Embed Size (px)
Citation preview

バイオインフォマティクス分⼦系統解析1
藤 博幸

本⽇の講義
(1) 分⼦系統解析の概要
(2) 犯罪捜査への応⽤

本⽇の講義
(1) 分⼦系統解析の概要
(2) 犯罪捜査への応⽤

分⼦系統解析の⼿続き
(1)相同配列の収集系統分類の場合は、オーソロガスな配列を収集
(2) 相同配列のマルチプルアラインメントの作成
(3) アラインメントから分⼦系統樹を構築

分⼦系統解析の⼿続き
(1)相同配列の収集系統分類の場合は、オーソロガスな配列を収集
例えばBLASTで検索して収集(2) 相同配列のマルチプルアラインメントの作成
例えばmafftで構築(3) アラインメントから分⼦系統樹を構築

p.17-18

分⼦時計の発⾒ (1)ライナス•ポーリングLinus Carl Pauling(1901-1994)量⼦化学者、⽣化学者
Pauling and Zuckerkandle (1962)
⼆つの⽣物のヘモグロビンのアミノ酸配列を⽐較し、その置換数を、化⽯から推定される、それら⽣物の分岐時期に対してプロット
近似的な直性関係が得られた。
化⽯がない⽣物でも、配列の⽐較から分岐年代を推定できる。
宮⽥隆 (2014) 「分⼦からみた⽣物進化」 講談社

>gi|57013850|sp|P69905.2|HBA_HUMAN Full=Hemoglobin alpha chainMVLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHFDLSHGSAQVKGHGKKVADALTNAVAHVDDMPNALSALSDLHAHKLRVDPVNFKLLSHCLLVTLAAHLPAEFTPAVHASLDKFLASVSTVLTSKYR
>gi|145301578|ref|NP_032244.2| hemoglobin subunit alpha MVLSGEDKSNIKAAWGKIGGHGAEYGAEALERMFASFPTTKTYFPHFDVSHGSAQVKGHGKKVADALANAAGHLDDLPGALSALSDLHAHKLRVDPVNFKLLSHCLLVTLASHHPADFTPAVHASLDKFLASVSTVLTSKYR
ヒトのヘモグロビンα (上段)とマウスのヘモグロビンα (下段)アミノ酸は1⽂字表記で表現

CLUSTAL format alignment by MAFFT L-INS-i (v7.221)
gi|57013850|sp| MVLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHFDLSHGSAQVKGHGgi|145301578|re MVLSGEDKSNIKAAWGKIGGHGAEYGAEALERMFASFPTTKTYFPHFDVSHGSAQVKGHG
**** **:*:******:*.*..*********** *************:***********
gi|57013850|sp| KKVADALTNAVAHVDDMPNALSALSDLHAHKLRVDPVNFKLLSHCLLVTLAAHLPAEFTPgi|145301578|re KKVADALANAAGHLDDLPGALSALSDLHAHKLRVDPVNFKLLSHCLLVTLASHHPADFTP
*******:**..*:**:*.********************************:* **:***
gi|57013850|sp| AVHASLDKFLASVSTVLTSKYRgi|145301578|re AVHASLDKFLASVSTVLTSKYR
**********************
アラインメント(alignment):相同な配列の対応するアミノ酸あるいは塩基を対応する位置に並べる操作、あるいは並べたもの

CLUSTAL format alignment by MAFFT L-INS-i (v7.221)
gi|57013850|sp| MVLSPADKTNVKAAWGKVGAHAGEYGAEALERMFLSFPTTKTYFPHFDLSHGSAQVKGHGgi|145301578|re MVLSGEDKSNIKAAWGKIGGHGAEYGAEALERMFASFPTTKTYFPHFDVSHGSAQVKGHG
**** **:*:******:*.*..*********** *************:***********
gi|57013850|sp| KKVADALTNAVAHVDDMPNALSALSDLHAHKLRVDPVNFKLLSHCLLVTLAAHLPAEFTPgi|145301578|re KKVADALANAAGHLDDLPGALSALSDLHAHKLRVDPVNFKLLSHCLLVTLASHHPADFTP
*******:**..*:**:*.********************************:* **:***
gi|57013850|sp| AVHASLDKFLASVSTVLTSKYRgi|145301578|re AVHASLDKFLASVSTVLTSKYR
**********************
19/142 = 0.1338028 ヒトとマウス 化⽯から約7500万年前に分岐

19/142 = 0.1338028 ヒトとマウス 化⽯から約7500万年前に分岐
分岐年代(化⽯から)7500
0.1338
アミノ酸の置換率
ヒト vs マウス

分岐年代(化⽯から)7500
0.1338
アミノ酸の置換率
ヒト vs マウス
様々な⽣物のペアについて同様のプロットを作成

分岐年代(化⽯から)7500
0.1338
アミノ酸の置換率
ヒト vs マウス
近似的な直線関係アミノ酸置換数は分岐年代に⽐例し⼀定のペースで置換している

分岐年代(化⽯から)7500
0.1338
アミノ酸の置換率
ヒト vs マウス
⽣物の進化の過程での分⼦の変化:分⼦進化(molecular Evolution)分⼦の変化が⼀定のペースを刻むこと:分⼦時計(molecular clock)変化の速度:分⼦進化速度(molecular evolutionary rate)
= 直線の傾き = 単位時間あたりのアミノ酸の置換数

分岐年代(化⽯から)7500
0.1338
アミノ酸の置換率
ヒト vs マウス
分⼦時計が成⽴していれば、化⽯がなくてもアミノ酸配列から分岐年代を推定できる。

分岐年代(化⽯から)7500
0.1338
アミノ酸の置換率
ヒト vs マウス
今、現存の⽣物Xと⽣物Yの分岐を⽰す化⽯はないが、ヘモグロビンαの置換率が0.3であったとすると、分岐年代を直線関係から推定できる。
0.3
XとYのヘモグロビンαのアミノ酸置換率

分岐年代(化⽯から)7500
0.1338
アミノ酸の置換率
ヒト vs マウス
今、現存の⽣物Xと⽣物Yの分岐を⽰す化⽯はないが、ヘモグロビンαの置換率が0.3であったとすると、分岐年代を直線関係から推定できる。
0.3
推定された分岐年代

分⼦時計の発⾒ (2)
宮⽥隆 (2014) 「分⼦からみた⽣物進化」 講談社
Dickerson (1971)
様々はタンパク質で分⼦時計が成⽴していること、タンパク質によって分⼦進化速度が違うことを発⾒
進化速度の違いは機能的制約の強さを反映している。
⽣物にとって機能的な重要性の⾼い分⼦は進化速度が遅く、それほどでもない分⼦は速く変化する。
||
p.17

分⼦時計の発⾒ (3)
宮⽥隆 (2014) 「分⼦からみた⽣物進化」 講談社
今⽇の分⼦進化学者は、すべての分⼦に対して、分⼦進化速度の⼀定性が成⽴するとは考えていない。• 分⼦によっては変動が激しく、分岐時間にも依存• 綱レベルの⽐較では近似的な⼀定性が認められるが、⽬、科、属、種などのレベルでは⼀定性が成⽴しないものも多い。
primates slow down, rodents speed up霊⻑類では進化速度は遅くなり、齧⻭類では速くなる傾向
がある。

分⼦系統樹の構築 (1)
Fitch and Margoliash (1967)
チトクローム c というタンパク質の配列を⽐べ、その置換数からほ乳類、⿃類、は⾍類、昆⾍、菌類を含む系統樹を構築した。これは、それ以前の形質の⽐較に基づく分類では不可能なものであった。

分⼦系統樹の構築 (2)分⼦系統樹は、これまでの分類を反映しており、これまでの分類と⾷い違う場合、分⼦系統樹が正しいことがしばしばあった。
特に”隠蔽種 (cryptic species)”の発⾒に⼒を発揮(⾒た⽬には区別がつかないが、DNAレベルでは全く異なるもの)例:ウーズは16S r RNAを利⽤して、界レベルでの隠蔽されていた古細菌を発⾒。
現在、分⼦系統学的⼿法は、系統分類の⼿法として広く受け⼊れられ、⼒を発揮している。
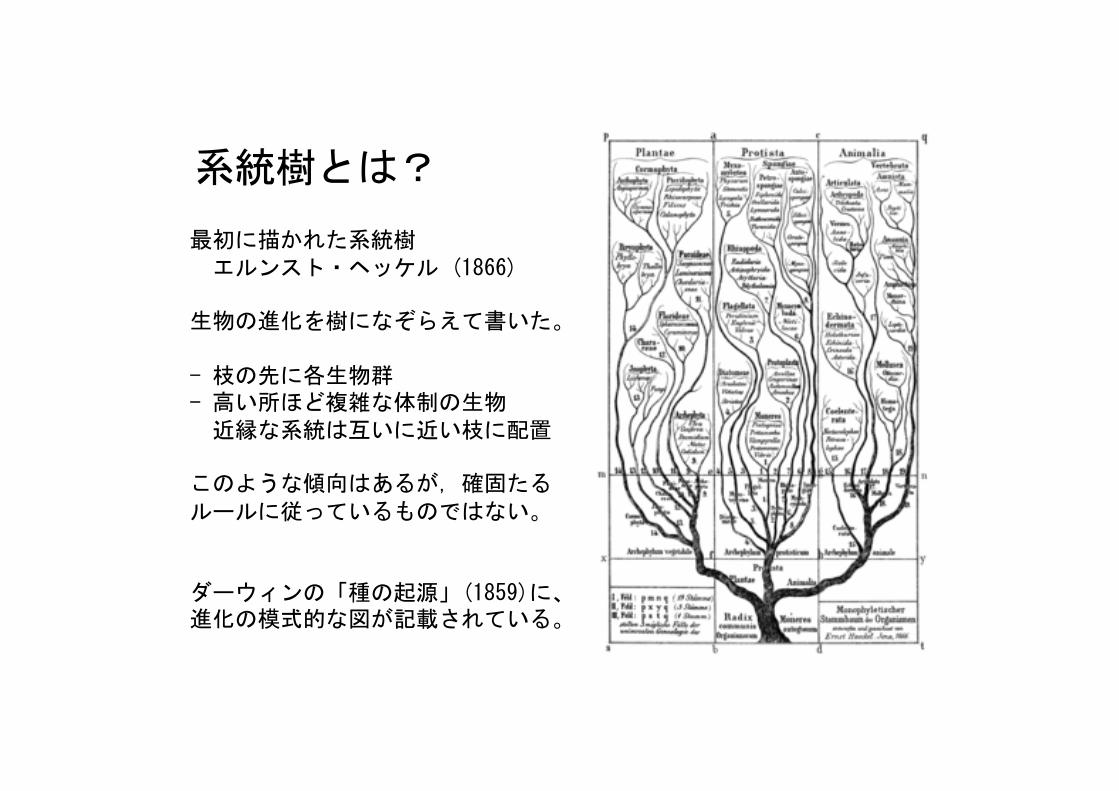
系統樹とは?
最初に描かれた系統樹エルンスト・ヘッケル (1866)
生物の進化を樹になぞらえて書いた。
- 枝の先に各生物群- 高い所ほど複雑な体制の生物近縁な系統は互いに近い枝に配置
このような傾向はあるが,確固たるルールに従っているものではない。
ダーウィンの「種の起源」(1859)に、進化の模式的な図が記載されている。

p.20-21

p.23-24

p.24 図2.4

p.24-25

p.25

相同(homologous, homolog)
orthologous, ortholog種分化に伴い分岐
paralogous, paralog遺伝⼦重複に伴い分岐
共通祖先から分岐

p. 25 図2.5

種系統樹と遺伝⼦系統樹
分⼦系統樹
種系統樹 (Species Tree)
遺伝⼦系統樹 (Gene Tree)
オーソロガスな遺伝⼦のみ使⽤⽣物の進化的関係を反映
パラロガスな遺伝⼦もオーソロガスな遺伝⼦も混在。遺伝⼦(タンパク質)の分⼦進化の歴史を反映

p.25-26

p.26 - 27

主な分子系統樹推定法
(1)距離行列法
(2)形質状態法●最節約法(Maximum Parsimony Method)
●統計的方法○最尤法(Maximum Likelihood Method)○ベイズ法

主な分子系統樹推定法
(1)距離行列法
(2)形質状態法●最節約法(Maximum Parsimony Method)
●統計的方法○最尤法(Maximum Likelihood Method)○ベイズ法

距離⾏列法
(1) アラインメントから全ての配列ペアの間の距離を計算
(2) 距離⾏列 (distance matrix) の構築
(3) 距離⾏列に基づき系統樹を構築

距離⾏列法
距離⾏列法として複数の系統樹構築法が提案されているが、ここでは現在最も広く利⽤されている近隣結合法(neighbor-joining method)について説明する

距離⾏列法
(1) アラインメントから全ての配列ペアの間の距離を計算
(2) 距離⾏列 (distance matrix) の構築
(3) (2)の距離⾏列に基づき系統樹を構築
(1), (2)は様々な距離⾏列法で共通(3)の部分が、距離⾏列法の⼿法により異なる。ただし、(1)には複数の⼿法がある。

距離⾏列法
(1) アラインメントから全ての配列ペアの間の距離を計算
(2) 距離⾏列 (distance matrix) の構築
(3) 距離⾏列に基づき系統樹を構築

p.21

最も簡単な配列間距離=p距離 (p distance)
Nヌクレオチドの⻑さの⼆つの配列で、Mサイトに差異がある場合p-distance = M / Np距離の問題点多重置換(復帰置換や平⾏置換を含む)
=同じサイトでの複数回の置換-------> 数回分の変化が隠されてしまう

p.21

p.22 図2.3

p.21-22

p.22

ピリミジン T, Cプリン A, G
トランジションピリミジン間あるいはプリン間の置換
トランスバージョンピリミジンとプリン間の置換
分⼦系統学への統計的アプローチ計算分⼦進化学Yang, Z著 藤、加藤、⼤安訳共⽴出版 より

分⼦系統学への統計的アプローチ計算分⼦進化学Yang, Z著 藤、加藤、⼤安訳共⽴出版 より

進化モデルを考え、観測値から真の距離を推定する。塩基の進化モデルの例JC69
全ての塩基は同じ速度で他の塩基に置換K80
トランジション(プリン間あるいはピリミジン間の置換)とトランスバージョン(プリンとピリミジン間の置換)では
異なる速度…この他にも多くの進化モデルが考えられ、距離の推定が⾏われている。
アミノ酸でも同様に多くの進化モデルが考えられており、観測値からの真の距離の推定が⾏われている。

マルチプルアラインメント種1種2種3種4種5
選択された進化モデル
種1と種2の距離d12
種1と種3の距離d12
.
.
.種4と種5の距離
d45
マルチプルアラインメント中の全ての配列のペアに対して、選択した進化モデルにしたがって、距離を計算

距離⾏列法
(1) アラインメントから全ての配列ペアの間の距離を計算
(2) 距離⾏列 (distance matrix) の構築
(3) 距離⾏列に基づき系統樹を構築

マルチプルアラインメント種1種2種3種4種5
選択された進化モデル
種1と種2の距離d12
種1と種3の距離d12
.
.
.種4と種5の距離
d45
マルチプルアラインメント中の全ての配列のペアに対して、選択した進化モデルにしたがって、距離を計算
種1と種2の距離d12
種1と種3の距離d12
.
.
.種4と種5の距離
d45
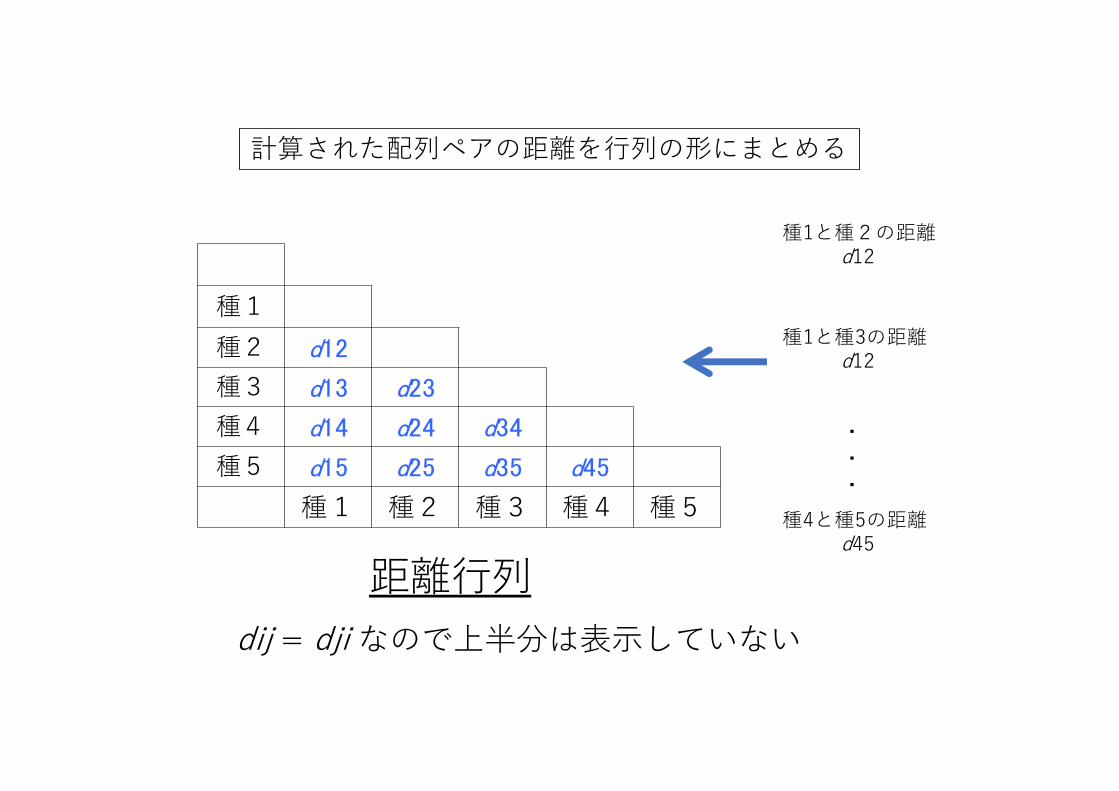
計算された配列ペアの距離を⾏列の形にまとめる
種1種2 d12
種3 d13 d23
種4 d14 d24 d34
種5 d15 d25 d35 d45
種1 種2 種3 種4 種5
距離⾏列dij = dji なので上半分は表⽰していない

距離⾏列法
(1) アラインメントから全ての配列ペアの間の距離を計算
(2) 距離⾏列 (distance matrix) の構築
(3) 距離⾏列に基づき系統樹を構築近隣結合法について説明

p.27

p.28

種1
種2
種3種4
種5
種1種2
種3種4
種5
種4種5
種1種2
種3
種1種3
種4種4
種5
.
.
.
.
.
.星状系統樹
任意の⼆つのOUTを組んだ全ての樹形を発⽣させる
それぞれの系統樹について、距離⾏列から枝の⻑さの総和を求める

種1種2
種3種4
種5 L1L2
L3L4
L6L5
枝の⻑さの総和とは
L1 + L2 + L3 + L4 + L5 + L6

距離⾏列からの枝の⻑さの求め⽅の基本的な考え⽅(配列3つのケースを例として)
種1 種2 種3
種1種2 d12
種3 d13 d23L3
L1 L2
L1 + L2 = d12L1 + L3 = d13L2 + L3 = d23
距離⾏列
d13 ‒ d23 + d122L1 =
L2 = d12 ‒ d13 + d23 2
L3 = d13 ‒ d12 + d23 2
系統樹と距離⾏列から連⽴⽅程式をつくる
⽅程式を解いてL1 + L2+L3を求める

種1種2
種3種4
種5
種4種5
種1種2
種3
種1種3
種4種4
種5
.
.
.
それぞれの系統樹について、距離⾏列から枝の⻑さの総和を求める
枝の⻑さの総和が最⼩になるペアリングを選択

種1種2
種3種4
種5
種4種5
種1種2
種3
種1種3
種4種4
種5
.
.
.
枝の⻑さの総和が最⼩のペアを選択することの意味
枝の⻑さは、距離(=サイトあたりの置換数)から計算される
枝の⻑さの総和が最⼩であるとは、距離⾏列から計算されるその系統樹の形の上で⽣じた置換の総数が最⼩であることを意味する

種1種2
種3種4
種5
種4種5
種1種2
種3
種1種3
種4種4
種5
.
.
.
種1種3
種4種4
種5
今、種1と種3のペアリングにおいて枝の⻑さの総和が最⼩になったする
種1と種2をOUTから外し、種1と種2をペアとした新たなOUTを考え距離⾏列を再構築

種2種4 d24
種5 d25 d45
種1-3 d2,1-3 d4,1-3 d5,1-3
種2 種4 種5 種1-3
距離⾏列の更新
d2,1-3= d12 + d23 ‒ d132
d4,1-3= d14 + d34 ‒ d132
d5,1-3= d15 + d35 ‒ d132

更新された距離⾏列をもとに、同じ処理を繰り返す。
OUTの数が3になったところで繰り返しを停⽌する。
上記の例であれば
種2 種4
種5 種1-3
種2 種5
種4 種1-3
種2 種1-3
種4 種5
の3つの樹形から枝の⻑さの総和が最⼩のものを選択種2と種5がペアをつくったものが最⼩であったとする。ここでOUT数が3となり、この結果の系統樹は次のようになる

種1種3
種2
種5種2
無根系統樹が得られる近隣結合法では進化速度の⼀定性は仮定されていないので、⼀つの内部節から分岐した外部節への枝の⻑さが異なることに注意

分⼦系統解析では、系統樹は無根系統樹として作成される。根の導⼊は、外群(outgroup)を⽤いてなされる
進化速度の⼀定性が成⽴する場合(=分⼦時計が成⽴している場合)を除き、注⽬するグループの根を決められない
多くの場合、分⼦時計の⼀定性は仮定できない
外群の利⽤、ブートストラップについては次回

最尤法・ベイズ推定プログラムPHYLIP (http://evolution.genetics.washington.edu/phyli.html)
最尤法、最節約法、距離⾏列法などPAUP* (http://paup.csit.fsu.edu)
最尤法、最節約法、距離⾏列法などMolphy (http://bioweb.pasteur.fr/seqanal/interfaces/prot.nucml.html)
最尤法RAxML (http://sco.h-its.org/exelixis/web/software/raxml/index.html)
最尤法MEGA (http://www.megasoftware.net)
最尤法MrBayes (http://mrbayes.csit.fsu.edu/index.php)
ベイズ推定 を導⼊した系統樹推定

本⽇の講義
(1) 分⼦系統解析の概要
(2) 犯罪捜査への応⽤

Maria Jones (20) ♀看護師既婚
Robert White (34) ♂消化器科の医者既婚
ルイジアナ州 ラファイエット
いわゆるW不倫
離婚 妻と離婚することを約束するが守らない

Maria Jones (20) ♀看護師
Robert White (34) ♂消化器科の医者既婚
・妻と離婚することを約束するが守らない
・異常に嫉妬深く、⽀配的(Mariaが他の男性を⾒ただけで、彼らを殺すなどという)
10年間つきあったが、ついに別れることを決⼼
1994年7⽉

Maria Jones (20) ♀看護師
Robert White (34) ♂消化器科の医者既婚
関係が悪化する前、定期的にビタミンB12の注射をしていた
1994年8⽉ 深夜にMariaが息⼦とベッドで眠っている所にRobertがやってきて、もう⼀度注射をしたいという
Mairaは疲れており、深夜であることから断ったが、注射されてしまうMairaは、何かがおかしいと感じた。これまで感じたことのない強い痛みを覚えた。

2週間後、Mairaのリンパ節が腫れ上がった(ウイルス感染を意味する)
12⽉の定期検診で、HIV陽性、HCV陽性であることが判明
MairaはRobertを疑い、1995年1⽉に警察に訴えた

当初、警察はMairaの訴えをまじめに捉えていなかった
しかし、(1) 1984 ‒ 1995の期間にMariaが関係を持った男性は全員 HIV陰性であった
(2) Mairaについて問い合わせた時に、Robertは嘘をついていた
(3) Robertの患者の記録の中に、8⽉初頭に2つの⾎液サンプルがとられているにも関わらず、適切な検査記録がないものがあることを警察が⾒つけた。⾎液サンプルの⼀⽅はAIDS患者からのもので、他⽅はC型肝炎の患者からのものであった。2⼈の患者はRobertから研究のために⾎液サンプルが欲しいといわれ、提供していた。

Robertの注射によって、MariaはHIVとHCVに感染した可能性が⾼い
しかし、検察はMairaのHIVが、他の⼈から感染したのではなく、Robertの患者のサンプルに由来するという確固とした証拠を必要とした
(1) Mariaから分離したHIV,(2) Robertの患者から分離したHIV,(3) 同じ地域の他のAIDS患者から分離したHIVで分⼦系統解析が⾏われた

配列データの⼊⼿法については、今回の資料の最後に書いてある。
今回のデータは既に論⽂になっており、NCBIから⼊⼿可能
今回は、mafftで作成したアラインメントデータ (hivpol.aln)を⽤いて、系統樹を構築する。

P1.BCM.RT ---------cccataagtcctattgaaactgtaccagtaaaattaaagccaggaatggatP4.BCM.RT ---------cccataagtcctattgaaactgtaccagtaaaattaaagccaggaatggatP7.BCM.RT ---------cccataagtcctattgaaactgtaccagtaaaattaaagccaggaatggatP3.MIC.RT ttaaattttcccataagtcctattgaaactgtaccagtaaaattaaagccaggaatggatP5.MIC.RT ttaaattttcccataagtcctattgaaactgtaccagtaaaattaaagccaggaatggatP2.BCM.RT ---------cccataagtcctattgaaactgtaccagtaaaattaaagccaggaatggatP1.MIC.RT ttaaattttcccataagtcctattgaaactgtaccagtaaaattaaagccaggaatggatP2.MIC.RT ttaaattttcccataagtcctattgaaactgtaccagtaaaattagagccaggaatggatP3.BCM.RT ---------cccataagtcctattgaaactgtaccagtaaaattaaagccaggaatggatP5.BCM.RT ---------cccataagtcctattgaaactgtaccagtaaaattaaagccaggaatggatV1.BCM.RT ---------cccataagtcctattgaaactgtaccagtaaaattaaagccaggaatggatV2.BCM.RT ---------cccataagtcctattgaaactgtaccagtaaaattaaagccaggaatggatP6.MIC.RT ttaaattttcccataagtcctattgaaactgtaccagtaaaattaaagccaggaatggatV1.MIC.RT ttaaattttcccataagtcctgttgaaactgtaccagtaaaattaaagccaggaatggatV2.MIC.RT ttaaattttcccataagtcctattgaaactgtaccagtaaaattaaagccaggaatggatP4.MIC.RT ttaaattttcccataagtcctattgaaactgtaccagtaaaattaaagccaggaatggatP6.BCM.RT ---------cccataagtcctattgaaactgtaccagtaaaattaaagccaggaatggatLA04.RT ---------cccattagtcctattgaaactgtaccagtaaaattaaagccaggaatggatLA21.RT ---------cccattagtcctattgraactgtaccagtaaaattaaagccaggaatggatLA24.RT ---------cccattagtcctattgaaactgtaccagtaaaattaaagccaggaatggatLA31.RT ---------cccattagtcctattgaaactgtaccagtaaaattaaagccaggaatggatLA10.RT ---------cccattagtcctattgaaactgtaccagtaaaattaaagccaggaatggat

準備したファイルでは、配列の名前を短くしてあります。
Vで始まる名前 victim = Mariaから分離されたHIVのRT
Pで始まる名前 patient=Robertの患者から分離されたHIVのRT
LAで始まる名前 Lafeyetteで⽣活するAIDS患者から分離されたHIVのRT
RT = Reverse Transcriptase 逆転写酵素

得られたアラインメントを使ってMEGAで系統樹を作成
(1) MEGAを起動してmafftで作成したアラインメントの読み込み
(2) MEGA形式へのデータの変換
(3) モデル選択
(1) 近隣結合法による系統樹の構築

得られたアラインメントを使ってMEGAで系統樹を作成
(1) MEGAを起動してmafftで作成したアラインメントの読み込み
(2) MEGA形式へのデータの変換
(3) モデル選択
(1) 近隣結合法による系統樹の構築

MEGAの起動
① 左下のスタートをクリック
②下部ウィンドウにMEGAと⼊⼒ ③MEGAのアイコンが出てくる
ので、クリックして起動

起動画⾯ 左上に注⽬

メニューバーのFileをクリック
Open A File/Sessionを選択

ファイル選択のウィンドウが表⽰される
前ページのファイル選択ウィンドウを拡⼤したもの
スクロールバーで表⽰位置を変更しながらファイルを探して選択

ファイルがおかれているフォルダを選択してクリック

ファイルを選択すると、File name ウィンドウにファイル名が現れるこの状態でOpenをクリック
①
②③

アラインメントを表⽰するウィンドウが表意される

得られたアラインメントを使ってMEGAで系統樹を作成
(1) MEGAを起動してmafftで作成したアラインメントの読み込み
(2) MEGA形式へのデータの変換
(3) モデル選択
(1) 近隣結合法による系統樹の構築

アラインメントを表⽰するウィンドウが表意される
Utilitiesをクリック

Convert to MEGA Format を選択

OKをクリック
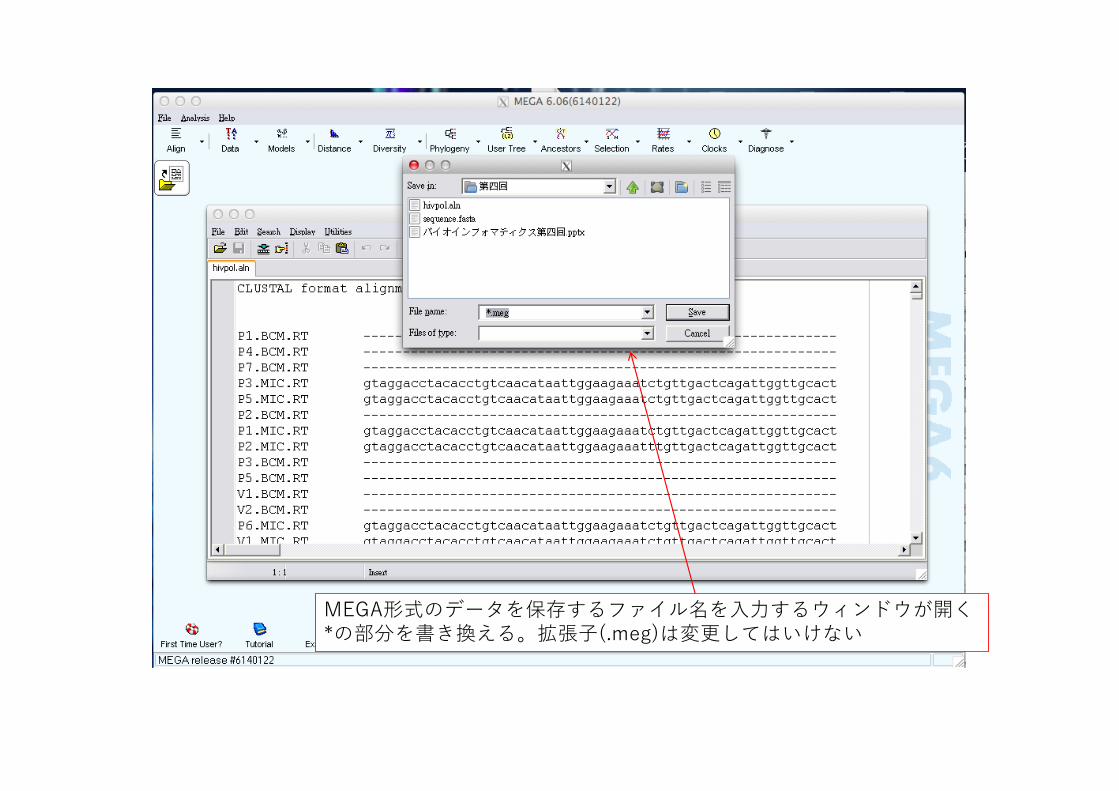
MEGA形式のデータを保存するファイル名を⼊⼒するウィンドウが開く*の部分を書き換える。拡張⼦(.meg)は変更してはいけない

前ページのファイル名⼊⼒ウィンドウを拡⼤ファイル名を*から書き換える

変換が終了したことを⽰すメッセージ。OKをクリック
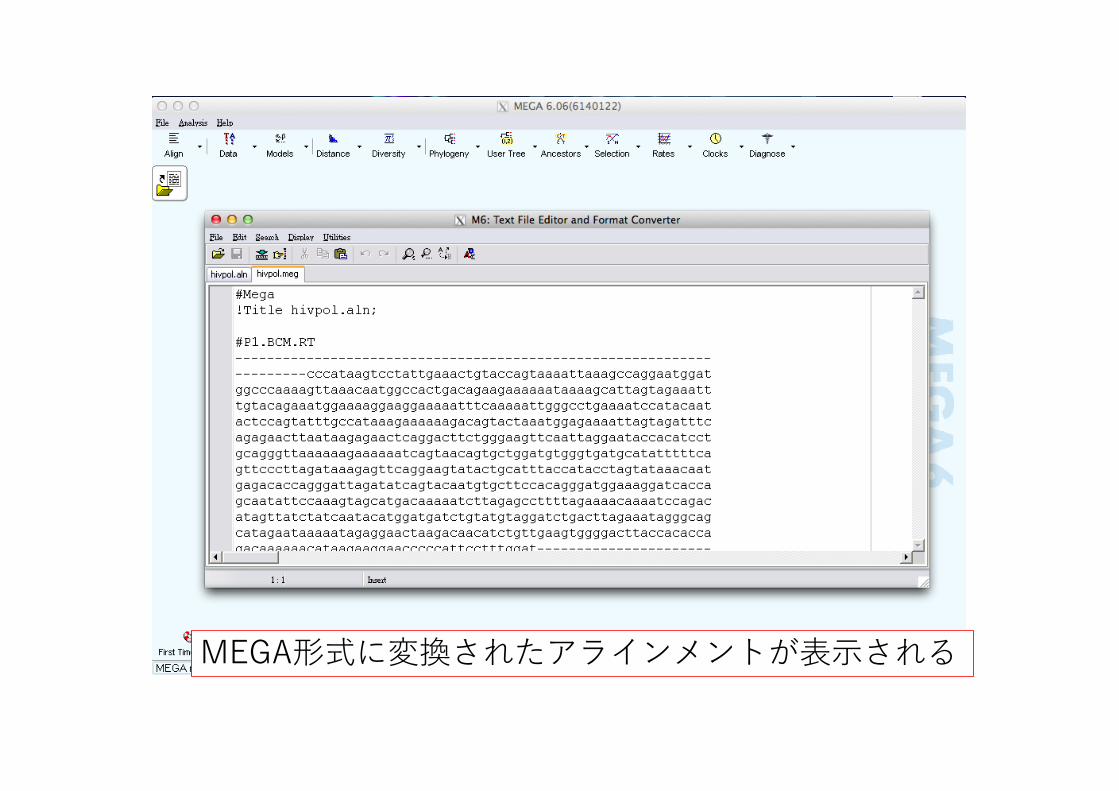
MEGA形式に変換されたアラインメントが表⽰される

得られたアラインメントを使ってMEGAで系統樹を作成
(1) MEGAを起動してmafftで作成したアラインメントの読み込み
(2) MEGA形式へのデータの変換
(3) モデル選択
(1) 近隣結合法による系統樹の構築

① Modelsをクリック
② Find Best DNA.Protein Models (ML)をクリック
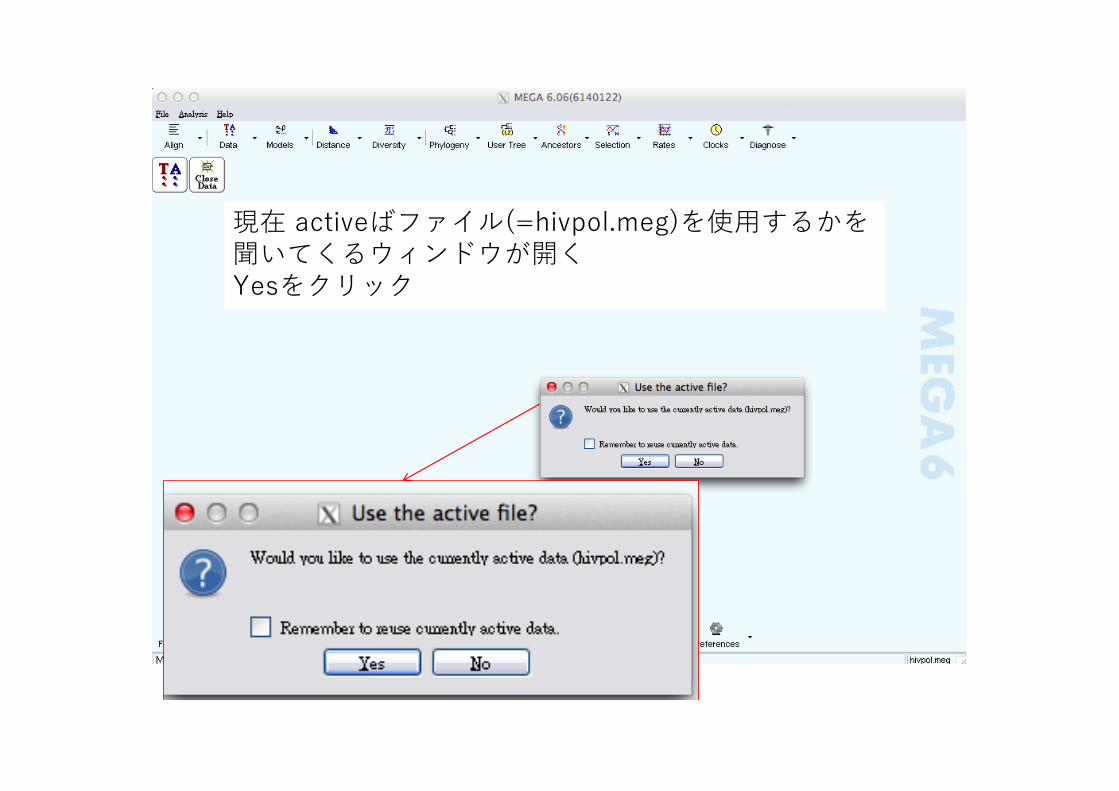
現在 activeばファイル(=hivpol.meg)を使⽤するかを聞いてくるウィンドウが開くYesをクリック

モデル選択の計算のオプション確認のウィンドウが開く⻩⾊の部分がAutomaticNucleotideComplete deletionになっていることを確認して、Computeをクリック
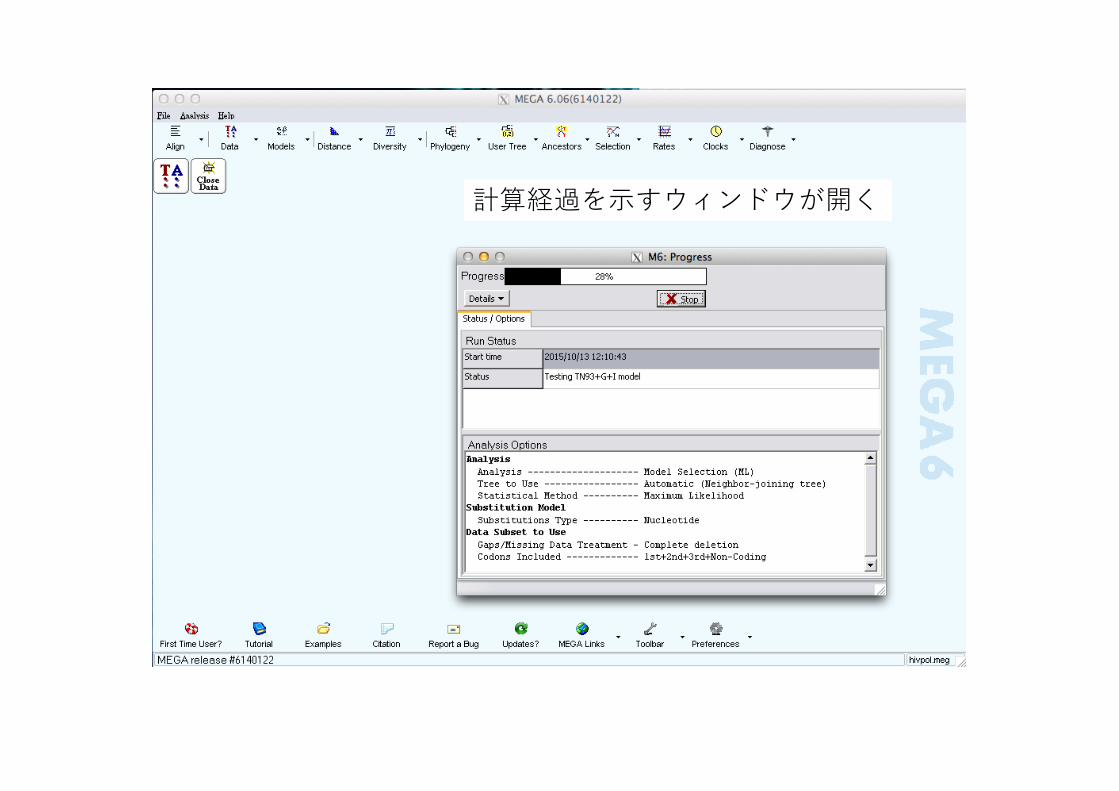
計算経過を⽰すウィンドウが開く

結果画⾯出⼒⾏がモデル列に情報量基準とパラメータが書かれている

BIC, AICc : 情報量基準この表がBICでソートされている情報量は⼩さい⽅が良い
lnL: 対数尤度⼤きい⽅が良い
BIC最⼩の T92+G モデルを今回使⽤することにする
このウィンドウは閉じる

得られたアラインメントを使ってMEGAで系統樹を作成
(1) MEGAを起動してmafftで作成したアラインメントの読み込み
(2) MEGA形式へのデータの変換
(3) モデル選択
(1) 近隣結合法による系統樹の構築とbootstrap解析
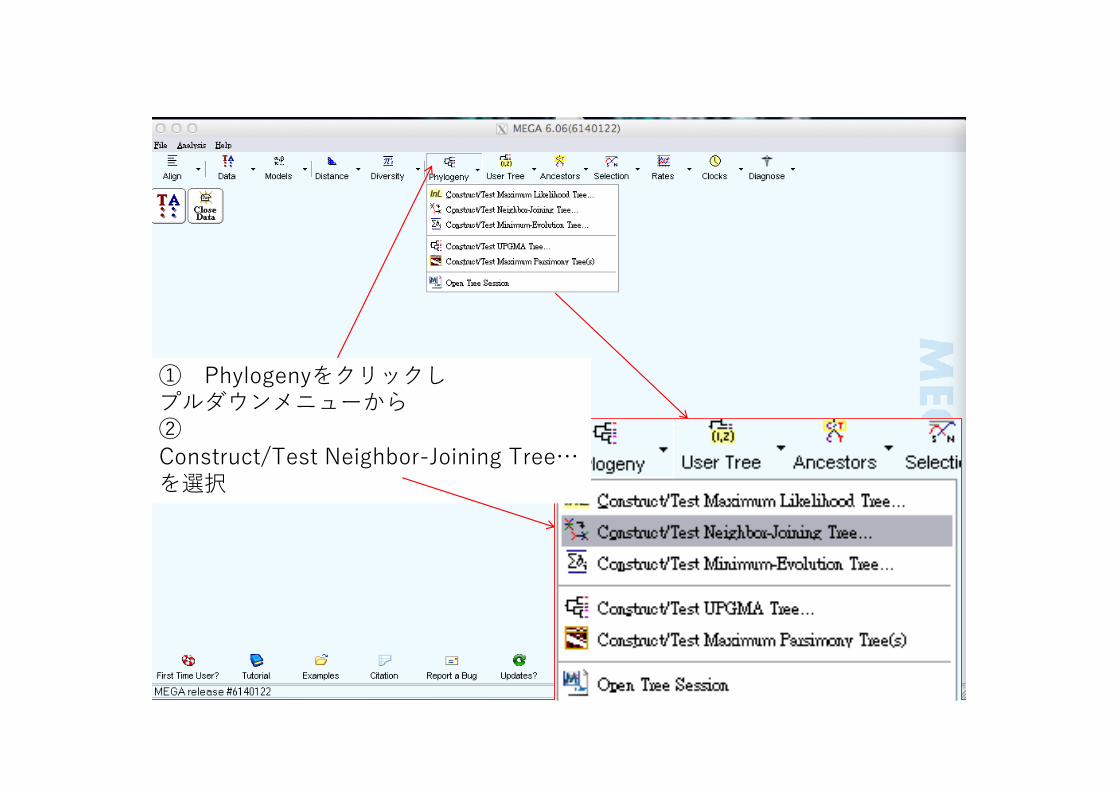
① Phylogenyをクリックしプルダウンメニューから②Construct/Test Neighbor-Joining Tree…を選択

現在activeなファイル(=hivpol.meg)を使⽤するかを問い合せるウィンドウが開くので、Yesをクリック

計算の設定を問い合わせるウィンドウが開く⻩⾊の部分が変更可能

Bootstrap法デフォルトのリサンプリング回数は500まんで、右端をクリックした時に現れる上下の⽮印の上向⽮印をクリックし1000にする。

Model/Methodは、デフォルトはNo. of differencesになっている
モデル選択の結果に従い、T92+Gに変更する。
Tamura 3-parameterモデルは1992に提案されており、これがT92に相当すると考えられるので、これを選択。

Tamura の3パラメータを選択するとRates among Sitesでは、GammaDistributedが⾃動的に選択されるこれがモデル選択の+Gの部分

Computeをクリックして計算
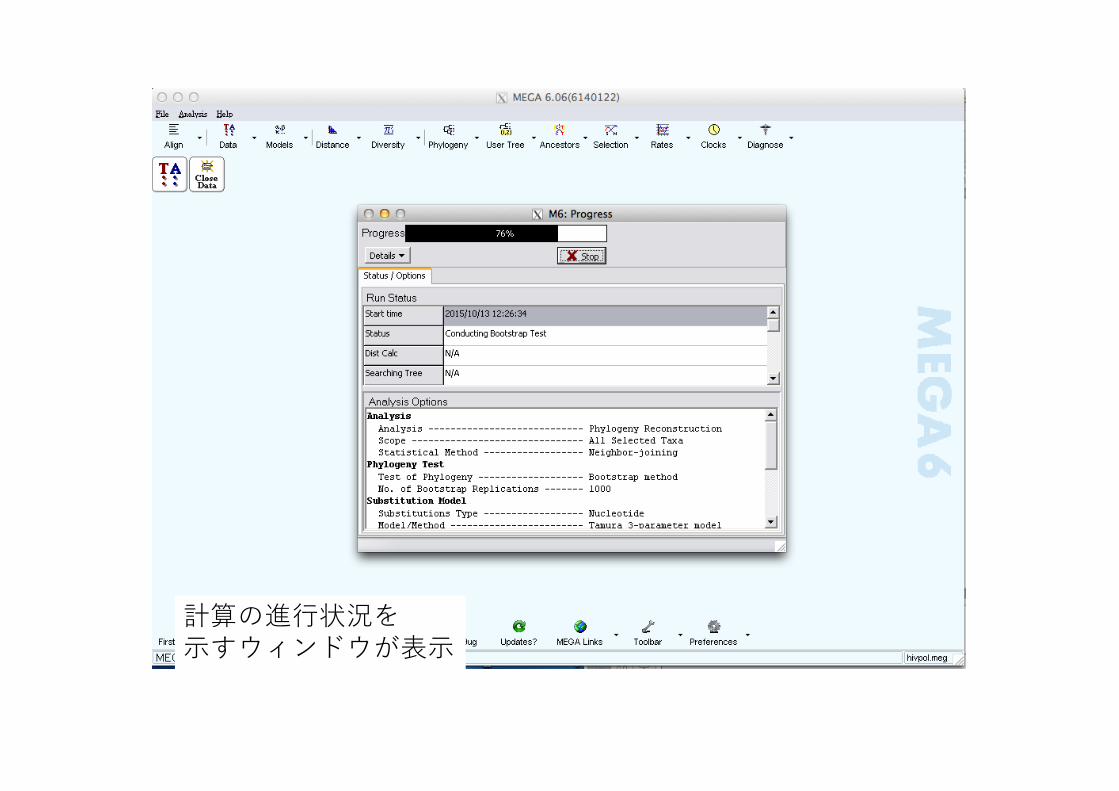
計算の進⾏状況を⽰すウィンドウが表⽰

計算が終わると系統樹が別のウィンドウに表⽰される。

Victim (Maria)から単離されたHIVは、Robertの患者から単離されたHIVに近縁(ただし、bootstrap support(bootstrap probabilityともよぶ)⼩さい)

デフォルトではOriginal Treeが表⽰されているBootstrap consensus treeのタブを選択

1000回のbootstrap サンプルのそれぞれについて構築された系統樹のコンセンサスが⽰される。系統樹の枝振り(トポロジー)についてのみコンセンサスが⽰されており、枝の⻑さには意味はない。
コンセンサスでもVictim由来HIVはPatient由来HIVに近い
再び、Original treeタブを選択

① Original treeタブを選択しオリジナルの系統樹を表⽰
② メニューバーのFileをクリック
③ Export Current Tree (Newick)を選択

Newick 形式のデータを保存するファイル名を聞いてくるので、Hivpol.nwkとファイル名を指定してSaveをクリック

hivpol.nwkをメモ帳で開く(((((((((((((((V1.MIC.RT:0.00183356,V2.MIC.RT:-0.00005991)0.8910:0.00358471,P6\.MIC.RT:-0.00000974)0.1770:0.00000487,V1.BCM.RT:-0.00000487)0.1240:0.00000487,P\5.BCM.RT:-0.00000487)0.1580:0.00000487,V2.BCM.RT:-0.00000649)0.6510:0.00179596,\P6.BCM.RT:-0.00002443)0.2560:0.00047535,(P3.MIC.RT:0.00247068,(P5.MIC.RT:0.0001\0360,(P4.BCM.RT:-0.00001486,(P1.BCM.RT:0.00180051,P7.BCM.RT:-0.00002890)0.3290:\0.00001486)0.6290:0.00167160)0.3700:0.00110497)0.1450:0.00042271)0.1300:0.00012\758,(P2.MIC.RT:0.00692434,(P3.BCM.RT:0.00000000,P4.MIC.RT:0.00000000)0.3170:0.0\0022865)0.3800:0.00123392)0.2950:0.00120878,P2.BCM.RT:0.00151719)0.1530:0.00021\449,LA32.RT:0.00555837)0.1380:0.00004296,(LA08.RT:0.00517567,LA05.RT:0.01117874\)0.4190:0.00175445)0.2030:0.00101235,P1.MIC.RT:0.00223222)0.3350:0.00231784,LA1\8.RT:0.00719763)0.0810:0.00037487,((((LA29.RT:0.01283766,LA06.RT:0.00724592)0.3\080:0.00136025,LA12.RT:0.00407447)0.1500:0.00100562,(LA28.RT:0.01213187,LA07.RT\:0.00795380)0.5010:0.00248453)0.0890:0.00048663,((LA10.RT:0.00771152,LA23.RT:0.\01441878)0.4210:0.00234077,((((LA04.RT:0.00992803,LA25.RT:0.01196780)0.2190:0.0\0075079,LA27.RT:0.00367005)0.1520:0.00156941,(LA22.RT:0.01275031,LA30.RT:0.0111\6664)0.2420:0.00081036)0.0260:0.00057137,((LA17.RT:0.00971516,LA13.RT:0.0103715\9)0.5080:0.00308458,(LA31.RT:0.00767816,(LA14.RT:0.01046118,(LA21.RT:0.00708465\,LA24.RT:0.00192401)0.8140:0.00438466)0.2290:0.00054839)0.1320:0.00076469)0.146\0:0.00116298)0.0090:0.00046111)0.0210:0.00077806)0.0200:0.00022654)0.1420:0.001\01897,LA16.RT:0.00625876)0.5290:0.00018712,(LA26.RT:0.00566221,LA02.RT:0.016280\61)0.5290:0.00268962);
Newick形式とは、系統樹の情報を、テキストとして記述したもの


合衆国の法廷ではじめて分⼦系統解析が利⽤されたのがこの事件
1998年、Robert Whiteは⼆級殺⼈について有罪判決をうけ現在50年の禁固刑に服している

モデル選択のモデルとは何か
距離の最尤推定とは何か
bootstrap support (bootstrap probablity)とは何か?
Newick 形式とは何か?

参考⽂献
Samuelsson, T. (2012) “Genomics and Bioinformatics- An Introduction to Programming Tools - “Cambridge Univ Press

今回の配列データの⼊⼿と、mafftによるマルチプルアラインメント

配列データはNCBIに登録されているAY156734 ‒ AY156907
配列を取得
Multi FASTA 形式のファイルで保存
mafft でmultiple alignment
MEGAで系統解析

NCBIをググる
クリック

① AY156734 を⼊⼒② Searchをクリック

前ページの⼊⼒ウィンドウを拡⼤したもの

Genesの中のPopSetをクリック

前ページのGenes部分を拡⼤
PopSetをクリック


前ページのトップを拡⼤ FASTA をクリック

Multi-Fasta形式で配列が表⽰される

前ページの図のトップを拡⼤
画⾯右上の Send to をクリック

Send to をクリックすると図のようなメニューが出てくるFileをチェックすると、下部のメニューが出てくるので最下段の Create File をクリック
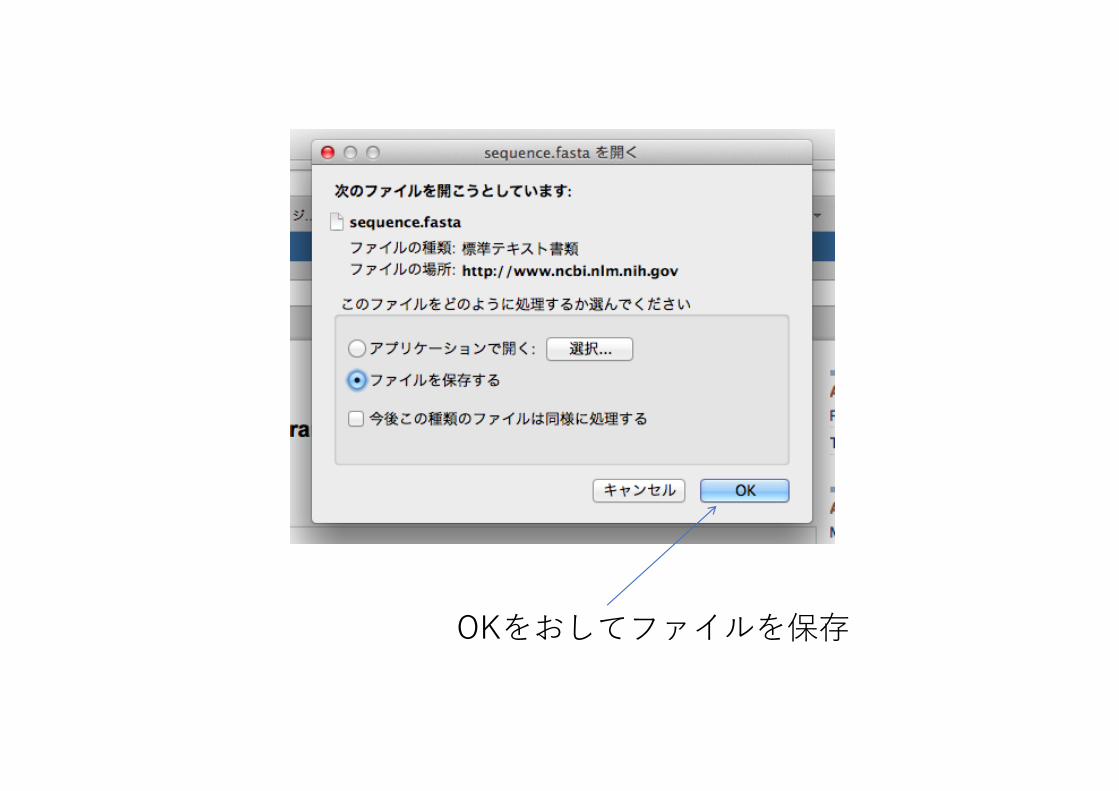
OKをおしてファイルを保存

>gi|24209939|gb|AY156734.1| HIV-1 clone P1.BCM.RT from USA reverse transcriptase (pol) gene, partial cdsCCCATAAGTCCTATTGAAACTGTACCAGTAAAATTAAAGCCAGGAATGGATGGCCCAAAAGTTAAACAATGGCCACTGACAGAAGAAAAAATAAAAGCATTAGTAGAAATTTGTACAGAAATGGAAAAGGAAGGAAAAATTTCAAAAATTGGGCCTGAAAATCCATACAATACTCCAGTATTTGCCATAAAGAAAAAAGACAGTACTAAATGGAGAAAATTAGTAGATTTCAGAGAACTTAATAAGAGAACTCAGGACTTCTGGGAAGTTCAATTAGGAATACCACATCCTGCAGGGTTAAAAAAGAAAAAATCAGTAACAGTGCTGGATGTGGGTGATGCATATTTTTCAGTTCCCTTAGATAAAGAGTTCAGGAAGTATACTGCATTTACCATACCTAGTATAAACAATGAGACACCAGGGATTAGATATCAGTACAATGTGCTTCCACAGGGATGGAAAGGATCACCAGCAATATTCCAAAGTAGCATGACAAAAATCTTAGAGCCTTTTAGAAAACAAAATCCAGACATAGTTATCTATCAATACATGGATGATCTGTATGTAGGATCTGACTTAGAAATAGGGCAGCATAGAATAAAAATAGAGGAACTAAGACAACATCTGTTGAAGTGGGGACTTACCACACCAGACAAAAAACATAAGAAGGAACCCCCATTCCTTTGGAT>gi|24209941|gb|AY156735.1| HIV-1 clone P2.BCM.RT from USA reverse transcriptase (pol) gene, partial cdsCCCATAAGTCCTATTGAAACTGTACCAGTAAAATTAAAGCCAGGAATGGATGGCCCAAAAGTTAAGCAATGGCCACTGACAGAAGAAAAAATAAAAGCATTAGTAGAAATTTGTACAGAAATGGAAAAGGAAGGAAAAATTTCAAAAATTGGGCCTGAAAATCCATACAATACTCCAGTATTTGCCATAAAGAAAAAAGACAGTACTAAATGGAGAAAATTAGTAGATTTCAGAGAACTTAATAAGAGAACTCAAGACTTCTGGGAAGTTCAATTAGGAATACCACATCCTGCAGGGTTAAAAAAGAAAAAATCAGTAACAGTGCTGGATGTGGGTGATGCATATTTTTCAGTTCCCTTAGATAAGGAGTTCAGGAAGTATACTGCATTTACCATACCTAGTATAAACAATGAGACACCAGGGATTAGATATCAGTACAATGTGCTTCCACAGGGATGGAAAGGATCACCAGCAATATTCCAAAGTAGCATGACAAAAATCTTAGAGCCTTTTAGAAAACAAAATCCAGACATAGTTATCTATCAATACATGGATGATTTGTATGTAGGATCTGACTTAGAAATAGGGCAGCATAGAATAAAAATAGAAGAACTAAGACAACATCTGTTGAAGTGGGGACTTACCACACCAGACAAAAAACATCAGAAGGAACCTCCATTCCTTTGGAT>gi|24209943|gb|AY156736.1| HIV-1 clone P3.BCM.RT from USA reverse transcriptase (pol) gene, partial cdsCCCATAAGTCCTATTGAAACTGTACCAGTAAAATTAAAGCCAGGAATGGATGGCCCAAAAGTTAAACAATGGCCACTGACAGAAGAAAAAATAAAAGCATTAGTAGAAATTTGTACAGAAATGGAAAAGGAAGGAAAAATTTCAAAGATTGGGCCTGAAAATCCATACAATACTCCAGTATTTGCCATAAAGAAAAAAAACAGTACTAGATGGAGAAAATTAGTAGATTTCAGAGAACTTAATAAGAGAACTCAAGACTTCTGGGAAGTTCAATTAGGAATACCACATCCTGCAGGGTTAAAAAAGAAAAAATCAGTAACAGTGCTGGATGTGGGTGATGCATATTTTTCAGTTCCCTTAGATAAAGAGTTCAGGAAGTATACTGCATTTACCATACCTAGTATAAACAATGAGACACCAGGGATTAGATATCAATACAATGTGCTTCCACAGGGATGGAAAGGATCACCAGCAATATTCCAAAGTAGCATGACAAAAATCTTAGAGCCTTTTAGAAAACAAAATCCAGACATAGTTATCTATCAATACATGGATGATCTGTATGTAGGATCTGACTTAGAAATAGGGCAGCATAGAATAAAAATAGAGGAACTAAGACAACATCTGTTGAAGTGGGGATTTATCACACCAGACGAAAAACACCAGAAGGAACCTCCATTCCGTTGGAT
ダウンロードされたファイルにはMulti-Fasta形式で塩基配列が含まれている

準備したファイルでは、配列の名前を短くしてあります。
Vで始まる名前 victim = Mariaから分離されたHIVのRT
Pで始まる名前 patient=Robertの患者から分離されたHIVのRT
LAで始まる名前 Lafeyetteで⽣活するAIDS患者から分離されたHIVのRT
RT = Reverse Transcriptase 逆転写酵素

ダウンロードしたファイルをMafftでアラインして、Clustal 形式のアラインメントを作成する。

Mafftを起動する
1 左下 スタート をクリック

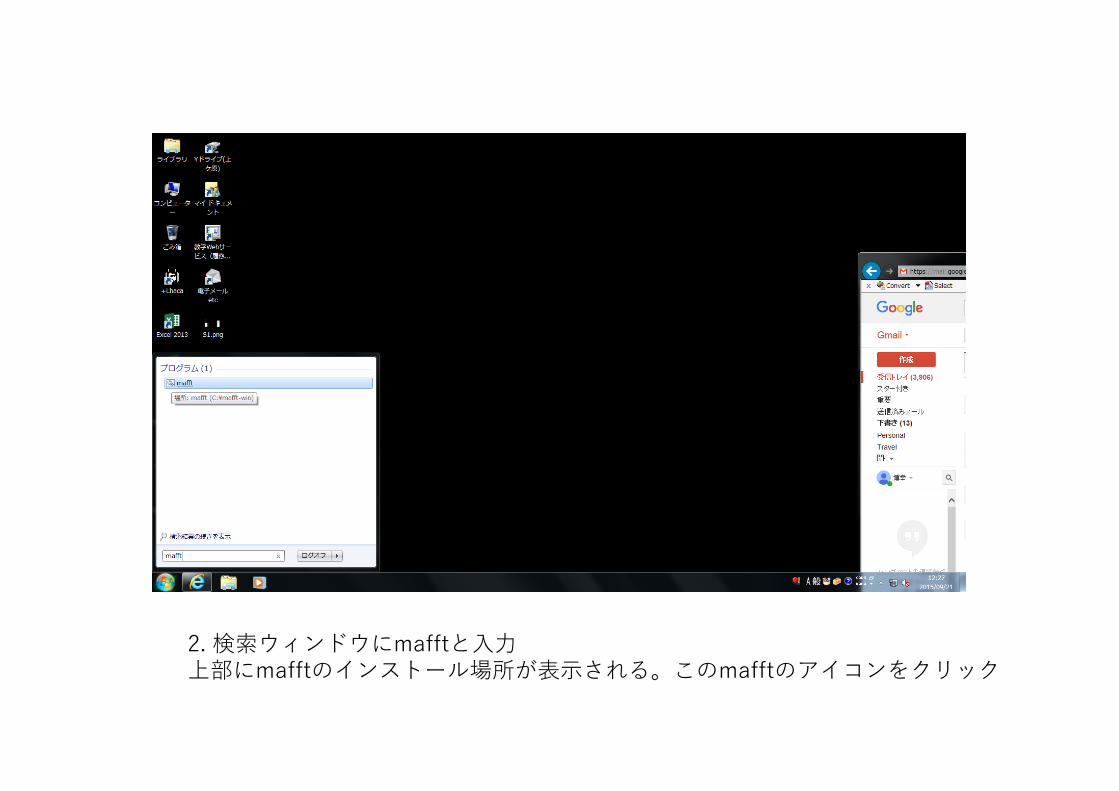
2. 検索ウィンドウにmafftと⼊⼒上部にmafftのインストール場所が表⽰される。このmafftのアイコンをクリック

1. このウィンドウにmafftと⼊⼒
2. 表⽰されたmafftをクリック

3. mafftの⼊⼒画⾯がたちあがる。

Input file? (fasta format)@ ここに⼊⼒ファイルを記⼊(次のようにする)

4. ⼊⼒ファイルを指定するために、multi-fasta formatのファイルが置かれたDirectoryを表⽰する。(ここからはWindows OS上での処理)左下のスタートをクリックし、出て来たパネル左上のドキュメントを選択

ドキュメントを選択
ファイルがドキュメントフォルダにある場合

5. ドキュメントdirectoryが表⽰される。Directoryからmafftのウィンドウにファイルをドラッグすると、ファイル名が⼊⼒される。ファイル名が⼊⼒されたらenterキーをおす。
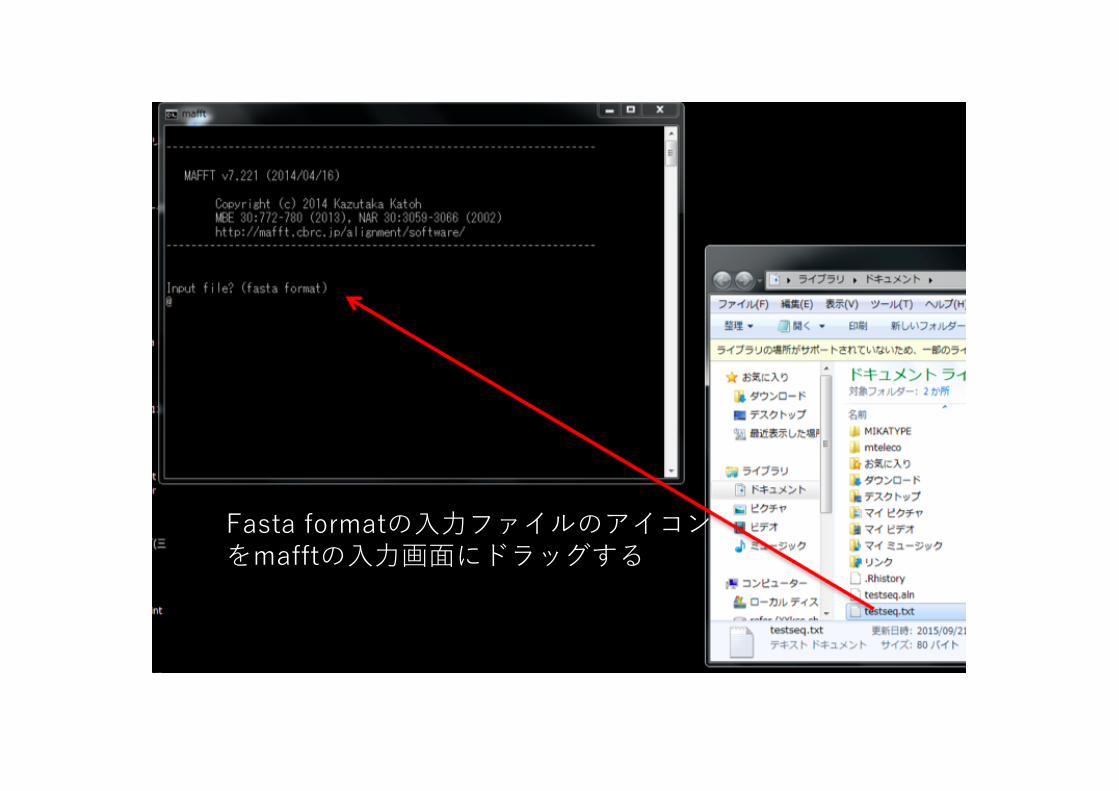
Fasta formatの⼊⼒ファイルのアイコンをmafftの⼊⼒画⾯にドラッグする

6. Outputすなわち、アラインメントを出⼒するファイル名を聞かれる、⼊⼒ファイル名を参考にZドライブ上のファイル(新規でも既存の者でも良い)を指定しEnterキーをおす。出⼒オプションを聞いてくるので2を指定する。Clustal形式/Fasta形式 Sorted Order/Input Order 説明はアラインメントを⾒ながら

1. ⼊⼒ファイルが
Z:\ファイル名の形で記⼊される
エンターキーをおす
2. Output file?@
とアラインメントの出⼒ファイルを聞いてくるのでZ:¥ファイル名としてドキュメントフォルダのファイル名を持つファイルに保存するようにしてエンターキーをおす
3. 6つのアウトプット形式が出⼒される

6つ出⼒形式1 Clustal format/ Sorted2 Clustal format / Input order3 Fasta format / Sorted4 Fasta format / Input Order5 Phylip format / Sorted6 Phylip format / Input Order
Clustal と Fastaは説明済みPhylipは系統解析の際に説明
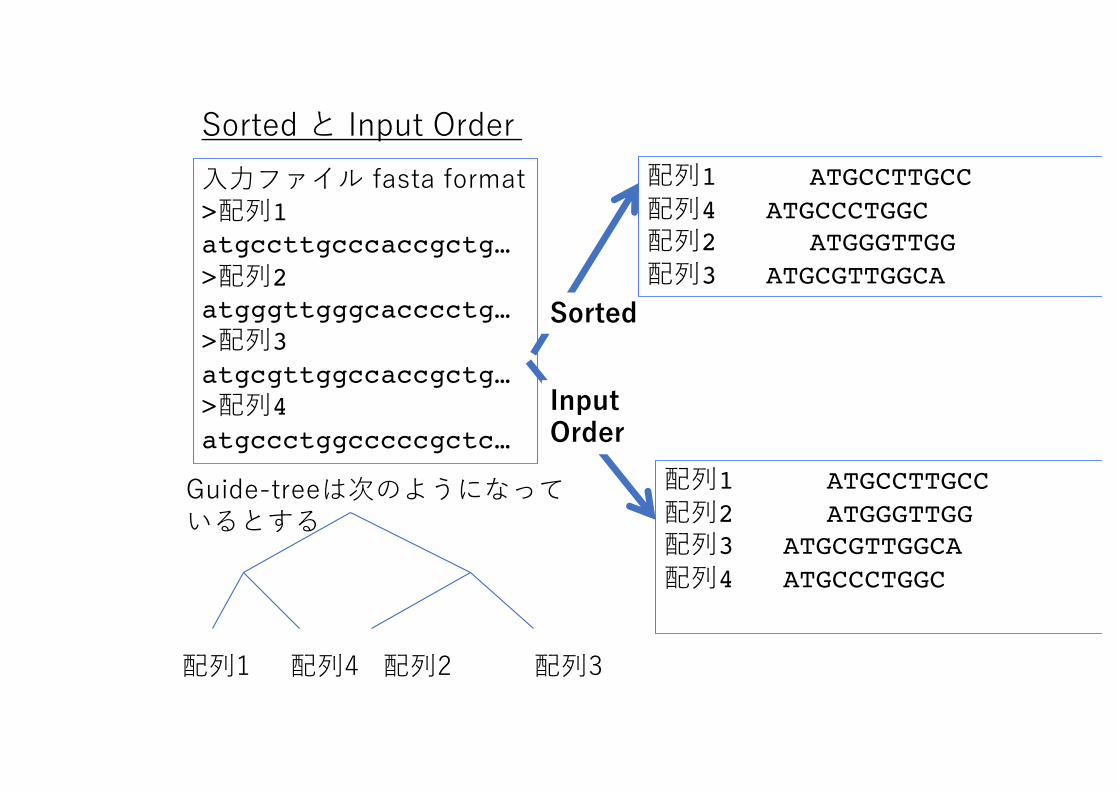
Sorted と Input Order ⼊⼒ファイル fasta format>配列1atgccttgcccaccgctg…>配列2atgggttgggcacccctg…>配列3atgcgttggccaccgctg…>配列4atgccctggcccccgctc…
Guide-treeは次のようになっているとする
配列1 配列4 配列2 配列3
Sorted
Input Order
配列1 ATGCCTTGCC配列4 ATGCCCTGGC配列2 ATGGGTTGG配列3 ATGCGTTGGCA
配列1 ATGCCTTGCC配列2 ATGGGTTGG配列3 ATGCGTTGGCA配列4 ATGCCCTGGC

7. アラインメントのオプションを聞いてくる。1の̶autoオプションを指定してenterautoオプション ⼩規模データ丁寧に、⼤規模データそれなりにアライン

アラインメントのオプションを聞いてくる(正確さ優先か、速度優先か)。1の̶autoオプションを指定してenterautoオプション ⼩規模データ丁寧に、⼤規模データそれなりにアライン

t7
8. 指定したファイルやオプションを、コマンドライン形式で確認してくる問題なければ Y を⼊⼒してenter
(前ページのウィンドウを拡⼤)

9. ウィンドウ中に、出⼒が表⽰(END)が表⽰された時点で、出⼒ファイルに書き込まれている。

(前ページのウィンドウを拡⼤)





























