Embed Size (px)
Citation preview

First principles study of the structure, electronic state and stability
of AlnAsCm cations
Ling Guo *, Hai-shun Wu
School of Chemistry and Material Science, Shanxi Normal University, Linfen 041004, China
Received 30 August 2005; accepted 12 October 2005
Available online 3 February 2006
Abstract
Structural and electronic properties of semiconductor binary microclusters AlnAsCm cations have been investigated using the B3LYP-DFT
method in the ranges of nZ1, 2 and mZ1–7. Full structural optimization, adiabatic ionization potentials calculation and frequency analysis are
performed with the basis of 6-311CG(d). The charged-induced structural changes in these cations have been discussed. The strong As–As bond is
also favored over Al–As bonds in the AlnAsCm cations in comparison with corresponding neutral cluster. With Asm forming the base, adding Al
atom(s) in different positions would find the stable structures of AlnAsCm cations quickly and correctly. AlAsC2 , AlAsC4 , and AlAsC6 are predicted to
be species with high stabilities and possible to be produced experimentally.
q 2006 Elsevier B.V. All rights reserved.
Keywords: AlnAsCCm cluster; Density functional theory; Stability
1. Introduction
The III–V semiconductor clusters have been the topic of
many experimental and theoretical studies [1–3]. A primary
driving force behind such studies is that III–V materials are of
great technological importance as they find applications in the
fabrication of fast microelectronic devices, small devices, and
light-emitting diodes. Consequently, a detailed study of the
properties of such clusters as a function of their sizes could
provide significant insight into the evolution from the
molecular level to the bulk. Despite the numerous experimental
investigations on the GaAs, InAs, InP, and more recently, the
AlP and GaP clusters, the literature contains very little on the
AlAs clusters. While ab initio calculations on properties of
AlxAsy clusters have been carried out by several groups [4–10].
Andreoni [4] calculated the structures, stability, and melting of
(AlAs)n (nZ2–5) using the Car-parrinello method. Quek et al.
[5] had reported tight binding molecular dynamics studies of
the structures of AlmAsn (mCn%13). Tozzini et al. [6]
presented extensive theoretical calculations of the geometric
and electronic properties of neutral and ionized AlAs fullerene-
like clusters of the type AlxAsxC4 with a number of atoms up to
52, on the basis of density functional theory. Costales et al. [7]
0166-1280/$ - see front matter q 2006 Elsevier B.V. All rights reserved.
doi:10.1016/j.theochem.2005.10.060
* Corresponding author.
E-mail address: [email protected] (L. Guo).
used density functional theory (DFT) to explore structural and
vibrational properties for (AlAs)n clusters up to six atoms,
finding the same behavior as in the Aluminum nitride clusters.
Archibong et al. [8] calculated the low-lying electronic states
of Al3As, AlAs3, and the corresponding anions at the B3LYP
and CCSD(T) levels using the 6-311CG(2df) one-particle
basis set. The adiabatic electron affinities, electron detachment
energies and harmonic vibrational frequencies of both the
anions and the neutral molecules are presented and discussed.
Feng et al. [9] reported MRSDCI study of the ground and
several low-lying excited states of Al2As3, Al3As2, and their
ions. Recently, Zhu [10] studied the spectroscopic properties
for Al2As, AlAs2, and their ions using density functional theory
(DFT:B3LYP) and complete active space multiconfiguration
self-consistent field (CASSCF) calculations.
Since the properties of clusters are unique, it is expected that
cluster assembled materials can have uncommon properties.
Studies on the electronic and geometric structures of clusters are
necessary. Theoretical calculations have been performed on
aluminum arsenide clusters by a number of methods for some
time. While most studies were devoted to the small neutral
clusters and theoretical investigations on cationic clusters are
few. In this work, we present a density function theory study for
semiconductor binary systems AlAsCm and Al2AsCm with mZ1–7.
We aim to provide more reliable ground-state geometries and
electronic states, relative orbital and total energies, HOMO–
LUMO gaps and theoretically calculated IR vibration frequen-
cies at the corresponding optimum structures. With Asm forming
Journal of Molecular Structure: THEOCHEM 760 (2006) 167–173
www.elsevier.com/locate/theochem
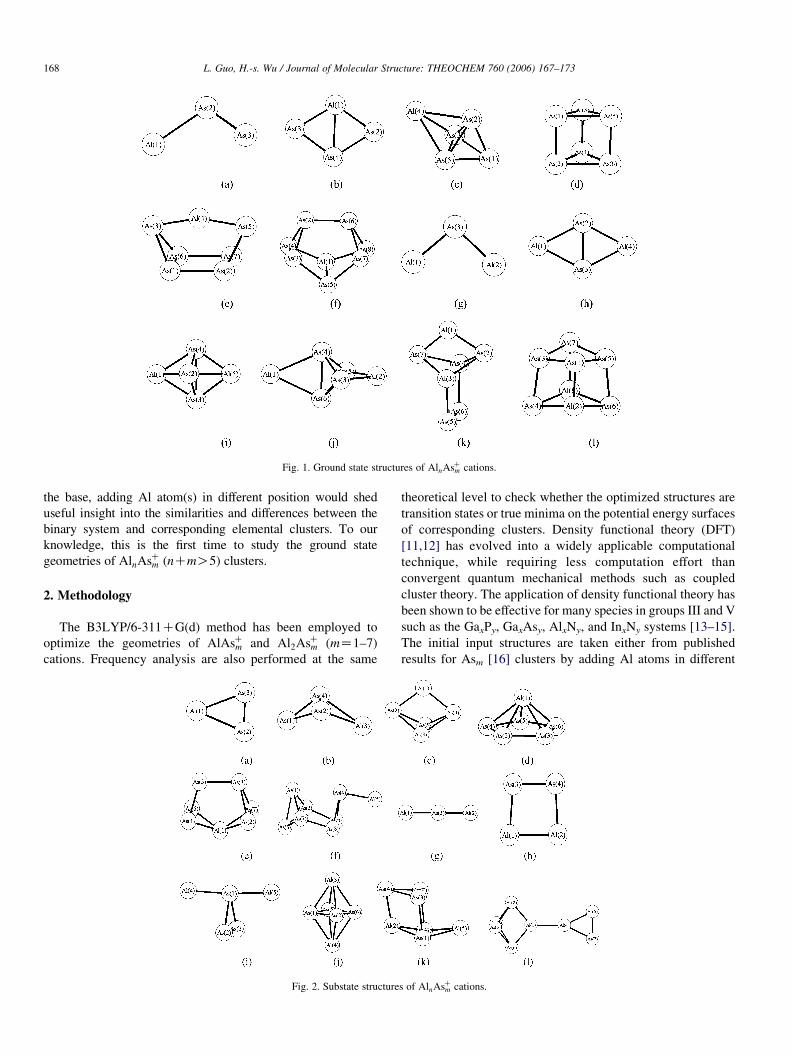
Fig. 1. Ground state structures of AlnAsCm cations.
L. Guo, H.-s. Wu / Journal of Molecular Structure: THEOCHEM 760 (2006) 167–173168
the base, adding Al atom(s) in different position would shed
useful insight into the similarities and differences between the
binary system and corresponding elemental clusters. To our
knowledge, this is the first time to study the ground state
geometries of AlnAsCm (nCmO5) clusters.
2. Methodology
The B3LYP/6-311CG(d) method has been employed to
optimize the geometries of AlAsCm and Al2AsCm (mZ1–7)
cations. Frequency analysis are also performed at the same
Fig. 2. Substate structure
theoretical level to check whether the optimized structures are
transition states or true minima on the potential energy surfaces
of corresponding clusters. Density functional theory (DFT)
[11,12] has evolved into a widely applicable computational
technique, while requiring less computation effort than
convergent quantum mechanical methods such as coupled
cluster theory. The application of density functional theory has
been shown to be effective for many species in groups III and V
such as the GaxPy, GaxAsy, AlxNy, and InxNy systems [13–15].
The initial input structures are taken either from published
results for Asm [16] clusters by adding Al atoms in different
s of AlnAsCm cations.

Table 1
Geometric parameters and electronic states of AlnAsCm clusters
Molecule Type L (A)
1A’ 1–2 2.867
2–3 2.1302A2 1–2 2.395
1–4 2.724
2–4 2.3981A’ 1–2 2.454
2–3 2.513
2–4 2.964
3–4 2.9712A’’ 1–2 2.569
L. Guo, H.-s. Wu / Journal of Molecular Structure: THEOCHEM 760 (2006) 167–173 169
positions, or the results reported for other III–V semiconductor
clusters, or arbitrarily constructed and fully optimized via the
Berny algorithm. For AlnAsCm clusters, the ground-state
structures are either relaxed within the geometries of
corresponding neutrals or distorted into new structures with
lower energies and much lower symmetries due to Jane–Teller
distortions. To determine the stability of the optimized
structures, harmonic vibration frequencies are further calcu-
lated with B3LYP functional. Some optimized geometries,
although low in energies, are found to be first order or even
higher order stationary points. All calculations are performed
with the GAUSSIAN 98 suite of programs [17].
1–3 2.4851–5 2.541
2–3 2.893
2–4 2.540
2–6 2.507
3–4 2.3831A’ 1–2 2.679
1–3 2.499
1–6 2.490
3–4 2.352
AlAsC7 ð2AÞ 1–4 2.396
1–5 2.510
3. Results and discussion
3.1. Geometry
The ground state and metastable state geometric sketch
figures of AlnAsCm (nZ1, 2; mZ1–7) optimized by B3LYP
method are shown in Figs. 1 and 2, respectively. Geometric
parameters are listed in Table 1.
1–8 2.472
2–3 2.456
2–4 2.523
2–6 2.573
3–4 2.555
3–5 2.544
5–7 2.500
6–7 2.510
6–8 2.443
Al2AsC(3B2) 1–3 2.3912B1u 1–2 2.569
2–3 2.3801A’1 1–2 2.499
2–3 2.655
Al2AsC4 ð2AÞ 1–3 2.513
1–6 2.583
2–3 2.582
1–4 2.5121A’ 1–2 2.376
1–3 2.764
2–3 2.547
2–4 2.591
3–5 2.429
3–7 2.547
4–7 2.591
5–6 2.2592B1 1–2 2.447
1–3 2.558
3–4 2.510
4–8 2.478
3.1.1. AlAsCmAlAsC. The optimized bond length of neutral AlAs is
2.33 A, somewhat shorter than the Costales’ result of 2.57 A
[7] at the GGA level of theory in conjunction with a double
numerical basis set supplemented with d polarization func-
tions. And the vibrational frequency (349 cmK1) is higher than
Costales’ result (282 cmK1) [8]. For the cationic case, the
ionize electron comes out from a bonding orbital predicting its
instability relative to the neutral monomer by 7.70 eV. It is also
manifested in an increase in the internuclear distance (2.54 A)
and a decrease in the frequency value (233 cmK1) indicating
that the bond in cationic state is weaker than the corresponding
one in the neutral monomer.
AlAsC2 . The present calculations predict an isosceles
triangles (C2v,2B2) ground state 1(a) for the equilibrium
geometry of AlAs2 molecule. Geometric sketch shows it to
be acute triangle with aAs–Al–AsZ47.28 and the Al–As, As–As
bond lengths are 2.749 and 2.202 A, which compare well with
Zhu’s values [10] of 48.68, 2.706 and 2.226, respectively,
optimized by the CASSCF calculation. The ground state of
AlAsC2 is the Cs (1A 0) geometry 1(a). The structure 2(a)
(C2v,1A1), which has the same geometry as the neutral AlAs2
cluster, is now a transition state with an imaginary frequency at
56i cmK1 and lying only 0.08 eV higher in energy.
AlAsC3 . A near degeneracy is found by previous ab initio and
density functional study [8] between the 1A1(C2v) and the1A 0(Cs) lowest states for AlAs3. The present calculation leads to
the same conclusion. The distorted tetrahedron 2(b) with Cs
symmetry is the ground state of neutral AlAs3. The local
minimum rhomboidal structure 1(b) with C2v symmetry and
another Cs (1A 0) structure with imaginary frequency lie 0.03
and 0.47 eV, respectively, higher in energy. The energy
ordering is different in the cations. Removing an electron
stabilizes the C2v (2A2) structure 1(b) with respect to the Cs
(2A 00) and Cs (2A 0) 2(b) form and their energy differences are
0.005 and 0.03 eV, respectively.
AlAsC4 . The most stable AlAs4 isomer is a tetrahedral As4
structure [16] with a two-fold Al atom bond to it. The structure
is very similar to 2(c). While the lowest-energy structure we
found for AlAsC4 is a distorted trigonal bipyramid (Cs) 1(c),
which can be derived from a tetrahedral As4 structure by

L. Guo, H.-s. Wu / Journal of Molecular Structure: THEOCHEM 760 (2006) 167–173170
capping an additional Al atom between atoms (2, 3, 5). At the
same time, bonding a two-fold Al atom to the tetrahedral As4
form, we obtain a substable isomer of AlAsC4 2(c) with C2v
symmetry lying 0.03 eV above the ground state, which has the
very similar geometry as that of the neutral AlAs4. Both AlAs4
and its cation have the same geometry as that of the AlP4 [18]
and GaAs4 [19], which is attributed to the fact that both of them
take a similar valence structure due to the same family in the
periodic table.
AlAsC5 . The ground state of neutral AlAs5 (C5v,1A1) 2(d) is
derived from the As5 [16] cluster by placing a five-fold Al atom
on the top. In the procession, the geometry of As5 is nearly
maintained and the five same As–As bond lengths are only
elonged by 0.6%. Removing an electron from the neutral
molecule yields a cationic AlAsC5 (C5v,2A1), which has the
same geometry as the neutral AlAs5. The distorted triangle
prism 1(d) lying only 0.03 eV below the 2(d) is a ground state
structure with Cs (2A 00) symmetry, which is built from
substitution of an As atom by an Al atom in the triangle
prism As6.
AlAsC6 . The face-capped triangle prism 2(e) is the ground
state of the neutral AlAs6 (C2v,2A2) in our present optimization.
It can be viewed as capping an additional Al atom on the square
face of the triangle prism As6. The As2–As4 and As6–As7 bonds
are broken in the capping procession. Removing an electron
produces the ground state structure of cationic AlAsC6 (Cs,1A 0)
1(e), which is different from the neutral isomer and derived
from a boat-shape As6 by adding of one two-fold Al atom
between atoms (1,5). Model 2(e) of AlAsC6 (C2v,1A1) is now a
transition state lying 0.39 eV above the ground state.
Removing an electron results that the four same Al–As bonds
(2.510 A) of 2(e) are elonged compared to the corresponding
Al–As bond lengths (2.490 A) in the neutral isomer, while the
average As–As bond length is contracted by about 0.41%. The
numbers of As–As bonds in 2(e) are less than those of 1(e),
which may be the reason for its less stable and indicates that the
As–As bonds play a more decisive role than the Al–As bonds in
the determination of the geometry and energy of AlAsC6 .
AlAsC7 . The present calculations consider a cuneane
structure 1(f) as the ground state of neutral AlAs7, which can
be derived from a square-face-capped triangle prism As7 by
adding an additional three-fold Al atom. The symmetry of As7
is changed from C2v to lower Cs (1A 0) symmetry of AlAs7 in the
procession. AlAsC7 with lower C1 (2A) symmetry takes the
similar geometry 1(f) as the neutral. Another Cs isomer 2(f) of
cationic AlAsC7 with the same electronic state as 1(f) lies
0.72 eV above the ground state, which contains a one-fold Al
atom.
3.1.2. Al2AsCm clusters
Al2AsC. The 2B2 state of triangle prevails as the ground state
of Al2As with the Al–As–Al apex angle of 92.18, and the Al–As
and Al–Al bond lengths of 2.351 and 3.384 A agree well with
Zhu’s [10] results. This global minimum exhibits the same
symmetry as that of the cationic Al2AsC (C2v,3B2) 1(g).
The comparison of geometries of the neutral and cation reveals
that the Al–Al (3.549 A) bond lengths in Al2AsC are elongated
with a more open Al–As–Al (95.88) bond angle, implying that
the Al–Al bond is further weaken upon ionization. The Al–As–
Al linear configuration of Al2AsC 2(g) is 0.21 eV less stable
than 1(g).
Al2AsC2 . Costales et al. [7] have investigated four different
linear configurations and a rhombic structure of Al2As2 at the
GGA/DNP level of theory, and we have calculated both the
singlet and triplet states of these and other isomers. We support
their prediction that the 1Ag electronic state with rhombus
equilibrium structure 1(h) is the ground state of Al2As2.
The As–As bond length is 2.308 A and is much shorter than the
Al–Al bond length of 4.793 A resulting in an acute As–Al–As
bond angle of 51.48 in the rhombus structure. Removing an
electron from it produces a cationic Al2AsC2 (2B1u) 1(h), which
has the same geometry as the neutral. In the rhombus cationic
dimmer, the loss of an electron results in the decrease of the
Al–As (from 2.660 to 2.569 A) bond distance, with a
corresponding increase in the As–As distances (from 2.308 to
2.380 A). The next lowest energy isomer is a trapezoidal form
2(h) with C2v (2A2) symmetry, which has an imaginary
frequency lying 0.79 eV above the ground state. The mode of
imaginary frequency shows a tendency for the rhombic ground
state.
Al2AsC3 . Balasubramanian et al. [9] studied different isomers
of neutral Al2As3 cluster. We have considered these and other
structures and support their results that the most
stable configuration is trigonal bipyramid geometry 1(i) with
D3h;ð2AÞ symmetry. The next structure in the energetic
ordering is the C2v (2A1) isomer 2(i) lying 0.42 eV above the
ground state. The ground state structure of Al2AsC3 ðD3h;1A
0
1)
1(i) is the same as that of the neutral molecular. The As–As
(2.655 A) bond length in the Al2AsC3 is elonged, while the Al–
Al (3.947 A) and Al–As (2.499 A) bond lengths are contracted
compared to the corresponding bond lengths of Al–Al
(4.213 A), Al–As (2.572 A) and As–As (2.557 A) in the
neutral ground state. Next low-lying Al2AsC3 isomer in the
energy ordering possesses (C2v1A1) geometry 2(i). This
structure is a transition state with imaginary frequency lying
0.63 eV above the ground-state structure discussed above.
Al2AsC4 . The lowest energy Al2As4 isomer is a slightly
distorted square bipyramid 2(j) with C2v (1A1) symmetry,
which is very similar to D4h point group. It can be derived from
the optimal structure of Al2As3 isomer 1(i) by capping an As
atom between two adjacent As atoms. The structure of the
Al2AsC4 1(j) is different from its neutral molecule. This global
minimum of Al2AsC4 (C1) can be obtained from the minimum
structure of neutral AlAs4 2(c) by capping an additional Al
atom between As3 and As5 atoms. The C2v (2B1) square
bipyramid 2(j) in the cationic isomers is now a transition state
lying only 0.06 eV higher in energy.
Al2AsC5 . The present calculations predict the Cs (2A 0) ground
state 1(k) for neutral Al2As5. Another Cs isomer 2(k) with the
same electronic state is located at 0.50 eV above the ground
state, which can be derived from the substable structure of
AlAs5 1(d) by capping an additional Al atom between atoms
(1,6). The energy ordering is preserved in the cation. Removing
an electron makes the Cs (1A 0) structure 1(k) still the global
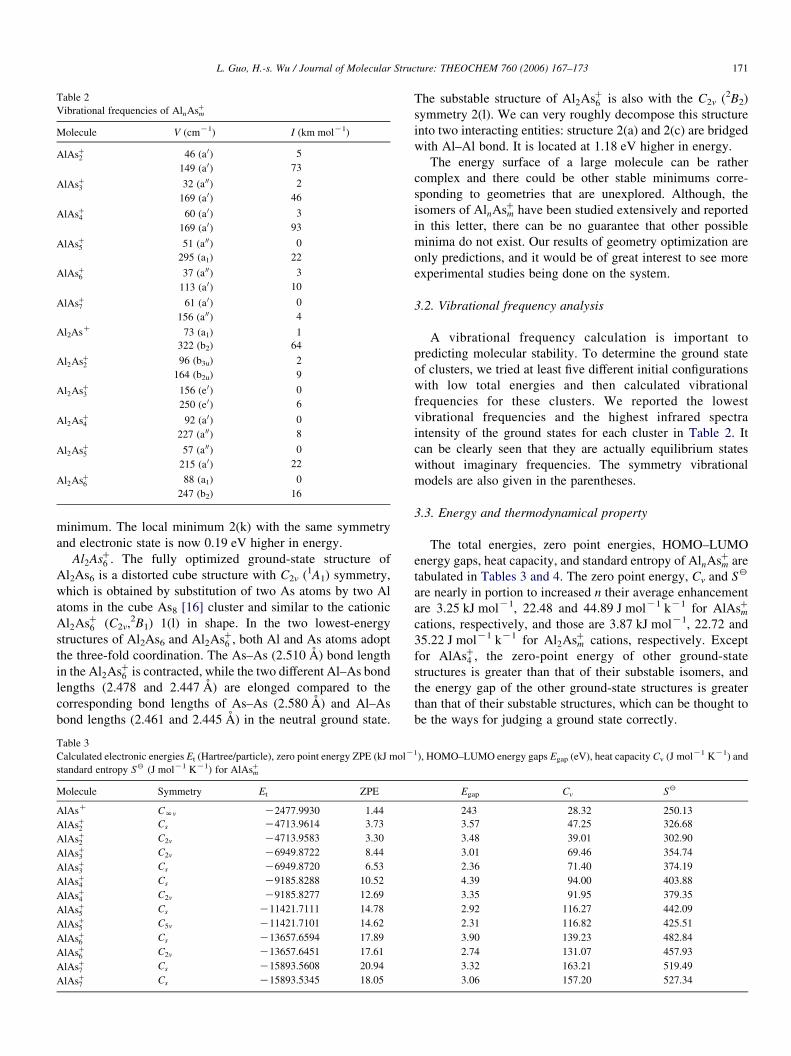
Table 2
Vibrational frequencies of AlnAsCm
Molecule V (cmK1) I (km molK1)
AlAsC2 46 (a 0) 5
149 (a 0) 73
AlAsC3 32 (a 00) 2
169 (a 0) 46
AlAsC4 60 (a 0) 3
169 (a 0) 93
AlAsC5 51 (a 00) 0
295 (a1) 22
AlAsC6 37 (a 00) 3
113 (a 0) 10
AlAsC7 61 (a 0) 0
156 (a 00) 4
Al2AsC 73 (a1) 1
322 (b2) 64
Al2AsC2 96 (b3u) 2
164 (b2u) 9
Al2AsC3 156 (e 0) 0
250 (e 0) 6
Al2AsC4 92 (a 0) 0
227 (a 00) 8
Al2AsC5 57 (a 00) 0
215 (a 0) 22
Al2AsC6 88 (a1) 0
247 (b2) 16
L. Guo, H.-s. Wu / Journal of Molecular Structure: THEOCHEM 760 (2006) 167–173 171
minimum. The local minimum 2(k) with the same symmetry
and electronic state is now 0.19 eV higher in energy.
Al2AsC6 . The fully optimized ground-state structure of
Al2As6 is a distorted cube structure with C2v (1A1) symmetry,
which is obtained by substitution of two As atoms by two Al
atoms in the cube As8 [16] cluster and similar to the cationic
Al2AsC6 (C2v,2B1) 1(l) in shape. In the two lowest-energy
structures of Al2As6 and Al2AsC6 , both Al and As atoms adopt
the three-fold coordination. The As–As (2.510 A) bond length
in the Al2AsC6 is contracted, while the two different Al–As bond
lengths (2.478 and 2.447 A) are elonged compared to the
corresponding bond lengths of As–As (2.580 A) and Al–As
bond lengths (2.461 and 2.445 A) in the neutral ground state.
Table 3
Calculated electronic energies Et (Hartree/particle), zero point energy ZPE (kJ molK
standard entropy S2 (J molK1 KK1) for AlAsCm
Molecule Symmetry Et ZPE
AlAsC CNv K2477.9930 1.44
AlAsC2 Cs K4713.9614 3.73
AlAsC2 C2v K4713.9583 3.30
AlAsC3 C2v K6949.8722 8.44
AlAsC3 Cs K6949.8720 6.53
AlAsC4 Cs K9185.8288 10.52
AlAsC4 C2v K9185.8277 12.69
AlAsC5 Cs K11421.7111 14.78
AlAsC5 C5v K11421.7101 14.62
AlAsC6 Cs K13657.6594 17.89
AlAsC6 C2v K13657.6451 17.61
AlAsC7 Cs K15893.5608 20.94
AlAsC7 Cs K15893.5345 18.05
The substable structure of Al2AsC6 is also with the C2v (2B2)
symmetry 2(l). We can very roughly decompose this structure
into two interacting entities: structure 2(a) and 2(c) are bridged
with Al–Al bond. It is located at 1.18 eV higher in energy.
The energy surface of a large molecule can be rather
complex and there could be other stable minimums corre-
sponding to geometries that are unexplored. Although, the
isomers of AlnAsCm have been studied extensively and reported
in this letter, there can be no guarantee that other possible
minima do not exist. Our results of geometry optimization are
only predictions, and it would be of great interest to see more
experimental studies being done on the system.
3.2. Vibrational frequency analysis
A vibrational frequency calculation is important to
predicting molecular stability. To determine the ground state
of clusters, we tried at least five different initial configurations
with low total energies and then calculated vibrational
frequencies for these clusters. We reported the lowest
vibrational frequencies and the highest infrared spectra
intensity of the ground states for each cluster in Table 2. It
can be clearly seen that they are actually equilibrium states
without imaginary frequencies. The symmetry vibrational
models are also given in the parentheses.
3.3. Energy and thermodynamical property
The total energies, zero point energies, HOMO–LUMO
energy gaps, heat capacity, and standard entropy of AlnAsCm are
tabulated in Tables 3 and 4. The zero point energy, Cv and S2
are nearly in portion to increased n their average enhancement
are 3.25 kJ molK1, 22.48 and 44.89 J molK1 kK1 for AlAsCmcations, respectively, and those are 3.87 kJ molK1, 22.72 and
35.22 J molK1 kK1 for Al2AsCm cations, respectively. Except
for AlAsC4 , the zero-point energy of other ground-state
structures is greater than that of their substable isomers, and
the energy gap of the other ground-state structures is greater
than that of their substable structures, which can be thought to
be the ways for judging a ground state correctly.
1), HOMO–LUMO energy gaps Egap (eV), heat capacity Cv (J molK1 KK1) and
Egap Cv S2
243 28.32 250.13
3.57 47.25 326.68
3.48 39.01 302.90
3.01 69.46 354.74
2.36 71.40 374.19
4.39 94.00 403.88
3.35 91.95 379.35
2.92 116.27 442.09
2.31 116.82 425.51
3.90 139.23 482.84
2.74 131.07 457.93
3.32 163.21 519.49
3.06 157.20 527.34
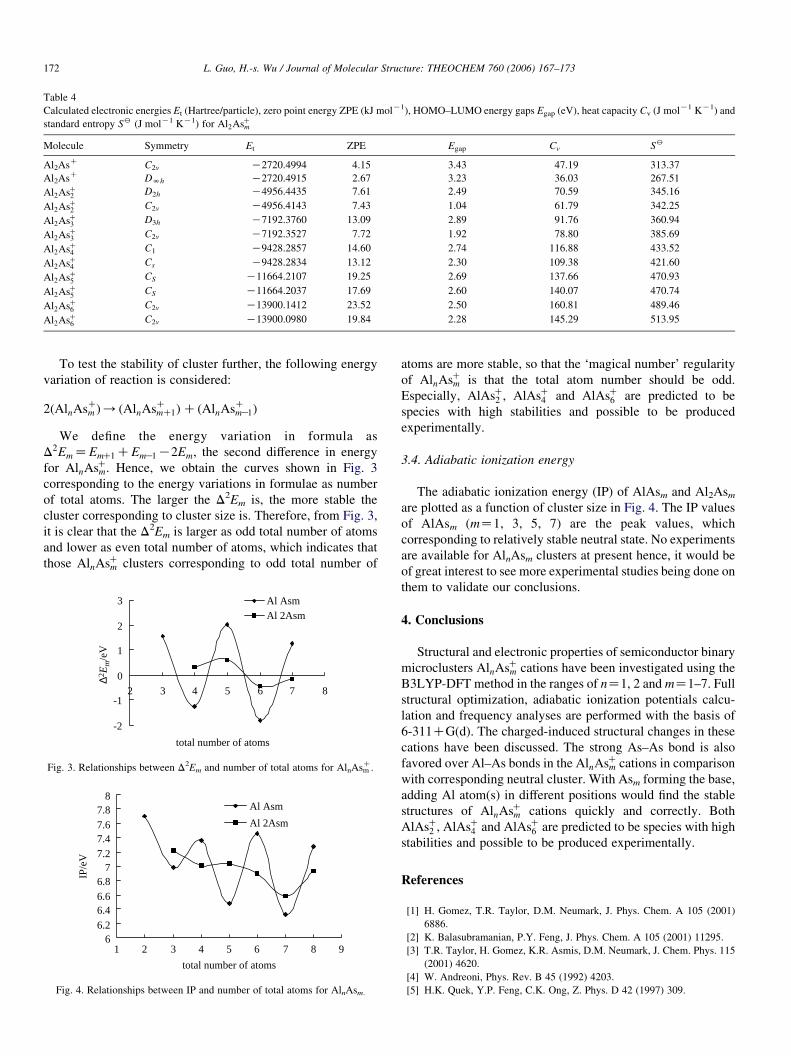
Table 4
Calculated electronic energies Et (Hartree/particle), zero point energy ZPE (kJ molK1), HOMO–LUMO energy gaps Egap (eV), heat capacity Cv (J molK1 KK1) and
standard entropy S2 (J molK1 KK1) for Al2AsCm
Molecule Symmetry Et ZPE Egap Cv S2
Al2AsC C2v K2720.4994 4.15 3.43 47.19 313.37
Al2AsC DNh K2720.4915 2.67 3.23 36.03 267.51
Al2AsC2 D2h K4956.4435 7.61 2.49 70.59 345.16
Al2AsC2 C2v K4956.4143 7.43 1.04 61.79 342.25
Al2AsC3 D3h K7192.3760 13.09 2.89 91.76 360.94
Al2AsC3 C2v K7192.3527 7.72 1.92 78.80 385.69
Al2AsC4 C1 K9428.2857 14.60 2.74 116.88 433.52
Al2AsC4 Cs K9428.2834 13.12 2.30 109.38 421.60
Al2AsC5 CS K11664.2107 19.25 2.69 137.66 470.93
Al2AsC5 CS K11664.2037 17.69 2.60 140.07 470.74
Al2AsC6 C2v K13900.1412 23.52 2.50 160.81 489.46
Al2AsC6 C2v K13900.0980 19.84 2.28 145.29 513.95
L. Guo, H.-s. Wu / Journal of Molecular Structure: THEOCHEM 760 (2006) 167–173172
To test the stability of cluster further, the following energy
variation of reaction is considered:
2ðAlnAsCm Þ/ ðAlnAsCmC1ÞC ðAlnAsCmK1Þ
We define the energy variation in formula as
D2EmZEmC1CEmK1K2Em, the second difference in energy
for AlnAsCm. Hence, we obtain the curves shown in Fig. 3
corresponding to the energy variations in formulae as number
of total atoms. The larger the D2Em is, the more stable the
cluster corresponding to cluster size is. Therefore, from Fig. 3,
it is clear that the D2Em is larger as odd total number of atoms
and lower as even total number of atoms, which indicates that
those AlnAsCm clusters corresponding to odd total number of
-2
-1
0
1
2
3
2 3 4 5 6 7 8
total number of atoms
∆2E
m/e
V
Al AsmAl 2Asm
Fig. 3. Relationships between D2Em and number of total atoms for AlnAsmC.
66.2
6.46.6
6.87
7.2
7.47.6
7.88
1 2 3 4 5 6 7 8 9
total number of atoms
IP/e
V
Al Asm
Al 2Asm
Fig. 4. Relationships between IP and number of total atoms for AlnAsm.
atoms are more stable, so that the ‘magical number’ regularity
of AlnAsCm is that the total atom number should be odd.
Especially, AlAsC2 , AlAsC4 and AlAsC6 are predicted to be
species with high stabilities and possible to be produced
experimentally.
3.4. Adiabatic ionization energy
The adiabatic ionization energy (IP) of AlAsm and Al2Asmare plotted as a function of cluster size in Fig. 4. The IP values
of AlAsm (mZ1, 3, 5, 7) are the peak values, which
corresponding to relatively stable neutral state. No experiments
are available for AlnAsm clusters at present hence, it would be
of great interest to see more experimental studies being done on
them to validate our conclusions.
4. Conclusions
Structural and electronic properties of semiconductor binary
microclusters AlnAsCm cations have been investigated using the
B3LYP-DFT method in the ranges of nZ1, 2 and mZ1–7. Full
structural optimization, adiabatic ionization potentials calcu-
lation and frequency analyses are performed with the basis of
6-311CG(d). The charged-induced structural changes in these
cations have been discussed. The strong As–As bond is also
favored over Al–As bonds in the AlnAsCm cations in comparison
with corresponding neutral cluster. With Asm forming the base,
adding Al atom(s) in different positions would find the stable
structures of AlnAsCm cations quickly and correctly. Both
AlAsC2 , AlAsC4 and AlAsC6 are predicted to be species with high
stabilities and possible to be produced experimentally.
References
[1] H. Gomez, T.R. Taylor, D.M. Neumark, J. Phys. Chem. A 105 (2001)
6886.
[2] K. Balasubramanian, P.Y. Feng, J. Phys. Chem. A 105 (2001) 11295.
[3] T.R. Taylor, H. Gomez, K.R. Asmis, D.M. Neumark, J. Chem. Phys. 115
(2001) 4620.
[4] W. Andreoni, Phys. Rev. B 45 (1992) 4203.
[5] H.K. Quek, Y.P. Feng, C.K. Ong, Z. Phys. D 42 (1997) 309.

L. Guo, H.-s. Wu / Journal of Molecular Structure: THEOCHEM 760 (2006) 167–173 173
[6] V. Tozzini, F. Buda, A. Fasolino, J. Phys. Chem. B 105 (2001)
12477.
[7] A. Costales, A.K. Kandalam, R. Franco, R. Pandey, J. Phys. Chem. B 106
(2002) 1940.
[8] E.F. Archibong, A. St-Amant, J. Phys. Chem. A 106 (2002) 7390.
[9] P.Y. Feng, D. Dai, K. Balasubramanian, J. Phys. Chem. A 104 (2000)
422.
[10] X. Zhu, J. Mol. Struct. (Theochem) 638 (2003) 99.
[11] (a) P. Hohenberg, W. Kohn, Phys. Rev. B 136 (1964) 864;
(b) W. Kohn, L. Sham, Phys. Rev. A 140 (1965) 1133.
[12] W. Kohn, A.D. Becke, R.G. Parr, J. Phys. Chem. 100 (1996) 12974.
[13] E.F. Achibong, A. St-Amant, Chem. Phys. Lett. 316 (2000) 151.
[14] S.K. Nayak, S.N. Khanna, P. Jena, Phys. Rev. B 57 (1998) 3787.
[15] A. Costales, R. Pandey, J. Phys. Chem. A 107 (2003) 192.
[16] P. Ballone, R.O. Jones, J. Chem. Phys. 100 (1994) 4941.
[17] M.J. Frisch, G.W. Trucks, H.B. Schlegel, G.E. Scuseria, M.A. Robb, J.R.
Cheeseman, V.G. Zakrzewski, J.A. Montgomery Jr., R.E. Stratmann, J.C.
Burant, S. Dapprich, J.M. Millam, A.D. Daniels, K.N. Kudin, M.C. Strain,
O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C.
Pomelli, C. Adamo, S. Clifford, J. Ochterski, G.A. Petersson, P.Y. Ayala,
Q. Cui, K. Morokuma, D.K. Malick, A.D. Rabuck, K. Raghava-chari, J.
B. Foresman, J. Cioslowski, J.V. Ortiz, B.B. Stefanov, G. Liu, A.
Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R.L. Martin, D.J. Fox,
T. Keith, M.A. Al-Laham, C.Y. Peng, A. Nanayakkara, C. Gonzalez, M.
Challacombe, P.M.W. Gill, B. Johnson, W. Chen, M.W. Wong, J.L.
Andres, C. Gonzalez, M. Head-Gordon, E.S. Replogle, J.A. Pople,
Computer Code GAUSSIAN98, Revision A.6, Gaussian Inc., Pittsburgh, PA,
1998.
[18] L. Guo, H. Wu, Z. Jin, Int. J. Mass Spectrom. 240 (2005) 149.
[19] P. Piquini, S. Canuto, A. Fazzio, Nanostruct. Mater. 10 (1998) 635.





























