Embed Size (px)
DESCRIPTION
Finding “the gene” for cystic fibrosis. Finding “the gene” for cystic fibrosis. Why is this in quotes? CF is not caused by a gene, it’s caused by multiple genes. CF is not caused by genetic factors. CF is not caused by a gene, it’s caused by a mutation. - PowerPoint PPT Presentation
Citation preview

Finding “the gene” for cystic fibrosis

Finding “the gene” for cystic fibrosis
Why is this in quotes?
A. CF is not caused by a gene, it’s caused by multiple genes.
B. CF is not caused by genetic factors.
C. CF is not caused by a gene, it’s caused by a mutation.


How to find genetic determinants of naturally
varying traits?

Genetic markers
Fig. 10.3
(microsatellite)

Genetic markers
Fig. 10.3
(microsatellite)
Table 11.1

Lots of benign variation between us.

How do you find polymorphisms?
Fig. 11.6
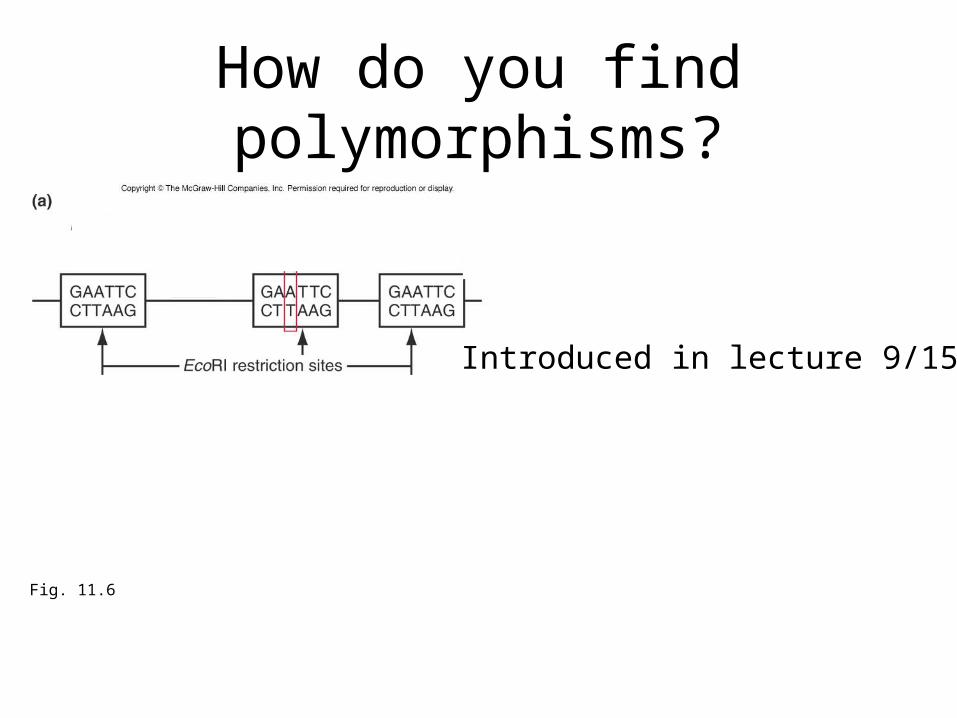
How do you find polymorphisms?
Fig. 11.6
Introduced in lecture 9/15.

How do you find polymorphisms?
Fig. 11.6

How do you find polymorphisms?
Fig. 11.6

How do you find polymorphisms?
Fig. 11.6

How do you find polymorphisms?
Fig. 11.6

How do you find polymorphisms?
Fig. 11.6

Hybrid mapping: location of probe
QuickTime™ and aTIFF (Uncompressed) decompressor
are needed to see this picture.
QuickTime™ and aTIFF (Uncompressed) decompressor
are needed to see this picture.
www3.mdanderson.org/depts/cellab/fish1.htm
mouse human/mouse hybrid

Hybrid mapping: location of probe
QuickTime™ and aTIFF (Uncompressed) decompressor
are needed to see this picture.
Back then, no technique to see 6kb
at cytological resolution.

Who cares about benign polymorphisms?
Remember Sturtevant?
Fig. 5.10

Who cares about benign polymorphisms?
We are going to do a two-point cross.
One of our genetic loci is represented by phenotype; the other is a DNA marker.

Mapping a disease locus
Fig. 11.A
(Autosomal dom)
A1
A2

Mapping a disease locus
Fig. 11.A
(Autosomal dom)phenotype (variation in locus 1)
A1
A2

Mapping a disease locus
Fig. 11.A
(Autosomal dom)phenotype (variation in locus 1)
marker genotype (variation in locus 2)
A1
A2

Mapping a disease locus
Fig. 11.A
(Autosomal dom)phenotype (variation in locus 1)
marker genotype (variation in locus 2)
How close are they in genetic distance?
A1
A2
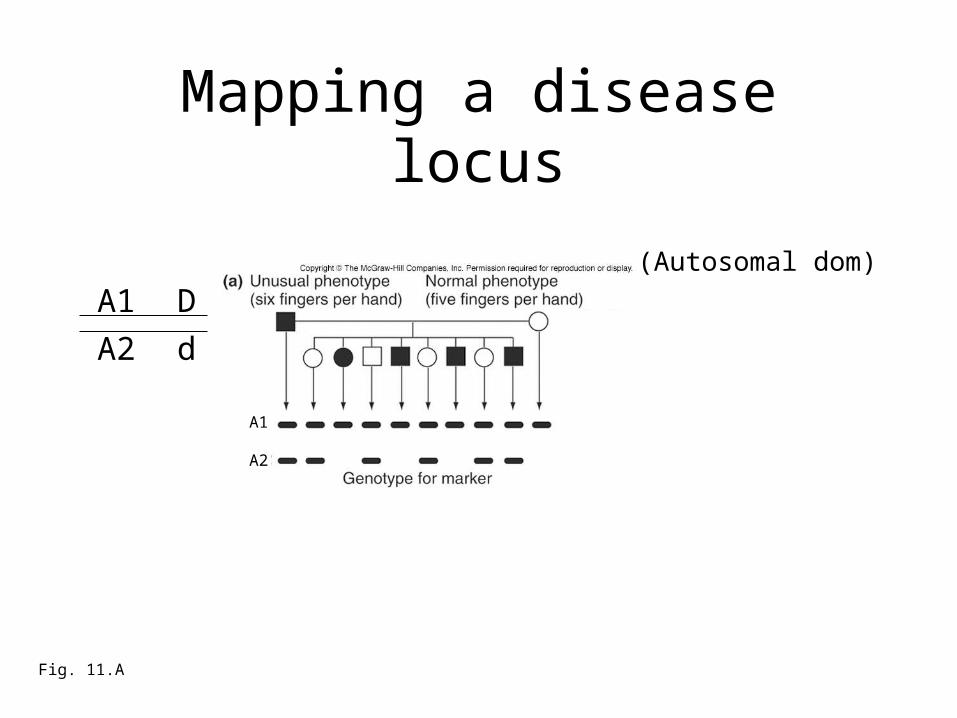
Mapping a disease locus
Fig. 11.A
A1 D
A2 d
(Autosomal dom)
A1
A2
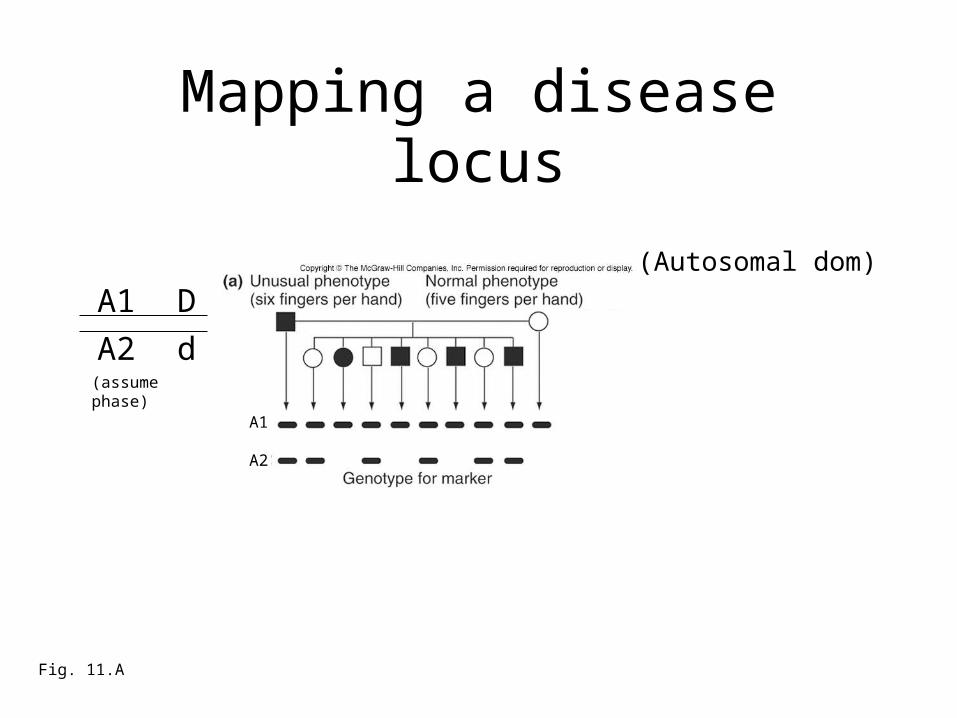
Mapping a disease locus
Fig. 11.A
A1 D
A2 d
(Autosomal dom)
A1
A2
(assume phase)

Mapping a disease locus
Fig. 11.A
A1 D
A2 d
A1 d
A1 d
A1
A2

Mapping a disease locus
Fig. 11.A
A1 D
A2 d
A1 d
A1 d
A2 d
A1
A2

Mapping a disease locus
Fig. 11.A
A1 D
A2 d
A1 d
A1 d
A1 D
A1
A2

Mapping a disease locus
Fig. 11.A
A1 D
A2 d
A1 d
A1 d
A2 d
A1
A2
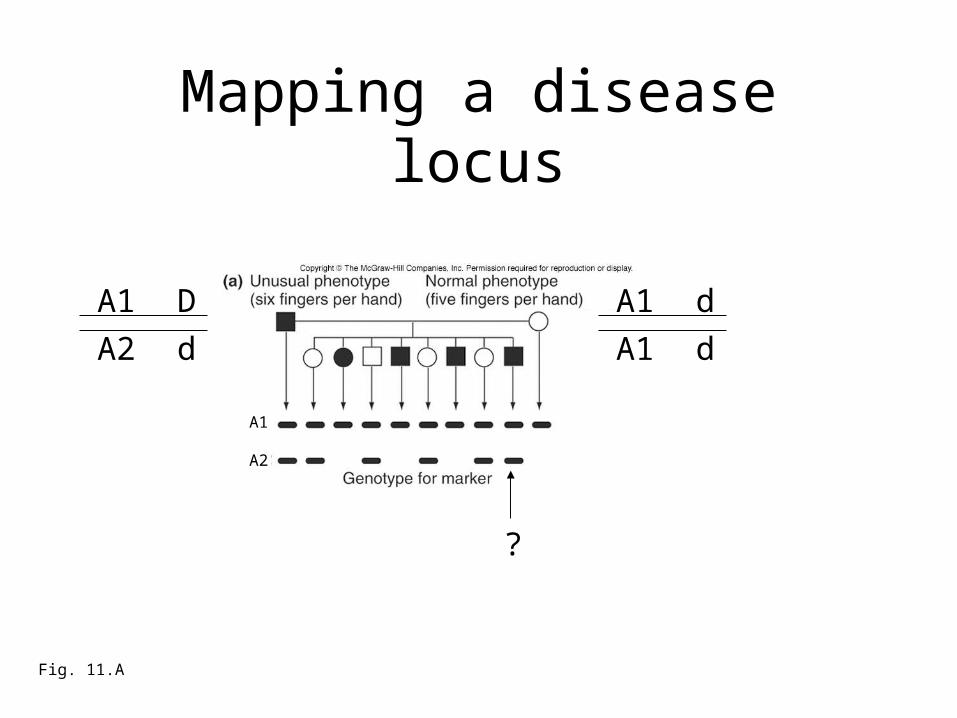
Mapping a disease locus
Fig. 11.A
A1 D
A2 d
A1 d
A1 d
?
A1
A2

Mapping a disease locus
Fig. 11.A
A1 d
A1 d
?
A1 D
A2 d(sperm)
A1
A2
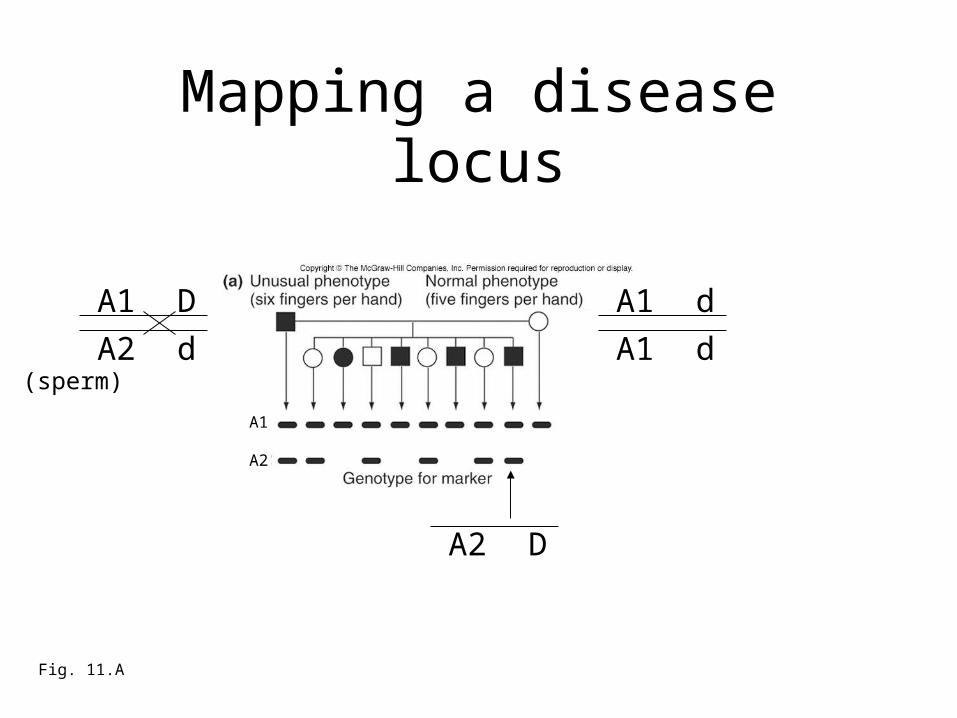
Mapping a disease locus
Fig. 11.A
A1 d
A1 d
A2 D
A1 D
A2 d(sperm)
A1
A2

Mapping a disease locus
Fig. 11.A
A1 d
A1 d
A1 D
A2 d
A1
A2
In total, 7 of the kids are non-recombinants and 1
is a recombinant.

Mapping a disease locus
Fig. 11.A
What is the apparent RF between the DNA marker and the disease mutation?
A. 1/10B. 1/8C. 1/20
A1
A2
In total, 7 of the kids are non-recombinants and 1
is a recombinant.

Mapping a disease locus
Fig. 11.A
What is the apparent RF between the DNA marker and the disease mutation?
1/8 = 12.5 m.u.
A1
A2
A. 1/10B. 1/8C. 1/20
In total, 7 of the kids are non-recombinants and 1
is a recombinant.

Why do I say “apparent RF?”
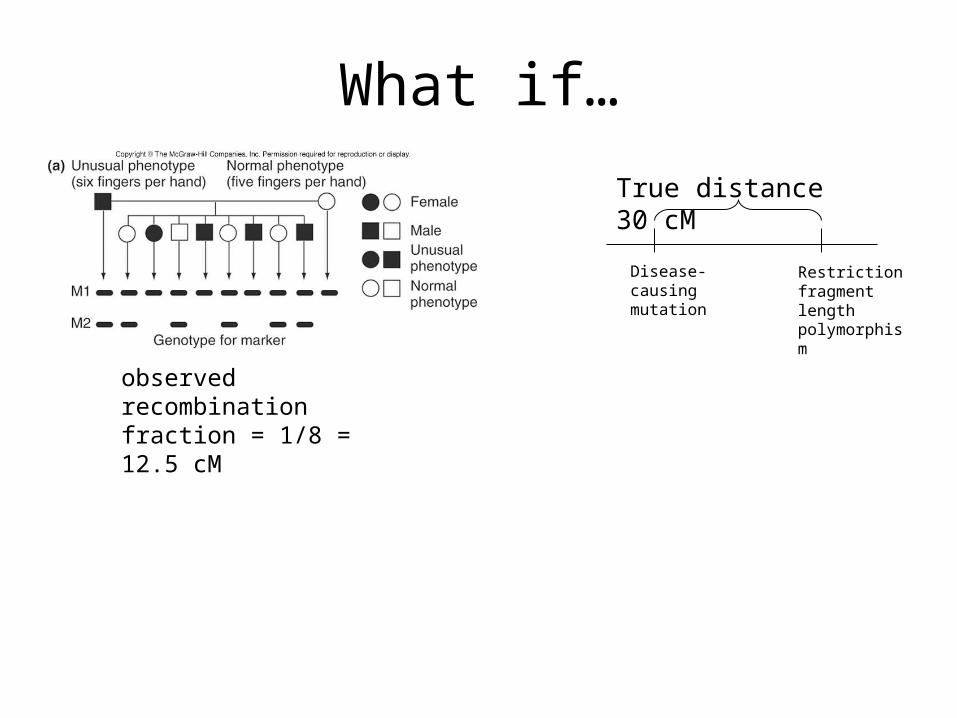
What if…
observed recombination fraction = 1/8 = 12.5 cM
Disease-causing mutation
Restriction fragment length polymorphism
True distance 30 cM

What if…
observed recombination fraction = 1/8 = 12.5 cM
Disease-causing mutation
Restriction fragment length polymorphism
True distance 30 cM
You could say this will never happen. But…

What if…
observed recombination fraction = 1/8 = 12.5 cM
Disease-causing mutation
Restriction fragment length polymorphism
True distance 30 cM
this is our observation

What if…
observed recombination fraction = 1/8 = 12.5 cM
Disease-causing mutation
Restriction fragment length polymorphism
True distance 30 cM
The observed number of recombinants is just a point estimate, with some error associated.
this is our observation

12 cM, 18 cM…who cares?
Further experiments need to find the causal variant, not just a marker. If distances are wrong, could be
hunting for years.

Mapping a disease locus
Fig. 11.A
We now know the mutation is near (linked to) the marker.
1/8 = 12.5 m.u.
A1
A2

Mapping a disease locus
We now know the mutation is near (linked to) the marker.
marker (known)
1/8 = 12.5 m.u.
A1
A2

Mapping a disease locus
We now know the mutation is near (linked to) the marker.
window containing causative mutation
1/8 = 12.5 m.u.marker (known)
A1
A2

Mapping a disease locus
1/8 = 12.5 m.u.
How significant?
A1
A2

Mapping a disease locus
1/8 = 12.5 m.u.
How significant?If RF = 0.5 (unlinked), would be like flipping a coin 8 times.
How likely would you be to get 7 heads and 1 tail?
A1
A2

If RF = 0.5 (unlinked), would be like flipping a coin 8 times.How likely would you be to get 7 heads and 1 tail?
How much MORE likely is a model of RF < 0.5?

If RF = 0.5 (unlinked), would be like flipping a coin 8 times.How likely would you be to get 7 heads and 1 tail?
How much MORE likely is a model of RF < 0.5?
For large cross between known parents, would use 2 to evaluate significance.
Here we can’t.

LOD scores
1 recomb, 7 non-recomb.
r = genetic distance between marker and disease locus
A1
A2

LOD scores
1 recomb, 7 non-recomb.
Odds = P(pedigree | r)
P(pedigree | r = 0.5)
r = genetic distance between marker and disease locus
A1
A2

LOD scores
1 recomb, 7 non-recomb.
Odds = P(pedigree | r)
P(pedigree | r = 0.5)
r = genetic distance between marker and disease locus
“How likely are the data given our model?”
A1
A2

LOD scores
k = 1 recomb, n = 7 non-recomb.
Odds = P(pedigree | r)
P(pedigree | r = 0.5)
r = genetic distance between marker and disease locus
Odds = (1-r)n • rk
0.5n • 0.5k
A1
A2

LOD scores
Odds = P(pedigree | r)
P(pedigree | r = 0.5)
r = genetic distance between marker and disease locus
Odds = (1-r)n • rk
0.5(total # meioses)
A1
A2
k = 1 recomb, n = 7 non-recomb.

LOD scores
Odds = P(pedigree | r)
P(pedigree | r = 0.5)
r = genetic distance between marker and disease locus
Odds = (1-r)n • rk
0.5(total # meioses)
We have an idea of true r, but it is imprecise.
k = 1 recomb, n = 7 non-recomb.
A1
A2

Remember?
observed recombination fraction = 1/8 = 12.5 cM
Disease-causing mutation
Restriction fragment length polymorphism
True distance 30 cM
The observed number of recombinants is just a point estimate, with some error associated.
this is our observation

LOD scores
Odds = P(pedigree | r)
P(pedigree | r = 0.5)
r = genetic distance between marker and disease locus
Odds = (1-r)n • rk
0.5(total # meioses)
k = 1 recomb, n = 7 non-recomb.
A1
A2
This formalism allows any r value. Let’s guess r = 0.3.

LOD scores
Odds = P(pedigree | r)
P(pedigree | r = 0.5)
r = genetic distance between marker and disease locus
Odds = (1-r)n • rk
0.5(total # meioses)
Odds = 0.77 • 0.31
0.58
k = 1 recomb, n = 7 non-recomb.
A1
A2
This formalism allows any r value. Let’s guess r = 0.3.

LOD scores
Odds = P(pedigree | r)
P(pedigree | r = 0.5)
r = genetic distance between marker and disease locus
Odds = (1-r)n • rk
0.5(total # meioses)
Odds = 0.77 • 0.31
0.58
= 6.325
k = 1 recomb, n = 7 non-recomb.
A1
A2
This formalism allows any r value. Let’s guess r = 0.3.

LOD scores
Odds = P(pedigree | r)
P(pedigree | r = 0.5)
r = genetic distance between marker and disease locus
Odds = (1-r)n • rk
0.5(total # meioses)
Odds = 0.77 • 0.31
0.58
= 6.325
Data >6 times more likely under LINKED hypothesis than under UNLINKED hypothesis.
k = 1 recomb, n = 7 non-recomb.
A1
A2

LOD scoresr odds
0.1 12.244
0.2 10.737
0.3 6.325
0.4 2.867
0.5 ??
Odds = P(pedigree | r)
P(pedigree | r = 0.5)
Odds = (1-r)n • rk
0.5(total # meioses)
k = 1 recomb, n = 7 non-recomb.

LOD scores
Odds = P(pedigree | r)
P(pedigree | r = 0.5)
Odds = (1-r)n • rk
0.5(total # meioses)
Odds at r=0.5?A. 2.5B. 0C. 1D. 10
r odds
0.1 12.244
0.2 10.737
0.3 6.325
0.4 2.867
0.5 ??

LOD scores
What’s the best (most likely) value of r?
A. 0.1B. 0.2C. 0.3D. 0.4E. 0.5
r odds
0.1 12.244
0.2 10.737
0.3 6.325
0.4 2.867
0.5 1

What problems will look like
1,2 1,2 1,1 1,2 1,1 1,2 1,1 1,2 1,2 1,1
A1
A2

What problems will look like
1,2 1,2 1,1 1,2 1,1 1,2 1,1 1,2 1,2 1,1

What problems will look like
1,2 1,2 1,1 1,2 1,1 1,2 1,1 1,2 1,2 1,1
Count number of recombinants, calculate odds.

Reading and chapter problems on web site.










