Embed Size (px)
Citation preview

European PharmacovigilanceChanges and Industry PerspectiveTeva Europe – Wendy Huisman, QPPV
4 Dec 2013
1

Why new EU legislation? Who plays what roles in European pharmacovigilance What are the important differences between old and new What is really new? Does it work?
What will be covered?
2

Why?
5% of all hospital admissions are for Adverse Drug Reactions (ADRs)
5% of all hospital patients suffer an ADR
ADRs are the 5th most common cause of hospital death
Estimated 197,000 deaths per year in EU from ADRs
EU societal cost of ADRs amounts to Euro 79 Billion per year
3

Background
A strengthened European safety monitoring system aims to reduce the number of ADRs
The European Commission began a review of the European system of safety monitoring in 2005 including an independent study sponsored by the European Commission
Extensive public consultation in 2006 and 2007 New legislation was adopted in December 2010 European legislation into practice in July 2012
Directive 2010/84/EU (amending 2001/83/EC) Regulation (EC) No 1235/2010 (amending 726/2004, 1394/2007)
Good Vigilance Practices guidance
4

Objectives
High level objectives – to strengthen and rationalise the community legislation with the overall objective of Better protection of public health Simplification of current rules and procedures Integrate benefit and risk Increased proactive planning Risk based and proportionate Engage patients and HCP Increase transparency
For patients and EU citizens, the new legislation has a number of goals; To inform them on the benefit – risk aspect of taking a medicine To provide the opportunity for them to report perceived problems with a
medicine through online reporting forms and take part in public hearings To raise awareness of safety issues through the creation of new websites
with information on medicine safety issues To increase public confidence in the safety monitoring system and the
positive risk benefit of medicines
5

Four main areas
Collection of key information on medicines Risk management plans Periodic safety update reports Post-authorisation safety and efficacy studies Electronic submission of information on medicines Adverse drug reaction reporting by patients and HCPs
Analysis and understanding of data information Strengthened signal detection in Eudravigilance Additonal monitoring of medicines Enhanced IT systems
6

Four main areas (cont.)
Regulatory action to safeguard public health Changes in scientific committees and decision-making Stregthened referral procedures
Communication with stakeholders Publishing of information on medicines Coordination of safety messages Public hearings
7

GVP guidances – continuous changes
MODULE I Pharmacovigilance Systems and their Quality Systems MODULE II Pharmacovigilance System Master File v2 MODULE III Pharmacovigilance Inspections MODULE IV Audits MODULE V Risk Management Systems MODULE VI Data Management of Individual Case Safety Reports MODULE VII Periodic Safety Update Reports MODULE VIII Post-Authorisation Safety Studies v2 MODULE IX Detection and Management of Signals and Information MODULE X Additional Monitoring MODULE XI Public Participation in Pharmacovigilance MODULE XII Continuous Pharmacovigilance, Ongoing Benefit-Risk Evaluation,
Regulatory Action and Planning of Public Communication MODULE XIII withdrawn MODULE XIV Referral Procedures for Safety Reasons MODULE XV Safety Communications MODULE XVI Tools, Educational Materials and Effectiveness Measurement for
Risk Minimisation draft
8

The different actors and their responsibilities
National competent authorities European Medicines Agency European commission
Regulatory network responsible for authorising and supervising medicinal products including the conduct of pharmacovigilance
Creation of the PRAC; Pharmacovigilance Risk Assessment Committee Monthly meetings starting september 2012
Pharmaceutical companies Tasks and responsibilities in pharmacovigilance
Healthcare professionals Reporting requirements, implement risk minimisation
Patients Reporting
9

Regulatory background in EU
Nationally authorised products
Centrally authorised products Responsibility for Committee on Medicinal products for Human Use
(CHMP) Adoption role for the European Commission
Products registered through Mutual Recognition (MRP) or Decentralised Procedures (DCP) Responsability for Coordination Group for Mutual Recognition and
Decentralised Procedures (CMDh)
10

11

12

Pharmacovigilance Risk Assessment Committee (PRAC) activities
Will advise the Committee on Medicinal products for Human Use (CHMP) and the Coordination Group for Mutual Recognition and Decentralised Procedures (CMDh) on safety issues
PRAC will cover all aspects of risk management and use of medicines including detection, minimisation and communication of risk and will consider this in the therapeutic context of the use of the medicine.
13
14

15

PRAC
16

EU QPPV and PSMF
Qualified Person for Pharmacovigilance (QPPV) Reside and operate in the Community Responsible for the establishment and maintenance of the
pharmacovigilance system “Oversight” and accountable
Pharmacovigilance System Master File (PSMF) Replaces the DDPS (detailed description of the pharmacovigilance
system) Single PSMF per PhV system To be kept up-to-date (availability in 7 days) Location determines the “European” inspection site
17

PSMF
Main modules QPPV Structure of the MAH Sources of safety data Systems and databases Pharmacovigilance processes Performance Quality system
Annexes CVs and job descriptions List of MAH and licenses Contact details List of SOPs Compliance data ICSRs, PSURs List of audits List of trials, market research, agreements, RMPs 18

Quality Management Systems (QMS)
To ensure the integrity of the companies global system. Is an integral part of the PhV system which should provide the necessary resources being attributed to the PhV activities
Company/QPPV oversight Set of obligations Monitoring of compliance Documented processes and procedures Resource management and audit trails
Periodically updated Regular audits
QMS helps to support risk management linking IB to DSUR to EU RMP to SmPC to new PBRER (PSUR)
19

Reporting of ICSRs
Eudravigilance – single point of reporting for the 28 EU members (when “functional” – estimated 2016)
Includes serious and non serious Includes patient reports (direct or indirect)
Transitional requirements Reporting still locally Reporting to EV Reporting to both Including non serious or not
Access policy Public MAH authorities
20

Eudravigilance and transparancy
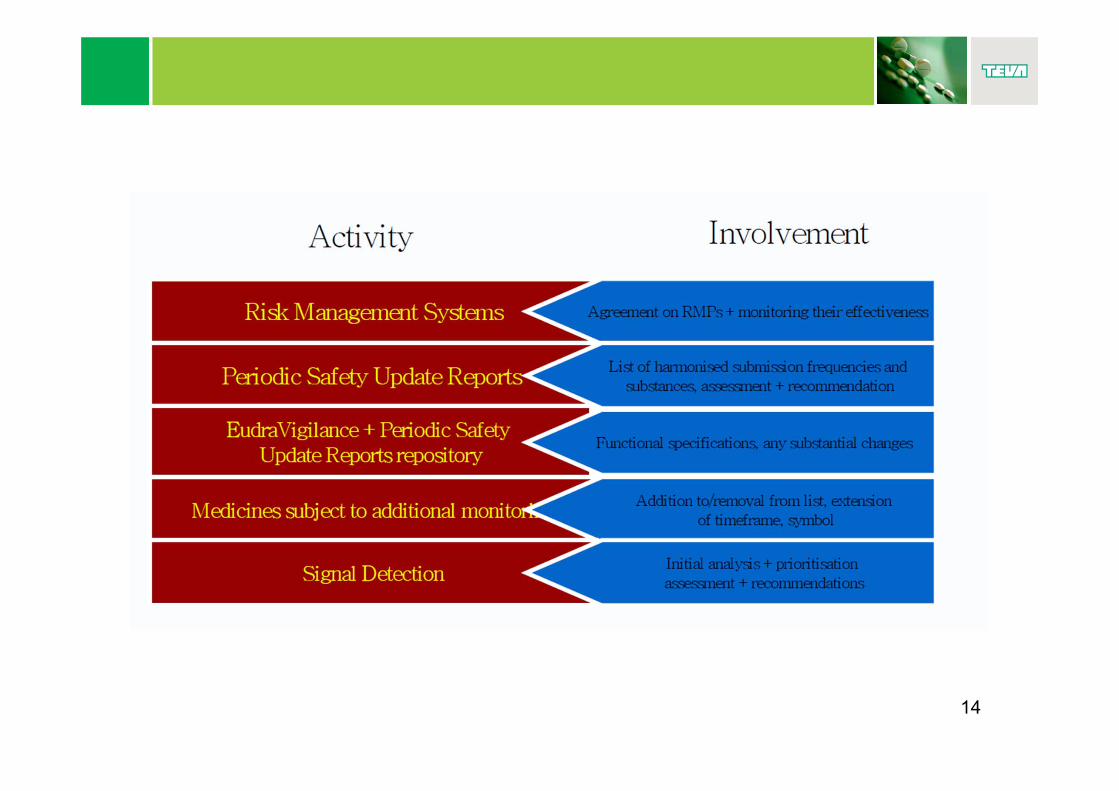
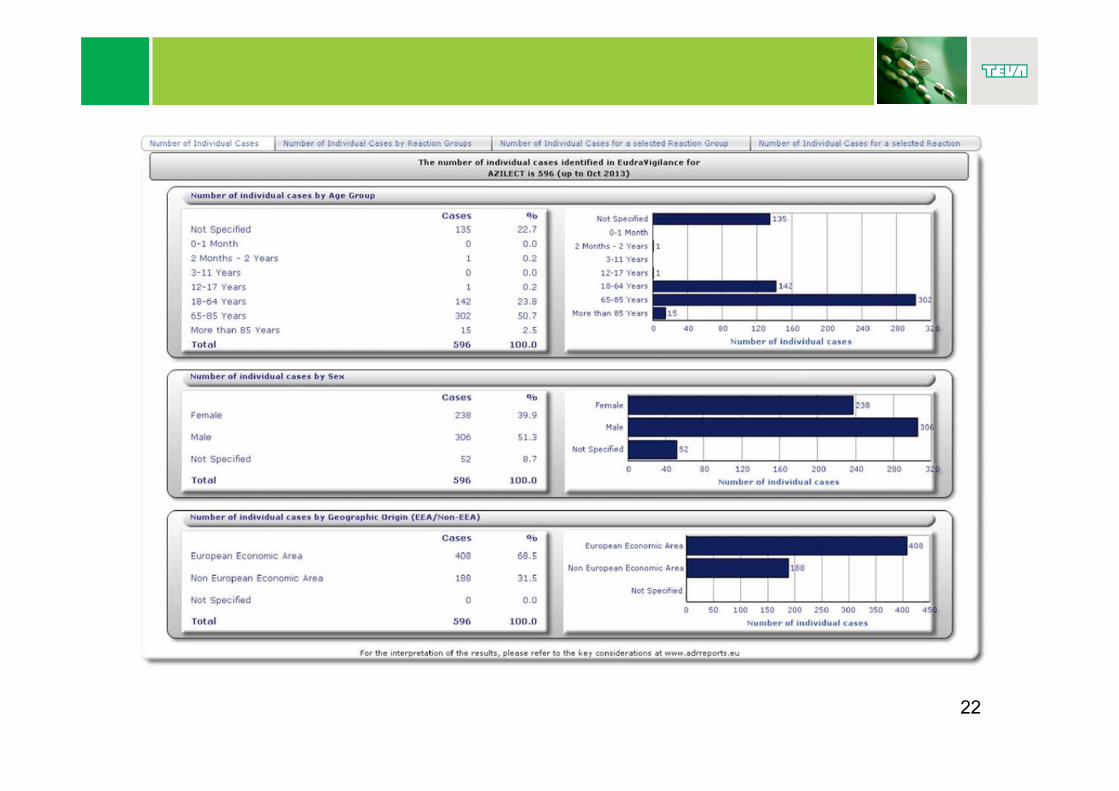
http://www.adrreports.eu/
21
22

23

Signal management
MAH responsible for quantitative and qualitative signal detection “own” signals from the safety database Literature PRAC recommendations
EMA/Member states responsible for signal detection in Eudravigilance
PRAC is responsible for the analysis, prioritisation and assessment of validated signals, resulting in a PRAC recommendation. These recommendations can cover any medicine with a valid marketing authorisation in the EU, including nationally and centrally authorised medicines.
24

PRAC and signal management
Causal relationship confirmed or considered likely? MAHs to provide further information update of the SmPC and the package leaflet.
PRAC recommendations to supply additional information are actionable directly by the MAHs concerned.
PRAC recommendations concerning centrally authorised medicines to the CHMP for endorsement.
PRAC recommendations concerning nationally authorised medicines to the CMDh.
MAHs are expected to take action according to the recommendations.
The Agency publishes PRAC recommendations every month. MAH to monitor the information
25

Risk Management Plans
Submitted with every dossier
Very resource intensive! Assessments not harmonised
Can also be imposed in PM phase Public summary
Several templates Full dossier Generics abbreviated template Hybrids, combinations, “Well Established Use” intermediate
template
26

Template
Part I Product(s) overview Part II Safety specification
Module SI Epidemiology of the indication(s) and target population(s) Module SII Non-clinical part of the safety specification Module SIII Clinical trial exposure Module SIV Populations not studied in clinical trials Module SV Post-authorisation experience Module SVI Additional EU requirements for the safety specification Module SVII Identified and potential risks Module SVIII Summary of the safety concerns
Part III Pharmacovigilance plan Part IV Plans for post-authorisation efficacy studies Part V Risk minimisation measures (including evaluation of the
effectiveness of risk minimisation measures) Part VI Summary of the risk management plan Part VII Annexes
27

Risk, potential risk, missing information
Early agreement needed about what are risks, potential risks and missing information.
Identified Risk An untoward occurrence for which there is adequate evidence of an association with the medicinal product of interest. Examples include: an adverse reaction adequately demonstrated in non-clinical
studies and confirmed by clinical data; an adverse reaction observed in well-designed clinical trials or
epidemiological studies for which the magnitude suggests a causal relationship;
an adverse reaction suggested by a number of well-documented spontaneous reports where causality is strongly supported (anaphylactic reactions, injection site reactions)
28

Risk, potential risk, missing information
Potential risk Some basis for suspicion of an association. Examples include: Preclinical tox findings not seen in clinical studies adverse events observed in clinical trials or epidemiological
studies for which the magnitude raises a suspicion of, but is not large enough to suggest a causal relationship
a signal arising from a spontaneous adverse reaction reporting an potential class effect
Missing information Information about the safety of a medicinal product which
represents a limitation of the safety data with respect to predicting the safety of the product in the marketplace.
Examples of missing information include populations not studied (e.g. pregnant women or patients with severe renal impairment) or where there is a high likelihood of off-label use.
29

Pharmacovigilance activities
Routine PhV PSUR Signal detection AE reporting + Specific adverse reaction
follow-up questionnaires
ADDITIONAL MEASURES
Post authorization Safety Studies (prospective, retrospective, RCT etc) DUS Registries
To identify or further characterize the safety risks post authorization
ROUTINE MEASURES

Risk minimisation activities
the summary of product characteristics (SmPC)
the package leaflet the labelling the pack size and design the legal (prescription) status of
the product
ADDITIONAL MEASURES Educational tools
targeting HCPs targeting patients (including
patient alert cards)
Controlled access programme
Other Prevention programmes (e.g.
pregnancy preventionprogramme)
DHPC
Appropriate measures to prevent or minimize known risks to patients
ROUTINE MEASURES

Effectiveness of the risk minimisation
To evaluate the effectiveness of risk minimisation measures two indicators should be considered: Process indicators
Gather evidence that the implementing steps of risk minimisation measures have been successful.
Provide insight into what extent the program has been executed as planned and whether the intended impacts on behaviour have been observed.
Outcome indicators Provide an overall measure of the level of risk control that has been
achieved with a risk minimisation measure. For example, where the objective of the intervention is to reduce the frequency and/or severity of an adverse reaction, the ultimate measure of success will be linked to this objective.
32

PSURs
Focus on Benefit – Risk Includes efficacy (benefit) data No routine linelistings No addendum or bridging reports Modular concept Signal and risk evaluation
Risk based schedules Generics waived Long term
ICH step 4 completed December 2012 New PSUR format to be used as of January 2013 New submission timelines Single assessment for NAPs in 2015
EURD lists published on the EMA portal 33

European Union Reference Date List
34

Post Authorisation Safety and Efficacy Studies
Definitions essential Observational study and PASS Organised data collection like Patient support or patient
discontinuation data
PASS All that investigates “safety” PRAC approval Protocol and abstract to be published
PAES May be imposed after MA is granted
35

Additional monitoring - ▼
▼ “This medicinal product is subject to additional monitoring. This will allow quick identification of new safety information. Healthcare professionals are asked to report any suspected adverse reactions. See section 4.8 for how to report adverse reactions.”
CAPs and NAPs in the following categories: medicines that contain a new active substance that was not
contained in any authorised medicine in the EU on 1 January 2011; biological medicines authorised after 1 January 2011 - this applies to
all biological medicines including biosimilars; medicines for which the marketing-authorisation holder is required to
carry out a post-authorisation safety study (PASS); medicines given conditional approval or authorised under
exceptional circumstances and medicines authorised with specific obligations on the recording or monitoring of suspected adverse drug reactions.
A product can get on the list any time during life cycle
36

Transparency - webportals
Public assessment reports SmPCs and PIL Lay summaries of the RMPs Lists of products ubject to addional monitoring Web based reporting forms for HCP and patients Minutes of CHMP and PRAC List of locations of the PSMF, protocols, abstracts Article 31 and 107
37

Authorities share findings with each other
Authorities inspect companies in countries out of their “jurisdiction”
PhV Inspections
38

39
A The Master slide structure
Teva UK Limited
Reference http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general
_content_000492.jsp&mid=WC0b01ac058033e8ad
40
http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/general/general_content_0
00492.jsp&mid=WC0b01ac058033e8ad

Does it work?
Main challenges to industry Managing the expectations Educate assessors “Oversight” – spider in the web Resources
41

Abbreviations
ADR – Adverse Drug Reaction AR – Assessment Report CA – Competent Authorities CAP – centrally Authorised Product CHMP –Committee on Medicinal products for Human Use CMDh - Coordination Group for Mutual Recognition and Decentralised Procedures CV – Curriculum Vitae DCP – Decentralised Procedure DHPC – Dear Health Care Profesional communication DSUR – development Safety Update Report DUS – Drug Utilisation Sstudy EC – European Commission EMA – European Medicines Agency EU – European Union EURD – European Union Reference Dates EV - Eudravigilance GVP – Good Vigilance Practices HCP – Health Care Professional IB – Investigational Brochure ICH – International Conference on Harmonisation ICSR – Individual Case Safety Report MAH – Marketing Authorisation Holder MS – Member State MRP – Mutual Recognition Procedure
42

Abbreviations (cont.)
NAP – nationally authorised Product RMP – Risk Management Plan MA – Marketign Authorisation MAH – Marketing Authorisation Holder MS – Member State MRP – Mutual Recognition Procedure NAP – nationally authorised Product RMP – Risk Management Plan PAES – Post authorisation Efficacy Study PASS – Post Authirisation Safety Study PhV - Pharmacovigilance PIL – Patient Information Leaflet PSUR – Periodic Safety Update Report PBRER – Periodic Benefit Risk Evaluation Report PM – Post Marketing PRAC – Pharmacovigilance Risk Assessment Committee PSMF – Pharmacovigilance System Master File RCT – Randomised Controlled Trial QMS – Quality management System QPPV – Qualified Person for Pharmacovigilance SmPC – Summary of Product Characteristics SOP – Standard Operating Procedure
43