Embed Size (px)
Citation preview

Exceptional Activity of a Pt–Rh–Ni Ternary NanostructuredCatalyst for the Electrochemical Oxidation of EthanolNina Erini,[a] Stefan Rudi,[a] Vera Beermann,[a] Paul Krause,[a] Ruizhi Yang,[b] Yunhui Huang,[c]
and Peter Strasser*[a, d]
1. Introduction
Direct ethanol fuel cells (DEFCs) have been a subject of inten-sive studies for fundamental and practical applications as alter-nate power sources (e.g. the development of compact porta-ble power as well as electric-vehicle range extenders operatingat high temperatures).[1] As a fuel, ethanol has high volumetricand gravimetric energy densities (8 kWh kg�1), good energy ef-ficiencies, and offers easy handling, storage, and transporta-tion, contrary to gaseous fuels.[2] Its low toxicity, biocompatibil-ity, and abundant availability in combination with its availabili-ty from renewable resources make ethanol an almost idealfuel. Its oxidation, however, is often incomplete, resulting ina number of by-products rather than CO2. This is attributed todifficulties with C�C bond cleavage in ethanol and, to someextent, to the formation of CO intermediates, leading to thepoisoning of the active sites on Pt catalysts.[3] Complete etha-nol electrooxidation to CO2 involves 12 electrons per molecule,whereas partial oxidation leads to by-products such as acetic
acid or acetaldehyde, which reduce the faradaic efficiency ofthe anodic reaction of DEFCs. Efforts to develop highly activeand selective electrocatalysts for the ethanol oxidation reaction(EOR) to CO2 have, therefore, concentrated on the addition ofco-catalysts to platinum.[4] Alloying Pt with highly oxophilictransition metals such as Rh, Ni, or Sn has been a promisingstrategy to modify the electrocatalytic surface properties of Ptin order to supply active oxygen-containing species, such asOH, that readily oxidize adsorbed molecular fragments whilereducing the cost of the catalyst considerably.[3a, 5] To achievethis goal, the oxophilic component is assumed to be presentas an oxide species at the surface of the operating catalyst.
Our past research focused on a promising family of EORnanocatalysts based on mixtures of Pt, Rh, and Sn.[3d, 6] In par-ticular, we uncovered and characterized the optimal 3D struc-tural arrangement of the atoms of the three components atthe surface and bulk of the final active catalyst to maximize ac-tivity and selectivity, finding a single-phase Rh-doped PtSnbulk Niggliite structure as the preferred and most catalyticallyactive nanocrystalline phase. On its surface, metallic Pt and Rhwere atomically mixed with a SnO2 phase, representing theactive surface-site ensembles. Although the ternary ensembleof the three metals led to high oxidation activities, it is alsoknown that nanocrystalline Pt and its Ni-based binary alloybelong to the most active catalysts for the catalytic oxidationof organic molecules.[7] We, therefore, developed the hypothe-sis that the partial random substitution of Pt with Rh in Pt–Nibulk alloys could lead to active EOR electrocatalysts, owing tothe formation of unique Pt–Rh–NiOx surface ensembles.
Mechanistic details of the Rh and Ni-promoted oxidation ofethanol on Pt are still not well understood. It is believed thatRh increases the yield of CO2 by promoting C�C bond cleav-age. However, there is reason to believe that this hypothesisdoes not represent a comprehensive picture of the role of Rh
[a] N. Erini, S. Rudi, V. Beermann, P. Krause, Prof. P. StrasserDepartment of Chemical EngineeringTechnical University of BerlinStraße des 17. Juni 124, 10623 Berlin (Germany)E-mail : [email protected]
[b] Prof. R. YangSchool of Energy, Soochow UniversityNo. 1 Shizi Street, Suzhou, Jiangsu 215006 (China)
[c] Prof. Y. HuangSchool of Materials Science and EngineeringHuazhong University of Science and Technology1037 Luoyu Road, Wuhan, 430074 (China)
[d] Prof. P. StrasserErtl Center for Electrochemistry and CatalysisGwangju Institute of Science and Technology500-712 Gwangju (South Korea)
Supporting Information for this article is available on the WWW underhttp://dx.doi.org/10.1002/celc.201402390.
Alloying Pt with highly oxophilic transition metals such as Rh,Ni, or Sn has been a promising strategy to modify the electro-catalytic surface properties of Pt in order to supply activeoxygen-containing species for ethanol electrooxidation. A new,highly active, ternary single-phased cubic PtRhNi/C nanoparti-cle electrocatalyst for the electrocatalytic oxidation of ethanol(EOR) is reported and its morphology (XRD and TEM), composi-tion (inductively coupled plasma optical emission spectrosco-
py), and electrochemical activity are discussed in comparisonwith the state-of-art PtRhSn/C electrocatalyst. The EOR activityof the PtRhNi/C material outperformed the benchmark PtRhSn/C material in acidic and alkaline media, showing high stability,especially in alkaline media. The higher intrinsic EOR activity ofthe Ni-containing electrocatalyst lends support to the notionthat surface NiOx is an excellent oxygenate-supplying catalystcomponent for the oxidation of ethanol.
ChemElectroChem 0000, 00, 0 – 0 � 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim1
These are not the final page numbers! ��These are not the final page numbers! ��
ArticlesDOI: 10.1002/celc.201402390
1 12 23 34 45 56 67 78 89 9
10 1011 1112 1213 1314 1415 1516 1617 1718 1819 1920 2021 2122 2223 2324 2425 2526 2627 2728 2829 2930 3031 3132 3233 3334 3435 3536 3637 3738 3839 3940 4041 4142 4243 4344 4445 4546 4647 4748 4849 4950 5051 5152 5253 5354 5455 5556 5657 57
in this family of catalysts. According to the bifunctional mecha-nism, the synergistic effect results from the fact that hydroussurface alpha-Ni(OH)x activates water molecules and providespreferential sites for OH species (H2O!OHads + H+ + e�) atlower potentials than Pt. Abundant�OHads species are necessa-ry to completely oxidize the CHx or CO surface fragments toCO2 (COads + OHads!CO2 + H+ + e�), avoiding electrode poison-ing.[8] Beyhan et al. studied the catalytic effect of Ni and Rhadded to a PtSn catalyst and the influence on the cleavage ofethanol with in situ FTIR spectroscopy, finding it to be morebeneficial for complete oxidation compared to the addition ofPd or Co.[3a] Soundararajan et al. synthesized a set of Pt andPt–Ni alloy nanoparticle catalysts through electrochemical dep-osition, showing that the Pt–Ni alloy formation leads to higheractivities in alkaline media for EOR.[9] Bonesi et al. investigatedhigh-surface-area carbon-supported binary PtSn and ternaryPtSnNi and PtSnRh catalysts for the EOR at elevated tempera-tures, concluding that the ternary catalysts show elevated ac-tivities.[10]
Assuming that Rh indeed facilitates the bond breaking, withSn or Ni being the responsible oxygen species provider, a com-bination of Pt, Rh, and Ni should result in an even higheractive EOR catalyst, but, to the best of our knowledge, therehave been no reports on ternary Pt–Rh–Ni catalyst systems forthe EOR to date. This is why, in this report, we prepared terna-ry single-phase PtRhNi/C nanoparticle catalysts and bench-marked their electrocatalytic activity and stability for the EORto the corresponding state-of-art Sn-containing electrocatalyst.We compared and contrasted their structure, morphology, andcatalytic performance and conclude that Ni offers great catalyt-ic performance benefits over Sn. We are confident that thisstudy will spark more research on the use of Ni in the oxida-tion of small organic molecules.
2. Results and Discussion
Two ternary electrocatalysts PtRhSn/C and PtRhNi/C were suc-cessfully synthesized and studied to investigate the influenceof Ni and Sn as oxygen-donating species for the EOR. Theatomic ratios of the as synthesized samples were characterizedby using inductively coupled plasma optical emission spectros-copy (ICP-OES), and the data are presented in Table 1. Both
catalysts were deliberately designed to have <10 % atomicmolar ratios in Rh with a molar Pt ratio of around 30 %, where-as the oxygen-donating species of Sn/Ni were designed tohave a molar ratio that was almost double that of Pt. Theweight loading of both catalysts was designed to be very simi-
lar. Additional experiments showed that the catalyst per-formance was insensitive to changes in the Rh content in the5–12 % range.
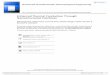
TEM images of the electrocatalysts (Figure 1) showed thatthe nanoparticles were well distributed across the carbon sup-port, with sizes ranging from 6 to 12 nm. Mean particle diame-ters were estimated from TEM-derived size distribution histo-grams and are inserted in the TEM graphs in Figure 1. PtRhNi/C nanoparticles showed an irregular, close to spherical shape,most likely because of the agglomeration of 2–3 particlesduring the synthesis. PtRhSn/C nanoparticles are spherical witha broad size range of 2–18 nm.
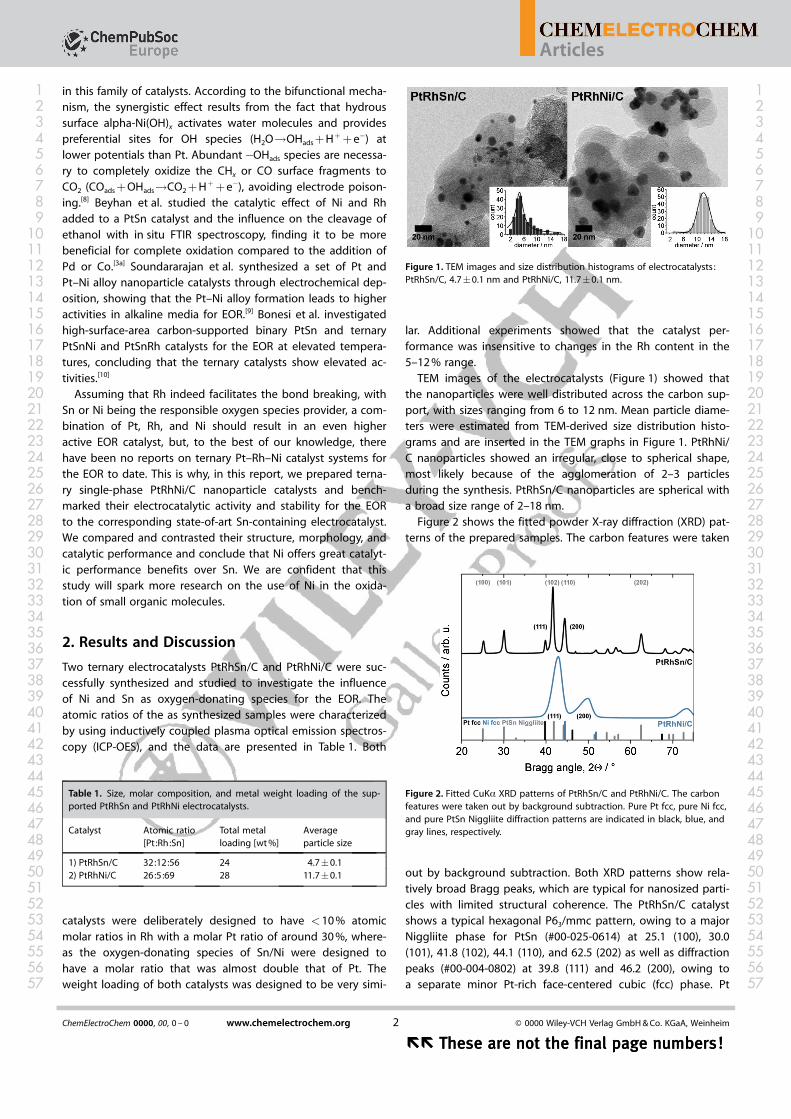
Figure 2 shows the fitted powder X-ray diffraction (XRD) pat-terns of the prepared samples. The carbon features were taken
out by background subtraction. Both XRD patterns show rela-tively broad Bragg peaks, which are typical for nanosized parti-cles with limited structural coherence. The PtRhSn/C catalystshows a typical hexagonal P63/mmc pattern, owing to a majorNiggliite phase for PtSn (#00-025-0614) at 25.1 (100), 30.0(101), 41.8 (102), 44.1 (110), and 62.5 (202) as well as diffractionpeaks (#00-004-0802) at 39.8 (111) and 46.2 (200), owing toa separate minor Pt-rich face-centered cubic (fcc) phase. Pt
Table 1. Size, molar composition, and metal weight loading of the sup-ported PtRhSn and PtRhNi electrocatalysts.
Catalyst Atomic ratio[Pt:Rh:Sn]
Total metalloading [wt %]
Averageparticle size
1) PtRhSn/C 32:12:56 24 4.7�0.12) PtRhNi/C 26:5:69 28 11.7�0.1
Figure 1. TEM images and size distribution histograms of electrocatalysts :PtRhSn/C, 4.7�0.1 nm and PtRhNi/C, 11.7�0.1 nm.
Figure 2. Fitted CuKa XRD patterns of PtRhSn/C and PtRhNi/C. The carbonfeatures were taken out by background subtraction. Pure Pt fcc, pure Ni fcc,and pure PtSn Niggliite diffraction patterns are indicated in black, blue, andgray lines, respectively.
ChemElectroChem 0000, 00, 0 – 0 www.chemelectrochem.org � 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim2
�� These are not the final page numbers!�� These are not the final page numbers!
Articles
1 12 23 34 45 56 67 78 89 9
10 1011 1112 1213 1314 1415 1516 1617 1718 1819 1920 2021 2122 2223 2324 2425 2526 2627 2728 2829 2930 3031 3132 3233 3334 3435 3536 3637 3738 3839 3940 4041 4142 4243 4344 4445 4546 4647 4748 4849 4950 5051 5152 5253 5354 5455 5556 5657 57
and Rh diffraction peaks cannot be resolved in XRD patterns,because they form a homogenous PtRh binary solid solutionover wide compositional ranges, as indicated by the Pt–Rhphase diagram (not shown here). Earlier studies suggested thatRh incorporates randomly into the Niggliite lattice, substitutingPt atoms.[6a, b] PtRhNi/C shows a single-phase fcc diffraction pat-tern; the diffraction peaks lay, in accordance with the atomicmixture of the three metals, between the reflexes of Pt, R,hand Ni. No reflexes for pure metallic Pt, Rh, or Ni can be ob-served, indicating alloy formation of all three metals. However,owing to the slightly non-symmetric diffraction peaks of thePtRhNi/C catalyst, the existence of a second fcc phase cannotbe completely excluded.
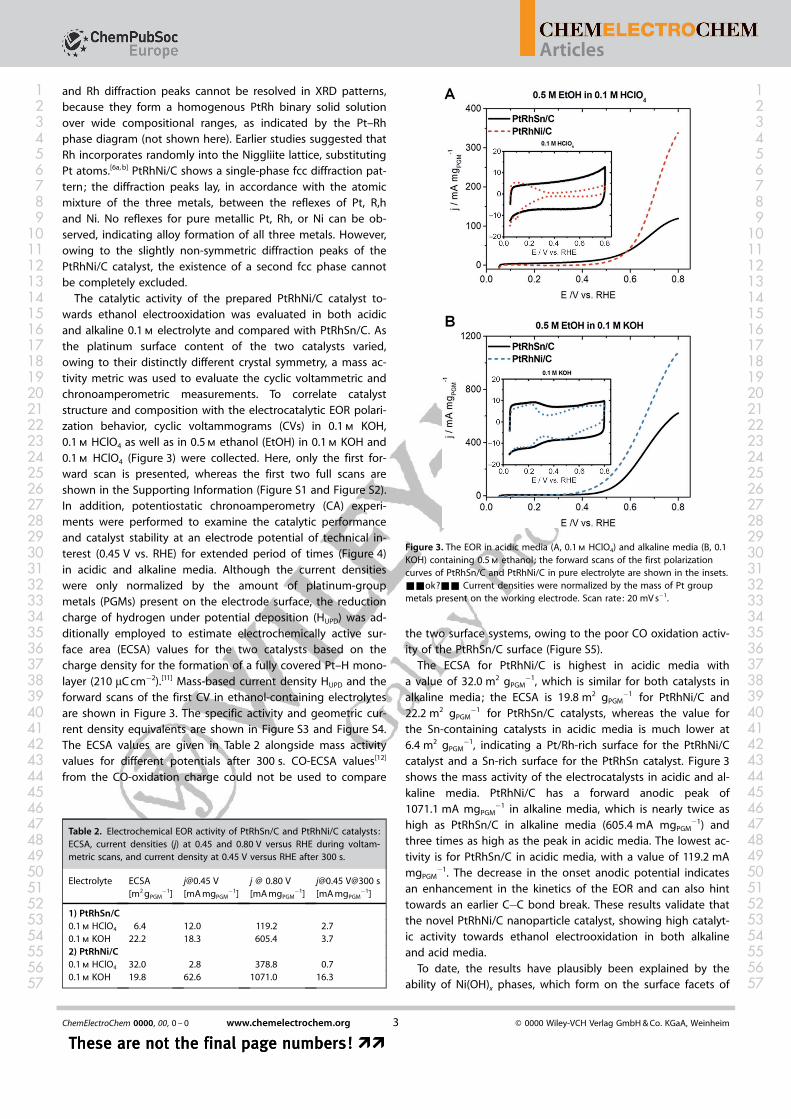
The catalytic activity of the prepared PtRhNi/C catalyst to-wards ethanol electrooxidation was evaluated in both acidicand alkaline 0.1 m electrolyte and compared with PtRhSn/C. Asthe platinum surface content of the two catalysts varied,owing to their distinctly different crystal symmetry, a mass ac-tivity metric was used to evaluate the cyclic voltammetric andchronoamperometric measurements. To correlate catalyststructure and composition with the electrocatalytic EOR polari-zation behavior, cyclic voltammograms (CVs) in 0.1 m KOH,0.1 m HClO4 as well as in 0.5 m ethanol (EtOH) in 0.1 m KOH and0.1 m HClO4 (Figure 3) were collected. Here, only the first for-ward scan is presented, whereas the first two full scans areshown in the Supporting Information (Figure S1 and Figure S2).In addition, potentiostatic chronoamperometry (CA) experi-ments were performed to examine the catalytic performanceand catalyst stability at an electrode potential of technical in-terest (0.45 V vs. RHE) for extended period of times (Figure 4)in acidic and alkaline media. Although the current densitieswere only normalized by the amount of platinum-groupmetals (PGMs) present on the electrode surface, the reductioncharge of hydrogen under potential deposition (HUPD) was ad-ditionally employed to estimate electrochemically active sur-face area (ECSA) values for the two catalysts based on thecharge density for the formation of a fully covered Pt–H mono-layer (210 mC cm�2).[11] Mass-based current density HUPD and theforward scans of the first CV in ethanol-containing electrolytesare shown in Figure 3. The specific activity and geometric cur-rent density equivalents are shown in Figure S3 and Figure S4.The ECSA values are given in Table 2 alongside mass activityvalues for different potentials after 300 s. CO-ECSA values[12]
from the CO-oxidation charge could not be used to compare
the two surface systems, owing to the poor CO oxidation activ-ity of the PtRhSn/C surface (Figure S5).
The ECSA for PtRhNi/C is highest in acidic media witha value of 32.0 m2 gPGM
�1, which is similar for both catalysts inalkaline media; the ECSA is 19.8 m2 gPGM
�1 for PtRhNi/C and22.2 m2 gPGM
�1 for PtRhSn/C catalysts, whereas the value forthe Sn-containing catalysts in acidic media is much lower at6.4 m2 gPGM
�1, indicating a Pt/Rh-rich surface for the PtRhNi/Ccatalyst and a Sn-rich surface for the PtRhSn catalyst. Figure 3shows the mass activity of the electrocatalysts in acidic and al-kaline media. PtRhNi/C has a forward anodic peak of1071.1 mA mgPGM
�1 in alkaline media, which is nearly twice ashigh as PtRhSn/C in alkaline media (605.4 mA mgPGM
�1) andthree times as high as the peak in acidic media. The lowest ac-tivity is for PtRhSn/C in acidic media, with a value of 119.2 mAmgPGM
�1. The decrease in the onset anodic potential indicatesan enhancement in the kinetics of the EOR and can also hinttowards an earlier C�C bond break. These results validate thatthe novel PtRhNi/C nanoparticle catalyst, showing high catalyt-ic activity towards ethanol electrooxidation in both alkalineand acid media.
To date, the results have plausibly been explained by theability of Ni(OH)x phases, which form on the surface facets of
Figure 3. The EOR in acidic media (A, 0.1 m HClO4) and alkaline media (B, 0.1KOH) containing 0.5 m ethanol; the forward scans of the first polarizationcurves of PtRhSn/C and PtRhNi/C in pure electrolyte are shown in the insets.&&ok?&& Current densities were normalized by the mass of Pt groupmetals present on the working electrode. Scan rate: 20 mV s�1.
Table 2. Electrochemical EOR activity of PtRhSn/C and PtRhNi/C catalysts:ECSA, current densities (j) at 0.45 and 0.80 V versus RHE during voltam-metric scans, and current density at 0.45 V versus RHE after 300 s.
Electrolyte ECSA[m2 gPGM
�1][email protected] V[mA mgPGM
�1]j @ 0.80 V[mA mgPGM
�1][email protected] V@300 s[mA mgPGM
�1]
1) PtRhSn/C0.1 m HClO4 6.4 12.0 119.2 2.70.1 m KOH 22.2 18.3 605.4 3.72) PtRhNi/C0.1 m HClO4 32.0 2.8 378.8 0.70.1 m KOH 19.8 62.6 1071.0 16.3
ChemElectroChem 0000, 00, 0 – 0 www.chemelectrochem.org � 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim3
These are not the final page numbers! ��These are not the final page numbers! ��
Articles
1 12 23 34 45 56 67 78 89 9
10 1011 1112 1213 1314 1415 1516 1617 1718 1819 1920 2021 2122 2223 2324 2425 2526 2627 2728 2829 2930 3031 3132 3233 3334 3435 3536 3637 3738 3839 3940 4041 4142 4243 4344 4445 4546 4647 4748 4849 4950 5051 5152 5253 5354 5455 5556 5657 57
Ni-containing bulk phases, to act as efficient oxygen-donatingentities.[7a, 13] Subbaraman et al. already demonstrated that theaddition of Ni(OH)2 to a platinum electrode surface manifestsa significant increase in hydrogen evolution reaction (HER) ac-tivity, owing to a bifunctional effect, where edges of Ni(OH)2
clusters promote the dissociation of water and the formationof intermediates, which can then adsorb on the nearby Pt sur-faces.[14] Consistent with this explanation is the reduced EORactivity at 0.45 V versus RHE in acid media, where Ni(OH)x
phases gradually leach from the top surface; in contrast, theEOR performance in alkaline media is greatly enhanced be-cause of the stability of Ni(OH)x phases under these conditions.The superior EOR activity of the PtRhNi catalyst at high overpo-tentials (+ 0.80 V vs. RHE) can be explained by the enhancedpresence of surface active oxygenates, such as OH, which helpsegregate Ni(OH)x to the surface solutions.[12, 13c, 15] Although thegeneral beneficial effect of Rh for C�C bond splitting has beenwell documented,[13c, 16] the comparison of PtRhSn/C andPtRhNi/C shows that the oxygen-donating effect of Ni versusSn appears to be enhanced and, thus, leads to higher activitiesfor the Ni-containing systems. The PtRhSn/C catalyst displayedthree times higher current densities at + 0.80 V in acidic media
than a similar PtRhSnO2/C catalyst[6f] and an earlier reportedPtRhSn catalyst with the same 3:1:4 ratio.[6c] Brouzgou et al.compared a variety of DEFC anode catalysts with increasing Ptloadings on the working electrode surfaces and establisheda certain region between 0.1–1.0 mA cmPt
[email protected] V versus RHEin which most of the reported catalyst to date belonged.[17]
PtRhSn/C already lies within the region (0.10 mA cmPt2) and
PtRhNi/C outperforms it (0.21 mA cmPt2), as does a previously
reported PtRhSnO2/C catalyst from Kowal et al. (0.19 mAcmPt
2).[3d] The PtRhNi/C catalyst also outperformed a binaryPtNi EOR electrocatalyst in alkaline media reported by Soun-darajan et al.[9]
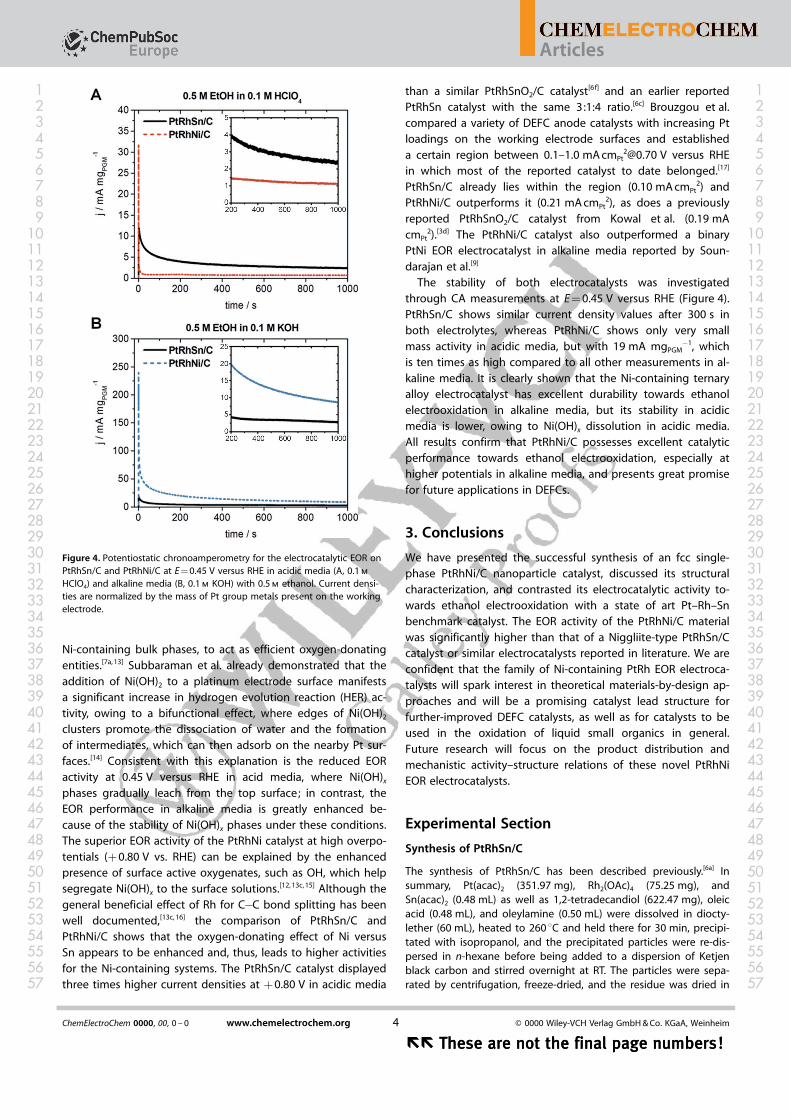
The stability of both electrocatalysts was investigatedthrough CA measurements at E = 0.45 V versus RHE (Figure 4).PtRhSn/C shows similar current density values after 300 s inboth electrolytes, whereas PtRhNi/C shows only very smallmass activity in acidic media, but with 19 mA mgPGM
�1, whichis ten times as high compared to all other measurements in al-kaline media. It is clearly shown that the Ni-containing ternaryalloy electrocatalyst has excellent durability towards ethanolelectrooxidation in alkaline media, but its stability in acidicmedia is lower, owing to Ni(OH)x dissolution in acidic media.All results confirm that PtRhNi/C possesses excellent catalyticperformance towards ethanol electrooxidation, especially athigher potentials in alkaline media, and presents great promisefor future applications in DEFCs.
3. Conclusions
We have presented the successful synthesis of an fcc single-phase PtRhNi/C nanoparticle catalyst, discussed its structuralcharacterization, and contrasted its electrocatalytic activity to-wards ethanol electrooxidation with a state of art Pt–Rh–Snbenchmark catalyst. The EOR activity of the PtRhNi/C materialwas significantly higher than that of a Niggliite-type PtRhSn/Ccatalyst or similar electrocatalysts reported in literature. We areconfident that the family of Ni-containing PtRh EOR electroca-talysts will spark interest in theoretical materials-by-design ap-proaches and will be a promising catalyst lead structure forfurther-improved DEFC catalysts, as well as for catalysts to beused in the oxidation of liquid small organics in general.Future research will focus on the product distribution andmechanistic activity–structure relations of these novel PtRhNiEOR electrocatalysts.
Experimental Section
Synthesis of PtRhSn/C
The synthesis of PtRhSn/C has been described previously.[6a] Insummary, Pt(acac)2 (351.97 mg), Rh2(OAc)4 (75.25 mg), andSn(acac)2 (0.48 mL) as well as 1,2-tetradecandiol (622.47 mg), oleicacid (0.48 mL), and oleylamine (0.50 mL) were dissolved in diocty-lether (60 mL), heated to 260 8C and held there for 30 min, precipi-tated with isopropanol, and the precipitated particles were re-dis-persed in n-hexane before being added to a dispersion of Ketjenblack carbon and stirred overnight at RT. The particles were sepa-rated by centrifugation, freeze-dried, and the residue was dried in
Figure 4. Potentiostatic chronoamperometry for the electrocatalytic EOR onPtRhSn/C and PtRhNi/C at E = 0.45 V versus RHE in acidic media (A, 0.1 m
HClO4) and alkaline media (B, 0.1 m KOH) with 0.5 m ethanol. Current densi-ties are normalized by the mass of Pt group metals present on the workingelectrode.
ChemElectroChem 0000, 00, 0 – 0 www.chemelectrochem.org � 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim4
�� These are not the final page numbers!�� These are not the final page numbers!
Articles
1 12 23 34 45 56 67 78 89 9
10 1011 1112 1213 1314 1415 1516 1617 1718 1819 1920 2021 2122 2223 2324 2425 2526 2627 2728 2829 2930 3031 3132 3233 3334 3435 3536 3637 3738 3839 3940 4041 4142 4243 4344 4445 4546 4647 4748 4849 4950 5051 5152 5253 5354 5455 5556 5657 57

a furnace under N2, oxidized under O2/N2, and calcined by heatingunder a H2/Ar atmosphere (see detailed information in previouspublication[12]).
Synthesis of PtRhNi/C
To prepare Pt26Rh5Ni69 alloy core–shell nanoparticles Ni(OAc)2·4 H2O(0.9 mmol), 1,2-tetradecanediol (1.2 mmol), oleylamine (0.3 mL),and oleic acid (0.3 mL) were dispersed in dibenzyl (50 mL) undera nitrogen atmosphere, stirred for 5 min at 80 8C, and heated to210 8C. After subsequent 30 min time periods, Pt(acac)2 (0.3 mmol),then Rh2(OAc)4 (0.03 mmol), and finally Pt(acac)2 (0.1351 mmol)were added; each was dissolved in 1,2-dichlorbenzene (3 mL).After a further 30 min, the reaction mixture was cooled rapidly andthe nanoparticles were precipitated with mixture of dichlorome-thane (5 mL) and ethanol (20 mL). Vulcan XC 72R (278.1 mg) wasdispersed in toluene ( 60 mL) and subjected to ultrasonication for5 min; then, pure ethanol (10 mL) was added, followed by the re-action mixture, and everything was stirred for 24 h (at RT). The par-ticles were separated by centrifugation, washed 3–4 times withethanol, freeze-dried, and the residue was heated in a furnaceunder an air atmosphere to 180 8C, at which it was held for 1 h toremove surfactants before being placed in a N2 atmosphere for30 min to remove any oxygen. Then, a H2/Ar (4 vol % H2) gas mix-ture was fed for 30 min at 180 8C to remove N2 before the temper-ature was raised to 240 8C to reduce any possible oxidized Ni onthe surface and to enhance the atomic mixing of Ni and Pt.
Physicochemical Characterization of Catalysts
The characterization and electrochemical testing protocol forPtRhSn/C has been described in detail elsewhere[6a] and is similarto that for PtRhNi/C. ICP-OES was used for compositional analysis,and this was performed by using a 715-ES inductively coupledplasma analysis system (Varian). The standard concentrations were2, 4.5, and 7.5 ppm for Pt and Ni; 3, 10, and 20 ppm for Sn; and0.7, 2.8, and 5.6 ppm for Rh. The chosen wavelengths for concen-tration determination were 203.646, 214.424, 265.495, and306.471 nm for Pt; 216.555, 221.648, 222.295, 222.486, 227.021,230.299, and 231.604 nm for Ni; 233.477, 246.103, 249.078,343.488, and 369.236 nm for Rh; and 189.9 nm for Sn. Transmissionelectron microscopy (TEM) and energy-dispersive X-ray (EDX) spec-troscopy were used to study the morphology and composition, re-spectively. A Cu grid (200 mesh) coated with a holey carbon filmwas impregnated with the sample solution and air-dried at 60 8C. AFEI TECNAI G2 20 S-TWIN microscope, equipped with a GATANMS794 P CCD detector and operated at 200 kV, was used. Themean particle size was determined from the TEM images by count-ing at least 200 particles. EDX data were collected for 120 s at anangle of 458 with respect to the sample holder. CuKa X-ray diffrac-tion (XRD) patterns were collected by using a D8 Advance diffrac-tometer (Bruker) equipped with a Lynx Eye Detector and KFL Cu2 K X-ray tube. The diffraction patterns were collected in a 20–8082q range with a step size of 0.001428 dwelling for 30 s for PtRhSn/C, and a step size of 0.058 dwelling for 10 s for PtRhNi/C at eachstep. The XRD patterns were analyzed by using the MDI Jade 8software package. Bragg reflex positions were compared with thereference XRD patterns (PDF data files, International Center for Dif-fraction Data). Electrochemical measurements were performed byusing polished glassy carbon working electrodes, which werecoated with 10 mL of the catalyst ink (PtRhSn/C: 6 mg electrocata-lyst powder, 2.0 mL ultrapure water, 0.5 mL isopropanol, 20 mL of5 wt % Nafion solution; PtRhNi/C: 8 mg electrocatalyst powder,
3.98 mL ultrapure water, 1.0 mL isopropanol, 20 mL of 5 wt %Nafion solution) and measured with a Biologic SP 150 potentiostatin an electrochemical glass cell with Pt wire as the counter elec-trode and saturated MMS (in HClO4) or saturated SCE (in KOH) asthe reference electrode. KOH and HClO4 electrolytes were freshlymade and purged with nitrogen prior to and during experiments.
Acknowledgements
The project on which this report is based was promoted withfunds from the Federal Ministry of Education under the promo-tional reference number 16N11929. Responsibility for the contentof this publication lies with the authors. We thank Annette Witte-brock for electrochemical measurements and Sçren Selve for TEMmeasurements. Partial financial support from the German Re-search Foundation (DFG) through grant STR 596/5–1 (“Shaped PtBimetallics”) is gratefully acknowledged.
Keywords: ethanol oxidation reaction · fuel cells · nickel ·platinum · rhodium
[1] a) E. Antolini, J. Power Sources 2007, 170, 1 – 12; b) H. Wang, Z. Jusys,R. J. Behm, Journal of Physical Chemistry B 2004, 108, 19413 – 19424;c) J. P. I. de Souza, S. L. Queiroz, K. Bergamaski, E. R. Gonzalez, F. C. Nart,The Journal of Physical Chemistry B 2002, 106, 9825 – 9830; d) J. J. Li-nares, T. A. Rocha, S. Zignani, V. A. Paganin, E. R. Gonzalez, Int. J. Hydro-gen Energy 2013, 38, 620 – 630.
[2] W. Zhou, Z. Zhou, S. Song, W. Li, G. Sun, P. Tsiakaras, Q. Xin, Applied Cat-alysis, B: Environmental 2003, 46, 273 – 285.
[3] a) S. Beyhan, J.-M. L�ger, F. Kadırgan, Appl. Catal. , B 2014, 144, 66 – 74;b) H. Hitmi, E. M. Belgsir, J. M. Leger, C. Lamy, R. O. Lezna, Electrochim.Acta 1994, 39, 407 – 415; c) T. Iwasita, E. Pastor, Electrochim. Acta 1994,39, 531 – 537; d) A. Kowal, M. Li, M. Shao, K. Sasaki, M. B. Vukmirovic, J.Zhang, N. S. Marinkovic, P. Liu, A. I. Frenkel, R. R. Adzic, Nat. Mater. 2009,8, 325 – 330; e) F. Colmati, E. Antolini, E. R. Gonzalez, Applied Catalysis B-Environmental 2007, 73, 106 – 115.
[4] a) S. Song, P. Tsiakaras, Applied Catalysis, B: Environmental 2006, 63,187 – 193; b) M. Oezaslan, F. Hasch�, P. Strasser, J. Electrochem. Soc. ,2012, 159, B444-B454; c) F. Hasch�, M. Oezaslan, P. Strasser, ChemCatCh-em 2011, 3, 1805 – 1813; d) M. Oezaslan, F. Hasch�, P. Strasser, J. Electro-chem. Soc. , 2012, 159, B394-B405; e) L. Gan, M. Heggen, R. O’Malley, B.Theobald, P. Strasser, Nano Lett. 2013, 13, 1131 – 1138.
[5] C.-T. Hsieh, J.-Y. Lin, J.-L. Wei, Int. J. Hydrogen Energy 2009, 34, 685 – 693.[6] a) N. Erini, R. Loukrakpam, V. Petkov, E. A. Baranova, R. Yang, D. Teschner,
Y. Huang, S. R. Brankovic, P. Strasser, ACS Catal. 2014, 4, 1859 – 1867;b) F. Colmati, E. Antolini, E. R. Gonzalez, J. Alloys Compd. 2008, 456,264 – 270; c) M. Li, A. Kowal, K. Sasaki, N. Marinkovic, D. Su, E. Korach, P.Liu, R. R. Adzic, Electrochim. Acta 2010, 55, 4331 – 4338; d) J. C. M. Silva,L. S. Parreira, R. F. B. De Souza, M. L. Calegaro, E. V. Spinac�, A. O. Neto,M. C. Santos, Appl. Catal. , B 2011, 110, 141 – 147; e) E. V. Spinac�, R. R.Dias, M. Brandalise, M. Linardi, A. O. Neto, Ionics 2010, 16, 91 – 95; f) A.Kowal, S. L. Gojkovic, K. S. Lee, P. Olszewski, Y. E. Sung, Electrochem.Commun. 2009, 11, 724 – 727; g) S. Garc�a-Rodr�guez, S. Rojas, M. A.PeÇa, J. L. G. Fierro, S. Baranton, J. M. L�ger, Applied Catalysis, B: Environ-mental 2011, 106, 520 – 528.
[7] a) C. H. Cui, L. Gan, M. Heggen, S. Rudi, P. Strasser, Nat. Mater. 2013, 12,765 – 771; b) F. Hasch�, M. Oezaslan, P. Strasser, J. Electrochem. Soc. ,2012, 159, B25-B34; c) S. Rudi, X. Tuaev, P. Strasser, Electrocatalysis 2012,3, 265 – 273.
[8] C.-T. Hsieh, J.-Y. Lin, J. Power Sources 2009, 188, 347 – 352.[9] D. Soundararajan, J. H. Park, K. H. Kim, J. M. Ko, Current Applied Physics
2012, 12, 854 – 859.[10] A. Bonesi, G. Garaventa, W. E. Triaca, A. M. Castro Luna, Int. J. Hydrogen
Energy 2008, 33, 3499 – 3501.
ChemElectroChem 0000, 00, 0 – 0 www.chemelectrochem.org � 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim5
These are not the final page numbers! ��These are not the final page numbers! ��
Articles
1 12 23 34 45 56 67 78 89 9
10 1011 1112 1213 1314 1415 1516 1617 1718 1819 1920 2021 2122 2223 2324 2425 2526 2627 2728 2829 2930 3031 3132 3233 3334 3435 3536 3637 3738 3839 3940 4041 4142 4243 4344 4445 4546 4647 4748 4849 4950 5051 5152 5253 5354 5455 5556 5657 57

[11] M. Shao, J. H. Odell, S.-I. Choi, Y. Xia, Electrochem. Commun. 2013, 31,46 – 48.
[12] S. Rudi, C. Cui, L. Gan, P. Strasser, Electrocatalysis 2014, 5, 408 – 418.[13] a) P. B. Balbuena, D. Altomare, N. Vadlamani, S. Bingi, L. A. Agapito, J. M.
Seminario, The Journal of Physical Chemistry A 2004, 108, 6378 – 6384;b) P. Novacek, P. Varga, Surf. Sci. 1991, 248, 183 – 192; c) C. Cui, M.Ahmadi, F. Behafarid, L. Gan, M. Neumann, M. Heggen, B. R. Cuenya, P.Strasser, Faraday Discuss. 2013, 162, 91 – 112.
[14] R. Subbaraman, D. Tripkovic, D. Strmcnik, K.-C. Chang, M. Uchimura,A. P. Paulikas, V. Stamenkovic, N. M. Markovic, Science 2011, 334, 1256 –1260.
[15] M. Ahmadi, F. Behafarid, C. Cui, P. Strasser, B. R. Cuenya, ACS Nano 2013,7, 9195 – 9204.
[16] a) M. Li, W. P. Zhou, N. S. Marinkovic, K. Sasaki, R. R. Adzic, Electrochim.Acta 2013, 104, 454 – 461; b) F. E. Teran, D. M. Santos, J. Ribeiro, K. B.Kokoh, Thin Solid Films 2012, 520, 5846 – 5850.
[17] A. Brouzgou, S. Q. Song, P. Tsiakaras, Appl. Catal. , B 2012, 127, 371 – 388.
Received: November 16, 2014Revised: January 16, 2015Published online on && &&, 2015
ChemElectroChem 0000, 00, 0 – 0 www.chemelectrochem.org � 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim6
�� These are not the final page numbers!�� These are not the final page numbers!
Articles
1 12 23 34 45 56 67 78 89 9
10 1011 1112 1213 1314 1415 1516 1617 1718 1819 1920 2021 2122 2223 2324 2425 2526 2627 2728 2829 2930 3031 3132 3233 3334 3435 3536 3637 3738 3839 3940 4041 4142 4243 4344 4445 4546 4647 4748 4849 4950 5051 5152 5253 5354 5455 5556 5657 57

ARTICLES
N. Erini, S. Rudi, V. Beermann, P. Krause,R. Yang, Y. Huang, P. Strasser*
&& –&&
Exceptional Activity of a Pt–Rh–NiTernary Nanostructured Catalyst forthe Electrochemical Oxidation ofEthanol
A perfect match? Platinum, rhodium,and nickel are combined to createa novel electrocatalyst with activity to-wards ethanol oxidation in alkaline andacidic media, supporting the notionthat surface NiOx is an excellent oxy-genate-supplying catalyst componentfor the complete oxidation of ethanol(see picture).
Strasser & co. study Pt-Rh-Ni ternary nano-catalyst for ethanol #electrooxidation @TUBerlin PR SPACE RE-
SERVED FOR IMAGE AND LINK
ChemElectroChem is piloting a social networking feature involving Twitter, an online microblogging service thatenables its users to send and read text-based messages of up to 140 characters, known as “tweets”. Please check thepre-written tweet in the galley proofs for accuracy. Should you or your institute have a Twitter account, please letus know the appropriate username (e.g., @ChemElectroChem), and we will do our best to include this informationin the tweet. This tweet will be posted to the journal's Twitter account upon online publication of your article.
ChemElectroChem 0000, 00, 0 – 0 www.chemelectrochem.org � 0000 Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim7
These are not the final page numbers! ��These are not the final page numbers! ��
1 12 23 34 45 56 67 78 89 9
10 1011 1112 1213 1314 1415 1516 1617 1718 1819 1920 2021 2122 2223 2324 2425 2526 2627 2728 2829 2930 3031 3132 3233 3334 3435 3536 3637 3738 3839 3940 4041 4142 4243 4344 4445 4546 4647 4748 4849 4950 5051 5152 5253 5354 5455 5556 5657 57


![Ternary Logic Gates and Ternary SRAM Cell ….pdf · According to blueprint of Weste & Harris in [4] for design of a binary SRAM, a ternary SRAM is constructed similarly. A ternary](https://img.pdfslide.net/doc/110x75/5a8290bb7f8b9aa24f8e2227/ternary-logic-gates-and-ternary-sram-cell-pdfaccording-to-blueprint-of-weste.jpg)