Embed Size (px)
Citation preview

Journal of Controlled Release 59 (1999) 361–376
Evaluation of colonic absorbability of drugs in dogs using a novelcolon-targeted delivery capsule (CTDC)
*Takashi Ishibashi , Kengo Ikegami, Hiroaki Kubo, Masao Kobayashi, Masakazu Mizobe,Hiroyuki Yoshino
Pharmaceutics Research Laboratory, Tanabe Seiyaku Co., Ltd., 16-89, Kashima 3-chome, Yodogawa-ku, Osaka 532, Japan
Received 23 July 1998; received in revised form 21 October 1998; accepted 8 December 1998
Abstract
A series of dog studies were performed to examine the in vitro / in vivo relationship of drug release behavior of the newlydeveloped colon-targeted delivery capsule (CTDC). The four kinds of CTDCs containing theophylline, each of which has adifferent in vitro dissolution lag time, were orally administered to four beagle dogs under fasted condition, and the onsettimes of drug absorption were compared. The CTDC with longer in vitro lag time had a later onset of drug absorption. It wasalso found that the time difference between the gastric emptying and the onset of drug absorption was almost equal to the invitro dissolution lag time of the capsule, suggesting a similar performance of CTDC in the gastrointestinal tract. From thecomparison to the absorption behavior of the colon arrival marker, i.e. sulfasalazine, it was proved that the CTDC with thelag time of 3 h can deliver the drug directly to the colon. This result implied that the CTDC can be used as a non-invasivemeans for assessing the regional absorbability of drugs in the gastrointestinal tract. To evaluate the absorbability of drugs inthe colon, three model drugs, theophylline (THEO), acetaminophen (ACET), and phenylpropanolamine hydrochloride (PPA)were directly delivered to the colons of beagle dogs using the CTDC with the lag time of about 3 h. The obtained relativebioavailabilities to the solution form were as high as 94.2%, 71.0%, and 91.5% for THEO, ACET and PPA, respectively,suggesting that the colonic absorbability of those drugs is essentially good. 1999 Elsevier Science B.V. All rightsreserved.
Keywords: Colon-targeted delivery capsule; Site-specific drug delivery; Beagle dog; Gastric emptying; Colonic bioavail-ability
1. Introduction ative colitis, Crohn’s disease, colon-cancer, etc.Another advantage of colon-targeting involves the
The colon-targeted delivery of drugs via the oral improved oral bioavailability of peptide or proteinroute has attracted much interest for the local drugs susceptible to acidic or enzymatic degradationtreatment of some colonic diseases, such as ulcer- in the stomach or small intestine [1]. The colon is
thought suitable as the absorption site for those drugsbecause of less activity of proteolysis [2].*Corresponding author. Tel.: 181-6-300-2788; fax: 181-6-300-
In the last decade, there has been much research2799.E-mail address: [email protected] (T. Ishibashi) aiming toward the colon-targeted delivery of drugs.
0168-3659/99/$ – see front matter 1999 Elsevier Science B.V. All rights reserved.PI I : S0168-3659( 99 )00005-X

362 T. Ishibashi et al. / Journal of Controlled Release 59 (1999) 361 –376
Various methodologies and technologies have been purpose is to evaluate the colonic absorbability ofproposed, such as pH-triggered [3,4], time-controlled some drugs with different solubilities using the[5–8], and microbially-controlled deliveries [9–11]. CTDC system.Nevertheless, it seems that only a few systems areapplicable for practical use in terms of the selectivityto the target site, the safety of the materials to be 2. Materials and methodsused, and the preparation techniques to be applied.
We recently developed a new capsule type colon- 2.1. Materialstargeted delivery system (colon-targeted deliverycapsule: CTDC) from a practical point of view, Theophylline (THEO; Shiratori Seiyaku, Japan)which can be manufactured using currently applic- and phenylpropanolamine hydrochloride (PPA; Alpsable pharmaceutical technologies and materials rec- Chemical Co., Japan) were used as model drugs.ognized as safe. The CTDC was designed by impart- Acetaminophen (ACET; Yamamoto Chemical Co.,ing a pH-sensing function and a time-release func- Japan) was used as both a model drug and antion to the conventionally used hard gelatin capsule, indicator determining the gastric emptying time ofso as to release the drug rapidly at the predetermined the capsule. Sulfasalazine (SAS; Sigma Chemicaltime after gastric emptying [12]. The lag time of the Co., Germany) was used as an indicator determiningCTDC can be controlled by the coating amount of arrival in the colon. Hard gelatin capsules (size [2;
Eudragit E, and the kind of organic acid as a pH 17.8 mm in length; 6.07 mm and 6.36 mm in widthadjusting agent [13]. Since the transit time of phar- of body and cap) were purchased from Warnermaceuticals in the small intestine is known to be less Lambert. Aminoalkylmethacrylate copolymer B
¨variable in humans, i.e. about 361 h [14], the colon- (Eudragit E 100, Rhom Pharm, Darmstadt, Ger-targeting could be achieved with the CTDC when the many) was used as the acid-soluble polymer, whichonset time of drug release is adjusted at 3 h or more. is soluble in low pH aqueous medium up to pH 5.
If the reliable site-specificity to the colon is Hydroxypropyl-methylcellulose (TC-5 , type EW,assured, the CTDC could be used to evaluate the Shin-etsu Chemical, Japan) was used as the hydro-colonic absorbability of drugs. As a matter of fact, philic polymer. Hydroxypropyl-methylcellulose ace-
the drug absorption from the colon is believed to be tate succinate (HPMC -AS, type MIF, Shin-etsu)generally poor because less aqueous fluid existed in was used as the enteric polymer. Ethylcellulose (10the lower part of the large intestine [15]. Therefore, cp grade, Shin-etsu) was used as the sealing agent ofwhen sustained-release formulations are designed, it the hard gelatin capsules. Succinic acid (Katayamais important to understand the rate or extent of the Chemical, Japan) was used as the pH-adjustingcolonic absorption of the drug. The simple and agent. 7-(2-Hydroxyethyl)-theophylline (Tokyorelevant methodology to evaluate properly the Kasei Kogyo, Japan) and phenethylamine hydrochlo-colonic absorbability of drugs under the intact phys- ride (Katayama) were used as the internal standards.iological condition, however, has not been offered to Tetragastrin was purchased from Mecto (Japan). Allformulators. The CTDC may be a useful means for other chemicals and solvents were of reagent grade.achieving this purpose, because the system enablesthe direct delivery of any type of drugs to the colon 2.2. Preparation of CTDC with different lag timewithout any invasive treatment.
The first purpose of the present study is to check if The powder mixture consisting of 20 mg ofthe CTDC can release the drugs at the targeted THEO, 50 mg of SAS and 100 mg of succinic acidposition in the gastrointestinal tract. Various CTDCs was filled into a hard gelatin capsule, and the joint ofwith different in vitro lag times were orally adminis- the capsule body and cap was sealed with a smalltered to beagle dogs. The in vitro / in vivo relation- amount of the 5% (w/w) ethylcellulose ethanolicship of drug release behavior is demonstrated, and solution. Four batches of the sealed capsules were
the in vitro lag time required for the colon-targeted spray-coated with different amounts of Eudragit Edrug delivery is estimated in dogs. The second (0, 11, 21, and 33 mg/capsule) to provide a different

T. Ishibashi et al. / Journal of Controlled Release 59 (1999) 361 –376 363
onset time of drug release. Each batch of the 2.4. In vitro release studyEudragit E-coated capsules was then coated with
TC-5 (15 mg/capsule) containing ACET (10 mg/ The release profiles of ACET, THEO, and PPAcapsule) as an intermediate layer, and finally with from the capsule were determined according to the
procedure described in the Japanese PharmacopoeiaHPMC -AS (250 mg/capsule) as the outmost en-XIII (the paddle method). The capsules were placedteric layer. The coating process was carried out using
in a vessel with 900 mL of the JP 1st-fluid (pH 1.2)a coating machine (Hicoater , Type HCT-mini,or 2nd-fluid (pH 6.8) at 3760.58C rotating at 100Freund Ind., Japan), and the formulas of the poly-rpm. The aliquots were removed periodically andmeric coating solutions for each layer are given inassayed for ACET, THEO, and PPA using the highTable 1 along with the standard operating conditions.performance liquid chromatography (HPLC) method,of which analytical conditions are given in Section
2.3. Preparation of CTDC for colonic 2.6.absorbability study Because of the different assay method applied, the
release profile of SAS was determined by a separateThe powder mixture consisting of three model run of the dissolution test. The capsules were placed
drugs (20 mg of THEO, 30 mg of PPA, and 60 mg of in a vessel with 900 mL of the JP 1st-fluid (pH 1.2)ACET), a colon arrival indicator (50 mg of SAS), or 2nd-fluid (pH 6.8) at 3760.58C rotating at 100and a pH-adjusting agent (100 mg of succinic acid) rpm. The released amount was periodically deter-was filled into a hard gelatin capsule (size [2). The mined by the spectrophotometric method.joint of the capsule body and cap was sealed with asmall amount of the 5% (w/w) ethylcellulose etha- 2.5. In vivo release /absorption studynolic solution. The sealed capsules were spray-
coated with Eudragit E (30 mg/capsule), then with The in vivo absorption studies were carried out TC-5 (15 mg/capsule), and finally with HPMC - using tetragastrin-controlled beagle dogs, in order to
AS (250 mg/capsule) using a coating machine minimize the intra-subject variance in gastric acidity.(Hicoater , Type HCT-mini). The formulas of the The healthy male beagle dogs, 11–12 kg, were used
polymeric coating solutions, and the operating con- in this study under fasted condition. Tetragastrin (10ditions of coating for each layer are described in mg/kg) was injected intramuscularly twice, 15 minTable 1. before and 45 min after oral administration of
Table 1Formulas of coating solution and standard operating conditions
Coating layer Acid-soluble layer Hydrophilic layer Enteric layer Composition of Eudragit E 5.0 TC-5 1.5 HPMC -AS 5.0
coating solution (w/w%) Ethanol 95.0 ACET 4.0 Talc 2.5Ethanol 23.0 Ethanol 55.8Water 71.5 Water 36.7
Operating conditions:Blower temperature (8C) 45 65 60Exhaust temperature (8C) 30 35 40
2Spray pressure (kg/cm ) 2 2 22Air flow rate (L/cm ) 30 30 30
Spray rate (g /min) 2.5 1.8 2.5Rotating speed of 40 40 40
coating pan (rpm)

364 T. Ishibashi et al. / Journal of Controlled Release 59 (1999) 361 –376
CTDC. The fasted dogs received no food, but had dichloromethane:diethylether (30:70) mixture for 10free access to water, for 18 h before the administra- min and centrifuged at 2000 rpm for 5 min. Then, 4tion. The CTDC was orally administered to the male mL of the organic layer was taken out and added tobeagle dogs along with 50 mL of water. Blood 0.5 mL of 0.5% phosphate aqueous. After shakingsamples (4 mL) were collected at every 1 h for the for 10 min, the mixture was centrifuged at 2000 rpmperiod from 0–14 h and again after 24 h. The plasma for 5 min. Five mL of cyclohexane were added to thewas obtained by centrifuging the blood samples at aqueous layer, and the mixture was shaken and3000 rpm for 5 min immediately after sampling. All centrifuged again under the above conditions. Thethe plasma samples were frozen at 2208C until organic layer was removed, and 200 mL of theanalysis. aqueous solution were analyzed by HPLC. The
The oral–caecal transit time was regarded as the HPLC system consisted of the same equipment astime required for the first appearance of sulfapyridine used in the assay of THEO, but the column used was(SP), a metabolite of SAS, in the plasma after a Hypersil 5-ODS (4.6 mm3250 mm; GL Science,administration [16,17]. Tokyo, Japan). The mobile phase, acetonitrile:0.2%
phosphate buffer containing 0.01 M sodium laurylsulfate (35:60), was pumped at 1.0 mL/min and2.6. Assay methodsmonitored at 270 nm; column temperature, 508C.
2.6.1. THEO, ACET and SPThe concentrations of THEO, ACET, and SP in
plasma were determined together by the HPLC 3. Results and discussionmethod. A 0.1 mL sample of 7-(2-hydroxyethyl)-theophylline containing aqueous solution (20 mg/ The fundamental structures of CTDC used in thismL) as the internal standard was added to 0.5 mL of study are depicted schematically in Fig. 1, and theplasma. Then the mixture was shaken with 5 mL of necessary information of individual capsules, regard-ethylacetate for 5 min prior to centrifugation at 2000 ing coating layers and drugs incorporated, are sum-rpm for 5 min. Four ml of the organic layer were marized in Table 2.taken out and evaporated to dryness at 408C under All the capsules commonly contain SAS as aN gas flow, and the residue was resolved in 0.5 mL2 colon-arrival indicator and succinic acid as a pH-of the mobile phase to make the sample solution for adjusting agent besides drugs. Except CTDC , the0analysis. Next, 200 mL of the sample solution were capsules are coated with a three-layered polymericloaded on to a HPLC system to determine the plasma film comprising the innermost acid-soluble layer
concentration level of the drugs. The HPLC con- (Eudragit E), the intermediate hydrophilic layer ditions applied were as follows: pump, L-6200 (TC-5 ), and the outermost enteric layer (HPMC -
(Hitachi, Japan); UV detection, SPD-10A AS). CTDC does not have the Eudragit E layer, so0(Shimadzu, Japan); column, KC-PAK SMA C18 this can be regarded as a sort of enteric-coated(4.63250 mm; Chemco Scientific, Osaka, Japan); capsule. For the three capsules, CTDC , CTDC and1 2mobile phase, acetonitrile: 0.01 M sodium acetate CTDC , the coating amounts of the acid-soluble3(pH 4) (1:20); flow rate, 0.8 mL/min; detection, layer is purposely different to vary the onset time ofwavelength at 254 nm; and column temperature, drug release after the gastric emptying. Except508C. CTDC , ACET is embedded in the hydrophilic layer4
as an indicator of the gastric emptying. The small2.6.2. PPA intestinal transit time can be estimated by monitoring
The concentration of PPA in plasma was de- the absorption of ACET and SP (a metabolite oftermined by the HPLC method. A 0.5 mL sample of SAS). Only CTDC contains three drugs, THEO,4
phenethylamine hydrochloride aqueous solution (5 ACET, and PPA, together in the capsule, which ismg/mL) as the internal standard was added to 0.5 used to compare the colonic absorbability of thosemL of plasma. The mixture was shaken with 5 mL of drugs.

T. Ishibashi et al. / Journal of Controlled Release 59 (1999) 361 –376 365
3.1. Drug delivery to the colon by the CTDC
3.1.1. In vitro release characteristicsCTDC , CTDC , CTDC , and CTDC were used0 1 2 3
in this study. The release profiles of THEO, SAS,and ACET from these capsules in the JP 1st-fluid(pH 1.2) and the JP 2nd-fluid (pH 6.8) are shown inFig. 2.
All the capsules did not release any drugs for over8 h in the 1st-fluid, suggesting the excellent acid-resistibility given by the enteric layer. The CTDC ,0
which has no acid-soluble layer, quickly releasedthree drugs in the 2nd-fluid, even though a smalldifference in release rate was observed between them(Fig. 2a), which may be due to the difference insolubility. Nevertheless, a relatively good coincidentin the release behavior of three drugs suggests that
either the capsule shell, the TC-5 layer, or theenteric layer does not function essentially as adissolution barrier, because each of those easilydissolves in the aqueous medium at pH 6.8. TheCTDC released ACET quickly after the dissolution1
test started, and about 60 min after that, THEO andFig. 1. Schematic representation of the fundamental structure of
SAS started to be released (Fig. 2b). This is thethe colon-targeted delivery capsule (CTDC). (a) Drug, (b) succinictypical dissolution characteristic of CTDC, and theacid, (c) hard gelatin capsule, (d) acid-soluble layer, (e) hydro-
philic layer, (f) enteric layer. observed delayed-release was brought about by the
Table 2Composition of the CTDC used in this study
Composition CTDC CTDC CTDC CTDC CTDC0 1 2 3 4
Capsule contentsDrugs THEO THEO THEO THEO THEO, ACET, PPAColon arrival indicator SAS SAS SAS SAS SASpH-adjusting agent Succinic acid Succinic acid Succinic acid Succinic acid Succinic acid
Coating layerAcid-soluble layer
Polymer – Eudragit E Eudragit E Eudragit E Eudragit E(Coating amount /capsule) (11 mg) (21 mg) (33 mg) (30 mg)
Hydrophilic layer Polymer TC-5 TC-5 TC-5 TC-5 TC-5
(Coating amount /capsule) (15 mg) (15 mg) (15 mg) (15 mg) (15 mg)Gastric emptying indicator ACET* ACET* ACET* ACET* –
Enteric layer Polymer HPMC -AS HPMC -AS HPMC -AS HPMC -AS HPMC -AS
(Coating amount /capsule) (250 mg) (250 mg) (250 mg) (250 mg) (250 mg)
THEO, theophylline (20 mg/capsule); ACET, acetaminophen (60 mg/capsule); PPA, phenylpropanolamine hydrochloride (30 mg/capsule);SAS, sulfasarazine (50 mg/capsule).*Used as the gastric emptying indicator (10 mg/capsule).

366 T. Ishibashi et al. / Journal of Controlled Release 59 (1999) 361 –376
Fig. 2. Release profiles of drugs from CTDC in the JP 1st-fluid (pH 1.2) and in the JP 2nd-fluid (pH 6.8); (a) CTDC , (b) CTDC , (c)0 1
CTDC , (d) CTDC ; h, j, ACET; n, m, THEO; s, d, SAS. Open symbols represent the release profiles in the JP 1st-fluid. Closed2 3
symbols represent the release profiles in the JP 2nd-fluid. Each data represents the mean6S.D. for six runs.
timed-corruption of the acid-soluble layer, which is 3.1.2. In vivo release studytriggered by the micro-environmental pH-change in In order to examine the drug release behavior ofthe capsule induced by the dissolution of succinic CTDC in vivo, and to estimate the small intestinalacid [13]. The CTDC and CTDC provide similar transit time, the dog study was performed. CTDC ,2 3 0
release profiles as CTDC , but it was noted that the CTDC , CTDC , and CTDC were orally adminis-1 1 2 3
dissolution lag time of THEO and SAS were extend- tered to four tetragastrin-treated beagle dogs undered in accordance with increasing the coating amount fasted condition. The plasma concentration profilesof the acid-soluble layer (Fig. 2c and d). The small of three drugs, i.e. THEO, ACET, and SP, weredifference in the starting time of dissolution between found to reflect well the in vitro release behavior ofTHEO and SAS could be attributed to the extremely CTDC. Individual plasma concentrations vs. timepoor water-solubility or water-repellent property of profiles of three drugs after oral administration ofSAS. This phenomenon can be improved by adding CTDC , CTDC , CTDC , and CTDC to four beagle0 1 2 3
the water soluble excipients or the disintegrate dogs are shown in Figs. 3–6, respectively.agents. From the results of this series of dissolution When CTDC was orally given to Dog 1, ACET,0
studies, the in vitro lag time of each capsule can be an indicator of gastric emptying, was found inregarded as 0.2 h, 1.3 h, 2.0 h, and 3.5 h for CTDC , plasma on the third-hour for the first time after the0
CTDC , CTDC , and CTDC , respectively. administration (Fig. 3). This suggests that the gastric1 2 3

T. Ishibashi et al. / Journal of Controlled Release 59 (1999) 361 –376 367
Fig. 3. Individual plasma concentration vs. time profiles of ACET, THEO, and SP after oral administration of CTDC to four beagle dogs.0
j, ACET (gastric emptying indicator); m, THEO; d, SP (colon arrival indicator).
emptying of the capsule occurred between 2 h and 3 dog, the first appearance of ACET in plasma wash. The first appearance of THEO in plasma was also observed at 4 h after administration. The first appear-observed in the third-hour, suggesting that THEO ance times (FAT) of THEO and SP were 5 h and 7was released at almost the same time as ACET, as h, respectively (Fig. 4). Attention was also paid toexpected. However, SP was detected in plasma at 6 h the change in the FAT of drugs in the CTDC and2
after the administration. SP is known as the major CTDC (Fig. 5 and Fig. 6). It was clearly found that,3
metabolite of SAS, most of which was converted with increasing the in vitro lag time, the difference infrom SAS by the bacterial degradation in the colon. the FAT became larger in the case between ACET,Since SAS cannot be absorbed from the intestine, SP and THEO and became closer in the case betweencan be regarded as a marker indicating the colon- THEO and SP.arrival of SAS. The above result, therefore, indicates In order to analyze the in vivo / in vitro relation-that the oral–caecal transit time of SAS was about 6 ship of CTDC in dogs more quantitatively, threeh, and the small intestinal transit time can be parameters, FAT , FAT , and FAT , whichACET THEO SP
estimated as about 3 h by subtracting the gastric are defined as the first appearance times of ACET,emptying time from the oral–caecal transit time. THEO, and SP in plasma, respectively, were calcu-
When the CTDC was administered to the same lated from the plasma concentration profiles of1

368 T. Ishibashi et al. / Journal of Controlled Release 59 (1999) 361 –376
Fig. 4. Individual plasma concentration vs. time profiles of ACET, THEO, and SP after oral administration of CTDC to four beagle dogs.1
j, ACET (gastric emptying indicator); m, THEO; d, SP (colon arrival indicator).
individual dogs. The mean values obtained from four values were found to vary from 5.3 h (CTDC ) to0
dogs are summarized in Table 3. 8.0 h (CTDC ) in accordance with the values of3
Since ACET is used as a marker of gastric FAT . The time difference between FAT andACET SP
emptying, the value of FAT corresponds to the FAT corresponds to the colon arrival time fromACET ACET
gastric emptying time in itself. Although the ob- the gastric emptying. This calculation provided quiteserved values of FAT implies that a certain closer values, such as 2.861.0 h for CTDC ,ACET 0
relation might exist between FAT and the in 2.760.5 h for CTDC , 3.560.5 h for CTDC , andACET 1 2
vitro lag time of CTDC, there was no statistically 3.060.6 h for CTDC , suggesting the relatively3
significant difference (P,0.05). In fact, considering constant small intestinal transit time in dogs. Thethe facts that one subject administered CTDC average value for all CTDCs was 3.0 h, which is0
provided a considerably short FAT of less than 1 almost coincident with the value reported by MizutaACET
h, and another subject administered CTDC provided et al. [17].3
an abnormally long FAT of 7 h, the apparent The value of FAT was found to be increasedACET THEO
difference should be judged as essentially negligible. with increasing the in vitro lag time of capsules,The value of FAT corresponds to the oral–caecal suggesting that all the capsules could release theSP
transit time in itself as mentioned before. The mean drug as designed even in the gastrointestinal tract in

T. Ishibashi et al. / Journal of Controlled Release 59 (1999) 361 –376 369
Fig. 5. Individual plasma concentration vs. time profiles of ACET, THEO, and SP after oral administration of CTDC to four beagle dogs.2
j, ACET (gastric emptying indicator); m, THEO; d, SP (colon arrival indicator).
dogs. The time difference between FAT and the colon. The time differences calculated for allACET
FAT means the onset time of theophylline CTDCs are compared in Fig. 8.THEO
release after the gastric emptying. The calculation It was clearly shown that the time differenceprovided 0.060.0 h, 1.060.0 h, 2.360.5 h, and decreased with increasing the in vitro lag time of2.760.6 h for CTDC , CTDC , CTDC , and CTDC. This means that the CTDC with a longer lag0 1 2
CTDC , respectively. In Fig. 7, all these values are time can deliver the drug to a deeper position in the3
plotted against the in vitro lag time of CTDCs, which small intestine. This result also indicates that, whenare obtained from Fig. 2. As was clearly shown, it the in vitro lag time of at least 3 h is given, thewas proven that there was a good correlation be- colonic delivery of drugs can be achieved in dogs.tween in vitro and in vivo drug release of the CTDC. This rather linear relation between the in vitro lag
The time difference between FAT and FAT time and the drug release site implies the possibilitySP THEO
should be a parameter indicating the release site in to use CTDC as the device for assessing the regionalthe small intestine even though it is still difficult to drug absorption in the gastrointestinal tract. Variousdefine the anatomical position exactly. When the experimental methods have been used to identify thetime difference is almost zero, it indicates that the specific regions where the maximum absorption andCTDC has released theophylline in the colon. The bioavailability of the drug are obtained in thesmaller time difference means a closer release site to gastrointestinal tract of animals or man. Examples of
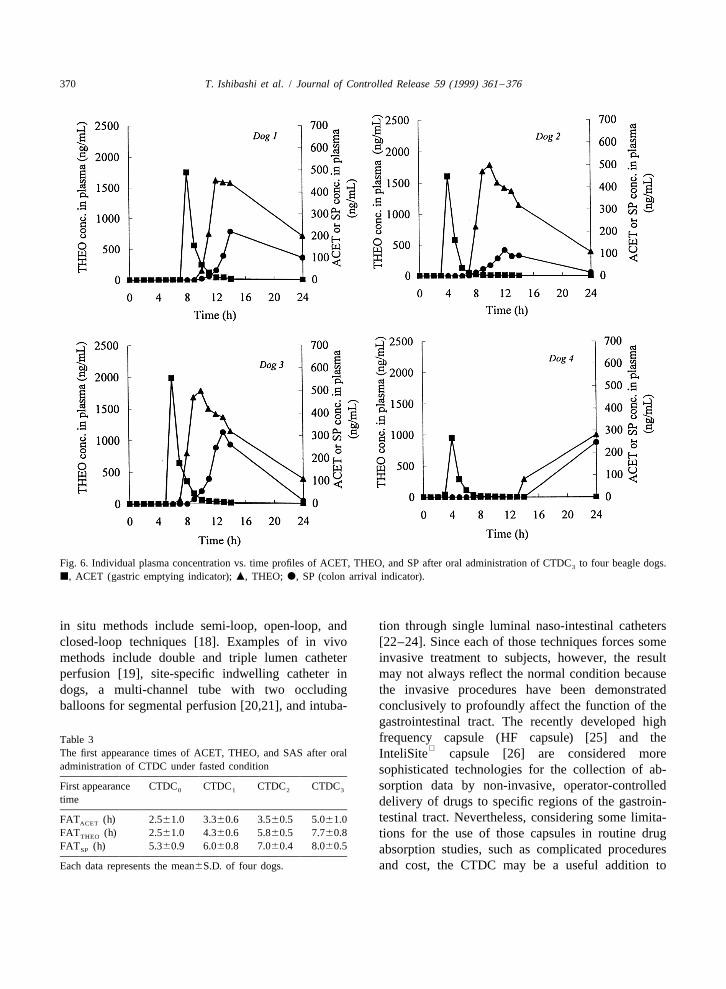
370 T. Ishibashi et al. / Journal of Controlled Release 59 (1999) 361 –376
Fig. 6. Individual plasma concentration vs. time profiles of ACET, THEO, and SP after oral administration of CTDC to four beagle dogs.3
j, ACET (gastric emptying indicator); m, THEO; d, SP (colon arrival indicator).
in situ methods include semi-loop, open-loop, and tion through single luminal naso-intestinal cathetersclosed-loop techniques [18]. Examples of in vivo [22–24]. Since each of those techniques forces somemethods include double and triple lumen catheter invasive treatment to subjects, however, the resultperfusion [19], site-specific indwelling catheter in may not always reflect the normal condition becausedogs, a multi-channel tube with two occluding the invasive procedures have been demonstratedballoons for segmental perfusion [20,21], and intuba- conclusively to profoundly affect the function of the
gastrointestinal tract. The recently developed highfrequency capsule (HF capsule) [25] and theTable 3
The first appearance times of ACET, THEO, and SAS after oral InteliSite capsule [26] are considered moreadministration of CTDC under fasted condition sophisticated technologies for the collection of ab-First appearance CTDC CTDC CTDC CTDC sorption data by non-invasive, operator-controlled0 1 2 3
time delivery of drugs to specific regions of the gastroin-testinal tract. Nevertheless, considering some limita-FAT (h) 2.561.0 3.360.6 3.560.5 5.061.0ACET
FAT (h) 2.561.0 4.360.6 5.860.5 7.760.8 tions for the use of those capsules in routine drugTHEO
FAT (h) 5.360.9 6.060.8 7.060.4 8.060.5SP absorption studies, such as complicated proceduresEach data represents the mean6S.D. of four dogs. and cost, the CTDC may be a useful addition to

T. Ishibashi et al. / Journal of Controlled Release 59 (1999) 361 –376 371
the JP 1st-fluid (pH 1.2) and the JP 2nd-fluid (pH6.8).
As was shown, all the drugs were not released forover 8 h in the 1st-fluid, whereas they were quicklyreleased after the lag time of about 3 h in the2nd-fluid. This release characteristic is quite suitableto evaluate the colonic absorbability of drugs, be-cause the drugs can be released directly on the dogcolon with this system.
The capsule was orally administered to threebeagle dogs under fasted condition. The aqueoussolution containing three drugs was also adminis-tered under the identical condition as the reference.The individual plasma concentration vs. time profiles
Fig. 7. In vivo/ in vitro correlation in the lag time of THEO of THEO, ACET, and PPA are compared with thoserelease from CTDC. In vitro lag time was directly determined by obtained from the aqueous form in Figs. 10–12,the dissolution study in the JP 2nd-fluid (pH 6.8). In vivo lag time
respectively.was estimated from FAT 2FAT . Each data of in vivo lagTHEO ACET
When the drugs were administered in the solutiontime and in vitro lag time represents the mean6S.D. for four runsand six runs, respectively. form, the blood concentration of each drug quickly
rose and attained the maximum level (C ) by 2 hmax
currently available non-invasive methodologies be- in all cases. On the other hand, when the CTDC wascause of its simple structure and ease of practical given, the onset of drug absorption was found to beuse. considerably delayed, up to 6 h or 7 h. It is also
noted that in the individual dog, the absorption of3.2. Drug absorbability in the colon three drugs occurred at the same time and the onset
time was almost coincident with that of SP (indicatedBased on the findings obtained in the first dog by an arrow in each profile). Necessary phar-
study, the CTDC , of which in vitro lag time was macokinetic parameters for discussion, calculated4
about 3 h, was used for the next dog study. This from the plasma drug concentration vs. time profiles,capsule contains three model drugs with different are listed in Table 4.solubilities, ACET, THEO, and PPA, and also it T is the time required to reach C , AUC ismax max 24h
contains SAS as the colon arrival indicator. Fig. 9 the area under the plasma concentration vs. timeshows the in vitro release profiles of those drugs in curve calculated from 0–24 h, and the relative
bioavailability was the percentage of the AUC of24h
CTDC to the AUC of the aqueous solution form24h
for each drug.The first appearance times of each drug in plasma
were all 6.360.5 h without any difference amongdrug species, and this value was very close to that ofSP (7.060.0 h) as mentioned above. These resultssuggest that the CTDC successfully delivered thedrugs to the caecum or proximal colon. The C ofmax
ACET was found to be 8236150 ng/mL whenadministered with CTDC, which was almost half ofthat obtained from the administration in the solutionform. In the cases of THEO and PPA, however, theFig. 8. Comparison of the time difference between FAT andTHEOC values were quite close to those of the solutionFAT for the CTDCs with different in vitro lag times. Each data maxSP
represents the mean6S.D. for four runs. form. The relative bioavailabilities of THEO and

372 T. Ishibashi et al. / Journal of Controlled Release 59 (1999) 361 –376
Fig. 9. Release profiles of drugs from CTDC in the JP 1st-fluid (pH 1.2) and in the JP 2nd-fluid (pH 6.8). (a) ACET and THEO, (b) PPA4
and SAS. h, j, ACET; n, m, THEO; x, ♦, PPA; s, d, SAS. Open symbols represent the release profiles in the JP 1st-fluid. Closedsymbols represent the release profiles in the JP 2nd-fluid. Each data represents the mean6S.D. for six runs.
Fig. 10. Individual plasma concentration vs. time profile of THEO after oral administration of solution and CTDC to beagle dogs. s,4
solution; d, CTDC . The arrow indicates the onset time of SP absorption.4

T. Ishibashi et al. / Journal of Controlled Release 59 (1999) 361 –376 373
Fig. 11. Individual plasma concentration vs. time profile of ACET after oral administration of solution and CTDC to beagle dogs. s,4
solution; d, CTDC . The arrow indicates the onset time of SP absorption.4
PPA were 94.2610.2% and 91.5622.6%, respective- likely reasons may be due to the poor absorbabilityly, for the CTDC administration, which is almost the of drugs from the colon. However, considering thesame level as those obtained from the solution form. fact that there is considerably less water in the colon,However, the relative bioavailability of ACET was the interference in the drug release from the deviceonly 71.066.0%, considerably lower than that of or in the diffusion of released drug to the mucosalsolution. This implies that the absorption of ACET surface should be a possible reason. In addition, thewould decrease in the colon. This is also supported bio-degradation by microflora in the colon could be aby the findings of Yamada et al., in which the possible reason for some drugs [30,31].absorption of ACET was relatively low from the The present study provided some fundamentalcolon, very poor from the stomach, and quite suffi- information to understand the colonic behavior of thecient from the jejunum and ileum [27]. released drugs in dogs, that is, the mucosal absorp-
It has been often pointed out that when drugs were tion of THEO and PPA was sufficient, no specificadministered in the sustained release dosage form, degradation occurred in the colon, and no significantthe relative bioavailability would be inevitably de- interference to the diffusion of the released drugcreased due to the poor absorption from the colon. In happened in the colon. Therefore, the most possiblefact, it was reported that the relative bioavailability reason for the commonly observed low bioavail-of THEO, ACET, and PPA in sustained release ability of those drugs in sustained release dosagedosage form decreased to 52% [28], 31.1% [27], and forms may be attributed to the considerable decrease78.8% [29], respectively, in dogs. One of the most in the drug release rate from the dosage form due to

374 T. Ishibashi et al. / Journal of Controlled Release 59 (1999) 361 –376
Fig. 12. Individual plasma concentration vs. time profile of PPA after oral administration of solution and CTDC to beagle dogs. s, solution;4
d, CTDC . The arrow indicates the onset time of SP absorption.4
Table 4The first appearance time and pharmacokinetic parameters of three drugs after oral administration of the aqueous solution form and CTDC tobeagle dogs under fasted condition
bDrug Dosage form FAT C T AUC Relativemax max 24hc(h) (ng /mL) (h) (ng h/mL) bioavailability
(%)
THEO Solution 060 26766123 1.360.6 25 59466067 100.0CTDC 6.360.5 26656117 9.760.5 23 66063890 94.2610.2
PPA Solution 060 456641 1.360.6 37516504 100.0CTDC 6.360.5 516681 9.760.5 33416510 91.5622.6
ACET Solution 060 1996626 0.560.0 37906430 100.0CTDC 6.360.5 8236150 8.760.5 27016436 71.066.0
aSP CTDC 7.060.0aColon arrival indicator.bThe first appearance time of drugs in plasma after oral administration.cThe percent of (AUC of CTDC/AUC of solution).Each data represents the mean6S.D. for three beagle dogs.

T. Ishibashi et al. / Journal of Controlled Release 59 (1999) 361 –376 375
Giordano, Time-dependent oral delivery systems for colona lower amount of fluids. The decreased bioavail-targeting, S.T.P. Pharm. Sci. 5 (1995) 83–88.ability of ACET may be caused by the first-pass
[8] I.R. Wilding, Scintigraphic evaluation of colonic deliveryeffect as was reported by Hirate et al. [32]. systems, S.T.P. Pharm. Sci. 5 (1995) 13–18.
From these results, this device is useful for [9] M. Saffran, G.S. Kumar, C. Savariar, J.C. Burnham, F.Williams, D.C. Neckers, A new approach to the oraldelivering the drugs to the colon and for estimatingadministration of insulin and other peptide drugs, Sciencethe regional drug absorption from the gastrointestinal233 (1979) 1081–1084.
tract. Moreover, this device might be useful for [10] S. Milojevic, J.M. Newton, J.H. Cummings, G.R. Gibson,anacidity or achylia gastrica if the enteric polymers R.L. Bothman, S.G. Ring, M.C. Allwood, M. Stockham,
Amylose, the new perspective in oral drug delivery to thedissolving at high pH like Eudragit S are used.human large intestine, S.T.P. Pharm. Sci. 5 (1995) 47–53.
[11] A. Rubinstein, D. Nakar, A. Sintov, Colonic drug delivery:enhanced release of indomethacin from cross-linked chon-droitin matrix in rat cecal content, Pharm. Res. 9(2) (1992)4. Conclusion276–278.
[12] T. Ishibashi, H. Hatano, H. Yoshino, M. Mizobe, M.Through the present in vivo dog studies, it was Kobayashi, In vivo drug release behavior in dogs from a new
proved that the CTDC had the potential to deliver the colon-targeted delivery system, J. Control. Release 57 (1999)45–53.drug to various positions in the gastrointestinal tract
[13] T. Ishibashi, H. Hatano, H. Yoshino, M. Mizobe, M.by adjusting the onset time of in vitro drug release inKobayashi, Design and evaluation of a new capsule-typethe JP 2nd-fluid (pH 6.8). It was also found that thedosage form for colon-targeted delivery of drugs, Int. J.
colon-targeted delivery of drugs can be achieved in Pharm. 168 (1998) 31–40.dogs, when the 3-h lag time is given to the CTDC. [14] S.S. Davis, The design and evaluation of controlled release
systems for the gastrointestinal tract, J. Control. Release 2This device is useful not only for the colon-targeted(1985) 27–38.delivery of drugs, but also for assessing the regional
[15] N. Katori, N. Aoyagi, T. Terao, Estimation of agitationdrug absorption from the gastrointestinal tract.intensity in the GI tract in humans and dogs based on invitro / in vivo correlation, Pharm. Res. 12(2) (1995) 237–243.
[16] M. Kennedy, P. Chinwah, D.N. Wade, A pharmacologicalReferences method of measuring mouth caecal transit time in man, Br. J.Clin. Pharm. 8 (1979) 372–373.
[1] P. Gruber, M.A. Longer, J.R. Robinson, Some biological [17] H. Mizuta, Y. Kawazoe, K. Haga, K. Ogawa, T. Yokobe,issues in oral controlled drug delivery, Adv. Drug. Del. Rev. Determination of small intestinal transit time in beagle dogs1 (1987) 1–18. using salicylazosulfapyridine, Yakugaku Zasshi 109 (1989)
[2] W.A. Ritschel, Targeting in the gastrointestinal tract: new 760–765.approaches, Methods Find. Exp. Clin. Pharmacol. 13 (1991) [18] N. Rouge, P. Buri, E. Doelker, Drug absorption sites in the313–336. gastrointestinal tract and dosage forms for site-specific
[3] M. Ashford, J.T. Fell, D. Attwood, P.J. Woodhead, An in delivery, Int. J. Pharm. 136 (1996) 117–139.vitro investigation into the suitability of pH-dependent [19] J.S. Fordtran, Segmental perfusion techniques, Gastroenterol-polymer for colonic targeting, Int. J. Pharm. 91 (1993) ogy 56 (1969) 987–989.
¨241–245. [20] L. Knutson, B. Odlind, R. Hallgren, A new technique for[4] R. Peeters, R. Kignet, Film-forming polymers for colonic segmental jejunal perfusion in man, Am. J. Gastroenterol.
drug delivery: I. Synthesis and physical and chemical 84(10) (1989) 1278–1284.¨¨ ¨properties of methyl derivatives of Eudragit S, Int. J. Pharm. [21] H. Lennernas, O. Ahrenstedt, R. Hallgren, L. Knutson, M.
94 (1993) 125–134. Ryde, K. Paalzow, Regional jejunal perfusion, a new in vivoapproach to study oral drug absorption in man, Pharm. Res.[5] T. Takaya, C. Ikeda, N. Imagawa, K. Niwa, K. Takada,9(10) (1992) 1243–1251.Development of a colon delivery capsule and the pharmaco-
[22] W.H. Barr, E.M. Zola, E.L. Candler, S. Hwang, A.V. Tendol-logical activity of recombinant human granulocyte colony-kar, R.S. Shamburek, B. Parker, D. Hilty, Differentialstimulating factor (rhG-CSF) in beagle dogs, J. Pharm.absorption of amoxicillin from the human small and largePharmacol. 47 (1995) 474–478.intestine, Clin. Pharmacol. Ther. 56(3) (1994) 279–285.[6] K. Niwa, T. Takaya, T. Morimoto, K. Takada, Preparation
[23] K.K.H. Chan, A. Buch, R.D. Glazer, V.A. John, W.H. Barr,and evaluation of a time-controlled release capsule made ofSite-differential gastrointestinal absorption of benazeprilethylcellulose for colon delivery of drugs, J. Drug Target. 3hydrochloride in healthy volunteers, Pharm. Res. 11(3)(1995) 83–89.(1994) 432–437.[7] A. Gazzaniga, C. Busetti, L. Moro, M.E. Sangalli, F.

376 T. Ishibashi et al. / Journal of Controlled Release 59 (1999) 361 –376
[24] N.Vidon, A. Pfeiffer, J. Godbillon, M. Rongier, S. Gauron, J. [29] R.C. DiLuccio, M.A. Hussain, D. CoffinBeach, G. Torosian,Hirtz, J.J. Bernier, J.P. Doubois, Evaluation of the gastric E. Shefter, A.R. Hurwitz, Polyvinyl alcohol–methyl acrylateabsorption and emptying of drugs under various pH con- copolymers as a sustained-release oral delivery system,ditions using a simple intubation method: application to Pharm. Res. 6(10) (1989) 844–847.diclofenac, Br. J. Clin. Pharm. 28(1) (1989) 121–124. ¨[30] P. Langguth, G. Breves, A. Stockli, H.P. Merkle, S.
[25] K.H. Antonin, Other methods in studying colonic drug Wolffram, Colonic absorption and bioavailability of theabsorption, in: P.R. Bieck (Ed.), Colonic Drug Absorption pentapeptide metkephamid in the rat, Pharm. Res. 11(11)and Metabolism, Marcel Dekker, New York, 1993, pp. 89– (1994) 1640–1645.107. [31] M. Kuroiwa, N. Inotsume, R. Iwaoku, M. Nakano, Degra-
[26] US Patent 5167626, 5170801. dation of drugs by gut microflora which may affect the[27] K. Yamada, A. Furuya, M. Akimoto, T. Maki, T. Suwa, H. bioavailability after oral administration, J. Pharm. Sci.
Ogata, Evaluation of gastrointestinal transit controlled-beagle 76(11) (1987) S79.dog as a suitable animal model for bioavailability testing of [32] J. Hirate, C.Y. Zhu, I. Horikoshi, V.O. Bhargava, First-passsustained-release acetaminophen dosage form, Int. J. Pharm. metabolism of acetaminophen in rats after low and high119 (1995) 1–10. doses, Biopharm. Drug Dispos. 11 (1990) 245–252.
[28] M. Turkoglu, W.A. Ritschel, A. Sakr, In vivo evaluation offluidized-bed coated pellets, Int. J. Pharm. 103 (1994) 115–118.



![WallFlex Colonic Stent - Boston Scientific- US · WallFlex ™ Colonic Stent Visualization Expertise in combining stent materials has resulted ... (BTS). “The WallFlex™ [Colonic]](https://img.pdfslide.net/doc/110x75/5ae601bc7f8b9a8b2b8ca931/wallflex-colonic-stent-boston-scientific-us-colonic-stent-visualization-expertise.jpg)

























