Embed Size (px)
Citation preview

PRENATAL DIAGNOSISPrenat Diagn 2008; 28: 447–449.Published online 8 April 2008 in Wiley InterScience(www.interscience.wiley.com) DOI: 10.1002/pd.1991
RESEARCH LETTER
Familial Beckwith–Wiedemann syndrome due to CDKN1Cmutation manifesting with recurring omphalocele
Antonio Percesepe1*, Emma Bertucci2, Paola Ferrari3, Licia Lugli3, Fabrizio Ferrari3, Vincenzo Mazza2 andAntonino Forabosco1
1Department of Medical Genetics, University of Modena, Italy2Department of Obstetrics and Gynecology, University of Modena, Italy3Department of Pediatrics, University of Modena, Italy
KEY WORDS: familial Beckwith-Wiedemann syndrome; omphalocele; prenatal diagnosis
Omphalocele is a midline anterior abdominal wall defectoccurring in about 1 in 5000 live births (Stoll et al.,2001). It is currently diagnosed by real-time ultrasonog-raphy at the end of the first trimester of pregnancy (Tanet al., 1996) and requires a general prenatal assessmentfor concurring malformations that have been reported in54% of the cases (Boyd et al., 1998). Associated mal-formations are diagnostic of genetic disorders in whichthe anterior wall defect is the principal sign, includinganeuploidies, full or partial (reviewed in Chen, 2007a),single gene disorders, neural tube defects, diaphragmaticdefects and syndromes of unknown aetiology (reviewedin Chen, 2007b). Recurrence in subsequent pregnanciesis rare.
If we exclude the aneuploidies that can be recog-nized non-specifically by performing prenatal karyotypicanalysis, syndromic cases require an accurate diagnosisfor a proper prognostic counselling. The most com-mon disorder associated with omphalocele is Beck-with–Wiedemann syndrome (BWS), a genetically het-erogeneous syndrome characterized, together with themidline abdominal wall defect, by prenatal overgrowth,macroglossia, hemi-hyperplasia, a distinct facial pheno-type and embryonal tumours (Cohen, 2005). Cases ofprenatal diagnosis of BWS have been reported and cri-teria for the ultrasonographic diagnosis have been given(Williams et al., 2005). The molecular diagnosis, how-ever, is still challenging due to the high heterogeneity ofthe syndrome, which can be due to defects in the 11p15.5locus gene cluster by several mechanisms, includingstructural chromosomal abnormalities, paternal isodis-omy, imprinting defects and deletions in either of thetwo imprinted gene domains, ICD1 and ICD2 (Sparagoet al., 2007). Of the BWS cases, 10 –15% are familialhaving been inherited with an autosomal dominant pat-tern, and about 40% of these are caused by inactivatingmutations in CDKN1C, a tumour-suppressor gene withmaternal expression (Engel et al., 2000; Cooper et al.,
*Correspondence to: Antonio Percesepe, Department of MedicalGenetics, University of Modena, Via del Pozzo, 71, 41100Modena, Italy. E-mail: [email protected]
2005). The remaining familial cases are partly accountedfor by inherited micro-deletions of both the imprintingcentres ICD1 and ICD2 (Sparago et al., 2007).
The heterogeneous molecular pathogenesis underliesa different clinical course of the disorder: for example,familial cases with CDKN1C mutations seem to havea more benign outcome compared to the sporadiccounterpart of BWS, more specifically a reduced risk ofWilms’ tumours (Cooper et al., 2005). This additionalinformation can be crucial for the parents in theirdecision to continue the pregnancy.
In the present report we characterize the prenatalfeatures of a case of familial BWS clinically suspecteddue to the recurrence of omphalocele in two consecutivepregnancies and molecularly confirmed with the findingof a novel mutation in the CDKN1C gene.
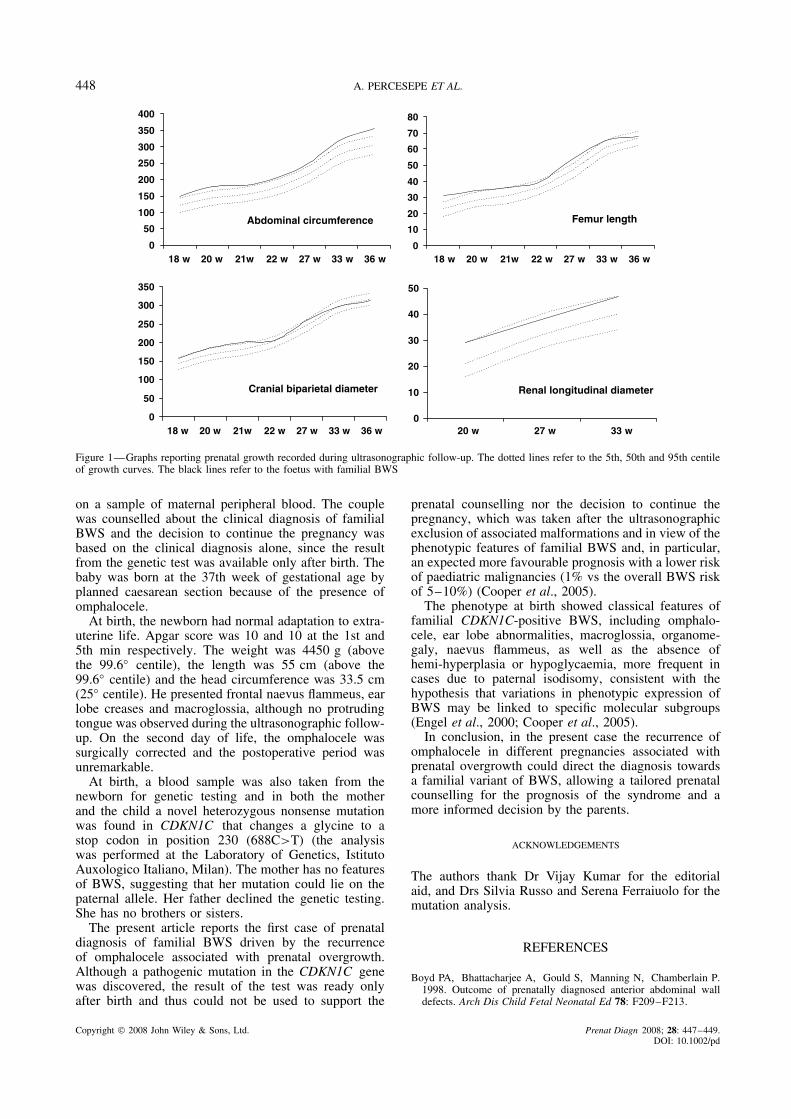
The proband, a 38-year-old healthy woman, wasreferred to us for prenatal counselling due to a diag-nosis of recurrent omphalocele at the 14th week ofpregnancy. She had a 20-week therapeutical abortion2 years previously because of an ultrasonographic diag-nosis of a massive omphalocele (6 cm in diameter),three earlier spontaneous abortions, all in the 6th weekof pregnancy and one healthy 5-year-old son. Prena-tal karyotypic analysis was performed through amnio-centesis at the 16th week of pregnancy resulting in anormal 46,XY karyotype. An accurate ultrasonographicfollow-up was started, reporting prenatal growth of theabdominal circumference, femur length, cranial bipari-etal diameter and kidney longitudinal diameter betweenthe 18th and the 36th week of pregnancy (Figure 1).Due to the growth above the 95◦ centile of the abdomi-nal circumference and of the femural length, associatedwith the presence of the omphalocele, a diagnosis ofBWS was postulated at the 20th week of pregnancy.The finding of omphalocele also in the previous preg-nancy pointed to a familial variant of BWS, althoughno growth parameters were available from the formercase. Because of the specific pathogenesis of the famil-ial cases, ascribed to the loss-of-function mutation of thematernally inherited CDKN1C gene, no invasive prena-tal diagnosis was proposed but a genetic test was offered
Copyright 2008 John Wiley & Sons, Ltd. Received: 27 November 2007Revised: 30 January 2008
Accepted: 19 February 2008Published online: 8 April 2008
448 A. PERCESEPE ET AL.
0
50
100
150
200
250
300
350
400
18 w 20 w 21w 22 w 27 w 33 w 36 w
Abdominal circumference
0
10
20
30
40
50
60
70
80
18 w 20 w 21w 22 w 27 w 33 w 36 w
Femur length
0
50
100
150
200
250
300
350
18 w 20 w 21w 22 w 27 w 33 w 36 w
Cranial biparietal diameter
0
10
20
30
40
50
20 w 27 w 33 w
Renal longitudinal diameter
Figure 1—Graphs reporting prenatal growth recorded during ultrasonographic follow-up. The dotted lines refer to the 5th, 50th and 95th centileof growth curves. The black lines refer to the foetus with familial BWS
on a sample of maternal peripheral blood. The couplewas counselled about the clinical diagnosis of familialBWS and the decision to continue the pregnancy wasbased on the clinical diagnosis alone, since the resultfrom the genetic test was available only after birth. Thebaby was born at the 37th week of gestational age byplanned caesarean section because of the presence ofomphalocele.
At birth, the newborn had normal adaptation to extra-uterine life. Apgar score was 10 and 10 at the 1st and5th min respectively. The weight was 4450 g (abovethe 99.6◦ centile), the length was 55 cm (above the99.6◦ centile) and the head circumference was 33.5 cm(25◦ centile). He presented frontal naevus flammeus, earlobe creases and macroglossia, although no protrudingtongue was observed during the ultrasonographic follow-up. On the second day of life, the omphalocele wassurgically corrected and the postoperative period wasunremarkable.
At birth, a blood sample was also taken from thenewborn for genetic testing and in both the motherand the child a novel heterozygous nonsense mutationwas found in CDKN1C that changes a glycine to astop codon in position 230 (688C>T) (the analysiswas performed at the Laboratory of Genetics, IstitutoAuxologico Italiano, Milan). The mother has no featuresof BWS, suggesting that her mutation could lie on thepaternal allele. Her father declined the genetic testing.She has no brothers or sisters.
The present article reports the first case of prenataldiagnosis of familial BWS driven by the recurrenceof omphalocele associated with prenatal overgrowth.Although a pathogenic mutation in the CDKN1C genewas discovered, the result of the test was ready onlyafter birth and thus could not be used to support the
prenatal counselling nor the decision to continue thepregnancy, which was taken after the ultrasonographicexclusion of associated malformations and in view of thephenotypic features of familial BWS and, in particular,an expected more favourable prognosis with a lower riskof paediatric malignancies (1% vs the overall BWS riskof 5–10%) (Cooper et al., 2005).
The phenotype at birth showed classical features offamilial CDKN1C-positive BWS, including omphalo-cele, ear lobe abnormalities, macroglossia, organome-galy, naevus flammeus, as well as the absence ofhemi-hyperplasia or hypoglycaemia, more frequent incases due to paternal isodisomy, consistent with thehypothesis that variations in phenotypic expression ofBWS may be linked to specific molecular subgroups(Engel et al., 2000; Cooper et al., 2005).
In conclusion, in the present case the recurrence ofomphalocele in different pregnancies associated withprenatal overgrowth could direct the diagnosis towardsa familial variant of BWS, allowing a tailored prenatalcounselling for the prognosis of the syndrome and amore informed decision by the parents.
ACKNOWLEDGEMENTS
The authors thank Dr Vijay Kumar for the editorialaid, and Drs Silvia Russo and Serena Ferraiuolo for themutation analysis.
REFERENCES
Boyd PA, Bhattacharjee A, Gould S, Manning N, Chamberlain P.1998. Outcome of prenatally diagnosed anterior abdominal walldefects. Arch Dis Child Fetal Neonatal Ed 78: F209–F213.
Copyright 2008 John Wiley & Sons, Ltd. Prenat Diagn 2008; 28: 447–449.DOI: 10.1002/pd

FAMILIAL BECKWITH–WIEDEMANN SYNDROME 449
Chen C-P. 2007a. Chromosomal abnormalities associated withomphalocele. Taiwan J Obstet Gynecol 46: 1–8.
Chen C-P. 2007b. Syndromes and disorders associated withomphalocele (III): single gene disorders, neural tube defects,diaphragmatic defects and other syndromes. Taiwan J ObstetGynecol 46: 111–120.
Cohen MM Jr. 2005. Beckwith-Wiedemann syndrome: historical,clinicopathological, and etiopathogenetic perspectives. Pediatr DevPathol 8: 287–304.
Cooper WN, Lunaria A, Evans GA, et al. 2005. Molecular subtypesand phenotypic expression of Beckwith–Wiedemann syndrome.Eur J Hum Genet 13: 1025–1032.
Engel JR, Smallwood A, Harper A, et al. 2000. Epigenotype-phenotype correlations in Beckwith-Wiedemann syndrome. J MedGenet 37: 921–926.
Sparago A, Russo S, Cerrato F, et al. 2007. Mechanisms causingimprinting defects in familial Beckwith-Wiedemann syndrome withWilms’ tumour. Hum Mol Genet 16: 254–264.
Stoll C, Alembik Y, Dott B, Roth MP. 2001. Risk factors incongenital abdominal wall defects (omphalocele and gastroschisi):a study in a series of 265,858 consecutive births. Ann Genet 44:201–208.
Tan KH, Kilby MD, Whittle MJ, Beattie BR, Booth IW, Botting BJ.1996. Congenital anterior abdominal wall defects in England andWales between 1987–93. Br Med J 313: 903–906.
Williams DH, Gauthier DW, Maizels M. 2005. Prenatal diagnosis ofBeckwith-Wiedemann syndrome. Prenat Diagn 25: 879–884.
Copyright 2008 John Wiley & Sons, Ltd. Prenat Diagn 2008; 28: 447–449.DOI: 10.1002/pd





























