Embed Size (px)
Citation preview

ELSEVIER Biochimica et Biophysica Acta 1253 (1995) 208-214
Biochi~ic~a et Biophysica AEta
Functional analysis of E. coli threonine dehydrogenase by means of mutant isolation and characterization
Yen-Wen Chen a,l, Eugene E. Dekker a,* , Ronald L. Somerville b a Department of Biological Chemistry, The University of Michigan, Ann Arbor, M148109-0606, USA
b Department of Biochemistry, Purdue University, West Lafayette, IN 47907, USA
Received 1 May 1995; accepted 4 August 1995
Abstract
The oxidation of L-threonine to 2-amino-3-ketobutyrate, as catalyzed by L-threonine dehydrogenase, is the first step in the major pathway for threonine catabolism in both eukaryotes and prokaryotes. Threonine dehydrogenase of E. coli has considerable amino-acid sequence homology with a number of Zn 2 +-containing, medium-chain alcohol dehydrogenases. In order to further explore structure/func- tion interrelationships of E. coli threonine dehydrogenase, 35 alleles of tdh that imparted a no-growth or slow-growth phenotype on appropriate indicator media were isolated after mutagenesis with hydroxylamine. Within this collection, 14 mutants had single amino-acid changes that were divided into 4 groups: (a) amino-acid changes associated with proposed ligands to Zn2÷; (b) a substitution of one of several conserved glycine residues; (c) mutations at the substrate or coenzyme binding site; (d) alterations that resulted in a change of charge near the active site. These findings uncover previously unidentified amino-acid residues that are important for threonine dehydrogenase catalysis and also indicate that the three-dimensional structure of tetrameric E. coli threonine dehydrogenase has considerable similarity with the dimeric horse liver alcohol dehydrogenase.
Keywords: Threonine dehydrogenase; Enzyme inactivation; Mutagenesis; Hydroxylamine
1. Introduction
Threonine dehydrogenase (EC 1.1.1.103) catalyzes the NAD+-dependent oxidation of L-threonine to 2-amino-3- ketobutyrate (or 2-amino-3-oxobutanoate). 2-Amino-3- oxobutanoate glycine-lyase (CoA-acetyla t ing) (EC 2.3.1.29), also called 2-amino-3-ketobutyrate lyase, cat- alyzes the second step in the threonine dehydrogenase- initiated pathway; by this reaction, glycine and acetyl CoA are formed via a CoA-dependent cleavage of 2-amino-3- ketobutyrate. The sequential action of these two enzymes constitutes the primary route for threonine degradation in both mammals [1,2] and bacteria [3,4]. In Escherichia coli, these reactions have the potential of being a part of an
Abbreviations: TDH, threonine dehydrogenase; LADH, horse liver alcohol dehydrogenase; YADH, yeast alcohol dehydrogenase; MADH, maize alcohol dehydrogenase; SDH, sheep liver sorbitol dehydrogenase; PAGE, polyacrylamide gel electrophoresis.
* Corresponding author. Fax: + 1 (313) 7634581. I Present address: Cardiovascular Research Institute, University of
California at San Francisco, 505 Parnassus Ave., Box 0130, San Fran- cisco, CA 94143, USA.
0167-4838/95/$09.50 © 1995 Elsevier Science B.V. All rights reserved SSDI 0 1 6 7 - 4 8 3 8 ( 9 5 ) 0 0 1 6 2 - X
efficient pathway for serine biosynthesis - - i.e., threonine glycine ~ serine [5]. Threonine dehydrogenase has been purified from ex-
tracts of chicken liver [6], pig liver [7], goat liver [8], and E. coli [4]. Whereas the enzyme from both chicken [6] and goat [8] liver is a single 88000 Da polypeptide, it is a homotetramer with subunits - 25 000 Da as isolated from pig liver. Threonine dehydrogenase from E. coli has been studied most extensively. We previously reported its pu- rification to homogeneity from extracts of a mutant of E. coli that is capable of growth on L-threonine as sole carbon source and which has highly elevated levels of the enzyme [4]. E. coli threonine dehydrogenase is a 148000 Da protein containing 4 apparently identical subunits. The enzyme from all four sources cited is specific for NAD ÷ or NAD+-analogs; the pH optimum observed in vitro, however, is in the range of 8.2-8.8 for the enzyme ob- tained from any one of the liver species, whereas the corresponding value for the E. coli dehydrogenase is 10.3. The E. coli enzyme is also unique in that its catalytic activity is stimulated 5- to 10-fold by added Cd 2+ or Mn 2+ ions [4,9-11].

Y.-W. Chen et al. / Biochimica et Biophysica Acta 1253 (1995) 208-214 209
The gene that encodes E. coli threonine dehydrogenase (tdh) has been cloned and sequenced [12]. The deduced primary structure of the enzyme has considerable homol- ogy with enzymes of ~Lhe Zn2+-containing medium-chain alcohol dehydrogenase family [12]. Horse liver alcohol dehydrogenase, whose 3-dimensional structure is known, typifies this family [13-15]; it contains 2 Zn 2÷ atoms per subunit, one of which is essential for activity ('catalytic' zinc) whereas the other stabilizes the structure ('structural' zinc). The catalytic Zn z+ atom is liganded to Cys-46, His-67, and Cys-174 as well as to a water molecule which is displaced by the hydroxyl group of the substrate during catalysis [16]. The structural Zn 2÷ atom, on the other hand, is bound by 4 closely spaced cysteine residues (Cys-97, 100, 103, and 111) located in a loop outside the active site. E. coli threonine dehydrogenase is also a Zn2+-containing enzyme which, as isolated in homoge- neous form, contains 1 Zn z+ atom per subunit [17].
While the general catalytic and structural properties of E. coli threonine dehydrogenase (TDH) were being deter- mined, parallel structure/function studies were pursued by selective chemical modification/inactivation techniques. Such studies established that 1 cysteine residue per subunit (i.e., Cys-38) is essential for catalytic activity and Mn 2÷ binding [11,17]; likewi,;e, chemical modification of 1 argi- nine (out of 16 total) per subunit causes inactivation. This Arg residue most likely is located in or near the cosub- strate binding pocket [18]. The specific role of amino-acid residues identified in this manner can then be explored by site-directed mutagenesis. This method, however, usually is most fruitful when prior information is available about 'essential' amino-acid residues. General mutant methodol- ogy, on the other hand, has the potential of disclosing the identity of all important amino-acid residues whether that be structural or catalytic. We report here the isolation and first characterization of a set of hydroxylamine-induced mutants of the E. coli ~dh gene.
2. Materials and methods
2.1. Chemicals, enzymes, and general materials
Hydroxylamine. HC1 was purchased from Mallinck- rodt. Sequenase Version 2.0 Sequencing Kit and NAD ÷ were obtained from United States Biochemical. Restriction endonucleases were purchased from New England Biolabs, Gibco Bethesda Research Laboratories, or Boehringer Mannheim Biochemicals. Sigma was the source of deoxy- ribonuclease I and ribonuclease A. Nitrocellulose mem- branes, goat anti-rabbit IgG(H + L) horseradish peroxidase conjugate and the hor:;eradish peroxidase color develop- ment reagent, 4-chloro-l-naphthol, were products of Bio- Rad Labs. All other chemicals were of the highest quality commercially available.
Oligodeoxynucleotides used as primers for double- stranded DNA sequencing were prepared with an auto- mated DNA synthesizer (Applied Biosystems, Inc.) by the Biochemical Research Core Facility at The University of Michigan. Structural analyses by computer modeling were done on an Evans and Sutherland computer graphics in- strument connected to a micro VAXII computer 15 using a Frodo software program.
A special growth medium, designated LRKTM(240 ), consisted of salts mix E of Vogel and Bonner [19] plus 240 mg per liter each of L-leucine, L-arginine, L-lysine, L- threonine, and L-methionine.
2.2. DNA preparations
Plasmid DNA was isolated by the alkaline lysis proce- dure of Ish-Horowicz and Burke [20]. Cells were trans- formed by the method of either Cohen et al. [21] or Chung and Miller [22].
2.3. Mutagenesis of plasmid pDR123 by NH20H
Random mutagenesis of the tdh gene was performed by the method of Humphreys et al. [23]. Plasmid pDR123 [amp r, tdh ÷ ] was incubated at 70°C for either 45 or 90 min with 0.4 M NH2OH-HC1 in 50 m M sodium phos- phate buffer (pH 6.0) containing 0.5 m M EDTA. Reaction mixtures were then dialyzed exhaustively at 4°C against 10 m M CaC12 to remove the NH2OH. HCI. The resulting material was subsequently transformed into E. coli SP1192 [tdh:: cat 1212 glyA A recA srl:: TnlO]. Plasmid-bearing cells were selected by their ampicillin resistant phenotype; transformants were screened for TDH-specific phenotypes by no-growth or slow-growth on medium containing an elevated level of L-threonine, i.e., LRKTM(240 ). The indi- vidual plasmid isolates obtained by this method were designated as pDR123YC-1, pDR123YC-2, etc. Each plas- mid was retransformed into SPl192 to ensure that the mutation(s) was/were in the plasmid.
2.4. Screening to eliminate mutants which did not form full-length threonine dehydrogenase
Overnight cultures of the plasmid-bearing strains (1 ml each) were harvested and resuspended in 200 /xl of 6% SDS containing 4% 2-mercaptoethanol. These suspensions were heated at 100°C for 5 min and then immediately cooled on ice. A mixture of deoxyribonuclease I and ribonuclease A (10 /xl, 2 mg /ml each) was subsequently added to the lysed cells and the solution kept on ice for 10 min. Such preparations were fractionated by a discontinu- ous SDS-PAGE system [24]; proteins separated on the gel were transferred electrophoretically onto a nitrocellulose membrane. The nitrocellulose membrane was then incu- bated with polyclonal antibodies elicited in rabbits against wild-type TDH. The antibody-containing serum was di-

210 Y.-W. Chen et al. / Biochimica et Biophysica Acta 1253 (1995) 208-214
luted 500-fold with 5% ( w / v ) powdered milk/phosphate- buffered saline which contained 8.5% NaCI in 10 m M sodium phosphate buffer (pH 7.4), and then incubated with the membrane at room temperature for at least 2 h. After washing out unabsorbed proteins, the membrane was probed for 2 h at room temperature with goat anti-rabbit IgG:horseradish peroxidase conjugate (1:3,000 dilution in 5% powdered milk/phosphate-buffered saline). Antibody bound to the membrane was detected in a solution contain- ing 20 /xl of H 2 0 2, 30 mg of 4-chloro-l-naphthol, 10 ml of methanol, and 50 ml of phosphate-buffered saline.
2.5. Structural characterization of plasmids containing tdh alleles
3.2. Analyses of cell extracts for the presence of full-length TDH
Since NH2OH reacts randomly with DNA causing GC AT mutations, it was predicted that NH2OH treatment
would cause the formation of a certain number of stop codons within the tdh gene. Cells with such mutations would also exhibit the Tdh- phenotype and be selected by the procedure used. Mutants of this nature were identified by immunoblotting with TDH polyclonal antibodies (see Section 2.4). Of the 35 isolates that showed no or slow growth on the LRKTM{240 ) medium, 6 had truncated forms of the TDH protein and were presumed to be nonsense mutations.
Plasmids used for DNA sequencing were isolated as described by Kraft et al. [25]. Four separate oligodeoxynu- cleotide primers, which were complementary to sequences within the tdh gene, separated by 255 base pairs (coordi- nates 1684-1700, 1942-1958, 2197-2213, and 2452-2468 in pDR123), were synthesized for double-stranded DNA sequencing. The sequencing procedure used was either that of Kraft et al. [25] or as recommended by the manufac- turer.
2.6. Assay for TDH activity of mutant proteins
TDH activity in crude extracts of cells containing the pDR123YC mutants was detected by the method previ- ously described as Assay I [4]. This assay colorimetrically measures the amount of aminoacetone that is formed by decarboxylation of 2-amino-3-ketobutyrate. Cell extracts were prepared by sonication [5 ml of an overnight culture resuspended in 4 ml of 50 m M Tris. HC1 buffer (pH 8.4), 1 m M 2-mercaptoethanol, and 0.02% NAN3], cell debris was removed by centrifugation, and aliquots of the super- natant fluid were tested for TDH activity.
3. Results and discussion
3.1. Isolation of TDH mutants
Ampicillin-resistant cells were replica plated onto LRKTM(240 ) medium. E. coli SP1192 is Tdh-; therefore, in order to utilize L-threonine for growth on LRKTM(240 ) medium, cells required functional TDH activity normally provided by plasmid pDR123 (tdh ÷, kbl-) . Cells that did not grow (or grew at a very slow rate) on this medium were assumed to have a mutation in the plasmid-encoded tdh gene. A total of 35 mutants were obtained by this selection method. Plasmid DNA prepared from these mu- tant cells was reintroduced into E. coli SPl192 to verify that the tdh mutations were plasmid-borne.
3.3. DNA sequence analysis of NHzOH-induced mutants
Of the remaining 29 mutants, 22 were characterized by DNA sequence analysis. Three of these were found to be duplicates of other mutants. Additionally, 4 mutants hav- ing slow-growth phenotypes had the wild-type tdh gene. Perhaps, one or more mutations had occurred in the tdh promoter but this was not explicitly verified. One other mutant had a change of CGC to CGT (both encode the amino-acid arginine) which caused no alteration in the primary structure of the enzyme. Each of the remaining 14 mutants contained single point mutations in the tdh gene.
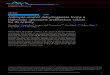
Fig. 1 presents an alignment of the amino-acid sequence of E. coli TDH with that of alcohol dehydrogenase from horse liver, yeast, and maize as well as of sheep liver sorbitol dehydrogenase, with which enzymes TDH has considerable homology. This figure shows conserved amino-acid residues in these enzymes that might be impor- tant in structure-function relationships of TDH and it also pinpoints the location of the NH2OH-induced mutations in TDH. The sites of these mutations are indicated by arrows together with the amino-acid changes that had occurred. As is evident, the mutations are distributed throughout the entire tdh gene with no apparent hot spots. With these results in hand, the known three-dimensional structure of LADH was used as a model in an attempt to evaluate what effect mutations at these specific sites might have on TDH function. Such structural analyses and comparisons are summarized in Tables 1 and 2 which list the site and nature of the amino-acid change in TDH, the correspond- ing amino-acid residue in LADH, and possible structural/catalytic implications as inferred from the crys- tal structure of LADH.
As is evident in Table 1, no TDH activity was detected in crude extracts of 7 of these mutants. Table 2, on the other hand, lists the amino-acid changes found in the remaining 7 mutants, crude extracts of which showed definite levels of TDH activity. The finding of some TDH-active mutants was not unexpected since the pheno- type of no- or slow-growth on the LRKTM(240 ) medium was the basis for mutant screening. For E. coli K-12, high

Y.-W. Chen et al. / Biochimica et Biophysica Acta 1253 (1995) 208-214 211
levels of TDH activity tend to be associated with the ability to utilize threonine as a carbon source [4,26]. The screening technique used here did not distinguish between those cells that formed partially active or completely inac-
tive enzyme. Hence, it was possible that some cells synthe- sized a form of TDH having a low level of activity that did not reach the threshold necessary for cell growth on LRKTMt240 ) medium.
T
TDII : MILLS KLKRE E G I ~ G ; I I ~ T-r-TvTRK'~IDVlIITNW SDH : AEPAAENLSLVVSGPGDLRLEN~rII~?~IEVZLI~HSVGI~SDVl- Y-W
YADH : SIPETGI~'TIFYESEGKLEYEDI~KP KAIELT~NVKYS GV~LH- - AW MADH: MATAGKVIKCR/IAVTWERGKP LS IEEVEVAP p Q ~ LY'ni~l ~V- - YFW ~DH: STAGKVIKC]~AVLWEEKKP F S IEEVEVAP p ~ T G I ~ R S D D H- -W
10 20 30 40 50
D ED ¢: H
TDH : D-EI~ ~'VVGIG(~r.vaz~ JuL~u~vbGEGHI-T~'H~RN~RG SDH: QGRI GD~GIIAS~D;VKVGSLVRHL~PG~RVAZ QPGA-P RQTDEFgKZ
YADH: HGDI~L-p TKLIPLVG~~~GDYAGIKWLNGS~IAg~Y~L MADB : EAK-GQ-Tp~w~tI~SVG~GVTDVAPGDKVLPVI~EG-E~KE~A/I~KS LADH : SGT-- L-~AGIIFd~A~v~ SX~EGVTTVRPGDKVIPLFTP ~
GO 70 80 90 100
S S
TDH: GRTHI~RNTTGVG- ..................... VNRP G~I~YLVIPAFNAFICZ SDH: GRYN/~PTIFFCA . . . . . TPPDDGNL~FCYEL
:¢ADH: ~'~ESN~ HADLSG- ..................... YTHDGSI~Q~TADAVQAAHI MADH : EES~LLRINVDRGVMIGDGKSRFTISGQP ~_EHFVGTS~HVGCIAEI LADH : PEGNF~.KNDLSMPRG-TMQDGTSRFTCRGI~ IHHFLGTSTFS(~TI'VVDE ISVAEX
II0 120 130 140 150 160
S DV
tt TDH : P~II SDDLAAZF -/II~GNAVI~AL S- - FD LV~DVLV~- ~TGI MAAAVAIGIVG SDH : I~NVTFEEGALI -E~PLSVGIIIACRR-AGVTL~II~'LVC~- ~X~'LVNL LAAKAMG
YADH : I~GTD LAEVAPVL~G I TVYKALKS-~WVAI SG~AGGLGS LAVQYAKAMG MADH : NPEAP LEKVCILS~ISTGLGATLNVAKPAKGSTVAIF~r--GAVGLAI K~IEGARLAG LADH : DAASPLEKVCLI G~GTSTGYGSAVKVAKVTQGSTCAVF~-GGVGLSVIMGCI~qAG
170 180 190 200 210
X
TDH : ~ 2 O ~ ~ I TRA~NVAEENLNDVMAELGZG'EG- - FDVG L ] ~ S G SDH : A A Q ~ " ~ L SASRLSKAKEVGADF I LE I SNESP EE IAKKVE GLLGSKP E~T IECTG
YADH: ¥-RVLGIDGGEGKEI~'IIS IGGEVFIDFTI~KD IVGAVLK-A~qGG-AHGVZNVSV MADE : ASRI I G~DII~~GCTEFVNP KD HD KPVQEVLIEL2NGG-VDRSV~: TG LADH : AARI I GVDINKDKF~TE C%IIP QDYKKP IQEVLTEMSN~-VDFS FE¢~ G
220 230 240 250 260 270
D D
TD H : APPAFR2MZ,DTMN- HGGRIAMLGIPPSZ:H- - S- ~D - -WTEVI FKGLF IICGIYGREM SDH: VETS IQAG IYATH - SGGTLVLVG[,GSEMT - - S-VP - - LVEAATREVD ~ ' V T - R Y -
YADH: SEAAIEASTRYVR-ANGI"TVLVG~AGAKCC~VFNQVVK . . . . S I S Z V G S T - V G - MADE : NVIqI~ISAFECVHDGWGVAVLV~T~HKI~QFKTHPI~/FLSEKTLKGTFFGNT-KP- LADH : RLDTMWEkLSCC(~EAYG~S~DSQNLSMNPMLLLSGRTWKGAIFGGF-KS"
280 290 300 310 320
TDH: FET~YI~IAA-LIQSGLDLSPZZTIIRFSIDDFQK~6~-S~tVZLSWD SDH: CNTHPMAI S~KSVNV]~L%zzsMmr LEKALEAFETSKKGL-GLXVMIKCDP SDQNP
YADH : NRADTREALDFFAR~.I~ZKVVGLSTLP--EIYEEI~KG~'V~YVVDTSK MADH : RTDLPNVVEMYMK]G~LEKF~FSEINTAIDLML~-SLRCXMRMED LADH : KDSVPELVADFMAKKFALDP LITIIV'LPFEKINEG]DLLRS~ - SIRTXLTF
330 340 350 350 370
Fig. l. Sequence alignment of E. coli threonine dehydrogenase and enzymes of the Zn2+-containing medium-chain alcohol dehydrogenase family. The amino acid changes in the hydroxylamine-induced mutants of threonine dehydrogenase are indicated by arrows immediately above the TDH sequence. Residue numbers refer to the amino acid sequence of LADH and correspond to the amino acid above the last digit of each number (e.g., residue 370 is Thr). Dashes signify gaps introduced to optimize homology. The underlined residues serve as Zn 2+ ligands in LADH. TDH = E. coli threonine dehydrogenase; SDH = sheep liver sorbitol dehydrogenase; YADH = yeast alcohol dehydrogenase; MADH = maize alcohol dehydrogenase; LADH = horse
liver alcohol dehydrogenase.

212 Y.-W. Chen et al. / Biochimica et Biophysica Acta 1253 (1995) 208 -214
Table 1 Inactive hydroxylamine-induced mutants of E. coli threonine dehydrogenase
Mutant Amino acid Corresponding Coding sequence in no. change in amino acid in wild type mutant
TDH a LADH a
Relevance to 3-D structure of LADH a
26 C38Y 46 TGC TAC 27 G62D 66 GGC G AC 18 E88K 92 GAA A AA 30 G 119S 144 GGC AGC 22 G171D 199 GGT GAT 12 G263D 293 GGT G AT 11 G285D 320 GGT G AT
Proposed Zn 2+ ligand Next to proposed Zn 2 + ligand; i.e. His-67 Near the Zn 2+ active site
NAD ÷ binding site (G-X-G-X-X-G)
Structural bend
a T D H = E. coli threonine dehydrogenase; LADH = horse liver alcohol dehydrogenase.
Based on alignments of the primary structure of E. coli TDH with alcohol dehydrogenases in the Zn2+-containing, medium-chain family and an analysis by computer com- parisons of the location of amino-acid changes in TDH mutants with the known 3-dimensional structure of horse liver alcohol dehydrogenase, the 14 single-site hydroxyl- amine-induced mutations of TDH were divided into 4 broad groups. Mutants in the first group affect the 3 residues that are likely to serve as ligands to the Zn 2+ atom in TDH - - i.e., Cys-38, His-63, and Asp-148. The change of Cys --* Tyr at position 38 (which corresponds to Cys-46, a known 'catalytic' Zn-ligand in LADH) in mu- tant #26 completely abolishes TDH activity. It was previ- ously reported [11,17] that selective alkylation of Cys-38 in E. coli TDH by iodoacetate abolishes all enzymatic activity. The isolation of mutant TDHC38Y by mutagene- sis with hydroxylamine further substantiates the central role of this residue in TDH catalysis. TDH mutants #13 (TDHP149S) and #27 (TDHG62D) each have an amino- acid change adjacent to two other possible Zn-ligands - - i.e., Asp-148 and His-63 (corresponding to the 'catalytic' Zn-ligands, Cys-174 and His-67, respectively, in LADH). Although the amino-acid residues changed in these two TDH mutants are not precisely the ones considered as possible ligands for Zn 2+, it is evident that substitution of an adjacent single amino-acid has a dramatic effect on TDH activity. More direct proof must still be obtained to
establish that these three amino acids (i.e., Cys-38, His-63, and Asp-148) are indeed Zn2+-ligands in E. coli TDH, but these new mutants focus further attention on these residues.
Each mutant of E. coli TDH in the second group has a change of one of four specific but different glycine residues, each of which is conserved in the primary structures of the homologous alcohol dehydrogenases (see Fig. 1); this group includes TDH-mutants #11, #12, #22, and #27 in Table 1. One other mutant, namely #6, also has a change of Gly ~ Asp at yet another site but this is not an invariant glycine residue in the other dehydrogenases. Where con- served glycine residues are changed in TDH (i.e., the four mutants G62D, G171D, G263D, and G285D), such muta- tions all result in the formation of an inactive form of the enzyme. As is known, glycine can adopt unusual confor- mational angles which disallow amino-acid residues hav- ing a side chain and as a consequence this residue is frequently found in protein loop regions between sec- ondary structural elements - - i.e., glycine residues are important in protein folding. The importance of glycine residues in structure-function relationships of the Zn 2÷- containing alcohol dehydrogenase family was examined by Eklund and Br'~ind6n [16], who compiled a list of residues conserved in all species. It was found that most conserved glycine residues occur in loop regions between secondary structures, especially in the coenzyme-binding domain which is made up of alternating fl-strands and c~-helices.
Table 2 Hydroxylamine-induced mutants of E. coli threonine dehydrogenase that have altered enzymatic activity
Mutant Amino acid Corresponding Coding sequence in No. change in amino acid in wild type mutant
TDH a LADH a
Relevance to 3-D structure of LADH a
6 G89D 93 GGC GAC 10 R97C 101 CGC TGC 15 R97H 101 CGC C AC 16 G114S 118 GGT AGT 13 P149S 175 CCC TCC 21 A 172V 200 GCA G TA 24 E238K 267 GAA A AA
Substrate binding pocket
Next to proposed Zn 2+ ligand; i.e., Cys-174 NAD + binding site (G-X-G-X-X-G) Near NAD + binding site
a TDH = E. coli threonine dehydrogenase; LADH = horse liver alcohol dehydrogenase.

Y.-w. Chen et al./ Biochimica et Biophysica Acta 1253 (1995) 208-214 213
According to Eklund ,'rod Br'5~ndrn, Gly-320 in LADH (corresponding to Gly-285 in wild-type TDH) is required at a position which constitutes a bend in the catalytic domain. Hence, it is likely that the Gly ~ Asp change in TDH mutant # 11 disrupts secondary structure and protein folding. Similar considerations probably apply to TDH mutant #27 (where Gly-62 in TDH corresponds to Gly-66 in LADH); Gly-66 of LADH is conserved in all the dehydrogenases in this family due to a lack of space in this area for amino-acid residues having a side chain. On the other hand, Gly-293 in LADH (corresponding to Gly-263 in native TDH, which is changed in mutant #12) is located at an internal region of the protein; the change to an Asp residue in this mutant could well disturb proper interac- tions in an internal hydrophobic region of the molecule. The loss of catalytic activity by TDH mutant #22 more specifically applies to the coenzyme binding site, involving the commonly seen sequence of -G-X-G-X-X-G- (where X can be any amino acid) which is known to be crucial for NAD ÷ binding. This mutant of TDH is considered in the next category.
TDH mutants in group three appear to affect the bind- ing of substrate or coenzyme; this includes TDH mutants #6, #21, #22, and #'.24. The 3-D structure of LADH shows that Phe-93 in this enzyme (which corresponds to Gly-89 in wild-type TDtt and Asp-89 in TDH mutant #6) is one of the residues that forms the substrate binding pocket. Likewise, the changes in TDH mutants #21 and #22 (Ala ~ Val and Gly ~ Asp, resp.), which correspond to residues 199 and 200 in LADH, are in the universally conserved sequence, -G-X-G-X-X-G-, at the NAD+-bind - ing site of these dehydmgenases. Although the first X in this sequence can be any amino acid, it is Ala in E. coli
TDH, SDH, and YADH and Leu in both MADH and LADH. This conserved ~unino-acid sequence constitutes a tight turn at the end of the first strand of a t-sheet and the beginning of an a-helix. The first Gly residue is essential for the tightness of the turn, the second allows the dinucleotide to bind without obstruction by an amino-acid side chain at this position, and the third Gly seems to provide space for close i[nteraction between the t-strands and a-helix [27]. The same amino-acid substitution that occurred in TDH mutan~I #22 (i.e., Gly ~ Asp) has also been' found in a mutant of alcohol dehydrogenase from Drosophila [28]. Although the enzyme from this source has little amino-acid sequence homology with the Zn 2÷- containing alcohol dehydrogenase family considered here, the insect mutant enzyme with the first Gly in the se- quence, -G-X-G-X-X-G-, replaced by Asp was catalyti- cally inactive. It was shown that alcohol dehydrogenase from this Drosophila mutant had a reduced affinity for the competitive inhibitor, 6-aminohexyl-5'-AMP, which sug- gests that coenzyme binding had been affected by the observed amino-acid substitution. Inactivation of TDH mu- tant #22 could very well be due to the same effect. This is consistent with our prelinfinary observation that TDH from
this mutant does not bind to the Cibacron blue F3G-A-based Reactive blue 2-Sepharose CL-2B column used in the routine purification of wild-type TDH [9]. The affinity interaction between this dye and many NAD +- or NADP+-dependent enzymes is generally believed to be due to the structural resemblance of Cibacron blue F3G-A to NAD +. Similar considerations may apply to TDH mu- tant #24 in which a change of Glu ~ Lys at position-238 also yields a form of TDH that has lost its affinity for Blue dextran-Sepharose. Glu-267 in LADH, which corresponds to Glu-238 in E. coli TDH, is located at the end of a /3-strand as part of the NAD+-binding domain.
The 6 remaining NH2OH-induced mutants of E. coli TDH (including #10, #12, #15, #16, #18 and #30) are grouped together, since no specific reason is currently apparent for their effect. An influence on subunit interac- tion is a possibility for mutant #30 which has a Gly --* Ser substitution at position-119 (corresponding to residue 144 in LADH). When the amino-acid sequences of the 5 homologous dehydrogenases listed in Fig. 1 are compared, the tetrameric enzymes, TDH, SDH, and YADH, are all seen to have a deletion in a region near this residue and all have a Gly residue at the position corresponding to 144 in LADH. The dimeric enzymes MADH and LADH, on the other hand, have no deletion in this region and both have a Ser residue at the position under consideration. According to the model constructed by Eklund et al. [29] for the structure of sheep liver SDH, this tetrameric enzyme is characterized by being slightly longer in the loop at posi- tions 55-60 (corresponding to positions 49-56 in E. coli TDH), lacks the loop at residues 119-139 (between posi- tions 114-115 in TDH), and is shorter in the region containing residues 305-315 (i.e., positions 272-280 in TDH). These regions are all located on one side of a given subunit and this side is probably involved in subunit interactions different from those found in the two homolo- gous dimeric enzymes. Such differences in SDH are also present in the primary structure of E. coli TDH. Perhaps, the conserved glycine residue at position l l9 in TDH serves a structurally important role in this region by main- taining a conformation appropriate for subunit association. In some of the other mutants of this group, the charge- charge (electrostatic) microenvironment of the enzyme might be affected by the loss (mutant #10, R97C), gain (mutant #12, G263D), or change (mutant #18, E88K) of charge by the amino-acid substitution that has occurred.
The results presented here, obtained by the technique of general mutagenesis, are the first describing many previ- ously unknown altered forms of E. coli TDH. Several new and interesting insights have been gained. The isolation of a new, inactive mutant - - i.e., #26, TDHC38Y - - by this method reinforces the unique role of Cys-38 in the func- tionality of this enzyme. Several nearby mutant sites (in mutants #13, # 18, and #27) that may participate in the binding of either Mn 2÷ or Zn 2÷, the change of an invari- ant glycine residue in mutant # 6 that quite likely is in the

214 Y.-W. Chen et al. / Biochimica et Biophysica Acta 1253 (1995) 208-214
substrate binding pocket, as well as other amino-acid residues (in mutants #21, #22, and #24) that relate to the NAD÷-binding domain can now be selectively modified and studied by site-directed mutagenesis. The same is true for some of the remaining mutants (i.e., #10, #15, and #16) where the observed activity effects could point to possible subunit interactions in this tetrameric protein. As more structural and catalytic data are obtained for TDH, it should be possible to make better and more meaningful correlations with excellent detailed insights gained in stud- ies with LADH, as exemplified by the recent results and conclusions of Ramaswamy et al. [30]. Finally, although determination of the 3-D structure of E. coli TDH awaits the availability of crystals suitable for X-ray analysis, it is apparent from these results that the known structure of dimeric LADH is quite similar in many respects to TDH and can truly serve as a provisional model to establish structure/function interrelationships of this important amino-acid-metabolizing enzyme. The preliminary report of Ramaswamy et al. [31] on the crystallization and some initial crystallographic data of YADH, a tetrameric protein, will also be significant and helpful in this regard.
Acknowledgements
This research was supported by Grants DK-03718 (to E.E.D.) and GM-22131 (to R.L.S.) from the National Institutes of Health, United States Public Health Service as well as by Grant MCB-9204829 (to E.E.D.) from the National Science Foundation. The authors thank Jill H. Zeilstra-Ryalls and Guoping Zhao for their advice and assistance.
References
[1] Bird, M.I. and Nunn, P.B. (1983) Biochem. J. 214, 687-694. [2] Dale, R.A. (1978) Biochim. Biophys. Acta 544, 496-503. [3] Komatsubara, S., Kurata, K., Kisumi, M. and Chibata, I. (1978) J.
Bacteriol. 135, 318-323. [4] Boylan, S.A. and Dekker, E.E. (1981) J. Biol. Chem. 256, 1809-
1815.
[5] Ravnikar, P.D. and Somerville, R.L. (1987)J. Bacteriol. 169, 2611- 2617.
[6] Aoyama, Y. and Motokawa, Y. (1981) J. Biol. Chem. 256, 127- 12373.
[7] Tressel, T., Thompson, R., Zieske, L.R., Menendez, M.I.T.S. and Davis, L. (1986) J. Biol. Chem. 261, 16428-16437.
[8] Ray, M. and Ray, S. (1985) J. Biol. Chem. 260, 5913-5918. [9] Craig, P.A. and Dekker, E.E. (1986) Biochemistry 25, 1870-1876.
[10] Craig, P.A. and Dekker, E.E. (1988) Biochim. Biophys. Acta 957, 222-229.
[11] Craig, P.A. and Dekker, E.E. (1990) Biochim. Biophys. Acta 1037, 30-38.
[12] Aronson, B.D., Somerville, R.L., Epperly, B.R. and Dekker, E.E. (1989) J. Biol. Chem. 264, 5226-5232.
[13] Br'~ind6n, C.-I., Eklund, H., NordstriSm, B., Boiwe, T., Si~derlund, G., Zeppezauer, E., Ohlsson, I. and .~keson, .~. (1973) Proc. Natl. Acad. Sci. U. S. A. 70, 2439-2442.
[14] Eklund, H., Nordstr~Sm, B., Zeppezauer, E., SiSderlund, G., Ohlsson, I., Boiwe, T. and Bf~d6n, C.-I. (1974) FEBS Lett. 44, 200-204.
[15] Schneider, G., Eklund, H., Cedergren-Zeppezauer, E. and Zeppeza- uer, M. (1983) Proc. Natl. Acad. Sci. U. S. A. 80, 5289-5293.
[16] Eklund, H. and Br~ind6n, C.-I. (1987) in Biological Macromolecules and Assemblies (Jurnak, F.A. and McPherson, A., Eds.) Vol. 3, pp. 73-142, John Wiley & Sons, New York.
[17] Epperly, B.R. and Dekker, E.E. (1991) J. Biol. Chem. 266, 6086- 6092.
[18] Epperly, B.R. and Dekker, E.E. (1989) J. Biol. Chem. 264, 18296- 18301.
[19] Vogel, H.J. and Bonner, D.M. (1956) J. Biol. Chem. 218, 97-106. [20] Ish-Horowicz, D. and Burke, J. (1981) Nucleic Acids Res. 9,
2989-2998. [21] Cohen, S.N., Chang, A.C.Y. and Hsu, L. (1972) Proc. Natl. Acad.
Sci. USA 69, 2110-2114. [22] Chung, C.T. and Miller, R.H. (1988) Nucleic Acids Res. 16, 3580. [23] Humphreys, G.O., Willshaw, G.A., Smith, H.R. and Anderson, E.S.
(1976) Mol. Gen. Genet. 145, 101-108. [24] Sch~igger, H. and von Jagow, G. (1987) Anal. Biochem. 166, 8-379. [25] Kraft, R., Tardiff, J., Krauter, K.S. and Leinwand, L.A. (1988)
BioTechniques 6, 544-547. [26] Chan, T.T. and Newman, E.B. (1981) J. Bacteriol. 145, 1150-1153. [27] Scrutton, N.S., Berry, A. and Perham, R.N. (1990) Nature 343,
38-43. [28] Thatcher, D.R. and Retzios, A. (1980) Protides of the Biological
Fluids 28, 157-160. [29] Eklund, H., Horjales, E., Jtirnvall, H., Bf~ind6n, C.-I. and Jeffery, J.
(1985) Biochemistry 24, 8005-8012. [30] Ramaswamy, S., Eklund, H. and Plapp, B.V. (1994) Biochemistry
33, 5230-5237. [31] Ramaswamy, S., Kratzer, D.A., Hershey, A.D., Rogers, P.H.,
Arnone, A., Eklund, H. and Plapp, B.V. (1994) J. Mol. Biol. 235, 777-779.