Embed Size (px)
Citation preview

Functional DNA Aptamers as Biotherapeutic Molecules
By
Erik William Orava
A thesis submitted in conformity with the requirements
for the degree of Doctor of Philosophy
Graduate Department of Pharmaceutical Sciences
University of Toronto
© Copyright by Erik William Orava, 2013

II
ABSTRACT
Functional DNA aptamers as biotherapeutic molecules
Erik William Orava
Doctor of Philosophy, 2013
Graduate Department of Pharmaceutical Sciences
University of Toronto
Aptamers are single-stranded oligonucleotides, DNA or RNA, which can bind to a
myriad of targets such as ions, peptides, proteins, drugs, organic and inorganic molecules
with high affinity and specificity. Aptamers are derived using combinatorial libraries
comprised of a variable region flanked by two primer regions used for a process termed
Systematic Evolution of Ligands by Exponential Enrichment (SELEX). The central
theme of my thesis was to use this technology to develop aptamers able to bind to
validated therapeutic targets, specifically the Tumour Necrosis Factor alpha (TNFα) and
Carcinoembryonic Antigen (CEA), and block their biological functions. As well, I
investigated the use of CEA and MUC1 binding aptamers as targeting agents to guide
and detect the delivery of contrast agent-loaded liposomes in tumour-bearing mice using
computed tomography (CT) imaging. Aptamer selections successfully identified a 25-
base aptamer (VR11) that can bind with high affinity and specificity to TNFα. VR11
blocked TNFα signaling, prevented apoptosis, reduced nitric oxide (NO) production in
cultured cells and was non-immunogenic when injected into C57BL/6 mice. As well,
aptamers were derived to the IgV-like N-domain of CEA. Two DNA aptamers were
isolated containing a 40-base variable region, N54 and N56, bearing anti- CEA
homotypic adhesive properties. These aptamers are not cytotoxic or immunogenic and

III
are able to prevent CEA-mediated homotypic and heterotypic cell adhesion events. In
addition, the pretreatment of murine cancer cells expressing CEA with these aptamers
prior to their intraperitoneal injection into C57BL/6 mice resulted in the prevention of
tumour foci formation. Finally, the in vivo targeting of nanoparticles such as pegylated
liposomes to tumour cells was enhanced by introducing tumour marker-specific DNA
aptamers on their surface. The CEA-specific aptamer N54 and a 40-base second
generation aptamer MUC1-VR1 that recognizes the tumour-associated mucin MUC1
were incorporated into liposomes containing the CT contrast agent Omnipaque350™ and
Cy5 to characterize their binding to CEA and MUC1-expressing cancer cells in vitro.
Pharmacokinetic studies also revealed that the incorporation of these aptamers into
pegylated liposomes significantly lenghthened their circulation half-lives to values that
parrallel that of untargeted pegylated liposomes.

IV
ACKNOWLEDGEMENTS
I would like to express my sincerest and deepest appreciation to Dr. Jean Gariépy for the
opportunity to work and study in his laboratory. I am particularly grateful for his
unwavering encouragement, support direction and persistent guidance over the 5 past
years. Without his motivation, creativity and assistance my reports, abstracts, patent
applications, manuscripts and this dissertation would not have been possible.
I would like to thank my committee members, Dr. Christine Allen, Dr. Robert
Macgregor, and Dr. Sachdev Sidhu for their time and expertise in supporting my graduate
studies. Their engagement and advice contributed greatly to the success I achieved
during my studies. I would like to acknowledge Dr. Chrstine Allen and former student
Mike Dunne and Huang Huang for their assistance in our CIHR Biotherapeutics
collaboration, Dr. Sachdev Sidhu and his lab member Nick Jarvik for their help with
surface plasmon resonance experiments as well as Dr. Robert Macgregor and Yuen Shek
for their advice and technical assistance for circular dichroism experiments.
In addition I would extend my greatest appreciation for my current lab members:
Arshiya, Aws, Amirul, Cailtin, Eric, Linda, Marzena, Nick, Nora and former lab
members: Andrew and Wei. A special thanks to Dr. Aws Abdul-Wahid for challenging me
and engaging in critical discussions that was instrumental in the development of CEA
aptamers. I wish everyone the best as they continue to pursue their future studies and
career paths.
This research would not have been possible without the financial assistance from
the Department of Pharmaceutical Sciences in the form of awards, bursaries and
scholarships, particularly the studentship award from the CIHR strategic training

V
program in biological therapeutics.
Lastly, I would like to thank Sarah and my family for their constant love and
support. I could not have pursued and accomplished my goals and attained my education
without them.

VI
Table of Contents
ABSTRACT................................................................................................................... II
ACKNOWLEDGEMENTS ........................................................................................ IV
LIST OF ABBREVIATIONS ...................................................................................... IX
LIST OF TABLES .................................................................................................... XIII
LIST OF FIGURES .................................................................................................. XIV
Chapter 1 : Introduction ....................................................................................................... 1
1.1. Aptamers as therapeutics ................................................................................... 2
1.1.1. The SELEX procedure: A rapid strategy to identify short single strand
synthetic oligonucleotides (aptamers) that recognize specific targets ......................... 3
1.1.2. Cell-SELEX ................................................................................................... 7
1.1.3. RNA versus DNA aptamers .......................................................................... 8
1.1. Aptamers as Inhibitors ....................................................................................... 9
1.2.1. The role of TNFα in disease progression ..................................................... 12
1.2.2. Inflammation and TNFα .............................................................................. 12
1.2.3. TNFα as a therapeutic target ........................................................................ 15
1.2. Aptamers can serve as intracellular delivery vehicles via their binding to
known cancer-associated surface antigens ................................................................ 15
1.3.1. CD33 ............................................................................................................ 18
1.3.2. Carcinoembryonic antigen (CEA) ............................................................... 21
1.3.3. CA15-3 antigen, MUC1 peptides and Tn antigens ...................................... 24
1.3. Aptamer-guided delivery of payloads into cancer cells ................................. 26
1.4.1. Aptamer-drug conjugates............................................................................. 28
1.4.2. Aptamer-protein conjugates......................................................................... 28
1.4.3. Aptamer-radionuclide conjugates ................................................................ 29
1.4.4. Aptamer-nanostructure conjugates .............................................................. 30
1.4.5. Challenges facing the in vivo use of aptamers ............................................ 32
1.4. Thesis Hypotheses ............................................................................................. 36
1.5. Specific Aims ...................................................................................................... 37
1.6. Chapter Overviews ........................................................................................... 38
Chapter 2 ............................................................................................................................ 39
2.1. Abstract .............................................................................................................. 40
2.2. Introduction ....................................................................................................... 41
2.1. Materials and Methods ..................................................................................... 43
2.3.1. Expression and purification of human TNFα. ............................................. 43
2.3.2. Aptamer selection screens. .......................................................................... 44
2.3.3. Aptamer-based enzyme linked binding assay.............................................. 45
2.3.4. NF-κB luciferase reporter assay. ................................................................. 45
2.3.5. Inhibition of TNFα induced cytotoxicity. .................................................... 46

VII
2.3.6. Surface plasmon resonance.......................................................................... 46
2.3.7. Determination of aptamer concentration and Circular Dichroism
Spectroscopy. ............................................................................................................. 47
2.3.8. Inhibition of TNFα induced NO production in macrophages. ..................... 48
2.3.9. Analysis of VR11 ability to activate an innate immune response. .............. 48
2.4. Results and Discussion ...................................................................................... 49
2.4.1. Identification of DNA aptamers directed at TNFα. ..................................... 49
2.4.2. DNA aptamer VR11 inhibits the binding of TNFα to its receptor. ............. 54
2.4.3. Structural features of DNA aptamer VR11. ................................................ 60
2.4.4. Immunogenicity of DNA aptamer VR11. ................................................... 64
Chapter 3 ............................................................................................................................ 66
Blocking the attachment of cancer cells in vivo with DNA aptamers displaying anti-
adhesive properties against the carcinoembryonic antigen ......................................... 66
3.0. Abstract .............................................................................................................. 67
3.1. Introduction ....................................................................................................... 68
3.2. Materials and Methods ..................................................................................... 71
3.2.1. Generation of recombinant CEA modules ................................................... 71
3.2.2. Aptamer selection and cloning .................................................................... 71
3.2.3. Aptamer-based inhibition of CEA homotypic interactions ......................... 72
3.2.4. Cells lines and growth conditions ................................................................ 73
3.2.5. Aptamer-based inhibition of homophilic cellular adhesion......................... 73
3.2.6. Aptamer binding to CEA on cells as measured by flow cytometry ............ 74
3.2.7. Inhibition of MC38.CEA tumour implantation ........................................... 75
3.2.8. Aptamer cytotoxicity assay.......................................................................... 75
3.2.9. Analysis of aptamer-based innate immune responses ................................. 76
3.2.10. Statistical methods and data analysis ........................................................... 76
3.3. Results ................................................................................................................ 77
3.3.1. Generation of DNA aptamers displaying inhibitory properties of CEA-
dependent homotypic adhesions ................................................................................. 77
3.3.2. Aptamers N54 and N56 inhibit homophilic cellular adhesion .................... 79
3.3.3. Aptamers N54 and N56 specifically recognize the N domain of CEA ....... 83
3.3.4. Addition of aptamers N54 and N56 to MC38.CEA cells reduces tumour
implantation in vivo ..................................................................................... 86
3.3.5. Aptamers specific to the CEA N domain are not cytotoxic and aptamer
N54 does not activate an innate immune response ...................................... 89
3.4. Discussion ........................................................................................................... 91
Chapter 4 ............................................................................................................................ 97
4.0. Abstract .............................................................................................................. 98
4.1. Introduction ....................................................................................................... 99
4.2. Materials and Methods ................................................................................... 103
4.2.1. MUC1 expression, purification and glycosylation .................................... 103
4.2.2. Identification of MUC1 binding aptamers using the SELEX process ....... 104
4.2.3. MUC1 Aptamer binding and internalization ............................................. 105
4.2.4. Binding kinetics of MUC1 aptamers determined by surface plasmon
resonance ................................................................................................... 105

VIII
4.2.5. Synthesis of Aptamer-PEG2000-DSPE conjugates ..................................... 106
4.2.6. Liposome preparation ................................................................................ 107
4.2.7. Efficiency of aptamer-DSPE-PEG insertion into liposomes ..................... 108
4.2.8. Aptamer-liposome characterization in vitro .............................................. 108
4.2.9. Physio-chemical characterization of aptamer-liposome formulations ...... 109
4.2.10. Aptamer-liposome pharmacokinetic profile .............................................. 110
4.2.11. Statistical methods and data analysis ......................................................... 110
4.3. Results .............................................................................................................. 111
4.3.1. Identification and characterization of MUC1-VR1, a MUC1-binding
aptamer ...................................................................................................... 111
4.3.2. Optimization of MUC1-VR1 and N54 aptamer-targeted liposomes ........ 114
4.3.3. Aptamer-Liposome internalization in MDA-MB-231 cells ...................... 117
4.3.4. Aptamer-Liposome physicochemical properties and stability in vivo ....... 119
4.4. Discussion ......................................................................................................... 120
Chapter 5 .......................................................................................................................... 124
5.1. Thesis Conclusions .......................................................................................... 125
5.2. Future Directions ............................................................................................ 127
5.2.1. Modification of aptamer VR11 for in vivo ................................................ 127
5.2.2. Determine the ability of aptamer N54 as a detection and targeting agent . 128
5.2.3. Determine the ability of MUC1 and CEA aptamer to target liposomes .... 128
References ................................................................................................................... 142

IX
LIST OF ABBREVIATIONS
99mTC Technetium-99m
ADCC antibody-dependent cell cytotoxicity
ALL acute lymphoblastic leukemia
AMD age-related macular degeneration
AML acute myeloid leukemia
BCL2 B-cell leukemia/lymphoma 2
BSA bovine serum albumin
C6 six carbon spacer
cApt control aptamer
CD circular dichroism
CD50 median curative dose
CDC complement-dependent cytotoxicity
CEA carcinoembryonic antigen
CEACAM carcinoembryonic antigen cell adhesion molecule
CH cholesterol
CI cell index
CT computed tomography
CTLA-4 cytotoxic T-cell antigen-4
Cy5 cyanine-5
DLS dynamic light scattering
DMARDs disease-modifying antirheumatic drugs

X
DMEM dulbecco’s modified eagle’s medium
DMSO Dimethyl sulfoxide
DPPC dipalmitoylphosphatidylcholine
DR5 death receptor 5
DSPE distearoyl-phosphatidyl ethanolamine
EDTA Ethylenediaminetetraacetic acid
ELISA enzyme-linked immunosorbent assay
EPR enhanced permeability and retention effect
FBS fetal bovine serum
FDA food and drug administration
FITC fluorescein isothiocyanate
FL full length
GPI glycosylphosphatidyl inositol
H3PO4 phosphoric acid
HBS hank’s buffered saline
HER2 human epidermal growth factor receptor 2
HER3 human epidermal growth factor receptor 3
hNE human neutrophil elastase
HPLC High performance liquid chromatography
HRP horseradish peroxidase
i.p. intraperitoneal
IFN-γ interferon-gamma
IgG immunoglobulin G
IgV Immunoglobulin variable

XI
IL-8 Interleukin 8
IPTG isopropyl β-D-thiogalactopyranoside
ka association rate
KCl potassium chloride
kD kilo dalton
KD dissociation constant
kd dissociation rate
KOH potassium hydroxide
LNA locked nucleic acid
LPS lipopolysaccharides
mAb monoclonal antibody
MAG2 mercapto-acetyl diglycine
MFI mean fluorescence intensity
MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenylttetrazolium bromide
MUC1 mucin 1
MWCO molecular weight cut off
NaCl Sodium chloride
NaOH sodium hydroxide
NF-κB nuclear factor kappa-light-chain-enhancer of activated B cells
Ni-NTA nickel-nitrilotriacetic acid
NO nitric oxide
NO2- nitrite ion
ODN oligodeoxynucleotide
OH hydroxide

XII
PBS phosphate buffered saline
PCR polymerase chain reaction
PEG polyethylene glycol
PLGA poly(lactic-co-glycolic acid)
PLK1 polo-like kinase 1
PSMA prostate-specific membrane antigen
PTK7 protein tyrosine kinase 7
RNA ribonucleic acid
SDS-PAGE sodium dodecyl sulfate polyacrylamide gel electrophoresis
SELEX systematic evolution of ligands by exponential enrichment
SPR surface plasmon resonance
SRB sulphorhodamine B
ssDNA single stranded deoxyribonucleic acid
TLR9 toll-like receptor 9
TMB 3,3’,5,5’-tetramethylbenzidine
TNFα tumour necrosis factor alpha
TNFβ tumour necrosis factor beta
VEGF vascular endothelial growth factor
VEGF vascular endothelial growth factor
VR variable region
α5β1 alpha-5-beta-1
β-gal beta-galactosidase

XIII
LIST OF TABLES
Table 1.1. Aptamers to targets of therapeutics interest ................................................... 10
Table 1.2. Advantages and limitations of aptamers versus antibodies. ............................ 11
Table 1.3. Examples of aptamers and their cargoes directed at internalized surface
markers on cancer cells ..................................................................................................... 17
Table 2.1. List of TNFα specific DNA aptamer sequences identified from the SELEX
screen as well as control aptamer [cApt(VR) and cApt(FL)] sequences used in the
present study. .................................................................................................................... 51
Table 4.1. Pharmacokinetics of different Omnipaque encapsulated non-targeted and
aptamer targeted liposome preparations in CD1 mice (n=5 per group). ........................ 119

XIV
LIST OF FIGURES
Figure 1.1. Isolation of aptamers using the SELEX procedure.. ........................................ 4
Figure 1.2. Overlapped crystal structures of a RNA and a DNA aptamer bound to the
protein thrombin.. ............................................................................................................... 6
Figure 1.3. Crystal structure of TNF alpha trimer created from the Protein Data Bank.. 14
Figure 1.4. A CD33-specific DNA aptamer binds to and is internalized by CD33+
myeloid leukemia cell lines.. ............................................................................................ 20
Figure 1.5. A CEA-specific DNA aptamer binds to and is imported into colon carcinoma
cells expressing human CEA.. .......................................................................................... 23
Figure 1.6. Proposed mechanisms of cellular entry and recycling of DNA aptamers
directed at aberrantly glycosylated mucin MUC1 present on the surface of epithelial
cancer cells........................................................................................................................ 25
Figure 1.7. Possible endocytic pathways taken by aptamer-cargoes.. .............................. 27
Figure 2.1. Binding of selected full-length (FL) and variable region (VR) DNA aptamers
to TNF and NfκB assays identify FL11 and VR11 as TNF inhibitors. ....................... 53
Figure 2.2. Aptamer VR11 inhibits TNF-induced cytotoxicity in murine fibroblasts. .. 55
Figure 2.3. Binding kinetics of TNF to aptamers VR11 and VR20 as determined by
surface plasmon resonance. .............................................................................................. 57
Figure 2.4. Inhibition of TNF-induced NO2- production in macrophages. ................... 59
Figure 2.5. Structural analysis of aptamers VR11 and VR20 by Circular Dichroism. ..... 63
Figure 2.6. Aptamer VR11 does not cause an innate immune response in vivo. ............. 65
Figure 3.1. Aptamers selected to bind to the CEA IgV-like N domain inhibit homotypic
adhesion events. ................................................................................................................ 78
Figure 3.2. Addition of aptamers specific to the CEA N domain inhibits homophilic
cellular adhesion. .............................................................................................................. 80
Figure 3.3. Minimal regions required for aptamer inhibition of homophilic cellular
adhesion. ........................................................................................................................... 82
Figure 3.4. Alignment of the IgV-like N-domains of CEACAM family members. ......... 84
Figure 3.5. Aptamer binding to CEA+ and CEA- cells using Cy5 labelled aptamers by
flow cytometry. ................................................................................................................ 85
Figure 3.6. Pre-treatment of CEA-expressing MC38.CEA cancer cells with CEA-specific

XV
aptamers reduces tumour implantation in vivo. ............................................................... 88
Figure 3.7. Aptamer N54 is noncytotoxic and does not activate innate immune responses.
. ......................................................................................................................................... 90
Figure 4.1. Proposed action of aptamer targeted liposomes accumulating at the site of the
tumour by virtue of the Enhanced permeability and Retention (EPR) effect and
internalzing into cells. ..................................................................................................... 102
Figure 4.2. Identification of MUC1-specific DNA aptamers and the ability of MUC1+
MDA-MB-231 cells to internalize FITC-labeled aptamer MUC1-VR1. ..................... 1133
Figure 4.3. Liposome internalization into CEA+/MUC1+ MDA-MB-231 cells is
dependent on the number of DNA aptamers present on the surface of liposomes. ........ 116
Figure 4.4. Binding and internalization of targeted liposome formulations in vitro. ..... 118

1
Chapter 1 : Introduction
A version of this chapter has been previously published:
Delivering cargoes into cancer cells using DNA aptamers targeting internalized
surface portals (Biochim Biophys Acta. 2010 Dec;1798(12):2190-200)
Erik W. Orava, Nenad Cicmil and Jean Gariépy
Author Contributions:
Erik Orava and Dr. Jean Gariépy wrote the review
Erik Orava contributed to Tables 1.3 and Figures 1.4.,1.5. and 1.6.
Nenad Cicmil contributed to Figures 1.2., 1.3. and 1.7.

2
1.1. Aptamers as therapeutics
Many classes of oligonucleotides such as siRNAs, microRNAs and antisense
oligonucleotides represent potential therapeutic agents in view of their ability to
selectively block the expression or transcription of genes and mRNAs inside diseased
cells. Unfortunately, their anionic character makes them cell-impermeant and thus will
not reach their intracellular targets unless they are conjugated or complexed to a cell-
penetrating peptide, a polymeric vector, a protein ligand (hormones, cytokines,
monoclonal antibodies), a nanoparticle or a liposome favoring their import into cells or
are delivered using a viral vector. A more recent and potentially simpler solution to this
challenge is to derive short synthetic oligonucleotides known as DNA and RNA aptamers
which themselves specifically bind to internalized surface markers (cellular portals) and
thus can act as delivery vehicles for therapeutic oligonucleotides and other therapeutic
cargoes. As well, these aptamers can be generated to bind to validated therapeutic targets
and act as antagonists (Keefe et al., 2010). Although in its infancy, the field of aptamer
development for therapeutic and diagnostic purposes is rapidly growing and their utility
is becoming even more evident (Soontornworajit and Wang, 2011; Yang et al., 2011)

3
1.1.1. The SELEX procedure: A rapid strategy to identify short single strand
synthetic oligonucleotides (aptamers) that recognize specific targets
Cancer cells typically harbor multiple oncogenic mutations leading to the aberrant
display and/or overexpression of molecular signatures on their surface. Classical
approaches to target such signatures have made use of peptides, proteins and mainly
antibodies. However, recent studies suggest that oligonucleotides known as aptamers can
be utilized in the same capacity. Aptamers are short single-stranded nucleic acid
oligomers (ssDNA or RNA) that can form specific and complex three-dimensional
structures which can bind with high affinity to specific targets. The term ‘aptamer’ is
derived from the Latin word aptus meaning “to fit” (Ellington and Szostak, 1990). Two
groups reported a PCR-based strategy termed SELEX (an acronym for Systematic
Evolution of Ligands by EXponential enrichment; (Tuerk and Gold, 1990)) to derive
aptamers that specifically recognized targets ranging from small molecules to large
proteins (Figure 1.1). SELEX is an iterative panning procedure where combinatorial
libraries composed of a random oligonucleotide element flanked by constant primer
regions are allowed to bind to an immobilized target. The bound oligonucleotides are
then recovered and amplified by PCR to generate a sub-library of aptamers able to
recognize a given target. The binding/amplification cycle is then repeated several times
on enriched pools of aptamers until one recovers ssDNA or RNA aptamers displaying
Kds in the nanomolar to picomolar range for their respective targets. So far, thrombin
represents the only protein that does not normally bind nucleic acids and for which
crystals structures of its complexes with aptamers have been obtained (Long et al., 2008;
Padmanabhan et al., 1993). Interestingly, the two available structures (thrombin
complexed to a DNA and a RNA aptamer) indicate that each aptamer binds to a distinct
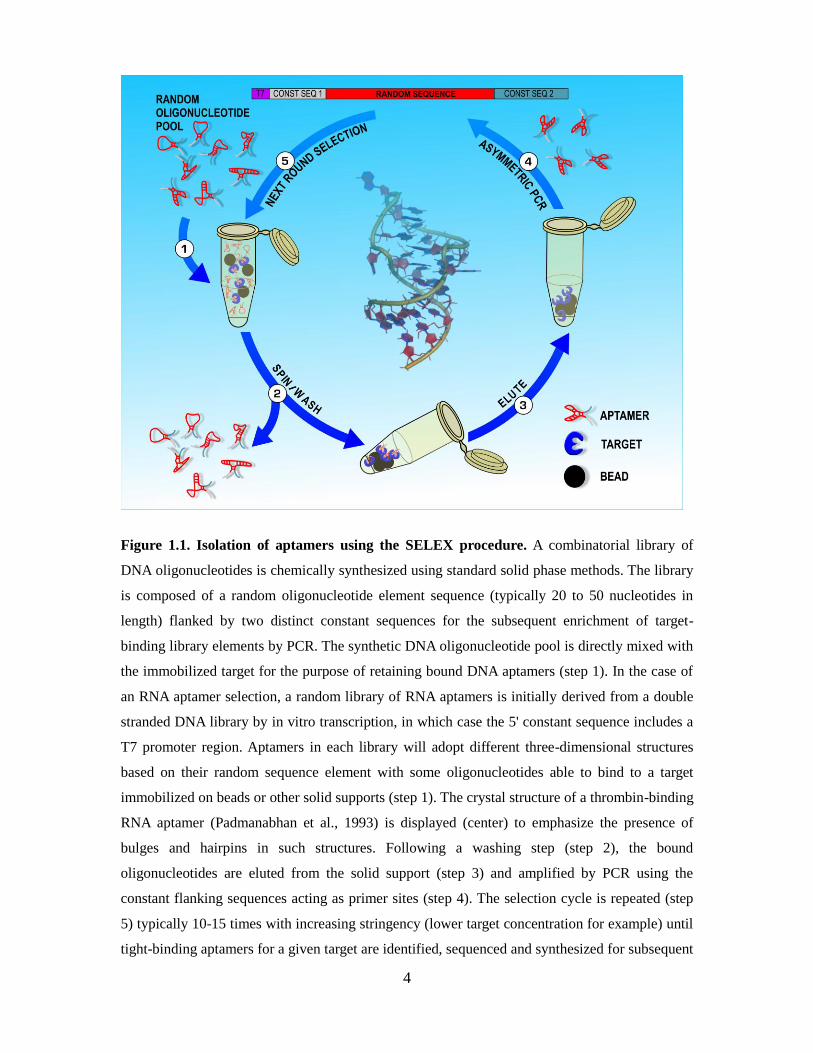
4
Figure 1.1. Isolation of aptamers using the SELEX procedure. A combinatorial library of
DNA oligonucleotides is chemically synthesized using standard solid phase methods. The library
is composed of a random oligonucleotide element sequence (typically 20 to 50 nucleotides in
length) flanked by two distinct constant sequences for the subsequent enrichment of target-
binding library elements by PCR. The synthetic DNA oligonucleotide pool is directly mixed with
the immobilized target for the purpose of retaining bound DNA aptamers (step 1). In the case of
an RNA aptamer selection, a random library of RNA aptamers is initially derived from a double
stranded DNA library by in vitro transcription, in which case the 5' constant sequence includes a
T7 promoter region. Aptamers in each library will adopt different three-dimensional structures
based on their random sequence element with some oligonucleotides able to bind to a target
immobilized on beads or other solid supports (step 1). The crystal structure of a thrombin-binding
RNA aptamer (Padmanabhan et al., 1993) is displayed (center) to emphasize the presence of
bulges and hairpins in such structures. Following a washing step (step 2), the bound
oligonucleotides are eluted from the solid support (step 3) and amplified by PCR using the
constant flanking sequences acting as primer sites (step 4). The selection cycle is repeated (step
5) typically 10-15 times with increasing stringency (lower target concentration for example) until
tight-binding aptamers for a given target are identified, sequenced and synthesized for subsequent

5
analyses.
region on the protein located on opposite sides of each other on the molecule (Figure
1.2). This finding suggests that the process of identifying aptamers using the SELEX
procedure does not necessarily favor a unique epitope on a given target. Specifically, the
DNA aptamer was shown to contact a region of thrombin that normally binds to
fibrinogen (exosite 1), while the RNA aptamer binds to a domain associated with heparin
binding (exosite 2)(Sheehan and Sadler, 1994). Interactions between these aptamers and
thrombin are mostly electrostatic since both of the exosites are positively charged
interfaces (Bode et al., 1992; Padmanabhan et al., 1993; Sheehan and Sadler, 1994).
These structural features highlight the fact that aptamers may recognize these targets
mostly through electrostatic interactions in contrast to dominant hydrophobic interactions
typically observed in protein-protein interactions. It also suggests that the number of
surface elements on a given target that could serve as recognized interfaces for aptamers
is finite and potentially predictable.
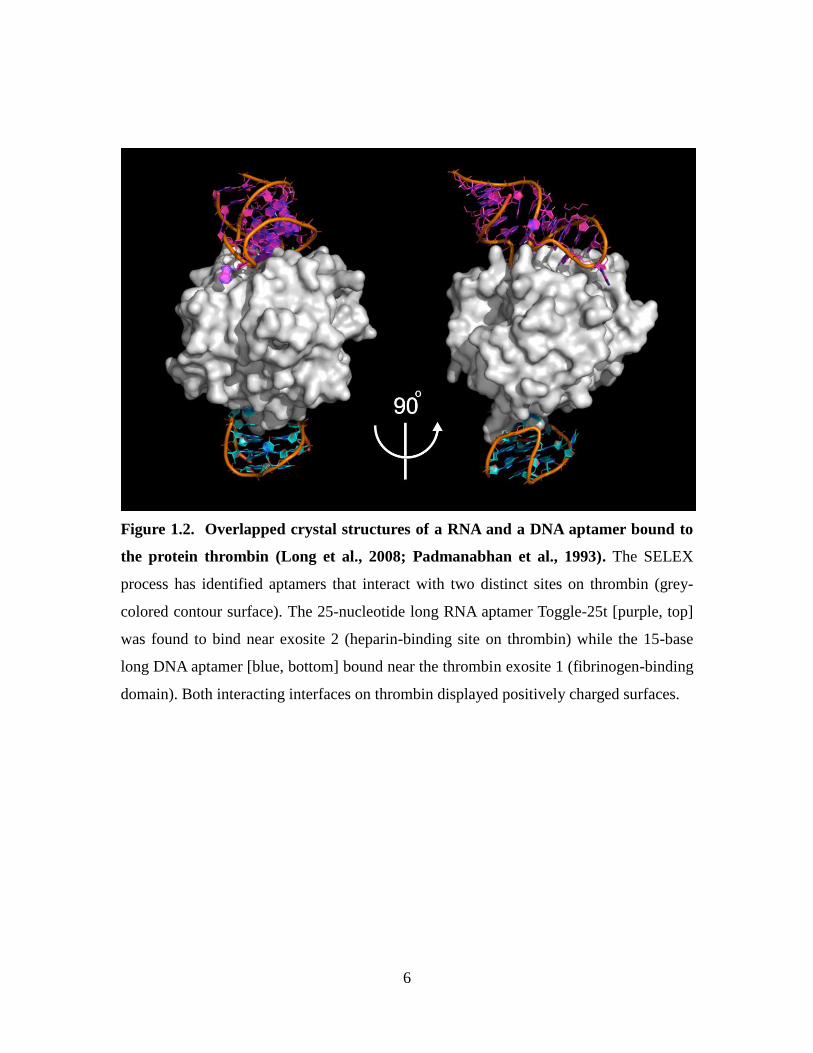
6
Figure 1.2. Overlapped crystal structures of a RNA and a DNA aptamer bound to
the protein thrombin (Long et al., 2008; Padmanabhan et al., 1993). The SELEX
process has identified aptamers that interact with two distinct sites on thrombin (grey-
colored contour surface). The 25-nucleotide long RNA aptamer Toggle-25t [purple, top]
was found to bind near exosite 2 (heparin-binding site on thrombin) while the 15-base
long DNA aptamer [blue, bottom] bound near the thrombin exosite 1 (fibrinogen-binding
domain). Both interacting interfaces on thrombin displayed positively charged surfaces.

7
1.1.2. Cell-SELEX
Aptamers can be selected against recombinant forms of surface markers and still
recognize the native forms on cells. Most notably are the aptamers to the MUC1 peptide,
the extracellular domain of prostate-specific membrane antigen (PSMA), cell adhesion
molecule P-selectin and protein tyrosine phosphotase 1B (PTP1B) (Ferreira et al., 2009;
Gutsaeva et al., 2011; Lupold et al., 2002; Townshend et al., 2010). However, the
recombinant form of proteins can be different in a physiological context resulting in a
loss of aptamer binding. A case in point is the RNA aptamers to the EGFRvIII
ectodomain which bound with high affinity in vitro however showed no binding to this
target displayed on cells (Liu et al., 2009b). A process termed Cell-SELEX (also called
Whole-Cell SELEX and cell-based SELEX) was recently devised to prevent the selection
of aptamers to protein targets lacking their native conformation in vitro (Ohuchi et al.,
2005; Zhang et al., 2010).
Established SELEX techniques required the immobilization of a protein or target
on an affinity sorbent such as beads, resins, membranes, columns or plates. Washing
steps, altering buffer solutions and reducing the amount of target represent SELEX
stringencies that can remove DNA aptamer species that non-specifically associate with a
given target from the selected pool of aptamers. In the Cell-SELEX procedure, non-
specific species are removed by including a counter selection stage using cells that do not
express any of the markers of interest. This step allows for a pool of aptamers to be
generated that most likely recognize unique surface markers on the cell line of interest
and possibly unkown or uncharacterized surface markers. A case in point is Cell-SELEX
derived aptamers that were able to distinguish between rat glioblastoma and microglial
cells (Blank et al., 2001). This approach may prove useful to derive aptamers as

8
biosensors to detect and discern populations of cells as well as deliver therapeutic cargos
into cells.
1.1.3. RNA versus DNA aptamers
A large number of RNA aptamers have now been reported against different targets. The
versatility of RNA molecules as functional ligands is well documented in regards to the
frequent occurrence of modified nucleotides within their structure, their base pairing
properties and their tendency to form intricate three-dimensional structures (Maas, 2011).
For instance, all natural riboswitches (which bind to small molecules) are RNA
molecules (Wakeman et al., 2007). The derivation and use of RNA aptamers does present
some important practical challenges. For instance, the SELEX process requires the
synthesis of random oligonucleotide libraries and the chemical synthesis of random RNA
oligonucleotide pools remains expensive. Therefore, an in vitro transcription step is
introduced in the SELEX procedure to obtain the initial RNA pool. Secondly, RNA
oligonucleotides are more susceptible to hydrolysis than their DNA counterparts and thus
their manipulation requires RNAse-free conditions.
DNA tertiary structures have been observed in nature (Chou et al., 2003). These
structures, rich in guanine, are found in telomeres and promoter regions (Neidle and
Parkinson, 2003; Siddiqui-Jain et al., 2002). Guanine-rich sequences form various G-
quadruplexes that appear to be major structural elements found in DNA aptamers as
exemplified in the thrombin DNA aptamer (Figure 1.2). Examples of DNA aptamers
have been reported and include an anti-HIV aptamer (Phan et al., 2005) and the anti-
nucleolin aptamer AS1411(Teng et al., 2007). Catalytically-active DNA aptamers have
also been derived using the SELEX approach (Liu et al., 2009a; Silverman, 2009). The

9
selection procedure for DNA aptamers is simpler than for RNA aptamers. Specifically,
inexpensive pools (libraries) of DNA oligonucleotides can be chemically synthesized and
contain only single stranded sequences as opposed to the initial double stranded pool of
DNA sequences required for the in vitro transcription step used for RNA-based aptamer
selection. Furthermore, reverse transcription is not required and an asymmetric PCR step
is sufficient to recover the sub-library of ligand-binding aptamers needed to proceed to
the next round of selection. In summary, the advantages of DNA aptamers stem from the
simpler enrichment procedure involved and the lower cost and stability of the final
aptamers while the benefit of selecting for RNA aptamers is the higher level of structural
diversity possible with RNA templates.
1.1. Aptamers as Inhibitors
In theory, aptamers that function as inhibitors in blocking a given protein from
interacting with another protein, receptor or ligand can be identified for the purpose of
treating any disease. Thus, disease-promoting or persisting protein-protein and protein-
ligand interactions can be disrupted (White et al., 2000). Most of the aptamers already
developed as inhibitors tend to act as antagonists in blocking proteins to their receptors.
Although there have been reports of aptamers that can acts as agonists, namely to HER3
and isoleucyl tRNA synthetase, these aptamers have not yet proved to have any clinical
or therapeutic advantage (Chen et al., 2003; Hale and Schimmel, 1996). Since the
invention of the SELEX process in the early 1990’s, researchers have been able to
identify aptamers that bind to currently validated therapeutic targets (Table 1.1). As well,
modifications of the SELEX approach have been reported that allow for the selection of
aptamers with higher affinities (by decreasing off-rates) which would allow the aptamers
10
to be bound to their targers and in the case of inhibitors, provide a prolonged blockage
(Keefe and Cload, 2008). Aptamers also possess considerable advantages to their
antibody counterparts with some of their limitations being overcome such as synthesis
costs and the optimization of protocols for their chemical modifications (Table 1.2).
Table 1.1. Aptamers to targets of therapeutics interest (Keefe et al., 2010).

11
Table 1.2. Advantages and limitations of aptamers versus antibodies (Keefe et al., 2010).

12
1.2.1. The role of TNFα in disease progression
Despite the importance of immune responses in controlling infections, it is now clear that
uncontrolled reactions for such responses are intimately associated with degenerative
diseases such as atherosclerosis, arthritis, encephalitis, and tumours (Feldmann et al.,
2005). The primary pro-inflammatory cytokine, tumour necrosis factor alpha (TNFα)
plays a critical regulatory role in enhancing these responses (Borish and Steinke, 2003)
In the past decade, disease modifying antirheumatic drugs (DMARDs) that target
underlying immune responses processes have improved treatment outcomes in patients
with chronic immune response disorders. Anti-TNF antibody-based therapies represent
the dominant categories of DMARDs (Feldmann et al., 1998; Maini et al., 1993). These
protein therapeutics remain expensive to produce and ~40% of patients still display
moderate to high levels of disease activity after treatment, suggesting a substantial need
for improved therapies (Mierau et al., 2007).
1.2.2. Inflammation and TNFα
While the cause of many chronic inflammatory diseases remains unknown, most are
characterized by dysregulation of cytokine networks, which often leads to the
overproduction of proinflammatory cytokines, causing a perturbation in the equilibrium
between pro- and anti-inflammatory cytokines (Feldmann et al., 2005). At the apex of
this cytokine network is the Tumour necrosis factor alpha (TNFα), which acts as a
primary trigger for the inflammatory response by promoting the synthesis of other
proinflammatory cytokines (Borish and Steinke, 2003; van den Berg, 2001). TNFα was
first identified for its ability to induce rapid haemorrhagic necrosis of experimental
cancers (Carswell et al., 1975). TNFα is a type 2 transmembrane protein with an

13
intracellular amino terminus. It is produced by many cell types, including macrophages,
monocytes, lymphocytes, keratinocytes and fibroblasts, in response to inflammation,
infection, injury and other environmental challenges. It is synthesized as a 26-kD
membrane bound protein (pro-TNFα) that is cleaved to release a soluble 17-kD TNFα
molecule. TNFα has the ability to signal as a membrane bound protein and the soluble
form is only active as a non-covalently associated homotrimer (Figure 1.3) (Hehlgans
and Pfeffer, 2005; Locksley et al., 2001). Sufficient levels of TNFα as well as other
mediators are critical for sustaining normal immune responses (Ehlers, 2005; Furst et al.,
2006; Wallis and Ehlers, 2005). TNFα can initiate host defence at the site of a local
injury but its sustained presence can also cause acute and chronic tissue damage (Beutler,
1999). Elevated levels of TNFα have been detected in patients with rheumatoid arthritis
(RA), psoriasis and Crohn’s disease (Komatsu et al., 2001; Mussi et al., 1997; Robak et
al., 1998). The evidence for the effects of sustained production of TNFα in inflammatory
and autoimmune diseases sparked investigations to develop TNFα antagonists as anti
inflammatory agents (Brennan et al., 1989; Feldmann et al., 1992).
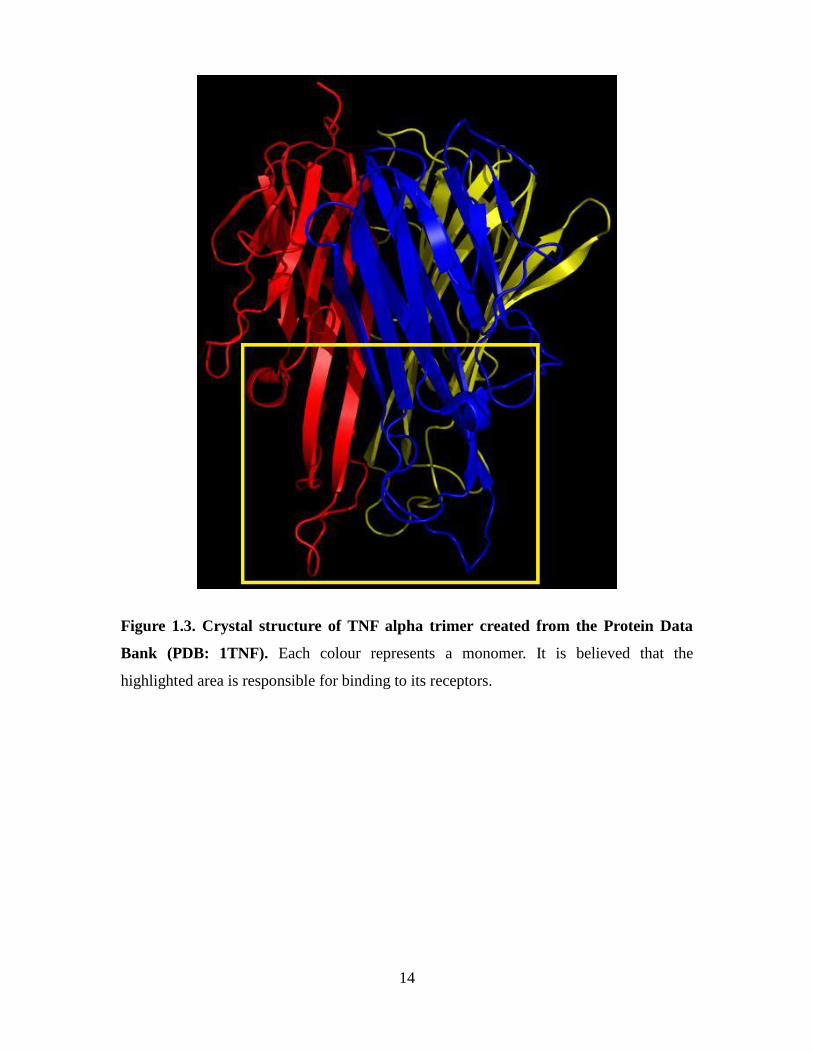
14
Figure 1.3. Crystal structure of TNF alpha trimer created from the Protein Data
Bank (PDB: 1TNF). Each colour represents a monomer. It is believed that the
highlighted area is responsible for binding to its receptors.

15
1.2.3. TNFα as a therapeutic target
The need for new therapies for the treatment of inflammatory diseases has arisen during
an era where clinicians have realized that chronic inflammatory conditions are far more
ominous than was previously thought. Inhibition of TNFα alone has been shown to
downregulate the proinflammatory cascade leading to two findings: the discovery of an
inter-linkage between different cytokines and the ability of one cytokine to orchestrate
inflammation (Feldmann et al., 2001). It was realized by the mid 1990’s that inhibitors
of TNFα would be successful therapeutic agents for a range of human chronic
inflammatory diseases (Feldmann and Maini, 2008). Although pro-inflammatory
cytokines aid in protecting the host against infectious agents, anti-TNFα protein
therapeutics can reduce systemic and local inflammation in patients with rheumatoid
arthritis (RA), psoriasis and Crohn’s disease. As of 2012, the global market for anti-
TNFα protein therapeutics was valued at US$26.5 billion with Humira, Remicade and
Enbrel representing the top 3 selling drugs in the world (2012).
1.2. Aptamers can serve as intracellular delivery vehicles via their binding to
known cancer-associated surface antigens
The main purpose of this review is to highlight the potential of membrane-impermeant
oligonucleotides to serve as intracellular delivery agents if they can be engineered to
target internalized surface markers on cancer cells. The best described surface
determinant used for this purpose (Table 1.3) has been the prostate-specific membrane
antigen (PSMA), a membrane protein overexpressed on the surface of prostate cancer
cells. PSMA is internalized by such cells via clathrin-coated pits (Chang et al., 1999; Liu
et al., 1998; Lupold et al., 2002; Ross et al., 2003). From a drug delivery perspective,

16
antibody studies have shown that the rate of PSMA internalization was promoted by the
binding of an antibody to its extracellular domain (Liu et al., 1998). The PSMA antigen is
also differentially expressed on prostate cancer cells with normal prostate cells
displaying an alternatively spliced cytosolic form of the protein while malignant cells
express the full length surface protein (Su et al., 1995). The extracellular domain of
PSMA served as a target for developing the first RNA aptamers known to bind a tumour-
associated antigen (Lupold et al., 2002). The selective delivery and uptake properties of
such aptamers by prostate cancer cells led to the subsequent design of an RNA chimera
incorporating a PSMA-specific aptamer (delivery vehicle) and a therapeutic siRNA that
targets Polo-like kinase 1 (PLK1) and BCL2. This RNA aptamer-siRNA construct was
shown to cause tumour regression in a xenograft model of prostate cancer (McNamara et
al., 2006). These findings suggested that by choosing appropriate internalized surface
markers on cancer cells, one may be able to develop aptamers that can serve as both cell
targeting agents and intracellular delivery vehicles. We will now focus our discussion on
recent evidence from our laboratory suggesting that DNA aptamers can indeed be
generated against membrane-bound tumour markers that are recycled inside cells.
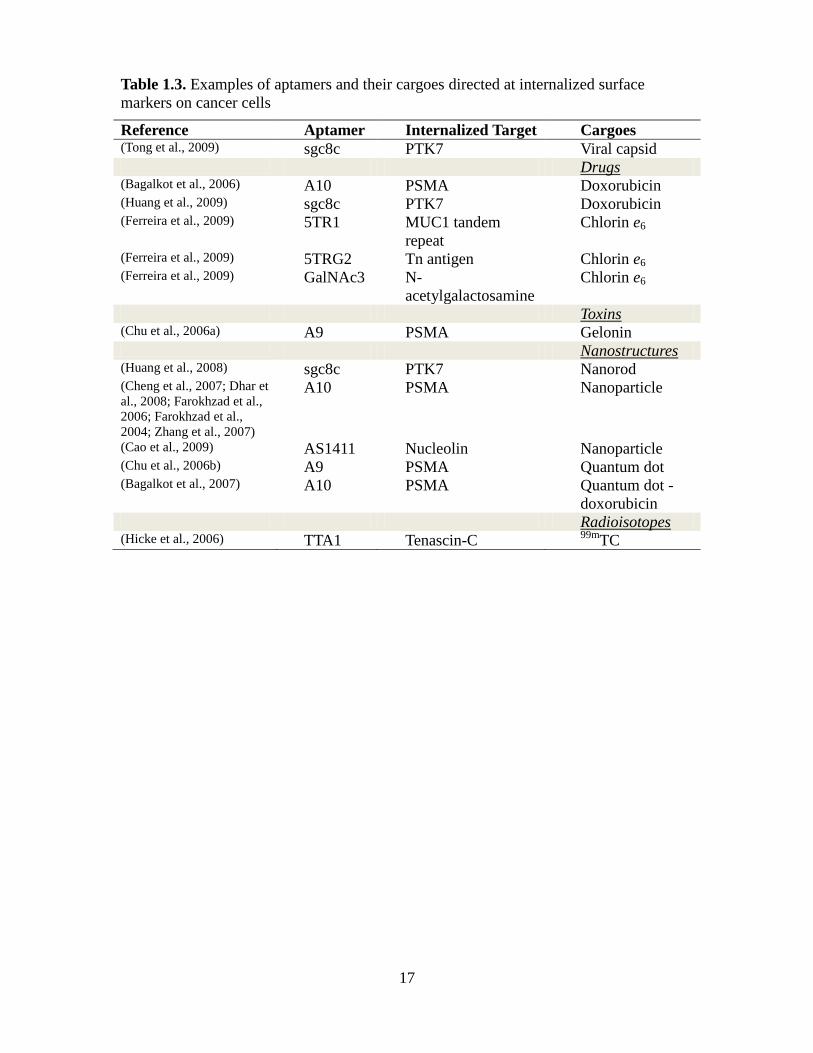
17
Table 1.3. Examples of aptamers and their cargoes directed at internalized surface
markers on cancer cells
Reference Aptamer Internalized Target Cargoes (Tong et al., 2009) sgc8c PTK7 Viral capsid Drugs (Bagalkot et al., 2006) A10 PSMA Doxorubicin (Huang et al., 2009) sgc8c PTK7 Doxorubicin (Ferreira et al., 2009) 5TR1 MUC1 tandem
repeat
Chlorin e6
(Ferreira et al., 2009) 5TRG2 Tn antigen Chlorin e6 (Ferreira et al., 2009) GalNAc3 N-
acetylgalactosamine
Chlorin e6
Toxins (Chu et al., 2006a) A9 PSMA Gelonin Nanostructures (Huang et al., 2008) sgc8c PTK7 Nanorod (Cheng et al., 2007; Dhar et
al., 2008; Farokhzad et al.,
2006; Farokhzad et al.,
2004; Zhang et al., 2007)
A10 PSMA Nanoparticle
(Cao et al., 2009) AS1411 Nucleolin Nanoparticle (Chu et al., 2006b) A9 PSMA Quantum dot (Bagalkot et al., 2007) A10 PSMA Quantum dot -
doxorubicin Radioisotopes (Hicke et al., 2006) TTA1 Tenascin-C
99mTC

18
1.3.1. CD33
The CD33 antigen is a 67-kDa type 1 transmembrane glycoprotein that belongs to the
superfamily of sialic acid-binding immunoglobulin-related lectins (siglecs; siglec-3)
(Freeman et al., 1995). CD33 is expressed on early multilineage hematopoietic
progenitors, myelomonocytic precursors, as well as more mature myeloid cells,
monocytes, macrophages and dendritic cells (Andrews et al., 1986; Andrews et al., 1983;
Griffin et al., 1984). Most adult and pediatric acute myeloid leukemia (AML) cases as
well as 15–25% of acute lymphoblastic leukemia (ALL) cases are CD33-positive
(Dinndorf et al., 1986; Putti et al., 1998; Scheinberg et al., 1989; Terstappen et al., 1992).
The presence of CD33 on AML blasts has led to the development of monoclonal
antibody treatments that have been approved for AML patients that have relapsed. One of
these anti-CD33 antibodies was conjugated to calicheamicin, a potent cytotoxic antibiotic
that cleaves double-stranded DNA at unique sites. The resulting antibody-drug conjugate
is commonly known as Gemtuzumab ozogamicin or Mylotarg (Wyeth Laboratories, PA,
USA) (Bernstein, 2000; Hamann et al., 2002). Antibody-bound CD33 has been shown to
be rapidly internalized by myeloid cells, a process that is largely modulated by its
cytoplasmic immunoreceptor tyrosine-based inhibitory motifs (van der Velden et al.,
2004; Walter et al., 2005). A 26% response rate has been observed for AML patients
treated in first relapse with gemtuzumab ozogamicin as a monotherapy with a median
disease-free-survival of 6·4 months in patients achieving complete remission
(Tsimberidou et al., 2006). Surprisingly, there is no major loss of surface CD33
expression on leukemic blasts at relapse after Gemtuzumab treatment suggesting that
alternate therapies targeting CD33-positive cell populations would be feasible and safe
(Chevallier et al., 2008; van der Velden et al., 2004). This finding would suggest the

19
development and use of smaller and less immunogenic CD33-specific aptamers carrying
less toxic cargoes than calicheamicin (hepatotoxicity) into CD33+ cells. As a proof-of-
concept, our group recently developed 25-base long synthetic DNA aptamers against a
recombinant form of CD33 to examine their ability to be internalized by myeloid
(CD33+) cell lines. As shown by flow cytometry and confocal microscopy (Figure 1.4),
one such CD33-specific Cy5-labeled DNA aptamer binds to (4°C) and is internalized
(37°C) by CD33+ cells within 90 minutes of exposing cells to this oligonucleotide. In
contrast, no binding or cellular uptake was observed for a control aptamer (25-base long
repeat of the sequence GATC) identically modified with a Cy5 probe exposed to the
same set of cell lines. Finally, neither aptamers bound to the CD33- cell line LP1. The
dissociation constant (Kd) of this monomeric CD33-specific aptamer was calculated to be
17.3 nM suggesting that it is only ~10 fold less avid for its target than modified forms of
the established bivalent-binding CD33-specific monoclonal antibody HuM195 (Chen et
al., 2006). These results suggest that DNA aptamers evolved to bind to the antigen CD33
can mimic the properties of anti-CD33 antibodies in terms of binding and being imported
into CD33-positive cells.
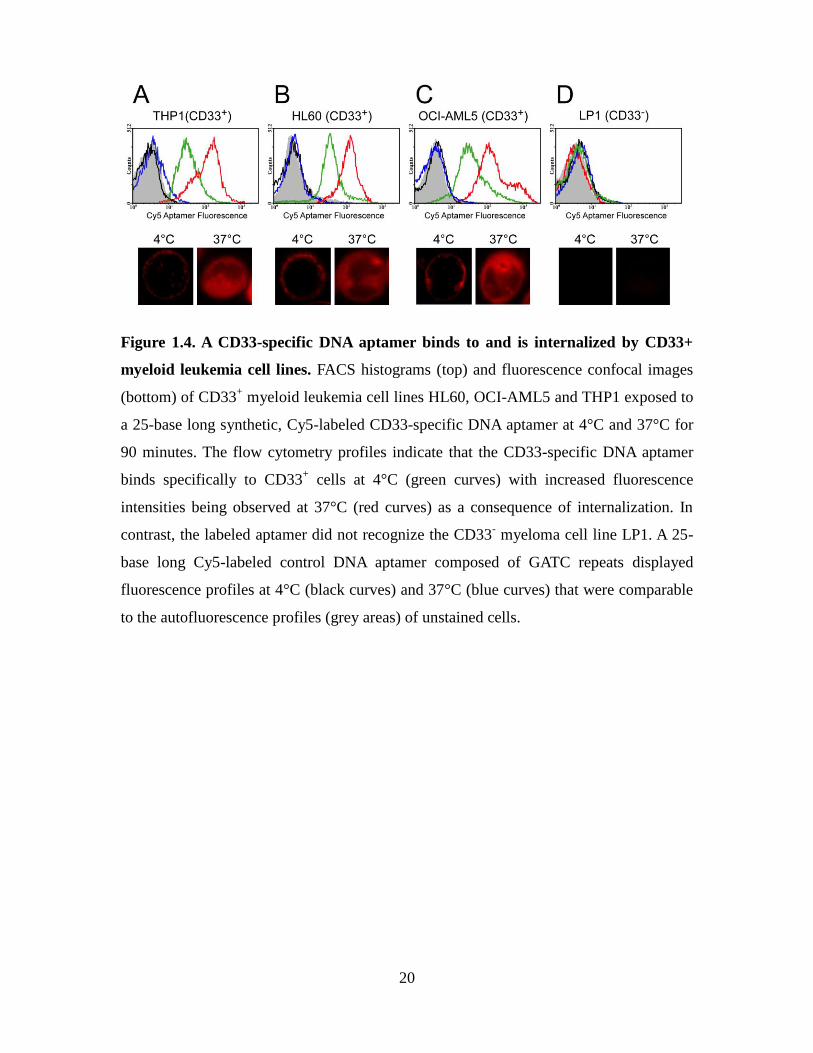
20
Figure 1.4. A CD33-specific DNA aptamer binds to and is internalized by CD33+
myeloid leukemia cell lines. FACS histograms (top) and fluorescence confocal images
(bottom) of CD33+ myeloid leukemia cell lines HL60, OCI-AML5 and THP1 exposed to
a 25-base long synthetic, Cy5-labeled CD33-specific DNA aptamer at 4°C and 37°C for
90 minutes. The flow cytometry profiles indicate that the CD33-specific DNA aptamer
binds specifically to CD33+ cells at 4°C (green curves) with increased fluorescence
intensities being observed at 37°C (red curves) as a consequence of internalization. In
contrast, the labeled aptamer did not recognize the CD33- myeloma cell line LP1. A 25-
base long Cy5-labeled control DNA aptamer composed of GATC repeats displayed
fluorescence profiles at 4°C (black curves) and 37°C (blue curves) that were comparable
to the autofluorescence profiles (grey areas) of unstained cells.

21
1.3.2. Carcinoembryonic antigen (CEA)
The human carcinoembryonic antigen (CEA) is a 180 kDa GPI-linked cell glycoprotein
and a member of an immunoglobulin cell adhesion molecule superfamily (CEACAMs).
CEA was originally identified as a surface marker on adenocarcinomas of the human
gastrointestinal tract as well as on cells of the fetal digestive system (Gold and Freedman,
1965a). Other CEACAM members have since been identified in an array of tumours
including breast, lung, pancreas, stomach, thyroid, ovaries and melanomas
(Hammarstrom, 1999). CEA is aberrantly overexpressed on the surface of colorectal
tumour cells in relation to normal colonic cells (Boucher et al., 1989). As the tumour
progresses and invades the basal lamina, elevated levels of CEA can be detected in sera.
For this reason, CEA has been used as a serum marker for recurrence of colorectal cancer
despite its low sensitivity and specificity (Goldstein and Mitchell, 2005). CEA has often
being referred to as a non-internalizing or as a shed antigen, yet studies have shown that
anti-CEA antibodies are endocytosed at a rate consistent with the metabolic turnover of
CEA (Ford et al., 1996; Schmidt et al., 2008; Shih et al., 1994; Stein et al., 1999) Anti-
CEA antibody targeted therapies have been reported to date (Behr et al., 2002;
Goldenberg, 2002). As in the case of antibody therapies aimed at solid tumours, poor
tumour penetration remains an issue and in the specific cases of high affinity CEA
antibodies, their rapid clearance due by free circulating antigen (Adams et al., 2001;
Graff and Wittrup, 2003). In order to assess the potential of CEA as an internalizing
antigen on cancer cells, DNA aptamers were developed specifically to recognize a
recombinant form of the N-terminal Ig domain of human CEA using the SELEX
approach. The binding of one such 25-base long DNA aptamer (and a control DNA
aptamer) to the mouse colon adenocarcinoma cell line MC-38 (CEA-) and its related cell

22
line transduced to express the human CEA gene, MC-38.cea (CEA+) (Robbins et al.,
1991) was monitored by flow cytometry. Specifically, these cells were incubated with a
Cy5 conjugated CEA-specific DNA aptamer at 4°C (surface binding only) and at 37°C
(binding and internalization). As shown in Figure 1.5, MC-38 (CEA-) MC38 cells
showed no significant binding of the CEA-specific aptamer at both temperatures (Figure
1.5B). In contrast, the CEA-specific aptamer strongly associated with the CEA-positive
cell line MC-38.cea, with a significant increase in mean fluorescence intensity being
observed after 2 hours at 37°C in relation to 4°C (Figure 1.5B). The higher fluorescence
signal observed at 37°C is attributed to the CEA aptamer being internalized during this
time period. The irrelevant Cy5-labeled DNA aptamer (control; GATC repeats) did not
bind to either cell lines at both temperatures. Thus, CEA may represent a powerful portal
for aptamer-directed conjugates to selectively reach and be imported into colon cancer
cells.
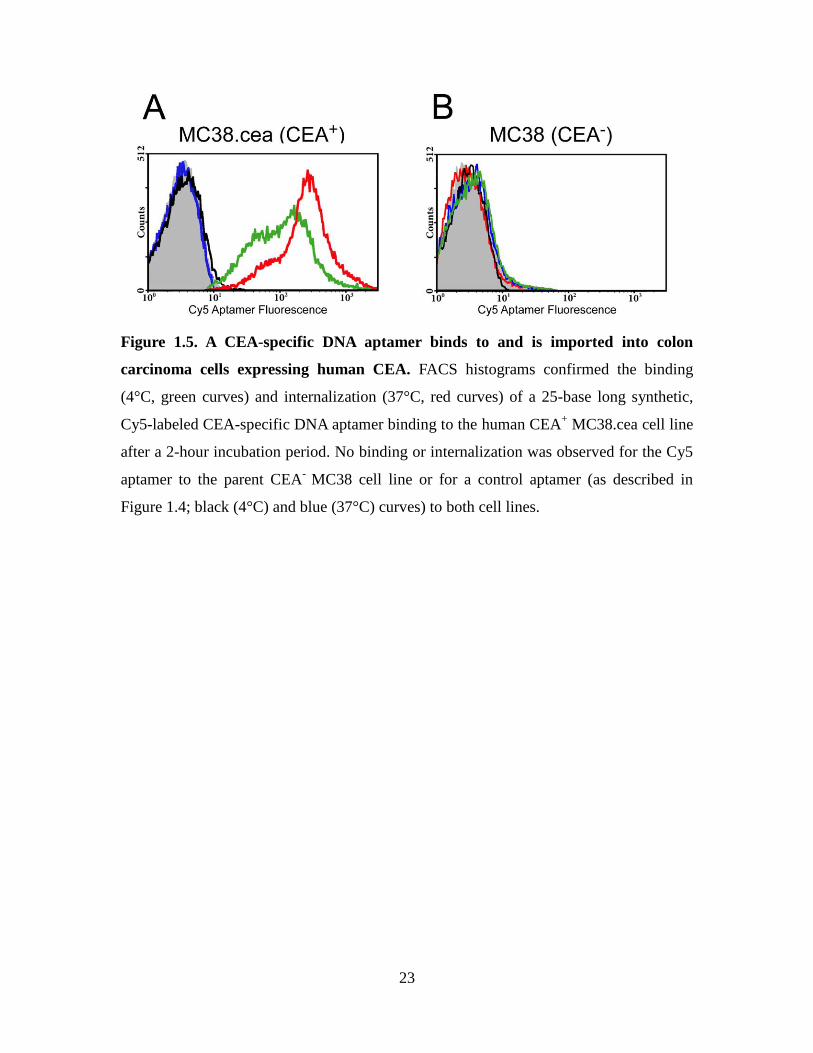
23
Figure 1.5. A CEA-specific DNA aptamer binds to and is imported into colon
carcinoma cells expressing human CEA. FACS histograms confirmed the binding
(4°C, green curves) and internalization (37°C, red curves) of a 25-base long synthetic,
Cy5-labeled CEA-specific DNA aptamer binding to the human CEA+ MC38.cea cell line
after a 2-hour incubation period. No binding or internalization was observed for the Cy5
aptamer to the parent CEA-
MC38 cell line or for a control aptamer (as described in
Figure 1.4; black (4°C) and blue (37°C) curves) to both cell lines.

24
1.3.3. CA15-3 antigen, MUC1 peptides and Tn antigens
The mucin MUC1 is a membrane glycoprotein that is highly expressed and is aberrantly
glycosylated [shortened O-glycans structures] in greater than 90% of all primary and
metastatic breast cancers (Hanisch and Muller, 2000; McGuckin et al., 1995; Spicer et
al., 1995; Taylor-Papadimitriou et al., 1999). The mucin MUC1 extracellular domain
largely consists of 30 to 100 copies of a 20-amino acid long tandem repeat (Gendler et
al., 1988). Serine and threonine residues within the tandem repeat represent sites of O-
glycosylation. The pattern of O-glycosylation at such sites is altered in cancer cells
giving rise to truncated short sugar chains known as the T, Tn and sialyl-Tn antigens as
well as exposing antigenic sites on the peptide chain itself (Hanisch et al., 1996). MUC1
peptide domains and its associated truncated carbohydrate epitopes are clinically referred
to as the CA15-3 antigen. Increasing serum levels of the CA15-3 antigen correlate with
poor prognosis. In terms of drug delivery, mucin MUC1 glycoforms are endocytosed and
recycled by cells in order to complete their glycosylation pattern prior to returning to the
cell surface (Altschuler et al., 2000; Ceriani et al., 1992; Henderikx et al., 2002; Litvinov
and Hilkens, 1993). Any ligands binding to such structures will thus be imported into
MUC1+ cells and in particular through Golgi compartments. Our group has recently
derived short 25-base long, synthetic DNA aptamers that specifically recognize either the
MUC1 peptide backbone or its Tn antigens (GalNAc sugars linked to serine and
threonine hydroxyl side chains on the MUC1 peptide tandem repeat) on epithelial cancer
cells with binding affinities (Kds) for their targets ranging from 18 to 85 nM (Ferreira et
al., 2009). Confocal microscopy and flow cytometry studies have shown that these
labeled aptamers circulate from the cell surface and into endosomal and Golgi
compartments upon binding to underglycosylated mucins (Figure 1.6). These DNA
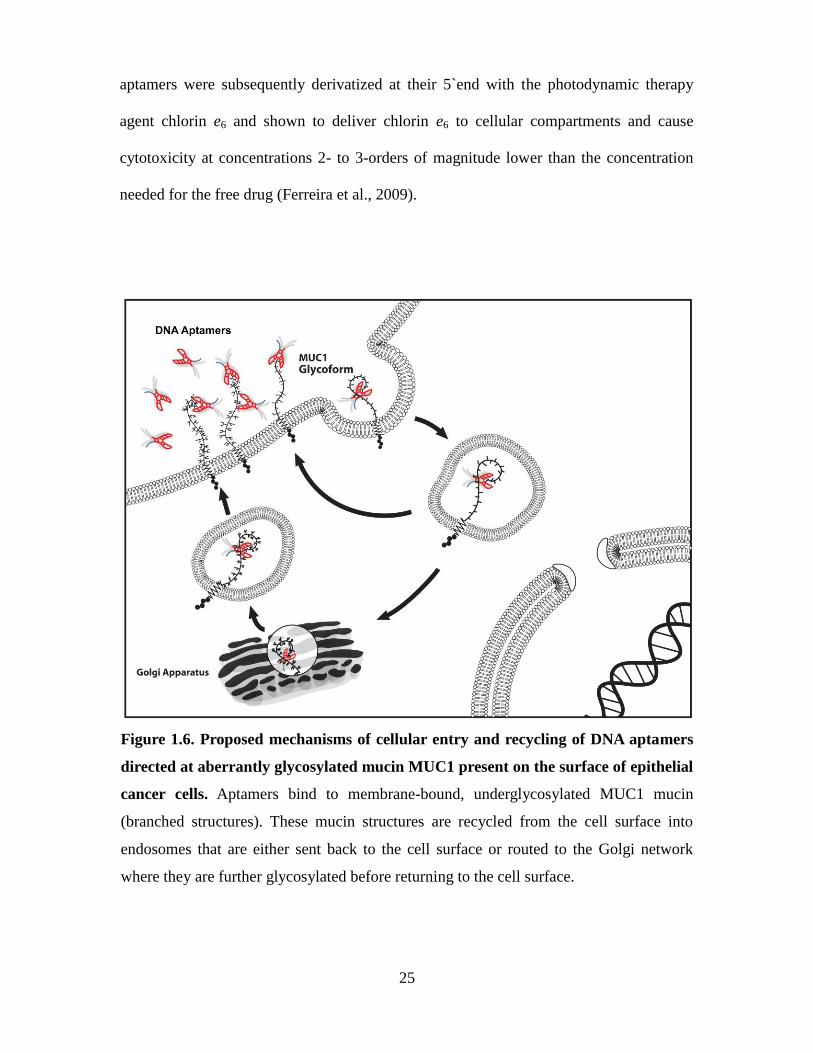
25
aptamers were subsequently derivatized at their 5`end with the photodynamic therapy
agent chlorin e6 and shown to deliver chlorin e6 to cellular compartments and cause
cytotoxicity at concentrations 2- to 3-orders of magnitude lower than the concentration
needed for the free drug (Ferreira et al., 2009).
Figure 1.6. Proposed mechanisms of cellular entry and recycling of DNA aptamers
directed at aberrantly glycosylated mucin MUC1 present on the surface of epithelial
cancer cells. Aptamers bind to membrane-bound, underglycosylated MUC1 mucin
(branched structures). These mucin structures are recycled from the cell surface into
endosomes that are either sent back to the cell surface or routed to the Golgi network
where they are further glycosylated before returning to the cell surface.

26
1.3. Aptamer-guided delivery of payloads into cancer cells
In theory, aptamers represent simpler antibody-like mimics in terms of their ability to
recognize tumour markers. Therapeutic agents can be directly coupled to aptamers or
packaged into particles modified with aptamers in order to exploit recycling pathways
associated with internalized cancer markers. However, the optimal efficacy of an
aptamer-based intracellular delivery agent will depend in part on the recycling properties
of their target and the possible induction of a receptor-mediated internalization event
upon binding to a surface marker. In addition, the intracellular routing of aptamers is
influenced by the abundance of the cell surface target itself, the macroscopic nature of
the aptamer conjugate being delivered (size and nature of the cargo) and the dominant
endocytic pathways associated with a given tumour cell type. The known cellular import
mechanisms that lead to the vesicular trafficking of ligands bound to cell surface
receptors are illustrated in Figure 1.7 and include (1) macropinocytosis and (2)
phagocytosis, distinguished by the size of their endocytic vesicles, (3) clathrin-mediated,
(4) caveolae (caveolin-based lipid rafts) and (5) clathrin-independent pathways. Recently
designed aptamer-cargoes complexes do exploit import pathways, although few studies
have explored their mode of cellular delivery. Most reported examples of internalized
aptamer conjugates (Table 1.3) have either made use of the RNA aptamers A9 and A10
directed at the prostate-specific membrane antigen (PSMA) or the DNA aptamer sgc8c
recognizing the tyrosine kinase 7 (PTK7).

27
Figure 1.7. Possible endocytic pathways taken by aptamer-cargoes. The nature and
size of a cargo, the membrane-cycling property and abundance of a targeted surface
portal on a given cell type as well as the number of portal-directed aptamers attached to a
cargo (valency) can influence which import mechanism(s) may dominate in routing an
aptamer-containing conjugate into cells. Large multivalent aptamer-targeted
nanoparticles, polymer aggregates and liposomes would favor actin-filament-mediated
uptake mechanisms such as macropinocytosis (1) or phagocytosis (2) while smaller
monomeric aptamer conjugates involving a drug, a radionuclide, an siRNA or a protein
as examples of payloads may enter cells via receptor-mediated events involving clathrin-
dependent (3), caveolin-dependent (4) and/or clathrin-independent (5) endocytic
pathways.

28
1.4.1. Aptamer-drug conjugates
Aptamer-drug conjugates have been constructed by chemically coupling a
chemotherapeutic drug to the aptamer via a linker or by intercalating the drug into the
aptamer folded structure creating a physical complex (Bagalkot et al., 2006; Huang et al.,
2009). The drug is then imported into target cells while reducing its toxicity towards
other cells (oligonucleotides including aptamers are cell-impermeant). Drugs can be
conjugated to aptamers during solid-phase synthesis or post-synthesis by incorporating
an amino or thiol group at one end of the oligonucleotide during their assembly. For
instance, doxorubicin, an anthracycline used in the treatment of various cancers, has been
coupled via an acid-labile hydrazone linker to a 41-nucleotide long tyrosine kinase 7
PTK7-specific DNA aptamer (sgc8c) to release the drug in endosomes (Willner et al.,
1993). This aptamer-drug conjugates has been shown to prevent the nonspecific
internalization of the drug as well as decrease its cellular toxicity towards non-target
cells. The conjugate is selectively internalized by CCFR-CEM cells (T-cell acute
lymphoblastic leukemia cells) with no apparent reduction in aptamer affinity for its target
(Huang et al., 2009). As mentioned in section 1.3.3, DNA aptamers targeting known
tumour-associated antigens such as mucin MUC1 peptides and mucin Tn antigens have
also been modified with a photodynamic therapy agent chlorin e6 and delivered to
epithelial cancer cells. These aptamer-chlorin e6 conjugates exhibited a >500-fold
increase in toxicity upon light activation as compared to the drug alone and were not
cytotoxic to cells lacking these mucin markers (Ferreira et al., 2009).
1.4.2. Aptamer-protein conjugates
Previous work with antibody-toxin conjugates has suggested that the most important
determinant of cellular cytotoxicity of immunotoxins is the efficiency of their import into

29
cells (Goldmacher et al., 1989). The coupling of aptamers to cytotoxic as well as
therapeutic proteins can facilitate them reaching their intracellular substrates. A case in
point is the anti-PSMA RNA aptamer (A9) conjugated to gelonin, a ribosome-
inactivating protein toxin. As mentioned in section 1.2, the prostate-specific membrane
antigen (PSMA) is internalized by prostate cancer cells and thus provides a portal for the
directed entry of the cytotoxic PSMA-specific aptamer-gelonin construct into such cells.
Gelonin is an enzyme that inactivates ribosomes when deposited in the cytosol of
intoxicated cells. The construct displayed a 600-fold increase in toxicity towards PSMA+
LNCaP cells as compared to non-PSMA-expressing PC3 cells and ~180-fold increase in
toxicity towards LNCaP cells relative to free gelonin (Chu et al., 2006a).
1.4.3. Aptamer-radionuclide conjugates
Few aptamers to date have been modified to incorporate radionuclides or metal chelators
with a view to image or kill cancer cells in vivo. Hicke and coworkers have reported the
introduction of the metal chelator mercapto-acetyl diglycine (MAG2) at the 5’ end of
TTA1, a Tenascin-C-specific aptamer (Hicke et al., 2001). TTA1 is a 40-nucleotide long
RNA aptamer that incorporates 2-fluoro-pyrimidines and binds to the protein Tenascin C
with a Kd of 5 nM (Hicke et al., 2001). Tenascin is a large, hexameric glycoprotein
associated with the extracellular matrix and is expressed during tissue remodeling events
linked to angiogenesis and tumour growth. The MAG2-containing TTA1 aptamer
chelates 99m
Tc and was used to determine its biodistribution in vivo in the context of nude
mice harboring a human glioblastoma U251 xenograft. 99m
Tc-TTA1 showed rapid blood
clearance and tumour uptake, reaching a tumour-to-blood ratio of 50 within 3 hours. In
addition, good scintigraphy images of a breast and glioblastoma tumour xenograft in
nude mice were recorded using this labeled aptamer (Hicke et al., 2006). The success of

30
this particular chelator-aptamer complex also highlighted the empirical nature of the
design process as an alternate choice of a chelator and radionuclide does result in
significant changes in the uptake and clearance patterns of this aptamer in vivo.
Nevertheless, the use of radiolabeled aptamers for imaging purposes in vivo is feasible.
1.4.4. Aptamer-nanostructure conjugates
The recent creation of aptamer conjugated nanostructures suggests that they may
represent a promising class of new agents for targeted cancer imaging and therapy. These
targeted structures include nanorods, quantum dots, as well as soft and hard
nanoparticles. Nanorods for example, can be viewed as an alternate scaffold for
assembling and immobilizing aptamers to nanomaterials in order to generate multivalent
conjugates. Huang and colleagues were able to show that up to 80 aptamers could be
covalently linked to the surface of Au-Ag nanorods via a 5’end thiol group introduced
into the structure of the fluorescein-labeled DNA aptamer sgc8c (section 1.4.1). The
avidity of the resulting aptamer-nanorods towards the tyrosine kinase 7 PTK7
transmembrane protein on CCFR-CEM cells was shown to be 26-fold higher than the
affinity of the unconjugated fluorescein-labeled aptamer sgc8c for the same cells. The
fluorescence intensity signal observed by flow cytometry was also 300-fold greater for
the aptamer-nanorod labelled cells than the signals observed for CCFR-CEM cells
labelled with the unconjugated fluorescein-labeled aptamer (Huang et al., 2008).
RNA aptamers directed at the prostate-specific membrane antigen (PSMA) have
been used in the design of numerous nanostructures. Streptavidin-coated quantum dots
(QD; semiconductor nanocrystals) have also been decorated with a biotinylated, 70-
nucleotide long PSMA-specific RNA aptamer termed A9 and the resulting conjugates
used for cellular imaging. Specifically, the photostability and small size of quantum dots

31
was shown to improve the visualization of PSMA-positive cells (LNCaP) as adherent cell
monolayers, in suspension preparations and embedded in a collagen matrix(Chu et al.,
2006b). Aptamer particles have also been designed to serve the dual purpose of acting as
a tumour-targeted agent and as a particle capable of controlled drug release. For example,
the FITC-labeled PSMA-specific RNA aptamer A10 was coupled to a poly(lactic acid)-
block-polyethylene glycol (PEG) copolymer nanoparticles that have been derivatized
with a terminal carboxylic acid functional group (PLA-PEG-COOH). Rhodamine-
labelled dextran was encapsulated (as a model drug) into these polymeric particles. The
nanoparticles including their cargo were selectively imported into PSMA-positive
LNCaP cells as confirmed by fluorescence microscopy (Farokhzad et al., 2004).
Farokhzad and colleagues subsequently loaded docetaxel, a chemotherapeutic drug into
the aptamer-conjugated nanoparticles and injected a single intratumoural dose of the
construct in nude mice harboring a LNCaP xenograft (Farokhzad et al., 2006).
Significant tumour regression was observed with no apparent immunogenicity. More
recently, the same aptamer-nanoparticle conjugates were loaded with docetaxel and
doxorubicin or with cisplatin although the overall improvement in survival in the treated
tumour-bearing animals was modest in relation to the non-aptamer targeted drug loaded
nanoparticles (Dhar et al., 2008; Zhang et al., 2007). Finally, the creation of a conjugate
composed of the PSMA-specific RNA aptamer A10-doxorubicin-quantum dot was
recently reported by Jon and Farokhzad groups (Bagalkot et al., 2007). Again, this
nanostructure is imported into PSMA+ LNCaP prostate cancer cells by PSMA-mediated
endocytosis. The construct offers the dual advantages of specifically delivering
doxorubicin intercalated into the A10 aptamer structure to prostate cancer cells as well as

32
imaging the delivery process through a FRET event arising from interactions of the
released doxorubicin and the QD itself (Bagalkot et al., 2007).
To date, liposomes remain one of the most successful drug delivery systems
(Kaneda, 2000). Liposome formulations of many of the most frequently prescribed
chemotherapeutic drugs have been approved and are currently used in clinical practice
(Wagner et al., 2006). Liposomes have been shown to increase the circulation time of
aptamers while these aptamers aid in targeting liposomes to their desired site of action
(Farokhzad et al., 2006; Willis et al., 1998). Liposomal drug delivery strategies have
focused on developing long-circulating liposomes that target areas of increased vascular
permeability via the enhanced permeation and retention (EPR) effect (Matsumura and
Maeda, 1986). The EPR effect however remains a passive tumour localization strategy
that can lead to detrimental systemic consequences and suboptimal antitumour efficacy
(Mamot et al., 2003; Woo et al., 2008). Aptamer-labeled liposomes can thus increase the
delivery of encapsulated therapeutic agents to cancer cells.
1.4.5. Challenges facing the in vivo use of aptamers
The concept of using aptamers as therapeutic agents was initially tested by selecting
aptamers to thrombin with a view to preventing blood clotting (Bock et al., 1992). The
rationale for creating thrombin-selective aptamers was to generate heparin mimics that
did not form complexes with platelet factor 4 which reacts with platelet-activating
antibodies leading to heparin-induced thrombocytopenia (Stribling et al., 2007). Larry
Gold`s group selected aptamers against the targeted HIV reverse transcriptase (Schneider
et al., 1995). Since virus transcriptases normally bind nucleic acids, they represent
excellent aptamer targets. Other parts of the virus are also being targeted by aptamers,
some of which are DNA aptamers (Chou et al., 2005; Zhou et al., 2009). In spite of their

33
large therapeutic potential, aptamer drugs are still not a commonplace treatment mostly
due to the previously mentioned challenges associated with translating small scale in
vitro laboratory experiments into medical practice. Currently, the only aptamer approved
by the FDA is Macugen (OSI Pharmaceuticals and Pfizer), an aptamer used to treat age-
related macular degeneration (AMD). Macugen is a PEGylated 29-nucleotide long RNA
aptamer with a modified backbone that significantly increases its circulating half-life (Ng
et al., 2006). Macugen recognizes the vascular endothelial growth factor isoform
VEGF165 but does not bind to VEGF121. In contrast, the antibody against VEGF
marketed by Genentech under the name Ranibizumab shows specificity towards both
isoforms (Kourlas and Abrams, 2007).
Aptamer structures can be evolved to recognize minor structural differences
within a given target and typically bind to their targets with affinities comparable to those
of antibodies (Conrad et al., 1994; Schneider et al., 1995). Practical advantages of
aptamers over antibodies include their lower mass, low cost of synthesis, long shelf-life
and consistent quality. However, aptamers do face challenges as potential therapeutic or
delivery agents. Firstly, nucleic acids are small, charged molecules. As such, they cannot
passively traverse a cell membrane. Secondly, oligonucleotides are rapidly degraded by
nucleases in plasma and cleared from circulation, resulting in short in vivo half-lives
(Chu et al., 2006a; White et al., 2000). Thirdly, oligonucleotides are typically not
immunogenic. Yet, immune responses mediated by Toll-like receptor family members
have been reported as exemplified by unmethylated CpG sequences (Wagner et al.,
2006). Solutions to these challenges are available. There are several approaches for
increasing the circulating time (half-life) of aptamers in plasma. One of them is
PEGylation, the process of conjugating polyethylene glycol (PEG) groups to such

34
molecules. The coupling of a cholesterol group or a cell-penetrating peptide can also
reduce their systemic clearance (Healy et al., 2004; Willis et al., 1998). Another approach
is by using chemically modified nucleotides shown to increase the half-life of aptamer
sequences by more than 40-fold (Peng and Damha, 2007). Such changes can be
introduced during the SELEX process by using modified nucleotides that are
incorporated by the T7 polymerase at the in vitro transcription step when RNA aptamers
are being selected. In the case of DNA aptamers, modified nucleotides are simply
introduced during library synthesis (Aurup et al., 1992; Latham et al., 1994). Possible
modifications compatible with the SELEX protocol include substitution of the 2' OH
group with a 2' fluoro or 2' amino group (Jellinek et al., 1995; Padilla and Sousa, 1999).
Besides the sugar component of the molecule, various groups such as aromatic and alkyl
moieties can be attached to the C5 position of UTP (Schoetzau et al., 2003). Other
modifications termed “post SELEX” have been introduced after a useful sequence is
identified (Eaton et al., 1997). One form of post-SELEX modification is Locked Nucleic
Acid (LNA) (Barciszewski et al., 2009). The LNAs can have one or more nucleotides
with a methylene linkage between the 2' oxygen and the 4' carbon, which results in the
“locked” conformation of the sugar. This modification provides an increased affinity for
the complementary strand, higher thermal stability, and resistance to nuclease
degradation (Vester and Wengel, 2004). Multivalency represents another factor that can
increase the avidity and potency of aptamers, as demonstrated by the oligomerization of
an RNA aptamer against the Drosophila protein B52 (Santulli-Marotto et al., 2003). The
tetravalent RNA aptamer recognizing the cytotoxic T-cell antigen-4 (CTLA-4) has also
shown a therapeutic advantage over its monomeric counterpart in prolonging the survival
of C57BL/6 mice implanted with the B16/F10.9 murine melanoma (McNamara et al.,

35
2008).
Among other aptamers selected to target tumour specific proteins, the first one to
enter clinical trials is an unmodified DNA aptamer termed AS1411 (Antisoma). It was
shown that its G rich sequence binds nucleolin present on the surface of cancer cells and
can inhibit NF-kB pathways (Bates et al., 1999; Girvan et al., 2006). This aptamer is
currently in Phase II clinical trials and shows activity towards many types of
hematological cancers (clinical trials.gov identifier NCT00512083; NCT00740441).
Interestingly, this 26-nucleotide long unmodified DNA aptamer is stable in serum, which
indicates that the sequence of the aptamers results in a three dimensional structure that is
not easily susceptible to nuclease degradation (Dapic et al., 2002). Thus, the need to
further modify DNA aptamers to increase their stability in vivo may not be necessary in
all cases.
Finally, Figure 1.7 outlines how aptamer-cargoes can reach several intracellular
vesicular compartments. The illustration is also meant to highlight the fact that the
cytosolic release of cargoes entrapped in vesicles remains an inefficient process and a
common challenge confronting other drug delivery strategies involving polymer
formulations, antibody conjugates and cell-penetrating peptides. Aptamer-targeted
cargoes such as radionuclides (acting within a cell diameter or via a bystander effect),
hydrophobic drugs, gold particles and liposomes may reach the cytosol or have their
therapeutic effect enhanced by simply residing or cycling through vesicles. Other
charged cargoes such as siRNAs, plasmids and proteins will be inefficiently released
from endosomal compartments and may require the use of endosomolytic agents.

36
1.4. Thesis Hypotheses
To date, many protein-based therapeutics have established themselves as the gold
standard for the treatment of an array of diseases. However, the use of proteins as
therapeutic agents comes with challenges such as immunogenicity [even for humanized
forms] and production costs. Many of the protein-based therapies involve the binding
and blocking of targets associated with the persistence or occurrence of a disease.
Therefore, new biotherapeutics are needed that exhibit reduced immunogenicity and are
less costly than proteins while acting as potent agonists or antagonists to clinically
relevent targets. My overall objective was to develop DNA aptamers able to target and
block the function of validated therapeutic targets or to act as specific delivery agents.
The hypotheses of this thesis were:
1) DNA aptamers can be developed to behave as inhibitors of TNFα activity.
2) DNA aptamers can be developed to act as anti-adhesives of carcinoembryonic
antigen (CEA).
3) DNA aptamers recognizing the unique cell surface biomarkers CEA and MUC1
can be used as targeting agents on liposomes containing a contrast agent for
Computed Tomography (CT) imaging of breast cancer tumours.

37
1.5. Specific Aims
To test these hypothese, the specifc aims were:
1) To develop a robust SELEX protocol that can be used to screen and identify DNA
aptamers to TNFα, CEA and MUC1.
2) Employ the SELEX methodology to select and identify DNA aptamers to TNFα
and evaluate their ability to block binding to the TNF receptor by monitoring the
inhibition of apoptosis, downstream signaling and reduction of nitric oxide (NO)
and characterize the level of immunogenicity.
3) Employ the SELEX methodology to select and identify DNA Aptamers to CEA
and characterize their ability to behave as anti-adhesive in blocking homotypic
binding and prevent peritoneal tumour foci formation.
4) To evaluate whether DNA aptamers to CEA and MUC1 can target liposomes
encapsulated with Omnipaque350™ to the breast cancer cell line MDA-MB-231
and characterize their effect on tumour accumulation and internalization.

38
1.6. Chapter Overviews
The studies addressing the specifc aims outlined are described in Chapters 2-4 of the
thesis. Chapter 2 describes the selection and identification of aptamer VR11 and
characterizes its ability to block TNFα-induced apoptosis in murine cells as well as
prevent the production of nitric oxide (NO) in macrophage cells. This study also showed
that aptamer VR11 has a high binding affinity and is specific to TNFα while exhibits no
immunogenicity in C57BL/6 mice. Chapter 3 demonstrates that DNA aptamers identified
to bind CEA with high affinity and specifically bound to the IgV-like N domain of CEA.
Two such aptamers prevented the adhesion properties of CEA expressing cells and
prevented the formation of peritoneal tumour foci whithout elicting an immune response.
Chapter 4 describes the characterization of CEA and MUC1 targeted liposomes
containing the contrast agent Omnipaque350™ and their ability to internalize in MDA-
MB-231 cells. As well, conjugation of these aptamers did not significantly change the
circulatory half-life of the liposomes formulations in mice. In Chapter 5 the overall
conclusions are presented and discussed and future research is proposed.

39
Chapter 2
A short DNA aptamer that recognizes TNF-alpha and blocks its activity
in vitro
A version of this chapter has been previously published:
A short DNA aptamer that recognizes TNF-alpha and blocks its activity in vitro
(ACS Chem Biol. 2013 Jan 18;8(1):170-8.)
Erik W. Orava, Nick Jarvik, Yuen Lai Shek, Sachdev S. Sidhu, Jean Gariépy
Author Contributions:
Erik Orava and Dr. Jean Gariépy designed the experiments, analyzed data and wrote the
paper.
Erik Orava contributed Table 2.1 and Figures 2.1, 2.2, 2.3A-B, 2.4, 2.6, 2.7, 2.8 and 2.9.
Nick Jarvik helped perform SPR experiments for Figure 2.3 C-G. in the laboratory of Dr.
Sachdev S. Sidhu
Yuen Lai Shek contributed by performing CD experiments for Figure 2.5.

40
2.1. Abstract
Tumour necrosis factor-alpha (TNF) is a pivotal component of the cytokine network
linked to inflammatory diseases. Protein-based, TNF inhibitors have proven to be
clinically valuable. Here, we report the identification of short, single stranded DNA
aptamers that bind specifically to human TNF. One such 25-base long aptamer, termed
VR11, was shown to inhibit TNF signalling as measured using NF-κB luciferase
reporter assays. This aptamer bound specifically to TNF with a dissociation constant of
7.0±2.1nM as measured by surface plasmon resonance (SPR) and showed no binding to
TNFβ. Aptamer VR11 was also able to prevent TNF-induced apoptosis as well as
reduce nitric oxide (NO) production in cultured cells for up to 24 hours. As well, VR11,
which contains a GC rich region, did not raise an immune response when injected
intraperitoneally into C57BL/6 mice when compared to a CpG oligodeoxynucleotide
(ODN) control, a known TLR9 ligand. These studies suggest that VR11 may represent a
simpler, synthetic scaffold than antibodies or protein domains upon which to derive
nonimmunogenic oligonucleotide-based inhibitors of TNF.

41
2.2. Introduction
Unregulated immune responses are intimately associated with degenerative diseases such
as atherosclerosis, arthritis, encephalitis, and tumours (Borish and Steinke, 2003;
Feldmann and Maini, 2001; van den Berg, 2001). The primary pro-inflammatory
cytokine, tumour necrosis factor alpha (TNF) plays a critical regulatory role in
enhancing these responses (Furst et al., 2006). In the past decade, disease-modifying
antirheumatic drugs (DMARDs) that target underlying immune responses processes have
improved disease outcomes in patients with chronic immune response disorders (Smolen
et al., 2007). Anti-TNF protein-based therapies (Enbrel, Humira, InfliximAb) have
emerged as a dominant category of DMARDs (Mount and Featherstone, 2005). However,
~ 40% of patients still display moderate to high levels of disease even after treatment
with protein therapeutics suggesting a substantial need for improved therapies (Mierau et
al., 2007).
TNF is a pro-inflammatory cytokine that is produced by an array of cell types
such as macrophages, monocytes, lymphocytes, keratinocytes and fibroblasts, in
response to inflammation, infection, injury and other environmental challenges (Carswell
et al., 1975). TNF is a type 2 transmembrane protein with an intracellular amino
terminus and is synthesized as a 26-kD membrane-bound protein (pro-TNF) that is
cleaved to release a soluble 17-kD TNF molecule. TNF has the ability to signal as a
membrane bound protein as well as a soluble cytokine. As a soluble cytokine, TNF is
only active as a non-covalently associated homotrimer (Hehlgans and Pfeffer, 2005;
Locksley et al., 2001). Sufficient levels of TNF as well as other mediators are critical
for sustaining normal immune responses. TNF can initiate host defence mechanisms to

42
local injury but can also cause acute and chronic tissue damage (Beutler, 1999; Feldmann
and Maini, 2008).
Despite advances in antibody and protein engineering, the major drawbacks of
protein-based TNF inhibitors are their immunogenicity arising from their chronic use
and their production costs resulting in expensive therapies for patients. Simpler,
synthetic, non-immunogenic classes of TNF inhibitors could be derived from short,
single-stranded nucleic acid oligomers (ssDNA or RNA) known as aptamers. These
molecules adopt a specific tertiary structure allowing them to bind to molecular targets
with high specificity and affinities comparable to that of monoclonal antibodies
(Ellington and Szostak, 1990). In some cases, aptamers will display functional properties
beyond just binding to their target. For instance, an aptamer to the inflammation factor
human neutrophil elastase (hNE) was shown to significantly reduce lung inflammation in
rats and displayed greater specificity for their target than an antielastase IgG control
(Bless et al., 1997). Examples of other aptamers exhibiting functional attributes include a
DNA aptamer to anti-HIV reverse transcriptase and RNA aptamers to the basic fibroblast
growth factor and vascular endothelial growth factor (Jellinek et al., 1994; Jellinek et al.,
1993; Schneider et al., 1995). Finally, a single-stranded DNA aptamer selected to bind to
thrombin has been shown to inhibit thrombin-catalyzed fibrin-clot formation in vitro
using either purified fibrinogen or human plasma (Bock et al., 1992).
In the present study, we constructed a 25-nucleotide variable region DNA library
to perform SELEX [Systematic Evolution of Ligands by Exponential enrichment] (Tuerk
and Gold, 1990) selections using recombinant TNF as our target. Several ssDNA
aptamers shown to specifically bind to TNF were further evaluated for their ability to
inhibit TNF signalling in several independent in vitro assays. These analyses led to the

43
identification of VR11, a DNA aptamer capable of inhibiting TNF functions in vitro.
2.1. Materials and Methods
2.3.1. Expression and purification of human TNF.
The expression plasmid (RCA-093) encoding a 157-amino acid TNFα his-tag
recombinant protein (residues 77-233) was obtained from Bioclone Inc. (San Diego,
CA). LB broth supplemented with kanamycin (50µg/ml) was inoculated with a glycerol
stock and grown at 37°C until the A600 reached 0.8. The expression of the TNFα fragment
was induced using a final concentration of 0.5 mM isopropyl β-D-thiogalactopyranoside
(IPTG) and incubated overnight. Soluble TNFα was purified by nickel affinity
chromatography under non-denaturing conditions. Cells were harvested by centrifugation
at 8,000 rpm for 30 min and resuspended for 1 hour in BugBuster Reagent (Novagen), 1
U Benzonase (Novagen) and a complete EDTA-free protease inhibitor tablet (Roche).
The lysate was then centrifuged at 8,000 rpm for 30 min and supernatant was loaded on a
Ni-NTA agarose column (Sigma-Aldrich) pre equilibrated with Buffer A (50 mM
phosphate buffer, pH 8, 300 mM NaCl, and 10 mM imidazole). After several washes with
buffer A the protein was eluted with 3 column volumes of Buffer B (50 mM phosphate
buffer, pH8, 300 mM NaCl and 250 mM imidazole). The eluate was concentrated by
ultrafiltration into a 100 mM Tris-HCl buffer (pH 6.0) using an Amicon ultrafiltration
unit (Millipore; 10 kDa MWCO). Purified TNFα was characterized by SDS-PAGE and
quantified using the Bradford assay (Bradford, 1976).To verify that the protein was
properly folded and functionally active, a cytotoxicity assay was performed using the
L929 cell line sensitized with Actinomycin D and compared to a commercial
recombinant human TNFpurchased from Bioclone Inc. (Appendix 3).

44
2.3.2. Aptamer selection screens.
The initial ssDNA library contained a central randomized sequence of 25 nucleotides
flanked by T3 and SP6 primer regions respectively. Briefly, the sequence of the starting
library was 5’ AAT TAA CCC TCA CTA AAG GG-(25N)-CTA TAG TGT CAC CTA
AAT CGTA. The forward primer 5’AAT TAA CCC TCA CTA AAG GG 3’ and reverse
primer 5’ TAC GAT TTA GGT GAC ACT ATA G 3’ were used for selection and cloning
(IDT Technologies, Inc.). A 25 nucleotide-long aptamer (cApt) comprised of AGTC
repeats [which has no predicted secondary structures] served as a control for all
experiments. To begin the selection, 50 nmol aliquot of the library representing ~3.0 ×
1016
molecules consisting of ~25 copies of each possible unique sequence was first
counter-selected against a 6-His peptide (HHHHHH) loaded onto Ni-NTA magnetic
agarose beads. The resulting sub-library was then exposed to 10 µg of His-tagged TNF
immobilized onto Ni-NTA beads suspended in Selection Buffer (PBS, 0.005% (v/v)
Tween-20) at 37°C for 1 hour. Unbound DNA sequences were washed away (PBS,
0.005% (v/v) Tween-20) and DNA-protein complexes were eluted from the recovered
beads using an imidazole buffer (PBS, 0.005% (v/v) Tween-20, 240 mM imidazole). The
ssDNA component was precipitated with sodium perchlorate/isopropanol and recaptured
using a silica membrane-based purification system (Qiagen Inc.). The DNA aptamers
were then amplified by asymmetrical PCR using a 100-fold excess of forward primer.
After every three subsequent rounds of selection, the amount of target was reduced in
half to increase the selection pressure to capture the tightest binding species. After 12
rounds of selection, the bound sequences were amplified by PCR, cloned into a pCR™4-
TOPO® TA vector (Invitrogen) and sequences were analyzed using BioEdit sequence
alignment editor software (Ibis Therapeutics).

45
2.3.3. Aptamer-based enzyme linked binding assay.
A 96-well ELISA microtiter-plate (BD Falcon) was coated overnight at 4°C with 100µl
of human TNF (10µg/ml) prepared in coating buffer (0.2 M carbonate/bicarbonate, pH
9.4). The plate was then washed 3 times with PBS-T (0.1M phosphate, 0.15M sodium
chloride, pH 7.0 containing 0.05% Tween-20) and blocked overnight at 4°C in 150 µl of
blocking solution (PBS-T + 1% (w/v) BSA). The plate was washed 3 times with PBS-T
and incubated overnight at 4°C with 100µl (10µg/ml) of 5’ biotinylated aptamers
dissolved in PBS, 0.005% (v/v) Tween-20. The plates were washed and incubated for 1
hour at 4°C with 100 µl of streptavidin-HRP (1:2000). Plates were then read on a plate
reader at 450 nm following washing and dispensing of 100µl of TMB substrate [1 TMB
tablet (Sigma-Aldrich Inc.) dissolved in 1ml of DMSO in 10 ml of 0.05M phosphate
citrate, pH 5.0 and 2 µl sodium peroxide] per well. Color development was stopped by
adding 50µl of 1M sulphuric acid per well.
2.3.4. NF-κB luciferase reporter assay.
An NF-κB luciferase reporter plasmid was transfected into cells as a marker of TNF
binding as previously described (Mamaghani et al., 2009). Briefly, 0.1μg/well TA-LUC
NF-κB and 10ng/well β-gal CMV were co-transfected to the cells. PANC1 (ATCC) cells
(1.0×104) and HEK293T (ATCC) (5.0×10
3) were seeded in a 96-well plate 24 hours
before the transfection steps. DNA was transfected into cells using Lipofectamine 2000
(Invitrogen Canada Inc.). Cells were incubated 4-6 hours in Opti-mem medium
(Promega) and transferred to 10% FBS DMEM overnight. Aptamers were added to the
medium (1 µM final concentration for PANC-1 cells or 2 µM, 200nM and 20nM for
HEK293T) 30 minutes prior to treating the transfected cells with recombinantly

46
expressed human TNF (100 ng/ml). Luciferase activity was measured after 6 hours
using the Dual-Light®
System luciferase assay from Applied Biosystems according to the
manufacturer's protocol and read on a Luminoskan Ascent luminometer (ThermoLab
Systems). The results were normalized to the values obtained for β-galactosidase activity.
2.3.5. Inhibition of TNF induced cytotoxicity.
L929 cells (ATCC) were seeded 24 hours before experiments in 96-well flat-bottom
microtiter plates at a density of 1.0×104
cells/well in DMEM medium containing 10%
FBS. Aptamers (2µM) or 20µg/ml anti-TNF mAb (R&D systems) were incubated with
human TNF for 2 hours in PBS prior to incubation with cells. Subsequently, aptamer-
TNF samples were added to the cells for 2 hours. Cells were then washed with warm
PBS and incubated in complete medium for another 48 hours. The viability of adherent
cells was subsequently determined using the sulforhodamine B assay (Skehan et al.,
1990).
2.3.6. Surface plasmon resonance.
All experiments were performed using the ProteOn XPR36 protein interaction array
system (Bio-Rad Laboratories, Inc.) and one ProteOn NLC sensor chip, coated with
NeutrAvidin for coupling of biotinylated molecules. Biotinylated aptamer VR11, VR20
and rApt(VR) were synthesized with a 5’ biotin with a standard C6 spacer and HPLC
purified (Integrated DNA Technologies, Inc.). Subsequently 2.5nM of aptamers were
diluted in PBS-T and injected for 30 sec at 30ul/min. Aptamer loading on the chip
yielded approximately 10–12 response units which represented ~1ng of aptamer. Human

47
TNF and TNFβ stock solutions were diluted as a series of two-fold dilutions ranging
from 2µM to 3.9nM in PBS-T (0.05% (v/v) Tween-20) pH 7.4 (Figure 3C-F). Protein
concentrations and a buffer control were injected in the analyte channel with a contact
time of 120 sec, dissociation time of 800 sec, and a flow rate of 100 µl/min. The ligand
channels were regenerated with a 30 sec injection of 1M H3PO4 followed by a 30 sec
injection of 1 M NaCl. All experiments were performed at 25°C and repeated in
triplicate. Sensorgrams were then double-referenced by subtracting the buffer response
and using the interspot reference. The sensorgrams were fitted globally to a 1:1 Langmuir
binding model and the kinetic parameters for the association (ka), dissociation rates (kd),
and binding constant (KD) were derived from the fitted curves.
2.3.7. Determination of aptamer concentration and Circular Dichroism
Spectroscopy.
The oligonucleotide concentration of each aptamer solution was determined from its
absorbance at 260 nm as measured at 25 °C with a Cary 300 Bio spectrophotometer
(Varian Canada, Inc.) using molar extinction coefficients of 244 400 M−1
cm−1
, 239 400
M−1
cm−1
and 251 800 M−1
cm−1
for VR11, VR20 and cApt respectively.. Circular
dichroism (CD) spectra were recorded at 25 °C using an Aviv model 62 DS
spectropolarimeter (Aviv Associates). The buffer used for CD measurements contained
10 mM phosphate, 0.1 mM EDTA (free acid) and 100 mM of either sodium or potassium
ions. Either KOH or NaOH was added to the buffers until pH 7 was reached and the ionic
strengths of the buffers were adjusted to the desired level by adding known amounts of
NaCl or KCl. Dry and desalted DNA aptamers were dissolved in the buffers until a
concentration of 0.03 mM was achieved. The DNA aptamer solutions were heated to

48
100°C for 5 minutes to transform the DNA aptamer into their unfolded forms and slowly
allowed to cool to 25°C. Ellipticity values [reported as θ] were measured from 320 nm to
200 nm in 1 nm increments using a quartz cuvette with an optical path length of 0.1 cm.
2.3.8. Inhibition of TNF induced NO production in macrophages.
Experiments were modified using a previous protocol (Ding et al., 1988). Briefly, RAW
264.7 cells (ATCC) were seeded at a density of 1.0 × 105
cells/well in a 12-well plate in
RPMI 1640 + 10% FBS. Cells were pre-treated for 1 hour with 2U/ml IFN-γ (Peprotech)
then treated with 100ng/ml of human TNF in 1ml of medium with aptamers VR11,
VR20 or the control aptamer cApt(VR) at a final concentration of 2µM or with 10µg/ml
anti-TNF mAb. Aliquots of the medium [100µl] were removed at each time point and the
NO2- level determined using the Griess reagent kit for nitrite determination (Invitrogen).
2.3.9. Analysis of VR11 ability to activate an innate immune response.
The aptamer VR11 and cApt were dissolved in sterilized saline at a concentration of 500
µg/ml and CpG ODN (5’-TCCATGACGTTCCTGACGTT-3’; type B murine, ODN
1826, Invivogen) was dissolved at a concentration of 50 µg/ml. Intraperitoneal injections
(200 µl) were administered in C57BL/6 mice. Untreated group received an injection of
200 µl of saline alone. Three hours after injection, mice were sacrificed and their serum
was collected for analysis. Protein cytokine concentrations were determined using the
DuoSet ELISA development kits for mouse CXCL1/KC (murine IL-8, R & D systems
Inc.) and murine TNF (R & D systems Inc.) as per manufacturer’s protocols. Animals
were kept under standard pathogen-free conditions at the Ontario Cancer Institute animal

49
facility. Experiments were performed in accordance with the rules and regulations of the
Canadian Council for Animal Care.
2.4. Results and Discussion
2.4.1. Identification of DNA aptamers directed at TNF.
Traditional treatments for chronic inflammation include NSAIDs, glucocorticoids,
cytostatic drugs and DMARDs. Due to the increased understanding of the molecular
pathology of several inflammatory disorders, immunosuppressants, such as those
directed at TNF, have moved to the forefront of anti-inflammatory drugs. The current
anti-TNF therapy market is dominated by protein therapeutics, namely etanercept
(Enbrel®), adalimumAb (Humira®) and infliximAb (Remicade®). These TNF
inhibitors have been shown to be therapeutically non-equivalent in terms of their
structure, binding to TNF and their therapeutic effects (van Vollenhoven, 2007). More
importantly, these protein-based therapeutics are large macromolecules that are costly to
produce, immunogenic when used in a chronic therapy setting, with close to 40% of
patients still showing disease symptoms even after treatment.
Using TNF as a target, we perform SELEX searches with a view to identify
short, synthetic DNA aptamers able to recognize this key cytokine and potentially mimic
the inhibitory action of an anti-TNF antibody (Figure 2.1). After 12 rounds of selection,
60 clones were sequenced and found to be enriched for 3 specific variable region (VR)
sequences. Specifically, the 25-base long sequences VR1, VR2, and VR6 accounted for
20%, 13% and 10% of the observed sequences respectively (Table 2.1). Aptamers VR11

50
and VR20 represented unique sequences identified in this search that also selectively
bound to TNF. Aptamer VR11 was the only aptamer that inhibited human TNF
functions on cells. Interestingly, VR11 incorporates the sequence 5’ – CAGTCGGCGA-
3’ which was identical to a region of aptamer VR12 with the exception of a single G base
insertion [5’ – CAGGTCGGCGA – 3’]. This 10-base long conserved span represents a
40% coverage of the variable region element of our library and despite the homology
between these two sequences, VR12 displayed no inhibitory activity. This finding
suggests that even a single base insertion can eliminate the functional properties of such
aptamers.
In order to identify and confirm that observed sequences were specifically
binding to TNF, 96-well ELISA microtitre plates were coated with human TNF. and
subsequently exposed to synthetic full-length (FL; a sequence that includes a specific VR
sequence as well as the two flanking primer regions) and variable region (VR) only DNA
aptamers harbouring a 5’ biotin group. Aptamer binding to TNF was confirmed with
streptavidin–HRP for 20 unique sequences tested (Figure 1A) with most of them showing
an ELISA signal at least double that of the control aptamers (cApt(VR), cApt(FL)).

51
Table 2.1. List of TNFα specific DNA aptamer sequences identified from the SELEX
screen as well as control aptamer [cApt(VR) and cApt(FL)] sequences used in the
present study.
Aptamer Sequence
VR1 ACAACCGACAAATTATCGCACTTAC
VR2 ATCACAGCGGGTACGAATGGCAGTG
VR4 ACGCTACGGGACTCTCAAACCGCC
VR5 AAGAACTGGCAGGCCGACCACCGGT
VR6 CGTCCGCTTTGAGTCTCGAAAAGGG
VR8 ATACCCATGGTACGACGGGCCATTC
VR11 TGGTGGATGGCGCAGTCGGCGACAA
VR12 CTCGTCAGTTCAGGTCGGCGATCAT
VR16 AGTGAGCGCTTAGTCTGCGCACTGG
VR20 TCCTCATATAGAGTGCGGGGCGTGT
cApt(VR) AGTCAGTCAGTCAGTCAGTCAGTCA
FL1 AATTAACCCTCACTAAAGGGACAACCGACAAATTATCGCACTTACCTATAGTGTCACCTAAATCGTA
FL2 AATTAACCCTCACTAAAGGGATCACAGCGGGTACGAATGGCAGTGCTATAGTGTCACCTAAATCGTA
FL4 AATTAACCCTCACTAAAGGGACGCTACGGGACTCTCAAACCGCCCTATAGTGTCACCTAAATCGTA
FL5 AATTAACCCTCACTAAAGGGAAGAACTGGCAGGCCGACCACCGGTCTATAGTGTCACCTAAATCGTA
FL6 AATTAACCCTCACTAAAGGGCGTCCGCTTTGAGTCTCGAAAAGGGCTATAGTGTCACCTAAATCGTA
FL8 AATTAACCCTCACTAAAGGGATACCCATGGTACGACGGGCCATTCCTATAGTGTCACCTAAATCGTA
FL11 AATTAACCCTCACTAAAGGGTGGTGGATGGCGCAGTCGGCGACAACTATAGTGTCACCTAAATCGTA
FL12 AATTAACCCTCACTAAAGGGCTCGTCAGTTCAGGTCGGCGATCATCTATAGTGTCACCTAAATCGTA
FL16 AATTAACCCTCACTAAAGGGAGTGAGCGCTTAGTCTGCGCACTGGCTATAGTGTCACCTAAATCGTA
FL20 AATTAACCCTCACTAAAGGGTCCTCATATAGAGTGCGGGGCGTGTCTATAGTGTCACCTAAATCGTA
cApt(FL) AATTAACCCTCACTAAAGGGAGTCAGTCAGTCAGTCAGTCAGTCACTATAGTGTCACCTAAATCGTA
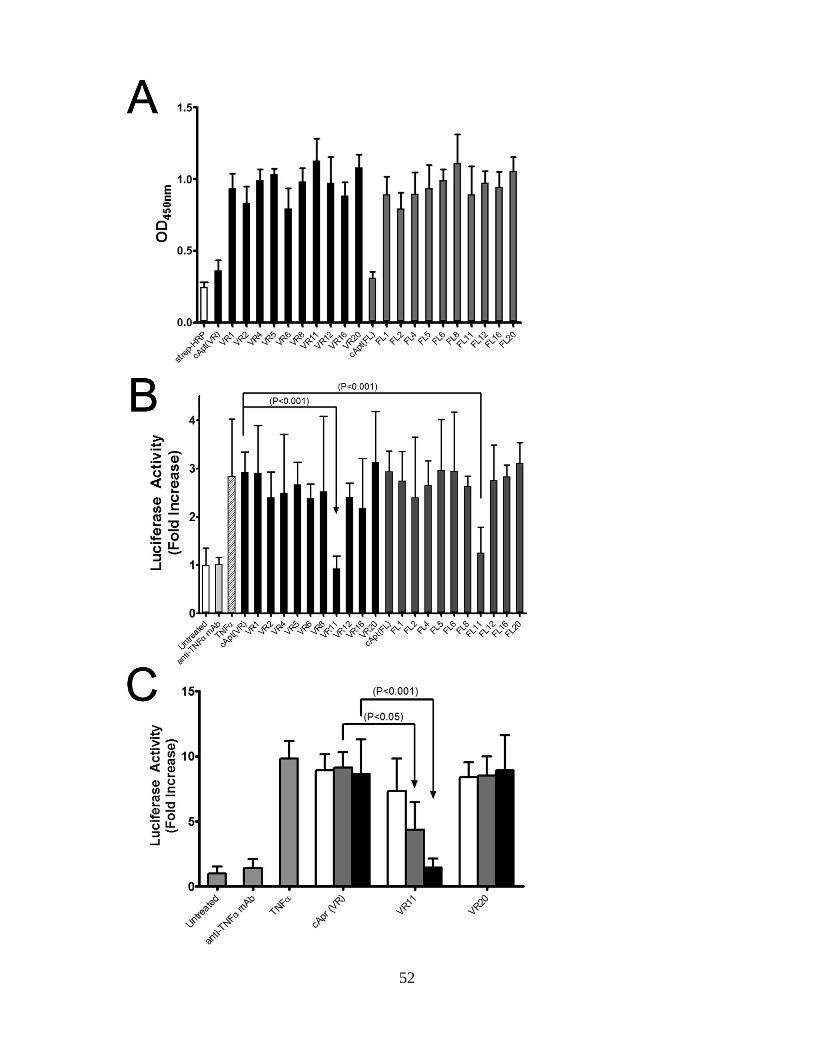
52

53
Figure 2.1. Binding of selected full-length (FL) and variable region (VR) DNA
aptamers to TNF and NfκB assays identify FL11 and VR11 as TNF inhibitors.
(A) Histogram of ELISA signals confirming the binding of biotinylated FL (grey bars)
and VR aptamers (black bars) to immobilized recombinant TNF. The control DNA
aptamers cApt(VR) and cApt (FL) gave ELISA signals comparable to wells treated with
streptavidin-HRP alone (open bar) (B) Histogram of fold increase in luciferase activity in
PANC-1 cells transiently transfected with a DNA plasmid construct expressing the
luciferase gene under the control of NfκB DNA binding promoter. Cells were treated
with TNF (100ng/ml), FL (dark grey bars) and VR aptamers (black bars) or an
inhibitory anti-TNF mAb (light grey bar). Only aptamer VR11 and its full-length
variant FL11 displayed inhibitory activities comparable to a control anti-TNF mAb. (C)
Histogram of TNF induced, NF-κB luciferase activity in HEK293T cells. Cells were
treated with 2 µM (black bars), 200 nM (grey bars) and 20 nM (white bars) of the control
aptamer cApt (VR), the TNF specific DNA aptamers VR11, VR20 as well as 20µg/ml
of the inhibitory anti-TNF mAb. Each histogram bar represents the average
enhancement in observed luciferase activity + SEM (n=9) normalized to that of untreated
samples.

54
2.4.2. DNA aptamer VR11 inhibits the binding of TNFto its receptor.
NF-κB (nuclear factor kappa-light-chain-enhancer of activated B cells) is a downstream
eukaryotic transcription factor that is activated by various intra and extra cellular
cytokines, including TNF (Sen and Baltimore, 1986). We selected a cell-based assay
using an NF-κB luciferase reporter assay in order to define aptamer sequences that
inhibited human TNF from binding to its receptor, leading to the loss of a cell
signalling event. Our preliminary screen was conducted using the human pancreatic
cancer cell line PANC-1 transiently transfected with the NF-κB luciferase reporter
(Mamaghani et al., 2009). The luciferase activity was measured 6 hours post-transfection
with only two DNA aptamers, namely VR11 and FL11, displaying an ability to inhibit
TNF-dependent NF-κB expression (Figure 2.1B). We subsequently used a more
sensitive TNF-mediated NF-κB assay employing HEK293T cells [higher transfection
efficiency] and confirmed the inhibitory ability of VR11 in a dose-dependent manner
(Figure 2.1C). The inhibition of TNF-mediated NF-κB signal was observed down to a
VR11 aptamer concentration of 200nM which represented a 35-fold molar excess of
aptamer over TNF. As expected, control aptamers VR20 and cApt(VR) did not show
any inhibition of TNF-mediated signalling suggesting that the inhibitory effects of
VR11 were not due to a concentration-dependent, non-specific binding event.
The L929 mouse fibroblast cell line was subsequently employed to define the
ability of aptamer VR11 to inhibit human TNF-induced cytotoxicity (Matthews et al.,
1987). The TNF CD50 value for L929 mouse fibroblast cells is approximately 3 nM
(Figure 2.2). When L929 mouse fibroblast cells were treated with human TNF in the
presence of aptamer VR11 or an inhibitory TNF mAb, the observed CD50 values were
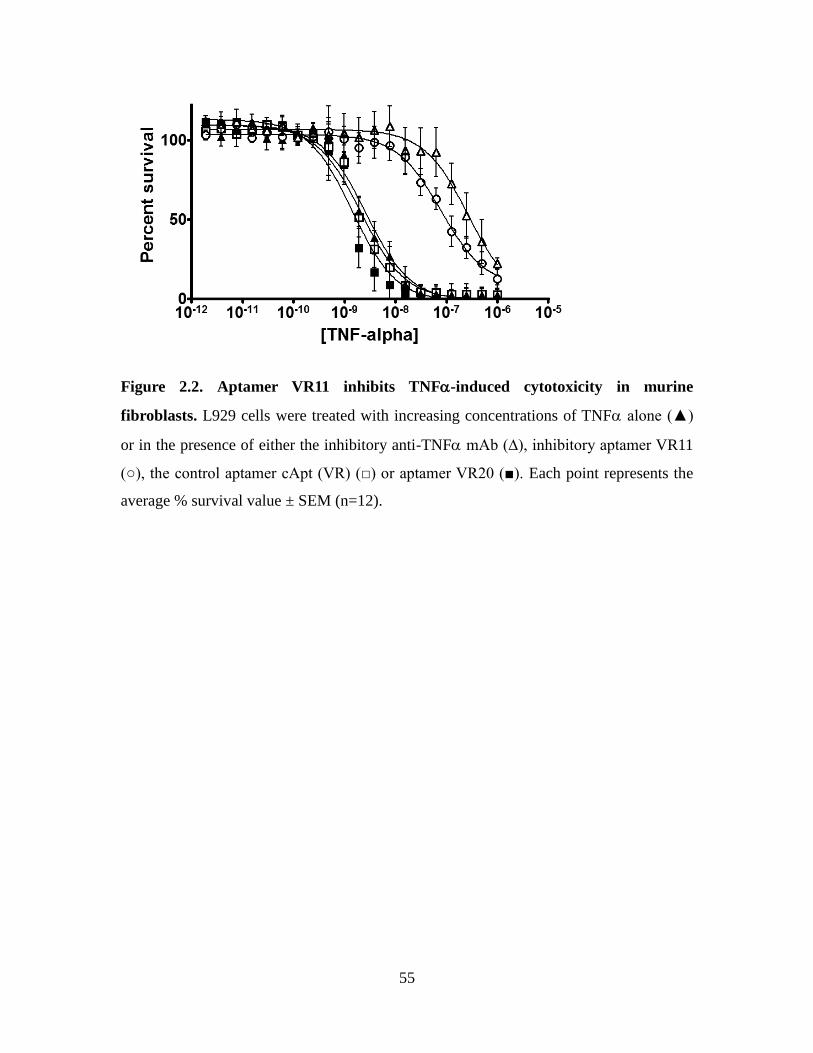
55
Figure 2.2. Aptamer VR11 inhibits TNF-induced cytotoxicity in murine
fibroblasts. L929 cells were treated with increasing concentrations of TNF alone (▲)
or in the presence of either the inhibitory anti-TNF mAb (∆), inhibitory aptamer VR11
(○), the control aptamer cApt (VR) (□) or aptamer VR20 (■). Each point represents the
average % survival value ± SEM (n=12).

56
shifted to approximately 100 nM and 260 nM respectively representing a 33-fold and 86-
fold decrease in toxicity (Figure 2.2). These CD50 values for aptamer VR11 and TNF
mAb, correspond to approximately a 24-fold and 5-fold molar excess over TNF
respectively. The control aptamer cApt(VR) as well as the TNF-specific DNA aptamer
VR20 showed no effect on the toxicity of TNF (Figure 2). Besides the removal of the 3’
terminal A base of VR11, the truncation of bases from either the 3’ or 5’ ends of VR11
resulted in the loss of its inhibitory activity towards human TNF (Appendix 1).
The binding properties of two TNF-binding DNA aptamers, namely VR11 and
VR20, were further characterized by surface plasmon resonance (SPR) with their binding
constants determined to be in the low nanomolar range [respective Kd values of
7.0±2.1nM and 8.7±2.9nM]. To assess the specificity of VR11 and VR20 for human
TNF, we also used human TNFβ as an analyte. TNFβ was chosen since it binds to TNF
receptors and is the closest known homolog to TNF having highly similar structures
(Figure 3B) despite having low sequence homology (Figure 2.3A). It was subsequently
observed that aptamers VR11 and VR20 showed no binding to TNFβ by SPR (Figure 3E
and 3F). One feature of aptamer VR11 which may contribute to its inhibitory function
may be its slow on- and off-rates (Figure 2.3). Once bound to TNF, VR11 was able to
block TNF-enhanced NO production in macrophages over a period of 24 hours (Figure
2.4B; as referred to as NO2- levels). Specifically, one of the consequences of unregulated
overproduction of TNF is the persistence of inflammation. Nitric oxide (NO) is a key
mediator of inflammation stimulated by TNF that is released by macrophages (Figure
4). We thus used the measurement of nitrite ions [NO2-] in solution as a marker of TNF-
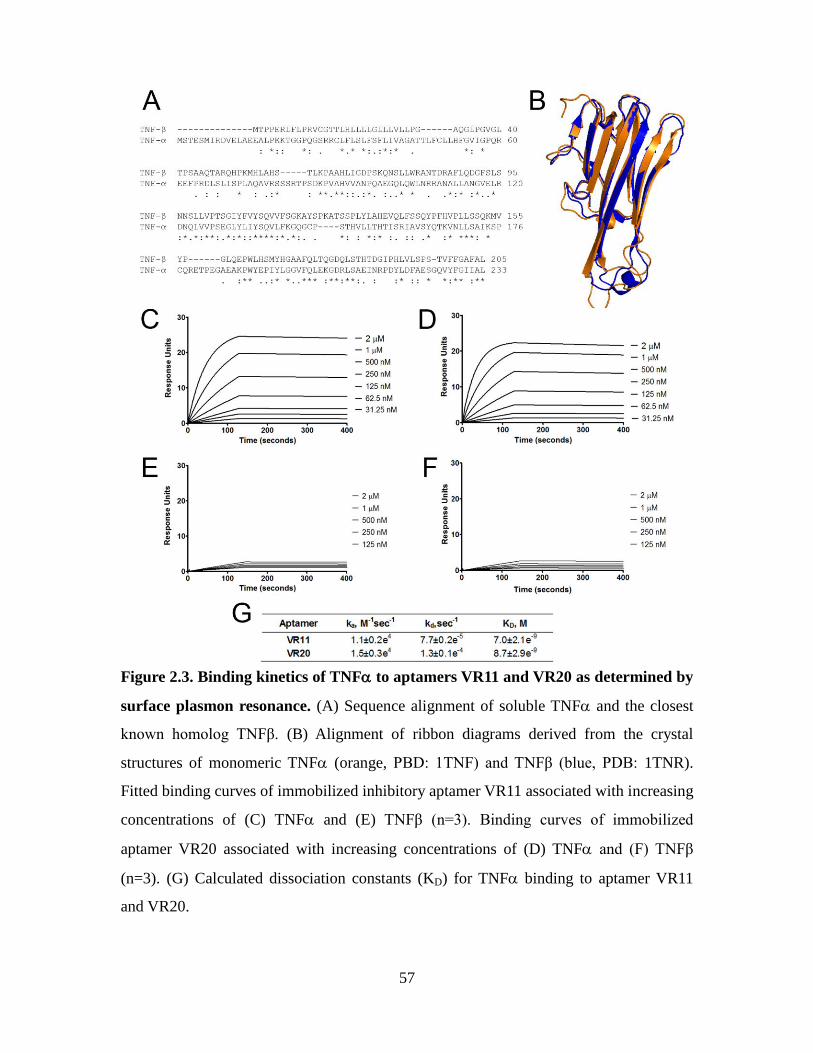
57
Figure 2.3. Binding kinetics of TNF to aptamers VR11 and VR20 as determined by
surface plasmon resonance. (A) Sequence alignment of soluble TNF and the closest
known homolog TNFβ. (B) Alignment of ribbon diagrams derived from the crystal
structures of monomeric TNF (orange, PBD: 1TNF) and TNFβ (blue, PDB: 1TNR).
Fitted binding curves of immobilized inhibitory aptamer VR11 associated with increasing
concentrations of (C) TNF and (E) TNFβ (n=3). Binding curves of immobilized
aptamer VR20 associated with increasing concentrations of (D) TNF and (F) TNFβ
(n=3). (G) Calculated dissociation constants (KD) for TNF binding to aptamer VR11
and VR20.

58
induced inflammatory signalling in the macrophage cell line RAW264.7. Human TNF
alone does not elicit the release of nitrite ions from macrophages. However, a synergistic
effect occurs when macrophages are concomitantly treated with interferon-gamma (IFN-
γ) and TNF (Paludan, 2000). At the 12 and 24 hour time periods, aptamer VR11 was
able to inhibit ~54% and ~45% of the TNF-dependent NO release signal respectively
(Figure 2.4). In comparison, the TNF mAb blocked ~79% and ~74% of the NO release
signal over the same two time periods (Figure 2.4). No inhibition of TNF-induced NO
release by RAW264.7 cells was observed when treated with control aptamers cApt(VR)
and VR20. This sequence thus represents a template that may be useful for modifications
that may yield a suitable product for use in vivo.
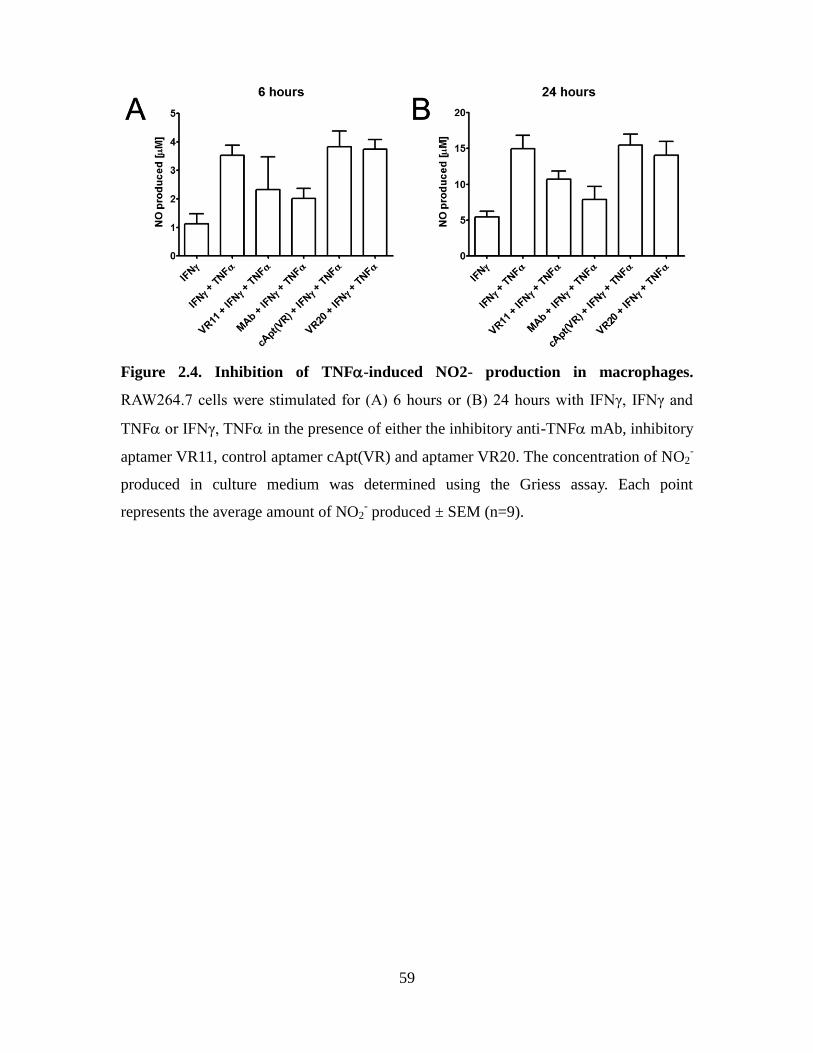
59
Figure 2.4. Inhibition of TNF-induced NO2- production in macrophages.
RAW264.7 cells were stimulated for (A) 6 hours or (B) 24 hours with IFNγ, IFNγ and
TNF or IFNγ, TNF in the presence of either the inhibitory anti-TNF mAb, inhibitory
aptamer VR11, control aptamer cApt(VR) and aptamer VR20. The concentration of NO2-
produced in culture medium was determined using the Griess assay. Each point
represents the average amount of NO2- produced ± SEM (n=9).

60
2.4.3. Structural features of DNA aptamer VR11.
The site targeted by VR11 on TNF was not defined in the present study. The epitope
recognized by the mAb to TNF has not been identified to date and as such this probe
could not be used to locate the VR11 binding site on TNF. In addition, the size and high
avidity of this anti-TNF mAb was able to block the binding of VR4, VR11 and VR20
TNF-directed aptamers to their target (see Appendix 2). A structural feature of aptamer
VR11 that may contribute to its inhibitory activity is its predicted G-quadruplex structure
as determined by the QGRS Mapper software (Kikin et al., 2006). G-quadruplexes arise
from the association of four G-bases into a cyclic Hoogsteen H-bonding arrangement.
Identifying G-quadruplex structures within aptamers has been the focus of recent interest
in view of the therapeutic efficacy of previously identified G-quadruplex forming ssDNA
and RNA (Bates et al., 1999; Marchand et al., 2002; Xu et al., 2001). A case in point is
the 15-mer DNA thrombin aptamer which folds into a G-quadruplex structure (Russo
Krauss et al., 2011). Structurally, VR11 incorporates 11 Guanine bases which accounts
for 44% of its sequence. G-quadruplexes typically occur when 3 or more consecutive G
bases are present within a sequence (Randazzo et al., 2012). However, the G-rich content
of VR11 sequence displays an 18-base long stretch containing 4 GG repeats (5’-
GGTGGATGGCGCAGTCGG -3’) which may stabilize the aptamer (Collie and
Parkinson, 2011). Interestingly, the DNA aptamer KS-B, specific for thrombin, also
incorporates 4 GG repeats within its sequence (GGTTGGTGTGGTTGG), and adopts an
intramolecular G-quadruplex structure (monomeric chair form) has proven from its
crystal structure (Macaya et al., 1993; Padmanabhan et al., 1993; Wang et al., 1993).
Thus, the projected G-quadruplex for VR11 may exist in solution. An intramolecular G-
quartet structure (Gilbert and Feigon, 1999; Williamson, 1994) within VR11 would

61
support our SPR data which suggest a 1:1 association of VR11 with TNF. The circular
dichroism spectra of VR11, VR20 and the control aptamer cApt were thus recorded in
order to monitor for the possible occurrence of a G-quadruplex structure within these
DNA aptamers. The sequence of VR20 harbours 4 consecutive G bases [Table 1; 5’-
CGGGGC-3’motif] and its CD spectrum displays a negative ellipticity band centered
around 240 nm as well as a positive band near 260 nm. This spectrum indicates that the
GGGG motif present within VR20 adopts a characteristic G quadruplex structure in
solution with a stacking pattern involving guanosines having identical glycosidic bond
angles giving rise to a four stranded parallel quadruplex structure typically observed for
the motif 5’-TGGGGT-’3 (Hardin et al., 1991; Hardin et al., 1992). The CD spectrum of
control aptamer cApt did not reveal any known spectral features indicative of G-
quadruplexes. As for VR11, its CD spectrum harbours a broad positive band around 290
nm suggesting that guanosines with different glycosidic bond angles within its sequence
are stacked in the presence of sodium or potassium (Figure 2.5A and 2.5B). Unlike the
CD spectrum observed for VR20, the VR11 spectrum does not provide conclusive proof
that the guanosine elements within its 25-bp sequence are arranged in a traditional G-
quadruplex structure. Nevertheless, a structural arrangement involving the stalking of
guanosine bases is present in VR11 and may stabilize its structure. Aptamer stability is
crucial for targeting TNF since the effectiveness of current protein-based therapies can
partly be attributed to their long circulating half-lives such as Etanercept which is linked
to the Fc portion of an IgG1 to help extend its half-life to 4-5 days or the PEGylation of
anti-TNF antibodies (Choy et al., 2002; Mohler et al., 1993). Recently, some of the most
common modifications of aptamers investigated have been their conjugation to
polyethylene glycol (PEG) groups for increased stability (Healy et al., 2004; Willis et al.,

62
1998). In addition, to increase the in vivo circulating half-life of DNA aptamers, modified
nucleotides could be introduced into the libraries during synthesis such as 2’ OH group
with 2’fluoro or 2’ amino, aromatic or alkyl moieties or introduction of Locked Nucleic
Acid (LNA) (Barciszewski et al., 2009; Schoetzau et al., 2003). Also, aptamer libraries
with longer variable regions may identify sequences with a higher G-rich content which
in turn could adopt different G-quadruplex structures and yield improved inhibitors to
TNF (Burge et al., 2006).
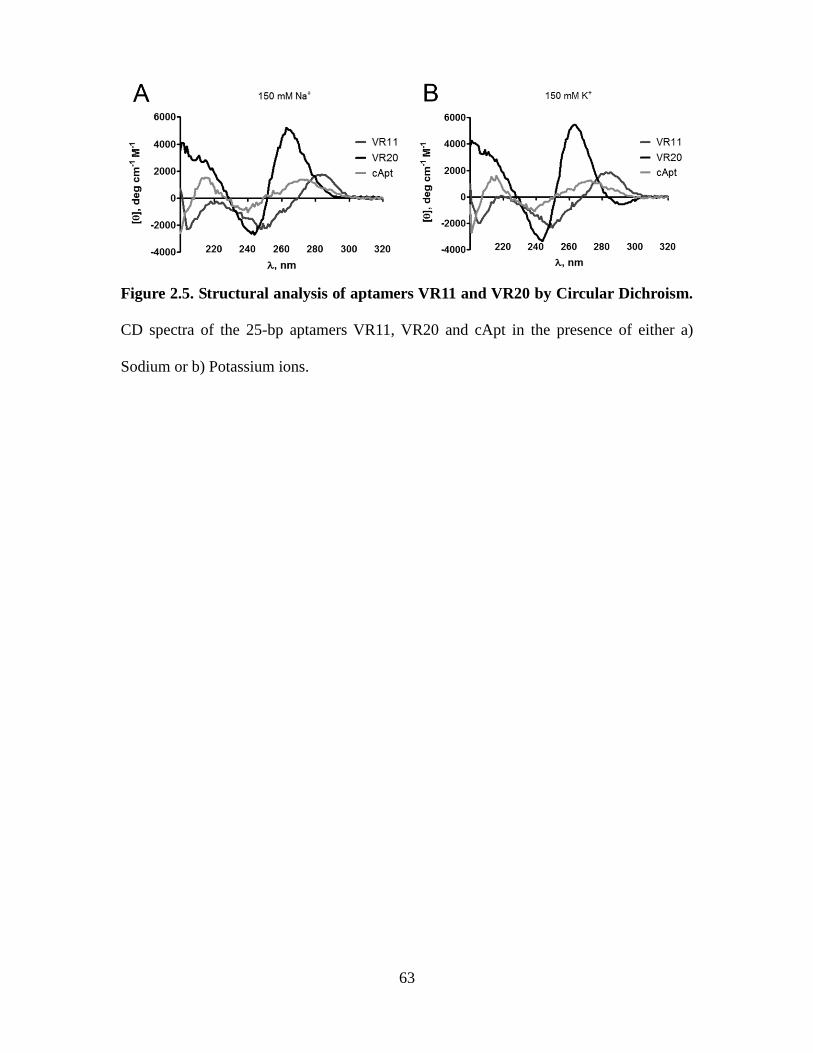
63
Figure 2.5. Structural analysis of aptamers VR11 and VR20 by Circular Dichroism.
CD spectra of the 25-bp aptamers VR11, VR20 and cApt in the presence of either a)
Sodium or b) Potassium ions.

64
2.4.4. Immunogenicity of DNA aptamer VR11.
One therapeutic obstacle that DNA aptamers may encounter is their possible activation of
an immune response similar to that of oligodeoxynucleotides containing CpG motifs.
These CpG motifs can promote a Th1 response by signaling through TLR9 which is
expressed on human B cells and plasmacytoid dendritic cells leading to an innate
immune response (Krieg, 2002; Krug et al., 2001). VR11 contains two possible CpG
motif sequences (Figure 2.6A). To test whether this DNA aptamer can induce an innate
immune response, we injected 100 µg of aptamer VR11 and cApt as well as 10 µg of a
known TLR9 ligand CpG ODN in the intraperitoneal cavity of C57BL/6 mice and
quantified the amount of TNF and mouse IL-8 present in their serum after 3 hours. The
CpG ODN positive control yielded a 35-fold increase in TNF and a 24-fold increase in
murine IL-8 serum levels while the control aptamer cApt (which contains no CpG
motifs) and the TNF-specific VR11 DNA aptamer yielded statistically non-significant
increases in both TNF and murine IL-8 cytokine levels as compared to untreated mice
(Figure 2.6B and 2.6C).
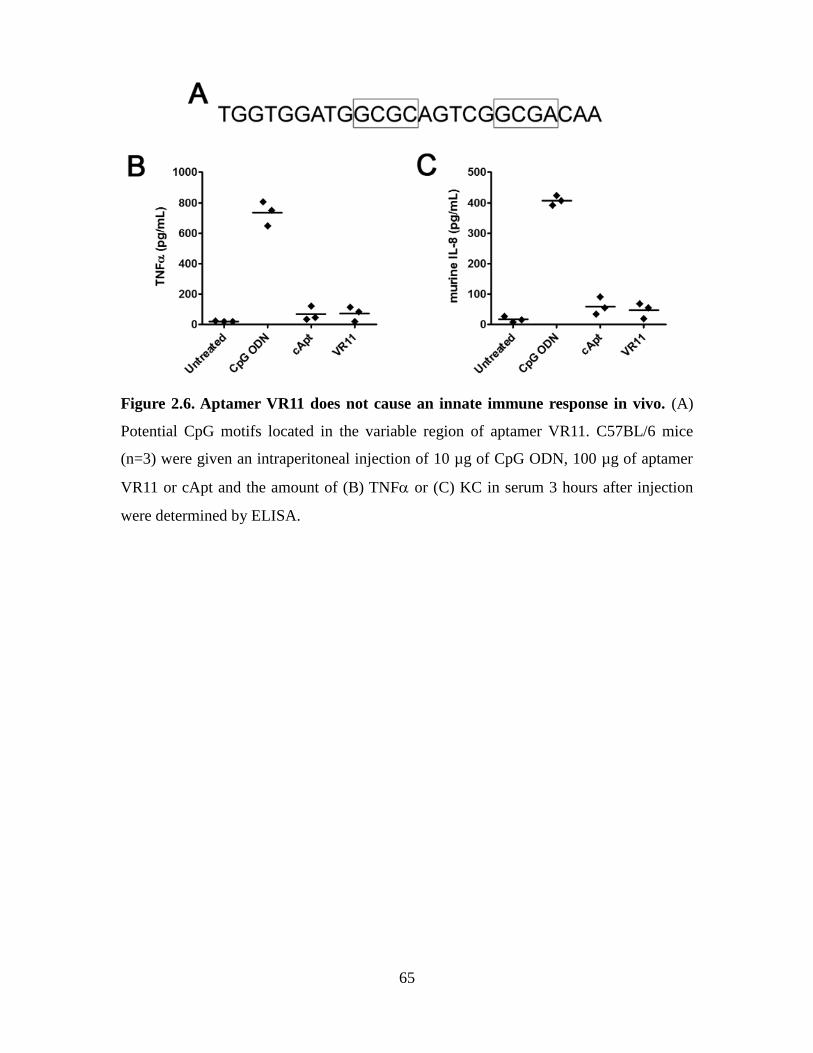
65
Figure 2.6. Aptamer VR11 does not cause an innate immune response in vivo. (A)
Potential CpG motifs located in the variable region of aptamer VR11. C57BL/6 mice
(n=3) were given an intraperitoneal injection of 10 µg of CpG ODN, 100 µg of aptamer
VR11 or cApt and the amount of (B) TNF or (C) KC in serum 3 hours after injection
were determined by ELISA.

66
Chapter 3
Blocking the attachment of cancer cells in vivo with DNA aptamers displaying anti-
adhesive properties against the carcinoembryonic antigen
A version of this chapter has been accepted for publication in the journal
Molecular Oncology
Blocking the attachment of cancer cells in vivo with anti-adhesive DNA aptamers
(Molecular Oncology 2013)
Erik W. Orava, Aws Abdul-Wahid, Eric H.B. Huang, Amirul Islam Mallick, Jean Gariépy
Author Contributions:
Erik Orava, Dr. Aws Abdul-Wahid and Dr. Jean Gariépy designed the experiments,
analyzed the data and wrote the paper.
Erik Orava contributed Figures 3.1, 3.2, 3.3, 3.4, 3.5 and 3.7.
Dr. Aws Abdul-Wahid contributed Figure 3.6
Eric Huang performed selection experiments that identify DNA aptamers in this project
as well as helped perform animal studies.
Dr. Amirul Islam Mallick helped perform animal studies.

67
3.0. Abstract
The formation of metastatic foci occurs through a series of cellular events, initiated by
the attachment and aggregation of cancer cells leading to the establishment of
micrometastases. We report the derivation of synthetic DNA aptamers bearing anti-
adhesive properties directed at cancer cells expressing the carcinoembryonic antigen
(CEA). Two DNA aptamers targeting the homotypic and heterotypic IgV-like binding
domain of CEA were shown to block the cell adhesion properties of CEA, while not
recognizing other IgV-like domains of CEACAM family members that share strong
sequence and structural homologies. More importantly, the pre-treatment of CEA-
expressing tumour cells with these aptamers prior to their intraperitoneal implantation
resulted in the prevention of peritoneal tumour foci formation. Taken together, these
results highlight the effectiveness of targeting the cell adhesion properties of cancer cells
with aptamers in preventing tumour implantation.

68
3.1. Introduction
Metastatic forms of cancer account for 90% of all cancer-related deaths (Sporn, 1996).
Cellular processes associated with tumour cell implantation and expansion of
micrometastases at sites distal from a primary tumour site are linked to altered growth
signals, deregulation of proliferative potential, evasion of apoptosis/anoikis and cell and
matrix adhesion events that create and support the formation of metastatic foci (Fidler,
2003; Hanahan and Weinberg, 2000). As such, surface molecules which mediate
intercellular adhesion represent candidate targets for engineering antiadhesive or
antiaggregative therapies. One such surface marker is the carcinoembryonic antigen
(CEA, CEACAM5 and CD66e), a member of the CEACAM family, an oncofetal antigen
over-expressed on the surface of breast, colon, lung and a range of other epithelial cancer
cells and an important cancer biomarker (Gold and Freedman, 1965b; Goldenberg et al.,
1976; Hammarstrom, 1999; Thompson et al., 1991). Under normal physiological
conditions, cells lining the colon express CEA in a polarized manner, with low levels of
this antigen being detected in the intestinal lumen and blood. In contrast, higher levels of
shed CEA are detected in the blood of 95% of patients with colorectal cancer (Chevinsky,
1991; Zhu et al., 2000). The deregulated overexpression of CEA has been linked to
tumour implantation and metastasis (Hammarstrom, 1999).
Membrane-bound CEA is comprised of a 108-amino acid IgV-like N domain
followed by 6 Ig-C like domains (A1, B1, A2, B2, A3 and B3) and a 27-amino acid C-
terminus region which includes a glycosylphosphatidyl inositol (GPI) anchor signal
sequence. The precise biological function of CEA has not been determined, but its
deregulated overexpression by cancer cells is associated with a vast array of functional
roles such as cooperating with novel oncogenes in cellular transformation, inhibition of

69
anoikis, differentiation inhibition via its GPI anchor, protection of tumour cells to
apoptotic stimuli, immunomodulation as well as functioning as an intercellular adhesion
molecule displaying both homotypic and heterotypic cell adhesion properties (Benchimol
et al., 1989; Jessup et al., 2004; Kitsuki et al., 1995; Ordonez et al., 2000; Screaton et al.,
2000; Screaton et al., 1997; Soeth et al., 2001).
A common denominator in CEA-dependent adhesion events is its IgV-like N-
domain which can lead to cellular aggregation through by binding to other N-domains
(defined as homotypic binding and homophilic interactions) or CEA IgC-like A3B3
domains on distinct tumour cells (defined as heterotypic binding and homophilic
interactions) or its association with extracellular markers (defined as heterophilic
interactions) such as its association with α5β1 integrins in binding to fibronectin (Jessup
et al., 1993a; Nicholson and Stanners, 2006; Taheri et al., 2000; Zhou et al., 1993).
Clinically, CEA is used to monitor patients with metastatic disease during active therapy,
as increasing levels of CEA in serum correlate with treatment failure and poor prognosis
(Duffy, 2006; Harris et al., 2007). Importantly, mounting a sustained antibody response
directed at an altered self form of the CEA N domain results in the prevention of tumour
implantation and formation of metastatic tumour foci in CEA transgenic mice (Abdul-
Wahid et al., 2012). This effect has been assigned to the blockage of CEA-dependent
adhesion properties by circulating antibodies as well as by immune mechanisms such as
antibody-dependent cell cytotoxicity (ADCC) and complement-dependent cytotoxicity
(CDC). Similarly, targeting the CEA IgV-like N domain with cyclised peptides or
monoclonal antibodies result in modest blockage of CEA-specific cell adhesion,
migration and invasion in vitro as well as impeding the metastatic potential in mouse
models (Blumenthal et al., 2005; Taheri et al., 2000; Zheng et al., 2011). These findings

70
suggest that CEA-specific, anti-adhesive agents may represent a successful treatment for
metastatic cancers linked to the overexpression of CEA.
Aptamers represent an emerging alternative to protein-based ligands. Specifically,
Aptamers are short single-stranded DNA or RNA oligonucleotides that adopt complex
secondary and tertiary structures that allow for their specific and high affinity binding to
a range of targets that include metal ions, proteins, bacterial cells and tumour cells
(Hamula et al., 2008; Hicke et al., 2001; Morris et al., 1998; Rajendran and Ellington,
2008). Aptamers are derived through an iterative selection process, termed systematic
evolution of ligands by exponential enrichment (SELEX), using a synthetic library
containing a randomized region of 25 to 80 nucleotides flanked by two constant regions
for PCR amplification (Tuerk and Gold, 1990). Alternatively, RNA aptamers are more
labile than DNA oligonucleotides and the cost as well as time required to perform RNA
SELEX searches are greater. More stable variants of RNA aptamers can be assembled
with a modified T7 polymerase to incorporate 2’-fluoro and 2’-O-Me nucleotides.
Our group has reported the expression and purification of a folded recombinant
form of the IgV-like N domain able to elicit an immune response as well as recapitulate
the binding property of glycosylated full length CEA with CEA-expressing cells and
purified human CEA from cancer patients. Importantly, the un-glycosylated form of the
CEA N domain represents a suitable target for identifying aptamers since this domain has
few putative glycosylation sites and that glycosylation of the N domain does not
contribute to the adhesive properties between CEA N domain molecules (Charbonneau
and Stanners, 1999; Krop-Watorek et al., 2002). We report the isolation of two functional
DNA aptamers selected to bind this recombinant form of the IgV-like N domain of CEA
and show its ability to block CEA-mediated cellular interactions and inhibit peritoneal

71
tumour nodule formation from CEA-expressing tumour cells in vivo.
3.2. Materials and Methods
3.2.1. Generation of recombinant CEA modules
Recombinant CEA (rCEA) modules N, FLAG-N and FLAG-A3B3 were expressed and
purified as previously reported (Abdul-Wahid et al., 2012). Briefly, recombinant CEA
domains were purified from inclusion bodies in E. coli under denaturing conditions using
urea (8M). The protein was subsequently purified by nickel-NTA chromatography and
treated with Detoxi-gel (endotoxin removing gel; Thermo Fisher Scientific Inc.) to
remove remaining traces of bacterial lipopolysaccharides (LPS).
3.2.2. Aptamer selection and cloning
The initial ssDNA library contained a central randomized sequence of 25 nucleotides
flanked by two primer regions with the sequence 5’ GAC GAT AGC GGT GAC GGC
ACA GAC G-(25N)-CGT ATG CCG CTT CCG TCC GTC GCT C 3’. The forward
primer 5’ GAC GAT AGC GGT GAC GGC ACA GAC G 3’ and reverse primer 5’ GAG
CGA CGG ACG GAA GCG GCA TAC G 3’ were used for selection and cloning (IDT
Technologies, Inc.). A 50 nmol aliquot of the library was first counter-selected against
Ni-NTA magnetic beads prior to selection against rCEA N in order to reduce non-
specifically bound DNA species. The resulting sub-library was then exposed to 10 µg of
His-tagged rCEA N domain immobilized onto Ni-NTA beads suspended in 1 mL of
Selection Buffer (150 mM NaCl, 50mM Tris pH 8.0) at 37°C for 1 hour. Unbound DNA
oligonucleotides were washed away with a 10-fold excess of selection buffer and DNA-

72
protein complexes were eluted from the recovered beads using an imidazole containing
buffer (Selection buffer with 240mM imidazole). The ssDNA component was
precipitated with sodium perchlorate/isopropanol and recaptured using a silica
membrane-based purification system (Qiagen Inc., Mississauga, Ontario). The DNA
aptamers were then amplified by asymmetrical PCR using a 10-fold excess of forward
primer. After every three subsequent rounds of selection, the amount of target was
reduced in half to increase the selection pressure to capture the tightest binding species.
After 12 rounds of selection, the bound sequences were amplified by PCR to produce
double stranded products, cloned into a pCR4-TOPO TA vector (Invitrogen) and
sequences were analyzed using BioEdit sequence alignment editor software (Ibis
Therapeutics, Carlsbad, USA).
3.2.3. Aptamer-based inhibition of CEA homotypic interactions
An enzyme-linked immunosorbent assay (ELISA)-based binding assay was employed to
identify aptamers capable of inhibiting homotypic interactions between FLAG-tagged
rCEA N domain and either rCEA A3B3 or rCEA N. Briefly, 96-well flat-bottomed Falcon
microtiter plates (Becton-Dickinson Biosciences, Franklin Lakes, NJ) were coated with
either N or A3B3 domain (1 µg/well in 100µl) in coating buffer (0.2 M
carbonate/bicarbonate, pH 9.4) at 37ºC. Plates were then blocked with BSA (1% in PBS)
and salmon sperm DNA (200 µl; 10 µg/mL) for 1 hour at room temperature then
aptamers were added (200 µl; 25 µg/ml, 1µM) overnight at 4 ºC in PBS-T (0.05%
Tween-20). Plates were then washed three times with PBS-T and FLAG-tagged rCEA N
domain was added (100 µl; 10 µg/ml, 670 nM) for 1 hour at room temperature after
being incubated with a given aptamer (100 µl; 25 µg/ml , 1µM) for 1 hour at room

73
temperature. After wash steps with PBS-T, the remaining bound FLAG-tagged rCEA N
was detected by incubating the plates for 1 hour at room temperature with horseradish
peroxidase (HRP) coupled anti-FLAG monoclonal antibody M2 (1:2500) dilution;
Sigma-Aldrich). All experiments were performed in quadruplicate.
3.2.4. Cells lines and growth conditions
The murine colonic carcinoma cell lines MC38.CEA and MC38 were kindly provided by
Dr. Jeffrey Schlom (National Cancer Institute, Bethesda, Maryland). The cervical
adenocarcinoma cell line HeLa (ATCC No CCL-2) as well as transfected cell lines
HeLaCEACAM1
, HeLaCEACAM3
, HeLaCEACAM5
, HeLaCEACAM6
and HeLaCEACAM8
used for
flow cytometry experiments were a gift from Dr. Scott Gray-Owen (University of
Toronto, Toronto, Canada). All cell lines were cultured at 37ºC, 5.0% CO2 in Dulbecco’s
modified Eagle’s medium supplemented with 10% fetal bovine serum, penicillin (100
U/mL) and dihydrostreptomycin (100 µg/mL).
3.2.5. Aptamer-based inhibition of homophilic cellular adhesion
The ability of aptamers to inhibit CEA-dependent cellular adhesion was measured in
real-time using an xCELLigence RTCA SP label-free, impedance-based cell sensing
device (Roche Applied Sciences, Laval, Canada). The inhibition of CEA-dependent
cellular adhesion was monitored using MC38.CEA and MC38 cells (2.5 × 104 cells per
well) grown in complete medium as described above. Cell suspensions were dispensed
alone, with aptamers (100 µl, 250 µg/ml, 10µM) or in the presence of the rCEA N
domain acting as a positive control (100 µl, 50 µg/ml, 3.3 µM) into wells of a 96-well

74
microtiter plate incorporating a sensor electrode array (E-plates) that had been precoated
with the rCEA N domain, rCEA A3B3 domain or BSA (1µg/well). Cell attachment was
quantified as a change in relative impedance, termed cell index (CI) (Matrone et al.,
2010). The adhesion of MC38.CEA cells in the absence of aptamers served as a positive
control. Data was collected after 3 hours to allow cells to fully adhere to protein-coated
plates but before the start of cell proliferation. All experiments were performed in
duplicate and were repeated three times.
3.2.6. Aptamer binding to CEA on cells as measured by flow cytometry
The binding specificity of aptamers N54 and N56 to the N domain of CEA was assessed
by flow cytometry using the cell lines MC38.CEA (CEA+) and MC38 (CEA
-) as well as
HeLa cells expressing different members of the CEACAM family. Aptamers N54, N56
and the control aptamer cApt were synthesized with a 5’end Cy5 fluorophore (IDT
Technologies, Inc., Coralville, Iowa). Cells were grown to mid-log phase and detached
using an enzyme-free EDTA based cell dissociation buffer (Sigma-Aldrich, St. Louis,
MO) washed with PBS (-CaCl2, -MgCl2) and resuspended at a concentration of 106
cells/mL in cold PBS. Aptamers were then added to 1.0×106 cells at a final concentration
of 200 nM in 1 mL. The expression of CEACAMs on HeLa transfected cells lines was
confirmed using a FITC-labeled polyclonal anti-CEACAM antibody (Gift from Dr.
Gray-Owen, University of Toronto, Toronto, Canada). CEA expression was confirmed
with a CEA-specific COL-1 antibody (Invitrogen Inc.). Aptamers and antibody were
allowed to bind for 2 hours at 4ºC. Cells were then washed three times in cold PBS and
subsequently analysed using a FACScan (BD Biosciences, Franklin Lakes, NJ).

75
3.2.7. Inhibition of MC38.CEA tumour implantation
For tumour implantation studies, 5.0 × 105 MC38.CEA cells were co-injected in the
intraperitoneal cavity of C57BL/6 mice with either aptamers N54, N56, N71, cApt or no
aptamer (saline) (200 µl; 2.5mg/mL, 100 µM). After 21 days, mice were sacrificed and
the number of nodules and their volumes were recorded following dissection, as
previously described (Abdul-Wahid et al., 2012). Specifically, the length and width of
tumour nodules were measured using microcallipers. Tumour volumes were calculated
using the modified formula where the volume of the tumour (mm3) equals [(x
2 × y)/2];
where x and y represent the transverse and longitudinal diameters of the tumour
respectively. Each group consisted of 3 females and 2 male mice. All animals were kept
under standard pathogen-free conditions at the Ontario Cancer Institute animal facility.
Experiments were performed under the approval of the local animal welfare committee
and in accordance with the rules and regulations of the Canadian Council for Animal
Care.
3.2.8. Aptamer cytotoxicity assay
MC38.CEA cells were seeded for 24 hours before cell viability experiments were
performed in 96-well flat-bottom microtiter plates at a density of 5.0×103
cells/well in
DMEM medium containing 10% FBS. Aptamers at a concentration of either 250 µg/ml
(10 µM) or 2.5 mg/ml (100 µM) were incubated with the cells in medium for 24 hours at
a volume of 100 µl. Cells were then washed with warm PBS and incubated in complete
medium for another 24 hours. The viability of adherent cells was subsequently
determined using a sulforhodamine B assay (Skehan et al., 1990). The absorbance of the
sulforhodamine B signal in each well was read at 570 nm using a plate reader. Each

76
experiment was performed in quadruplicate and repeated three times.
3.2.9. Analysis of aptamer-based innate immune responses
The aptamers N54 (200 µl; 500 µg/ml, 20µM) and cApt (200 µl; 500 µg/ml, 20µM) as
well as a TLR9 ligand CpG ODN (5’-TCCATGACGTTCCTGACGTT-3’; type B
murine, ODN 1826, Invivogen, CA; (200 µl; 50 µg/ml, 500nM) were dissolved in sterile
USP saline. These oligonucleotides (200µl) were administered intraperitoneally to 6-8
weeks old C57BL/6 mice. An untreated group of mice received an injection of 200 µl of
saline alone. Three hours after injection, mice were sacrificed and their serum was
collected for analysis. Serum IL-8 and TNFα concentrations were determined using the
DuoSet ELISA development kits for mouse CXCL1/KC (murine IL-8, R & D systems
Inc.) and murine TNFα (R & D systems Inc.) as outlined by the manufacturer.
3.2.10. Statistical methods and data analysis
Data sets derived from individual groups of mice were compared using student’s t-test
and grouped data sets were analyzed by ANOVA. Statistical analyzes and graphs were
assembled using GraphPad PRISM (version 5.01, GraphPad software for Science, San
Diego, CA). P values ≤ 0.05 were considered significant unless otherwise indicated.
Flow cytometry statistics were analyzed using WinMDI (version 2.8, Windows multiple
document interface for flow cytometry, Scripps Research Institute).

77
3.3. Results
3.3.1. Generation of DNA aptamers displaying inhibitory properties of CEA-
dependent homotypic adhesions
Short, 25-base long DNA aptamer sequences specifically recognizing a recombinant
protein coding for residues 1-132 of mature CEA (referred to as rCEA N domain) were
identified by the SELEX approach (Figure 3.1). Specifically, twelve iterative rounds of
PCR-based selection were performed yielding six unique DNA aptamer sequences
(Figure 1A). These six sequences, labelled N54, N56, N57, N59, N65 and N71, as well
as a control aptamer (cApt) were synthesized with their primer regions and subsequently
tested using an ELISA-based assay to assess their ability to directly block the binding of
the IgV-like N domain to either rCEA IgC-like A3B3 domains (Figure 3.1B) or to itself (to
rCEA N domain; Figure 1C). Aptamers N54 and N56 were the only aptamers found to
display inhibitory properties of both types of binding events, where N54 inhibited 39% of
the rCEA N→rCEA A3B3 signal and 45% of the rCEA N→rCEA N signal relative to
control wells (in the absence of aptamer. Aptamer N56 inhibited 32% of the signal
associated with either homotypic interactions (Figure 3.1B and 3.1C). Interestingly,
aptamer N65 shares a nearly identical sequence to N54 with the exception of two bases
flanking the ends of the sequence and a C to T substitution in the sequence [5’ GCTGAC
3’]. This finding suggests that the antiadhesive property of N54 is sensitive to even a
single base change in its sequence.
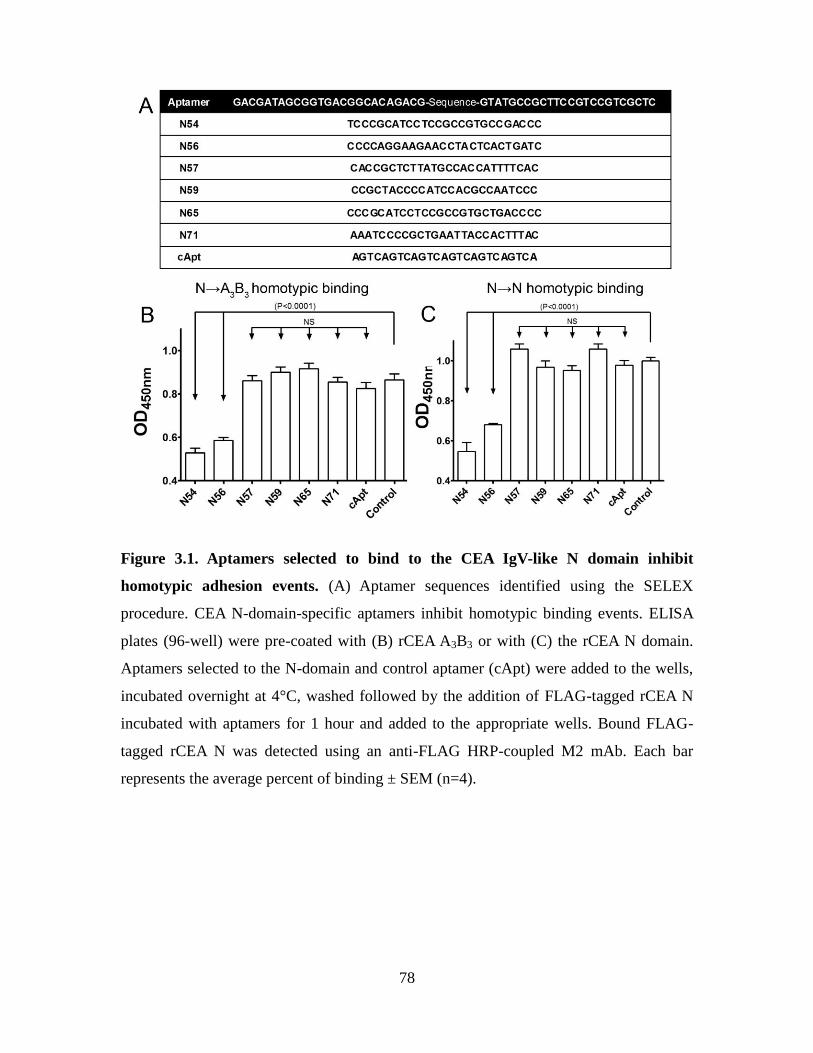
78
Figure 3.1. Aptamers selected to bind to the CEA IgV-like N domain inhibit
homotypic adhesion events. (A) Aptamer sequences identified using the SELEX
procedure. CEA N-domain-specific aptamers inhibit homotypic binding events. ELISA
plates (96-well) were pre-coated with (B) rCEA A3B3 or with (C) the rCEA N domain.
Aptamers selected to the N-domain and control aptamer (cApt) were added to the wells,
incubated overnight at 4°C, washed followed by the addition of FLAG-tagged rCEA N
incubated with aptamers for 1 hour and added to the appropriate wells. Bound FLAG-
tagged rCEA N was detected using an anti-FLAG HRP-coupled M2 mAb. Each bar
represents the average percent of binding ± SEM (n=4).

79
3.3.2. Aptamers N54 and N56 inhibit homophilic cellular adhesion
CEA-dependent homophilic interactions are a prerequisite to the expansion of metastatic
tumour foci. As such, the inhibitory capacity of aptamers N54 and N56 was determined
using the CEA-expressing murine cell line MC38.CEA and its parental CEA-negative
cell line MC38. MC38.CEA cells adhere to the wells of impedence-based plates (E-
plates) pre-coated with the rCEA N and A3B3 domains, but only weakly to wells coated
with BSA (Figure 3.2A). Addition of aptamers N54 and N56 resulted in a loss of 59%
and 49% of the signal arising from homophilic cellular adhesion between the
immobilized CEA N domain and MC38.CEA cells respectively after 3 hours. This time
point was chosen as it was observed that cells were able to fully adhere without the
presence of inhibitors (Appendix 4). Similarly, a 45% and 51% decrease in signal was
observed for MC38.CEA cells interacting with the immobilized rCEA A3B3. In contrast,
the control aptamers cApt and N71 showed no significant inhibition of homophilic
interactions (Figure 3.2A). As a positive control, the soluble rCEA N domain was pre-
incubated with MC38.CEA cells and resulted in a 59% and 44% loss of binding signal of
these cells to the immobilized rCEA N or A3B3 domain respectively. As expected,
aptamer treatments as well as BSA and rCEA N domain showed no ability to inhibit
MC38 cells from adhering to rCEA N- or BSA-coated plates (Figure 3.2B).
Subsequently, the inhibitory effects of aptamers N54 and N56 (as 75-base long aptamers)
on cell adhesion were titrated in a dose-dependent manner (Figure 3.2C). A significant
inhibition of MC38.CEA adhesion was observed starting at an aptamer concentration of 1
µM. Control aptamers cApt and N71 showed no effect suggesting that the inhibition of
homophilic cellular adhesion by aptamers N54 and N56 was not due to a concentration
dependent non-specific effect.
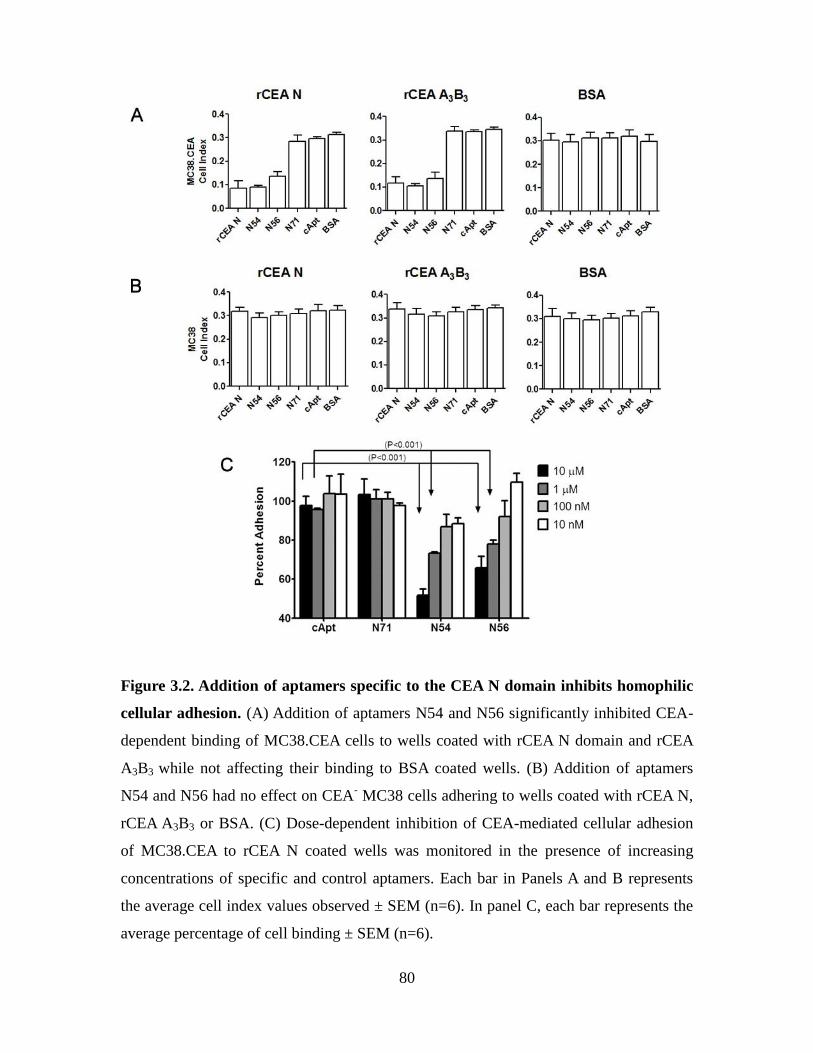
80
Figure 3.2. Addition of aptamers specific to the CEA N domain inhibits homophilic
cellular adhesion. (A) Addition of aptamers N54 and N56 significantly inhibited CEA-
dependent binding of MC38.CEA cells to wells coated with rCEA N domain and rCEA
A3B3 while not affecting their binding to BSA coated wells. (B) Addition of aptamers
N54 and N56 had no effect on CEA- MC38 cells adhering to wells coated with rCEA N,
rCEA A3B3 or BSA. (C) Dose-dependent inhibition of CEA-mediated cellular adhesion
of MC38.CEA to rCEA N coated wells was monitored in the presence of increasing
concentrations of specific and control aptamers. Each bar in Panels A and B represents
the average cell index values observed ± SEM (n=6). In panel C, each bar represents the
average percentage of cell binding ± SEM (n=6).

81
To further characterize the inhibitory properties of aptamers N54 and N56, we
constructed series of truncated forms to determine the minimal binding regions required
to retain their inhibition of CEA-dependent homophilic adhesion (Figure 3.3A). Aptamer
N54 did not retain its ability to inhibit cellular adhesion after a total of 18 bases were
removed from both ends of its sequence (aptamer N54-57; Figure 3B). Interestingly,
although full length N56 was not as effective as N54 in inhibiting MC38.CEA cell
adherence (36% compared to 48% inhibition respectively), N56 did retain its inhibitory
ability when truncated down to 32 bases with no significant decrease in the adherence of
cells as compared to the full length sequence (Figure 3.3B). Further truncations of N56
however yielded inactive inhibitors of cell adhesion.
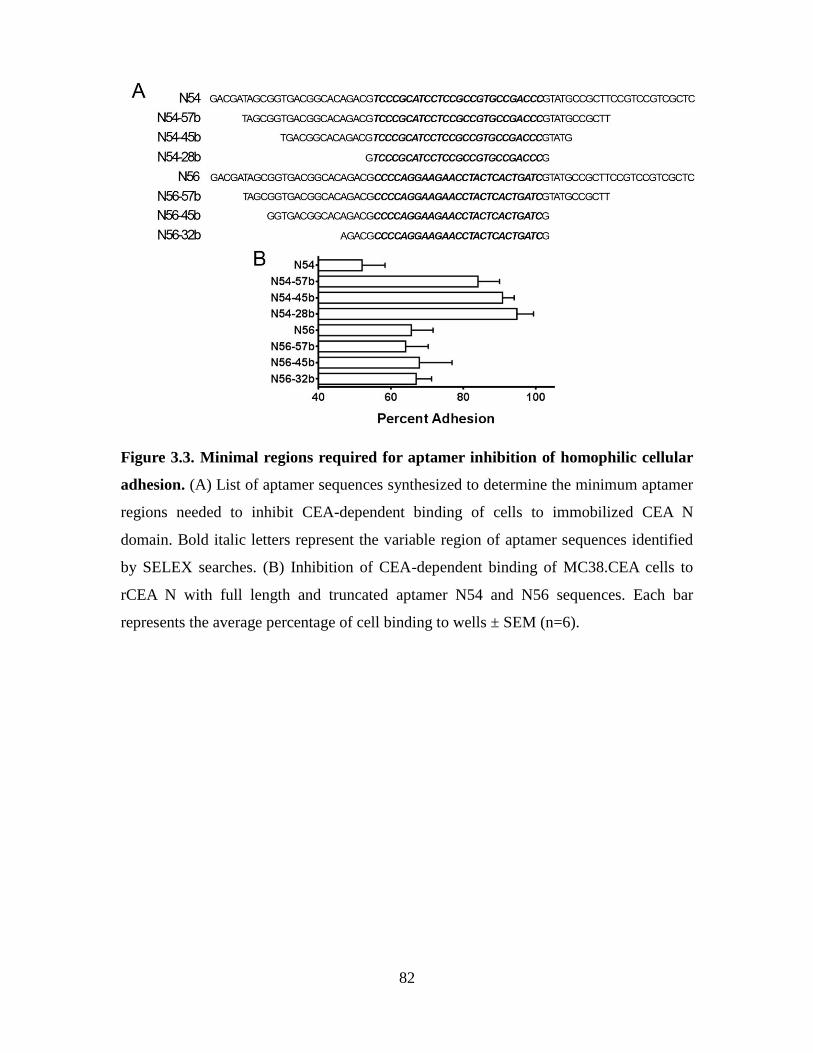
82
Figure 3.3. Minimal regions required for aptamer inhibition of homophilic cellular
adhesion. (A) List of aptamer sequences synthesized to determine the minimum aptamer
regions needed to inhibit CEA-dependent binding of cells to immobilized CEA N
domain. Bold italic letters represent the variable region of aptamer sequences identified
by SELEX searches. (B) Inhibition of CEA-dependent binding of MC38.CEA cells to
rCEA N with full length and truncated aptamer N54 and N56 sequences. Each bar
represents the average percentage of cell binding to wells ± SEM (n=6).

83
3.3.3. Aptamers N54 and N56 specifically recognize the N domain of CEA
The IgV-like N domain of CEA is homologous in sequence to that of other CEACAM
members. Specifically, the alignment of CEACAM1, CEACAM3, CEACAM5 (CEA),
CEACAM6, and CEACAM8 IgV-like N domain primary structures indicate that 61% of
residues along their sequences are identical with up to 84 % of residues being similar
(Figure 3.4A). In addition, the known structures of the CEACAM1, CEACAM 5 and
CEACAM8 N domains also indicate that these IgV-like N domains adopt the same
folded structure (Figure 3.4B) (Fedarovich et al., 2006; Korotkova et al., 2008).
Accordingly, the ability of aptamers N54 and N56 to specifically recognize the N domain
of CEA and not IgV-like N domains of related CEACAMs was assessed by monitoring
the binding of Cy5-labelled aptamers to CEACAM+ and CEACAM
- cells by flow
cytometry. Specifically, HeLa cells were stably transfected to express CEACAM1,
CEACAM3, CEACAM5, CEACAM6 or CEACAM8.
Analysis of the CEACAM-expressing HeLa cells as well as the CEA+
MC38.CEA cells, CEA- HeLa and MC38 cells demonstrated that aptamers N54 and N56
specifically bound to MC38.CEA and HeLaCEACAM5
cells while the irrelevant cApt
control aptamer showed no binding to any of the cells tested (Figure 3.5). The FITC-
labeled polyclonal anti-CEACAM antibody confirmed the expression of individual
CEACAMs in all transfected HeLa cell lines and the CEA antibody COL-1 confirmed
the presence of CEA (Figure 3.5). Aptamer N56 binding to MC38.CEA and HeLaCEACAM5
resulted in a ~7-fold increase in mean fluorescence signal intensity (Figure 3.5B,D).
Aptamer N54 binding to MC38.CEA and HeLa CEACAM5
resulted in a greater increase in
binding as shown by a ~ 16-fold and ~14-fold increase in mean fluorescence intensities
respectively (Figure 3.5B,D). Similar binding patterns were seen for the endogenously
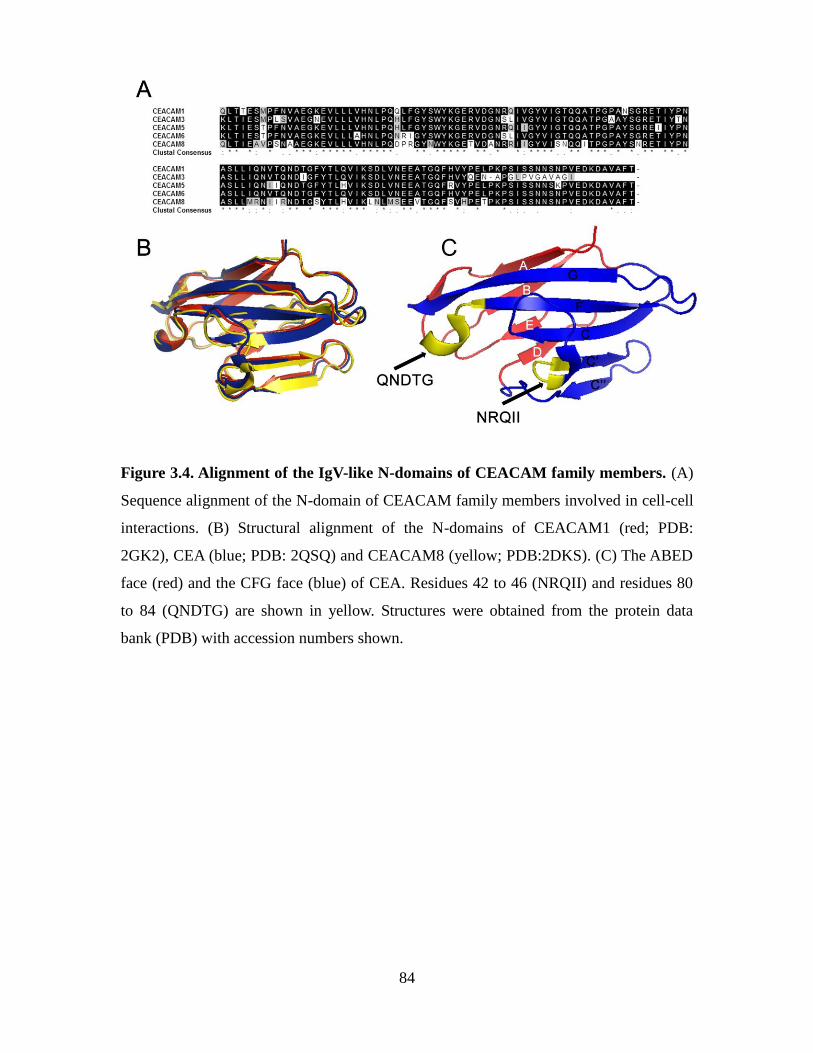
84
Figure 3.4. Alignment of the IgV-like N-domains of CEACAM family members. (A)
Sequence alignment of the N-domain of CEACAM family members involved in cell-cell
interactions. (B) Structural alignment of the N-domains of CEACAM1 (red; PDB:
2GK2), CEA (blue; PDB: 2QSQ) and CEACAM8 (yellow; PDB:2DKS). (C) The ABED
face (red) and the CFG face (blue) of CEA. Residues 42 to 46 (NRQII) and residues 80
to 84 (QNDTG) are shown in yellow. Structures were obtained from the protein data
bank (PDB) with accession numbers shown.
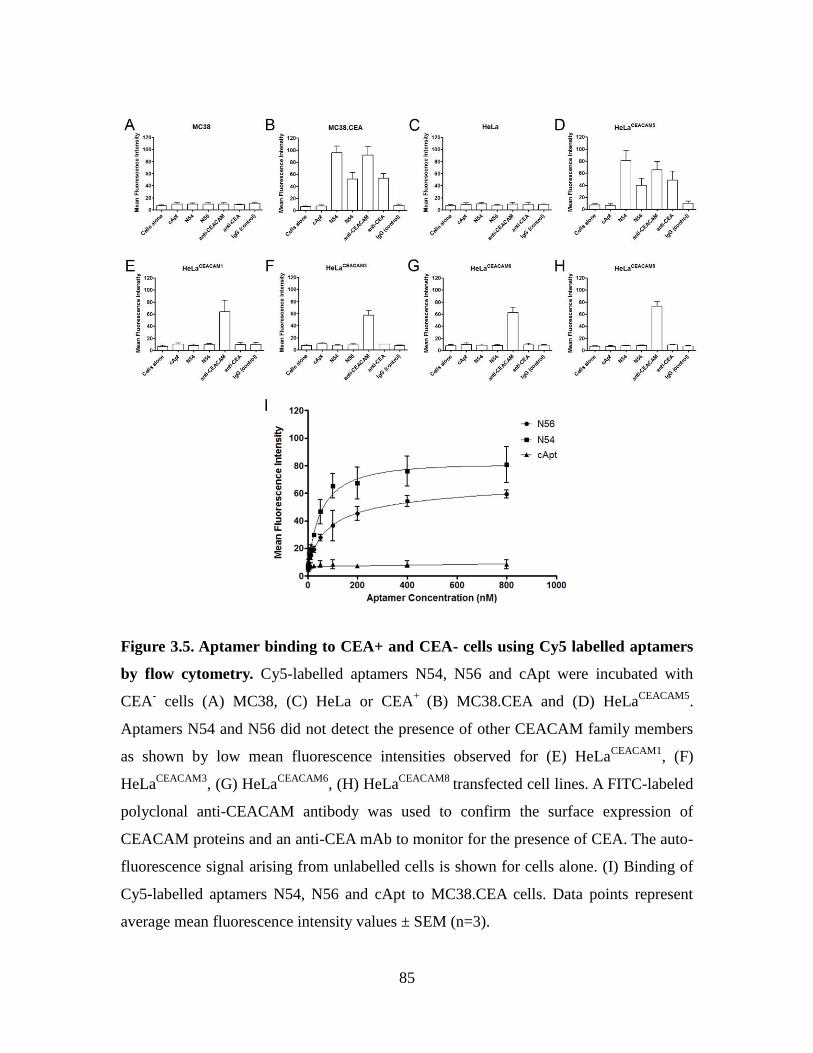
85
Figure 3.5. Aptamer binding to CEA+ and CEA- cells using Cy5 labelled aptamers
by flow cytometry. Cy5-labelled aptamers N54, N56 and cApt were incubated with
CEA- cells (A) MC38, (C) HeLa or CEA
+ (B) MC38.CEA and (D) HeLa
CEACAM5.
Aptamers N54 and N56 did not detect the presence of other CEACAM family members
as shown by low mean fluorescence intensities observed for (E) HeLaCEACAM1
, (F)
HeLaCEACAM3
, (G) HeLaCEACAM6
, (H) HeLaCEACAM8
transfected cell lines. A FITC-labeled
polyclonal anti-CEACAM antibody was used to confirm the surface expression of
CEACAM proteins and an anti-CEA mAb to monitor for the presence of CEA. The auto-
fluorescence signal arising from unlabelled cells is shown for cells alone. (I) Binding of
Cy5-labelled aptamers N54, N56 and cApt to MC38.CEA cells. Data points represent
average mean fluorescence intensity values ± SEM (n=3).

86
CEA expressing cell lines MCF-7, HT29 and BxPC3 (Appendix 5). The binding of Cy5-
labeled aptamers N54 and N56 to MC38.CEA cells was also determined as a function of
concentration (Figure 3.5, panel I). Using a single site binding model, it was calculated
that aptamers N54 and N56 display binding constants (Kd) of 45 ± 11 nM and 78 ± 24
nM respectively to their CEA target on MC38.CEA cells. Together, these findings
suggest that the derived N54 and N56 aptamer sequences specifically bind their cognate
target with high affinity.
3.3.4. Addition of aptamers N54 and N56 to MC38.CEA cells reduces tumour
implantation in vivo
The ability of aptamers N54 and N56 to inhibit CEA-dependent tumour implantation and
subsequent metastasis was addressed by monitoring their ability to interfere with the
implantation of murine MC38.CEA tumour cells in the peritoneal cavity of C57BL/6
mice. Briefly, murine MC38.CEA cells were pretreated for 30 minutes at 37°C with 500
µg (200 µl of a 100 µM concentration) of control aptamers cApt, N71, inhibitory
aptamers N54, N56 or untreated and co-injected directly into the peritoneal cavity of
mice (Figure 3.6A). Mice were then euthanized after 21 days to assess tumour
implantation by recording the number of tumour nodules as well as their volumes. Post-
mortem analyses of the dissected mice showed that tumour masses were limited to the
peritoneal cavity, and that tumour nodules were numerous in the control animal groups
given either no aptamer, cApt or N71 (Figure 3.6B,C). In contrast, implanted MC38.CEA
cells treated with aptamers N54 and N56 generated significantly fewer tumour nodules
(Figure 3.6B). Specifically, aptamer N56 reduced tumour implantation as seen by an
average decrease of 48% of cumulative tumour volume while N54 had a significant

87
decrease of 57% compared to untreated mice (Figure 3.6C). Control aptamers cApt and
N71 showed no significant decrease in tumour implantation in relation with the control
group that was just implanted with MC38.CEA cells (untreated). The inhibitory effect of
aptamers N54 and N56 was even more dramatic in terms of implantation when tumour
nodules were enumerated and compared to control groups (Figure 3.6D). Specifically, in
the group pretreated with aptamer N54, four of five mice did not develop a secondary
tumour nodule compared to untreated groups which had an average of ~6 tumour
nodules. Mice treated with control aptamer cApt and N71 showed no significant decrease
in the number of tumour foci when compared to the aptamer-untreated group.
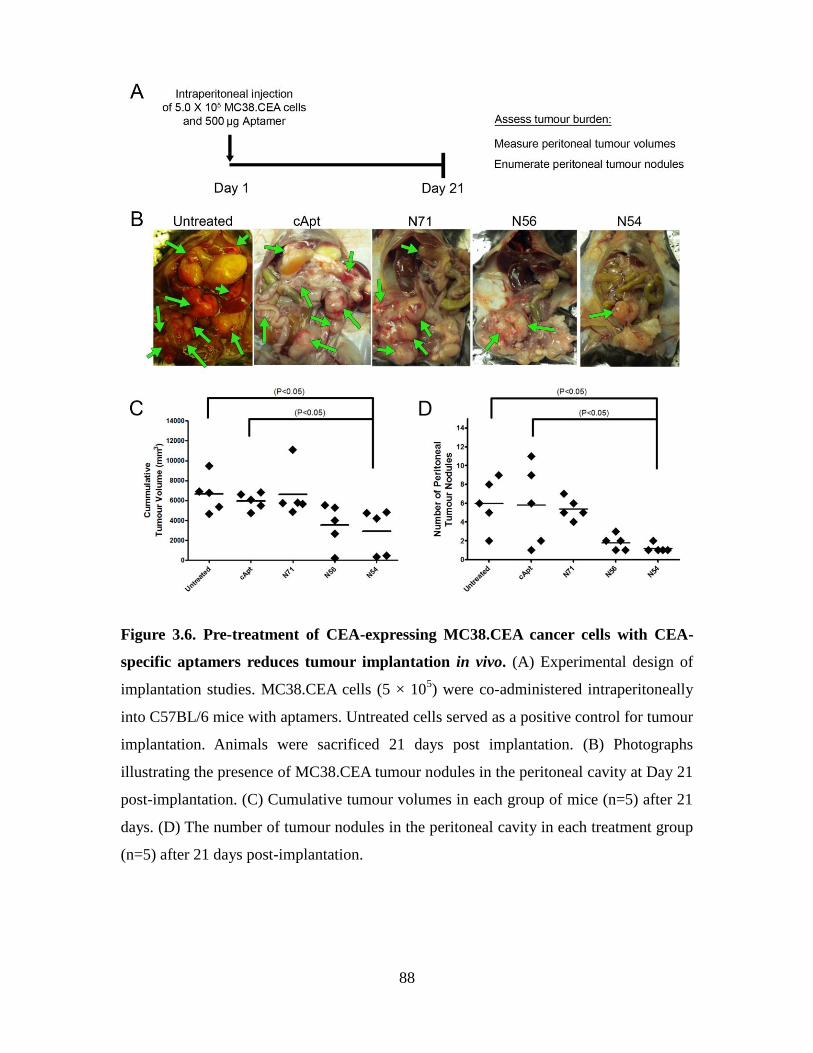
88
Figure 3.6. Pre-treatment of CEA-expressing MC38.CEA cancer cells with CEA-
specific aptamers reduces tumour implantation in vivo. (A) Experimental design of
implantation studies. MC38.CEA cells (5 × 105) were co-administered intraperitoneally
into C57BL/6 mice with aptamers. Untreated cells served as a positive control for tumour
implantation. Animals were sacrificed 21 days post implantation. (B) Photographs
illustrating the presence of MC38.CEA tumour nodules in the peritoneal cavity at Day 21
post-implantation. (C) Cumulative tumour volumes in each group of mice (n=5) after 21
days. (D) The number of tumour nodules in the peritoneal cavity in each treatment group
(n=5) after 21 days post-implantation.

89
3.3.5. Aptamers specific to the CEA N domain are not cytotoxic and aptamer N54
does not activate an innate immune response
Cytotoxicity studies on MC38.CEA cells treated with aptamers and rCEA N were
conducted using the sulforhodamine B cell viability assay to determine if the decrease in
tumour implantation seen was due to a cytotoxic effect of the aptamers (Matthews et al.,
1987). MC38.CEA cells were incubated in DMEM complete media for 24 hours in the
presence of either 250 µg/ml of aptamer (100 µl; 10 µM, same as used for in vitro
experiments) or 2.5 mg/ml (100 µl; 100 µM), which represented the concentration of
aptamer pretreated MC38.CEA cells prior to tumour implantation (Figure 3.7A). None of
the aptamers incubated with MC38.CEA cells led to a loss of cell viability at these
concentrations, which suggests that the reduction in tumour implantation observed for
N54 and N56 was not due to aptamer-based cytotoxicity towards MC38.CEA cells.
In view of its potency as an inhibitor of CEA-mediated cell adhesion, we also
tested whether aptamer N54 could induce an innate immune response similar to
oligodeoxynucleotides containing CpG motifs, which promote Th1 responses by
signaling through TLR9. Mice received an i.p. injection of 100 µg (200 µl; 20 µM) of
control aptamer (cApt), aptamer N54 or 10 µg (100 µl; 500 nM) of a known TLR9 ligand
CpG ODN and the amount of TNFα and mouse IL-8 present in their serum was
quantified after 3 hours. Activation of TLR9 with the CpG ODN increased the serum
levels of mouse IL-8 by ~45-fold while aptamers cApt N54 yielded statistically non-
significant increases in serum IL-8 (Figure 3.7B). Similar results were observed in terms
of increased production of serum TNFα by CpG ODN but not by aptamers cApt and N54
(Figure 7C). These results suggest that N54 does not activate an in vivo innate immune
response associated with the secretion of inflammatory cytokines.
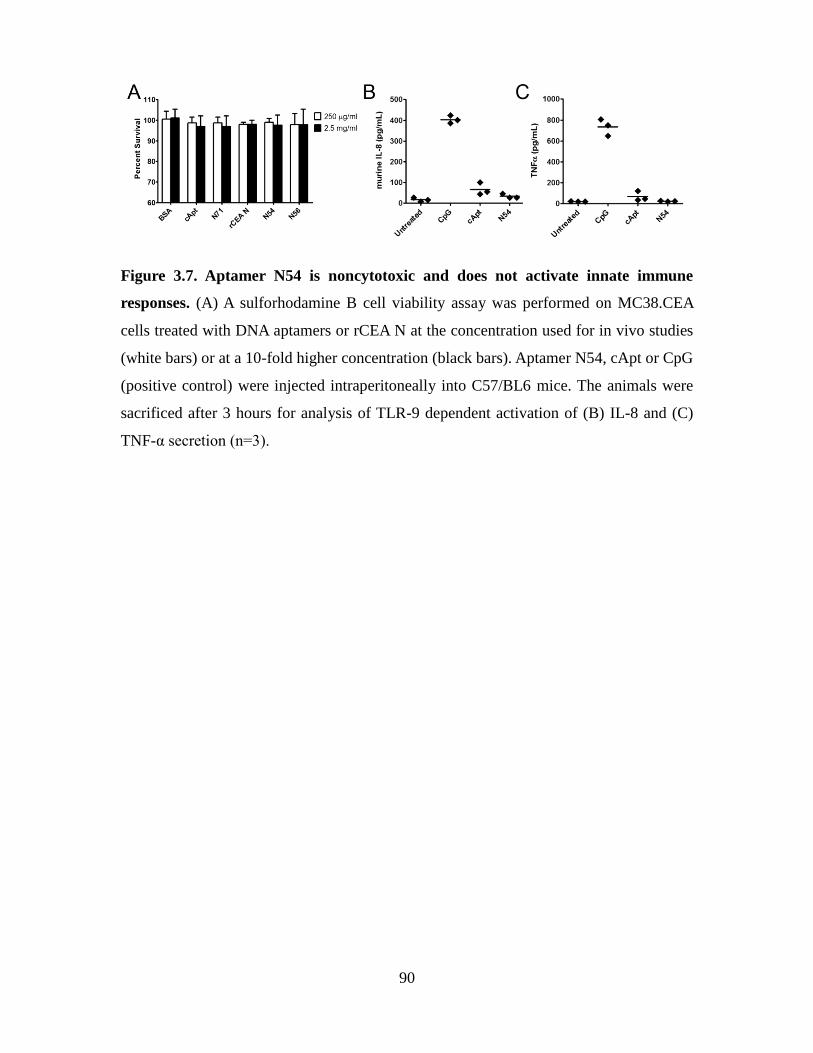
90
Figure 3.7. Aptamer N54 is noncytotoxic and does not activate innate immune
responses. (A) A sulforhodamine B cell viability assay was performed on MC38.CEA
cells treated with DNA aptamers or rCEA N at the concentration used for in vivo studies
(white bars) or at a 10-fold higher concentration (black bars). Aptamer N54, cApt or CpG
(positive control) were injected intraperitoneally into C57/BL6 mice. The animals were
sacrificed after 3 hours for analysis of TLR-9 dependent activation of (B) IL-8 and (C)
TNF-α secretion (n=3).

91
3.4. Discussion
Cancer cells typically display molecular signatures on their surface which have been
exploited more recently as targets for aptamers to chaperone therapeutic cargos into cells
(Orava et al., 2010). In the present study, functional DNA aptamers were developed
against the key homotypic binding domain of CEA generated from E. coli. We selected
DNA aptamers, as opposed to RNA aptamers, because they are more stable [less
susceptible to hydrolysis], less expensive and more easily derived by SELEX approaches
than RNA aptamers. Functional aptamers acting as mimics of molecules linked to
cellular signalling pathways have been reported and in some instances shown to block
events as diverse as angiogenesis, thrombosis, viral replication and inflammatory
responses (Bless et al., 1997; Bock et al., 1992; Boiziau et al., 1999; Chou et al., 2005;
Muller et al., 2009; Ng et al., 2006; Paborsky et al., 1993; Schneider et al., 1995). In the
context of adhesion, several aptamers have previously been reported which bind to a
group cell surface glycoproteins known as selectins (Huang et al., 1997; Schmidmaier
and Baumann, 2008).
Specifically, a phosphorothioate-modified aptamer to E-selectin named ESTA-1
bound with nanomolar affinity and inhibited over 75% of sialyl lewis X positive cells
from adhering to endothelial cells overexpressing E-selectin in vitro (Mann et al., 2010).
As well, an RNA aptamer to P-selectin known as ARC5690 which contained 2’-fluoro
pyrimidine and 2’-methoxy purines inhibited the adhesion of sickle red blood cells and
leukocytes to endothelial cells by 90% and 80% respectively in a sickle cell disease
model in vivo (Gutsaeva et al., 2010). Furthermore, a DNA aptamer to L-selectin
inhibited L-selectin-mediated rolling of lymphocyte and neutrophils on activated
endothelial cells in vitro as well as blocking lymphocyte trafficking to the lymph nodes in

92
vivo (Hicke et al., 1996). The present study highlights functional DNA aptamers able to
specifically inhibit CEA-expressing tumour cells from forming metastastic tumour foci.
Importantly, CEA plays a key role in tumour progression and the establishment of
metastatic foci by CEA-expressing tumours (Berinstein, 2002). CEA has been shown to
function as an intercellular adhesion molecule, as a function of reciprocal homophilic
binding between N and A3 domains (Jessup et al., 1993a; Taheri et al., 2000; Zhou et al.,
1993), an attribute that contributes to its tumourigenicity (Camacho-Leal and Stanners,
2008; Samara et al., 2007). Specifically, its involvement in homotypic and heterotypic
interactions correlates with the level of implantation and proliferation of CEA-expressing
tumours at distal sites such as the lungs, liver and peritoneal cavity (Asao et al., 1991;
Samara et al., 2007; Zhou et al., 1993; Zimmer and Thomas, 2001). CEA molecules bind
in a homotypic manner by virtue of the interaction between their N and A3 domains of
opposing CEA molecules, events which results in the formation of a network of
homophilic cellular contacts between CEA-expressing cells causing cell aggregation,
implantation and invasion of organs (Hostetter et al., 1990; Jessup et al., 1993b; Zhou et
al., 1993; Zimmer and Thomas, 2001). CEA has also been shown to heterotypically
interact with additional binding partners (Benchimol et al., 1989; Oikawa et al., 1989).
We hypothesized that blocking the homotypic binding of CEA would represent an
effective strategy for inhibiting its adhesive behaviour.
Using the CEA IgV-like N domain as a target for aptamer selection using the
SELEX process, we identified two unique 25-base long aptamers, termed N54 and N56
that possess the ability to inhibit CEA-mediated homotypic interactions (Figure 3.1). Of
the two aptamers, N54 displayed a moderately greater ability to inhibit these interactions
that was comparable to tumour-neutralizing antisera derived from mice vaccinated with

93
the rCEA N domain (Abdul-Wahid et al., 2012). Addition of N54 significantly inhibited
the binding of murine CEA-expressing MC38.CEA cells to wells coated with rCEA N
domain yet had no effect on CEA- MC38 cells adhering to plates coated with rCEA N
(Figure 3.2A and 3. 2B). These results demonstrate that aptamers N54 and N56 are able
to effectively block the homotypic interaction between the CEA N domains and CEA N
domain to rCEA A3B3.
Aptamers N54 and N56 differ greatly in the minimal regions needed to inhibit
homophilic cellular adhesion. Aptamer N54 requires both its primer and variable regions
as the deletion of a total of 18 bases from its 3`and 5` ends resulted in a loss of its
inhibition of CEA-mediated cell adhesion from ~50% to ~16% relative to the full length
sequence (Figure 3A and 3B). However, aptamer N56 can be truncated from 75 bases
down to 32 bases while retaining its inhibitory function (Figure 3.3A and 3.3B). Further
deletions to N56 however resulted in a loss of its inhibition of CEA cell adhesion
function.
Several members of the CEACAM family have been shown to be involved in
cell-cell interactions sharing a high level of sequence identity within their N domain with
CEA. Yet, aptamers N54 and N56 were able to uniquely bind to the N domain of CEA
(Figures 3.4 and 3.5). Structurally, the N domain of CEACAM1, CEAMCAM3, CEA,
CEACAM6 and CEACAM8 adopt an identical Ig V-like fold displaying defined faces.
The N domain of CEACAMs displays two faces: an ABED face and an opposite CFG
face (Korotkova et al., 2008; Taheri et al., 2000). The CFG interface of CEACAMs has
been shown to mediate CEACAM-CEACAM interactions (Figure 3.4 C) (Markel et al.,
2004). Furthermore, peptides to residues 42 to 46 (NRQII) and residues 80 to 84
(QNDTG) on CEA were found to modestly block CEA-mediated cellular aggregation

94
(Taheri et al., 2000) at concentrations >150-fold higher than aptamers N54 and N56.
Both of these peptide sequences are found on the CFG face of CEA (Taheri et al., 2000).
Interestingly, the peptide NRQII corresponding to residues 42 to 46 is unique to CEA
while the sequence QNDTG is found in CEACAM1 and CEACAM6. In view of the
specificity of aptamers N54 and N56 in binding to CEA, it would suggest that these
aptamers might interact with residues in the vicinity of peptide NRQII on the CFG face
of the CEA N domain.
One issue facing the use DNA aptamers as therapeutics is their short half-life in
vivo due to nuclease degradation and their rapid clearance through the kidneys (based on
their low molecular weights). The circulation half-lives of aptamers can be increased by
conjugating them to polyethylene glycol (PEG) moieties. As well, the stability in serum
can be increased by substituting nucleotides with modifications of either sugar residues
(eg. 2’ OH group with 2’-amino, 2’-fluoro 2’-O-methyl), the phosphate (eg.
Phosphorothioate) or of the base (2’-thiopyrimidine, methyl or trifluoromethyl) (Healy et
al., 2004; Latham et al., 1994; Mayer, 2009; Schoetzau et al., 2003; Willis et al., 1998).
Also, Locked Nucleic acids (LNA) have been introduced within their structures to
increase stability (Barciszewski et al., 2009). However, the in vivo tumour implantation
mouse model used in this study to assess the ability of aptamers N54 and N56 to inhibit
the adhesion and proliferation of murine MC38.CEA cells in the peritoneal cavity of
mice were performed with no modification to the aptamer structure. Both of these
aptamers were effective in inhibiting tumour implantation in the peritoneal model (Figure
3.6B). Interestingly, an RNA aptamer to CEA has recently been reported that prevents
hepatic metastasis (Lee et al., 2012). However, this RNA aptamer differs from the present
DNA apamers N54 and N56 in two key aspects. First, this RNA aptamer prevents the

95
binding of CEA to death receptor 5 (DR5), thus contributing to the prevention hepatic
metastasis by inducing anoikis. In contrast, aptamers N54 and N56 are non-cytotoxic
towards MC38.CEA cells and the treatment of MC38.CEA cells with aptamers N54 and
N56 did not lead to growth arrest/reduction (SRB cell viability assay) in relation to
untreated cells (Figure 3.7A). Secondly, the reported RNA aptamer to CEA was shown to
bind the PELPK motif present at residues 108-122 of CEA. This motif is present on
CEACAM1 and CEACAM 6: two broadly-expressed CEACAMs on normal tissues,
suggesting that this aptamer may not be specific for CEA (possible off-target effects). In
contrast, aptamers N54 and N56 showed specific binding to CEA suggesting that their
anti-adhesive effects focus on the specific inhibition of homotypic CEA interactions
(Figure 3.4). Importantly, the murine cell line MC38.CEA used for both in vitro and in
vivo studies, does not express other human CEACAM members on its surface. In view of
the specificity of aptamer N54 and N56 for the N domain of CEA only, these aptamers
are not expected to inhibit possible homophilic cellular interactions involving other
CEACAM members present on tumour cells.
Finally, DNA aptamers are generally considered to be non-immunogenic (Foy et
al., 2007; Yu et al., 2009). However, to confirm that aptamer N54, as an example, did not
generate an inflammatory innate immune response as seen for CpG ODN (a ligand for
TLR-9), mice were given via an intraperitoneal injection, a bolus of either an irrelevant
aptamer (cApt), aptamer N54 or a positive control CpG ODN, and their sera analyzed
after 3 hours for the production of inflammatory cytokines IL-8 and TNFα (Figure 3.7B
and C). As projected, aptamer N54 did not generate a serum increase in CpG ODN-
associated cytokines suggesting that this aptamer is non-immunogenic.
In summary, this study reports the identification of two DNA aptamers that are

96
able to specifically recognize the N domain of the cancer-associated antigen CEA and
block its homophilic adhesive properties. These aptamers specifically bound to the IgV-
like N domain of CEA, with a dissociation constant in the nanomolar range and
significantly inhibited tumour implantation of murine MC38.CEA cells by virtue of their
antiadhesive properties. As well, aptamer N54 displayed no cytotoxicity towards
MC38.CEA cells and did not trigger a TLR-9 dependent innate immune response.

97
Chapter 4
Aptamer-targeted liposomes for the computed tomography (CT)
imaging of breast cancer tumours
Erik W. Orava, Michael Dunne, Jinzi Zheng, Huang Huang, Christine Allen,
Jean Gariépy
Author Contributions:
Erik Orava, Dr. Jean Gariépy and Dr. Christine Allen designed experiments, analyzed the
data and prepared a manuscript.
Erik Orava contributed Figure 4.1, 4.2, 4.3, 4.4
Michael Dunne and Dr. Jinzi Zheng helped prepare the liposomes used in this study as
well as perform the animal imaging study at STTARR (Table 4.1).
Huang Huang was involved in preliminary experiments in characterizing aptamer
liposome conjugation.

98
4.0. Abstract
The in vivo targeting of nanoparticles such as pegylated liposomes to tumour cells can be
enhanced by introducing tumour marker-specific DNA aptamers on their surface. We
have recently generated DNA aptamers that specifically recognize either the mucin
MUC1 protein tandem repeat (40-base long MUC1-VR) or the N domain of the
carcinoembryonic antigen CEA (75-base long N54), both markers being aberrantly and
abundantly expressed on the surface of a broad range of epithelial cancer cells (breast,
colon, lung, ovarian, pancreatic and prostate). These DNA aptamers were conjugated via
an amino group to cyanur-PEG2000-DPSE and incorporated into preformed liposomes
(~50 aptamers/liposome) containing the contrast agent Omnipaque 350™. The level of
incorporation of aptamer was quantified using Cy5-labeled aptamers and found to be ~
95%. MUC1 and CEA surface antigens are internalized by cancer cells at different rates.
We report that a previously developed and characterized multimodal pegylated
liposomes loaded with the contrast agent for use in computed tomography (CT) imaging
modified to display these aptamers can either increase the binding to cells and
internalization. In vivo studies in mice have shown that the incorporation of these
aptamers on the liposome surface does not significantly alter the pharmacokinetic profile
of these nanoparticles. Current studies are underway to determine the extent of tumour
accumulation and internalization of the MUC1 and CEA targeted liposomes using an
MDA MB 231 orthotopic tumour model.

99
4.1. Introduction
A number of approaches are being employed to increase the selective toxicity of
anticancer agents. The discovery and application of single pathway-based cancer-targeted
drugs have showed some clinical promise, although their overall antitumour efficacy
being often temporary (Hanahan and Weinberg, 2011). One approach that is being
extensively investigated is the identification and use of interactome-based drugs to
interfere with newly identified cancer specific pathways (Roukos, 2012). Multimodal,
nanomaterial-based formulations offer the potential of delivering anticancer drugs and
contrast agents in a more directed fashion for guided in vivo imaging and therapeutic
approaches (Huang et al., 2012).
In theory, the best approach to improve the quality of life in patients with cancer
is to detect diseases as early as possible so that the timely initiation of therapy can
maximize the efficacy of the treatment. In the past decade, the field of nanomaterial-
based technologies has made fundamental advancements in the detection of diseases in
early stages as well as the delivery of drugs (Ryu et al., 2012; Taruttis and Ntziachristos,
2012). Magnetic nanoparticles incorporating inorganic nanocrystals as their magnetic
cores and composed of metals, alloys and metal oxides have been reported (Laurent et
al., 2008). The surfaces of these nanoparticles have also been modified with layers of
organic polymer or inorganic metal or oxide surfaces making them suitable for the
conjugation to biomolecules (Berry et al., 2003) with the resulting nanoparticles being
used for magnetic resonance imaging (MRI) and drug delivery (Fang and Zhang, 2009;
Veiseh et al., 2010). Metal nanoparticles have also been used by generating gold and
silver nanoparticles, which display excellent plasmonic properties advancing the use of
surface-enhanced Raman-spectroscopy (Ando et al., 2011; Sathuluri et al., 2011). Silica-

100
based nanoparticles have also been useful for encapsulating proteins such as the green
fluorescence protein (Cai et al., 2011) for cellular applications, in light of their nonporous
structure which protects GFP in terms of photo-stability and their versatile surface for
conjugating targeting moieties (Qhobosheane et al., 2001; Wittenberg and Haynes, 2009).
Hydrogels represent another category of nanomaterials composed of crosslinked polymer
structures that can retain large amounts of water or biological fluids within their matrix
and have found numerous medical and pharmaceutical applications due to their ability to
provide a slow controlled release of their cargo (Kashyap et al., 2005; Miyata et al.,
1999). Carbon-based nanomaterials, such as carbon nanotubes, fullerenes and recently
graphene and graphene oxide have shown promise in the field of cellular imaging and
drug delivery (Hong et al., 2012; Liu et al., 2011; Peng et al., 2010). More recently,
studies have shown these compounds are toxic at higher concentrations as MTT assays
failed to predict toxicity of graphene oxide and graphene sheets because of the reduction
of MTT by graphene resulting in a false positive signal (Liao et al., 2011). These
nanomaterials have been investigated due to their beneficial characteristics such as their
high surface area, mechanical strength, electrical and thermal conductivity and
photoluminescence. As such they have shown promise as sensors but also for imaging
applications (Ajayan, 1999).
Liposomes have had the greatest clinical impact as they have been used as a drug
delivery system for over 40 years (Bangham et al., 1965). These lipid-based
nanomaterials allow for retention of their cargo until they reach their intended target
organ. They can be modified with the addition of polyethyleneglycol (PEG), to enhance
their circulation time (Gregoriadis, 1995) and will accumulate at newly vascularized
tumour sites as a consequence of the enhanced permeability and retention (EPR) effect

101
(Maeda, 2001). At least twelve liposome-based drugs have now been approved for
clinical use the US Food and Drug Administration (FDA), four of which are for the
treatment of cancers and over twenty more liposomal formulations are currently in
clinical trials (Chang and Yeh, 2012). Importantly, antibodies, peptides, aptamers and
small molecular weight ligands have been introduced on the surface of nanoparticles to
target them to diseased tissues (Kumar et al., 2012).
In the present study, we report the incorporation of aptamer-lipid conjugates into
pegylated liposomes loaded with a contrast agent with a view to guide their delivery and
imaging of MDA -MB-231tumour xenografts implanted in athymic CD-1 mice (Figure
4.1). Aptamers are short single-stranded DNA or RNA oligonucleotides selected using a
PCR-based in vitro iterative process termed SELEX (Ellington and Szostak, 1990; Tuerk
and Gold, 1990). The identified aptamers bind to their target with high specificity and
affinity. DNA aptamers have been derived that can behave as sensors, therapeutics,
regulators of cellular processes or as cellular delivery agents (Holthoff and Bright, 2007;
Kaur and Roy, 2008; Orava et al., 2010; Toulme et al., 2004). DNA aptamers, unlike
antibodies, are not immunogenic, are easy to synthesize and to couple to nanoparticles,
and can be reversibly denatured (Yang et al., 2011). There are currently a dozen aptamers
in clinical trials with the anti-VEGF aptamer Pegaptanib (Macugen) approved since 2004
for age-related macular degeneration (Bunka et al., 2010; D'Amico et al., 2006).
Aptamer-functionalized nanomaterials are now being investigated in multiple fields of
research (Xing et al., 2012; Yang et al., 2011).
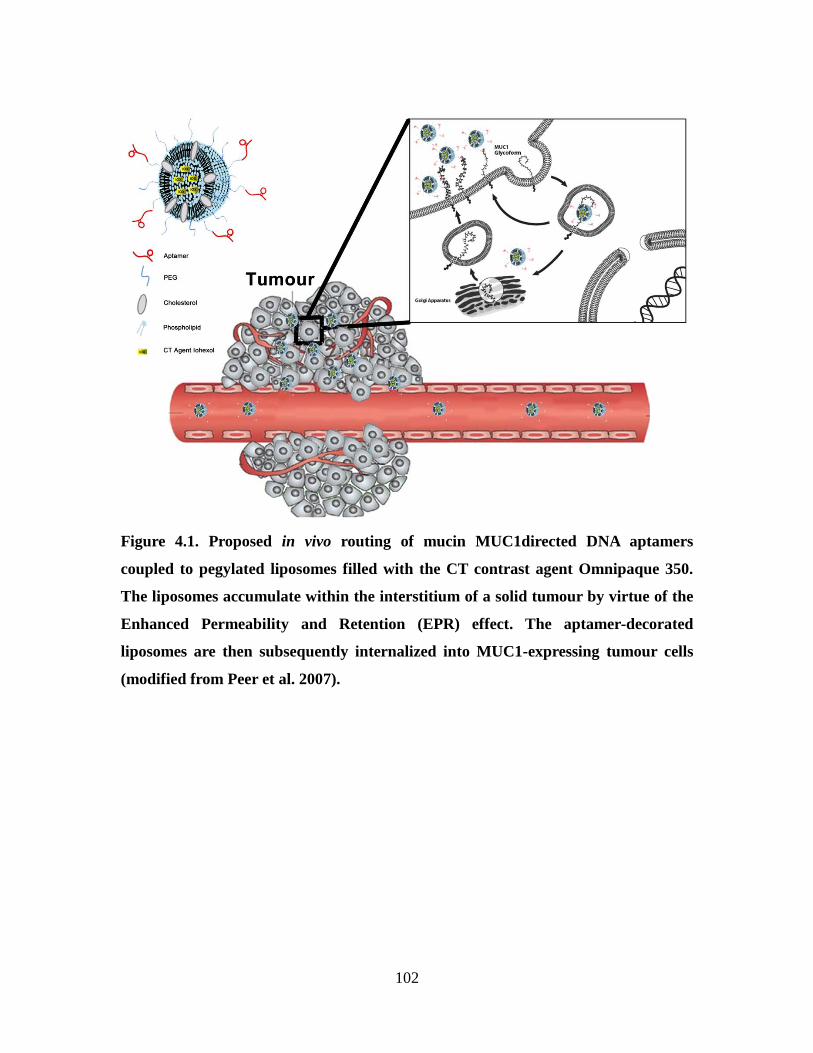
102
Figure 4.1. Proposed in vivo routing of mucin MUC1directed DNA aptamers
coupled to pegylated liposomes filled with the CT contrast agent Omnipaque 350.
The liposomes accumulate within the interstitium of a solid tumour by virtue of the
Enhanced Permeability and Retention (EPR) effect. The aptamer-decorated
liposomes are then subsequently internalized into MUC1-expressing tumour cells
(modified from Peer et al. 2007).

103
The DNA aptamers N54 and MUC1-VR used in the present study were respectively
derived against two established tumour markers, namely the carcinoembryonic antigen
(CEA) and the mucin MUC1. CEA is a 180 kDa GPI-linked cell glycoprotein that is also
a member of the immunoglobulin cell adhesion molecule superfamily (CEACAMs)(Gold
and Freedman, 1965a). It is aberrantly over-expressed on the surface of colon, breast,
lung and other epithelial cancer cells (Hammarstrom, 1999). Antibodies against CEA are
slowly internalized by CEA-expressing cells at a rate comparable to their metabolic
turnover rate suggesting that CEA may be a suitable target for tumour targeting (Schmidt
et al., 2008). The derivation of the CEA-specific aptamer N54 was described in Chapter
3. In contrast to CEA, the mucin MUC1 membrane glycoprotein is highly expressed and
aberrantly glycosylated in more than 90% of primary and metastatic breast cancers
(McGuckin et al., 1995) with mucin MUC1 underglycosylated forms such as the Tn
antigen being rapidly endocytosed by cells (Altschuler et al., 2000). Our group has
previously identified aptamers to the Tn antigen of MUC1 (Ferreira et al., 2009). A
second generation, 40-base long DNA aptamer MUC1-VR1 was derived for the present
study.
4.2. Materials and Methods
4.2.1. MUC1 expression, purification and glycosylation
A recombinant mucin MUC1 peptide composed of five 20-residue long MUC1 tandem
repeats was expressed in E.coli and purified to homogeneity by Ni-NTA chromatography
as previously reported (Brokx et al., 2003). The purity and sequence of the MUC1
peptide were confirmed by SDS-PAGE and mass spectrometry. The peptide was stored at

104
-20°C. Fifteen GalNac sugars (Tn antigens) were enzymatically introduced on the
hydroxyl side chain group of serines and threonines within each MUC1 tandem repeat
using a recombinant secreted form of the human glycosyltransferase ppGalNAc-T1
expressed in Pichia pastoris as previously described (Brokx et al, 2003). The level of
glycosylation was confirmed by SDS PAGE and mass spectrometry (Appendix 6).
4.2.2. Identification of MUC1 binding aptamers using the SELEX process
The initial ssDNA library contained a central randomized sequence of 40 nucleotides
flanked by two primer regions with the sequence 5’ GAC GAT AGC GGT GAC GGC
ACA GAC G-(25N)-CGT ATG CCG CTT CCG TCC GTC GCT C 3’. The forward
primer 5’ GAC GAT AGC GGT GAC GGC ACA GAC G 3’ and reverse primer 5’ GAG
CGA CGG ACG GAA GCG GCA TAC G 3’ were used for selection and cloning (IDT
Technologies, Inc.). A 50 nmol aliquot of the library was first counter-selected against
Ni-NTA agarose beads prior to selection against the GalNAc-modified MUC1 peptide
(Tn antigen) in order to remove DNA species that non-specifically bound to Ni-NTA
agarose itself. The resulting sub-library was then exposed to 5 µg of His-tagged
glycosylated MUC1 peptide dissolved in 200 µl of PBS, 10 µg/ml salmon sperm DNA
and 10 µg/ml BSA (selection buffer) for 30 min then added to a column containing 200
µl of Ni-NTA agarose beads (Qiagen Inc.). Unbound DNA sequences were washed away
with a 10-fold column volume excess of the selection buffer. DNA-protein complexes
were eluted from the beads with selection buffer containing 240mM imidazole. The
ssDNA component was precipitated with sodium perchlorate/isopropanol and recaptured
using a silica membrane-based purification system (Qiagen Inc., Mississauga, Ontario).
The DNA aptamers were then amplified by asymmetrical PCR using a 10-fold excess of

105
forward primer. After every second round of selection, the amount of target (Tn antigen
MUC1 peptide) was reduced in half to capture the tightest binding aptamers against the
MUC1 target. The bound sequences were amplified by PCR after 10 rounds of selection
to produce double stranded products, cloned into a pCR4-TOPO TA vector (Invitrogen)
and sequences were analyzed using BioEdit sequence alignment editor software (Ibis
Therapeutics, Carlsbad, USA).
4.2.3. MUC1 Aptamer binding and internalization
To determine if the identified MUC1 aptamers were able to specifically bind MUC1 on
MUC1+
cells, FITC-labeled MUC1-VR1, MUC1-VR4, MUC1-VR7 and MUC1-VR
were synthesized (IDT technologies) and incubated for 1 hour in 1 ml of DMEM + 10%
FBS with the human breast cancer cell line (MUC1+) MDA-MB-231 (Gilbey et al., 2004;
McGuckin et al., 1995) or the MUC1- human cell line HeLa at a density of 10
6 cells/ml at
4°C or 37°C with or without the addition of trypan blue to quench aptamer fluorescence.
Cells were subsequently placed on ice, washed three times with cold PBS (-MgCl2/-
CaCl2) and analyzed by flow cytometry for the binding of fluorescent MUC1 aptamers
(FACScan, BD Biosciences, Franklin Lakes, NJ). Experiments were repeated in triplicate
and averages were reported as mean fluorescence intensity + S.E.M.
4.2.4. Binding kinetics of MUC1 aptamers determined by surface plasmon
resonance
All experiments were performed using the ProteOn XPR36 surface plasmon resonance
instrument (Bio-Rad Laboratories, Inc.). The amine reactive ProteOn GLC sensor chip
was washed and activated according to manufacturer’s protocols. MUC1 and

106
glycosylated MUC1 peptides were diluted to 100 µg/ml in PBS-T (0.05% (v/v) Tween-
20), pH 7.4 and injected for 120 seconds at 30 µl/min. After the loading phase of protein
~100 and 150 RUs of protein was bound respectively. Control aptamer (cApt) used for
subsequent experiments was a 40 base sequence of AGTC repeats, 5’ AGT CAG TCA
GTC AGT CAG TCA GTC AGT CAG TCA GTC AGT C 3’. Subsequently aptamers
MUC1-VR1, MUC1-FL1 and cApt were diluted as a series of two-fold dilutions ranging
from 2µM to 3.9nM in PBS-T (0.05% (v/v) Tween-20) pH 7.4. Aptamer concentrations
and a buffer control were injected in the analyte channel with a contact time of 200 sec,
dissociation time of 800 sec, and a flow rate of 100 µl/min. The ligand channels were
regenerated with a 30 sec injection of 1M H3PO4 followed by a 30 sec injection of 1 M
NaCl. All experiments were performed at 25°C and repeated in duplicate. Sensorgrams
were then double-referenced by subtracting the buffer response and using the interspot
reference. The sensorgrams were fitted globally to a 1:1 Langmuir binding model and the
kinetic parameters for the association (ka), dissociation rates (kd), and binding constant
(KD) were derived from the fitted curves (MUC1-VR1 and MUC1-FL1 fitted to MUC1
and glycosylated MUC1 had χ2
> 0.93).
4.2.5. Synthesis of Aptamer-PEG2000-DSPE conjugates
Aptamers were synthesized with a C6 spacer 5’ Amino group (IDT Technologies) and
incubated in a 1:1 molar ratio with a cyanur-PEG2000-DPSE (Avanti Polar Lipids) in 0.1M
boric acid (pH 8.8) for 24 hours. Unconjugated aptamer was separated by dialysis
(MWCO 13 000) in 0.01M HEPES buffer (pH 7.4). The concentration of DNA in the
samples was measured at 260/280 nm using a nanodrop spectrophotometer. The coupling
yield of aptamers to cyanur-PEG2000-DPSE was 80%.

107
4.2.6. Liposome preparation
The liposomes were prepared as previously described (Zheng et al., 2006). Briefly,
DPPC, cholesterol, and DSPE-PEG2000 were dissolved in ethanol (250mM) at 70C at a
percent molar ratio of 55:40:5 DPPC:CH:DSPE-PEG2000. Omnipaque® (300mg/ml of
Iodine, GE Healthcare, Mississauga, ON) was added to yield a final lipid concentration
of 100 mM after ethanol removal and the mixture was kept at 70C for 6 h with
intermittent vortexing. The resulting multilamellar vesicles were then extruded at 70C
through a 10-mL Lipex Extruder (Northern Lipids Inc, Vancouver, British Columbia,
Canada) with five passages through two stacked 200 nm pore size polycarbonate filters
and five passages through two stacked 80 nm polycarbonate filters (Nucleopore; Track-
Etch Membrane, Whatman Inc., Clifton, NJ). Unencapsulated contrast agent was
removed by dialysis overnight (MWCO 8000) against HEPES buffered saline (HBS, pH
7.4). The liposome formulation was then concentrated to a final iodine concentration of
40mg/mL using a MidJet membrane separation system with a 750,000 NMWC MidGee
cross flow cartridge (GE Healthcare, Mississauga, ON). Targeted and untargeted
liposomes were generated by adding a specific aptamer-PEG2000-DSPE conjugate
(~2.5mg/250mM of lipid resulting in ~50 aptamers per liposome assuming a
homogenous size distribution) or a control PEG2000-DSPE conjugate to a liposome
suspension and each mixture was stirred at 37C for 5 hours following by stirring at
room temperature for 18 hours.

108
4.2.7. Efficiency of aptamer-DSPE-PEG insertion into liposomes
Aptamers were synthesized with a 5’ Cy5 fluorophore and a 3’ amino group (IDT
Technologies) and conjugated to cyanur-PEG2000-DPSE as previously described. The
fluorescence signal of Cy5-labeled aptamer-PEG2000-DPSE solutions (excitation 625 nm;
emission 670 nm) was measured prior to adding Cy5-aptamer-PEG2000-DPSE (from 30
µg to 2mg) to 25 mM of lipids for 30 minutes at 37°C then overnight at room
temperature (Ishida et al., 1999; Moreira et al., 2002). Unincorporated Cy5-aptamer-
PEG2000-DPSE conjugates were removed by dialysis (MWCO 30,000) in 0.01M HEPES
buffer (pH 7.4). The fluorescence signal of aliquots from each liposome formulation was
then measured and compared to our standard curve. Close to 95% of each evaluated Cy5-
aptamer-PEG2000-DPSE conjugate was incorporated into liposomes. Assuming that a 100
nm diameter [d] liposome contains ~80,000 lipids, the total number of lipids [Ntot] in
solution was calculated as follows Ntot = 17.69 × [ (d/2)2 + (d/2 – 5)
2 (Pidgeon and Hunt,
1981) with the number of liposomes (Nlipo) estimated to be equal to (Mlipid × NA) / (Ntot ×
1000) wherein Mlipid is the molar concentration of lipid and NA is Avogadro’s number
(6.02×1023
mol−1
). The estimated average number of aptamers per liposome was then
calculated by dividing the total number of aptamers by the total number of liposomes
assuming a homogeneous distribution of the aptamers on the liposomes and the near
complete integration of the Cy5-aptamer-PEG2000-DPSE conjugate added (as discussed
above).
4.2.8. Aptamer-liposome characterization in vitro
The human breast cancer cell lines MCF-7 and MDA-MB-231 as well as the human
cervical adenocarcinoma cell line HeLa, murine fibroblast cell line L929 and rat

109
gliosarcoma cell line 9L were cultured at 37ºC, 5.0% CO2 in Dulbecco’s modified Eagle’s
medium supplemented with 10% fetal bovine serum, penicillin (100 U/mL) and
dihydrostreptomycin (100 µg/mL). MDA-MB-231 or HeLa cells at a final density of 107
cells/ml in 100 µl of DMEM were treated to a final concentration of 20 µM of either Tn
antigen-directed, CEA-directed, cApt-directed aptamer-liposomes or non-targeted
liposomes for 1 hour at 4°C to reduce internalization. The cells were then washed three
times with cold PBS and analyzed by flow cytometry. For aptamer-liposome
concentration studies, MDA-MB-231 and HeLa cells at a final density of 107 cells/ml in
100 µl of DMEM, were exposed to increasing two-fold concentrations (from 5-80 µM)
of aptamer-liposomes displaying ~50 aptamers per liposome or to non-targeted
liposomes for 1 hour at 4°C.
For internalization studies, MDA-MB-231 or HeLa cells [107 cells/ml in 100 µl
DMEM] were incubated with 20 µM aptamer-liposomes (~50 aptamers/liposome) or
non-targeted liposomes at 4°C or 37°C, for 0.5, 1, 2, 4, 8 and 24 hours. All studies were
repeated in triplicate and averages are represented as mean fluorescence intensity ±
S.E.M.
4.2.9. Physio-chemical characterization of aptamer-liposome formulations
The hydrodynamic particle diameter and zeta potential of aptamer-liposome formulations
as well as non-targeted liposomes were determined by dynamic light scattering (DLS)
using a Zetasizer nano ZS (Malvern Instruments Ltd.).

110
4.2.10. Aptamer-liposome pharmacokinetic profile
The liposome preparations (0.35g/kg iodine and 1.8g/kg lipid) were injected into athymic
female CD-1 mice via the tail vein. The animals were subsequently anaesthetized using
2% isoflurane and full body, 16-second anatomical micro-CT scans (GE Locus Ultra
micro-CT, GE Healthcare, Waunakee, WI) were recorded prior to liposome
administration and at 5 min, 2h, 4h, 8h, 24h, 48h and 72 hours post-injection intervals.
CT images were acquired at 80kVp and 70mA with a voxel size of 0.15mm x 0.15mm x
0.15mm and a field of view of 0.57cm x 0.57cm x 10.5cm.
CT images were analyzed using MicroView v2.2 (GE Healthcare, Waunakee, WI).
Briefly, the mean iodine concentrations in selected organs were determined through
contouring the desired region on multiple 2D slices to generate a 3D volume and
calculating the mean voxel intensity (expressed in CT attenuation Hounsfield Units, or
HU) for each volume. Iodine concentrations in blood were determined by contouring the
aorta.
4.2.11. Statistical methods and data analysis
Data sets derived from individual groups of mice were compared using student’s t-test
and grouped data sets were analyzed by ANOVA. Statistical analyses and graphs were
assembled using GraphPad PRISM (version 5.01, GraphPad software for Science, San
Diego, CA). P values ≤ 0.05 were considered significant unless otherwise indicated.
Flow cytometry statistics were analyzed using WinMDI (version 2.8, Windows multiple
document interface for flow cytometry, Scripps Research Institute).

111
4.3. Results
4.3.1. Identification and characterization of MUC1-VR1, a MUC1-binding
aptamer
The target used to identify MUC1-binding DNA aptamers generated using a SELEX
approach, was a recombinant MUC1 peptide corresponding to five consecutive 20-
residue long tandem repeats of the mucin MUC1 protein extracellular domain. This
peptide was enzymatically modified with the human glycosyltransferase ppGalNAc-T1
to harbor up to 15 GalNAc sugars known as Tn antigens (Brokx et al. 2006). Forty
recovered oligonucleotide inserts from the SELEX search were sequenced yielding 37
DNA aptamer sequences that could be regrouped into 4 unique sequences (Figure 4.2A).
In particular, MUC1-VR1 (40-base long variable region) and MUC1-FL1 (primers and
the same 40-base long variable region) accounted for 86% of identified aptamer
sequences. All 4 DNA aptamer sequences were then synthesized with a 5’ biotin group to
confirm their binding to the MUC1 Tn antigen using a modified ELISA protocol
(Appendix 7) (Orava et al., 2012). The ELISA binding assay confirmed that of the 4
identified sequences, 3 of the 40-base aptamers specifically bound to the Tn antigen
MUC1 peptide, namely MUC1-VR1, MUC1-VR4 and MUC1-7.
The ability and specificity of these aptamers to bind to Tn antigen-containing mucin
MUC1 on the surface of cancer cells was subsequently assessed using FITC-labeled
MUC1 aptamers as well as the FITC-labeled CEA-binding aptamer N54 (5’ GAC GAT
AGC GGT GAC GGC ACA GAC GTC CCG CAT CCT CCG CCG TGC CGA CCC
GTA TGC CGC TTC CGT CCG TCG CTC 3’; Orava et al. 2013). The MUC1-VR1, -
VR4 and -VR7 aptamers all bound to the CEA+/MUC1
+ cell lines MDA MB 231 (Gilbey
et al., 2004; McGuckin et al., 1995) and MCF-7 while none of these fluorescent aptamers

112
recognized CEA-/MUC1
- cell lines 9L, L929 and HeLa (Appendix 8 and 9). MUC1
expression on cells was confirmed using the anti-MUC1 M23 antibody (Linsley et al.,
1988) and a FITC-labeled mouse anti-human secondary antibody (Appendix 10).
Although MUC1-VR1, MUC1-VR4 and MUC1-7 bound to Tn antigen MUC1 peptide
using ELISA-based method and flow cytometry showed binding to MUC1+
cells, only
MUC1-VR1 and its full length MUC1-FL1 homolog showed binding to the MUC1
peptide and its Tn antigen MUC1 variant by SPR. The binding constant of MUC1-VR
and MUC1-FL aptamers were determined by surface plasmon resonance by coupling
unglycosylated MUC1 peptide and the Tn antigen-containing MUC1 peptide variant to
an amine reactive chip. Of the three aptamers tested, aptamer MUC1-VR1 showed no
significant difference in binding affinity between the MUC1 peptide and Tn antigen form
(410 ± 130 nM and 380 ± 90 nM). Interestingly, the MUC1-FL1 aptamer was ~7-more
avid to the MUC1 peptide relative to the Tn antigen-labeled MUC1 peptide (230 ± 60
nM vs. 1600 ± 400 nM) (Figure 4.2 B). Since there was no significant difference between
the binding constants of MUC1-VR1 and MUC1-FL1 to the Tn antigen-labeled MUC1
peptide we chose to continue our studies using the smaller shorter MUC1-VR1 aptamer.
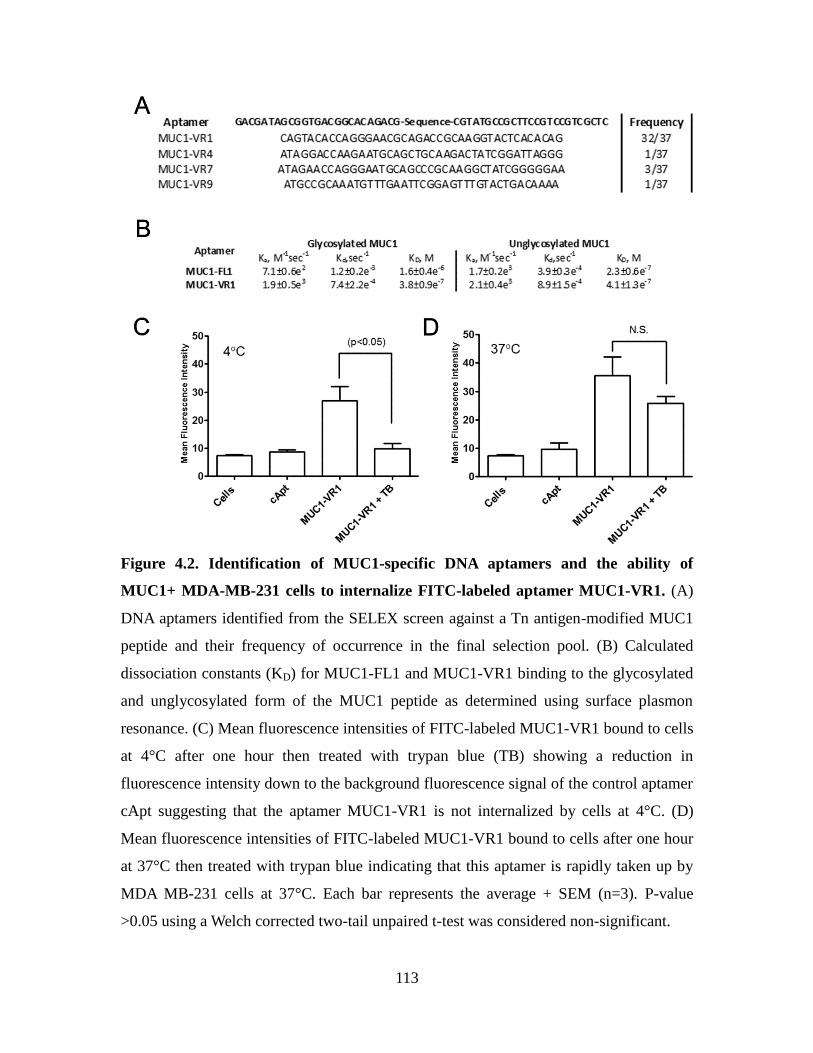
113
Figure 4.2. Identification of MUC1-specific DNA aptamers and the ability of
MUC1+ MDA-MB-231 cells to internalize FITC-labeled aptamer MUC1-VR1. (A)
DNA aptamers identified from the SELEX screen against a Tn antigen-modified MUC1
peptide and their frequency of occurrence in the final selection pool. (B) Calculated
dissociation constants (KD) for MUC1-FL1 and MUC1-VR1 binding to the glycosylated
and unglycosylated form of the MUC1 peptide as determined using surface plasmon
resonance. (C) Mean fluorescence intensities of FITC-labeled MUC1-VR1 bound to cells
at 4°C after one hour then treated with trypan blue (TB) showing a reduction in
fluorescence intensity down to the background fluorescence signal of the control aptamer
cApt suggesting that the aptamer MUC1-VR1 is not internalized by cells at 4°C. (D)
Mean fluorescence intensities of FITC-labeled MUC1-VR1 bound to cells after one hour
at 37°C then treated with trypan blue indicating that this aptamer is rapidly taken up by
MDA MB-231 cells at 37°C. Each bar represents the average + SEM (n=3). P-value
>0.05 using a Welch corrected two-tail unpaired t-test was considered non-significant.

114
The internalization of FITC-labeled MUC1-VR1 aptamer into cells was measured
by FACS analysis at 37°C (surface binding and internalized components) and 4°C
(surface binding only), after a 1-hour incubation period. The mean fluorescence
intensities (MFI) from aptamer bound to the surface of cells was quenched with trypan
blue prior to FACS experiments. Cells treated with trypan blue at 4°C showed a
significant decrease in fluorescence signal when compared to MUC1-VR1 with no
quenching but no such difference in MFIs was observed for the control FITC-labeled
aptamer cApt which does not bind the MDA MB 231. Importantly, cells treated with
trypan blue after binding and internalization at 37°C showed no significant decrease in
MFI when compared to cells treated with MUC1-VR1 alone suggesting that the aptamer
was being internalized into target cells.
4.3.2. Optimization of MUC1-VR1 and N54 aptamer-targeted liposomes in vitro
Cy5-labeled MUC1-VR1, N54 and cApt (control) aptamers were conjugated via their 3’
end amino group to cyanur-PEG2000-DPSE. The resulting conjugates were incorporated
into preformed pegylated liposomes. Greater than 95% of the Cy5-labeled aptamer-PEG
lipid constructs were inserted into liposomes up to a density of 320 aptamers per
liposome (Appendix 11) (Moreira et al., 2002). As well, the binding of Cy-5 labeled
aptamer N54 was confirmed by FACS on MCF-7 and MDA-MB-231 cells before
incorporation (Appendix 12). At the lowest aptamer concentration tested (10 aptamers
per liposome) there was ~2-fold increase in MFIs of MDA-MB-231 cells treated with
MUC1-VR1 and N54 as compared to the control aptamer (Figure 4.3A). At a
concentration of ~40 aptamers per liposome, MUC1-VR1- and N54-modified liposomes
respectively showed a 7-fold and 5-fold increase in cell surface-associated fluorescence

115
intensities as compared to cApt liposomes.
The effect of aptamer-liposomes concentration on cell surface association to
MDA-MB-231 and HeLa cells at 4°C was monitored by FACS analysis using a loading
of 50 Cy5-labeled aptamers per liposome. At the lowest concentration tested of 5 µM, the
MUC1-VR1 and N54 liposomes showed ~3-fold and 2.5-fold increase in MFIs as
compared to the cApt and non-targeted liposomes on CEA+/MUC1
+ MDA-MB-231 cells
(Figure 4.3C). Interestingly, at concentrations at or above 20 µM, there was a significant
increase in MUC1-VR1 liposomes binding (MFIs) to CEA+/MUC1
+ MDA-MB-231 cells
as compared to the N54-tagged liposomes. As expected, no significant difference in MFIs
was observed between aptamer groups on CEA-/MUC1
- HeLa cells although a small
increase in nonspecific binding (MFIs) occurred for non-targeted liposomes at
concentrations ≥ 40 µM (Figure 4.3D).
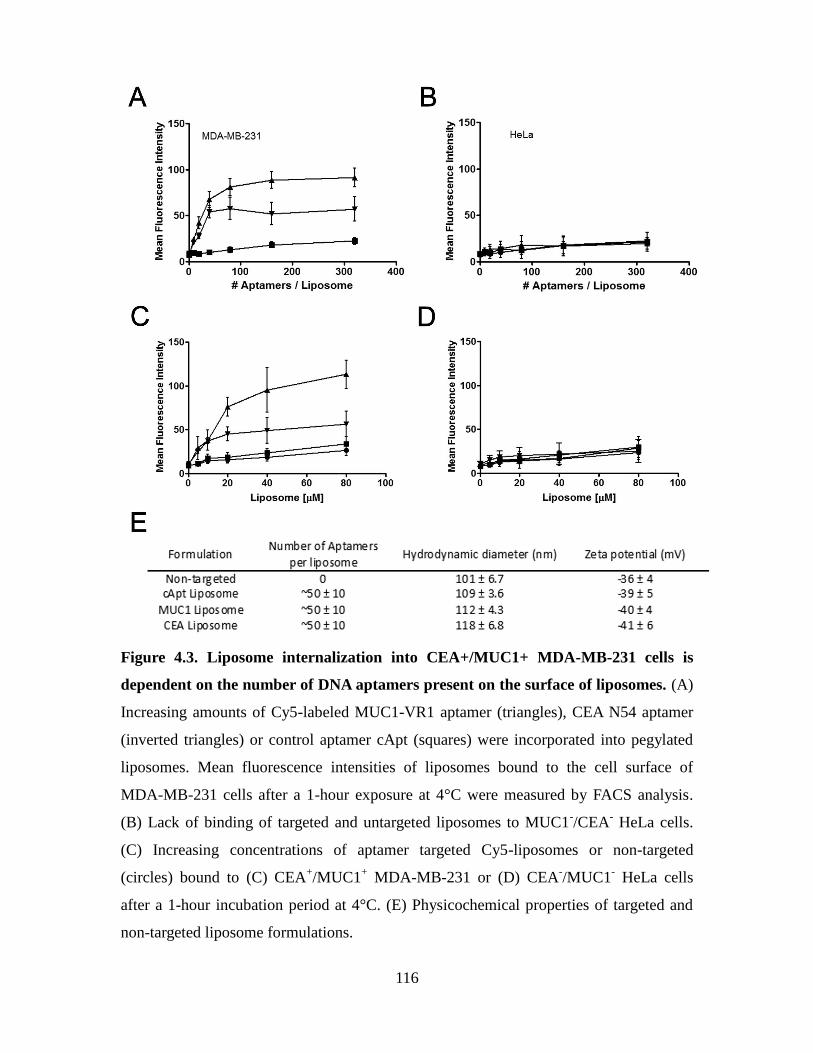
116
Figure 4.3. Liposome internalization into CEA+/MUC1+ MDA-MB-231 cells is
dependent on the number of DNA aptamers present on the surface of liposomes. (A)
Increasing amounts of Cy5-labeled MUC1-VR1 aptamer (triangles), CEA N54 aptamer
(inverted triangles) or control aptamer cApt (squares) were incorporated into pegylated
liposomes. Mean fluorescence intensities of liposomes bound to the cell surface of
MDA-MB-231 cells after a 1-hour exposure at 4°C were measured by FACS analysis.
(B) Lack of binding of targeted and untargeted liposomes to MUC1-/CEA
- HeLa cells.
(C) Increasing concentrations of aptamer targeted Cy5-liposomes or non-targeted
(circles) bound to (C) CEA+/MUC1
+ MDA-MB-231 or (D) CEA
-/MUC1
- HeLa cells
after a 1-hour incubation period at 4°C. (E) Physicochemical properties of targeted and
non-targeted liposome formulations.

117
4.3.3. Aptamer-Liposome internalization in MDA-MB-231 cells
Cellular uptake and recycling of surface markers takes place at 37°C. The effect of
temperature on the binding and cellular uptake of Cy5-labeled MUC1-VR1, N54 or
cApt-tagged aptamer-liposomes into MDA-MB-231 and HeLa cells was monitored by
FACS analysis at 4°C (surface binding) and 37°C (binding and internalization) using a
loading of 50 Cy5-labeled aptamers per liposome. At 4°C, both N54- and MUC1-VR1
liposomes reached a maximum fluorescence signal within the first 2 hours of incubation
with MDA-MB-231 cells with no significant difference in MFI being observed after a
24-hour incubation period (Figure 4.4A). The same aptamer liposomes did not
significantly bind to MUC1-/CEA
- HeLa cells even after 24 hours (Figure 4.4C). At
37°C, fluorescence signals associated with both N54- and MUC1-VR1 liposomes
increase with time although the uptake of MUC1-VR1 was significantly more rapid
reaching a plateau at 4 hours as opposed to the CEA N54-labeled liposome preparation
which led to increased MFI values only after 24 hours (Figure 4.4B). Their uptake levels
into HeLa cells at 37°C were comparable to that of untargeted liposomes (Figure 4.4D).
Although comparable to fluorescence values observed at 4°C, the nonspecific uptake of
cApt-labeled or untargeted liposomes by MDA-MB-231 and HeLa cells showed a slight
increase in MFI values with time at 37°C (Panels 4.4B and 4.4D).
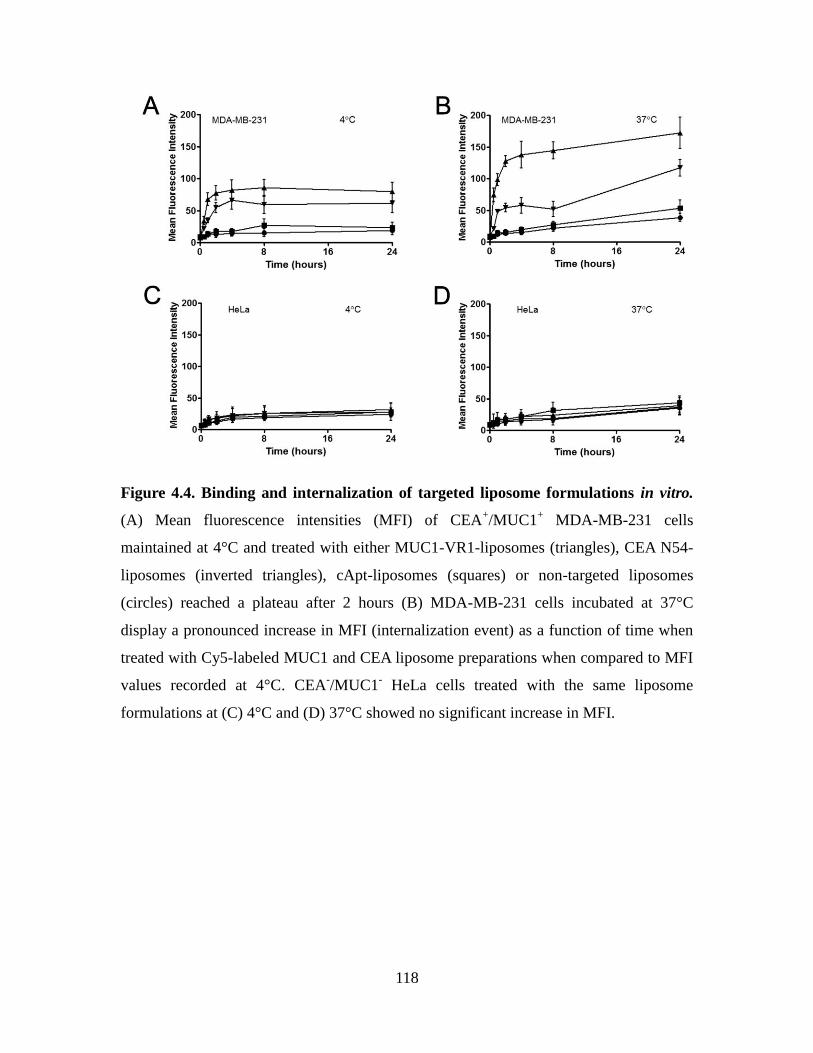
118
Figure 4.4. Binding and internalization of targeted liposome formulations in vitro.
(A) Mean fluorescence intensities (MFI) of CEA+/MUC1
+ MDA-MB-231 cells
maintained at 4°C and treated with either MUC1-VR1-liposomes (triangles), CEA N54-
liposomes (inverted triangles), cApt-liposomes (squares) or non-targeted liposomes
(circles) reached a plateau after 2 hours (B) MDA-MB-231 cells incubated at 37°C
display a pronounced increase in MFI (internalization event) as a function of time when
treated with Cy5-labeled MUC1 and CEA liposome preparations when compared to MFI
values recorded at 4°C. CEA-/MUC1
- HeLa cells treated with the same liposome
formulations at (C) 4°C and (D) 37°C showed no significant increase in MFI.

119
4.3.4. Aptamer-Liposome physicochemical properties and stability in vivo
Liposome preparations formulated with aptamers were analyzed by light scattering
[Zetasizer] to define their hydrodynamic diameter and zeta-potential in relation to non-
targeted liposome formulations. The incorporation of MUC1-VR1 and N54 into
liposomes resulted in ~10% and ~20% larger liposome diameter as expected from the
fact that the CEA aptamer N54 has 35 more bases than the 40-base long MUC1-VR1
aptamer (Figure 4.3E). The incorporation of aptamers decreased the surface potential of
the liposomes by ~10% as compared to non-targeted liposomes although these changes
were not statistically significant.
To determine whether the incorporation of aptamers MUC1-VR1, N54 and cApt
would significantly affect the stability of the liposomes, preparations were injected via
the tail vein into CD1 mice and imaged at time intervals of 0, 0.5, 1, 2, 4, 8, 24, 48 and
72 hours. Images were examined at the different time points and the descending aorta
was contoured to determine the amount of liposomes in the blood by quantifying the
amount of contrast agent. The results showed that there was no significant difference in
the elimination half-life of the aptamer-liposomes compared to the non-targeted
formulation suggesting that the incorporation of the DNA aptamers did not alter the
stability of the liposomes (student’s t-test, p>0.05) (Table 4.1).
Table 0.1. Pharmacokinetics of different Omnipaque encapsulated non-targeted and
aptamer targeted liposome preparations in CD1 mice (n=5 per group).
T1/2 (h) in blood # Aptamers per liposome Aptamer size
Non-targeted Liposome 39.10 ± 6.22 0 N/A
cApt Lipsome 38.44 ± 4.03 ~50±10 40 b (~13 kDa)
MUC1 Liposome 35.90 ± 2.35 ~50±10 40 b (~13 kDa)
CEA Liposome 37.69 ± 3.12 ~50±10 75 b (~24.6kDa)

120
4.4. Discussion
The targeting of nanoparticles and nanomaterials with aptamers offers the
potential to increase their accumulation at tumour sites by binding specifically to unique
cancer markers.
For instance, the aptamer TTA1 directed at Tenascin-C, an extracellular matrix
protein that is expressed during tissue remodeling and notably tumour growth was
radiolabeled with 99M
-Tc and reported to display good tumour penetration and a high
tumour-to-blood ratio most likely due to its rapid clearance (Hicke et al., 2006).
Aptamers have recently been derived to the mucin MUC1 protein (Ferreira et al., 2009);
a well-known tumour marker that is overexpressed and underglycosylated on epithelial
cancers. Studies have shown that MUC1-directed aptamers are capable of delivering
cytotoxic agents or as targeting agents in the context of PLGA nanoparticles in vitro (Hu
et al., 2012; Yu et al., 2011). The biodistribution of a PEGylated MUC1 aptamer as well
as their ability to target quantum-dot-doxorubicin conjugates in vivo studies have also
been reported (Da Pieve et al., 2012; Savla et al., 2011). To date, no studies have shown
the utility of an aptamer recognizing CEA as a targeting agent.
In the present studies, we show that a loading of >80 MUC1-VR1 or CEA N54
aptamers per liposome is not essential for the maximal in vitro uptake of such
formulations into the CEA+/MUC1
+ human breast cancer MDA-MB-231 cells (Figure
4.3A). This level of aptamer loading is comparable to the density of 34-60 VEGF
aptamers per liposome for 50 nm nanoparticles reported in the past (Willis et al., 1998)
but less than the loading of 500 aptamers per liposome required to achieve a pronounced
targeting effect for an aptamer directed at E-selectin (Mann et al., 2011).
We did observe that the MUC1 aptamer MUC1-VR1 had a greater amount of cell

121
association as compared to our CEA N54 aptamer (Figure 4.4A). Since the CEA aptamer
had a higher reported binding constant than MUC1-VR1 for its target (~45 nM vs. ~400
nM), the lower amount of cell association may reflect a lower level of CEA expression
on the cell surface of MDA-MB-231 cells or could be that surface CEA is being shed
from cells.
Importantly, the rates of internalization of CEA and mucin MUC1 by MDA-MB-
231 cells showed significant differences. At 37°C, the MUC1-VR1 aptamer displays an
estimated internalization rate of <1 hour which is consistent with the published recycling
rate of MUC1 on cells (Altschuler et al., 2000) (Figure 4.4B). However, MDA-MB-231
cells significantly accumulate CEA N54-tagged liposomes over a period of 24 hours
(Figure 4.4B). There was no significant difference in mean fluorescence intensities at 8
hours between 4°C and 37°C exposure of CEA-targeted liposomes to MDA-MB-231
cells, suggesting that the CEA liposomes were being internalized after this time point.
This finding is consistent with the metabolic turnover rate of CEA on cells of between
~10 – 17 hours using monoclonal antibodies to CEA (Schmidt et al., 2008). As expected,
no significant difference in cell-associated fluorescence was observed for CEA-/ MUC1
-
HeLa cells treated with MUC1-VR1- or N54-liposomes at either 4°C or 37°C as
compared to the non-targeted and cApt (control) liposome (Figure 4.4C, 4.4D).
These aptamer-targeted pegylated liposomes were loaded with the contrast agent
and injected intravenously (tail vein) into mice to establish their circulatory half-life in
the blood over a period of 72 hours. CT results confirmed that the incorporation of
aptamers into pegylated liposomes did not significantly alter their stability and
circulation half-life (Table 4.1). Interestingly, the non-targeted liposomes were
considered to possess a very negative zeta potential (-36 ± 4 mV) and it was thought the

122
incorporation of aptamers would make the formulation significantly more negative.
However, it was found that the average zeta potential of these pegylated liposome
preparations were not statistically different, being comparable to that of non-targeted
pegylated liposomes (Figure 4.3E).
Although tumour accumulation studies have not yet been completed, the affinity
difference in these aptamers and their respective targets on cancer cells should allow for
an interesting comparative study. Recently, studies using HER2-specific antibodies as
targeting agents demonstrated that differing avidities can affect the level of tumour
penetration and internalization in solid tumours (Rudnick et al., 2011). This finding
would suggest that MUC1-VR1 aptamer liposomes may accumulate to a greater degree
in the tumour than untargeted liposome formulations. As well, it was shown in a
comparative study that the in vitro internalization and in vivo distribution of fast-
internalizing and slow-internalizing receptors showed a significant difference in tumour
accumulation patterns (Bryan et al., 2005). This study would suggest that targeting a non-
or slow-internalizing receptor such as CEA might yield better tumour accumulation than
a rapidly internalizing receptor such as MUC1. As well, characterizing the ability of
these aptamers to target this liposome formulation encapsulating a contrast agent for
imaging would allow an easy transition to engineering similar liposomes with a variety
of chemotherapeutic drugs.
We have shown in vitro that CEA and MUC1 aptamer targeted pegylated
liposomes are able to bind and be internalized by CEA+/MUC1
+ MDA-MB-231 cells but
not by MUC1-/CEA
- HeLa cells. The incorporation of these aptamers into pegylated
liposomes did not affect their stability in vivo or their circulatory half-life of ~ 36 hours.
Studies are currently underway to determine that biodistribution and internalizing rates of

123
these aptamer-targeted liposomes in vivo.

124
Chapter 5
Overall Thesis Conclusions and Future Research

125
5.1. Thesis Conclusions
The main focus of my thesis was to derive functional aptamers to targets that could
functionally inhibit their function in vitro and in vivo. The rationale for pursuing such an
objective was that therapeutic targets such as TNFα and CEA can have a therapeutic
advantage in blocking binding to their cognate receptors as well as their adhesive
properties. In chapter 2, I described how one can successfully identify a DNA aptamer
able to block TNFα and confirmed this in vitro by monitoring the blocking of the
downstream signaling in NfKB (Figure 2.1) as well as blocking TNFα induced
cytotoxicity (Figure 2.2). The inhibitory aptamer VR11 and aptamer VR20 both bound to
TNFα with low nanomolar affinities and did not bind to the closest known homolog
TNFβ (Figure 2.3). Aptamer VR11 was also able to reduce TNFα dependant NO
production in macrophages (Figure 2.4). Circular dichroism spectroscopy also suggests
that VR11 does not fold into a G-quadruplex structure however there may exist a
secondary structure that aids in binding and blocking TNFα (Figure 2.5). Lastly, it was
also determined that VR11 does induce an innate immune response via TLR9 in
C57BL/6 mice despite harbouring two possible CpG motifs (Figure 2.6).
In Chapter 3, the homotypic binding domain (N domain) of the human
carcinoembryonic antigen (CEA) was selected as a target for aptamer searches since
cancer cells expressing CEA have been shown to aggregate and promote tumour foci
formation. There is a need to develop new concepts and therapies that can halt or control
the establishment or expansion of secondary tumour foci. Thus, the identification of
aptamers able to block such intercellular interactions could in theory halt the formation
of tumour metastasis. CEA represents a logical target for designing new therapies as it is
over-expressed on many epithelial cancer tissues and serves key roles in cellular

126
aggregation processes and attachment to extracellular matrix elements. We initially
identified aptamers, specifically N54 and N56, were able to block the homotypic and
heterotypic binding of recombinant N and A3B3 domains (Figure 3.1). Subsequently we
confirmed the ability of these aptamers to block the adhesion of the CEA expressing
murine cell line MC38.CEA to rCEA and r A3B3 immoblized on wells (Figure 3.2). The
truncations of aptamer N54 resulted in a loss of anti-adhesive properties while aptamer
N56 could be truncated to a 32-base sequence without any loss of its inhibitory properties
relative to the full-length sequence (Figure 3.3). Despite a high level of homology
between the N domain of CEACAMs (Figure 3.4), Cy5-labelled aptamers N54 and N56
did not bind by FACS to other CEACAMs known to be involved in cellular adhesion
processes suggesting these aptamers are specific to CEA (CEACAM5) (Figure 3.5). Most
importantly, it was shown that pretreatment of MC38.CEA cells with aptamer N54 and
N56 prevented the tumour implantation in an intraperitoneal model (Figure 3.6). This
was not due to an induction of apoptosis or an innate immune response caused by the
aptamers (Figure 3.7). This study provided the first, direct evidence that aptamer-based,
CEA-directed, anti-adhesive strategies can block metastatic foci formation in vivo.
Since DNA aptamers are small molecules displaying short circulating half lives in
vivo, I explored in Chapter 4, the design and construction of long-circulating pegylated
liposomes decorated on their surface with aptamers. DNA aptamers directed at two well-
known tumour markers, namely CEA and the mucin protein MUC1 were derivatized
with a pegylated lipid tail and inserted into pegylated liposomes commonly used to
deliver imaging agents and therapeutic drugs to tumour xenografts in
immunocompromised mice. I explored the design of pegylated liposomes decorated with
aptamers. In 2012, approximately 26 percent of all cancer cases in Canadian women

127
were breast cancer and represents the second leading cause of cancer deaths and third
leading cause of death. This year there will be an estimated 22700 more Canadian
women diagnosed with breast cancer however breast cancer deaths have decreased nearly
40 percent since its peak in 1986 partially attributed to more advanced screaning
technology and improved treatments (Jemal et al., 2008). Targeting unique cancer marker
such as CEA and MUC1 represents a novel strategy to increase detection of breast cancer
tumours by delivering contrast agent encapsulated liposomes. We have shown in vitro
that these aptamers can recognize several breast cancer cell lines and appear to have
effect on binding to CEA-/MUC
- cells. These aptamers also had no effect on the
pharmacokinetic profile of the previously reported liposome formulations. Our data
suggest that these formulations may have the ability to increase tumour accumulation or
internalization into breast cancer cells.
5.2. Future Directions
5.2.1. Modification of aptamer VR11 for in vivo
Single-stranded DNA aptamers themselves cannot be used in in vivo studies that require
them to circulate longer without modifications to stabilize them from degradation.
Without modification previous studies have shown that the circulation time of these
aptamers are only a few minutes (Healy et al., 2004). It has previously been shown that
modifications such as conjugation to polyethylene glycol (PEG) groups allow for the
prolonged circulation time of aptamers in mice (Boomer et al., 2005). This study
demonstrated that DNA aptamers, when conjugated to a 20kDa or 40kDa PEG group
were able to achieve therapeutic concentrations at the sites of inflammation. Another

128
mode of prolonging circulation is by conjugation to liposomes by conjugating the
aptamers to PEG-lipid groups and inserting into preformed liposomes (as described in
Chapter 4). In the case of the TNFα aptamer VR11, the VR11 conjugated liposomes
would significantly increase the circulatory half-life of the aptamer as well as passively
target them to the sites of inflammation (Boomer et al., 2005)
5.2.2. Determine the ability of aptamer N54 as a detection and targeting agent
Our study has shown that the aptamer N54 and to a lesser extent aptamer N56 are able to
block the adhesive properties of CEA. Our animal data also showed that pretreating
colon cancer cells expressing CEA and no other CEACAMs with these aptamers was
able to prevent the formation of tumour foci in vivo. This suggested that CEA homotypic
binding is an important event in the attachment and establishment of tumour foci.
However, CEA has been shown to interact with other CEACAMs and these interactions
may contribute to the metastatic potential of cancer cells. As such, a treatment targeting
only CEA in a combination therapy could be advantageous in preventing metastatis
during chemotherapy. These aptamers may also be used in pathology for detecting CEA-
positive tumours in tissue biopsies or as a delivery agent to target therapeutic cargos to
CEA-expressing cancers.
5.2.3. Determine the ability of MUC1 and CEA aptamer to target liposomes
A major challenge associated with chemotherapeutic agents remains their toxicity
towards normal tissues. This challenge limits their use to suboptimal doses and
ultimately leads to disease re-occurrence. Therapeutic cargos linked to antibodies and

129
proteins are being developed to specifically deliver chemotherapeutic agents to cancer
cells (Dillman, 2011). Yet, protein-guided therapies come with major limiting factors
including their size, cost and immunogenicity. Accordingly, simpler targeting agents are
needed to focus the delivery of useful cargos to cancer cells. Since their inception in
1990, short DNA/RNA aptamers have been developed to recognize therapeutically
important molecular targets such as VEGF, thrombin and HIV. More importantly,
aptamers can serve as cellular delivery vehicles by targeting cell surface markers that are
internalized by cancer cells, allowing for the intracellular localization of therapeutic
cargoes. Aptamers can be rapidly developed through SELEX screens, are easily
synthesized, are typically non-immunogenic and are readily amenable to modifications
leading to increased circulation times and stability. Aptamers directed at internalized
surface markers can be conjugated directly to drugs, RNA/DNA, radionuclides, proteins
and nanostructures to serve as tumour selective diagnostic and therapeutic agents. Future
studies will reveal the potential of CEA and MUC1 aptamers as targeting agents and their
ability to increase the accumulation and internalization of drugs in tumours.
Biodistribution analysis from imaging studies and examination of tumours by FACS
should determine if either of these formulations is able to increase the amount of
liposomes internalized into cancer cells. These anticipated results would suggest that
aptamers may enhance the therapeutic potential of drug-loaded liposomes such as
Doxil®.
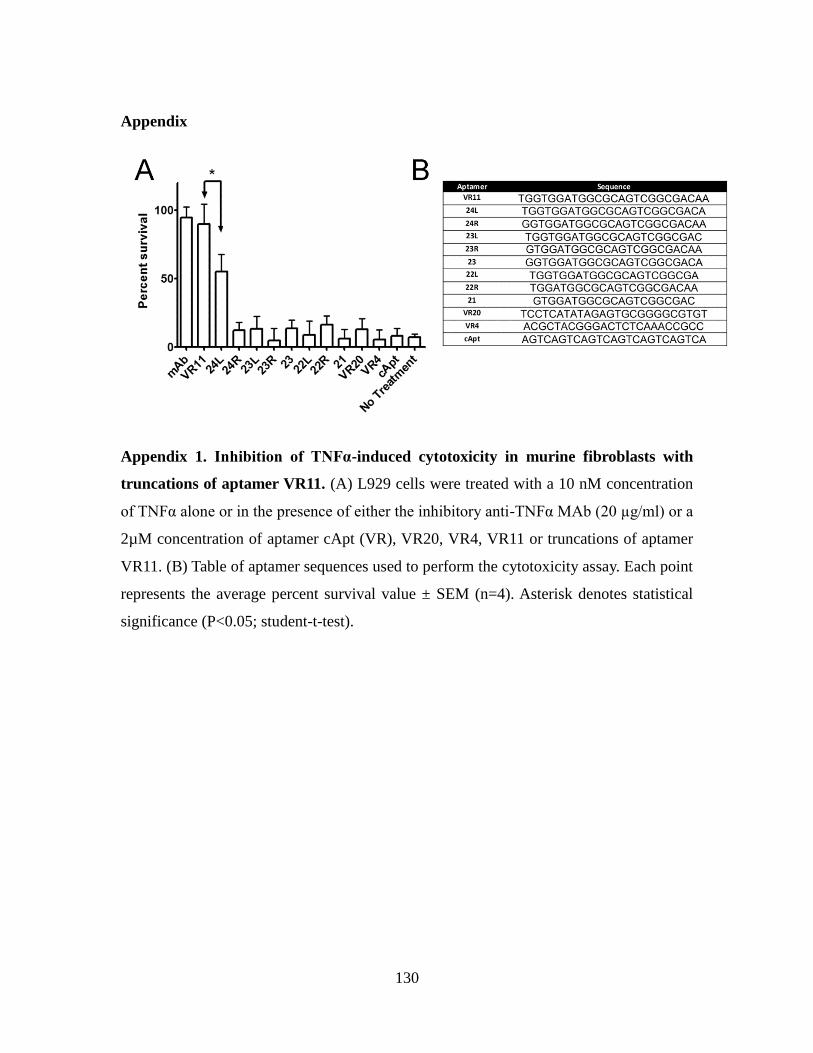
130
Appendix
Appendix 1. Inhibition of TNFα-induced cytotoxicity in murine fibroblasts with
truncations of aptamer VR11. (A) L929 cells were treated with a 10 nM concentration
of TNFα alone or in the presence of either the inhibitory anti-TNFα MAb (20 µg/ml) or a
2µM concentration of aptamer cApt (VR), VR20, VR4, VR11 or truncations of aptamer
VR11. (B) Table of aptamer sequences used to perform the cytotoxicity assay. Each point
represents the average percent survival value ± SEM (n=4). Asterisk denotes statistical
significance (P<0.05; student-t-test).
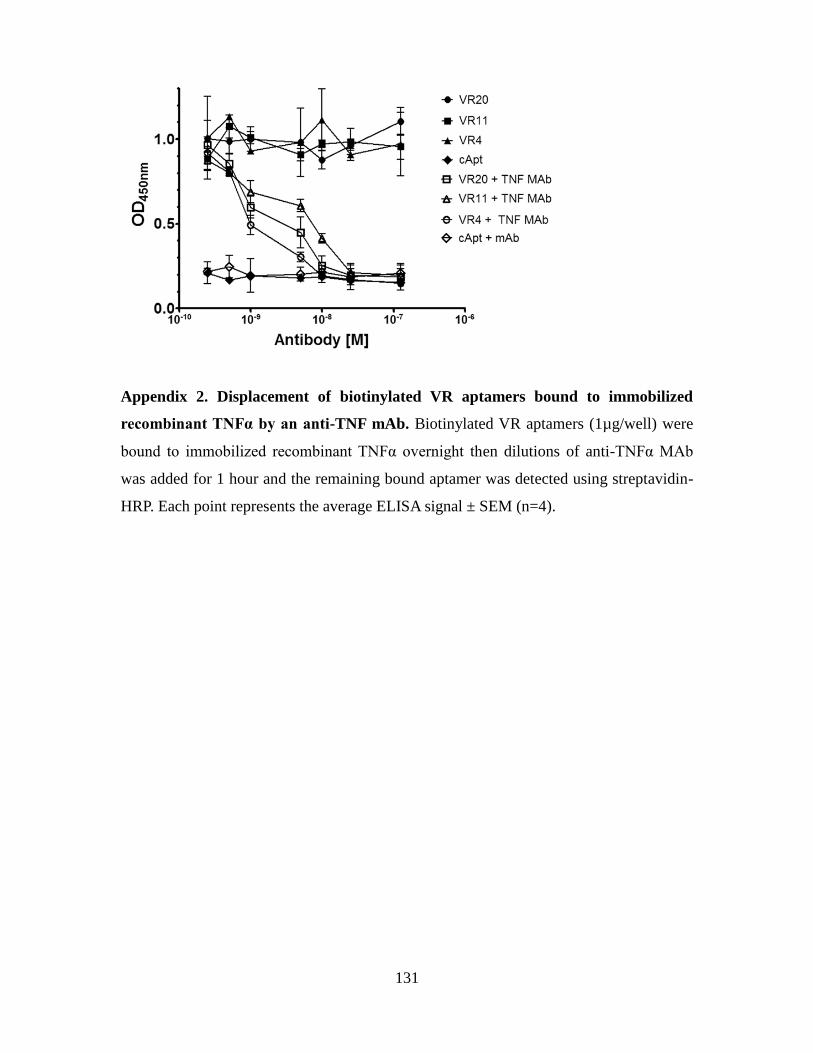
131
Appendix 2. Displacement of biotinylated VR aptamers bound to immobilized
recombinant TNFα by an anti-TNF mAb. Biotinylated VR aptamers (1µg/well) were
bound to immobilized recombinant TNFα overnight then dilutions of anti-TNFα MAb
was added for 1 hour and the remaining bound aptamer was detected using streptavidin-
HRP. Each point represents the average ELISA signal ± SEM (n=4).
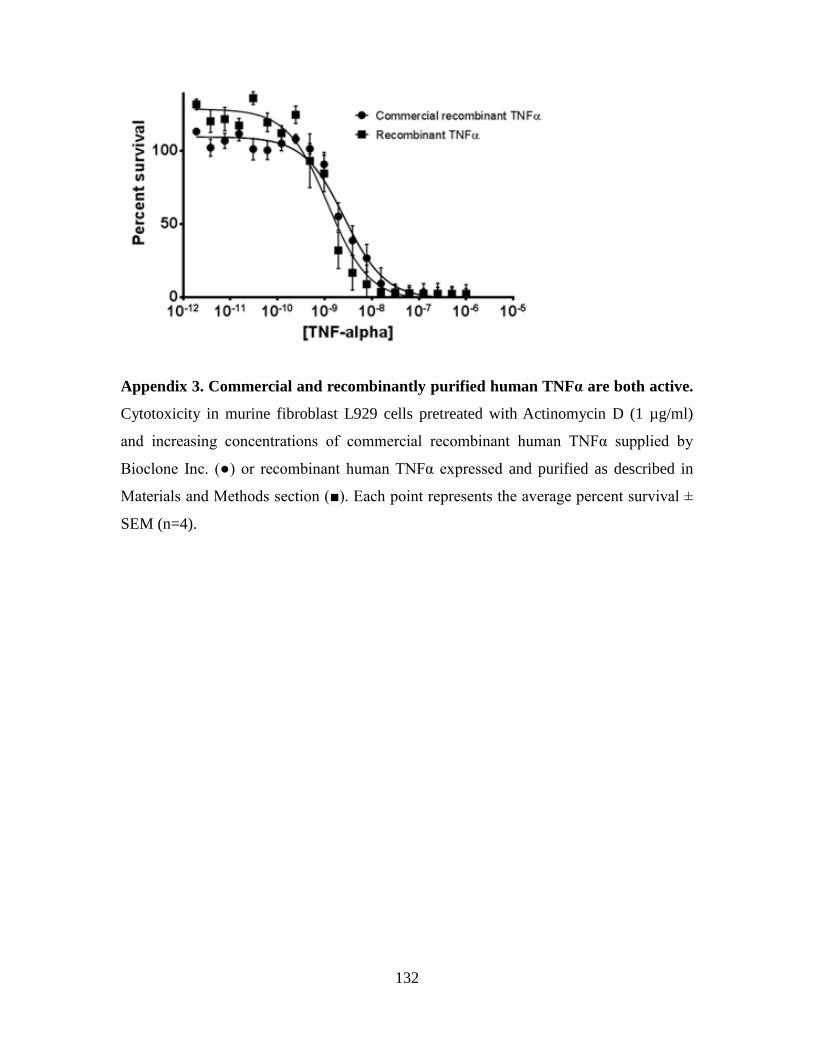
132
Appendix 3. Commercial and recombinantly purified human TNFα are both active.
Cytotoxicity in murine fibroblast L929 cells pretreated with Actinomycin D (1 µg/ml)
and increasing concentrations of commercial recombinant human TNFα supplied by
Bioclone Inc. (●) or recombinant human TNFα expressed and purified as described in
Materials and Methods section (■). Each point represents the average percent survival ±
SEM (n=4).
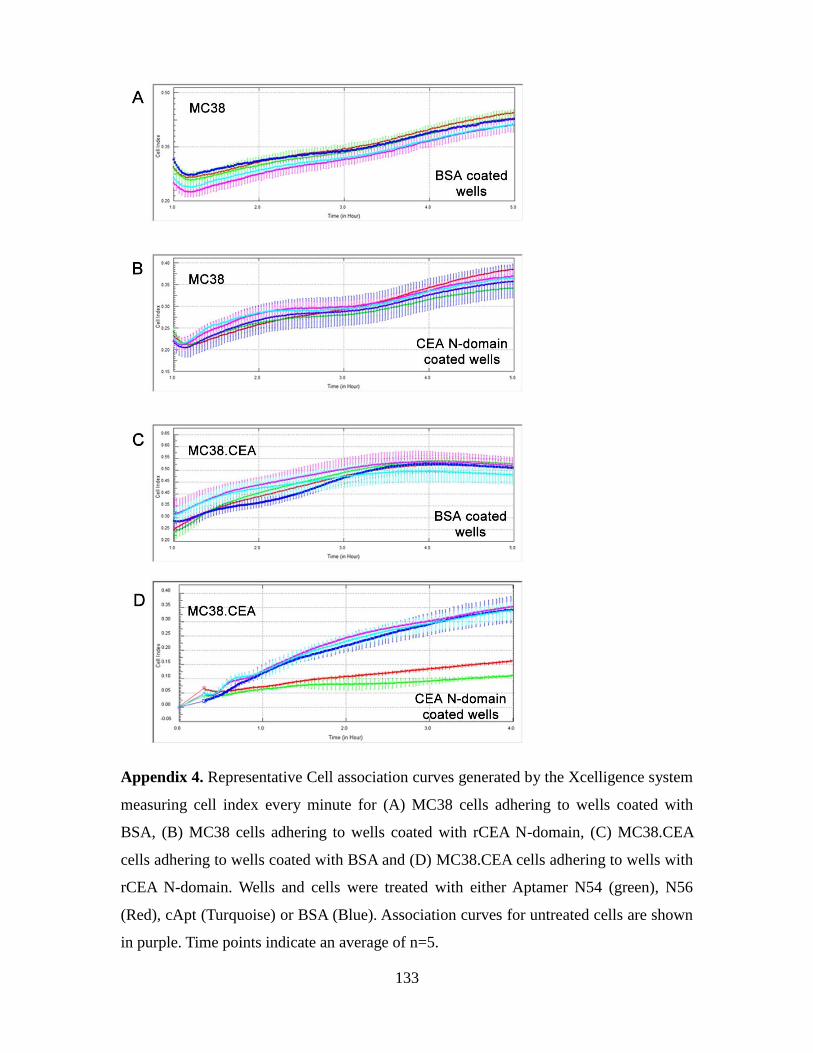
133
Appendix 4. Representative Cell association curves generated by the Xcelligence system
measuring cell index every minute for (A) MC38 cells adhering to wells coated with
BSA, (B) MC38 cells adhering to wells coated with rCEA N-domain, (C) MC38.CEA
cells adhering to wells coated with BSA and (D) MC38.CEA cells adhering to wells with
rCEA N-domain. Wells and cells were treated with either Aptamer N54 (green), N56
(Red), cApt (Turquoise) or BSA (Blue). Association curves for untreated cells are shown
in purple. Time points indicate an average of n=5.
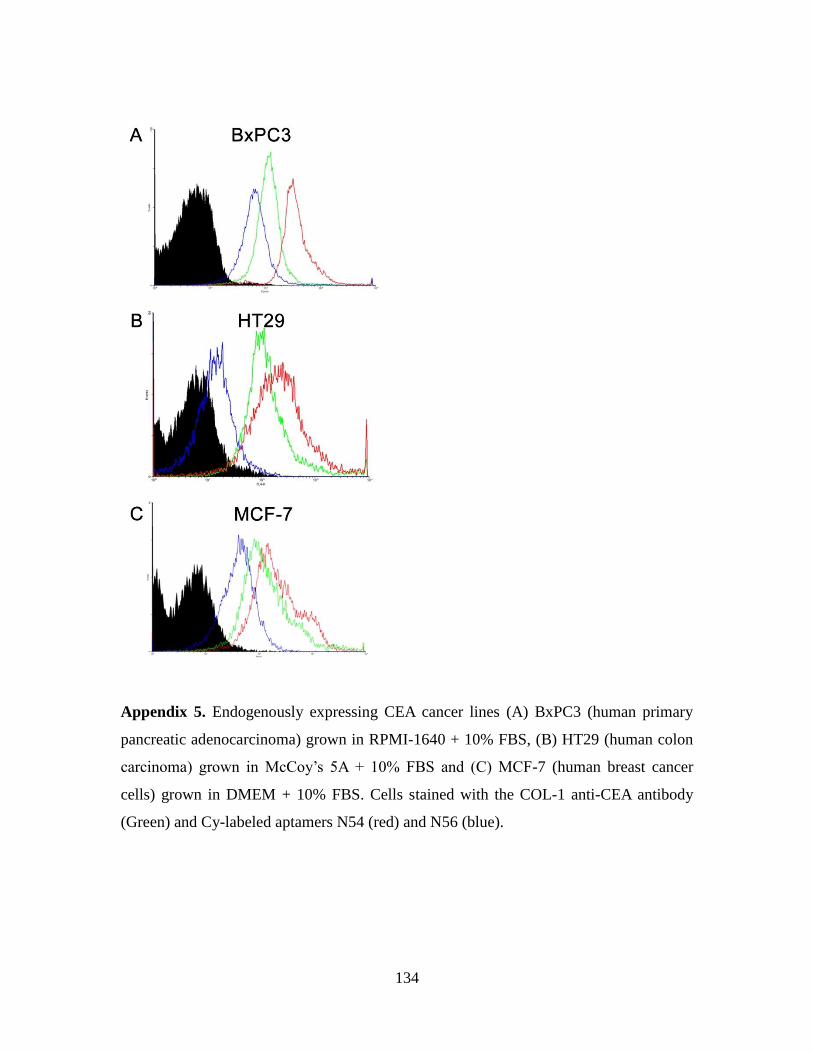
134
Appendix 5. Endogenously expressing CEA cancer lines (A) BxPC3 (human primary
pancreatic adenocarcinoma) grown in RPMI-1640 + 10% FBS, (B) HT29 (human colon
carcinoma) grown in McCoy’s 5A + 10% FBS and (C) MCF-7 (human breast cancer
cells) grown in DMEM + 10% FBS. Cells stained with the COL-1 anti-CEA antibody
(Green) and Cy-labeled aptamers N54 (red) and N56 (blue).

135
Appendix 6. 4-12% SDS PAGE gel confirming the purity of Ni-NTA purified MUC1
peptide and the mass shift confirming the glycosylation of the MUC1 peptide.
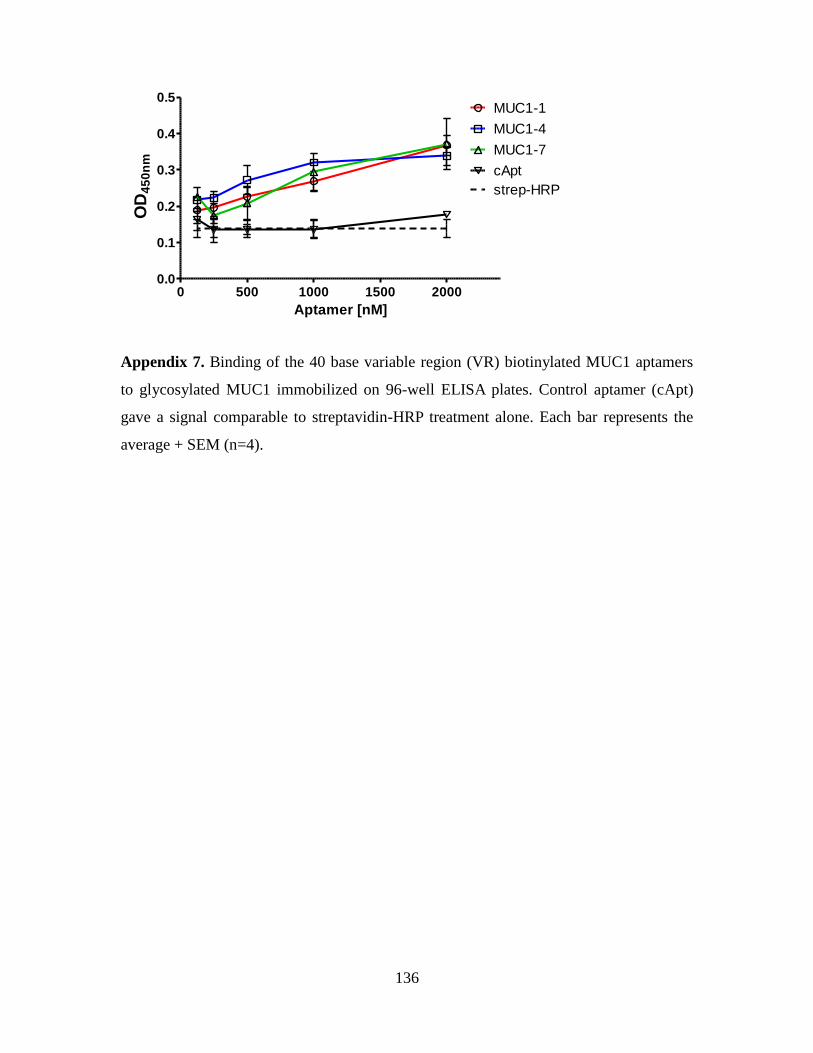
136
0 500 1000 1500 20000.0
0.1
0.2
0.3
0.4
0.5MUC1-1
MUC1-4
MUC1-7
cApt
strep-HRP
Aptamer [nM]
OD
45
0n
m
Appendix 7. Binding of the 40 base variable region (VR) biotinylated MUC1 aptamers
to glycosylated MUC1 immobilized on 96-well ELISA plates. Control aptamer (cApt)
gave a signal comparable to streptavidin-HRP treatment alone. Each bar represents the
average + SEM (n=4).
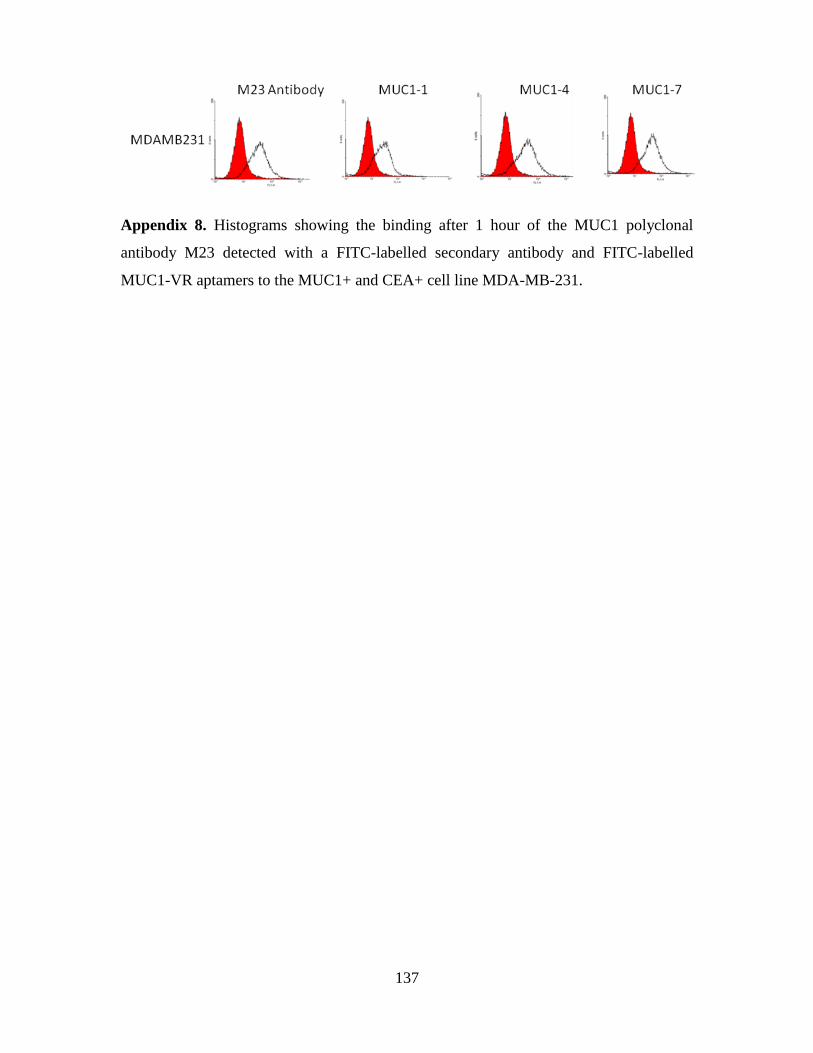
137
Appendix 8. Histograms showing the binding after 1 hour of the MUC1 polyclonal
antibody M23 detected with a FITC-labelled secondary antibody and FITC-labelled
MUC1-VR aptamers to the MUC1+ and CEA+ cell line MDA-MB-231.
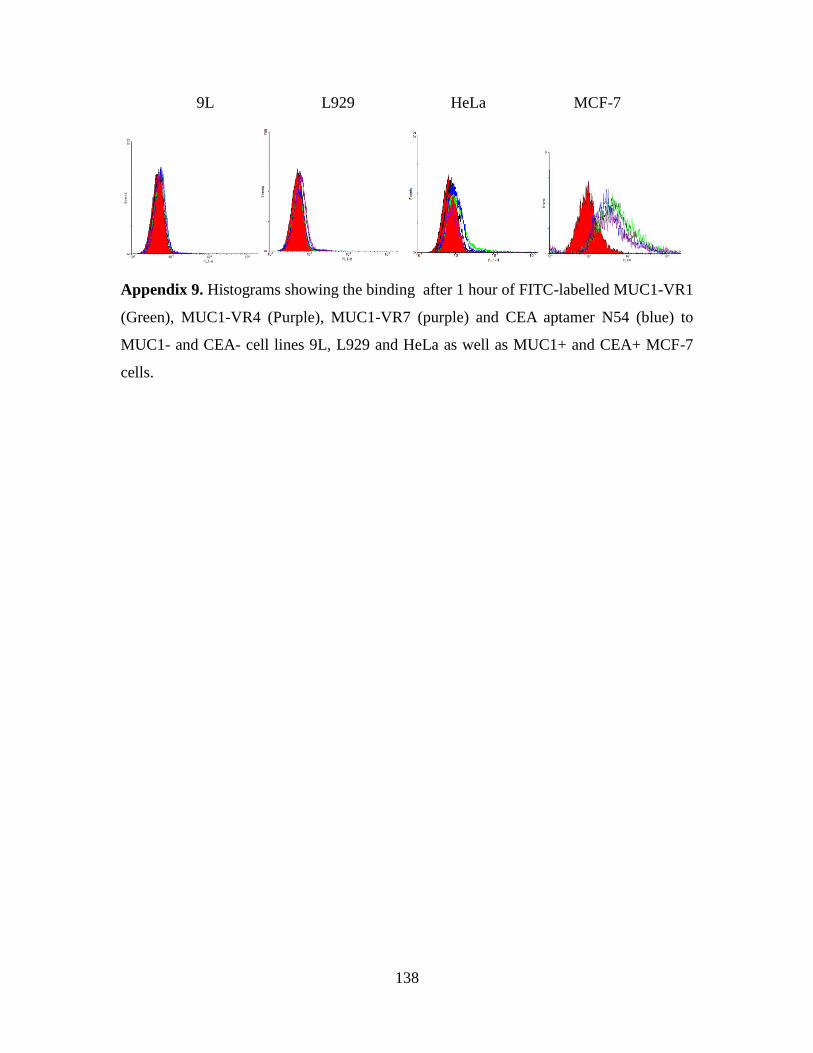
138
9L L929 HeLa MCF-7
Appendix 9. Histograms showing the binding after 1 hour of FITC-labelled MUC1-VR1
(Green), MUC1-VR4 (Purple), MUC1-VR7 (purple) and CEA aptamer N54 (blue) to
MUC1- and CEA- cell lines 9L, L929 and HeLa as well as MUC1+ and CEA+ MCF-7
cells.
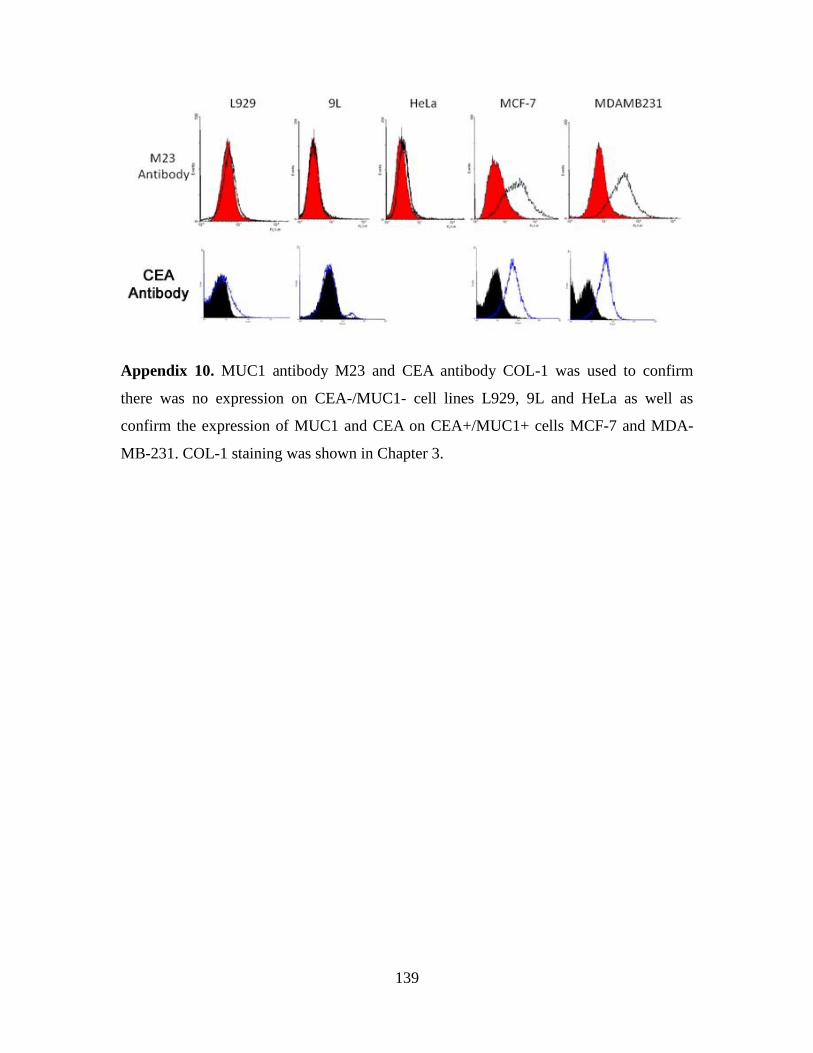
139
Appendix 10. MUC1 antibody M23 and CEA antibody COL-1 was used to confirm
there was no expression on CEA-/MUC1- cell lines L929, 9L and HeLa as well as
confirm the expression of MUC1 and CEA on CEA+/MUC1+ cells MCF-7 and MDA-
MB-231. COL-1 staining was shown in Chapter 3.
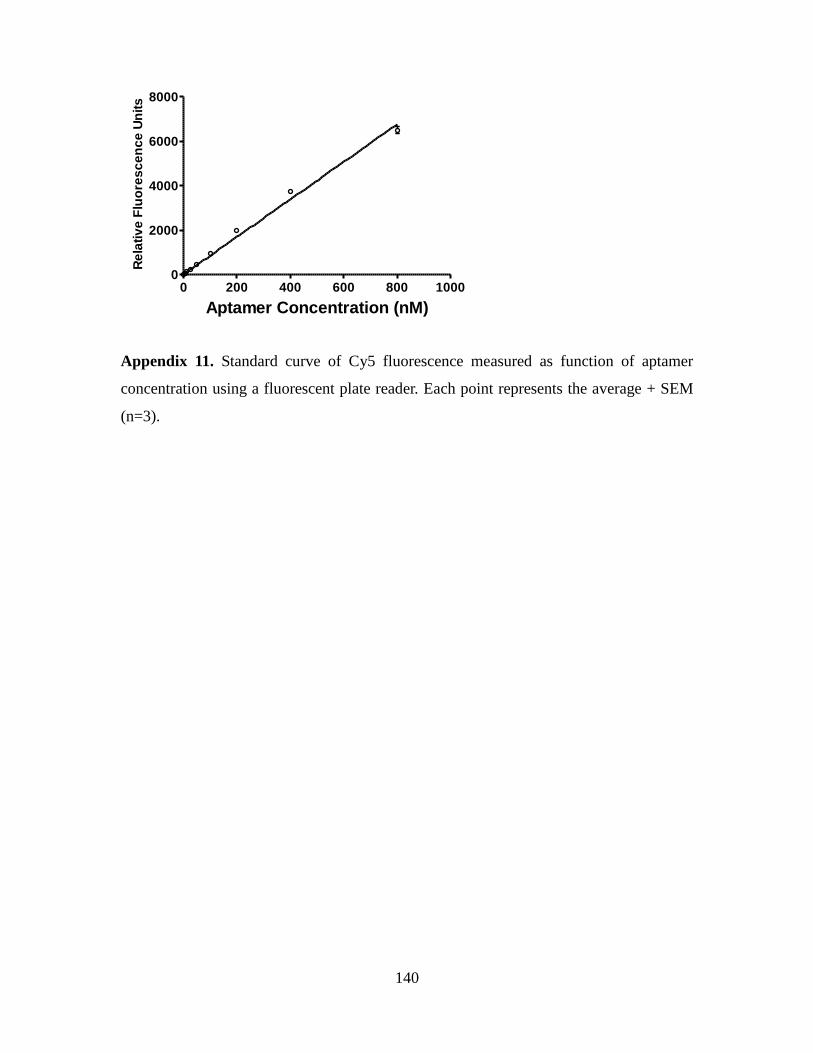
140
0 200 400 600 800 10000
2000
4000
6000
8000
Aptamer Concentration (nM)
Re
lati
ve
Flu
ore
sc
en
ce
Un
its
Appendix 11. Standard curve of Cy5 fluorescence measured as function of aptamer
concentration using a fluorescent plate reader. Each point represents the average + SEM
(n=3).
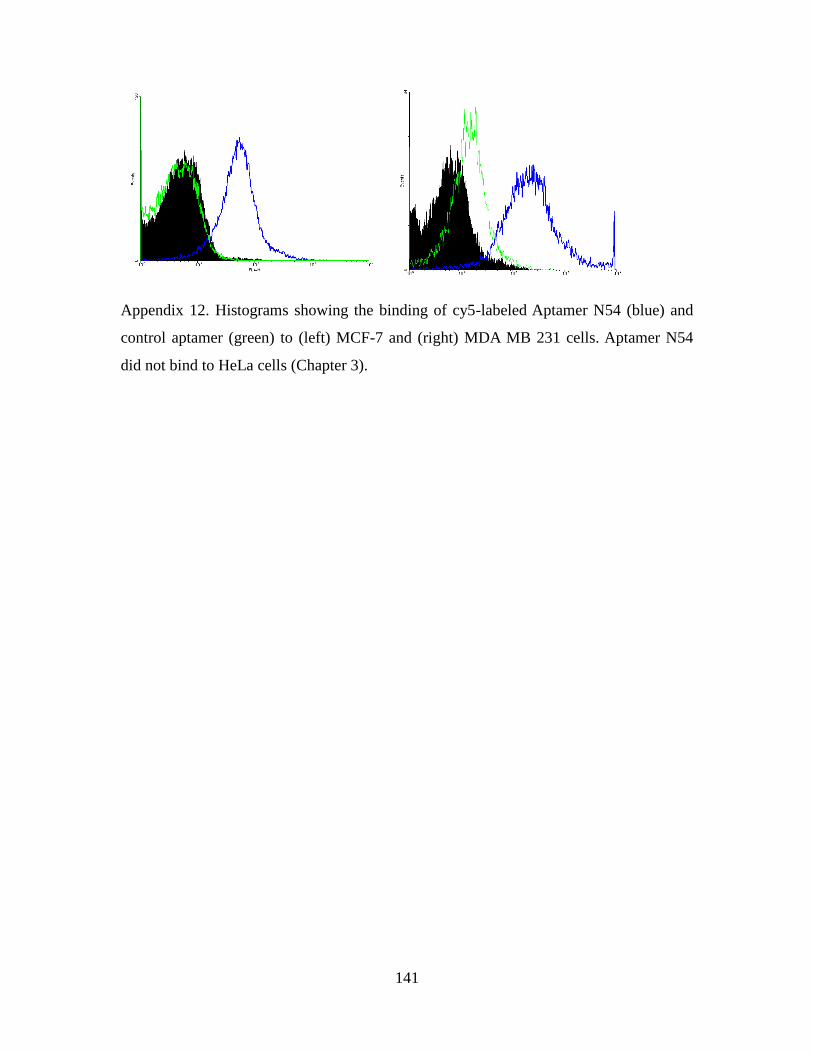
141
Appendix 12. Histograms showing the binding of cy5-labeled Aptamer N54 (blue) and
control aptamer (green) to (left) MCF-7 and (right) MDA MB 231 cells. Aptamer N54
did not bind to HeLa cells (Chapter 3).

142
References
2012. Biologic drugs set to top 2012 sales. Nat Med 18, 636.
Abdul-Wahid, A., Huang, E.H., Lu, H., Flanagan, J., Mallick, A.I., Gariepy, J., 2012. A
focused immune response targeting the homotypic binding domain of the
carcinoembryonic antigen blocks the establishment of tumor foci in vivo. Int J Cancer.
Adams, G.P., Schier, R., McCall, A.M., Simmons, H.H., Horak, E.M., Alpaugh, R.K.,
Marks, J.D., Weiner, L.M., 2001. High affinity restricts the localization and tumor
penetration of single-chain fv antibody molecules. Cancer Res 61, 4750-4755.
Ajayan, P.M., 1999. Nanotubes from Carbon. Chem Rev 99, 1787-1800.
Altschuler, Y., Kinlough, C.L., Poland, P.A., Bruns, J.B., Apodaca, G., Weisz, O.A.,
Hughey, R.P., 2000. Clathrin-mediated endocytosis of MUC1 is modulated by its
glycosylation state. Mol Biol Cell 11, 819-831.
Ando, J., Fujita, K., Smith, N.I., Kawata, S., 2011. Dynamic SERS imaging of cellular
transport pathways with endocytosed gold nanoparticles. Nano Lett 11, 5344-5348.
Andrews, R.G., Takahashi, M., Segal, G.M., Powell, J.S., Bernstein, I.D., Singer, J.W.,
1986. The L4F3 antigen is expressed by unipotent and multipotent colony-forming cells
but not by their precursors. Blood 68, 1030-1035.
Andrews, R.G., Torok-Storb, B., Bernstein, I.D., 1983. Myeloid-associated differentiation
antigens on stem cells and their progeny identified by monoclonal antibodies. Blood 62,
124-132.
Asao, T., Fukuda, T., Yazawa, S., Nagamachi, Y., 1991. Carcinoembryonic antigen levels
in peritoneal washings can predict peritoneal recurrence after curative resection of gastric
cancer. Cancer 68, 44-47.
Aurup, H., Williams, D.M., Eckstein, F., 1992. 2'-Fluoro- and 2'-amino-2'-
deoxynucleoside 5'-triphosphates as substrates for T7 RNA polymerase. Biochemistry
31, 9636-9641.
Bagalkot, V., Farokhzad, O.C., Langer, R., Jon, S., 2006. An aptamer-doxorubicin
physical conjugate as a novel targeted drug-delivery platform. Angew Chem Int Ed Engl
45, 8149-8152.
Bagalkot, V., Zhang, L., Levy-Nissenbaum, E., Jon, S., Kantoff, P.W., Langer, R.,
Farokhzad, O.C., 2007. Quantum dot-aptamer conjugates for synchronous cancer
imaging, therapy, and sensing of drug delivery based on bi-fluorescence resonance
energy transfer. Nano Lett 7, 3065-3070.
Bangham, A.D., Standish, M.M., Watkins, J.C., 1965. Diffusion of univalent ions across
the lamellae of swollen phospholipids. J Mol Biol 13, 238-252.
Barciszewski, J., Medgaard, M., Koch, T., Kurreck, J., Erdmann, V.A., 2009. Locked
nucleic acid aptamers. Methods Mol Biol 535, 165-186.
Bates, P.J., Kahlon, J.B., Thomas, S.D., Trent, J.O., Miller, D.M., 1999. Antiproliferative
activity of G-rich oligonucleotides correlates with protein binding. J Biol Chem 274,
26369-26377.
Behr, T.M., Liersch, T., Greiner-Bechert, L., Griesinger, F., Behe, M., Markus, P.M.,
Gratz, S., Angerstein, C., Brittinger, G., Becker, H., Goldenberg, D.M., Becker, W., 2002.
Radioimmunotherapy of small-volume disease of metastatic colorectal cancer. Cancer
94, 1373-1381.
Benchimol, S., Fuks, A., Jothy, S., Beauchemin, N., Shirota, K., Stanners, C.P., 1989.
Carcinoembryonic antigen, a human tumor marker, functions as an intercellular adhesion

143
molecule. Cell 57, 327-334.
Berinstein, N.L., 2002. Carcinoembryonic antigen as a target for therapeutic anticancer
vaccines: a review. J Clin Oncol 20, 2197-2207.
Bernstein, I.D., 2000. Monoclonal antibodies to the myeloid stem cells: therapeutic
implications of CMA-676, a humanized anti-CD33 antibody calicheamicin conjugate.
Leukemia 14, 474-475.
Berry, C.C., Wells, S., Charles, S., Curtis, A.S., 2003. Dextran and albumin derivatised
iron oxide nanoparticles: influence on fibroblasts in vitro. Biomaterials 24, 4551-4557.
Beutler, B.A., 1999. The role of tumor necrosis factor in health and disease. J Rheumatol
Suppl 57, 16-21.
Blank, M., Weinschenk, T., Priemer, M., Schluesener, H., 2001. Systematic evolution of a
DNA aptamer binding to rat brain tumor microvessels. selective targeting of endothelial
regulatory protein pigpen. J Biol Chem 276, 16464-16468.
Bless, N.M., Smith, D., Charlton, J., Czermak, B.J., Schmal, H., Friedl, H.P., Ward, P.A.,
1997. Protective effects of an aptamer inhibitor of neutrophil elastase in lung
inflammatory injury. Curr Biol 7, 877-880.
Blumenthal, R.D., Hansen, H.J., Goldenberg, D.M., 2005. Inhibition of adhesion,
invasion, and metastasis by antibodies targeting CEACAM6 (NCA-90) and CEACAM5
(Carcinoembryonic Antigen). Cancer Res 65, 8809-8817.
Bock, L.C., Griffin, L.C., Latham, J.A., Vermaas, E.H., Toole, J.J., 1992. Selection of
single-stranded DNA molecules that bind and inhibit human thrombin. Nature 355, 564-
566.
Bode, W., Turk, D., Karshikov, A., 1992. The refined 1.9-A X-ray crystal structure of D-
Phe-Pro-Arg chloromethylketone-inhibited human alpha-thrombin: structure analysis,
overall structure, electrostatic properties, detailed active-site geometry, and structure-
function relationships. Protein Sci 1, 426-471.
Boiziau, C., Dausse, E., Yurchenko, L., Toulme, J.J., 1999. DNA aptamers selected
against the HIV-1 trans-activation-responsive RNA element form RNA-DNA kissing
complexes. J Biol Chem 274, 12730-12737.
Boomer, R.M., Lewis, S.D., Healy, J.M., Kurz, M., Wilson, C., McCauley, T.G., 2005.
Conjugation to polyethylene glycol polymer promotes aptamer biodistribution to healthy
and inflamed tissues. Oligonucleotides 15, 183-195.
Borish, L.C., Steinke, J.W., 2003. 2. Cytokines and chemokines. J Allergy Clin Immunol
111, S460-475.
Boucher, D., Cournoyer, D., Stanners, C.P., Fuks, A., 1989. Studies on the control of gene
expression of the carcinoembryonic antigen family in human tissue. Cancer Res 49, 847-
852.
Bradford, M.M., 1976. A rapid and sensitive method for the quantitation of microgram
quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72,
248-254.
Brennan, F.M., Chantry, D., Jackson, A., Maini, R., Feldmann, M., 1989. Inhibitory
effect of TNF alpha antibodies on synovial cell interleukin-1 production in rheumatoid
arthritis. Lancet 2, 244-247.
Brokx, R.D., Revers, L., Zhang, Q., Yang, S., Mal, T.K., Ikura, M., Gariepy, J., 2003.
Nuclear magnetic resonance-based dissection of a glycosyltransferase specificity for the
mucin MUC1 tandem repeat. Biochemistry 42, 13817-13825.
Bryan, J.N., Jia, F., Mohsin, H., Sivaguru, G., Miller, W.H., Anderson, C.J., Henry, C.J.,
Lewis, M.R., 2005. Comparative uptakes and biodistributions of internalizing vs.

144
noninternalizing copper-64 radioimmunoconjugates in cell and animal models of colon
cancer. Nucl Med Biol 32, 851-858.
Bunka, D.H., Platonova, O., Stockley, P.G., 2010. Development of aptamer therapeutics.
Curr Opin Pharmacol 10, 557-562.
Burge, S., Parkinson, G.N., Hazel, P., Todd, A.K., Neidle, S., 2006. Quadruplex DNA:
sequence, topology and structure. Nucleic Acids Res 34, 5402-5415.
Cai, Z., Ye, Z., Yang, X., Chang, Y., Wang, H., Liu, Y., Cao, A., 2011. Encapsulated
enhanced green fluorescence protein in silica nanoparticle for cellular imaging.
Nanoscale 3, 1974-1976.
Camacho-Leal, P., Stanners, C.P., 2008. The human carcinoembryonic antigen (CEA)
GPI anchor mediates anoikis inhibition by inactivation of the intrinsic death pathway.
Oncogene 27, 1545-1553.
Cao, Z., Tong, R., Mishra, A., Xu, W., Wong, G.C., Cheng, J., Lu, Y., 2009. Reversible
cell-specific drug delivery with aptamer-functionalized liposomes. Angew Chem Int Ed
Engl 48, 6494-6498.
Carswell, E.A., Old, L.J., Kassel, R.L., Green, S., Fiore, N., Williamson, B., 1975. An
endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci U S A
72, 3666-3670.
Ceriani, R.L., Chan, C.M., Baratta, F.S., Ozzello, L., DeRosa, C.M., Habif, D.V., 1992.
Levels of expression of breast epithelial mucin detected by monoclonal antibody BrE-3
in breast-cancer prognosis. Int J Cancer 51, 343-354.
Chang, H.I., Yeh, M.K., 2012. Clinical development of liposome-based drugs:
formulation, characterization, and therapeutic efficacy. Int J Nanomedicine 7, 49-60.
Chang, S.S., Reuter, V.E., Heston, W.D., Bander, N.H., Grauer, L.S., Gaudin, P.B., 1999.
Five different anti-prostate-specific membrane antigen (PSMA) antibodies confirm
PSMA expression in tumor-associated neovasculature. Cancer Res 59, 3192-3198.
Charbonneau, J., Stanners, C.P., 1999. Role of carbohydrate structures in CEA-mediated
intercellular adhesion. Cell Adhes Commun 7, 233-244.
Chen, C.H., Chernis, G.A., Hoang, V.Q., Landgraf, R., 2003. Inhibition of heregulin
signaling by an aptamer that preferentially binds to the oligomeric form of human
epidermal growth factor receptor-3. Proc Natl Acad Sci U S A 100, 9226-9231.
Chen, P., Wang, J., Hope, K., Jin, L., Dick, J., Cameron, R., Brandwein, J., Minden, M.,
Reilly, R.M., 2006. Nuclear localizing sequences promote nuclear translocation and
enhance the radiotoxicity of the anti-CD33 monoclonal antibody HuM195 labeled with
111In in human myeloid leukemia cells. J Nucl Med 47, 827-836.
Cheng, J., Teply, B.A., Sherifi, I., Sung, J., Luther, G., Gu, F.X., Levy-Nissenbaum, E.,
Radovic-Moreno, A.F., Langer, R., Farokhzad, O.C., 2007. Formulation of functionalized
PLGA-PEG nanoparticles for in vivo targeted drug delivery. Biomaterials 28, 869-876.
Chevallier, P., Robillard, N., Ayari, S., Guillaume, T., Delaunay, J., Mechinaud, F., Avet-
Loiseau, H., Mohty, M., Harousseau, J.L., Garand, R., 2008. Persistence of CD33
expression at relapse in CD33(+) acute myeloid leukaemia patients after receiving
Gemtuzumab in the course of the disease. Br J Haematol 143, 744-746.
Chevinsky, A.H., 1991. CEA in tumors of other than colorectal origin. Semin Surg Oncol
7, 162-166.
Chou, S.H., Chin, K.H., Wang, A.H., 2003. Unusual DNA duplex and hairpin motifs.
Nucleic Acids Res 31, 2461-2474.
Chou, S.H., Chin, K.H., Wang, A.H., 2005. DNA aptamers as potential anti-HIV agents.
Trends Biochem Sci 30, 231-234.

145
Choy, E.H., Hazleman, B., Smith, M., Moss, K., Lisi, L., Scott, D.G., Patel, J., Sopwith,
M., Isenberg, D.A., 2002. Efficacy of a novel PEGylated humanized anti-TNF fragment
(CDP870) in patients with rheumatoid arthritis: a phase II double-blinded, randomized,
dose-escalating trial. Rheumatology (Oxford) 41, 1133-1137.
Chu, T.C., Marks, J.W., 3rd, Lavery, L.A., Faulkner, S., Rosenblum, M.G., Ellington,
A.D., Levy, M., 2006a. Aptamer:toxin conjugates that specifically target prostate tumor
cells. Cancer Res 66, 5989-5992.
Chu, T.C., Shieh, F., Lavery, L.A., Levy, M., Richards-Kortum, R., Korgel, B.A.,
Ellington, A.D., 2006b. Labeling tumor cells with fluorescent nanocrystal-aptamer
bioconjugates. Biosens Bioelectron 21, 1859-1866.
Collie, G.W., Parkinson, G.N., 2011. The application of DNA and RNA G-quadruplexes
to therapeutic medicines. Chem Soc Rev 40, 5867-5892.
Conrad, R., Keranen, L.M., Ellington, A.D., Newton, A.C., 1994. Isozyme-specific
inhibition of protein kinase C by RNA aptamers. J Biol Chem 269, 32051-32054.
D'Amico, D.J., Masonson, H.N., Patel, M., Adamis, A.P., Cunningham, E.T., Jr., Guyer,
D.R., Katz, B., 2006. Pegaptanib sodium for neovascular age-related macular
degeneration: two-year safety results of the two prospective, multicenter, controlled
clinical trials. Ophthalmology 113, 992-1001 e1006.
Da Pieve, C., Blackshaw, E., Missailidis, S., Perkins, A.C., 2012. PEGylation and
biodistribution of an anti-MUC1 aptamer in MCF-7 tumor-bearing mice. Bioconjug
Chem 23, 1377-1381.
Dapic, V., Bates, P.J., Trent, J.O., Rodger, A., Thomas, S.D., Miller, D.M., 2002.
Antiproliferative activity of G-quartet-forming oligonucleotides with backbone and sugar
modifications. Biochemistry 41, 3676-3685.
Dhar, S., Gu, F.X., Langer, R., Farokhzad, O.C., Lippard, S.J., 2008. Targeted delivery of
cisplatin to prostate cancer cells by aptamer functionalized Pt(IV) prodrug-PLGA-PEG
nanoparticles. Proc Natl Acad Sci U S A 105, 17356-17361.
Dillman, R.O., 2011. Cancer immunotherapy. Cancer Biother Radiopharm 26, 1-64.
Ding, A.H., Nathan, C.F., Stuehr, D.J., 1988. Release of reactive nitrogen intermediates
and reactive oxygen intermediates from mouse peritoneal macrophages. Comparison of
activating cytokines and evidence for independent production. J Immunol 141, 2407-
2412.
Dinndorf, P.A., Andrews, R.G., Benjamin, D., Ridgway, D., Wolff, L., Bernstein, I.D.,
1986. Expression of normal myeloid-associated antigens by acute leukemia cells. Blood
67, 1048-1053.
Duffy, M.J., 2006. Serum tumor markers in breast cancer: are they of clinical value? Clin
Chem 52, 345-351.
Eaton, B.E., Gold, L., Hicke, B.J., Janjic, N., Jucker, F.M., Sebesta, D.P., Tarasow, T.M.,
Willis, M.C., Zichi, D.A., 1997. Post-SELEX combinatorial optimization of aptamers.
Bioorg Med Chem 5, 1087-1096.
Ehlers, S., 2005. Tumor necrosis factor and its blockade in granulomatous infections:
differential modes of action of infliximab and etanercept? Clin Infect Dis 41 Suppl 3,
S199-203.
Ellington, A.D., Szostak, J.W., 1990. In vitro selection of RNA molecules that bind
specific ligands. Nature 346, 818-822.
Fang, C., Zhang, M., 2009. Multifunctional Magnetic Nanoparticles for Medical Imaging
Applications. J Mater Chem 19, 6258-6266.
Farokhzad, O.C., Cheng, J., Teply, B.A., Sherifi, I., Jon, S., Kantoff, P.W., Richie, J.P.,

146
Langer, R., 2006. Targeted nanoparticle-aptamer bioconjugates for cancer chemotherapy
in vivo. Proc Natl Acad Sci U S A 103, 6315-6320.
Farokhzad, O.C., Jon, S., Khademhosseini, A., Tran, T.N., Lavan, D.A., Langer, R., 2004.
Nanoparticle-aptamer bioconjugates: a new approach for targeting prostate cancer cells.
Cancer Res 64, 7668-7672.
Fedarovich, A., Tomberg, J., Nicholas, R.A., Davies, C., 2006. Structure of the N-
terminal domain of human CEACAM1: binding target of the opacity proteins during
invasion of Neisseria meningitidis and N. gonorrhoeae. Acta Crystallogr D Biol
Crystallogr 62, 971-979.
Feldmann, M., Brennan, F.M., Foxwell, B.M., Taylor, P.C., Williams, R.O., Maini, R.N.,
2005. Anti-TNF therapy: where have we got to in 2005? J Autoimmun 25 Suppl, 26-28.
Feldmann, M., Brennan, F.M., Williams, R.O., Cope, A.P., Gibbons, D.L., Katsikis, P.D.,
Maini, R.N., 1992. Evaluation of the role of cytokines in autoimmune disease: the
importance of TNF alpha in rheumatoid arthritis. Prog Growth Factor Res 4, 247-255.
Feldmann, M., Charles, P., Taylor, P., Maini, R.N., 1998. Biological insights from clinical
trials with anti-TNF therapy. Springer Semin Immunopathol 20, 211-228.
Feldmann, M., Maini, R.N., 2001. Anti-TNF alpha therapy of rheumatoid arthritis: what
have we learned? Annu Rev Immunol 19, 163-196.
Feldmann, M., Maini, R.N., Bondeson, J., Taylor, P., Foxwell, B.M., Brennan, F.M.,
2001. Cytokine blockade in rheumatoid arthritis. Adv Exp Med Biol 490, 119-127.
Feldmann, M., Maini, S.R., 2008. Role of cytokines in rheumatoid arthritis: an education
in pathophysiology and therapeutics. Immunol Rev 223, 7-19.
Ferreira, C.S., Cheung, M.C., Missailidis, S., Bisland, S., Gariepy, J., 2009. Phototoxic
aptamers selectively enter and kill epithelial cancer cells. Nucleic Acids Res 37, 866-876.
Fidler, I.J., 2003. The pathogenesis of cancer metastasis: the 'seed and soil' hypothesis
revisited. Nat Rev Cancer 3, 453-458.
Ford, C.H., Tsaltas, G.C., Osborne, P.A., Addetia, K., 1996. Novel flow cytometric
analysis of the progress and route of internalization of a monoclonal anti-
carcinoembryonic antigen (CEA) antibody. Cytometry 23, 228-240.
Foy, J.W., Rittenhouse, K., Modi, M., Patel, M., 2007. Local tolerance and systemic
safety of pegaptanib sodium in the dog and rabbit. J Ocul Pharmacol Ther 23, 452-466.
Freeman, S.D., Kelm, S., Barber, E.K., Crocker, P.R., 1995. Characterization of CD33 as
a new member of the sialoadhesin family of cellular interaction molecules. Blood 85,
2005-2012.
Furst, D.E., Wallis, R., Broder, M., Beenhouwer, D.O., 2006. Tumor necrosis factor
antagonists: different kinetics and/or mechanisms of action may explain differences in
the risk for developing granulomatous infection. Semin Arthritis Rheum 36, 159-167.
Gendler, S., Taylor-Papadimitriou, J., Duhig, T., Rothbard, J., Burchell, J., 1988. A highly
immunogenic region of a human polymorphic epithelial mucin expressed by carcinomas
is made up of tandem repeats. J Biol Chem 263, 12820-12823.
Gilbert, D.E., Feigon, J., 1999. Multistranded DNA structures. Curr Opin Struct Biol 9,
305-314.
Gilbey, A.M., Burnett, D., Coleman, R.E., Holen, I., 2004. The detection of circulating
breast cancer cells in blood. J Clin Pathol 57, 903-911.
Girvan, A.C., Teng, Y., Casson, L.K., Thomas, S.D., Juliger, S., Ball, M.W., Klein, J.B.,
Pierce, W.M., Jr., Barve, S.S., Bates, P.J., 2006. AGRO100 inhibits activation of nuclear
factor-kappaB (NF-kappaB) by forming a complex with NF-kappaB essential modulator
(NEMO) and nucleolin. Mol Cancer Ther 5, 1790-1799.

147
Gold, P., Freedman, S.O., 1965a. Demonstration of Tumor-Specific Antigens in Human
Colonic Carcinomata by Immunological Tolerance and Absorption Techniques. J Exp
Med 121, 439-462.
Gold, P., Freedman, S.O., 1965b. Specific carcinoembryonic antigens of the human
digestive system. J Exp Med 122, 467-481.
Goldenberg, D.M., 2002. Targeted therapy of cancer with radiolabeled antibodies. J Nucl
Med 43, 693-713.
Goldenberg, D.M., Sharkey, R.M., Primus, F.J., 1976. Carcinoembryonic antigen in
histopathology: immunoperoxidase staining of conventional tissue sections. J Natl
Cancer Inst 57, 11-22.
Goldmacher, V.S., Scott, C.F., Lambert, J.M., McIntyre, G.D., Blattler, W.A., Collnhson,
A.R., Stewart, J.K., Chong, L.D., Cook, S., Slayter, H.S., et al., 1989. Cytotoxicity of
gelonin and its conjugates with antibodies is determined by the extent of their
endocytosis. J Cell Physiol 141, 222-234.
Goldstein, M.J., Mitchell, E.P., 2005. Carcinoembryonic antigen in the staging and
follow-up of patients with colorectal cancer. Cancer Invest 23, 338-351.
Graff, C.P., Wittrup, K.D., 2003. Theoretical analysis of antibody targeting of tumor
spheroids: importance of dosage for penetration, and affinity for retention. Cancer Res
63, 1288-1296.
Gregoriadis, G., 1995. Engineering liposomes for drug delivery: progress and problems.
Trends Biotechnol 13, 527-537.
Griffin, J.D., Linch, D., Sabbath, K., Larcom, P., Schlossman, S.F., 1984. A monoclonal
antibody reactive with normal and leukemic human myeloid progenitor cells. Leuk Res
8, 521-534.
Gutsaeva, D.R., Parkerson, J.B., Yerigenahally, S.D., Kurz, J.C., Schaub, R.G., Ikuta, T.,
Head, C.A., 2010. Inhibition of cell adhesion by anti-P-selectin aptamer: a new potential
therapeutic agent for sickle cell disease. Blood 117, 727-735.
Gutsaeva, D.R., Parkerson, J.B., Yerigenahally, S.D., Kurz, J.C., Schaub, R.G., Ikuta, T.,
Head, C.A., 2011. Inhibition of cell adhesion by anti-P-selectin aptamer: a new potential
therapeutic agent for sickle cell disease. Blood 117, 727-735.
Hale, S.P., Schimmel, P., 1996. Protein synthesis editing by a DNA aptamer. Proc Natl
Acad Sci U S A 93, 2755-2758.
Hamann, P.R., Hinman, L.M., Beyer, C.F., Lindh, D., Upeslacis, J., Flowers, D.A.,
Bernstein, I., 2002. An anti-CD33 antibody-calicheamicin conjugate for treatment of
acute myeloid leukemia. Choice of linker. Bioconjug Chem 13, 40-46.
Hammarstrom, S., 1999. The carcinoembryonic antigen (CEA) family: structures,
suggested functions and expression in normal and malignant tissues. Semin Cancer Biol
9, 67-81.
Hamula, C.L., Zhang, H., Guan, L.L., Li, X.F., Le, X.C., 2008. Selection of aptamers
against live bacterial cells. Anal Chem 80, 7812-7819.
Hanahan, D., Weinberg, R.A., 2000. The hallmarks of cancer. Cell 100, 57-70.
Hanahan, D., Weinberg, R.A., 2011. Hallmarks of cancer: the next generation. Cell 144,
646-674.
Hanisch, F.G., Muller, S., 2000. MUC1: the polymorphic appearance of a human mucin.
Glycobiology 10, 439-449.
Hanisch, F.G., Stadie, T.R., Deutzmann, F., Peter-Katalinic, J., 1996. MUC1 glycoforms
in breast cancer--cell line T47D as a model for carcinoma-associated alterations of 0-
glycosylation. Eur J Biochem 236, 318-327.

148
Hardin, C.C., Henderson, E., Watson, T., Prosser, J.K., 1991. Monovalent cation induced
structural transitions in telomeric DNAs: G-DNA folding intermediates. Biochemistry
30, 4460-4472.
Hardin, C.C., Watson, T., Corregan, M., Bailey, C., 1992. Cation-dependent transition
between the quadruplex and Watson-Crick hairpin forms of d(CGCG3GCG).
Biochemistry 31, 833-841.
Harris, L., Fritsche, H., Mennel, R., Norton, L., Ravdin, P., Taube, S., Somerfield, M.R.,
Hayes, D.F., Bast, R.C., Jr., 2007. American Society of Clinical Oncology 2007 update of
recommendations for the use of tumor markers in breast cancer. J Clin Oncol 25, 5287-
5312.
Healy, J.M., Lewis, S.D., Kurz, M., Boomer, R.M., Thompson, K.M., Wilson, C.,
McCauley, T.G., 2004. Pharmacokinetics and biodistribution of novel aptamer
compositions. Pharm Res 21, 2234-2246.
Hehlgans, T., Pfeffer, K., 2005. The intriguing biology of the tumour necrosis
factor/tumour necrosis factor receptor superfamily: players, rules and the games.
Immunology 115, 1-20.
Henderikx, P., Coolen-van Neer, N., Jacobs, A., van der Linden, E., Arends, J.W.,
Mullberg, J., Hoogenboom, H.R., 2002. A human immunoglobulin G1 antibody
originating from an in vitro-selected Fab phage antibody binds avidly to tumor-
associated MUC1 and is efficiently internalized. Am J Pathol 160, 1597-1608.
Hicke, B.J., Marion, C., Chang, Y.F., Gould, T., Lynott, C.K., Parma, D., Schmidt, P.G.,
Warren, S., 2001. Tenascin-C aptamers are generated using tumor cells and purified
protein. J Biol Chem 276, 48644-48654.
Hicke, B.J., Stephens, A.W., Gould, T., Chang, Y.F., Lynott, C.K., Heil, J., Borkowski, S.,
Hilger, C.S., Cook, G., Warren, S., Schmidt, P.G., 2006. Tumor targeting by an aptamer. J
Nucl Med 47, 668-678.
Hicke, B.J., Watson, S.R., Koenig, A., Lynott, C.K., Bargatze, R.F., Chang, Y.F.,
Ringquist, S., Moon-McDermott, L., Jennings, S., Fitzwater, T., Han, H.L., Varki, N.,
Albinana, I., Willis, M.C., Varki, A., Parma, D., 1996. DNA aptamers block L-selectin
function in vivo. Inhibition of human lymphocyte trafficking in SCID mice. J Clin Invest
98, 2688-2692.
Holthoff, E.L., Bright, F.V., 2007. Molecularly templated materials in chemical sensing.
Anal Chim Acta 594, 147-161.
Hong, B.J., Compton, O.C., An, Z., Eryazici, I., Nguyen, S.T., 2012. Successful
stabilization of graphene oxide in electrolyte solutions: enhancement of
biofunctionalization and cellular uptake. ACS Nano 6, 63-73.
Hostetter, R.B., Augustus, L.B., Mankarious, R., Chi, K.F., Fan, D., Toth, C., Thomas, P.,
Jessup, J.M., 1990. Carcinoembryonic antigen as a selective enhancer of colorectal
cancer metastasis. J Natl Cancer Inst 82, 380-385.
Hu, Y., Duan, J., Zhan, Q., Wang, F., Lu, X., Yang, X.D., 2012. Novel MUC1 aptamer
selectively delivers cytotoxic agent to cancer cells in vitro. PLoS One 7, e31970.
Huang, Y., He, S., Cao, W., Cai, K., Liang, X.J., 2012. Biomedical nanomaterials for
imaging-guided cancer therapy. Nanoscale 4, 6135-6149.
Huang, Y.F., Chang, H.T., Tan, W., 2008. Cancer cell targeting using multiple aptamers
conjugated on nanorods. Anal Chem 80, 567-572.
Huang, Y.F., Shangguan, D., Liu, H., Phillips, J.A., Zhang, X., Chen, Y., Tan, W., 2009.
Molecular assembly of an aptamer-drug conjugate for targeted drug delivery to tumor
cells. Chembiochem 10, 862-868.

149
Huang, Y.W., Baluna, R., Vitetta, E.S., 1997. Adhesion molecules as targets for cancer
therapy. Histol Histopathol 12, 467-477.
Ishida, T., Iden, D.L., Allen, T.M., 1999. A combinatorial approach to producing
sterically stabilized (Stealth) immunoliposomal drugs. FEBS Lett 460, 129-133.
Jellinek, D., Green, L.S., Bell, C., Janjic, N., 1994. Inhibition of receptor binding by
high-affinity RNA ligands to vascular endothelial growth factor. Biochemistry 33, 10450-
10456.
Jellinek, D., Green, L.S., Bell, C., Lynott, C.K., Gill, N., Vargeese, C., Kirschenheuter,
G., McGee, D.P., Abesinghe, P., Pieken, W.A., et al., 1995. Potent 2'-amino-2'-
deoxypyrimidine RNA inhibitors of basic fibroblast growth factor. Biochemistry 34,
11363-11372.
Jellinek, D., Lynott, C.K., Rifkin, D.B., Janjic, N., 1993. High-affinity RNA ligands to
basic fibroblast growth factor inhibit receptor binding. Proc Natl Acad Sci U S A 90,
11227-11231.
Jemal, A., Siegel, R., Ward, E., Hao, Y., Xu, J., Murray, T., Thun, M.J., 2008. Cancer
statistics, 2008. CA Cancer J Clin 58, 71-96.
Jessup, J.M., Kim, J.C., Thomas, P., Ishii, S., Ford, R., Shively, J.E., Durbin, H.,
Stanners, C.P., Fuks, A., Zhou, H., et al., 1993a. Adhesion to carcinoembryonic antigen
by human colorectal carcinoma cells involves at least two epitopes. Int J Cancer 55, 262-
268.
Jessup, J.M., Laguinge, L., Lin, S., Samara, R., Aufman, K., Battle, P., Frantz, M.,
Edmiston, K.H., Thomas, P., 2004. Carcinoembryonic antigen induction of IL-10 and IL-
6 inhibits hepatic ischemic/reperfusion injury to colorectal carcinoma cells. Int J Cancer
111, 332-337.
Jessup, J.M., Petrick, A.T., Toth, C.A., Ford, R., Meterissian, S., O'Hara, C.J., Steele, G.,
Jr., Thomas, P., 1993b. Carcinoembryonic antigen: enhancement of liver colonisation
through retention of human colorectal carcinoma cells. Br J Cancer 67, 464-470.
Kaneda, Y., 2000. Virosomes: evolution of the liposome as a targeted drug delivery
system. Adv Drug Deliv Rev 43, 197-205.
Kashyap, N., Kumar, N., Kumar, M.N., 2005. Hydrogels for pharmaceutical and
biomedical applications. Crit Rev Ther Drug Carrier Syst 22, 107-149.
Kaur, G., Roy, I., 2008. Therapeutic applications of aptamers. Expert Opin Investig Drugs
17, 43-60.
Keefe, A.D., Cload, S.T., 2008. SELEX with modified nucleotides. Curr Opin Chem Biol
12, 448-456.
Keefe, A.D., Pai, S., Ellington, A., 2010. Aptamers as therapeutics. Nat Rev Drug Discov
9, 537-550.
Kikin, O., D'Antonio, L., Bagga, P.S., 2006. QGRS Mapper: a web-based server for
predicting G-quadruplexes in nucleotide sequences. Nucleic Acids Res 34, W676-682.
Kitsuki, H., Katano, M., Morisaki, T., Torisu, M., 1995. CEA-mediated homotypic
aggregation of human colorectal carcinoma cells in a malignant effusion. Cancer Lett 88,
7-13.
Komatsu, M., Kobayashi, D., Saito, K., Furuya, D., Yagihashi, A., Araake, H., Tsuji, N.,
Sakamaki, S., Niitsu, Y., Watanabe, N., 2001. Tumor necrosis factor-alpha in serum of
patients with inflammatory bowel disease as measured by a highly sensitive immuno-
PCR. Clin Chem 47, 1297-1301.
Korotkova, N., Yang, Y., Le Trong, I., Cota, E., Demeler, B., Marchant, J., Thomas, W.E.,
Stenkamp, R.E., Moseley, S.L., Matthews, S., 2008. Binding of Dr adhesins of

150
Escherichia coli to carcinoembryonic antigen triggers receptor dissociation. Mol
Microbiol 67, 420-434.
Kourlas, H., Abrams, P., 2007. Ranibizumab for the treatment of neovascular age-related
macular degeneration: a review. Clin Ther 29, 1850-1861.
Krieg, A.M., 2002. CpG motifs in bacterial DNA and their immune effects. Annu Rev
Immunol 20, 709-760.
Krop-Watorek, A., Klopocki, A.G., Czerwinski, M., Lisowska, E., 2002. Adhesive
properties of carcinoembryonic antigen glycoforms expressed in glycosylation-deficient
Chinese hamster ovary cell lines. Acta Biochim Pol 49, 273-283.
Krug, A., Rothenfusser, S., Hornung, V., Jahrsdorfer, B., Blackwell, S., Ballas, Z.K.,
Endres, S., Krieg, A.M., Hartmann, G., 2001. Identification of CpG oligonucleotide
sequences with high induction of IFN-alpha/beta in plasmacytoid dendritic cells. Eur J
Immunol 31, 2154-2163.
Kumar, P., Gulbake, A., Jain, S.K., 2012. Liposomes a vesicular nanocarrier: potential
advancements in cancer chemotherapy. Crit Rev Ther Drug Carrier Syst 29, 355-419.
Latham, J.A., Johnson, R., Toole, J.J., 1994. The application of a modified nucleotide in
aptamer selection: novel thrombin aptamers containing 5-(1-pentynyl)-2'-deoxyuridine.
Nucleic Acids Res 22, 2817-2822.
Laurent, S., Forge, D., Port, M., Roch, A., Robic, C., Vander Elst, L., Muller, R.N., 2008.
Magnetic iron oxide nanoparticles: synthesis, stabilization, vectorization,
physicochemical characterizations, and biological applications. Chem Rev 108, 2064-
2110.
Lee, Y.J., Han, S.R., Kim, N.Y., Lee, S.H., Jeong, J.S., Lee, S.W., 2012. An RNA aptamer
that binds carcinoembryonic antigen inhibits hepatic metastasis of colon cancer cells in
mice. Gastroenterology 143, 155-165 e158.
Liao, K.H., Lin, Y.S., Macosko, C.W., Haynes, C.L., 2011. Cytotoxicity of graphene
oxide and graphene in human erythrocytes and skin fibroblasts. ACS Appl Mater
Interfaces 3, 2607-2615.
Linsley, P.S., Brown, J.P., Magnani, J.L., Horn, D., 1988. Monoclonal antibodies reactive
with mucin glycoproteins found in sera from breast cancer patients. Cancer Res 48,
2138-2148.
Litvinov, S.V., Hilkens, J., 1993. The epithelial sialomucin, episialin, is sialylated during
recycling. J Biol Chem 268, 21364-21371.
Liu, H., Rajasekaran, A.K., Moy, P., Xia, Y., Kim, S., Navarro, V., Rahmati, R., Bander,
N.H., 1998. Constitutive and antibody-induced internalization of prostate-specific
membrane antigen. Cancer Res 58, 4055-4060.
Liu, J., Cao, Z., Lu, Y., 2009a. Functional nucleic acid sensors. Chem Rev 109, 1948-
1998.
Liu, K., Zhang, J.J., Wang, C., Zhu, J.J., 2011. Graphene-assisted dual amplification
strategy for the fabrication of sensitive amperometric immunosensor. Biosens
Bioelectron 26, 3627-3632.
Liu, Y., Kuan, C.T., Mi, J., Zhang, X., Clary, B.M., Bigner, D.D., Sullenger, B.A., 2009b.
Aptamers selected against the unglycosylated EGFRvIII ectodomain and delivered
intracellularly reduce membrane-bound EGFRvIII and induce apoptosis. Biol Chem 390,
137-144.
Locksley, R.M., Killeen, N., Lenardo, M.J., 2001. The TNF and TNF receptor
superfamilies: integrating mammalian biology. Cell 104, 487-501.
Long, S.B., Long, M.B., White, R.R., Sullenger, B.A., 2008. Crystal structure of an RNA

151
aptamer bound to thrombin. RNA 14, 2504-2512.
Lupold, S.E., Hicke, B.J., Lin, Y., Coffey, D.S., 2002. Identification and characterization
of nuclease-stabilized RNA molecules that bind human prostate cancer cells via the
prostate-specific membrane antigen. Cancer Res 62, 4029-4033.
Maas, S., 2011. Base modification RNA editing: information recoding on the fly. Semin
Cell Dev Biol 23, 243.
Macaya, R.F., Schultze, P., Smith, F.W., Roe, J.A., Feigon, J., 1993. Thrombin-binding
DNA aptamer forms a unimolecular quadruplex structure in solution. Proc Natl Acad Sci
U S A 90, 3745-3749.
Maeda, H., 2001. The enhanced permeability and retention (EPR) effect in tumor
vasculature: the key role of tumor-selective macromolecular drug targeting. Adv Enzyme
Regul 41, 189-207.
Maini, R.N., Brennan, F.M., Williams, R., Chu, C.Q., Cope, A.P., Gibbons, D., Elliott,
M., Feldmann, M., 1993. TNF-alpha in rheumatoid arthritis and prospects of anti-TNF
therapy. Clin Exp Rheumatol 11 Suppl 8, S173-175.
Mamaghani, S., Patel, S., Hedley, D.W., 2009. Glycogen synthase kinase-3 inhibition
disrupts nuclear factor-kappaB activity in pancreatic cancer, but fails to sensitize to
gemcitabine chemotherapy. BMC Cancer 9, 132.
Mamot, C., Drummond, D.C., Greiser, U., Hong, K., Kirpotin, D.B., Marks, J.D., Park,
J.W., 2003. Epidermal growth factor receptor (EGFR)-targeted immunoliposomes
mediate specific and efficient drug delivery to EGFR- and EGFRvIII-overexpressing
tumor cells. Cancer Res 63, 3154-3161.
Mann, A.P., Bhavane, R.C., Somasunderam, A., Liz Montalvo-Ortiz, B., Ghaghada, K.B.,
Volk, D., Nieves-Alicea, R., Suh, K.S., Ferrari, M., Annapragada, A., Gorenstein, D.G.,
Tanaka, T., 2011. Thioaptamer conjugated liposomes for tumor vasculature targeting.
Oncotarget 2, 298-304.
Mann, A.P., Somasunderam, A., Nieves-Alicea, R., Li, X., Hu, A., Sood, A.K., Ferrari,
M., Gorenstein, D.G., Tanaka, T., 2010. Identification of thioaptamer ligand against E-
selectin: potential application for inflamed vasculature targeting. PLoS One 5.
Marchand, C., Pourquier, P., Laco, G.S., Jing, N., Pommier, Y., 2002. Interaction of
human nuclear topoisomerase I with guanosine quartet-forming and guanosine-rich
single-stranded DNA and RNA oligonucleotides. J Biol Chem 277, 8906-8911.
Markel, G., Gruda, R., Achdout, H., Katz, G., Nechama, M., Blumberg, R.S., Kammerer,
R., Zimmermann, W., Mandelboim, O., 2004. The critical role of residues 43R and 44Q
of carcinoembryonic antigen cell adhesion molecules-1 in the protection from killing by
human NK cells. J Immunol 173, 3732-3739.
Matrone, M.A., Whipple, R.A., Thompson, K., Cho, E.H., Vitolo, M.I., Balzer, E.M.,
Yoon, J.R., Ioffe, O.B., Tuttle, K.C., Tan, M., Martin, S.S., 2010. Metastatic breast
tumors express increased tau, which promotes microtentacle formation and the
reattachment of detached breast tumor cells. Oncogene 29, 3217-3227.
Matsumura, Y., Maeda, H., 1986. A new concept for macromolecular therapeutics in
cancer chemotherapy: mechanism of tumoritropic accumulation of proteins and the
antitumor agent smancs. Cancer Res 46, 6387-6392.
Matthews, N., Neale, M.L., Jackson, S.K., Stark, J.M., 1987. Tumour cell killing by
tumour necrosis factor: inhibition by anaerobic conditions, free-radical scavengers and
inhibitors of arachidonate metabolism. Immunology 62, 153-155.
Mayer, G., 2009. The chemical biology of aptamers. Angew Chem Int Ed Engl 48, 2672-
2689.

152
McGuckin, M.A., Walsh, M.D., Hohn, B.G., Ward, B.G., Wright, R.G., 1995. Prognostic
significance of MUC1 epithelial mucin expression in breast cancer. Hum Pathol 26, 432-
439.
McNamara, J.O., 2nd, Andrechek, E.R., Wang, Y., Viles, K.D., Rempel, R.E., Gilboa, E.,
Sullenger, B.A., Giangrande, P.H., 2006. Cell type-specific delivery of siRNAs with
aptamer-siRNA chimeras. Nat Biotechnol 24, 1005-1015.
McNamara, J.O., Kolonias, D., Pastor, F., Mittler, R.S., Chen, L., Giangrande, P.H.,
Sullenger, B., Gilboa, E., 2008. Multivalent 4-1BB binding aptamers costimulate CD8+
T cells and inhibit tumor growth in mice. J Clin Invest 118, 376-386.
Mierau, M., Schoels, M., Gonda, G., Fuchs, J., Aletaha, D., Smolen, J.S., 2007. Assessing
remission in clinical practice. Rheumatology (Oxford) 46, 975-979.
Miyata, T., Asami, N., Uragami, T., 1999. A reversibly antigen-responsive hydrogel.
Nature 399, 766-769.
Mohler, K.M., Torrance, D.S., Smith, C.A., Goodwin, R.G., Stremler, K.E., Fung, V.P.,
Madani, H., Widmer, M.B., 1993. Soluble tumor necrosis factor (TNF) receptors are
effective therapeutic agents in lethal endotoxemia and function simultaneously as both
TNF carriers and TNF antagonists. J Immunol 151, 1548-1561.
Moreira, J.N., Ishida, T., Gaspar, R., Allen, T.M., 2002. Use of the post-insertion
technique to insert peptide ligands into pre-formed stealth liposomes with retention of
binding activity and cytotoxicity. Pharm Res 19, 265-269.
Morris, K.N., Jensen, K.B., Julin, C.M., Weil, M., Gold, L., 1998. High affinity ligands
from in vitro selection: complex targets. Proc Natl Acad Sci U S A 95, 2902-2907.
Mount, C., Featherstone, J., 2005. Rheumatoid arthritis market. Nat Rev Drug Discov 4,
11-12.
Muller, J., Isermann, B., Ducker, C., Salehi, M., Meyer, M., Friedrich, M., Madhusudhan,
T., Oldenburg, J., Mayer, G., Potzsch, B., 2009. An exosite-specific ssDNA aptamer
inhibits the anticoagulant functions of activated protein C and enhances inhibition by
protein C inhibitor. Chem Biol 16, 442-451.
Mussi, A., Bonifati, C., Carducci, M., D'Agosto, G., Pimpinelli, F., D'Urso, D., D'Auria,
L., Fazio, M., Ameglio, F., 1997. Serum TNF-alpha levels correlate with disease severity
and are reduced by effective therapy in plaque-type psoriasis. J Biol Regul Homeost
Agents 11, 115-118.
Neidle, S., Parkinson, G.N., 2003. The structure of telomeric DNA. Curr Opin Struct Biol
13, 275-283.
Ng, E.W., Shima, D.T., Calias, P., Cunningham, E.T., Jr., Guyer, D.R., Adamis, A.P.,
2006. Pegaptanib, a targeted anti-VEGF aptamer for ocular vascular disease. Nat Rev
Drug Discov 5, 123-132.
Nicholson, T.B., Stanners, C.P., 2006. Specific inhibition of GPI-anchored protein
function by homing and self-association of specific GPI anchors. J Cell Biol 175, 647-
659.
Ohuchi, S.P., Ohtsu, T., Nakamura, Y., 2005. A novel method to generate aptamers
against recombinant targets displayed on the cell surface. Nucleic Acids Symp Ser (Oxf),
351-352.
Oikawa, S., Inuzuka, C., Kuroki, M., Matsuoka, Y., Kosaki, G., Nakazato, H., 1989. Cell
adhesion activity of non-specific cross-reacting antigen (NCA) and carcinoembryonic
antigen (CEA) expressed on CHO cell surface: homophilic and heterophilic adhesion.
Biochem Biophys Res Commun 164, 39-45.
Orava, E.W., Cicmil, N., Gariepy, J., 2010. Delivering cargoes into cancer cells using

153
DNA aptamers targeting internalized surface portals. Biochim Biophys Acta 1798, 2190-
2200.
Orava, E.W., Jarvik, N., Shek, Y.L., Sidhu, S.S., Gariepy, J., 2012. A short DNA aptamer
that recognizes TNF-alpha and blocks its activity in vitro. ACS Chem Biol.
Ordonez, C., Screaton, R.A., Ilantzis, C., Stanners, C.P., 2000. Human carcinoembryonic
antigen functions as a general inhibitor of anoikis. Cancer Res 60, 3419-3424.
Paborsky, L.R., McCurdy, S.N., Griffin, L.C., Toole, J.J., Leung, L.L., 1993. The single-
stranded DNA aptamer-binding site of human thrombin. J Biol Chem 268, 20808-20811.
Padilla, R., Sousa, R., 1999. Efficient synthesis of nucleic acids heavily modified with
non-canonical ribose 2'-groups using a mutantT7 RNA polymerase (RNAP). Nucleic
Acids Res 27, 1561-1563.
Padmanabhan, K., Padmanabhan, K.P., Ferrara, J.D., Sadler, J.E., Tulinsky, A., 1993. The
structure of alpha-thrombin inhibited by a 15-mer single-stranded DNA aptamer. J Biol
Chem 268, 17651-17654.
Paludan, S.R., 2000. Synergistic action of pro-inflammatory agents: cellular and
molecular aspects. J Leukoc Biol 67, 18-25.
Peng, C., Hu, W., Zhou, Y., Fan, C., Huang, Q., 2010. Intracellular imaging with a
graphene-based fluorescent probe. Small 6, 1686-1692.
Peng, C.G., Damha, M.J., 2007. G-quadruplex induced stabilization by 2'-deoxy-2'-
fluoro-D-arabinonucleic acids (2'F-ANA). Nucleic Acids Res 35, 4977-4988.
Phan, A.T., Kuryavyi, V., Ma, J.B., Faure, A., Andreola, M.L., Patel, D.J., 2005. An
interlocked dimeric parallel-stranded DNA quadruplex: a potent inhibitor of HIV-1
integrase. Proc Natl Acad Sci U S A 102, 634-639.
Pidgeon, C., Hunt, C.A., 1981. Calculating number and surface area of liposomes in any
suspension. J Pharm Sci 70, 173-176.
Putti, M.C., Rondelli, R., Cocito, M.G., Arico, M., Sainati, L., Conter, V., Guglielmi, C.,
Cantu-Rajnoldi, A., Consolini, R., Pession, A., Zanesco, L., Masera, G., Biondi, A.,
Basso, G., 1998. Expression of myeloid markers lacks prognostic impact in children
treated for acute lymphoblastic leukemia: Italian experience in AIEOP-ALL 88-91
studies. Blood 92, 795-801.
Qhobosheane, M., Santra, S., Zhang, P., Tan, W., 2001. Biochemically functionalized
silica nanoparticles. Analyst 126, 1274-1278.
Rajendran, M., Ellington, A.D., 2008. Selection of fluorescent aptamer beacons that light
up in the presence of zinc. Anal Bioanal Chem 390, 1067-1075.
Randazzo, A., Spada, G.P., Silva, M.W., 2012. Circular Dichroism of Quadruplex
Structures. Top Curr Chem.
Robak, E., Sysa-Jedrzejewska, A., Dziankowska, B., Torzecka, D., Chojnowski, K.,
Robak, T., 1998. Association of interferon gamma, tumor necrosis factor alpha and
interleukin 6 serum levels with systemic lupus erythematosus activity. Arch Immunol
Ther Exp (Warsz) 46, 375-380.
Robbins, P.F., Kantor, J.A., Salgaller, M., Hand, P.H., Fernsten, P.D., Schlom, J., 1991.
Transduction and expression of the human carcinoembryonic antigen gene in a murine
colon carcinoma cell line. Cancer Res 51, 3657-3662.
Ross, J.S., Sheehan, C.E., Fisher, H.A., Kaufman, R.P., Jr., Kaur, P., Gray, K., Webb, I.,
Gray, G.S., Mosher, R., Kallakury, B.V., 2003. Correlation of primary tumor prostate-
specific membrane antigen expression with disease recurrence in prostate cancer. Clin
Cancer Res 9, 6357-6362.
Roukos, D.H., 2012. Disrupting cancer cells' biocircuits with interactome-based drugs: is

154
'clinical' innovation realistic? Expert Rev Proteomics 9, 349-353.
Rudnick, S.I., Lou, J., Shaller, C.C., Tang, Y., Klein-Szanto, A.J., Weiner, L.M., Marks,
J.D., Adams, G.P., 2011. Influence of affinity and antigen internalization on the uptake
and penetration of Anti-HER2 antibodies in solid tumors. Cancer Res 71, 2250-2259.
Russo Krauss, I., Merlino, A., Giancola, C., Randazzo, A., Mazzarella, L., Sica, F., 2011.
Thrombin-aptamer recognition: a revealed ambiguity. Nucleic Acids Res 39, 7858-7867.
Ryu, J.H., Koo, H., Sun, I.C., Yuk, S.H., Choi, K., Kim, K., Kwon, I.C., 2012. Tumor-
targeting multi-functional nanoparticles for theragnosis: new paradigm for cancer
therapy. Adv Drug Deliv Rev 64, 1447-1458.
Samara, R.N., Laguinge, L.M., Jessup, J.M., 2007. Carcinoembryonic antigen inhibits
anoikis in colorectal carcinoma cells by interfering with TRAIL-R2 (DR5) signaling.
Cancer Res 67, 4774-4782.
Santulli-Marotto, S., Nair, S.K., Rusconi, C., Sullenger, B., Gilboa, E., 2003. Multivalent
RNA aptamers that inhibit CTLA-4 and enhance tumor immunity. Cancer Res 63, 7483-
7489.
Sathuluri, R.R., Yoshikawa, H., Shimizu, E., Saito, M., Tamiya, E., 2011. Gold
nanoparticle-based surface-enhanced Raman scattering for noninvasive molecular
probing of embryonic stem cell differentiation. PLoS One 6, e22802.
Savla, R., Taratula, O., Garbuzenko, O., Minko, T., 2011. Tumor targeted quantum dot-
mucin 1 aptamer-doxorubicin conjugate for imaging and treatment of cancer. J Control
Release 153, 16-22.
Scheinberg, D.A., Tanimoto, M., McKenzie, S., Strife, A., Old, L.J., Clarkson, B.D.,
1989. Monoclonal antibody M195: a diagnostic marker for acute myelogenous leukemia.
Leukemia 3, 440-445.
Schmidmaier, R., Baumann, P., 2008. ANTI-ADHESION evolves to a promising
therapeutic concept in oncology. Curr Med Chem 15, 978-990.
Schmidt, M.M., Thurber, G.M., Wittrup, K.D., 2008. Kinetics of anti-carcinoembryonic
antigen antibody internalization: effects of affinity, bivalency, and stability. Cancer
Immunol Immunother 57, 1879-1890.
Schneider, D.J., Feigon, J., Hostomsky, Z., Gold, L., 1995. High-affinity ssDNA
inhibitors of the reverse transcriptase of type 1 human immunodeficiency virus.
Biochemistry 34, 9599-9610.
Schoetzau, T., Langner, J., Moyroud, E., Roehl, I., Vonhoff, S., Klussmann, S., 2003.
Aminomodified nucleobases: functionalized nucleoside triphosphates applicable for
SELEX. Bioconjug Chem 14, 919-926.
Screaton, R.A., DeMarte, L., Draber, P., Stanners, C.P., 2000. The specificity for the
differentiation blocking activity of carcinoembryonic antigen resides in its
glycophosphatidyl-inositol anchor. J Cell Biol 150, 613-626.
Screaton, R.A., Penn, L.Z., Stanners, C.P., 1997. Carcinoembryonic antigen, a human
tumor marker, cooperates with Myc and Bcl-2 in cellular transformation. J Cell Biol 137,
939-952.
Sen, R., Baltimore, D., 1986. Inducibility of kappa immunoglobulin enhancer-binding
protein Nf-kappa B by a posttranslational mechanism. Cell 47, 921-928.
Sheehan, J.P., Sadler, J.E., 1994. Molecular mapping of the heparin-binding exosite of
thrombin. Proc Natl Acad Sci U S A 91, 5518-5522.
Shih, L.B., Goldenberg, D.M., Xuan, H., Lu, H.W., Mattes, M.J., Hall, T.C., 1994.
Internalization of an intact doxorubicin immunoconjugate. Cancer Immunol Immunother
38, 92-98.

155
Siddiqui-Jain, A., Grand, C.L., Bearss, D.J., Hurley, L.H., 2002. Direct evidence for a G-
quadruplex in a promoter region and its targeting with a small molecule to repress c-
MYC transcription. Proc Natl Acad Sci U S A 99, 11593-11598.
Silverman, S.K., 2009. Deoxyribozymes: selection design and serendipity in the
development of DNA catalysts. Acc Chem Res 42, 1521-1531.
Skehan, P., Storeng, R., Scudiero, D., Monks, A., McMahon, J., Vistica, D., Warren, J.T.,
Bokesch, H., Kenney, S., Boyd, M.R., 1990. New colorimetric cytotoxicity assay for
anticancer-drug screening. J Natl Cancer Inst 82, 1107-1112.
Smolen, J.S., Aletaha, D., Koeller, M., Weisman, M.H., Emery, P., 2007. New therapies
for treatment of rheumatoid arthritis. Lancet 370, 1861-1874.
Soeth, E., Wirth, T., List, H.J., Kumbhani, S., Petersen, A., Neumaier, M., Czubayko, F.,
Juhl, H., 2001. Controlled ribozyme targeting demonstrates an antiapoptotic effect of
carcinoembryonic antigen in HT29 colon cancer cells. Clin Cancer Res 7, 2022-2030.
Soontornworajit, B., Wang, Y., 2011. Nucleic acid aptamers for clinical diagnosis: cell
detection and molecular imaging. Anal Bioanal Chem 399, 1591-1599.
Spicer, A.P., Rowse, G.J., Lidner, T.K., Gendler, S.J., 1995. Delayed mammary tumor
progression in Muc-1 null mice. J Biol Chem 270, 30093-30101.
Sporn, M.B., 1996. The war on cancer. Lancet 347, 1377-1381.
Stein, R., Juweid, M., Mattes, M.J., Goldenberg, D.M., 1999. Carcinoembryonic antigen
as a target for radioimmunotherapy of human medullary thyroid carcinoma: antibody
processing, targeting, and experimental therapy with 131I and 90Y labeled MAbs.
Cancer Biother Radiopharm 14, 37-47.
Stribling, W.K., Slaughter, T.F., Houle, T.T., Sane, D.C., 2007. Beyond the platelet count:
heparin antibodies as independent risk predictors. Am Heart J 153, 900-906.
Su, S.L., Huang, I.P., Fair, W.R., Powell, C.T., Heston, W.D., 1995. Alternatively spliced
variants of prostate-specific membrane antigen RNA: ratio of expression as a potential
measurement of progression. Cancer Res 55, 1441-1443.
Taheri, M., Saragovi, U., Fuks, A., Makkerh, J., Mort, J., Stanners, C.P., 2000. Self
recognition in the Ig superfamily. Identification of precise subdomains in
carcinoembryonic antigen required for intercellular adhesion. J Biol Chem 275, 26935-
26943.
Taruttis, A., Ntziachristos, V., 2012. Translational optical imaging. AJR Am J Roentgenol
199, 263-271.
Taylor-Papadimitriou, J., Burchell, J., Miles, D.W., Dalziel, M., 1999. MUC1 and cancer.
Biochim Biophys Acta 1455, 301-313.
Teng, Y., Girvan, A.C., Casson, L.K., Pierce, W.M., Jr., Qian, M., Thomas, S.D., Bates,
P.J., 2007. AS1411 alters the localization of a complex containing protein arginine
methyltransferase 5 and nucleolin. Cancer Res 67, 10491-10500.
Terstappen, L.W., Safford, M., Konemann, S., Loken, M.R., Zurlutter, K., Buchner, T.,
Hiddemann, W., Wormann, B., 1992. Flow cytometric characterization of acute myeloid
leukemia. Part II. Phenotypic heterogeneity at diagnosis. Leukemia 6, 70-80.
Thompson, J.A., Grunert, F., Zimmermann, W., 1991. Carcinoembryonic antigen gene
family: molecular biology and clinical perspectives. J Clin Lab Anal 5, 344-366.
Tong, G.J., Hsiao, S.C., Carrico, Z.M., Francis, M.B., 2009. Viral capsid DNA aptamer
conjugates as multivalent cell-targeting vehicles. J Am Chem Soc 131, 11174-11178.
Toulme, J.J., Di Primo, C., Boucard, D., 2004. Regulating eukaryotic gene expression
with aptamers. FEBS Lett 567, 55-62.
Townshend, B., Aubry, I., Marcellus, R.C., Gehring, K., Tremblay, M.L., 2010. An RNA

156
aptamer that selectively inhibits the enzymatic activity of protein tyrosine phosphatase
1B in vitro. Chembiochem 11, 1583-1593.
Tsimberidou, A.M., Giles, F.J., Estey, E., O'Brien, S., Keating, M.J., Kantarjian, H.M.,
2006. The role of gemtuzumab ozogamicin in acute leukaemia therapy. Br J Haematol
132, 398-409.
Tuerk, C., Gold, L., 1990. Systematic evolution of ligands by exponential enrichment:
RNA ligands to bacteriophage T4 DNA polymerase. Science 249, 505-510.
van den Berg, W.B., 2001. Anti-cytokine therapy in chronic destructive arthritis. Arthritis
Res 3, 18-26.
van der Velden, V.H., Boeckx, N., Jedema, I., te Marvelde, J.G., Hoogeveen, P.G.,
Boogaerts, M., van Dongen, J.J., 2004. High CD33-antigen loads in peripheral blood
limit the efficacy of gemtuzumab ozogamicin (Mylotarg) treatment in acute myeloid
leukemia patients. Leukemia 18, 983-988.
van Vollenhoven, R.F., 2007. Switching between anti-tumour necrosis factors: trying to
get a handle on a complex issue. Ann Rheum Dis 66, 849-851.
Veiseh, O., Gunn, J.W., Zhang, M., 2010. Design and fabrication of magnetic
nanoparticles for targeted drug delivery and imaging. Adv Drug Deliv Rev 62, 284-304.
Vester, B., Wengel, J., 2004. LNA (locked nucleic acid): high-affinity targeting of
complementary RNA and DNA. Biochemistry 43, 13233-13241.
Wagner, A., Platzgummer, M., Kreismayr, G., Quendler, H., Stiegler, G., Ferko, B.,
Vecera, G., Vorauer-Uhl, K., Katinger, H., 2006. GMP production of liposomes--a new
industrial approach. J Liposome Res 16, 311-319.
Wakeman, C.A., Winkler, W.C., Dann, C.E., 3rd, 2007. Structural features of metabolite-
sensing riboswitches. Trends Biochem Sci 32, 415-424.
Wallis, R.S., Ehlers, S., 2005. Tumor necrosis factor and granuloma biology: explaining
the differential infection risk of etanercept and infliximab. Semin Arthritis Rheum 34, 34-
38.
Walter, R.B., Raden, B.W., Kamikura, D.M., Cooper, J.A., Bernstein, I.D., 2005.
Influence of CD33 expression levels and ITIM-dependent internalization on gemtuzumab
ozogamicin-induced cytotoxicity. Blood 105, 1295-1302.
Wang, K.Y., Krawczyk, S.H., Bischofberger, N., Swaminathan, S., Bolton, P.H., 1993.
The tertiary structure of a DNA aptamer which binds to and inhibits thrombin determines
activity. Biochemistry 32, 11285-11292.
White, R.R., Sullenger, B.A., Rusconi, C.P., 2000. Developing aptamers into
therapeutics. J Clin Invest 106, 929-934.
Williamson, J.R., 1994. G-quartet structures in telomeric DNA. Annu Rev Biophys
Biomol Struct 23, 703-730.
Willis, M.C., Collins, B.D., Zhang, T., Green, L.S., Sebesta, D.P., Bell, C., Kellogg, E.,
Gill, S.C., Magallanez, A., Knauer, S., Bendele, R.A., Gill, P.S., Janjic, N., 1998.
Liposome-anchored vascular endothelial growth factor aptamers. Bioconjug Chem 9,
573-582.
Willner, D., Trail, P.A., Hofstead, S.J., King, H.D., Lasch, S.J., Braslawsky, G.R.,
Greenfield, R.S., Kaneko, T., Firestone, R.A., 1993. (6-Maleimidocaproyl)hydrazone of
doxorubicin--a new derivative for the preparation of immunoconjugates of doxorubicin.
Bioconjug Chem 4, 521-527.
Wittenberg, N.J., Haynes, C.L., 2009. Using nanoparticles to push the limits of detection.
Wiley Interdiscip Rev Nanomed Nanobiotechnol 1, 237-254.
Woo, J., Chiu, G.N., Karlsson, G., Wasan, E., Ickenstein, L., Edwards, K., Bally, M.B.,

157
2008. Use of a passive equilibration methodology to encapsulate cisplatin into preformed
thermosensitive liposomes. Int J Pharm 349, 38-46.
Xing, H., Wong, N.Y., Xiang, Y., Lu, Y., 2012. DNA aptamer functionalized
nanomaterials for intracellular analysis, cancer cell imaging and drug delivery. Curr Opin
Chem Biol 16, 429-435.
Xu, X., Hamhouyia, F., Thomas, S.D., Burke, T.J., Girvan, A.C., McGregor, W.G., Trent,
J.O., Miller, D.M., Bates, P.J., 2001. Inhibition of DNA replication and induction of S
phase cell cycle arrest by G-rich oligonucleotides. J Biol Chem 276, 43221-43230.
Yang, L., Zhang, X., Ye, M., Jiang, J., Yang, R., Fu, T., Chen, Y., Wang, K., Liu, C., Tan,
W., 2011. Aptamer-conjugated nanomaterials and their applications. Adv Drug Deliv Rev
63, 1361-1370.
Yu, C., Hu, Y., Duan, J., Yuan, W., Wang, C., Xu, H., Yang, X.D., 2011. Novel aptamer-
nanoparticle bioconjugates enhances delivery of anticancer drug to MUC1-positive
cancer cells in vitro. PLoS One 6, e24077.
Yu, D., Wang, D., Zhu, F.G., Bhagat, L., Dai, M., Kandimalla, E.R., Agrawal, S., 2009.
Modifications incorporated in CpG motifs of oligodeoxynucleotides lead to antagonist
activity of toll-like receptors 7 and 9. J Med Chem 52, 5108-5114.
Zhang, L., Radovic-Moreno, A.F., Alexis, F., Gu, F.X., Basto, P.A., Bagalkot, V., Jon, S.,
Langer, R.S., Farokhzad, O.C., 2007. Co-delivery of hydrophobic and hydrophilic drugs
from nanoparticle-aptamer bioconjugates. ChemMedChem 2, 1268-1271.
Zhang, Y., Chen, Y., Han, D., Ocsoy, I., Tan, W., 2010. Aptamers selected by cell-SELEX
for application in cancer studies. Bioanalysis 2, 907-918.
Zheng, C., Feng, J., Lu, D., Wang, P., Xing, S., Coll, J.L., Yang, D., Yan, X., 2011. A
novel anti-CEACAM5 monoclonal antibody, CC4, suppresses colorectal tumor growth
and enhances NK cells-mediated tumor immunity. PLoS One 6, e21146.
Zheng, J., Perkins, G., Kirilova, A., Allen, C., Jaffray, D.A., 2006. Multimodal contrast
agent for combined computed tomography and magnetic resonance imaging applications.
Invest Radiol 41, 339-348.
Zhou, H., Fuks, A., Alcaraz, G., Bolling, T.J., Stanners, C.P., 1993. Homophilic adhesion
between Ig superfamily carcinoembryonic antigen molecules involves double reciprocal
bonds. J Cell Biol 122, 951-960.
Zhou, J., Swiderski, P., Li, H., Zhang, J., Neff, C.P., Akkina, R., Rossi, J.J., 2009.
Selection, characterization and application of new RNA HIV gp 120 aptamers for facile
delivery of Dicer substrate siRNAs into HIV infected cells. Nucleic Acids Res 37, 3094-
3109.
Zhu, M.Z., Marshall, J., Cole, D., Schlom, J., Tsang, K.Y., 2000. Specific cytolytic T-cell
responses to human CEA from patients immunized with recombinant avipox-CEA
vaccine. Clin Cancer Res 6, 24-33.
Zimmer, R., Thomas, P., 2001. Mutations in the carcinoembryonic antigen gene in
colorectal cancer patients: implications on liver metastasis. Cancer Res 61, 2822-2826.





























