
un cromatógrafo de gases marca HP modelo 5890 con las características reportadas en la Tabla
3.15. Este equipo esta constituido por dos detectores: un detector de ionización de llama (FID
siglas en inglés) y un detector de conductividad térmica (TCD siglas en Inglés). El FID determina y
cuantifica los hidrocarburos presentes en la corriente y el TCD hace lo mismo pero con el ácido

PROCEDIMIENTOS EXPERIMENTALES
57
sulfídrico e hidrógeno. Con estos dos detectores se cuantifica totalmente la corriente gaseosa que
pasa a través del cromatógrafo.
Tabla 3.15. Características del equipo utilizado para la cromatografía de gases.
Equipo
Columnas
Detectores Front : Poraplot Q, 25m, 0.32 mm, 10 um (Chrompack) Hayesep Q, 80/100,
1900 1A-Q01
Front TCD Cromatógrafo de
Gases HP 5890 Back : Al2O3 / KCl , 50 m, 0.32 mm, 5
um (Chrompack) Front TCD Back FID
El cromatógrafo de gases utiliza el programa de temperatura mostrado en la Figura 3.16. En esta
Figura se puede observar que la temperatura inicial es de 313 K (40°C), mantenida a 300 s
(5 minutos), para después incrementarse a una velocidad de 0.25 K/s (15°C/min), hasta alcanzar
la temperatura final de 473 K (200°C) la cual permanecerá así durante 5 minutos hasta detenerse
el programa y llegar a la temperatura inicial.
40 ºC
5 min15 ºC/min
200 ºC
5min
40 ºC
5 min15 ºC/min
200 ºC
5min
Fig. 3.16. Programa de temperatura utilizado en la determinación de la composición de las corrientes gaseosas.
Las válvulas multipuerto del cromatógrafo se accionan con el programa descrito en la Tabla 3.16.
La representación gráfica de las válvulas del cromatógrafo puede observarse en la Figura 3.17, en
esta Figura se presenta la secuencia a seguir para analizar las corrientes gaseosas. El gas se
inyecta en la válvula V-1, en donde una parte del gas se elimina como purga para limpiar la línea,
pasa a la válvula V-2 la cual envía el gas al detector TCD. Al tiempo 75 s (1.25 minutos) se inyecta
a la válvula V-3 para una limpieza antes de ser inyectado al detector FID hasta la válvula V-4.
Las características de la pureza y presión de operación de los gases utilizados en la operación del
cromatógrafo suministrados por Infra Air Products están reportadas en la Tabla 3.17.
Tabla 3.16. Funcionamiento de las válvulas del cromatógrafo
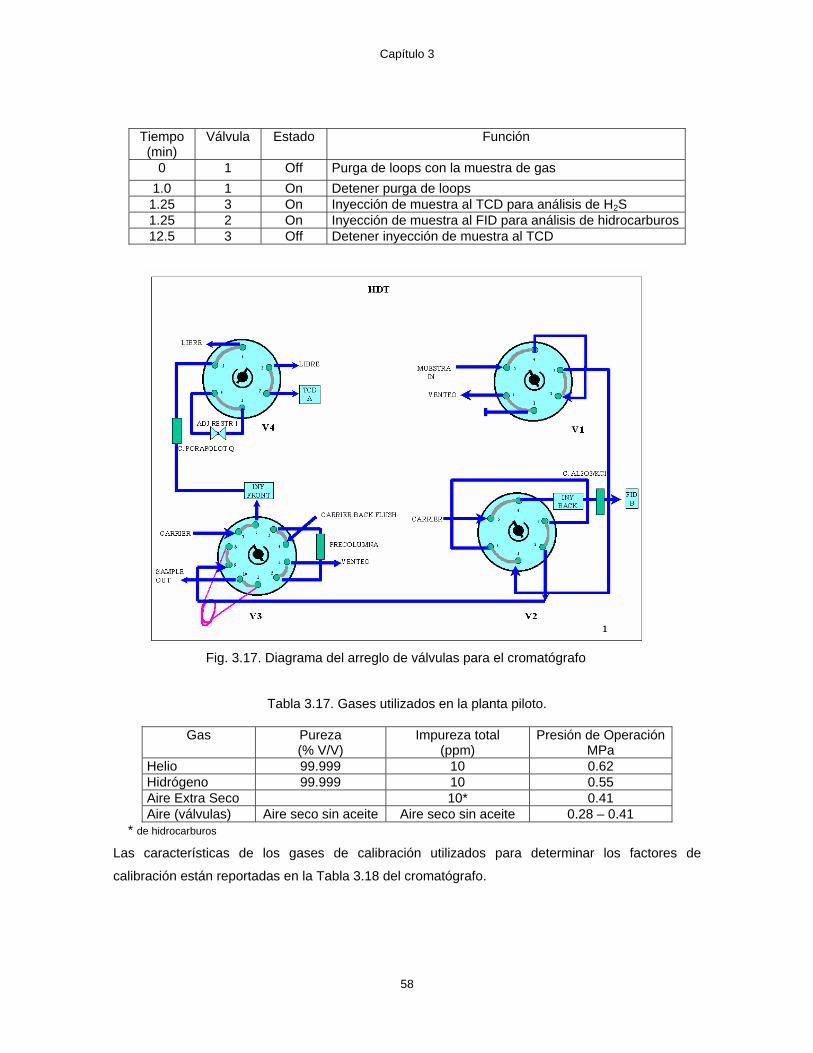
Capítulo 3
58
Tiempo (min)
Válvula Estado Función
0 1 Off Purga de loops con la muestra de gas 1.0 1 On Detener purga de loops 1.25 3 On Inyección de muestra al TCD para análisis de H2S 1.25 2 On Inyección de muestra al FID para análisis de hidrocarburos12.5 3 Off Detener inyección de muestra al TCD
Fig. 3.17. Diagrama del arreglo de válvulas para el cromatógrafo
Tabla 3.17. Gases utilizados en la planta piloto.
Gas Pureza (% V/V)
Impureza total (ppm)
Presión de Operación MPa
Helio 99.999 10 0.62 Hidrógeno 99.999 10 0.55 Aire Extra Seco 10* 0.41 Aire (válvulas) Aire seco sin aceite Aire seco sin aceite 0.28 – 0.41
* de hidrocarburos
Las características de los gases de calibración utilizados para determinar los factores de
calibración están reportadas en la Tabla 3.18 del cromatógrafo.
PROCEDIMIENTOS EXPERIMENTALES
59
Tabla 3.18. Características de los gases para cromatografía realizada en planta piloto.
Compuestos Utilizados Marca
Mezcla Grado Certificado de H2S al 1%, Balance N2
Praxair
Mezcla Grado Certificado de H2S al 5%, Balance N2
Praxair
Mezcla Grado Certificado de H2S al 6%, Balance H2
U.S. Gas Standards INC.
Mezcla estándar de hidrocarburos (% mol)
Air Products
La composición de la mezcla de hidrocarburos utilizada como estándar en el área de planta piloto
se muestra en Tabla 3.19 siguiente:
Tabla 3.19. Composición de la mezcla de hidrocarburos
Componente
% mol
Componente
% mol
Metano 16.80 Trans-2-Buteno 3.96 Etano 11.74 1-Buteno 3.96 Etileno 4.00 i-Butileno 3.96 Propano 13.65 Cis-2-Buteno 3.92 Propileno 5.89 i-Pentano 1.98 i-Butano 11.72 n-Pentano 1.98 Propadieno 1.79 1,3-Butadieno 5.81 n-Butano 7.84 Hexano 0.99
La cantidad inyectada de hidrocarburos y ácido sulfhídrico fue de 0.4 mL/min considerando un
tiempo de purga de 60 s (1min). Además se utilizaron dos mezclas estándares de H2S: La mezcla
de H2S al 3% se preparo en un embolo (1.0 L) a partir de la mezcla al 6% H2S utilizando N2 como
disolvente. La mezcla de H2S al 0.5% se preparo en un embolo (1.0 L) a partir de la mezcla al 1%
H2S utilizando N2 como disolvente.


ANÁLISIS DE LOS DATOS EXPERIMENTALES
61
4. ANÁLISIS DE LOS DATOS EXPERIMENTALES En este capítulo se establece la metodología para la validación de la información experimental
obtenida en la planta piloto, continuando con la caracterización de las corrientes de entrada y
salida para conocer el nivel de hidrodesulfuración del DBT utilizado como compuesto modelo y
también la composición de los productos de reacción tanto en la fase líquida como en las fases
gaseosa y flasheada. Un análisis estadístico se realiza para conocer la dependencia entre las
variables de operación y finalmente se define el tipo de expresión de velocidad que representa la
trayectoria de reacción de la hidrodesulfuración .
4.1. METODOLOGÍA DE ANÁLISIS DE LAS CORRIENTES DE LA PLANTA PILOTO
En esta sección se considera el análisis de las corrientes de entrada y salida de la planta piloto del
hidrotratamiento de destilados medios, con el objeto de validar cada experimento a través del
balance de los átomos involucrados en el procesamiento.
La Figura 4.1 muestra esquemáticamente la planta piloto utilizada en la hidrodesulfuración de
dibenzotiofeno (DBT). En esta Figura se muestran las corrientes de entrada y salida en donde se
observa que es posible conocer los flujos de DBT, hidrógeno y aceite mineral a la entrada del
reactor. El efluente del reactor previamente pasa a la sección de separación en donde se obtienen
3 corrientes: la de gas amargo (rica en hidrocarburos ligeros, ácido sulfhídrico e hidrógeno), el
líquido flasheado (libre de hidrógeno) y el gas flasheado (ácido sulfhídrico e hidrógeno).
Estas corrientes de entrada y salida se caracterizan a fin de conocer por un lado el nivel de
hidrodesulfuración desde la corriente líquida y por otro la composición de H2S y gases ligeros
distribuidos en las corrientes gaseosa y de gas flasheado.
FASE GAS La corriente gaseosa esta constituida de un gas amargo el cual consiste de una mezcla de
hidrocarburos, ácido sulfhídrico e hidrógeno. La composición y flujo de esta corriente se calcula a
través de las mediciones en la planta y el análisis cromatográfico el cual se realiza con los
detectores de conductividad térmica TCD e ionización de llama FID.
El análisis en el TCD determina la composición de hidrógeno y H2S, mientras que la composición
de los hidrocarburos ligeros se determina por medio del FID. Una vez conocida esta composición y
con la ayuda del simulador de procesos PRO-II utilizando una ecuación de estado (Peng-Robinson)

Capítulo 4
62
se determina el peso molecular y densidad de esta corriente. De esta manera el flujo volumétrico
medido en las corrientes de la planta se puede convertir a flujo másico y/o molar. Una vez
conocidos estos flujos molares globales es posible, calcular el flujo molar para cada componente
dentro de la mezcla utilizando la siguiente expresión:
FyiF gggi = 4.1
Figura 4.1 Diagrama Esquemático de la planta piloto
PRODUCTO LÍQUIDO Esta corriente la constituye la mezcla de hidrocarburos del aceite mineral, el dibenzotiofeno y los
aromáticos provenientes de la reacción de hidrodesulfuración. La determinación de la composición
de los primeros se realiza a través de la destilación simulada en un crómatografo de gases,
mientras que para dibenzotiofeno y los aromáticos se utiliza cromatografía en fase líquida CLAE.
Nuevamente con la composición de esta corriente y su flujo volumétrico medido en la planta piloto
es posible calcular el peso molecular y densidad para posteriormente determinar el flujo másico y/o
molar. Una vez realizada esta operación es posible calcular el correspondiente flujo molar para
cada componente en la mezcla de reacción por:

ANÁLISIS DE LOS DATOS EXPERIMENTALES
63
FxF 2l2l2i
2li = 4.2
GAS FLASHEADO
El corriente de gas flasheado esta constituida principalmente por hidrógeno con pequeñas
cantidades de hidrocarburos ligeros y ácido sulfihídrico, su composición se determina a través del
análisis cromatográfico con los detectores de conductividad térmica y de ionización de llama. El
análisis en el detector de ionización de llama produce la composición de los hidrocarburos ligeros
mientras que la composición de hidrógeno y ácido sulfihídrico se determina en el detector de
conductividad térmica. La composición de los hidrocarburos ligeros puede ser calculada utilizando
la siguiente ecuación:
FxF 1l1li
1li = 4.3
DETERMINACIÓN DEL FLUJO MOLAR TOTAL DE CADA COMPONENTE
El flujo molar total de cada hidrocarburo e hidrógeno en el efluente del reactor se determina como
la suma del flujo molar de cada hidrocarburo e hidrógeno calculado de las corrientes de gas, líquido
y gas flasheado:
FFFF gi
2li
1lii ++= 4.4
BALANCE DE ÁTOMOS DE HIDRÓGENO, AZUFRE Y CARBONO A manera de realizar una verificación rigurosa de los resultados experimentales, un balance de
átomos de carbono, hidrógeno y azufre se calculo para cada experimento entre las corrientes de
entrada y salida. Estos balances se basan en los flujos molares de cada componente. El calculo de
estos balances se realiza utilizando las siguientes ecuaciones:

Capítulo 4
64
FcFcFc lj
NCOM
2jj
gj
NCOM
2jj
oj
NCOM
2jj ∑+∑=∑
=== 4.5
FSFSFS lj
NCOM
2jj
gj
NCOM
2jj
oj
NCOM
2jj ∑+∑=∑
=== 4.6
)FF(2FhFhFhF2 g2H
l2H
lj
NCOM
2jj
gj
NCOM
2jj
oj
NCOM
2jj
o2H +⋅+∑+∑=∑+⋅
=== 4.7
La relación porcentual de los flujos molares a la salida y entrada de átomos de carbono, hidrógeno
y azufre se calcula utilizando las siguientes ecuaciones:
Fc
FcFc%100BAL
0j
NCOM
2jj
lj
NCOM
2jj
gj
NCOM
2jj
C∑
∑+∑⋅=
=
== 4.8
Fs
FsFs%100BAL
0j
NCOM
2jj
lj
NCOM
2jj
gj
NCOM
2jj
S∑
∑+∑⋅=
=
== 4.9
FhF2
)FF(2FhFh%100BAL
0j
NCOM
2jj
02H
g2H
l2H
lj
NCOM
2jj
gj
NCOM
2jj
H∑+⋅
+⋅+∑+∑⋅=
=
== 4.10
De manera ideal los valores de BALC, BALS y BALH deberían de estar alrededor del 100 %, sin
embargo, valores entre 95 y 105 % pueden ser tomados como aceptables.
4.2. ANALISIS DE LAS CORRIENTES A LA SALIDA DE LA PLANTA PILOTO
El análisis de los resultados experimentales de la hidrodesulfurización del DBT se realizó en dos
etapas, la primera tuvo por objetivo validar los resultados que se obtuvieron en la planta piloto de
reciente construcción tomando como base al dibenzotiofeno como compuesto modelo, y el aceite
mineral NS-85 como disolvente, cuyas características se encuentran reportadas en el informe
interno del IMP de la construcción de la planta piloto [Arzate E. y Muñoz Arroyo J.A., 2003]. Para la
ANÁLISIS DE LOS DATOS EXPERIMENTALES
65
experimentación se utilizaron condiciones de operación similares a las reportadas por el grupo de
Froment G. F., 1996 (T = 553 K y P = 7.84 MPa a diferentes LHSV).
El objetivo de la segunda etapa fue realizar el hidrotratamiento de los compuestos modelo a
condiciones de operación cercanas a las industriales a fin de reunir información experimental para
ser utilizada en el desarrollo de un modelo cinético para la hidrodesulfuración de dibenzotiofeno
como compuesto modelo.
La Tabla 4.1 muestra un resumen de las condiciones de operación utilizadas en la etapa I, así
como también, el balance de materia y la evaluación de los experimentos vía el balance de átomos
entre la carga y productos.
El sistema de reacción (reactor Robinson Mahoney) presenta a la salida tres corrientes de
productos (ver Figura 4.1), una corriente líquida constituida principalmente por dibenzotiofeno no
reaccionado, bifenilo, ciclohexilbenceno, diciclohexilo, H2S en pequeñas cantidades y el disolvente
(aceite mineral), la corriente de gas dada por el gas amargo rico en hidrógeno, H2S e hidrocarburos
ligeros y una corriente de gas flasheado constituido también principalmente por hidrógeno, H2S,
hidrocarburos ligeros y una pequeña fracción de hidrocarburos más pesados.
Tabla 4.1. Hidrotratamiento de dibenzotiofeno: a diferentes LHSV, P=7.84 MPa y T=553 K , balance de materia y % de error en el balance de átomos
entre la carga y productos
No. Experimento 1 2 3 Temperatura K 553 553 553
Presión MPa 7.84 7.84 7.84
LHSV h-1 5.20 6.20 8.20 H2/HC mol/mol 1.30 1.33 1.33 Flujo de Carga µKg/seg /(g/h) 25 / 92 15.8 / 57 25 / 92 Flujo de H2 entrada µKg/seg /(g/h) 0.234 / 0.85 0.2 / 0.72 0.22 / 0.78 Flujo Total de entrada µKg/seg (g/h) 25.8 / 92 15.8 / 57 25.8 / 92 Flujo Efluente líquido µKg/seg (g/h) 25.8 / 92 15.6 / 56 25.3 / 91 Flujo de Gas Amargo µKg/seg (g/h) 0.36 / 1.29 0.30 / 1.07 0.31 / 1.11 Flujo Flash Gas nKg/seg (g/h) 33.3 / 0.12 44.4 / 0.16 33.3 / 0.12 Flujo Total de Salida µKg/seg (g/h) 25.8 / 93 15.8 / 57 25 / 92 Error en masa % 4.81 0.84 0.59 Error en átomos de C % 1.95 -2.17 -1.44 Error en átomo de H % 5.06 -4.15 2.48 Error en átomos de S % 4.66 3.7 0.88
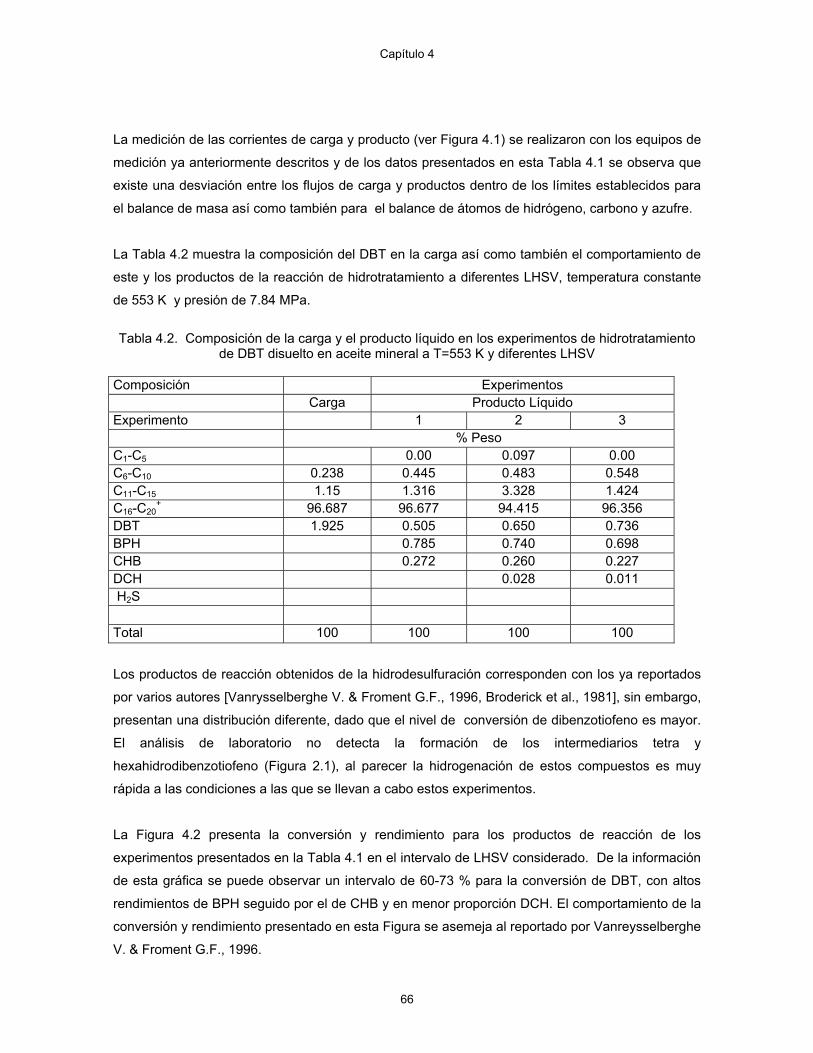
Capítulo 4
66
La medición de las corrientes de carga y producto (ver Figura 4.1) se realizaron con los equipos de
medición ya anteriormente descritos y de los datos presentados en esta Tabla 4.1 se observa que
existe una desviación entre los flujos de carga y productos dentro de los límites establecidos para
el balance de masa así como también para el balance de átomos de hidrógeno, carbono y azufre.
La Tabla 4.2 muestra la composición del DBT en la carga así como también el comportamiento de
este y los productos de la reacción de hidrotratamiento a diferentes LHSV, temperatura constante
de 553 K y presión de 7.84 MPa.
Tabla 4.2. Composición de la carga y el producto líquido en los experimentos de hidrotratamiento
de DBT disuelto en aceite mineral a T=553 K y diferentes LHSV
Composición Experimentos Carga Producto Líquido Experimento 1 2 3 % Peso C1-C5 0.00 0.097 0.00 C6-C10 0.238 0.445 0.483 0.548 C11-C15 1.15 1.316 3.328 1.424 C16-C20
+ 96.687 96.677 94.415 96.356 DBT 1.925 0.505 0.650 0.736 BPH 0.785 0.740 0.698 CHB 0.272 0.260 0.227 DCH 0.028 0.011 H2S Total 100 100 100 100
Los productos de reacción obtenidos de la hidrodesulfuración corresponden con los ya reportados
por varios autores [Vanrysselberghe V. & Froment G.F., 1996, Broderick et al., 1981], sin embargo,
presentan una distribución diferente, dado que el nivel de conversión de dibenzotiofeno es mayor.
El análisis de laboratorio no detecta la formación de los intermediarios tetra y
hexahidrodibenzotiofeno (Figura 2.1), al parecer la hidrogenación de estos compuestos es muy
rápida a las condiciones a las que se llevan a cabo estos experimentos.
La Figura 4.2 presenta la conversión y rendimiento para los productos de reacción de los
experimentos presentados en la Tabla 4.1 en el intervalo de LHSV considerado. De la información
de esta gráfica se puede observar un intervalo de 60-73 % para la conversión de DBT, con altos
rendimientos de BPH seguido por el de CHB y en menor proporción DCH. El comportamiento de la
conversión y rendimiento presentado en esta Figura se asemeja al reportado por Vanreysselberghe
V. & Froment G.F., 1996.
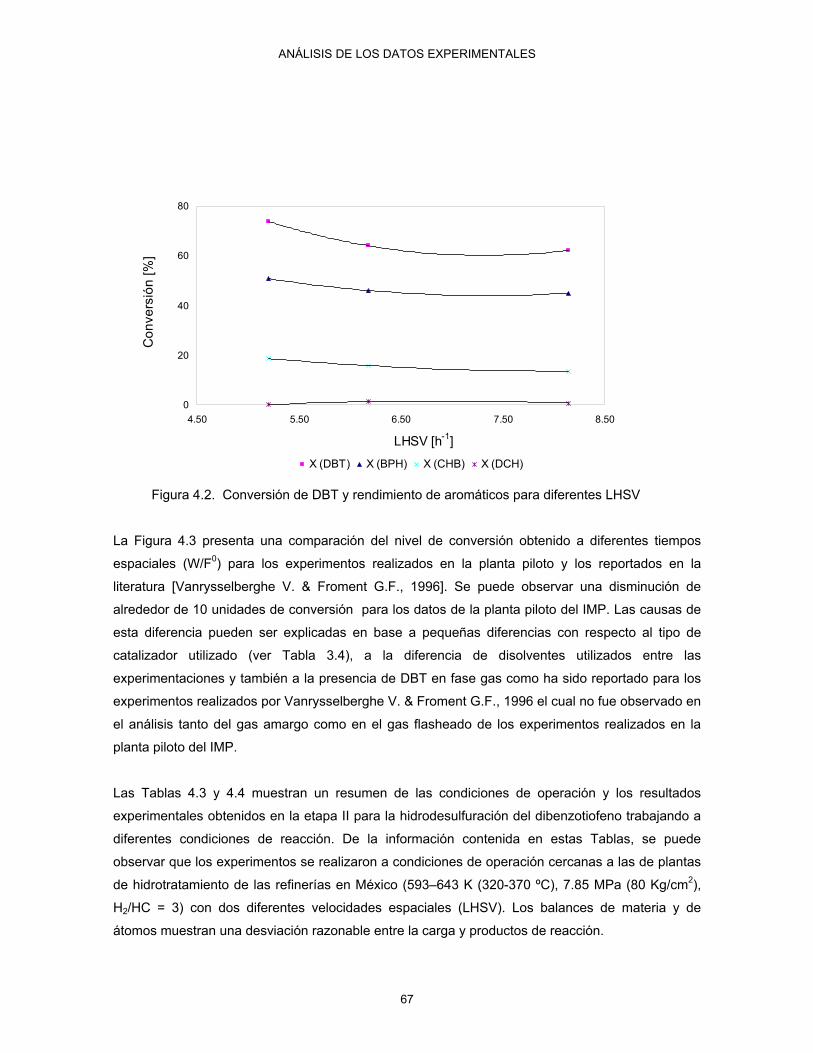
ANÁLISIS DE LOS DATOS EXPERIMENTALES
67
0
20
40
60
80
4.50 5.50 6.50 7.50 8.50
LHSV [h-1]
Con
vers
ión
[%]
X (DBT) X (BPH) X (CHB) X (DCH)
Figura 4.2. Conversión de DBT y rendimiento de aromáticos para diferentes LHSV
La Figura 4.3 presenta una comparación del nivel de conversión obtenido a diferentes tiempos
espaciales (W/F0) para los experimentos realizados en la planta piloto y los reportados en la
literatura [Vanrysselberghe V. & Froment G.F., 1996]. Se puede observar una disminución de
alrededor de 10 unidades de conversión para los datos de la planta piloto del IMP. Las causas de
esta diferencia pueden ser explicadas en base a pequeñas diferencias con respecto al tipo de
catalizador utilizado (ver Tabla 3.4), a la diferencia de disolventes utilizados entre las
experimentaciones y también a la presencia de DBT en fase gas como ha sido reportado para los
experimentos realizados por Vanrysselberghe V. & Froment G.F., 1996 el cual no fue observado en
el análisis tanto del gas amargo como en el gas flasheado de los experimentos realizados en la
planta piloto del IMP.
Las Tablas 4.3 y 4.4 muestran un resumen de las condiciones de operación y los resultados
experimentales obtenidos en la etapa II para la hidrodesulfuración del dibenzotiofeno trabajando a
diferentes condiciones de reacción. De la información contenida en estas Tablas, se puede
observar que los experimentos se realizaron a condiciones de operación cercanas a las de plantas
de hidrotratamiento de las refinerías en México (593–643 K (320-370 ºC), 7.85 MPa (80 Kg/cm2),
H2/HC = 3) con dos diferentes velocidades espaciales (LHSV). Los balances de materia y de
átomos muestran una desviación razonable entre la carga y productos de reacción.
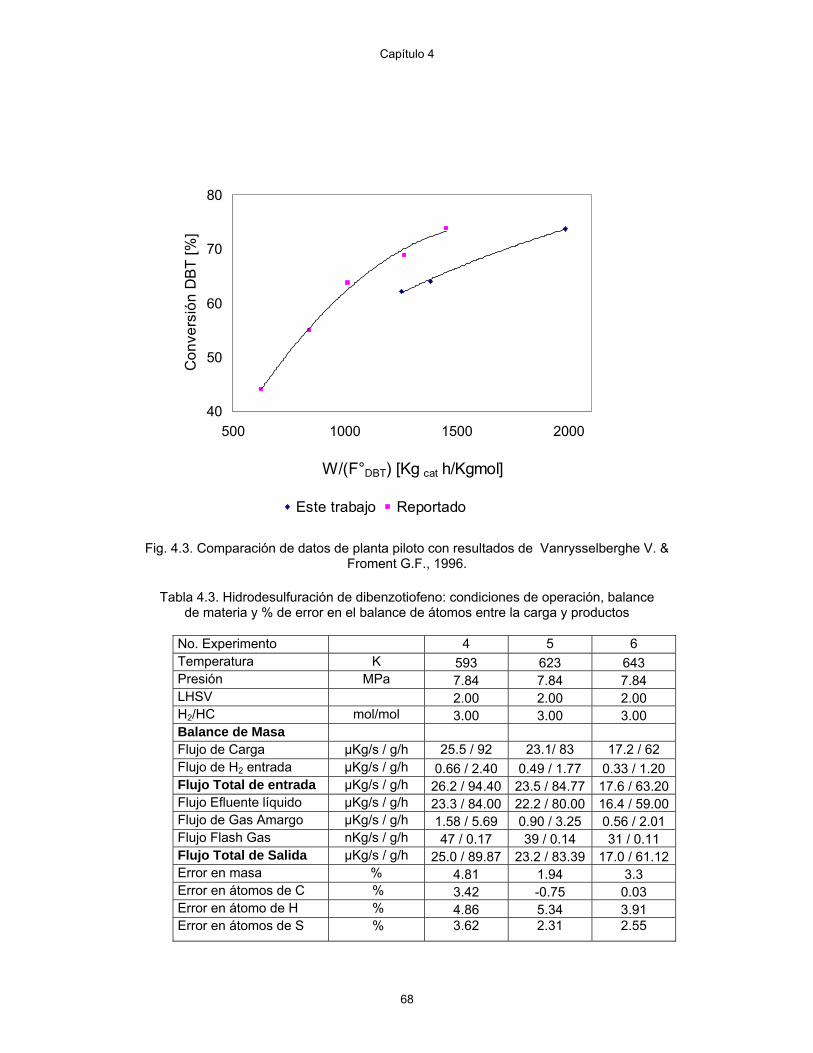
Capítulo 4
68
40
50
60
70
80
500 1000 1500 2000
W/(F°DBT) [Kg cat h/Kgmol]
Con
vers
ión
DBT
[%]
Este trabajo Reportado
Fig. 4.3. Comparación de datos de planta piloto con resultados de Vanrysselberghe V. & Froment G.F., 1996.
Tabla 4.3. Hidrodesulfuración de dibenzotiofeno: condiciones de operación, balance
de materia y % de error en el balance de átomos entre la carga y productos
No. Experimento 4 5 6 Temperatura K 593 623 643 Presión MPa 7.84 7.84 7.84 LHSV 2.00 2.00 2.00 H2/HC mol/mol 3.00 3.00 3.00 Balance de Masa Flujo de Carga µKg/s / g/h 25.5 / 92 23.1/ 83 17.2 / 62 Flujo de H2 entrada µKg/s / g/h 0.66 / 2.40 0.49 / 1.77 0.33 / 1.20 Flujo Total de entrada µKg/s / g/h 26.2 / 94.40 23.5 / 84.77 17.6 / 63.20Flujo Efluente líquido µKg/s / g/h 23.3 / 84.00 22.2 / 80.00 16.4 / 59.00Flujo de Gas Amargo µKg/s / g/h 1.58 / 5.69 0.90 / 3.25 0.56 / 2.01 Flujo Flash Gas nKg/s / g/h 47 / 0.17 39 / 0.14 31 / 0.11 Flujo Total de Salida µKg/s / g/h 25.0 / 89.87 23.2 / 83.39 17.0 / 61.12Error en masa % 4.81 1.94 3.3 Error en átomos de C % 3.42 -0.75 0.03 Error en átomo de H % 4.86 5.34 3.91 Error en átomos de S % 3.62 2.31 2.55

ANÁLISIS DE LOS DATOS EXPERIMENTALES
69
Tabla 4.4. Hidrodesulfuración de dibenzotiofeno: condiciones de operación, balance de materia y % de error en el balance de átomos entre la carga y productos
No. Experimento 7 8 9 Temperatura K (°C) 593 623 643 Presión MPa (Kg/Cm2) 7.84 7.84 7.84 LHSV 3.00 3.00 3.00 H2/HC mol/mol 3.00 3.00 3.00 Balance de Masa Flujo de Carga µKg/s / g/h 18.1 / 65 23.1/ 83 17.5 / 63 Flujo de H2 entrada µKg/s / g/h 0.40 / 1.45 0.48 / 1.74 0.47 / 1.49 Flujo Total de entrada µKg/s / g/h 18.5 / 66 23.5 / 84 17.9 / 64 Flujo Efluente líquido µKg/s / g/h 17.5 / 63 22.5 / 81 16.9 / 61 Flujo de Gas Amargo µKg/s / g/h 0.41 / 1.48 0.41 / 1.48 0.60 / 2.15 Flujo Flash Gas nKg/s / g/h 55 / 0.20 11 / 0.04 50 / 0.18 Flujo Total de Salida µKg/s / g/h 18.0 / 64 23.2 / 83 17.6 / 63 Error en masa % 2.65 1.60 1.78 Error en átomos de C % 0.44 -0.47 0.53 Error en átomo de H % 3.23 0.51 -3.16 Error en átomos de S % -2.36 1.31 -1.50
En las Tablas 4.5 y 4.6 se muestran las composiciones de la carga y los productos de reacción
para los experimentos 4 a 9, las cuales corresponden a las condiciones de operaciones reportadas
en las Tablas 4.3 y 4.4, esto es, a intervalos de temperatura de 593 a 643 K y dos diferentes LHSV
(2,3). La comparación de estas Tablas muestra claramente que a una mayor temperatura y una
disminución en el LHSV, la conversión del DBT se incrementa. También puede observarse el
comportamiento que tiene las reacciones consecutivas del bifenilo y el ciclohexilbenceno, al
aumentar y disminuir su composición con respecto a la temperatura.
Para estos resultados experimentales, la Figura 4.4 muestra el comportamiento de la conversión
del DBT y el rendimiento de los productos como una función de la temperatura de reacción. De la
información contenida en esta Figura se observa que a baja temperatura el principal producto de la
hidrodesulfuración de dibenzotiofeno corresponde al bifenilo (BPH), seguido por el
ciclohexilbenceno (CHB) y por lo que respecta al biciclohexilo (BCH) este no se produce. Un
aumento en la temperatura provoca que el BPH disminuya y este se convierta a CHB promoviendo
también la formación de DCH. El comportamiento observado en esta Figura corresponde a las
reacciones consecutivas mostradas en el esquema de reacción de la Figura 2.1. El nivel de
conversión de DBT se incrementa con la temperatura alcanzándose la máxima a 643 K (370 ºC).

Capítulo 4
70
0
20
40
60
80
100
530 550 570 590 610 630 650Temperatura (K)
Con
vers
ión
[%]
DBT BPH CHB DCH
Figura 4.4. Comportamiento del DBT y los productos de la HDS con la temperatura, a un LHSV de 2, ∗ DBT, ∆ BPH, ♦ CHB, ■ DCH
Tabla 4.5. Composición de la carga y el producto líquido en los experimentos de hidrotratamiento de DBT disuelto en aceite mineral
Composición Experimentos Carga Producto Líquido Experimento 4 5 6 % Peso C1-C10 0.238 0.4920 1.006 1.969 C11-C15 1.15 1.4460 2.239 4.238 C16-C20+ 96.687 96.6345 95.049 92.624 DBT 2.186 0.1250 0.084 0.007 BPH 0.5930 0.529 0.262 CHB 0.5930 0.721 0.419 BCH 0.1150 0.37 0.481 H2S 0.0015 0.002 0.003 Total 100 100 100 100
Con respecto a las corrientes gaseosas (gas amargo y gas flasheado), las Tablas 4.7 y 4.8
muestran la composición de estas para la etapa II. Los datos reportados en estas Tablas
corresponden con la composición de H2S, H2 e hidrocarburos ligeros en un intervalo de
temperatura entre 593- 643 K (320-370°C) para dos LHSV respectivamente. Los datos de estas
Tablas, no muestran una clara tendencia del comportamiento del H2, H2S y gases ligeros. El
hidrógeno se mantiene en un rango de composición de alrededor del 97 y 80% para el gas amargo
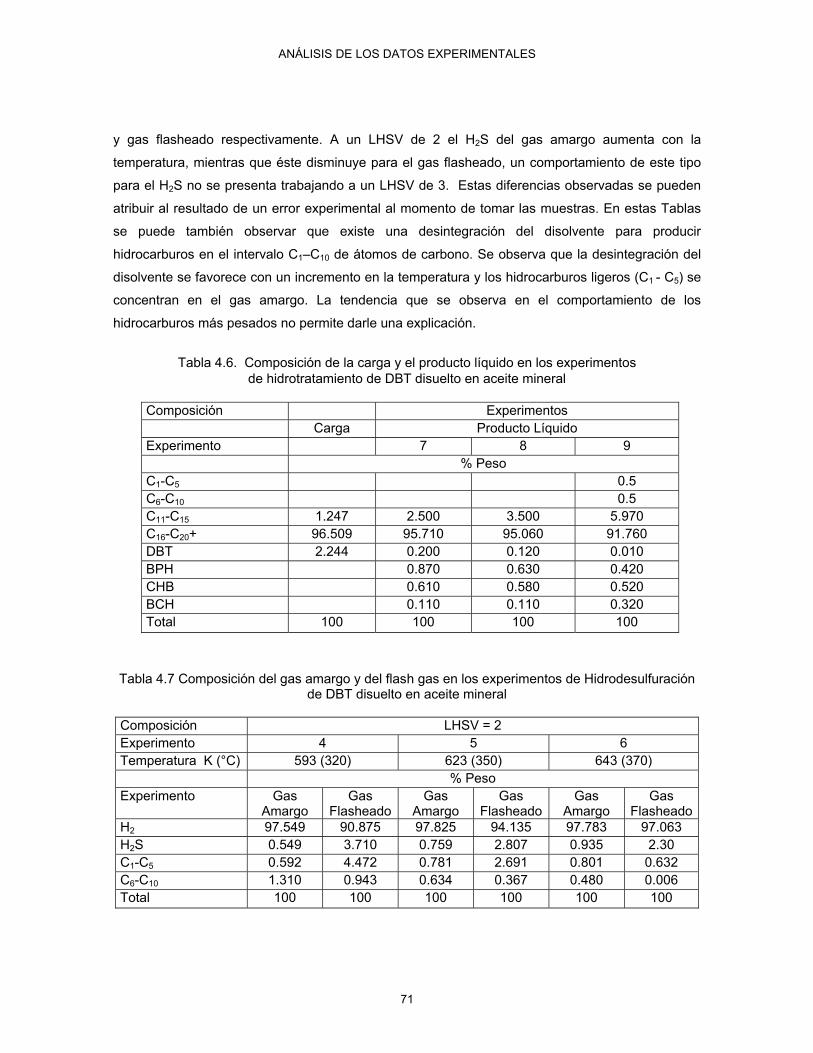
ANÁLISIS DE LOS DATOS EXPERIMENTALES
71
y gas flasheado respectivamente. A un LHSV de 2 el H2S del gas amargo aumenta con la
temperatura, mientras que éste disminuye para el gas flasheado, un comportamiento de este tipo
para el H2S no se presenta trabajando a un LHSV de 3. Estas diferencias observadas se pueden
atribuir al resultado de un error experimental al momento de tomar las muestras. En estas Tablas
se puede también observar que existe una desintegración del disolvente para producir
hidrocarburos en el intervalo C1–C10 de átomos de carbono. Se observa que la desintegración del
disolvente se favorece con un incremento en la temperatura y los hidrocarburos ligeros (C1 - C5) se
concentran en el gas amargo. La tendencia que se observa en el comportamiento de los
hidrocarburos más pesados no permite darle una explicación.
Tabla 4.6. Composición de la carga y el producto líquido en los experimentos
de hidrotratamiento de DBT disuelto en aceite mineral
Composición Experimentos Carga Producto Líquido Experimento 7 8 9 % Peso C1-C5 0.5 C6-C10 0.5 C11-C15 1.247 2.500 3.500 5.970 C16-C20+ 96.509 95.710 95.060 91.760 DBT 2.244 0.200 0.120 0.010 BPH 0.870 0.630 0.420 CHB 0.610 0.580 0.520 BCH 0.110 0.110 0.320 Total 100 100 100 100
Tabla 4.7 Composición del gas amargo y del flash gas en los experimentos de Hidrodesulfuración
de DBT disuelto en aceite mineral
Composición LHSV = 2 Experimento 4 5 6 Temperatura K (°C) 593 (320) 623 (350) 643 (370) % Peso Experimento Gas
Amargo Gas
FlasheadoGas
Amargo Gas
FlasheadoGas
Amargo Gas
FlasheadoH2 97.549 90.875 97.825 94.135 97.783 97.063 H2S 0.549 3.710 0.759 2.807 0.935 2.30 C1-C5 0.592 4.472 0.781 2.691 0.801 0.632 C6-C10 1.310 0.943 0.634 0.367 0.480 0.006 Total 100 100 100 100 100 100
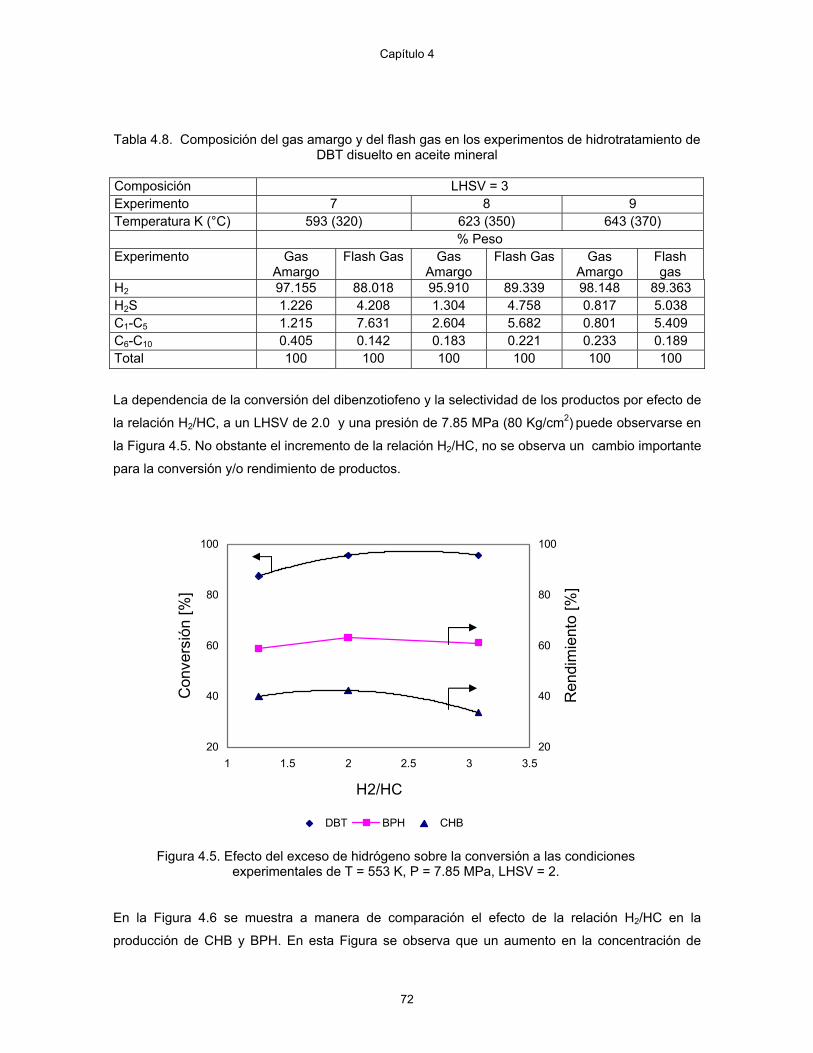
Capítulo 4
72
Tabla 4.8. Composición del gas amargo y del flash gas en los experimentos de hidrotratamiento de DBT disuelto en aceite mineral
Composición LHSV = 3 Experimento 7 8 9 Temperatura K (°C) 593 (320) 623 (350) 643 (370) % Peso Experimento Gas
Amargo Flash Gas Gas
Amargo Flash Gas Gas
Amargo Flash gas
H2 97.155 88.018 95.910 89.339 98.148 89.363 H2S 1.226 4.208 1.304 4.758 0.817 5.038 C1-C5 1.215 7.631 2.604 5.682 0.801 5.409 C6-C10 0.405 0.142 0.183 0.221 0.233 0.189 Total 100 100 100 100 100 100
La dependencia de la conversión del dibenzotiofeno y la selectividad de los productos por efecto de
la relación H2/HC, a un LHSV de 2.0 y una presión de 7.85 MPa (80 Kg/cm2) puede observarse en
la Figura 4.5. No obstante el incremento de la relación H2/HC, no se observa un cambio importante
para la conversión y/o rendimiento de productos.
20
40
60
80
100
1 1.5 2 2.5 3 3.5
H2/HC
Con
vers
ión
[%]
20
40
60
80
100
Ren
dim
ient
o [%
]
DBT BPH CHB
Figura 4.5. Efecto del exceso de hidrógeno sobre la conversión a las condiciones experimentales de T = 553 K, P = 7.85 MPa, LHSV = 2.
En la Figura 4.6 se muestra a manera de comparación el efecto de la relación H2/HC en la
producción de CHB y BPH. En esta Figura se observa que un aumento en la concentración de

ANÁLISIS DE LOS DATOS EXPERIMENTALES
73
hidrógeno en el sistema de reacción provoca un aumento en la conversión del DBT. La producción
del BPH en todos los casos es mayor que la producción del CHB. La Figura 4.6 muestra las
selectividades del CHB y del BPH en la hidrodesulfuración del DBT con respecto a la concentración
de hidrógeno.
20
40
60
80
1 1.5 2 2.5 3 3.5H2/Hc
Xi /
XDB
T
CHB BPH
Figura 4.6. Selectividad para la hidrogenación del ciclohexilbenceno y bifenilo en función de la relación H2/HC a las condiciones experimentales de T = 553 K, P = 7.85 MPa, LHSV = 2.
Una mayor selectividad hacia la producción de BPH puede ser observada en esta Figura dentro del
intervalo de relación H2/HC considerado. Este resultado es el esperado, dado que con
catalizadores a base CoMo/Al2O3 la reacción de hidrodesulfuración vía hidrogenólisis (DDS) se
favorece en comparación con las reacciones de hidrogenación.
4.3. ANÁLISIS ESTADÍSTICO DE LOS RESULTADOS
De los resultados obtenidos en la experimentación se realizó el análisis estadístico de las variables
que interactúan. La secuencia del análisis consistió en primero revisar los factores significativos
que afectan la conversión y el rendimiento de los productos. Por lo que se consideraron primero los
resultados de la etapa experimental II, en la cual se trabaja a moderadas condiciones de operación
(baja temperatura) obteniéndose niveles medios de la conversión de dibenzotiofeno.
Para obtener la panorámica de cada una de las variables independientes en la conversión del DBT
y los rendimientos de los productos (XBPH y XCHB), se realizó un análisis preliminar univariable de
esta etapa, para una presión de 7.85 MPa y un LHSV de 2.0, cuyos valores se reportan en el

Capítulo 4
74
apéndice D. En este análisis se determinó la significancia entre cada par de variables. Esto es, la
relación H2/Hc en la conversión del dibenzotiofeno XDBT y en los rendimientos de los productos XBPH
y XCHB. Para conocer la influencia de la temperatura en el rendimiento de los productos se realizó
un análisis multivariable. Para realizar este se utilizaron los datos experimentales de la etapa I
considerando una variación de la temperatura a diferentes LHSV y relaciones H2/Hc (ver apéndice
D).
4.4. ANÁLISIS CUANTITATIVO DEL COMPORTAMIENTO DE LA VELOCIDAD DE REACCIÓN
En el proceso de hidrotrodesulfuración de DBT se llevan a cabo por dos diferentes trayectorias de
reacciones que son la hidrogenólisis y la hidrogenación. Estas reacciones ocurren en dos
diferentes sitios activos como ya ha sido reportado por Broderick D. H. and Gates B.C., 1981,
Vanrysselberghe V. et al., 1996. En base al esquema de reacción de la Figura 2.1 la velocidad neta
de producción Ri para el BPH y CHB en los dos sitios activos σ y τ puede definirse de la siguiente
manera:
rrR BPHDBTBPH τσ ,, −= 4.11
rrrR CHBBPHDBTCHB τττ ,,, −+= 4.12
Utilizando el mismo esquema de reacción la velocidad total de desaparición del dibenzotiofeno se
define de la siguiente manera:
rrR DBTDBTDBT τσ ,, += 4.13
Los valores experimentales de Ri son directamente obtenidos de la conversión y rendimiento
experimental Xi :
FXR
DBT
ii W °= 4.14
Los valores de la velocidad de reacción Ri se muestran en la Tabla 4.9, calculados a una
temperatura de 553 K, una presión de 7.85 MPa y H2/Hc igual a 3.0.

ANÁLISIS DE LOS DATOS EXPERIMENTALES
75
Tabla 4.9. Velocidades de reacción de los productos
LHSV (h -1)
W / F°DBT X DBT XBPH XCHB XDCH RDBT RBPH RCHB RDCH
8.15 1252 62.21 44.95 13.43 0.63 0.05 0.04 0.01 0.0005 6.18 1382 64.00 46.00 15.75 1.31 0.05 0.03 0.01 0.0010 5.20 1982 73.77 50.77 18.62 0.00 0.04 0.03 0.01 0.0000
La Figura 4.7 muestra la tendencia que tiene la velocidad de reacción con respecto al reciproco del
espacio – velocidad molar, F DBTW 0/ . Esta Figura confirma el comportamiento esperado para la
velocidad de remoción del DBT al disminuir el valor de F DBTW 0/ (menor LHSV). Una mayor
cantidad de catalizador permite obtener un mayor nivel de conversión.
0.030
0.035
0.040
0.045
0.050
0.055
1200 1400 1600 1800 2000 2200
W/FDBT [Kg cat h/Kgmol]
rDB
T [K
gmol
/Kg
cat]
Figura 4.7. Comportamiento de la velocidad de reacción del dibenzotiofeno como función del
espacio-velocidad molar
Al pasar de los años, se han hecho muchos intentos para determinar las expresiones de la
velocidad de reacción heterogénea como una función de la concentración de los reactivos. Un
cambio importante que se ha observado es que, estas velocidades de reacción heterogéneas no
se ajustan a una simple relación estequiométrica. Para reacciones en fase gas, las velocidades de
reacción son frecuentemente proporcionales a las concentraciones de los reactivos a algunas
potencias simples. Sin embargo, en reacciones catalíticas heterogéneas se siguen ecuaciones de

Capítulo 4
76
velocidad mucho más complejas. Es común para una reacción catalítica permanecer constante,
aumentar o disminuir cuando la concentración de algunos de los reactivos se incrementa. Esto es
un poco diferente para reacciones en fase gas, donde la velocidad generalmente se incrementa
con el aumento de la concentración del reactivo.
En la Figura 4.8 se muestra la rapidez de reacción para la aparición del BPH con respecto a la
concentración del DBT y BPH dado que estos compuestos intervienen en la trayectoria de
reacción. En esta Figura se observa que la rapidez de reacción se incrementa al aumentar la
concentración del reactivo y/o producto; sin embargo llega a un valor máximo y comienza a
disminuir. Este comportamiento permite visualizar que la forma de la expresión de la velocidad de
reacción, es del tipo Langmuir-Hinshelwood-Hougen-Watson. En este tipo de expresiones se
involucra un numerador y un denominador. Para el primero se incluye el término relativo a la
cinética intrínseca, mientras que en el segundo se toman en cuenta los parámetros de adsorción y
un exponente (causa del cambio de pendiente) que indica el número de sitios activos involucrados
en la reacción química.
0.020
0.025
0.030
0.035
0.040
0 0.001 0.002 0.003 0.004 0.005 0.006 0.007
Concentración [Kgmol/m3]
r BP
H [K
gmol
/Kg c
at]
C(DBT) C(BPH)
Figura 4.8. Comportamiento de la velocidad de reacción del BPH con respecto a la concentración del DBT y BPH @ T= 553 K, P = 7.85 MPa y H2/Hc = 3.0.

ANÁLISIS DE LOS DATOS EXPERIMENTALES
77
El comportamiento que sigue la rapidez de reacción del CHB con respecto a si mismo, DBT y BPH
se muestra en la Figura 4.9. De los datos de esta Figura se observa que la rapidez de reacción del
CHB presenta un comportamiento lineal tanto con el reactivo DBT como también para los
productos BPH y CHB. Sin embargo no ocurre de la misma manera para el mismo CHB, ya que la
rapidez de reacción disminuye al aumentar la concentración de este compuesto. De estos
resultados se puede inferir que también la expresión de velocidad para representar la rapidez de
reacción del CHB es de tipo Langmuir-Hinshelwood-Hougen-Watson.
0.90
1.00
1.10
1.20
0 1 2 3 4 5 6 7
Concentración x 103 [Kgmol/m3]
r CH
B x 1
02 [Kgm
ol/K
g cat
]
C(CHB) C(DBT) C(BPH)
Figura 4.9. Comportamiento de la velocidad de reacción del CHB con respecto a la concentración del CHB, DBT y BPH @ T = 553 K, P = 7.85 MPa y H2/Hc = 3.0.
Finalmente si analizamos la velocidad del DCH, con respecto a la concentración del DBT, BPH,
CHB y el mismo DCH (Figura 4.10), únicamente existe una disminución de la rapidez de reacción
cuando se incrementa la concentración de CHB y DCH dentro del esquema de reacción. El
comportamiento observado nos indica que la expresión de velocidad para representar la trayectoria
de reacción de este compuesto también es de tipo Langmuir-Hinshelwood-Hougen-Watson dado
que estos compuestos son los que intervienen directamente en el mecanismo de reacción. En esta
Figura no se observa una influencia importante por la adsorción del DBT y BPH en la velocidad de
reacción de la producción del DCH.
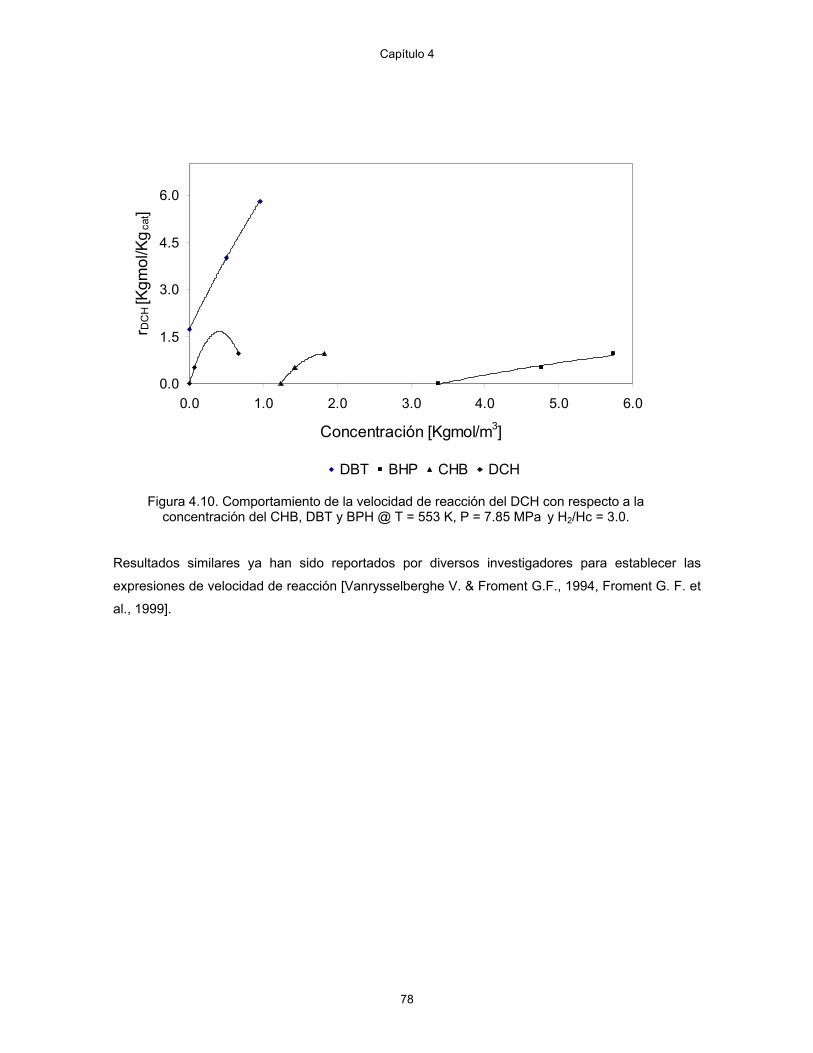
Capítulo 4
78
0.0
1.5
3.0
4.5
6.0
0.0 1.0 2.0 3.0 4.0 5.0 6.0
Concentración [Kgmol/m3]
r DC
H [K
gmol
/Kg c
at]
DBT BHP CHB DCH
Figura 4.10. Comportamiento de la velocidad de reacción del DCH con respecto a la concentración del CHB, DBT y BPH @ T = 553 K, P = 7.85 MPa y H2/Hc = 3.0.
Resultados similares ya han sido reportados por diversos investigadores para establecer las
expresiones de velocidad de reacción [Vanrysselberghe V. & Froment G.F., 1994, Froment G. F. et
al., 1999].

MODELAMIENTO CINÉTICO DE LOS DATOS EXPERIMENTALES
79
5. MODELAMIENTO DE LOS DATOS EXPERIMENTALES
5.1. Modelamiento Cinético de la Hidrodesulfuración de Dibenzotiofeno En un sistema físico el fenómeno que influye en este puede ser representado por una función
matemática. Una reacción química no es la excepción, ya que posee un mecanismo verdadero que
describe cada detalle microscópico de la reacción. En la práctica por supuesto la descripción
completa de este mecanismo no puede ser obtenido, pero una aproximación para éste puede ser
desarrollada. Por ejemplo, en un estudio fundamental del sistema de reacción catalítica
heterogénea, esto puede implicar el hacer uso de una propiedad promedio, tal como el tamaño
promedio del poro, del tamaño de la partícula del metal dispersado en el soporte o un promedio del
estado adsorbido de las moléculas.
Con respecto al modelamiento cinético se requiere información experimental precisa que
represente el esquema de reacción del proceso catalítico. Por lo que es posible obtener una
expresión matemática apropiada que represente la velocidad de reacción. La forma definitiva de
esta representación involucra la estimación de los parámetros para diversas expresiones de la
velocidad las cuales son el resultado del análisis de los diferentes mecanismos involucrados en el
esquema de reacción. Una vez definida la expresión de velocidad, se puede realizar de manera
precisa la estimación de parámetros cinéticos y de adsorción involucrados en esta.
La especificación del modelo de velocidad de reacción, se puede realizar en base a dos tipos
principales de expresiones de la velocidad de reacción. El primero de ellos representado por la ley
de acción de masas [Kittrell, J.R., 1996].
bB
aA CCkr = 5.1
y un modelo de tipo hiperbólico dado por las expresiones del tipo Langmuir-Hinshelwood-Hougen-
Watson [Kittrell, J.R, 1996]:
BBAA
BA
CKCKCCkr++
=1 5.2

Capítulo 5
80
La ecuación (5.1) establece que la velocidad de reacción es proporcional a la fuerza impulsora que
provoca la reacción química (concentración, presión parcial del (os) componente (s)). Esta
ecuación ha sido utilizada en muchos sistemas de reacción, particularmente aquellos de
considerable complejidad, considerando que la reacción se realiza en la superficie exterior de la
partícula catalítica y en el estado estacionario.
Este tipo de modelos ha sido aplicado a reacciones en fase gas (teoría de Lindermann), reacciones
enzimáticas (mecanismo de Michaelis–Menten), y reacciones gaseosas en superficies sólidas
[Kittrell, J.R. 1996]. Sin embargo este tipo de expresiones tiene ciertas limitaciones particularmente
en la interpretación de los datos experimentales que permitan explicar de manera fundamental los
efectos de los diferentes parámetros y condiciones a los que se lleva a cabo la reacción química.
Para incluir estos efectos se hace necesario utilizar ecuaciones de velocidad de reacción del tipo
Langmuir-Hinshelwood-Hougen-Watson (ec. 5.2).
En caso del hidrotratamiento de destilados intermedios, la hidrodesulfuración conforma un sistema
complejo de reacciones las cuales al ser bien modeladas pueden utilizarse en la formulación de un
modelo matemático el cual pueda aplicarse como una herramienta en la operación y normalización
de un proceso. Sin embargo el desarrollo de tal modelo no es una tarea fácil. Un programa
experimental debe llevarse a cabo tomando en cuenta una completa descripción de la composición
de la alimentación y de los productos hidrotratados a través del análisis químico para proporcionar
información segura y que permita establecer un modelo cinético confiable que describa la red de
reacción compleja generada [Steijns M. & Froment G. F., 1981, Baltanas et al., 1983, Froment G.
F., 1987].
Actualmente, el modelamiento cinético del proceso de hidrotratamiento se ha enfocado al uso de
agrupamiento de los compuestos involucrados en el sistema complejo de reacción, junto con
ecuaciones del tipo Langmuir-Hinshelwood-Hougen-Watson determinadas en base al mecanismo
de reacción [Broderick D. H. & Gates B.C., 1981, Vanrysselberghe V. & Forment G.F., 1996]. La
descripción de este proceso se ha basado en la simplificación de la red de reacción involucrando
uno o algunos de los compuestos modelo para representar la familias de hidrocarburos que existen
en cargas de destilados intermedios: parafinas, aromáticos, nitrogenados y compuestos azufrados,
[Girgis M. J. & Gates B.C., 1991, Broderick D. H. & Gates B.C., 1981, Vanrysselberghe V. &
Froment G.F., 1996].
Un modelo cinético detallado considera pasos de reacción individuales, incorporando un
gigantesco número de coeficientes cinéticos y de adsorción, aún cuando únicamente se procesan

MODELAMIENTO CINÉTICO DE LOS DATOS EXPERIMENTALES
81
compuestos modelo [Muñoz Arroyo. J. A., 2000]. Sin embargo las reacciones catalíticas
pertenecen a un limitado conjunto de familias las cuales resultan de agrupar compuestos
independientes por el número de átomos de carbón para los compuestos alifáticos y por reactividad
para los compuestos azufrados, nitrogenados y aromáticos contenidos en la alimentación del
proceso. Como consecuencia el número requerido de coeficientes cinéticos y de adsorción se
reduce a un conjunto limitado de coeficientes para los compuestos en la alimentación.
En un esfuerzo por mantener la naturaleza fundamental del modelo se requiere de un análisis
detallado de la alimentación cuando se utiliza un disolvente junto con la molécula modelo a ser
estudiada. A través de los años las técnicas químicas para el análisis de mezclas complejas se han
mejorado. La identificación del disolvente (mezcla compleja) por componente todavía no ha sido
resuelta. Consecuentemente se aplican algunas técnicas de agrupamiento, en las cuales los
compuestos se agrupan ya sea en base al número de átomos de carbón, el grado de substitución
y/o estructura molecular a manera de retener el carácter fundamental de los coeficientes de
velocidad [Froment G. F., Depauw Guy A. & Vanrysselberghe V., 1994].
Shafi R. & Hutching G.J., 2000, establecieron el estado del arte de la hidrodesulfuración del
dibenzotiofeno utilizando la evidencia experimental de este proceso, el cual concuerda con el
mecanismo propuesto de reacción utilizando dos sitios catalíticos. En uno de ellos se lleva a cabo
la hidrogenación del anillo aromático y en el otro la reacción de eliminación del azufre
(hidrogenólisis) produciendo el H2S (como ya se menciono en el capítulo 2 de este trabajo). Estos
autores también mencionan que Kabe et al., 1991, estudiaron la hidrodesulfuración del
dibenzotiofeno con diferentes disolventes que van desde el n-heptano, xileno, decalina y tetralina,
para un determinado intervalo de condiciones experimentales (553 K, 7.85 MPa y una relación
molar H2/Hc de 3.0) indicando que la reacción de hidrodesulfuración se lleve a cabo en fase vapor
como resultado de un análisis termodinámico.
En base a los comentarios antes mencionados, diversos factores deben ser considerados para la
estimación de los parámetros cinéticos y de adsorción del proceso catalítico de la
hidrodesulfuración, tratando de representar la mezcla compleja con un disolvente y compuestos
modelo que permitan acercarse lo más posible a la operación del proceso industrial. De esta
manera para tomar en cuenta la caracterización y propiedades fisicoquímicas de los destilados
intermedios en México (mezcla de hidrocarburos alifáticos, aromáticos, nitrogenados y azufrados)
en la experimentación se considero utilizar aceite mineral de tipo parafínico con hidrocarburos
desde C7 a C32 . A esta mezcla se le adiciono dibenzotiofeno como compuesto modelo azufrado en
un rango de 2 - 2.5 % peso (Tabla 4.2). En este estudio no se consideraron los efectos tanto de los

Capítulo 5
82
compuestos aromáticos como de los nitrogenados, ya que debido a su estructura química existe
cierta competencia en la adsorción sobre los sitios catalíticos que modifican de cierta manera la
velocidad de reacción. En la Figura 5.1 se muestra un esquema del manejo tanto de las fases
como de los productos involucrados en la hidrodesulfuración de dibenzotiofeno en un reactor tipo
Robinson Mahoney (reactor en 3 fases). En esta Figura se puede observar que las corrientes de
salida del reactor Robinson Mahoney pueden ser trasladados a la operación industrial
representándolos a través de corrientes en fases gas y líquida obtenidas por medio del calculo del
equilibrio de fases del contenido del Robinson Mahoney [Mahoney J. A., Robinson K. K. & Myers E.
C., 1978]. Esta determinación se realizó por medio de un cálculo en el simulador comercial PRO-
II™, utilizando la ecuación de estado de Peng-Robinson, la cual tiene incluido los parámetros de
interacción entre el hidrógeno y los hidrocarburos presentes en la mezcla.
Flash T y P delreactor
Fase Gaseosa
Fase líquida
Gas amargo
Gas flasheado
Producto líquido
Flash T y P delreactor
Fase Gaseosa
Fase líquida
Gas amargo
Gas flasheado
Producto líquido
Figura 5.1. Determinación del equilibrio de fases de la mezcla de reacción presente en el
reactor Robinson Mahoney
Además de las reacciones de hidodesulfuración en el reactor experimental fue posible observar la
reacción de hidrodesintegración, trabajando en el intervalo de condiciones de operación
mencionadas en la Tabla 5.2. Esta efecto se verificó mediante los resultados a la salida del reactor
(Tabla 3.10 y 3.11), obteniendo moléculas que van desde el C1 al C6. De esta manera el número
total de compuestos considerados en el modelamiento es de 41 (Tabla 5.1). Para facilitar el manejo
del total de compuestos en la estimación de parámetros se practico la agrupación de estos,
considerando que los hidrocarburos alifáticos tienen el mismo comportamiento cinético dentro del
sistema. Esto es, se consideraron los hidrocarburos ligeros y pesados en el intervalo desde C1 a
C19 y C20 a C32 como pseudo-compuestos ligero y pesado respectivamente, de manera individual el
dibenzotiofeno que es el representante de los compuestos azufrados y sus productos de reacción
(bifenilo, ciclohexilbenceno, diciclohexilo y el ácido sulfhídrico), así como también el hidrógeno.

MODELAMIENTO CINÉTICO DE LOS DATOS EXPERIMENTALES
83
Tabla 5.1. Agrupamiento de compuestos para el modelamiento
Compuestos base
Agrupamiento de Compuestos
Dibenzotiofeno Sin agrupar
Bifenilo Sin agrupar
Ciclohexilbenceno Sin agrupar
Diciclohexilo Sin agrupar
Aceite Mineral Ligero del C1 a C19 Pseudo Compuesto Ligero
Aceite Mineral Pesado desde C20 a C32 Pseudo Compuesto Pesado
Ácido Sulfhídrico Sin agrupar
Hidrógeno Sin agrupar
5.2. CONDICIONES EXPERIMENTALES
Para la estimación de los parámetros cinéticos para el hidrotratamiento en fase líquida de la mezcla
de aceite mineral y dibenzotiofeno se consideraron las condiciones de reacción mostradas en la
Tabla 5.2.
Tabla 5.2 Condiciones de Reacción consideradas para la Estimación de Parámetros
Parámetro Unidades Rango
Temperatura K 553-643
Presión MPa 8.15
Relación H2/Hc mol/mol 3.0
LHSV hr-1 1-2
La estimación de parámetros fue llevada a cabo a temperatura constante y a una sola presión y
diferentes relaciones H2/Hc.
La composición de la alimentación al sistema de reacción ya fue reportada en la Tabla 4.1 la cual
esta constituida de una mezcla de parafinas en el rango de C7 a C32, y Dibenzotiofeno con una
composición en el rango de 2-2.5 % peso.

Capítulo 5
84
5.3. PROCEDIMIENTO DE ESTIMACIÓN DE PARAMETROS.
La Figura 2.1 muestra el mecanismo de reacción del proceso de hidrodesulfuración del
dibenzotiofeno el cual fue tomado como base en la estimación de los parámetros cinéticos y de
adsorción involucrados en las expresiones de velocidad de reacción. Este mecanismo considera
dos trayectorias de reacción para la eliminación del azufre. La reacción de hidrogenólisis en donde
el azufre se elimina del DBT para formar BPH y H2S, y la reacción de hidrogenación del anillo
aromático del DBT para después producir el CHB y H2S. El CHB puede seguir hidrogenándose
hasta obtener el DCH.
La velocidad neta de reacción Ri, para los productos de la reacción de hidrodesulfuración,
derivadas del esquema anterior se pueden calcular de la siguiente manera:
τσ ,, BPHDBTBPH rrR −= 5.3
rCHBBPHDBTCHB rrrR ,,, −+= ττ 5.4
rCHBBCH rR ,= 5.5
τς ,,2 DBTDBTSH rrR += 5.6
La velocidad neta de desaparición del DBT e hidrógeno están dadas por:
τσ ,, DBTDBTDBT rrR += 5.7
CHBDBTDBTH rrrR *3*5*2 ,,2++= τσ 5.8
Los valores experimentales de Ri se calcularon de manera directa de los valores experimentales
de la conversión (Xi), utilizando el balance de materia en el reactor de flujo mezclado Robinson
Mahoney:
oDBT
ii FW
xR/
= 5.9
Para la estimación de parámetros primeramente se tomo como base el trabajo reportado por
Vanrysselberghe V. & Froment G.F., 1996 en el cual no se tiene un efecto importante del disolvente
utilizado en la experimentación (mezcla de parafinas normales C10-C14). Estos autores reportan que

MODELAMIENTO CINÉTICO DE LOS DATOS EXPERIMENTALES
85
el paso limitante de las velocidades de reacción para el DBT, BPH, CHB esta dado por la reacción
en la superficie de la partícula catalítica. Bajo este contexto se realizó un análisis cualitativo de los
datos experimentales de este trabajo para determinar si el paso limitante de la reacción es el
mismo que el reportado por Vanrysselberghe V. & Froment G.F., 1996. El análisis de los datos
experimentales se realizó utilizando la metodología reportada por Fogler H.S., 2001.
La Figura 5.2 muestra el comportamiento de la velocidad de reacción (calculado con la información
experimental y la ecuación 5.9) como una función de la concentración de cada una de los
compuestos involucrados en el proceso de hidrodesulfuración del DBT. Esta Figura muestra que el
paso controlante en este proceso esta dado por la reacción en la superficie dado que el
comportamiento de la velocidad para los compuestos (DBT, BPH, CHB, DCH) involucrados
presentan una función polinomial con respecto a la concentración.
0.0
1.0
2.0
3.0
4.0
5.0
6.0
0.0 10.0 20.0 30.0 40.0
Concentración i x103, Kgmol/m3
r i x
104 K
gmol
/Kg c
at h
0.00
0.10
0.20
0.30
0.40
r i x
104 K
gmol
/Kg c
at h
DBT BPH CHB DCH
Figura 5.2. Comportamiento de las velocidades de reacción experimentales con las concentraciones para el DBT, BPH, CHB, DCH
Basado en estos resultados se consideraron en detalle las etapas de adsorción y reacción química
utilizando dos diferentes sitios catalíticos (σ, τ ) de manera similar al trabajo ya reportado por
Vanrysselberghe V. & Froment G.F., 1996.

Capítulo 5
86
La reacción de hidrogenólisis del DBT para producir BPH y H2S se lleva a cabo en el sitio σ :
σσ ⋅⇔+ DBTDBT
σσ ⋅⇔+ HH 222
σσσσσ +⋅+⋅→⋅+⋅ SBPHHDBT 2
σσσσ 22 2 +⋅⇔⋅+⋅ SHHS
σσ +⇔⋅ BPHBPH
σσ +⇔⋅ BPHSH 2
La reacción de hidrogenación del DBT en THDBT y HHDBT en el sitio τ siguiendo a la reacción de
hidrogenólisis para producir el CHB y H2S en el sitio σ es:
ττ ⋅⇔+ DBTDBT
ττ ⋅⇔+ HH 222
ττττ 22 +⋅⇔⋅+⋅ DHDBTHDBT
ττττ 22 +⋅⇔⋅+⋅ THDBTHDHDBT
ττ +⇔⋅ THDBTTHDBT
ττττ 22 +⋅⇔⋅+⋅ HHDBTHTHDBT
ττ +⇔⋅ HHDBTHHDBT
σσ ⋅⇔+ THDBTTHDBT
σσ ⋅⇔+ HHDBTHHDBT
σσ ⋅⇔+ HH 222
σσσσσ +⋅+⋅→⋅+⋅ SPHCHHTHDBT 2
σσσσ 22 +⋅⇔⋅+⋅ CHBHPHCH
σσσσσ +⋅+⋅→⋅+⋅ SCHBHHHDBT 2
σσσσ 22 2 +⋅⇔⋅+⋅ SHHS
σσ +⇔⋅ CHBCHB
σσ +⇔⋅ SHSH 22

MODELAMIENTO CINÉTICO DE LOS DATOS EXPERIMENTALES
87
Por otro lado el mecanismo de reacción de hidrogenación de BPH para producir CHB en los sitios
τ esta dado por las siguientes etapas:
ττ ⋅⇔+ BPHBPH
ττ ⋅⇔+ HH 222
ττττ 22 +⋅→⋅+⋅ PHCHDHBPH
ττττ 22 +⋅⇔⋅+⋅ PHCHHPHCHD
ττττ 22 +⋅⇔⋅+⋅ CHBHPHCH
ττ +⇔⋅ CHBCHB
El mecanismo de la reacción de hidrogenación del CHB formado para producir DCH en los sitios τ
esta dado de la siguiente manera:
ττ ⋅⇔+ CHBCHB
ττ ⋅⇔+ HH 222
ττττ 22 +⋅→⋅+⋅ CHCHDHCHB
ττττ 22 +⇔+ ... CHCHHCHCHD
ττττ 22 +⇔+ ... BCHHCHCH
ττ +⇔⋅ BCHBCH
Este mecanismo no considera la competición del disolvente en la adsorción sobre los sitios activos
de la partícula catalítica, sin embargo dado a que en la experimentación se utilizó un aceite mineral
que contiene compuestos parafínicos que van desde C1 hasta C32 y las moléculas más grandes
que a pesar de estar en concentraciones muy bajas pueden competir, se decidió realizar el análisis
de los mecanismos de reacción anteriores incluyendo el efecto en la adsorción del disolvente. Para
introducir el efecto de la matriz se tomo en consideración lo siguiente:
A las condiciones de reacción experimentales es mínima la hidrodesintegración que sufre
el aceite mineral.
El aceite mineral se considera como un inerte que compite en la adsorción de los sitios
activos sin participar en las reacciones de hidrodesulfuración del dibenzotiofeno.

Capítulo 5
88
El aceite mineral se involucra como un pseudocompuesto que se adsorbe ocupando sitios
activos del catalizador.
En base a las hipótesis anteriores se incluye en los mecanismos de las reacciones de la
hidrogenólisis e hidrogenación la siguiente etapa relativa al aceite mineral:
σσ ⋅⇔+ AMAM
ττ .AMAM ⇔+
Puesto que la adsorción del aceite mineral está en equilibrio, la concentración de sitios ocupados
en la superficie se puede calcular de la siguiente manera:
VAMAMAM CCKC =⋅τ 5.10.a
VAMAMAM CCKC =σ. 5.10.b
La concentración total de sitios activos se modifica de la siguiente manera:
SAMSDCHSCHBSBPHSDBTvT CCCCCCC ⋅⋅⋅⋅⋅ +++++= 5.11
Las ecuaciones de velocidad de reacción derivadas del mecanismo de hidrogenación e
hidrogenolisis reportado por Vanrysselberghe V. & Froment G.F., 1996 se modifican de la siguiente
manera:
)( ,,,,,
,,,, 3
AMPAMSHSHBPHBPH2HHDBTDBT
2HDBTDBTHDBTDBT
CKCKCKCKCK1CCKKk
r22 σσσσσ
σσσσ
+++++= 5.12
)( ,,,,
,,,, 3
AMPAMBPHBPH2HHDBTDBT
2HDBTDBTHDBTDBT CKCKCKCK1
CCKKkr
ττττ
ττττ
++++= 5.13
)( ,,,
,,,, 3
AMPAMPBPHBPH2HHDBTDBT
2HBPHBPHHBPHBPH CKCKCKCK1
CCKKkr
++++=
τττ
ττττ 5.14

MODELAMIENTO CINÉTICO DE LOS DATOS EXPERIMENTALES
89
)( ,,,
,,,, 3
AMPAMPBPHBPH2HHDBTDBT
2HCHBCHBHCHBCHB CKCKCKCK1
CCKKkr
++++=
τττ
ττττ 5.15
Una vez establecidas las expresiones de velocidad de reacción, es posible realizar el balance de
materia respectivo para reactivos y productos del proceso de hidrodesulfuración.
Para un reactor de flujo mezclado el balance de materia produce un sistema de ecuaciones
algebraicas no lineales donde la solución permite calcular el flujo molar de los compuestos en la
mezcla líquida a la salida del reactor. La expresión matemática del balance de materia por
componente y experimento es la siguiente (Froment G. F. & Bischoff K. 1990):
0W)C,P,(TRFF hhhhkh,o
kh,l2
kh, =−− 5.16
En esta expresión la velocidad de reacción esta dada por los modelos ya establecidos en los que
se involucra un conjunto de parámetros cinéticos ki y parámetros de adsorción iK , además de la
temperatura Th, de la presión total Ph y la concentración de los componentes y
pseudocomponentes en la fase líquida hC .
Los flujos molares experimentales para cada componente en la corriente de salida del líquido 2lkhF ,
para la hth observación se determinan del cálculo del equilibrio líquido – vapor de la corriente de
salida.
Para realizar la estimación de los parámetros cinéticos y de adsorción de los modelos de velocidad
de reacción, se utilizan diferentes alternativas. En la Figura 5.3 se muestra un esquema de la
metodología de estimación de los parámetros. Se requiere contar con el valor de los flujos molares
experimentales de cada uno de los compuestos a la entrada y a la salida del reactor (balance de
materia). El flujo molar de salida total consiste de tres flujos molares: Fase Gas FG, Líquido
Flasheado F12 y el Gas Falseado Fl1 (ver Figura 4.1) de donde se calculo el equilibrio de fases.
Cuando se calcula este equilibrio es posible conocer las concentraciones de los compuestos en la
fase líquida. Esta concentración se calculó utilizando la metodología del calculo del volumen molar
de la mezcla (Reid R.C., Prausnitz J.M. and Sherwood T.K., 1977), también se utilizó el simulador
de procesos PRO -II™ y la ecuación de estado de Peng-Robinson la cual ya tiene incorporada una
corrección de los parámetros de interacción del hidrógeno y los hidrocarburos. Debe también
especificarse el modelo cinético que será tomado en cuenta para la determinación. Una vez

Capítulo 5
90
calculadas las concentraciones con ayuda del modelo cinético y las suposiciones iniciales se
procede a realizar la optimización, para determinar los valores de las constantes cinéticas y de
adsorción. Si estas, satisfacen la función objetivo en el mínimo dado entre el valor calculado y el
experimental del flujo molar en la corriente líquida de salida del reactor, entonces se ha encontrado
la solución al problema, en caso contrario, se requerirá hacer algún ajuste a la suposición inicial de
los coeficientes cinéticos para continuar con el procedimiento de optimización.
VALORES INICIALES
CALCULO DE LA CONCENTRACIÓN
EN LA FASE LÍQUIDA
MODELO DEL REACTOR DE FLUJO MEZCLADO
MinF
(OBJETIVO)
AJUSTE DE LA SUPOSICIÓN INICIAL
ECUACIÓN DE ARRENIUS PARA K´s ??OBTENCIÓN DE K´s
I
CALCULO DEL BALANCE DE MATERIA
DATOS EXPERIMENTALES
FIN
VALORES INICIALES
CALCULO DE LA CONCENTRACIÓN
EN LA FASE LÍQUIDA
MODELO DEL REACTOR DE FLUJO MEZCLADO
MinF
(OBJETIVO)
AJUSTE DE LA SUPOSICIÓN INICIAL
ECUACIÓN DE ARRENIUS PARA K´s ??OBTENCIÓN DE K´s
I
CALCULO DEL BALANCE DE MATERIA
DATOS EXPERIMENTALES
FIN
Figura 5.3. Metodología de la estimación de los parámetros cinéticos y de adsorción para el proceso de hidrodesulfuración del DBT
En la optimización se propusieron dos formas de la función objetivo. Una es la propuesta por
Muñoz Arroyo J.A., 2000 como la suma de los cuadrados de los residuales de los pesos de los
flujos molares experimentales y calculados por la ecuación del reactor de flujo mezclado perfecto:
2nob
h12
kh12
khnrespk
kcomp FFkS )()( ,,∑ −∑ σ= 5.17

MODELAMIENTO CINÉTICO DE LOS DATOS EXPERIMENTALES
91
en donde los factores de peso kσ se calculan utilizando la siguiente relación (Martens G. G. et al.,
2000)
∑ ∑∑
=
−
=
−
== nresp
k
nobs
hhk
nobs
h hkk
F
F
1
1
1
12
1
112,
)(
)(σ 5.18
Otra definición de la función objetivo utilizada en este trabajo es propuesta por Vanrysselberghe V.
& Froment G.F., 1996 la cual tiene la siguiente forma:
∑ ∑∑=
=
=
=
=
=
−−=vh
hik
ni
iikihih
vk
k
hk RRRRS1 11
))(()( σθ 5.19
Para esta ecuación los factores de peso son definidos de manera semejante a los propuestos por
Martens G.G., 2000, pero utilizando las velocidades de reacción experimentales, como sigue:
∑ ∑∑
=
−
=
−
== nresp
k
nobs
hhk
nobs
h hkk
R
R
1
1
1
1
1 ,
)(
)(σ 5.20
La minimización de las funciones objetivo (ecuaciones 5.17 y 5.19) se realizó usando los métodos
Rosenbrock y Marquardt (Rosenbrock H.H., 1966; Marquardt D.W., 1963), el utilizado por el
software Gams (Conopt), para la solución de ecuaciones no lineales y el propuesto en Excel, el
cual fue desarrollado en la Universidad Leon Lasdon de Austin (Texas) y la Universidad Allan
Waren de Cleveland (Ohio). La determinación se llevo a cabo con la finalidad de comparar el
comportamiento que tienen estos métodos para alcanzar la convergencia al mínimo solucionando
el sistema de ecuaciones no lineal resultado del balance de materia.
5.4. RESULTADOS DEL MODELADO CINÉTICO
La estimación de parámetros se realizó utilizando la información experimental, el intervalo de
condiciones de reacción y los modelos de Broderick D.H. & Gates B.C., 1981, Vanrysselberghe V.
& Froment G.F., 1996 y la modificación propuesta en este estudio.

Capítulo 5
92
Primeramente se probó el modelo cinético propuesto por Broderick D.H. & Gates B.C., 1981
(ecuaciones 2.1 y 2.2) utilizando las constantes cinéticas propuestas por los autores. La
determinación de los parámetros del modelo se realizó minimizando la función objetivo dada por la
ecuación 5.19 utilizando el solver establecido en Excel. La Tabla 5.3 muestra los parámetros
cinéticos y de adsorción estimados para este modelo. Como puede observarse en ambos grupos
de la Tabla tanto la calculada como la reportada, la tendencia de la constante cinética es de
acuerdo a lo esperado para el proceso de hidrodesulfuración, es decir a un aumento de
temperatura la constante cinética de velocidad de reacción tiende a aumentar. Por otro lado las
constantes de adsorción de las especies presentes en la reacción, tendrán un efecto contrario al
obtenido por el cinético, es decir, a un aumento de temperatura la constante de adsorción será
menor o puede en un momento determinado no presentar cambio, lo cual significará que la
constante relativa a ese compuesto no presenta cambio con una modificación a la temperatura de
reacción, tal es el caso del H2S para el modelo reportado por Broderick D.H. & Gates B.C., 1981.
La constante de adsorción del hidrógeno calculada muestra un comportamiento diferente al
reportado por estos autores.
El valor del parámetro calculado esta muy por abajo del valor reportado por estos autores.
Diferencias importantes pueden también observarse para los valores de los parámetros cinéticos y
de adsorción, reportados y calculados en este trabajo, consecuentemente este modelo y método
de optimización fueron descartados para representar el proceso de hidrodesulfuración de
dibenzotiofeno.
Un procedimiento similar fue seguido para analizar el modelo propuesto por Vanrysselberghe V.
and Froment G. F., 1996 (Ecs. 2.3 a 2.6). Como ya ha sido comentado este modelo, no considera
el efecto del disolvente utilizado en la experimentación dentro de las ecuaciones de velocidad.
Primeramente se realizó el análisis de la tendencia que guardan los resultados experimentales con
el modelo para determinar el comportamiento de los parámetros cinéticos y de adsorción con la
temperatura. La Figura 5.4 muestra este comportamiento que concuerda con las ecuaciones de
Arrhenius y Van’ t Hoff.
Después se realizó la determinación de estos parámetros, con el mismo modelo sin considerar el
efecto del disolvente y la información experimental obtenida en este trabajo. La minimización de la
función objetivo (Ec. 5.19) se realizó utilizando los métodos de optimización incluidos en el
programa Excel, la programación lineal utilizando GAMS™ y el método de Rosenbrock programado
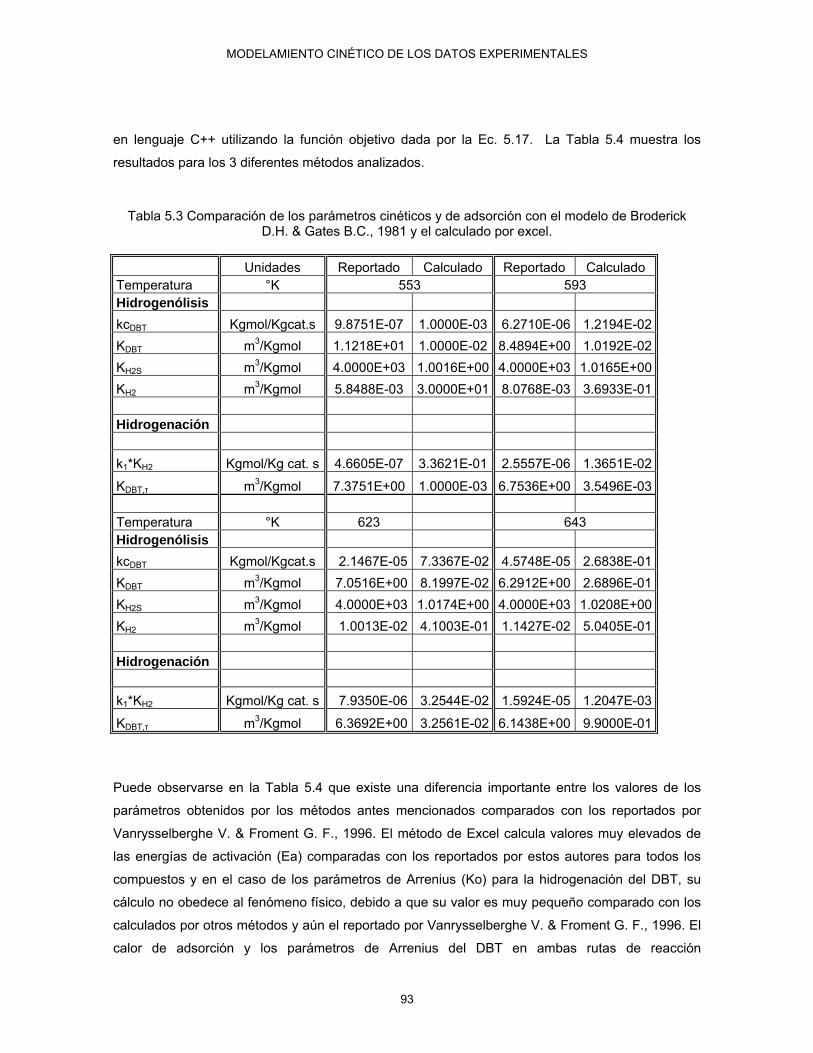
MODELAMIENTO CINÉTICO DE LOS DATOS EXPERIMENTALES
93
en lenguaje C++ utilizando la función objetivo dada por la Ec. 5.17. La Tabla 5.4 muestra los
resultados para los 3 diferentes métodos analizados.
Tabla 5.3 Comparación de los parámetros cinéticos y de adsorción con el modelo de Broderick D.H. & Gates B.C., 1981 y el calculado por excel.
Unidades Reportado Calculado Reportado Calculado Temperatura °K 553 593 Hidrogenólisis kcDBT Kgmol/Kgcat.s 9.8751E-07 1.0000E-03 6.2710E-06 1.2194E-02 KDBT m3/Kgmol 1.1218E+01 1.0000E-02 8.4894E+00 1.0192E-02 KH2S m3/Kgmol 4.0000E+03 1.0016E+00 4.0000E+03 1.0165E+00 KH2 m3/Kgmol 5.8488E-03 3.0000E+01 8.0768E-03 3.6933E-01 Hidrogenación k1*KH2 Kgmol/Kg cat. s 4.6605E-07 3.3621E-01 2.5557E-06 1.3651E-02
KDBT,τ m3/Kgmol 7.3751E+00 1.0000E-03 6.7536E+00 3.5496E-03 Temperatura °K 623 643 Hidrogenólisis kcDBT Kgmol/Kgcat.s 2.1467E-05 7.3367E-02 4.5748E-05 2.6838E-01 KDBT m3/Kgmol 7.0516E+00 8.1997E-02 6.2912E+00 2.6896E-01 KH2S m3/Kgmol 4.0000E+03 1.0174E+00 4.0000E+03 1.0208E+00 KH2 m3/Kgmol 1.0013E-02 4.1003E-01 1.1427E-02 5.0405E-01 Hidrogenación k1*KH2 Kgmol/Kg cat. s 7.9350E-06 3.2544E-02 1.5924E-05 1.2047E-03
KDBT,τ m3/Kgmol 6.3692E+00 3.2561E-02 6.1438E+00 9.9000E-01
Puede observarse en la Tabla 5.4 que existe una diferencia importante entre los valores de los
parámetros obtenidos por los métodos antes mencionados comparados con los reportados por
Vanrysselberghe V. & Froment G. F., 1996. El método de Excel calcula valores muy elevados de
las energías de activación (Ea) comparadas con los reportados por estos autores para todos los
compuestos y en el caso de los parámetros de Arrenius (Ko) para la hidrogenación del DBT, su
cálculo no obedece al fenómeno físico, debido a que su valor es muy pequeño comparado con los
calculados por otros métodos y aún el reportado por Vanrysselberghe V. & Froment G. F., 1996. El
calor de adsorción y los parámetros de Arrenius del DBT en ambas rutas de reacción

Capítulo 5
94
(hidrogenólisis e hidrogenación) calculadas utilizando el método de Excel muestra valores más
pequeños que aquellos reportados por estos autores. Para el BPH y el CHB el orden de magnitud
reportado para estos parámetros es similar. Sin embargo, para el hidrógeno el resultado es menor
al ya reportado. Una posible explicación a esta diferencia pudiese darse por el método de cálculo
de las derivadas de las ecuaciones de los balances de materia las cuales utilizan el método de
aproximaciones sucesivas.
-1.00
0.00
1.00
2.00
3.00
4.00
5.00
1.50 1.55 1.60 1.65 1.70 1.75 1.80 1.85
1/T x 103 [K]
Ln K
i [K
mol
/K ca
t h]
-4.00
-3.00
-2.00
-1.00
0.00
1.00
2.00
3.00
4.00
Ln k
c i [K
mol
/Kca
t.h
K(DBT,σ) K(DBT,τ) K(H2S,σ)K(H2,σ) K(BPH,σ) K(H2,τ)K(BPH,τ) kc(DBT,σ) kc(DBT,τ )
Figura 5.4. Comportamiento de las constantes cinéticas y de adsorción reportadas por Vanrysselberghe V. & Froment G. F., 1996
La comparación de los parámetros cinéticos y de adsorción calculados por los métodos de
Rosenbrock y GAMS muestra un orden de magnitud muy semejante, sin embargo estos valores
comparados con los reportados por Vanrysselberghe V. & Froment G. F., 1996 muestran
diferencias significativas. Puede observarse en la Tabla 5.4 que la energía de activación para el
DBT en la reacción de hidrogenólisis, GAMS y Rosenbrock muestra valores mayores a los
reportados en la literatura. No obstante estos resultados, el valor de la energía de activación para
la producción de BPH se invierte. El valor estimado de la constante de adsorción del hidrógeno en

MODELAMIENTO CINÉTICO DE LOS DATOS EXPERIMENTALES
95
Tabla 5.4 Resultados de la optimización de parámetros cinéticos y de adsorción considerando el modelo de Vanrysselberghe V.& Froment G.F., 1996
Optimización de los parámetros cinéticos y de adsorción sin considerar el efecto del disolvente Ea ó ∆H (Kjoule/Kmol) Ko (m3/Kmol)
Reportado Calculado en este trabajo Reportado Calculado en este trabajo Parámetros Vanrysselberghe
V. & Froment G.F.
GAMS Método de Rosenbrock Excel
Vanrysselberghe V. & Froment
G.F. GAMS Método de
Rosenbrock Excel
kc(DBT),σ 122770 182368 314810 139299 2.44E+10 1.4200E+16 3.66E+25 3.9274E+12kc(DBT),τ 186190 77714 132359 70609 3.41E+23 3.2217E+07 3.87E+12 6.5936E-07kc(BPH),τ 255714 135641 155430 429040 3.41E+23 4.9857E+11 7.50E+13 1.0896E+38kc(CHB),τ - 87900 119214 285500 - 1.5125E+09 6.12E+10 1.1212E+25 K(DBT),σ - 112851 270247 438984 7.57E+01 1.3880E-10 2.77E-25 2.1913E-35K(H2),σ 113232 243221 170944 7549 3.36E-11 1.3206E-19 8.60E-22 1.7138E+00K(BPH),σ 48214 29006 47674 354 3.85E-04 1.9411E-02 6.61E-04 1.1272E+01K(H2S),σ 105670 117965 41886 2977 1.47E-08 2.1864E-09 1.53E-03 5.5896E+00 K(DBT),τ 76840 55912 261625 2466 2.50E-07 2.8804E-04 3.99E-24 1.2176E+00K(H2),τ 142693 216707 237207 3044 1.40E-15 6.8406E-22 6.22E-24 6.4913E-02K(BPH),τ 37899 97370 49028 60543 4.97E-04 1.1226E-08 8.40E-04 1.5067E-05K(CHB),τ - 90968 53889 63217 - 1.4518E-27 3.23E-04 7.6945E-06

Capítulo 5
96
la reacción de hidrogenólisis por ambos métodos es mayor comparado con el ya reportado, sin
embargo, para el BPH el orden de magnitud es similar calculado con el método de Rosenbrock. En
general los valores estimados para las constantes cinéticas y de adsorción en la reacción de
hidrogenación en ambos métodos para todos los compuestos es menor con respecto los valores
obtenidos para la reacción de hidrogenólisis. Este hecho permite establecer que el catalizador de
CoMo dirige la hidrodesulfuración a este último tipo de reacción.
En la Tabla 5.5 se muestran los resultados de la optimización realizada considerando el efecto
competitivo del disolvente para la adsorción en los sitios activos del catalizador (el cual ya fue
discutido anteriormente), modificando el modelo de Vanrysselberghe V. & Froment G. F.,1996
(Ecs. 5.12 a 5.15). De los resultados mostrados en esta Tabla se puede observar que la energía de
activación y calor de adsorción calculadas utilizando el método de Excel tienden a alejarse de los
valores de los reportados por estos autores. No así para aquellos valores calculados con los
métodos de GAMS y el de Rosenbrock, dado que estos se acercan a los valores ya reportados.
Estos resultados indican que el disolvente compite por los sitios activos del catalizador y
consecuentemente modifica la velocidad de reacción.
Al explorar el calor de adsorción calculado para la hidrodesulfuración por la ruta de la reacción de
hidrogenólisis, se puede observar que el método de Rosenbrock determina valores mayores a los
ya reportados y el método de GAMS invierte estos valores. Es claro que existen dos procesos
diferentes para alcanzar el mínimo en la optimización de la función objetivo. Los calores de
adsorción para la hidrodesulfuración por la ruta de hidrogenación muestran un comportamiento
semejante al ya observado por la ruta de hidrogenólisis, sin embargo se observa un valor mayor de
los calores de adsorción calculados por el método de GAMS comparadas con aquellas
determinadas por el método de Rosenbrock. Estos resultados confirman que estos métodos utilizan
dos rutas diferentes para alcanzar el mínimo de la función objetivo.
Por otro lado el disolvente muestra una competencia importante para alcanzar los sitios activos del
catalizador. Este efecto se puede evidenciar ya que los valores de los parámetros de adsorción
asociadas al disolvente muestran un incremento con un cambio en la temperatura reflejado en el
calor de adsorción.

MODELAMIENTO CINÉTICO DE LOS DATOS EXPERIMENTALES
97
Tabla 5.5 Resultados de la optimización de parámetros cinéticos y de adsorción considerando el efecto del disolvente
en el modelo de Vanrysselberghe and Froment [1996]
Optimización de los parámetros cinéticos y de adsorción considerando el efecto del disolvente Ea ó ∆H (Kjoule/Kmol) Ko (m3/Kmol)
Reportado Calculado en este trabajo Reportado Calculado en este trabajo Parámetros Vanrysselberghe
V. & Froment G.F.
GAMS Rosenbrock Excel Vanrysselberghe
V. & Froment G.F.
GAMS Rosenbrock Excel
kc(DBT),σ 122770 55455 107904 270085 2.44E+10 1.8049E+06 5.23E+09 2.9623E+24kc(DBT),τ 186190 36101 132049 593099 3.41E+23 2.5387E+04 9.18E+11 4.6681E+50kc(BPH), τ 255714 15248 126312 376143 3.41E+23 5.7409E+01 5.78E+11 3.7445E+32kc(CHB),τ - 62401 92988 278192 - 3.6020E+06 3.48E+08 7.5910E+24 K(DBT),σ - 60240 464160 260873 7.57E+01 8.3405E-05 5.34E-41 1.4337E-21K(H2),σ 113232 163536 114290 264240 3.36E-11 1.1459E-14 2.9815E-11 7.4326E-22K(BPH),σ 48214 154249 48388 79851 3.85E-04 5.9966E-14 3.73E-04 3.9334E-06K(H2S),σ 105670 162671 34912 115811 1.47E-08 6.6966E-13 1.4851E-02 6.5815E-09K(AM),σ - 68481 1652 236719 - 4.5700E-06 7.60E+00 1.2842E-19 K(DBT),τ 76840 68270 29613 267125 2.50E-07 4.0186E-05 1.08E-02 4.7535E-22K(H2),τ 142693 312813 143382 494590 1.40E-15 1.2280E-28 2.35E-13 3.2666E-44K(BPH),τ 37899 230816 269686 106458 4.97E-04 6.8337E-22 8.41E-23 1.1861E-08K(CHB),τ - 279139 394 49270 - 2.0162E-26 9.28E+00 7.7884E-04K(AM),τ - 180040 248 94177 - 3.2426E-16 1.2993E+01 1.5544E-07

Capítulo 5
98
0.0
1.0
2.0
3.0
4.0
5.0
6.0
0.0 1.0 2.0 3.0 4.0 5.0 6.0
RDBT x 104 exp [Kgmol/Kg cat/hr]
RD
BT
x 10
4 cal
[Kgm
ol/K
g ca
t/hr]
Figura 5.5.a Diagrama de paridad de la velocidad de reacción calculada vs experimental del DBT
obtenida por el método de Gams
0.0
1.0
2.0
3.0
4.0
5.0
0.0 1.0 2.0 3.0 4.0 5.0
R BPH x 104 exp [Kgmol/Kg cat /hr]
R B
PH x
104 c
al [K
gmol
/Kg
cat/h
r]
Figura 5.5.b Diagrama de paridad de la velocidad de reacción del BPH calculada vs la
experimental obtenidas por el método de Gams

MODELAMIENTO CINÉTICO DE LOS DATOS EXPERIMENTALES
99
0.0
0.4
0.8
1.2
1.6
0.0 0.4 0.8 1.2 1.6
R CHB x 10 4exp[Kgmol/Kg cat /hr]
RC
HB x
10
4 cal [
Kgm
ol/K
g ca
t/hr]
Figura 5.5.c Diagrama de paridad de la velocidad de reacción del CHB calculada vs la
experimental obtenidas por el método de Gams
0.0
0.5
1.0
1.5
2.0
0.0 0.2 0.4 0.6 0.8 1.0 1.2 1.4 1.6 1.8 2.0
R DCH x 10 5exp[Kgmol/Kg cat /hr]
R D
CH x
10
5 cal [
Kgm
ol/K
g ca
t /hr
]
Figura 5.5.d Diagrama de paridad de la velocidad de reacción del DCH calculada vs la
experimental obtenidas por el método de Gams A manera de comparación de los resultados obtenidos y los valores experimentales se prepararon
las gráficas de paridad tomando en cuenta las dos funciones objetivo utilizadas en la optimización

Capítulo 5
100
(Ecs. 5.17 y 5.19). Las Figuras 5.5.a-d muestran la comparación del comportamiento obtenido por
el método de GAMS. De la información de estas Figuras se observa que existe una buena
aproximación en la estimación de los parámetros cinéticos y de adsorción, no obstante que existe
cierta dispersión en algunas de las Figuras. Para el DBT, CHB y el DCH, la diferencia de los datos
experimentales con los calculados por este método no es significativa, sin embargo para el BPH si
se presenta. Las gráficas de paridad obtenidas del cálculo del flujo molar por medio de los
parámetros estimados por el método de Rosenbrock se muestran en las Figuras 5.6.a-d. Los
resultados mostrados en estas Figuras evidencian una buena aproximación de los parámetros
cinéticos y de adsorción calculados por Rosenbrock.
Se sabe de la literatura abierta que la hidrodesulfuración utilizando catalizadores a base de CoMo
facilitan esta reacción utilizando el mecanismo de la hidrogenólisis, por lo cual los resultados
obtenidos por el método de Rosenbrock evidencian este mecanismo.
De los métodos de optimización utilizados en este trabajo, el método de Rosenbrock proporciona
una mejor aproximación de los parámetros estimados en comparación con los reportados por
Vanrysselberghe V. & Froment G. F.,1996, tomando en cuenta que existe una adsorción
competitiva por parte del aceite mineral en la mezcla de reacción.
0.0
1.0
2.0
3.0
4.0
5.0
6.0
7.0
0.0 1.0 2.0 3.0 4.0 5.0 6.0 7.0
Flujo Molar Experimental [x 105 mol/h]
Fluj
o M
olar
Cal
cula
do [
x10 5
m
ol/h
]
Figura 5.6.a Gráfica de paridad para el flujo molar calculado vs experimental a T = 553K y
P = 7.85 MPa obtenido por el método de Rosenbrock
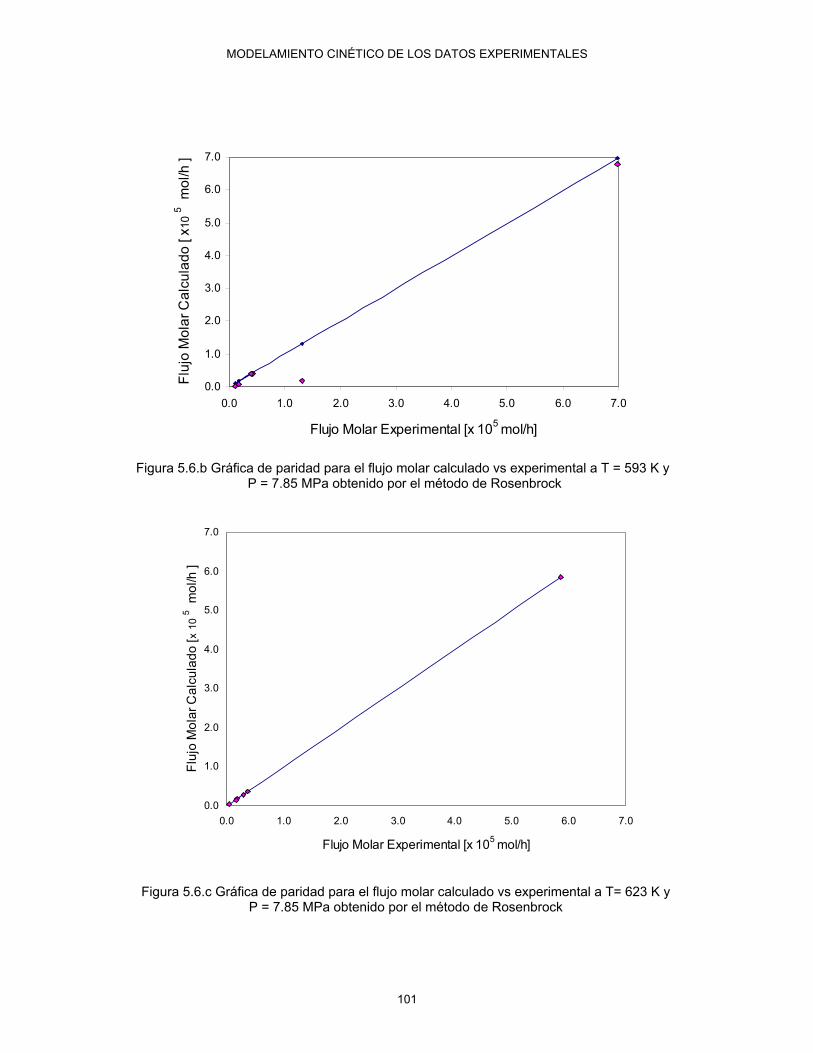
MODELAMIENTO CINÉTICO DE LOS DATOS EXPERIMENTALES
101
0.0
1.0
2.0
3.0
4.0
5.0
6.0
7.0
0.0 1.0 2.0 3.0 4.0 5.0 6.0 7.0
Flujo Molar Experimental [x 105 mol/h]
Fluj
o M
olar
Cal
cula
do [
x 10
5 m
ol/h
]
Figura 5.6.b Gráfica de paridad para el flujo molar calculado vs experimental a T = 593 K y
P = 7.85 MPa obtenido por el método de Rosenbrock
0.0
1.0
2.0
3.0
4.0
5.0
6.0
7.0
0.0 1.0 2.0 3.0 4.0 5.0 6.0 7.0
Flujo Molar Experimental [x 105 mol/h]
Fluj
o M
olar
Cal
cula
do [
x 10
5 m
ol/h
]
Figura 5.6.c Gráfica de paridad para el flujo molar calculado vs experimental a T= 623 K y
P = 7.85 MPa obtenido por el método de Rosenbrock
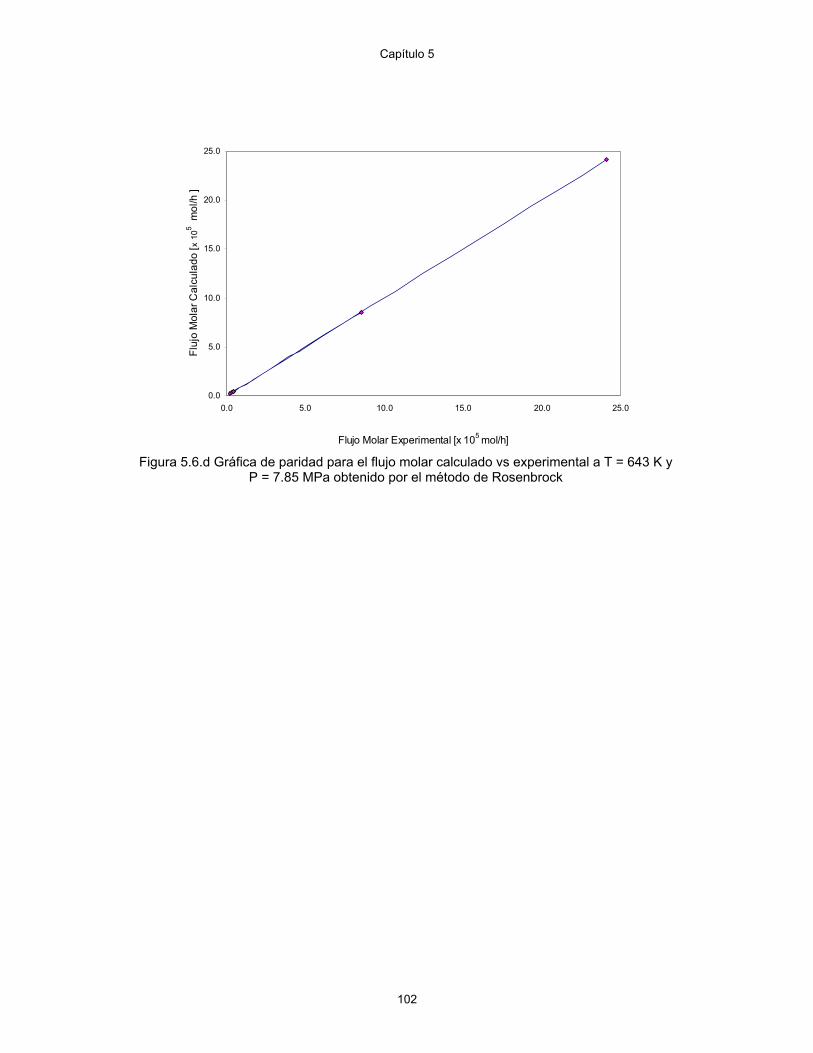
Capítulo 5
102
0.0
5.0
10.0
15.0
20.0
25.0
0.0 5.0 10.0 15.0 20.0 25.0
Flujo Molar Experimental [x 105 mol/h]
Fluj
o M
olar
Cal
cula
do [
x 10
5 m
ol/h
]
Figura 5.6.d Gráfica de paridad para el flujo molar calculado vs experimental a T = 643 K y
P = 7.85 MPa obtenido por el método de Rosenbrock

CONCLUSIONES
103
6. CONCLUSIONES
El desarrollo de un modelo cinético confiable en la hidrodesulfuración de destilados intermedios
que permita explorar el comportamiento de un reactor industrial ha sido trabajo de muchos grupos
de investigación. El IMP tratando de explicar el comportamiento de las cargas industriales y de
cumplir con la legislación ambiental ha incursionado también en está área de investigación de tal
manera que actualmente se tiene construida y en operación una planta piloto que permite realizar
el hidrotratamiento de compuestos modelo y/o carga reales, para obtener información experimental
precisa para ser utilizada en el desarrollo de un modelo cinético.
La determinación de la composición de la carga y productos de reacción, así como también del
disolvente utilizado en la experimentación se debió al desarrollo de dos nuevas técnicas analíticas.
De esta manera se realizó la validación de la operación de la planta piloto en un intervalor amplio
de condiciones de operación. La confiabilidad de los datos experimentales ha sido confirmada en
base al balance de masa y de átomos entre las corrientes de entrada y salida del sistema de
reacción.
El análisis estadístico del comportamiento de las variables del proceso ha permitido establecer la
dependencia que existe entre ellas y cuantificar el efecto de la temperatura con respecto a la
presión, la relación H2/Hc y el LHSV.
En el intervalo de condiciones de operación y de acuerdo al comportamiento de los productos con
la temperatura y el LHSV se confirmó que el esquema de reacción propuesto es el adecuado para
representar el proceso de hidrodesulfuración del DBT.
En el establecimiento del modelo cinético se ha destacado el efecto de adsorción del disolvente en
las expresiones de velocidad, dado que existe competencia en la adsorción con los compuestos
reactivos en la mezcla de reacción. En base a esta consideración y tomando como base los
mecanismos y expresiones de velocidad de reacción ya publicados en literatura fue posible
establecer un nuevo modelo cinético incluyendo el efecto de la mezcla compleja de reacción que
puede ser utilizado más adelante en el modelamiento del reactor de la planta industrial.
La consideración del efecto del disolvente en las expresiones de velocidad modifica notablemente
la energía de activación (Ea) en la ruta de hidrogenólisis, facilitando la representación de la

Capítulo 6
104
reacción de la hidrogenólisis de manera más precisa, dado que la comparación de los valores de
las energías de activación entre las reacciones de hidrogenólisis e hidrogenación incluyendo el
disolvente permite que la primera se vea favorecida de acuerdo a lo esperado con catalizadores de
CoMo/Al2O3.
La magnitud de las velocidades dentro del esquema de reacción confirma la selectividad
preferencial de la formación del BPH por la reacción de hidrogenólisis seguida por la de CHB y más
difícilmente la de DCH.
Se obtuvo una mejor aproximación de la función objetivo utilizando los flujos molares de los
compuestos a la salida del reactor comparada con la que maneja velocidades de reacción
atribuyéndolo a un mayor orden de magnitud en el flujo molar.

RECOMENDACIONES
105
RECOMENDACIONES
Del trabajo experimental se ha observado que el consumo de sustancias químicas que representan
a los compuestos modelo es muy alto utilizando el tamaño del reactor actual (un litro), dando como
resultado que cada experimento sea muy costoso. Por tal motivo una planta con un reactor
Robinson Manohey de tamaño más pequeño ayudaría notablemente a bajar los costos de la
experimentación.
La experiencia adquirida tanto en la operación de la planta piloto, así como también en el análisis
de los resultados experimentales permite establecer las siguientes recomendaciones en un trabajo
experimental futuro:
• El efecto de la presión en el proceso de hidrodesulfuración afecta de manera directa a la
conversión de los compuestos azufrados, de tal manera que un nuevo diseño de
experimentos debe incluir a la presión como una de las variables independientes
importantes del proceso.
• La determinación analítica del H2S deberá realizarse inmediatamente después que cada
una de las muestras sea obtenida, debido a que la presión parcial de este compuesto es
muy baja y se pierde con facilidad. Un cambio pequeño en las cantidades de este
compuesto influye de manera significativa en el balance de átomos entre entradas y
salidas de la planta piloto.
Se conoce de la literatura abierta y de la experiencia industrial y de laboratorio que la
hidrodesulfuración se ve impedida por la presencia de compuestos nitrogenados y/o aromáticos,
por lo que es recomendable realizar un estudio de la influencia de estos compuestos sobre el
proceso.
La hidrodesulfuración de dibenzotiofeno reportada en este trabajo no permite establecer las
condiciones de operación requeridas para obtener el nivel de azufre fijado por las regulaciones
ambientales nacionales y/o internacionales. De tal manera que se recomienda que el trabajo
experimental incluya a los compuestos refractarios contenidos en la cargas reales (4,6-
dimetildibenzotiofeno, 4-metildibenzotiofeno). Sin embargo, el alto costo de estos compuestos
puros impide trabajar con estos como compuestos modelo. Este hecho dirige el trabajo
experimental considerando el seguimiento del comportamiento de estos compuestos en cargas
reales.


107
REFERENCIAS 1. Arzate E., Figueroa Y., Del Rio H., Informe Técnico D.1029-TCM9.1.1 Determinación y
Reactividad de los Compuestos de Azufre presentes en Destilados Intermedios, Instituto
Mexicano del Petróleo (Programa de Tratamiento de Crudo Maya), (2001), Diciembre.
2. Arzate E., Muñoz J. A., Ramírez V., Cuarenta J. J., Morales A., García L. M., Abrego A. S.,
Chávez A., Bones L. R., Informe Técnico D.1029-TCM9.9, Construcción, prueba y arranque
de una planta piloto para estudios cinéticos en el hidrotratamiento de destilados intermedios.
3. Babich I. V. & Moulijn, J. A., Science and Technology of novel processes for deep
desulfurization of oil refinery streams: a review, Fuel, 82 (2003) 607-631
4. Bataille F., Lemberton J. L., Michaud Philippe, Pérot G., Vrinart M., Lemaire M., Shulz E.
Breysse M. Kasztelan S., Alkyldibenzothiophenes Hydrodesulfurization-Promoter Effect,
Reactivity, and Reaction Mechanism, J. Catal., 191 (2000) 409-422.
5. Bej S. K., Performance Evaluation of Hydroprocessing Catalysts- A Review of Experimental
Techniques, Energy & Fuels, 16 (2002) 3, 774-784
6. Berger R. J., Hugh Stitt, E., Marin G. B., Kapteijn F., Moulijn J. A., EUROKIN, Chemical
Reaction Kinetics in Practice, CATTECH, 5 (2001) 1, 30
7. Bos A. N. R., Lefferts L., Marin G. B., Steijns M. H. G. M., Kinetic Research on
Heterogeneously catalysed Processes: a Questionnaire on the State of Art in Industry, Appl.,
Catal. A:, 160 (1997) 185-190
8. Broderick D. H., High-pressure reaction chemistry and kinetics studies of hydrodesulfurization
of dibenzothiophene catalyzed by sulfided CoO-MoO3/γ-Al2O3, Ph D Thesis of University of
de Delaware, may, 1980
9. Broderick, D. H. and Gates, B. C., Hydrogenolysis and Hydrogenation of Dibenzothiophene
Catalyzed by Sulfided CoO-MoO3/γ-Al2O3 : Reaction Kinetics., AICHE J., 27 (1981) 663.
10. Daly F. P., Hydrodesulfurization of Benzothiophene over CoO-MoO3-Al2O3, J. Catal., 51
(1978) 221 11. Depauw G. A. and Froment G.F., Molecular analysis of the sulphur componts in a light cycle
oil of a catalytic cracking unit by gas chromatography with mass spectrometric and atomic
emission detection, Journal of Chromatography A, 761 (1997) 231-247
12. Edvinsson, R. and Irandoust, S., Hydrodesulfurization of Dibenzothiophene in a Monolithic
Catalyst Reactor., Ind. Eng. Chem. Res., 32 (1993) 391
13. Fogler H. S., “Elementos de Ingeniería de las Reacciones Químicas”, Prentice Hall, Tercera
edición, 2001
14. Froment G. F. and Bischoff K. B., Chemical Reactor Analysis and Design, Wiley Series in
Chemical Engineering, 2nd Edition, 1990

REFERENCIAS
108
15. Froment G. F., Depauw Guy A. and Vanrysselberghe V., Kinetic Modeling and Reactor
simulation in Hydrodesulfurization of Oil fractions., Ind. Eng. Chem. Res. 33 (1994) 2975-
2988.
16. Froment, G. F. Kinetic Modeling of Acid-Catalyzed oil refining Process., Cat. Today, 52
(1999) 153-163.
17. Gams, (http://www.gams.com/solvers/solvers.htm)
18. Geneste P., Amblard P., Bonnet M., Graffin P., Hydrodesulfurization of Oxidized Sulfur
Componets in Benzothiophene, Methyl Benzothiophene and Dibenzothiophene Series, J.
Catal., 61 (1980) 115
19. Girgis, M. J. and Gates B. C., Reactivities, reaction networks, and kinetics in high-pressure
catalytic hydroprocessing, Ind. Eng. Chem. Res., 30 (1991) 2021- 2058
20. Hattiangadi U, Spoor M, Pal SC. Petr Tech Quart (2000) 4:35
21. Houalla M., Nag N. K., Sapre A. V., Broderick, D. H. and Gates, B. C., Hydrodesulfurization of
Dibenzothiophene Catalyzed by Sulfided CoO-MoO3/γ-Al2O3 : Reaction Kinetics., AICHE J.,
24 (1978) 6, 1015.
22. Hougen O.A.,Watson K.M., Chemical Process Principles, Part Three, John Wiley& Sons, Inc,
1966.
23. Huss A., Production of Low Sulfur Gasoline and Diesel Fuel: Tier2 and Beyon, Refining
Process Services Inc., August, (2001) 15-17
24. Instituto Mexicano del Petróleo, Manual de Operación de Catalizadores IMP-DSD para
Hidrotratamiento, Primera Edición, 1994.
25. Kittrell, J.R. Mathematical Modeling of Chemical Reactions, Adv. Chem. Eng., 8 (1996) 97
26. Koehloefl, K., Development of industrial solid catalysts, Chem. Eng. Technol., 24 (2001) 3
27. Koltai T., Macaud M., Guevara A. E. Schulz, Lemaiare M. Bacaud R. and Vrinat M.,
Comparative inhibiting effect of polycondensed aromatics and nitrogen compounds on the
hydrodesulfurization of alkyldibenzothiophenes, App. Catal. A:, 231 (2002) 253-261.
28. Korsten H., Hoffmann U., Three-phase reactor model for hydrotreating in pilot trickle-bed
reactors, AICHE J., 42 (1996) 5, 1350
29. Krishnamyrthy S. Panvelkar S. Shah Y. T., Hydrodeoxigenation of Dibenzofuran and Related
Compounds, AICHE J., 27 (1981) 994
30. Laredo S. C. G., De los Reyes H. J. A., Cano D. J. L. and Castillo M. J. J., Inhibition effects of
nitrogen compounds on the hydrodesulfurization of dibenzothiophene., Appl. Catal. 207
(2001) 103-112.
31. Larivé J.F., Low Sulfur Fuels in Europe: An Analysis, Hydrocarbon Engineering, 6 (2001) 4,
15

REFERENCIAS
109
32. Le page J. F., Cosyns J., Courty P., Freund E., Franck J. P., Jacquin Y., Juguin B., Marcilly
C., Martino G., Miquel J. , Montarnal R., Sugier A., Van Landeghem H. Applied
Heterogeneous Catalysis: Design, Manufacture, Use of Solid Catalysts, Technip
Publications, Paris, 1987
33. Mahoney J. A., Robinson K. K. And Myers E. C., Catalyst Evaluation with the Gradientless
Reactor, Chemtech, December, 8 (1978) 758
34. Manual de Operación de Catalizadores IMP-DSD para Hidrotratamiento, Instituto Mexicano
del Petróleo, Primera Edición, 1994
35. Marquardt D.W., An algorithm for least-squares estimation of Nonlinear Parameters, J. Soc
Indust. Appl. Math, 11(1963) 2
36. Marroquín R. O., Valencia L. J., Modelo del Reactor de Hidrotratamiento de Diesel, Informe
Técnico D.-1029-TCM9.2, Programa de Tratamiento de Crudo Maya, Diciembre de 2002.
37. Martens G. G., Marin G. B., Martens J. A., Jacobs P. A. and Baron G. V., A fundamental
kinetic model for hydrocracking of C8 to C12 alkanes on Pt/US-Y zeolites, J. Catal., 195 (2000)
253-267
38. Martínez M. C., Obtención de Diesel a partir del hidrotratamiento de mezclas de gasóleo
ligero primario y de aceite cíclico ligero, Tesis de Maestría, IPN-ESIQIE, septiembre 2001.
39. Mendenhall W., Sincich T., Probabilidad y Estadística para Ingeniería y Ciencias. Prentice
Hall Hispanoamericana, S.A., 4ª. Edición. 1997.
40. Mijoin J., Pérot G., Bataille F., Lemberton J. L, Breysse M., Kasztelan S., Mechanistic
considerations on the involvement of of dihydrointermediates in the hydrodesulfurization of
dibenzothiophene-type compounds over molybdenum sulphide catalysts, Catal. Letters, 71
(2001) 3-4, 139-145.
41. Miller J. T., Hineman M. F., Non first-order. Hydrodenitrogenation Kinetics of Quinoline, J.
Catal. 85 (1984) 117
42. Miller RB, Macris A, Gentry AR. Petr Tech Quart 2001;2,69
43. Muñoz Arroyo J. A., Hydrocracking and isomerization of n-paraffin mixtures and a
hydrotreated gasoil on Pt/ZSM-22: confirmation of pore mouth and key-lock catalysis in liquid
phase, Ph Thesis, Universiteit Gent, Belgium, 2000
44. Prada R., Future trends in the refining catalyst market, Applied Catalysis A: General, 261
(2004) 247–252
45. Reid R. C., Prausnitz J. M. and Sherwood T. K., The Properties of Gases & Liquids, Mc Graw
Hill 3rd Edition, 1977
46. Rosenbrock H. H. and Storey C. Computational Techniques for Chemical Engineers,
Pergamon Press 1966.

REFERENCIAS
110
47. Santes V., Herbert J., Cortez M.T., Zarate R., Díaz L., Narayana P., Aouine M., Vrinat,
Catalytic hydrotreating of heavy gasoil FCC feed on alumina–titania-supported NiMo
catalysts, Applied Catalysis A: General, 281 (2005) 121–128
48. Shafi R. and Hutchings J. G., Hydrodesulfurization of hindered dibenzothiphenes: an
overview, Cat. Today, 59 (2000) 423-442.
49. Statterfield C. N., Yang S. H., Hydrodenitrogenation of Quinoline in a Trickle Bed Reactor,
Ind. Eng. Chem. Process Des. Dev., 23 (1984) 11
50. Taller de validación de Métodos Analíticos, Centro Educacional Analítica, Agilent
Technologies. Method Validation. www.labcompliance.com/methods/meth_val.htm
51. Tikhonov, G. F., Shestov G. K., Temkin O. N., Flid R. M., Gradientless Reactor Suitable for
Studying the Kinetics of Liquid Phase Reactions in Gas-Liquid Systemas, Kinetika Kataliz, 7
(1986) 5, 914, Sep-Oct,.
52. Tsamatsoulis D., Papayannakos N., Investigation of intrinsic hydrodesulphurization kinetics of
a VGO in a trickle bed reactor with backmixing effects Chem. Eng. Sci., 53 (1998) 3449
53. Van Parijs I. A. and Froment G. F., The influence of intraparticle diffusional in naphtha
hydrodesulfurization., I. CHEM. E. Symposium Series 87, (1984) 319-327.
54. Van Parijs I (a). A., Hosten L. H. and Froment G. F., Kinetics of Hydrodesulfurization on a
CoMo/γ-Al2O3 Catalyst: 2. Kinetics of the hydrogenolysis of Thiophene., Ind. Eng. Chem.
Prod. Res. Dev. 25 (1986) 431.
55. Van Parijs I (b). A., Hosten L. H. and Froment G. F., Kinetics of Hydrodesulfurization on a
CoMo/γ-Al2O3 Catalyst: 2. Kinetics of the hydrogenolysis of Benzothiophene., Ind. Eng.
Chem. Prod. Res. Dev. 25 (1986) 437.
56. Vanrysselberghe V. and Froment G. F., Hydrodesulfurization of Dibenzothiphene on a
CoMo/Al2O3 Catalyst: reaction Network and Kinetics., Ind. Eng. Chem. Res. 35, (1996) 3311-
3318.
57. Vanrysselberghe V., Le Gall R., and Froment G. F., Hydrodesulphurization of 4-
Methyldibenzothiphene and 4, 6- Dimethyldibenzothiphene on a CoMO/Al2O3 Catalyst:
Reaction Network and Kinetics., Ind. Eng. Chem. Res. 37, (1998) 1235-1242.
58. Vanrysselberghe V. and Froment G. F., Kinetic Modeling of Hydrodesulfurization of Oil
Fractions: Light Cycle Oil., Ind. Eng. Chem. Res. 37, (1998) 4231-4240.
59. Vradman L., Landau M. V., Herskowitz, M., Deep desulfurization of diesel fuels: Kinetic
modeling of model compounds in trickle-bed, Catal. Today, 48 (1999) 41
60. Whitehurst D. D., Isoda T. and Mochida I., Present State of the art and future Challenges in
the Hydrodesulfurization of Polyaromatic Sulfur Compounds., Adv. in Catal., 42, (1998) 345-
467

REFERENCIAS
111
61. Wiwel P. , Knudsen, K., Zeuthen P. and Whitehurst D., Assessing Compositional Changes of
Nitrogen Compounds during Hydrotreating of Typical Diesel Range Gas Oils Using a Novel
Preconcentration Technique Coupled with Gas Chromatography and Atomic Emission
Detection, Ind. Eng. Chem. Res.,39 ( 2000) 533-540
62. Xiaoliang Ma, Kinya S. and Isao M., Hydrodesulfurization Reactivities of Various Sulfur
Compounds in Diesel Fuel. Ind. Eng. Chem. Res., 33 (1994) 218-222.
63. Yui S. M., Sanford E. C. Mild hydrocracking of bitumen-derived coker and hydrocracker
heavy gas oils: kinetics, product yields, and product properties, Ind. Eng. Chem. Res., 28 (1989) 1278
64. US EPA Clean Air Act Tier2; 1999


APÉNDICES


APÉNDICE A
OPERACIÓN DE LA PLANTA PILOTO


APÉNDICE A
A.1
A. OPERACIÓN DE LA PLANTA PILOTO
La confiabilidad en la obtención de datos experimentales radica principalmente en la operación del
equipo experimental de planta piloto, sin embargo el manejo del catalizador, el arranque y
estabilidad de la planta piloto así como también la medición y toma de muestras son aspectos
importantes a considerar en este apéndice.
A.1. PROCEDIMIENTO DE ACTIVACIÓN DEL CATALIZADOR
La activación del catalizador tiene como propósito fundamental convertir los óxidos ú oxisulfuros
metálicos de Mo, Co, y Ni, contenidos en el catalizador a sulfuros metálicos, los cuales constituyen
la especie catalíticamente activa. El mejoramiento continuo en la formulación, distribución de
centros activos y área superficial de los catalizadores de hidrotratamiento, ha promovido el
desarrollo de métodos de activación y el uso de compuestos orgánicos de azufre empleados como
agentes sulfhidrantes [Shafi R. & Hutchings J. G, 2000].
En la activación fuera del reactor, se distribuye azufre sobre el catalizador mediante un sistema de
impregnación rotatorio empleando para ello compuestos órgano sulfurados mezclados en una
solución con un solvente alifático ligero.
La concentración de la solución se ajusta de acuerdo al volumen de poro y al contenido metálico,
para obtener el nivel de azufre deseado en el catalizador. El catalizador presulfhidrado no es tóxico
y puede ser manejado en condiciones atmosféricas, tiene un color oscuro debido a la presencia de
hidrocarburos, los cuales son eliminados durante la activación con hidrógeno.
Aunque comúnmente se habla de catalizadores presulfhidrados esta aseveración es incorrecta ya
que ninguna de las tecnologías actuales convierte totalmente los óxidos metálicos a sulfuros
metálicos sino a compuestos intermedios llamados oxisulfuros metálicos. Cuando estos
compuestos se calientan en una atmósfera de hidrógeno, se rompen y forman los sulfuros
metálicos directamente. El término presulfurizar se refiere sólo al depósito de azufre en la
superficie del catalizador y no a la formación de la especie catalíticamente activa; por ello, es más
apropiado hablar de catalizadores presulfurizados que de catalizadores presulfhidrados.
En la activación del catalizador, los oxisulfuros metálicos se transforman en sulfuros metálicos de

APÉNDICE A
A.2
acuerdo a las siguientes reacciones:
MoOxSy + χ H2 → MoS2 + χ H2O A.1
9 CoOxSy + χ H2 → Co9S8 + χ H2O A.2
El sulfhidrado se debe efectuar en presencia de hidrógeno para favorecer el desplazamiento del
oxígeno y formar agua, de lo contrario, la tendencia será a formar óxidos menores o metales puros,
los que difícilmente son convertidos a sulfuros metálicos. La activación de un catalizador
presulfurizado se realiza sin necesidad de adicionar una cantidad extra de azufre al catalizador y
puede llevarse a cabo en fase gas, fase líquida o en fase mixta. La activación se hace patente por
la presencia de una exoterma en el reactor y la formación de agua, las cuales crean un peligro
potencial de daño al catalizador ya que pueden causar una distribución anómala de los sulfuros
metálicos o modificar la estructura del soporte
La activación se realiza a la presión y tipo de carga típicas de la unidad. La temperatura en el
reactor no debe exceder 563 K antes de que el catalizador haya sido activado. Si se excediera
563 K antes de la activación del catalizador, se causaría una reducción de metales, lo cual ocurre
en presencia de H2 a temperaturas mayores de 573 K y tiempos prolongados y que trae como
consecuencia la necesidad de oxidar nuevamente el catalizador ya que no es posible activarlo a
partir del estado reducido.
Durante la activación, se recomienda registrar las temperaturas de entrada y salida del reactor,
presión, flujos de carga y gas de recirculación, contenidos de azufre en carga y producto y
contenido de ácido sulfhídrico en el gas amargo.
El reactor Robinson Mahoney contiene una canasta donde el catalizador se deposita. Esta canasta
se empaca y carga con el catalizador IMP-DSD-14 (Co-Mo/Al2O3) con un tamaño de partícula entre
800 -1000 µM asegurando con esto una mínima resistencia al transporte de masa y calor. La
cantidad de catalizador cargado corresponderá con el LHSV de acuerdo a la corrida en el diseño
de experimentos. La canasta ya preparada se introduce al reactor el cual se cierra herméticamente
de acuerdo a las instrucciones del manual del equipo.
• Antes de iniciar la etapa de activación y después de haber cargado el catalizador nuevo

APÉNDICE A
A.3
presulfurizado, se establece la presión a 0.981 MPa en el regulador de presión (PCV 61) y se
comienza la alimentación de H2 (50 % del valor máximo), revisando la hermeticidad de la
planta.
• En este punto de arranque se enciende el agitador magnético y se fija su velocidad a 1500
rpm.
• Una vez que no existan fugas en el sistema se continúa el incremento de la presión hasta
3.9 MPa a través del regulador de presión (PCV 61).
• Manteniendo el mismo nivel de flujo de H2 comenzar con el drenado de agua formada por las
reacciones A.1 – A.2 en el separador (FA-02).
Aumentar la temperatura del reactor y equipos periféricos corriente abajo y corriente arriba
hasta un valor de 513 K a razón de 303 K/h y mantenerla durante una hora. Purgar
frecuentemente el agua en el separador de alta presión y alta temperatura (FA-02).
A lo largo de toda la etapa de activación, registrar temperaturas de entrada y salida del
reactor, presión, flujo de gas, contenido de H2S en el gas.
Revisar el contenido de H2S en el gas amargo y el volumen de agua purgada en el separador
de productos, hasta que estos hayan disminuido.
Una vez alcanzada la condición anterior se establece el flujo de hidrógeno al nivel del plan de
experimentación.
Manteniendo el nivel de temperatura y flujo de H2, se introduce carga líquida con referencia al
espacio velocidad 1.2 h-1. Esperar hasta que las condiciones de operación se estabilicen.
A.2. PROCEDIMIENTO DE ARRANQUE DE LA PLANTA PILOTO El arranque de la planta piloto se lleva a cabo ajustando la temperatura, presión y flujo de de las
corrientes de alimentación a los valores típicos dentro del plan de experimentación.

APÉNDICE A
A.4
Las líneas del gas producido que conectan al cromatógrafo de gases deben de ser calentadas
a la temperatura de 498 K a manera de evitar cualquier condensación que provoque problemas
en la operación del cromatógrafo de gases y afecta el análisis de la corriente respectiva.
Una vez que la presión y temperaturas han sido alcanzadas, el flujo de hidrógeno y la
alimentación líquida se fijan a los valores del experimento en turno. Al mismo tiempo la
velocidad de agitación se fija a 1500 rpm con el objeto de mantener la recirculación de la
mezcla liquido-gas en la canasta de catalizador.
A.3. ESTABILIDAD, MEDICIÓN Y TOMA DE MUESTRAS
De acuerdo a las características del reactor Robinson-Mahoney es necesario esperar un
determinado tiempo de estabilización el cual depende del valor del flujo volumétrico del
experimento en turno, sin embargo es recomendable comenzar la experimentación con el flujo
mayor con el objeto de que el reactor sea llenado más rápidamente y por consiguiente el tiempo de
estabilización se reduzca, este tiempo puede estar en el rango entre 46.8 – 900 Ks (13 - 25
horas).
La mezcla líquido-vapor que sale del reactor se separa en el equipo de alta presión y temperatura
FA-02. Por el domo de este equipo se obtiene una corriente gaseosa rica en H2 y H2S, mientras
que por el fondo se obtiene una corriente líquida con gases disueltos.
La mezcla gaseosa conteniendo pequeñas gotas de mezcla líquida se separan en el equipo de alta
presión y baja temperatura FA-03. El líquido remanente se mezcla con la corriente líquida del fondo
del equipo FA-02.
La mezcla gaseosa obtenida en el domo del FA-03 se envía al equipo cromatográfico para
determinar su composición. Este equipo tiene instalados detectores de conductividad térmica para
determinar la composición de H2 y uno fotométrico de flama para cuantificar el H2S producido.
El separador FA-02 tiene un controlador de nivel adaptado a una válvula la cual se abre cuando
se alcanza el nivel calibrado de líquido. La mezcla liquido-vapor sufre una pérdida de presión.
Como consecuencia el gas disuelto en el líquido se separa de la mezcla. El vapor y líquido
separados se recolectan y se miden en la bureta y probeta respectivamente. Varias mediciones se

APÉNDICE A
A.5
realizan hasta que el estado estacionario del sistema de reacción es alcanzado.
El estado estacionario puede ser visualizado en el momento en que las mediciones de volumen de
líquido y de vapor se repitan y el rango de desviación entre una y otra no rebase el 5%.
Una vez alcanzado el estado estacionario es necesario recuperar al menos 2 muestras durante las
subsecuentes mediciones para determinar la composición tanto de líquido como del vapor. La toma
de muestra líquida se debe realizar cuando esta se haya estabilizado (temperatura de 433 K y
presión atmosférica). La muestra líquida se debe almacenar en un frasco ámbar y rotularlo,
indicando el número de balance para cada uno de ellos.
Finalmente se reporta la composición de gas, líquido y vapor así como también los flujos tanto de
la carga como de los productos para realizar el cálculo de balance de átomos y/o balance de
materia.


APÉNDICE B
CALIBRACIÓN DE LOS EQUIPOS DE MEDICIÓN


APÉNDICE B
B.1
B CALIBRACIÓN DE LOS EQUIPOS DE MEDICIÓN Una de las primeras actividades que deben llevarse a cabo dentro de la obtención de los datos
experimentales en una planta piloto es la calibración de los equipos de medición que serán
utilizados en la medición de las condiciones de operación, entrada y salida de la planta. Con esta
actividad se obtendrá la precisión de los equipos de medición tanto para gases como para líquidos.
Este parámetro influirá directamente en el error global que se calcula en el balance de masa de la
planta. A continuación se detallan las características de cada uno de estos equipos y se reportan
las desviaciones, rangos de aplicabilidad e intervalos de confianza que se obtuvieron en esta.
B.1 CONTROLADORES Y MEDIDORES DE FLUJO MÁSICO B.1.1. Controladores para el Flujo de Carga Líquida Para obtener un balance de masa confiable en la planta piloto (HDS-1) localizada en el área I del
IMP, se considero que el sistema de control debe tener suficiente exactitud para obtener errores
globales menores o iguales al 5%, lo cual estadísticamente significa que los valores de los
parámetros obtenidos en este rango serán confiables. Primeramente se realizó una calibración de
las bombas utilizadas para la alimentación líquida a la planta, las cuales manejan diferentes flujos
cada una: GA-01 Milton Roy y GA-01-A MiniPump.
En la Figura B.1 se puede observar la relación entre el flujo másico y el % de carrera de la bomba
Milton Roy a condiciones de presión de 0.77 bar (585 mm Hg) y temperatura de 294 K. En esta
gráfica se puede observar una desviación importante para un flujo de aproximadamente 50 µKg/s
(180 g/h). Para esta misma bomba la Tabla B.1 muestra un resumen de la calibración en % peso y
volumen con sus respectivas desviaciones estándar. De esta información se observan las mayores
desviaciones a los valores más bajos y altos del flujo. No obstante estos resultados, esta bomba
trabajando a un flujo de 27.7חM3/s (100 ml/h) es muy inestable. Consecuentemente el manejo de
flujos de carga alrededor de este rango se utiliza la bomba MiniPump. Por lo que dependiendo del
flujo calculado del experimento a realizarse se utiliza una u otra bomba.

APÉNDICE B
B.2
Bomba tipo diafragma Curva de calibración en peso
0
100
200
300
400
500
600
700
17 22 27 32 37 42
% de carrera
Prom
edio
de F
lujo g
/h
Figura B.1. Calibración de la bomba GA-01Milton Roy en volumen
Tabla B.1 Calibración de la Bomba Milton Roy
% Carrera
Variables de Prueba
18.30 21.40 29.60
25.90 41.10 35.20
Flujo Promedio µKg/s(g/h)
5.83 (21.0)
28.75 (103.5)
50.83 (183.0)
45.0 (162.0)
158.88 (572.0)
117.36 (422.50)
Promedio M3/s(ml/h)ח
7.22 (26.00)
33.89 (122.0)
61.39 (221.0)
54.17 (195.0)
188.89 (680.0)
140.28 (505.0)
Desviación Estándar Peso 7.39 5.51 6.0 1.63 5.29 13.70 Volumen 8.33 7.66 6.0 2.0 4.00 17.09
La gráfica de calibración de la bomba MiniPump puede observarse en la Figura B.2, de los datos
de esta Figura se obtiene un coeficiente de Pearson muy cercano a 1, lo cual indica que existe una
funcionalidad casi lineal del porcentaje de carrera y el flujo en el rango de 20 a 170 ml/h.
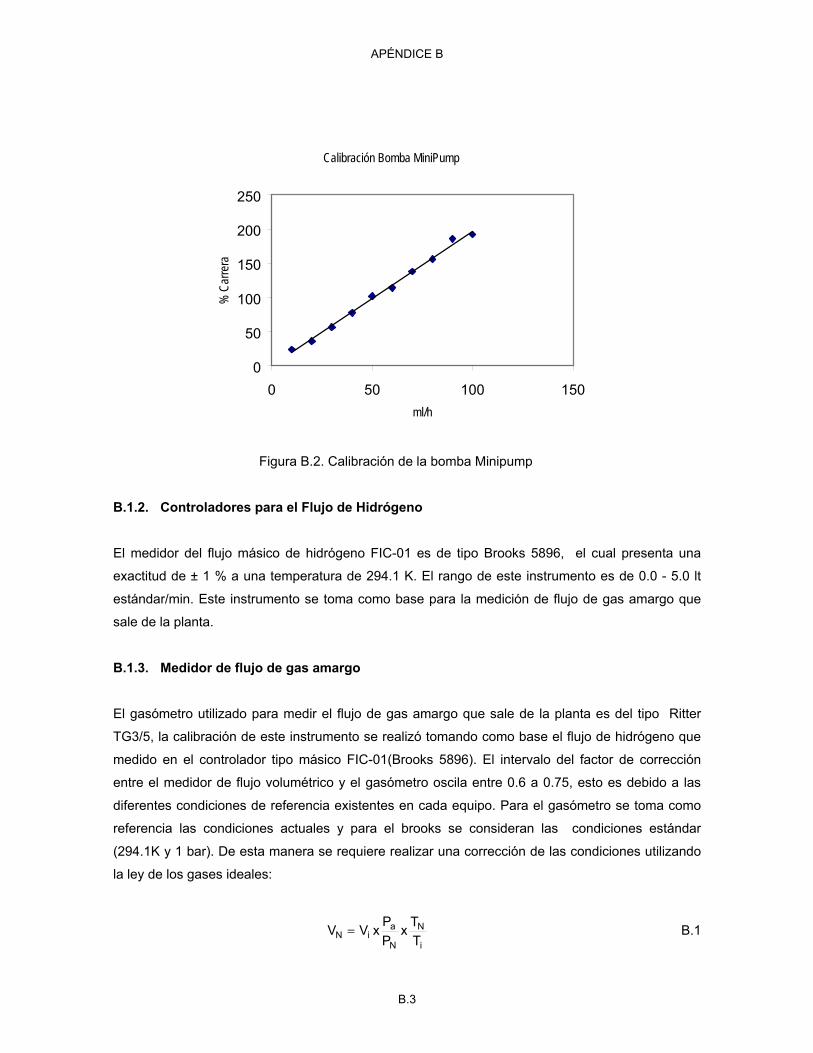
APÉNDICE B
B.3
Calibración Bomba MiniPump
0
50
100
150
200
250
0 50 100 150ml/h
% C
arre
ra
Figura B.2. Calibración de la bomba Minipump
B.1.2. Controladores para el Flujo de Hidrógeno
El medidor del flujo másico de hidrógeno FIC-01 es de tipo Brooks 5896, el cual presenta una
exactitud de ± 1 % a una temperatura de 294.1 K. El rango de este instrumento es de 0.0 - 5.0 lt
estándar/min. Este instrumento se toma como base para la medición de flujo de gas amargo que
sale de la planta.
B.1.3. Medidor de flujo de gas amargo El gasómetro utilizado para medir el flujo de gas amargo que sale de la planta es del tipo Ritter
TG3/5, la calibración de este instrumento se realizó tomando como base el flujo de hidrógeno que
medido en el controlador tipo másico FIC-01(Brooks 5896). El intervalo del factor de corrección
entre el medidor de flujo volumétrico y el gasómetro oscila entre 0.6 a 0.75, esto es debido a las
diferentes condiciones de referencia existentes en cada equipo. Para el gasómetro se toma como
referencia las condiciones actuales y para el brooks se consideran las condiciones estándar
(294.1K y 1 bar). De esta manera se requiere realizar una corrección de las condiciones utilizando
la ley de los gases ideales:
i
N
N
aiN T
TxPPxVV = B.1

APÉNDICE B
B.4
La Figura B.3 muestra la medición del flujo del gas amargo durante un período de 50 hrs. De los
datos de esta Figura se puede observar una desviación importante en la medición del flujo con
respecto al valor medio, por lo que es necesario una calibración frecuente de este equipo.
15.0
18.0
21.0
24.0
27.0
30.0
0.0 20.0 40.0 60.0
Hrs
Lt/hrs
Fig. B.3. Estabilidad para la medición del Flujo de gas Amargo

APENDICE C
CALIBRACIÓN DEL CROMATÓGRAFO DE
GASES


APÉNDICE C
C.1
Metodología de calibración para el cromatógrafo de gases
Para determinar la exactitud de una mezcla de varios componentes, se realizó el análisis de una
muestra con concentraciones conocidas (material de referencia certificado) y se compararon los
valores medidos con los valores verdaderos.
De acuerdo a la metodología que existe en plantas piloto del IMP se realizan seis repeticiones del
estándar de la mezcla de interés.
Para el análisis cuantitativo de cada componente de la mezcla a analizar se consideraron los
criterios estadísticos siguientes: la media, la desviación estándar, el coeficiente de variación,
intervalo de confianza y porcentaje de recuperación los cuales son definidos por Mendenhall W. &
Sincich T., 1997. La desviación estándar (S) y la media (M) de los resultados obtenidos al introducir
la mezcla estándar están reportados en la Tabla C.1. El criterio de aceptación para el coeficiente
de variación es menor o igual al 2 %. De los datos de la Tabla C.2 se observa que este criterio se
cumple en los compuestos ligeros (C1-nC4). Para moléculas más pesadas existe una ligera
desviación a este criterio.
Al comparar la composición estimada con respecto a la composición de referencia se observa que
esta no es mayor al 0.001 %. El intervalo de confianza (IC) para las concentraciones de cada
componente esta reportado también en esta Tabla, además se observa que la recuperación (R) es
muy cercana al 100 %. El coeficiente de variación de la mezcla analizada es de 0.1016 lo cual
indica que la determinación de la concentración por componente es muy aproximada por este
método.
La Figura C.1 muestra la calibración de H2S considerando los resultados de área vs concentración.
Los datos de presentados se ajustan adecuadamente a una línea recta con un coeficiente de
correlación (R = 0.984), es decir, el área del pico es directamente proporcional a la concentración.
La desviación de los tiempos de retención con respecto a la media (Tabla C.1) para cada
compuesto es menor de 0.1% indicando una buena precisión del método cromatográfico y un
análisis cualitativo aceptable de los componentes de la mezcla. Para él calculo del intervalo de
confianza se utilizan las tablas de distribución de Student con 5 grados de libertad , t0.025 = 2.57.
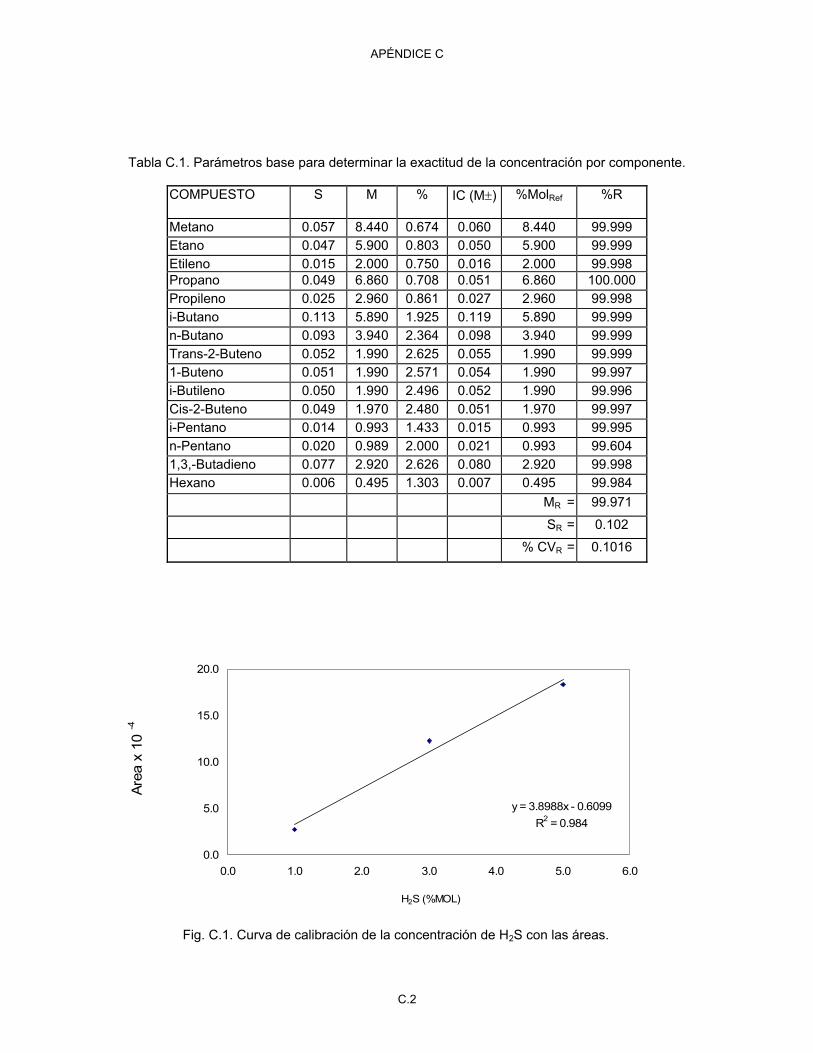
APÉNDICE C
C.2
Tabla C.1. Parámetros base para determinar la exactitud de la concentración por componente.
COMPUESTO S M % IC (M±)
%MolRef %R
Metano 0.057 8.440 0.674 0.060 8.440 99.999 Etano 0.047 5.900 0.803 0.050 5.900 99.999 Etileno 0.015 2.000 0.750 0.016 2.000 99.998 Propano 0.049 6.860 0.708 0.051 6.860 100.000 Propileno 0.025 2.960 0.861 0.027 2.960 99.998 i-Butano 0.113 5.890 1.925 0.119 5.890 99.999 n-Butano 0.093 3.940 2.364 0.098 3.940 99.999 Trans-2-Buteno 0.052 1.990 2.625 0.055 1.990 99.999 1-Buteno 0.051 1.990 2.571 0.054 1.990 99.997 i-Butileno 0.050 1.990 2.496 0.052 1.990 99.996 Cis-2-Buteno 0.049 1.970 2.480 0.051 1.970 99.997 i-Pentano 0.014 0.993 1.433 0.015 0.993 99.995 n-Pentano 0.020 0.989 2.000 0.021 0.993 99.604 1,3,-Butadieno 0.077 2.920 2.626 0.080 2.920 99.998 Hexano 0.006 0.495 1.303 0.007 0.495 99.984
MR = 99.971
SR = 0.102
% CVR = 0.1016
y = 3.8988x - 0.6099R2 = 0.984
0.0
5.0
10.0
15.0
20.0
0.0 1.0 2.0 3.0 4.0 5.0 6.0
H2S (%MOL)
Area
x 1
0 -4
Fig. C.1. Curva de calibración de la concentración de H2S con las áreas.

APÉNDICE C
C.3
Tabla C.2. Repetibilidad del tiempo de retención.
IC COMPUESTO S M %CV Límite Inferior Límite Superior
Metano 0.001 3.467 0.024 3.466 3.467 Etano 0.002 3.886 0.042 3.885 3.888 Etileno 0.004 4.486 0.084 4.482 4.490 Propano 0.006 6.054 0.092 6.048 6.060 Propileno 0.007 8.896 0.083 8.888 8.903 i-Butano 0.006 10.309 0.059 10.302 10.315 n-Butano 0.008 10.844 0.073 10.835 10.852 Trans-2-Buteno 0.010 13.155 0.078 13.145 13.166 1-Buteno 0.009 13.293 0.068 13.284 13.303 i-Butileno 0.009 13.685 0.066 13.675 13.695 Cis-2-Buteno 0.010 14.036 0.070 14.025 14.046 i-Pentano 0.009 14.958 0.063 14.948 14.968 n-Pentano 0.010 15.390 0.066 15.380 15.401 1,3,-Butadieno 0.009 15.790 0.055 15.780 15.799 Hexano 0.010 19.331 0.053 19.320 19.342 La desviación de las áreas con respecto a la media (Tabla C.3) para cada compuesto es menor de
2.6 %, tomando como base al coeficiente de variación. Esto indica una buena precisión del método
cromátografico y un análisis cuantitativo aceptable de los componentes de la mezcla. Para él
cálculo de esta desviación se utilizan las tablas de distribución de Student con 5 grados de
libertad, t 0.025 = 2.57.
Tabla C.3. Desviación de las áreas con respecto a la media.
IC COMPUESTO S M %CV Límite inferior Límite superior
Metano 1192 176918 0.674 175667 178169 Etano 1909 237839 0.803 235837 239842 Etileno 608 81189 0.749 80551 81827 Propano 2955 417217 0.708 414116 420317 Propileno 1544 179335 0.861 177715 180955 i-Butano 9087 471941 1.925 462407 481474 n-Butano 8715 368758 2.363 359614 377901 Trans-2-Buteno 4094 155979 2.625 151684 160275 1-Buteno 4104 159676 2.570 155370 163981 i-Butileno 3886 155632 2.497 151555 159709 Cis-2-Buteno 3876 156344 2.479 152277 160411 i-Pentano 1449 101063 1.434 99543 102583 n-Pentano 1982 101670 1.949 99591 103750 1,3,-Butadieno 5975 227477 2.627 221208 233745 Hexano 768 58950 1.303 58144 59756

APÉNDICE C
C.4
Considerando los resultados presentados en las Tablas C.1, C.2 y C.3 se puede considerar con un
95% de certeza que el valor estimado de la concentración, áreas y tiempos de retención para cada
componente se encuentran dentro del intervalo de confianza.

APENDICE D
ANÁLISIS ESTADÍSTICO DE LOS DATOS
EXPERIMENTALES

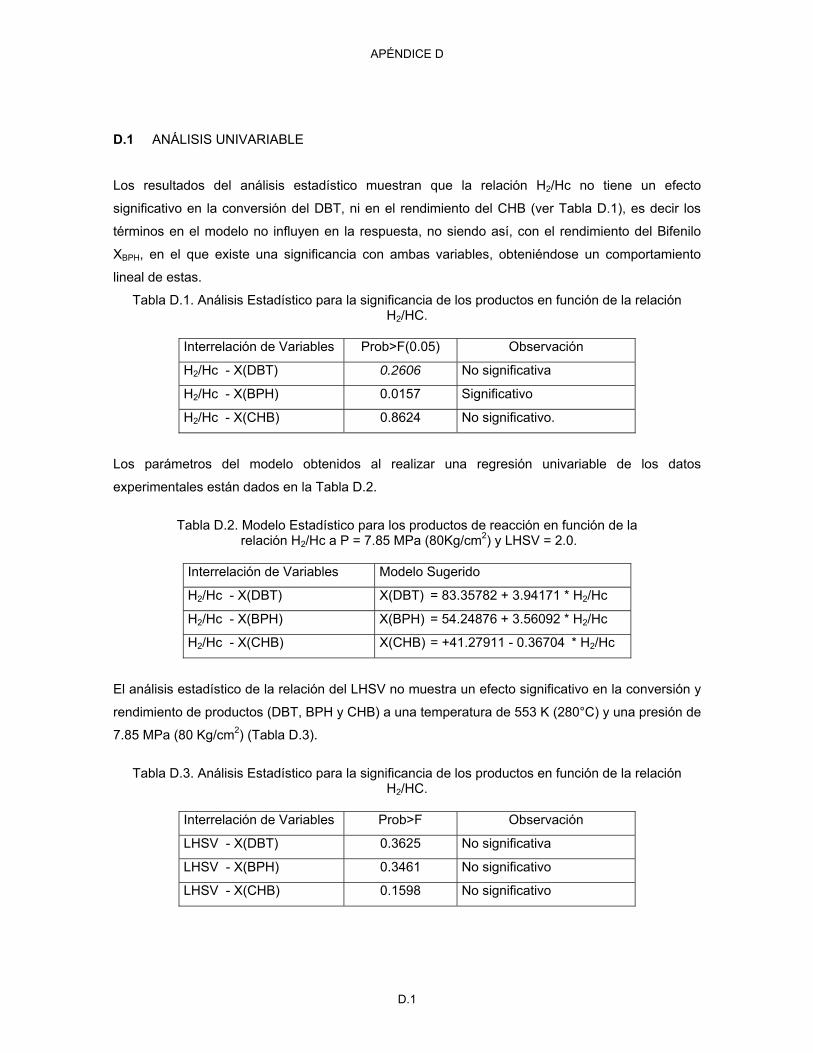
APÉNDICE D
D.1
D.1 ANÁLISIS UNIVARIABLE
Los resultados del análisis estadístico muestran que la relación H2/Hc no tiene un efecto
significativo en la conversión del DBT, ni en el rendimiento del CHB (ver Tabla D.1), es decir los
términos en el modelo no influyen en la respuesta, no siendo así, con el rendimiento del Bifenilo
XBPH, en el que existe una significancia con ambas variables, obteniéndose un comportamiento
lineal de estas.
Tabla D.1. Análisis Estadístico para la significancia de los productos en función de la relación H2/HC.
Interrelación de Variables Prob>F(0.05) Observación
H2/Hc - X(DBT) 0.2606 No significativa
H2/Hc - X(BPH) 0.0157 Significativo
H2/Hc - X(CHB) 0.8624 No significativo.
Los parámetros del modelo obtenidos al realizar una regresión univariable de los datos
experimentales están dados en la Tabla D.2.
Tabla D.2. Modelo Estadístico para los productos de reacción en función de la
relación H2/Hc a P = 7.85 MPa (80Kg/cm2) y LHSV = 2.0.
Interrelación de Variables Modelo Sugerido
H2/Hc - X(DBT) X(DBT) = 83.35782 + 3.94171 * H2/Hc
H2/Hc - X(BPH) X(BPH) = 54.24876 + 3.56092 * H2/Hc
H2/Hc - X(CHB) X(CHB) = +41.27911 - 0.36704 * H2/Hc
El análisis estadístico de la relación del LHSV no muestra un efecto significativo en la conversión y
rendimiento de productos (DBT, BPH y CHB) a una temperatura de 553 K (280°C) y una presión de
7.85 MPa (80 Kg/cm2) (Tabla D.3).
Tabla D.3. Análisis Estadístico para la significancia de los productos en función de la relación
H2/HC.
Interrelación de Variables Prob>F Observación
LHSV - X(DBT) 0.3625 No significativa
LHSV - X(BPH) 0.3461 No significativo
LHSV - X(CHB) 0.1598 No significativo

APÉNDICE D
D.2
Para este caso la relación de las variables resulta en un modelo lineal cuyos parámetros se
encuentran reportados en la Tabla D.4.
Tabla D.4. Modelo Estadístico para los productos de reacción en función de la relación LHSV a
P = 7.85 MPa (80Kg/cm2) y H2/Hc = 1.33
Interrelación de Variables Modelo Sugerido
LHSV - X(DBT) X(DBT) = 89.40367 -3.49407 * LHSV
LHSV - X(BPH) X(BPH) = 58.77956 –1.77254 * LHSV
LHSV - X(CHB) X(CHB) = 26.85747-1.67768 * LHSV
Un primer resultado del análisis anterior muestra que la influencia de la relación H2/HC es pequeña
en comparación con el LHSV trabajando a 553 K (280 °C). No obstante estos resultados, la
influencia de la temperatura en el rendimiento de productos del hidrotratamiento es determinante,
consecuentemente se requiere un análisis multivarible, en el que también participe la temperatura a
manera de establecer la influencia de la combinación de estos tres parámetros. De esta manera se
realizó el análisis estadístico de los datos experimentales de la etapa I, considerando una variación
de la temperatura a diferentes LHSV y diferentes relaciones H2/HC.
El análisis estadístico consideró la conversión del DBT en función de las tres variables:
Temperatura, LHSV, H2/Hc. Los datos experimentales corresponden con los ya presentados en la
Tabla 4.7. El análisis estadístico muestra que existe una dependencia lineal de la conversión del
DBT con la temperatura y el LHSV. La dependencia con respecto a la relación H2/HC es no
significativa. Sin embargo, el rango de prueba para estos dos últimos factores se podría considerar
constante. El análisis estadístico involucra también la determinación de un modelo que incluye la
interrelación de cada parámetro involucrado y la conversión del DBT. Los resultados de este
análisis involucrando los parámetros del modelo lineal son reportados en la Tabla D.5.
Tabla D.5. Modelo lineal multivariable en función de la temperatura, LHSV y H2/HC
Variables Modelo
X(DBT) – [Temperatura, LHSV, H2/Hc] X(DBT) = 213.38568 + 0.14936 * Temperatura
- 45.73087 * LHSV - 25.41125 * H2/Hc
En la Figura D.1.a, se muestra la relación obtenida al mantener la relación H2/HC a un valor
constante de 3.0, el cuál corresponde con el reportado en planta industrial. El comportamiento
típico de este parámetro muestra que la disminución del LHSV y el incremento de la temperatura
da como resultado un mayor nivel de conversión para el DBT. El comportamiento que tiene la
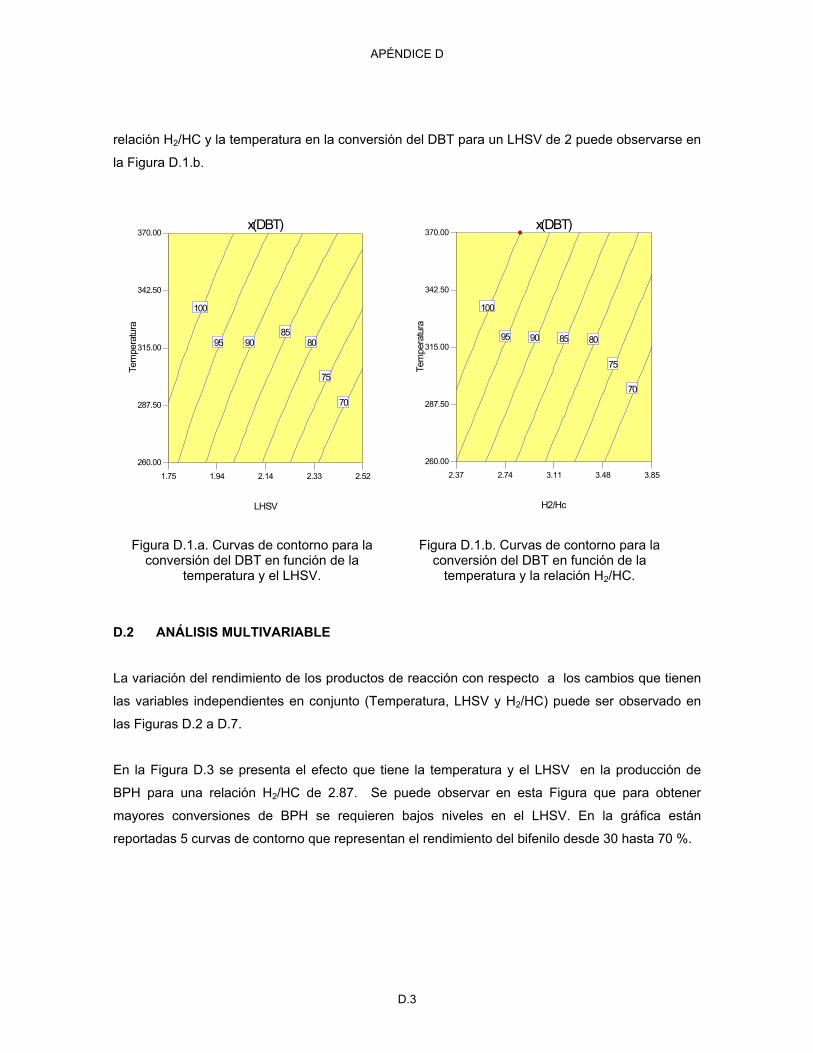
APÉNDICE D
D.3
relación H2/HC y la temperatura en la conversión del DBT para un LHSV de 2 puede observarse en
la Figura D.1.b.
x(DBT)
LHSV
Tem
pera
tura
1.75 1.94 2.14 2.33 2.52
260.00
287.50
315.00
342.50
370.00
70
75
8085
9095
100
x(DBT)
H2/Hc
Tem
pera
tura
2.37 2.74 3.11 3.48 3.85260.00
287.50
315.00
342.50
370.00
70
75
80859095
100
Figura D.1.a. Curvas de contorno para la conversión del DBT en función de la
temperatura y el LHSV.
Figura D.1.b. Curvas de contorno para la conversión del DBT en función de la
temperatura y la relación H2/HC.
D.2 ANÁLISIS MULTIVARIABLE
La variación del rendimiento de los productos de reacción con respecto a los cambios que tienen
las variables independientes en conjunto (Temperatura, LHSV y H2/HC) puede ser observado en
las Figuras D.2 a D.7.
En la Figura D.3 se presenta el efecto que tiene la temperatura y el LHSV en la producción de
BPH para una relación H2/HC de 2.87. Se puede observar en esta Figura que para obtener
mayores conversiones de BPH se requieren bajos niveles en el LHSV. En la gráfica están
reportadas 5 curvas de contorno que representan el rendimiento del bifenilo desde 30 hasta 70 %.

APÉNDICE D
D.4
x(BPH)
LHSV
Tem
pera
tura
1.75 1.90 2.04 2.19 2.33
260.00
287.50
315.00
342.50
370.00
30
40506070
Figura D.2. Curvas de contorno para el rendimiento de bifenilo en función de la temperatura y el
LHSV
La gráfica de contorno para el rendimiento de BPH en función de la relación H2/HC para un LHSV
de 2 se presenta en la Figura D.3. En esta Figura puede observarse que se requiere un mayor
consumo de Hidrógeno para obtener un mayor rendimiento de bifenilo.
x(BPH)
H2/Hc
Tem
pera
tura
2.30 2.52 2.75 2.98 3.20260.00
287.50
315.00
342.50
370.00
30
405060
7080
Figura D.3. Curva de contorno para el rendimiento de bifenilo en función de la temperatura y el H2/Hc
La Figura D.4 muestra las líneas de contorno del rendimiento del CHB como una función de la
temperatura y el LHSV a una relación H2/HC de 3.0. En esta Figura se observa no existe un

APÉNDICE D
D.5
cambio significativo del rendimiento del CHB con respecto al LHSV, las líneas de contorno del
rendimiento de CHB no muestran cambios importantes al cambio del LHSV.
x(CHB)
LHSV
Tem
pera
tura
1.50 1.75 2.00 2.25 2.50260.00
287.50
315.00
342.50
370.00
20
25
30
35
40
Figura D.4. Curvas de contorno para el rendimiento de Ciclohexilbenceno en función de la
temperatura y el LHSV.
Dado que este producto es el resultado de la reacción de hidrogenación de DBT, existe un mayor
consumo de hidrógeno. En la Figura D.5 a un LHSV de 2.0 puede observarse que un incremento
en el rendimiento de CHB requiere una mayor relación H2/Hc así como también una mayor
temperatura.
La Figura D.6 muestra las curvas de contorno del rendimiento del diciclohexilo (DCH) como una
función de la temperatura y el LHSV para una relación H2/HC constante de 3.0. Esta Figura
muestra un comportamiento diferente para el rendimiento de CHB y BPH mostrado en las Figuras
D.2 y D.4. El DCH solo puede ser obtenido a severas condiciones de reacción, esto es a altas
temperaturas y bajos LHSV`s. Estos resultados concuerdan con aquellos presentados por
Vanrysselberghe V. & Froment G.F. 1996, donde el DCH se obtiene trabajando a alta temperatura
y a bajos valores de LHSV.
El comportamiento del rendimiento a DCH con respecto a la relación H2/Hc es similar al observado
para el BPH y el CHB. La Figura D.7 muestra este comportamiento a un LHSV constante de 2.0. La
relación H2/Hc y Temperatura son proporcionales al rendimiento del diciclohexilo.

APÉNDICE D
D.6
x(CHB)
H2/Hc
Tem
pera
tura
2.00 2.50 3.00 3.50 4.00
260.00
287.50
315.00
342.50
370.00
20
25
30
35
40
Figura D.5.Curvas de contorno para el rendimiento de Ciclohexilbenceno en función de la
temperatura y la relación H2/Hc.
X(DCH)
LHSV
Tem
pera
tura
1.50 1.73 1.95 2.17 2.40260.00
287.50
315.00
342.50
370.00
5
10
1520
2530
35
Figura D.6.Curvas de contorno para el diciclohexilo en función de la temperatura y el LHSV.
La metodología para saber si un modelo es adecuado, es probar si el valor calculado de F excede
al punto porcentual α (probabilidad) de distribución F. Si esto es cierto hay una probalibidad de 1-α
que el modelo es inadecuado por lo que el modelo debe ser rechazado, de otra forma se acepta.

APÉNDICE D
D.7
X(DCH)
H2/Hc
Tem
pera
tura
1.50 2.13 2.75 3.38 4.00260.00
287.50
315.00
342.50
370.00
5101520253035
Figura D.7. Curvas de contorno para el diciclohexilo en función de la temperatura y
la relación H2/Hc.
En la Tabla D.6 están reportados los valores de los coeficientes para cada función. Estos
parámetros fueron obtenidos por minimización de la función objetivo la cual contiene todas las
respuestas experimentales de la planta piloto. El número de replicas experimentales es tal que el
cuadrado de la suma del error y el modelo seleccionado debe ser probado por medio de la prueba
de distribución F con ∑ −−− v)s(pnv n 1 y con ∑ − v)s(n 1 grados de libertad.
Tabla D.6. Modelos para representar los productos de reacción en función de la temperatura, LHSV y H2/Hc
Variable Modelo
Dibenzotiofeno X(DBT) = -150.61500 + 0.24476 * Temperatura + 36.33886 * LHSV +
28.64198 * H2/Hc
Bifenilo X(BPH) = -277.47197 -0.051841*Temperatura + 79.71786 * LHSV +
54.91897 * H2/Hc
Ciclohexilbenceno X(CHB) = 0.62828 + 0.094305 * Temperatura - 5.10930 * LHSV +
4.34768 * H2/Hc
Diciclohexilo X(DCH) = -182.81879+1.07167 * Temperatura + 87.70071 * LHSV –
26.40687 * H2/Hc – 0.38422 * Temperatura * LHSV
Para las funcionalidades reportadas en este apéndice, el valor de F se situa en el intervalo de 95%
de confianza, por lo que el modelo es adecuado.





























