Embed Size (px)
Citation preview

Evidence For Multiple Cell Death Pathways during Development ofExperimental Cytomegalovirus Retinitis in Mice with Retrovirus-Induced Immunosuppression: Apoptosis, Necroptosis, and Pyroptosis
Hsin Chiena and Richard D. Dixa,b
Department of Biology, Viral Immunology Center, Georgia State University, Atlanta, Georgia, USA,a and Department of Ophthalmology, Emory University School ofMedicine, Atlanta, Georgia, USAb
AIDS-related human cytomegalovirus (HCMV) retinitis remains a major ophthalmologic problem worldwide. Although thissight-threatening disease is well characterized clinically, many pathogenic issues remain unresolved, among them a basic under-standing of the relative roles of cell death pathways during development of retinal tissue destruction. Using an established modelof experimental murine cytomegalovirus (MCMV) retinitis in mice with retrovirus-induced immunosuppression (MAIDS), weinitially investigated MCMV-infected eyes for evidence of apoptosis-associated molecules in mice with MAIDS of 4 weeks’(MAIDS-4) and 10 weeks’ (MAIDS-10) duration, which were resistant and susceptible to retinal disease, respectively, but whichharbored equivalent amounts of infectious MCMV. Whereas MCMV-infected eyes of MAIDS-4 mice showed little evidence ofapoptosis-associated molecules, MCMV-infected eyes of MAIDS-10 mice showed significant amounts of tumor necrosis factoralpha (TNF-�), TNF receptors 1 and 2, active caspase 8, active caspase 3, TNF-related apoptosis-inducing ligand (TRAIL),TRAIL-R(DR5), Fas, and Fas ligand mRNAs and/or proteins, all detected at peak amounts prior to development of most severeretinal disease. Immunohistochemical staining showed macrophages, granulocytes (neutrophils), Müller cells, and microglialcells as TNF-� sources. Remarkably, quantification of apoptosis by terminal deoxynucleotidyltransferase-mediated dUTP-biotinnick end labeling (TUNEL) assay suggested that apoptosis contributed minimally to retinal disease in MCMV-infected eyes ofMAIDS-10 mice. Subsequent studies demonstrated that MCMV-infected eyes of MAIDS-10 mice, but not MAIDS-4 mice,showed evidence of significant increases in molecules associated with two additional cell death pathways, necroptosis (receptor-interacting protein 1 [RIP1] and RIP3 mRNAs) and pyroptosis (caspase 1, interleukin 1� [IL-1�], and IL-18 mRNAs). We con-clude that apoptosis, necroptosis, and pyroptosis participate simultaneously during MAIDS-related MCMV retinitis, and all mayplay a role during AIDS-related HCMV retinitis.
AIDS-related human cytomegalovirus (HCMV) retinitis is aslowly progressive retinal disease of betaherpesvirus origin
that historically caused vision loss and blindness in up to 30% ofAIDS patients (48). However, with the development of active an-tiretroviral therapy (ART) to manage HIV infection directly, theincidence of AIDS-related HCMV retinitis has fallen significantlyin recent years. Nonetheless, this sight-threatening disease re-mains an ophthalmologic problem worldwide, affecting HIV-in-fected patients who do not have access to ART or who fail torespond to ART (39). AIDS-related HCMV retinitis is also notlimited to HIV/AIDS patients and can develop in patients who areimmunosuppressed for solid-organ or bone marrow transplanta-tion, albeit at a lower incidence (44).
We have therefore continued our investigations of the patho-genesis of AIDS-related HCMV retinitis using a well-character-ized experimental mouse model of murine cytomegalovirus(MCMV) retinitis that develops in C57BL/6 mice with MAIDS(14), a murine retrovirus-induced immunodeficiency syndromethat remarkably mimics HIV-induced AIDS in humans (37, 56).During the course of a previous investigation on the pathogenesisof MAIDS-related MCMV retinitis (15), we reported thatMCMV-infected eyes of mice with MAIDS of 4 weeks’ duration(MAIDS-4 mice) exhibited retinal folding and proliferation ofretinal pigment epithelium (RPE) in response to subretinal virusinfection, but without development of retinal necrosis. In sharpcontrast, �100% of the MCMV-infected eyes of mice withMAIDS of 10 weeks’ duration (MAIDS-10 mice) exhibited severe
retinal necrosis. Further investigation revealed that MCMV-in-fected eyes of MAIDS-4 and MAIDS-10 animals harbored equiv-alent amounts of infectious MCMV. Since MCMV-infected eyesof MAIDS-4 mice failed to develop retinal necrosis despite largeamounts of infectious virus also found to be associated with con-sistent development of retinal necrosis in MCMV-infected eyes ofMAIDS-10 mice, we concluded that virus infection alone is notsufficient for the onset and progression of the severe retinal de-struction observed in our experimental model of AIDS-relatedHCMV retinitis. We therefore hypothesized that cell death path-ways might contribute to MAIDS-related MCMV retinitis andelected to focus initially on tumor necrosis factor alpha (TNF-�)-induced apoptosis as a pathogenic mechanism whereby retinaltissue destruction ensues following MCMV infection of mice withMAIDS of 10 weeks’ duration.
TNF-� is a proinflammatory cytokine that induces diverse cel-lular responses ranging from apoptosis to the activation of anti-apoptotic genes involved in inflammation and acquired immuneresponses (7, 59). TNF-� signal transduction is accomplished
Received 21 May 2012 Accepted 19 July 2012
Published ahead of print 25 July 2012
Address correspondence to Richard D. Dix, [email protected].
Copyright © 2012, American Society for Microbiology. All Rights Reserved.
doi:10.1128/JVI.01275-12
October 2012 Volume 86 Number 20 Journal of Virology p. 10961–10978 jvi.asm.org 10961
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 31
Jan
uary
202
2 by
103
.207
.7.5
7.

through two distinct receptors, TNF receptor 1 (TNFR1) and TNFreceptor 2 (TNFR2) (3, 70). Binding of TNF-� to TNFR1 results inactivation of a caspase cascade involving active (cleaved) caspase 8and active (cleaved) caspase 3 that leads to apoptosis and celldeath. In contrast, binding of TNF-� to TNFR2 more commonlyresults in activation of cellular genes involved in the inflammatoryprocess (7, 59). More importantly, a subset of these genes may alsoact to suppress TNF-�-induced apoptosis, thereby giving TNF-�an antiapoptotic role that can lead to cell survival (7, 59). Pro-duced mainly by activated macrophages, TNF-� can also be pro-duced by a wide variety of cell types, including neutrophils andlymphoid cells (2, 13, 69).
Mondino and coworkers (55) initially provided evidence thatTNF-� is expressed within the ocular compartment during AIDS-related HCMV retinitis. A similar observation was made later byHofman and Hinton (45), who suggested infiltrating macro-phages as a possible source for TNF-�. We subsequently reportedsignificantly elevated levels of TNF-� within whole MCMV-in-fected eyes of mice with MAIDS which were susceptible to retinalnecrosis compared with levels in MCMV-infected eyes of normalmice resistant to retinal necrosis (15). A more thorough investi-gation of the role of TNF-� during experimental MCMV retinitisin methylprednisolone acetate-immunosuppressed BALB/c micewas then provided by several studies from Atherton’s laboratory(6, 74, 76). Following an initial study by Bigger et al. (6), whodemonstrated that apoptosis (as measured by terminal deoxy-nucleotidyltransferase-mediated dUTP-biotin nick end labeling[TUNEL] assay) increased during MCMV-induced retinal dis-ease, Zhou et al. (76) concluded that the TNFR1/TNF-� pathwayis indeed involved in induction of apoptosis and onset of retinaldisease but that TNF-� can also play an antiapoptotic role duringdisease progression as suggested by Zhang et al. (74). Given theavailability of our mouse model of MAIDS-related MCMV retini-tis (14) and our observation that MAIDS-4 mice and MAIDS-10mice exhibit strikingly different outcomes following subretinalMCMV infection despite eyes of both groups harboring equiva-lent amounts of infectious virus (15), we elected to explore TNF-�-induced apoptosis relative to onset and evolution of cytomeg-alovirus retinal disease from a different experimental perspective.We report herein that while apoptosis induced by TNFR1/TNF-�as well as TNF-related apoptosis-inducing ligand (TRAIL) andFas/Fas ligand (FasL) may collectively contribute to MAIDS-re-lated MCMV retinitis, apoptosis overall surprisingly contributesminimally to MCMV-induced retinal pathology in the setting ofretrovirus-induced immunosuppression. Additional investiga-tion of MCMV-infected eyes collected from MAIDS-10 mice dur-ing progression of MAIDS-related MCMV retinitis revealed mol-ecules unique to two additional cell death pathways, necroptosis(31, 43) and pyroptosis (5). Taken together, our results suggestthat multiple cell death pathways, perhaps operating simultane-ously, contribute to the pathogenesis of MAIDS-related MCMVretinitis and possibly AIDS-related HCMV retinitis.
MATERIALS AND METHODSAnimals. Adult female wild-type C57BL/6 mice, adult female mice defi-cient in TNF-� (B6.129S6-Tnf tm1Gkl/J), adult female mice deficient inTNFR1 (B6.129-Tnfrsf1atm1Mak/J), and adult female mice deficient inTNFR2 (B6.129S2-Tnfrsf1btm1Mwm/J) were purchased from Jackson Lab-oratory (Bar Harbor, ME). Adult female wild-type BALB/c mice werepurchased from Taconic Farms (Germantown, NY). All mice were main-
tained on alternative 12-h light-dark cycles and allowed access to food anddrink ad libitum. All experiments were performed with strict adherence toNational Institutes of Health guidelines, and all procedures were per-formed in accordance with the Association for Research in Vision andOphthalmology Resolution on the Use of Animals in Research.
Viruses. Stocks of the Smith strain of MCMV were prepared in mousesalivary glands of BALB/c mice as described previously (16). Mice wereinfected intraperitoneally with 102 to 103 PFU of MCMV containedwithin a 0.2-ml volume. Fourteen days later, the salivary glands wereremoved aseptically, homogenized (10% [wt/vol]) in Dulbecco’s modi-fied Eagle’s medium (DMEM) containing 10% fetal bovine serum, andclarified by centrifugation, and 0.5-ml aliquots of the supernatants werestored in liquid N2. Quantification of virus stocks was determined onmonolayers of mouse embryo fibroblasts grown in DMEM. A fresh ali-quot of MCMV stock was thawed and used for a single experiment.
Stocks of murine retrovirus (LP-BM5 murine leukemia virus[MuLV]) were prepared by using SC-1/LP-BM5 MuLV cells obtainedfrom the AIDS Research and Reference Reagent Program, Division ofAIDS, National Institute of Allergy and Infectious Diseases, National In-stitutes of Health. Approximately 106 SC-1/LP-BM5 MuLV cells weremixed with approximately 106 uninfected SC-1 cells and maintained for 6days in DMEM. The cells were then scraped into the medium, pelleted bycentrifugation, resuspended in phosphate-buffered saline, and stored at�70°C. Prior to use, the suspension was thawed and clarified by centrif-ugation to remove cellular debris.
Induction of MAIDS. MAIDS was induced in wild-type C57BL/c miceand in various groups of knockout mice (B6.129S6-Tnf tm1Gkl/J, B6.129-Tnfrsf1atm1Mak/J, and B6.129S2-Tnfrsf1btm1Mwm/J) by intraperitoneal in-jection of 1.0 ml of the LP-BM5 MuLV preparation (56). The inoculumcontained approximately 5 � 103 to 5 � 104 infectious murine retrovi-ruses. Mice with MAIDS of 4 weeks’ duration (MAIDS-4) and 10 weeks’duration (MAIDS-10) were used in these studies.
Experimental mouse model of MCMV retinitis. AIDS-relatedHCMV retinitis is thought to originate from virus that invades the retinafrom the blood during systemic infection (46). Intravenous or intraperi-toneal injection of immunosuppressed mice with MCMV to produce sys-temic MCMV infection fails to infect the neurosensory retina and causeretinal disease, although virus infects the choroid, RPE, and ciliary body ofthe eye (13, 32). Consequently, several laboratories (1, 16, 32, 49) rou-tinely have induced experimental MCMV retinitis in mice by supraciliaryand subretinal MCMV injection, a procedure that induces reproducibleretinal disease in mice with histopathologic features similar to those foundin AIDS-related HCMV retinitis (1, 16). The left eyes of all mice in thepresent study were therefore subjected to subretinal injection with ap-proximately 104 PFU of MCMV contained within a 2-�l volume ofDMEM following dilation of both eyes and administration of anesthesiaas described previously (16). The right (contralateral) eyes of all mice wereinjected subretinally with DMEM alone and served as controls.
Histopathologic analysis and evaluation of retinitis. At 3, 6, or 10days following subretinal MCMV infection, eyes were carefully removedfrom euthanized animals, fixed in 10% buffered formalin, and subjectedto frozen sectioning using a Shandon Cryotome (Thermo Scientific,Rochester, NY) to yield 8-�m sections. Sections were stained with hema-toxylin and eosin, examined by light microscopy, and scored for fre-quency and severity of disease as described previously (16).
Recovery of infectious MCMV. Whole MCMV-infected eyes fromMAIDS-4 and MAIDS-10 animals were collected at 10 days after subreti-nal injection and frozen at �70°C. At the time of quantitative plaqueassay, eyes were thawed, homogenized individually in 1.0 ml of coldDMEM, and clarified by centrifugation. Tenfold dilutions of the resultingsupernatant were inoculated in duplicate onto monolayers of mouse em-bryo fibroblasts contained within six-well plates, allowed to adsorb for 1 hat 37°C, overlaid with 2% methylcellulose-containing DMEM, and incu-bated for 6 days at 37°C in a humidified CO2 atmosphere. Individual
Chien and Dix
10962 jvi.asm.org Journal of Virology
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 31
Jan
uary
202
2 by
103
.207
.7.5
7.

plaques were counted with an inverted light microscope. Results for wholeeyes were reported as PFU/ml/eye.
Quantitative real-time reverse transcriptase PCR (RT-PCR) assay.Whole MCMV-infected eyes and whole contralateral mock-infected eyes(controls) collected at 3, 6, and 10 days after subretinal injection werestored in RNAlater solution (Ambion, Austin, TX) at �70°C prior toanalysis. Upon analysis, whole eyes were thawed at room temperature for5 min and individually homogenized in 1 ml of TRIzol reagent (Invitro-gen Life Technologies, Carlsbad, CA) using a 2-ml Ten Broeck tissuegrinder (Wheaton, Millville, NJ). Total RNA was extracted from each eyehomogenate using the PureLink total RNA purification system (Invitro-gen Life Technologies) according to the manufacturer’s instructions, andtotal RNA amounts for each sample were determined using a SmartSpec3000 spectrometer (Bio-Rad Laboratories, Hercules, CA). After the totalRNA concentration was normalized for each sample, cDNA synthesis wasperformed using the SuperScript III first-strand synthesis system (Invit-rogen) according to the manufacturer’s instructions. Detection and quan-tification of gene expression for each gene of interest were performedusing specific primers and the SYBR green PCR master mix (AppliedBiosystems, Foster City, CA), and products were detected and quantifiedusing an ABI Prism 7500 real-time PCR instrument coupled with se-quence detection system software (Applied Biosystems). Primers used fordetection and quantification of transcripts for TNF-�, TNFR1, TNFR2,active (cleaved) caspase 8, active (cleaved) caspase 3, TRAIL, TRAIL-R(DR5), Fas, FasL, caspase 9, cytochrome c, apoptotic protease-activatingfactor 1 (Apaf-1), receptor-interacting protein 1 (RIP1), RIP3, interleukin1� (IL-1�), IL-6, IL-18, caspase 1, and glyceraldehyde-3-phosphate dehy-drogenase (GAPDH) were purchased from Qiagen Inc., Valencia, CA.Primers used for detection and quantification of transcripts for MCMV-specific genes for immediate-early 1 (IE1) protein and glycoprotein H(gH) have been described previously (17, 64).
Western blot analysis. Whole MCMV-infected eyes and whole con-tralateral mock-infected (control) eyes stored at �70°C were thawed andindividually homogenized in a protease-inhibitor cocktail containedwithin phosphate-buffered saline (Sigma, St. Louis, MO). Standard West-ern blot analysis was performed for detection of TNF-�, active (cleaved)caspase 8, active (cleaved) caspase 3, and GAPDH proteins using rabbitanti-mouse TNF-� antibody (1:1,000) (Millipore, Temecula, CA), rabbitanti-mouse active caspase 8 antibody (1:1,000) (Sigma), rabbit anti-mouse active caspase 3 antibody (1:1,000) (Sigma), or rabbit anti-mouseGAPDH antibody (1:3,000) (Sigma) as primary antibodies, respectively.Following incubation with ImmunoPure goat anti-rabbit IgG antibody(heavy plus light chains [H�L]) (Thermo Scientific, Pittsburgh, PA) assecondary antibody, the resulting nitrocellulose membrane (Bio-Rad,Hercules, CA) was treated with enhanced chemiluminescence (ECL)Western blot detection reagents (GE Healthcare, Piscataway, NJ) and ex-posed to BioMax light film (Kodak, Rochester, NY).
TUNEL assay. TUNEL assay was used for detection and quantificationof apoptotic cells (33, 57, 58) in tissue sections of whole MCMV-infectedand contralateral mock-infected (control) eyes recovered from animals at3, 6, and 10 days after subretinal MCMV injection. Briefly, tissue sectionson L-lysine-treated glass microscope slides were treated with permeabili-zation solution (0.1% Triton X-100 in 0.1% sodium citrate buffer) for 5min on ice, washed in phosphate-buffered saline, and subjected to theTUNEL assay using the fluorescein in situ cell death detection kit (RocheDiagnostics Corporation, Indianapolis, IN) followed by counterstainingwith 4=,6-diamidino-2-phenylindole (DAPI) using Vectorshield mount-ing medium (Vector Laboratories, Burlingame, CA). Tissue sections wereinspected and photographed for TUNEL-positive cells using a NikonEclipse 50i microscope equipped with an X-cite Series 120 EPi-fl illumi-nator (Nikon, Japan).
Quantification of TUNEL-positive cells for tissue sections from indi-vidual MCMV-infected eyes (n � 5 mice per group) and mock-infectedeyes (n � 5 mice per group) was accomplished by subjecting every 5thsection of 30 serial sections per eye to TUNEL assay and DAPI counter-
staining as described above. For each section per eye subjected to TUNELassay and DAPI counterstaining (i.e., 6 sections per eye), 3 random rep-resentative regions within a 400� microscope field were counted forTUNEL-positive cells versus the total number of cells per microscope fieldas determined by DAPI-stained nuclei. Results were expressed as the av-erage percentage of TUNEL-positive cells per total number of cells persection per eye. One positive control and two negative controls were in-cluded for each experiment. The positive control consisted of sections ofmock-infected eyes reacted with DNase I (New England BioLabs, Ipswich,MA) (500 U/ml in 50 mM Tris-HCl, pH 7.5, containing 1 mg/ml bovineserum albumin) for 10 min at room temperature to induce DNA strandbreakage in all cells of each eye section prior to performance of the TUNELassay as described above and according to the manufacturer’s instruc-tions. Negative controls consisted of sections of mock-infected eyes re-acted with 50 mM Tris-HCl, pH 7.5, containing 1 mg/ml bovine serumalbumin in the absence of DNase I for 10 min at room temperature priorto performance of the TUNEL assay.
Immunohistochemical staining. Whole MCMV-infected eyes andcontralateral whole mock-infected (control) eyes were carefully removedfrom mice (n � 5 mice per group) at 3, 6, and 10 days after subretinalinjection and immediately fixed with 10% neutralized formalin at 4°C forat least 5 days. Individual eyes were frozen in OCT compound (Tissue-Tek, Torrance, CA) and serially sectioned onto L-lysine-coated glass mi-croscope slides using a Shandon Cryotome SME cryostat instrument(Thermo Electron Corporation, Pittsburgh, PA), resulting in �30 sec-tions of 8-mm thickness per eye. All eye tissue sections were stored at�20°C. Prior to immunohistochemical staining, eye tissue sections wererehydrated for 10 min at room temperature in 10 mM sodium citrateretrieval solution, washed with phosphate-buffered saline, and reactedwith 5% normal goat serum containing 0.2% Triton X-100 for 30 min atroom temperature. Immunohistochemical staining for detection andidentification of TNF-�-producing retinal cells was accomplished by in-cubating sections with fluorescein isothiocyanate (FITC)-conjugated ratanti-mouse TNF-� antibody (1:200) (eBioscience, San Diego, CA) at 4°Covernight in a humidified atmosphere. Control sections were incubatedwith FITC-conjugated rat anti-mouse antibody of matched IgG1 isotype(1:500) (eBioscience). Identification of TNF-�-producing retinal cellswas accomplished by incubation with a second cell-specific antibody. Fol-lowing three 5-min washes with phosphate-buffered saline, retinal sec-tions initially reacted with anti-TNF-� antibody were again incubated at4°C overnight in a humidified atmosphere with chicken anti-mouse glialfibrillary acidic protein (GFAP) antibody (1:500) (Abcam, Cambridge,MA) for identification of Müller cells, goat anti-mouse Iba-1 antibody(1:100) (Santa Cruz Biotechnology, Santa Cruz, CA) for identification ofretinal microglial cells, or rabbit anti-mouse Ly-6G antibody (1:100)(Santa Cruz Biotechnology) for identification of infiltrating granulocytes(neutrophils). Following three additional 5-min washes with phosphate-buffered saline, the retinal sections subsequently were incubated at roomtemperature for 1 h in a humidified atmosphere with donkey anti-chickenDyLight-594-labeled antibody (1:100) (Jackson ImmunoResearch, WestGrove, PA) for visualization of TNF-�-producing, GFAP-positive Müllercells, goat anti-rabbit DyLight-594-labeled antibody (1:100) (Jackson Im-munoResearch) for visualization of TNF-�-producing, Iba-1-positiveretinal microglial cells, or goat anti-rabbit DyLight-594-labeled antibody(1:100) (Jackson ImmunoResearch) for visualization of TNF-�-produc-ing, infiltrating granulocytes (neutrophils). For visualization of TNF-�-producing, infiltrating macrophages, retinal sections were incubated withphycoerythrin (PE)-Texas red-conjugated rat anti-mouse F4/80 antibody(1:100) (Invitrogen) and rabbit anti-mouse TNF-� antibody (1:500) (Ab-cam) at 4°C overnight in a humidified atmosphere, followed by incuba-tion with goat anti-rabbit DyLight-488 (1:100) (Jackson ImmunoRe-search) for 1 h at room temperature. All antibody-treated retinal sectionswere ultimately washed three times for 5 min each with phosphate-buff-ered saline, mounted with medium containing DAPI (Vectorshield; Vec-
Cell Death Pathways and MCMV Retinitis
October 2012 Volume 86 Number 20 jvi.asm.org 10963
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 31
Jan
uary
202
2 by
103
.207
.7.5
7.
tor Laboratories), and examined and photographed by fluorescence mi-croscopy.
Statistical analysis. Morphometric data for individual lesions ineach eye were averaged to provide one value per eye. The mean andstandard error of the mean for each group was calculated, and P valueswere determined using Student’s t test and the Wilcoxon rank sumtest. P values of �0.05 were considered significant for all forms ofstatistical analysis used.
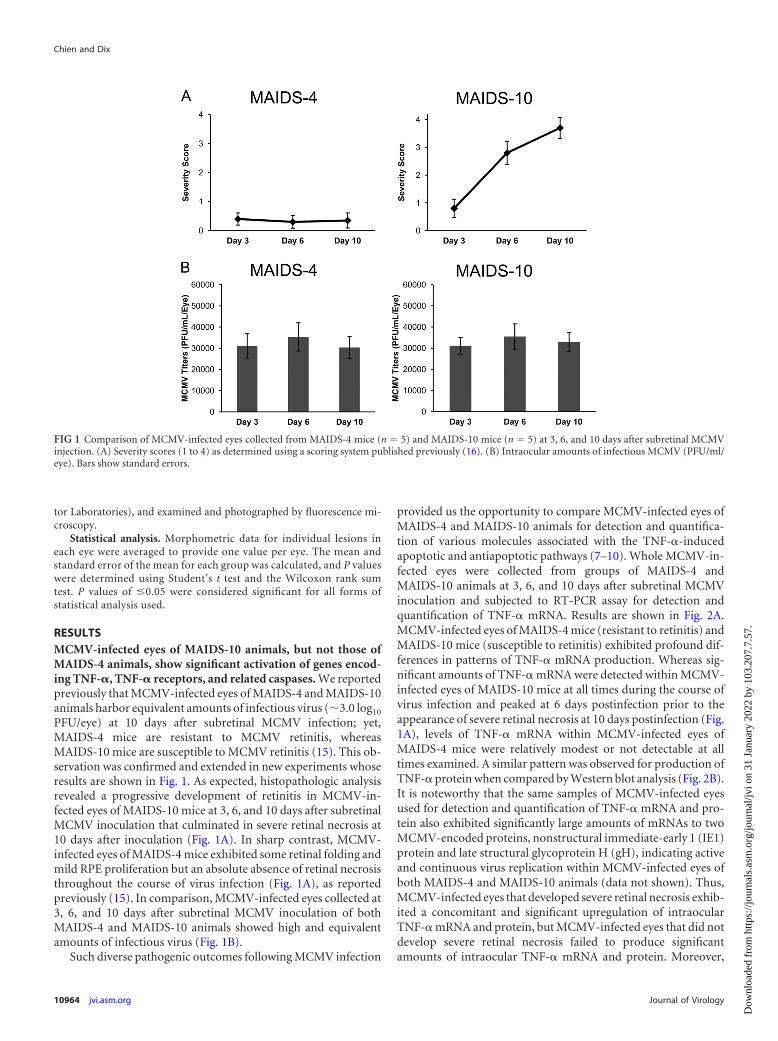
RESULTSMCMV-infected eyes of MAIDS-10 animals, but not those ofMAIDS-4 animals, show significant activation of genes encod-ing TNF-�, TNF-� receptors, and related caspases. We reportedpreviously that MCMV-infected eyes of MAIDS-4 and MAIDS-10animals harbor equivalent amounts of infectious virus (�3.0 log10
PFU/eye) at 10 days after subretinal MCMV infection; yet,MAIDS-4 mice are resistant to MCMV retinitis, whereasMAIDS-10 mice are susceptible to MCMV retinitis (15). This ob-servation was confirmed and extended in new experiments whoseresults are shown in Fig. 1. As expected, histopathologic analysisrevealed a progressive development of retinitis in MCMV-in-fected eyes of MAIDS-10 mice at 3, 6, and 10 days after subretinalMCMV inoculation that culminated in severe retinal necrosis at10 days after inoculation (Fig. 1A). In sharp contrast, MCMV-infected eyes of MAIDS-4 mice exhibited some retinal folding andmild RPE proliferation but an absolute absence of retinal necrosisthroughout the course of virus infection (Fig. 1A), as reportedpreviously (15). In comparison, MCMV-infected eyes collected at3, 6, and 10 days after subretinal MCMV inoculation of bothMAIDS-4 and MAIDS-10 animals showed high and equivalentamounts of infectious virus (Fig. 1B).
Such diverse pathogenic outcomes following MCMV infection
provided us the opportunity to compare MCMV-infected eyes ofMAIDS-4 and MAIDS-10 animals for detection and quantifica-tion of various molecules associated with the TNF-�-inducedapoptotic and antiapoptotic pathways (7–10). Whole MCMV-in-fected eyes were collected from groups of MAIDS-4 andMAIDS-10 animals at 3, 6, and 10 days after subretinal MCMVinoculation and subjected to RT-PCR assay for detection andquantification of TNF-� mRNA. Results are shown in Fig. 2A.MCMV-infected eyes of MAIDS-4 mice (resistant to retinitis) andMAIDS-10 mice (susceptible to retinitis) exhibited profound dif-ferences in patterns of TNF-� mRNA production. Whereas sig-nificant amounts of TNF-� mRNA were detected within MCMV-infected eyes of MAIDS-10 mice at all times during the course ofvirus infection and peaked at 6 days postinfection prior to theappearance of severe retinal necrosis at 10 days postinfection (Fig.1A), levels of TNF-� mRNA within MCMV-infected eyes ofMAIDS-4 mice were relatively modest or not detectable at alltimes examined. A similar pattern was observed for production ofTNF-� protein when compared by Western blot analysis (Fig. 2B).It is noteworthy that the same samples of MCMV-infected eyesused for detection and quantification of TNF-� mRNA and pro-tein also exhibited significantly large amounts of mRNAs to twoMCMV-encoded proteins, nonstructural immediate-early 1 (IE1)protein and late structural glycoprotein H (gH), indicating activeand continuous virus replication within MCMV-infected eyes ofboth MAIDS-4 and MAIDS-10 animals (data not shown). Thus,MCMV-infected eyes that developed severe retinal necrosis exhib-ited a concomitant and significant upregulation of intraocularTNF-� mRNA and protein, but MCMV-infected eyes that did notdevelop severe retinal necrosis failed to produce significantamounts of intraocular TNF-� mRNA and protein. Moreover,
FIG 1 Comparison of MCMV-infected eyes collected from MAIDS-4 mice (n � 5) and MAIDS-10 mice (n � 5) at 3, 6, and 10 days after subretinal MCMVinjection. (A) Severity scores (1 to 4) as determined using a scoring system published previously (16). (B) Intraocular amounts of infectious MCMV (PFU/ml/eye). Bars show standard errors.
Chien and Dix
10964 jvi.asm.org Journal of Virology
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 31
Jan
uary
202
2 by
103
.207
.7.5
7.
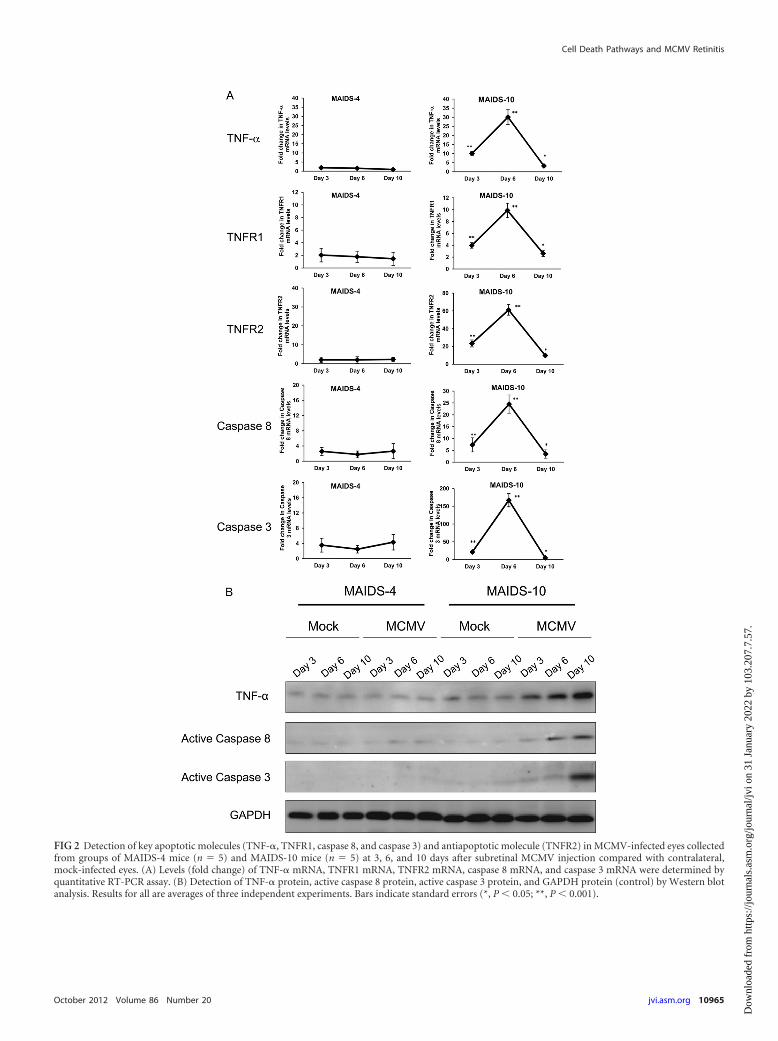
FIG 2 Detection of key apoptotic molecules (TNF-�, TNFR1, caspase 8, and caspase 3) and antiapoptotic molecule (TNFR2) in MCMV-infected eyes collectedfrom groups of MAIDS-4 mice (n � 5) and MAIDS-10 mice (n � 5) at 3, 6, and 10 days after subretinal MCMV injection compared with contralateral,mock-infected eyes. (A) Levels (fold change) of TNF-� mRNA, TNFR1 mRNA, TNFR2 mRNA, caspase 8 mRNA, and caspase 3 mRNA were determined byquantitative RT-PCR assay. (B) Detection of TNF-� protein, active caspase 8 protein, active caspase 3 protein, and GAPDH protein (control) by Western blotanalysis. Results for all are averages of three independent experiments. Bars indicate standard errors (*, P 0.05; **, P 0.001).
Cell Death Pathways and MCMV Retinitis
October 2012 Volume 86 Number 20 jvi.asm.org 10965
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 31
Jan
uary
202
2 by
103
.207
.7.5
7.

outcome was independent of active and continuous intraocularvirus replication.
Since TNF-� is known to drive apoptosis and to play an anti-apoptotic role via the distinct receptors TNFR1 and TNFR2, re-spectively (3, 70), we compared MCMV-infected eyes ofMAIDS-4 and MAIDS-10 animals for detection and quantifica-tion of TNFR1 and TNFR2 mRNA levels at 3, 6, and 10 days aftersubretinal MCMV inoculation. Results shown in Fig. 2A paral-leled those found for TNF-�. Little to no TNFR1 mRNA orTNFR2 mRNA was detected within MCMV-infected eyes ofMAIDS-4 mice at all times investigated after subretinal virus in-oculation. In sharp contrast, MCMV-infected eyes of MAIDS-10mice showed increased levels of mRNA to both TNFR1 andTNFR2 at day 3 postinfection that significantly increased andpeaked at day 6 postinfection prior to the appearance of severeretinal necrosis at day 10 postinfection, when TNFR1 and TNFR2mRNA levels decreased dramatically. Although a significant in-crease in apoptosis-related TNFR1 was expected concomitantwith the appearance of retinal pathology, the 60-fold increase inTNFR2 mRNA was unexpected, suggesting that TNF-�-drivenantiapoptotic forces also operate to a far greater extent than orig-inally appreciated during MAIDS-related MCMV retinitis.
The TNF-�-induced apoptotic pathway involves key proteinsthat include active (cleaved) caspase 8 and active (cleaved) caspase3 (3, 7, 59, 70). If TNF-�-induced apoptosis is involved in onsetand progression of retinal disease during MAIDS-related MCMVretinitis, we should detect evidence for upregulation of active(cleaved) caspase 8 and active (cleaved) caspase 3 within MCMV-infected eyes during development of retinitis. To confirm this pre-diction, the eyes of MAIDS-10 mice were inoculated subretinallywith MCMV and collected 3, 6, and 10 days later for quantificationof active (cleaved) caspase 8 and active (cleaved) caspase 3 mR-NAs. Both molecules were detected in significantly large amounts(Fig. 2A) within MCMV-infected eyes in a pattern relative to virusinfection that mimicked production for TNF-�, TNFR1, andTNFR2 mRNAs. Specifically, peak levels of the active forms ofcaspase 8 and caspase 3 mRNAs were observed at day 6 afterMCMV infection and prior to the appearance of severe retinalnecrosis. Evidence that caspase 8 and caspase 3 mRNAs weretranslated into active (cleaved) caspase 8 and caspase 3 proteinswas provided by Western blot analysis of MCMV-infected eyes ofMAIDS-10 mice collected at days 3, 6, and 10 after subretinal virusinoculation (Fig. 2B). Since MCMV-infected eyes of MAIDS-4mice resistant to retinal disease failed to show significant increasesin TNF-�, TNFR1, and TNFR2 mRNAs, it was not surprising tofind that these eyes also did not contain significantly largeamounts of active caspase 8 and caspase 3 proteins.
Apoptosis may also be induced during MAIDS-relatedMCMV retinitis by TRAIL and Fas/FasL cell death pathways butnot by the apoptosome complex. Other cell death pathways havebeen identified that culminate in apoptosis (19), among themTRAIL and its receptors TRAIL-R(DR4) and TRAIL-R(DR5) (20,34), the Fas/FasL cell death pathway (10, 18, 20, 71), and the mi-tochondrion-associated apoptosome, a multimeric complex con-sisting of cytochrome c, Apaf-1, and caspase 9 (11, 42). Whereasthe TRAIL cell death pathway is TNF-� dependent (and thereforecaspase 8 dependent), the apoptosis-inducing Fas/FasL pathwayand the apoptosome are TNF-� independent. Since we initiallyinvestigated the TNF-�/TNFR1-induced apoptotic pathway rela-tive to the onset and progression of MAIDS-related MCMV reti-
nitis (Fig. 2), we also compared MCMV-infected eyes fromMAIDS-4 and MAIDS-10 animals for detection and quantifica-tion of transcripts specific for key molecules associated with theseother apoptotic pathways. As expected, significant amounts ofTRAIL and TRAIL-R(DR5) mRNAs as well as Fas and FasLmRNAs were detected in MCMV-infected eyes of MAIDS-10mice (susceptible to retinitis), with peak amounts seen at 6 daysafter subretinal MCMV infection (Fig. 3A and B). In comparison,MCMV-infected eyes of MAIDS-4 mice (resistant to retinitis)failed to show production of significant amounts of mRNAs toTRAIL, TRAIL-R(DR5), Fas, and FasL at all times examined. Toour surprise, however, was the finding that MCMV-infected eyesof both MAIDS-4 mice and MAIDS-10 mice consistently showedno detectable production or less than a 2-fold production ofmRNAs to cytochrome c, Apaf-1, and caspase 9 (Fig. 3C), all keymolecules associated with apoptosome-induced apoptosis. Thus,whereas the TNF-�-associated TRAIL and Fas/FasL apoptotic celldeath pathways appear to be stimulated during the pathogenesisof MAIDS-related MCMV retinitis, the apoptosome apparentlyplays no role in MCMV-induced retinal tissue destruction duringMAIDS.
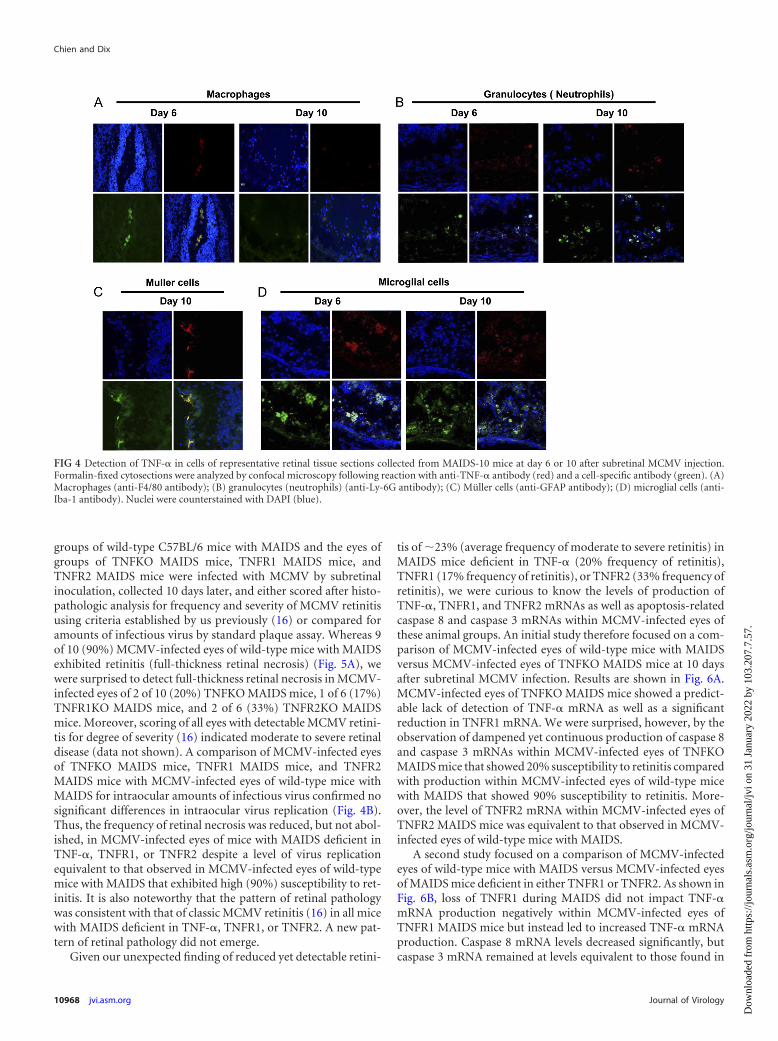
Several cell populations within retinal tissue of MCMV-in-fected eyes of MAIDS-10 animals serve as sources for TNF-�production. Previous clinical studies (45) and experimental stud-ies (76) have suggested that infiltrating macrophages may serve asa potential source for TNF-� production during development ofcytomegalovirus retinitis in immunosuppressed hosts. To con-firm and extend these findings using our MAIDS model of MCMVretinitis, frozen sections of MCMV-infected eyes collected fromMAIDS-10 mice at 10 days postinfection were investigated usingantibodies against individual cell-specific markers to identifyTNF-�-producing cells among candidate cell populations withinareas of retinal tissues showing severe pathology. Specifically, ret-inal sections were assessed using antibodies against F4/80 antigenas a marker for infiltrating macrophages, LY-6G antigen as amarker for infiltrating granulocytes (neutrophils), GFAP antigenas a marker for resident Müller cells, and Iba-1 antigen as a markerfor resident microglial cells. Results are shown in Fig. 4. In agree-ment with previous findings from other laboratories (45, 76),TNF-� production was detected in infiltrating macrophages (Fig.4A) within retinal tissues of MCMV-infected eyes with retinitis. Inaddition, TNF-� production was also detected in infiltrating gran-ulocytes (neutrophils) (Fig. 4B) as well as resident Müller cells(Fig. 4C) and resident microglial cells (Fig. 4D). Thus, many di-verse cell types may serve as possible sources for TNF-� produc-tion during MAIDS-related MCMV retinitis. These include infil-trating proinflammatory cells (macrophages and neutrophils) aswell as resident retinal cells (Müller cells and microglial cells).
MCMV retinitis is reduced, but detectable, in TNF-��/�
mice with MAIDS, TNFR1�/� mice with MAIDS, andTNFR2�/� mice with MAIDS. To define with greater precisionthe contribution of TNF-�-induced apoptosis toward onset andprogression of MCMV retinitis during MAIDS, we inducedMAIDS of 10 weeks’ duration in C57BL/6 mice deficient in TNF-�(TNFKO MAIDS mice), TNFR1 (TNFR1KO MAIDS mice), orTNFR2 (TNFR2KO MAIDS mice). All TNF-��/� mice,TNFR1�/� mice, and TNFR2�/� mice infected with the immu-nosuppressive murine retrovirus mixture (LP-BM5 MuLV) ex-hibited physical and immunologic characteristics (16) consistentwith the development of MAIDS (data not shown). The eyes of
Chien and Dix
10966 jvi.asm.org Journal of Virology
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 31
Jan
uary
202
2 by
103
.207
.7.5
7.
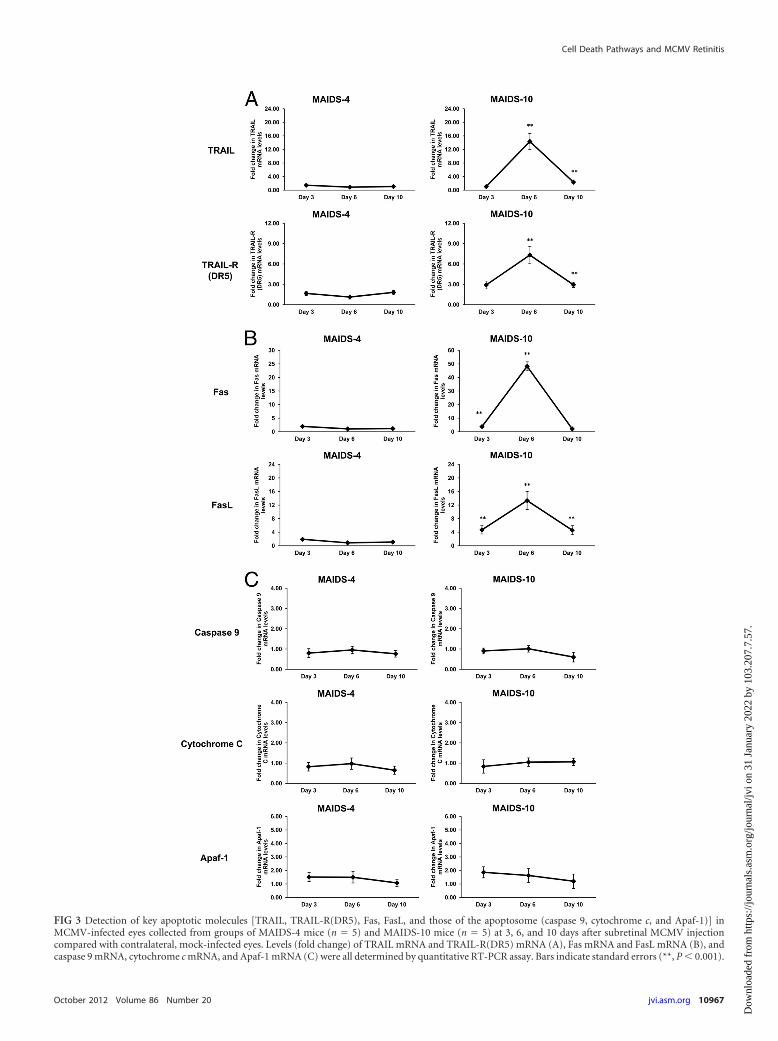
FIG 3 Detection of key apoptotic molecules [TRAIL, TRAIL-R(DR5), Fas, FasL, and those of the apoptosome (caspase 9, cytochrome c, and Apaf-1)] inMCMV-infected eyes collected from groups of MAIDS-4 mice (n � 5) and MAIDS-10 mice (n � 5) at 3, 6, and 10 days after subretinal MCMV injectioncompared with contralateral, mock-infected eyes. Levels (fold change) of TRAIL mRNA and TRAIL-R(DR5) mRNA (A), Fas mRNA and FasL mRNA (B), andcaspase 9 mRNA, cytochrome c mRNA, and Apaf-1 mRNA (C) were all determined by quantitative RT-PCR assay. Bars indicate standard errors (**, P 0.001).
Cell Death Pathways and MCMV Retinitis
October 2012 Volume 86 Number 20 jvi.asm.org 10967
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 31
Jan
uary
202
2 by
103
.207
.7.5
7.
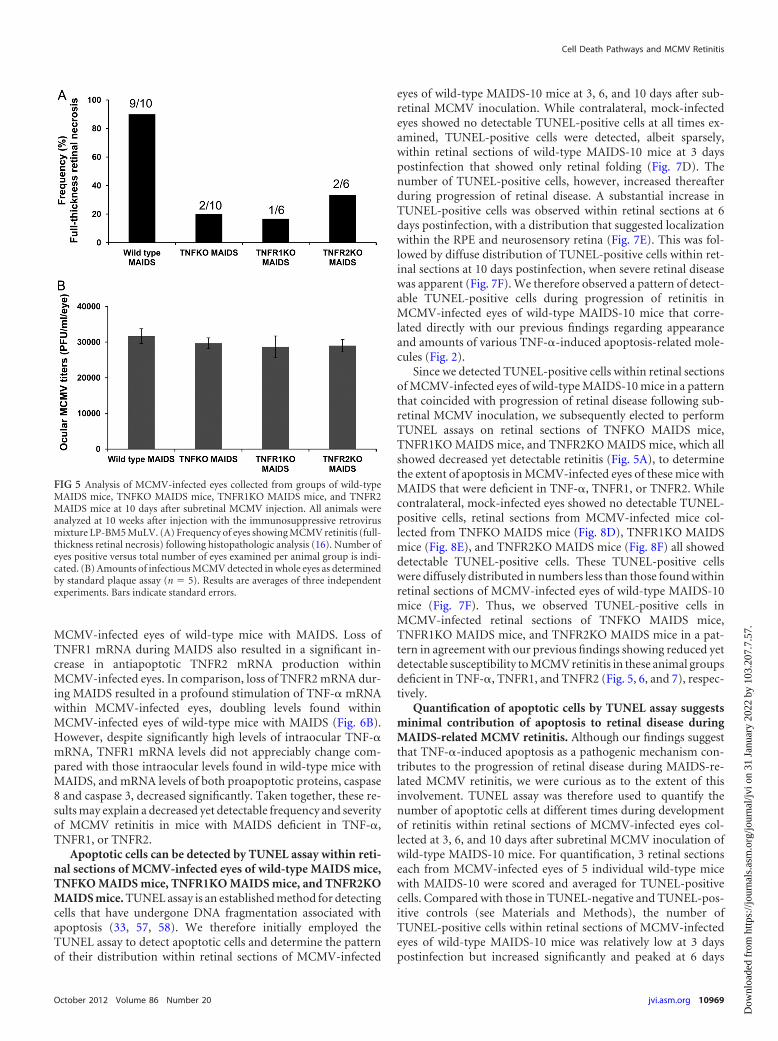
groups of wild-type C57BL/6 mice with MAIDS and the eyes ofgroups of TNFKO MAIDS mice, TNFR1 MAIDS mice, andTNFR2 MAIDS mice were infected with MCMV by subretinalinoculation, collected 10 days later, and either scored after histo-pathologic analysis for frequency and severity of MCMV retinitisusing criteria established by us previously (16) or compared foramounts of infectious virus by standard plaque assay. Whereas 9of 10 (90%) MCMV-infected eyes of wild-type mice with MAIDSexhibited retinitis (full-thickness retinal necrosis) (Fig. 5A), wewere surprised to detect full-thickness retinal necrosis in MCMV-infected eyes of 2 of 10 (20%) TNFKO MAIDS mice, 1 of 6 (17%)TNFR1KO MAIDS mice, and 2 of 6 (33%) TNFR2KO MAIDSmice. Moreover, scoring of all eyes with detectable MCMV retini-tis for degree of severity (16) indicated moderate to severe retinaldisease (data not shown). A comparison of MCMV-infected eyesof TNFKO MAIDS mice, TNFR1 MAIDS mice, and TNFR2MAIDS mice with MCMV-infected eyes of wild-type mice withMAIDS for intraocular amounts of infectious virus confirmed nosignificant differences in intraocular virus replication (Fig. 4B).Thus, the frequency of retinal necrosis was reduced, but not abol-ished, in MCMV-infected eyes of mice with MAIDS deficient inTNF-�, TNFR1, or TNFR2 despite a level of virus replicationequivalent to that observed in MCMV-infected eyes of wild-typemice with MAIDS that exhibited high (90%) susceptibility to ret-initis. It is also noteworthy that the pattern of retinal pathologywas consistent with that of classic MCMV retinitis (16) in all micewith MAIDS deficient in TNF-�, TNFR1, or TNFR2. A new pat-tern of retinal pathology did not emerge.
Given our unexpected finding of reduced yet detectable retini-
tis of �23% (average frequency of moderate to severe retinitis) inMAIDS mice deficient in TNF-� (20% frequency of retinitis),TNFR1 (17% frequency of retinitis), or TNFR2 (33% frequency ofretinitis), we were curious to know the levels of production ofTNF-�, TNFR1, and TNFR2 mRNAs as well as apoptosis-relatedcaspase 8 and caspase 3 mRNAs within MCMV-infected eyes ofthese animal groups. An initial study therefore focused on a com-parison of MCMV-infected eyes of wild-type mice with MAIDSversus MCMV-infected eyes of TNFKO MAIDS mice at 10 daysafter subretinal MCMV infection. Results are shown in Fig. 6A.MCMV-infected eyes of TNFKO MAIDS mice showed a predict-able lack of detection of TNF-� mRNA as well as a significantreduction in TNFR1 mRNA. We were surprised, however, by theobservation of dampened yet continuous production of caspase 8and caspase 3 mRNAs within MCMV-infected eyes of TNFKOMAIDS mice that showed 20% susceptibility to retinitis comparedwith production within MCMV-infected eyes of wild-type micewith MAIDS that showed 90% susceptibility to retinitis. More-over, the level of TNFR2 mRNA within MCMV-infected eyes ofTNFR2 MAIDS mice was equivalent to that observed in MCMV-infected eyes of wild-type mice with MAIDS.
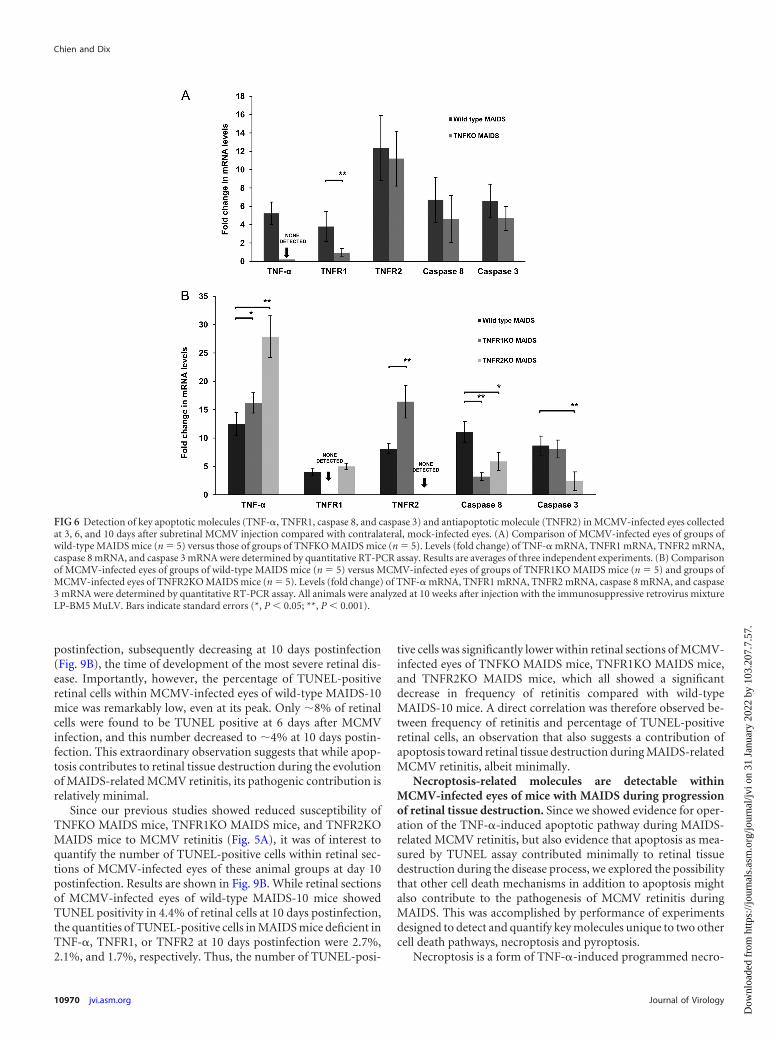
A second study focused on a comparison of MCMV-infectedeyes of wild-type mice with MAIDS versus MCMV-infected eyesof MAIDS mice deficient in either TNFR1 or TNFR2. As shown inFig. 6B, loss of TNFR1 during MAIDS did not impact TNF-�mRNA production negatively within MCMV-infected eyes ofTNFR1 MAIDS mice but instead led to increased TNF-� mRNAproduction. Caspase 8 mRNA levels decreased significantly, butcaspase 3 mRNA remained at levels equivalent to those found in
FIG 4 Detection of TNF-� in cells of representative retinal tissue sections collected from MAIDS-10 mice at day 6 or 10 after subretinal MCMV injection.Formalin-fixed cytosections were analyzed by confocal microscopy following reaction with anti-TNF-� antibody (red) and a cell-specific antibody (green). (A)Macrophages (anti-F4/80 antibody); (B) granulocytes (neutrophils) (anti-Ly-6G antibody); (C) Müller cells (anti-GFAP antibody); (D) microglial cells (anti-Iba-1 antibody). Nuclei were counterstained with DAPI (blue).
Chien and Dix
10968 jvi.asm.org Journal of Virology
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 31
Jan
uary
202
2 by
103
.207
.7.5
7.
MCMV-infected eyes of wild-type mice with MAIDS. Loss ofTNFR1 mRNA during MAIDS also resulted in a significant in-crease in antiapoptotic TNFR2 mRNA production withinMCMV-infected eyes. In comparison, loss of TNFR2 mRNA dur-ing MAIDS resulted in a profound stimulation of TNF-� mRNAwithin MCMV-infected eyes, doubling levels found withinMCMV-infected eyes of wild-type mice with MAIDS (Fig. 6B).However, despite significantly high levels of intraocular TNF-�mRNA, TNFR1 mRNA levels did not appreciably change com-pared with those intraocular levels found in wild-type mice withMAIDS, and mRNA levels of both proapoptotic proteins, caspase8 and caspase 3, decreased significantly. Taken together, these re-sults may explain a decreased yet detectable frequency and severityof MCMV retinitis in mice with MAIDS deficient in TNF-�,TNFR1, or TNFR2.
Apoptotic cells can be detected by TUNEL assay within reti-nal sections of MCMV-infected eyes of wild-type MAIDS mice,TNFKO MAIDS mice, TNFR1KO MAIDS mice, and TNFR2KOMAIDS mice. TUNEL assay is an established method for detectingcells that have undergone DNA fragmentation associated withapoptosis (33, 57, 58). We therefore initially employed theTUNEL assay to detect apoptotic cells and determine the patternof their distribution within retinal sections of MCMV-infected
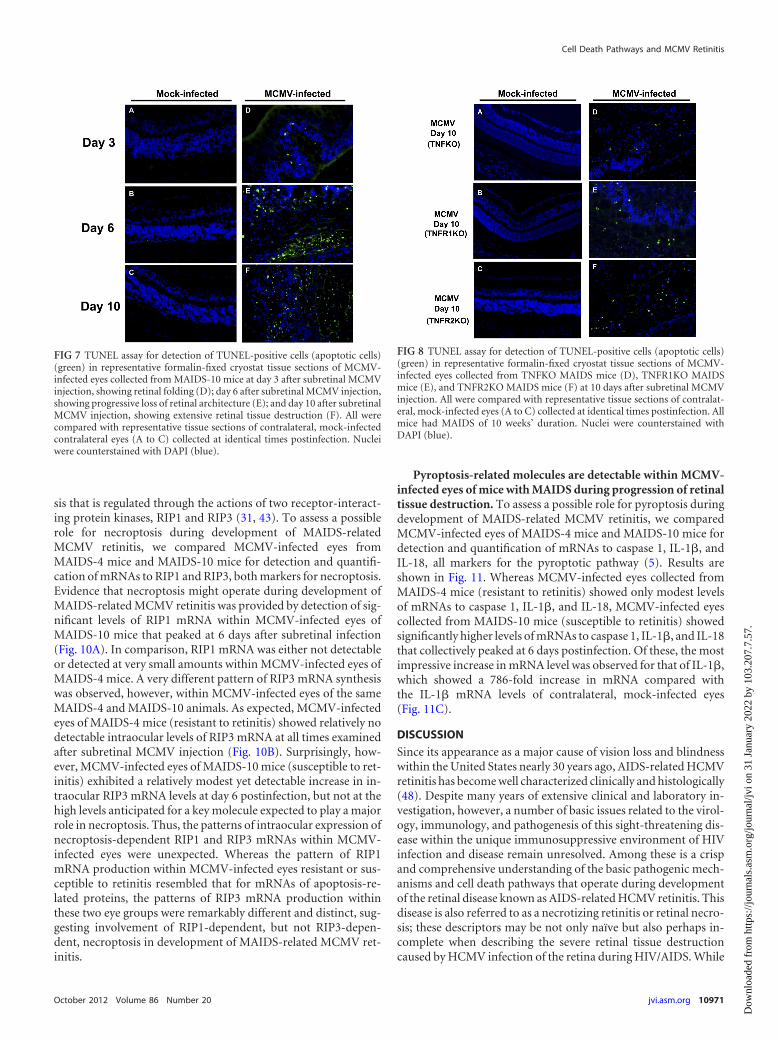
eyes of wild-type MAIDS-10 mice at 3, 6, and 10 days after sub-retinal MCMV inoculation. While contralateral, mock-infectedeyes showed no detectable TUNEL-positive cells at all times ex-amined, TUNEL-positive cells were detected, albeit sparsely,within retinal sections of wild-type MAIDS-10 mice at 3 dayspostinfection that showed only retinal folding (Fig. 7D). Thenumber of TUNEL-positive cells, however, increased thereafterduring progression of retinal disease. A substantial increase inTUNEL-positive cells was observed within retinal sections at 6days postinfection, with a distribution that suggested localizationwithin the RPE and neurosensory retina (Fig. 7E). This was fol-lowed by diffuse distribution of TUNEL-positive cells within ret-inal sections at 10 days postinfection, when severe retinal diseasewas apparent (Fig. 7F). We therefore observed a pattern of detect-able TUNEL-positive cells during progression of retinitis inMCMV-infected eyes of wild-type MAIDS-10 mice that corre-lated directly with our previous findings regarding appearanceand amounts of various TNF-�-induced apoptosis-related mole-cules (Fig. 2).
Since we detected TUNEL-positive cells within retinal sectionsof MCMV-infected eyes of wild-type MAIDS-10 mice in a patternthat coincided with progression of retinal disease following sub-retinal MCMV inoculation, we subsequently elected to performTUNEL assays on retinal sections of TNFKO MAIDS mice,TNFR1KO MAIDS mice, and TNFR2KO MAIDS mice, which allshowed decreased yet detectable retinitis (Fig. 5A), to determinethe extent of apoptosis in MCMV-infected eyes of these mice withMAIDS that were deficient in TNF-�, TNFR1, or TNFR2. Whilecontralateral, mock-infected eyes showed no detectable TUNEL-positive cells, retinal sections from MCMV-infected mice col-lected from TNFKO MAIDS mice (Fig. 8D), TNFR1KO MAIDSmice (Fig. 8E), and TNFR2KO MAIDS mice (Fig. 8F) all showeddetectable TUNEL-positive cells. These TUNEL-positive cellswere diffusely distributed in numbers less than those found withinretinal sections of MCMV-infected eyes of wild-type MAIDS-10mice (Fig. 7F). Thus, we observed TUNEL-positive cells inMCMV-infected retinal sections of TNFKO MAIDS mice,TNFR1KO MAIDS mice, and TNFR2KO MAIDS mice in a pat-tern in agreement with our previous findings showing reduced yetdetectable susceptibility to MCMV retinitis in these animal groupsdeficient in TNF-�, TNFR1, and TNFR2 (Fig. 5, 6, and 7), respec-tively.
Quantification of apoptotic cells by TUNEL assay suggestsminimal contribution of apoptosis to retinal disease duringMAIDS-related MCMV retinitis. Although our findings suggestthat TNF-�-induced apoptosis as a pathogenic mechanism con-tributes to the progression of retinal disease during MAIDS-re-lated MCMV retinitis, we were curious as to the extent of thisinvolvement. TUNEL assay was therefore used to quantify thenumber of apoptotic cells at different times during developmentof retinitis within retinal sections of MCMV-infected eyes col-lected at 3, 6, and 10 days after subretinal MCMV inoculation ofwild-type MAIDS-10 mice. For quantification, 3 retinal sectionseach from MCMV-infected eyes of 5 individual wild-type micewith MAIDS-10 were scored and averaged for TUNEL-positivecells. Compared with those in TUNEL-negative and TUNEL-pos-itive controls (see Materials and Methods), the number ofTUNEL-positive cells within retinal sections of MCMV-infectedeyes of wild-type MAIDS-10 mice was relatively low at 3 dayspostinfection but increased significantly and peaked at 6 days
FIG 5 Analysis of MCMV-infected eyes collected from groups of wild-typeMAIDS mice, TNFKO MAIDS mice, TNFR1KO MAIDS mice, and TNFR2MAIDS mice at 10 days after subretinal MCMV injection. All animals wereanalyzed at 10 weeks after injection with the immunosuppressive retrovirusmixture LP-BM5 MuLV. (A) Frequency of eyes showing MCMV retinitis (full-thickness retinal necrosis) following histopathologic analysis (16). Number ofeyes positive versus total number of eyes examined per animal group is indi-cated. (B) Amounts of infectious MCMV detected in whole eyes as determinedby standard plaque assay (n � 5). Results are averages of three independentexperiments. Bars indicate standard errors.
Cell Death Pathways and MCMV Retinitis
October 2012 Volume 86 Number 20 jvi.asm.org 10969
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 31
Jan
uary
202
2 by
103
.207
.7.5
7.
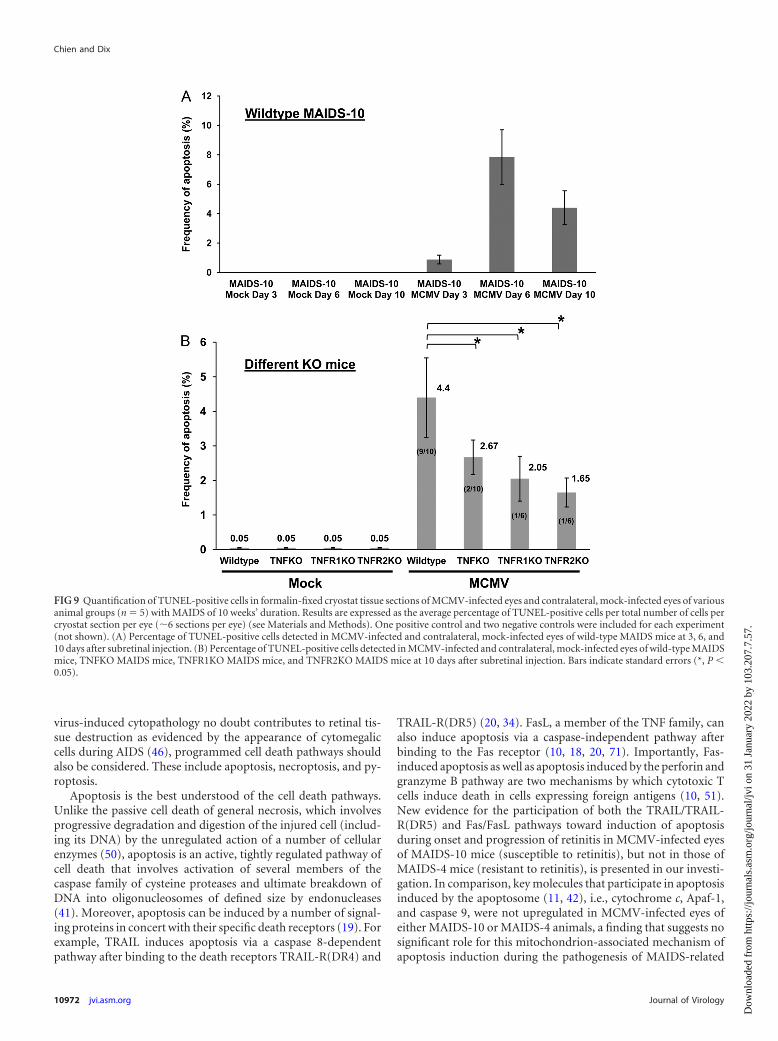
postinfection, subsequently decreasing at 10 days postinfection(Fig. 9B), the time of development of the most severe retinal dis-ease. Importantly, however, the percentage of TUNEL-positiveretinal cells within MCMV-infected eyes of wild-type MAIDS-10mice was remarkably low, even at its peak. Only �8% of retinalcells were found to be TUNEL positive at 6 days after MCMVinfection, and this number decreased to �4% at 10 days postin-fection. This extraordinary observation suggests that while apop-tosis contributes to retinal tissue destruction during the evolutionof MAIDS-related MCMV retinitis, its pathogenic contribution isrelatively minimal.
Since our previous studies showed reduced susceptibility ofTNFKO MAIDS mice, TNFR1KO MAIDS mice, and TNFR2KOMAIDS mice to MCMV retinitis (Fig. 5A), it was of interest toquantify the number of TUNEL-positive cells within retinal sec-tions of MCMV-infected eyes of these animal groups at day 10postinfection. Results are shown in Fig. 9B. While retinal sectionsof MCMV-infected eyes of wild-type MAIDS-10 mice showedTUNEL positivity in 4.4% of retinal cells at 10 days postinfection,the quantities of TUNEL-positive cells in MAIDS mice deficient inTNF-�, TNFR1, or TNFR2 at 10 days postinfection were 2.7%,2.1%, and 1.7%, respectively. Thus, the number of TUNEL-posi-
tive cells was significantly lower within retinal sections of MCMV-infected eyes of TNFKO MAIDS mice, TNFR1KO MAIDS mice,and TNFR2KO MAIDS mice, which all showed a significantdecrease in frequency of retinitis compared with wild-typeMAIDS-10 mice. A direct correlation was therefore observed be-tween frequency of retinitis and percentage of TUNEL-positiveretinal cells, an observation that also suggests a contribution ofapoptosis toward retinal tissue destruction during MAIDS-relatedMCMV retinitis, albeit minimally.
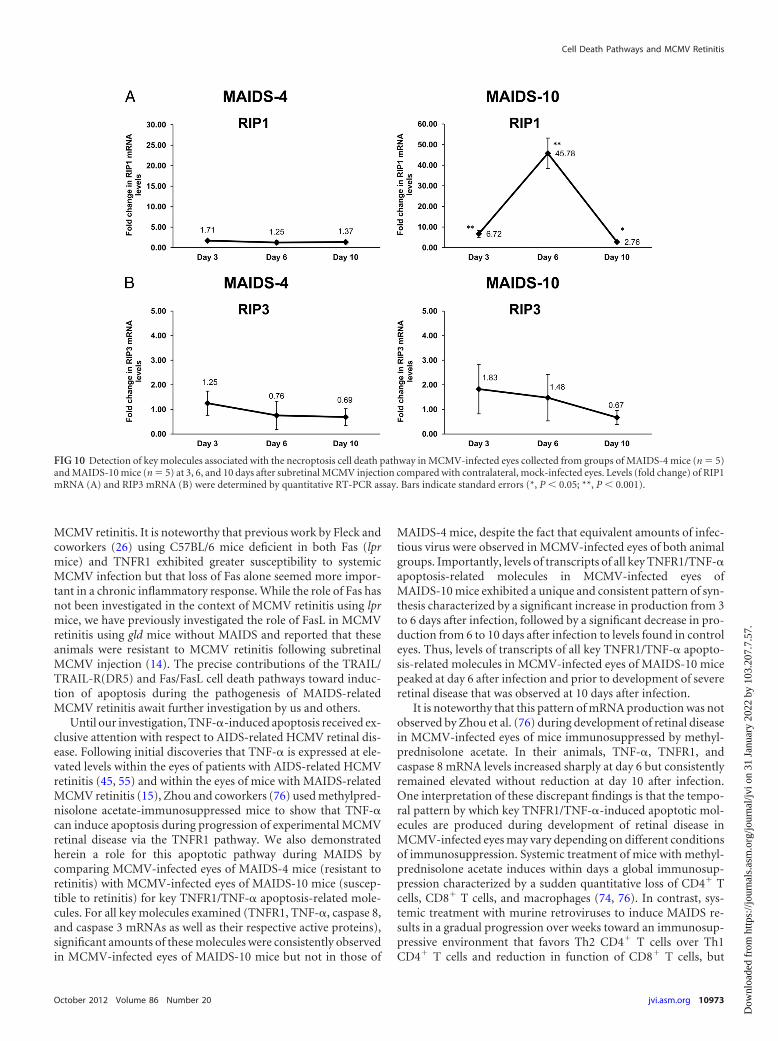
Necroptosis-related molecules are detectable withinMCMV-infected eyes of mice with MAIDS during progressionof retinal tissue destruction. Since we showed evidence for oper-ation of the TNF-�-induced apoptotic pathway during MAIDS-related MCMV retinitis, but also evidence that apoptosis as mea-sured by TUNEL assay contributed minimally to retinal tissuedestruction during the disease process, we explored the possibilitythat other cell death mechanisms in addition to apoptosis mightalso contribute to the pathogenesis of MCMV retinitis duringMAIDS. This was accomplished by performance of experimentsdesigned to detect and quantify key molecules unique to two othercell death pathways, necroptosis and pyroptosis.
Necroptosis is a form of TNF-�-induced programmed necro-
FIG 6 Detection of key apoptotic molecules (TNF-�, TNFR1, caspase 8, and caspase 3) and antiapoptotic molecule (TNFR2) in MCMV-infected eyes collectedat 3, 6, and 10 days after subretinal MCMV injection compared with contralateral, mock-infected eyes. (A) Comparison of MCMV-infected eyes of groups ofwild-type MAIDS mice (n � 5) versus those of groups of TNFKO MAIDS mice (n � 5). Levels (fold change) of TNF-� mRNA, TNFR1 mRNA, TNFR2 mRNA,caspase 8 mRNA, and caspase 3 mRNA were determined by quantitative RT-PCR assay. Results are averages of three independent experiments. (B) Comparisonof MCMV-infected eyes of groups of wild-type MAIDS mice (n � 5) versus MCMV-infected eyes of groups of TNFR1KO MAIDS mice (n � 5) and groups ofMCMV-infected eyes of TNFR2KO MAIDS mice (n � 5). Levels (fold change) of TNF-� mRNA, TNFR1 mRNA, TNFR2 mRNA, caspase 8 mRNA, and caspase3 mRNA were determined by quantitative RT-PCR assay. All animals were analyzed at 10 weeks after injection with the immunosuppressive retrovirus mixtureLP-BM5 MuLV. Bars indicate standard errors (*, P 0.05; **, P 0.001).
Chien and Dix
10970 jvi.asm.org Journal of Virology
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 31
Jan
uary
202
2 by
103
.207
.7.5
7.
sis that is regulated through the actions of two receptor-interact-ing protein kinases, RIP1 and RIP3 (31, 43). To assess a possiblerole for necroptosis during development of MAIDS-relatedMCMV retinitis, we compared MCMV-infected eyes fromMAIDS-4 mice and MAIDS-10 mice for detection and quantifi-cation of mRNAs to RIP1 and RIP3, both markers for necroptosis.Evidence that necroptosis might operate during development ofMAIDS-related MCMV retinitis was provided by detection of sig-nificant levels of RIP1 mRNA within MCMV-infected eyes ofMAIDS-10 mice that peaked at 6 days after subretinal infection(Fig. 10A). In comparison, RIP1 mRNA was either not detectableor detected at very small amounts within MCMV-infected eyes ofMAIDS-4 mice. A very different pattern of RIP3 mRNA synthesiswas observed, however, within MCMV-infected eyes of the sameMAIDS-4 and MAIDS-10 animals. As expected, MCMV-infectedeyes of MAIDS-4 mice (resistant to retinitis) showed relatively nodetectable intraocular levels of RIP3 mRNA at all times examinedafter subretinal MCMV injection (Fig. 10B). Surprisingly, how-ever, MCMV-infected eyes of MAIDS-10 mice (susceptible to ret-initis) exhibited a relatively modest yet detectable increase in in-traocular RIP3 mRNA levels at day 6 postinfection, but not at thehigh levels anticipated for a key molecule expected to play a majorrole in necroptosis. Thus, the patterns of intraocular expression ofnecroptosis-dependent RIP1 and RIP3 mRNAs within MCMV-infected eyes were unexpected. Whereas the pattern of RIP1mRNA production within MCMV-infected eyes resistant or sus-ceptible to retinitis resembled that for mRNAs of apoptosis-re-lated proteins, the patterns of RIP3 mRNA production withinthese two eye groups were remarkably different and distinct, sug-gesting involvement of RIP1-dependent, but not RIP3-depen-dent, necroptosis in development of MAIDS-related MCMV ret-initis.
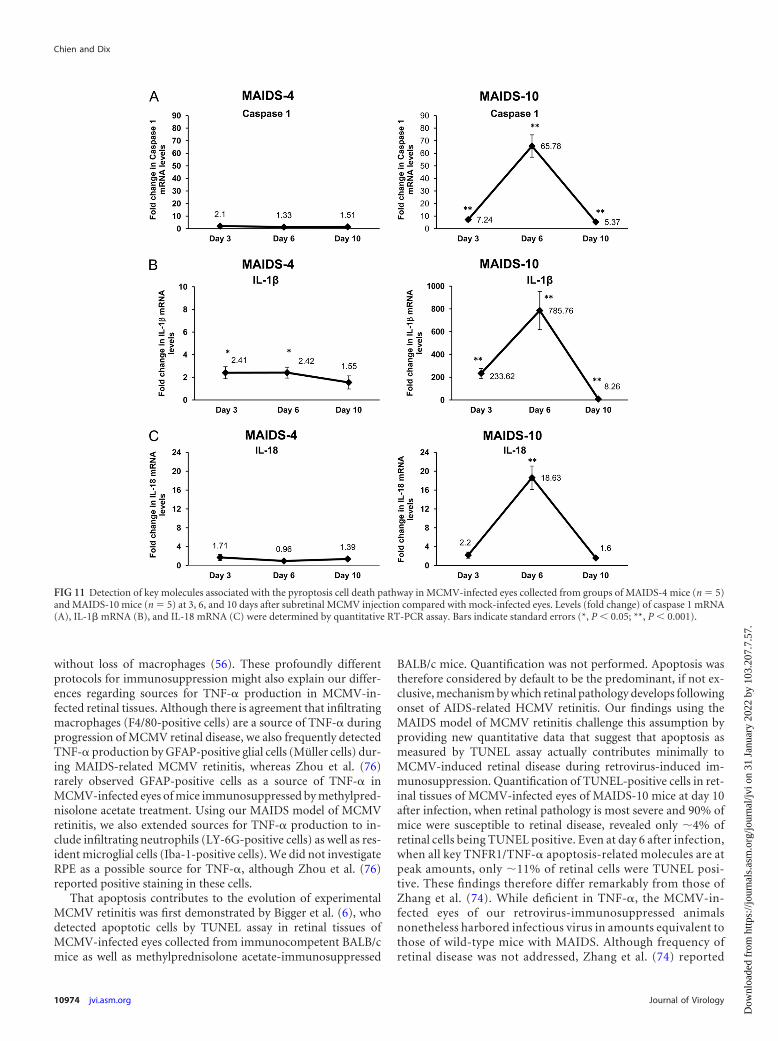
Pyroptosis-related molecules are detectable within MCMV-infected eyes of mice with MAIDS during progression of retinaltissue destruction. To assess a possible role for pyroptosis duringdevelopment of MAIDS-related MCMV retinitis, we comparedMCMV-infected eyes of MAIDS-4 mice and MAIDS-10 mice fordetection and quantification of mRNAs to caspase 1, IL-1�, andIL-18, all markers for the pyroptotic pathway (5). Results areshown in Fig. 11. Whereas MCMV-infected eyes collected fromMAIDS-4 mice (resistant to retinitis) showed only modest levelsof mRNAs to caspase 1, IL-1�, and IL-18, MCMV-infected eyescollected from MAIDS-10 mice (susceptible to retinitis) showedsignificantly higher levels of mRNAs to caspase 1, IL-1�, and IL-18that collectively peaked at 6 days postinfection. Of these, the mostimpressive increase in mRNA level was observed for that of IL-1�,which showed a 786-fold increase in mRNA compared withthe IL-1� mRNA levels of contralateral, mock-infected eyes(Fig. 11C).
DISCUSSION
Since its appearance as a major cause of vision loss and blindnesswithin the United States nearly 30 years ago, AIDS-related HCMVretinitis has become well characterized clinically and histologically(48). Despite many years of extensive clinical and laboratory in-vestigation, however, a number of basic issues related to the virol-ogy, immunology, and pathogenesis of this sight-threatening dis-ease within the unique immunosuppressive environment of HIVinfection and disease remain unresolved. Among these is a crispand comprehensive understanding of the basic pathogenic mech-anisms and cell death pathways that operate during developmentof the retinal disease known as AIDS-related HCMV retinitis. Thisdisease is also referred to as a necrotizing retinitis or retinal necro-sis; these descriptors may be not only naïve but also perhaps in-complete when describing the severe retinal tissue destructioncaused by HCMV infection of the retina during HIV/AIDS. While
FIG 8 TUNEL assay for detection of TUNEL-positive cells (apoptotic cells)(green) in representative formalin-fixed cryostat tissue sections of MCMV-infected eyes collected from TNFKO MAIDS mice (D), TNFR1KO MAIDSmice (E), and TNFR2KO MAIDS mice (F) at 10 days after subretinal MCMVinjection. All were compared with representative tissue sections of contralat-eral, mock-infected eyes (A to C) collected at identical times postinfection. Allmice had MAIDS of 10 weeks’ duration. Nuclei were counterstained withDAPI (blue).
FIG 7 TUNEL assay for detection of TUNEL-positive cells (apoptotic cells)(green) in representative formalin-fixed cryostat tissue sections of MCMV-infected eyes collected from MAIDS-10 mice at day 3 after subretinal MCMVinjection, showing retinal folding (D); day 6 after subretinal MCMV injection,showing progressive loss of retinal architecture (E); and day 10 after subretinalMCMV injection, showing extensive retinal tissue destruction (F). All werecompared with representative tissue sections of contralateral, mock-infectedcontralateral eyes (A to C) collected at identical times postinfection. Nucleiwere counterstained with DAPI (blue).
Cell Death Pathways and MCMV Retinitis
October 2012 Volume 86 Number 20 jvi.asm.org 10971
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 31
Jan
uary
202
2 by
103
.207
.7.5
7.
virus-induced cytopathology no doubt contributes to retinal tis-sue destruction as evidenced by the appearance of cytomegaliccells during AIDS (46), programmed cell death pathways shouldalso be considered. These include apoptosis, necroptosis, and py-roptosis.
Apoptosis is the best understood of the cell death pathways.Unlike the passive cell death of general necrosis, which involvesprogressive degradation and digestion of the injured cell (includ-ing its DNA) by the unregulated action of a number of cellularenzymes (50), apoptosis is an active, tightly regulated pathway ofcell death that involves activation of several members of thecaspase family of cysteine proteases and ultimate breakdown ofDNA into oligonucleosomes of defined size by endonucleases(41). Moreover, apoptosis can be induced by a number of signal-ing proteins in concert with their specific death receptors (19). Forexample, TRAIL induces apoptosis via a caspase 8-dependentpathway after binding to the death receptors TRAIL-R(DR4) and
TRAIL-R(DR5) (20, 34). FasL, a member of the TNF family, canalso induce apoptosis via a caspase-independent pathway afterbinding to the Fas receptor (10, 18, 20, 71). Importantly, Fas-induced apoptosis as well as apoptosis induced by the perforin andgranzyme B pathway are two mechanisms by which cytotoxic Tcells induce death in cells expressing foreign antigens (10, 51).New evidence for the participation of both the TRAIL/TRAIL-R(DR5) and Fas/FasL pathways toward induction of apoptosisduring onset and progression of retinitis in MCMV-infected eyesof MAIDS-10 mice (susceptible to retinitis), but not in those ofMAIDS-4 mice (resistant to retinitis), is presented in our investi-gation. In comparison, key molecules that participate in apoptosisinduced by the apoptosome (11, 42), i.e., cytochrome c, Apaf-1,and caspase 9, were not upregulated in MCMV-infected eyes ofeither MAIDS-10 or MAIDS-4 animals, a finding that suggests nosignificant role for this mitochondrion-associated mechanism ofapoptosis induction during the pathogenesis of MAIDS-related
FIG 9 Quantification of TUNEL-positive cells in formalin-fixed cryostat tissue sections of MCMV-infected eyes and contralateral, mock-infected eyes of variousanimal groups (n � 5) with MAIDS of 10 weeks’ duration. Results are expressed as the average percentage of TUNEL-positive cells per total number of cells percryostat section per eye (�6 sections per eye) (see Materials and Methods). One positive control and two negative controls were included for each experiment(not shown). (A) Percentage of TUNEL-positive cells detected in MCMV-infected and contralateral, mock-infected eyes of wild-type MAIDS mice at 3, 6, and10 days after subretinal injection. (B) Percentage of TUNEL-positive cells detected in MCMV-infected and contralateral, mock-infected eyes of wild-type MAIDSmice, TNFKO MAIDS mice, TNFR1KO MAIDS mice, and TNFR2KO MAIDS mice at 10 days after subretinal injection. Bars indicate standard errors (*, P 0.05).
Chien and Dix
10972 jvi.asm.org Journal of Virology
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 31
Jan
uary
202
2 by
103
.207
.7.5
7.
MCMV retinitis. It is noteworthy that previous work by Fleck andcoworkers (26) using C57BL/6 mice deficient in both Fas (lprmice) and TNFR1 exhibited greater susceptibility to systemicMCMV infection but that loss of Fas alone seemed more impor-tant in a chronic inflammatory response. While the role of Fas hasnot been investigated in the context of MCMV retinitis using lprmice, we have previously investigated the role of FasL in MCMVretinitis using gld mice without MAIDS and reported that theseanimals were resistant to MCMV retinitis following subretinalMCMV injection (14). The precise contributions of the TRAIL/TRAIL-R(DR5) and Fas/FasL cell death pathways toward induc-tion of apoptosis during the pathogenesis of MAIDS-relatedMCMV retinitis await further investigation by us and others.
Until our investigation, TNF-�-induced apoptosis received ex-clusive attention with respect to AIDS-related HCMV retinal dis-ease. Following initial discoveries that TNF-� is expressed at ele-vated levels within the eyes of patients with AIDS-related HCMVretinitis (45, 55) and within the eyes of mice with MAIDS-relatedMCMV retinitis (15), Zhou and coworkers (76) used methylpred-nisolone acetate-immunosuppressed mice to show that TNF-�can induce apoptosis during progression of experimental MCMVretinal disease via the TNFR1 pathway. We also demonstratedherein a role for this apoptotic pathway during MAIDS bycomparing MCMV-infected eyes of MAIDS-4 mice (resistant toretinitis) with MCMV-infected eyes of MAIDS-10 mice (suscep-tible to retinitis) for key TNFR1/TNF-� apoptosis-related mole-cules. For all key molecules examined (TNFR1, TNF-�, caspase 8,and caspase 3 mRNAs as well as their respective active proteins),significant amounts of these molecules were consistently observedin MCMV-infected eyes of MAIDS-10 mice but not in those of
MAIDS-4 mice, despite the fact that equivalent amounts of infec-tious virus were observed in MCMV-infected eyes of both animalgroups. Importantly, levels of transcripts of all key TNFR1/TNF-�apoptosis-related molecules in MCMV-infected eyes ofMAIDS-10 mice exhibited a unique and consistent pattern of syn-thesis characterized by a significant increase in production from 3to 6 days after infection, followed by a significant decrease in pro-duction from 6 to 10 days after infection to levels found in controleyes. Thus, levels of transcripts of all key TNFR1/TNF-� apopto-sis-related molecules in MCMV-infected eyes of MAIDS-10 micepeaked at day 6 after infection and prior to development of severeretinal disease that was observed at 10 days after infection.
It is noteworthy that this pattern of mRNA production was notobserved by Zhou et al. (76) during development of retinal diseasein MCMV-infected eyes of mice immunosuppressed by methyl-prednisolone acetate. In their animals, TNF-�, TNFR1, andcaspase 8 mRNA levels increased sharply at day 6 but consistentlyremained elevated without reduction at day 10 after infection.One interpretation of these discrepant findings is that the tempo-ral pattern by which key TNFR1/TNF-�-induced apoptotic mol-ecules are produced during development of retinal disease inMCMV-infected eyes may vary depending on different conditionsof immunosuppression. Systemic treatment of mice with methyl-prednisolone acetate induces within days a global immunosup-pression characterized by a sudden quantitative loss of CD4� Tcells, CD8� T cells, and macrophages (74, 76). In contrast, sys-temic treatment with murine retroviruses to induce MAIDS re-sults in a gradual progression over weeks toward an immunosup-pressive environment that favors Th2 CD4� T cells over Th1CD4� T cells and reduction in function of CD8� T cells, but
FIG 10 Detection of key molecules associated with the necroptosis cell death pathway in MCMV-infected eyes collected from groups of MAIDS-4 mice (n � 5)and MAIDS-10 mice (n � 5) at 3, 6, and 10 days after subretinal MCMV injection compared with contralateral, mock-infected eyes. Levels (fold change) of RIP1mRNA (A) and RIP3 mRNA (B) were determined by quantitative RT-PCR assay. Bars indicate standard errors (*, P 0.05; **, P 0.001).
Cell Death Pathways and MCMV Retinitis
October 2012 Volume 86 Number 20 jvi.asm.org 10973
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 31
Jan
uary
202
2 by
103
.207
.7.5
7.
without loss of macrophages (56). These profoundly differentprotocols for immunosuppression might also explain our differ-ences regarding sources for TNF-� production in MCMV-in-fected retinal tissues. Although there is agreement that infiltratingmacrophages (F4/80-positive cells) are a source of TNF-� duringprogression of MCMV retinal disease, we also frequently detectedTNF-� production by GFAP-positive glial cells (Müller cells) dur-ing MAIDS-related MCMV retinitis, whereas Zhou et al. (76)rarely observed GFAP-positive cells as a source of TNF-� inMCMV-infected eyes of mice immunosuppressed by methylpred-nisolone acetate treatment. Using our MAIDS model of MCMVretinitis, we also extended sources for TNF-� production to in-clude infiltrating neutrophils (LY-6G-positive cells) as well as res-ident microglial cells (Iba-1-positive cells). We did not investigateRPE as a possible source for TNF-�, although Zhou et al. (76)reported positive staining in these cells.
That apoptosis contributes to the evolution of experimentalMCMV retinitis was first demonstrated by Bigger et al. (6), whodetected apoptotic cells by TUNEL assay in retinal tissues ofMCMV-infected eyes collected from immunocompetent BALB/cmice as well as methylprednisolone acetate-immunosuppressed
BALB/c mice. Quantification was not performed. Apoptosis wastherefore considered by default to be the predominant, if not ex-clusive, mechanism by which retinal pathology develops followingonset of AIDS-related HCMV retinitis. Our findings using theMAIDS model of MCMV retinitis challenge this assumption byproviding new quantitative data that suggest that apoptosis asmeasured by TUNEL assay actually contributes minimally toMCMV-induced retinal disease during retrovirus-induced im-munosuppression. Quantification of TUNEL-positive cells in ret-inal tissues of MCMV-infected eyes of MAIDS-10 mice at day 10after infection, when retinal pathology is most severe and 90% ofmice were susceptible to retinal disease, revealed only �4% ofretinal cells being TUNEL positive. Even at day 6 after infection,when all key TNFR1/TNF-� apoptosis-related molecules are atpeak amounts, only �11% of retinal cells were TUNEL posi-tive. These findings therefore differ remarkably from those ofZhang et al. (74). While deficient in TNF-�, the MCMV-in-fected eyes of our retrovirus-immunosuppressed animalsnonetheless harbored infectious virus in amounts equivalent tothose of wild-type mice with MAIDS. Although frequency ofretinal disease was not addressed, Zhang et al. (74) reported
FIG 11 Detection of key molecules associated with the pyroptosis cell death pathway in MCMV-infected eyes collected from groups of MAIDS-4 mice (n � 5)and MAIDS-10 mice (n � 5) at 3, 6, and 10 days after subretinal MCMV injection compared with mock-infected eyes. Levels (fold change) of caspase 1 mRNA(A), IL-1� mRNA (B), and IL-18 mRNA (C) were determined by quantitative RT-PCR assay. Bars indicate standard errors (*, P 0.05; **, P 0.001).
Chien and Dix
10974 jvi.asm.org Journal of Virology
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 31
Jan
uary
202
2 by
103
.207
.7.5
7.

more TUNEL-positive cells in retinal tissues of MCMV-in-fected TNF-��/� mice immunosuppressed by methylpred-nisolone acetate treatment than in MCMV-infected eyes ofwild-type mice treated with the immunosuppressive drug.Once again, differences in immunosuppressive protocols mayaccount for this difference in findings.
At least two events, possibly working in concert, could explainour observation that apoptosis contributes minimally to retinaldisease during MAIDS-related MCMV retinitis. First, while thesignaling pathway initiated by TNFR1 is stimulated during devel-opment of MCMV retinal disease during systemic immunosup-pression, whether it is drug-induced or retrovirus-induced, thesignaling pathway(s) initiated by TNFR2 may also be stimulatedduring MCMV infection of eyes of mice with MAIDS as evidencedby significant upregulation of TNFR2 mRNA levels in a patternidentical to that observed for TNFR1 mRNA. The precise conse-quence(s) of TNFR2 stimulation on the pathogenesis of MAIDS-related MCMV retinal disease remains uncertain since the signal-ing pathway(s) initiated by TNFR2 is less well characterized thanthe TNFR1-initiated signaling pathway that stimulates apoptosis.Nevertheless, there is general agreement that TNFR2 appears tosignal outcomes both shared with and opposite to that of TNFR1and, unlike TNFR1, may independently mediate signals that pro-mote tissue repair and angiogenesis (7, 59). Thus, intraocular in-crease in TNF-� production during the course of MCMV retinaldisease development may place into motion two opposing effects,one promoting the death of retinal cells by apoptosis and the otherpromoting survival of retinal cells despite stimulation of TNFR1/TNF-�-induced apoptosis. A similar antiapoptotic role forTNF-� was suggested by Zhang et al. (74), although these workersdid not investigate TNFR2 directly during the course of experi-mental MCMV retinal disease development. Thus, TNF-� mightpromote retinal cell survival during MAIDS-related MCMV reti-nitis via the TNFR2 signaling pathway(s) and counteract retinalcell death by apoptosis induced by the TNFR1 signaling pathway.Second, several genes encoding suppressors of apoptosis havebeen identified in the genomes of HCMV and MCMV (reviewedby Brune) (8). Of these, the most attention has been given to theHCMV gene open reading frame (ORF) UL37x1, encoding vMIA(29), and the MCMV gene m38.5, encoding vIBO (52). Both geneproducts are thought to inhibit apoptosis at the mitochondriallevel, possibly by functioning as cellular antiapoptotic proteinsBcl-x and Bax, respectively (8). Zhang et al. (74) suggested a sim-ilar mitochondrion-targeting antiapoptotic role for TNF-� dur-ing development of MCMV retinal disease in methylprednisoloneacetate-immunosuppressed mice. Thus, we envision two counter-active forces at play during the evolution of MCMV retinal infec-tion, one promoting apoptosis through the TNFR1/TNF-� path-way and the other protecting against apoptosis through thecombined efforts of the TNFR2/TNF-� pathway and MCMV-en-coded antiapoptotic gene products. We further postulate that ret-inal tissue destruction is actually minimized by the apoptotic celldeath pathway as antiapoptotic forces counteract and dampenTNFR1/TNF-�-induced apoptosis.
If TNFR1/TNF-�-induced apoptosis were the exclusive celldeath pathway by which retinal tissue destruction evolves duringMAIDS-related MCMV retinitis, we would predict that mice withMAIDS deficient in TNF-� or TNFR1 would remain resistant toMCMV retinitis. This was not our finding. MCMV-infected eyesof mice with MAIDS deficient in TNF-� (TNFKO MAIDS mice)
or deficient in TNFR1 (TNFR1KO MAIDS mice) continued toexhibit susceptibility to retinitis, albeit at a reduced frequency.This observation prompted us to investigate other cell death path-ways that might also operate in combination with apoptosis dur-ing MAIDS-related MCMV retinitis, focusing our attention onnecroptosis and pyroptosis.
Necroptosis (31, 43) is a specialized TNF-�-induced caspase-independent programmed cell death process that results in earlymembrane and organelle swelling, followed by cell lysis (necrosis)via regulated signal transduction pathways mediated by receptor-interacting protein (RIP) kinases, especially when caspase 8 activ-ity is compromised (23, 35, 54). RIP1, a serine-threonine proteinkinase, promotes necroptosis through mitochondrial membranepermeabilization that is mediated by a Bcl-2 family member pro-tein, Bmf (43). During apoptosis, RIP1 is cleaved and inactivatedby caspase 8 (22). However, when caspase 8 is inhibited or cannotbe activated efficiently, RIP1 is thought to mediate necroptosisthrough expression of another receptor-interacting protein ki-nase, RIP3, that leads to reactive oxygen species production andnecrotic cell death (12, 38, 75). While the precise interaction be-tween RIP1 and RIP3 during development of necroptosis remainsunclear, RIP3, for now, appears to be a key regulator of RIP1kinase activation (30), and its level of expression correlates withresponsiveness to programmed necrosis (38). Necroptosis hasbeen shown recently to operate in the eye during experimentalretinal detachment-induced photoreceptor death in rats whenapoptosis is prevented by caspase inhibition (65), and during neu-ronal death associated with experimental retinal ischemia in rats(62). In the present investigation, we obtained new evidence thatnecroptosis may also operate during progression of MAIDS-re-lated MCMV retinitis. Specifically, we observed a significant anddramatic upregulation of RIP1 mRNA levels within MCMV-in-fected eyes of MAIDS-10 mice (susceptible to retinitis) but notwithin those of MAIDS-4 mice (resistant to retinitis). Moreover,the pattern of RIP1 mRNA synthesis was similar to that observedfor key molecules involved in the TNFR1/TNF-� apoptotic path-way; i.e., peak amounts were observed at day 6 after infection andprior to the appearance of severe retinal disease at day 10. Surpris-ingly, however, a similar pattern of synthesis was not observed formRNA of RIP3, another necroptosis-related molecule. MCMV-infected eyes of both MAIDS-10 and MAIDS-4 animals showedonly minimal increases of RIP3 mRNA throughout the course ofdisease development. This remarkable finding suggests that ifRIP1-mediated programmed necrosis contributes to retinal tissuedestruction during MAIDS-related MCMV retinitis, it does sowithout the need for significant upregulation of RIP3 mRNA.Present studies are oriented toward further understanding thisunexpected observation.
As with apoptosis (8, 29, 52), MCMV also encodes a gene prod-uct that serves to modulate necroptosis during infection. Uptonand coworkers (66) initially demonstrated that the M45 protein ofMCMV suppresses necroptotic cell death in vitro by interactionwith RIP1 via a RIP homotypic interaction motif (RHIP). Sup-pression of necroptosis is thought to take place soon after infec-tion since M45 is a structural tegument protein that is delivered tocells upon virion entry, and therefore, it would be available forinteraction with the RHIP modulator of RIP1 for immediate sup-pression of necroptosis. Moreover, since M45 is required for pro-tection from MCMV-induced cell death in endothelial cells andmacrophages (9), M45-mediated suppression of necroptosis may
Cell Death Pathways and MCMV Retinitis
October 2012 Volume 86 Number 20 jvi.asm.org 10975
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 31
Jan
uary
202
2 by
103
.207
.7.5
7.

also represent an important cell tropism determinant. UL45 (36),the HCMV homologue to MCMV M45, awaits investigation todetermine if it, too, possesses an analogous function with respectto necroptotic suppression. More recent in vitro work by Upton etal. (67) has suggested that induction of necroptosis followingMCMV infection may be RIP3 mediated and proceed indepen-dently of RIP1 but that MCMV can nonetheless inhibit RIP3-mediated necroptosis through a virus M45-encoded inhibitor ofRIP activation, vIRA. DNA-dependent activator of interferon reg-ulatory factors (DAI), a cytosolic double-stranded DNA sensorthat activates beta interferon expression associated with the innateimmune response (63, 72), appears to interact with RIP3 to me-diate MCMV-induced necroptosis (68). Whether these in vitrofindings regarding MCMV infection and necroptosis can be du-plicated in vivo using our MAIDS model of MCMV retinitis re-mains to be determined.
Pyroptosis (5) is an inflammatory process of caspase 1-depen-dent cell death that is morphologically and mechanistically dis-tinct from apoptosis and necroptosis. Cells undergoing pyroptosisdevelop pores within their plasma membranes of sufficient size toallow a net increase in osmotic pressure, water influx, cell swelling,osmotic lysis, and eventually the release of intracellular contentsthat promote inflammation, including release of the inflamma-tory cytokines IL-18 and IL-1�. While pyroptosis results in cleav-age of chromosomal DNA, this cleavage does not result in theoligonucleosomal fragments observed during apoptosis (4, 25,73), and unlike the nuclear fragmentation observed during apop-tosis, nuclear integrity is maintained during pyroptosis. Evidencefor involvement of pyroptosis during development of MAIDS-related MCMV retinitis was provided by detection of significantamounts of mRNAs to three key molecules involved in the pyrop-tosis pathway, i.e., caspase 1, IL-18, and IL-1�. Importantly, thesekey pyroptosis-related molecules were detected in large amountswithin the MCMV-infected eyes of MAIDS-10 mice but notwithin those of MAIDS-4 mice, and their patterns of synthesisduring the course of MCMV retinal disease were identical to thoseof key molecules associated with apoptosis or necroptosis. Re-markably, levels of IL-1� mRNA within eyes of MCMV-infected
MAIDS-10 mice were increased nearly 800-fold compared withthose in mock-infected control eyes from MAIDS-10 mice, al-though the precise contribution of this IL-1� release to pyropto-sis-associated inflammation compared with IL-1� release duringcaspase 1-independent inflammation (53) remains unclear.
Caspase 1 activity and the maturation of IL-1� and IL-18 areregulated by a multiprotein complex known as the AIM2 inflam-masome (21, 27, 28, 40, 47, 61). Rathinam and coworkers (60, 61)recently showed that the AIM2 inflammasome regulates caspase1-dependent processing of pro-IL-1� and pro-IL-18 in responseto double-stranded DNA in dendritic cells and macrophages butdoes not mediate type 1 interferon responses to double-strandedDNA, instead providing a negative regulation. Using Aim2-defi-cient mice, these investigators subsequently demonstrated a cen-tral role for AIM2 in regulation of caspase 1-dependent matura-tion of IL-1� and IL-18 as well as pyroptosis in response tosystemic MCMV infection (60). Since pilot studies by us detectedAIM2-associated inflammasome molecules in MCMV-infectedeyes of MAIDS-10 mice but not MAIDS-4 mice (data not shown),we therefore predict that the AIM2 inflammasome plays a centralrole in regulating pyroptosis during development of MAIDS-re-lated MCMV retinitis.
In summary, our findings provide new evidence for the partic-ipation of at least three cell death pathways during the onset andprogression of MAIDS-related MCMV retinitis: apoptosis,necroptosis, and pyroptosis (Table 1). Our study therefore servesto broaden our understanding of the pathogenesis of AIDS-re-lated HCMV retinitis. In fact, our findings suggest that multipleprogrammed cell death pathways are involved in the developmentof this sight-threatening retinal disease in patients with HIV/AIDS, perhaps operating simultaneously within individual retinalcells to cause intermediate forms of cell death. Whether theTUNEL assay used by us and others (6, 76) as an in situ marker forapoptosis during experimental MCMV retinitis serves also as amarker for necroptosis and/or pyroptosis remains unclear andawaits further investigation. We suggest, however, that theTUNEL assay will no longer serve as a reliable in situ marker forapoptosis exclusively, and future investigations to detect and
TABLE 1 Comparison of cell death pathways relative to cytomegalovirus infection
Cell deathpathway Signaling pathway (key molecules) Consequence Proinflammatory?
Virus-specificmechanism(s) fordownregulation
Apoptosis TNF-�, TNFR1, subset of caspases(caspases 8 and 3)
Formation of apoptotic bodies (phagocytosis) No Engagement of TNFR2antiapoptotic pathway
MCMV m38.5 (vIBO)HCMV UL37x1 (vMIA)
TRAIL, TRAIL-R(DR5) MCMV gene (?)HCMV gene (?)
Fas, FasL MCMV gene (?)HCMV gene (?)
Necroptosis Protein kinases Release of intracellular contents (inflammation) Yes MCMV M45RIP1 HCMV UL45RIP3
Pyroptosis Caspase 1 Release of cytokines (inflammation) Yes MCMV gene (?)IL-1� HCMV gene (?)IL-18AIM2 inflammasome
Chien and Dix
10976 jvi.asm.org Journal of Virology
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 31
Jan
uary
202
2 by
103
.207
.7.5
7.

quantify cell death pathways within virus-infected tissues willtherefore use immunostaining strategies that target and visualizekey pathway-dependent molecules.
ACKNOWLEDGMENTS
We thank the Biology Core Facilities, Department of Biology, GeorgiaState University, for assistance with real-time RT-PCR analysis and DNAsequencing.
This work was supported in part by NIH/NEI grant EY010568, NIH/NEI core grant P30EY006360, and Fight for Sight, Inc.
REFERENCES1. Atherton SS, Newell CK, Kanter MY, Cousins SW. 1991. Retinitis in
euthymic mice following inoculation of murine cytomegalovirus(MCMV) via the supraciliary route. Curr. Eye Res. 10:667– 677.
2. Bacci S, et al. 2008. Smooth muscle cells, dendritic cells and mast cells aresources of TNFalpha and nitric oxide in human carotid artery atheroscle-rosis. Thromb. Res. 122:657– 667.
3. Baud V, Karin M. 2001. Signal transduction by tumor necrosis factor andits relatives. Trends Cell Biol. 11:372–377.
4. Bergsbaken T, Cookson BT. 2007. Macrophage activation redirects Ye-rsinia-infected host cell death from apoptosis to caspase-1-dependent py-roptosis. PLoS Pathog. 3:e161. doi:10.1371/journal.ppat.0030161.
5. Bergsbaken T, Fink SL, Cookson BT. 2009. Pyroptosis: host cell deathand inflammation. Nat. Rev. Microbiol. 7:99 –109.
6. Bigger JE, Tanigawa M, Zhang M, Atherton SS. 2000. Murine cytomeg-alovirus infection causes apoptosis of uninfected retinal cells. Invest. Oph-thalmol. Vis. Sci. 41:2248 –2254.
7. Bradley JR. 2008. TNF-mediated inflammatory disease. J. Pathol. 214:149 –160.
8. Brune W. 2011. Inhibition of programmed cell death by cytomegalovirus.Virus Res. 157:144 –150.
9. Brune W, Menard C, Heesemann J, Koszinowski UH. 2001. A ribonu-cleotide reductase homolog of cytomegalovirus and endothelial cell tro-pism. Science 291:303–305.
10. Brunner T, et al. 2003. Fas (CD95/Apo-1) ligand regulation in T cellhomeostasis, cell-mediated cytotoxicity and immune pathology. Semin.Immunol. 15:167–176.
11. Chinnaiyan AM. 1999. The apoptosome: heart and soul of the cell deathmachine. Neoplasia 1:5–15.
12. Cho YS, et al. 2009. Phosphorylation-driven assembly of the RIP1-RIP3complex regulates programmed necrosis and virus-induced inflamma-tion. Cell 137:1112–1123.
13. Dix RD. 1998. Systemic murine cytomegalovirus infection of mice withretrovirus-induced immunodeficiency results in ocular infection but notretinitis. Ophthalmol. Res. 30:295–301.
14. Dix RD, Cousins SW. 2004. AIDS-related cytomegalovirus retinitis: les-sons from the laboratory. Curr. Eye Res. 29:91–101.
15. Dix RD, Cousins SW. 2004. Susceptibility to murine cytomegalovirusretinitis during progression of MAIDS: correlation with intraocular levelsof tumor necrosis factor-� and interferon-. Curr. Eye Res. 29:173–180.
16. Dix RD, Cray C, Cousins SW. 1994. Mice immunosuppressed by murineretrovirus infection (MAIDS) are susceptible to cytomegalovirus retinitis.Curr. Eye Res. 13:587–595.
17. Duan Y, Atherton SS. 1996. Immunosuppression induces transcriptionof murine cytomegalovirus glycoprotein H in the eye and at non-ocularsites. Arch. Virol. 141:411– 423.
18. Ehrenschwender M, Wajant H. 2009. The role of FasL and Fas in healthand disease. Adv. Exp. Med. Biol. 647:64 –93.
19. Elmore S. 2007. Apoptosis: a review of programmed cell death. Toxicol.Pathol. 35:495–516.
20. Ferguson TA, Griffith TS. 2007. The role of Fas ligand and TNF-relatedapoptosis-inducing ligand (TRAIL) in the ocular immune response.Chem. Immunol. Allergy 92:140 –154.
21. Fernandes-Alnemri T, Yu Datta J-WP, Wu J, Alnemri ES. 2009. AIM2activates the inflammasome and cell death in response to cytoplasmicDNA. Nature 458:509 –513.
22. Festjens N, Vanden Berghe T, Cornelis S, Vandenabeele P. 2007. RIP1,a kinase on the crossroads of a cell’s decision to live or die. Cell DeathDiffer. 14:400 – 410.
23. Festjens N, Vanden Berghe T, Vandenabeele P. 2006. Necroptosis, a
well-orchestrated form of cell demise: signaling cascades, important me-diators and concomitant immune response. Biochim. Biophys. Acta 1757:1371–1387.
24. Fink SL, Cookson BT. 2005. Apoptosis, pyroptosis, and necroptosis:mechanistic description of dead and dying eukaryotic cells. Infect. Im-mun. 73:1907–1926.
25. Fink SL, Cookson BT. 2006. Caspase-1-dependent pore formation dur-ing pyroptosis leads to osmotic lysis of infected host macrophages. Cell.Microbiol. 8:1812–1825.
26. Fleck M, et al. 1998. Apoptosis mediated by Fas but not tumor necrosisfactor receptor 1 prevents chronic disease in mice infected with murinecytomegalovirus. J. Clin. Invest. 102:1431–1443.
27. Franchi L, Eigenbrod T, Munoz-Planillo R, Nunez G. 2009. The inflam-masome: a caspase-1-activation platform that regulates immune re-sponses and disease pathogenesis. Nat. Immunol. 10:241–247.
28. Franchi L, Munoz-Plannilo R, Nunez G. 2012. Sensing and reacting tomicrobes through the inflammasomes. Nat. Immunol. 13:325–332.
29. Galluzzi L, Brenner C, Morselli E, Touat Z, Kroemer G. 2008. Viralcontrol of mitochondrial apoptosis. PLoS Pathog. 4(5):e10000018. doi:10.1371/journal.ppat.1000018.
30. Galluzzi L, Kepp O, Kroemer G. 2009. RIP kinases initiate programmednecrosis. J. Mol. Cell Biol. 1:8 –10.
31. Galluzzi L, Kroemer G. 2008. Necroptosis: a specialized pathway of pro-grammed necrosis. Cell 135:1161–1163.
32. Gao E-R, Yu X-H, Lin P-C, Zhang H, Kaplan HJ. 1995. Intraocular viralreplication after systemic murine cytomegalovirus infection requires im-munosuppression. Invest. Ophthalmol. Vis. Sci. 36:2322–2327.
33. Gavrieli Y, Sherman Y, Ben-Sasson SA. 1992. Identification of pro-grammed cell death in situ via specific labeling of nuclear DNA fragmen-tation. J. Cell Biol. 119:493–501.
34. Golstein P. 1997. Cell death: TRAIL and its receptors. Curr. Biol. 7:R750 –R753.
35. Golstein P, Kroemer G. 2007. Cell death by necrosis: towards a moleculardefinition. Trends Biochem. Sci. 32:37– 43.
36. Hahn G, et al. 2002. The human cytomegalovirus ribonucleotide reduc-tase homolog UL45 is dispensable for growth in endothelial cells, as deter-mined by a BAC-cloned clinical isolate of human cytomegalovirus withpreserved wild-type characteristics. J. Virol. 76:9551–9555.
37. Hartley JW, Fredrickson TN, Yetter RA, Makino M, Morse HC III.1989. Retrovirus-induced murine acquired immunodeficiency syndrome:natural history of infection and differing susceptibility of inbred mousestrains. J. Virol. 63:1223–1231.
38. He S, et al. 2009. Receptor interacting protein kinase-3 determines cel-lular necrotic response to TNF-alpha. Cell 137:1100 –1111.
39. Heiden D, et al. 2007. Cytomegalovirus retinitis: the neglected disease ofthe AIDS pandemic. PLoS Med. 4:e334. doi:10.1371/journal-.pmed.0040334.
40. Henao-Mejia J, Elinav E, Strowig T, Flavell RA. 2012. Inflammasomes:far beyond inflammation. Nat. Immunol. 13:321–324.
41. Hengartner MO. 2000. The biochemistry of apoptosis. Nature 407:770 –776.
42. Hill MM, Adrain C, Duriez PJ, Creagh EM, Martin SJ. 2004. Analysis ofthe composition, assembly kinetics and activity of native Apaf-1 apopto-somes. EMBO J. 23:2134 –2145.
43. Hitomi J, et al. 2008. Identification of a molecular signaling network thatregulates a cellular necrotic cell death pathway. Cell 135:1311–1323.
44. Ho M. 1994. Advances in understanding cytomegalovirus infections aftertransplantation. Transplant. Proc. 26:7–11.
45. Hofman F, Hinton DR. 1992. Tumor necrosis factor-� in the retina ofacquired immune deficiency syndrome. Invest. Ophthalmol. Vis. Sci. 33:1829 –1835.
46. Holland GN, Tufail A, Jordan MC. 1996. Cytomegalovirus disease, p1088 –1130. In Pepose JS, Holland GN, Wilhelmus KR (ed), Ocular infec-tions and immunity. C. V. Mosby Co., Inc., St. Louis, MO.
47. Hornung V, et al. 2009. AIM2 recognizes cytosolic dsDNA and forms acaspase-1-activating inflammasome with ASC. Nature 458:514 –518.
48. Jabs DC. 2011. Cytomegalovirus retinitis and the acquired immunodefi-ciency syndrome. Bench to bedside: LXVII Edward Jackson MemorialLecture. Am. J. Ophthalmol. 151:198 –216.
49. Kercher L, Mitchell BM. 2000. Immune transfer protects severely immu-nosuppressed mice from murine cytomegalovirus retinitis and reducesviral load in ocular tissue. J. Infect. Dis. 182:652– 661.
Cell Death Pathways and MCMV Retinitis
October 2012 Volume 86 Number 20 jvi.asm.org 10977
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 31
Jan
uary
202
2 by
103
.207
.7.5
7.

50. Kumar V, Abbas AK, Faustro N. 2005. Robbins and Contran pathologicbasis of disease, 7th ed. Elsevier Sanders, Philadelphia, PA.
51. Lowin B, Peitsch MC, Tschopp J. 1995. Perforin and granzymes: crucialeffector molecules in cytolytic T lymphocyte and natural killer cell-mediated cytotoxicity. Curr. Top. Microbiol. Immunol. 198:1–24.
52. McCormick AI, Meiering CD, Smith GB, Mocarski GB. 2005. Mito-chondrial cell death suppressors carried by human and murine cytomeg-alovirus confer resistance to proteasome inhibitor-induced apoptosis. J.Virol. 79:12205–12217.
53. Miwa K, et al. 1998. Caspase-1-independent IL-1beta release and inflam-mation induced by the apoptosis inducer Fas ligand. Nat. Med. 4:1287–1292.
54. Mocarski E, Upton JW, Kaiser WJ. 2012. Viral infection and the evolu-tion of caspase 8-regulated apoptotic and necrotic death pathways. Nat.Rev. Immunol. 12:79 – 88. [Epub ahead of print.] doi:10.1038/nri3131.
55. Mondino BJ, Sidikaro Y, Mayer FJ, Sumner HL. 1990. Inflammatorymediators in the vitreous humor of AIDS patients with retinitis. Invest.Ophthalmol. Vis. Sci. 31:798 – 804.
56. Mosier DE, Yetter RA, Morse HC III. 1985. Retroviral induction of acutelymphoproliferative disease and profound immunosuppression in adultC57BL/6 mice. J. Exp. Med. 161:766 –784.
57. Negoescu A, Guillermet C, Lorimier P, Brambilla E, Labat-Moleur F.1998. Importance of DNA fragmentation in apoptosis with regard toTUNEL specificity. Biomed. Pharmacother. 52:252–258.
58. Negoescu A, et al. 1996. In situ apoptotic cell labeling by TUNEL method:improvement and evaluation on cell preparations. J. Histochem. Cy-tochem. 44:959 –968.
59. Rahman MM, McFadden G. 2006. Modulation of tumor necrosis factor bymicrobial pathogens. PLoS Pathog. 2:e4. doi:10.1371/journal.ppat.0020004.
60. Rathinam VAK, et al. 2010. The AIM2 inflammasome is essential forhost-defense against cytosolic bacteria and DNA viruses. Nat. Immunol.11:395– 402.
61. Rathinam VAK, Vanaja SK, Fitzgerald KA. 2012. Regulation of inflam-masome signaling. Nat. Immunol. 13:333–342.
62. Rosenbaum DM, et al. 2010. Necroptosis, a novel form of caspase-independent cell death, contributes to neuronal damage in a retinal isch-emia-reperfusion injury model. J. Neurosci. Res. 88:1569 –1576.
63. Takaoka A, et al. 2007. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor andan activator of innate immune response. Nature 448:501–505.
64. Tang-Feldman YJ, et al. 2006. Use of quantitative real-time PCR (qRT-PCR) to measure cytokine transcription and viral load in murine cyto-megalovirus infection. J. Virol. Methods 131:122–129.
65. Trichonas G, et al. 2010. Receptor interacting protein kinases mediateretinal detachment-induced photoreceptor necrosis and compensatefor inhibition of apoptosis. Proc. Natl. Acad. Sci. U. S. A. 107:21695–21700.
66. Upton JW, Kaiser WJ, Mocarski ES. 2008. Cytomegalovirus M45 celldeath suppression requires receptor-interacting protein (RIP) homotypicinteraction motif (RHIM)-dependent interaction with RIP1. J. Biol.Chem. 283:16966 –16970.
67. Upton JW, Kaiser WJ, Mocarski ES. 2010. Virus inhibition of RIP-dependent necrosis. Cell Host Microbe 7:302–313.
68. Upton JW, Kaiser WJ, Mocarski ES. 2012. DAI/ZBP1/DLM-1 complexeswith RIP3 to mediate virus-induced programmed necrosis that is targetedby murine cytomegalovirus vIRA. Cell Host Microbe 11:1– 8.
69. Vassalli P. 1992. The pathophysiology of tumor necrosis factors. Annu.Rev. Immunol. 10:411– 452.
70. Wajant H, Pfizenmaier K, Scheuich P. 2003. Tumor necrosis factorsignaling. Cell Death Differ. 10:45– 65.
71. Wallach D, et al. 1999. Tumor necrosis factor receptor and Fas signalingmechanisms. Annu. Rev. Immunol. 17:331–367.
72. Wang Z, et al. 2008. Regulation of innate immune responses by DAI(DLM-1/ZBP1) and other DNA-sensing molecules. Proc. Natl. Acad. Sci.U. S. A. 105:5477–5482.
73. Watson PR, et al. 2000. Salmonella enterica serovars Typhimurium andDublin can lyse macrophages by a mechanism distinct from apoptosis.Infect. Immun. 68:3744 –3747.
74. Zhang M, Covar J, Marshall B, Dong Z, Atherton SS. 2011. Lack ofTNF-� promotes caspase-3-independent apoptosis during murine cyto-megalovirus retinitis. Invest. Ophthalmol. Vis. Sci. 52:1800 –1808.
75. Zhang DW, et al. 2009. RIP3, an energy metabolism regulator thatswitches TNF-induced cell death from apoptosis to necrosis. Science 325:332–336.
76. Zhou J, Zhang M, Atherton SS. 2007. Tumor necrosis factor-�-inducedapoptosis in murine cytomegalovirus retinitis. Invest. Ophthalmol. Vis.Sci. 48:1691–1700.
Chien and Dix
10978 jvi.asm.org Journal of Virology
Dow
nloa
ded
from
http
s://j
ourn
als.
asm
.org
/jour
nal/j
vi o
n 31
Jan
uary
202
2 by
103
.207
.7.5
7.





























