Embed Size (px)
Citation preview

©
New Phytologist
(2003)
159
: 1–10
www.newphytologist.com
1
Forum
Blackwell Publishing Ltd.
Commentary
Functional genomics of plant–pathogen interactions
The ability to cause plant disease is a complex trait thatoccurs in only a small subset of bacterial and fungal species.Understanding the developmental and physiologicaladaptations of pathogens that allow them to invade plants,colonize tissues, and subvert plant metabolism is therefore aconsiderable challenge. A task that will be equally demandingwill be developing an understanding of the molecular basisof pathogen recognition by plants, which underlies theevolution of disease resistance. Functional genomic analysisis poised to revolutionize our understanding of these complexbiological systems (Brown, 2003; Wren, 2000), and reviewsin this
Special Issue
of
New Phytologist
highlight recentadvances in the application of functional genomics to thestudy of plant–pathogen interactions. These illustrate veryeffectively how organisms that were in the past experi-mentally intractable can now be investigated in far greaterdetail than was previously possible, and also highlightsimilarities and differences in comparison with the situationin other plant–microbe interactions, including legumes/nitrogen-fixing bacteria (Colebatch
et al.
, 2002; Sprent,2002) and mycorrhizas (Franken & Requena, 2001; Marsh& Schultze, 2001; Martin, 2001; Tunlid & Talbot, 2002)
The philosophy – reversing reductionism
Functional genomics stems from the availability of genomesequence information from an organism. The generation ofgenome sequence data from two model plant species,
Arabidopsis
and rice, a number of phytopathogenic bacteriaand most recently a phytopathogenic fungus, has providedthe raw information necessary for these new approaches. So,what is functional genomics and why does it offer suchpromise? A philosophical definition might be, ‘a holistic orsystems-based approach to studying information flow withina cell’ (Brown, 2003). This is in marked contrast tomolecular biology which has been a profoundly reductionistdiscipline, investigating the individual actions of genes in anorganism and allowing metabolic or signalling pathways tobe characterised in an empirically determined step-by-stepmanner. A second, more practical definition of functionalgenomics would be ‘the application of high throughputmethods using automated technologies to biology allowingfunctional analysis of the genome, proteome and meta-bolome of an organism’ (see Box 1; also Colebatch
et al
.,
2002; Tunlid, 2003). Genomics therefore embraces theinherent complexity of biological systems, allowing insighton the interplay of a large number of gene products and theconsequences of this communication to the physiology ofa cell. The reviews presented here give a glimpse of whatcan be achieved by applying functional genomics to plant–microbe interactions and the type of studies that are likely tobe carried out in forthcoming years.
Obtaining the information: genome analysis of pathogenic species
The first step in functional genomics obviously requiresgeneration of a high quality DNA sequence. The reviewby Mitchell
et al
. (pp. 53–61) shows the progress madetowards generating a full genome sequence of the patho-genic ascomycete
Magnaporthe grisea
. This fungus, whichcauses a devastating disease of cultivated rice, is widelystudied because of the relative ease with which it can begenetically manipulated and the fascinating developmentalbiology that it displays (Tucker & Talbot, 2001). Theauthors describe how a physical map of the
M. grisea
genome has been generated by anchoring short sequencereads from the end of BAC clones of a large insert genomiclibrary of the fungus. In this way they have provided theskeleton outline upon which a full genome sequence,generated using information from a whole genome shotgunapproach at the Whitehead Institute, can be constructed.The second draft assembly of the
M. grisea
sequence isalready available (http://www-genome.wi.mit.edu/annotation/fungi/magnaporthe) and is in the process of beingannotated. The recent description of the first genomesequence of a filamentous fungus,
Neurospora crassa
, hasprovided a glimpse of the treats in store in such a resource(Galagan et al., 2003). The 10 000 or so identified geneshave already revealed significant differences in signaltransduction apparatus in
N. crassa
compared with therelatively closely related yeasts,
Saccharomyces cerevisiae
and
Schizosaccharomyces pombe
, and the presence of an extensivearray of secondary metabolic pathways. The
N. crassa
genome also contains the smallest number of duplicatedgenes in any eukaryotic genome, a sign of the extensive arrayof mechanisms – including, most notably, repeat-inducedpoint mutations – that prevent duplicated gene sequencesbeing tolerated in the fungus, probably as a defence againstinvading intracellular parasites, such as viruses or otherforms of foreign DNA. The
M. grisea
genome sequencecontains some of the same features, but analysis so faralready indicates important differences in gene number,

Commentary
www.newphytologist.com
©
New Phytologist
(2003)
159
: 1–10
Forum2
secondary metabolism and extracellular enzyme productionthat might be clues to the traits that determine its success asa pathogen. Dean and coworkers emphasise the need forbioinformatic resources and have established a central datarepository for all forms of gene functional analysis of thefungus, which will serve the international community thatworks on the fungus. Similar resources to allow comparativeanalysis of phytopathogenic fungi are also being establishedand will allow the whole phytopathogen research communityto benefit from the
M. grisea
genomic information (Soanes
et al
., 2002).The use of bioinformatics to identify genes of interest in a
pathogenic microorganism is also described in the review inthis issue by Bos
et al
. (pp. 63–72). They have used theavailable expressed sequence tag collections for the potatolate blight oomycete pathogen
Phytophthora infestans
veryeffectively to identify secreted proteins that may act asvirulence factors, or as a consequence of such a role havebecome recognised as avirulence gene products by theproducts of plant resistance genes. An algorithm calledPexfinder was devised which allows putative signal peptidesto be identified in raw cDNA sequences. This was used toselect potential secreted proteins, which were then expressedin host plants using the Potato virus X expression system.This has enabled identification of a group of necrosis-inducing peptides in
P. infestans
that can now be studied in a
systematic manner. Development of gene silencing strategiesfor
P. infestans
will provide an effective route for gene func-tional analysis in an organism that was previously difficultto study using genetics (van West
et al
., 1999). In a novelextension of their strategy, Bos
et al.
have identified poly-morphic sequences in this group of genes putatively encodinglow molecular weight secreted proteins and used linkagedisequilibrium to determine if they correspond to knownavirulence gene loci, or whether they represent novelavirulence gene specificities for which resistance genes haveyet to be identified.
Dissecting the process of plant infection
Plant diseases caused by fungi normally start with a pro-pagule landing on the leaf or root surface of a compatiblehost plant. Many fungal pathogens produce specialisedinfection structures to breach the outer cuticles of their planthosts, while other locate natural openings such as stomata.The development of infection structures such as appressoriahas proceeded rapidly in model species such as
M. grisea
,and related fungi that produce melanin pigmented appres-soria such as
Colletotrichum
spp. (Tucker & Talbot, 2001).In these species, appressorium formation is regulated by acyclic AMP response pathway and requires a mitogen-activated protein kinase (MAPK) encoded by the
PMK1
Box 1 Applying functional genomic analysis techniques to plant pathogens
Functional genomic analysis demands systematic, large-scale analysis of gene function in an organism. The genome of an organism represents the entire gene complement and is therefore a context-independent store of biological information about a given cell or organism. Making precise and predictable changes in the genome is the hallmark of molecular genetics and has now been applied in a more systematic manner to large sets of genes. In the case of plant pathogenic bacteria, methods such as transposon mutagenesis and signature tagged mutagenesis have allowed relatively high throughput forward genetics to be carried out, providing collections of nonpathogenic mutants for subsequent characterisation (Hensel et al., 1995; Titarenko et al., 1997). Phytopathogenic and mutualistic fungal species have proved more recalcitrant to molecular genetic manipulation, however, largely because of the inefficiency (or in some cases impossibility) of introducing DNA into these organisms by transformation. This has precluded complementation cloning and has meant that reverse genetic approaches have predominated in the study of pathogenic fungi. The well studied model phytopathogenic fungi Ustilago maydis and Magnaporthe grisea have predominantly been studied using this approach (Kronstad, 1997; Tucker & Talbot, 2001). New high throughput insertional mutagenesis approaches using, for example, T-DNA tagging by Agrobacterium-mediated transformation, are now providing the mutant collection necessary for a more intensive survey of the genomes of these species. An alternative method for gene analysis in pathogenic fungi relies on in vitro mutagenesis of cloned gene sequences using a bacterial transposon (Tn5, Tn7 or an equivalent). This method, called TAG-KO, allows a transposable element to be introduced into a gene library, providing a set of templates for both genome sequence analysis and targeted gene disruption. This process is highly scalable and has been carried out in a systematic manner in the private sector to investigate fungal virulence and select effective antifungal drug targets (Hamer et al., 2001).
Analysis of the transcriptome – the mRNA pool of a cell and thus a highly context-dependent and ever-changing form of biological information – can be carried out using a number of methods such as expressed sequence tag (EST) sequencing, microarray analysis, and serial analysis of gene expression (SAGE) (for a discussion of transcriptome analysis in mycorrizas, see Nehls, pp. 5–7 in this issue). All of these methods have now been applied to phytopathogens and have begun to reveal the major changes in gene expression that accompany plant infection (Rauyaree et al., 2001; Thomas et al., 2002) and the establishment of mycorrhizal interactions, highlighted in this issue (Peter et al. pp. 117–129). Proteomic analysis to explore the protein complement of a cell, another highly context-dependent and dynamic source of biological information, is also under way and promises to identify many of the proteins that are differentially produced during host interactions and disease generation. This form of analysis will be accompanied by systemic analysis of low molecular weight compounds (metabolomics) that represent the most dynamic form of information in a cell.

Commentary
©
New Phytologist
(2003)
159
: 1–10
www.newphytologist.com
Forum 3
gene in
M. grisea
(Xu & Hamer, 1996). This signallingcascade leads to cellular differentiation and production ofappressoria that develop turgor and subsequently breach thecuticle using, predominantly, physical force (Thines
et al
.,2000). Less is known, however, regarding the moleculargenetics of appressorium development in obligate pathogenicfungi such as the rusts and powdery mildews because of theirrelative intractability to investigation, although some geneticcomponents are clearly shared (Bindslev
et al
., 2001). In soilborne fungi such as
Fusarium oxysporum
, which has a broadhost range and is responsible for many economicallyimportant crop diseases, a wide variety of both forward andreverse genetic approaches have been carried out, asreviewed by Recorbet
et al
. (pp. 73–91). They highlighthow comparative analysis of
F. oxysporum
with fungi suchas
M. grisea
and the corn pathogen
Cochliobolus carbonum
has been made possible by carrying out a logical reversegenetic series, but in contrast how insertional mutagenesis isoffering fresh insights into the attributes required for thissoilborne fungus to be a successful pathogen.
Following infection of plants, biotrophic fungi such as therusts and powdery mildews elaborate specialised intra-cellular structures, called haustoria – the review in this issue byVoegele & Mendgen (pp. 93–100) showcases how a multi-disciplinary approach utilising high resolution microscopy,differential gene expression studies and biochemistry canprovide very significant advances in our understanding ofthese specialised structures. Long thought to be simplefeeding structures, Voegele & Mendgen show that haustoriafulfil other biosynthetic functions and may act to suppressplant defences and to alter the flow of metabolites withinplants, dramatically altering the sink–source relationships inthe plant in favour of the invading pathogen.
Host defence mechanisms
The host of course, is far from passive during plant infectionand launches an orchestrated set of cellular and biochemicalresponses to pathogen invasion. Plants have a set of con-stitutive chemical barriers to infection, including preformedantimicrobial compounds such as saponins and cyanogenicglucosides. The biosynthesis and biological function of thesecompounds is described in the review by Osbourn
et al
.(pp. 101–108). They emphasise the potential for defencecompounds to act in a variety of different ways to protectplants from pathogen attack. The use of genomic resourcesand gene functional analysis is also well documented in theongoing investigation into the pathway for saponin bio-synthesis in oats and its potential for providing engineeredprotection from root pathogens in other cereal crops. Arecent study also shows that saponin detoxification, acounter-attacking process carried out by some pathogenicfungi during plant infection, may not simply inactivatepreformed antimicrobial compounds, but additionally the
degradation products generated by saponin detoxificationmay themselves act to suppress inducible plant defenceresponses (Bouarab
et al
., 2002). This highlights the factthat seemingly straightforward enzymatic functions withpredictable substrates can lead to very unpredictable cellularconsequences in the context of plant–microbe interactions.
Cultivar-specific resistance, resulting from the recognitionof a pathogen-associated molecular pattern (an avirulencegene product), by the product of a plant resistance gene, canoften result in a form of localised cell death called the hyper-sensitive reaction, which is very widely studied and showsparallels (and important differences) with programmed celldeath, which occurs in metazoan development. The com-plexity of the hypersensitive response makes it a good targetfor genomic analysis using transcriptional profiling andproteomic analysis. An example of how gene expressionprofiling can lead to identification of novel regulatory genenetworks can be seen by the study of systemic acquiredresistance carried out using microarrays in
Arabidopsis
(Maleck
et al
., 2000). This study shows how genomics canrapidly define hitherto unrecognised associations betweengenes in a manner which fully embraces the complexity ofthe cellular response, while also clarifying the transcriptionalresponse into a subset of regulatory networks that can indi-vidually be subjected to further experimentation. A review inthis issue by Dale Walters pp. 109–115 also considers the poten-tial involvement of polyamine catabolism in the generationof hydrogen peroxide prior to hypersensitive cell death andthe biochemical mechanism by which this might take place.
The way ahead
The potential for investigating plant–microbe interactionsusing genomic approaches is very clear and it seems likelythat genome sequence will become available for a number ofphytopathogenic and mutualistic fungal species in the nextfew years. Harnessing this information effectively andlearning lessons in how systematically to analyse genes in ahigh throughput manner (Oliver, 2002) will be the keychallenges for the future.
Acknowledgements
Reviews highlighted here emerged from the 10th
NewPhytologist
Symposium, ‘Functional Genomics of Plant–Microbe Interactions’, held in Nancy, France in October2002.
Nicholas J. Talbot
School of Biological Sciences, University of Exeter,Washington Singer Laboratories,
Exeter EX4 4QG, United Kingdom(email [email protected])

Commentary
www.newphytologist.com
©
New Phytologist
(2003)
159
: 1–10
Forum4
References
Bindslev L, Kershaw MJ, Talbot NJ, Oliver RP. 2001.
Complementation of the cAMP-dependent protein kinase A mutant of
Magnaporthe grisea
by
Blumeria graminis pka-c
gene: functional genetic analysis of an obligate biotrophic fungus.
Molecular Plant–Microbe Interactions
14
: 1368–1375.
Bos JIB, Torto TA, Ochwo M, Armstrong M, Whisson SC, Birch PRJ, Kamoun S. 2003
. Intraspecific comparative genomics to identify avirulence genes from
Phytophthora
.
New Phytologist
159: 63–72.
Bouarab K, Melton R, Peart J, Baulcombe D, Osbourn A. 2002.
A saponin-detoxifying enzyme mediates suppression of plant defences.
Nature
418
: 889–892.
Brown SM. 2003.
Essentials of medical genomics
. Hoboken NJ, USA: John Wiley and Sons.
Colebatch G, Trevaskis B, Udvardi M. 2002.
Functional genomics: tools of the trade.
New Phytologist
153
: 27–36.
Colebatch G, Trevaskis B, Udvardi M. 2002.
Symbiotic nitrogen fixation in the postgenomics era.
New Phytologist
153
: 37–42.
Franken P, Requena N. 2001.
Analysis of gene expression in arbuscular mycorrizas: new approaches and challenges.
New Phytologist
150
: 517–523.
Galagan JE, Calvo SE, Borkovich KA, Selker EU, Read ND, Jaffe D, FitzHigh W
et al.
2003.
The genome sequence of the filamentous fungus
Neurospora crassa
.
Nature
422
: 859–868.
Hamer L, Adachi K, Montenegro-Chamorro MV, Tanzer MM, Mahanty SK, Lo C, Tarpey RW, Skalchunes AR, Heiniger RW, Frank SA, Darveaux BA, Lampe DJ, Slater TM, Ramamurthy L, DeZwaan TM, Nelson GH, Shuster JR, Woessner J, Hamer JE. 2001.
Gene discovery and gene function assignment in filamentous fungi.
Proceedings of the National Academy of Sciences, USA
98
: 5110–5115.
Hensel M, Shea JE, Gleeson C, Jones MD, Dalton E, Holden DW. 1995.
Simultaneous identification of bacterial virulence genes by negative selection.
Science
269
: 400–403.
Kronstad JW. 1997.
Virulence and cAMP in smuts, blasts and blights.
Trends in Plant Sciences
2
: 193–199.
Maleck K, Levine A, Eulgem T, Morgan A, Schmid J, Lawton KA, Dangl JL, Dietrich RA. 2000.
The transcriptome of
Arabidopsis thaliana
during systemic acquired resistance.
Nature Genetics
26
: 403–410.
Marsh JF, Schultze M. 2001.
Analysis of arbuscular mycorrizas using symbiosis-defective plant mutants.
New Phytologist
150
: 525–532.
Martin F. 2001.
Frontiers in molecular mycorrhizal research – genes, loci, dots and spins.
New Phytologist
150
: 499–505.
Mitchell TK, Thon MR, Jeong JS, Brown D, Deng J, Dean RA. 2003.
The rice blast pathosystem as a case study for the development of new tools and raw materials for genome analysis of fungal plant pathogens.
New Phytologist
159
: 53–61.
Nehls U. 2003.
Ectomycorrhizal development and function – transcriptome analysis.
New Phytologist 159: 5–7.Oliver SG. 2002. Functional genomics: lessons from yeast.
Philosophical Transaction of the Royal Society B 357: 17–23.Osbourn AE, Qi X, Townsend B, Qin B. 2003. Dissecting plant
secondary metabolism – constitutive chemical defences in cereals. New Phytologist 159: 101–108.
Peter M, Courty P-E, Kohler A, Delaruelle C, Martin D, Tagu D, Frey-Klett P, Duplessis S, Chalot M, Podila G, Martin F. 2003. Analysis of expressed sequence tags from the ectomycorrhizal basidiomycetes Laccaria bicolor and Pisolithus microcarpus. New Phytologist 159: 117–129.
Rauyaree P, Choi W, Fang E, Blackmon B, Dean RA. 2001. Genes expressed during early stages of rice infection with the rice blast fungus Magnaporthe grisea. Molecular Plant Pathology 2: 347–354.
Recorbet G, Steinberg C, Olivain C, Edel V, Trouvelot S, Alabouvette C, Dumas-Gaudot E, Gianinazzi S. 2003. Wanted: pathogenesis-related marker molecules for Fusarium oxysporum. New Phytologist 159: 73–91.
Soanes DM, Skinner W, Keon J, Hargreaves J, Talbot NJ. 2002. Functional genomics of pathogenic fungi and development of bioinformatic resources. Molecular Plant–Microbe Interactions 15: 421–427.
Sprent J. 2002. Knobs, knots and nodules – the renaissance in legume symbiosis research. New Phytologist 153: 2–6.
Thines E, Weber RWS, Talbot NJ. 2000. MAP kinase and protein kinase A-dependent mobilization of triacylglycerol and glycogen during appressorium turgor generation by Magnaporthe Grisea. Plant Cell 12: 1703–1718.
Thomas SW, Glaring MA, Rasmussen SW, Kinane JT, Oliver RP. 2002. Transcript profiling in the barley mildew pathogen Blumeria graminis by serial analysis of gene expression (SAGE). Molecular Plant–Microbe Interactions 15: 847–856.
Titarenko E, LopezSolanilla E, GarciaOlmedo F, RodriguezPalenzuela P. 1997. Mutants of Ralstonia (Pseudomonas) solanacearum sensitive to antimicrobial peptides are altered in their lipopolysaccharide structure and are avirulent in tobacco. Journal of Bacteriology 179: 6699–6704.
Tucker SL, Talbot NJ. 2001. Surface attachment and pre-penetration stage development by plant pathogenic fungi. Annual Review of Phytopathology 39: 385–417.
Tunlid A. 2003. Exploring plant–microbe interactions using DNA microarrays. New Phytologist 158: 235–238.
Tunlid A, Talbot NJ. 2002. Genomics of parasitic and symbiotic fungi. Current Opinion in Microbiology 5: 513–519.
Voegele RT, Mendgen K. 2003. Rust haustoria: nutrient uptake and beyond. New Phytologist 159: 93–100.
Walters D. 2003. Resistance to plant pathogens: possible roles for free polyamines and polyamine catabolism. New Phytologist 159: 109–115.
van West P, Kamoun S, van ‘T, Klooster JW, Govers F. 1999. Internuclear gene silencing in. Phytophthora Infestans Molecular Cell 3: 339–348.
Wren BW. 2000. Microbial genome analysis: insights into virulence, host adaptation and evolution. Nature Genetics 1: 30–39.
Xu JR, Hamer JE. 1996. MAP kinase and cAMP signalling regulate infection structure formation and pathogenic growth in the rice blast fungus Magnaporthe grisea. Genes and Development 10: 2696–2706.
Key words: functional genomics, Magnaporthe grisea, plant–pathogen interactions, transcriptome, mycorrhiza.159CommentaryCommentary

Commentary
© New Phytologist (2003) 159: 1–10 www.newphytologist.com
Forum 5
Ectomycorrhizal development and function – transcriptome analysis
Differential gene expression during ectomycorrhizal develop-ment has been investigated intensely during the past decade,revealing a first glance of events physiologically importantfor the development and function of this symbiosis (Martin,2001). These investigations have enabled the formulation ofdifferent working hypotheses that could explain structuraland functional adaptations observed during plant–fungalinteractions (Martin et al., 1999; Ditengou & Lapeyrie,2000; Nehls et al., 2001). Nevertheless, all hypotheses arebased on the rather small number of genes that have beeninvestigated. To overcome this limitation, a number of EST-projects have been initiated using a small set of ectomy-corrhizal plant and fungal systems (Colebatch et al., 2002;Tunlid, 2003). In this issue (pp. 117–129), Peter et al. describethe generation and analysis of large EST sets obtained fromtwo different ectomycorrhizal fungi, Laccaria bicolor andPisolithus microcarpus (Cairney, 2002; Martin et al., 2002).
‘To complete the picture of fungal gene expression
during ectomycorrhizal development and function a
genome project for at least one of the well-established
model fungi is essential’
Models – two trees, many fungi
Poplar and birch are the two angiosperm partners mainlyused for ectomycorrhizal plant–fungal EST projects. Inaddition to their value for studying ectomycorrhizas, bothplants are handled as model trees to address questions thatare difficult or impossible to investigate in Arabidopsis (e.g.wood formation, seasonal nutrient cycling and storage; seeChaffey, 2002). Thus, for both trees, transformation ispossible and large EST collections from different organs(including roots – Kohler et al., 2003) are already available.In addition, a genome project has been initiated for poplar,which will be finished by the end of this year (News inbrief, 2002) allowing genome-wide expression analysis. Oncompletion, and then with ectomycorrhizas obtained from
both natural sites and well defined lab conditions, ectomy-corrhizal research with these angiosperms will speed up.Compared with angiosperms, model gymnosperms (such asPicea or Pinus spp.) lag behind – they are more difficult tohandle, there is no reliable method of transformation andgenome projects are yet to be initiated.
While only a limited number of tree genera (though withlarge economical and ecological importance) are able toform ectomycorrhizas, several thousand different fungalspecies, mainly basidiomycetes, can undergo symbiosis. Toreflect this enormous biodiversity found in forest ecosystems,a larger number of model fungi has been established, con-taining pioneer as well as intermediate- and late-stage fungi(Amanita muscaria, Hebeloma cylindrosporum, Laccaria bicolor,Paxillus involutus, Pisolithus microcarpus, Suillus bovinus, Tuberborchii). To get an initial set of genes for broad-range inves-tigation of gene expression during development and func-tion of ectomycorrhizas, several EST-projects have recentlybeen initiated (T. Johansson et al. pers. comm.; Lacourtet al., 2002; U. Nehls et al. unpublished; Podila et al.,2002; Peter et al., 2003; C. Plassard et al. pers. comm.;Voiblet et al., 2001) using nonmycorrhized hyphae as wellas mycorrhizas of different ectomycorrhizal model fungi astheir sources.
Laccaria and Pisolithus
In the research by Peter et al. (pp. 117–129 in this issue), thesequences of L. bicolor were obtained from nonmycorrhizalhyphae cultivated under two different carbon regimes, andthose of P. microcarpus from nonmycorrhizal hyphae grownin a rich carbon source as well as from ectomycorrhizas. Atotal of 905 and 806 unique transcripts, respectively, wereanalyzed, representing about 10% of the expected genes ofthese fungi.
A very large number of these genes (60% of the Laccariatranscripts and 50% of those of Pisolithus) did not reveal anysequence homology in existing databases. This rather largeproportion of new genes is typical for EST projects offilamentous fungi, where usually 50–65% of the sequencesobtained represent unknown genes (Skinner et al., 2001)compared with only 20–25% for plant ESTs (Ronninget al., 2003).
When the 20 most abundant ESTs of each fungus,representing 19.8% of the L. bicolor and 31.7% of theP. microcarpus dataset, were compared, only one homologousEST of each set could be found in the dataset of the otherfungus. The total percentage of TCs (tentative consensussequences) shared between L. bicolor and P. microcarpustissues was about 5% (based on nucleotide sequencesimilarity). A similar low percentage of shared TCs was alsofound when the EST sets of either L. bicolor or P. microcarpuswere compared with those of two saprophytic fungi, Agaricusbisporus and Pleurotus ostreatus.

Commentary
www.newphytologist.com © New Phytologist (2003) 159: 1–10
Forum6
The high percentage of unknown genes as well as thelow percentage of homology between different datasets ofbasidiomycetes illustrates the limitations for ectomycor-rhizal research. By contrast to ascomycetes, where a numberof well investigated models and thus large EST datasets aswell as genome sequences exist (Saccharomyces, Neurospora,Aspergillus, Magnaporthe), only a very few basidiomycetes(e.g. Ustilago, Cryptococcus) have been studied in closerdetail. Thus, compared to the large biodiversity found in thefungal kingdom, only a relatively small number of fungalDNA sequences in general and basidiomycete sequences inparticular are available in databases and could be used forhomology analysis. In addition, fungal genes are much morevariable in their DNA sequence than plant genes, frequentlymaking it difficult to identify proteins of homologousfunction in different species.
In silico transcript profiling
For large EST sets, digital analysis of gene expression(in silico transcript profiling) can be performed by comparingthe number of ESTs for a given gene within EST populationsobtained from cells after different treatments (Ewing et al.,1999). Peter et al. compared in silico transcript-profilingfor the 20 most abundant ESTs of both datasets withexpression studies obtained by macro-array hybridization.Five of the 20 most abundant ESTs of L. bicolor and eight ofP. microcarpus were also detected as highly expressed by thearray technique. However, several transcripts identified bymicroarray analysis as highly expressed in both samples(different C-regimes for L. bicolor and nonmycorrhizal vsmycorrhizal hyphae for P. microcarpus) were found only inone of the two EST libraries. These data indicate that in silicoprofiling is robust enough to detect the most abundanttranscripts; however, they also show that results of digitalprofiling should be very carefully interpreted, as they areoften subject to technical limitations (e.g. a limited numberof ESTs). Therefore, unless verified by other techniques(microarray analysis, Northern blot, RT-PCR), the dataobtained by in silico transcript profiling can only provide aninitial glimpse about gene expression.
Conclusion
Even though several independent EST projects are now inprogress, only a limited number of fungal genes can bediscovered by this strategy due to redundancy (which increaseswith sample number) in the DNA sequences obtained. Inconclusion therefore for the establishment of a detailedpicture of fungal gene expression during ectomycorrhizaldevelopment and function, which will be available in thenear future for one of the angiosperm models (poplar), agenome project for at least one of the well-established modelfungi is essential.
Uwe Nehls
Eberhard-Karls-Universität, Physiologische Ökologieder Pflanzen, Auf der Morgenstelle
1, 72076 Tübingen, Germany(email [email protected])
References
Cairney JWG. 2002. Pisolithus – death of the pan-global super fungus. New Phytologist 153: 199–201.
Chaffey NJ. 2002. Why is there so little research into the cell biology of the secondary vascular system of trees. New Phytologist 153: 213–223.
Colebatch G, Trevaskis B, Udvardi M. 2002. Functional genomics: tools of the trade. New Phytologist 153: 27–36.
Ditengou FA, Lapeyrie F. 2000. Hypaphorine from the ectomycorrhizal fungus Pisolithus tinctorius counteracts activities of indole-3-acetic acid and ethylene but not synthetic auxins in eucalypt seedlings. Molecular Plant–Microb Interactions 13: 151–158.
Ewing RM, Kahla AB, Poirot O, Lopez F, Audic S, Claverie JM. 1999. Large-scale statistical analyses of rice ESTs reveal correlated patterns of gene expression. Genome Research 9: 950–959.
Kohler A, Delaruelle C, Martin D, Encelot N, Martin F. 2003. The poplar root transcriptome: analysis of 7000 expressed sequence tags. FEBS Letters 542: 37–41.
Lacourt I, Duplessis S, Abba S, Bonfante P, Martin F. 2002. Isolation and characterization of differentially expressed genes in the mycelium and fruit body of Tuber borchii. Applied Environmental Microbiology 68: 4574–4582.
Martin F. 2001. Frontiers in molecular mycorrhizal research – genes, loci, dots and spins. New Phytologist 150: 499–505.
Martin F, Diez J, Dell B, Delaruelle C. 2002. Phylogeography of the ectomycorrhizal Pisolithus species as inferred from nuclear ribosomal DNA ITS sequences. New Phytologist 153: 345–357.
Martin F, Laurent P, de Carvalho D, Voiblet C, Balestrini R, Bonfante P, Tagu D. 1999. Cell wall proteins of the ectomycorrhizal basidiomycete Pisolithus tinctorius: Identification, function, and expression in symbiosis. Fungal Genetics and Biology 27: 161–174.
Nehls U, Mikolajewski S, Magel E, Hampp R. 2001. The role of carbohydrates in ectomycorrhizal functioning: gene expression and metabolic control. New Phytologist 150: 533–541.
News in brief. 2002. Geneticists branch out to sequence poplar genome. Nature 415: 725.
Peter M, Courty P-E, Kohler A, Delaruelle C, Martin D, Tagu D, Frey-Klett P, Duplessis S, Chalot M, Podila G, Martin F. 2003. Analysis of expressed sequence tags from the ectomycorrhizal basidiomycetes Laccaria bicolor and Pisolithus microcarpus. New Phytologist 159: 117–129.
Podila GK, Zheng J, Balasubramanian S, Sundaram S, Hiremath S, Brand JH, Hymes MJ. 2002. Fungal gene expression in early symbiotic interactions between Laccaria bicolor and red pine. Plant Soil 244: 117–128.

Letters
© New Phytologist (2003) 159: 1–10 www.newphytologist.com
Forum 7
Ronning CM, Stegalkina SS, Ascenzi RA, Bougri O, Hart AL, Utterbach TR, Vanaken SE, Riedmüller SB, White JA, Cho J, Pertea GM, Lee Y, Karamycheva S, Sultana R, Tsai J, Quackenbush J, Griffiths HM, Restrepo S, Smart CD, Fry WE, van der Hoeven R, Tanksley S, Zhang P, Jin H, Yamamoto ML, Baker BJ, Buell RC. 2003. Comparative analyses of potato expressed sequence tag libraries. Plant Physiology 131: 419–429.
Skinner W, Keon J, Hargreaves J. 2001. Gene information for fungal plant pathogens from expressed sequences. Current Opinions in Microbiology 4: 381–386.
Tunlid A. 2003. Exploring plant–microbe interactions using DNA microarrays. New Phytologist 158: 235–238.
Voiblet C, Duplessis S, Encelot N, Martin F. 2001. Identification of symbiosis-regulated genes in Eucalyptus globulus–Pisolithus tinctorius ectomycorrhiza by differential hybridization of arrayed cDNAs. Plant Journal 25: 181–191.
Key words: transcriptome, Pisolithus, Laccaria, ectomycorrhiza, genomics.
Letters
Ergosterol and fatty acids for biomass estimation of mycorrhizal fungi
Ergosterol has recently been used as a biomass indicator tocompare the growth of different arbuscular mycorrhizal(AM) fungi (Hart & Reader, 2002a,b). Here, we show thatergosterol is not a suitable biochemical marker for estimatingthe biomass of AM fungi and that the comparison ofbiomass between different fungal taxa is very difficult usingany kind of currently available biochemical marker.
Because they are usually degraded rapidly after cell deathand because membrane area is assumed to be well correlatedwith the biovolume of microbial cells (Tunlid & White,1992), membrane compounds, such as sterols, are attractivebiomass indicators of microorganisms in environmentalsamples. Furthermore, sterols seem to represent a ratherconstant part of the fungal biomass, constituting somewherebetween 5 and 15 mg g−1 in most fungal groups (Weete &Gandhi, 1996). In particular, ergosterol is specific to thefungal kingdom (Weete & Gandhi, 1996) and occurs mainlyas a membrane constituent. Ergosterol has been used toindicate the fungal biomass in soil (Grant & West, 1986;Frostegård & Bååth, 1996), pathogenic fungi in roots(Bindler et al., 1988), fungi in cereal grains (Seitz et al., 1972),saprophytic fungi in decaying plant material (Newell et al.,1988) and ectomycorrhizal fungi in roots (Salmanowicz &Nylund, 1988; Wallander et al., 1997) and soil (Ek et al.,1994; Ekblad et al., 1995).
The occurrence of ergosterol is generally restricted to themore advanced fungal taxa, while the more primitive taxacontain other sterols (Weete & Gandhi, 1996). Thus, it isthe dominating sterol in ascomycetes and basidiomycetes.By contrast, the picture is rather more complex within thephylum Zygomycota where members of Mucorales containergosterol, while Mortierella contain desmosterol, but noergosterol (Weete & Gandhi, 1999). In a similar way, mostmembers of the newly identified phylum Glomeromycota(Schüssler et al., 2001), fungal obligate symbionts formingarbuscular mycorrhizas (AM), seem to contain sterols otherthan ergosterol. No ergosterol was detected in several studiesin which gas chromatography-mass spectrometry (GC-MS)analysis was carried out on spores or extraradical myceliumof either Glomus or Acaulospora species (Beilby & Kidby,1980; Beilby, 1980; Nordby et al., 1981; Grandmougin-Ferjani et al., 1999; Fontaine et al., 2001) or mature sporesof Gigaspora margarita (Grandmougin-Ferjani et al., 1999).However, Frey et al. (1992, 1994) identified ergosterol inroots colonised by Glomus intraradices using GC-MS, but notin noncolonised roots, and they proposed the ergosterolcontent in extraradical hyphae of this fungus to be 0.063 mgper g mycelium. More recently, Fujiyoshi et al. (2000) foundthat the mycelium collected around roots colonised byGigaspora margarita had 0.63 mg ergosterol per g of myc-elium. Nevertheless, neither of the former two studies wascarried out under in vitro conditions, and thus ergosterol fromcontaminating fungi could hardly be avoided. The slightest con-tamination may have a significant effect on the results becauseof the high ergosterol content in many saprophytic fungi.
Despite the fact that ergosterol has been shown to beabsent in AM fungi on several occasions, high performance
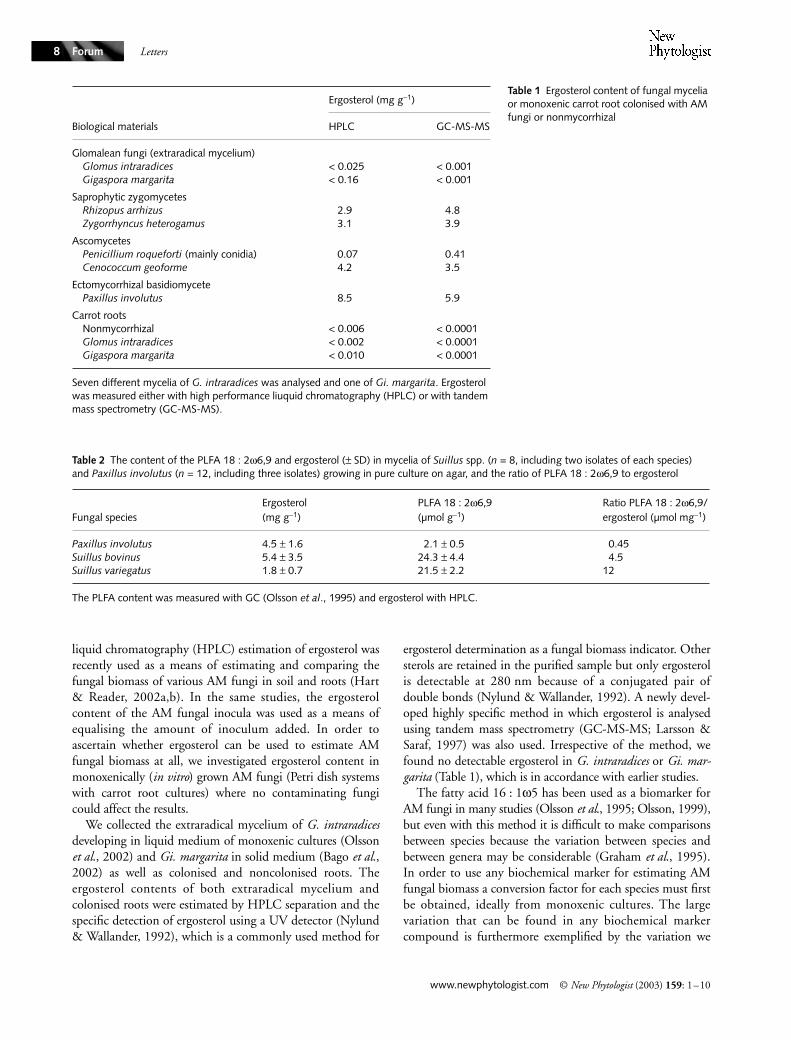
Letters
www.newphytologist.com © New Phytologist (2003) 159: 1–10
Forum8
liquid chromatography (HPLC) estimation of ergosterol wasrecently used as a means of estimating and comparing thefungal biomass of various AM fungi in soil and roots (Hart& Reader, 2002a,b). In the same studies, the ergosterolcontent of the AM fungal inocula was used as a means ofequalising the amount of inoculum added. In order toascertain whether ergosterol can be used to estimate AMfungal biomass at all, we investigated ergosterol content inmonoxenically (in vitro) grown AM fungi (Petri dish systemswith carrot root cultures) where no contaminating fungicould affect the results.
We collected the extraradical mycelium of G. intraradicesdeveloping in liquid medium of monoxenic cultures (Olssonet al., 2002) and Gi. margarita in solid medium (Bago et al.,2002) as well as colonised and noncolonised roots. Theergosterol contents of both extraradical mycelium andcolonised roots were estimated by HPLC separation and thespecific detection of ergosterol using a UV detector (Nylund& Wallander, 1992), which is a commonly used method for
ergosterol determination as a fungal biomass indicator. Othersterols are retained in the purified sample but only ergosterolis detectable at 280 nm because of a conjugated pair ofdouble bonds (Nylund & Wallander, 1992). A newly devel-oped highly specific method in which ergosterol is analysedusing tandem mass spectrometry (GC-MS-MS; Larsson &Saraf, 1997) was also used. Irrespective of the method, wefound no detectable ergosterol in G. intraradices or Gi. mar-garita (Table 1), which is in accordance with earlier studies.
The fatty acid 16 : 1ω5 has been used as a biomarker forAM fungi in many studies (Olsson et al., 1995; Olsson, 1999),but even with this method it is difficult to make comparisonsbetween species because the variation between species andbetween genera may be considerable (Graham et al., 1995).In order to use any biochemical marker for estimating AMfungal biomass a conversion factor for each species must firstbe obtained, ideally from monoxenic cultures. The largevariation that can be found in any biochemical markercompound is furthermore exemplified by the variation we
Biological materials
Ergosterol (mg g−1)
HPLC GC-MS-MS
Glomalean fungi (extraradical mycelium)Glomus intraradices < 0.025 < 0.001Gigaspora margarita < 0.16 < 0.001
Saprophytic zygomycetesRhizopus arrhizus 2.9 4.8Zygorrhyncus heterogamus 3.1 3.9
AscomycetesPenicillium roqueforti (mainly conidia) 0.07 0.41Cenococcum geoforme 4.2 3.5
Ectomycorrhizal basidiomycetePaxillus involutus 8.5 5.9
Carrot rootsNonmycorrhizal < 0.006 < 0.0001Glomus intraradices < 0.002 < 0.0001Gigaspora margarita < 0.010 < 0.0001
Seven different mycelia of G. intraradices was analysed and one of Gi. margarita. Ergosterol was measured either with high performance liuquid chromatography (HPLC) or with tandem mass spectrometry (GC-MS-MS).
Table 2 The content of the PLFA 18 : 2ω6,9 and ergosterol (± SD) in mycelia of Suillus spp. (n = 8, including two isolates of each species) and Paxillus involutus (n = 12, including three isolates) growing in pure culture on agar, and the ratio of PLFA 18 : 2ω6,9 to ergosterol
Fungal speciesErgosterol(mg g−1)
PLFA 18 : 2ω6,9(µmol g−1)
Ratio PLFA 18 : 2ω6,9/ergosterol (µmol mg−1)
Paxillus involutus 4.5 ± 1.6 2.1 ± 0.5 0.45Suillus bovinus 5.4 ± 3.5 24.3 ± 4.4 4.5Suillus variegatus 1.8 ± 0.7 21.5 ± 2.2 12
The PLFA content was measured with GC (Olsson et al., 1995) and ergosterol with HPLC.
Table 1 Ergosterol content of fungal mycelia or monoxenic carrot root colonised with AM fungi or nonmycorrhizal

Letters
© New Phytologist (2003) 159: 1–10 www.newphytologist.com
Forum 9
found in the content of the phospholipid fatty acid(PLFA) 18 : 2ω6,9 between Suillus spp. and Paxillus involutus,while both fungi contained similar amounts of ergosterol(Table 2).
We conclude that: ergosterol cannot be used as biomassindicator for glomalean fungi; and regardless of whichbiochemical marker is used, it is very difficult to makecomparisons of biomass between different species becausethere are always taxonomic-based differences in content ofany signature compound. Ideally, a specific conversionfactor would have to be developed for each taxa when theaim is to compare the growth of different species.
Acknowledgements
We thank Christina Pehrsson and Custodia Cano for tech-nical assistance, and Erland Bååth for valuable suggestions.
Pål Axel Olsson1,*, Lennart Larsson2, Bert Bago3,Håkan Wallander1 and Ingrid M. van Aarle1
1Department of Microbial Ecology, Ecology Building,Lund University, SE-223 62 Lund, Sweden;
2Department of Medical Microbiology, Division ofBacteriology, Lund University, Sölvegatan 23, SE 223 62
Lund, Sweden; 3Centro de Investigaciones sobreDesertification (CSIC/UV/GV), Valencia, Spain
(*Author for correspondence tel +46 46 2229614;fax +46 46 2224158;
email [email protected])
References
Bago B, Zipfel W, Williams RC, Jun J, Arreola R, Pfeffer PE, Lammers PJ, Shachar-Hill Y. 2002. Translocation and utilization of fungal storage lipid in the arbuscular mycorrhizal symbiosis. Plant Physiology 128: 108–124.
Beilby JP. 1980. Fatty acid and sterol composition of ungerminated spores of the vesicular-arbuscular mycorrhizal fungus, Acaulospora laevis. Lipids 15: 949–952.
Beilby JP, Kidby DK. 1980. Sterol composition of ungerminated and germinated spores of the vesicular-arbuscular mycorrhizal fungus, Glomus caledonius. Lipids 15: 375–378.
Bindler GN, Piadé JJ, Schulthess D. 1988. Evaluation of selected steroids as chemical markers of past or presently ocurring fungal infections on tobacco. Beiträge Zur Tabakforschung International 14: 127–134.
Ek H, Sjögren M, Arnebrant K, Söderström B. 1994. Extramatricel mycelial growth, biomass allocation and nitrogen uptake in ectomycorrhizal systems in response to collembolan grazing. Applied Soil Ecology 1: 155–169.
Ekblad A, Wallander H, Carlsson R, Huss-Danell K. 1995. Fungal biomass in roots and extramatrical mycelium in relation to macronutrients and plant biomass of ectomycorrhizal Pinus sylvestris and Alnus incana. New Phytologist 131: 443–451.
Fontaine J, Grandmougin-Ferjani A, Hartmann M-A, Sancholle M. 2001. Sterol biosynthesis by the arbuscular mycorrhizal fungus Glomus intraradices. Lipids 36: 1357–1363.
Frey B, Buser HR, Schüepp H. 1992. Identification of ergosterol in vesicular-arbuscular mycorrhizae. Biology and Fertility of Soils 13: 229–234.
Frey B, Vilarino A, Schüepp H, Arines J. 1994. Chitin and ergosterol content of extraradical and intraradical mycelium of the vesicular-arbuscular mycorrhizal fungus Glomus intraradices. Soil Biology and Biochemistry 26: 711–717.
Frostegård Å, Bååth E. 1996. The use of phospholipid fatty acid analysis to estimate bacterial and fungal biomass in soil. Biology and Fertility of Soils 22: 59–65.
Fujiyoshi M, Nakatsubo T, Ogura S, Horikoshi T. 2000. Estimation of mycelial biomass of arbuscular mycorrhizal fungi associated with the annual legume Kummerowia striata by ergosterol analysis. Ecological Research 15: 121–131.
Graham JH, Hodge NC, Morton JB. 1995. Fatty acid methyl ester profiles for characterization of glomalean fungi and their endomycorrhizae. Applied and Environmental Microbiology 61: 58–64.
Grandmougin-Ferjani A, Dalpé Y, Hartmann M-A, Laurelle F, Sancholle M. 1999. Sterol distribution in arbuscular mycorrhizal fungi. Phytochemistry 50: 1027–1031.
Grant WD, West AW. 1986. Measurement of ergosterol, diaminopimelic acid and glucosamine in soil: evaluation as indicators of microbial biomass. Journal of Microbiological Methods 6: 47–53.
Hart MM, Reader RJ. 2002a. Taxonomic basis for variation in the colonization strategy of arbuscular mycorrhizal fungi. New Phytologist 153: 335–344.
Hart MM, Reader RJ. 2002b. Host plant benefit from association with arbuscular mycorrhizal fungi: variation due to differences in size of mycelium. Biology and Fertility of Soils 36: 357–366.
Larsson L, Saraf A. 1997. Use of gas chromatography-ion trap tandem mass spectrometry for the detection and characterization of microorganisms in complex samples. Molecular Biotechnology 7: 279–287.
Newell SY, Arsuffi TL, Fallon RD. 1988. Fundamental procedures for determining ergosterol content of decaying plant material by liquid chromatography. Applied and Environmental Microbiology 54: 1876–1879.
Nordby HE, Nemec S, Nagy S. 1981. Fatty acid and sterols associated with Citrus root mycorrhizae. Journal of Agricultural Food Chemistry 29: 396–401.
Nylund J-E, Wallander H. 1992. Ergosterol analysis as a means of quantifying mycorrhizal biomass. In: Norris JR, Read DJ, Varma AK, eds. Methods in microbiology, Vol. 24. London, UK: Academic Press, 77–88.
Olsson PA. 1999. Signature fatty acids provide tools for determination of distribution and interactions of mycorrhizal fungi in soil. FEMS Microbiology Ecology 29: 303–310.
Olsson PA, Bååth E, Jakobsen I, Söderström B. 1995. The use of phospholipid and neutral lipid fatty acids to estimate biomass of arbuscular mycorrhizal fungi in soil. Mycological Research 99: 623–629.
Olsson PA, Van Aarle IM, Allaway WG, Ashford AE, Rouhier H. 2002. Phosphorus effects on metabolic processes in monoxenic arbuscular mycorrhiza cultures. Plant Physiology 130: 1162–1171.

Letters
www.newphytologist.com © New Phytologist (2003) 159: 1–10
Forum10
Salmanowicz B, Nylund J-E. 1988. High performance liquid chromatography determination of ergosterol as a measure of ectomycorrhizae infection in Scots pine. European Journal of Forest Pathology 18: 291–298.
Schüssler A, Schwarzott D, Walker C. 2001. A new fungal phylum, the Glomeromycota: phylogeny and evolution. Mycological Research 105: 1413–1421.
Seitz LM, Mohr HF, Burroughs R, Sauer DB. 1972. Ergosterol as an indicator of fungal invasion in grains. Cereal Chemistry 54: 1207–1217.
Tunlid A, White DC. 1992. Biochemical analysis of biomass, community structure, nutritional status, and metabolic activity of microbial communities in soil. Soil biochemistry, Vol. 7. New York, NY, USA: Dekker, 229–262.
Wallander H, Massicotte HB, Nylund J-E. 1997. Seasonal variation in protein, ergosterol and chitin in five morphotypes pf Pinus sylvestris L. ectomycorrhizae in a mature swedish forest. Soil Biology and Biochemistry 29: 45–53.
Weete JD, Gandhi SR. 1996. Biochemistry and molecular biology of fungal sterols. In: Brambl R, Marzluf G, eds. The mycota III biochemistry and molecular biology. Berlin, Germany: Springer, 421–438.
Weete JD, Gandhi SR. 1999. Sterols and fatty acids of the Mortierellaceae: taxonomic implications. Mycologia 91: 642–649.
Key words: ergosterol, fatty acids, biomass estimation, mycorrhiza.