Embed Size (px)
Citation preview

Fc-Gamma Receptor Reactivityin Nonorgan-Specific Autoimmune Diseases
Véronique Durand,1 Armelle Lamour,1
Valérie Devauchelle,2 Pierre Youinou,*,1
and Christophe Jamin1
1Laboratory of Immunology, Institut de Synergie des Sciences et de la Santé;and 2Department of Rheumatology, Brest University Medical
School Hospital, Brest, France
IntroductionReceptors for the Fc portion of IgG (FcγRs) belong to the Ig gene
superfamily. They are expressed on immune effector cells, and by link-ing antibody specificity and effector cell function, they associate cellu-lar and humoral immunity. They might be markedly good candidatesfor an implication in the pathogenesis of autoimmune disorders. Aftera brief description of the structures and functions of membrane andsoluble FcγRs, we discuss their putative role in autoimmune diseasestogether with the presence of anti-FcγR autoantibodies.
Structure and Functions of Fc�Rs
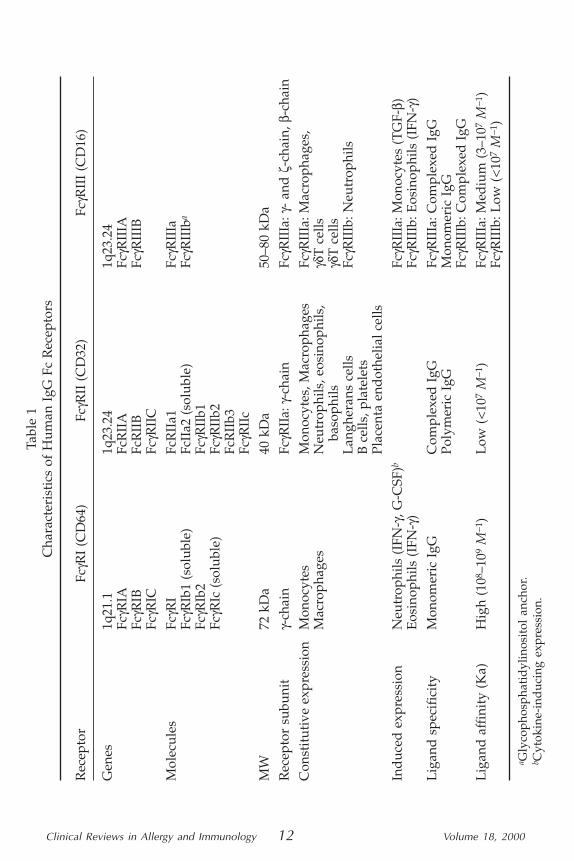
Fc�R StructuresThere are three classes of FcγR: FcγRI (CD64), FcγRII (CD32), and
FcγRIII (CD16) encoded by eight genes mapped on chromosome 1,permitting expression of nine membrane-associated and three solubleFcγR molecules (Table 1).
FcγRI is a 72 kDa glycoprotein that binds monomeric IgG withhigh affinity. This receptor is expressed by monocytes and macro-phages and can be induced on polymorphonuclear cells (PMN) andeosinophils following IFNγ or G-CSF stimulation. It is a transmembrane
Clinical Reviews in Allergy and Immunology© Copyright 2000 by Humana Press Inc.1080–0549/00/011–026 $14.00
Clinical Reviews in Allergy and Immunology 11 Volume 18, 2000
*Author to whom all correspondence and reprint requests should be addressed.
Clinical Reviews in Allergy and Immunology 12 Volume 18, 2000
Tabl
e 1
Cha
ract
eris
tics
of
Hum
an I
gG F
c R
ecep
tors
Rec
epto
rFc
γRI
(CD
64)
FcγR
II (
CD
32)
FcγR
III
(CD
16)
Gen
es1q
21.1
1q23
.24
1q23
.24
FcγR
IAFc
RII
AFc
γRII
IAFc
γRIB
FcR
IIB
FcγR
IIIB
FcγR
ICFc
γRII
CM
olec
ules
FcγR
IFc
RII
a1Fc
γRII
IaFc
γRIb
1 (s
olub
le)
FcII
a2 (
solu
ble)
FcγR
IIIb
a
FcγR
Ib2
FcγR
IIb1
FcγR
Ic (
solu
ble)
FcγR
IIb2
FcR
IIb3
FcγR
IIc
MW
72 k
Da
40 k
Da
50–8
0 kD
aR
ecep
tor
subu
nit
γ-ch
ain
FcγR
IIa:
γ-ch
ain
FcγR
IIIa
:γ-
and
ζ-c
hain
,β-c
hain
Con
stit
utiv
e ex
pres
sion
Mon
ocyt
esM
onoc
ytes
, Mac
roph
ages
FcγR
IIIa
: Mac
roph
ages
,M
acro
phag
esN
eutr
ophi
ls, e
osin
ophi
ls,
γδT
cel
lsba
soph
ilsγδ
T c
ells
Lan
gher
ans
cells
FcγR
IIIb
: Neu
trop
hils
B c
ells
, pla
tele
tsPl
acen
ta e
ndot
helia
l cel
lsIn
duc
ed e
xpre
ssio
nN
eutr
ophi
ls (
IFN
-γ, G
-CSF
)bFc
γRII
Ia: M
onoc
ytes
(T
GF-
β)E
osin
ophi
ls (
IFN
-γ)
FcγR
IIIb
: Eos
inop
hils
(IF
N-γ
)L
igan
d s
peci
fici
tyM
onom
eric
IgG
Com
plex
ed I
gGFc
γRII
Ia: C
ompl
exed
IgG
Poly
mer
ic I
gGM
onom
eric
IgG
FcγR
IIIb
: Com
plex
ed I
gGL
igan
d a
ffin
ity
(Ka)
Hig
h (1
08–1
09M
−1)
Low
(<
107
M−1
)Fc
γRII
Ia: M
ediu
m (
3–10
7M
−1)
FcγR
IIIb
: Low
(<
107
M−1
)
a Gly
coph
osph
atid
ylin
osit
ol a
ncho
r.b C
ytok
ine-
ind
ucin
g ex
pres
sion
.

receptor with an extracellular region consisting of three Ig-like domain,the third proximal-membrane of which is unique because it is notfound in the other class of FcγRs (Fig. 1). Three genes encode for fourdifferent isoforms using only one exon for the combined transmem-brane/cytoplasme region. Whereas the FcγRIA gene encodes FcγRIa, asingle molecule with three Ig-like domains, FcγRIB gene encodes twotranscripts, a full-length containing a stop codon in the latter exon(FcγRIb1 coding for a soluble molecule with two Ig-like domains), andone that lack this stop codon (FcγRb2 coding for a transmembrane mole-cule with two Ig-like domains). FcγRIC gene encodes only for a solubleform of FcγRIc, also owing to a stop codon in the extracellular domain (1).
FcγRII is a 40 kDa glycoprotein that binds complexed IgG or poly-meric form with low affinity. This receptor is constitutively broadlyexpressed (Table 1). This receptor is a transmembrane molecule withtwo Ig-like domains in the extracellular region. Three genes encodefor six isoforms, specifically using three exons encoding the cyto-plasmic tail, differing by their cytoplasmic region of variable lengths(Fig. 1). Four transmembrane isoforms—FcγRIIa1, RIIb1, RIIb3, andRIIc—are thus expressed, and a fifth isoform, a soluble form FcγRIIa2,may be generated by an alternate RNA splicing.
Furthermore, additional polymorphisms (2) are present on theFcγRIIa transmembrane form resulting from a single amino aciddifference at position 131: either an Arg (FcγRIIa-R131) or a histidine(FcγRII-H131) within the second Ig-like domain (Fig. 2). It is also note-worthy that the cytoplasmic region of FcγRIIa contains three immuno-receptor tyrosine activation motifs (ITAMs) that are important to itscellular-induced activation, whereas that of FcγRIIb contains a uniqueimmunoreceptor tyrosine inhibition motif (ITIM) that plays an impor-tant inhibitory-signaling function.
FcγRIII is a glycoprotein ranging between 50–80 kDa owing to ex-tensive glycosylation. This receptor is constitutively expressed on macro-phages, K/NK and γδT cells as a transmembrane molecule and on PMNas a glycosyl phosphatidylinositol (GPI) anchor, both with two Ig-likedomains. These two forms may be induced on monocytes by TGFβstimulation and on eosinophils after IFNγ stimulation, respectively(Table 1). These receptors bind complexed IgG with low affinity and areencoded by two genes. FcγRIIIA gene encodes the transmembrane form(FcγRIIIa) with a Phe at position 203 and FcγRIIIB gene encodes theGPI-anchor form (FcγRIIIb) owing to a Ser at position 203 (Fig. 1). Asoluble form resulting from proteolytic cleavage for both isoforms hasbeen detected in serum; the level of this, however, is derived mainly, ifnot wholly, from the FcγRIIIb molecule.
FcγRs and Autoimmune Diseases 13
Clinical Reviews in Allergy and Immunology Volume 18, 2000
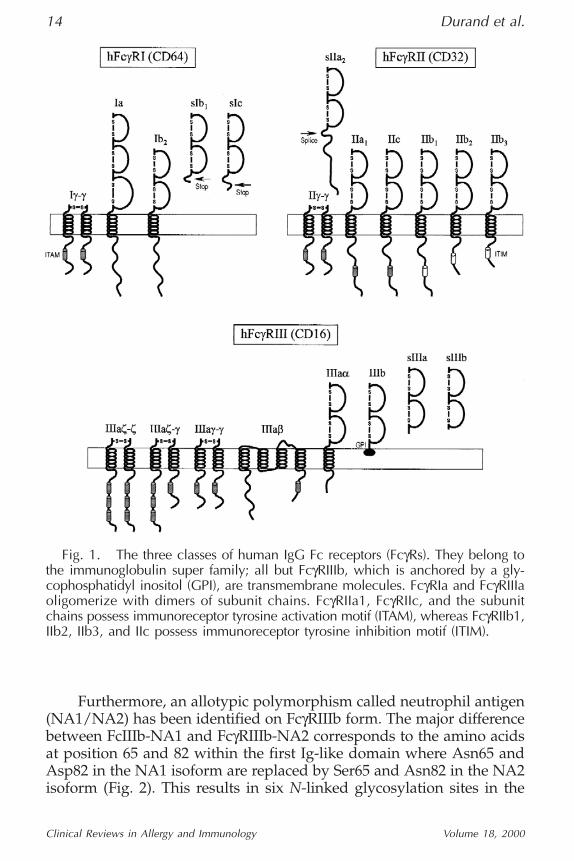
Furthermore, an allotypic polymorphism called neutrophil antigen(NA1/NA2) has been identified on FcγRIIIb form. The major differencebetween FcIIIb-NA1 and FcγRIIIb-NA2 corresponds to the amino acidsat position 65 and 82 within the first Ig-like domain where Asn65 andAsp82 in the NA1 isoform are replaced by Ser65 and Asn82 in the NA2isoform (Fig. 2). This results in six N-linked glycosylation sites in the
14 Durand et al.
Clinical Reviews in Allergy and Immunology Volume 18, 2000
Fig. 1. The three classes of human IgG Fc receptors (FcγRs). They belong tothe immunoglobulin super family; all but FcγRIIIb, which is anchored by a gly-cophosphatidyl inositol (GPI), are transmembrane molecules. FcγRIa and FcγRIIIaoligomerize with dimers of subunit chains. FcγRIIa1, FcγRIIc, and the subunitchains possess immunoreceptor tyrosine activation motif (ITAM), whereas FcγRIIb1,IIb2, IIb3, and IIc possess immunoreceptor tyrosine inhibition motif (ITIM).

NA2 isoform instead of four in NA1 and consistently explains the dif-ferences in molecular weights. In addition, the NB1 polymorphism isalso biallelic. Its epitope, which is not a carbohydrate, is expressed bya GPI-linked glycoprotein of 56–62 kDa on PMN (3), likely FcγRIIIb. Itis noteworthy that a new alloantigen as been characterized recently onFcγRIIIb. Named SH, this phenotype results from an Ala78Asp amino-acid substitution and seems to be shared by the NA2 form (4).
All FcγRs belong to the family of multichain immune recogni-tion receptors (MIRRs), and most of FcγRs exist as hetero-oligomericcomplexes. The ligand-binding α-chain can associate with signalingcomponents called FcRγ-, β- or ζ-chains, which form disulfide-linked
FcγRs and Autoimmune Diseases 15
Clinical Reviews in Allergy and Immunology Volume 18, 2000
Fig. 2. The human IgG Fc receptor (FcγR) polymorphism. FcγRIIa can be dis-tinguished by two allotypic forms. They differed by a single amino acid at posi-tion 131: either an arginine (FcγRIIa R131) or histidine (FcγRIIa H131). FcγRIIIbpossesses two allotypic isoforms differentiated by the substitution of two aminoacid: either an asparagine 65 and an apartic acid 82 (FcγRIIIb NA1) or serine 65and asparagine 82 (FcγRIIIb-NA2) leading to two additional N-linked glycosyla-tion sites (six instead of four in the NA1 isoform). Moreover, a new alloantigenSH resulting from Ala78Asp substitution has been identified recently as beingshared by the NA2 form.

dimers (γ-γ, ζ-ζ, γ-ζ) and bear a unique ITAM in their cytoplasmicdomain. Stable expression of FcγRIa requires its association with aγ-γ-chain homodimer. FcγRIIa is also capable of interacting with γ-γ-chain dimers, modulating its functional capacity. FcγRIIIa associateswith γ-γ-chain, ζ-ζ-chain homodimers as well as γ-ζ heterodimers,but also with a β-chain on most cells (Fig. 1).
Fc�R FunctionsFcγR-induced biological responses depend on the nature of the ef-
fector cell and the transmembrane and intracellular tail of the receptor.Moreover, MIRRs may interact with other molecule through the trans-membrane region in order to influence functional responses. Thus, onB-cells, crosslinking of FcγRIIb and the B-cell receptor downmodulatesantibody secretion. On PMN, FcγRIIa-mediated phagocytosis in en-hanced by FcγRIIIb cross-linking, whereas complement receptor 3(CR3) acts as a signaling partner for FcγRIIIb (reviewed in ref. 5).Moreover, it should be noted that PMN, which exclusively expressesFcγRIIIb, plays an important role in inflammation and clearance of im-mune complexes (IC) after activation. Following crosslinking of FcγRs,protein tyrosine kinase (PTK) src family associate and are activated, in-ducing the tyrosine phosphorylation of the ITAM. In turn, PTKs-sykfamily interact with their SH2 domains to the phosphorylated ITAMand are activated, initiating a cascade of biochemical events such as ac-tivation of phospholipase Cγ, Ca2+ mobilization, and phosphoinositideturnover, resulting in biological responses. These include phagocyto-sis, superoxide production and respiratory burst, antibody-dependentcellular cytotoxicity, mediator release, platelet activation, enhanced-antigen presentation, regulation of immunoglobulin secretion, and in-fluence on lymphoid and myeloid differentiation.
Moreover, it has been observed that the polymorphic forms ofFcγRs may differ in ligand binding. Thus, individuals expressingFcγRIIaR131 have been named high responders, whereas those ex-pressing FcγRIIaH131 were named low responders regarding their ca-pacity to bind murine IgG1. Clinically the response to therapeuticmurine monoclonal antibodies (MAbs) and the risk of infection withencapsulated organisms have been linked to this polymorphism (1).Furthermore, it has been observed that neutrophils from NA2/NA2homozygous individuals are less efficient in phagocytosis of opsonizedmicroorganisms than neutrophils from NA1/NA1 homozygous.
Soluble Fc�RsConcerning the soluble forms of FcγRs, it has been shown that
they may be generated by three different mechanisms (1). Soluble
16 Durand et al.
Clinical Reviews in Allergy and Immunology Volume 18, 2000

FcγRIb1 and FcγRIc are produced because of the presence of stopcodons in their extracellular domain, soluble FcγRIIa2 are generated fol-lowing alternate mRNA splicing and soluble FcγRIIIa, and FcγRIIIb arethe result of proteolytic cleavage. It is noteworthy that FcγRIIIb can beshed from PMN upon activation (6). Their functions have been re-viewed (7); they might act as IgG-binding factors. Moreover, solu-ble FcγRs are able to downregulate the antigen-presenting capacity ofantigen-presenting cells, to inhibit the proliferative response of B-cellsas well as the antigen or LPS-induced antibody production. Recentlysoluble FcγRIIIb has been shown to induce activation of monocytesthrough a CR3-dependent interaction, resulting in production of IL6and IL8 (8). These data suggest that soluble form of FcγRs can trigger andregulate immune functions following interaction with their ligands.
Fc�R and Autoimmune Diseases
Relationship Between Fc�Rs, anti-Fc�R Autoantibodies,and Clinical Settings
Because FcγRs are at the crossroads between humoral and cellu-lar immune responses, clinical involvements of these molecules willconcern not only host defense against microorganisms, but also hema-tologic disorders and autoimmune diseases. The following discussionwill focus on the latter pathophysiological situations.
In the case of systemic lupus erythematosus (SLE), patients werereported to have gene defects in FcγRIIIb (9). However, Sylvestre et al.(10) suggested a role for FcγRs in SLE patients during inflammationand tissue destruction, and from our own data (Durand, unpublishedobservations), no alteration of FcγRIIIb expression on SLE PMN cellscould be observed.
Furthermore, abnormal clearance of IgG-sensitized erythrocyteshas been observed in SLE patients. Because the numbers of FcγRs onSLE blood cells are normal, the defect in IC-clearance may be ascribedto an altered signal transduction. Several observations reported thatSLE patients presented higher frequency of FcγRIIa R/R131 or FcγRIIaR/H131 than FcγRIIa H/H131 allotypes when compared to healthycontrols, and markedly when SLE is associated with nephritis (2). Theimpaired IC-clearance therefore may be linked also to the expressionof particular FcγR polymorphism.
It is known that Wegener’s granulomatosis and systemic vasculi-tis are associated with anti-neutrophil cytoplasmic antibodies (ANCA),which are able to activate PMN through the binding of FcγRIIa. It hasbeen reported that superoxide production was increased following ac-tivation and mainly when PMN expressed FcγRIIa R/R131 phenotype.
FcγRs and Autoimmune Diseases 17
Clinical Reviews in Allergy and Immunology Volume 18, 2000

These data likewise support the idea that FcγRIIa allotypic polymor-phism may allow susceptibility for ANCA-induced inflammation (11).
Recently, studies have been focused on the detection of autoanti-bodies against FcγRs in patients with autoimmune disorders. Indeed,for the past decade, it has been shown that FcγRs and their allotypicpolymorphisms are targets of anti-neutrophil autoantibodies (12,13).Furthermore, anti-neutrophil antibodies have been found in a variety ofclinical settings associated with autoimmune neutropenia (14). Besides,it has been suggested that neonatal alloimmune neutropenia may be theresult of the transmission of either anti-FcγRIIIb antibodies from motherswho lack FcγRIIIb to their babies or of the transmission of anti-SH allo-antibodies from mother to the fetus (4). At the same time, reports (15)demonstrated the presence of anti-FcγR antibodies in the serum of pa-tients with SLE, progressive systemic sclerosis, and primary Sjögren’ssyndrome (pSS). These autoantibodies were found to be of IgG and IgMisotypes and directed against FcγRs of each class as determined byELISA tests with recombinant truncated murine FcγRs (16). In our ownstudies using ELISA assay with complete recombinant human FcγR, wefound the presence of anti-FcγRIIIb autoantibodies in the serum of 44%of SLE patients (Table 2) (Durand et al. unpublished observations).
We further explored the anti-FcγRs characteristics by studying thepresence of these autoantibodies in the serum of pSS patients. Usingthe same ELISA test (Table 2), we found that 45% of the pSS patientscontained anti-FcγRIIIb autoantibodies, 47% were of IgM isotype, 20%of IgG isotype, and 33% contained both isotype. Correlations havebeen done between the ELISA-positive sera and clinical settings (17) toshow that the frequency of IgG and IgM autoantibodies was higher inpatients with extraglandular manifestations (nonerosive arthritis, Ray-naud’s phenomenon and lung involvement) compared with patientswithout such complications or with renal disease. Moreover, the levelof autoantibodies correlated with the presence of IgA-containing ICand was higher in patients expressing the DR3 allele, which has beenassociated with the reduced clearance of IC in pSS patients (18). Incontrast, there was no relationship between autoantibodies activityand the PMN count, the level of serum IgG, or the degree of neutro-penia. Furthermore, using immunofluorescence (IIF) techniques (19),we observed that only 15% of pSS patients disclosed autoantibodies inthe serum recognizing membrane FcγRs. Among these positive sera,60% contained autoantibodies of IgG isotype, 30% contained both IgGand IgM isotype, whereas only 10% contained autoantibodies ofIgG and IgA isotype. In the description of the presence of anti-FcγRsautoantibodies, Boros et al. (15) hypothesized that they may affect theclearance of IC and phagocytosis. Since we found that anti-FcγRIIIb arepredominantly detected in pSS sera with IC, these data support the
18 Durand et al.
Clinical Reviews in Allergy and Immunology Volume 18, 2000

possibility that anti-FcγR autoantibodies may be responsible for the de-fective reticulo-endothelial system FcγR specific clearance describedin pSS patients. These data also suggest that anti-FcγRIIIb auto-antibodies may be an acquired additional factor in pSS further com-promising IC binding in individuals sharing HLA-DR3 alloantigen.Finally, correlation of anti-FcγRIIIb activity with IC led us to hypothe-size that IC might be responsible for the shedding of FcγRIIIb, since it islikely that IC and anti-FcγRIII autoantibodies might activate PMN, thusreflecting extension of the disease with production of autoantibodies.
We also evaluated the presence of anti-FcγR autoantibodies in theserum and synovial fluid (SF) of rheumatoid arthritis (RA) patientswith the same ELISA test. We found that 36% of these patients pre-sented anti-FcγRIIIb autoantibodies in their sera (Table 2) and 27% inthe corresponding SF (20). We further observed that 33% of positivesera contained antibodies of IgM isotype, 42% of IgG isotype, and25% contained both isotype. In the SF, 33% contained IgM autoanti-bodies, 45% were of IgG isotype, and 22% contained both isotype.
In order to ascribe a role for the anti-FcγR autoantibodies, we alsoevaluated the presence of soluble FcγRs.
Soluble Fc�Rs and Anti-Fc�R AutoantibodiesSoluble FcγRs were first described in human serum more than 10
years ago (21), and later in other body fluids (22). It has been demon-strated that this soluble receptor originate from PMN (6), upon stim-ulation (23) or following an apoptotic process (24). Although elevated
FcγRs and Autoimmune Diseases 19
Clinical Reviews in Allergy and Immunology Volume 18, 2000
Table 2Presence of Anti-FcγRIIIb Autoantibodies and Cell-Free FcγRIIIb in the Serumof Systemic Lupus Erythematosus (SLE), Primary Sjögren’s Syndrome (pSS),Rheumatoid Arthritis (RA) Patients and Controls, and in the Synovial Fluids
(SF) of RA and Non-RA Patientsa
Anti-FcγRIIIb autoantibodies Cell-free FcγRIIIb
Positive Negative Detectable NondetectablePatients (%) (%) (%) (%)
SLE 44 56 22 78pSS 45 55 35 65RA 36 64 67 33RA (SF) 27 73 45 55Control 6 94 2 98Control (SF) 0 100 15 85
aDetection was performed by ELISA tests using recombinant nonglycosylated humanFcγRIIIb and sandwich assay, respectively.

levels have been found in serum from HIV-infected patients (25), thepathophysiologic role of this soluble receptor is still unknown. It hasbeen demonstrated that soluble FcγRs were capable of inhibiting theArthus reaction (26), suggesting a role in type III hypersensitivity. Thiswas strengthened by the fact that FcγRIII-deficient mice as well as FcRγ-chain-deficient mice had reduced Arthus reaction (10,27). It should benoted that FcγRs also have a role in type II hypersensitivity disorderslike autoimmune hemolytic anemia and idiopathic thrombocytopeniapurpura. Indeed, in FcR(-chain-deficient mice, anti-platelet antibodieswere unable to induce thrombocytopenia (28). We studied the presenceof soluble FcγRIIIb in autoimmune disorders and looked for a possiblecorrelation with the presence of anti-FcγRIIIb autoantibodies.
We found soluble FcγRIIIb by a sandwich ELISA test in the serumof patients from SLE, pSS, and RA. In a recent study, we observed that22% of SLE patients had elevated soluble FcγRIIIb (Table 2), whereasnone of the control sera showed elevated levels of cell-free FcγRIIIb (29).Furthermore 22% of SLE patients were associated with renal involve-ment, and showed reduced levels of soluble FcγRIIIb compared withpatients without renal involvement. This observation suggested thatrenal failure is less frequent when cell-free FcγRIIIb in elevated. More-over 54% of patients displayed lymphopenia and had lower level ofcell-free FcγRIIIb compared to those SLE patients without lymphopeniasuggesting likewise that lymphopenia is more frequent when level ofsoluble FcγRIIIb is low. It is noteworthy that all other clinical and bio-logical features of SLE we studied did not correlated with cell-free Fc-γRIIIb level. In contrast to pSS and RA patients for whom the level ofcell-free FcγRb was higher when visceral complications occured, nosuch negative prognosis was associated with elevated soluble FcγRIIIb.
In pSS patients, 35% of sera contained detectable level of FcγRIIIbcompared to 8% of the controls (Table 2). When phenotyped, we ob-served that the proportion of FcγRIIIb-carrying PMN cells was de-creased in patients with high level of soluble FcγRIIIb (30). We furtherstudied PMN functions of these patients. Chemotaxis, but not phago-cytosis, adherence, bactericidal activity or chemiluminescence, was sig-nificantly reduced in pSS patients compared with controls (Table 3).However, regarding patients with cell-free FcγRIIIb, we also found a di-minished adherence and chemotaxis compared with pSS patients with-out detectable soluble FcγRIIIb. In contrast to the presence of anti-FcγRautoantibodies, no relationship could be found between the presence ofcell-free FcγRIIIb and the prevalence of HLA-DR3 antigen, mainlyowing to the small population of patients studied in the experiments.
When studying RA patients, we found that 67% of sera containedelevated level of soluble FcγRIIIb compared with 0% in control sera(Table 2). Furthermore, 45% of SF had elevated cell-free FcγRIIIb,
20 Durand et al.
Clinical Reviews in Allergy and Immunology Volume 18, 2000

whereas only 15% of SF from non-RA patients were also positive. Sincethe concentration of SF FcγRIIIb was higher than that of serum, it hasbeen suggested that cell-free FcγRIIIb may be related to inflammatoryprocess (22). It is noteworthy that patients displaying a high level ofsoluble FcγRIIIb in serum also displayed a high level in SF. As awhole, there was a strong correlation between the concentration ofcell-free FcγRIIIb in sera and SF. When phenotyped, density of FcγRI-IIb on PMN cells in blood and SF was lower in RA patients than incontrols achieving, like pSS patients, and inverse correlation betweenFcγRIIIb density on PMN surface and the level of soluble FcγRIIIb inblood as well as SF. We noted, in contrast, an increased expression ofCD35 and CD11b—which is associated with PMN activation—onPMN from blood and SF of RA patients compared with controls. Theincreased expression of activation markers was correlated to the levelof cell-free FcγRIIIb, suggesting that the soluble form of FcγRs mightbe released following activation of the cells in inflammatory sites.
Correlation studies were done to evaluate the relationship be-tween anti-FcγRIIIb autoantibodies and soluble FcγRIIIb. In pSS pa-tients, there were correlations between the levels of cell-free FcγRIIIband the titers of IgG and IgM autoantibodies (31). Anti-FcγRIIIb auto-antibodies levels were higher in patients with activated PMN andhigh levels of soluble FcγRIIIb. As a whole, we hypothesize that ICmight activate PMN, which might be responsible for the shedding ofFcγRIIIb, thus reflecting extension of the disease with production of anti-FcγRIIIb autoantibodies. A similar correlation can be made in the ser-um and SF of RA patients. We found that the level of cell-free FcγRIIIbin the serum and SF correlated with the amount of IgG and IgM anti-FcγRIIIb autoantibodies. One may also speculate that IgG-containing ICinduce PMN activation followed by the shedding of FcγRIIIb. These
FcγRs and Autoimmune Diseases 21
Clinical Reviews in Allergy and Immunology Volume 18, 2000
Table 3Detection of Cell-Free FcγRIIIb and Correlation with FχgRIIIb
Positive Cells and PMN Functionsa
pSS patients Controls
Cell-free FcγRIIIb + − + −Individuals (%) 35% 65% 9% 91%FcγRIIIb + PMN 87% 87% ND 98%Chemotaxis Highly reduced Reduced ND NormalAdherence Reduced Normal ND NormalPhagocytosis Normal Normal ND NormalBactericidal ability Normal Normal ND NormalChemiluminescence Normal Normal ND Normal
aCell-free FcγRIIIb was detected using a sandwich ELISA test.bND, not determined.

soluble receptors could then self-aggregate or bind to their ligand andthus become immunogenic. Consequently, anti-FcγRIIIb autoantibodieswould be produced. Yet, the pathogenic role of anti-FcγRs is still spec-ulative. When bound to the membrane FcγRs, they may inhibit thebinding of IC and thus their clearance. Alternatively, they may acti-vate PMN function normally induced by IC and thereby trigger inap-propriate degranulation, promoting tissue damage in inflammatorysites. When bound to cell-free FcγRs, they may inhibit the physiolog-ical effects of soluble FcγRs mainly on the control of immune responses.
Antigenic Specificities of Anti-Fc�R AntibodiesElicited data allowed us to distinguish different populations of
anti-FcγRIIIb autoantibodies. Indeed, using IIF technique (31), wedemonstrated the presence of autoantibodies recognizing membraneFcγRs on PMN surface. An ELISA test with a recombinant nonglyco-sylated human FcγRIIIb showed that some ELISA positive sera werenegative in the IIF assay. From these experiments and together withothers (Durand et al. unpublished observations), we identified threepopulations of patients (Table 4). Seven to 23% of autoimmune pa-tients contained anti-FcγRIIIb autoantibodies in their sera detectedboth in IIF assay and ELISA (IIF+/ELISA+ sera), 11 to 24% of sera con-tained autoantibodies detectable only in IIF technique (IIF+/ELISA-sera), and 11 to 32% of sera were anti-FcγRIIIb positive only by ELISA(IIF-/ELISA+ sera). The major difference between the two tests is that,the recombinant FcγRIIIb is a non-glycosylated form in the ELISA tech-nique in contrast to the natural membrane FcγRIIIb. However, whenisolated and bound onto ELISA plates, IIF+/ELISA-sera are able torecognize the natural receptor suggesting that epitopes are cryptic atthe surface or that changes occur after shedding.
In order to ascribe particular epitopes to the three autoantibod-ies-identified populations, sera from pSS patients were first tested bythe IIF technique on PMN expressing different polymorphismsNA1±/NA2±/NB1± (32). As shown in Table 5, the positivity re-mained unchanged when NB1+ or NB1− PMN were used as targetcells. This suggest that the NB1+/NB1− system is not important forthe binding of anti-FcγRs on membrane FcγRs. However, whereas allsera reacted with NA2− (NA1+) PMN, we found that 50% of the seradid not bind to the NA1− (NA2+) PMN. These data further suggestthat membrane-antigenic targets of anti-FcγR autoantibodies com-prised the NA1/NA2 system. Markedly, the NA2 isoform is not aspecific epitope of the autoantibodies, but the two additional glyco-sylation sites might hide antigenic targets of the NA1 system at leastfor the group II sera. It should be noted that these autoantibodies are
22 Durand et al.
Clinical Reviews in Allergy and Immunology Volume 18, 2000

not specifically directed to FcγRIII class since 60% also recognize theFcγRII class and 10% also FcγRII and FcγRI classes (31).
Moreover, though group II sera recognize the natural glycosy-lated NA1+ isoform of FcγRIIIb even when extracted from PMN andcoated onto plates (19), they do not bind further to this receptor whenit has been previously deglycosylated with N-glycanase treatment(Table 5), in contrast to the group I sera. All together, these experi-ments strongly suggest that group I autoantibodies recognize a nongly-cosylated peptidic region independent on the glycosylation sites of theNA1 and NA2 isoforms. The group II autoantibodies likely target aglyscosylated domain of the NA1 isoform that may be hidden by the twoadditional glycosylations sites of the NA2 isoform. Finally, the groupIII autoantibodies might bind a nonglycosylated region of FcγRIIIbthat may be masked by the glycosylation sites of the NA1 isoform.
The pathological implications of anti-FcγR autoantibodies are notyet clearly understand. Our observations let us to think that in auto-immune disorders, they might be produced consequently to the shed-ding of soluble FcγRs which is the result of PMN activation likely byIC. In turn, on the one hand they probably influence the PMN FcγR-dependent functions, and this may be part of the group I and IIautoantibodies population. On the other hand, they will be able tomodulate soluble FcγRs biological functions, and likely this might bepart of the group III anti-FcγR autoantibodies. Our preliminary ex-periments strengthened these hypothesis. We demonstrated that groupI and group II autoantibodies are capable of decreasing adhesivenessof PMN on endothelial cells by lowering CD11b density on PMN sur-face. Moreover, in these experiments, we also observed that sponta-neous apoptosis of PMN cells was delayed associated with decreased
FcγRs and Autoimmune Diseases 23
Clinical Reviews in Allergy and Immunology Volume 18, 2000
Table 4Distribution of Anti-FcγR Autoantibodies in Sera from Systemic Lupus
Erythematosus (SLE), Primary Sjögren’s Syndrome (pSS), Rheumatoid Arthritis(RA) Patients and Controls Between Indirect Immunofluorescence (IIF),
and ELISA Detection Methodsa
Assay Individuals
Population IIF ELISA SLE (%) pSS (%) RA (%) Controls (%)
Group I + + 9 9 7 0Group II + − 11 11 24 0Group III − + 32 32 20 6Group IV − − 48 48 49 94
aThe IIF technique was performed on NA1+NA2+NB1+ PMN cells and the ELISA testusing recombinant nonglycosylated human FcγRIIIb.

Clinical Reviews in Allergy and Immunology 24 Volume 18, 2000
Tabl
e 5
Spec
ific
itie
s an
d I
mpo
rtan
ce o
f th
e G
lyco
syla
tion
Sit
es in
the
NA
1 Is
ofor
m o
f A
nti-
FcγR
Aut
oant
ibod
ies
from
Pri
mar
y Sj
ögre
n’s
Synd
rom
e (p
SS)
Pati
ents
a
EL
ISA
ass
ay
IIF
assa
yG
lyco
syla
ted
Non
glyc
osyl
ated
pSS
pati
ents
NA
1+N
A2+
NB
1+N
A1+
NA
2+N
B1+
NA
1+N
A1+
NB
1+N
A2+
NA
2+N
B1+
NA
1+ F
cγR
IIIb
NA
1+ F
cγR
IIIb
Gro
up I
++
++
++
Gro
up I
I+
++
−+
−G
roup
III
−−
−−
−±
a Spe
cifi
city
of
anti
-Fcγ
Rs
auto
anti
bod
ies
was
tes
ted
in
the
ind
irec
t im
mun
oflu
ores
cenc
e (I
IF)
assa
y us
ing
PMN
cel
ls f
rom
don
ors
expr
essi
ng d
iffer
-en
t al
loty
pic
poly
mor
phis
m,
and
the
im
port
ance
of
glyc
osyl
atio
n fo
r an
ti-F
cγR
s ac
tivi
ty w
as t
este
d b
y E
LIS
A t
est
usin
g no
rmal
gly
cosy
late
d o
rN
-gly
cana
se-t
reat
ed F
cγR
IIIb
ext
ract
ed f
rom
NA
1+N
A1+
PM
N c
ells
.

chemiluminescence. This strongly suggests that group I and II autoanti-bodies, by prolonging the life of PMN cells and having a low level offunctional capacities, will favor chronicity of the diseases. Furthermore,we observed that group III autoantibodies are able to inhibit the bind-ing of soluble FcγRs, further suggested that they may control solubleFcγR-dependent immune reactions. Therefore the group III autoantibod-ies is also likely able to influence the dysregulated-immune responsesobserved in autoimmune disorders.
References1. van de Winkel JGJ, Capel JA. Human IgG Fc receptor heterogeneity: molecular
espects and clinical implications. Immunol Today 1993; 14:215–221.2. Rascu A, Repp R, Westerdaal NAC, Kalden JR, van de Winkel JGJ. Clinical rel-
evance of Fcγ receptor polymorphisms. Ann New York Acad Sci 1997;815:282–295.
3. Goldschmeding R, van Dalen CM, Faber N, Calafat J, Huizinga TWJ, van derSchoot CE, et al. Further characterization of the NB 1 antigen as a variably ex-pressed 56–62 kD GPI-linked glycoprotein of plasma membranes and specificgranules of neutrophils. Br J Haematol 1992; 81:336–345.
4. Bux J, Stein EL, Bierling P, Fromont P, Clay M, Stronceck D, Santoso S. Charac-terization of a new alloantigen (SH) on the human neutrophil Fcγreceptor IIIb.Blood 1997; 89:1027–1034.
5. Deo YM, Graziano RF, Repp R, van de Winkel JGJ. Clinical significance of IgG Fcreceptors and FcγR-directed immunotherapies. Immunol Today 1997; 18:127–135.
6. Huizinga TWJ, van der Schoot CE, Jost C, Klaassen R, Kleijer M, von dem BorneAEGKr, et al. The PI-linked receptor FcRIII is released on stimulation of neu-trophils. Nature 1988; 333:667–669.
7. Galon J, Paulet P, Galinha A, Lorès P, Bonnerot C, Jami J, et al. Soluble Fcγ re-ceptors: interaction with ligands and biological consequences. Int Rev Immunol1997; 16:87–111.
8. Galon J, Gauchat JF, Mazières N, Spagnoli R, Storkus W, Lötze M, et al. SolubleFcg receptor type III (FcγRIII, CD16) triggers cell activation through interactionwith complement receptors. J Immunol 1996; 157:1184–1192.
9. Clarck MR, Liu L, Clarkson SB, Ory PA, Goldstein IM. An abnormality of thegene that encodes neutrophil Fc receptor III in a patient with systemic lupuserythematosus. J Clin Invest 1990; 86:341–346.
10. Sylvestre DL, Ravetch JV. Fc receptors initiate the Arthus reaction: redefiningthe inflammatory cascade. Science 1994; 265:1095–1098.
11. Porges AJ, Redecha PB, Kimberly WT, Csernock E, Gross WL, Kimberly RP.Anti-neutrophil cytoplasmic antibodies engage and activate human neutrophilsvia FcγRIIa. J Immunol 1994; 153:1271–1280.
12. Rothko K, Kickler TS, Clay ME, Johnson RJ, Stroncek DF. Immunoblotting char-acterization of neutrophil antigenic targets in autoimmune neutropania. Blood1989; 74:1698–1703.
13. Bux J, Robertz-Vaupel GM, Glasmacher A, Dengler HJ, Mueller-Eckhardt C. Au-toimmune neutropenia due to NA1 specific antibodies in primary biliary cir-rhosis. Br J Haematol 1991; 77:121–126.
14. Shastri KA, Logue GL. Autoimmune neutropenia. Blood 1993; 81:1984–1988.15. Boros P, Muryoi T, Spiera H, Bona C, Unkeless JC. Autoantibodies directed
against different classes of FcγR are fond in sera of autoimmune patients. J Im-munol 1993; 150:2018–2024.
FcγRs and Autoimmune Diseases 25
Clinical Reviews in Allergy and Immunology Volume 18, 2000

16. Boros P, Odin A, Chen J, Unkeless JC. Specificity and class distribution of FcγR-specific autoantibodies in patients with autoimmune disease. J Immunol 1994;152:302–305.
17. Lamour A, Le Corre R, Pennec YL, Youinou P. The presence of anti-Fc gamma re-ceptor autoantibodies is related to the clinical presentation of primary Sjögren’ssyndrome. J Rgeumatol 1995; 22:2241–2245.
18. Salmon JE, Kimberly RP, Gibofski A, Fotino M. Altered phagocytosis by mono-cytes from HLA-DR2 and DR3-positive healthy adults in Fc-gamma receptor. JImmunol 1986; 136:3625–3630.
19. Lamour A, Le Corre R, Pennec YL, Cartron J, Youinou P. Heterogeneity of neu-trophil antibodies in patients with primary Sjögren’s syndrome. Blood 1995;86:3553–3559, 1995.
20. Lamour A, Baron B, Soubrane C, Cartron J, Khayat D, Adler Y, et al. Anti-Fcgamma receptor III autoantibody is associated with soluble receptor in rheuma-toid arthritis serum and synovial fluid. J Autoimmun 1995; 8:249–265.
21. Khayat D, Geffrier C, Yoon S, Scigliano E, Soubrane C, Weil M, et al. Solublecirculating Fcγ receptors in human serum. A new ELISA assay for specific andquantitative detection. J Immunol Methods 1987; 100:235–241.
22. Fleit HB, Kobasiuk CD, Daly C, Furie R, Levy PC, Webster RO. A soluble formof FcγRIII is present in human serum and other body fluids and is elevated atsites of inflammation. Blood 1992; 79:2721–2728.
23. Huizinga TWJ, de Haas M, Kleijers M, Huijens JN, Roos D, von dem BorneAEGKr. Soluble FcγRIII in human plasma originates from releases by neu-trophils. J Clin Invest 1990; 86:416–423.
24. Homburg CHE, de Haas M, von dem Borne AEGKr, Verhoeven AJ, Reutel-ingsperger CPM, Roos D. Human neutrophils lose their surface FcγRIII and ac-quire annexin V binding sites during apoptosis in vitro. Blood 1995; 85:532–540.
25. Khayat D, Soubrane C, Andrieu JM, Visonneau S, Eme D, Tourani JM, et al.Changes of soluble CD16 levels in HIV-infected patients; correlation with clini-cal and biological prognostic factors. J Infect Dis 1989; 161:431–435.
26. Ierino FL, Powell MS, McKenzie FC, Hogarth PM. Recombinant soluble FcγRII:production, characterization, and inhibition of the Arthus reaction J Exp Med1993; 178:1617–1628.
27. Hazenbos WLW, Gessner JE, Hofhuis FMA. Impaired IgG-dependent anaphy-laxis and Arthus reaction in Fc gamma RIII (CD16) deficient mice. Immunity1996; 5:181–188.
28. Clynes R, Ravetch JV. Cytotoxic antibodies trigger inflammation through Fc re-ceptors. Immunity 1995; 3:21–26.
29. Hutin P, Lamour A, Pennec YL, Soubrane C, Dien G, Khayat D, Youinou P. Cell-free Fc-gamma receptor III in sera from patients with systemic lupus erythe-matosus: correlation with clinical and biological features. Int Arch AllergyImmunol 1994; 103:23–27.
30. Lamour A, Soubrane C, Ichen M, Pennec YL, Khayat D, Youinou P. Fc-gammareceptor III shedding by polymorphonuclear cells in primary Sjögren’s syn-drome. Eur J Clin Invest 1993; 23:97–101.
31. Lamour A, Le Corre R, Soubrane C,Khayat D, Youinou P. Anti-Fc gamma re-ceptor autoantibodies from patients with Sjögren’s syndrome do not react withnative receptor on human polymorphonuclear leukocytes. J Autoimmun 1996;9:181–191.
32. Lamour A, Le Corre R, Jamin C, Soubrane C, Khayat D, Youinou P. Novel en-zyme-linked immunosorbent assay for the detection of anti-Fc gamma receptorautoantibodies. Clin Diag Lab Immunol 1996; 3:315–320, 1996.
26 Durand et al.
Clinical Reviews in Allergy and Immunology Volume 18, 2000





























