Embed Size (px)
Citation preview

Synthesis of Heterocyclic Compounds by Intramolecular
Cyclization of Nitrile Imines
By
Eyad Ahmad Mohammed Younes
Supervisor
Dr. Ahmad Q. Hussein, Prof.
This Dissertation was Submitted in Partial Fulfillment of the Requirements for theMaster’s Degree of Science in Chemistry
Faculty of Graduate StudiesUniversity of Jordan
May, 2010

Synthesis of Heterocyclic Compounds by Intramolecular
Cyclization of Nitrile Imines
Examination Committee Signature
Dr. Ahmad Q. Hussein, Chairman..……………… Prof. of Organic chemistry
Dr. Mustafa M. El- Abadelah, Member……………….. Prof. of Organic Chemistry
Dr. Raed A. Al-Qawasmeh, Member ..………………Assoc. Prof. of Organic Chemistry
Dr. Rajab Abu-El-Halawa, Member ..………………Prof. of Organic Chemistry
(Al Al-Bayt University)
ii

DEDICATION
To My
Parents
Brothers and Sisters
And
All My Friends
iii

Acknowledgements
I want to express my deepest appreciation and sincere gratitude to Prof.
Ahmad Q. Hussein for his direct supervision, guidance and encouragement during
the course of this work.
Thanks are also due to Prof. Mustafa El- Abadelah, Dr. Raed Al-Qawasmeh, and
Prof. Rajab Abu-El-Halawa, (members of the examining committee) for their
valuable suggestion and comments.
Thanks are also due to Dr. Jalal A. Zahra and Mss. Fatima for obtaining the NMR
spectral data.
Thanks are also due to Areej Jaber for obtaining the HRMS spectral data.
Special thanks also to my friends; Anas Rasras, Obada S. Abdel Rahman, and all
my friends for their encouragement and help.
iv

Contents
Examination Committee ii
Dedication iii
Acknowledgements iv
List of Contents v
List of figures viii
List of abbreviations ix
Abstract x
1. Introduction 1
I.1 1,3-Dipoles 1
1.2 Nitrile Imines 1
1.2.1 Genration of Nitrile Imines 2
I.2.2 Methods of Synthesis of Hydrazonyl Halides 3
I.2.3 Reactions of Nitrile Imines 5
II. Purpose of the present work 6
III. Results and discussion 10
III.1 Synthesis of 1,2,4-Benzotriazepines 10
III.2 Synthesis of Pyrazolo[1,5-a]indoles 21
III.3 Synthesis of pyrazolo[1,5-a]quinoxalin-4)5H(-ones 23
III.4 Synthesis of pyrazolo[1,5-a]quinoxalines 25
III.5 Synthesis of 1,4-dihydro-5H-1,2,4-benzodiazepin-5-one 35 28
v

Iv Experimental 32
IV.1 Materials and equipments 32
IV.2.1 preparation of compounds 2)a-d( 33
IV.2.2 Preparation of 4-substituted-1,2-diamine 35
IV.2.3 Preparation of compounds )3a-d( 36
IV.2.4Preparation of compounds )5a-c(
39
IV.2.5 Preparation of compounds )13a-c( 41
IV.2.6 Prepration of compounds )14a-c( 44
IV.2.7 Preparation of compound )17( 47
IV.2.8 Preparation of compound )18( 48
IV.2.9 Preparation of compound )23( 48
IV.2.10 Preparation of compound )26( 49
IV.2.11 Preparation of compound )27( 49
IV.2.12 Preparation of compounds )34a-c( 50
IV.2.13 Preparation of compounds )35a-c (
References55
Appendix NMR Spectra 60
vi

LIST OF FIGURES
LIST OF SCHEMES
NUMBER
FIGURE CAPTION PAGE
1 Nitrilium Betaines 1
2 Resonance structures of nitrile imine 1
3 COSY-45 spectrum for compound 5b 12
4 HMQC spectrum for compound 5b 12
5 IR spectrum for compound 5a 13
6 IR spectrum for compound 3b 16
NUMBER
SCHEME CAPTION PAGE
1 Halogenation of N-aroyl aryl –N/-arylhydrazines 3
2 Japp-Klingmann Reaction 3
3 Nucleophilic followed by intramolecular cyclization 5
4 Cyclization routs of nitrile imine 21 8
5 Cyclization routs of nitrile imine 32 9
6 Preparation of 1,2,4-Benzotriazepines 5a-c 10
7 Attempted to cyclization 3d 14
8 Preparation of 3a-c 15
9 Preparation of hydrazonoyl chlorides 2a-d 16
10 Preparation of 4-substituted-1,2-diaminobenzene 17
11 Attempted to preparation of compound 8 18
12 Formation of compound 17 21
13 Possible cyclization routes for nitrile imine 21 24
14 Attempt to synthesize hydrazonoyl chloride 20 25
15 Attempted to diazotization of compound 26 27
16 preparation of hydrazonoyl chloride 27 28
vii

List of Abbreviations
COSY : Correlated Spectroscopy.
2D : Two Dimensional.
DEPT : Distortionless Enhancement by Polarization Transfer.
EIMS : Electron Impact Mass Spectrum.
HMQC : Heteronuclear Multiple Quantum Coherence.
HRMS : High Resolution Mass Spectrometry.
IR : Infrared Spectroscopy.
NMR : Nuclear Magnetic Resonance.
TLC : Thin Layer Chromatography.
viii

Synthesis of Heterocyclic Compounds by Intramolecular
Cyclization of Nitrile Imines
By
Eyad Ahmad Mohammed Younes
Supervisor
Dr. Ahmad Q. Hussein
ABSTRACT
This thesis describes the synthesis of heterocyclic compounds through intramolecular
cyclization of nitrile imine derivatives, carrying suitable functional groups. Thus, the
two-stage cyclization of nitrile imines derived from hydrazonoyl chlorides 2b-d with 1,2-
diaminobenzene gave the new [1,2,4]benzotriazepine derivatives 5.
Hydrazonoyl chloride 14 could not, however, be converted to the corresponding nitrile
imine, in an attempt to synthesize pyrazolo[1,5-a]indoles 16 through analogous
intramolecular cycloaddition.
An attempt to synthesize pyrazolo[1,5-a]quinoxalin-4)5H(-one 22 by intramolecular 1,3-
dipolar cycloaddition of the nitrile imine 21 was unsuccessful since the key diazotization
reaction led to benzotriazole 23, and not to the desired hydrazonoyl chloride 20.
Reduction of compounds 34, with Zn / acetic acid, led directly to the cyclized products,
namely 1,4-benzodiazepine-5-ones 35.
ix

I. Introduction
I.1 1,3-Dipoles
A 1,3-Dipole 1 is a three-atom system which can be a variety of combinations of carbon,
nitrogen, oxygen or sulfur atoms. It has an electron sextet at one end that carries a
positive charge, and a negative charge, together with a lone pair, at the other end
)Huisgen, 1961(.
One class of 1,3-dipoles are nitrile betaines. Three types of nitrile betaines are shown in
Figure 1, namely nitrile ylides, nitrile imines, and nitrile oxides )Houk, et al., 1973(.
Figure 1. Nitrilium Betaines.
I.2 Nitrile Imines
Nitrile imines are typical 1,3-dipolar species, that can be represented by the resonance
structures shown in Figure 2, )Huisgen, et al., 1961( )Huisgen, 1968(.
Figure 2. Resonance structures of nitrile imine.

Nitrile imines are thermally unstable, and are usually generated in situ from their
precursors in the presence of a suitable reactant )Shawali and Parkanyi, 1980(.
I.2.1 Generation of Nitrile Imines
1. Elimination of hydrogen halide from hydrazonoyl halides by a base )Shawali and
Parkanyi, 1980( )Yung Hong and Baldwin, 1968(:
R C
X
N N
H
ArBase
HX R C N N Ar
2. Thermolysis or photolysis of 2,5-disubstituted tetrazoles )Padwa, et al., 1978(:
3. Photolysis of sydnones )Angadiyavar and George, 1971(:
4. Thermolysis of 1,3,4-oxadiazolin-5-ones (Wentrup, et al., 1978(:
I.2.2 Methods of Synthesis of Hydrazonoyl Halides
1. Halogenation of N-aroyl aryl –N/-arylhydrazines Scheme 1, with phosphorus
pentachloride
2

)Leandro and Marilena, 1983(, thionyl chloride )Polumbrik, et al., 1980( or
triphenylphosphine / carbon tetrachloride reagent )Hassaneen, et al., 1987(:
Scheme 1. Halogenation of N-aroyl aryl –N/-arylhydrazines.
2. Halogenation of aldehyde arylhydrazones )Chattway and Adamson, 1931(:
This method has the disadvantage that halogenation of the activated aryl ring may also
take place. However, this may be avoided if a strong electron-withdrawing group )EWG(,
such a nitro group, is present at the aryl nucleous.
3. Diazo-coupling with activated -halo-methinyl compounds, a reaction known as Japp-
Klingmann Reaction )Shawali and Osman, 1971(. This method involves coupling of a
diazonium salt with -halomethinyl compounds, activated by electron-withdrawing
groups, in basic aqueous media giving high yields )80-90%( of the corresponding
hydrazonoyl halides )Scheme 2).
Scheme 2. Japp-Klingmann Reaction.
3

I. 2. 3 Reactions of Nitrile Imines
Nitrile imines undergo the following general types of reaction modes:
1. 1,3-Dipolar cycloaddition reactions.
Being versatile 1,3-dipoles, nitrile imines undergo neat 1,3-dipolar cycloadition reactions
with a large variety of dipolarophiles .Alkenes )Kornet and Chu, 1981(, alkynes )Molteni,
2004(, nitriles )Molteni and Del Buttero, 2005(, and thiones )Pocar, et al., 1975( are
examples of such dipolarophiles, which react neatly with nitrile imines, thus providing
synthetic routes to certain five-membered nitrogen-containing heterocyclic systems,
according to the following general equation:
Ar
NHN
Cl
R
X Y+ BaseY
NN
XR
Ar
Of particular interest, are intramolecular 1,3-dipolar cycloaddition reactions of nitrile
imines incorporating a suitably-located dipolarophilic moiety. Cyclization of such
derivatives would provide access to annulated multi-ring heterocyclic systems: )Padwa,
et al., 1978( )Bruched, et al., 1982(.
2. Nucleophilic addition reactions.
Nitrile imines react with various nucleophiles )Nu(, eventually leading to 1,3-
nucleophilic addition products, which are essentially hydrazone derivatives. Typical
4

nucleophiles are amines )Trimarco and Lastr, 1976(, hydrazines )Ferwanah and
Awadallah, 2005(, alcohols )Rowe, 1991(, and thiols )Zahra, et al., 2005(.
3. Nucleophilic addition reactions followed by intramolecular cyclization.
Subsequent intramolecular cyclization may take place, following the initial nucleophilic
addition reaction, if the reacting nucelophile incorporates an electrophilic center,
suitably-located with respect to the developing nucleophilic NH terminus of the initially
formed adduct (Scheme 3). This normally leads to various heterocyclic products.
Scheme 3. Nucleophilic addition to nitrile imine followed by intramolecular cyclization.
5

The following are examples of such reaction modes:
6

II. Purpose of the present work
The present study aims at exploring the synthetic applications of intramolecular
cyclization reactions, involving appropriate nitrile imine derivatives, towards the
synthesis of certain heterocyclic systems.
Particularly, cyclizations of the following derivatives will be investigated, with the aim,
in each case, to achieve a synthetic route to the indicated heterocyclic system.
1. Intramolecular cyclization of the dihydroquinoxaline 3, obtained through
nucleophilic addition of 1,2-diaminobenzene onto the nitrile imine, generated by
dehydrohalogenation of hydrazonoyl chloride 2, into pyrazino[2,1-c]
[1,2,4]benzotriazepine-1,6)2H,11H(-dione 5.
2. Intramolecular 1,3-dipolar cycloaddition of the nitrile imine moiety in compounds of
type 15 with the C=C bond in the )–CO-C=C-( group to pyrazolo[1,5-a]indol-4-one
derivatives.
7

3. Intramolecular 1,3-dipolar cycloaddition of the nitrile imine moiety in compounds of
the type 21 with a C=C bond in a )–NH-CO-C=C-( group to pyrazolo[1,5-a]quinoxalin-
4)5H(-one )Scheme 4, path a(, competing with the formation of a 1,2,4-benzotriazine
derivative, resulting from intramolecular nucleophilic addition of the nitrogen in )-NH-
CO-( to the nitrile imine moiety )Scheme 4, path b(.
Scheme 4. Cyclization routes of hydrazonoyl chloride 21
4. Intramolecular 1,3-dipolar cycloaddition of the nitrile imine moiety in compounds of
type 27 with the C=C bond in the )–N=C-C=C-( group to pyrazolo[1,5-a]quinoxaline
ring system.
8

5. Intramolecular 1,3-dipolar cycloaddition of the nitrile imine moiety in compounds of
type 32 with the C≡N bond in the )–NH-C-C≡N( moiety to 4,5-dihydro-6H-
[1,2,4]triazolo[1,5-a][1,4]benzodiazepin-6-ones )Scheme 5, path b(, competing with the
formation of 5H-1,2,4-benzotriazepin-5-ones, which might result from the
intramolecular nucleophilic addition of the )-NH-( to the nitrile imine
)Scheme 5, path a(.
Scheme 5. Cyclization routes of nitrile imine 32.
9

III. Results and discussion
III.1 Synthesis of 1,2,4-Benzotriazepines 5a-c
These compounds were obtained in good yields, in the present study, by the action of the
coupling agent, carbonyldiimidazole )CDI(, on the quinoxaline-2-one hydrazones 3a-c
(Scheme 6). The structures of compounds 5 were confirmed by 1H NMR, 13C NMR, IR
and high resolution mass spectroscopy )HRMS(.
It is worthmentioning, however, that the latter compounds 5a-c were insoluble in common
organic solvents, such as alcohols, chloroform, and THF, and exhibited only limited
solubility in dimethylformamide )DMF(, from which they have been recrystallized.
Consequently, their NMR spectra )DMSO( showed rather weak signals, and compound 5c
was insoluble even in DMSO, hence, no NMR spectrum could be obtained for this
particular compound.
5 R1 R2
a H H
b Cl H
c H NO2
Scheme 6. Preparation of 1,2,4-Benzotriazepines 5a-c.
10

The latter condensation reaction leading to compounds 5a-c proceeds through
nucleophilic attack by the amidrazone NH at the carboxyl group, activated by CDI, as
shown in the following equation.
In agreement with the assigned structures, the 1H NMR )DMSO-d6( spectra of compounds
5a-b show two broad singlets )2H( in the region 10 - 14 ppm for the NH protons, as well
as a broad multiplet at 6.9-7.8 ppm for the aromatic protons.
The following correlation of protons were observed in COSY experiment for compound 5b
(Figure 3), as a representative example:
H-11 )7.12-7.18( is correlated to H-10 )δ 7.74-7.78(., H-6' )δ 7.34-7.40( is correlated to
both H-5') δ 7.74-7.78( and H-7' )δ 7.56-7.61(.
HMQC experiment of the same compound (Figure 4) showed the following correlation
between protons and carbons: H-11 )δ 7.12-7.18( is correlated to C-11 )δ 122.3(., H-6' and
H-8' )δ 7.34-7.40( are correlated to C-6' and C-8' )δ 116.0 and δ9124.4( respectively., H-8
)δ 7.54 ( is correlated to C-8 )δ 122.9(., H-7') δ 7.61( is correlated to C-7' )δ 131.1(., H-10
and H-5' )δ 7.74-7.78 ( are correlated to C-10 )δ 125.(and C-5' )δ 128.7(
11

Figure 3. COSY-45 spectrum for compound 5b.
Figure 4. HMQC spectrum for compound 5b
12

The IR spectra for compounds 5 show, in addition to the C=N and C=O bond stretching
modes at about 1550 cm-1 and 1690 cm-1, respectively, sharp N-H stretching at 3225 cm-1,
but no broad absorption in the 3600-3200 cm-1 for an OH group which means that the
compounds 3 cyclized to 5.
458.
1851
7.98
586.
1761
7.08
674.
2775
1.87
830.
7887
6.59
919.
83
1057
.88
1118
.14
1247
.62
1296
.13
1335
.44
1365
.43
1422
.58
1470
.80
1478
.19
1556
.22
1635
.73
1691
.69
2832
.74
2902
.80
2960
.03
3076
.62
3225
.87
10
15
20
25
30
35
40
45
50
55
60
65
70
75
80
85
90
95
100
%T
500 1000 1500 2000 2500 3000 3500 4000
Wavenumbers (cm-1)
Figure 5. IR spectrum for compound 5a.
In contrast to the behaviour of the quinoxaline derivatives 3a-c, prolonged treatment )2
days, room temperature( of the quinoxaline derivative 3d, obtained from the condensation
of 1,2-diaminobenzene with the hydrazonoyl chloride 2d, with CDI did not lead to the
formation of any detectable reaction products, and the reaction mixture revealed only
starting materials upon TLC examination. This may be attributed to tautomerization of 3d
to 3d´, whereby the quinoxaline nucleous becomes fully aromatized. However, it remains
unclear, why the latter compound did not undergo CDI-catalysed cyclization leading to the
formation of a 5-membered pyrazole ring system 4 )Scheme 7).
13

i( Et3N, EtOH, reflux.
Scheme 7. Attempted cyclization of 3d.
The starting hydrazones 3a-c were synthesized from the reaction of the corresponding
1,2-diaminobenzene with the appropriate hydrazonyl chloride 2a-c in presence of
triethylamine as the dehydrohalogenating base. This reaction involves nucleophilic
addition of the diamine, through one of its amino groups, onto the electrophilic carbon of
the nitrile imine, generated from the hydrazonoyl chloride by the action of triethylamine.
The intermediate, thus formed, undergoes subsequent condensation, involving the second
amino group of the diamine and the ester moiety belonging to the nitrile imine, to furnish
the corresponding 1,2,4-benzotriazepine derivative 5a-c.
14

3a: R1 = R2 = X = H3b: R1 = Cl., R2 = X = H3c: R1 = X = H., R2 = NO2
Scheme 8. Preparation of compounds 3a-c.
The 1H NMR spectra for compounds 3 show multiplets at 6.7-7.9 ppm for the aromatic
protons, and broad singlets at 9.2-9.9, 9.6-10.0, and at 12.3-13 ppm due, respectively, to
the NH-1', NH5', and the COOH protons. No signal was observed for the ester OMe group,
which appears as a singlet at 3.5-3.8 ppm the 1H NMR spectra of the starting compounds 2.
The 13C NMR spectra revealed, in addition to the correct number of the aromatic carbon
atoms, one signal at 169 ppm, assigned to the CO2H carbon, and another signal at 131 ppm
for the C=N carbon. In addition, the CH3 protons signal in compound 3a gave rise to a
singlet at 2.47, and the CH3 carbon signal appeared at 21.56 ppm.
The IR spectra )KBr( of compounds 3 showed broad absorption bands in the region 3500
cm-1 and 3350 cm-1 corresponding to the OH and the N-H bond stretching, respectively.
The C=O stretching appeared at about 1660 cm-1. A representative spectrum is given in
Figure 6.
15

462.
9149
4.8752
3.04
565.
6358
7.95
626.
1669
8.16
754.
33
848.
1390
0.46
1038
.71
1153
.00
1220
.08
1261
.76
1301
.94
1337
.16
1367
.58
1434
.32
1463
.33
1498
.56
1573
.37
1611
.81
1664
.07
2837
.55
2896
.36
2977
.16
3341
.88
0
10
20
30
40
50
60
70
80
90
100
%T
500 1000 1500 2000 2500 3000 3500 4000
Wavenumbers (cm-1)
Figure 6. IR spectrum for compound 3b.
The hydrazonoyl chlorides 2 are accessible via the Japp-Klingmann reaction between the
diazonium salt generated from anthranilic acid and chloroacetylacetone or ethyl
chloroacetoacetate at 0 oC in the presence of sodium acetate as a base (Scheme 9(.
2 R1 R2 Ra H H OCH3
b Cl H OCH3
c H NO2 OCH3
d H H CH3
Scheme 9. Preparation of hydrazonoyl chlorides 2a-d.
16

The letter reaction resulted in high yields )85-90%( of the products, 2a-d, as yellow
crystalline solids which were characterized by spectroscopic techniques, and by
comparison with literature data.
Whereas 1,2- diaminobenzene 6a was obtained commercially, the remaining substituted
1,2-diaminobenzenes have been prepared from the corresponding N-acetyl-2-
nitroanilines through reduction of the nitro group with zinc powder in dichloromethane
and acetic acid, and subsequent hydrolysis of the protecting N-acetyl group )Scheme 10),
following literature procedure for similar compounds )Warnecke and Kratz, 2007(.
)i( HNO3 / H2SO4, 0-5 oC., )ii( HCl + EtOH, 3 hr reflux., )iii( Zn /AcOH. ) 6b: X = Cl., 6c: X = Br(
Scheme 10. Preparation of 4-substituted-1,2-diaminobenzene.
The diamines prepared in this way were characterized through comparison with literature
data. It is worth noting that the 1,2-diaminobenzenes are quite susceptible to oxidation.
Therefore, they should be stored in the refrigerator under an inert atmosphere, and the
reactions involving the use of these compounds are also carried out under an inert
17

atmosphere of nitrogen gas, and in the presence of an antioxidant, such as butylated
hydroxytoluene )BHT( or Butylated hydroxyanisole )BHA( to minimize oxidation.
Still, when 4-substitued 1,2-diaminobenzenes 6b,c were allowed to react for 12 hours
with the hydrazonoyl chloride under the same conditions employed before, and in the
presence of BHT or BHA as antioxidants, they suffered from excessive oxidation, so that
the mixture developed a dark black coloration. Preliminary examination of the reaction
mixture by TLC revealed the formation of a complex mixture of unidentified and
practically inseparable products.
In contrast to 1,2-diaminobenzene, 1,2-ethylenediamine gave only the acyclic addition
product 7 upon reaction with the hydrazonoyl chloride 2b )Scheme 11). Attempted
cyclization of this compound to the desired cylic derivative 8, through prolonged
refluxing )12 hours( in various solvents )ethanol, DMF, DMSO, and acetic acid( was
unccessful. This might be attributed to its insolubility in all common solvents.
i( TEA ,THF/ 0 oC, 1 hr.
Scheme 11. Attempted preparation of compound 8.
18

The chemistry of benzotriazepine heterocycles is a subject of continuing interest due to
their diverse applications in pharmacological fields )Chumako, et al., 2006(. Some of
these derivatives are known to have useful biological properties, such as anti-
inflammatory )Fernandez, et al., 2004(, anticonvulsant and antothrombotic )Vlasiuk, et
al., 200(, nervous system related disorders and certain types of cancer )Kaur and Talele,
2008( )McDonald, et al., 2007(. Compounds of type 9 have been reported as CCK2
Antagonists )McDonald, et al., 2006(., whreas indazolo-triazolo-benzotriazepines 10
have shown anti-inflammatory effects )Fernandez, et al., 2003(.
Naphthofuran benzotriazepines 11 have been used as antibacterial and antifungal agents
) Nagaraja, et al., 2003(, and 1,3,4-benzotriazepines 12 showed activity on central
nervous system and HIV virus )El Messaoudi, et al., 2002(.
19

Only few methods have been reported for the construction of the 1,2,4-benzotriazepine
ring system. The following are some examples:
III.2 Attemted Synthesis of Pyrazolo[1,5-a]indoles (16)
A synthetic strategy that seemed plausible for the synthesis of the title heterocyclic system
is based on intramolecular 1,3-dipolarcycloaddition of the nitrile imine of type 15, which
may be generated in situ from the corresponding chlorohydrazonoyl chlorides 14.
It is found, in the present study, however, that the latter hydrazonoyl chlorides 14a-c
remained unchanged upon treatment with triethylamine for 48 hours at room temperature.
No appreciable change could be detected even after prolonged heating )24 hours reflux( in
CHCl3, or in pyridine )8 hours reflux(, in the presence of triethylamine.
20

The resistance of of this hydrazonoyl chloride to dehydrohalogenation might be attributed
to the involvement of the acidic N-H proton in H-bonding with both neighbouring C=O
groups )Scheme 12(, causing this proton to be deeply buried, and therefore unaccessible for
abstraction by triethylamine.
However, when the dehydrohalogenation reaction of the hydrazonoyl chloride 14a was
conducted at higher temperature )refluxing dioxane., boiling point 101 °C ( for 12 hours in
the presence of excess triethylamine, TLC examination indicated the formation of a
complex mixture, from which a major product could be isolated, though in low yield, after
workup of the reaction mixture. The structure of this product proved to be 17, rather than
than the desired heterocycle 16 (Scheme 12(, as evidenced from its 1H NMR, 13C NMR,
and high resolution mass spectroscopy.
Scheme 12. Formation of compound 17.
21

Apparently, the formation of compound 17 suggests that the nucleophilic attack by
triethylamine at the chlorine-carrying carbon atom of the hyrazonoyl chloride is favoured
over the abstraction of the N-H proton. This lends further support to the former suggestion
concerning the involvement of the N-H in hydrogen bonding with the two carbonyl groups
in compound 14a )Scheme 12(.
The hydrazonoyl chlorides 14a-c were prepared via Japp-Klingmann's reaction of the
appropriate diazonium salts, derived from the appropriate aminochalcones 13a-c, and
chloroacetylacetone as shown in the following equation:
NH
O
Ar
N
)i(
)ii(
NH2
O
Ar
Ac
Cl
13(a-c) 14(a-c)
i( NaNO2 / HCl, 0-4°C, 30 min .ii( CH3)CO(CH)Cl()CO(CH3, CH3COONa, 2 hr.
14 a b cAr 4-MeOC6H4 4-Cl C6H4 4-F C6H4
The 1H NMR )CDCl3-d1( spectra for compounds 14a-c showed a singlet at 2.5 ppm )3H(
assigned to the methyl protons of the acetyl group, while the NH proton appeared as a
broad singlet )1H( at about 12.7 ppm. The aromatic protons gave a multiplet in the region
7.1-8.0 ppm. In addition to the signals arising from the aromatic carbon atoms, the 13C-
NMR spectra of these compounds revealed two signals at 25.5 and 191 ppm for the CH3
and C=O carbon atoms of the acetyl group, respectively, and one signal at 144 ppm
assigned to the C=N carbon atom.
22

Aminochalcones 13a-c are accessible through condensation of o-aminoacetophenone with
the appropriate aldehyde in the presence of catalytic amount of sodium hydroxide
)Donnelly and Farrell, 1989( as illustrated in the following reaction:
NH2
O
NH2
O
CH3 Ar
+ EtOH
NaOHArCHO
13 a b c
Ar 4-MeOC6H4 4-Cl C6H4 4-F C6H4
Compounds 13a-c, prepared in this way were obtained in almost quantitative yields, and
their structures were confirmed through their spectral data )1H NMR, 13C NMR, and
HRMS(.
The 1H NMR )CDCl3-d1( spectra for compounds 13a-c showed the two vinylic hydrogens
as two doublets in the region 7.50-7.70 and 7.80-7.90 ppm, the NH2 as broad singlet at 6.3
ppm, and the aromatic ring protons appeared as a multiplet in the range 7.00-7.90 ppm.
In the 13C NMR spectra of these compounds, the carbon of the C=O group appeared at 191
ppm, and the methoxy carbon in compound 13a appeared at 55.4 ppm.
The DEPT experiments indicated the correct multiplicities of the various carbon atoms,
whereby DEPT-90 revealed the presence of eight CH groups.
III.3 Attempted synthesis of pyrazolo[1,5-a]quinoxalin-4(5H)-ones
Another target heterocyclic system, which seemed likely to be accessible through
intramolecular 1,3-dipolar cycloaddition of the nitrile imine moiety with a suitably-
located dipolarophile, is the pyrazolo[1,5-a]quinoxalin-4)5H(-one nucleous 22. It seemed
23

plausible, that the nitrile imine 21, which may be generated by dehydrohalogenation of
the corresponding hydrazonoyl chloride 20, would undergo 1,3-dipolar intramolecular
cycloaddition, involving the nitrile imine moiety and the carbon-carbon double bond, to
give the desired heterocyclic system )Scheme 13, path a(.
Scheme 13. Possible cyclization routes for nitrile imine 21.
Alternatively, competitive nucleophilic addition of the weakly nucleophilic amide
nitrogen to the nitrile imine functionality )Figure 10, path b(, would constitute a
valuable synthetic route to 1,2,4-benzotriazine ring system, of which several derivatives
have pharmaceutical applications, encompassing anti-cancer, and other biological
activities )Neunhoeffer, 1996(.
The starting compounds 18 could be obtained through monoacylation of 1,2-
diaminobenzene. Examination of the literature )Sánchez, et al., 2008(, however, revealed
the infeasibility of such transformation, as it usually leads exclusively to the formation of
the quinoxaline derivative 19. Only if a strong electron-withdrawing group, such as -NO2,
is present, the mono-acylation derivative could then be obtained.
24

Therefore, 4-nitro-1,2-diaminobenzene was employed for the condensation with maleic
anhydride, and the product )compound 18), was obtained in almost quantitative yield,
following literature procedure )Sánchez, et al., 2008(.
Diazotization of compound 18 with nitrous acid at 0 oC , followed by treatment with
chloroacetylacetone, in the conventional manner employed before for Japp-Klingman's
coupling reactions, in an attempt to synthesize the corresponding hydrazonoyl chloride 20,
did not, however, lead to the desired product. The only product isolated from the reaction
mixture was the 5-nitro-1H-benzo[d][1,2,3]triazole 23 (Scheme 14), which was identified
by 1H NMR, IR, HRMS, and by comparison of its m.p. with literature value )Korepin, et
al., 2003(.
i( NaNO2 / HCl, 0-4°C, 30 min .
Scheme 14. Attempted synthesis of hydrazonoyl chloride 20.
III.4 Attempted Synthesis of pyrazolo[1,5-a]quinoxalines:
25

One kind of hydrazonoyl chlorides, which is structurally related to 20, is compound 24,
which seems to be a candidate that could undergo intramolecular 1,3-dipolar cycloaddition,
eventually leading to the formation of the pyrazoloquinoxaline heterocyclic system 25, as
depicted in the following equation:
The hydrazonoyl chloride 24 may be synthesized by Japp-Klingmann's coupling of the
diazonium salt derived from the 1,2-diaminobenzene derivative, which is accessible
through condensation of an α,β-unsaturated aromatic aldehyde with 1,2-diaminobenzene
according to a literature procedure )Ried and Stahlhofen, 1957(.
It is worth noting here, that similar condensations using aliphatic unsaturated aldehydes
follow a different path eventually leading to benzodiazobicycloheptene )Ried and
Stahlhofen, 1957(.
In the present work, 1,2-diaminobenzene was condensed with cinnamaldehyde, giving the
corresponding condensation product 26 in excellent yield. The structure of 26 was
confirmed by comparison of its spectral data and melting point with literature )Ried, and
Stahlhofen, 1957(.
26

Diazotization of the latter compound in the usual manner, in an attempt to prepare the
diazonium salt required for the synthesis of the desired hydrazonoyl chloride 27, was
found however to be accompanied with unforeseen complications. Soon after dissolving
compound 26 in cold HCl )0o C(, the reactin mixture acquired a dark red to black
coloration. Work-up of the reaction mixture in the usual manner gave a product that
proved to be the benzimidazole derivative 28 (Scheme 15), as evidenced by its 1H NMR,
IR, HRMS, and further confirmed through comparison of its melting point with that
reported in the literature )Hassaneen, et al., 1982(.
Scheme 15. Attempted diazotization of compound 26.
An alternative promising route to the hydrazonoyl chloride 27 was therefore undertaken. It
involves simultaneous reductive condensation of of the 2-nitrohydrazonoyl chloride 29
27

with cinnamaldehyde (Scheme 16). The relatively mild, and neutral conditions employed
for such reacion would leave the hydrazonoyl chloride functionality intact, since
dehydrohalogenation of the latter functionality normally takes place only under basic
reaction conditions.
Scheme 16. Preparation of hydrazonoyl chloride 27.
Thus, treatement of a solution of the hydrazonoyl chloride 29 with tin)II( chloride /
methanol )Al-Said and Al-Qaisi, 2006( in the presence of cinnamaldehyde, gave the
desired condensation product 27.
III.5 Synthesis of 2-amino-3,4-dihydro-5H-1,4-benzodiazepin-5-one 35a-c.
Nitrile imines of type 32, which may be generated from the precursor hydrazonoyl
chloride 31 by the action of a base, have two potentially reactive sites available for the
nitrile imine functionality to undergo intramolecular reaction with, namely the nucleophilic
NH and the dipolarophilic C≡N bond.
28

Cyclization of nitrile imine 32 involving the NH would yield the 2-amino-3,4-dihydro-5H-
1,2,4-benzotriazepin-5-ones )Scheme 16, path a(, while 1,3-dipolar cycloaddition of the
nitrile imine with the C=C bond would lead to the 4,5-dihydro-6H-[1,2,4]triazolo[1,5-a]
[1,4]benzodiazepin-6-ones )Scheme 17, path b(.
Scheme 17. Possible cyclization routes of nitrile imine 32.
The hydrazonoyl chlorides 31 could be obtained from corresponding arylamine
derivatives 30 through the Japp-Klingmann's reaction.
The required arylamines 30 may be synthesized through condensation of the α-
aminonitrile 33 with 2-nitrobenzoyl chloride, and subsequent reduction of the nitro
group.
29

It is found in the present study, that mild reduction of 34, with Zn / acetic acid, gives the
corresponding 2-amino-1,4-benzodiazepine-5-ones 35. Apparently, the latter products are
formed via intramolecular nucleophilic addition of the ortho-amino group, formed
through the reduction reaction, to the nitrile )-C≡N( functionality.
35 a b c
R1 / R2 )CH2(5 Ph / H p-MeC6H4 / H
Benzodiazepines exhibit a wide variety of biological activities, so that some of them are
in use as muscle relaxants, anticonvulsants, sedative-hypnotics and anxiolytics
)Sternbach, 1979(, while others are cholecystokinin A and B antagonists )Bock, et al.,
1989(.
1H NMR, 13C- NMR, IR, and HRMS spectra of these compounds are in agreement with
the proposed structure. Unlike their acyclic precursors 34, which exhibit an IR absorption
30

at 2230 cm-1, characteristic of the cyanide group, the IR spectra of compounds 35a-c
show no absorption in the region 2100-2400 cm-1, confirming the involvement of the –
C≡N in the cyclization process. The IR spectra of the latter compounds exihibit two
strong absorption bands at 3470 cm-1 and 3370 cm-1, assigned to the NH2 group
)symmetric and asymmetric stretching modes(, as well as absorptions at about 1630, and
at 3250-3290 cm-1, assigned, respectively, to the amide C=O, and N-H stretching.
The 1H NMR )CDCl3-d1( spectra of compounds 35a-c showed a broad singlet at 5.6 ppm
)2H( corresponding to the NH2 protons, whlile the amide proton appeared as broad singlet
at about 6.00 ppm, and the C-3 proton in compounds 35b,c appeared as a doublet )1H( at
6.2 – 6.3 ppm. In the 13C NMR spectra of these compounds, the carbon of the amide
appeared at 168 ppm, and that of the C=N group at 149 ppm.
31

IV. Experimental
IV.1 Materials and equipments
Derivatives of 2-aminobenzoic acids, and 1,1'-carbonyldiimidazole )CDI( from Aldrich,
1,2-diaminobenzene from Fluka, THF was dried over Na wire for 24 h before use.
Melting points were measured on Harris melting point apparatus and are uncorrected.
1H- and 13C NMR spectra were measured on a Bruker DPX-300 instrument. Chemical
shifts are expressed in ppm with reference to TMS as internal standard. Infrared spectra
)IR( were recorded, as potassium bromide )KBr( discs, on a Nicolet Impact-400 FT-IR
spectrophotometer. High resolution mass spectra )HRMS( were measured in positive / or
negative ion mode by Electrospray Ionization )ESI( on a Bruker Apex IV instrument.
The samples were dissolved in acetonitrile, diluted in spray solution )methanol/water 1:1
v/v + 0.1% formic acid( and infused using a syringe pump with a flow rate of 2 uL/min.
External calibration was conducted using Arginine cluster in a mass range m/z 175-871.
32
IV.2.1 Preparation of compounds 2a-d
To a cold solution of 2-aminobenzoic acid )0.1 mol( aqueous hydrochloric acid )80 ml,
5M( was added dropwise a solution of cold sodium nitrite )7.6 g, 0.12 mol(in 20 mL of
water.The solution was stirred at )0 – 5 °C( for 20 – 30 min. A cold solution of methyl
chloroacetylacetone or chloroacetoacetate )0.1 mol( and 16.4 g sodium acetate )0.2 mol(
in 200 ml of ethanol was then added in one portion. The mixture was stirred for 20 min,
diluted with cold water )100 ml(, and the solid product was collected by filtration,
washed with water and recrystalized from ethanol.
2-(2-(1-Chloro-2-oxopropylidene)hydrazinyl)benzoic acid 2a
Yield, 91%, yellow solid, m.p 243-245 oC.
IR (KBr) v: 3213, 3091, 1740, 1697, 1668 cm-1.
HRMS (ESI): m/z calculated for [M-H]- 239.02289., found 239.02289
1H NMR )300 MHz, DMSO-d6, in ppm(: 2.47) s, 3H, CH3(, 7.08-7.13 )t, J = 15.03 Hz ,
H-5 (, 7.62-7.67 )t, J = 15.81 Hz , H-4 (, 7.74-7.77 )t, J = 8.27 Hz , H-3 (, 7.95-7.97 )d, J
= 9.30Hz , H-6 (.
13C NMR )75 MHz, DMSO-d6, in ppm(: δ = 25.9 )CH3(, 113.9 )C-1(, 114.9 )C-3(, 122.3
)C-5(, 126.9 )C-2(, 131.9)C-6(, 135.4)C-4(, 144.3)C1'), 170.0) CO2H), 188.4)C-2').
33

2-(2-(1-Chloro-2-methoxy-2-oxoethylidene)hydrazinyl)benzoic acid 2b
Yield, 86%, yellow solid, m.p 209-210 °C
)Lit. m.p 215-216 °C(( Zahra, et al., 2008(
4-Chloro-2-(2-(1-chloro-2-methoxy-2-oxoethylidene)hydrazinyl)benzoic acid 2c
Yield, 94%, yellow solid, m.p 262-263 °C
IR (KBr) v: 3152, 2953, 1727, 1678, 1639, 1581 cm-1.
HRMS (ESI): m/z calculated for [M-H]- 288.97883, found 288.97884.
1H NMR )300 MHz, DMSO-d6, in ppm( 3.35)s, 3H, OCH3(, 7.11-7.14 )dd, J = 10.66
Hz , H-5 (, 7.54-7.57 )d, J = 1.97 Hz , H-3 (, 7.91-7.96 )d, J = 12.99 Hz , H-6( .
34

13C NMR )75 MHz, DMSO-d6, in ppm): δ = 54.2 )OCH3(, 112.7 )C-1(, 113.9 )C-3(,
119.4 )C-4(, 121.9 )C-5(, 133.9)C-6(, 139.8 )C-2(, 145.5 )C-1'), 159.6 )C-2'),169.3
) CO2H).
2-(2-(1-Chloro-2-methoxy-2-oxoethylidene)hydrazinyl)-5-nitrobenzoic acid 2d
Yield, 85%, yellow solid, mp 244-245 °C.
IR (KBr) v: 3482, 3367, 1681, 1622 cm-1.
HRMS (ESI): m/z calculated for [M-H]- 300.00178 found 300.00289
1H NMR )300 MHz, DMSO-d6, in ppm): δ = 3.83)s, 3H, OCH3(, 7.50-7.53 )d, J = 9.29
Hz , H-3(, 8.29-8.33 )d d , J = 11.95 Hz , H-6 (, 8.54-8.55 )d, J = 2.66 Hz , H-4( .
13C NMR )75 MHz, DMSO-d6, in ppm): δ = 54.3 )OCH3(, 113.2 )C-1(, 115.2 )C-3(, 121.9
)C-5(, 127.6 )C-6(, 129.9)C-4(, 140.7 )C-2(, 148.5 )C-1'), 159.3 )C-3'), 168.3 )CO2H).
IV.2.2. Preparation of 4-substituted-1,2-diamines 6b-c
N-)4- substituted phenyl( acetamide (65 mmol( was dissolved in concentrated sulfuric
acid )36.5 ml(, concentrated nitric acid )34.0 ml( was then added dropwise to the reaction
mixture at 0-5 ºC during 1 hr with stirring. Stirring was continued for additional 1 hr. The
reaction mixture was then poured over ice-water )200 mL( and the resulting yellow
precipitate was collected, and washed with water.
35

Hydrolysis the acetamide group: a solution of N-)4- substituted -2-nitrophenyl(
acetamide )43 mmol( in concentrated hydrochloric acid )20 mL( and ethanol )80 mL(
was refluxed for 3 hr. The reaction mixture was then cooled and poured onto ice-water
)120 mL(. The resulting orange precipitate was collected, washed with water and dried.
Reduction to 4- substituted 1,2-diaminobenzene: 32.4 mmol of the nitro aniline and zinc
powder )10 g( were suspended in dichloromethane )300 mL( in a double-necked flask
equipped with a condenser. To the stirred mixture was added acetic acid )18.4 mL, 0.32
mol( dissolved in dichloromethane )100 mL( dropwise within 30 min and stirring was
continued for 2 h. Excessive zinc powder was filtered off and the filtrate was washed
with saturated NaHCO3 solution )3 x150 mL(, dried over MgSO4. The solvent was
evaporated and the brown product collected by filtration and recrystalized from
chloroform / petroleum ether.
4-Chlorobenzene-1,2-diamine 6b
Yield, 43%, m.p 75-76 °C )Lit. m.p 76-78 °C( )Knoboloch, 1985(.
4-Bromobenzene-1,2-diamine 6c
Yield, 52%, m.p 64-67 °C )Lit. m.p 65-69 °C( )Indusegaram, et al., 2003(.
IV.2.3. Preparation of compounds 3a-d
36

The hydrazonoyl chloride 2 )5 mmol(, 1,2-diaminobenzenes )5 mmol( and triethylamine
)6 mmol( were refluxed in ethenaol for 12 h, after cooling, the precipitated solid was
collected by filtration, washed with water and recrystalized from DMF / water.
2-[2-(3-Methylquinoxalin-2-yl)hydrazinyl]benzoic acid 3a
Yield, 73%, yellow solid, m.p 259-263 ºC
IR (KBr): v 3292, 1672, 1587, 1528, 1490. cm-1.
HRMS (ESI): m/z calculated for [M-H]- 293.10439, found 293.10440.
1H NMR )300 MHz, DMSO-d6, in ppm( δ= 2.47)s, CH3(, 6.71-7.08 )t, J=14.89 Hz, H-5(,
7.08-7.10 )d, J=8.34 Hz, H-6(, 7.33-.46 )m, H-4, H-3, H8', H9'(, 7.75-7.88 )dd, H-7', H-
10'(, 9.23 )s, NH-1'(, 9.44 )s, NH-5'(, 13.00 )s, CO2H(.
13C NMR )75 MHz, DMSO-d6, in ppm( δ = 21.5 )CH3(, 112.2 )C-1(, 113.2 )C-3(, 117.7
)C-5(, 125.1 )C-8'(, 126.3 )C-10'(, 128.1 )C-7'(, 129.2 )C-9'(, 131.8 )C-6(, 134.7 )C-4(,
137.4 )C-6'(, 140.6 )C-11'(, 145.9 )C-2(, 145.9 )C-4'(, 151.1 )C-4'(, 152.0 )C-3'(,
169.9 )CO2H(.
2-[2-(3-Oxo-3,4-dihydroquinoxalin-2(1H)-ylidene)hydrazinyl]benzoic acid 3b
37

Yield, 53%, yellow solid, m.p 294-295 ºC.
IR (KBr) v: 3341, 1664 , 1611 , 1573 , 1498 cm-1
HRMS (ESI): m/z calculated for [M-H]- 295.08366, found 295.08366.
1H NMR )300 MHz, DMSO-d6, in ppm(: 6.71-6.99 )t, J=14.60 Hz, H-5(, 7.06-7.39 )m,
H-6, H-4, H-3, H-8', H-9' , H-10'(, 7.84-7.86 )d, J= 7.82, H-7'(, 9.29 )s, NH-1'(, 9.65 )s,
NH-2'(, 12.32 )s, CO2H(.
13C NMR )75 MHz, DMSO-d6, in ppm(: δ = 111.9 )C-1(, 113.0 )C-3(, 115.4 )C-5(,
117.1)C-8'(, 123.7 )C-10'(, 124.8 )C-7'(, 125.4 )C-9'(, 129.0 )C-6'(, 131.7 )C-6(, 132.8
)C-11'(, 134.7 )C-4(, 149.9 )C-2(, 151.3 )C-3(, 151.8 )C-4'(, 169.9 )CO2H(.
4-Chloro-2-[2-(3-oxo-3,4-dihydroquinoxalin-2(1H)-ylidene)hydrazinyl]benzoic acid 3c
Yield, 85%, yellow solid, m.p 317-318 ºC.
IR (KBr) v: 3328, 3288, 1670 , 1598, 1492 cm-1
38

HRMS (ESI): m/z calculated for [M+H]+ 331.05924, found 331.05924.
1H NMR )300 MHz, DMSO-d6, in ppm(: 7.34-7.48 )m, H-10', H-8', H-9', H-3, H-7',H-5(,
7.88-7.90 )d, J= 6.45 Hz, H-6(, 9.65 )s, NH-1'(, 10.09 )s, NH-5'(, 12.36)s, CO2H(.
13C NMR )75 MHz, DMSO-d6, in ppm(: δ = 111.1 )C-3(, 115.4 )C-10'(, 116.4 )C-9'(,
123.6 )C-6(, 124.6 )C-5(, 125.4 )C-7'(, 129.1 )C-6'(, 133.0 )C-8'(, 134.0 )C-11'(, 137.0
)C-4(, 149.9 )C-2(, 151.3 )C-3(, 152.1 )C-4'(, 172.0 )CO2H(.
5-Nitro-2-[2-(3-oxo-3,4-dihydroquinoxalin-2(1H)-ylidene)hydrazinyl]benzoic acid 3d
Yield, 85%, yellow to red solid, m.p 168-169 ºC.
IR (KBr) v: 3547, 3178, 1693, 1644, 1591 cm-1
HRMS (ESI): m/z calculated for [M-H] - 340.06874, found 340.06874.
1H NMR )300 MHz, DMSO-d6, in ppm(: 6.78-8.15 )m, H-10', H-8', H-9', H-3, H-7'(,
8.40-843 )d, J=7.55 Hz, H-4(, 8.68 )s, H-6(.
13C NMR )75 MHz, DMSO-d6, in ppm(: δ = 113.9 )C-3(, 115.5 )C-8'(, 123.7)C-10'(,
124.4 )C-7'(, 125.7 )C-9'(, 128.5 )C-6(, 132.5 )C-11'(, 137.0 )C-5(, 149.5 )C-2(,151.4 )C-
4'(, 169.1 )CO2H(.
IV.2.4. Prepration of compounds 5a-c
39

CDI )0.41 g, 2.5mmol( was added to a cooled )0 oC( and stirred solution of hydrazones
3b-d )2mmol( in 30 ml dry THF, and the resulting mixture was further stirred at room
temp for 8 h. The reaction mixture was then immediately treated with 20 ml cold water,
most of the THF was evaporated. The precipitate solid formed was collected by filtration
and washed with water and recrystalized from DMF / water.
Quinoxalino[2,1-c][1,2,4]benzotriazepine-1,6(2H,11H)-dione 5a
Yield, 82%, yellow solid, m.p 285-287 ºC )d(.
IR (KBr) v : 3225 , 1691 , 1635 , 1556 cm-1 .
HRMS (ESI): ): m/z calculated for [M+Na]+ 301.069596, found 301.069597
1H NMR )300 MHz, DMSO-d6( δ= 7.14-7.19 ) br, t, H-10 (, 7.33-7.39)m, H-8' , H-6' and
H-11( , 7.65-7.64 )m,H-7' and H-8(, 7.72-7.78 )m, H-5 ' and H-9(.
13C NMR )75 MHz, DMSO-d6( δ = 113.4 )C-8(, 115.9 )C-6'(, 116.9 )C-6(, 122.0 )C-11(,
124.1 )C-9(, 124.4 )C-10(128.7 )C-8'(, 130.9 )C-3'(, 131.2 )C-5'(, 132.5 )C-4'(, 133.5
)C-7'(,146.6 )C-7(, 148.1 )C-3(, 151.6 )C-1'(, 161.9 )C-5(.
9-Chloro-quinoxalino[2,1-c][1,2,4]benzotriazepine-1,6(2H,11H)-dione 5b
40

Yield, 78%, yellow solid, m.p 305-308 ºC.
IR (KBr v: 3011.89, 2968.08, 1740.62, 1698.32 cm-1.
HRMS (ESI): m/z calculated for [M+Na]+ 335.03062, found 335.03062.
1H NMR )300 MHz, DMSO-d6(: δ = 7.10-7.18 )d, J=8.17Hz, H-11(, 7.34-7.40 )m, H-8',
H-6'(, 7.45 )s, H-8(, 7.56-7.61 )t, J=15.167Hz, H-7'(, 7.74-7.78 )m, H-5 ' and H-10(.
13C NMR )75 MHz, DMSO-d6( δ = 113.00 )C-8(, 115.50 )C-6(, 116.08 )C-6'(, 122.37
)C-11(, 124.48 )C-10(, 125.93 )C-8'(, 128.97 )C-5'(, 130.99 )C-3'(, 131.22 )C-7'(, 132.61
)C-4'(, 138.43 )C-9(, 146.14 )C-7(, 148.65 )C-3(, 151.58 )C-1'(, 160.84 )C-5(.
8-Nitro-quinoxalino[2,1-c][1,2,4]benzotriazepine-1,6(2H,11H)-dione 5c
Yield, 72%, yellow solid, m.p 315-316 ºC
IR (KBr) v : 3082, 2954, 2907, 1696, 1657 cm-1
HRMS (ESI): ): m/z calculated for [M-H]- 322.05817, found 322.05818
41

IV.2.5. Prepration ofcompounds 13a-c
2'-Aminoacetophenone )10 mmol( was added to a solution of benzaldehyde derivative
)11 mmol( in ethanol )100 mL( containing sodium hydroxide )0.5 g( and stirred at room
temperature for 12 h. Tthe resulting precipitate was collected by filtration and
recrystalized from chloroform.
(E)-1-(2-Aminophenyl)-3-(4-methoxyphenyl)prop-2-en-1-one 13a
Yield, 89%, yellow solid, m.p 90-91 ºC.
IR (KBr) v :3475, 3308, 1644, 1610 cm-1
HRMS (ESI): m/z calculated for [M+H]+ 254.1175, found 254.12
1H NMR )300 MHz, CDCl3-d1(: δ 3.84 )s, 3H, OCH3(, 6.30 )brs, NH2(, 6.67-6.72 )m, H-
3'' and H-5'(, 6.92-6.95 )d, J = 8.72 Hz , H-3' and H-5'( , 7.26 )m, H-4''(,7.48-7.53 )d,
J=15.49 Hz, H-2(, 7.58-7.61 )d, J= 8.73 Hz, H-2' and H-6' (, 7.70-7.75 )d, J = 15.49 Hz,
H-6''(, 7.85-7.87 )d, J= 8.33Hz, H-3(.
42

13C NMR )75 MHz, CDCl3-d1(: δ = 55.4 )OCH3(, 114.3 )C-3' and C-5'( , 115.1 )C-3''(,
117.3 )C-2(, 119 .3 )C-5''(, 120.8 )C-6''(, 128.0 )C-1''( ,130.0 )C-2' and C-6'(, 134.1 )C-4''(,
142.8 )C-3(, 161.3 )C-4'(, 191.8 )C-1(.
(E)-1-(2-Aminophenyl)-3-(4-chlorophenyl)prop-2-en-1-one 13b
Yield, 93%, yellow solid, m.p 82-84 ºC.
IR (KBr) v : 3475, 3308, 1644, 160 cm-1 .
HRMS (ESI): m/z calculated for [M+H]+ 258.0680, found 258.07
1H NMR )300 MHz, CDCl3-d1(: δ = 6.36 )brs, NH2(, 6.66-6.71 )m, H3'' and H5''(, 7.26-
7.32 )m, H4''(, 7.35-7.38 )d, J = 8.51 Hz , H-3' and H-5' (, 7.53-7.69 )m, H-2' , H-6' and H-
2(, 7.65-7.70 )d ,J = 15.57 Hz, H-6''(, 7.82-7.85 )dd, J = 1.26 and 1.42 Hz, H-3(.
13C NMR )75 MHz, CDCl3-d1(: δ = 115.9 )C-3''(, 117.4 )C-2(, 118.8 )C-5''(,123.5 )C-6''(,
129.1 )C-3' and C-5'(, 129.4 )C-2' and C-6'(, 131.0 )C-4'(, 133.8 ) C-1''(, 135.9 )C-1' and
C-4'(, 141.46)C-3(, 151.15 )C-2(, 191.36 )C-1(.
43

(E)-1-(2-Aminophenyl)-3-(4-fluorophenyl)prop-2-en-1-one 13c
Yield, 87%, yellow solid, m.p 68-69 ºC.
IR (KBr) v : 3462, 3309, 1646, 1613 cm-1
HRMS (ESI): m/z calculated for [M+H]+ 242.0975, found 242.10.
1H NMR )300 MHz, CDCl3-d1(: δ = 6.35 )brs, NH2(, 6.67-6.72 )m, H3'' and H5''(, 7.07-
7.13 )m, H-3' and H-5' (, 7.26-7.32 )m, H-2(, 7.51-7.83 )m, H-2', H-6' and H-6''(
)dd, J= 1.19 and 1.39 Hz, H-3(.
13C NMR )75 MHz, CDCl3-d1(: δ = 115.9 )C-3' and C-5'(, 116.2 )C-3'' (, 117.3 )C-2(,
118 .9 )C-5''(, 122.8 )C-6''(, 130.0-130.1 )C-2' and C-6'(, 131.5 )C1' (, 134.4 )C-4''(, 141.6
)C3(, 151.1 )C2''(, 191.4 )C-1(.
IV.2.6. Prepration of compounds 14a-c
To a cold solution of 0.67 g chloroacetylacetone )5 mmol ( and 3.11 g sodium acetate
in 50 mL ethanol was added 13 )5 mmol( in cold aqueous hydrochloric acid )10 mL,
3M(. To this solution was added dropwise a cold solution of sodium nitrite )0.37 g, 55
mol( and stirring was continued for additional 30 min at )0 – 5 °C(. To this solution of
arene diazonium chloride was then added of chloroacetylacetone at )-8 °C( in one portion
44

with vigorous stirring. Stirring was continued for 3-4 h. The reaction mixture was then
dilute with cold water )50 mL(, and the solid product was collected by filtration, washed
with water and recrystalized from chloroform.
N'-(2-((E)-3-(4-Methoxyphenyl)acryloyl)phenyl)-2-oxopropanehydrazonoyl chloride
14a
Yield, 81%, dark yellow solid, m.p 190-192 ºC.
IR (KBr) v: 3356, 1686, 1638 cm-1 .
HRMS (ESI): m/z calculated for [M+Na]+ 379.081, found 379.08.
1H NMR )300 MHz, CDCl3-d1(: δ = 2.58 )s, COCH3(, 3.84 )s, OCH3(, 6.92-6.963 )d,
J=11.63 Hz, H-3' and H-5' (, 7.08-7.14 )m, H5''(, 7.50-7.63 )m, H-2' , H-6' , H-2 and H-
4''(, 7.793-7.88 )m, H-3'' and H-6''(, 8.01-8.04 )d, J=9.36, H-3(, 12.76 )br, NH(.
13C NMR )75 MHz, CDCl3-d1(: δ = 25.5 )COCH3(, 55.5 )OCH3(, 114.5 )C-3' and C-5'(,
115.1 )C-3''(, 119.1 )C-2(, 120.9 )C-1'(, 121.4 )C-5''(, 130.5 )C-2' and C-6'(, 130.7 )C-4''(,
134.8 )C-3(, 144.7 )C=N(, 161.9 )C-4'(,188.8 )C-1(, 192.0 )COCH3(.
45

N'-(2-((E)-3-(4-Chlorophenyl)acryloyl)phenyl)-2-oxopropanehydrazonoyl chloride
14b
Yield, 87%, dark yellow solid, m.p 199-201 ºC.
IR (KBr): v 3432.50, 1694.90, 1641.77 cm-1.
HRMS (ESI): m/z calculated for [M+Na]+ 383.4166, found 383.03.
1H NMR )300 MHz, CDCl3-d1(: δ = 2.57)s, COCH3(, 7.08-7.14 )m, H5''(,
7.39-7.45 )d, J=8.49 Hz, H-3' and H-5' (, 7.59-7.67 )m, H-2', H-6', H-2 and H-4''(, 7.82-
7.87 )m, H-3'' and H-6''(, 8.02-8.05 )d, J= 8.09 Hz, H-3(, 12.70 )br, NH(.
13C NMR )75 MHz, CDCl3-d1(: δ = 25.55 )COCH3(, 115.28 )C-3''(, 120.61 )C-1'' (,
121.48 )C-2(, 122.09 )C-5''(, 129.39 )C-3' and C-5'(, 129.76 )C-2' and C-6'(, 130.84 )C-1'(,
133.20 )C-4''(, 135.24 )C-4'(, 136.82 )C-2''(, 144.02 )C-3), 144.96 )C=N(, 188.77 )C-1(,
191.89 )COCH3(.
46

N'-(2-((E)-3-(4-fluorophenyl)acryloyl)phenyl)-2-oxopropanehydrazonoyl chloride
14c
Yield., 83%, dark yellow solid, m.p 181-182 ºC.
IR (KBr) v :3075.20, 1685.96, 1642.25, 1582.04 cm-1
HRMS (ESI): m/z calculated for [M+Na]+ 367.0511, found 367.06
1H NMR )300 MHz, CDCl3-d1(: δ = 2.59 )s, COCH3(, 7.09-7.15 )m, H5'', H3' and H5'(,
7.56-7.67 )H-2, H-3'', H-4'', H-6''(, 7.81-8.02 )H-2' and H-6' ( ,
8.02-8.04 )d, J= 8.05 Hz, H3(, 12.70 )br, NH(.
13C NMR )75 MHz, CDCl3-d1(: δ = 25.54 )COCH3(, 115.24 )C-3''(, 116.14 and 116.44
)C-3' and C-5'(, 121.31 )C-2(, 121.34 )C-5''(, 121.48 )C-8''(, 127.98 )C-1''(, 130.52 and
130.63 )C-2' and C-6'(, 130.82 )C-1'(, 13096 )C-4''(, 131.00 )C-2''(, 144.19 )C-3), 144.91
)C=N(, 162.58-165.93 )d, C-4'(, 188.75)C-1(, 191.91)COCH3(.
47

IV.2.7. Preparation of N'-{2-[(2E)-3-(4-methoxyphenyl)prop-2-enoyl]phenyl}-N,N-diethyl-2-oxopropanehydrazonamide 17
The hydrazonoyl chloride 14a )5 mmol(, and triethylamine )25 mmol (were refluxed in
1,4-dioxan for 12 h , the resulting red yellow product was purified on TLC using
chloroform as mobile phase .
Yield, 60%, m.p
IR (KBr) v: 3193, 1674, 1641 cm-1
HRMS (ESI): m/z calculated for [M+Na]+ 416.1925, found 416.20.
1H NMR )300 MHz, CDCl3-d1(: δ = 2.57 )s, COCH3(, 7.08-7.14 )m, H-5''(, 7.39-7.45 )d,
J= 8.49 Hz, H-3' and H-5' (, 7.59-7.67 )m, H-2', H-6', H-2 and H-4''(, 7.82-7.87 )m, H3''
and H6''(, 8.02-8.05 )d, J= 8.09 Hz, H-3(, 12.70 )br, NH(.
13C NMR )75 MHz, CDCl3-d1(: 13.9 )CH3(, 26.1 )COCH3(, 45.7 )CH2(, 55.4 )OCH3(,
114.4 )C-3' and C-5'(, 115.1 )C-3''(, 119.4 )C-5''(, 120.4 )C-2(, 127.7 )C-1'(, 130.2 )C-2'
and C-6'(, 130.7 )C-6''(, 134.4 )C-4''(, 143.9 )C-3(, 145.3 )C-2''(, 145.5 )C=N(,161.6 )C-
4'(, 191.8 )C-1(, 169.0 )COCH3(.
48

IV.2.8. Preparations of (2Z)-4-[(2-amino-5-nitrophenyl)amino]-4-oxobut-2-enoic acid 18
To a solution of maleic anhydride )1.18 g, 12 mmol( in THF )20 mL( was added a
solution of 4-nitro-1,2-diaminobenzene )1.54 g, 10 mmol( in THF (40 mL)
The mixture was stirred at room temperature for 12 hr, the solid product
was collected by filtration, washed by water to give a dark yellow solid.
Yield, 94% , m.p 188-190 °C )Lit. m.p 185-187 °C( )Sánchez, et al., 2008(.
IV.2.9. Preparations of 5-nitro-1H-benzo[d][1,2,3]triazole 23
Compound 18 )5 mmol( was dissolved in cold aqueous hydrochloric acid )10 mL, 3M(.
To this solution was added dropwise a solution of sodium nitrite )0.37 g, 55mol( with
efficient stirring at 0 – 5 °C, chloroacetylacetone was added to this soloution with
vigorous stirring, and the resulting a yellow mixture was stirred further for 3-4 h. The
reaction mixture was then dilute with cold water )50 mL( the solid product was collected
by filtration and washed well with water.
49

Yield, %, yellow solid, m.p 189-191 ºC. )Lit. m.p 187-190 °C( (Korepin, et al., 2003(.
HRMS (ESI): m/z calculated for [M-H]- 163.0339, found 163.0261.
1H NMR )300 MHz, DMSO-d6, in ppm): δ = 8.05-8.08 )d, J= 9.05 Hz, H-2(, 8.26-8.30
)dd, J= 11.10 Hz, H-3(, 8.93 )s, H-5, 16.50 )s, br, NH(.
IV.2.10. Preparation of N1-((E)-3-phenylallylidene)benzene-1,2-diamine( 26)
A solution of cinnamaldehyde )10 mmol( and 1,2-diaminobenzenes )10 mmol( in
methanol )20 mL( was stirred at room temperature for 12 hr, the solid
product was collected by filtration and washed with water to give a yellow solid.
Yield, 88%, m.p.
50

IV.2.11. Preparation of 2-Oxo-N-(2-{[(2E)-3-phenylprop-2-en-1-ylidene]amino}phenyl)propanehydrazonoyl chloride 27
Compound 29 )1.6 g, 5 mmol( was dissolved in methanol )10 mL( containing
SnCl2.2H2O )2 g( and the reaction mixture was refluxed for 3 h. Then solvent was
evaporated under reduced pressure. The residue was dissolved in ethyl acetate )20 mL(.
Saturated aqueous solution of NaHCO3 )10 mL( was added with stirring, followed by
addition of celite. The mixture was stirred at room temperature for 4 h, then filtreted.
The organic layer was seprated and the solvent was evaporated under reduced pressure.
The resulting solid was collected by filtration, washed by water, yellow solid was
collected and recrystalized from methanol.
Yield, 70%, m.p 178-179.
IR (KBr) v: 3281, 1685, 1589 cm-1
HRMS (ESI): m/z calculated for [M-H]- 324.09091, found 324.09090.
1H NMR )300 MHz, CDCl3-d1(: δ = 2.58 )s,CH3(, 7.182-7.614 )m, H-5, H-6, H-7, H-8,
H-9, H-3', H-4', H-2'', H-3'', H-4'', H-5'', H-6''(, 8.46-8.37 )d, J= 7.80 Hz, H-2''(.
51

13C NMR )75 MHz, CDCl3-d1(: δ = 25.3 )CH3(, 114.0 )C-5(, 116.6 )C-3'(,123.0 )C-7 (,
126.3 )C-1''(, 127.7 )C -2'' and C-6''(, 128.1 )C-8(, 128.4 )C-4'(, 128.9 )C -3'' and C-5''(,
160.2 )C-2'(, 168.6 )COCH3(.
IV.2.12. Preparation of compounds 34a-c
2-Nitrobenzoyl chloride )10 mmol( was added to a solution of the amino nitrile
derivative, prepared acording to literature procedure )Hussein, et al., 1994(, )11 mmol( in
dry THF )50 mL( containing TEA )12 mmol( at 0 oC. Tthe resulting mixture was stirred
for 30 minutes a room temperature, the salt was filtered off and THF was evaporated at
reduced pressure. The remaining solid was collected and recrystalized from
methanol/water.
N-(1-Cyanocyclohexyl)-2-nitrobenzamide 34a
Yield, 94%, yellow solid, m.p 152-154 ºC.
IR (KBr) v: 3216, 2239, 1656, 1639 cm-1
HRMS (ESI): m/z calculated for [M-H]- 272.10406, found 272.10406.
52

1H NMR )300 MHz, CDCl3-d1(: δ = 1.25-2.42)m, 10H, , H-2' , H-3', , H-4' , H-5' , and H-
6'(, 6.07) brs, NH(, 7.49-7.60)m, H-4 and H-5(, 7.65-7.71)m, H-6(, 8.03-8.06)d, J=9.82
Hz, H-3(.
13C NMR )75 MHz, CDCl3-d1(: δ = 22.15 )C-3' and C-5'(, 24.6 )C-4'(, 35.0 )C-2' and C-
6'(, 52.4 )C-1' (, 119.2 )CN(, 124.5 )C-3(, 128.9 )C-6(, 130.8 )C-4(, 131.8 )C-1(, 134.0
)C-5(, 145.9 )C-2(, 165.6 )CONH(.
N-(Cyano(phenyl)methyl)-2-nitrobenzamide 34b
Yield, 74%, yellow solid, m.p 154-156 ºC.
IR (KBr) v : 3245.88, 3035.51, 2242.86, 1648.88 cm-1.
HRMS (ESI): m/z calculated for [M+Na]+ 304.06926, found 304.06926.
1H NMR )300 MHz, CDCl3-d1(: δ = 6.25-6.28 )d, J= 8.16 Hz, H-1'(, 6.70 )brs, NH(,
7.26-7.68 )H-3', H-7', H-5', H-4', H-6', H-4, H-5 and H-6(, 80.5-8.06 )d, J= 8.03 Hz, H-3(.
13C NMR )75 MHz, CDCl3-d1(: δ = 44.6 )C-1' (, 116.8 )CN(, 124.7 )C-3(, 127.2 )C-4' and
C-6'(, 128.7 )C-5'(, 129.4 )C-3' and C-7'(, 129.7 )C-6(, 131.1 )C-4(, 133.9 )C-2'(, 134.1
)C-5(, 146.0 )C-2(, 165.6 )CONH(.
53
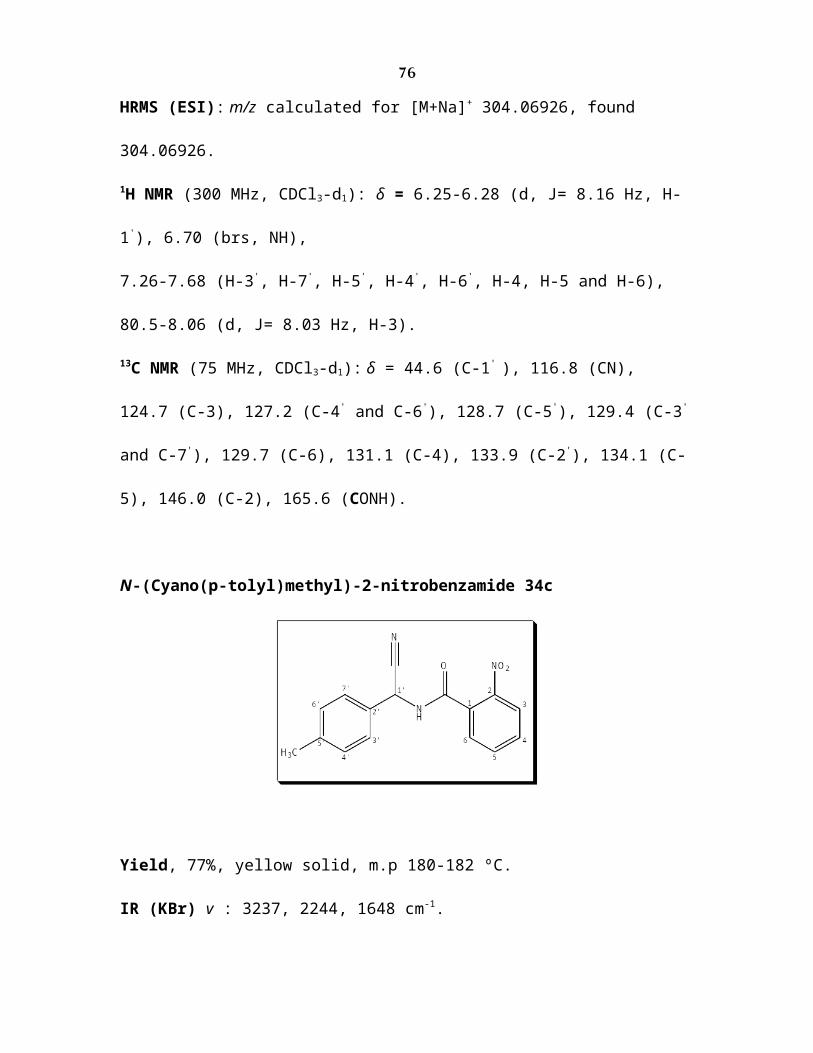
N-(Cyano(p-tolyl)methyl)-2-nitrobenzamide 34c
Yield, 77%, yellow solid, m.p 180-182 ºC.
IR (KBr) v : 3237, 2244, 1648 cm-1.
HRMS (ESI): m/z calculated for [M+Na]+ 318.08491, found 318.08491.
1H NMR )300 MHz, CDCl3-d1(: δ = 2.34)s,CH3(, 6.18-6.28)d, J= 8.14 Hz, H-1'(, 6.67)s,
brs, NH(, 7.22-7.72)H-3', H-7' , H-5' , H-4', H-6', H-4 , H-5 and H-6(, 8.06-8.09)d, J =9.2
Hz, H-3(.
13C NMR )75 MHz, CDCl3-d1(: δ =21.2 )CH3(, 44.4 )C-1' (, 117.0 )CN(, 124.7 )C-3(,
127.1 )C-4' and C-6'(, 128.7 )C-5'(, 129.4 )C-3' and C-7'(, 130.1 )C-6(, 131.1 )C-4(, 165.7
)CONH(.
IV.2.13.Preparation of compounds 35a-c
The nitro compound 34a-c )3 mmol( and zinc powder )1.00 g( were suspended in
dichloromethane )30 mL( in a double-necked flask equipped with a condenser. To the
stirred mixture was added acetic acid )2 mL( dissolved in dichloromethane )10 mL(
dropwise within 5 min. Stirring was continued for 1 h. Excessive zinc powder was
filtered off and the filtrate was washed with saturated NaHCO3 solution )3x15
54
mL(, dried over MgSO4, and evaporated. The product was collected by filtration and
recrystalized from methanol.
2-Aminospiro[1,4-benzodiazepine-3,1'-cyclohexan]-5(4H)-one 35a
Yield, 85%, white solid, m.p 148-150 ºC.
IR (KBr) v: 3479, 3377, 3294, 1632, 1616 cm-1
HRMS (ESI): m/z calculated for [M+Na]+ 266.12638, found 266.12638.
1H NMR )300 MHz, CDCl3-d1(: δ = 1.25-2.42 )m, 10H, H-13, H-12, H-14, H-12 and H-
16(, 5.59 )brs, NH2(, 6.06 ) brs, NH(, 6.60-6.38 )m, H-9 and H-11(, 7.19-7.30 )m, H-8
and H-10(.
13C NMR )75 MHz, CDCl3-d1(: δ = 22.1 )C-13 and C-15'(, 24.6 )C-14(, 35.6 )C-12 and
C-16(, 51.6 )C-14 (, 114.2 )C-6(, 116.5 )C-11(, 117.6 )C-9(, 119.9 )C-7(, 127.2 )C-8(,
133.1 )C-10(, 149.3 )C-2(, 168.5 )C-5(.
55

2-Amino-3-phenyl-3,4-dihydrobenzo[e][1,4]diazepin-5-one 35b
Yield, 83%, white solid, m.p 167-169 ºC.
IR (KBr) v: 3487, 3377, 3252, 1629, 1583 cm-1.
HRMS (ESI): m/z calculated for [M+Na]+ 274.0950, found 274.0951.
1H NMR )300 MHz, CDCl3-d1(: δ =5.61 )brs, NH2(, 6.25-6.27 )d, J= 8.06 Hz, H-3(, 6.56
)brs, NH(, 6.60 )m, H-2' and H-6'(, 7.24-7.31)m, H-4'(, 7.47-7.57 )m, H-3', H-5', H-11, H-
9, H-10 and H-8(.
13C NMR )75 MHz, CDCl3-d1(: δ = 44.3 )C-3(, 113.4 )C-6(, 116.7 )C-1(, 117.5 )C-1' (,
117.6 )C-4'(, 127.0 )C-2' and C-6'(, 127.0 )C-3' and C-5'(,133.4 )C-10(, 149.5 )C-2(, 168.1
)C-5(.
2-Amino-3,4-dihydro-3-p-tolylbenzo[e][1,4]diazepin-5-one 35c
56

Yield, 83%, white solid, m.p 167-169 ºC.
IR (KBr) v: 3452.15, 3362.65, 3235.58, 1637.21 cm-1.
HRMS (ESI): m/z calculated for [M+Na]+ 288.110733, found 288.110733.
1H NMR )300 MHz, CDCl3-d1(: δ = 2.36 )s, CH3(, 5.62 )brs, NH2(, 6.16-6.19 )d, J= 7.29
Hz, H-3(, 6.58-6.70 )m, NH, H-2' and H-6'(, 7.210-7.43 )m, H-4', H-3', H-5', H-11, H-9,
H-10 and H-8(.
13C NMR )75 MHz, CDCl3-d1(: δ = 21.2 )CH3(, 44.6 )C-3(, 113.3 )C-6(, 116.6 )C-1(,
117.6 )C-1' (, 117.8 )C-4'(, 127.1 )C-2' and C-6'(, 130.1 )C-3' and C-5'(,133.4 )C-10(,
149.4 )C-2(, 168.2 )C-5(.
57

V. References
Abdel-Jalil, R;Voelter,W. and Stol, R. )2005(. Microwave-assisted synthesis of1-aryl-3-acetyl-1,4,5,6-tetrahydrobenzimidazo[1,2-d][1,2,4]triazine: first example of a novel ring system. Tet. Lett., 46 ,1725–1726.
Alexandre, F; Berecibar, A; Wrigglesworth, R; Perreux, L; Guillon, J; Le´ger, J; Thie´ry,V. and Besson, T. )2005(. Synthesis of novel 1,3,4-benzotriazepine derivatives from 4-oxo-3,1-benzoxazine and 3,1-benzothiazine-2-carbonitriles. Tetrahedron, 61, 8288–8294.
Al-Said, N; and Al-Qaisi, L. )2006(.A Biomimetic Approach to Quinazolino[3,2-d][1,4]benzodiazepine Ring System: The First Total Synthesis of Asperlicin D. Acta Chim. Slov, 53, 204–209.
Angadiyavar, C. and George, M. )1971(. Photochemical Cycloadditions of 1,3-Dipolar Systems Additions of N,C-Diphenylsydnone and 2,5-Diphenyltetrazole. J. Org. Chem., 36,1589-1594.
Bock, M; Dipardo, R; Evans, B; Rittle, K; Whitter, W; Veber, D; Anderson, P; Freidinger, R. )1989(. Benzodiazepine gastrin and brain cholecystokinin receptor ligands; L-365,260. J. Med. Chem., 32, 13-16.
Bruched, L; Del Buttero, P; Garanti, L and Zecchi, G.)1982(,Sterochemical and Mechanisic Aspect of the Base-promoted Cyclization of o-vinylphenylhydrazonyl Chlorides under Phasetranfer Condition .J. Chem. Soc. Perkin Trans.I, 2041-2044.
Chattway, F. and Adamson, A. )1931(. The Action of Bromine and ofChlorine upon 2 : 4-Dinitrobenxaldehyde-phenyland -p-tolyl –hydrazones, J. Chem. Soc., 2792-2796.
Chumakov, Y; Simonov, Y; Bocelli,G; Gdaniec,M; Vlassiuk, S; and Pavlovsky, V.)2006(. Molecular and crystal structure of 1,2-dihydro-3H-1,3,4-benzotrazepines. Chem. Heterocycl. Comp., 42, No. 7, 20, 907-913.
Donnelly, J. and Farrell, D. )1989(. The Chemistry of 2’-Amino Analogues of 2’-Hydroxychalcone and Its Derivatives. J. Am. Chem. Soc., 55, 1757-1761.
El-Baih, F; Koraa, M. and Al-Hazimi, G. )2006(. Synthesis of some pyrimido[5,4-c]cinnoline and triazepino[6,5-c]cinnoline derivatives. Int. J. Appl. Chem, 2,103-114.
El Messaoudi, T; El Messaoudi, I; Zair, T; Hasanoui, A; Esseffar, M. and Lavergene, J. )2002(. Synthesis of New)1,2,4(Triazol and )1,2,4 oxadiazol-)1,3,4( Benzotrazpines. Synth. Comm., 32, 1815-1820.
58

Fernandez, P; Guille´n, M; Goma, F; Aller, E; Molina, P. and Alcaraz, M. )2004(. A novel cyclo-oxygenase-2 inhibitor modulates catabolic and antiinflammatory mediators in osteoarthritis. Biochem. Pharmacol., 68 ,417–421.
Fernandez, P; Guill, M. and Ubeda, A. )2003(. A novel indazolo-triazolo-benzotriazepine exerts anti-inflammatory effects by inhibition of cyclooxygenase-2 activity and nitric oxide synthase-2 expression . Naunyn-Schmiedeberg's Arch. Pharmacol., 368, 26–32.
Ferwanah, A. and Awadallah, A. )2005(. Reaction of Nitrilimines and Nitrile Oxides with Hydrazines,Hydrazones and Oximes. Molecules, 10, 492-507.
Hassaneen, H; Abdelhamid, A; Shawali, A; Pagni, R. )1982(. A study of the effect of nitro group in the synthesis of pyrazoles and thiadiazolines from hydrazidoyl halides. Heterocycles.19, 319-326.
Hassaneen, H; Mousa, H. and Shawali, A. )1987( .Chemistry of C-heteroarylnitrilimines. Synthesis and cycloaddition reactions of N-phenyl-C-)2-thienyl(nitrilimine .J. Heterocycl. Chem., 24, 1665-1668.
Houk, K; Sims, J; Duke, R; Strozier, R. and George, J. )1973(. Frontier Molecular Orbitals of 1,3 Dipoles and Dipolarophiles. J. Am. Chem. Soc., 95, 7287-7301.
Hussein, A; El-abadelah, M. and Farwanah, A.)1994(. Heterocyclic from Nitrilimine.Dirasat(University of Jordan), 21B, 71-78.
Huisgen, R. )1961(. 1 , 3 -Dipolar additions. Proceedings of the Robert A. Welch Foundation Conference on Chemical Research , 4, 61-86.
Huisgen, R. )1968(. On the Mechanism of 1,3-Dipolar Cycloadditions. J. Org. Chem. 33, 1968-22 97.
Indusegaram, S; Katsifis, A; Ridley, D; Vonwiller, S.)2003(. Nitrogen versus Oxygen Group Protection in Hydroxypropylbenzimidazoles. Aust. J. Chem., 56, 819-827.
Kaur, K. and T, Talele. )2008(. 3D QSAR studies of 1,3,4-benzotriazepine derivatives as CCK2 receptor antagonists. J. Mol. Graphics Modell, 27, 409–420.
Korepin, A; Galkin, P; Perepelkina, E; Glushakova, N; Lodygina V; Eremenko,I; Nefedov, S. and Eremenkoa,L.)2003(. New N_nitrosoamines2. Transformations of tetrahydro_1,3_oxazines into nitrates of N_nitrosoamino alcohols. Russ. Chem. Bull, 52, 2214-2220.
Kornet, M; and Chu, J. )1981(. Synthesis of 1 - methyl - and 1 - ethoxycarbonyl - 2 - phenylcarbamoylpiperidazines as potential anticonvulsant agents . J. Heterocycl. Chem., 18, 293-295.
59

Knoboloch, W. )1985(. Pharmacologically active benzimidazoles. III. Synthesis of substituted benzimidazoles with potential anti-tumor activity. Chem. Ber., 91, 2557 – 2561.
Leandro, B. and Marilena, G. )1983(. Indazoles and dihydrophthalizines from N-phenylhydrazidoyl chlorides . J. Heterocycl. Chem., 20, 225-228.
Leiby, R. and Heindel, N. )1976(. Synthesis of 3,4-Dihydro- and 1,4-Dihydro-5H-l,3,4-benzotriazepin-5-ones. J. Org. Chem.,41, 2736-2739
McDonald, I; Austin, C; Buck, I Dunstone, D; Griffin, E; Harper, E; Hull, R; Kalindjian, S; Linney, I; Low, C; Pether, M; Spencer, J; Wright, P; Adatia, T. and Bashall, A.)2006(. Novel, Achiral 1,3,4-Benzotriazepine Analogues of 1,4-Benzodiazepine-Based CCK2 Antagonists That Display High Selectivity over CCK1 Receptors. J. Med. Chem., 49, 2253-2261.
McDonald, I; Black, J; Buck, I; Dunstone, D; Griffin, E; Harper, E; Hull, R;Kalindjian, S; Lilley, E; Linney, I; Pether, M; Roberts, S; Shaxted, M; Spencer, J; Steel, K; Sykes, D; Walker, M; Watt, G; Wright,L; Wright, P and Xun, W.)2007(. Optimization of 1,3,4-Benzotriazepine-Based CCK2 Antagonists to Obtain Potent, Orally Active Inhibitors of Gastrin-Mediated Gastric Acid Secretion. J. Med. Chem., 50, 3101-3112.
Molteni, G; and Del Buttero, P. )2005( . Nitrilimine cycloadditions to the cyano group in aqueous media . Heterocycles, 65, 1183-1188.
Molteni, G. )2004( . Synthesis of the new pyrazolo[4,3-c]pyrrolizine skeleton via intramolecular nitrilimine cycloaddition. Heterocycles, 63,1423-1428.
Nagaraja, G; Kumaraswam, M; Vaidya, V. and Mahadevan, K .)2006(. Microwave assisted synthesis of naphtho[2,1-b]furan-1, 3, 4-benzotriazepines: a potent antimicrobial agent. Arkivoc, 211-219.
Neunhoeffer, H. )1996(.1,2,4-Triazines and their benzo derivatives. Comp. Heterocycl. Chem., 6, 507-573, 1177-1307.
Nikulin, N; Artamonova, T. and Koldobskii ,G .)2003(. 3H-1,3,4-BenzoandPyrido[6,7-b][1,3,4]triazepines from 5-Aryltetrazoles.Synthesis and Chemical Properties. Russ. J. Org. Chem., 39, 1525-1529.
Padwa, A; Nahm, S and Sato, E . )1978(. Intramolecular 1,3-Dipolar Cycloaddition Reactions of Alkenyl-Substituted Nitrile Imines. J. Org. Chem., 43, 1664-1671.
Pocar, D; Rossi, L and Trimarco, P. )1975(. Reactions of β-keto thioacid anilides with acyl chloride aryl-hydrazones. J. Heterocycl. Chem., 12, 401-403.
60

Polumbrik,O; Ryabokon,I and Markovskii , L)1980(. Perfluorophenyl-containing verdazyl radicals. Khim. Geterotsikl. Soed., 16, 882-885.
Ried, W; and Stahlhofen, P. )1957(. Heterocyclic seven-membered ring systems. V. Reaction of o -phenylenediamine with α,β -unsaturated carbonyl compounds. Chem. Ber. 90, 815- 824.
Rowe, J. )1991(. Mechanisms of Nucleophilic Attack at Carbon-Nitrogen Double Bonds. The Solvolysis of Substituted Benzohydrazonoyl Halides in Aqueous Binary Mixtures. Aust. J. Chem., 44, 463 – 468.
Sánchez, N; Coronado, R; Peralta, R; Hueso, A; Ruiz, S and Parra,A.)2008(. Novel synthesis of 1-alkyl-4-tosyl-3-carboxymethyl-1,2,3,4-tetrahydroquinoxalin-2-ones. Arkivoc, )v(, 187-199.
Shawali ,A. and Osman, A. )1971(. Synthesis and reactions of phenylcarbamoylarylhydrazidic chlorides.Tetrahedron, 27, 2517-2528.
Shawali, A. and Parkanyi, C. ) 1980(. Hydrazidoyl Halides in the Synthesis of Heterocycles. J. Org. Chem., 17, 833-854.
Sternbach, L.)1979(.The benzodiazepine story. J. Med. Chem., 22, 1-7.
Trimarco, P; and Lastr, C. )1976(. Synthesis of 3-arylamino-4)3H(quinazolinone derivatives from 1-acetyl- or 1-ethoxycarbonylmethylene-2-arylhydrazines. J. Heterocycl. Chem., 13, 913-915.
Vlasiuk, S; Pavlovsky, V; Andronatie, S; Gdaniec, M and Simonov, Y.)200(.7-Bromo-5-Phenyl-1,2-DiHydro-3H-1,3,5-Benztriazepin. Chem Heterocycl Comp, 36, 1077-1085.
Warnecke, A. and Kratz, F. ) 2007(. 2,4-Bis)hydroxymethyl(aniline as a Building Block for Oligomers with Self-Eliminating and Multiple Release Properties. J. Org. Chem.,73, 1546-1552.
Wentrup, C; Damerius, A. and Reichen, W. )1978(. Intramolecular Cyclization of Nitrile Imines.Synthesis of Indazoles, Fluorenes, and Aza Analogues. J. Org. Chem., 43, 2037-2041.
Yung Hong, S. and Baldwin, J. )1968(. Kinetics of the thermal decomposition of 2,5-diaryltetrazoles. Tetrahedron, 24, 3787.
Zahra, J; Abu Thaher, B; El-Abadelah, M. and Boese, R. )2005( . 3-Mercaptopropionic acid–nitrile imine adducts. An unprecedented cyclization into 1,3,4-thiadiazol-2)3H(-ones and -2)3H(-thiones. Org. Biomol. Chem., 3, 2259-2603.
61

Zahra, J; El-Abadelah, M; Abu Thaher, B; Laufer, S. and Boes, R. )2008(. Facile Synthesis of Model Indazolo[2,1-c][1,3,4]benzotriazepin-5,13-diones. Heterocycles, 75, 2989-3004.
62

APPENDIX
NMR SPECTRA
Figure 12. 1H NMR spectrum for compound 2a
63
Figure 13. 13C NMR spectrum for compound 2a
Figure 14. DEPT 135 spectrum for compound 2a
64

Figure 15. 1H NMR spectrum for compound 2c
Figure 16. 13C NMR spectrum for compound 2c
65

Figure 17. DEPT 135 spectrum for compound 2c
Fig18. 1H NMR spectrum for compound 2d
66

Figure 19. 13C NMR spectrum for compound 2d
Figure 20. DEPT 135 spectrum for compound 2d
67

Figure 21. 1H NMR spectrum for compound 3a
Figure 22. 13C NMR spectrum for compound 3a
68

Figure 23. DEPT 135 spectrum for compound 3a
Figure 24. 1H NMR spectrum for compound 3b
69

Figure 25. 13C NMR spectrum for compound 3b
Figure 26. DEPT 135 spectrum for compound 3b
70

Figure 27. 1H NMR spectrum for compound 3c
Figure 28. 13C NMR spectrum for compound 3c
71

200 180 160 140 120 100 80 60 40 20 ppmFigure 29. DEPT 135 spectrum for compound 3c
Figure 30. 1H NMR spectrum for compound 3d
72

Figure 35. 13C NMR spectrum for compound 3d
-20220 200 180 160 140 120 100 80 60 40 20 0 ppmFigure 36. DEPT 135 spectrum for compound 3d
73

Figure 37. 1H NMR spectrum for compound 5a.
2030405060708090100110120130140150160170180 ppm
115120125130135 ppm
Figure 38. 13C NMR spectrum for compound 5a.
74

-20220 200 180 160 140 120 100 80 60 40 20 0 ppm
125 ppm
Figure 39. DEPT 135 spectrum for compound 29d
234567891011121314151617 ppm
1214 ppm
7.27.47.67.8 ppm
Figure 40. 1H NMR spectrum for compound 5b.
75

2030405060708090100110120130140150160170180190200 ppm
Figure 41. 13C NMR spectrum for compound 5b.
9095100105110115120125130135140 ppmFigure 42. DEPT 135 spectrum for compound 5b
76

Figure 43. 1H NMR spectrum for compound 13a.
2030405060708090100110120130140150160170180190200210 ppm Figure 44. 13C NMR spectrum for compound 13a.
77

170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10 ppmFigure 45. DEPT 135 spectrum for compound 13a
10 9 8 7 6 5 4 3 2 1 ppm
6.16.26.36.46.56.66.76.86.97.07.17.27.37.47.57.67.77.87.98.0 ppm
Figure 46.1H NMR spectrum for compound 13b
78

405060708090100110120130140150160170180190200210 ppm
Figure 47. 13C NMR spectraum for compound 13b.
9095100105110115120125130135140145150155160165170175 ppmFigure 48. DEPT 135 spectrum for compound 13b.
79

12 11 10 9 8 7 6 5 4 3 2 1 0 ppm
6.26.36.46.56.66.76.86.97.07.17.27.37.47.57.67.77.87.98.0 ppm
Figure 49. 1H NMR spectrum for compound 13c.
210 200 190 180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10 ppm
Figure 50. 13C NMR spectrum for compound 13c
80
100105110115120125130135140145150 ppm
Figure 51. DEPT 135 spectrum for compound 13c
17 16 15 14 13 12 11 10 9 8 7 6 5 4 3 2 1 0 ppm
7.07.27.47.67.88.0 ppm
Figure 52.1H NMR spectrum for compound 14a.
81

2030405060708090100110120130140150160170180190200210220 ppm
Figure 53. 13C NMR spectrum for compound 14a
180 170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10 ppmFigure 54. DEPT 135 spectrum for compound 14a.
82

17 16 15 14 13 12 11 10 9 8 7 6 5 4 3 2 1 0 ppm
7.07.27.47.67.88.08.2 ppm
Figure 55. 1H NMR spectrum for compound 14b.
30405060708090100110120130140150160170180190200210 ppm Figure 56. 13C NMR spectrum for compound 14b.
83

30405060708090100110120130140150160170 ppmFigure 57. DEPT 135 spectrum for compound 14b
17 16 15 14 13 12 11 10 9 8 7 6 5 4 3 2 1 0 ppm
7.07.27.47.67.88.0 ppm
Figure 58. 1H NMR spectrum for compound 14c
84

200 180 160 140 120 100 80 60 40 20 ppm Figure 59. 13C NMR spectrum for compound 14c.
170 160 150 140 130 120 110 100 90 80 70 60 50 40 30 20 10 ppm
116118120122124126128130 ppm
Figure 60. DEPT 135 spectrum for compound 14c
85

17 16 15 14 13 12 11 10 9 8 7 6 5 4 3 2 1 0 ppm
8.08.59.0 ppm
Figure 70. 1H NMR spectrum for compound 23.
Figure 71. 1H NMR spectrum for compound 27.
86

Figure 72. 13C NMR spectrum for compound 27..
Figure 73. 1H NMR spectrum for compound 34a.
87

Figure 74. 13C NMR spectrum for compound 34a
Figure 75. DEPT 135 spectrum for compound 34a.
88

Figure 76. 1H NMR spectrum for compound 34b.
Figure 77. 13C NMR spectrum for compound 34b.
89

Figure 78. DEPT 135 spectrum for compound 34b.
Figure 79. 1H NMR spectrum for compound 35a.
90

Figure 80. 13C NMR spectrum for compound 35a.
Figure 81. DEPT 135 spectrum for compound 35a.
91

Figure 82. 1H NMR spectrum for compound 35b.
Figure 83. 13C NMR spectrum for compound 35b..
92

Figure 84. DEPT 135 spectrum for compound 35b
Figure 85. 1H NMR spectrum for compound 35c.
93

Figure 86. 13C NMR spectrum for compound 35c.
94

نايترايل اليمينات الذاتي التحلق بواسطة متجانسة غير حلقية مركبات تحضير
اعداديونس محمد احمد اياد
المشرفحسين قاسم احمد الدكتور االستاذ
الملخص
ايمينات من مختلفة لمشتقات الذاتي التحلق خالل من متجانسة غير حلقية مركبات تحضيرمن, جديدة مشتقات اصطناع تم حيث . 5بنزوترايزبين- 4,3,1نايترايل
كلورايد اندول 14هيدروزوينل بيرزولو لتحضير محاولة في امين النيترايل الي يتحول .16لم
كوينكزالين بيرزولو مركبات تحضير محاولة تنجح لم الحظ التحلق 22لسوء خالل منامين لنيترايل الذاتي
كلورايد, 21 هيدروزوينل تحضير محاولة عند بنزين تريزولو مركب انتج .20حيث
مركبات الزنك, / 34اختزال االسيتيك بواسطة مركبات, حمض مباشرة -4,1اعطىامين 35بنزودايزبين النيترايل الى الوصول خالله من تحضيره المرغوب االمين تعطي ولم
32. بنزودايازبين, ترايزولو من مشتقة مركبات ليعطي داخليا يتحلق ان المحتمل من والذي
وطيف المغناطيسي الرنين طيف على باالعتماد تشخيصها تم الجديدة المركبات جميع. الكتلة ومطياف الحمراء تحت االشعة
95





























