Embed Size (px)
Citation preview

Next Generation Sequencing: Next Generation Sequencing:
technical issuestechnical issues
Francesca Ariani, PhD
Medical Genetics, University of Siena

Advances in knowledge have been driven by the Advances in knowledge have been driven by the
advent of new technologiesadvent of new technologies……
1977

Advances in knowledge have been driven by the Advances in knowledge have been driven by the
advent of new technologiesadvent of new technologies……
2005
A paradigm shift respect to Sanger sequencing that has allowed scaling-up by orders of magnitudes…

NGS: method of the year 2007NGS: method of the year 2007
It has fundamentaly alterated genomics research and allowed investigators to conduct experiments that were previously not
technically feasible or affordable

James WatsonJames Watson’’s genome sequenced at high speeds genome sequenced at high speed
2008

NGS technologyNGS technology
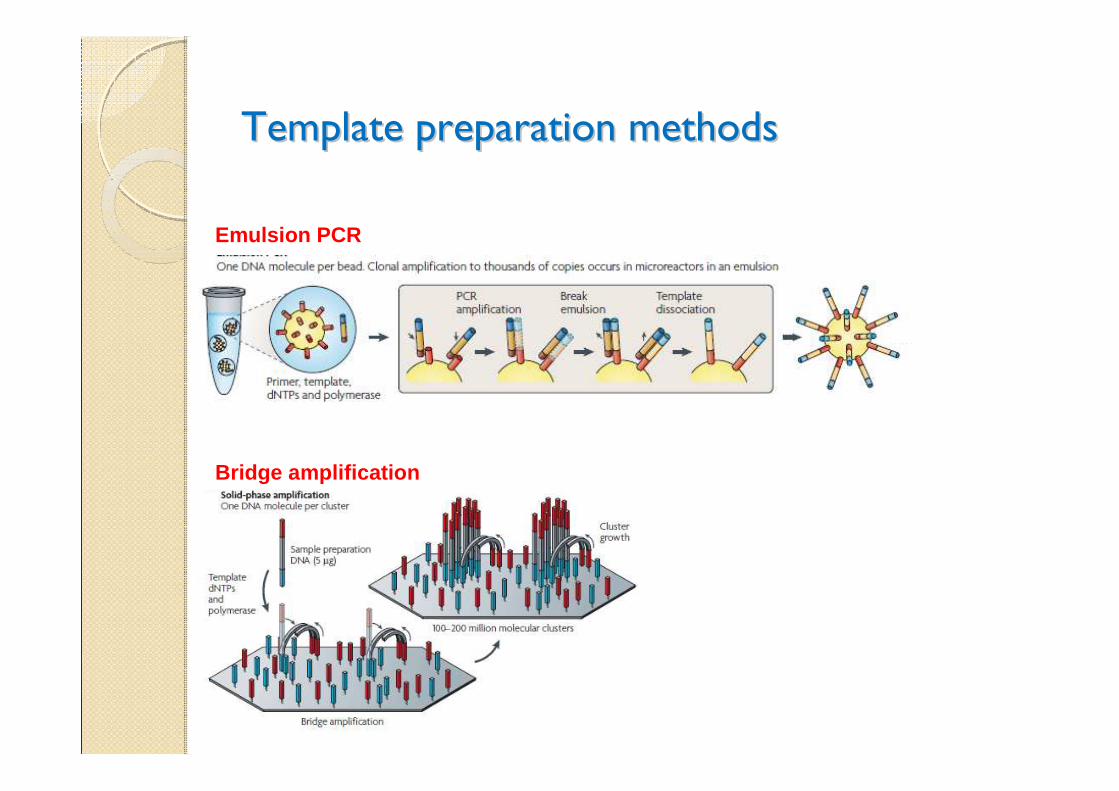
Template preparation methodsTemplate preparation methods
Emulsion PCR
Bridge amplification
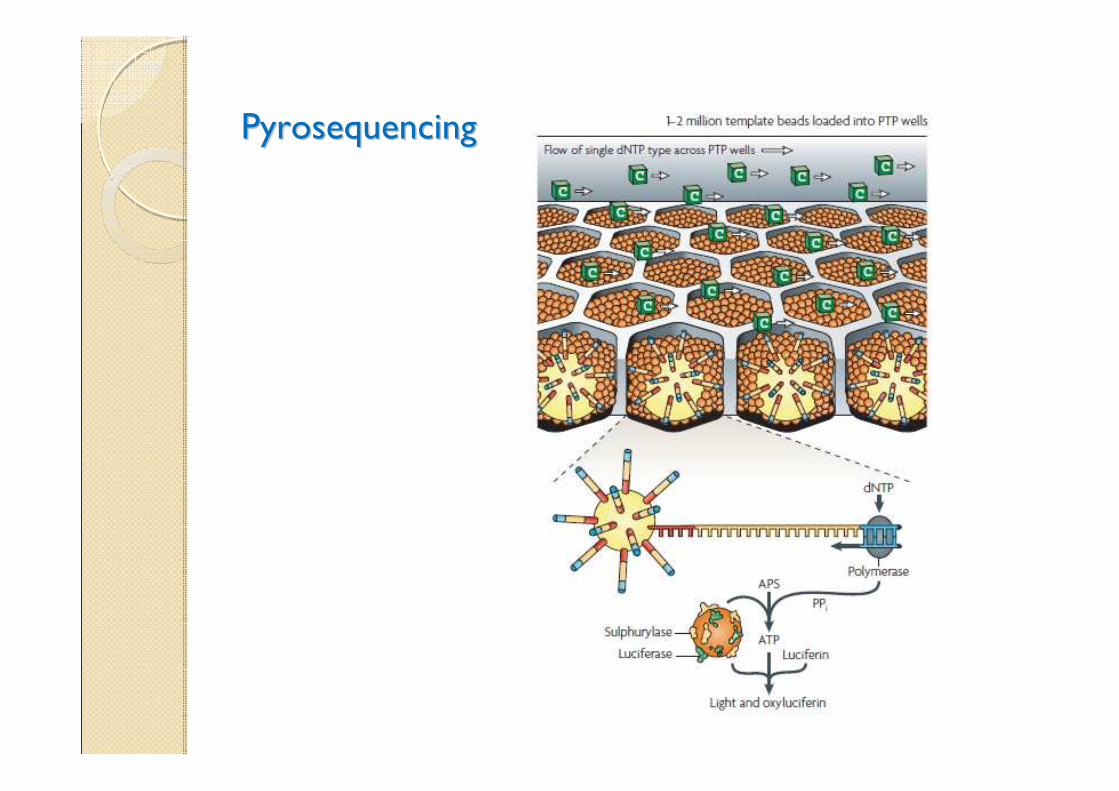
PyrosequencingPyrosequencing
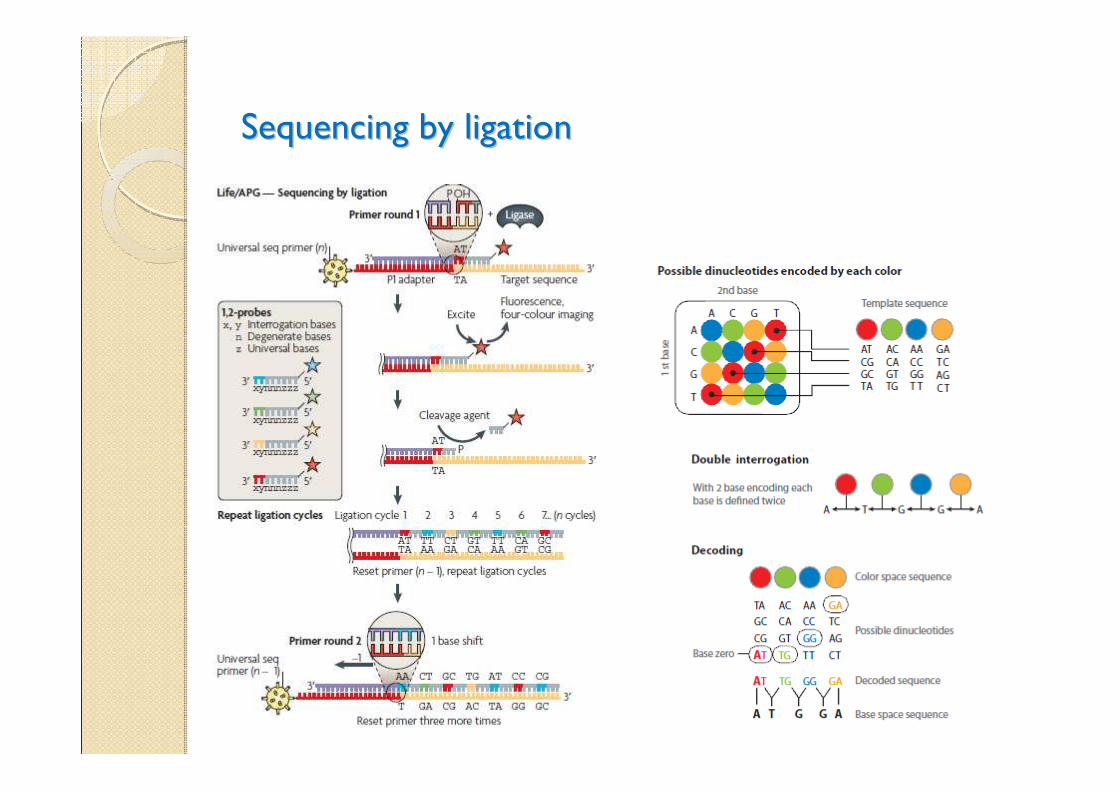
Sequencing by ligationSequencing by ligation
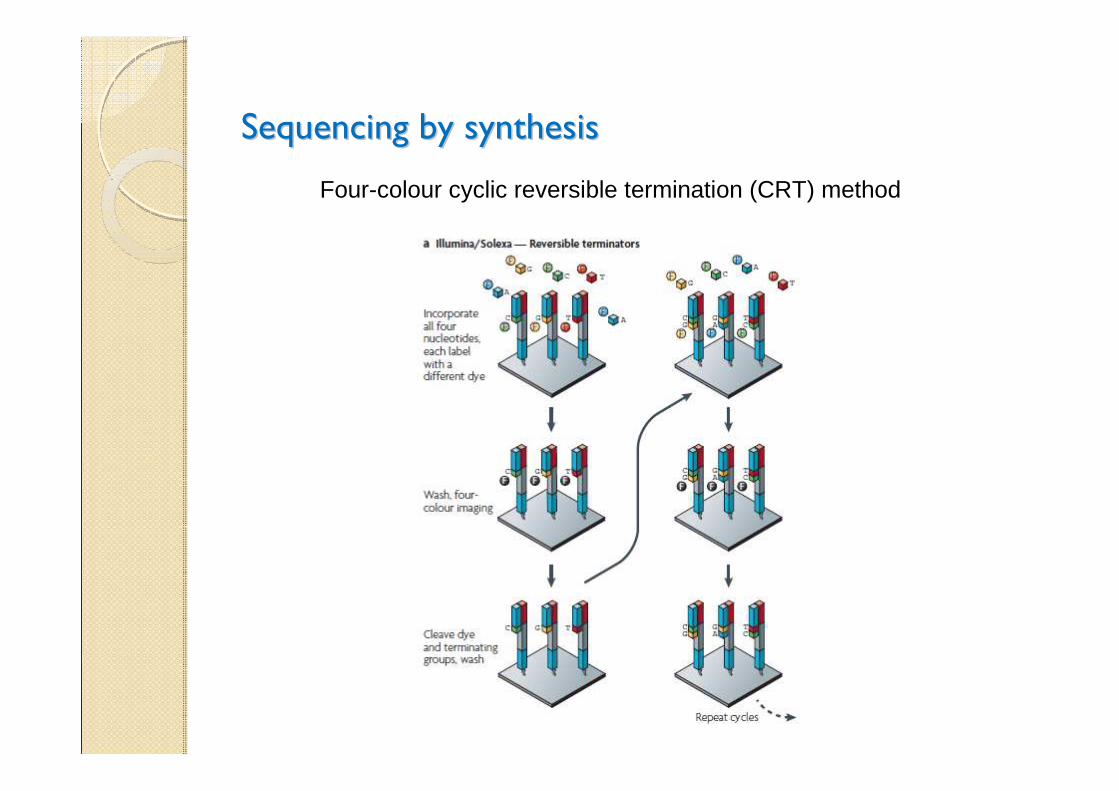
Sequencing by synthesisSequencing by synthesis
Four-colour cyclic reversible termination (CRT) method

Whole Exome Sequencing (WES)Whole Exome Sequencing (WES)
More cost-efficient sequencing strategies have been developed to study ~1% of our genome that is protein-coding (the exome), by using various capturing approaches to enrich before NGS
Protein coding genes harbor 85% of the mutations with large effects on disease-related traits.
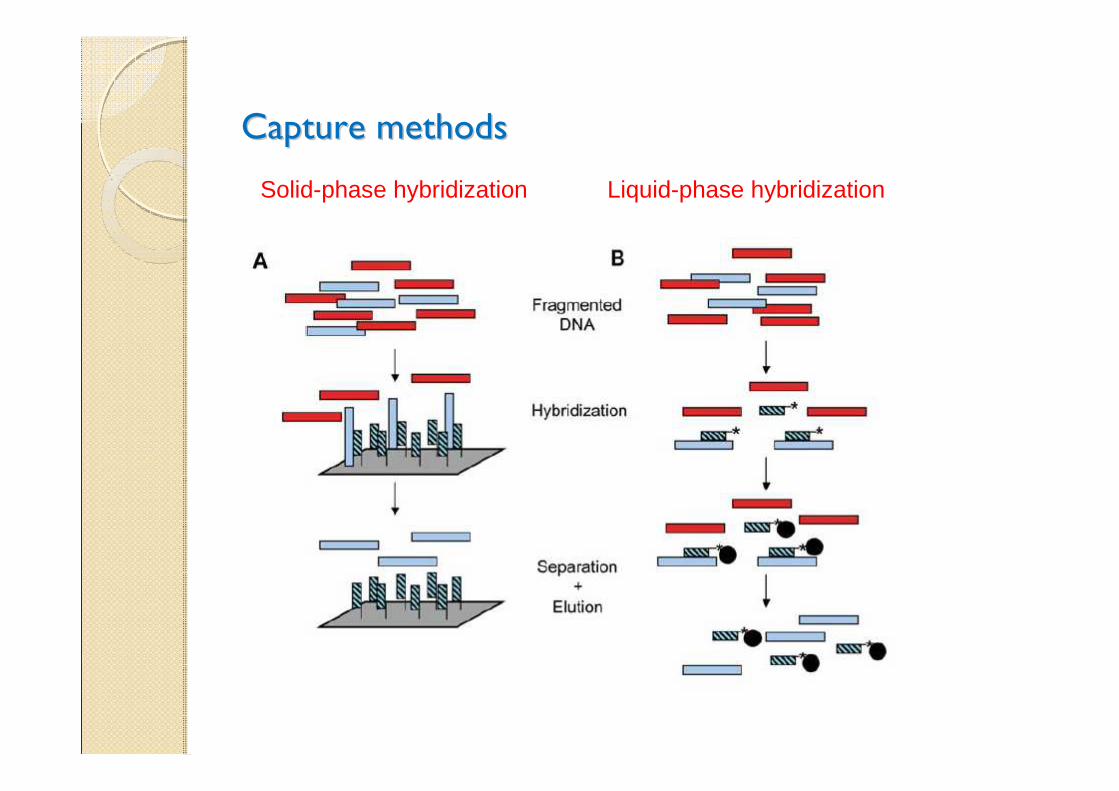
Capture methodsCapture methods
Solid-phase hybridization Liquid-phase hybridization

Challenges: data analysisChallenges: data analysis
Typically, 20.000-50.000 variants identified per sequenced exome
need for filtering!!!

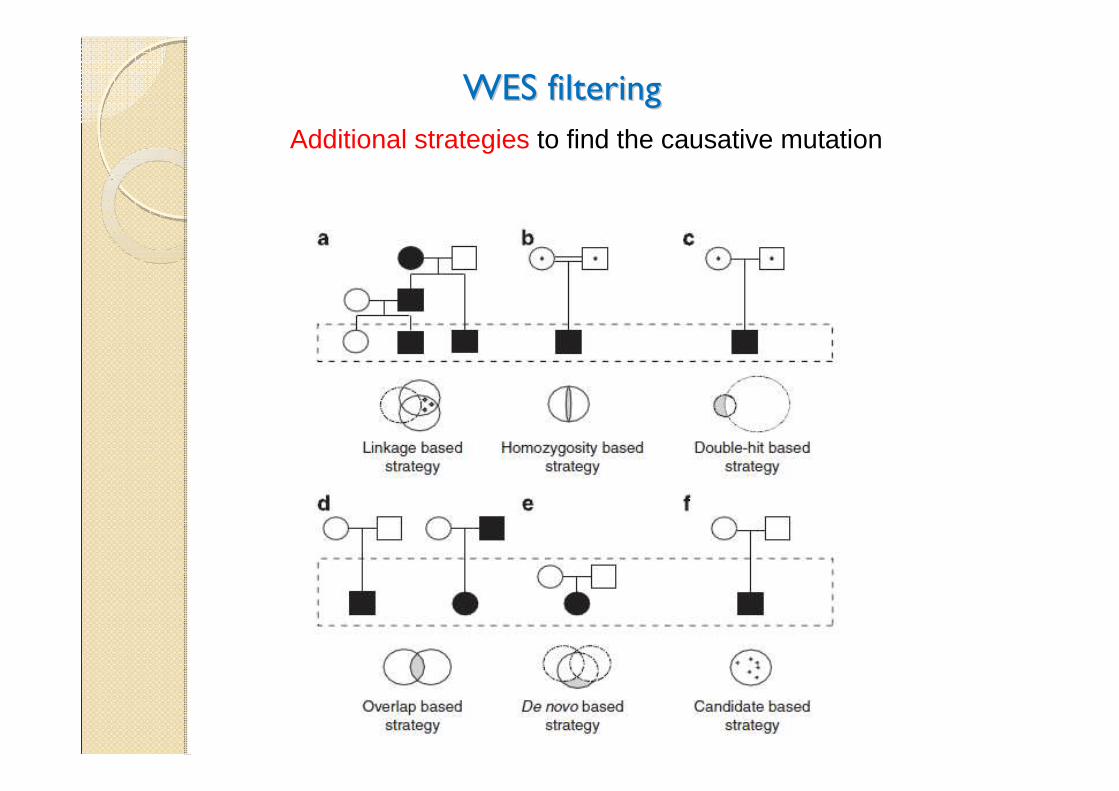
WES filteringWES filtering
20.000-50.000 variants
Quality criteria(Coverage, % of reads showing the variant…)
Exonic and splice site variants
Affecting protein sequence
Excluding known variants (dbSNP, published studies, in-house databases)
5.000
150-500
WES filteringWES filtering
Additional strategies to find the causative mutation

These startegies donThese startegies don’’t always workt always work……
“Current challenges in exome or genome-based analysis of Mendelian disorders.” Jay Shendure, pltform presentation, ASHG 2012

Data analysis: Data analysis:
pathogenecity assessment of nonsynonimous variantspathogenecity assessment of nonsynonimous variants
“Current challenges in exome or genome-based analysis of Mendelian disorders”. Jay Shendure, pltform presentation, ASHG 2012

Data analysis: Data analysis:
pathogenecity assessment of nonsynonimous variantspathogenecity assessment of nonsynonimous variants
“Current challenges in exome or genome-based analysis of Mendelian disorders”. Jay Shendure, pltform presentation, ASHG 2012

PolyPhenPolyPhen--2 2
((PolyPolymorphism morphism PhenPhenotyping program version 2) otyping program version 2) http://genetics.bwh.harvard.edu/pph2/index.shtmlhttp://genetics.bwh.harvard.edu/pph2/index.shtml
Probably
damaging
TRINARY PREDICTOR
Possible
damaging
Benign
FN: 31%FP: 9%

SIFT (SIFT (SSorting orting IIntolerant ntolerant FFrom rom TTolerantolerant))
http://sift.bii.ahttp://sift.bii.a--star.edu.sg/star.edu.sg/
Tolerated Not tolerated
BINARY PREDICTOR
FN: 31%FP: 20%
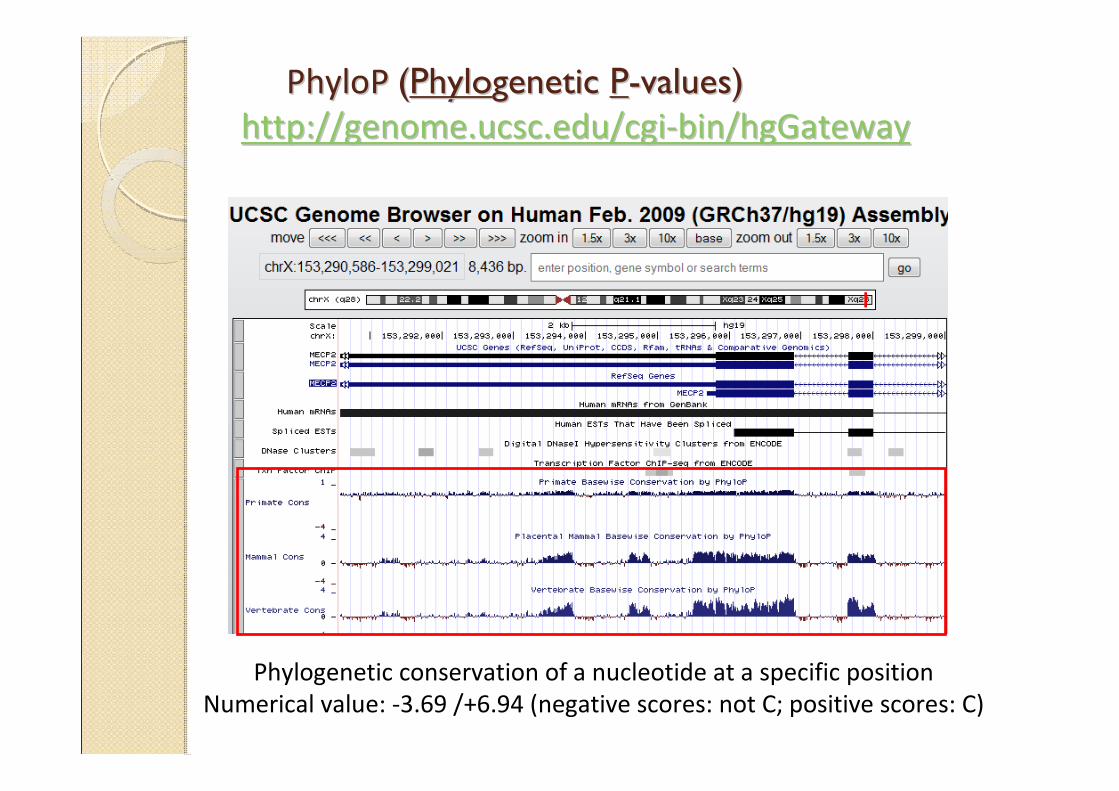
PhyloP PhyloP ((PhyloPhylogenetic genetic PP--valuesvalues))
http://genome.ucsc.edu/cgihttp://genome.ucsc.edu/cgi--bin/hgGatewaybin/hgGateway
Phylogenetic conservation of a nucleotide at a specific position
Numerical value: -3.69 /+6.94 (negative scores: not C; positive scores: C)
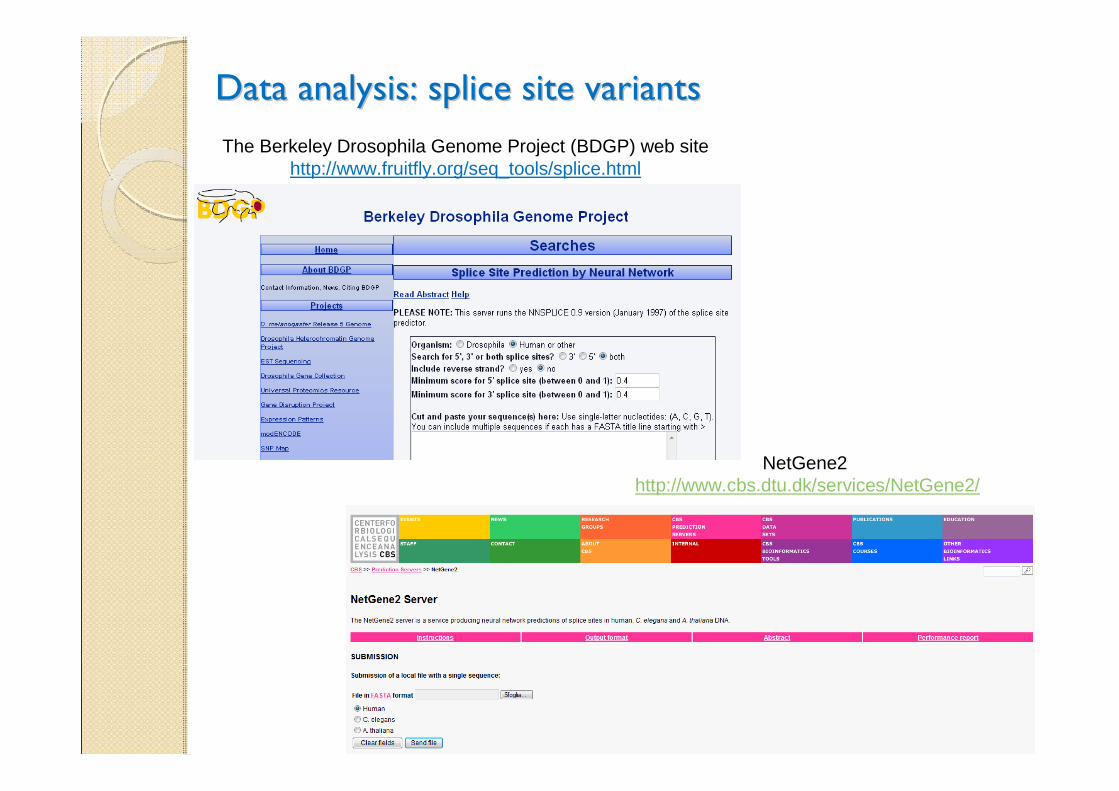
Data analysis: splice site variantsData analysis: splice site variants
The Berkeley Drosophila Genome Project (BDGP) web site http://www.fruitfly.org/seq_tools/splice.html
NetGene2http://www.cbs.dtu.dk/services/NetGene2/

NGS technology applied to Alport syndrome (ATS):NGS technology applied to Alport syndrome (ATS):
• to improve diagnosis
• to identify new disease genes
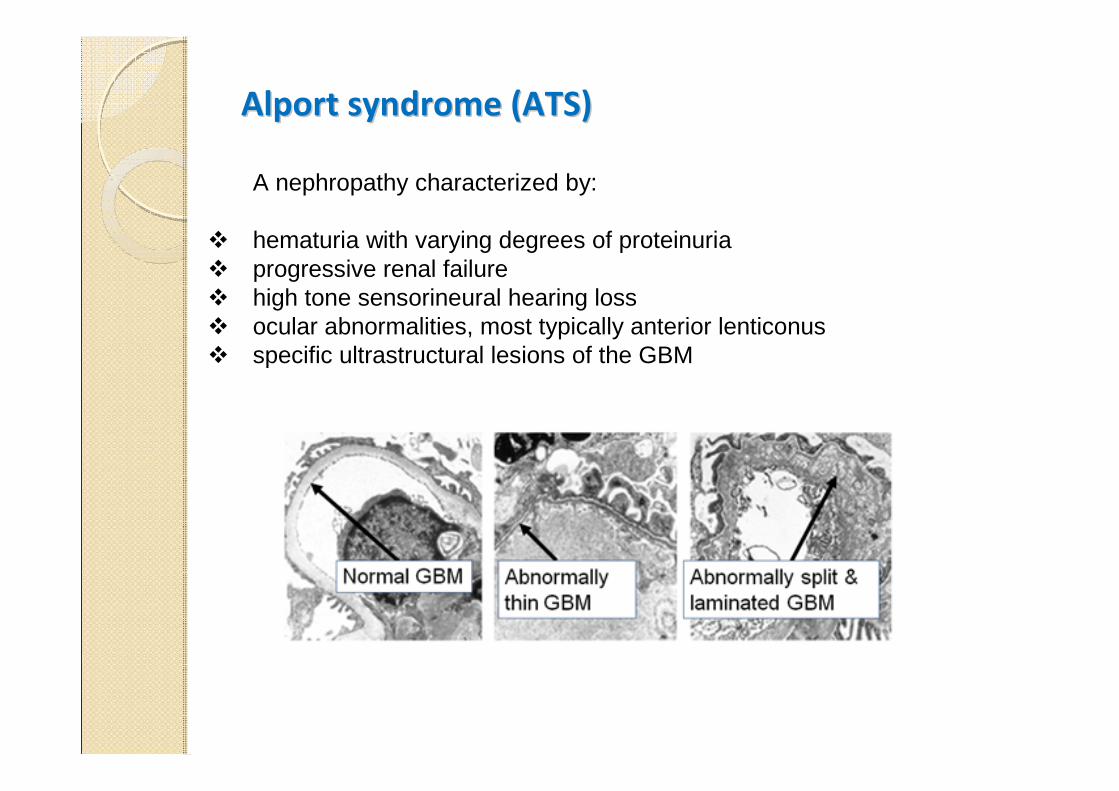
Alport syndrome (ATS)Alport syndrome (ATS)
A nephropathy characterized by:
� hematuria with varying degrees of proteinuria� progressive renal failure � high tone sensorineural hearing loss � ocular abnormalities, most typically anterior lenticonus� specific ultrastructural lesions of the GBM
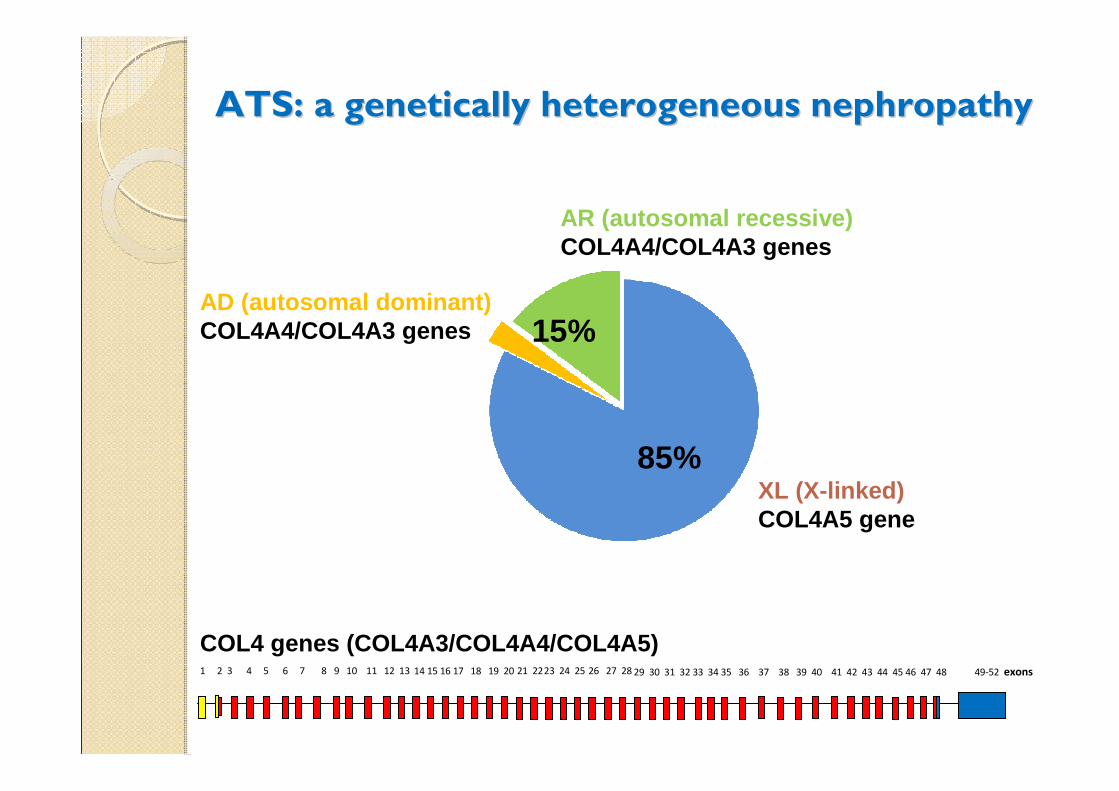
ATS: a ATS: a genetically heterogeneous nephropathygenetically heterogeneous nephropathy
XL (X-linked) COL4A5 gene
AR (autosomal recessive) COL4A4/COL4A3 genes
AD (autosomal dominant) COL4A4/COL4A3 genes 15%
85%
1 2 3 4 5 6 7 8 14 15 16 17 18 19 20 21 2223 24 25 26 27 28 29 30 31 32 33 34 35 36 37 38 39 40 41 42 43 44 45 46 47 48 49-52 exons9 10 11 12 13
COL4 genes (COL4A3/COL4A4/COL4A5)

ATS is also a clinically ATS is also a clinically heterogeneous conditionheterogeneous condition
• X-linked form : males severely affected with persistent hematuria, proteinuria, constant progression to ESRD and high incidence of hearing loss and ocular anomalies. Females usually show only urinary abnormalities.
• AR form : phenotype identical to the X-linked form, but females are severely affected as males. Parents may be completely asymptomatic or may have isolated microhematuria.
• AD form : also associated to a high risk to develop renal failure but atan older age. Extra-renal manifestations are rarely observed.
High inter- and intra-familial phenotypic variability !
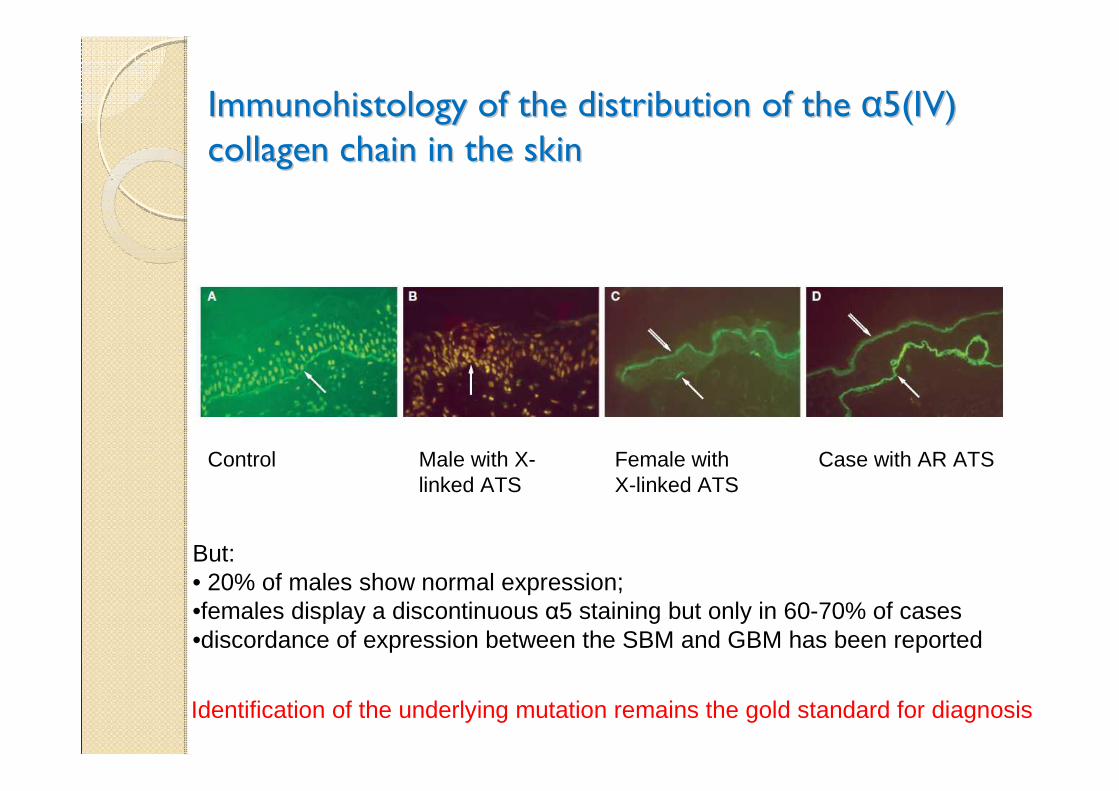
Immunohistology of the distribution of the Immunohistology of the distribution of the αα5(IV) 5(IV)
collagen chain in the skin collagen chain in the skin
Control Male with X-linked ATS
Female with X-linked ATS
Case with AR ATS
But:• 20% of males show normal expression;•females display a discontinuous α5 staining but only in 60-70% of cases•discordance of expression between the SBM and GBM has been reported
Identification of the underlying mutation remains the gold standard for diagnosis

2010: NGS protocol for ATS diagnosis 2010: NGS protocol for ATS diagnosis
Method coupling selective amplification to the Roche DNA
sequencing platform(454 junior Sequencer)
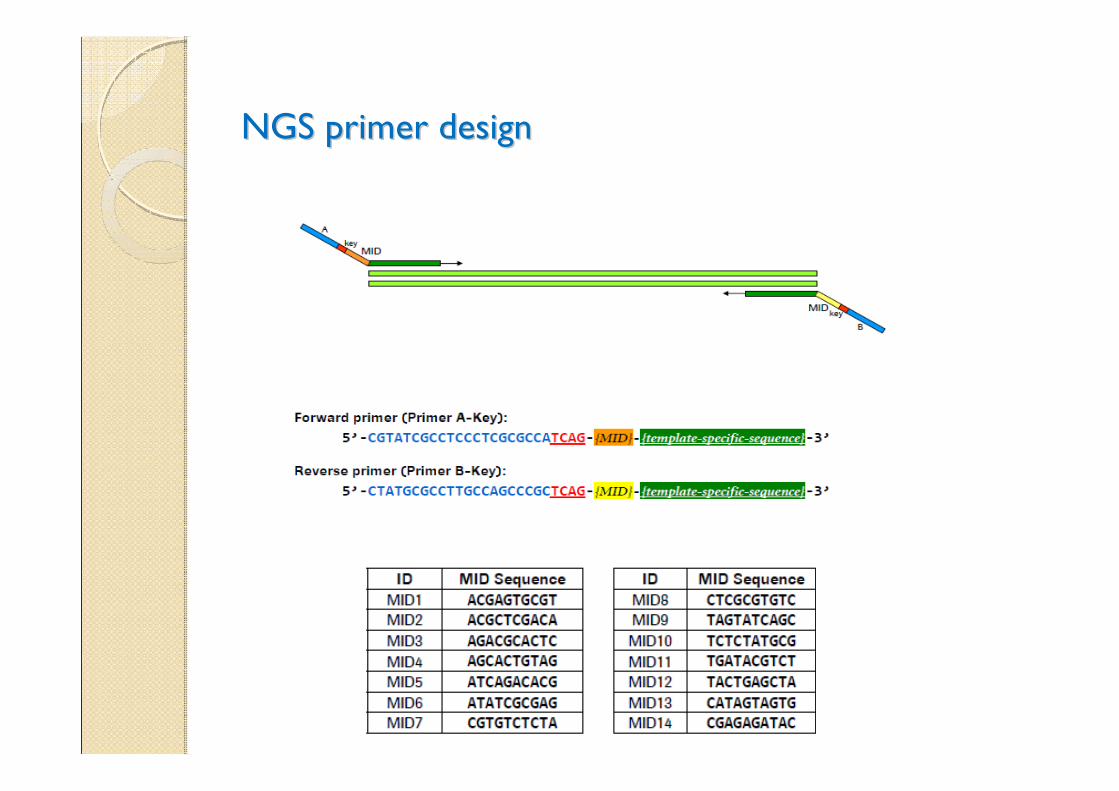
NGS primer designNGS primer design
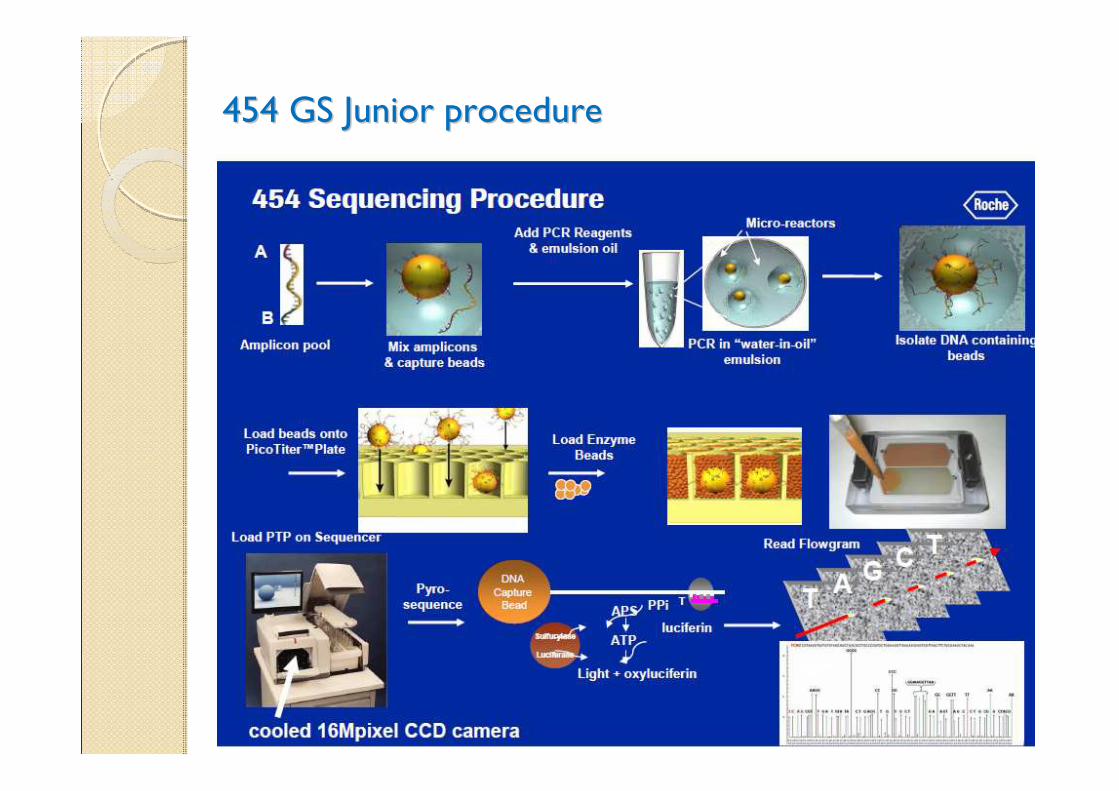
454 GS Junior procedure454 GS Junior procedure
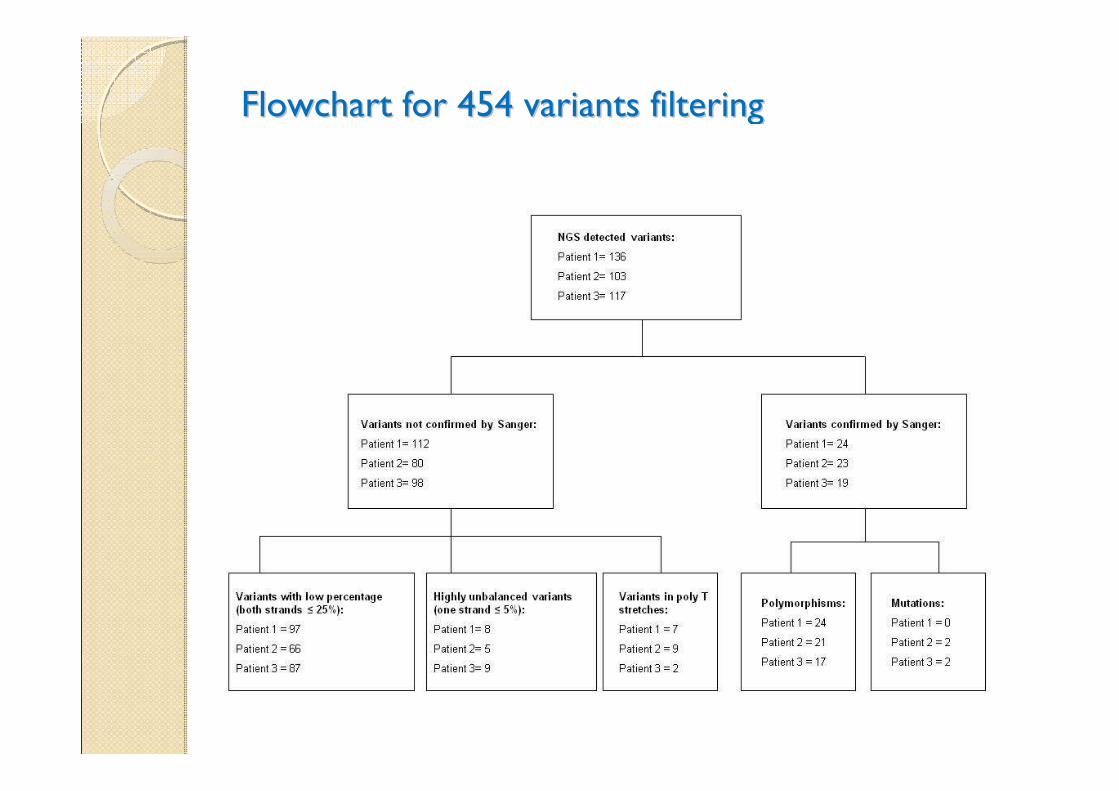
Flowchart for 454 variants filteringFlowchart for 454 variants filtering
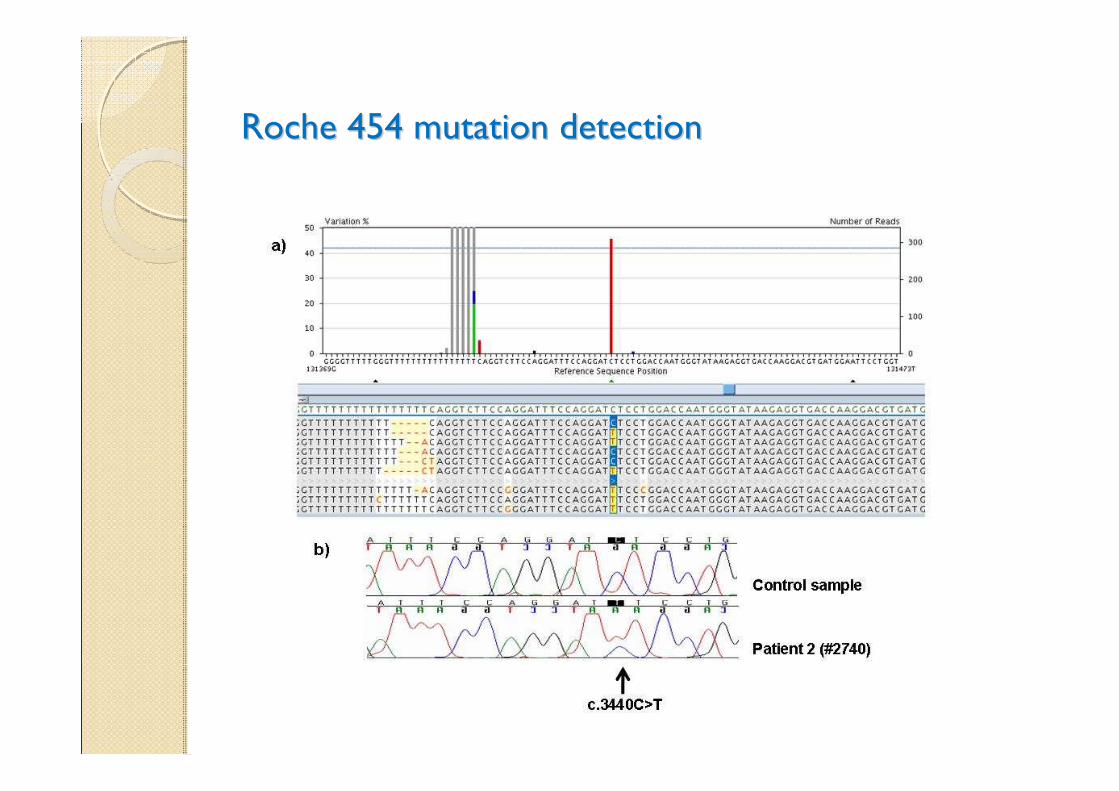
Roche 454 mutation detectionRoche 454 mutation detection
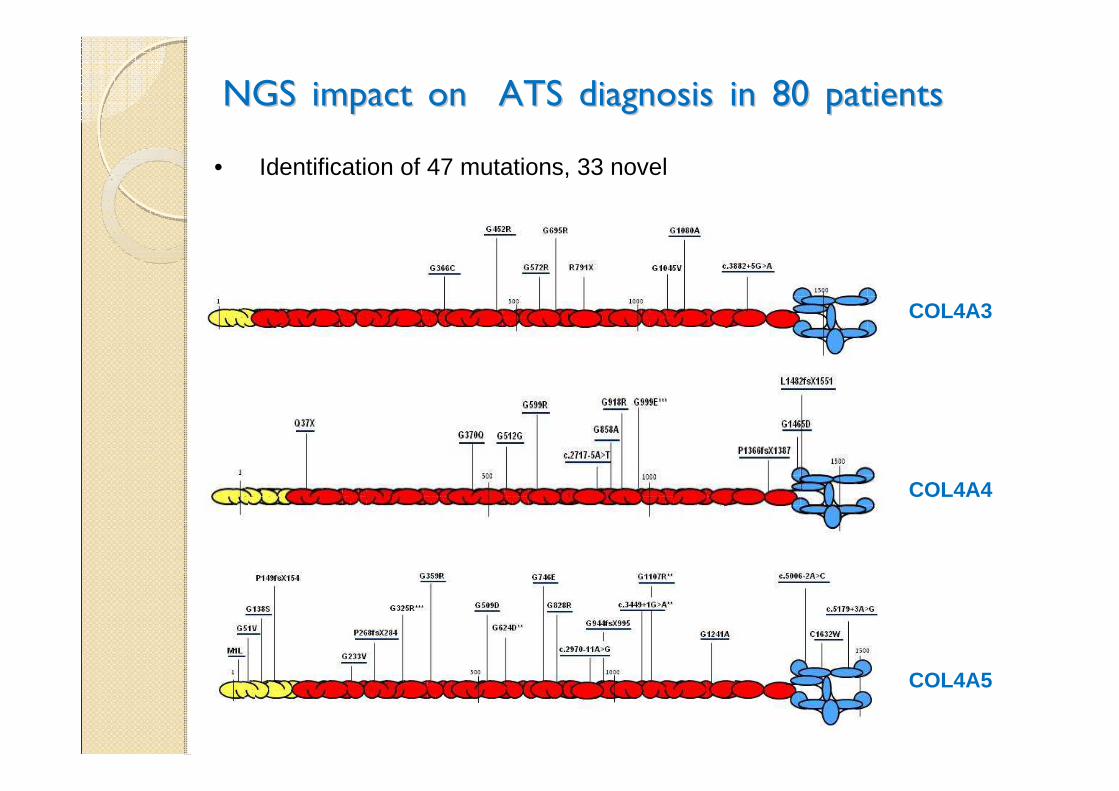
NGS impact on ATS diagnosis in 80 patientsNGS impact on ATS diagnosis in 80 patients
COL4A3
COL4A4
COL4A5
• Identification of 47 mutations, 33 novel
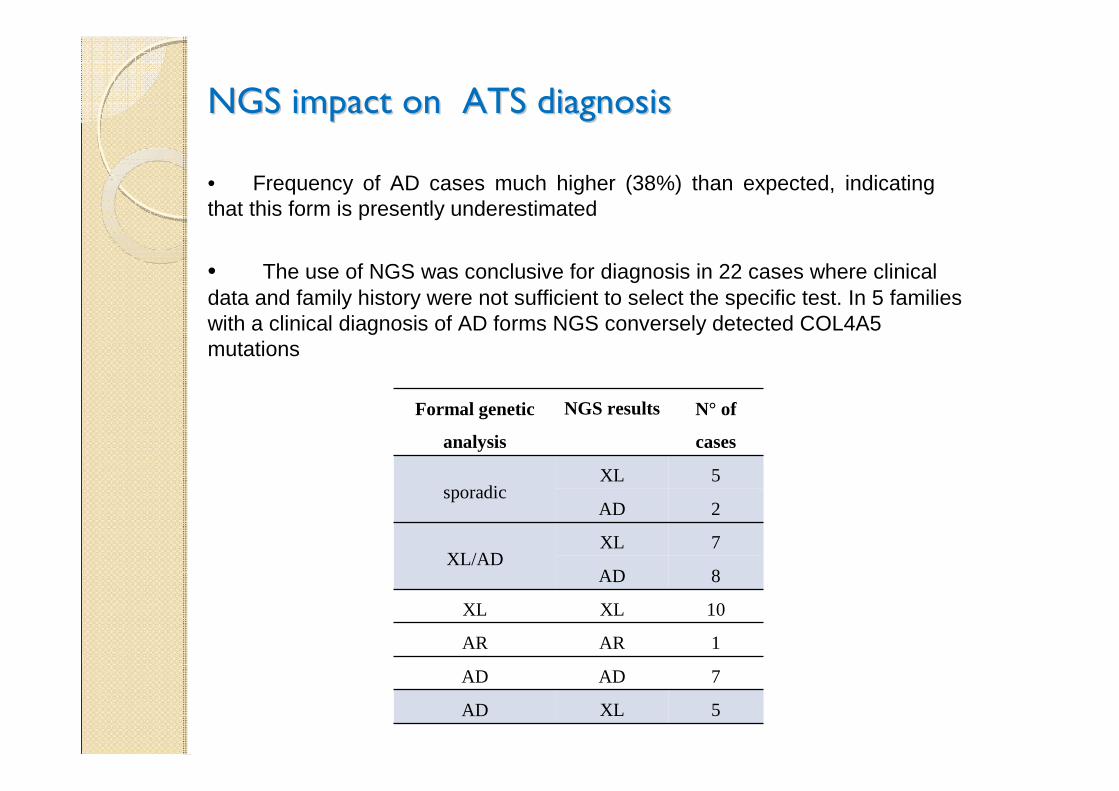
• The use of NGS was conclusive for diagnosis in 22 cases where clinical data and family history were not sufficient to select the specific test. In 5 families with a clinical diagnosis of AD forms NGS conversely detected COL4A5 mutations
• Frequency of AD cases much higher (38%) than expected, indicating that this form is presently underestimated
NGS impact on ATS diagnosisNGS impact on ATS diagnosis
Formal genetic
analysis
NGS results N° of
cases
sporadicXL 5
AD 2
XL/ADXL 7
AD 8
XL XL 10
AR AR 1
AD AD 7
AD XL 5

NGS impact on ATS diagnosisNGS impact on ATS diagnosis
Submitted to Nephrology Dialysis Transplantation
• Importantly, this test will allow a rapid and cost effective diagnosis also in oligosymptomatic ATS children and this is extremely important since early treatment with ACE inhibitors has been proven to delay renal failure and to improve life expectancy in a time-dependent manner
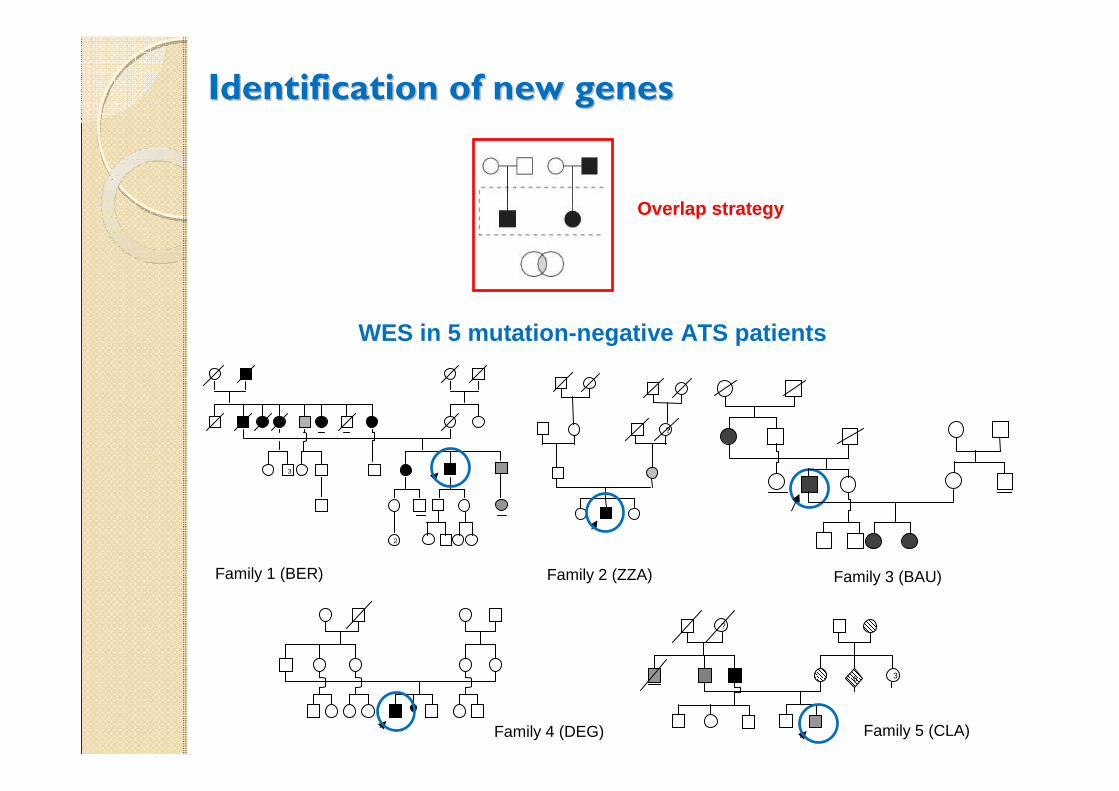
Identification of new genesIdentification of new genes
Overlap strategy
Family 1 (BER)
2
3
?
?
6 3
Family 2 (ZZA) Family 3 (BAU)
Family 4 (DEG) Family 5 (CLA)
WES in 5 mutation-negative ATS patients

Prof. V. Nigro
WES in 5 mutationWES in 5 mutation--negative ATS patientsnegative ATS patients
Illumina platform
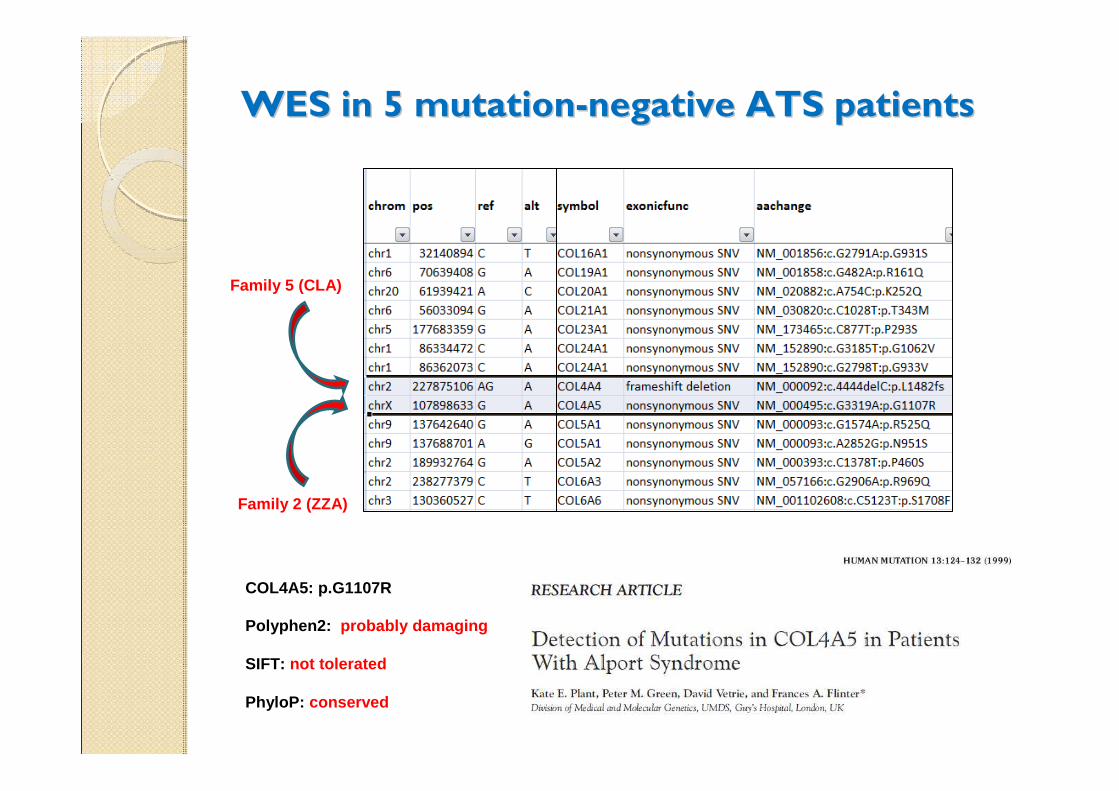
WES in 5 mutationWES in 5 mutation--negative ATS patientsnegative ATS patients
Family 5 (CLA)
Family 2 (ZZA)
Polyphen2: probably damaging
SIFT: not tolerated
PhyloP: conserved
COL4A5: p.G1107R
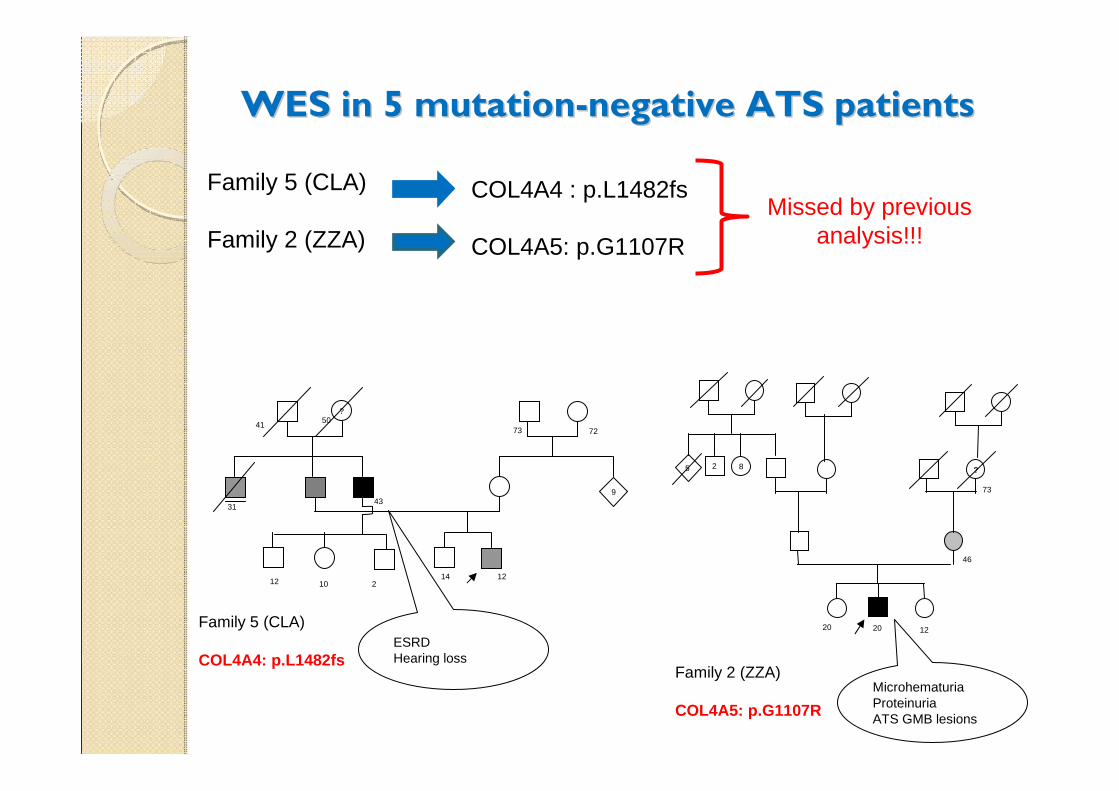
WES in 5 mutationWES in 5 mutation--negative ATS patientsnegative ATS patients
Family 5 (CLA)
Family 2 (ZZA)
COL4A4 : p.L1482fs
COL4A5: p.G1107R
Missed by previous analysis!!!
?
14 12
9
73 72
3143
12 10 2
4150
Family 5 (CLA)
COL4A4: p.L1482fsESRDHearing loss
Family 2 (ZZA)
COL4A5: p.G1107R
?
20 12
73
5 2 8
20
46
Microhematuria ProteinuriaATS GMB lesions
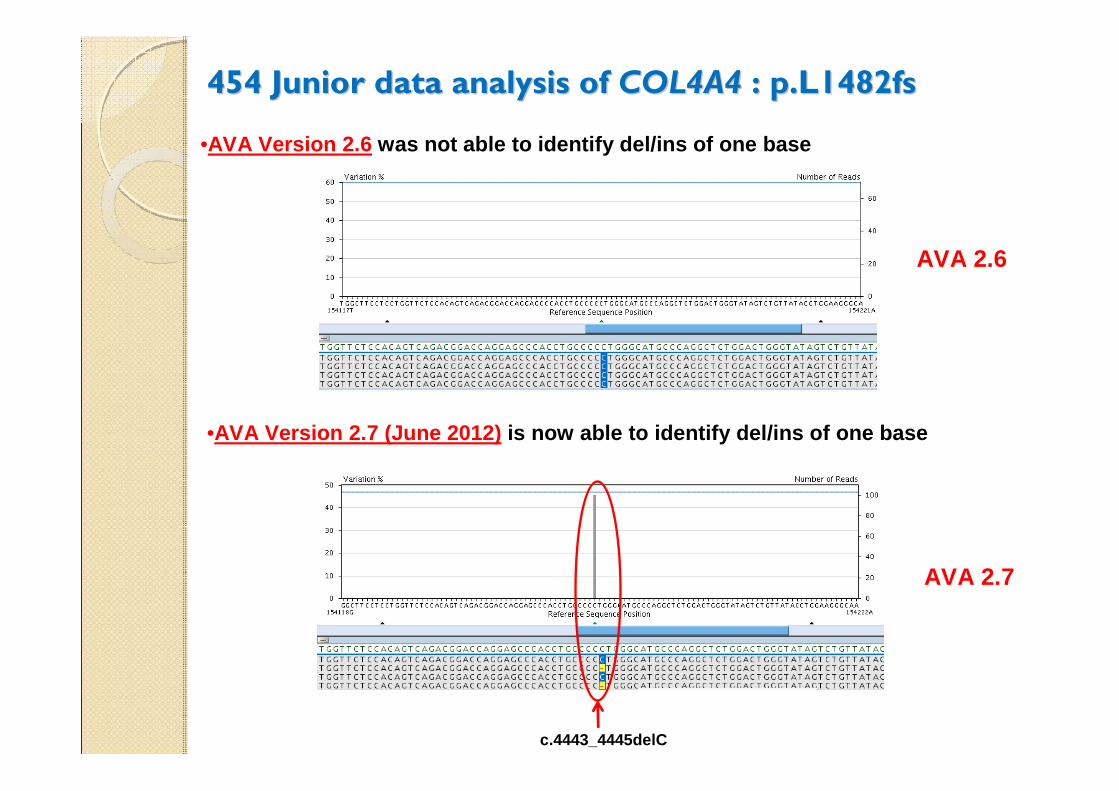
454 Junior data analysis of 454 Junior data analysis of COL4A4COL4A4 : p.L1482fs : p.L1482fs
c.4443_4445delC
•AVA Version 2.7 (June 2012) is now able to identify del/ins of one base
AVA 2.7
•AVA Version 2.6 was not able to identify del/ins of one base
AVA 2.6
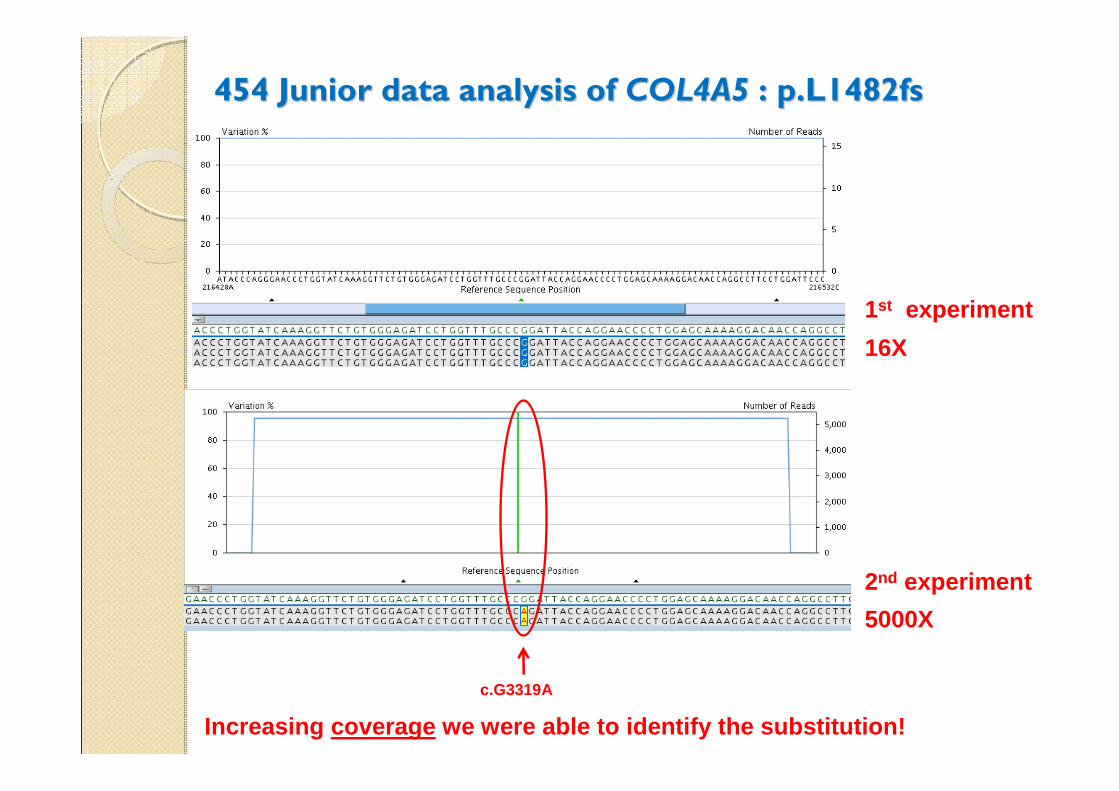
454 Junior data analysis of 454 Junior data analysis of COL4A5COL4A5 : p.L1482fs : p.L1482fs
16X
1st experiment
5000X
2nd experiment
Increasing coverage we were able to identify the substitution!
c.G3319A
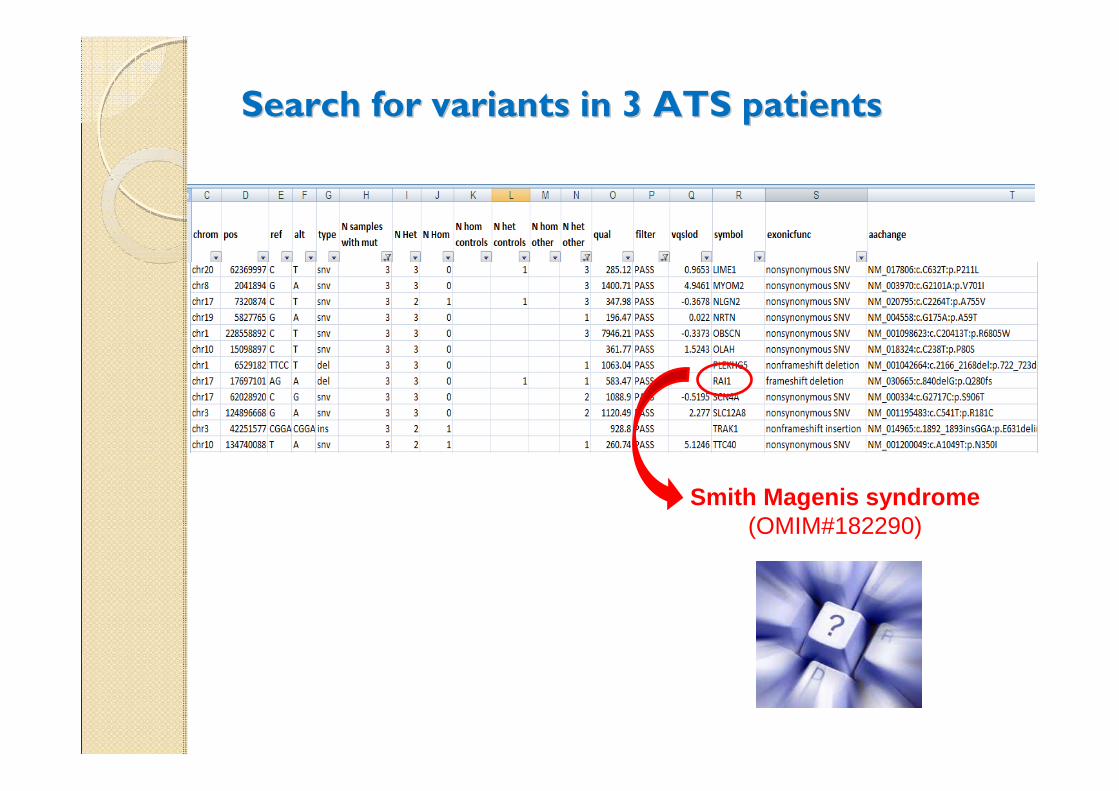
Search for variants in 3 ATS patients Search for variants in 3 ATS patients
Smith Magenis syndrome(OMIM#182290)
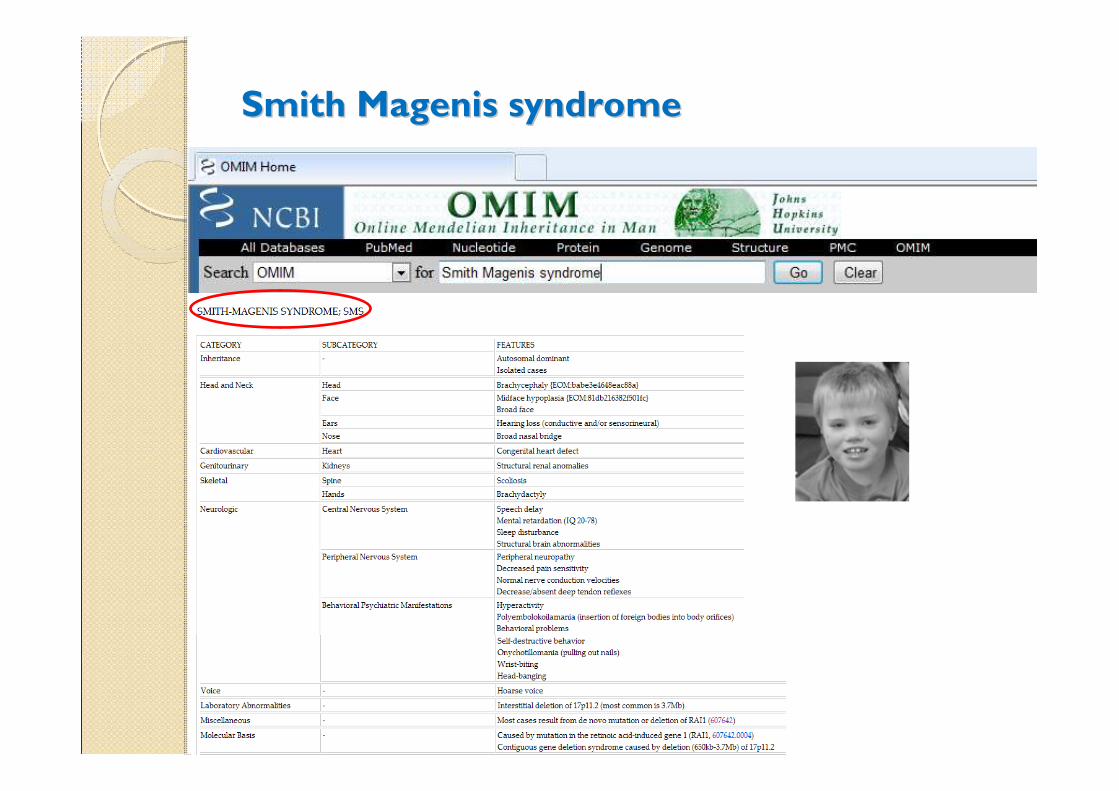
Smith Magenis syndromeSmith Magenis syndrome

Variants in 3 ATS patients: Variants in 3 ATS patients: RAI1 RAI1
(c.840delG) (c.840delG)
Prof. M. Zollino Lab (Medical Genetics, Hospital "A. Gemelli“, Roma) did not confirmed RA1 variant by Sanger sequencing
FALSE POSITIVE RESULTS of WES!!!
Probably due to a region of CAG repeats causing misalignment!
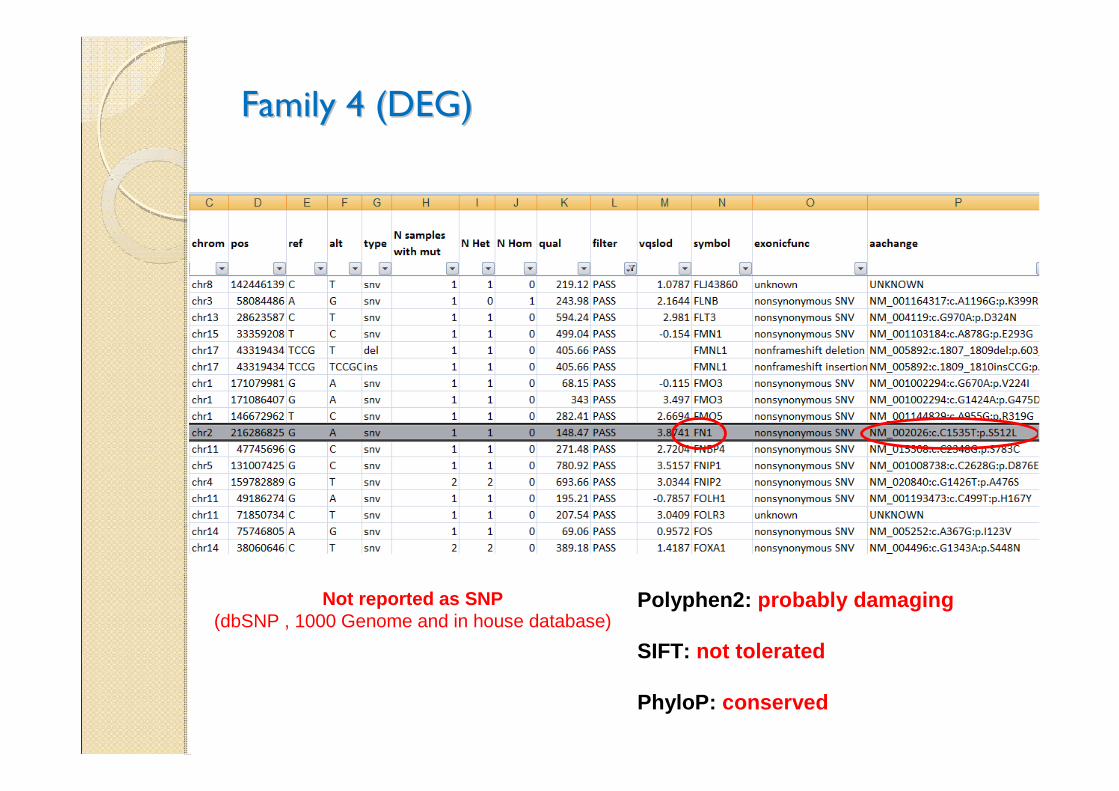
Family 4 (DEG)Family 4 (DEG)
Polyphen2: probably damaging
SIFT: not tolerated
PhyloP: conserved
Not reported as SNP(dbSNP , 1000 Genome and in house database)
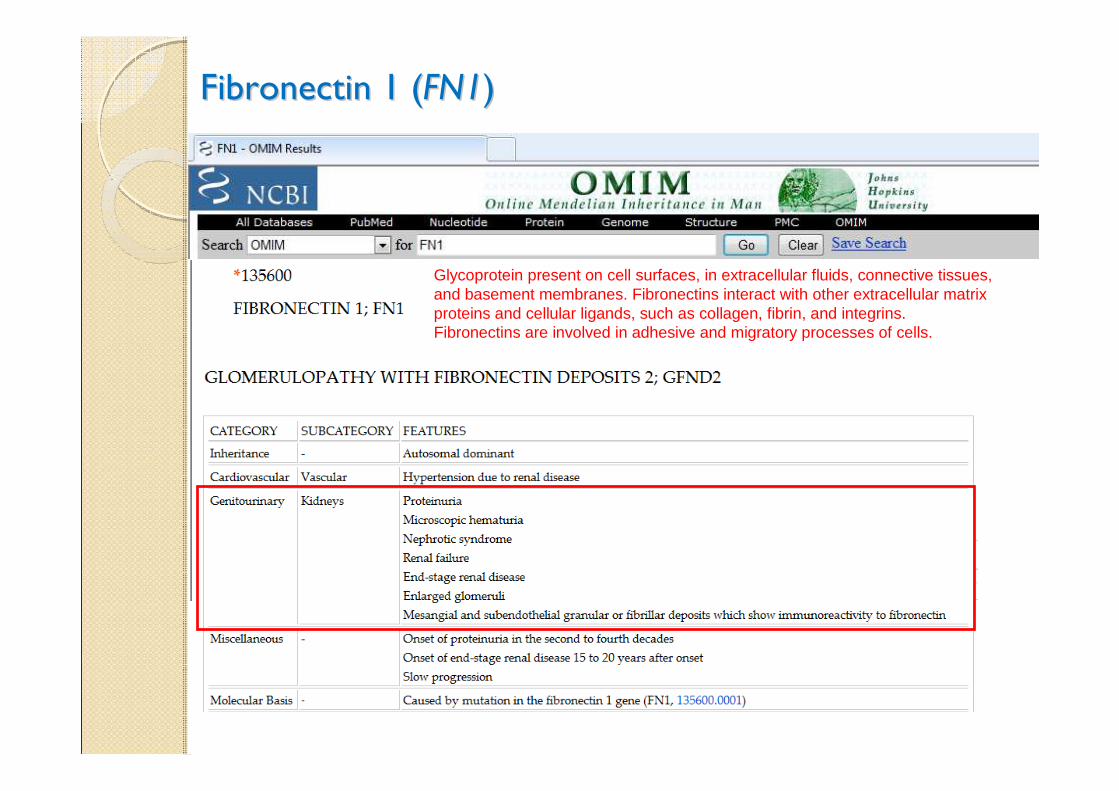
Fibronectin 1 (Fibronectin 1 (FN1FN1))
Glycoprotein present on cell surfaces, in extracellular fluids, connective tissues, and basement membranes. Fibronectins interact with other extracellular matrix proteins and cellular ligands, such as collagen, fibrin, and integrins. Fibronectins are involved in adhesive and migratory processes of cells.
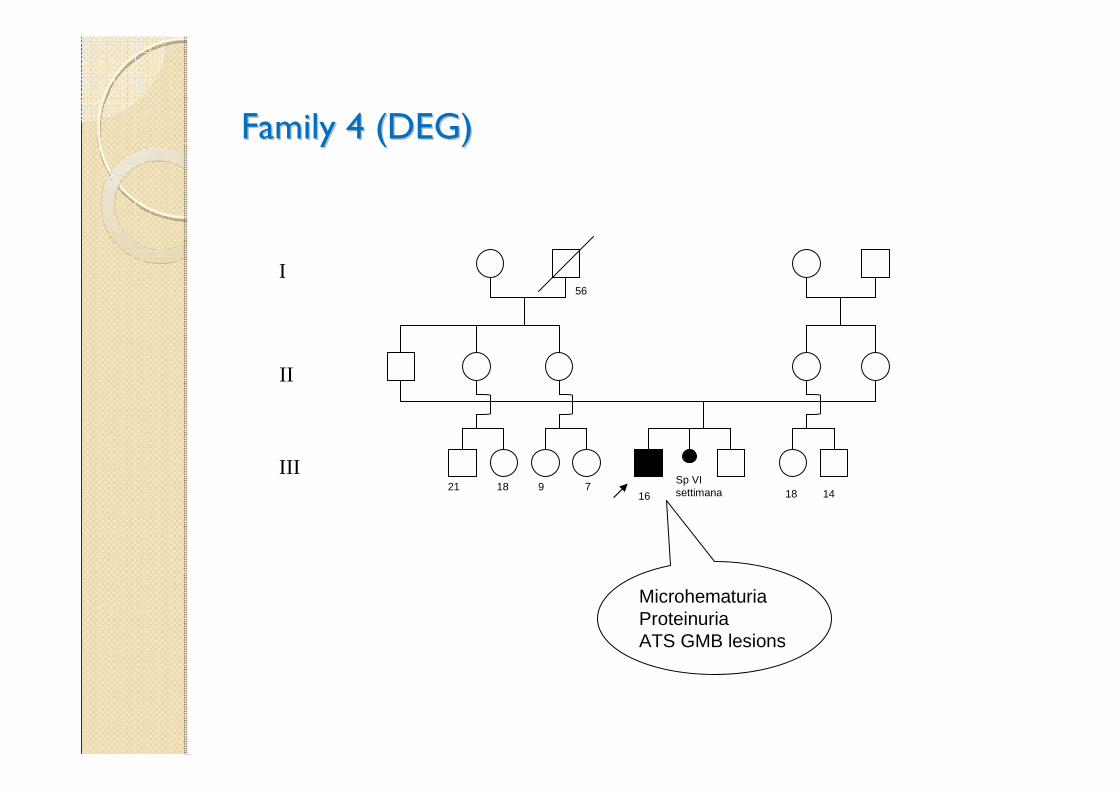
Family 4 (DEG)Family 4 (DEG)
Sp VI settimana21 18 9 7
56
18 1416
MicrohematuriaProteinuriaATS GMB lesions
I
II
III
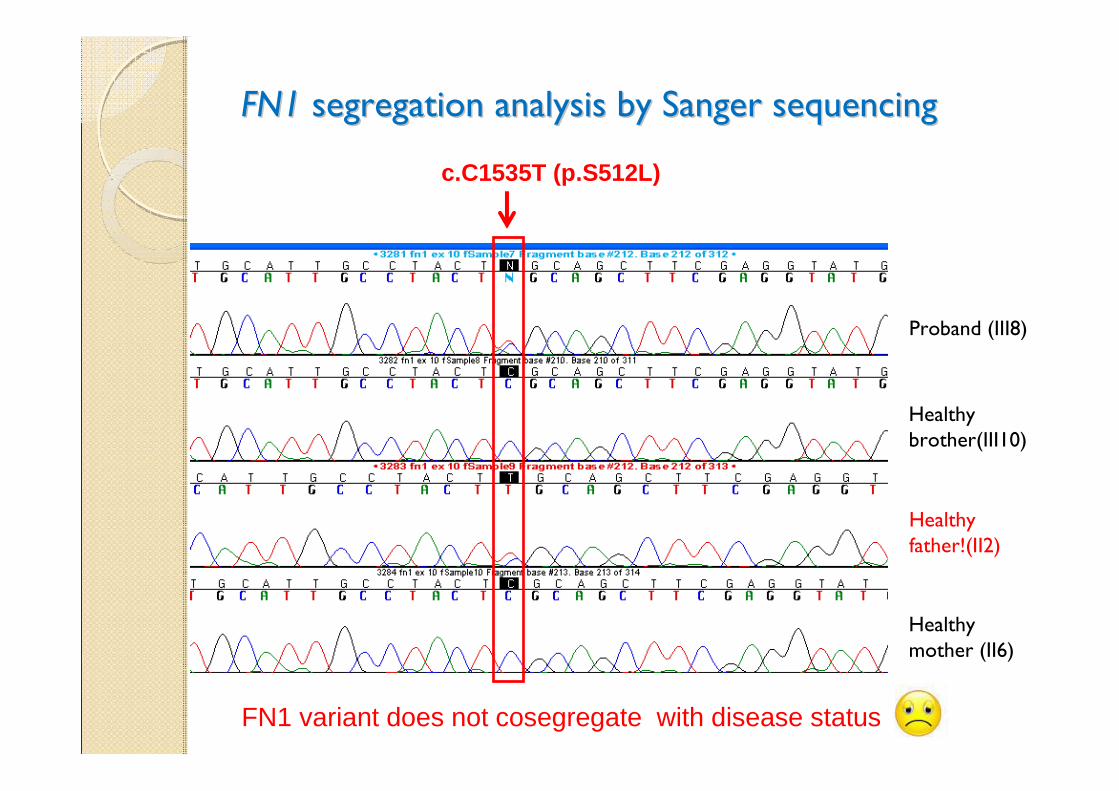
FN1FN1 segregation analysis by Sanger sequencing segregation analysis by Sanger sequencing
Proband (III8)
Healthy father!(II2)
c.C1535T (p.S512L)
FN1 variant does not cosegregate with disease status
Healthy mother (II6)
Healthy brother(III10)
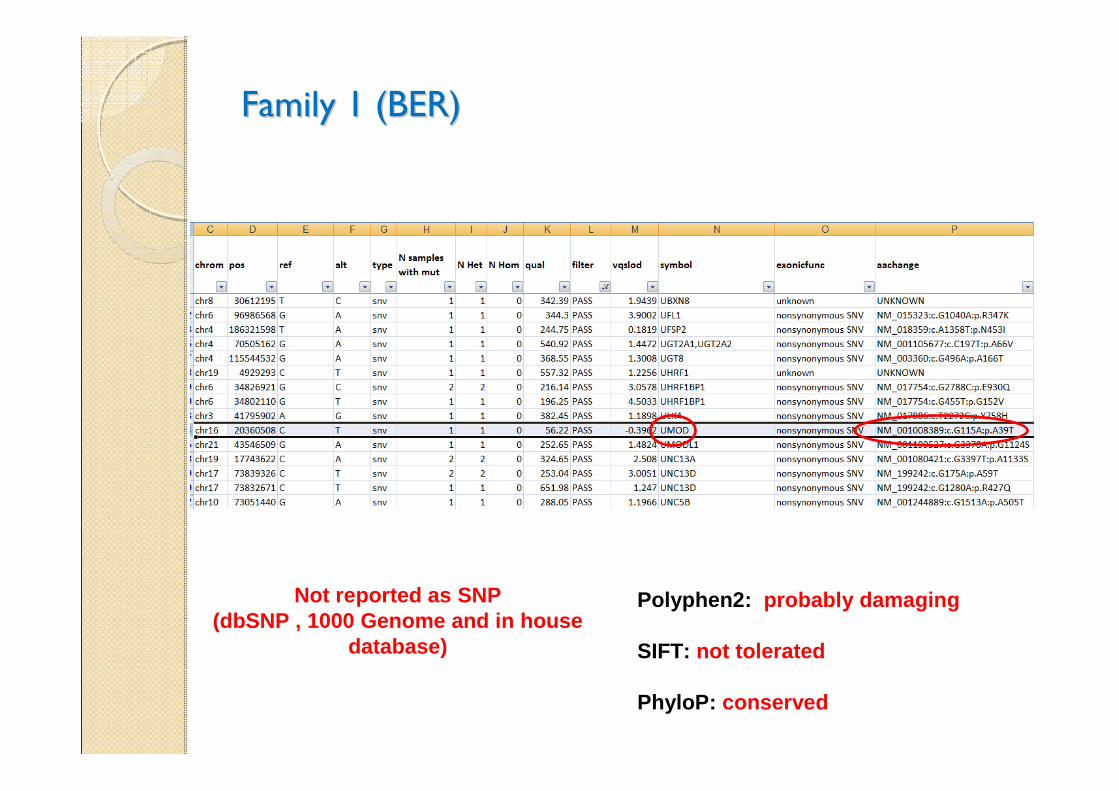
Family 1 (BER)Family 1 (BER)
Polyphen2: probably damaging
SIFT: not tolerated
PhyloP: conserved
Not reported as SNP(dbSNP , 1000 Genome and in house
database)
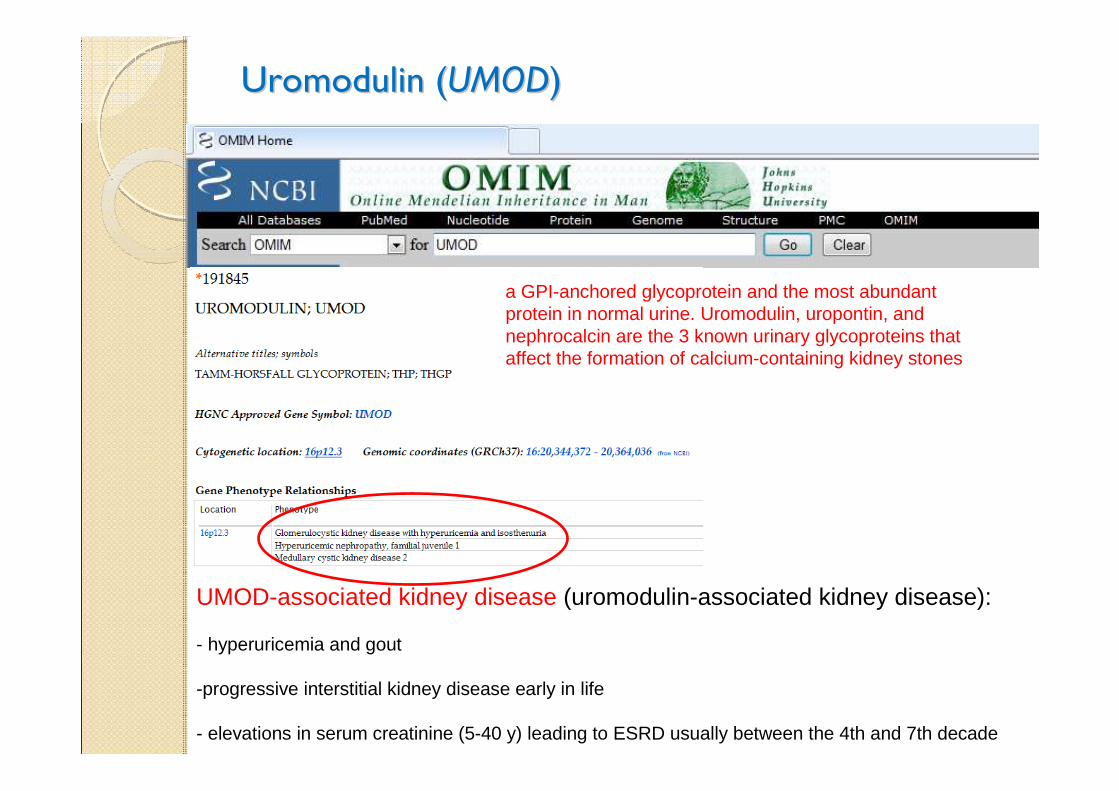
Uromodulin (Uromodulin (UMODUMOD))
a GPI-anchored glycoprotein and the most abundant protein in normal urine. Uromodulin, uropontin, and nephrocalcin are the 3 known urinary glycoproteins that affect the formation of calcium-containing kidney stones
UMOD-associated kidney disease (uromodulin-associated kidney disease):
- hyperuricemia and gout
-progressive interstitial kidney disease early in life
- elevations in serum creatinine (5-40 y) leading to ESRD usually between the 4th and 7th decade
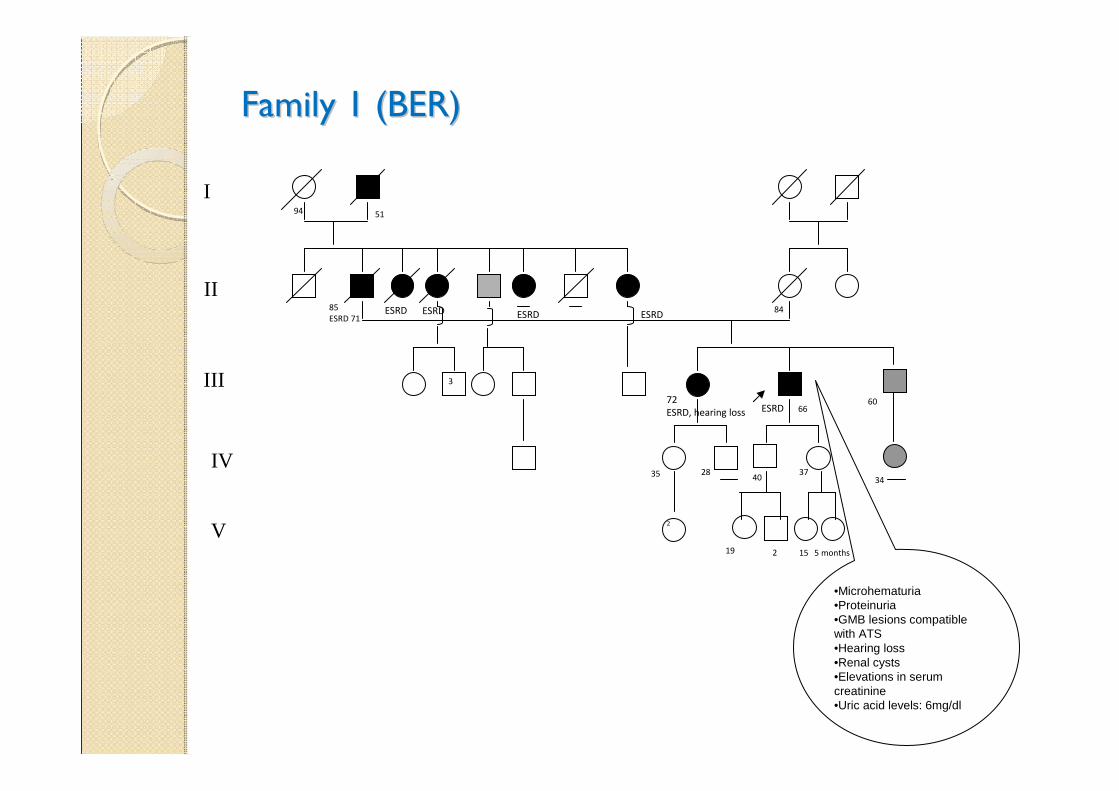
Family 1 (BER)Family 1 (BER)
72
ESRD, hearing loss
35 28
2
66
40
19
37
15
85
ESRD 71
5194
84ESRD ESRD
3
ESRD
60
2 5 months
34
ESRD
ESRD
•Microhematuria•Proteinuria•GMB lesions compatible with ATS•Hearing loss•Renal cysts •Elevations in serum creatinine•Uric acid levels: 6mg/dl
I
II
III
IV
V
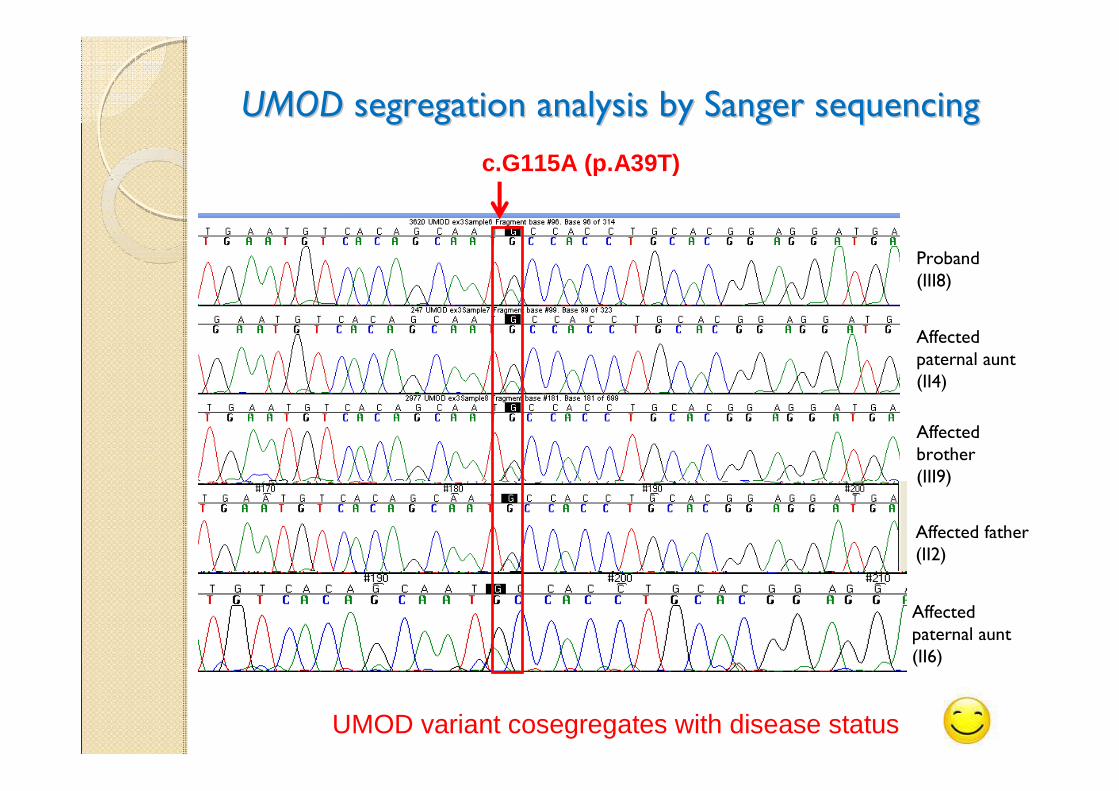
UMODUMOD segregation analysis by Sanger sequencing segregation analysis by Sanger sequencing
Proband(III8)
Affected brother(III9)
Affected paternal aunt (II4)
UMOD variant cosegregates with disease status
c.G115A (p.A39T)
Affected father (II2)
Affected paternal aunt (II6)

Family BER: Family BER: UMODUMOD atypical phenotypeatypical phenotype
Prof. Mario De Marchi’s lab. (Medical Genetics, Uni versity of Torino) :
“….. the UMOD gene variant that you have identified is known to us for a long time… This is not a classical mutation (not involving a Cysteine��) of UMOD gene and for this reason we are doing to further investigations, in collaboration with the Consortium of the Medullary Cystic Disease…
Also from the clinical point of view is not a typical diagnosis of uromodulin-associated disease and the phenotype described in your family is very similar to that observed in our family.
It would be interesting to know the geographical origin of your familyto see if it is a founder effect, we have available the microsatellite region adjacent to UMOD and we can investigate a possible founder effect of the mutation…”

Family 3 (BAU)Family 3 (BAU)
Polyphen2: probably damaging
SIFT: not tolerated
PhyloP: conserved
Not reported as SNP(dbSNP , 1000 Genome and in house database)
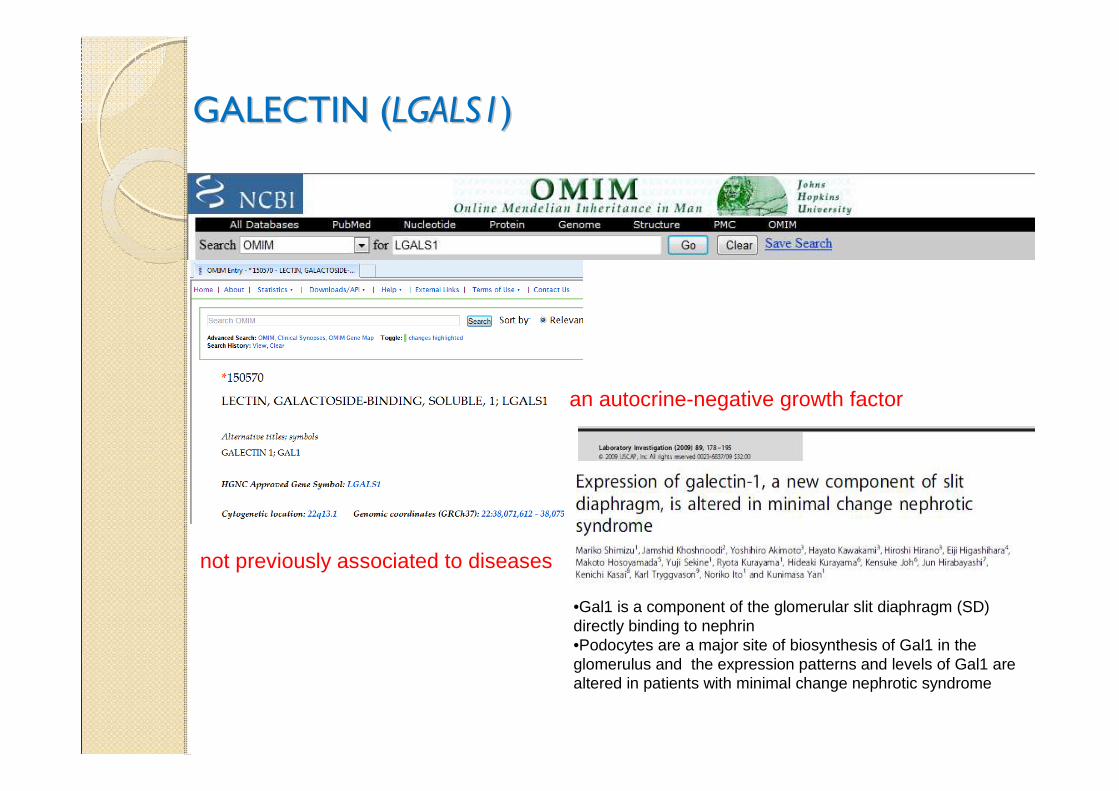
GALECTIN (GALECTIN (LGALS1LGALS1))
an autocrine-negative growth factor
not previously associated to diseases
•Gal1 is a component of the glomerular slit diaphragm (SD) directly binding to nephrin•Podocytes are a major site of biosynthesis of Gal1 in the glomerulus and the expression patterns and levels of Gal1 are altered in patients with minimal change nephrotic syndrome
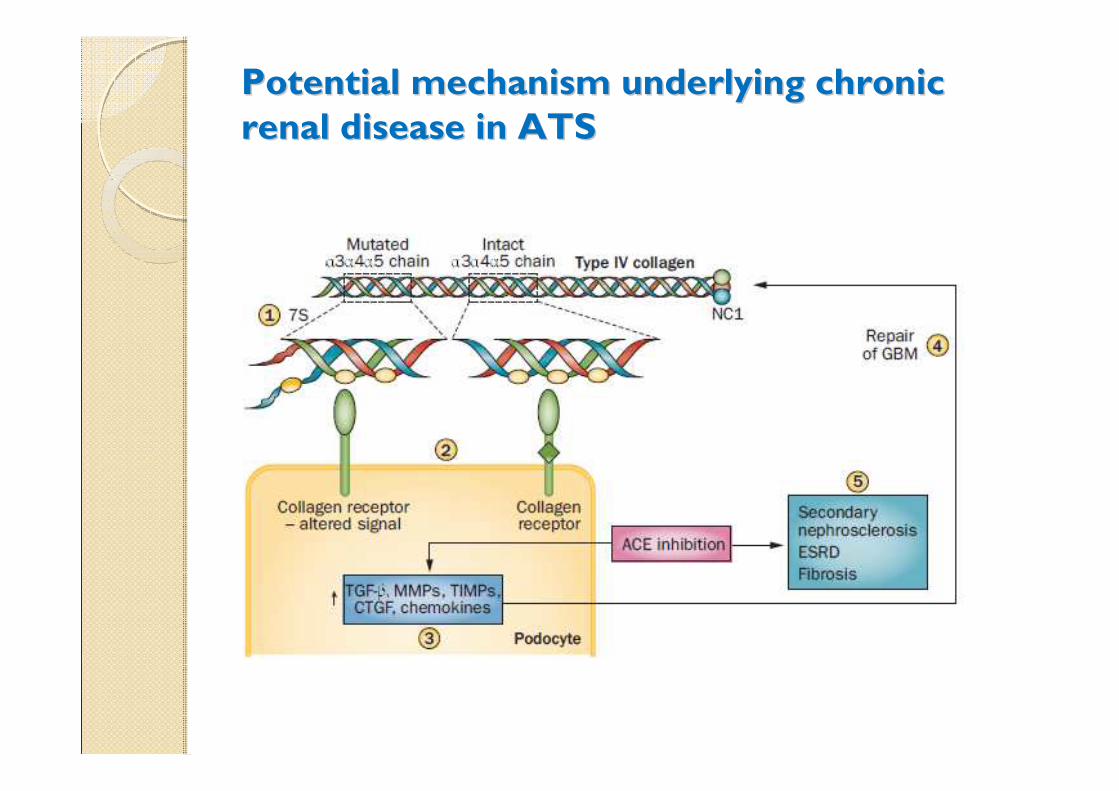
Potential mechanism underlying chronic Potential mechanism underlying chronic
renal disease in ATSrenal disease in ATS
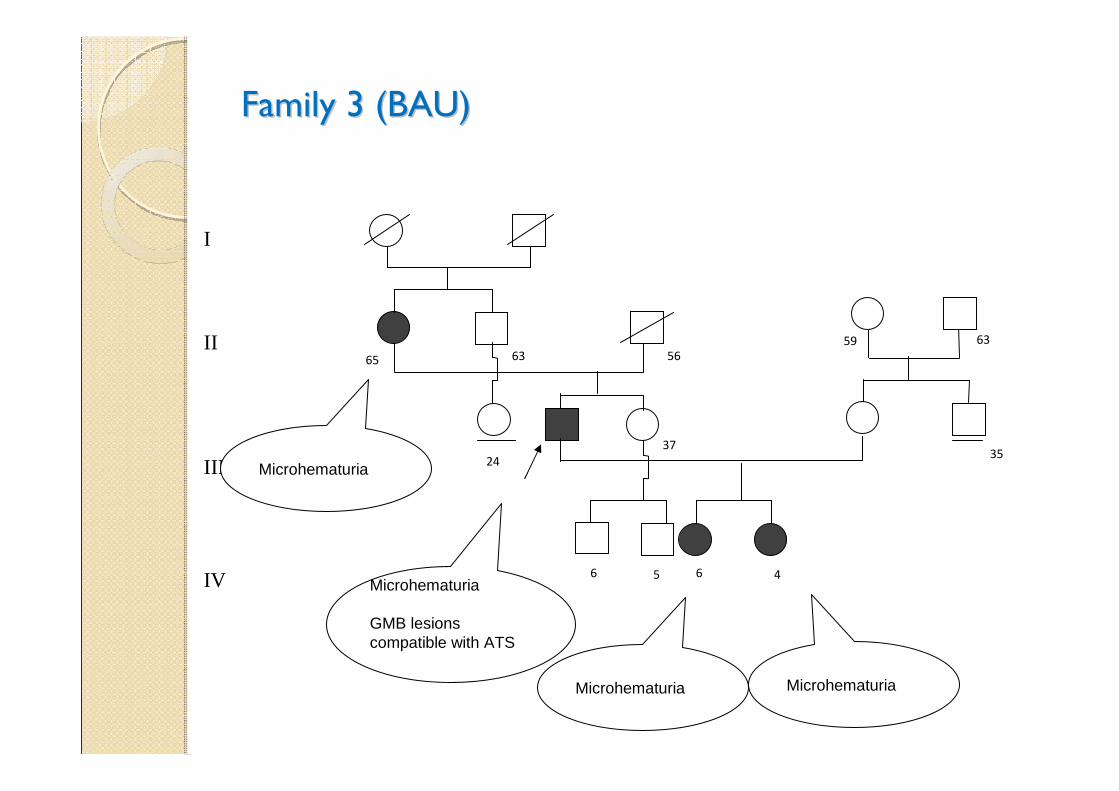
Family 3 (BAU)Family 3 (BAU)
MicrohematuriaMicrohematuria
Microhematuria
GMB lesions compatible with ATS
I
II
III
IV
Microhematuria
65 56
37
6359
63
24
6 5 6 4
35
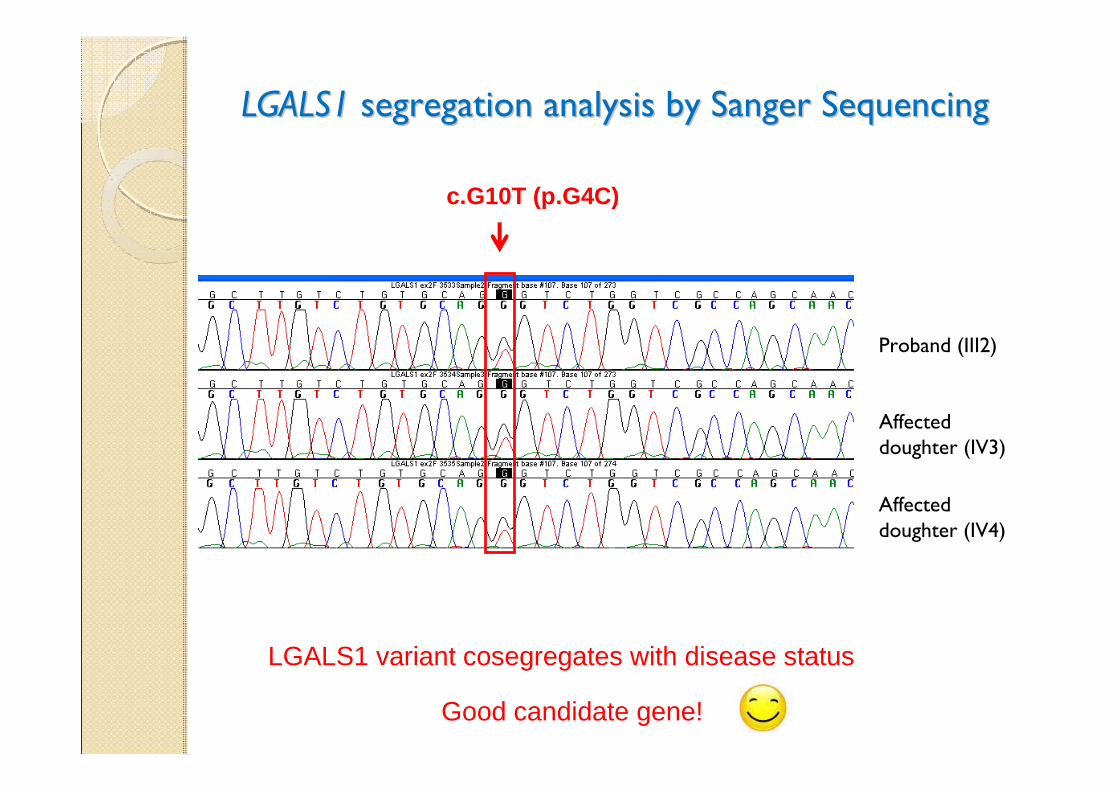
LGALS1LGALS1 segregation analysis by Sanger Sequencing segregation analysis by Sanger Sequencing
Proband (III2)
Affected doughter (IV3)
LGALS1 variant cosegregates with disease status
Affected doughter (IV4)
c.G10T (p.G4C)
Good candidate gene!

ConclusionsConclusions
•• NGS technology is improving NGS technology is improving diagnosis and patientsand patients’’managementmanagement
•• it also provides an exciting opportunity to solve it also provides an exciting opportunity to solve ““thousandsthousands”” of of Mendelian and nonMendelian and non--Mendelian disordersMendelian disorders
•• This will allow to better understand the pathogenetic This will allow to better understand the pathogenetic mechanisms of diseases and to design therapeutic strategiesmechanisms of diseases and to design therapeutic strategies
•• Going forward there will be enormous opportunity and Going forward there will be enormous opportunity and challenges in the lab and in the clinicchallenges in the lab and in the clinic

AcknowledgmentsAcknowledgments
Medical Genetics, University of Siena
Dr. Laura Dosa
Dr. Caterina Lo Rizzo
Dr. Chiara Fallerini
Dr. Laura Bianciardi
Medical Genetics, University of Torino Prof. Mario De Marchi, Prof. Daniela Giachino
TigemProf. Vincenzo NigroDr. Margherita Mutarelli
"Cell lines and DNA bank of Rett Syndrome, X-linked mental retardation and other genetic diseases” biobank” supported by TELETHON grant GTB07001
A donation in favour of “Graziano and Marco Laurini”

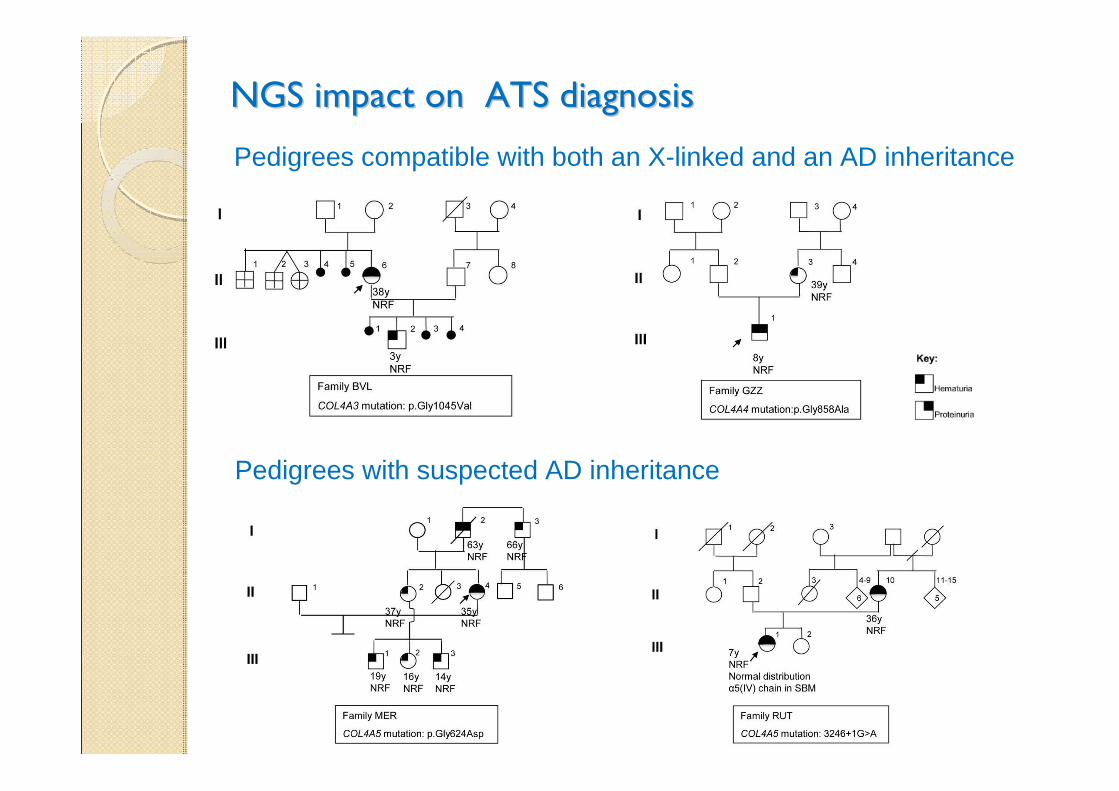
NGS impact on ATS diagnosisNGS impact on ATS diagnosis
Pedigrees compatible with both an X-linked and an AD inheritance
Pedigrees with suspected AD inheritance





























