Embed Size (px)
Citation preview

| 1
FDA Harmonization with ISO 13485:2016 and MDSAP
Implementation
Kimberly TrautmanMedical Device, IVD and Combination Product Expert
UGA and FDA Co-SponsoredMedical Device Regulations Conference

| 2
KIM TRAUTMAN, M.S.Medical Device, IVD and Combination Product Expert
EXPERIENCEKimberly A. Trautman is an experienced Medical Devices, InVitro Diagnostics, and Combination Product Expert with over 30 years of experience. She worked at the US Food and Drug Administration (FDA) for 24 years and continues to work with Regulatory Agencies around the globe. Industry experience as well as regulatory agency experience. Demonstrated history of working collaboratively with industry, regulators and patient groups for the betterment of public health. Executes several medical device regulatory services and developed a formal Education/Training business. Established an Authorized Medical Device Single Audit Program (MDSAP) Auditing Organization and a new Notified Body for EU IVDR/MDR Designation.
Expert in global medical device regulations, wrote and harmonized the current US FDA Quality System Regulation and was on the international authoring group of ISO 13485 since inception. Conceived and developed the Medical Device Single Audit Program and its consortium of five Global Regulators. Twenty-year veteran of the Global Harmonization Tasks Force (GHTF) and foundational member of the International Medical Device Regulators Forum (IMDRF).
EDUCATIONM.S. of Biomedical & Medical Engineering, University of Virginia, Charlottesville, VAB.Sc. of Molecular Cell Biology and Engineering Sciences, Pennsylvania State University, State College, PA
| 3
Key Takeaways
Understand where there are similarities and differences in the two QMS requirements
Explore the FDA required process of rulemaking
Discuss how FDA might use some aspects of MDSAP and where they will likely need to still do their own types of inspections

| 4
US FDA Proposed Rule for 21 CFR 820
for Revisions to Harmonize with ISO
13485:2016

| 5
21 CFR 820 Revisions
Purpose of the Initiative
“Givens” – Definitions and Ties
Some Major Areas for Change

| 6
21 CFR 820 Revisions to Harmonize with ISO 13485:2016
➢Harmonization and convergence initiatives over the years
➢White House Agenda to reduce regulatory burden
➢Extension of success of MDSAP
➢OMB Rule Making - Comment and Reviews in 2020
➢After FINAL Rule published US FDA Transition – 3 years mostly for FDA!

| 7
© NSF HEALTH SCIENCES
GIVENS
21 CFR 820 Revisions to Harmonize with ISO 13485:2016
➢ Some definitions will be unique to 820
❖ Component❖ Finished device❖Manufacturer❖ Remanufacturer
➢ Links to UDI and 803

| 8
21 CFR 820 Revisions to Harmonize with ISO 13485:2016
AREAS FOR CHANGE
➢Utilization of risk management practices and activities throughout the lifecycle of the medical device;
➢Purchasing controls or supply chain management to include external as well as internal suppliers;

| 9
21 CFR 820 Revisions to Harmonize with ISO 13485:2016
AREAS FOR CHANGE
➢ Stronger delineation of roles and responsibilities for functions under applicable regulatory requirements to include Manufacturers, Authorized Representatives, and Importers or Distributors;
➢ QS regulation’s labeling requirements, where there are more pronounced differences between the regulation and ISO 13485:2016. 820.120 Device labelling, prescriptive nature could be softened

| 10
21 CFR 820 Revisions to Harmonize with ISO 13485:2016
❖U.S. FDA 21 CFR mapping to the applicable regulatory requirement references in ISO 13485:2016
❖Bi-directional
| 11
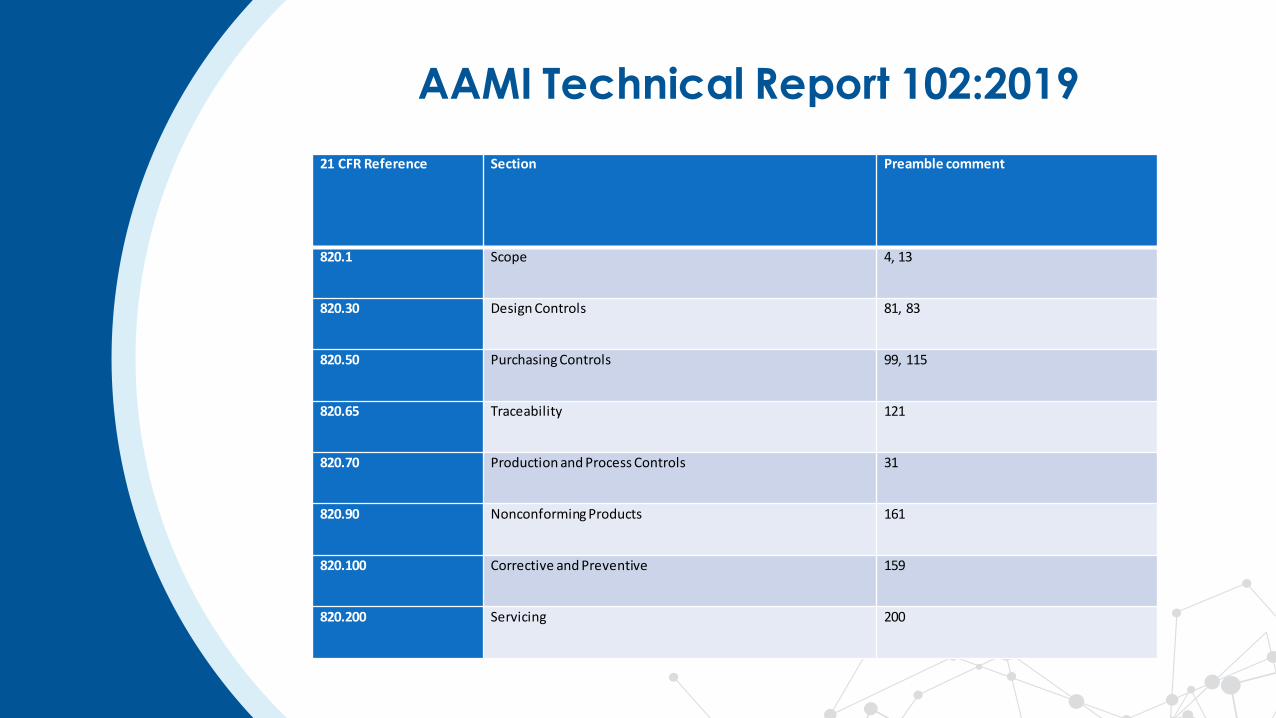
AAMI Technical Report 102:2019
21 CFR Reference Section Preamble comment
820.1 Scope 4, 13
820.30 Design Controls 81, 83
820.50 Purchasing Controls 99, 115
820.65 Traceability 121
820.70 Production and Process Controls 31
820.90 Nonconforming Products 161
820.100 Corrective and Preventive 159
820.200 Servicing 200

| 12
21 CFR 820 Revisions to Harmonize with ISO 13485:2016
CAPA VS IMPROVEMENT PROCESS
The concept of “CAPA” – Corrective Action and Preventive Action has progressed and been refined within both ISO 13485 and ISO 9001 over the past two decades, since the QS regulation was published in 1996. The QS regulation based off the ISO 9001:1994 standard has the combined concepts of analysis, corrective action and preventive action all in one section 820.100, which does not separate out many important aspects of these critical processes.

| 13
21 CFR 820 Revisions to Harmonize with ISO 13485:2016
AAMI TECHNICAL REPORT 102:2019
Table 1: QS regulation and risk related preamble comments
The terminology used in the QS regulation is only one component of the risk management process as it includes risk analysis, risk evaluation, risk control, and production and post-production information collection and review. Although the terminology in the QS regulation is not encompassing of the entire risk management process, the expectation is that full risk management activities occur across the entire medical device lifecycle.

| 14
Requirements for Risk Management

| 15
Risk Management
International Standard
ISO 14971: 2019

| 1616
PREVIOUS
Table of Contents ComparisonRisk Management
1 Scope
2 Terms and definitions
3 General requirements for risk management
3.1 Risk management process
3.2 Management responsibilities
3.3 Qualification of personnel
3.4 Risk management plan
3.5 Risk management file
4 Risk analysis
4.1 Risk analysis process
4.2 Intended use and identification of characteristics related to the safety of the medical device
4.3 Identification of hazards
4.4 Estimation of the risk(s) for each hazardous situation
4.5 Risk estimation
5 Risk evaluation
1 Scope
2 Normative references
3 Terms and definitions
4 General requirements for risk management system
4.1 Risk management process
4.2 Management responsibilities
4.3 Competence of personnel
4.4 Risk management plan
4.5 Risk management file
5 Risk analysis
5.1 Risk analysis process
5.2 Intended use and reasonably foreseeable misuse
5.3 Identification of characteristics related to safety
5.4 Identification of hazards and hazardous situations
5.5 Risk estimation
6 Risk evaluation
7 Risk control
7.1 Risk control option analysis
7.2 Implementation of risk control measures
7.3 Residual risk evaluation
7.4 Benefit-risk analysis
7.5 Risks arising from risk control measures
7.6 Completeness of risk control
8 Evaluation of overall residual risk
9 Risk management review
10 Production and post-production activities
10.1 General
10.2 Information collection
10.3 Information review
10.4 Actions
6 Risk control
6.1 Risk reduction
6.2 Risk control option analysis
6.3 Implementation of risk control measures
6.4 Residual risk evaluation
6.5 Risk/benefit analysis
6.6 Risks arising from risk control measures
6.7 Completeness of risk control
7 Evaluation of overall residual risk acceptability
8 Risk management report
9 Production and post-production information

| 17
ISO/TR 24971:2020 ISSUED JUNE 2020Medical devices — Guidance on the application of ISO 14971Summary
➢ This document provides guidance on the development, implementation and maintenance of a risk management system for medical devices according to ISO 14971:2019.
➢ The risk management process can be part of a quality management system, for example one that is based on ISO 13485:2016, but this is not required by ISO 14971:2019. Some requirements in ISO 13485:2016 (Clause 7 on product realization and 8.2.1 on feedback during monitoring and measurement) are related to risk management and can be fulfilled by applying ISO 14971:2019.
Risk Management

| 18
Influencers

| 19
ISO 13485 and ISO Guide 83 or
High Level Structure

| 20
2
0
© NSF HEALTH SCIENCES
ISO 13485 and ISO Guide 83 or High Level Structure
REVISED ANNEX SL APPENDIX 2 (HLS)Appendix 2 of Annex SL provides the high-level structure (HLS) and identical text for management system standards and common core management system terms and definitions.

| 21
ISO 13485 and ISO Guide 83 or High Level Structure
ISO Technical Committee 210’s Position
“ISO/TC 210 is very supportive of a common approach for management system standards and is happy toparticipate in the discussions of TF14 and JTCG on revision of the current texts in Annex L. ISO/TC 210 has the following considerations that force us to vote negatively:”

| 22
ISO 13485 and ISO Guide 83 or High Level Structure
ISO Technical Committee 210’s Position
“1. Firstly, there is the semantic discussion on terminology around "risk." ISO/TC 210 is convinced that the definition attached to the term "risk" in Annex L is too remote from the understanding of "risk" with the general public, and not in line with the regulatory definition of that same term applicable to medical devices in many jurisdictions. It has repeatedly been made clear that such terminology is not acceptable in a standard supposed to be used in regulatory context for the sector ISO/TC 210 serves. Nevertheless, TF14 decided to not provide sufficient leeway.”

| 23
© NSF HEALTH SCIENCES
ISO Technical Committee 210’s Position
ISO 13485 and ISO Guide 83 or High Level Structure
“2. Secondly, it is realised that there are provisions in Annex L for sector specific requirements and for deviations from the core text. Unfortunately, sector specific material is only allowed as additional text inthe mandatory structure and normative text. Deviations from the core text are subject to approval of a deviation report. ISO/TC 210 considers that the normative text in Annex L deviates more from the sector specific needs, both terminology and requirements, than reasonably can be covered in deviation reports.”

| 24
ISO 13485 and ISO Guide 83 or High Level Structure
“ISO/TC 210 is committed to operate within the context of the ISO/IEC Directives to the extent that ISO ensures that ISO/TC 210 can continue to serve its users and international regulatory authorities with a standard –ISO 13485- that over the past 3 decades has established a very large, and loyal customer base, and that continues to play a major role in global harmonisation of regulatory practice.”
ISO Technical Committee 210’s Position

| 25
All management systemstandards (including ISO 13485) in the future will have the same high level structure, identical core text, as well ascommon terms and definitions.
Whilst the high level structurecannot be changed, sub-clausesand discipline-specific text canbe added.
Unless deviations are approved!
ISO 13485 and ISO Guide 83 or High Level Structure

| 26
ISO Guide 83 or High Level Structure –Approved with Minor Revisions

| 27
ISO 13485 and ISO Guide 83 or High Level Structure
Major issue is the potential conflict on the Definition of “Risk” which in ISO 14971:2019 differs from ISO 31000:2018
➢ISO 31000:2018 definition of Risk "effect of uncertainty on objectives" ... thus causing the word "risk" to refer to positive consequences of uncertainty, as well as negative ones.
➢ISO 14971:2019 definition of Risk “combination of the probability of occurrence of harm and the severity of that harm”

| 28
EU MDR and EU IVDREU CEN/TR 17223:2018

| 29
Feedback (ISO 13485:2016, 8.2.1)
As one of the measurements of the performance of the quality
management system, the organization shall gather and monitor
information relating to whether the organization has met customer
requirements. Methods for obtaining and using this information
shall be documented.
The organization shall document procedures for the feedback
process, which shall include the provisions to gather data from
production, as well as post-production activities.
The information gathered in the feedback process shall serve as
potential input into risk management for monitoring and
maintaining the product requirements, as well as product
realization or improvement processes.
If applicable regulatory requirements require the organization to
gain specific experience from post-production activities, the review
of this experience shall form part of the feedback process.
Post-Market Surveillance

| 30PMS Requirements – The PMS System
Data gathered by the manufacturer's post-market surveillance system shall in particular be used:
• to update the benefit-risk determination and to improve the risk management;
• to update the design and manufacturing information, the instructions for use and the labelling;
• to update the clinical evaluation;
• to update the summary of safety and clinical performance;
• for the identification of needs for preventive, corrective or field safety corrective action;
• for the identification of options to improve the usability, performance and safety of the device;
• when relevant, to contribute to the post-market surveillance of other devices; and
• to detect and report trends.
EN ISO 14971
PMS per MDD
EN ISO 13485
PMS per MDD
MEDDEV 2.12/1
New
Current Practice
MDD/ MEDDEV 2.7/1
EN ISO 14971
MDR Article 32
[ Regulation (EU) 2017/745 Article 83 ]

| 31
EU CEN/TR 17223

| 32
EU CEN/TR 17223
This Technical Report has been prepared to provide guidance on the relationship between EN ISO 13485:2016 (Medical devices – Quality management systems –Requirements for regulatory purposes) and the requirements in the European Regulations on Medical Devices (MDR)-Regulation (EU) 2017/745 - and in vitro Diagnostic Medical Devices (IVDR) -Regulation (EU) 2017/746.

| 33
Relationship between the European Regulations for Medical Devices and in vitro Diagnostic Medical Devices and the clauses of EN ISO 13485
Table 1 shows the relationship between the clauses of EN ISO 13485 and the requirements of the European Regulations on Medical Devices (Regulation (EU) 2017/745), together with commentary on the extent to which the requirements of the standard cover the specific details in the Regulation.
Table 2 shows the relationship between the clauses of EN ISO 13485 and the requirements of the European Regulations on in vitro Diagnostic Medical Devices (Regulation (EU) 2017/746), together with commentary on the extent to which the requirements of the standard cover the specific details in the Regulation.
EU CEN/TR 17223

| 34
International Medical Device
Regulators Forum (IMDRF)
Medical Device Single Audit
Program (MDSAP)

| 35
IMDRF MDSAP:Harmonization Efforts FDA Needs to Consider due to International Convergence Effects since 2011

| 36
IMDRF and MDSAP
➢The International Medical Device Regulators Forum (IMDRF) recognized the value in developing a global approach to auditing and monitoring the manufacturing of medical devices to ensure safe medical devices
➢The IMDRF, at its inaugural meeting in Singapore in 2012, identified a Work Group to develop specific documents for advancing the concept of the Medical Device Single Audit Program (MDSAP)

| 37
Therapeutic Goods
Administration (TGA)
Agência Nacional de Vigilância
Sanitária (ANVISA)
Health Canada(HC)
U.S. Food and Drug
Administration (FDA)
Pharmaceuticals and Medical
Devices Agency (PMDA)
The International Consortium of countries for MDSAP:

| 38| 38
MDSAP International Consortium
➢ 2014 Added Observers:
• World Health Organization (WHO) Diagnostic Prequalification Program
• European Union
➢ 2019 Added Affiliates:
• ANMAT – Argentina’s National Administration of Drugs, Foods and Medical Devices
• Republic of Korea’s Ministry of Food and Drug Safety

| 39
MDSAP Audit Criteria
The MDSAP audit process was designed and developed to ensure a single audit will provide efficient yet thorough coverage of the quality management system requirements:
➢ISO 13485:2016
➢Brazilian Good Manufacturing Practices (ANVISA RDC 16)
➢Japanese requirements (MHLW MO 169)
➢FDA’s Quality System Regulation (21 CFR Part 820)

| 40
MDSAP Audit Criteria
AND other specific requirements of medical device regulatory authorities participating in the Pilot MDSAP program such as:
➢ registration
➢ licensing
➢ adverse event reporting and more

| 41
Thank you for participating!
Q&A





























