Embed Size (px)
Citation preview

RESEARCH PAPER
Evolution of ConAl clusters and chemisorption of hydrogenon ConAl clusters
Ling Guo
Received: 7 March 2012 / Accepted: 25 May 2012
� Springer Science+Business Media B.V. 2012
Abstract The growth behavior of ConAl (n = 1–15)
and the chemisorptions of hydrogen on the ground
state geometries have been studied using the density
functional theory (DFT) within the generalized gradi-
ent approximation (GGA). The growth pattern for
ConAl is Al-substituted Con?1 clusters, and it keeps
the similar frameworks of the most stable Con?1
clusters except for n = 2, 3, and 6. The Al atom
substitutes the surface atom of the Con?1 clusters for
n B 13. Starting from n = 14, the Al atom completely
falls into the center of the Co-frame. The dissociation
energy, the second-order energy differences, and the
HOMO–LUMO gaps indicate that the magic numbers
of the calculated ConAl clusters are 7, 9, and 13,
corresponding to the high symmetrical structures. To
my knowledge, this is the first time that a systematic
study of chemisorption of hydrogen on cobalt alumi-
num clusters. The twofold bridge site is identified to be
the most favorable chemisorptions site for one hydro-
gen adsorption on ConAl (n = 1–6, 8, 10), and two
hydrogen adsorption on ConAl (n = 1–7), while
threefold hollow site is preferred for one hydrogen
adsorption on ConAl (n = 7, 9, 11–15) and two
hydrogen adsorption on ConAl (n = 8–10, 12–15)
clusters. The ground state structure of two hydrogen
adsorption on Co11Al is exceptional. In general, the
binding energy of both H and 2H of ConAl (n = 1–12)
is found to increase with the cluster size. And the result
shows that large binding energies of the hydrogen
atoms and large fragmentation energies for Co11AlH
and Co12AlH make these species behaving like magic
clusters.
Keywords ConAl cluster � Hydrogenated cobalt
aluminum cluster � Stability � Electronic properties
Introduction
Bulk phase bimetallic systems provide a matter of
increasing interest in pure and applied materials
sciences and traditional fields of physics and chemis-
try. In catalytic chemistry and chemical engineering,
real catalysts mainly consist of a heterometallic or
bimetallic system, which can profoundly enhance
reactivity and selectivity. Thus, to get a deeper
understanding of the microscopic behavior of these
species, the study of bimetallic or so-called alloy
clusters provides a suitable tool, because cluster
science enables one to investigate chemical and
physical properties starting from a single atom or
molecule toward bulk phase as a function of size.
Therefore, in the last decades, a number of studies of
bimetallic clusters and diatomic molecules have been
performed (Lu et al. 2011; Zhao et al. 2011; Laguna
et al. 2010; Zanti and Peeters 2010).
L. Guo (&)
School of Chemistry and Material Science, Shanxi
Normal University, Linfen 041004, China
e-mail: [email protected]
123
J Nanopart Res (2012) 14:957
DOI 10.1007/s11051-012-0957-7

Among the candidate systems to have been con-
sidered, the bimetallic cobalt aluminum clusters have
been the topic of some experimental and theoretical
studies (Nonose et al. 1989; Menezes and Knickelbein
1991, 1993; Behm et al. 1994; Pramann et al. 2001).
Several years ago, Nonose et al. (1989) performed
chemisorptions reactivity studies of neutral AlnCom
(n [ m) and ConAlm (n [ m) clusters toward H2 using
a fast flow reactors. In that study, they found that the
doping of Con clusters with only one Al atom reveals a
remarkable increase of hydrogen chemisorptions rates
compared with pure Con clusters. On the other hand,
pure Aln clusters do not adsorb hydrogen, which is
comparable to Al bulk phase behavior. Menezes and
Knickelbein (1991, 1993) succeeded in a comprehen-
sive investigation of the size dependence of ionization
energies of these clusters. These IE studies show that
the electronic shell structure of AlnCo and AlnCo2
clusters remains similar to that of pure Aln clusters.
Behm et al. (1994) have performed resonant two-
photon ionization spectroscopy on small diatomic
AlCo aluminides. Pramann and co-workers (2001)
have measured the photoelectron spectra of small
mass-selected aluminum-rich AlnCo- (n = 8–17) and
cobalt-rich ConAlm- clusters (n = 6, 8, 10; m = 1, 2) at
photon energies of 3.49 eV with the aid of a magnetic
bottle photoelectron spectrometer.
Hydrogen adsorption on metal surfaces and clusters
is a widely studied subject that provides the opportu-
nity to gain a basic understanding of the complicated
nature of many interesting problem, such as hydrogen
embitterment of metals, catalytic processes, hydrogen
storage, etc. Technological advancement in recent
decades enables us to study and control systems
having dimensions of a few atoms. Hydrogen adsorp-
tion on such nanosystems, for example on clusters,
gives an atomic perspective of the process (Huda and
Kleinman 2006; Dhilip Kumar et al. 2009; Varano
et al. 2010). In this study, our main goal is to explore
the sequential growth of small ConAl clusters with
n = 1–15 and study hydrogen molecule adsorption
and its effect on the geometric and electronic proper-
ties of ConAl (n = 1–15) clusters. And to obtain
further insights on the nature of chemisorption, the
extensive calculations of chemisorption of H2 and
sequential hydrogen loading on the energetically
stable ConAl (n = 1–15) clusters are studied. A
detailed picture of chemisorption of H2 on ConAl
(n = 1–15) nanoclusters based on an analysis of
energies, stability, fragmentation behavior, and bond-
ing nature is presented. So the understanding of the
adsorption of H2 molecule on cobalt aluminum
clusters could give useful insight on hydrogen inter-
action with other alloy clusters.
The article is organized as follows: A brief account
of the computational methodology is given in ‘‘Meth-
odology’’ section, followed by a detailed presentation
and discussion of the structures of different size ConAl
(n = 1–15) in ‘‘Results and discussion’’ section. In the
following section, I present the structures obtained
after adsorption of one and two atomic hydrogens.
These will provide the understanding of the interac-
tional nature and the magic behavior of hydrogen with
cobalt aluminum clusters. A summary of my findings
and conclusions are given in Summary section.
Methodology
Our calculations are carried out using density func-
tional theory (DFT) implemented in the DMol3
package (Delley 1990). We perform all-electron
spin-unrestricted calculations, using double numerical
plus polarization basis set (DNP) and the Beck’s
exchange functional and Lee–Yang–Parr correlation
functional (BLYP) (Lee et al. 1988) within general-
ized gradient approximation (GGA). The direct
inversion in iterative subspace (DIIS) approach is
used to speed up SCF convergence. I also applied
thermal smearing to the orbital occupation to speed it
up. The value of smearing is 0.005 hartree. For the
accurate calculations, I have chosen an octupole
scheme for the multipolar expansion of the charge
density and coulomb potential. The grid for numerical
integration is set as ‘‘fine.’’ The convergence criteria
for energy, energy gradient, and displacement are
1 9 10-5 Hartree, 2 9 10-3 Hartree/A, and 5 9
10-3 A, respectively.
To test the reliability of our calculation, the Co2 and
Al2 dimers are calculated. The results are summarized
in Table 1. From Table 1, it is obvious that the
calculated bond length (2.41 A), averaged binding
energy (0.25 eV/atom), and ionization potential
(6.50 eV) of Co2 with BLYP method are closest to
the experimental values (Kant and Strauss 1964; Hales
et al. 1994) of 2.31 A, 1.69 ± 0.26 eV, and 6.42 eV,
respectively. It should also be noted that the BLYP
functional has been successfully applied to several
Page 2 of 14 J Nanopart Res (2012) 14:957
123

other TM cluster systems, including Tin clusters
(Wang et al. 2004), Nin cluster (Xie et al. 2005), and
Lan clusters (Zhang and Shen 2004). Additionally, the
bond length (2.51 A), averaged binding energy
(0.94 eV/atom), and ionization potential (5.82 eV)
of Al2 (triplet state) are obtained, which are in good
agreement with the experimental values (Rosen 1970)
of 2.56 A, 0.77 ± 0.15 eV, and 6.20 ± 0.20 eV,
respectively. Therefore, the calculation method
employed is reliable and accurate enough.
Results and discussions
Structure and properties of the ConAl (n = 1–15)
clusters
In cluster physics, one of the most fundamental
problems is to determine the ground state geometry.
Accordingly, I first study the ground state structures of
the ConAl (n = 1–15) clusters, which are shown in
Fig. 1. Symmetries (sym), HOMO–LUMO gaps,
atomic averaged binding energy (Eb), the dissociation
energy (DE), the second-order energy differences
(D2E), vertical ionization potential (VIP), and vertical
electron affinities (VEA) for the most stable ConAl
(n = 1–15) clusters are all shown in Table 2. For
proper comparison, the structures of Con (2–16)
clusters have also been shown in Fig. 1 and Table 3.
For the CoAl dimer (Fig. 1[1a]) with C?v symme-
try, the optimized results indicate that the spin singlet
state is lower in total energy than the triplet and quintet
isomers by 0.55 eV and 1.80 eV, respectively. Fur-
thermore, the binding energy per atom (Eb) of the
CoAl dimer is 0.391 eV/atom larger than that of the
Co2 (Fig. 1[1a0]) one (0.25 eV/atom), the bond
lengths of CoAl and Co2 dimers are 2.454 and
2.410 A, respectively. For the Co3 cluster, three
structures (C2v, D3h, and D?h) have been discussed.
The linear structure (D?h, Fig. 1[2a0]) is found to be
the ground state, which is in agreement with the study
of Ma et al. (2006), who adopted the BLYP exchange–
correlation functional in the study. The ground state
Co2Al cluster is a spin doublet acute-angle triangular
structure (Fig. 1[2a]) with C2v symmetry, in which the
Al atom is at the apex, the Co–Co distance (2.388 A) is
a little smaller than that of the Co2 dimer (2.410 A),
and bond length of the Co–Al is 2.480 A. Five
different initial geometries have been considered for
the tetramer: tetrahedron, square, rhombus, planar
Y-like, and linear structures. A plane rhombus (C2v,
Fig. 1[3a0]) appears to be the most stable structure,
which is consistent with the calculations of Ma et al.
(2006). The ground state Co3Al cluster is a spin quartet
tetrahedral configuration (C3v, Fig. 1[3a]). For Co5,
many different initial geometries such as pyramid,
bipyramid, and capped tetrahedron, have been tried.
The triangular bipyramid structure (Fig. 1[4a0]) is
found to be the most stable structure with D3h
symmetry. The present result is in agreement with
the previous calculation (Ma et al. 2006). For Co4Al, a
spin septet distorted triangle bipyramid structure
(C2v, Fig. 1[4a]) has been proven to be most stable.
The capped trigonal bipyramid, octahedron, and
pentagonal pyramid structures have been studied to
search the ground state for Co6 cluster. A distortional
octahedral structure (Oh, Fig. 1[5a0]) is found to be the
ground state. It has an average bond length of 2.551 A.
The present result is in agreement with previous
theoretical studies (Ma et al. 2006). For Co5Al, the
ground state structure is a spin state of ten and slightly
distorted octahedron configuration with Cs symmetry,
where an Al atom is at the vertex position. In the case
of Co7, a capped octahedron, a pentagonal bipyramid,
a tri-capped tetrahedron, and a bicapped triangular
bipyramid structure have been considered. The capped
Table 1 Calculated bond lengths, averaged binding energies, and vertical ionization potential and experiment results
Co2 (this work) Experimentala,b Al2 (this work) Experimentalc
Bond length (A) 2.41 2.31 2.51 2.56
Eb(eV/atom) 0.25 1.69 ± 0.26 0.94 0.77 ± 0.15
Ionization potential (eV) 6.50 6.42 5.82 6.20 ± 0.20
a Kant and Strauss (1964)b Hales et al. (1994)c Rosen (1970)
J Nanopart Res (2012) 14:957 Page 3 of 14
123

octahedron (C3v, Fig. 1[6a0]) appears as the most
stable structure. The pentagonal bipyramid (C5v,
Fig. 1[6a]) appears as the most stable structure of
Co6Al. For Co8, three different geometries of bicapped
octahedron, capped pentagonal bipyramid, and tri-
capped triangular bipyramid have been studied. The
bicapped octahedron with D2d symmetry (Fig. 1[7a0])
is found to be most stable structure. For the Co9
cluster, the distortion tricapped octahedron is found to
be the most stable structure (D3h, Fig. 1[8a0]). As the
clusters grow from Co10 to Co16, the new addition
always occurs at a site where interactions with more
atoms are available to form the close-packed structure.
Their symmetries are D4d (Co10, Fig. 1[9a0]), D4d
(Co11, Fig. 1[10a0]), C5v (Co12, Fig. 1[11a0]), Ih
(Co13, Fig. 1[12a0]), C3v (Co14, Fig. 1[13a0]), Cs
(Co15, Fig. 1[14a0]), and Cs (Co16, Fig. 1[15a0]),
respectively. The lowest energy structures of ConAl
(n = 7–13) are found to be as Al substituted the
surface atom of Con?1. Furthermore, the most stable
ConAl (n = 7–13) clusters always keep the same
frameworks as Con?1 clusters. For example, the
ground state geometry of Co7Al (Fig. 1[7a]) with Cs
symmetry is a bicapped octahedron configuration
similar to that of the Co8 configuration. For Co8Al
cluster, the lowest energy isomer (Fig. 1[8a]) with Cs
symmetry is capped tetragonal antiprism structure and
can be regarded as substitutional structure of Co9. The
1a0 1a 2a0 2a 3a0 3a
4a0 4a 5a0 5a 6a0 6a
7a0 7a 8a0 8a 9a0 9a
10a0 10a 11a0 11a 12a0 12a
13a0 013a 14a 14a 15a0 15a
Fig. 1 The calculated lowest energy structures of Con?1 and ConAl (n = 1–15) clusters
Page 4 of 14 J Nanopart Res (2012) 14:957
123
ground state geometries of Co9Al, Co10Al, and Co11Al
are spin 16 structure with C2v symmetry (Fig. 1[9a]),
spin 17 structure with C4v symmetry (Fig. 1[10a]), and
spin 20 structure with C5v symmetry (Fig. 1[11a]),
respectively. For Co12Al, the lowest energy structure
is the icosahedral configuration (Fig. 1[12a]) with C5v
symmetry, where the Al atom is at vertex position.
Co13Al (Fig. 1[13a]) takes the C3v structure as its
ground state. Starting from n = 14, the Al atom
completely falls into the center of the Co-frame. And
ConAl (n = 14 and 15) (Fig. 1[14a, 15a]) takes the Cs
structures as their ground states, respectively.
We now discuss the relative stability of ConAl
clusters by computing the energy that is indicative of
the stability. We compute the atomization or binding
energy (Eb) per atom, the dissociation energy (DE),
and the second-order energy differences (D2E) as,
respectively,
Eb ConAl½ � ¼ nE Co½ � þ E Al½ � � E ConAl½ �ð Þnþ 1ð Þ ; ð1Þ
DE ConAl½ � ¼ E Con�1Al½ � þ E Co½ � � E ConAl½ �; ð2Þ
D2E ConAl½ � ¼ E Conþ1Al½ � þ E Con�1Al½ �� 2E ConAl½ �: ð3Þ
In general, the Eb increases as the cluster size
grows. Small humps or dips for the specific size of
clusters signify their relative stabilities. The Eb of the
ConAl clusters (shown in Fig. 2) is calculated using
the Eq. 1, where E(Co), E(Al), and E(ConAl) represent
the energies of a Co atom, an Al atom, and the total
energy of the ConAl cluster, respectively. For com-
parison, we also plot the Eb of the host Con cluster,
Eb[Con] = (nE[Co] - E[Con])/n, in Fig. 2. As seen in
the figure, the average binding energies of the most
ConAl clusters are higher than those of the pure Con
clusters (except for n = 14). It indicates that the doped
Al atom in the Con clusters contributes to strengthen
the stabilities of the ConAl framework. The averaged
binding energy of ConAl clusters generally increases
with increasing size; it is concluded that these clusters
continue to gain energy during the growth process. In
addition, the comparison of Con with the BE curve for
ConAl clusters shows that the small clusters of ConAl
are strongly bound. As the cluster grows in size, the
difference between the BE curves of ConAl clusters
and pure Con clusters steadily diminishes, indicating
that the bonding in doped clusters is essentially similar
to that in pure clusters.
In cluster physics, the dissociation energy (DE) and
the second-order energy differences (D2E) are sensi-
tive quantities that reflect the relative stability of the
investigated clusters. The DE shows the energy that
one atom is separated from the host clusters. The
Table 2 Symmetries (sym), HOMO–LUMO gaps, atomic
averaged binding energy (Eb), the dissociation energy (DE),
the second-order energy differences (D2E), vertical ionization
potential (VIP), vertical electron affinities (VEA) (all in eV),
and the mean nearest-neighbor bond lengths between Al and
Co atoms (dAl–Co (A)) for the most stable ConAl (n = 1–15)
clusters
Cluster sym gap Eb DE D2E VIP VEA dAl–Co (A)
CoAl C?v 1.909 0.391 6.930 0.585 2.454
Co2Al C2v 0.745 0.674 1.241 -0.046 6.699 0.863 2.486
Co3Al C3v 0.504 0.827 1.284 0.196 6.634 0.917 2.538
Co4Al C2v 0.356 0.879 1.088 0.063 6.539 0.901 2.438
Co5Al Cs 0.473 0.904 1.026 -0.661 6.519 1.328 2.480
Co6Al C5v 0.824 1.015 1.687 -0.049 6.324 1.064 2.530
Co7Al Cs 0.375 1.105 1.736 0.800 6.221 1.227 2.416
Co8Al C4v 0.028 1.087 0.936 -1.037 6.221 1.257 2.549
Co9Al C2v 0.430 1.175 1.973 0.952 6.133 1.358 2.489
Co10Al C4v 0.441 1.161 1.020 -1.064 5.891 1.396 3.043
Co11Al C5v 0.334 1.238 2.084 -0.142 5.989 1.377 2.471
Co12Al C5v 0.338 1.310 2.171 -2.465 5.948 1.442 2.604
Co13Al C3v 0.593 1.530 4.637 3.557 5.859 1.687 2.562
Co14Al Cs 0.135 1.516 1.080 -0.882 5.676 2.000 2.652
Co15Al Cs 0.266 1.544 1.962 5.565 1.997 2.501
J Nanopart Res (2012) 14:957 Page 5 of 14
123
Table 3 Structures, binding energies of various clusters obtained using the BLYP-DFT method
Cluster Location of H Total BE (eV) BE of H (eV) dAl–Co (A) dAl–H (A) DCo–H (A)
1a CoAlH b(Co, Al) 3.029 2.248 2.430 1.910 1.600
1b CoAlH2 b(Co, Al) 5.516 4.735 2.425 1.870 1.630
2a Co2AlH b(Co, Co) 4.547 2.526 2.480 1.687
2b Co2AlH2 n-b,b(Co, Al) (Co, Co) 6.993 4.972 2.485 1.900 1.670
2c Co2AlH2 n-b,b(Co, Co) (Co, Co) 6.822 4.801 2.490 1.706
2d Co2AlH2 n-b,b(Co, Al) (Co, Al) 6.670 4.649 2.460 1.870 1.645
3a Co3AlH b(Co, Co) 6.057 2.751 2.497 1.687
3b Co3AlH h(Co, Co, Co) 5.929 2.623 2.529 1.791
3c Co3AlH2 n-b,b(Co, Co) (Co, Co) 8.591 5.285 2.383 1.605
3d Co3AlH2 o-b,b(Co, Co) (Al, Co) 8.343 5.037 2.517 1.745 1.693
3e Co3AlH2 n-b,h(Co, Co)(Co, Co, Co) 8.335 5.029 2.530 1.762
4a Co4AlH b(Co, Co) 7.652 3.257 2.545 1.689
4b Co4AlH2 n-b,b(Co, Co) (Co, Co) 10.278 5.883 2.565 1.690
4c Co4AlH2 o-b,b(Co, Co) (Co, Co) 10.112 5.717 2.555 1.701
5a Co5AlH b(Co, Co) 9.257 3.837 2.468 1.695
5b Co5AlH h(Co, Co, Co) 9.256 3.836 2.545 1.780
5c Co5AlH2 n-b,b(Co, Co) (Co, Co) 11.845 6.424 2.498 1.695
5d Co5AlH2 n-h,h(Co, Co, Co) (Co, Co, Co) 11.668 6.247 2.468 1.777
5e Co5AlH2 n-b,b(Co, Co) (Al, Co) 11.638 6.217 2.440 1.880 1.647
6a Co6AlH b(Co, Co) 11.051 3.943 2.532 1.690
6b Co6AlH h(Co, Co, Co) 10.925 3.817 2.506 1.787
6c Co6AlH2 n-b,b(Co, Co) (Co, Co) 13.739 6.331 2.512 1.695
6d Co6AlH2 n-h,h(Co, Co, Co) (Co, Co, Co) 13.309 6.201 2.512 1.788
7a Co7AlH h(Co, Co, Co) 12.678 3.834 2.553 1.790
7b Co7AlH b(Co, Co) 12.648 3.805 2.565 1.690
7c Co7AlH2 n-b,b(Co, Co) (Co, Co) 15.347 6.504 2.545 1.695
7d Co7AlH2 o-b,b(Co, Co) (Co, Co) 15.102 6.259 2.560 1.700
8a Co8AlH b(Co, Co) 14.419 4.639 2.560 1.703
8b Co8AlH h(Co, Co, Co) 14.403 4.623 2.560 1.795
8c Co8AlH2 n-h,h(Co, Co, Co) (Co, Co, Co) 16.920 7.140 2.560 1.797
8d Co8AlH2 n-h,h(Co, Co, Co) (Co, Co, Co) 16.882 7.102 2.560 1.800
8e Co8AlH2 n-b,b(Co, Co) (Co, Co) 16.798 7.018 2.560 1.693
9a Co9AlH h(Co, Co, Co) 16.229 4.476 2.550 1.790
9b Co9AlH2 n-h,h(Co, Co, Co) (Co, Co, Co) 18.771 7.018 2.550 1.790
9c Co9AlH2 n-h,h(Co, Co, Co) (Co, Co, Co) 18.730 6.977 2.558 1.790
10a Co10AlH b(Co, Co) 17.432 4.659 2.700 1.720
10b Co10AlH2 n-h,h(Co, Co, Co) (Co, Co, Co) 20.626 7.853 2.705 1.800
10c Co10AlH2 n-b,b(Co, Co) (Co, Co) 20.050 7.277 2.705 1.691
11a Co11AlH h(Co, Co, Co) 19.979 5.121 2.605 1.807
11b Co11AlH2 n-h,b(Co, Co, Co) (Co, Co) 22.564 7.706 2.611 1.774
11c Co11AlH2 n-h,h(Co, Co, Co) (Co, Co, Co) 22.520 7.662 2.600 1.793
12a Co12AlH h(Co, Co, Co) 22.194 5.165 2.627 1.797
12b Co12AlH2 n-h,h(Co, Co, Co) (Co, Co, Co) 24.733 7.704 2.623 1.803
12c Co12AlH2 o-h,h(Co, Co, Co) (Co, Co, Co) 24.675 7.646 2.613 1.810
Page 6 of 14 J Nanopart Res (2012) 14:957
123

D2E is often directly compared with the relative
abundances determined in mass spectroscopy exper-
iments. They are defined as Eqs. 2 and 3, where
E(ConAl), E(Con?1Al), E(Con-1Al), and E(Co) rep-
resent the total energies of the most stable ConAl,
Con?1Al, and Con-1Al clusters and a Co atom,
respectively. As shown in Fig. 3, particularly prom-
inent maxima of D2E are found at n = 7, 9, and 13,
indicating higher stability than their neighboring
clusters. It is observed that, for the ConAl cluster, the
DE of Co7Al (1.736 eV), Co9Al (1.973 eV), and
Co13Al (4.637 eV) clusters are higher than other
clusters. And the HOMO–LUMO gaps of Co7Al
(0.375 eV), Co9Al (0.430 eV), and Co13Al (0.593 eV)
shown in Table 2 are larger than those of their
neighboring clusters. Thus, we can conclude that the
magic clusters are found at n = 7, 9, and 13 for ConAl.
It is worth pointing out that the relative stability of the
Co13Al cluster in terms of the calculated dissociation
energy and the second-order difference of cluster
energy are both the strongest among all different sized
clusters. The high stability of Co13Al cluster might
stem from its highly symmetric geometry.
Experimentally, the electronic structure is probed
through measurements of ionization potentials, elec-
tron affinities, polarizabilities, etc. Therefore, we also
study these quantities to understand their evolution
with size. These quantities are determined within
BLYP for the lowest energy structures obtained within
the same scheme.
The vertical ionization potential (VIP) is calculated
as the self-consistent energy difference between the
cluster and its positive ion with the same geometry.
The VIP is plotted in Fig. 4 as a function of cluster
size. In general, the VIP decreases for n = 1–15. Also
shown in Fig. 4 are the VIPs of pure Co clusters. These
have also been calculated at the BLYP level of theory,
with structures optimized at the same level of theory.
The comparison of the two curves shows that replac-
ing one Co in Con cluster with Al, to give Con-1Al,
results in the approximate values of VIPs for most
clusters except for CoAl and Co2Al, which have
smaller VIPs than the corresponding Con clusters.
-3.2-2.4-1.6-0.8
00.81.62.43.2
44.85.6
n
Ene
rgy/
eV
ΔE
Δ2E
0 2 4 6 8 10 12 14 16
Fig. 3 Size dependence of the second-order energy difference
and the dissociation energy of the lowest energy ConAl
(n = 2–15) clusters
Table 3 continued
Cluster Location of H Total BE (eV) BE of H (eV) dAl–Co (A) dAl–H (A) DCo–H (A)
13a Co13AlH h(Co, Co, Co) 23.886 2.465 2.562 1.800
13b Co13AlH2 n-h,h(Co, Co, Co) (Co, Co, Co) 26.425 5.004 2.560 1.808
14a Co14AlH h(Co, Co, Co) 25.547 2.801 2.543 1.806
14b Co14AlH2 n-h,h(Co, Co, Co) (Co, Co, Co) 27.984 5.238 2.533 1.801
15a Co15AlH h(Co, Co, Co) 27.208 2.500 2.511 1.810
15b Co15AlH2 n-h,h(Co, Co, Co) (Co, Co, Co) 29.663 4.955 2.509 1.810
dAl–Co, dAl–H, and dCo–H are the mean nearest-neighbor bond lengths between Al and Co atoms, Al and H atoms, and Co and H atoms.
Location of H is represented by symbols n, o, b, t, and h which mean neighboring, opposite, bridge, top, and hollow site, respectively
0.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
1.8
Eb/e
V
n
ConAl
Con+1
0 2 4 6 8 10 12 14 16
Fig. 2 Size dependence of average binding energy of the
lowest energy ConAl and Con?1 clusters
J Nanopart Res (2012) 14:957 Page 7 of 14
123

We have also calculated vertical electron affinities
(VEA) for these clusters (see Fig. 5) by assuming the
geometry for the charged cluster to be the same as for
the neutral one. Figure 5 shows that the VEA values of
the ConAl clusters increase as the number of atoms
increase except at n = 5 with a local maximum. A
comparison of the VEAs of ConAl clusters and pure
Co clusters shows that the most ConAl have the higher
values of VEAs than those of Con. It is interesting to
point out that the large-sized clusters usually have
smaller VIPs and larger VEAs as compared with the
small-sized clusters, implying that it is much more
difficult to ionize the smaller clusters than the larger
ones but much easier to attach an electron to the larger
clusters than to the smaller ones.
Hydrogen on ConAl (n = 1–15)
The optimized geometries for the adsorption of one
and two H atoms on small ConAl (n = 1–15) clusters
are shown in Fig. 6. In Table 4, structures, binding
energies of various ConAlHm (n = 1–15; m = 1, 2)
clusters obtained using the BLYP-DFT method are
displayed for the isomers shown in Fig. 6. In Table 4,
total BE is the binding energy of ConAlHm, using the
following equation:
Eb ConAlHm½ � ¼ nE Co½ � þ E Al½ � þ mE H½ �� E ConAlHm½ � ð4Þ
and BE of H is the difference of the binding energy of
ConAlHm and ConAl.
For the chemisorptions of one or two H on the
ConAl cluster, there are three possible adsorption sites:
onefold on top, twofold edge, and threefold hollow
site. The calculation result shows that the twofold edge
and threefold hollow adsorption configurations are
energetically most stable.
It is seen that the BE of H on CoAlH (4.735 eV) is
different to that of H on CoAl (2.248 eV). This shows
that both CoAlH and CoAlH2 have the different
stability.
The ground state corresponding to Co2AlH cluster
(Fig. 6[2a]) is a spin singlet with a Co–H bond length
of 1.687 A and the H atom takes the bridge adsorption
with the Co and Co atoms. The Cs isomer (Fig. 6[2b])
with two H atoms bridging in Co, Co atoms and Co, Al
atoms is found for the most stable geometry of
Co2AlH2 cluster. Other optimized geometries are also
considered for this cluster, for example, occupied
different places of Al, Co atoms (Fig. 6[2c]) and Co,
Co atoms (Fig. 6[2d]). None of them are more stable
than the ground state structure.
Two optimized geometries are found for Co3AlH
cluster, both of the same multiplicity, doublets.
Interaction of H twofold edge site of Co (Fig. 6[3a])
with Cs symmetry is favorable as compared with a
threefold on the hollow of Co3Al with C3v symmetry
(Fig. 6[3b]) by 0.128 eV. The BE is small (2.751 eV)
that it is easy to make further interaction with
hydrogen atom. To confirm this, I carried out calcu-
lation on Co3AlH2. Three configurations for H are
studied: (i) where both H atoms are bridging with two
Co atoms (Fig. 6[3c]), (ii) where one H atom is
bridging with two Co atoms and another one with Co
and Al atoms (Fig. 6[3d]), and (iii) where one H atom
0 2 4 6 8 10 12 14 160.2
0.4
0.6
0.8
1.0
1.2
1.4
1.6
1.8
2.0
2.2
Ver
tical
ele
ctro
n af
fini
ty/e
V
n
ConAl
Con+1
Fig. 5 Size dependence of VEAs of the lowest energy ConAl
and Con?1 clusters
5.2
5.4
5.6
5.8
6.0
6.2
6.4
6.6
6.8
7.0
7.2
7.4
7.6V
ertic
al I
oniz
atio
n Po
tent
ial/e
V
n
ConAl
Con+1
0 2 4 6 8 10 12 14 16
Fig. 4 Size dependence of VIPs of the lowest energy ConAl
and Con?1 clusters
Page 8 of 14 J Nanopart Res (2012) 14:957
123

1a 1b 2a 2b 2c 2d
3a 3b 3c 3d 3e
4a 4b 4c
5a 5b 5c 5d 5e
6a 6b 6c 6d
7a 7b 7c 7d
8a 8b 8c 8d8e
9a 9b 9c 10a 10b 10c
11a 11b 11c 12a 12b 12c
13a 13b 14a 14b 15a 15b
Fig. 6 Relaxed structures
of ConAlHm (n = 1–15;
m = 1, 2). Black, gray, and
white balls are used for Co,
Al, and H, respectively
J Nanopart Res (2012) 14:957 Page 9 of 14
123

is bridging with two Co atoms and another one is in the
hollow site of Co, Co, Co atoms making the Cs
structures as shown in Fig. 6[3e]. The energy differ-
ence of the first two of these is 0.248 eV (Table 4).
Also the Cs structure 3e lies 0.256 eV higher in energy
than the 3c structure. The BE for 2H is 5.285 eV
(Table 4) and it shows that interaction between two
hydrogens on Co3Al is not attractive. This energy is
higher than the dissociation energy of H2 (4.60 eV).
Accordingly, hydrogen is likely to be dissociated on
Co3Al. The distance between two hydrogens on Co3Al
in the lowest energy state is 3.139 A as compared with
the bond length of 0.75 A in H2. Therefore, two
hydrogens are in a dissociated configuration. It is
worth mentioning that the thermodynamics of
hydrogen uptake and release (physical adsorption) is
actually ruled by the combined effect of adsorption
enthalpy and entropy. But it is necessary to indicate
that it is only favored by the enthalpy change for the
chemical adsorption. Entropy effects may not favour
this reaction.
The Co4AlH is a spin quartet and its structure, the
same as the small clusters Co2Al and Co3Al, prefers
the bridge site of Co, Co atoms (Fig. 6[4a]). In the case
of two H on Co4Al, the structure with two H atoms
taking twofold bridge site on two neighboring Co, Co
atoms (Fig. 6[4b]) is the ground state. And the
structure with two H atoms on the bridge site of
opposite Co, Co atoms (Fig. 6[4c]) is the low-lying
one.
Table 4 Fragmentation
energies of ConAlHm
clusters with the product
Con-pAlHm-q, p = 1 and 2,
and q = 1
All the values mean the
parent cluster has a larger
binding energy than the sum
of the BE of the products
Cluster (ConAlHm) Con-1AlHm ?Co Con-2AlHm ?Co2 Con-1AlHm-1 ?CoH
2a Co2AlH 1.518 2.074 1.576
2b Co2AlH2 1.478 2.234 1.774
3a Co3AlH 1.510 2.539 1.845
3c Co3AlH2 1.597 2.585 1.853
4a Co4AlH 1.595 2.615 2.155
4b Co4AlH2 1.687 2.795 2.030
5a Co5AlH 1.605 2.710 2.672
5c Co5AlH2 1.567 2.765 2.003
6a Co6AlH 1.793 2.909 3.440
6c Co6AlH2 1.894 2.972 2.291
7a Co7AlH 1.627 2.931 3.380
7c Co7AlH2 1.608 3.012 2.106
8a Co8AlH 1.742 2.879 3.385
8c Co8AlH2 1.573 2.691 2.052
9a Co9AlH 1.810 3.061 4.295
9b Co9AlH2 1.850 2.933 2.161
10a Co10AlH 1.203 2.523 3.489
10b Co10AlH2 1.856 3.216 2.207
11a Co11AlH 2.547 3.260 5.015
11b Co11AlH2 1.937 3.303 2.942
12a Co12AlH 2.215 4.272 5.146
12b Co12AlH2 2.169 3.616 2.563
13a Co13AlH 1.693 3.418 4.667
13b Co13AlH2 1.693 3.372 2.041
14a Co14AlH 1.660 2.863 1.690
14b Co14AlH2 1.559 2.762 1.908
15a Co15AlH 1.663 2.833 2.272
15b Co15AlH2 1.679 2.748 1.927
Page 10 of 14 J Nanopart Res (2012) 14:957
123

Similar to ConAl discussed earlier, one H is most
favorable on a bridge site of Co, Co atoms (Fig. 6[5a])
in Co5Al cluster, and it is with a binding energy of only
0.001 eV stronger than that of the threefold hollow
adsorption of Co, Co, Co atoms (Fig. 6[5b]). The BE
(3.837 eV) of H on Co5Al is also one of the smallest
among all the clusters studied. Accordingly, Co5AlH
should have small abundance. Two H prefer the bridge
sites of adjacent Co, Co atoms (Fig. 6[5c]). Isomers
with two H on the threefold hollow site of Co, Co, Co
atoms (Fig. 6[5d]) and two H on the different bridge
sites of Al, Co and Co, Co atoms (Fig. 6[5e]) have
0.177 and 0.207 eV higher energies, respectively.
The BE (3.943 eV) of H on the bridge site of two
Co, Co atoms of Co6Al (Fig. 6[6a]) is the ground state
structure of Co6AlH. The fragmentation energy (see
below) is also small and this gives further support for
the instability of Co6AlH. Accordingly, it may not
have large abundances. And the structure (Fig. 6[6b])
with hydrogen on a hollow site of Co, Co, Co atoms
lies only 0.125 eV high in energy. For two hydrogen
atoms on Co6Al, several configurations are studied.
These include bridge of Co, Co atoms in the neigh-
boring triangle (Fig. 6[6c]), hollow of Co, Co, Co
atoms in the adjacent triangle (Fig. 6[6d]) The calcu-
lated BE’s given in Table 4. The most favorable
adsorption sites of Co6AlH2 are structure 6c. The two
H have a similar configuration as in ConAlH2 (n = 3,
4, 5). The BE for 2H is 6.331 eV and it shows that
interaction between two hydrogen on Co6Al is more
attractive than clusters discussed earlier. This energy
is also higher than the dissociation energy of H2
(4.60 eV). Accordingly, hydrogen is likely to be
dissociated on Co6Al. The distance between two
hydrogens on Co6Al in the lowest energy state is very
long as compared with the bond length of 0.75 A in
H2. Therefore, two hydrogens are also in a dissociated
state on Co6Al.
One hydrogen adsorption of Co7Al is favorable on
the hollow of the Co, Co, Co atoms (Fig. 6[7a]). The
BE (3.834 eV) of H on Co7Al is one of the largest
among all the clusters studied. Isomer with H on the
bridge site of Co, Co atoms (Fig. 6[7b]) is 0.03 eV
higher in energy. The small HOMO–LUMO gap
(0.681 eV) is likely to make further interaction of
hydrogen with this cluster energetically not so favor-
able. To confirm this, some calculations are carried out
on Co7AlH2. Several initial configurations are con-
sidered for two hydrogens. These include H atoms on
the bridge of adjacent Co, Co atoms in the lower part
of the Co7Al cluster (Fig. 6[7c]). This has the lowest
energy. The HOMO–LUMO gap is lower (0.807 eV)
and the addition of one more hydrogen to Co7Al leads
to a gain of 6.504 eV, an increase of more than
2.67 eV in the BE of H as compared with one
hydrogen on Co7Al. The other calculated position for
two hydrogens on Co7Al is one on bridge of two
distant Co atoms (Fig. 6[7d]). The energy difference
of the isomers (Fig. 6[7c, 7d]) is 0.24 eV.
Co8Al has Cs symmetry (Fig. 6[8a]). I can very
roughly decompose this structure into two interacting
entities. That is the structure Co4Al and Co4 are
bridged with Co–Co bonds. I anticipate that, similar to
Co4Al, this cluster would not favor to react with one
hydrogen. Indeed I find that, the BE of H is 4.639 eV
different from Co4Al of 2.107 eV. One H is favorable
on the bridge site of Co, Co atoms of Co8Al
(Fig. 6[8a]). Structure Fig. 6[8b] with H atom on a
hollow site of Co, Co, Co atoms is only 0.02 eV less
stable. Therefore, the interaction depends very sensi-
tively on the electronic and atomic structures of
clusters. Adsorption of two hydrogen is studied on a
few selected sites which included two neighboring
faces with H atoms on the hollow Co, Co, Co atoms
(Fig. 6[8c]), the two H atoms on the hollow sites of
adjacent Co, Co, Co atoms (Fig. 6[8d]), and two
bridge sites of neighboring Co, Co atoms in the Co8Al
cluster (Fig. 6[8e]). We find that the 8c isomer has the
lowest energy. The BE of this isomer is 7.140 eV,
which is larger than the ConAl cluster discussing
earlier.
H adsorption on hollow Co, Co, Co atoms
(Fig. 6[9a]) is the most stable structure of Co9AlH
with a binding energy of 4.476 eV. The ground state
corresponding to Co9AlH2 cluster is the geometry
Fig. 6[9b] with an average Al–Co bond length of
2.550 A, which is the same as Co9AlH. Two H prefer
the hollow sites of adjacent Co, Co, Co atoms. Two H
adsorption on distant hollow site of Co, Co, Co atoms
(Fig. 6[9c]) is a substable structure with a binding
energy of only 0.04 eV less than the ground state.
One H is favorable on the bridge site of Co, Co
atoms of Co10Al (Fig. 6[10a]). Structure Fig. 6[10b]
with two H atoms on a neighboring hollow site of Co,
Co, Co atoms is the ground state corresponding to
Co10AlH2 cluster. The structure with two H atoms on
the bridge of Co, Co atoms of the neighboring
triangles Fig. 6[10c] is 0.576 eV less stable.
J Nanopart Res (2012) 14:957 Page 11 of 14
123
H adsorption on hollow Co, Co, Co atoms of
Co11Al (Fig. 6[11a]) is the most stable structure of
Co11AlH with a binding energy of 5.121 eV. And the
lowest energy structure for Co11AlH2 clusters is the
structure Fig. 6[11b] with two neighboring H atoms
adding on the hollow site of Co, Co, Co atom and the
bridge site of Co, Co atom. Structure Fig. 6[11c] with
two H atoms on a neighboring hollow site of Co, Co,
Co atoms is only 0.044 eV less stable.
Adsorption of single hydrogen on the hollow site of
Co, Co, Co atoms (Fig. 6[12a]) is considered to be the
ground state structure of Co12AlH. The BE (5.165) of
H on Co12Al is the largest among all the clusters
above. However, the HOMO–LUMO gap is small
(0.308 eV). The small HOMO–LUMO gap is likely to
make further interaction of hydrogen with this cluster
energetically so favorable. To confirm this, calcula-
tions on Co12AlH2 are carried out. Several initial
configurations are considered for two hydrogens.
These include two H atoms on the hollow sites of
Co, Co, Co atoms in the adjacent Fig. 6[12b] and
opposite triangle faces Fig. 6[12c] of Co12Al. The
isomer Fig. 6[12b] is 0.058 eV more stable than the
Fig. 6(12c) isomer.
For ConAlH (n = 13–15), the ground states reveal
hollow site H bonding to the Co, Co, Co atoms
(Fig. 6[13a, 14a, 15a]). Hollow site on the neighboring
Co, Co, Co triangles is found for the ground state
structures of ConAlH2 (n = 13–15) (Fig. 6[13b, 14b,
15b]).
Stability and fragmentation behavior
To check the stability of the lowest energy isomers,
vibrational frequencies for selected clusters have been
calculated using the BLYP/DNP level of theory. It is
found that the lowest energy isomers of all kinds of
clusters discussed earlier have all real frequencies and
are, therefore, stable. Figure 7 shows the plot of the BE
of one and two H atoms on ConAl clusters. The BE is
large for H with n = 8, 11, and 12 of ConAlH atoms.
And, for 2H, clusters with n = 5, 8, and 10 have higher
BE’s. The stability of these complexes is further
studied from the fragmentation energies (Table 5).
Channels with Con-1AlHm, Con-2AlHm, or Con-1-
AlHm-1 molecule being one of the fragments have
been studied. It is noted that in all these processes, the
fragmentation energy is the largest for Co12AlH and
therefore, I expect it to be among the most stable
species. Also the fragmentation energy for Co11AlH is
next largest for all these channels, suggesting it to be
other stable cluster.
Bonding nature
To understand the bonding nature of hydrogen on
cobalt aluminum clusters, the bond lengths in both
hydrogenated clusters and pure cobalt aluminum
clusters are discussed. From Table 2, one finds that
the Al–Co bond lengths increase generally as the size
of the cluster increases. The Al–Co, Al–H, and Co–H
bond lengths for the adsorption on ConAl clusters are
in the range of 2.383–2.705, 1.745–1.910, and
1.600–1.810 A, respectively. Thus, one can conclude
that these bond lengths evolve very slowly with cluster
size. In addition, the nearly constant value for Co–H
and Al–H bond lengths on different clusters at specific
adsorption sites suggests the similar nature of bonding
of H in different clusters. From Table 4 and Fig. 6, one
can also see that one hydrogen adsorption on ConAl
(n = 1–6, 8, 10) and two hydrogen adsorption on
ConAl (n = 1–7) take the twofold bridge site as their
most favorable chemisorptions site. While threefold
hollow site is preferred for one hydrogen adsorption
on ConAl (n = 7, 9, 11–15) and two hydrogen
adsorption on ConAl (n = 8–10, 12–15) clusters.
The ground state structure of two hydrogen adsorption
on Co11Al is an exception.
0 2 4 6 8 10 12 14 160
1
2
3
4
5
6
7
8
9
Bin
ding
Ene
rgy
of H
ydro
gen
/eV
n
ConAlH
ConAlH
2
Fig. 7 Binding energies of H (down) and 2H (up) atoms on
ConAl clusters
Page 12 of 14 J Nanopart Res (2012) 14:957
123
Summary
The geometrical structures, stabilities, VIPs, VEAs of
the ConAl and Con?1(n = 1–15) clusters and the
adsorption behaviors, binding energies and fragmen-
tation energies of ConAlHm(n = 1–15, m = 1,2) have
been investigated by the GGA functional at the DNP
level. The results are summarized as follows:
(1) The optimized geometries show that Al atom
substitutes the surface atom of the Con?1 clusters
for n B 13 and it keeps the similar frameworks
of the most stable Con?1 clusters except for
n = 2, 3, and 6. Starting from n = 14, the Al
atom completely falls into the center of the Co-
frame.
(2) The dissociation energy, the second-order
energy differences, and the HOMO–LUMO gaps
of the most stable ConAl clusters indicate that the
magic numbers are n = 7, 9, and 13, corre-
sponding to the high symmetrical structures.
(3) The result on hydrogen interaction with cobalt
aluminum clusters has been presented. Hydrogen
undergoes chemisorptions and interacts strongly
with cobalt aluminum clusters. One hydrogen
adsorption on ConAl (n = 1–6, 8, 10) and two
hydrogen adsorption on ConAl (n = 1–7) take
the twofold bridge site as their most favorable
chemisorptions site. While threefold hollow site
is preferred for one hydrogen adsorption on
ConAl (n = 7, 9, 11–15) and two hydrogen
adsorption on ConAl (n = 8–10, 12–15) clusters.
The ground state structure of two hydrogen
adsorption on Co11Al is exceptional.
(4) There is a slight increase in the mean Al–Co
bond lengths after H adsorption on the lowest
energy sites of the most ConAl clusters. In
addition, the nearly constant value for Co–H and
Al–H bond lengths on different clusters at
specific adsorption sites suggests the similar
nature of bonding of H in different clusters. In
general, the binding energy of H and 2H of
ConAl (n = 1–12) are both found to increase
with the cluster size. And the result shows that
large binding energies of the hydrogen atoms and
large fragmentation energies for Co11AlH and
Co12AlH make these species behaving like
magic clusters.
Acknowledgments This work was financially supported by
the National Natural Science Foundation of China (Grant No.
20603021), Youth Foundation of Shanxi (Grant No.
2007021009), and the Youth Academic Leader of Shanxi.
References
Behm JM, Brugh DJ, Morse MD (1994) Spectroscopic analysis
of the open 3d subshell transition metal aluminides: AlV,
AlCr, and AlCo. J Chem Phys 101(8):6487–6499
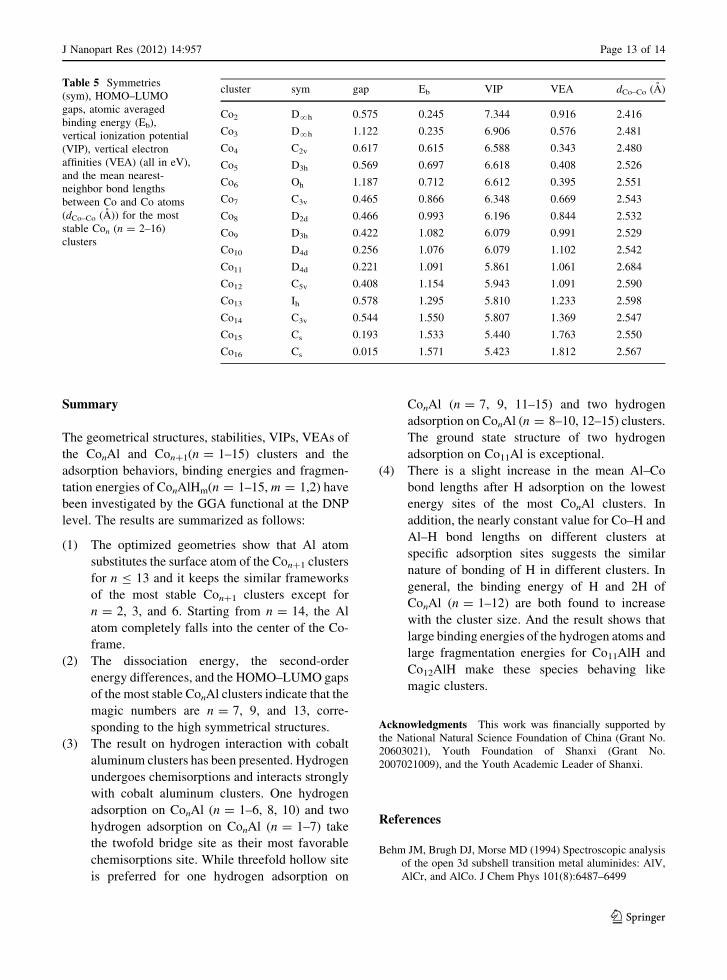
Table 5 Symmetries
(sym), HOMO–LUMO
gaps, atomic averaged
binding energy (Eb),
vertical ionization potential
(VIP), vertical electron
affinities (VEA) (all in eV),
and the mean nearest-
neighbor bond lengths
between Co and Co atoms
(dCo–Co (A)) for the most
stable Con (n = 2–16)
clusters
cluster sym gap Eb VIP VEA dCo–Co (A)
Co2 D?h 0.575 0.245 7.344 0.916 2.416
Co3 D?h 1.122 0.235 6.906 0.576 2.481
Co4 C2v 0.617 0.615 6.588 0.343 2.480
Co5 D3h 0.569 0.697 6.618 0.408 2.526
Co6 Oh 1.187 0.712 6.612 0.395 2.551
Co7 C3v 0.465 0.866 6.348 0.669 2.543
Co8 D2d 0.466 0.993 6.196 0.844 2.532
Co9 D3h 0.422 1.082 6.079 0.991 2.529
Co10 D4d 0.256 1.076 6.079 1.102 2.542
Co11 D4d 0.221 1.091 5.861 1.061 2.684
Co12 C5v 0.408 1.154 5.943 1.091 2.590
Co13 Ih 0.578 1.295 5.810 1.233 2.598
Co14 C3v 0.544 1.550 5.807 1.369 2.547
Co15 Cs 0.193 1.533 5.440 1.763 2.550
Co16 Cs 0.015 1.571 5.423 1.812 2.567
J Nanopart Res (2012) 14:957 Page 13 of 14
123

Delley B (1990) An all-electron numerical method for solving
the local density functional for polyatomic molecules.
J Chem Phys 92(1):508–517
Dhilip Kumar TJ, Tarakeshwar P, Balakrishnan N (2009)
Geometric and electronic structures of hydrogenated tran-
sition metal (Sc, Ti, Zr) clusters. Phys Rev B 79(20):
205415-1-11
Hales DA, Su CX, Lian L, Armentrout BP (1994) Collisionin-
duced dissociation of Con? (n = 2–18) with Xe: bond
energies of cationic and neutral cobalt clusters, dissociation
pathways, and structures. J Chem Phys 100(2):1049–1057
Huda MN, Kleinman L (2006) Hydrogen adsorption and dis-
sociation on small platinum clusters: an electronic structure
density functional study. Phys Rev B 74(19):195407-1-7
Kant A, Strauss B (1964) Dissociation energies of diatomic
molecules of the transition elements. II. Titanium, chro-
mium, manganese, and cobalt. J Chem Phys 41(12):
3806–3808
Laguna A, Lasanta T, Lopez-de-Luzuriaga JM, Monge M,
Naumov P, Olmos ME (2010) Combining aurophilic
interactions and halogen bonding to control the lumines-
cence from bimetallic gold silver clusters. J Am Chem Soc
132(2):456–457
Lee C, Yang W, Parr RG (1988) Development of the Colle-
Salvetti correlation-energy formula into a functional of the
electron density. Phys Rev B 37(2):785–789
Lu C, Kuang XY, Lu ZW, Mao AJ, Ma YM (2011) Determi-
nation of structures, stabilities, and electronic properties
for bimetallic cesium-doped gold clusters: a density func-
tional theory study. J Phys Chem A 115(33):9273–9281
Ma QM, Xie Z, Wang J, Liu Y, Li YC (2006) Structures, sta-
bilities and magnetic properties of small Co clusters. Phys
Lett A 358(4):289–296
Menezes WJC, Knickelbein MB (1991) Bimetallic clusters
of cobalt and aluminum: ionization potentials versus
reactivity, and the importance of geometric structure.
Chem Phys Lett 183(5–6):357–362
Menezes WJC, Knickelbein MB (1993) The evolution of elec-
tronic structure in AlnCom. Z Phys D 26:322–325
Nonose S, Sone Y, Onodera K, Sudo S, Kaya K (1989) Reac-
tivity study of alloy clusters made of aluminum and some
transition metals with hydrogen. Chem Phys Lett 164(4):
427–432
Pramann A, Nakajima A, Kaya K (2001) Photoelectron spec-
troscopy of bimetallic aluminum cobalt cluster anions:
comparison of electronic structure and hydrogen chemi-
sorption rates. J Chem Phys 115(12):5404–5410
Rosen B (1970) Spectroscopic data relative to diatomic mole-
cules. Pergamon, Oxford
Varano A, Henry DJ, Yarovsky I (2010) DFT study of H
adsorption on magnesium-doped aluminum clusters. J Phys
Chem A 114(10):3602–3608
Wang SY, Yu JZ, Mizuseki H, Yan JA (2004) First-principles
study of the electronic structures of icosahedral
TiN(N = 13,19,43,55) clusters. J Chem Phys 120(18):
8463–8468
Xie Z, Ma QM, Liu Y, Li YC (2005) First-principles study of the
stability and Jahn-Teller distortion of nickel clusters. Phys
Lett A 342(5):459–467
Zanti G, Peeters D (2010) DFT study of bimetallic palladium
gold clusters PdnAum of low nuclearities (n ? m \14).
J Phys Chem A 114(38):10345–10356
Zhang DB, Shen J (2004) Ground state, growth, and electronic
properties of small lanthanum clusters. J Chem Phys
120(11):5104–5109
Zhao YR, Kuang XY, Zheng BB, Li YF, Wang SJ (2011)
Equilibrium geometries, stabilities, and electronic proper-
ties of the bimetallic M2-doped Aun (M = Ag, Cu;
n = 1–10) clusters: comparison with Pure Gold clusters.
J Phys Chem A 115(5):569–576
Page 14 of 14 J Nanopart Res (2012) 14:957
123
![Gui-Xian Ge Hong-Xia Yan Qun Jing...metals. Axel Pramann et al. [22] studied hydrogen chemisorption rates and electronic structures of small Nb nAl-clusters by photoelectron spectroscopic](https://img.pdfslide.net/doc/110x75/60d56d38b748071e692c180a/gui-xian-ge-hong-xia-yan-qun-jing-metals-axel-pramann-et-al-22-studied-hydrogen.jpg)




![MODELLING OF CESIUM CHEMISORPTION UNDER NUCLEAR … 2019 - Final... · chemisorption procesimprovement of s, the Cs chemisorption model Nakajima et alby . [17] is then described](https://img.pdfslide.net/doc/110x75/6079e6b83b443d67370b949a/modelling-of-cesium-chemisorption-under-nuclear-2019-final-chemisorption.jpg)