Embed Size (px)
Citation preview

www.wjpr.net Vol 7, Issue 16, 2018. 800
Peddi et al. World Journal of Pharmaceutical Research
GENERIC DRUG REGISTRATION AND REGULATORY
REQUIREMENTS IN EUROPEAN COUNTRIES
Meghana Peddi*, M. V. Nagabhushanam, D. Nagarjuna Reddy, Brahmaiah
Bonthagarala and G. Ramakrishna
Department of Pharmaceutical Management and Regulatory Affairs, Hindu College of
Pharmacy, Amaravathi Road, Guntur, Andhra Pradesh, India-522002.
ABSTRACT
The aim of this document is to examine methodological challenges by
comparing generic drug registration and regulatory requirements in
European countries. European countries are fast growing and emerging
markets in pharmaceutical sector. Increasing patent expirations and the
need for less expensive drugs is fuelling the growth of generics market
in the region. However, there are some challenges to be met in order to
sustain the growth. Hence the objective of present study is to analyse
overall aspects like the requirements for registration of generics in the
following 28 “EMA” (European medicines agency) countries, Drug
registration procedure for the approval of generic drug in these
countries, Mark out the differences in the requirements to register
generic drug product in these countries. The primary purpose of the
rules governing generic drugs in Europe is to safeguard to public
health. It is the role of public regulatory authorities to ensure that
pharmaceutical companies comply with regulations. There are legislations that require drugs
to be developed, tested, trailed, and manufactured in accordance to the guidelines so that they
are safe and patient’s well - being is protected. The regulatory submissions in the EU, in the
world continue to have significant differences. When compared to others the EU approval
process is typical and contain more data to be summarized for the dossier submission.
KEYWORDS: Generic drug registration, Regulatory requirements, European medicines
agency (EMA), European countries.
World Journal of Pharmaceutical Research SJIF Impact Factor 8.074
Volume 7, Issue 16, 800-815. Research Article ISSN 2277– 7105
Article Received on
22 June 2018,
Revised on 12 July 2018,
Accepted on 02 August 2018,
DOI: 10.20959/wjpr201816-13108
*Corresponding Author
Meghana Peddi
Department of
Pharmaceutical
Management and
Regulatory Affairs, Hindu
College of Pharmacy,
Amaravathi Road, Guntur,
Andhra Pradesh, India-
522002.

www.wjpr.net Vol 7, Issue 16, 2018. 801
Peddi et al. World Journal of Pharmaceutical Research
1. INTRODUCTION
GENERIC DRUGS
A generic drug (generic drugs, short: generics) is a drug defined as "a drug product that is
comparable to a brand/reference listed drug product in dosage form, strength, quality and
performance characteristics, and intended use.
Generic drugs are labeled with the name of the manufacturer and the adopted name
(nonproprietary name) of the drug.
A generic drug must contain the same active ingredients as the original formulation. The
generic drug have same rigid standards as the innovator drug.
Contains the same active ingredients as the innovator drug
Be identical in strength, dosage form, and route of administration
Have the same use indications
Be bioequivalence
Current Status of Generics in Europe
Europe, with its fast-growing, young population and uninsured majority represent great
opportunity for generics in the pharmaceutical industry.
Flag of Europe
Generic market shares vary from country to country, presenting differing oppurtunities for
generic manufacturers, see Figure 1, but in general are increasing across the regions.
Increasing use of generics in the region is expected due to healthcare reforms – making drugs
available to more of the population – and governmental cost-containment strategies. The
expanding middle class is also expected to drive demand for medicines, and especially
generics, in the region.
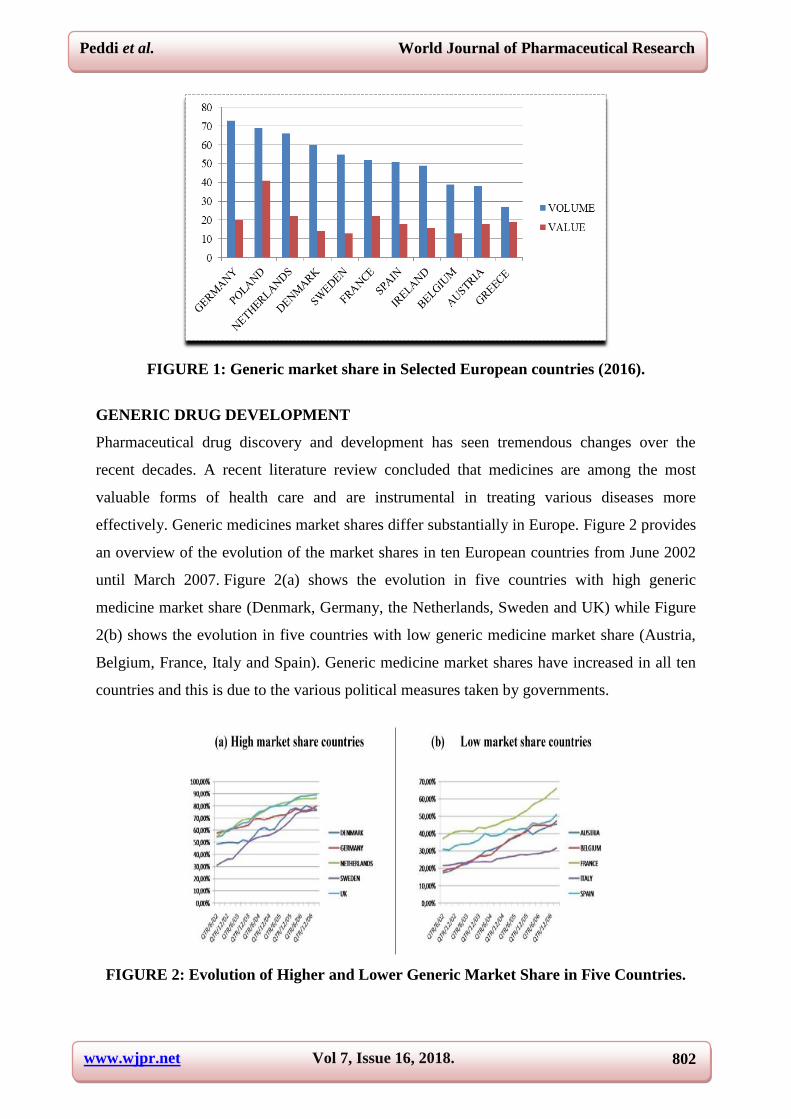
www.wjpr.net Vol 7, Issue 16, 2018. 802
Peddi et al. World Journal of Pharmaceutical Research
FIGURE 1: Generic market share in Selected European countries (2016).
GENERIC DRUG DEVELOPMENT
Pharmaceutical drug discovery and development has seen tremendous changes over the
recent decades. A recent literature review concluded that medicines are among the most
valuable forms of health care and are instrumental in treating various diseases more
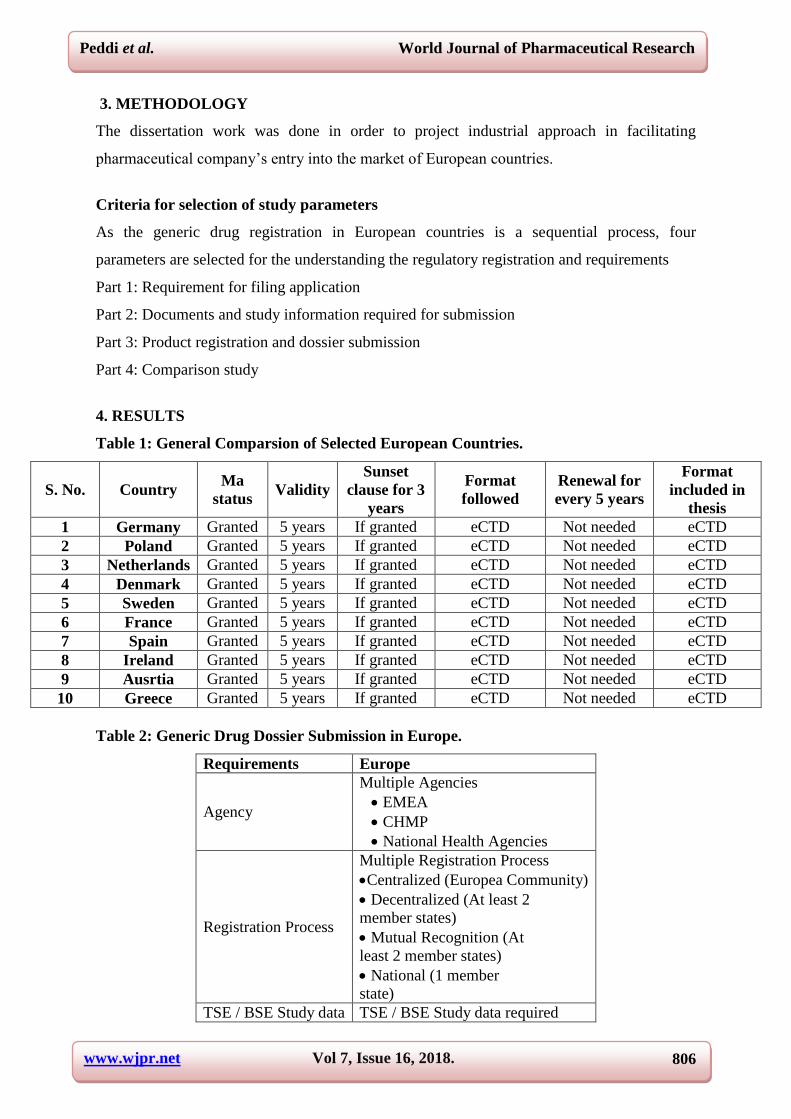
effectively. Generic medicines market shares differ substantially in Europe. Figure 2 provides
an overview of the evolution of the market shares in ten European countries from June 2002
until March 2007. Figure 2(a) shows the evolution in five countries with high generic
medicine market share (Denmark, Germany, the Netherlands, Sweden and UK) while Figure
2(b) shows the evolution in five countries with low generic medicine market share (Austria,
Belgium, France, Italy and Spain). Generic medicine market shares have increased in all ten
countries and this is due to the various political measures taken by governments.
FIGURE 2: Evolution of Higher and Lower Generic Market Share in Five Countries.

www.wjpr.net Vol 7, Issue 16, 2018. 803
Peddi et al. World Journal of Pharmaceutical Research
INTRODUCTION TO EMA
The European Medicines Evaluation Agency (here after referred to as EMA) is a
decentralised agency of the European Union (EU). The Management Board is the European
Medicines Agency's integral governance body. The Agency is responsible for the scientific
evaluation, supervision and safety monitoring of medicines developed by pharmaceutical
companies for use in the EU. EMA protects public and animal health in 28 EU Member
States, as well as the countries of the European Economic Area, by ensuring that all
medicines available on the EU market are safe, effective and of high quality. EMA serves a
market of over 500 million people living in the EU.
All parties are linked by an IT network EudraNet. (EUDRANET, the European
TelecommunicationNetworkinPharmaceuticals (European Union Drug Regulating Authoritie
s Network), is an IT platform to facilitate the exchange of information between regulatory
partners and industry during submission and evaluation of applications).
Organization chart of the European Medicines Agency
ELECTRONIC COMMON TECHNICAL DOCUMENT (eCTD)
The European countries established the electronic common technical document (eCTD) as
their format for submissions. It is a standard derived from ICH CTD. The eCTD is defined as
an interface for industry to agency transfer of regulatory information while at the same time

www.wjpr.net Vol 7, Issue 16, 2018. 804
Peddi et al. World Journal of Pharmaceutical Research
taking into consideration the facilitation of the creation, review, lifecycle management and
archival of the electronic submission.
STRUCTURE OF eCTD
Figure 3: Structure of eCTD.
eCTD application
An eCTD application may comprise a number of sequences. In the EU an eCTD application
may comprise several dosage forms and strengths, all under one invented product name.
Some review tools describe such a collection as a dossier.
EGA (European Generic Medicine Association)
The EGA was established in 1993. The EGA is the official representative body of the
European generic and biosimilar pharmaceutical industry, which is at the forefront of
Providing high-quality affordable medicines to millions of Europeans and stimulating
Competitiveness and innovation in the pharmaceutical sector.
Generic Drug Registration in Europe
Registration procedure of drugs in EU
National Procedure
Centralized Procedure
Decentralized Procedure
Mutual Recognition Procedure
Marketing Authorization based on approval received from above procedures
1. Repeat use application

www.wjpr.net Vol 7, Issue 16, 2018. 805
Peddi et al. World Journal of Pharmaceutical Research
2. Duplicate use application
Dossier requirements for generic drug application in Europe
The requirements for the dossier for the European countries are in principle very similar to
requirements for the ICH countries.
Dossier for generic drug filling shall be submitted in the form of CTD in Europe. Generic
Drugs are approved under MAA (Marketing Authorization Application).
Types of Applications
Generic Application – According to Article 10 (I) of Directive 2001/83/EC, the applicant is
not required to provide the results of Pre clinical tests and Clinical tests if he can demonstrate
that the medicinal product is a generic medicinal product of a reference medicinal product
which is or has been authorized for not less than 8 years in a Member State or a Community.
A medicinal product is defined as a generic medicinal product if it has –
The same qualitative and quantitative composition in active substance as that of reference
product.
The same pharmaceutical form as the reference product.
And whose Bio-equivalence with the reference medicinal product has been Demonstrated
with appropriate Bio-availability studies.
2. SCOPE OF THE STUDY
European countries are fast growing and emerging markets in pharmaceutical sector.
Increasing patent expirations and the need for less expensive drugs is fuelling the growth of
generics market in the region. However, there are some challenges to be met in order to
sustain the growth. Hence the objective of present study is to analyze overall aspects about:
The requirements for registration of generics in the following 28 “EMA” (European
medicines agency) countries.
Drug registration procedure for the approval of generic drug in these countries.
Mark out the differences in the requirements to register genric drug product in these
countries.
www.wjpr.net Vol 7, Issue 16, 2018. 806
Peddi et al. World Journal of Pharmaceutical Research
3. METHODOLOGY
The dissertation work was done in order to project industrial approach in facilitating
pharmaceutical company’s entry into the market of European countries.
Criteria for selection of study parameters
As the generic drug registration in European countries is a sequential process, four
parameters are selected for the understanding the regulatory registration and requirements
Part 1: Requirement for filing application
Part 2: Documents and study information required for submission
Part 3: Product registration and dossier submission
Part 4: Comparison study
4. RESULTS
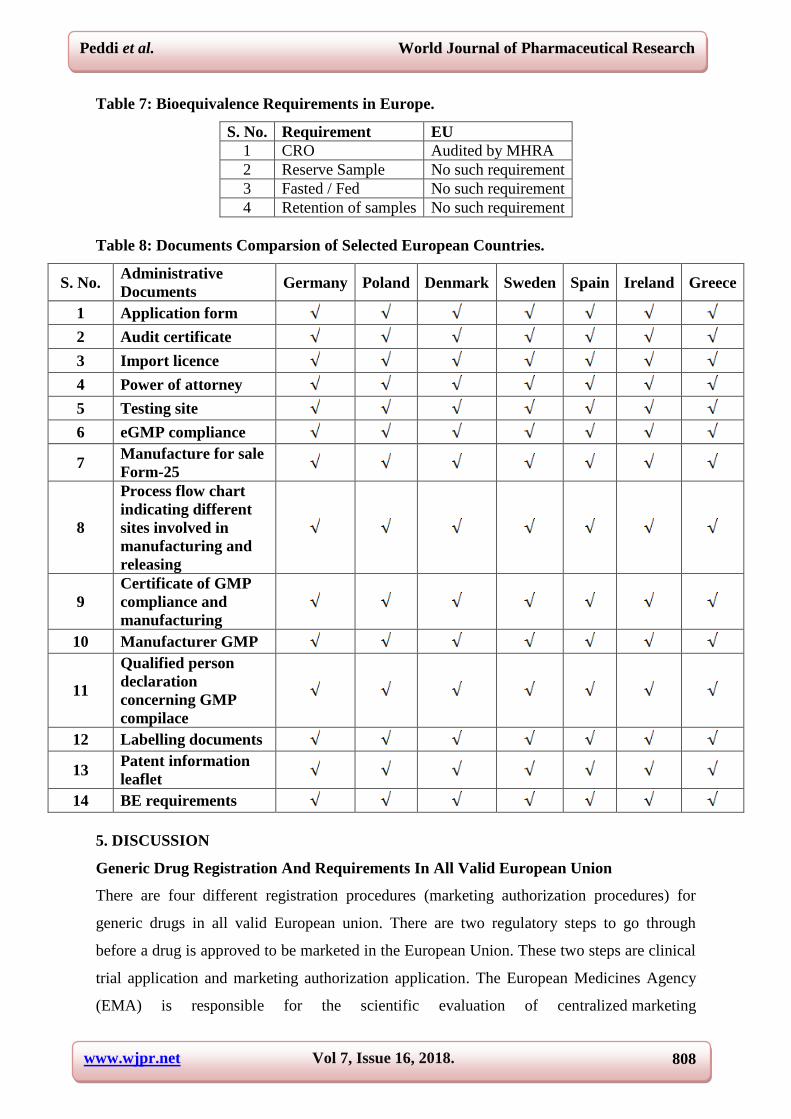
Table 1: General Comparsion of Selected European Countries.
S. No. Country Ma
status Validity
Sunset
clause for 3
years
Format
followed
Renewal for
every 5 years
Format
included in
thesis
1 Germany Granted 5 years If granted eCTD Not needed eCTD
2 Poland Granted 5 years If granted eCTD Not needed eCTD
3 Netherlands Granted 5 years If granted eCTD Not needed eCTD
4 Denmark Granted 5 years If granted eCTD Not needed eCTD
5 Sweden Granted 5 years If granted eCTD Not needed eCTD
6 France Granted 5 years If granted eCTD Not needed eCTD
7 Spain Granted 5 years If granted eCTD Not needed eCTD
8 Ireland Granted 5 years If granted eCTD Not needed eCTD
9 Ausrtia Granted 5 years If granted eCTD Not needed eCTD
10 Greece Granted 5 years If granted eCTD Not needed eCTD
Table 2: Generic Drug Dossier Submission in Europe.
Requirements Europe
Agency
Multiple Agencies
EMEA
CHMP
National Health Agencies
Registration Process
Multiple Registration Process
Centralized (Europea Community)
Decentralized (At least 2
member states)
Mutual Recognition (At
least 2 member states)
National (1 member
state)
TSE / BSE Study data TSE / BSE Study data required

www.wjpr.net Vol 7, Issue 16, 2018. 807
Peddi et al. World Journal of Pharmaceutical Research
Application MAA
Stability data
The stability data for
accelerated studies are
submitted for complete 6
months at the time of original
Submission.
Approval time 12 months
Pharmacopeias BP/Ph. Eur
Process
Validation required
Braille code Braille code is required on labelling
Post
approval changes
Post variation in the approved drug
Type 1A variation
Type 1B variation
Type II variation
Table 3: Administrative Requirements in Europe.
S. No. Requirement Europe
1 Application MAA
2 Debarment classification Not required
3 Number of copies 1
4 Approval Timeline 12 months
5 Fees 10-20 lakhs
6 Presentation eCTD
Table 4: Finished Product Control Requirements in Europe.
S. No. Requirement EU
1 Justification ICH Q6A
2 Assay 95-105%
3 Disintegration Required
4 Color Identification Required
5 Water Content Not required
Table 5: Manufacturing & Control Requirements in Europe.
S. No. Requirement EU
1 Number of batches 3
2 Packaging Required
3 Process Validation Required
4 Batch Size Minimum of 1,00,000 Units
Table 6: Stability Requirements in Europe.
S. No. Requirement EU
1 Number of batches 2
2 Condition 25/60 - 40/75
3 Date & Time of Submission 6 Months Accelerate & 6 Months long term
4 Container orientation Do not address
5 Clause Vol 4 EU Guidelines for medicinal products
6 QP Certification Required
www.wjpr.net Vol 7, Issue 16, 2018. 808
Peddi et al. World Journal of Pharmaceutical Research
Table 7: Bioequivalence Requirements in Europe.
S. No. Requirement EU
1 CRO Audited by MHRA
2 Reserve Sample No such requirement
3 Fasted / Fed No such requirement
4 Retention of samples No such requirement
Table 8: Documents Comparsion of Selected European Countries.
S. No. Administrative
Documents Germany Poland Denmark Sweden Spain Ireland Greece
1 Application form 2 Audit certificate 3 Import licence 4 Power of attorney 5 Testing site 6 eGMP compliance
7 Manufacture for sale
Form-25
8
Process flow chart
indicating different
sites involved in
manufacturing and
releasing
9
Certificate of GMP
compliance and
manufacturing
10 Manufacturer GMP
11
Qualified person
declaration
concerning GMP
compilace
12 Labelling documents
13 Patent information
leaflet
14 BE requirements
5. DISCUSSION
Generic Drug Registration And Requirements In All Valid European Union
There are four different registration procedures (marketing authorization procedures) for
generic drugs in all valid European union. There are two regulatory steps to go through
before a drug is approved to be marketed in the European Union. These two steps are clinical
trial application and marketing authorization application. The European Medicines Agency
(EMA) is responsible for the scientific evaluation of centralized marketing

www.wjpr.net Vol 7, Issue 16, 2018. 809
Peddi et al. World Journal of Pharmaceutical Research
authorization applications (MAA). Once granted by the European Commission, the
centralized marketing authorization is valid in all European Union (EU) Member States,
Iceland, Norway and Liechtenstein.
Procedures for application for a marketing authorization
Centralized procedure
National procedure
Mutual recognition procedure
Decentralized procedure
Centralized Procedure (CP)
Figure 4: Summary of Centralized Procedure.
National Procedure (NP)
Generally, this procedure is no longer used nowadays. If an applicant wishes to obtain a
license in one Member State (MS) an application must be made to the national Competent
Authority (CA) which then issues a national license. With the exception of products granted a
marketing authorization under the centralized procedure as set out above, all products are
granted marketing authorizations on a country-by-country basis by the competent authorities
in each Member State. Such marketing authorizations permit the holder to market the product
www.wjpr.net Vol 7, Issue 16, 2018. 810
Peddi et al. World Journal of Pharmaceutical Research
in question in the Member State concerned, subject to any restrictions or requirements that
accompany the authorization.
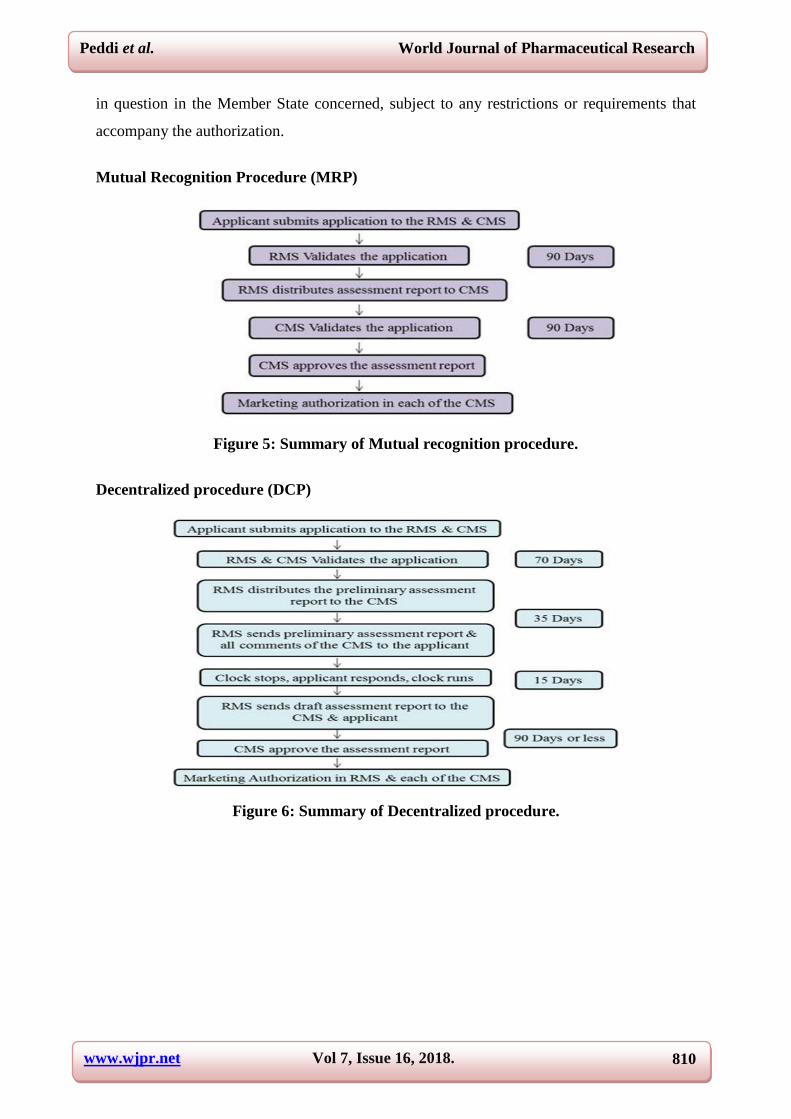
Mutual Recognition Procedure (MRP)
Figure 5: Summary of Mutual recognition procedure.
Decentralized procedure (DCP)
Figure 6: Summary of Decentralized procedure.
www.wjpr.net Vol 7, Issue 16, 2018. 811
Peddi et al. World Journal of Pharmaceutical Research
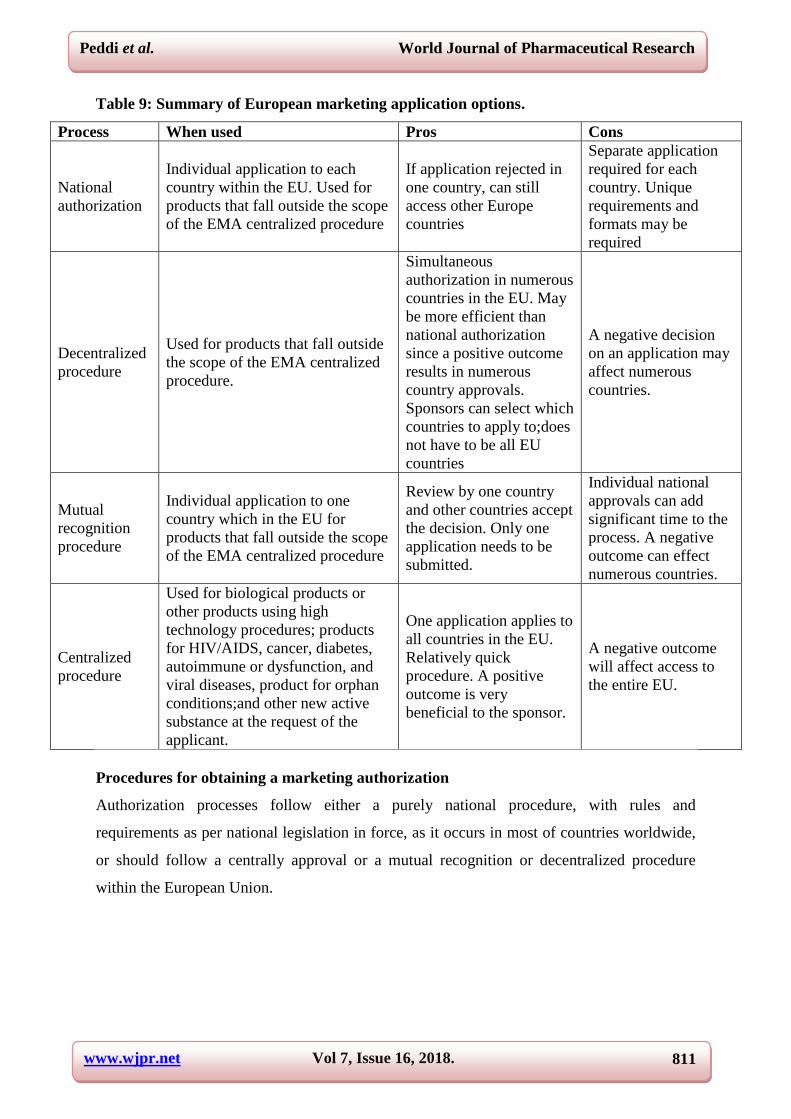
Table 9: Summary of European marketing application options.
Process When used Pros Cons
National
authorization
Individual application to each
country within the EU. Used for
products that fall outside the scope
of the EMA centralized procedure
If application rejected in
one country, can still
access other Europe
countries
Separate application
required for each
country. Unique
requirements and
formats may be
required
Decentralized
procedure
Used for products that fall outside
the scope of the EMA centralized
procedure.
Simultaneous
authorization in numerous
countries in the EU. May
be more efficient than
national authorization
since a positive outcome
results in numerous
country approvals.
Sponsors can select which
countries to apply to;does
not have to be all EU
countries
A negative decision
on an application may
affect numerous
countries.
Mutual
recognition
procedure
Individual application to one
country which in the EU for
products that fall outside the scope
of the EMA centralized procedure
Review by one country
and other countries accept
the decision. Only one
application needs to be
submitted.
Individual national
approvals can add
significant time to the
process. A negative
outcome can effect
numerous countries.
Centralized
procedure
Used for biological products or
other products using high
technology procedures; products
for HIV/AIDS, cancer, diabetes,
autoimmune or dysfunction, and
viral diseases, product for orphan
conditions;and other new active
substance at the request of the
applicant.
One application applies to
all countries in the EU.
Relatively quick
procedure. A positive
outcome is very
beneficial to the sponsor.
A negative outcome
will affect access to
the entire EU.
Procedures for obtaining a marketing authorization
Authorization processes follow either a purely national procedure, with rules and
requirements as per national legislation in force, as it occurs in most of countries worldwide,
or should follow a centrally approval or a mutual recognition or decentralized procedure
within the European Union.

www.wjpr.net Vol 7, Issue 16, 2018. 812
Peddi et al. World Journal of Pharmaceutical Research
Steps involved in obtaining an EU marketing authorization
Submission of eligibility request
Notification of intention to submit an application
Appointment of rapporteurs
Pre-submission meeting
Reconfirmation of communicated submission date
Submission and validation of application
Scientific evaluation
CHMP scientific opinion
European commission decision
Dossier to be submitted
The EMEA requires from the applicant: One full copy of the dossier (modules 1-5 according
to the EU-CTD format), including the applicant’s part of the Active Substance Master File, if
any; Two additional copies of Modules 1 and 2 including the draft summary of product
characteristics, labelling and package leaflet in English; One electronic copy of module 1
and 2 (at least 2.1-2.5) in WORD. In addition, applicants must submit the dossier to both the
Rapporteur and the Co-Rapporteur in parallel to the EMEA. Otherwise there may be a delay
in the start of the procedure because of the time lapse between the validation by the EMEA
and the confirmation from the Rapporteur and the Co-Rapporteur that they have received the
dossiers.
Validation by the EMA
During validation the EMA Product Team Lead (PTL) consults the Rapporteur and Co-
Rapporteur, on the need for action relating to matters such as GMP inspection, ad-hoc expert
groups, Scientific Advisory Groups, GCP inspections, and completeness of data.

www.wjpr.net Vol 7, Issue 16, 2018. 813
Peddi et al. World Journal of Pharmaceutical Research
Positive outcome of the validation
Negative outcome of the validation
Types of applications
The type of application may vary according to status of the active ingredient.
Thus, if the application concerns a new active ingredient (new active substance, new
chemical entity, new molecular entity), one talks about a full application.
Generic Application – According to Article 10 (I) of Directive 2001/83/EC, the applicant is
not required to provide the results of Pre clinical tests and Clinical tests if he can demonstrate
that the medicinal product is a generic medicinal product of a reference medicinal product
which is or has been authorized for not less than 8 years in a Member State or a Community.
A medicinal product is defined as a generic medicinal product if it has:
The same qualitative and quantitative composition in active substance as that of reference
product.
The same pharmaceutical form as the reference product.
And whose Bio-equivalence with the reference medicinal product has been demonstrated
with appropriate Bio-availability studies.
6. CONCLUSION
The primary purpose of the rules governing generic drugs in Europe is to safeguard to public
health. It is the role of public regulatory authorities to ensure that pharmaceutical companies
comply with regulations. There are legislations that require drugs to be developed, tested,
trailed, and manufactured in accordance to the guidelines so that they are safe and patient’s
well - being is protected. The regulatory submissions in the EU, in the world continue to have
significant differences. When compared to others the EU approval process is typical and
contain more data to be summarized for the dossier submission. All OECD countries see the
development of generic markets as a good opportunity to increase efficiency in
pharmaceutical spending but many do not fully exploit the potential of generics. Some of the
differences in generic uptake can be explained by market structures, notably the number of
off-patent medicines, and by prescribing practices, but generic uptake also very much
depends on policies implemented by countries. It also compares the registration procedure,
regulatory pathways for the registration of drugs in EU. The EU requires to select from any
one of the marketing authorization procedure.

www.wjpr.net Vol 7, Issue 16, 2018. 814
Peddi et al. World Journal of Pharmaceutical Research
To succeed with multinational registrations, a sponsor must:
Identify key target markets for submissions.
Understand important regional differences.
Find the right local resources to avoid regulatory pitfalls.
Create a robust CTD (Common Technical Document).
7. REFERENCE
1. European Commission. Guidance on elements required to support the significant clinical
benefit in comparison with existing therapies of a new therapeutic indication in order to
benefit from an extended (11 years) marketing protection period, 2007. Available
from: http://ec.europa.eu/health/files/eudralex/vol-2/c/guideline_14-11-2007_en.pdf
2. European Commission Competition, EU Sector inquiry, 8 July 2009. Available
from: http://ec.europa.eu/competition/sectors/pharmaceuticals/inquiry/communication_en
3. Tschabitscher D, Platzer P, Baumgärtel C, Müllner M. Generic drugs: quality, efficacy,
safety and interchangeability. Wien Klin Wochenschr, 2008; 120: 63-9.
4. American Medical Association. Summaries and recommendations of Council on
Scientific Affairs Reports. Generic drugs (CSA Rep 6, A-02). 2002 Annual Meeting of
the American Medical Association, 2002; 13-4.
5. Henney JE. Review of generic bioequivalence studies from the food and drug
administration. JAMA, 1999; 282: 1995.
6. G. Sai Hanuja, B. Sai Kumari, M.V. Nagabhushanam, D. Nagarjuna Reddy, Brahmaiah
Bonthagarala, Regulatory Requirements for Registration of Generic Drugs in “BRICS”
Countries, International Journal of Pharmaceutical Science and Health Care, ISSN 2249 –
5738, November-December 2016; 6(6): 20-40.
7. L Evangeline, NVN Mounica, V Sharmila Reddy, MV Ngabhushanam, D Nagarjuna
Reddy and Brahmaiah Bonthagarala, Regulatory process and ethics for clinical trials in
India (CDSCO)- A Review, The Pharma Innovation Journal, ISSN (E): 2277- 7695, ISSN
(P): 2349-8242, 2017; 6(4): 165-169.
8. B. Sai Kumari, G. Sai Hanuja, M.V. Nagabhushanam, D. Nagarjuna Reddy, Brahmaiah
Bonthagarala, Current Regulatory Requirements for Registration of Medicines,
Compilation and Submission of Dossier in Australian Therapeutic goods Administration,
International Journal of Advanced Scientific and Technical Research, ISSN 2249-9954,
November-December 2016; 6(6): 144-157.

www.wjpr.net Vol 7, Issue 16, 2018. 815
Peddi et al. World Journal of Pharmaceutical Research
9. Shaik Salman Basha, S. M. Shakeel, M. V. Nagabhushanam, D. Nagarjuna Reddy,
Brahmaiah Bonthagarala, The Assesment of Current Regulatory Guidelines for
Biosimilars- A Global Scenario, World Journal of Pharmaceutical Research, ISSN 2277–
7105; 6(1): 351-369.
10. S.M. Shakeel, Shaik Salman Basha, M.V. Nagabhushanam, D. Nagarjuna Reddy,
Brahmaiah Bonthagarala, Comparision of Regulataory Requirements for Generic Drugs
Dossier Submission in United States and Canada, International Journal of Pharmaceutical
Science and Health Care, ISSN 2249 – 5738, November-December 2016; 6(6): 1-19.
11. Srinivasan R. Indian pharmaceutical industry: Evaluation of current scenario and future
trends. [Last accessed on 2012 Jun 10]. Available from: http://www.tejas-
iimb.org/interviews/13.php.





























