Embed Size (px)
Citation preview

PRENATAL DIAGNOSISPrenat Diagn 2008; 28: 862–864.Published online 22 July 2008 in Wiley InterScience(www.interscience.wiley.com) DOI: 10.1002/pd.2058
RESEARCH LETTER
Fetal anemia and hydrops associated with homozygosityfor hemoglobin Quong Sze
Can Liao, Jian Li and Dong-Zhi Li*Prenatal Diagnostic Center, Guangzhou Maternal & Neonatal Hospital, Guangzhou Women and Children’s Medical CareCenter, Guangzhou Medical College, Guangzhou, Guangdong, China
KEY WORDS: α-thalassemia; hemoglobin Quong Sze; hydrops fetalis
The α-thalassemias are the most common inheriteddisorders of hemoglobin (Hb) synthesis in southernChina with a prevalence rate of 8.53% (Xu et al., 2004).These disorders arise from deletions or mutations (orboth) of α-globin genes, of which there are four inthe normal genome. The severest form of α-thalassemiais Hb Bart’s hydrops fetalis, in which all four α-globin genes are missing, and the affected individualsare, with rare exceptions, lethal in utero or shortlyafter birth. Occasionally, hydrops fetalis can be causedby Hb H disease, a condition called Hb H hydropsfetalis syndrome (Chan et al., 1997; Lorey et al., 2001;Viprakasit et al., 2002; Li et al., 2005). In this report,we describe a case of fetal anemia and hydrops resultingfrom homozygous Hb Quong Sze (QS), which is rare.
A Chinese couple presented at our center for the firstprenatal visit at 10 weeks of gestation. This was thesecond pregnancy. The first pregnancy, 8 years previ-ously, was uneventful, and the daughter was healthy.The mother’s medical history was nonsignificant, andshe denied having taken any medications. Because theinformation about their previous thalassemia testing wasunavailable, blood samples were obtained from the cou-ple for screening. Hematological studies suggested thatboth parents had an α-thalassemia trait (Table 1). Gap-polymerase chain reaction (PCR) was performed todetect the three common Chinese α-globin gene dele-tion mutations (−SEA, −α3.7, and −α4.2), and this failedto detect any of these alleles in the parents. A com-mon nondeletional α-thalassemia in Chinese, Hb con-stant spring (CS), was also excluded in the couple byusing high-performance liquid chromatography (HPLC)(Tangvarasittichai et al., 2005). The couple was thencounseled regarding the possible clinical outcomes. Itwas explained that the chance of having an offspringwith severe α-thalassemia was remote, since they werenot carriers of the common deletional and nondeletionaldefects.
A scan at 12 weeks demonstrated a normal nuchaltranslucency (NT) measurement (1.5 mm). However,
*Correspondence to: Dong-Zhi Li, Prenatal Diagnostic Center,Guangzhou Maternal & Neonatal Hospital, Renminzhong Road402, Guangzhou, Guangdong 510 180, China.E-mail: [email protected]
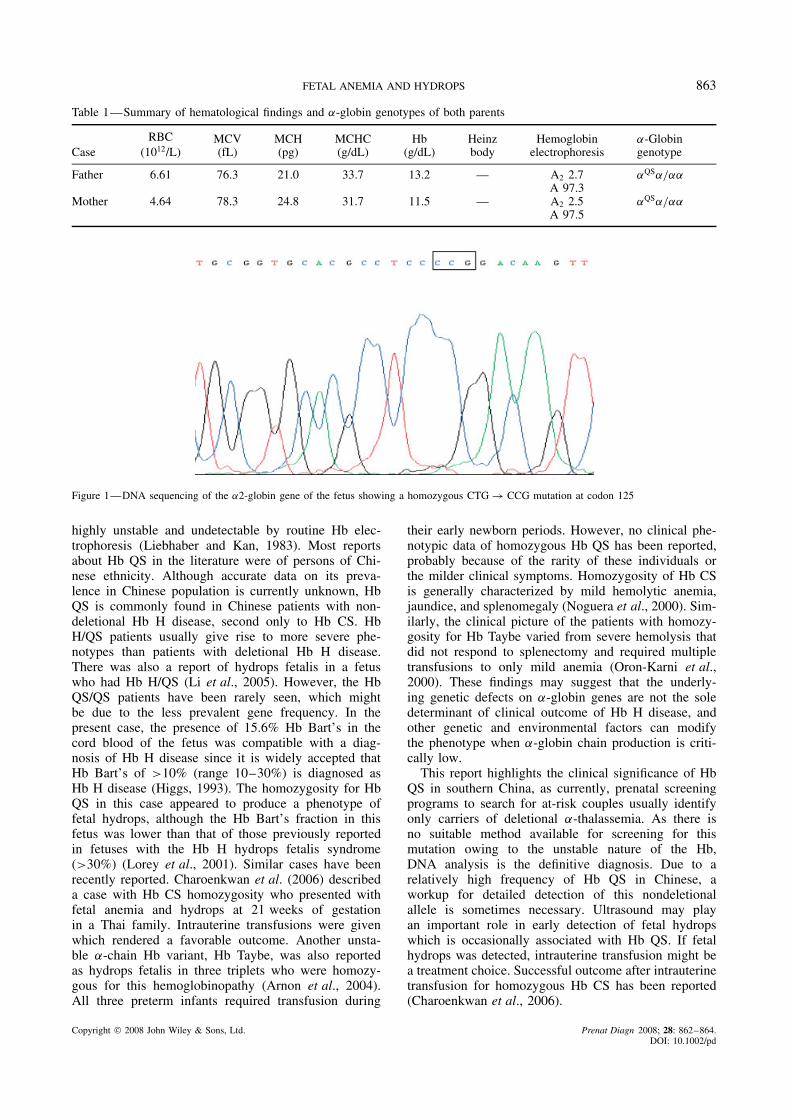
a routine anatomic scan at 21 weeks of gestationshowed that the fetus had cardiomegaly with a cardiac: thoracic area ratio of 0.43 (normal range 0.25–0.35;cardiomegaly is defined as >0.35) (Huhta, 2005), peri-cardial effusion (2.3 mm), and enlarged placenta (6 cm).The fetal middle cerebral artery peak systolic veloc-ity (MCA-PSV) was 50 cm/s (>1.55 MoM), whichsuggested severe fetal anemia (Mari et al., 2002). Nostructural abnormality was identified. Maternal investi-gations, including Rh and ABO blood group, Kleihauerand screening for the TORCH, syphilis, and parvovirusinfections were all negative. The decision to samplefetal blood was made, and cord blood was obtained bycordocentesis. An automated complete blood count con-firmed severe anemia in the fetus, with Hb level being7.0 g/dL (0.55–0.65 MoM) (Mari, 2000). On the bloodsmear, the red blood cells (RBC) were microcytic andhypochromic, with mild anisopoikilocytosis, basophilicstippling and markedly increased nucleated RBC. Thewhite blood cells and platelet morphology was normal.Hb analysis by agarose gel electrophoresis showed thefollowing hematological findings: Hb Bart’s 15.6, Hb(A + Portland) 5.4, Hb F 79.0 and Hb A2 0%. The fetalblood group was B, Rh(D) positive, whereas the mater-nal blood group was AB, Rh(D) positive. The fetal kary-otype was normal female. These results suggested thediagnosis of Hb H disease, and it was concluded that thefetus was most probably homozygous for a nondeletionalα+ –thalassemia mutation. DNA sequencing of the α-globin genes showed that both parents were hemoglobinQuong Sze (Hb QS) carriers and the fetus was a HbQS homozygote (Figure 1). The couple was counseledagain about the prognosis of the fetus if no treatmentmeasure was taken. It was explained that there was riskof progressing to classical hydrops fetalis in the laterperiod of gestation, and transfusion might be neededafter birth. However, the exact natural course of the dis-ease was uncertain because of the absence of data inthe literature. After serious consideration, they decidedto terminate the pregnancy. The female phenotypicallynormal fetus weighed 330 g, with an enlarged placentaweighing 150 g. The couple declined an autopsy.
The Hb QS is a nondeletional α-thalassemia with themissense mutation at codon 125 of the α2-globin gene(CTG → CCG, or Leu → Pro). This Hb variant is
Copyright 2008 John Wiley & Sons, Ltd. Received: 14 March 2008Revised: 27 May 2008
Accepted: 16 June 2008Published online: 22 July 2008
FETAL ANEMIA AND HYDROPS 863
Table 1—Summary of hematological findings and α-globin genotypes of both parents
CaseRBC
(1012/L)MCV(fL)
MCH(pg)
MCHC(g/dL)
Hb(g/dL)
Heinzbody
Hemoglobinelectrophoresis
α-Globingenotype
Father 6.61 76.3 21.0 33.7 13.2 — A2 2.7 αQSα/ααA 97.3
Mother 4.64 78.3 24.8 31.7 11.5 — A2 2.5 αQSα/ααA 97.5
Figure 1—DNA sequencing of the α2-globin gene of the fetus showing a homozygous CTG → CCG mutation at codon 125
highly unstable and undetectable by routine Hb elec-trophoresis (Liebhaber and Kan, 1983). Most reportsabout Hb QS in the literature were of persons of Chi-nese ethnicity. Although accurate data on its preva-lence in Chinese population is currently unknown, HbQS is commonly found in Chinese patients with non-deletional Hb H disease, second only to Hb CS. HbH/QS patients usually give rise to more severe phe-notypes than patients with deletional Hb H disease.There was also a report of hydrops fetalis in a fetuswho had Hb H/QS (Li et al., 2005). However, the HbQS/QS patients have been rarely seen, which mightbe due to the less prevalent gene frequency. In thepresent case, the presence of 15.6% Hb Bart’s in thecord blood of the fetus was compatible with a diag-nosis of Hb H disease since it is widely accepted thatHb Bart’s of >10% (range 10–30%) is diagnosed asHb H disease (Higgs, 1993). The homozygosity for HbQS in this case appeared to produce a phenotype offetal hydrops, although the Hb Bart’s fraction in thisfetus was lower than that of those previously reportedin fetuses with the Hb H hydrops fetalis syndrome(>30%) (Lorey et al., 2001). Similar cases have beenrecently reported. Charoenkwan et al. (2006) describeda case with Hb CS homozygosity who presented withfetal anemia and hydrops at 21 weeks of gestationin a Thai family. Intrauterine transfusions were givenwhich rendered a favorable outcome. Another unsta-ble α-chain Hb variant, Hb Taybe, was also reportedas hydrops fetalis in three triplets who were homozy-gous for this hemoglobinopathy (Arnon et al., 2004).All three preterm infants required transfusion during
their early newborn periods. However, no clinical phe-notypic data of homozygous Hb QS has been reported,probably because of the rarity of these individuals orthe milder clinical symptoms. Homozygosity of Hb CSis generally characterized by mild hemolytic anemia,jaundice, and splenomegaly (Noguera et al., 2000). Sim-ilarly, the clinical picture of the patients with homozy-gosity for Hb Taybe varied from severe hemolysis thatdid not respond to splenectomy and required multipletransfusions to only mild anemia (Oron-Karni et al.,2000). These findings may suggest that the underly-ing genetic defects on α-globin genes are not the soledeterminant of clinical outcome of Hb H disease, andother genetic and environmental factors can modifythe phenotype when α-globin chain production is criti-cally low.
This report highlights the clinical significance of HbQS in southern China, as currently, prenatal screeningprograms to search for at-risk couples usually identifyonly carriers of deletional α-thalassemia. As there isno suitable method available for screening for thismutation owing to the unstable nature of the Hb,DNA analysis is the definitive diagnosis. Due to arelatively high frequency of Hb QS in Chinese, aworkup for detailed detection of this nondeletionalallele is sometimes necessary. Ultrasound may playan important role in early detection of fetal hydropswhich is occasionally associated with Hb QS. If fetalhydrops was detected, intrauterine transfusion might bea treatment choice. Successful outcome after intrauterinetransfusion for homozygous Hb CS has been reported(Charoenkwan et al., 2006).
Copyright 2008 John Wiley & Sons, Ltd. Prenat Diagn 2008; 28: 862–864.DOI: 10.1002/pd

864 C. LIAO ET AL.
REFERENCES
Arnon S, Tamary H, Dgany O, et al. 2004. Hydrops fetalis associatedwith homozygosity for hemoglobin Taybe (α 38/39 THR deletion)in newborn triplets. Am J Hematol 76: 263–266.
Chan V, Chan VW, Tang M, Lau K, Todd D, Chan TK. 1997.Molecular defects in Hb H hydrops fetalis. Br J Haematol 96:224–228.
Charoenkwan P, Sirichotiyakul S, Chanprapaph P, et al. 2006.Anemia and hydrops in a fetus with homozygous hemoglobinconstant spring. J Pediatr Hematol Oncol 28: 827–830.
Higgs DR. 1993. Alpha-thalassemias. Baillieres Clin Haematol Blood6: 117–150.
Huhta JC. 2005. Fetal congestive heart failure. Semin Fetal NeonatalMed 10: 542–552.
Li DZ, Liao C, Li J, Xie XM, Huang YN, Wu QC. 2005. HemoglobinH hydrops fetalis syndrome resulting from the association of the−SEA deletion and the αQuong Szeα mutation in a Chinese woman.Eur J Haematol 75: 259–261.
Liebhaber SA, Kan YW. 1983. Alpha-Thalassemia caused by anunstable alpha-globin mutant. J Clin Invest 71: 461–466.
Lorey F, Charoenkwan P, Withowska HE, et al. 2001. Hb H hydropsfoetalis syndrome: a case report and review of literature. BrJ Haematol 115: 72–78.
Mari G. 2000. Noninvasive diagnosis by Doppler ultrasonography offetal anemia due to maternal red-cell alloimmunization. N EnglJ Med 342: 9–14.
Mari G, Detti L, Oz U, Zimmerman R, Duerig P, Stefos T. 2002.Accurate prediction of fetal hemoglobin by Doppler ultrasonog-raphy. Obstet Gynecol 99: 589–593.
Noguera NI, Gonzalez FA, Ropero P, Anguita E, Milani AC, Ville-gas A. 2000. Homozygous Constant Spring: the first case describedin the west. Haematologica 85: 667–668.
Oron-Karni V, Filon D, Shifrin Y, et al. 2000. Diversity of alpha-globin mutations and clinical presentation of alpha-thalassemia inIsrael. Am J Hematol 65: 196–203.
Tangvarasittichai O, Jeenapongsa R, Sitthiworanan C, Sang-uansermsri T. 2005. Laboratory investigations of Hb ConstantSpring. Clin Lab Haematol 27: 47–49.
Viprakasit V, Green S, Height S, Ayyub H, Higgs DR. 2002. Hb Hhydrops fetalis syndrome associated with the interaction of twocommon determinants of α thalassaemia (−MED/αTSaudiα). BrJ Haematol 117: 759–762.
Xu XM, Zhou YQ, Luo GX, et al. 2004. The prevalence and spectrumof alpha and beta thalassaemia in Guangdong Province: implicationsfor the future health burden and population screening. J Clin Pathol57: 517–522.
Copyright 2008 John Wiley & Sons, Ltd. Prenat Diagn 2008; 28: 862–864.DOI: 10.1002/pd





























