Embed Size (px)
Citation preview

Journal of Virological Methods 152 (2008) 49–55
Contents lists available at ScienceDirect
Journal of Virological Methods
journa l homepage: www.e lsev ier .com/ locate / jv i romet
Evidence for novel viruses by analysis of nucleic acids in virus-like particlefractions from Ambrosia psilostachya
Ulrich Melchera,∗ , Vijay Muthukumara , Graham B. Wileyb , Byoung Eun Minc , Michael W. Palmerd ,Jeanmarie Verchot-Lubicze, Akhtar Ali f, Richard S. Nelsonc, Bruce A. Roeb,Vaskar Thapad, Margaret L. Piercea
a Department of Biochemistry & Molecular Biology, 246 NRC, Oklahoma State University, Stillwater, OK 74078, USAb Department of Chemistry & Biochemistry, University of Oklahoma, Norman, OK 73019, USAc Plant Biology Division, Samuel Roberts Noble Foundation, Inc., Ardmore, OK 73401, USA
K 740
t manned rocedeservve spnce o
in tw
d Department of Botany, Oklahoma State University, Stillwater, OK 74078, USAe Department of Entomology & Plant Pathology, Oklahoma State University, Stillwater, Of Department of Biological Science, University of Tulsa, Tulsa, OK 74104, USA
Article history:Received 23 December 2007Received in revised form 14 May 2008Accepted 19 May 2008Available online 17 July 2008
Keywords:
a b s t r a c t
To test the hypothesis thasequence nucleic acids cloof the efficiency of the prat the Tallgrass Prairie Prfractions from six of twelwas obtained for the preseviral species were found
MetagenomicsBiodiversityBadnavirusFlexiviridaePlant virusAmbrosia
population varied from less thaknown viruses. Thus, the analyknowledge of the universe of v
1. Introduction
The number of viral species associated with plants is likely tobe much larger than the number of species recognized currently(Wren et al., 2006). Support for the suggestion comes from theobservation that the vast majority of viral species are the ones thatcaught society’s attention through their induction of noticeable dis-ease symptoms in economically valuable plants. Although dwarfedby the number of studies on viruses in crop species, surveys fornovel viruses in plant populations from non-managed ecosystemshave been reported (Bodaghi et al., 2004; Fraile et al., 1997; Ooiand Yahara, 1999; Raybould et al., 1999). In these studies, inci-
∗ Corresponding author. Tel.: +1 405 744 6210; fax: +1 405 744 7799.E-mail addresses: [email protected] (U. Melcher),
[email protected] (V. Muthukumar), [email protected](G.B. Wiley), [email protected] (B.E. Min), [email protected](M.W. Palmer), [email protected] (J. Verchot-Lubicz), [email protected] (A.Ali), [email protected] (R.S. Nelson), [email protected] (B.A. Roe),[email protected] (V. Thapa), [email protected] (M.L. Pierce).
0166-0934/$ – see front matter © 2008 Elsevier B.V. All rights reserved.doi:10.1016/j.jviromet.2008.05.030
78, USA
y viruses remain to be discovered in plants, a procedure was developed toandomly from virus-like particle fractions of plant homogenates. As a testure we targeted Ambrosia psilostachya, western ragweed, plants growinge of northeastern Oklahoma. Amplifiable nucleic acid was found in theecimens and sequences were characterized from four of them. Evidencef viruses belonging to two families (Caulimoviridae, Flexiviridae). Multipleo of the four specimens and their level within the isolated nucleic acidn 1–37%. None of the sequences were derived from reported sequences ofsis of nucleic acid from virus-like particles is a useful tool to expand ouriruses to non-cultivated species.
© 2008 Elsevier B.V. All rights reserved.
dence rates of infection of single plant species with single viralspecies cover a wide range, but are frequently between 30 and70%. Previously unknown viruses have been discovered in non-cultivated plants because those plants exhibited novel symptoms(Ooi and Yahara, 1999; Robertson, 2005). No reports surveying fornovel viruses associated with plants in non-managed ecosystemsregardless of the presence or absence of symptoms were found.
Metagenomic approaches (Casas and Rohwer, 2007; Edwardsand Rohwer, 2005; Riesenfeld et al., 2004; Xu, 2006) have been usedto study viruses not associated with host organisms, primarily inaqueous environments (Angly et al., 2006; Breitbart et al., 2004)and to identify viruses in mammalian systems (Delwart, 2007).The Plant Virus Biodiversity and Ecology project was formed toapply metagenomic strategies to begin to fill the gap in knowl-edge of viruses of non-cultivated plants (Wren et al., 2006). TheTallgrass Prairie Preserve of Osage County, Oklahoma, a site ownedand minimally managed by The Nature Conservancy (Hamilton,2007), was chosen as the site for sampling of plant viruses in nat-ural ecosystems. The preserve contains more than 700 species ofplants (Palmer, 2007).

50 U. Melcher et al. / Journal of Virologic
Table 1Location and sampling dates of Ambrosia psilostachya plants with amplifiable ornon-amplifiable material in VLP-VNA fractions
Plant no. Eastinga Northinga Date sampled
No amplification05TGP00303 4086000 730000 2005-06-1405TGP00361 4082000 731000 2005-06-1605TGP00387 4071000 734000 2005-06-2105TGP00426 4074000 731000 2005-06-2205TGP00408 4078000 728000 2005-06-2205TGP00417 4076000 730000 2005-06-22
Amplification05TGP00100 4080602 730202 2005-05-1305TGP00295 4074000 734000 2005-06-0805TGP00312 4086000 731000 2005-06-1405TGP00321 4079000 734000 2005-06-1505TGP00379 4075000 732000 2005-06-2105TGP00432 4079000 733000 2005-06-23
a Sector 14, using NAD 27.
In this project, a virus-like particle-viral nucleic acid extraction(VLP-VNA) metagenomic method to discover viruses was assem-bled from threes established techniques. In this method, differentialcentrifugation of plant homogenates for virus isolation (Lane, 1986,1992; Robertson, 2005) was used to obtain an ultracentrifugal pelletexpected to contain any virions present in the sample. Nucleic acidswere then extracted from the virus-like particle fraction by a mod-ification of a procedure designed to recover DNA from Cauliflowermosaic virus (CaMV) particles (Gardner and Shepherd, 1980). Thisprocedure was adopted because CaMV virions are known to beamong the most recalcitrant to viral nucleic acid extraction tech-niques (Hull, 1978). The resulting nucleic acid fraction was thenamplified by a sequence-independent method originally devisedfor diagnosis of human infection by respiratory viruses (Wanget al., 2002), and the amplification products cloned in a plas-mid vector and the resulting plasmids submitted to nucleotidesequencing. This report describes the application of the VLP-VNAmethod to Ambrosia psilostachya DC., western ragweed, an abun-dant plant species in the Tallgrass Prairie Preserve and the abilityof this method to detect the presence of sequences from at leastfour probable viral species associated with A. psilostachya. None ofthese sequences has been described previously and all representsequences from recognizable members of known viral taxa. Onevirus, a probable member of the Flexiviridae, has a novel genome
structure suggestive of a genus in this family, not described previ-ously.2. Materials and methods
2.1. Plant sampling
Samples (Table 1) of twelve A. psilostachya plants were takenin late spring and in summer of 2005 from different locationsin the Tallgrass Prairie Preserve. Voucher specimens also wereobtained and were deposited at the Oklahoma State UniversityHerbarium. Samples in individual plastic bags were brought to thelaboratory on wet ice, weighed, aliquoted and placed in storage at−80 ◦C.
2.2. Virus-like particle preparation
Plant material (100 mg) and 12–14 sterile glass beads (diameter,2.5 mm) were placed in a 1.5-ml self-standing screw cap micro-tube (United Scientific Products), followed by addition of 750 �l of
al Methods 152 (2008) 49–55
ice-cold 0.1 M sodium citrate pH 6.5 (citrate buffer) and 6.3 �l of0.25 M iodoacetamide (Lane, 1986, 1992). After 10 min on ice, sam-ples were disrupted at 4600 rpm for 3 min in a Mini-bead Beater(Biospec Products, Bartlesville, OK). After 15 min of centrifugationat 12,000 × g at room temperature, 137.5 �l of each supernatantwas transferred to a sterile 1.5-ml microcentrifuge tube followedby the addition of 12.5 �l of 33.3% (v/v) of Triton X-100. Mixtureswere transferred to 7 × 20-mm cellulose propionate ultracentrifugetubes (Beckman cat. no. 342303) and were underlaid with 50 �l of20% (w/v) sucrose solution in citrate buffer (sucrose-citrate). Tubeswere subjected to ultra-centrifugation at 70,000 × g for 45 minat room temperature, in a Beckman Ti 42.2 rotor. Supernatantswere removed by aspiration. Pellets were resuspended in 200 �lof 0.5× citrate buffer and the suspension was centrifuged for 10min at 8000 × g at room temperature. Supernatants (150 �l) weresubjected to ultracentrifugation over a sucrose-citrate cushion at150,000 × g for 65 min at room temperature. Pellets containingvirus-like particles (VLP) were resuspended in 250 �l of 0.1 MTris–HCl pH 7.5, 2.5 mM MgCl2.
2.3. Nucleic acid isolation from virus-like particles
Nucleic acids were isolated from the VLP-containing pelletsusing a modification of the procedure for CaMV (Gardner andShepherd, 1980). Two �l of 1 mg/ml (w/v) DNase I (Sigma) wereadded to 200 �l of each VLP preparation After a 10 min incubationat 37 ◦C, 4 �l 0.5 M EDTA, 50 �l of 2.5 mg/ml Proteinase K (Invit-rogen) and 12.5 �l of 20% sodium dodecyl sulfate were added tothe samples and the mixtures incubated at 65 ◦C for 30 min. Afteraddition of 6 �l 5 M NaCl, suspensions were extracted with 300 �lof buffer saturated phenol (MP Biomedicals, Inc.). After centrifuga-tion at 12,000 × g for 5 min, the aqueous phases were transferred toclean tubes and extracted with diethyl ether. Linear polyacrylamide(4 �l 5 mg/ml; Ambion) was added to the solutions followed byaddition of 95% ethanol for overnight precipitation at −20 ◦C. Pre-cipitates were pelleted, washed with 75% ethanol and dried. Finalpellets (VLP-VNA) were dissolved in 30 �l of 10 mM Tris–HCl pH7.5, 1 mM EDTA, 10 mM NaCl.
2.4. Nucleic acid amplification
Nucleic acid was amplified essentially as described previously(Wang et al., 2002). In short, during round A, reverse transcription
′
was undertaken with a primer containing a degenerate 3 end, fol-lowed by second strand synthesis using Sequenase (USB). In roundB, the products of round A were used as templates in a standardPCR reaction. In the work reported here, CCTGAATTCGGATCCTC-CNNNNNNNNNNNN (primer A12; Integrated DNA Technologies,Inc.) served as round A primer and CCTGAATTCGGATCCTCC (primerB) as round B primer. PCR in round B was limited to 30 cycles.Aliquots of the amplified products were subjected to 2% agarosegel electrophoresis in 40 mM Tris acetate, 1 mM EDTA.2.5. Cloning of amplified products
Aliquots of the amplified PCR products were joined into thepCR 2.1-TOPO vector (Invitrogen) according to the manufacturer’sinstructions. The transformed cell populations were separated into200- and 100-�l aliquots. The former were transferred to 100 mlLuria-Bertani (LB) broth containing kanamycin (50 �g/ml). Aftergrowth at 37 ◦C for 16–18 h, 15% glycerol stocks were prepared andstored at −80 ◦C. The 100-�l aliquots were subjected to appropri-ate dilution and spread on LB agar plates containing kanamycin(50 �g/ml) and X-gal (40 mg/ml) for overnight incubation at 37 ◦C.

rological Methods 152 (2008) 49–55 51
U. Melcher et al. / Journal of Vi2.6. Plasmid preparation and analysis
Plasmids were isolated from culture aliquots by using a QIAprepSpin Miniprep Kit protocol (Qiagen). Presence and sizes of insertswere evaluated after Alw N1restriction digestion by gel elec-trophoresis in a 0.8% agarose gel. Observation of a distinct 2.3-kbpband (vector-derived) and a smear of DNA from about 1.6 kbp tolarger than 2.3 kbp (insert-containing) together with a predomi-nance of white colonies on the test plate was taken as evidence ofsuccessful cloning.
2.7. Sequence analysis
The 15% glycerol stock of bacterial growth that contained plas-mids having large inserts (>500 bp) was sent to the Advanced Centerfor Genome Technology (ACGT), University of Oklahoma at Nor-man, for sequencing. A 10−4 serial dilution of the glycerol stockwas plated on LB-agar media containing 0.1 mg/ml ampicillin andincubated at 37 ◦C overnight. A random library of 384 colonies waspicked into a 384 plate containing 80 �l of Terrific Broth (TB) sup-plemented with 100 �g of ampicillin. The inoculated plates thenwere incubated in a 37 ◦C, oxygenated shaker for 24 h. Two �laliquots from these plates were used as inoculants for a second setof 384-well plates that also were incubated in a 37 ◦C, oxygenatedshaker for 24 h. After incubation, the plates were centrifuged to pel-let the cells and the media were decanted. The plasmids then wereisolated using a modified cleared lysate procedure that was auto-mated on a Zymark SciClone robot (Bodenteich et al., 1993; Roe,2004).
The sequencing reactions were carried out as described pre-viously (Bodenteich et al., 1993; Chissoe et al., 1995; Roe, 2004)using Amersham ET terminator sequencing reaction mixes at 1:16dilutions for 60 cycles in a Perkin-Elmer Cetus DNA Thermocycler9600. Unincorporated terminators and nucleotides were removedvia ethanol precipitation and the DNA was dissolved in 0.1 mMEDTA. The samples then were sequenced on an ABI 3730 capil-lary DNA sequencer. The chromatograms then were transferred toa Sun Workstation cluster where they then were base called andassembled using Phred and Phrap, respectively (Ewing and Green,1998; Ewing et al., 1998). Virus-related sequences were depositedin GenBank on 7 January 2008 under accession numbers EU362846through EU362854.
Assembled contigs were analyzed using BLAST with bothnucleotide vs. nucleotide (BLASTn) and translated nucleotide vs.
protein (BLASTx) queries (Altschul et al., 1997). Analysis in each casewas against the Genbank non-redundant databases for nucleotides(nt) and proteins (nr), respectively, using an e-value cutoff of 10−3.The top 5 hits against each database then were reported for eachcontig. The 3′ end region of the Flexiviridae member was amplifiedfrom the VLP-VNA and cloned using a SMART RACE cDNA Amplifica-tion kit (Clontech, USA) with a Flexivirus-specific forward primer(5′-CTA AGC ATG ACT ACT GGC CCA ACA CT-3′) according to themanufacturer’s instructions.The top five BLASTn and BLASTx search results for eachsequence were examined to determine a tentative assignment ofthe sequence to a source (e.g. plant, bacterial, fungal, animal, virus(including retroelements), or other). Sequences without significantsimilarities in the databases were classified as source “uncertain”.These sequences were used as queries of the nucleotide sequencedatabase (nt) by tBLASTx. For viral taxa selected based on prelim-inary evidence of their possible presence in the sample, proteinsequences encoded by known members of the taxa were collectedand used as queries in a tBLASTn search of insert sequences.
Several tests of the validity of viral hits were performed. In thereciprocal search approach, the best nr match at the amino acid
Fig. 1. Analysis of products of random amplification of VLP-VNA from selectedA. psilostachya specimens 05TGP00303 (lane2), 05TGP00426 (lane3), 05TGP00361(lane 4), 05TGP00312 (lane 5), 05TGP00321 (lane 6), 05TGP00379 (lane 7) and a notemplate control (lane 8). 1 kB plus marker ladder with lowest band representing100 bp (lane 1).
sequence level in the above BLASTx, tBLASTx or tBLASTn searcheswas used as a query of the protein database. Only the matchingresidues were used. The range of E-values for hits to sequencesfrom the same viral species, from other members of the nexthigher taxon, and of unrelated sequences were recorded. If theE-value of the original search was within the range of values forother members of the taxon and below the lowest E-value forunrelated sequences, the subject sequence was deemed to belongto a virus of that taxon. In most cases, aligned nucleotides (incases of good conservation) or amino acids were used to gen-erate phylogenetic trees. Both parsimony and distance methodsas available in the Phylip package (Felsenstein, 1989) were used.
Branching of the subject sequence within the clade defining thetaxon was taken as evidence supporting the assignment. Finally,in some cases, the significance score of the alignment of the sub-ject sequence with other members of the taxon was determined(Melcher, 1990).3. Results
3.1. Amplification
The ability to amplify material from the VLP-VNA fraction of12 A. psilostachya plants through PCR is shown in Table 1. A clearvisual distinction could be made after agarose gel electrophoresisand ethidium bromide staining between samples producing visiblesmears of stained DNA and those in which smears were absent orextremely faint (Fig. 1). Amplification was obtained in 6 (50%) ofthe plants.
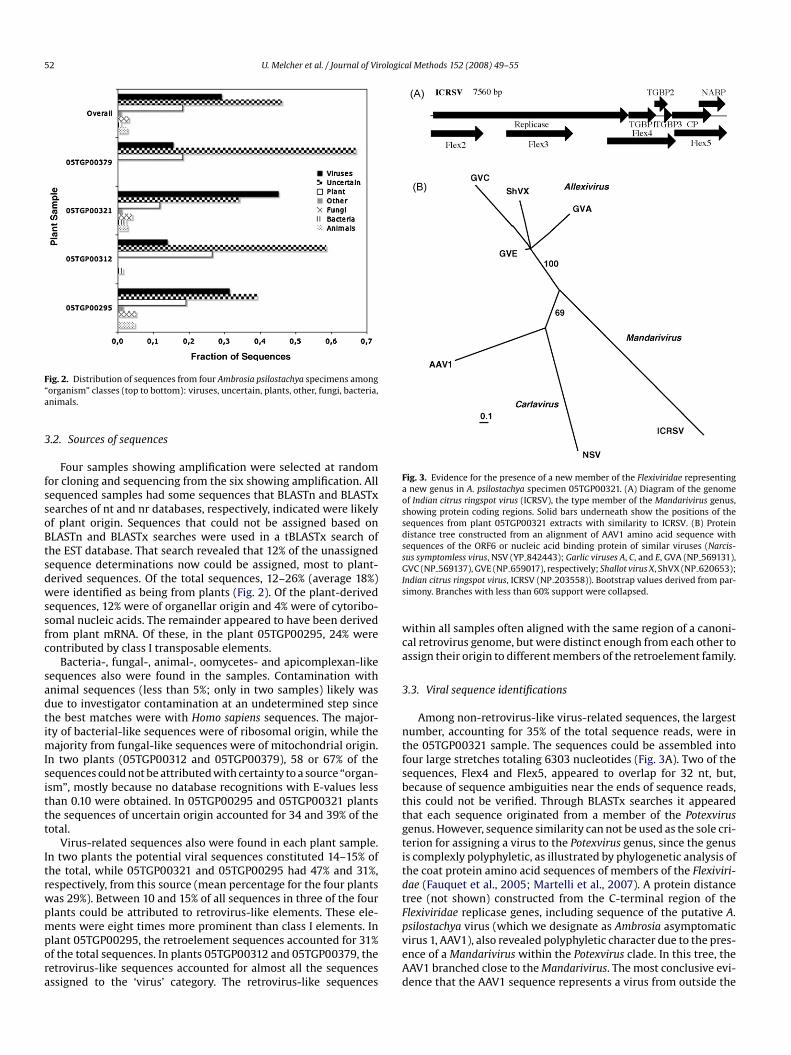
52 U. Melcher et al. / Journal of Virological Methods 152 (2008) 49–55
Fig. 2. Distribution of sequences from four Ambrosia psilostachya specimens among“organism” classes (top to bottom): viruses, uncertain, plants, other, fungi, bacteria,animals.
3.2. Sources of sequences
Four samples showing amplification were selected at randomfor cloning and sequencing from the six showing amplification. Allsequenced samples had some sequences that BLASTn and BLASTxsearches of nt and nr databases, respectively, indicated were likelyof plant origin. Sequences that could not be assigned based onBLASTn and BLASTx searches were used in a tBLASTx search ofthe EST database. That search revealed that 12% of the unassignedsequence determinations now could be assigned, most to plant-derived sequences. Of the total sequences, 12–26% (average 18%)were identified as being from plants (Fig. 2). Of the plant-derivedsequences, 12% were of organellar origin and 4% were of cytoribo-somal nucleic acids. The remainder appeared to have been derivedfrom plant mRNA. Of these, in the plant 05TGP00295, 24% werecontributed by class I transposable elements.
Bacteria-, fungal-, animal-, oomycetes- and apicomplexan-likesequences also were found in the samples. Contamination withanimal sequences (less than 5%; only in two samples) likely was
due to investigator contamination at an undetermined step sincethe best matches were with Homo sapiens sequences. The major-ity of bacterial-like sequences were of ribosomal origin, while themajority from fungal-like sequences were of mitochondrial origin.In two plants (05TGP00312 and 05TGP00379), 58 or 67% of thesequences could not be attributed with certainty to a source “organ-ism”, mostly because no database recognitions with E-values lessthan 0.10 were obtained. In 05TGP00295 and 05TGP00321 plantsthe sequences of uncertain origin accounted for 34 and 39% of thetotal.Virus-related sequences also were found in each plant sample.In two plants the potential viral sequences constituted 14–15% ofthe total, while 05TGP00321 and 05TGP00295 had 47% and 31%,respectively, from this source (mean percentage for the four plantswas 29%). Between 10 and 15% of all sequences in three of the fourplants could be attributed to retrovirus-like elements. These ele-ments were eight times more prominent than class I elements. Inplant 05TGP00295, the retroelement sequences accounted for 31%of the total sequences. In plants 05TGP00312 and 05TGP00379, theretrovirus-like sequences accounted for almost all the sequencesassigned to the ‘virus’ category. The retrovirus-like sequences
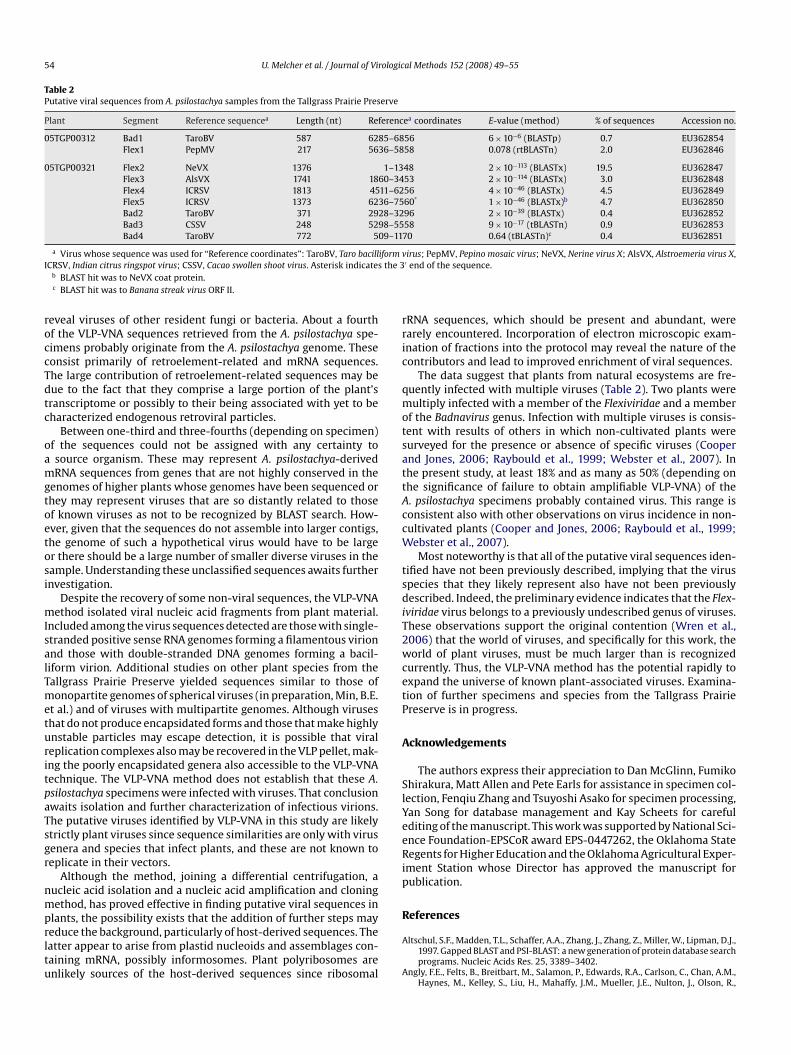
Fig. 3. Evidence for the presence of a new member of the Flexiviridae representinga new genus in A. psilostachya specimen 05TGP00321. (A) Diagram of the genomeof Indian citrus ringspot virus (ICRSV), the type member of the Mandarivirus genus,showing protein coding regions. Solid bars underneath show the positions of thesequences from plant 05TGP00321 extracts with similarity to ICRSV. (B) Proteindistance tree constructed from an alignment of AAV1 amino acid sequence withsequences of the ORF6 or nucleic acid binding protein of similar viruses (Narcis-sus symptomless virus, NSV (YP 842443); Garlic viruses A, C, and E, GVA (NP 569131),GVC (NP 569137), GVE (NP 659017), respectively; Shallot virus X, ShVX (NP 620653);Indian citrus ringspot virus, ICRSV (NP 203558)). Bootstrap values derived from par-simony. Branches with less than 60% support were collapsed.
within all samples often aligned with the same region of a canoni-cal retrovirus genome, but were distinct enough from each other toassign their origin to different members of the retroelement family.
3.3. Viral sequence identifications
Among non-retrovirus-like virus-related sequences, the largestnumber, accounting for 35% of the total sequence reads, were inthe 05TGP00321 sample. The sequences could be assembled intofour large stretches totaling 6303 nucleotides (Fig. 3A). Two of thesequences, Flex4 and Flex5, appeared to overlap for 32 nt, but,because of sequence ambiguities near the ends of sequence reads,this could not be verified. Through BLASTx searches it appearedthat each sequence originated from a member of the Potexvirusgenus. However, sequence similarity can not be used as the sole cri-terion for assigning a virus to the Potexvirus genus, since the genusis complexly polyphyletic, as illustrated by phylogenetic analysis ofthe coat protein amino acid sequences of members of the Flexiviri-dae (Fauquet et al., 2005; Martelli et al., 2007). A protein distancetree (not shown) constructed from the C-terminal region of theFlexiviridae replicase genes, including sequence of the putative A.psilostachya virus (which we designate as Ambrosia asymptomaticvirus 1, AAV1), also revealed polyphyletic character due to the pres-ence of a Mandarivirus within the Potexvirus clade. In this tree, theAAV1 branched close to the Mandarivirus. The most conclusive evi-dence that the AAV1 sequence represents a virus from outside the

rological Methods 152 (2008) 49–55 53
U. Melcher et al. / Journal of ViPotexvirus genus is the coding potential for a polypeptide overlap-ping, in a different reading frame, the 3′ portion of the coat proteinORF and continuing beyond the position of the coat protein ter-mination codon. The position of this ORF, not found in potexvirusgenomes, is homologous to an ORF for a nucleic acid binding pro-tein encoded by the genome of Indian citrus ringspot virus (ICRSV),a Mandarivirus. In addition, the AAV1 predicted C-terminal halfamino acid sequence is similar, as judged by BLASTp search, tothat of a smaller ORF in genomes of the Allexivirus genus. Parsi-mony and distance phylogenetic methods, using the amino acidsequences of regions common to the proteins from Mandari-, Carla-and Allexivirus genera gave identical topologies (Fig. 3B). The AAV1ORF branched, with modest bootstrap support, with the Carlavirusprotein and did not branch with the similar length Mandarivirusprotein suggesting that the AAV1 ORF is from yet another genuswithin the Flexiviridae.
Analysis of BLASTx search results also identified a contig (Bad2,Table 2) from 05TGP00321 consisting of two sequence reads as orig-inating from a Badnavirus genus. Its position relative to the genomeof the type member of the genus is shown in Fig. 4A. A phyloge-netic tree using nucleotide sequences of Bad2 (Fig. 4B) showedthat the putative A. psilostachya badnavirus branched togetherwith Taro bacilliform virus within the Badnavirus clade, confirm-ing that the virus belongs to that genus. BLASTx search analysisalso revealed another badnaviral sequence (Bad1, four reads) fromsample 05TGP00312. Its phylogenetic location on a neighbor join-ing tree, as sister to Taro bacilliform virus, was identical to that ofthe 05TGP00321 Bad2 (not shown).
Supplemental strategies resulted in the identification of twoother sequences as related to Badnavirus and Flexiviridae viruses.The tBLASTn approach with Badnavirus protein query sequencesrevealed two sequences from 05TGP00321, Bad3 and Bad4. Bad3was detected with E = 9 × 10−17 by Cacao swollen shoot virussequences. Bad4 was detected with E = 0.68 by part of ORFII-predicted sequences from several badnaviruses. ORF II is highlydivergent among badnaviruses and, in most badnaviruses, overlapsat its 5′ end with the very 3′ end of the similarly poorly conservedORF I. The contig (Bad4, Table 2) whose sequence was detected bytBLASTn search had a single uninterrupted reading frame, with thetBLASTn-detected sequence near its 3′ end. Interestingly, the aminoacid sequence predicted from the contig resembled the C-terminalsequences of badnavirus ORFs I in its N-terminal half (significancescore = 2.8) and the N-terminal sequences of badnavirus ORFs II inits C-terminal half (significance score = 5.6). The similarities imply
that the badnavirus from which the sequence was derived can pro-duce the ORF II polypeptide product both by de novo translationinitiation and by producing an ORF I-II fusion polypeptide (Fig. 4A).The tBLASTn search with sequences of Potexvirus proteinsrevealed a sequence (Flex1, Table 2) in the collection from05TGP00312 that could code for the N-terminus of the coat proteinof a potexvirus relative. The significance score for the alignmentof the predicted amino acid sequence with the top tBLASTn searchhit was 4.9, close to the 6.7 score obtained for significance of thealignment of the Pepino mosaic virus segment with the correspond-ing segments of coat proteins of other Flexiviridae members, butsufficiently below it to preclude a definite assignment. Neither thenucleotide sequence nor the predicted amino acid sequence fromthis sample closely matched the sequence identified for a similarsegment from the AAV1 from 05TGP00321 (Flex4, Table 2).
A precise number for viruses identified in these four plants can-not be given (Table 2). The evidence for the presence of two differentmembers of the Flexiviridae in plants 05TGP00312 and 05TGP00321was strong since their divergent virus-like sequences were froma common region within the Flexiviridae genome. However, forthe samples containing badnavirus-like sequences there was no
Fig. 4. Evidence for the presence of one or more new member of the Badnavirusgenus in A. psilostachya specimens 05TGP00311 and 05TGP00321. (A) Diagram ofgenome of Taro bacilliform virus (linearized at the origin of reverse transcription)showing protein coding regions. Solid bars underneath show the genomic positionsof the sequences from plant specimen extracts with similarity to Taro bacilliformvirus (TBV) the location of three segments from badnaviruses recovered from twoAmbrosia psilostachya plants (05TGP00312 and 05TGP00321). The leftmost segmentwas identified by tBLASTn search using badnavirus protein sequences as query whilethe other two were identified by BLASTx of the general database. (B) Protein distancephylogenetic tree of badnaviruses based on sequences related to Bad2/321: Sugar-cane bacilliform virus (SCBV IN, NC 003031), Banana streak virus (BSV, NC 008018),Banana streak OL virus (BSV OL, NC 003381), Banana streak Mysore virus (BSV Mysore,NC 006955), Commelina yellow mottle virus (CoYMV, NC 001343), Kalanchoe top-spotting virus (KTSV, NC 004540), Cacao swollen shoot virus (CSSV, NC 001574), Tarobacilliform virus (TBV, NC 004450), Dracaena mottle virus (DaMV, NC 008034), Citrus
yellow mosaic virus (CYMV, NC 003382), virus from A. psilostachya (05TGP00321),Rice tungro bacilliform virus (RTBV, NC 001914). RTBV was assigned as outgroup.Bootstrap values derived from parsimony. Branches with less than 60% support werecollapsed.region where these sequences overlapped within the archetypi-cal badnavirus genome and thus, the presence of more than onebadnavirus in these plants cannot be ascertained without furtherinvestigation.
4. Discussion
The application of the VLP-VNA isolation procedure to four A.psilostachya specimens yielded strong evidence for the presence ofat least four viruses. That contigs of two of these putative virusesdid not overlap leaves open the possibility that there could be asmany as nine actual viruses (Table 2). However, the results showthat virions are not the only components of the VLP fraction thatcontain nucleic acids. Fungal and bacterial sequences also werepresent in the VLP-VNA fractions. Since bacteria and fungi may con-tain viruses, application of VLP-VNA to other plant specimens may
rologic
erve
ferenc
85–6836–58
1–1360–3411–6236–7528–3298–5509–11
iforms the 3
54 U. Melcher et al. / Journal of Vi
Table 2Putative viral sequences from A. psilostachya samples from the Tallgrass Prairie Pres
Plant Segment Reference sequencea Length (nt) Re
05TGP00312 Bad1 TaroBV 587 62Flex1 PepMV 217 56
05TGP00321 Flex2 NeVX 1376Flex3 AlsVX 1741 18Flex4 ICRSV 1813 45Flex5 ICRSV 1373 62Bad2 TaroBV 371 29Bad3 CSSV 248 52Bad4 TaroBV 772 5
a Virus whose sequence was used for “Reference coordinates”: TaroBV, Taro bacillICRSV, Indian citrus ringspot virus; CSSV, Cacao swollen shoot virus. Asterisk indicate
b BLAST hit was to NeVX coat protein.c BLAST hit was to Banana streak virus ORF II.
reveal viruses of other resident fungi or bacteria. About a fourthof the VLP-VNA sequences retrieved from the A. psilostachya spe-cimens probably originate from the A. psilostachya genome. Theseconsist primarily of retroelement-related and mRNA sequences.The large contribution of retroelement-related sequences may bedue to the fact that they comprise a large portion of the plant’stranscriptome or possibly to their being associated with yet to becharacterized endogenous retroviral particles.
Between one-third and three-fourths (depending on specimen)of the sequences could not be assigned with any certainty toa source organism. These may represent A. psilostachya-derivedmRNA sequences from genes that are not highly conserved in thegenomes of higher plants whose genomes have been sequenced orthey may represent viruses that are so distantly related to thoseof known viruses as not to be recognized by BLAST search. How-ever, given that the sequences do not assemble into larger contigs,the genome of such a hypothetical virus would have to be largeor there should be a large number of smaller diverse viruses in thesample. Understanding these unclassified sequences awaits furtherinvestigation.
Despite the recovery of some non-viral sequences, the VLP-VNAmethod isolated viral nucleic acid fragments from plant material.Included among the virus sequences detected are those with single-stranded positive sense RNA genomes forming a filamentous virionand those with double-stranded DNA genomes forming a bacil-liform virion. Additional studies on other plant species from theTallgrass Prairie Preserve yielded sequences similar to those of
monopartite genomes of spherical viruses (in preparation, Min, B.E.et al.) and of viruses with multipartite genomes. Although virusesthat do not produce encapsidated forms and those that make highlyunstable particles may escape detection, it is possible that viralreplication complexes also may be recovered in the VLP pellet, mak-ing the poorly encapsidated genera also accessible to the VLP-VNAtechnique. The VLP-VNA method does not establish that these A.psilostachya specimens were infected with viruses. That conclusionawaits isolation and further characterization of infectious virions.The putative viruses identified by VLP-VNA in this study are likelystrictly plant viruses since sequence similarities are only with virusgenera and species that infect plants, and these are not known toreplicate in their vectors.Although the method, joining a differential centrifugation, anucleic acid isolation and a nucleic acid amplification and cloningmethod, has proved effective in finding putative viral sequences inplants, the possibility exists that the addition of further steps mayreduce the background, particularly of host-derived sequences. Thelatter appear to arise from plastid nucleoids and assemblages con-taining mRNA, possibly informosomes. Plant polyribosomes areunlikely sources of the host-derived sequences since ribosomal
al Methods 152 (2008) 49–55
ea coordinates E-value (method) % of sequences Accession no.
56 6 × 10−6 (BLASTp) 0.7 EU36285458 0.078 (rtBLASTn) 2.0 EU362846
48 2 × 10−113 (BLASTx) 19.5 EU36284753 2 × 10−114 (BLASTx) 3.0 EU36284856 4 × 10−46 (BLASTx) 4.5 EU36284960* 1 × 10−46 (BLASTx)b 4.7 EU36285096 2 × 10−39 (BLASTx) 0.4 EU36285258 9 × 10−17 (tBLASTn) 0.9 EU36285370 0.64 (tBLASTn)c 0.4 EU362851
virus; PepMV, Pepino mosaic virus; NeVX, Nerine virus X; AlsVX, Alstroemeria virus X,′ end of the sequence.
rRNA sequences, which should be present and abundant, wererarely encountered. Incorporation of electron microscopic exam-ination of fractions into the protocol may reveal the nature of thecontributors and lead to improved enrichment of viral sequences.
The data suggest that plants from natural ecosystems are fre-quently infected with multiple viruses (Table 2). Two plants weremultiply infected with a member of the Flexiviridae and a memberof the Badnavirus genus. Infection with multiple viruses is consis-tent with results of others in which non-cultivated plants weresurveyed for the presence or absence of specific viruses (Cooperand Jones, 2006; Raybould et al., 1999; Webster et al., 2007). Inthe present study, at least 18% and as many as 50% (depending onthe significance of failure to obtain amplifiable VLP-VNA) of theA. psilostachya specimens probably contained virus. This range isconsistent also with other observations on virus incidence in non-cultivated plants (Cooper and Jones, 2006; Raybould et al., 1999;Webster et al., 2007).
Most noteworthy is that all of the putative viral sequences iden-tified have not been previously described, implying that the virusspecies that they likely represent also have not been previouslydescribed. Indeed, the preliminary evidence indicates that the Flex-iviridae virus belongs to a previously undescribed genus of viruses.These observations support the original contention (Wren et al.,2006) that the world of viruses, and specifically for this work, theworld of plant viruses, must be much larger than is recognizedcurrently. Thus, the VLP-VNA method has the potential rapidly toexpand the universe of known plant-associated viruses. Examina-
tion of further specimens and species from the Tallgrass PrairiePreserve is in progress.Acknowledgements
The authors express their appreciation to Dan McGlinn, FumikoShirakura, Matt Allen and Pete Earls for assistance in specimen col-lection, Fenqiu Zhang and Tsuyoshi Asako for specimen processing,Yan Song for database management and Kay Scheets for carefulediting of the manuscript. This work was supported by National Sci-ence Foundation-EPSCoR award EPS-0447262, the Oklahoma StateRegents for Higher Education and the Oklahoma Agricultural Exper-iment Station whose Director has approved the manuscript forpublication.
References
Altschul, S.F., Madden, T.L., Schaffer, A.A., Zhang, J., Zhang, Z., Miller, W., Lipman, D.J.,1997. Gapped BLAST and PSI-BLAST: a new generation of protein database searchprograms. Nucleic Acids Res. 25, 3389–3402.
Angly, F.E., Felts, B., Breitbart, M., Salamon, P., Edwards, R.A., Carlson, C., Chan, A.M.,Haynes, M., Kelley, S., Liu, H., Mahaffy, J.M., Mueller, J.E., Nulton, J., Olson, R.,

rologic
U. Melcher et al. / Journal of ViParsons, R., Rayhawk, S., Suttle, C.A., Rohwer, F., 2006. The marine viromes offour oceanic regions. PLoS Biol. 4, 2121–2131.
Bodaghi, S., Mathews, D.M., Dodds, J.A., 2004. Natural incidence of mixed infec-tions and experimental cross protection between two genotypes of Tobacco mildgreen mosaic virus. Phytopathology 94, 1337–1341.
Bodenteich, A., Chissoe, S., Wang, Y.F., Roe, B.A., 1993. Shotgun cloning as the strat-egy of choice to generate templates for high-throughput dideoxynucleotidesequencing. In: Venter, J.C. (Ed.), Automated DNA Sequencing and Analysis Tech-nique. Academic Press, London, pp. 42–50.
Breitbart, M., Felts, B., Kelley, S., Mahaffy, J.M., Nulton, J., Salamon, P., Rohwer, F.,2004. Diversity and population structure of a near-shore marine-sediment viral
community. Proc. R. Soc. Lond. Ser. B-Biol. Sci. 271, 565–574.Casas, V., Rohwer, F., 2007. Phage metagenomics. Methods Enzymol. 421, 259–268.
Chissoe, S.L., Bodenteich, A., Wang, Y.F., Wang, Y.P., Burian, D., Clifton, S.W., Crab-tree, J., Freeman, A., Iyer, K., Jian, L., Ma, Y., McLaury, H.-J., Pan, H.-Q., Sarhan,O.H., Toth, S., Wang, Z., Zhang, G., Heisterkamp, N., Groffen, J., Roe, B.A.,1995. Sequence and analysis of the human ABL gene, the BCR gene, andregions involved in the Philadelphia chromosomal translocation. Genomics 27,67–82.
Cooper, I., Jones, R.A.C., 2006. Wild plants and viruses: under-investigated ecosys-tems. Adv. Virus Res. 67, 1–47.
Delwart, E.L., 2007. Viral metagenomics. Revi. Med. Virol. 17, 115–131.Edwards, R.A., Rohwer, F., 2005. Viral metagenomics. Nat. Rev. Microbiol. 3, 504–510.Ewing, B., Green, P., 1998. Base-calling of automated sequencer traces using phred
II. Error probabilities. Genome Res. 8, 186–194.Ewing, B., Hillier, L., Wendl, M.C., Green, P., 1998. Base-calling of automated
sequencer traces using phred I. Accuracy assessment. Genome Res. 8, 175–185.Fauquet, C.M., Mayo, M.A., Maniloff, J., Desselberger, U., Ball, L.A., 2005. Virus Taxon-
omy. Elsevier Academic Press, Amsterdam.Felsenstein, J., 1989. PHYLIP-Phylogeny Inference Package (Version 3.2). Cladistics 5,
164–166.Fraile, A., Escriu, F., Aranda, M.A., Malpica, J.M., Gibbs, A.J., Garcia-Arenal, F., 1997.
A century of tobamovirus evolution in an Australian population of Nicotianaglauca. J. Virol. 71, 8316–8320.
Gardner, R.C., Shepherd, R.J., 1980. A procedure for rapid isolation and analysis ofcauliflower mosaic virus DNA. Virology 106, 159–161.
Hamilton, R.G., 2007. Restoring heterogeneity on the Tallgrass Prairie Preserve:applying the fire-grazing interaction model. In: Masters, R.E., Galley, K.E.M.
al Methods 152 (2008) 49–55 55
(Eds.), Proceedings of the 23rd Tall Timbers Fire Ecology Conference: Fire inGrassland and Shrubland Ecosystems. Tall Timbers Research Station, Tallahassee.
Hull, R., 1978. The DNA of plant DNA viruses. In: Hall, T.C., Davies, J.W. (Eds.), NucleicAcids in Plants, vol. II. CRC Press, Boca Raton, pp. 3–29.
Lane, L.C., 1986. Propagation and purification of RNA plant viruses. Methods Enzy-mol. 118, 687–696.
Lane, L.C., 1992. A general method of detecting plant viruses. In: Maramorosch, K.(Ed.), Plant Diseases of Viral, Viroid, Mycoplasma and Uncertain Etiology. West-view Press, Boulder, pp. 1–15.
Martelli, G.P., Adams, M.J., Kreuze, J.F., Dolja, V.V., 2007. Family Flexiviridae: a casestudy in virion and genome plasticity. Annu. Rev. Phytopathol. 45, 73–100.
Melcher, U., 1990. Similarities between putative transport proteins of plant viruses.J. Gen. Virol. 71, 1009–1018.
Ooi, K., Yahara, T., 1999. Genetic variation of geminiviruses: comparison betweensexual and asexual host plant populations. Mol. Ecol. 8, 89–97.
Palmer, M.W., 2007. Vascular flora of the Tallgrass Prairie Preserve. Castanea 71,235–246.
Raybould, A.F., Maskell, L.C., Edwards, M.L., Cooper, J.I., Gray, A.J., 1999. The preva-lence and spatial distribution of viruses in natural populations of Brassicaoleracea. New Phytol. 141, 265–275.
Riesenfeld, C.S., Schloss, P.D., Handelsman, J., 2004. Metagenomics: genomic analysisof microbial communities. Annual Rev. Genet. 38, 525–552.
Robertson, N.L., 2005. A newly described plant disease complex involving two dis-tinct viruses in a native Alaskan lily Streptopus amplexifolius. Can. J. Bot. 83,1257–1267.
Roe, B.A., 2004. Shotgun library construction for DNA sequencing. In: Zhao, S., Stodol-sky, M. (Eds.), Methods in Molecular Biology, vol. 255. Humana Press Inc., Totowa,New Jersey, pp. 171–187.
Wang, D., Coscoy, L., Zylberberg, M., Avila, P.C., Boushey, H.A., Ganem, D., DeRisi, J.L.,2002. Microarray-based detection and genotyping of viral pathogens. Proc. Natl.Acad. Sci. U.S.A. 99, 15687–15692.
Webster, C.G., Coutts, B.A., Jones, R.A.C., Jones, M.G.K., Wylie, S.J., 2007. Virus impactat the interface of an ancient ecosystem and a recent agroecosystem: studies onthree legume-infecting potyviruses in the southwest Australian floristic region.Plant Pathol. 56, 729–742.
Wren, J.D., Roossinck, M.J., Nelson, R.S., Scheets, K., Palmer, M.W., Melcher, U., 2006.Plant virus biodiversity and ecology. PLoS Biol. 4, e80.
Xu, J.P., 2006. Microbial ecology in the age of genomics and metagenomics: concepts,tools, and recent advances. Mol. Ecol. 15, 1713–1731.