Embed Size (px)
Citation preview

Fixation-dependent organization of core histonesfollowing DNA fluorescent in situ hybridizationMichael J. Hendzel, David P. Bazett-Jones
Departments of Anatomy and Medical Biochemistry, Faculty of Medicine, University of Calgary,3330 Hospital Dr. N.W., Calgary, Alberta, Canada T2N 4N1
&misc:Received: 19 November 1996; in revised form: 10 January 1997 / Accepted: 10 January 1997
&p.1:Abstract. We have evaluated the effects of differentDNA denaturation protocols commonly used in DNAfluorescent in situ hybridization (FISH) experiments onchromatin structure using indirect immunofluorescence.The use of antibodies to acetylated histones H3 and H4demonstrates that the different procedures differ consid-erably in their extent of histone displacement. Proce-dures involving paraformaldehyde fixation were foundto be compatible with the structural preservation of ace-tylated chromatin organization by indirect immunofluo-rescence. These results provide a basis for interpretingDNA FISH experiments aimed at determining chromatinorganization of individual loci.
Introduction
DNA fluorescence in situ hybridization (FISH) is a pow-erful tool for analysing large-scale organization of wholechromosomes, regions of chromosomes, and individualgenes (Zirbel et al. 1993; Xing et al. 1993, 1995; Yokotaet al. 1995). The technique has the unique potential tooffer information on the organization of individual geneloci within the interphase nucleus and the metaphasechromosome, particularly since the spatial arrangementof individual gene loci in mammalian interphase nucleiis poorly understood. A commonly invoked architecturefor the chromatin of transcriptionally active genes, de-rived from biochemical evidence (see van Holde 1989;Wolffe 1995), is the loop domain. The loop-domainmodel envisions the chromatin of transcriptionally activegenes as a decondensed structure maintained in a loopconfiguration through stable contacts with an underlyingnuclear skeleton and is consistent with the transmissionelectron microscopy (TEM) observations of the tran-scribing Balbiani ring genes (Daneholt 1992). This open
architecture should be amenable to analysis by DNAFISH for relatively large genes that might be anticipatedto produce a hybridization signal with a length greaterthan 1µm. Where the length of a gene locus is consider-able, probes located many kilobases apart can be simul-taneously visualized, their relative distances measured,and compaction ratios estimated (e.g. Lawrence et al.1990). To apply these techniques to the study of inter-phase chromatin organization, however, it is important toestablish that the relatively harsh DNA denaturation pro-tocols involved in preparing the specimen do not signifi-cantly alter chromatin organization.
We have addressed this issue by characterizing thedistribution of acetylated histones H3 and H4. If localchromatin organization is not perturbed during DNA de-naturation, then the immunofluorescence patterns gener-ated for these two chromatin proteins should not be sig-nificantly different from those of cells that have beenfixed but not denatured. Analysis of the well-defined pat-terns of these chromatin proteins within interphase nucleigenerates a very sensitive assay for alterations in chroma-tin structure during preparation of cells. Moreover, sincethe antibodies recognize acetylated histone species thatare highly enriched in transcriptionally active chromatin,these probes are particularly sensitive to alterations in thestructure of this functionally important class of genes. Inthis paper we show that the amount of chromatin disrup-tion that occurs depends on the FISH protocol that isused. Not surprisingly, some protocols proved totally un-satisfactory for such a study. However, cells prefixed withparaformaldehyde, followed by DNA denaturation withNaOH in 70% ethanol, consistently maintained native or-ganization of chromatin at the resolution provided by in-direct immunofluorescence microscopy.
Materials and methods
Cell culture. &p.2:HeLa cells were grown in Joklik Minimal EssentialMedium in the presence of 10% fetal bovine serum (Gibco). Cellswere then plated into eight-well plates with a single glass coverslipin each well. Typically cells were allowed to adhere overnight.
Edited by:P.B. Moens
Correspondence to:D.P. Bazett-Jones(e-mail: [email protected])&/fn-block:
Chromosoma (1997) 106:114–123
© Springer-Verlag 1997
115
Immunofluorescence. &p.2:Coverslips were inverted onto 30µl drops ofdiluted antibody applied to a Parafilm square. The coverslips wereincubated for 30 min, washed in PBS, and then incubated withsecondary antibodies in an identical fashion. Cells were finallywashed in PBS and then mounted with 90% citifluor (a commer-cially available anti-fading reagent) in PBS in the presence of4′,6-diamidino-2-phenylindole (DAPI).
Cell fixation, DNA denaturation, and mock hybridization. &p.2:Cellswere pretreated in one of three ways. (1) Cells were fixed with 4%paraformaldehyde in PBS for 5 min at room temperature and thenmembranes were removed by extraction with 0.1% Triton X-100
in PBS for 5 min. (2) Cells were fixed in –20°C methanol for5 min. (3) Cells were incubated in 75 mM KCl, 20 mM HEPES,pH 7.5 for 15 min at 0°C and then fixed with 3:1 methanol:aceticacid for 10 min at room temperature. Cells were then rinsed threetimes with PBS and denatured with either 70% formamide in2×SSC at 70°C for 2 min or 0.2 M NaOH, 70% ethanol at roomtemperature for 5 min (1×SSC is 0.15 M NaCl, 0.015 M sodiumcitrate.) For mock hybridizations (RNA FISH protocol), cellswere fixed with 1% paraformaldehyde in PBS, membranes wereremoved with 0.1% Triton X-100 in PBS, and then coverslipswere incubated for 3.5 h in hybridization mix (1 part 20×SSC, 1part BSA, 1 part double-distilled H2O, and 2 parts 50% dextran
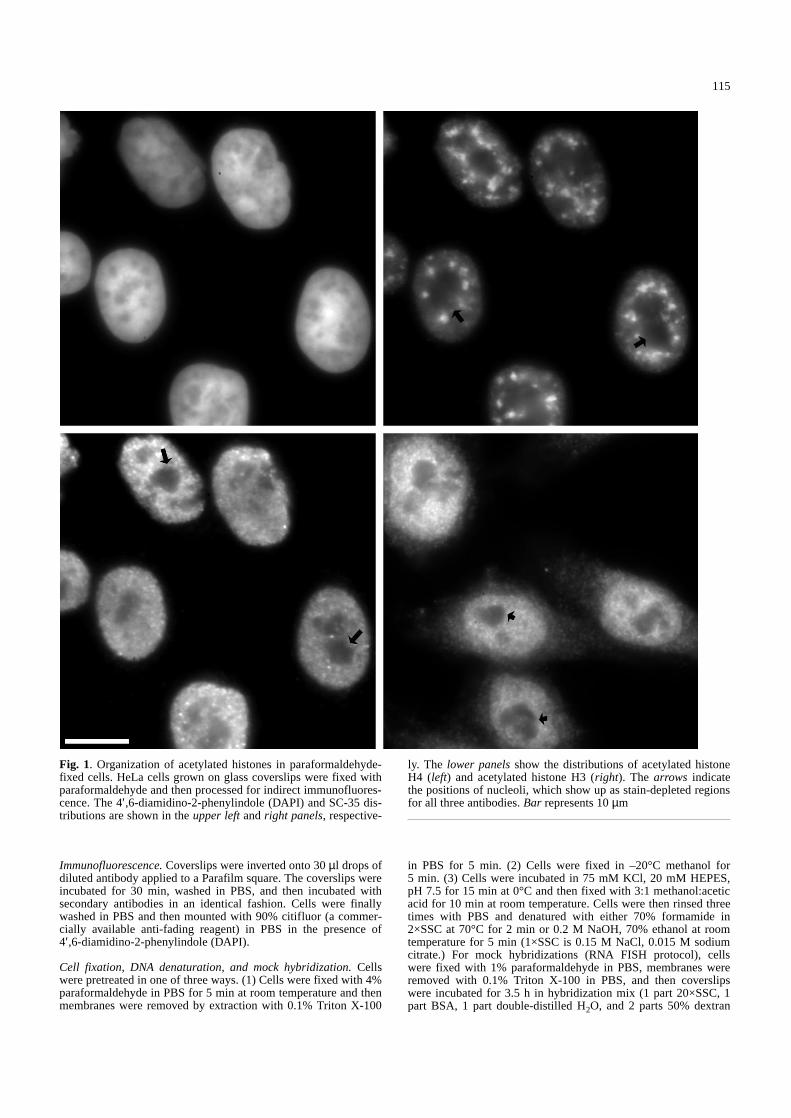
Fig. 1. Organization of acetylated histones in paraformaldehyde-fixed cells. HeLa cells grown on glass coverslips were fixed withparaformaldehyde and then processed for indirect immunofluores-cence. The 4′,6-diamidino-2-phenylindole (DAPI) and SC-35 dis-tributions are shown in the upper leftand right panels, respective-
ly. The lower panelsshow the distributions of acetylated histoneH4 (left) and acetylated histone H3 (right). The arrows indicatethe positions of nucleoli, which show up as stain-depleted regionsfor all three antibodies. Bar represents 10µm&/fig.c:
116
sulphate) as described by Johnson et al. (1991). Cells were thenrinsed with PBS and stained with antibodies for indirect immuno-fluorescence.
Antibodies. &p.2:SC-35 was obtained from the cell culture medium of amouse B-cell line producing the monoclonal antibody (ATCC).SC-35 was used undiluted and its subnuclear distribution has beenpreviously characterized (Spector et al. 1991). The antibodies rec-ognizing acetylated H4 are those described by Lin et al. (1989).The “penta” serum used has been shown in immunoprecipitationexperiments specifically to recognize acetylated histone H4 withinnative chromatin isolated from HeLa cells (Perry et al. 1993;
Annunziato et al. 1995). The antibody recognizing acetylated his-tone H3 was prepared as a peptide-specific antibody, targetingacetylated lysines 9 and 14. Sequencing studies of N-termini ofH3 in HeLa cells show that lysine 14 is the first lysine acetylated,whereas lysine 9 is utilized fourth, appearing predominantly intetra-, but not tri-, di-, or monoacetylated H3 (Thorne et al. 1990).Consistent with the expected specificity, the antibody recognizespredominantly tetra- and penta-acetylated histone H3 in HeLacells (Boggs et al., 1996). The secondary antibodies used were aCy-3-conjugated goat anti-rabbit antibody (Chemicon), and a flu-orescein isothiocyanate-conjugated goat anti-mouse (IgA, M, andG) antibody (Sigma).
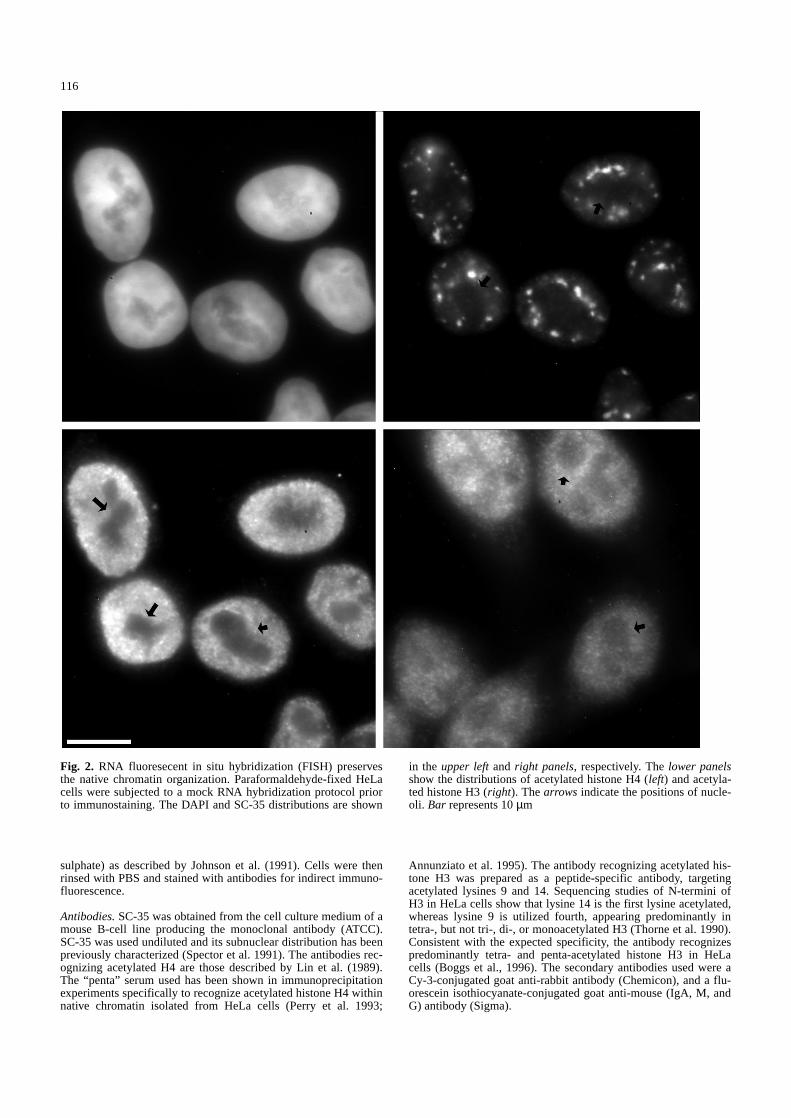
Fig. 2. RNA fluoresecent in situ hybridization (FISH) preservesthe native chromatin organization. Paraformaldehyde-fixed HeLacells were subjected to a mock RNA hybridization protocol priorto immunostaining. The DAPI and SC-35 distributions are shown
in the upper leftand right panels, respectively. The lower panelsshow the distributions of acetylated histone H4 (left) and acetyla-ted histone H3 (right). The arrows indicate the positions of nucle-oli. Bar represents 10µm&/fig.c:
117
Results
Evaluation of the effects of RNA FISHon nuclear structure
The RNA FISH technique has been important in theevaluation of nuclear structures associated with RNAprocessing and transcription (e.g. Xing et al. 1993; John-son et al. 1995). In order to evaluate the preservation ofnuclear ultrastructure following RNA FISH, we exam-ined the labelling of the chromatin-specific and SC-35
antigens following this procedure. Control cells thatwere fixed with paraformaldehyde in situ and thenstained are shown in Fig. 1. Figure 2 shows the organiza-tion of DNA (upper left), SC-35 (upper right), acetylatedhistone H3 (lower left), and acetylated histone H4 (lowerright) following a mock RNA in situ hybridization pro-cedure. The organization of each antigen is identical tothat of control cells (compare with Fig. 1). The antibod-ies to acetylated histones produce a nuclear staining pat-tern that is both punctate and fibrillar. The DAPI distri-bution (top left) also appears similar to that of control
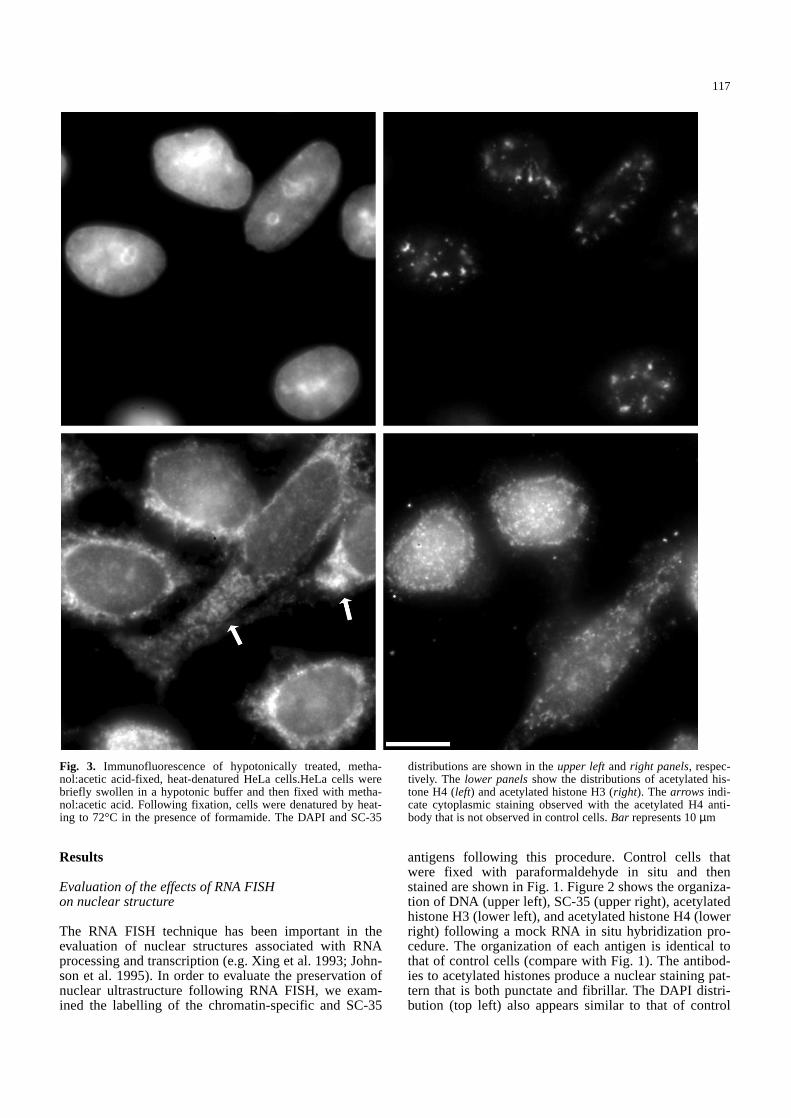
Fig. 3. Immunofluorescence of hypotonically treated, metha-nol:acetic acid-fixed, heat-denatured HeLa cells.HeLa cells werebriefly swollen in a hypotonic buffer and then fixed with metha-nol:acetic acid. Following fixation, cells were denatured by heat-ing to 72°C in the presence of formamide. The DAPI and SC-35
distributions are shown in the upper leftand right panels, respec-tively. The lower panelsshow the distributions of acetylated his-tone H4 (left) and acetylated histone H3 (right). The arrows indi-cate cytoplasmic staining observed with the acetylated H4 anti-body that is not observed in control cells. Bar represents 10µm&/fig.c:
118
cells. The SC-35 pattern produces a characteristic nucle-ar speckle pattern and is also not distinguishable fromthat of control cells.
Evaluation of fixation and DNA denaturation protocolsfor preservation of acetylated histone staining patterns
The denaturation of DNA that is essential for DNA insitu hybridization might be incompatible with the preser-vation of the fine structure of chromatin. The evidence
that is generally presented that organization is main-tained during DNA FISH protocols is limited to compar-ing DAPI staining patterns in control and hybridizedsamples (e.g. Yokota et al. 1995; Clemson et al. 1996).Unfortunately, only very crude assessments of preserva-tion can be obtained using DAPI. For example, follow-ing the denaturation protocol, the DNA appears to belargely confined to the boundaries of the nucleus ormetaphase chromosome (Yokota et al. 1995; Clemson etal. 1996). We have used the antibody probes to acetyla-ted histones to determine whether acetylated histones
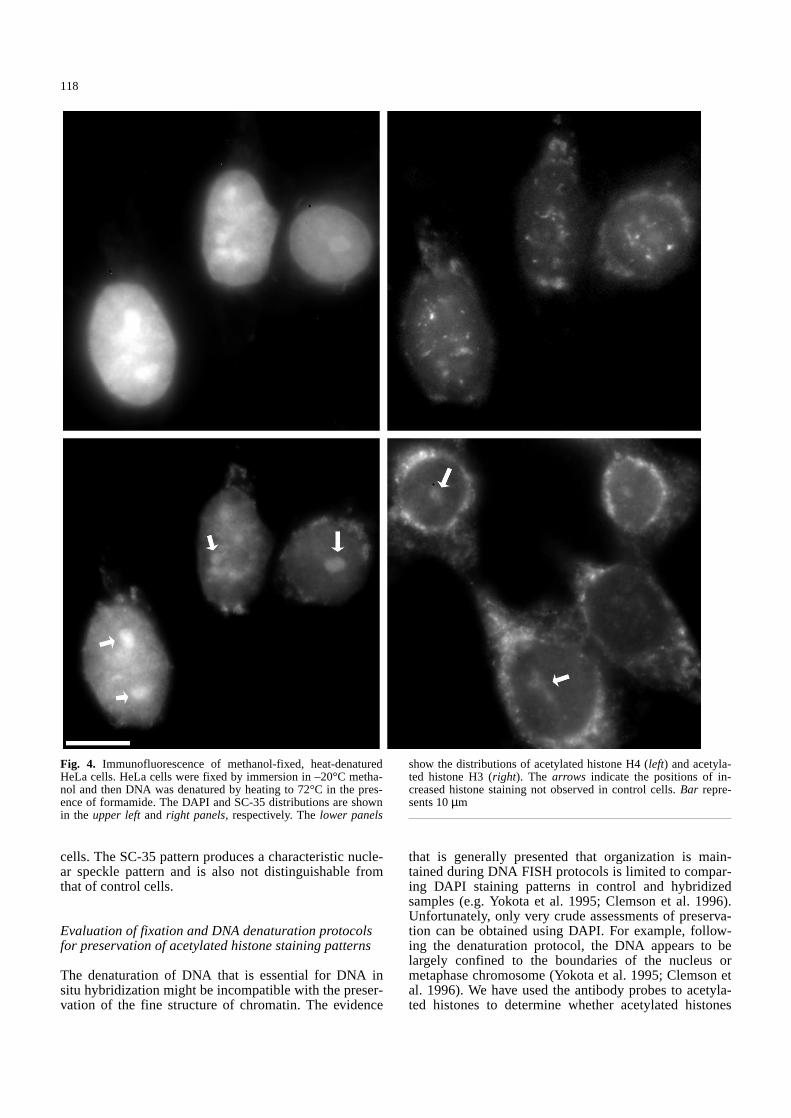
Fig. 4. Immunofluorescence of methanol-fixed, heat-denaturedHeLa cells. HeLa cells were fixed by immersion in –20°C metha-nol and then DNA was denatured by heating to 72°C in the pres-ence of formamide. The DAPI and SC-35 distributions are shownin the upper leftand right panels, respectively. The lower panels
show the distributions of acetylated histone H4 (left) and acetyla-ted histone H3 (right). The arrows indicate the positions of in-creased histone staining not observed in control cells. Bar repre-sents 10µm&/fig.c:
119
dissociate from chromatin during denaturation. Thiswould be indicative of a loss of nucleosomal structureand would undermine confidence in using these proto-cols to study intragenic organization.
Three types of fixation protocols were used in combi-nation with two different DNA denaturation protocols.There was considerable variability in the immunofluores-cent staining patterns between the different protocols(Figs. 3–7). Hypotonic swelling followed by fixation inmethanol:acetic acid (Yokota et al. 1995) appeared com-pletely to dissociate acetylated histones. The results are
shown for formamide denaturation (Fig. 3). Denaturationwith NaOH/ethanol produced similar results (not shown).Staining was observed within the cytoplasm for both an-tibodies (see arrows in Fig. 3). In control cells, the pentaantibody never produces extranuclear staining, whereasthe antibody to acetylated H3 typically shows strong nu-clear labelling and a low level of cytoplasmic labelling.Using methanol fixation, neither the formamide nor theNaOH/ethanol denaturation protocol produced chromatinthat maintained its acetylated histone content (Figs. 4, 5).Methanol fixation alone (data not shown) produces re-
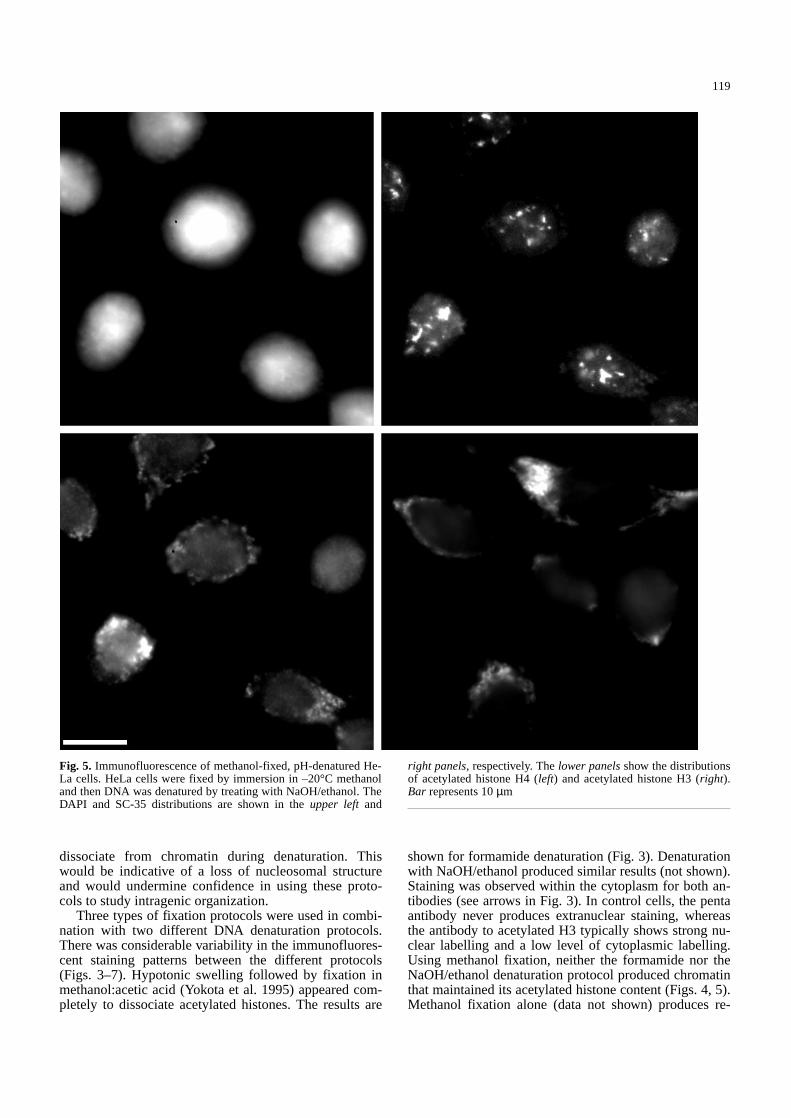
Fig. 5. Immunofluorescence of methanol-fixed, pH-denatured He-La cells. HeLa cells were fixed by immersion in –20°C methanoland then DNA was denatured by treating with NaOH/ethanol. TheDAPI and SC-35 distributions are shown in the upper left and
right panels, respectively. The lower panelsshow the distributionsof acetylated histone H4 (left) and acetylated histone H3 (right).Bar represents 10µm&/fig.c:
120
sults similar to paraformaldehyde fixation of control cells(Fig. 1). Immunofluorescent reactivity of acetylated his-tone epitopes was predominantly cytoplasmic (comparelower panels of Figs. 4 and 5 with Figs. 1 and 2). It wasalso common to observe the staining of nucleoli and re-gions of heterochromatin with these antibodies using theabove fixation/denaturation protocols (arrows in Fig. 4;unpublished observations). This observation indicatesthat there is the potential for significant reorganization ofacetylated histones during DNA FISH and this varies in aprotocol-dependent fashion. When the other markers for
the integrity of gross nuclear organization are used, DAPIand SC-35 staining patterns are similar to those of con-trol cells (compare upper panels of Figs. 3–5 with Fig. 1).For example, the DAPI staining pattern in Fig. 4 is notsignificantly different from that of control cells. Similar-ly, the SC-35 staining pattern in Fig. 5 cannot be distin-guished from that of control cells.
Paraformaldehyde fixation was found to be essential tomaintain native or near-native chromatin organization. Inparaformaldehyde-fixed NaOH/ethanol-denatured cells,staining of acetylated H4 was exclusively nuclear and
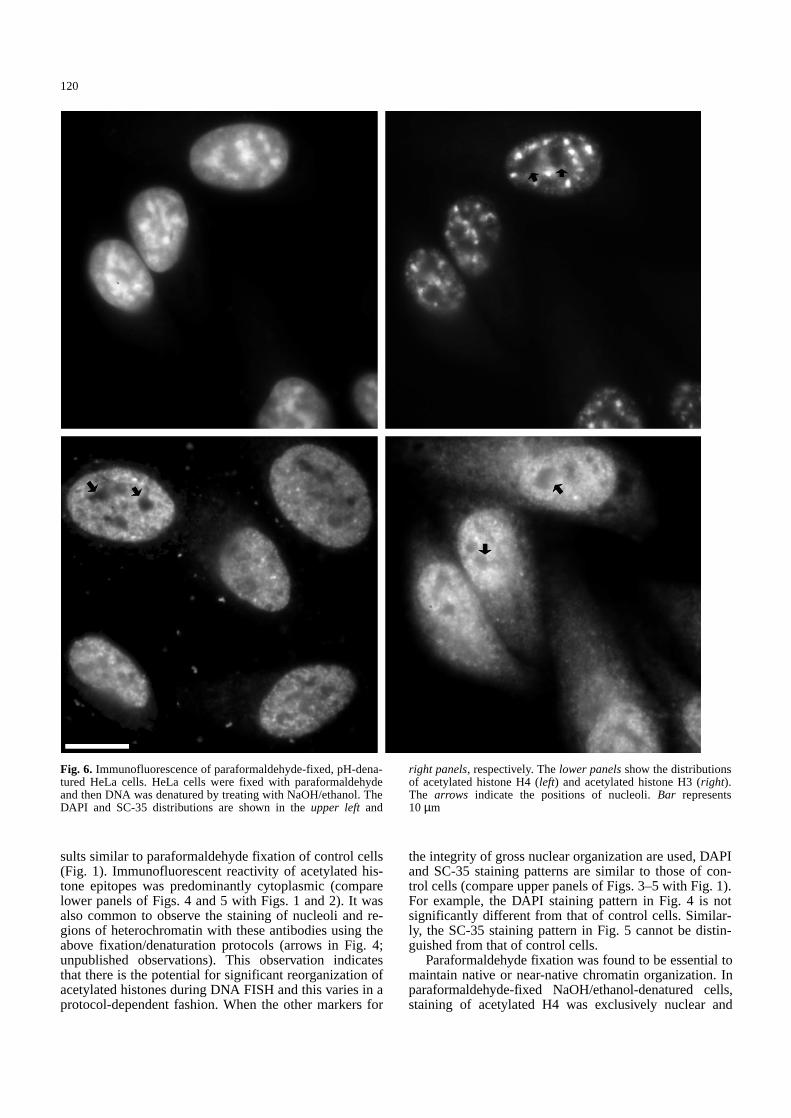
Fig. 6. Immunofluorescence of paraformaldehyde-fixed, pH-dena-tured HeLa cells. HeLa cells were fixed with paraformaldehydeand then DNA was denatured by treating with NaOH/ethanol. TheDAPI and SC-35 distributions are shown in the upper left and
right panels, respectively. The lower panelsshow the distributionsof acetylated histone H4 (left) and acetylated histone H3 (right).The arrows indicate the positions of nucleoli. Bar represents10 µm&/fig.c:
121
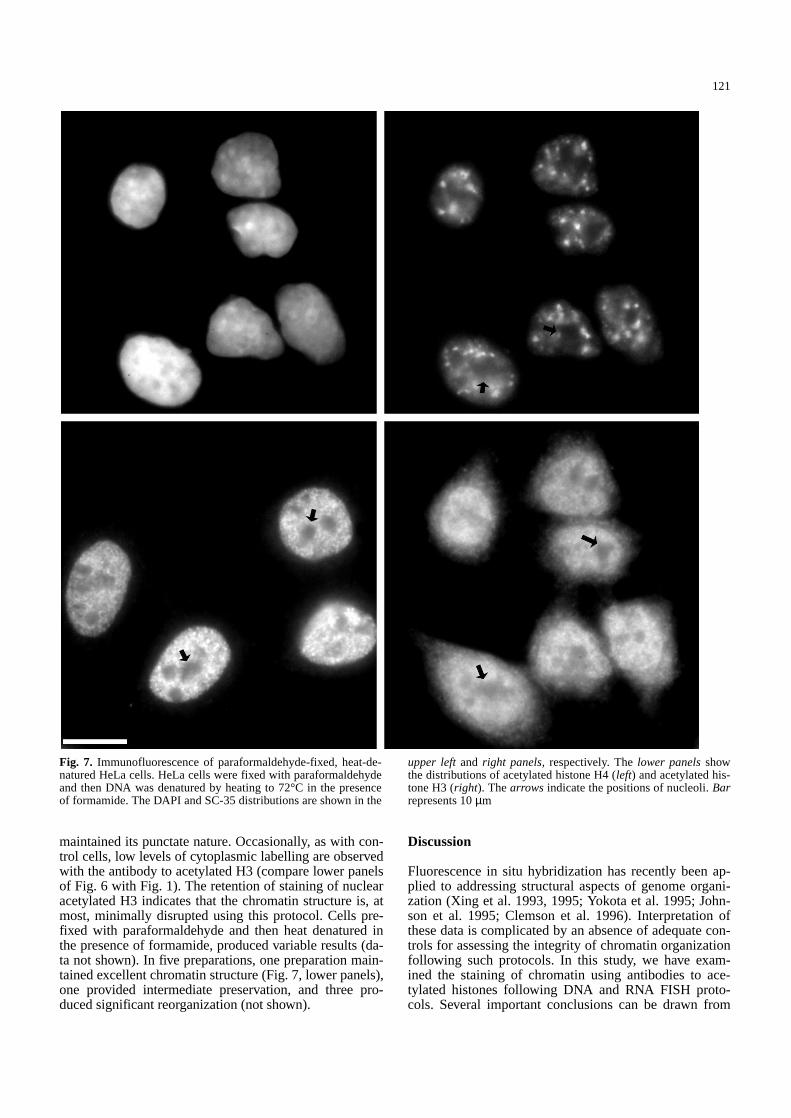
maintained its punctate nature. Occasionally, as with con-trol cells, low levels of cytoplasmic labelling are observedwith the antibody to acetylated H3 (compare lower panelsof Fig. 6 with Fig. 1). The retention of staining of nuclearacetylated H3 indicates that the chromatin structure is, atmost, minimally disrupted using this protocol. Cells pre-fixed with paraformaldehyde and then heat denatured inthe presence of formamide, produced variable results (da-ta not shown). In five preparations, one preparation main-tained excellent chromatin structure (Fig. 7, lower panels),one provided intermediate preservation, and three pro-duced significant reorganization (not shown).
Discussion
Fluorescence in situ hybridization has recently been ap-plied to addressing structural aspects of genome organi-zation (Xing et al. 1993, 1995; Yokota et al. 1995; John-son et al. 1995; Clemson et al. 1996). Interpretation ofthese data is complicated by an absence of adequate con-trols for assessing the integrity of chromatin organizationfollowing such protocols. In this study, we have exam-ined the staining of chromatin using antibodies to ace-tylated histones following DNA and RNA FISH proto-cols. Several important conclusions can be drawn from
Fig. 7. Immunofluorescence of paraformaldehyde-fixed, heat-de-natured HeLa cells. HeLa cells were fixed with paraformaldehydeand then DNA was denatured by heating to 72°C in the presenceof formamide. The DAPI and SC-35 distributions are shown in the
upper leftand right panels, respectively. The lower panelsshowthe distributions of acetylated histone H4 (left) and acetylated his-tone H3 (right). The arrows indicate the positions of nucleoli. Barrepresents 10µm

122
this study. First, the organization of SC-35 is maintainedin RNA FISH and all DNA FISH protocols with the ex-ception of those involving hypotonic swelling and metha-nol:acetic acid fixation prior to in situ hybridization. Thisresult indicates that organization of interchromatin gran-ule clusters is unaffected by most procedures. Second,RNA FISH protocols preserve chromatin architecture atthis level of resolution. This suggests that the positionalinformation on the relative localization of specific pre-mRNAs (e.g. Xing et al. 1993, 1995) reflects the nativeorganization of the nucleus. Third, paraformaldehyde fix-ation is essential for the maintenance of chromatin orga-nization during DNA denaturation. However, we find thatparaformaldehyde-fixed cells denatured by elevated tem-perature show preparation to preparation variability, butoften yield excellent preparations. The NaOH/ethanol de-naturation protocol, when preceded by paraformaldehydefixation, was the only procedure in which normal stainingpatterns were consistently preserved.
Current evidence indicates that there is at least one ad-ditional level of organization that is an intermediate be-tween the 30 nm fibre and the metaphase chromosome(Manuelidis 1990). This organization into a fibre with re-ported dimensions of between 130 and 250 nm is pre-dominant within the interphase nucleus (Manuelidis andChen 1990; Belmont and Bruce 1994). Moreover, this fi-bre can be disrupted during hypotonic swelling (Belmontet al. 1989). Under many isolation conditions, the 30 nmfibre is the only recognizable higher order organizationof interphase chromatin (Belmont et al. 1989; Giannascaet al. 1993). In vitro evidence indicates that the core his-tone tails drive the formation of the interfibre interactionsthat establish this organization from 30 nm chromatin(Schwarz et al. 1996). Similarly, chromosome structurehas also been shown to be sensitive to buffer and fixationconditions (Dietrich 1986; Belmont et al. 1989).
Given evidence that a higher order chromatin fibreexists in interphase nuclei and that its structure is depen-dent upon the integrity of the core histones, our resultsindicate that extreme care must be taken in choosingconditions for DNA FISH when a relatively local organi-zation of the genome is being investigated. For example,experimental evidence has been presented for the organi-zation of DNA in approximately 2 Mb loops (Yokota etal. 1995). The observations of Yokota et al. (1995) werepredominantly made on hypotonically swollen, metha-nol:acetic acid-fixed material. These were found to bethe worst conditions for the retention of the core hi-stones on the chromatin. Although it is possible that thecontacts responsible for the large loop structure aremaintained under these conditions, it is highly likely thatthe chromatin structure is affected in ways that could in-fluence results at this scale. Similarly, the recent investi-gations of the fine structure of interphase chromosomes(Eils et al. 1996; Kurz et al. 1996) involved paraformal-dehyde fixation but heat/formamide DNA denaturation.In these studies, the fine structure of the surface of inter-phase chromosomal territories was investigated. The or-ganization of the interphase chromosomal surface do-mains, which were shown to be enriched in coding se-quences, might be significantly altered by core histone
displacement, which might occur with this denaturationprotocol.
Organization of individual genes, which generally en-compass several kilobases, is expected to be particularlysensitive to DNA FISH methodology. The well-estab-lished enhanced nuclease sensitivity of actively tran-scribing genes, for example, indicates a less condensedchromatin structure that, because of fewer local spatialconstraints than chromatin contained in more condensedfibres, could undergo significant rearrangement uponcore histone dissociation. DNA FISH indicates a surpris-ingly compact organization of transcribed loci (Xing etal. 1993, 1995; Kurz et al. 1996) and certainly this ques-tion is worthy of further experimentation using DNAFISH methods. These studies will need to be performedunder carefully chosen conditions with internal morpho-logical controls, such as core histone antibodies, if confi-dence is to be placed in the interpretation. In this re-spect, it is important to note that at least some geneshave been demonstrated by TEM to decondense belownucleosomal organization in transcribing regions (Dane-holt 1992).
Increasingly, it is becoming apparent that organiza-tion at levels above the linear sequence content of theDNA is important in genomic regulation. New questionsare being asked that involve complex organizations bet-ter addressed with cytological than biochemical meth-ods. DNA FISH is potentially a powerful tool for ad-dressing many of these questions as they relate to ge-nomic organization. This study demonstrates that DNAFISH is compatible with the maintenance of chromatinstructure if cells are prefixed with paraformaldehyde.This study also demonstrates a clear need to co-monitorthe preservation of interphase chromatin structure withantibody probes to histones when the purpose of a DNAFISH study is to evaluate the structure of interphasegenes and their organization. Our results further indicatethat DAPI staining does not provide sufficient detail toevaluate preservation of chromatin structure followingthese protocols.
&p.2:Acknowledgements.We thank Maryse Fillion for excellent techni-cal assistance. We also thank Dr. C. David Allis (University ofRochester) for the gift of antisera to acetylated H3 and H4. Thiswork was supported by operating grants from the Medical Re-search Council of Canada.
References
Annunziato AT, Eason MB, Perry CA (1995) Relationship be-tween methylation and acetylation of arginine-rich histonesin cycling and arrested HeLa cells. Biochemistry 34:2916–2924
Belmont AS, Bruce K (1994) Visualization of G1 chromosomes: afolded, twisted, supercoiled chromonema model of interphasechromosome structure. J Cell Biol 127:287–302
Belmont AS, Braunfeld MB, Sedat JW, Agard DA (1989) Large-scale chromatin structural domains within mitotic and inter-phase chromosomes in vivo and in vitro. Chromosoma 98:129–143
Boggs BA, Connors B, Sobel RE, Chinault AC, Allis CD (1996)Reduced levels of histone H3 acetylation on the inactive Xchromosome in human females. Chromosoma 105:303–309

Perry CA, Dadd CA, Allis CD, Annunziato AT (1993) Analysis ofnucleosome assembly and histone exchange using antibodiesspecific for acetylated H4. Biochemistry 32:13605–13614
Schwarz PM, Felthauser A, Fletcher TM, Hansen JC (1996) Re-versible oligonucleosome self-association: dependence on di-valent cations and core histone tail domains. Biochemistry 35:4009–4015
Spector DL, Fu X-D, Maniatis T (1991) Associations betweendistinct pre-mRNA splicing components and the cell nucleus.EMBO J 11:3467–3481
Thorne AW, Kmiciek D, Mitchelson K, Sautiere P, Crane-Robin-son C (1990) Patterns of histone acetylation. Eur J Biochem193:701–713
van Holde KE (1989) Chromatin. Springer Berlin Heidelberg NewYork
Wolffe AP (1995) Chromatin structure and function, 2nd edn.Academic Press, San Diego, Calif
Xing Y, Johnson CV, Dobner PR, Lawrence JB (1993) Higher lev-el organization of individual gene transcription and RNAsplicing. Science 259:1326–1330
Xing Y, Johnson CV, Moen PT Jr, McNeil JA, Lawrence JB(1995) Nonrandom gene organization: structural arrangementsof specific pre-mRNA transcription and splicing with SC-35domains. J Cell Biol 131:1635–1647
Yokota H, van den Engh G, Hearst JE, Sachs RK, Trask BJ (1995)Evidence for the organization of the chromatin in megabasepair-sized loops arranged along a random walk path in thehuman G0/G1 interphase nucleus. J Cell Biol 130:1239–1249
Zirbel RM, Mathieu UR, Kurz A, Cremer T, Lichter P (1993) Evi-dence for a nuclear compartment of transcription and splicinglocated at chromosome domain boundaries. Chromosome Res1:93–106
123
Clemson CM, McNeill JA, Willard HR, Lawrence JB (1996) XistRNA paints the inactive X chromosome at interphase: evi-dence for a structural association. J Cell Biol 132:259–275
Daneholt B (1992) The transcribed template and the transcriptionloop in Balbiani rigns. Cell Biol Int Rep 16:709–715
Dietrich AJJ (1986) The influence of fixation on the morphologyof mitotic chromosomes. Can J Genet Cytol 28:536–539
Eils R, Dietzel S, Bertin E, Schrock E, Speicher MR, Ried T,Robert-Nicoud M, Cremer C, Cremer T (1996) Three-dimen-sional reconstruction of painted human interphase chromo-somes: active and inactive X chromosome territories havesimilar volumes but differ in shape and surface structure. JCell Biol 135:1427–1440
Giannasca PJ, Horowitz RA, Woodcock CL (1993) Transitionsbetween in situ and isolated chromatin structure. J Cell Sci105:551–561
Johnson CV, Singer RH, Lawrence JB (1991) Fluorescent detec-tion of nuclear RNA and DNA: implication for genome orga-nization. Methods Cell Biol 35:73–99
Kurz A, Lampel S, Nickolenko JE, Bradl J, Benner A, Zirbel RM,Cremer T, Lichter P (1996) Active and inactive genes localizepreferentially in the periphery of chromosome territories. JCell Biol 135:1195–1205
Lawrence JB, Singer RH, McNeil JA (1990) Interphase and meta-phase resolution of different distances within the human dys-trophin gene. Science 249:928–932
Lin R, Leone JW, Cook RG, Allis CD (1989) Anitobodies specificto acetylated histones document the existence of deposition-and transcription-related histone acetylation in Tetrahymena. JCell Biol 108: 1577–1588
Manuelidis L (1990) A view of interphase chromosomes. Science250:1533–1540
Manuelidis L, Chen TL (1990) A unified model of eukaryoticchromosomes. Cytometry 11:8–25





























