Embed Size (px)
Citation preview
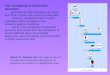
Eukaryotic genomes are complex 3D structures comprised of modified and unmodified DNA, RNA and
many types of interacting proteins
• Most DNA is wrapped around a “histone core”, to form nucleosomes• The classical histone protein complexes bind very tightly to DNA and prevent
association with other proteins• Modifications of the classical histones, or their replacement with unusual histone
types under certain conditions, can “loosen” the interaction with DNA, allowing access to transcription factors, RNA polymerase, and other proteins
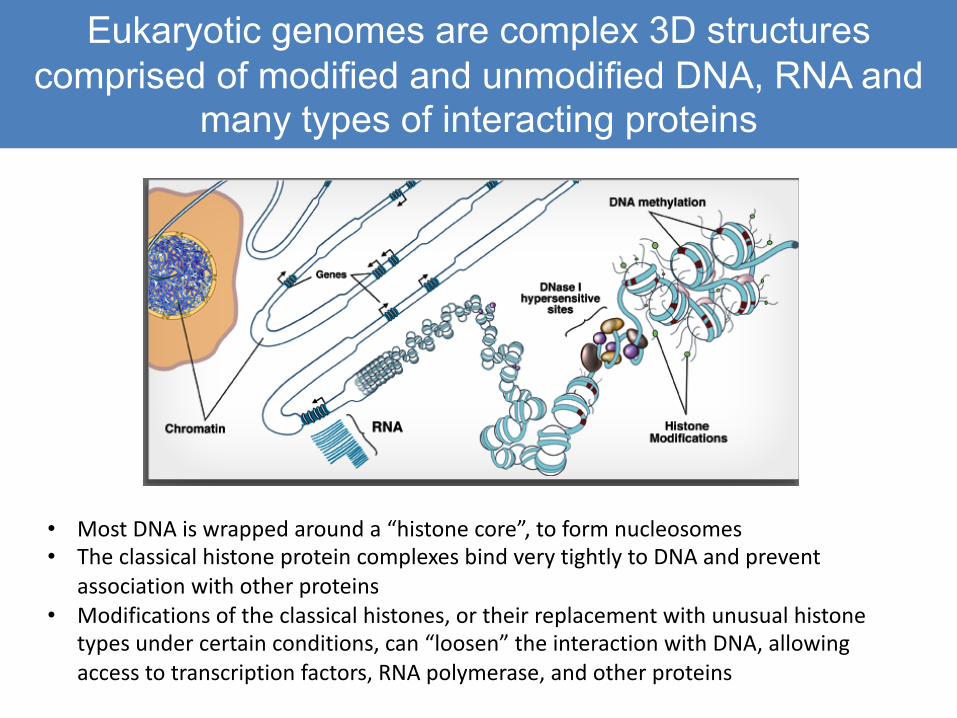
Regulatory elements can be far away: most enhancers interact with promoters and each other through long-range
chromatin loops• Regulatory elements are essentially
“docking sites” for specific types of DNA-binding proteins
– Transcription factors, TATA-binding factors, and others
• These proteins serve to attract co-factors, which then mediate protein: protein interactions across chromatin loops
• Very long range interactions are common in vertebrates, less so in invertebrate species with lower coding:nocoding ratios
• ChIP with an antibody that binds to “E” DNA will bring down “P” DNA as well
• Proteins are crosslinked very efficiently to each other, as well as to DNA, by formaldehyde treatment
• When crosslinking is reverse the complex falls apart, and Both DNA fragments are released independently
• Only one sequence binds to the TF!• Common issue in analysis of ChIP
Shear chromatin(Sonication or restriction enzyme)
TF

How to find the regulatory needles in the haystack?
• Vertebrate genomes are mostly non-coding– ~2% coding; ~5% noncoding and evolutionarily conserved (at the DNA
sequence alignment level)– Conservation has been used to identify important functional elements,
but not all functional elements are conserved at the level that DNA sequence alignments can detect
– Furthermore the important question is: which elements are accessible in a particular cell type at a particular time and in a particular state?

Focusing on accessible chromatin
• Even well conserved motifs cannot be accessed in closed regions of chromatin
accessible
Not accessible e.g. H3K9Me3, H3K27Me3
e.g. H3K27Ac
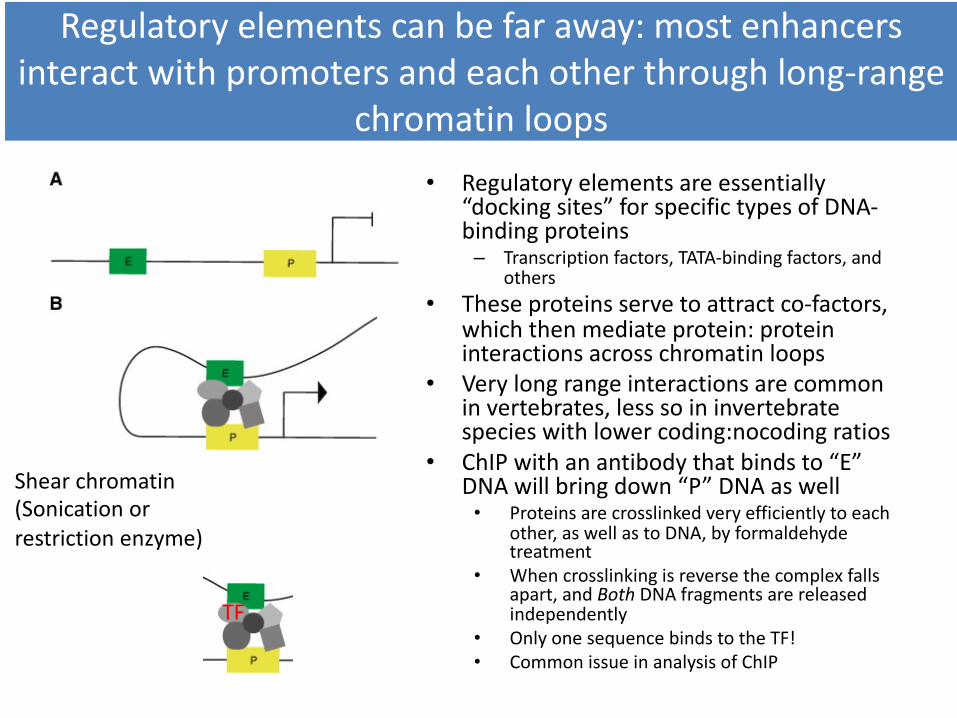
All four histones in the tetramer have “tails” that can be modified in various ways, but the most consequential modifications, with respect to transcriptional activity,
appear to involve methylation or acetylation of Lysines(K) in histone H3
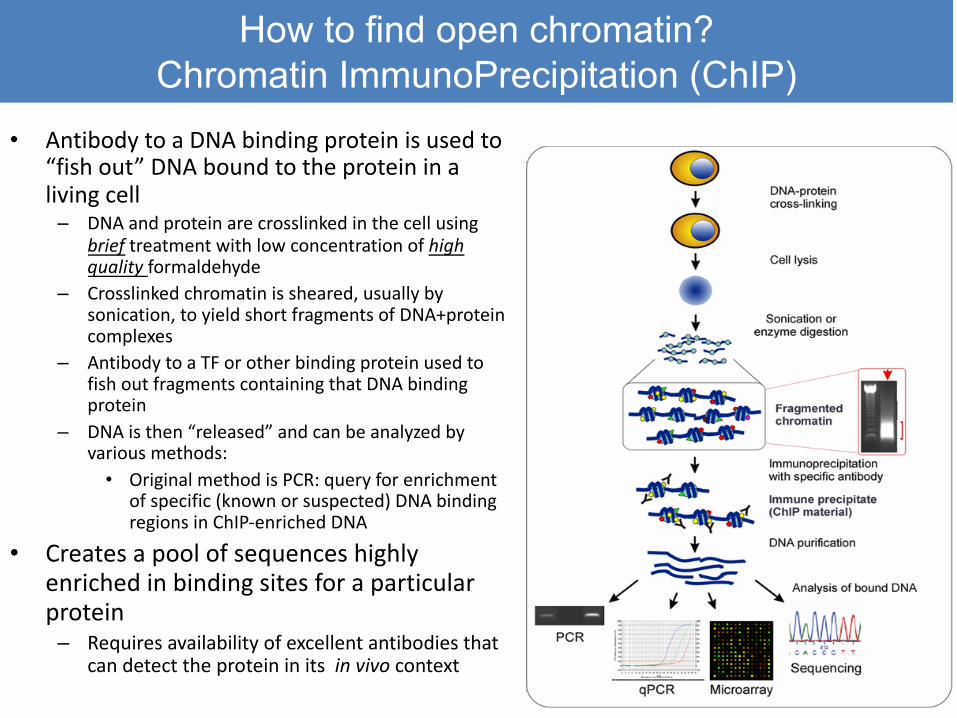
How to find open chromatin? Chromatin ImmunoPrecipitation (ChIP)
• Antibody to a DNA binding protein is used to “fish out” DNA bound to the protein in a living cell
– DNA and protein are crosslinked in the cell using brief treatment with low concentration of high quality formaldehyde
– Crosslinked chromatin is sheared, usually by sonication, to yield short fragments of DNA+proteincomplexes
– Antibody to a TF or other binding protein used to fish out fragments containing that DNA binding protein
– DNA is then “released” and can be analyzed by various methods:
• Original method is PCR: query for enrichment of specific (known or suspected) DNA binding regions in ChIP-enriched DNA
• Creates a pool of sequences highly enriched in binding sites for a particular protein
– Requires availability of excellent antibodies that can detect the protein in its in vivo context
A basic ChIP-like approach can be used to map nucleic acid:protein interactions of virtually any type
• Histone modifications:• Secondary interactions (no direct linkage to DNA)
– Histone modifying proteins, such as SWI/SNF, histone deacetylases, histone methylases
– Cofactors that bind to TFs at particular sites, and that stablizechromatin loops
– Proteins that link chromatin to nuclear matrix• RNA polymerase and elongation factors, to find
promoters and active sites of transcription• Proteins involved in DNA recombination, repair, and
replication• All of these methods require highly specific and efficient
antibodies (which are rare!)
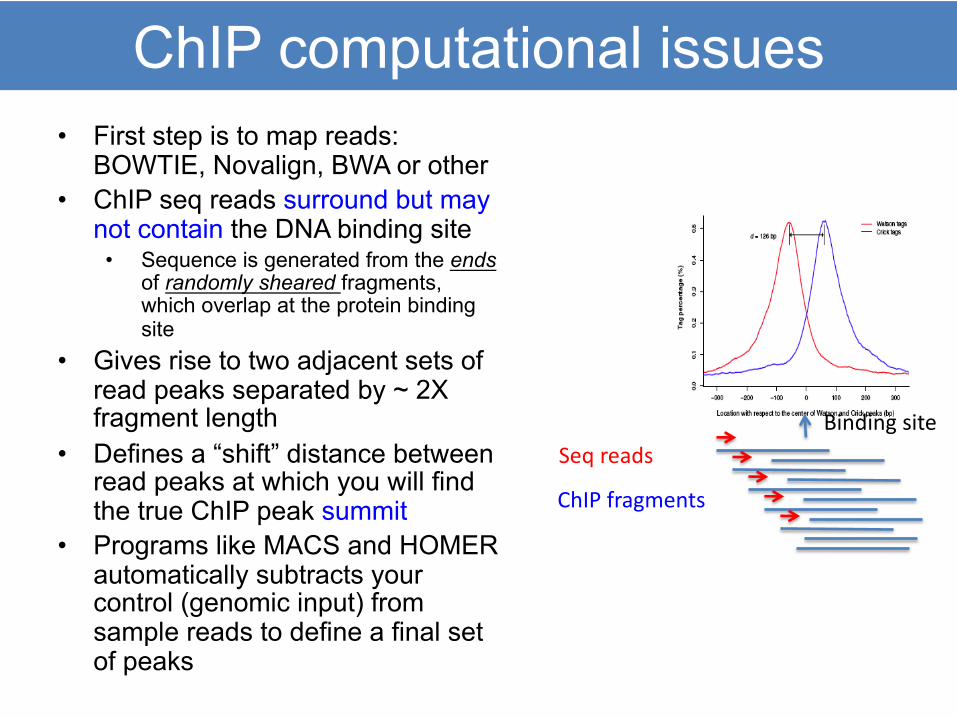
ChIP computational issues• First step is to map reads:
BOWTIE, Novalign, BWA or other• ChIP seq reads surround but may
not contain the DNA binding site• Sequence is generated from the ends
of randomly sheared fragments, which overlap at the protein binding site
• Gives rise to two adjacent sets of read peaks separated by ~ 2X fragment length
• Defines a “shift” distance between read peaks at which you will find the true ChIP peak summit
• Programs like MACS and HOMER automatically subtracts your control (genomic input) from sample reads to define a final set of peaks
Binding site
ChIP fragments
Seq reads

ChIP Analytical challenges• Genomic neighborhoods
– Shear efficiency is not really “random”• Some genomic regions are fragile and sensitive• Some regions are protected from shear or degradation
– Other artifacts• Centromeres: repeat sequences that are not all represented in the
genome sequence build• Polymorphic regions, and e.g. regions that are amplified in cell line
DNA• Repeats: most programs cannot manage sequence reads that are
not mapped uniquely• Peak width
– Transcription factors are typically sharp peaks; chromatin marks are more diffuse
• The best tools permit the user to modify these parameters– MACS ( Xiaole Liu Lab; Zhang et al, 2008; Feng et al. Nature
Methods 2102) is a user-friendly and widely used tool– HOMER, a highly versatile tool with many different annotation
features and high sensitivity (Chris Benner, http://homer.salk.edu/homer/ngs/)

Analyzing ChIP data
• User-friendly tools– MACS:
• ‘Model based” peak detection, is sensitive to peak enrichment and background
• Zhang et al, Genome Biology 2008, Feng et al. 2012, Nat Procols PMID: 22936215 (Xiaole Liu lab);
• MACS1 is best for sharp peaks (TFs); will break diffuse peaks into smaller regions
• MACS2 is designed to allow broad- or sharp-peak detection– HOMER (http://homer.salk.edu/homer)
• Can be easily tweaked for more sensitive peak detection• Comes packaged wiith a rich set of peak annotation tools• Tools for DNAse-seq, High-C, differential ChIP analysis and many more
– Both tools permit generation of “wiggle files” or similar that can be viewed in the UCSC browser
• Looking at your data is a very important step! Peak finders can miss peaks that you can easily see by eye!
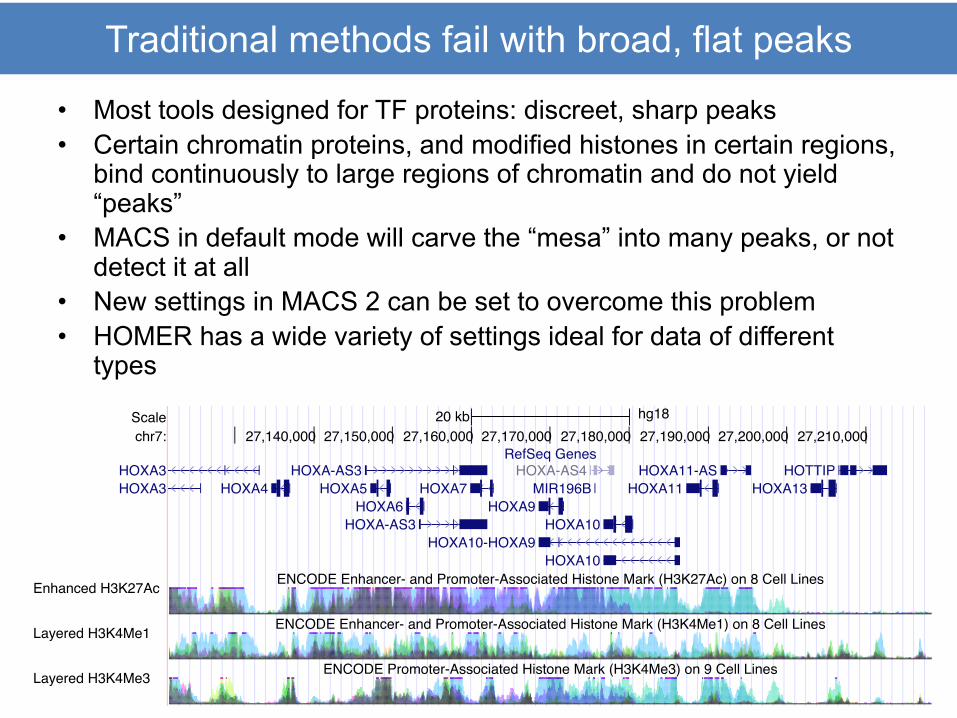
Traditional methods fail with broad, flat peaks
• Most tools designed for TF proteins: discreet, sharp peaks• Certain chromatin proteins, and modified histones in certain regions,
bind continuously to large regions of chromatin and do not yield “peaks”
• MACS in default mode will carve the “mesa” into many peaks, or not detect it at all
• New settings in MACS 2 can be set to overcome this problem• HOMER has a wide variety of settings ideal for data of different
types
Scalechr7:
DNase Clusters
Txn Factor ChIP
20 kb hg1827,140,000 27,150,000 27,160,000 27,170,000 27,180,000 27,190,000 27,200,000 27,210,000
RefSeq Genes
ENCODE Enhancer- and Promoter-Associated Histone Mark (H3K27Ac) on 8 Cell Lines
ENCODE Enhancer- and Promoter-Associated Histone Mark (H3K4Me1) on 8 Cell Lines
ENCODE Promoter-Associated Histone Mark (H3K4Me3) on 9 Cell Lines
NHGRI Catalog of Published Genome-Wide Association Studies
ENCODE Digital DNaseI Hypersensitivity Clusters
ENCODE Transcription Factor ChIP-seq
HOXA3HOXA3 HOXA4
HOXA-AS3HOXA5
HOXA6HOXA-AS3
HOXA7
HOXA10-HOXA9
HOXA9
HOXA-AS4MIR196B
HOXA10
HOXA10
HOXA11HOXA11-AS
HOXA13HOTTIP
rs4722672
Enhanced H3K27Ac
Layered H3K4Me1
Layered H3K4Me3

ChIP analysis workflow
FASTQC -> BOWTIE -> Peak finder (MACS or HOMER)This same workflow and tools can be used for a variety of methods
e.g. Methyl DIP, ATAC-seq, DNase seq
Downstream analysis:
Mapping peaks to nearby genes (and perhaps, DEGs)Identifying enriched motifs
For your factorFor co-binding factors
Overlapping with other genome featurese.g. open chromatin, known binding sites, etc.
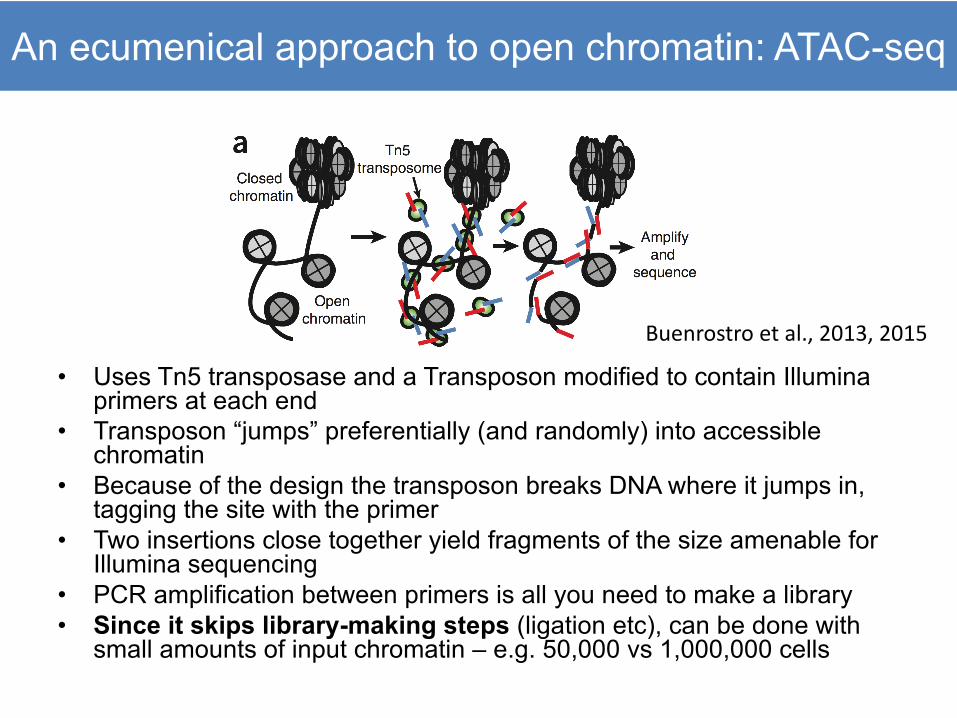
An ecumenical approach to open chromatin: ATAC-seq
• Uses Tn5 transposase and a Transposon modified to contain Illuminaprimers at each end
• Transposon “jumps” preferentially (and randomly) into accessible chromatin
• Because of the design the transposon breaks DNA where it jumps in, tagging the site with the primer
• Two insertions close together yield fragments of the size amenable for Illumina sequencing
• PCR amplification between primers is all you need to make a library• Since it skips library-making steps (ligation etc), can be done with
small amounts of input chromatin – e.g. 50,000 vs 1,000,000 cells
Buenrostro et al., 2013, 2015
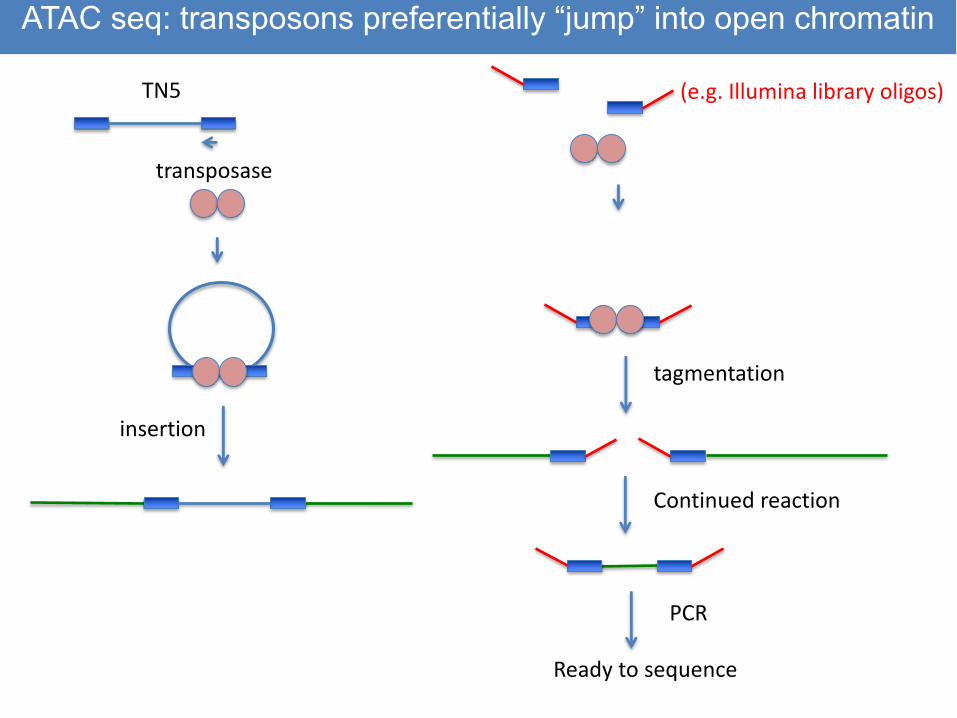
TN5
transposase
insertion
(e.g. Illumina library oligos)
tagmentation
Continued reaction
PCR
Ready to sequence
ATAC seq: transposons preferentially “jump” into open chromatin
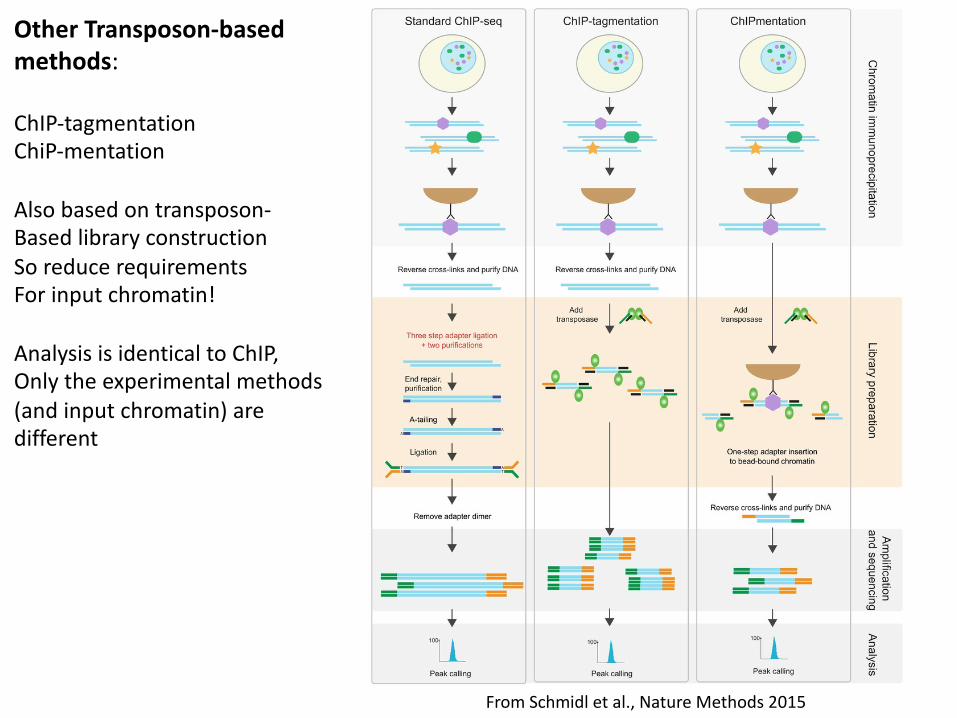
From Schmidl et al., Nature Methods 2015
Other Transposon-based methods:
ChIP-tagmentationChiP-mentation
Also based on transposon-Based library constructionSo reduce requirementsFor input chromatin!
Analysis is identical to ChIP,Only the experimental methods(and input chromatin) are different

Issues related to Tagmentation Protocols
• Ratio of DNA: transposase– Has to be adjusted for each cell type and
chromatin prep• Need even fragmentation to avoid bias, and small
enough fragments, in general, for illumina• Need to avoid making fragments too small• Bias observed in DNA: controls are complicated
• Solution in “ChiPmentation”– Tagmentation while DNA is still protected by the
antibody and cross-linked chromatin, still on the bead
• Protects from over-tagmentation, this allowing a full digestion without fear of losing the DNA
• Allows the protocol to work over a 25X range of DNA: transposon and lessens worries about time

Current summary
• ATAC-seq and H3K27Ac ChIP win the day– Simple technology, can be completed with
relatively low input and low sequencing reads– Excellent kits are available for beginners, and
many sequencing centers will do the work for a fee
– Methods work for all species and cell types– Robust computational tools are readily
available

Issues related to Tagmentation Protocols
• Ratio of DNA: transposase– Has to be adjusted for each cell type and
chromatin prep• Need even fragmentation to avoid bias, and small
enough fragments, in general, for illumina• Need to avoid making fragments too small• Bias observed in DNA: controls are complicated
• Solution in “ChiPmentation”– Tagmentation while DNA is still protected by the
antibody and cross-linked chromatin, still on the bead
• Protects from over-tagmentation, this allowing a full digestion without fear of losing the DNA
• Allows the protocol to work over a 25X range of DNA: transposon and lessens worries about time

How to tie back to 3D structure?Probing 3-dimensional chromatin structure with conformation capture
from Wit and de Laat, 2012
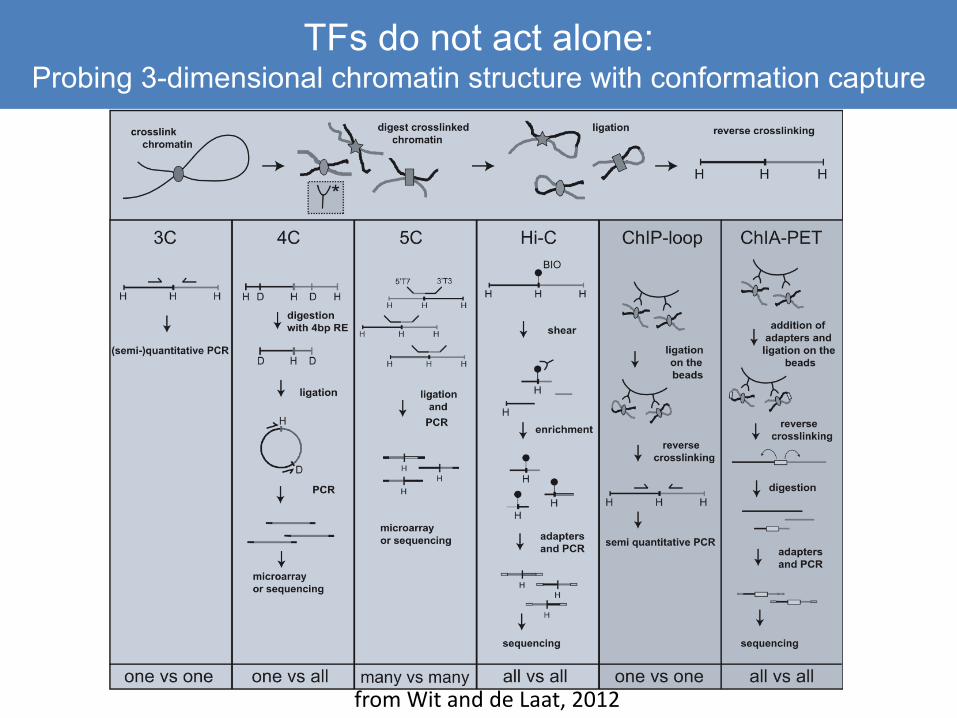
TFs do not act alone:Probing 3-dimensional chromatin structure with conformation capture
from Wit and de Laat, 2012

Requires analysis methods that are different from ChIP
• Provides the essential “big picture” view, since it is otherwise impossible to predict long-range enhancer-enhancer or enhancer-promoter interactions
• Sequenced fragments contain a bit of DNA from two distant regions– Data need to be trimmed and mapped to allow non-contiguous sequences
• Long-distant contacts are numerous, and each contact point is relatively rare: peaks are small are require deep sequencing
– For most of these methods, restriction enzymes are used to shear, not sonication, and your endpoints may be spread over a restriction fragment
– Analytical methods create a restriction map of your viewpoint region in 4C, and bin reads to those fragments
• Hi C kits are now readily available and quite reliable, giving a whole-genome view of interactions
– Lots of interactions and lots of noise! Computational issues are tricky– All 3D methods require deep sequencing and paired-end reads

Summary and Overview
• Many user-friendly methods and analytical tools are available to identify active elements in large genomes
• The issue is finding out “who is talking to whom?”– Enhancers can be shared by multiple genes– Alternative promoters for the same gene can have
very different regulatory partners– Position relative to the TSS is not a reliable indicator
in large vertebrate genomes– 3D methods are necessary to tie enhancers and
promoters together• Fortunately, 3D genomic interaction tools are
becoming easier and more cost-effective so are accessible to virtually any lab!