Embed Size (px)
Citation preview

ACTAUNIVERSITATIS
UPSALIENSISUPPSALA
2008
Digital Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Medicine 395
Genetic Risk Factors for SystemicLupus Erythematosus
From Candidate Genes to Functional Variants
ANNA-KARIN ABELSON
ISSN 1651-6206ISBN 978-91-554-7336-5urn:nbn:se:uu:diva-9367

���������� �������� �� ������ �������� � �� �������� ������� � ������������������������������ ��� ������������� ��� ��� ������� �������� �������� !� ���" ��#$%#& '� �(� ������ ' ���� ' )(����(� *+������ ' ,������-. /(� �������� 0��� ��������� � 1����(.
��������
2����� 234. ���". 5����� ���� +����� '� ������� 6���� 1���(�������. +��7������� 5��� � +������ 8������. 2��� ����������� ���������. ���������� � ���� ����� � � ������� ���� ������� �� �� ������� � � ����� $9&. !#��. ������. : ;< 9="39#3&&>3=$$!3&.
/(� ��� ' �(�� �(���� (�� ��� � �����'� ������ ������� �(�� ������� �(� �������������� '� ������� 6���� 1���(������� * 61-� � �������� ������� ������ �� � ������ �����������0�� ������ ������ �� ��������� '�����.+��� ��''���� �������� ���� 0��� �������� �(���( ��''���� ����������� �� 0��� �������
'� �������� 0��( 61 � � ������� � ���������( ��� ' �(� �������� ���(����� '�(�� �������. /0 ' �(��� ����� ����� �� ������ �������� � � ���� �� ���� ����'����� '� 61 � �(� ������� 1����� �� 6��� 2������ ��������. : �0 �(�� �������� �� �!"! � �(��� �������� � �� �������3�����'�� �''����. /(� "#$% ��� �������������� � �(� ��� ���� ��������� ���������� 0��( �������� ��������� 0�� ���������� � ����( �(��� 0��( � 0��� ���� � 5���� �������� �� �������� � 0����(./(� ��������� ������� �� (��(�� ������ �������� ' �(� �!"! ���������� '������� 0�� �'����� � ��� �� �(���. :����������� �(� ������� ������� �(� ������� ' ������� �0 �������� ���� �������% �(� '����� ���������� �� � ��������� ������� <)� 0���(� ������� � ���������� ' <��(�� 1����� �������� �� �(� ���� 0�� ��������� � ���������� '�� ��(�� 1���� �� 6��� 2������. ?� ��� ����� �(������'����� ' � ��� �������������� ���. /(� &"'(� ���� ����� � ���''�� ����������� � ;3���� ��������� ����� '������ ������� �''����� ������� ������ 0(��(��� ��������� � ��� ����������� �(��� '�� 1���� �� 6��� 2������./(��� ������� �'��� �(� �������� ' ���������� ��������� ���0�� ������ ������� ��
61� 0(��( ��� ��� �� ������ � ��� ��������. /(� ������� ��� ���������� � ������������ ' (����������� 0(��� ��� ���� '����� ���� (��� �������� �''��� � ��''������������.
( �)���* ������� 6���� 1���(�������� 61� �������� ������ ������ �������� ������
"����(���� "+ ���, � ���� �� � - � ���� ��� �������, .��+ �/��+����� �, ����������� �����, �0�$#�1# �������, �) � �
@ 2�34��� 2���� ���"
: < #!!��!: ;< 9="39#3&&>3=$$!3&��%�%��%��%����39$!= *(���%AA��.��.��A������B��C��%�%��%��%����39$!=-

Till min familj


List of papers
This thesis is based on the following papers, which are referred to in the text by their roman numerals.
I Abelson AK, Johansson CM, Kozyrev SV, Kristjansdottir H, Gunnars-
son I, Svenungsson E, Jönsen A, Lima G, Scherbarth HR, Gamron S, Allievi A, Palatnik SA, Alvarellos A, Paira S, Graf C, Guillerón C, Ca-toggio LJ, Prigione C, Battagliotti CG, Berbotto GA, García MA, Per-andones CE, Truedsson L, Steinsson K, Sturfelt G, Pons-Estel B; Ar-gentinean Collaborative Group, Alarcón-Riquelme ME. No evidence of association between genetic variants of the PDCD1 ligands and SLE Genes and Immunity 2007 Jan;8(1):69-74.
II Sánchez E, Abelson AK, Sabio JM, González-Gay MA, Ortego-
Centeno N, Jiménez-Alonso J, de Ramón E, Sánchez-Román J, López-Nevot MA, Gunnarsson I, Svenungsson E, Sturfelt G, Truedsson L, Jönsen A, González-Escribano MF, Witte T; The German SLE Study Group, Alarcón-Riquelme ME, Martín J. Association of a CD24 gene polymorphism with susceptibility to sys-temic lupus erythematosus. Arthritis and Rheumatism 2007 Sep;56(9):3080-6.
III Kozyrev SV*, Abelson AK*, Wojcik J, Zaghlool A, Linga Reddy MV,
Sanchez E, Gunnarsson I, Svenungsson E, Sturfelt G, Jönsen A, Truedsson L, Pons-Estel BA, Witte T, D'Alfonso S, Barizzone N, Danieli MG, Gutierrez C, Suarez A, Junker P, Laustrup H, González-Escribano MF, Martín J, Abderrahim H, Alarcón-Riquelme ME. Functional variants in the B-cell gene BANK1 are associated with systemic lupus erythematosus. Nature Genetics 2008 Feb;40(2):211-6.

IV Abelson AK*, Delgado-Vega AM*, Kozyrev SV*, Sánchez E*, Velázquez-Cruz R, Eriksson N, Wojcik J, Linga Reddy MV, Lima G, D’Alfonso S, Migliaresi S, Baca V, Orozco L, Witte T, Ortego-Centeno
N and the AADEA group, Abderrahim H, Pons-Estel BA, Gutiérrez C, Suárez A, González-Escribano MF, Martín J and Alarcón-Riquelme ME. STAT4 associates with SLE through two independent effects that correlate with gene expression and act additively with IRF5 to in-crease risk Annals of the Rheumatic Diseases, in press.
* These authors contributed equally to the work The articles were reprinted with permission of the publisher: I, III Nature Publishing group II Copyright� 2007 Wiley-Liss, Inc., a subsidiary of John Wiley
& Sons Inc.

Contents
Introduction...................................................................................................11 Genetic variation ......................................................................................11 Medical genetics.......................................................................................13
Mendelian disorders.............................................................................13 Complex disorders ...............................................................................15
Identification of susceptibility genes........................................................16 Linkage analysis ..................................................................................16 Association studies ..............................................................................17 Genome-wide association studies........................................................19 Candidate-gene approach.....................................................................20 Animal models.....................................................................................21 Complicating factors............................................................................21 Functional mutations ...........................................................................22
Systemic Lupus Erythematosus................................................................22 Pathology .............................................................................................22 Aetiology and pathogenesis .................................................................24 Genetic studies of SLE ........................................................................26
Present investigation .....................................................................................31 Aim...........................................................................................................31 Material and methods ...............................................................................31
Patients and controls ............................................................................31 Genotyping ..........................................................................................32 Statistical analysis................................................................................33
Paper I: No evidence of association between genetic variants of the PDCD1 ligands and SLE..........................................................................34
Background..........................................................................................34 Results and discussion .........................................................................34
Paper II: Association of a CD24 gene polymorphism with susceptibility to systemic lupus erythematosus...................................................................36
Background..........................................................................................36 Results and discussion .........................................................................36
Paper III: Functional variants in the B-cell gene BANK1 are associated with systemic lupus erythematosus...........................................................38
Background..........................................................................................38

Results and discussion .........................................................................39 Paper IV: STAT4 associates with SLE through two independent effects that correlate with gene expression and act additively with IRF5 to increase risk .............................................................................................41
Background..........................................................................................41 Results and discussion .........................................................................42
Concluding remarks ......................................................................................46
Acknowledgements.......................................................................................48
References.....................................................................................................51

Abbreviations
ACR American College of Rheumatology BANK1 B-cell scaffold protein with Ankyrin repeats 1 bp Base pairs BLK B-Lymphocyte tyrosine Kinase CNP Copy Number Polymorphism CNV Copy Number Variation DNA Deoxyribonucleic Acid EBV Epstein-Barr Virus FBAT Family-Based Association Test FCGR (FcγR) Fc Gamma (γ) Receptor GWAS Genome-Wide Association Study HLA Human Leukocyte Antigen HHRR Haplotype-based Haplotype Relative Risk HWE Hardy-Weinberg Equilibrium IL-12 Interleukin-12 IFN Interferon IRF5 Interferon Regulatory Factor 5 ITGAM Integrin Alpha M IP3R Inositol 1,4,5-triphosphate Receptor kb Kilobasepairs LD Linkage Disequilibrium LOD Logarithm of Odds MS Multiple Sclerosis NK cells Natural Killer cells PCR Polymerase Chain Reaction PTPN22 Protein Tyrosine Phosphatase Non-receptor 22 RFLP Restriction Fragment Length Polymorphism SLE Systemic Lupus Erythematosus SNP Single Nucleotide Polymorphism STAT4 Signal Transducer and Activator of Transcription 4 TDT Transmission Disequilibrium Test TNF Tumour Necrosis Factor


11
Introduction
Since prehistoric times it has been known that offspring resemble their par-ents and that some traits are inherited. This knowledge was used for refining plants and animals by breeding long before the underlying mechanisms were known. In the mid 19th century, important steps towards the understanding of these genetic mechanisms were taken by the Augustinian monk Gregor Mendel. His now famous experiments with pea plant crossings revealed that the inheritance of certain traits, such as petal colour, is a discrete process following distinct laws1,2. These laws are now known as Mendel’s laws of inheritance. A series of discoveries during the early 20th century then lead to the identification of DNA as the carrier of genetic information, and in 1953, the structure of DNA as a double helix was determined by James Watson and Francis Crick3. Since then, the increase of genetic knowledge has been explosive. A recent milestone in genetic history was the publication of the first draft of the entire human sequence in the year 20014,5. It is now known that the human genome consists of more than 3.25 billion base pairs (bp) organised into 23 chromosome pairs, comprising approximately 20,000-25,000 protein-coding genes. Furthermore, large international efforts have contributed to the field by mapping a great proportion of the genetic varia-tion that exists6. The knowledge of the human genetic sequence, in combina-tion with the continuous development of new, increasingly efficient and cost-effective tools for sequencing and genotyping, has provided valuable tools for geneticists of today. With the help of these tools, we are continuing to discover genetic causes to diseases and to unravel the mysteries of human biology.
The aim of this thesis has been to identify genetic variants that increase the susceptibility for Systemic Lupus Erythematosus (SLE), an autoimmune disease caused by a complex interplay between various genetic and envi-ronmental factors. Five different candidate genes are selected through differ-ent strategies, and are analysed for association with SLE in an attempt to distinguish some of the underlying mechanisms of this disease.
Genetic variation The genetic composition is roughly 99.9% identical between humans, but that still leaves millions of base pairs that differ7. These genetic variations

12
vary in type and size, ranging from differences in single nucleotides to du-plications of large segments. Most sequence differences have no effect, but some contribute to variation in appearance, risk of disease and response to the environment.
Single Nucleotide Polymorphisms The most prevalent type of genetic variation is the Single Nucleotide Poly-morphism (SNP). Almost 15 million SNPs are currently registered in NCBI’s SNP database, which gives an average of one SNP every 220 bp in the genome (www.ncbi.nlm.nih.gov/SNP; build 128). As understood by the name, SNPs are differences in single nucleotides, and usually a minor allele frequency of at least 1% is used as a definition. Those with lower frequen-cies are generally regarded as ‘rare mutations’.
SNPs may have functional importance, e.g. by altering amino acid se-quence of a protein. They are relatively cheap and easy to genotype, and have therefore been extensively studied in medical genetics.
Indels Insertion/deletions, or indels, comprising one or a few bp are the second most common type of variation. More than 2 million are listed in the NCBI database (www.ncbi.nlm.nih.gov/SNP; build 128). However, since relatively few efforts have been made to identify new indels, it is believed that many more are yet to be found. It has been estimated that indels represent around 16-25% of all human variation8. Similar to SNPs, indels can affect the phe-notype by altering important genetic sequences.
Repetitive sequences A substantial part of the genome consists of repetitive sequences of varying lengths, with varying numbers of repeat units. There are several classes of repetitive sequences, which can be tandemly repeated or interspersed. Mi-crosatellites, and perhaps also minisatellites, are the types most extensively studied in human genetics.
Microsatellites, also known as Short Tandem Repeats, are tandemly re-peated sequences of one to five, sometimes six, bp per repeat unit. Their high degree of polymorphism, in combination with high abundance dis-persed across the whole genome, has made microsatellites useful markers in forensics as well as medical genetics.
Minisatellites are slightly longer tandem repeats of about 10-100 bp, which are found dispersed in the genome and clustered at the telomeres. Their qualities resemble those of microsatellites, and they are therefore used in similar types of analyses, although to a lesser degree. Tandem repeats of longer sequences are called satellite DNA or megasatellites, and can be sev-eral kb long.

13
Copy-Number Variations Copy-Number Variations (CNVs) are usually defined as DNA segments longer than 1 kb that are present at variable copy numbers in comparison with a reference genome9,10. The more common CNVs, with frequencies >1% are also referred to as Copy-Number Polymorphisms (CNPs). CNVs include insertions, deletions, duplications and complex multi-site variants, and are widely distributed throughout the human genome. CNVs have re-ceived relatively little attention compared with SNPs and smaller inser-tions/deletions. However, several studies have recently been published, re-vealing a high proportion of copy-number variable regions in the human genome10-12. Notably, the CNV regions have been shown to have higher nucleotide content per genome than SNPs10. The importance of CNVs is further emphasised by their colocalisation with known genes and other func-tional elements. Currently (October 2008), 7332 genes are overlapped by CNVs according to the Database of Genomic Variants11,13. These variants may disrupt genes or alter gene dosage and thereby influence gene expres-sion and phenotype. Several diseases have recently been associated with variations in copy-number of genes14-18.
Medical genetics Medical genetics is the science of inherited diseases. It is a genetic subdisci-pline that involves analysis of the connection between inherited variations and human disorders.
Mendelian disorders
Gregor Mendel, mentioned earlier in the introduction, studied pea plants in the 19th century and discovered that some traits follow distinct rules of in-heritance. For example, he discovered that each individual has two ‘factors’ for each trait, one from each parent, which may or may not contain the same information. The ‘factor’ variants are called alleles, and an individual with two identical alleles is called homozygous, whereas one carrying two differ-ent alleles is called heterozygous. Mendel also observed that for many traits, there is one dominant and one recessive allele. For example, pink petal col-our is dominant over white, so that a pea plant that is heterozygous for that trait will express the pink colour. Similarly, there are human diseases that follow Mendel’s laws of inheritance.
In dominant disorders (Figure 1a), the disease allele is dominant over the healthy allele, so that heterozygous individuals will express the disease. If the causative mutation is located on an autosomal (i.e. non-sex determin-ing) chromosome, the disease is termed autosomal dominant. Achondropla-

14
sia (a common form of dwarfism) and Huntington’s disease are examples of autosomal dominant disorders.
In recessive disorders (Figure 1b), the healthy allele is dominant and the disease allele is recessive. Individuals that are homozygous for the healthy allele as well as those who are heterozygous will then be healthy, and only those who are homozygous for the disease allele will express the disease. Two examples of autosomal recessive disorders are cystic fibrosis and albi-nism.
If a disease allele is located on the X-chromosome, it is termed X-linked. Recessive X-linked diseases (Figure 1c), such as haemophilia or colour blindness, mainly affect men. Since men only have one X-chromosome, they will always express the disease if they have the disease allele. Women, on the other hand, have two X-chromosomes and thus require two copies of the disease allele to develop the disease.
These diseases that follow Mendel’s laws of inheritance are often referred to as Mendelian diseases. Most of them are due to the mutation of a single gene, resulting in disruption or an altered functionality of the protein. A cer-tain degree of allelic heterogeneity is usually present, where several different mutations within the same gene may cause the disease. There can also be locus heterogeneity, where mutations of different genes give rise to the same disease. However, the common feature of these genetic variants is their high penetrance. The term genetic penetrance denotes the degree to which a ge-netic variant is displayed in the phenotype. For example, a disease polymor-phism with 95% penetrance will lead to disease in 95% of the cases.
Mitochondrial disorders Genetic diseases can also be caused by mutations in the mitochondrial
genome. Mutations within this genome can give rise to various disorders, such as Leber Hereditary Optic Neuropathy (LHON) and Maternally Inher-ited Diabetes and Deafness (MIDD)19. Since the mitochondria are maternally inherited in humans, the diseases are transmitted from mother to child.

15
Figure 1. Genetic disorders. a) Autosomal dominant disorder: individual with healthy alleles (H) on both chromosomes is healthy, individual with both H and disease (D) alleles is affected, individual with two D alleles is also affected. b) Autosomal recessive disorder: individuals with two H alleles or with both H and D alleles are healthy, individual with two D alleles is affected. c) X-linked recessive disorder: male with H allele is healthy, male with D allele is affected, females are affected as by an autosomal recessive disorder. d) Complex disease: a combination of various genetic risk factors ( R) on different chromosome pairs and environmental factor causes the disease.
Complex disorders
A complex disorder can be defined as a disease with an unclear aetiology, or one that displays familial aggregation, but the pattern of inheritance is not consistent with Mendelian disease models. Instead, the disease is caused by a combination of several genetic and environmental risk factors (Figure 1d), each only making a small contribution to the overall heritability. The con-ferred increase in risk for a genetic variant is often only two-fold or less. In other words, the penetrance is very low and may be affected by interactions with other genetic factors or require a certain environment to be expressed. Diabetes mellitus, bipolar disorder, schizophrenia and many types of cancer belong to this category of diseases, as well as SLE.

16
The proportion of genetic versus environmental causes varies between dif-ferent complex disorders. The genetic component of a disease can be meas-ured by the level of familial aggregation, denoted λ (lambda). This value specifies the difference in disease prevalence for relatives of an affected individual compared with the general population. The higher the λ value, the higher the degree of familial aggregation. To some extent, the λ value also reflects the degree of genetic causes to the disease. However, in addition to sharing genetic background, family members usually also share many envi-ronmental factors, which may also affect the λ value. An alternative measure of the genetic component of a disease is the comparison of concordance rates in monozygotic and dizygotic twins. Twin pairs usually share the same envi-ronment, but whereas monozygotic twins are genetically identical, dizygotic twins only share around half of their genetic composition. Therefore, if monozygotic twins have a significantly higher concordance (i.e. both twins are affected) than dizygotic, it is an indication that the disease has a strong genetic component.
Identification of susceptibility genes There are many different approaches to identifying genetic predisposing factors behind a disease. The methods can be broadly divided into two main categories: linkage studies and association studies. The basic difference be-tween the two methods is that linkage analysis studies how the inheritance of a disease follows certain chromosomal regions in a family, whereas associa-tion analysis compares allele frequencies of affected individuals with a set of controls. A brief overview of different strategies for identification of risk genes is given here below.
Linkage analysis
Adjacent genetic features tend to be inherited together more frequently than those located further apart on the chromosome. The distance between two genetic loci correlates with the probability of them being separated by mei-otic recombination. This coinheritance of closely located loci is the basis for linkage analysis. Genetic linkage is measured by the fraction of recombina-tion between loci, where unlinked genes show 50% recombination.
A genome-wide scan for linkage can be performed, studying how chro-mosomal segments co-segregate with a disease in one or several families. In such studies, a large amount of genetic markers, usually microsatellites, spread across the whole genome are genotyped in the families. Linkage is then calculated for each marker based on the degree of concurrence between alleles and disease within each family.

17
Linkage can be measured by a LOD score, which is the logarithm of the odds that two loci are linked with a recombination fraction less than 0.5, compared with the likelihood of independent assortment. There are different opinions on the threshold for significant linkage, but LOD scores above 3.3 are often considered significant in a genome-wide analysis20. This is equiva-lent to a p value of 5×10-5, which corresponds to a 5% probability of associa-tion by chance in a genome-wide scan.
Linkage analysis is a method with high power for finding rare variants with high penetrance21. It has therefore been very successful for mapping Mendelian diseases, which, by definition, are caused by such genetic vari-ants. Several factors for complex diseases have also been found through linkage analysis, although it is in general a less powerful method for detect-ing common variants with low penetrance21,22.
Association studies
Genetic association describes the co-occurrence of a phenotypic trait, e.g. a disease, with a genetic trait, often an allele of a certain SNP. In an associa-tion study, one may for example investigate if the frequency of an allele is higher in a set of patients compared with healthy individuals. When associa-tion between an allele and a disease is found, the association may reflect a functional effect of the associated allele. However, the associated allele often has no function, but is in linkage disequilibrium (described below) with the causative mutation.
There are two main categories of association studies: case/control and family-based studies. Case/control studies simply compare the frequencies of the allele of interest in a group of patients (cases) and a group of healthy individuals (controls). Family-based studies usually analyse which alleles are inherited from healthy parents to an affected child. The chromosomes that are not inherited are then used as the ‘healthy’ control set, and are compared with the allele frequencies in the affected children.
Linkage disequilibrium There is often a certain degree of correlation between alleles of different polymorphisms located within the same chromosomal region. This correla-tion is referred to as Linkage Disequilibrium (LD). For example, two adja-cent SNPs may have the alleles C/T and A/G, respectively. If they are com-pletely independent of one another with random segregation of the alleles, the frequency (f) of the TA haplotype will be: f (TA) = f (T) × f (A). Any deviation from this formula indicates LD between the SNPs (Figure 2). The extreme case would be if only two haplotypes are observed, for example TA and CG. This means that the two SNPs are perfect proxies of each other, and are said to be in complete LD.
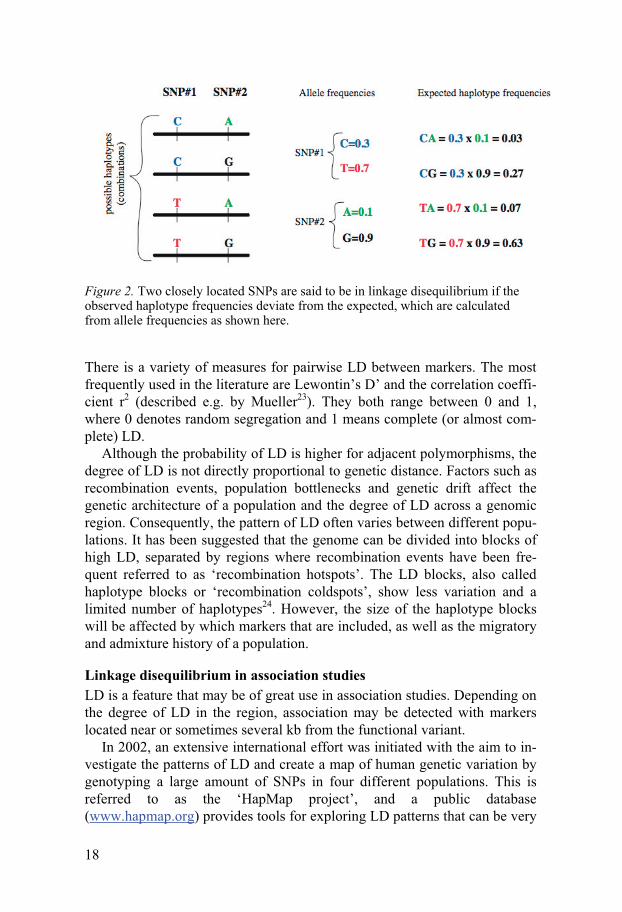
18
Figure 2. Two closely located SNPs are said to be in linkage disequilibrium if the observed haplotype frequencies deviate from the expected, which are calculated from allele frequencies as shown here.
There is a variety of measures for pairwise LD between markers. The most frequently used in the literature are Lewontin’s D’ and the correlation coeffi-cient r2 (described e.g. by Mueller23). They both range between 0 and 1, where 0 denotes random segregation and 1 means complete (or almost com-plete) LD.
Although the probability of LD is higher for adjacent polymorphisms, the degree of LD is not directly proportional to genetic distance. Factors such as recombination events, population bottlenecks and genetic drift affect the genetic architecture of a population and the degree of LD across a genomic region. Consequently, the pattern of LD often varies between different popu-lations. It has been suggested that the genome can be divided into blocks of high LD, separated by regions where recombination events have been fre-quent referred to as ‘recombination hotspots’. The LD blocks, also called haplotype blocks or ‘recombination coldspots’, show less variation and a limited number of haplotypes24. However, the size of the haplotype blocks will be affected by which markers that are included, as well as the migratory and admixture history of a population.
Linkage disequilibrium in association studies LD is a feature that may be of great use in association studies. Depending on the degree of LD in the region, association may be detected with markers located near or sometimes several kb from the functional variant.
In 2002, an extensive international effort was initiated with the aim to in-vestigate the patterns of LD and create a map of human genetic variation by genotyping a large amount of SNPs in four different populations. This is referred to as the ‘HapMap project’, and a public database (www.hapmap.org) provides tools for exploring LD patterns that can be very

19
useful in association studies6. An important application of HapMap data is the possibility to select ‘tag SNPs’, which represent several haplotypes within a region of interest. Since many of the adjacent SNPs will be corre-lated, only a certain number of them need to be genotyped to gain informa-tion regarding the whole region. In many cases, a long haplotype block can be tagged by a single SNP, thus capturing all variation within that block by a single genotyping experiment. However, sceptics have raised concerns that too wide assumptions are made based on the limited dataset of the HapMap project. There are several potential confounding factors, such as allelic het-erogeneity, ethnic admixture, and the correlation between a small sample set and over-estimation of the degree of LD. If not fully evaluated, such factors may give rise to incorrect conclusions25.
Genome-wide association studies
The recent advances in genotyping techniques, which enable fast genotyping of large amounts of markers at a low cost, in combination with the deposi-tion of millions of SNPs into databases and the mapping of LD patterns by the HapMap project6, have laid the foundation for genome-wide association studies (GWAS). This large-scale approach, where a dense set of markers (usually SNPs) across the genome is genotyped, is a promising new tool for detecting genetic risk factors for complex diseases.
Similar to genome-wide scans for linkage, GWAS have no a priori hy-pothesis of the location of susceptibility variants. However, whereas linkage analysis has been successful in the location of rare variants with strong ef-fects, GWAS has a higher power to detect common variants with moderate effects on disease risk21. Since it has been argued that common complex diseases are caused mainly by common variants with moderate effects26, a powerful association study seems to be an adequate method in the search for such risk variants. Indeed, in the past few years, numerous disease-susceptibility loci have been identified through GWAS27-30.
Since only a fraction of all genomic variation is genotyped in a GWA scan, risk variants will only be detected if they are among the genotyped markers or if they are in LD with these markers. There are many indications that the genome contains long segments of strong LD, which enables detec-tion of a large proportion of genetic variants with this approach. However, the power to descry an association decreases with the degree of LD, and those variants that lie outside LD block structures (approximately 1% of all SNPs31) will not be detected unless they are directly genotyped.
In a GWA study, a substantial number of tests are performed, which con-sequently leads to a great number of markers associated by chance. It is therefore necessary to perform some type of correction for multiple testing in order to separate the wheat from the chaff. Risch and Merikangas21 sug-gested a conservative p-value threshold of 5×10-8, which is equivalent to

20
p=0.05 after Bonferroni correction for 1 million independent tests. However, the Bonferroni method is perhaps not completely relevant since many of the markers are in LD and therefore not independent. Other, less stringent meth-ods for correction have also been suggested, such as permutation testing, or a multi-stage approach, with a relatively liberal threshold in the first step (to minimise the loss of true signals), followed by thorough studies in additional datasets32.
As mentioned earlier, the power of GWAS to detect common susceptibil-ity variants is high. This is of course on condition that the number of patients and controls is sufficiently high. Numbers in the thousands are often re-quired for true signals of moderate size to be distinguished in the noise of spurious signals27,33. Considering the large amount of data generated in a GWAS, careful thought must be put into both study design and data analysis. Biases and errors can be created in every step of the procedure, including sample collection, genotyping and statistical evaluation. Even if careful pre-cautions are taken and the results are closely scrutinised, a great number of false positive results are likely to emerge. It is therefore essential to replicate each finding in other cohorts.
Candidate-gene approach
A candidate-gene study is a hypothesis-based search, in which a gene is se-lected based on its function and biological context. A candidate gene is sim-ply a gene for which there is indication of a role in the studied disease or trait.
The analysis of a candidate gene often starts with an association study of common variants, usually SNPs, that represent as much as possible of the variation within the gene. By genotyping these ‘tag SNPs’, clues can be given to where in the gene a putative association lies. The association study can also incorporate polymorphisms based on a potential functional effect, such as missense mutations or alterations of putative regulatory sites for the gene. If it is suspected that the candidate gene contains functional variants that are previously unknown, it may be necessary to resequence the gene in a selected number of patients and controls. This resequencing may include the entire gene and its promoter region, or may be focused on selected regions, such as exons, splice sites and sequences that are conserved between species and thus may have regulatory importance.
When comparing the candidate-gene approach and the genome-wide ap-proach, both have their pros and cons. Genome-wide scanning for linkage or association has the advantage of being hypothesis-free. This means that in-formation of the disease pathogenesis is not required before study initiation, and completely new pathways may be discovered. The candidate-gene ap-proach, on the other hand, has a major economic advantage, and allows a

21
dense study of the gene in question with fewer requirements for multiple-testing corrections.
Animal models
An animal that has a disease or symptoms similar to a human condition is called an animal model. These animals can be valuable tools in identifying genetic risk factors behind the human disease in question. It is common to study inbred strains of mice or rats that spontaneously develop a disease, or strains that are susceptible to induction of the disease, e.g. by immunisation with an auto-antigen. Similar methods can be used as in human genetics: susceptibility regions can be identified by linkage analysis, and association studies of candidate genes can be performed. In animals, the reversed ap-proach is also possible, where a genetic sequence is modified and the pheno-typic consequences are studied.
Studying animal models may have many advantages over human subjects. Much of the variability that exists in humans with complex diseases can be eliminated by the use of inbred strains with a common genetic background, which are housed in a controlled environment. The breeding possibilities in animals are also major benefits, enabling crossings of interesting specimen and creating large amounts of offspring, which increases the statistical power and the genetic resolution. There are also experiments that cannot be per-formed on human test subjects for ethical reasons, such as gene knock-out. Although these animals are just models of the human condition, this type of studies can yield valuable complementary information.
Complicating factors
In all types of genetic studies, there are factors that complicate the matter and that may give rise to inaccurate results. For complex diseases, there is often substantial heterogeneity on several levels, including the phenotype, the risk genes involved, and even different risk variants within the same gene. There may also be epistatic (gene-gene interaction) effects, where the risk of an allele only is expressed when combined with another genetic fac-tor. An additional aggravating factor in the study of complex traits is that most risk variants seem to convey only a modest effect, which demands large datasets to reach statistical significance.
There are numerous factors and events that can skew the statistics of a genetic study and give rise to false positive (or false negative) results. For example, an imprecise phenotype or differences in diagnosis of a disease may create a heterogeneous patient group. Population stratification, with different ethnic backgrounds of the studied individuals, can also be a major cause for spurious signals in case/control studies. However, family-based studies are generally protected from the effects of ethnic admixture owing to

22
their use of internal controls. Bias can also be caused by inaccurate sample handling or technical errors, such as incorrect genotyping.
In other words, biases and errors can be created in every step of a study. Especially in large-scale studies such as GWAS, precaution is required to prevent the true associations from drowning in a sea of spurious signals.
Functional mutations
A major reason for studying genetic risk factors for complex diseases is to achieve a better insight into the pathological processes of the disease. How-ever, proceeding from an associated marker to a deeper understanding of functional, causative variants and their means of action is complicated. A wide range of possible processes that affect the gene product may be in-volved. There could be polymorphisms that affect protein structure, such as altered amino acid sequences or splicing variants. There is also the possibil-ity of changes in gene expression levels or localisation. Such changes could be caused by alterations in various types of regulatory sequences, such as binding sites for transcription factors or other regulatory elements, or affect epigenetic factors such as DNA methylation sites.
Bioinformatic tools that may assist in the interpretation of association data are available, where information from several databases is combined, including LD, gene expression, gene structure and other features34. However, there will most likely be a significant development of such tools in the fu-ture, as well as improvement of experimental approaches.
Systemic Lupus Erythematosus
Pathology
SLE is a chronic autoimmune disease associated with a multitude of symp-toms and a vast array of autoantibodies. The clinical manifestations range from rashes, fatigue, fever, arthritis and lack of various blood cells, to thrombosis, serositis, nephritis, seizures and psychosis. The autoantibodies are mainly directed against various nuclear components, such as double-stranded DNA or histones, but antibodies against cytoplasmic, cell-membrane or extracellular molecules are also frequently observed35. The disease severity can also differ significantly between the patients. Some have relatively mild symptoms and require little or no medical treatment, whereas others are severely affected by chronic inflammation of multiple internal organs and require aggressive treatment with high-dose corticosteroids and cytostatic drugs. Due to the heterogeneity of this disease, the American Col-lege of Rheumatology (ACR) has established 11 criteria for classification36,37 (Table 1). Fulfilment of at least four criteria is sometimes a requirement for

23
diagnosis, although the ACR criteria were constructed mainly for classifica-tion purposes. A large study of European patients identified the most com-mon clinical manifestations as arthritis (48.1% of patients), the characteristic malar rash (31.1%) and renal disorder (27.9%)38.
Table 1. The American College of Rheumatology’s criteria for classification of SLE, revised in 1982 and 1997.
Criterion Definition
Malar rash Fixed erythema, flat or raised, over the malar eminences, tending to spare the nasolabial folds
Discoid rash Erythematosus raised patches with adherent keratotic scaling and fol-licular plugging; atrophic scarring may occur in older lesions
Photosensitivity Skin rash as a result of unusual reaction to sunlight, by patient history or physician observation
Oral ulcers Oral or nasopharyngeal ulceration, usually painless, observed by a physician
Arthritis Nonerosive arthritis involving 2 or more peripheral joints, characterised by tenderness, swelling or effusion
Serositis a) b)
Pleuritis (convincing history of pleuritic pain or rub heard by a physi-cian or evidence of pleural effusion) OR Pericarditis (documented by ECG or rub or evidence of pericardial effusion)
Renal disorder a) b)
Persistent proteinurea > 0.5 g/day, or greater than 3+ if quantification not performed OR Cellular casts: red cell, hemoglobin, granular, tubular or mixed
Neurologic disorder
a) b)
Seizures (in the absence of offending drugs or known metabolic de-rangements) OR Psychosis (in the absence of offending drugs or known metabolic de-rangements)
Haematologic disorder
a) b) c) d)
Haemolytic anaemia with reticulocytosis OR Leukopenia (<4000/mm3 on 2 or more occasions) OR Lymphopenia (<1500/mm3 on 2 or more occasions) OR Thrombocytopenia (<100,000/mm3 in the absence of offending drugs)
Immunologic disorder
a) b) c)
Antibody to native DNA in abnormal titre OR Antibody to Sm nuclear antigen OR Antiphospholipid antibodies - based on 1) abnormal serum level of IgG or IgM anticardiolipin antibodies, 2) positive test result for lupus antico-agulant using a standard method, or 3) a false-positive serologic test for syphilis for at least 6 months confirmed by Treponema pallidum immo-bilisation or fluorescent treponemal antibody absorption test
Anti-nuclear antibodies
Abnormal titre of antinuclear antibody by immunofluorescence or equivalent assay, in the absence of drugs known to be associated with ‘drug-induced lupus’ syndrome

24
SLE affects about 0.03% of populations with European ancestry, but the prevalence varies with several factors, such as gender, age, ethnicity and time period studied. About 90% of the SLE patients are women, and the peak age of onset is during childbearing years. Populations of African and Asian decent have a significantly higher prevalence than those with Euro-pean ancestry, and the general prevalence has increased in the recent years, probably due to improved diagnosis and better survival rates for SLE pa-tients39-41.
Aetiology and pathogenesis
SLE is classified as a complex disease, where the cause is believed to be a complicated interaction between various genetic and environmental factors. There are, however, a few examples of rare monogenic forms of SLE, with complete deficiencies of early complement components, such as C1q42, or defects in the TREX1 gene43.
The genetic component of SLE The disease has a clear genetic component, as shown by an increased risk to develop the disease for relatives of SLE patients44. The risk of disease has been estimated to be 20 times higher if you have a sibling with SLE45. Twin studies also indicate a significant genetic contribution: the concordance rate is 24-69% in monozygotic twins, but only 2-3% in dizygotic twins46-48.
A number of genetic risk factors for SLE have been identified, and re-search within this field has been successful especially in the last few years. The genes associated with SLE have various functions important for regula-tion of the immune system. In combination, these genetic variants will lead to exaggerated and prolonged immune reactions and sub-optimal suppres-sion of reactions to self, which results in increased susceptibility for the dis-ease. Genetic studies of SLE are further discussed in the next section.
Epigenetics, which refers to inheritable modifications not affecting the DNA sequence, have also been suggested in SLE pathogenesis, especially hypomethylation of DNA in T cells, leading to dysregulation49.
Environmental factors Although twin studies clearly imply a genetic component in SLE aetiology, the concordance rates are still relatively low, indicating involvement of envi-ronmental factors. Various infectious agents, especially the Epstein-Barr Virus (EBV) causing mononucleosis, have been connected with the disease. Possible mechanisms behind this association are antibody cross-reactivity between viral antigens and self antigens, and immortalisation of self-reactive B cells by EBV 50-53.
UV radiation also appears to be a risk factor. SLE patients are often ex-tremely photosensitive, but UV light could also trigger disease onset54. There

25
is also a long list of medications inducing lupus-like symptoms. Drug-induced lupus erythematosus often has a long latency period, where symp-toms emerge months or sometimes years after starting on the medication55. Other examples of environmental factors implicated with risk of SLE are exposure to high levels of silica dust56, diet (especially alfalfa sprouts, which contain high amounts of L-canavanine57) and smoking58. There are also some indications that heavy metals, solvents, pollutants, pesticides and hair dyes could be risk factors54, although this may need confirmation in larger studies.
Gender bias and hormonal effects The female prevalence of the disease, with post-puberty onset and disease activity sometimes affected by menstrual cycle or pregnancy, suggest that there may be hormonal effects involved. Indeed, the female sex hormone oestrogen has been implicated with SLE in several ways. It has been shown to repress tolerance of self, and to have various effects on B and T cells, as well as other leukocytes59-63. However, many studies of oestrogen effect on SLE have had inconclusive results. Some studies have found association of oral contraceptives with slight increase in risk of SLE64-66. During preg-nancy, some studies67,68 but not all69,70 have observed an increase in flares and disease activity. In men with SLE, lower testosterone levels and higher oestrogen levels have been noted71. Interestingly, men with Klinefelter’s syndrome (XXY instead of the normal XY) are more prone to SLE, which could suggest a gene dosage effect72.
In conclusion, there is no simple explanation of the gender bias observed in SLE, although several sex hormones in combination with X chromosome genes are likely to be involved.
Generalised SLE pathogenesis A likely course of events in the development of SLE could be the follow-
ing: An immune response is triggered by an environmental factor, such as an EBV infection53, or possibly by a self-antigen73. Antigens are taken up by antigen-presenting cells, which present peptides to T cells. Activated T cells stimulate B cells to produce autoantibodies, and further contribution to B- and T-cell stimulation is provided by various accessory molecules and cyto-kines. An inadequate tolerance system fails to repress the autoreactivity. Increasing amounts of immune complexes are formed, and an impaired clearing of these complexes causes deposition in tissues, where they give rise to inflammation via complement activation74. In some cases, autoanti-bodies may cause symptoms without involvement of the complement sys-tem, for example by binding of different types of blood cells, and thereby cause haematologic disorder. This model is illustrated in Figure 3.
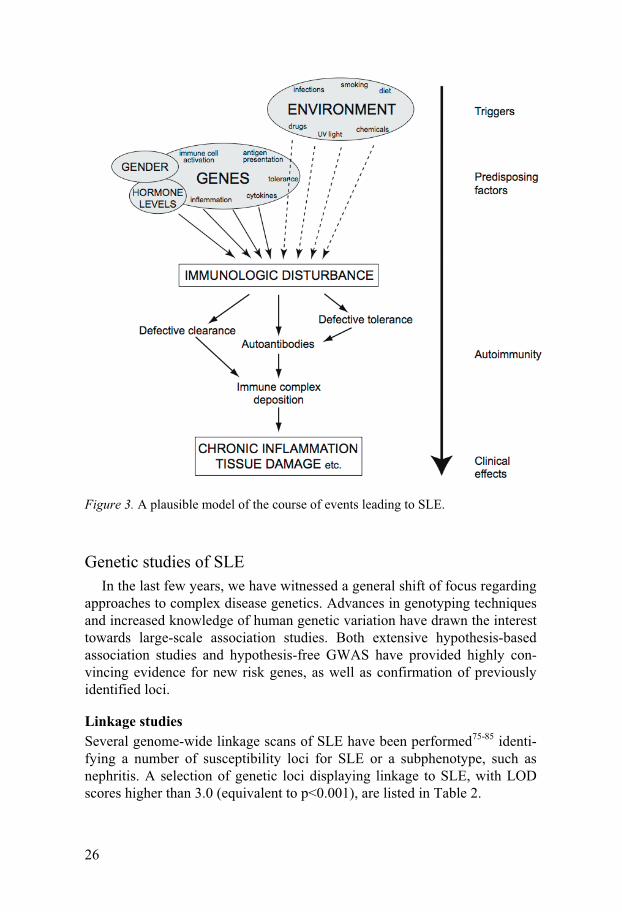
26
Figure 3. A plausible model of the course of events leading to SLE.
Genetic studies of SLE
In the last few years, we have witnessed a general shift of focus regarding approaches to complex disease genetics. Advances in genotyping techniques and increased knowledge of human genetic variation have drawn the interest towards large-scale association studies. Both extensive hypothesis-based association studies and hypothesis-free GWAS have provided highly con-vincing evidence for new risk genes, as well as confirmation of previously identified loci.
Linkage studies Several genome-wide linkage scans of SLE have been performed75-85 identi-fying a number of susceptibility loci for SLE or a subphenotype, such as nephritis. A selection of genetic loci displaying linkage to SLE, with LOD scores higher than 3.0 (equivalent to p<0.001), are listed in Table 2.

27
Table 2. Susceptibility regions with LOD scores >3.0 (or p<0.001) identified in genome-wide scans for linkage with SLE. Chromosomal re-gion
LOD or p value1
References
1q22-24 3.37; 4.41 Moser82; Olson84 1q41 3.50 Moser82 1q44 3.33 Shai85 2q37 4.24; 4.1 Lindqvist81; Olson84 4p15-13 3.20 Lindqvist81 4p16-15 3.84; 3.28 Gray-McGuire78; Olson84 6p11-p21 4.19 Gaffney77 12p12-11 3.98 Olson84 12q24 4.39 Nath83 16q13 3.85; 3.06 Gaffney77, Nath83 16p12-16q12 P=0.00017 Lee86 (meta-analysis) 17p12-q11 3.49 Johansson79 17p13 4.41 Olson84 18q22-23 P=0.0003 Cantor75 19q13 3.16 Johansson79 1 LOD scores or p values as given in the original articles. Different methods were used, and
the values are therefore only roughly comparable between studies.
Although several of the susceptibility loci have been replicated, none of them has been reproduced in all studies. Two meta-analyses both confirm the HLA region on chromosome 6p21 as the strongest region, and report linkage to one additional region each, namely 16p12.3-16q12.286 (significant linkage) and 20p11-q13.1387 (suggestive linkage), respectively.
There are several mouse models for SLE, such as MRL, BXSB and the F1 hybrid of NZW/NZW88. A number of murine linkage regions have been identified, for example Sle1, Sle2, and Sle3, which are syntenic with the hu-man linkage loci 1q23, 9p22, and 19q13, respectively81,89.
Several of the findings from linkage studies have lead to the identification of interesting susceptibility genes. For example, the human 1q22-23 region contains genes encoding receptors for IgG, which have been associated with SLE90, chromosome 2q37 harbours the associated PDCD1 gene91, and re-cently, the strong association of ITGAM was identified through fine-mapping of a linkage region on chromosome 16p1192.
Associated genes A great number of association studies have been performed for SLE, some with more convincing results than others. In general, associations detected in larger cohorts that have been replicated in independent samples and/or sup-ported by functional studies, can be considered as more convincing.
Thus far, three large GWA studies of SLE have been published28-30. A handful of genes have reached genome-wide significance in all three studies, which must be regarded as extremely convincing of a role in SLE aetiology.

28
The combined result of the three GWAS confirms the already well-established association of the HLA region, as well as the recently identified strong association of IRF5 and STAT4. In addition, the novel risk genes BLK and ITGAM are identified.
The HLA region on the short arm of chromosome 6 is a genetic locus that
is distinguished in most contexts. The extended HLA region contains at least 252 expressed genes and is unique for its level of polymorphism, LD and cluster of genes that are important in the immune system93. The region has shown highly convincing association with numerous diseases, including SLE. This association has been known for over 30 years94, and it continues to be replicated. Recent GWAS and meta-analyses of linkage screens indi-cate that the HLA region contains the greatest genetic risk factors in SLE susceptibility28-30,86,87,95. However, the nature of this locus, with its high level of LD and great variability, makes it difficult to identify the causative gene. The most consistent associations seem to be with MHC class II genes, and for example, haplotypes including the alleles DRB1*1501/DQB1*0602, DRB1*0801/DQB1*0402, and DRB1*0301/DQB1*0201 have shown con-vincing association96,97. In addition to the genes of the MHC complex, the region also contains several highly interesting genes that have been impli-cated with SLE, such as Tumour Necrosis Factor α and β (TNFα and β)98,99 and complement components, such as C4A, C4B and C2100,101. It is not unlikely that several independent effects exist within this region, which is indicated by results of the SLEGEN GWA study29.
The association of the gene encoding the IRF5 (Interferon Regulatory
Factor 5) transcription factor was first identified in an association screen of genes related to type I interferon (IFN)102. An independent study then identi-fied a strongly associated haplotype containing functional SNPs affecting expression levels and splicing of IRF5103. High expression of type I IFN and IFN-inducible genes, the so-called ‘IFN signature’, is commonly observed in SLE patients104, suggesting an important role in the pathogenesis. There are also many reports of SLE as a consequence of IFN-α treatment in cancer patients105.
The gene encoding the STAT4 transcription factor was first identified as
a risk factor for rheumatoid arthritis in an association study of a linkage re-gion. The same study also found association to SLE with the same risk hap-lotype106. In addition to the three GWA studies, this association was recently confirmed by two independent studies, including paper IV in this thesis28-
30,107. STAT4 plays an important role in the differentiation of Th1 cells and IFN-γ production, and it has also been reported to mediate type I IFN signal-ling108. The identification of IRF5 and STAT4 as SLE risk genes supports the

29
hypothesis that the type I interferon pathway is central to the disease patho-genesis.
The ITGAM gene (Integrin Alpha M, also called CD11b) is localised in a
linkage region for SLE (16p11), and association was identified in a fine-mapping study of the region92. Association was also detected in the three GWAS studies, which were published simultaneously with the linkage-region fine-mapping or shortly after28-30. ITGAM encodes a subunit of the complement receptor 3 (CR3, also Mac-1), which is expressed mainly on neutrophils, macrophages and dendritic cells. The receptor is important in the regulation of many immunologic functions, including iC3b-mediated phagocytosis and leukocyte adhesion and emigration from the bloodstream via interactions with ICAM-1 and ICAM-2109.
Association with the BLK gene (B lymphoid tyrosine kinase) was de-
tected as one of the most significant associations with SLE in all three GWA studies28-30. A risk allele localised between BLK and the gene C8orf13 was associated with reduced expression of the BLK, but also with increased ex-pression of C8orf1330. It is thereby implied that both genes may constitute risk factors for SLE. BLK is a B-cell specific member of the Src family of tyrosine kinases, and may thus influence proliferation and differentiation of B cells110. The function of C8orf13 is still unknown.
In addition to the five genes with triple genome-wide significance de-
scribed above, there are several other genes displaying convincing associa-tion with SLE in several datasets. PTPN22 (protein tyrosine phosphatase non-receptor 22), which was first identified as a risk gene for type I diabe-tes111 and shortly after for Rheumatoid Arthritis and Grave’s disease112,113, has been associated with SLE in several large association studies, including one of the GWAS29,114,115. PTPN22 encodes a protein involved in down-regulation of T-cell activation, and the risk allele results in an amino acid substitution interfering with the interactions of this protein with the protein kinase CSK112.
The Fcγ receptor genes (FcγR), which encode low-affinity receptors for IgG antibodies, have been extensively studied in connection with autoim-mune diseases. A cluster of five FcγR genes is located on chromosome 1q23, a region with reported linkage to SLE82,84. High degree of sequence similar-ity between the genes has made analysis of this region difficult. Early studies have sometimes reported conflicting results for these genes, however, the most consistent association seems to be with a missense mutation in FCGR2A90. This allele was also significantly associated in a GWA study29. Interestingly, copy-number variation of the FCGR3B gene has also been associated with SLE14,15.

30
Protective and risk haplotypes within the gene encoding TNFSF4 (TNF Super-Family 4, also OX40 ligand) were recently identified in several large case-control and family-based cohorts116. TNFSF4 is expressed on antigen-presenting cells, and binding of the OX40 receptor mediates T-cell prolifera-tion and differentiation into memory T cells117.
Other genes that have been associated in several cohorts and supported by functional data are for example PDCD191 and CTLA-4118, both inhibitory receptors present on T cells, and the recently identified association with BANK1 encoding a B-cell scaffold protein (paper III). Genetic and func-tional data have also indicated roles for the tyrosine kinase LYN29,119 (which interacts with BANK1), the MBL-2 gene encoding the complement activator MBL120, and the cytokine IL-10121,122.
It will also be very interesting to follow further studies of genes that were identified as potential risk factors for SLE by reaching genome-wide signifi-cance in only one of the GWA studies. These genes include XKR, ATG5, ICA1, SCUBE129 and TNFAIP328.
Table 3. A selection of genes associated with SLE. Gene Chromosomal
location References
HLA region 6p21 Reviewed by Fernando95 IRF5 7q32 Sigurdsson102, Graham103 STAT4 2q32 Remmers106 ITGAM 16p11 Nath92, GWAS28-30 BLK, C8orf13 8p23-p22 GWAS28-30 PTPN22 1p13 Kyogoku114 FCGRs 1q23 Reviewed by Brown90 TNFSF4 1q25 Graham116 PDCD1 2q37 Prokunina91 CTLA-4 2q33 Ahmed118 BANK1 4q24 Paper III MBL-2 10q11-q21 Reviewed by Monticielo120 IL-10 1q31-32 IL-10122
31
Present investigation
Aim The studies presented here aim to identify genetic risk factors for the auto-immune disease Systemic Lupus Erythematosus.
Material and methods
Patients and controls
Several different sets of patients, families and healthy controls have been studied in the present investigation. The same samples have often been in-cluded in several studies. However, due to variation in access of samples, the number in each set may differ between studies. An overview of the case-control datasets is presented in Table 4. In addition, 149 Swedish and 90 Mexican trios, and 10 Icelandic multicase families were studied in Paper I. All patients fulfil at least four of the ACR classification criteria36,37, and have given their informed consent to participate in the studies. The population controls were matched for ethnicity, and individuals with parents or grand-parents with other ancestry were excluded.
Table 4. Number of cases and controls analysed in the four papers of this thesis, indicated by their roman numbers.
Cohort No of patients No of controls
I II III IV I II III IV
Sweden 152 310 279-464 448 247 352-515
Denmark 84 Spain 696 678-799 390 539 457-542 620 Germany 257 384 247 317 374 220 Argentina 288 288 171 288 372 171 Mexico adult
231 250
Mexico paediatric
321 383
Italy 286 221 252 207

32
Genotyping
There is a broad range of methods available for identifying an individual’s genotype at a certain genetic position. In the papers on which this thesis is based, four different methods were used for SNP genotyping: sequencing, restriction fragment length polymorphism (RFLP), TaqMan SNP genotyp-ing, and a high-density oligonucleotide SNP array.
Since direct sequencing yields the complete genetic sequence of the frag-ment you wish to analyse, it can be used to identify new genetic variations as well as obtain genotypes of known SNPs. It is also easy to assess the quality of each assay, so that uncertain genotypes can be excluded. Sometimes this method is referred to as resequencing, since the general sequence already is known, and the aim is to find any deviations from this sequence in your sample of interest.
RFLP genotyping, which uses a restriction enzyme that will cleave one allele but not the other, is a simpler method that was used for genotyping one of the SNPs in paper I. Depending on the enzyme and the sequence analysed, the reliability of this assay will vary. Therefore, the accuracy of the assay must be verified by another genotyping method, such as sequencing.
The TaqMan SNP genotyping assay123 provided by Applied Biosystems was used for genotyping most of the SNPs in all four papers. The method is based on hybridisation of allele-specific fluorescent probes to your sample during a Polymerase Chain Reaction (PCR). The probe contains a reporter dye at the 5’ end and a quencher dye at the 3’ end. During the PCR reaction, the probe anneals to the DNA. As elongation proceeds, the probe is cleaved whereupon reporter and quencher are separated, resulting in increased fluo-rescence of the reporter. Since each assay contains two allele-specific probes with different reporter dyes, the genotype of each sample can be calculated as the relative fluorescence of these two dyes. The genotypes are determined automatically by computer software. This genotyping assay is significantly faster than the two methods described above, due to a single-step setup in the lab and the automated genotype calling.
In papers III and IV, genotypes of some of the SNPs were obtained from a collaboration where a GWAS was performed using the Affymetrix 100k GeneChip®. This type of high-density oligonucleotide SNP array contains probes for almost 100,000 SNPs, allowing them to be genotyped simultane-ously. The procedure includes restriction enzyme digestion of the DNA, PCR amplification, fragmentation, biotin labelling, hybridisation to the array with allele-specific probes, addition of streptavidin-conjugated fluorophores, detection of fluorescence, and automated allele calling. SNP array is a high-throughput genotyping method, although the specificity and sensitivity may be lower compared with other methods.

33
Statistical analysis
There is a wide range of statistical tests within the field of association stud-ies, each with strengths and weaknesses. The two main categories of associa-tion analysis are case-control and family-based studies. Comparing cases with healthy controls using a χ2 (chi square) test or a similar statistic is per-haps the most commonly used approach. As mentioned previously, it is im-portant to use ethnically homogenous cohorts in case-control studies, to avoid spurious association due to population stratification.
Family-based association studies were developed in order to circumvent the problem of population stratification. Many of the family-based methods are variations of the Transmission-Disequilibrium Test (TDT)124, which studies cases with unaffected parents, and compares allele frequencies in the group of transmitted versus non-transmitted alleles. TDT is not affected by population stratification, but can be biased by inclusion of incomplete fami-lies or by reconstruction of parental genotypes from their offspring125. The Haplotype-Based Haplotype Relative Risk test (HHRR)126 is based on the same principle, but is considered more powerful than TDT, which only uses information from heterozygous parents. HHRR includes both homozygous and heterozygous parents, which increases the power, especially when a rare allele is analysed. There are also tests that can incorporate all types of pedi-grees, such as the Family-Based Association Test (FBAT), which compares the observed and expected allele frequencies of affected individuals127.
In the present study, case-control cohorts were primarily analysed using χ2 of 2×2 contingency tables. In paper I, the trios were analysed by HHRR, since many of the SNPs had low minor allele frequencies and the statistical power of TDT would not be sufficient. We also applied the FBAT test to the same material.
There are several different methods for studying genotypic effects. In pa-pers II-IV in this thesis, we have used the Unphased software128, where ho-mozygosity for the non-risk allele was set as a reference with odds ratio=1.
Testing for Hardy-Weinberg Equilibrium (HWE) can serve as a rough quality control. The HWE test analyses the relation between allelic and genotypic frequencies, which should be correlated under certain conditions, such as unlimited population sizes, random mating and no selection or mi-gration. Although these criteria will never be fully met, an approximate HWE should be expected unless genotyping errors or other confounding factors have caused skewed distributions in genotype frequencies. However, if association is strong it may create deviations from HWE in the patient cohort. Thus, deviations from HWE in the patient group may reflect true association, whereas in controls, it could indicate a bias129,130.
A meta-analysis is an approach that combines the results of several differ-ent datasets. In case-control studies, this is a chance to analyse an overall effect across several populations, but still keep the cohorts as separate units.

34
Different statistical methods exist, where some analyse fixed effects and assume homogeneity between the strata, whereas other methods analyse random effects and can be applied to heterogeneous cohorts. An initial analysis of homogeneity between the cohorts can therefore be necessary. The studies in this thesis have applied the Breslow-Day test for homogeneity, Mantel-Haenszel meta-analysis of homogeneous strata, and DerSimonian-Laird test for heterogeneous strata, all of which were implicated in the StatsDirect software.
Paper I: No evidence of association between genetic variants of the PDCD1 ligands and SLE
Background
In this study, we analyse SNPs in the genes of the PDCD1 ligands, PD-L1 (CD274) and PD-L2 (CD273), for association with SLE. The importance of the PDCD1 pathway in peripheral tolerance has been demonstrated by sev-eral studies (reviewed e.g. by Keir et al131). Furthermore, a regulatory poly-morphism in the PDCD1 receptor gene had previously been identified as a risk factor for SLE91, and suggestive linkage had been found to a marker located close to the genes of the two PDCD1 ligands81. We therefore consid-ered the PD-L1 and PD-L2 genes as interesting candidates for susceptibility to SLE.
Results and discussion
When this study was initiated, very few SNPs were known in these genes. We therefore sequenced samples from patients and controls in order to iden-tify polymorphisms in the exons and in regions of potential regulatory im-portance. Later, as more SNPs became available in the databases, we also selected a SNP that altered the binding site of an important transcription factor. In total, 23 SNPs were then analysed in Swedish trios using two dif-ferent family-based tests for association: HHRR and FBAT. The HHRR test found eight of these SNPs to be associated to SLE (p<0.05), whereas the FBAT test did not find any association.
Due to the conflicting results of the two statistical tests, we analysed the eight ambiguously associated SNPs in three additional cohorts: Mexican trios and case-control cohorts from Sweden and Argentina. No association was found in either of the replication sets, with the exception of one allele that was found in 2.0% of patients and 4.5% in controls in the Argentinean cohort (p=0.0157). However, for several reasons we believe that this is very likely to be a false positive result. First, the risk of type I error is greater for a rare allele than for a common132. Second, since several SNPs now have been
35
analysed in four different cohorts, the risk of spurious association has in-creased. If correction for multiple testing is applied, the association disap-pears. Third, this Argentinean cohort contains some elements of admixture between European and Amerindian ancestry, which further increases the risk of false positive results133.
Allele frequencies in the Swedish trios and case-control cohort are shown in Table 5. Comparison of allele frequencies (transmitted with patient al-leles, and non-transmitted with control alleles) indicates that bias may have been introduced.
Table 5. Allele frequencies in the PD-L1 and PD-L2 genes. SNPs with rs- or ENSSNP-numbers can be found in databases. Minor allele frequencies are shown. SNP Location Transm
alleles (HHRR) N=298
Patient alleles N=294
Non-transm alleles (HHRR) N=78-144
Control alleles N=896
PD-L1
L1x11 -85 0.3% Nt 1.9% Nt rs10815225 -62 9.3% 13.0% 19.8% 9.8% rs7866740 -29 7.3% 6.2% 15.3% 7.6% rs17742278 5965 30.1% Nt 27.6% Nt rs1411262 8862 30.6% Nt 27.6% Nt rs10815226 9148 23.0% Nt 22.9% Nt rs7041009 12686 30.3% Nt 25.8% Nt ENSSNP5915018 15529 17.5% 15.1% 31.8% 16.6% rs41303227 17244 6.3% 5.9% 14.2% 7.0% rs2297136 17399 55.3% 45.9% 40.3% 46.7% rs4143815 17701 27.4% Nt 32.2% Nt rs10118693 33683 29.7% Nt 36.7% Nt C57:2 33799 0.8% Nt 0.0% Nt
PD-L2
rs16923189 74 26.1% Nt 29.1% Nt C91:1 7641 0.7% Nt 0.9% Nt C91:2 7667 0.7% Nt 0.0% Nt rs12001295 7690 3.4% 5.6% 10.1% 5.6% rs7870226 19447 33.5% Nt 38.2% Nt C103:2 19500 0.4% Nt 0.0% Nt C103:3 19512 1.5% Nt 2.9% Nt rs6476989 45002 0.4% Nt 0.0% Nt rs7854413 47138 5.1% 9.2% 10.7% 9.3% rs7852996 59290 3.6% 8.6% 11.1% 8.2% Nt= not tested. Transm=transmitted. Number of non-transmitted alleles varies with the num-ber of informative trios for the SNP, and is therefore given as a range.
The lack of association in the replication cohorts, including the Swedish cases and controls, indicates that the association seen in the HHRR analysis was a false positive result. The allele frequencies differ markedly between the non-transmitted alleles from the HHRR analysis and the healthy popula-

36
tion controls. This indicates that bias of some sort has been introduced in the HHRR analysis of the Swedish trios, and the FBAT result is more likely to reflect the true picture.
Power estimations show that the investigated cohorts should have enough power to detect association of alleles with relative risks above 1.5. We ana-lysed 23 SNPs spread across the PD-L1 and PD-L2 genes, and do not find cogent evidence of association, which suggests that these genes are not risk factors for SLE. The possibility of a risk factor that is not in linkage disequi-librium with any of the SNPs analysed can, however, not be excluded.
A study published shortly after paper I reported association to SLE with the SNP rs7854303 in PD-L2 in 164 patients and 160 controls from Tai-wan134. This SNP is not polymorphic in any of our Swedish or Icelandic cohorts as verified by sequencing, and may thus be an Asian-specific risk factor.
Paper II: Association of a CD24 gene polymorphism with susceptibility to systemic lupus erythematosus
Background
CD24 (or Heat-Stable Antigen) is a glycosyl phosphatidylinositol (GPI)–linked protein which is expressed on the surface of various cell types, such as activated T cells, B cells, mature granulocytes, macrophages, and den-dritic cells135-138. The biologic function of CD24 is unclear, although a role in the activation and differentiation of B cells has been indicated139, as well as in activation of both CD4+ and CD8+ T cells through a CD28-independent costimulatory pathway135,137,140. CD24 has also been shown to modulate the interaction between Very Late Activation antigen 4 (VLA-4) and Vascular Cell Adhesion Molecule 1 (VCAM-1)141. These adhesion molecules are im-portant in lymphocyte costimulation in specific tissues and sites of inflam-mation in SLE patients142.
The CD24 gene maps to a linkage region for SLE and other autoimmune diseases on chromosome 6q21–2581,82. Association with Multiple Sclerosis (MS) was detected for the SNP rs8734 encoding an amino acid substitution from alanine to valine (A57V). The same study also reported a higher ex-pression of CD24-57V compared with CD24-57A on T cells, as determined by flow cytometry143.
Results and discussion
In this study, we analyse if the A57V polymorphism in CD24 is associated with SLE in three independent cohorts from Spain, Germany and Sweden. In the Spanish cohort, the CD24-57V allele was associated with increased risk
37
for SLE. There was also an increased risk associated with the V/V homozy-gous genotype when compared with the A/A genotype. In the German co-hort, a similar trend was observed, with higher frequencies of the V-allele and the V/V-genotype in the patients than in the controls. This difference, however, did not reach statistical significance. In the Swedish cohort, no differences between patients and controls were observed. Data from these analyses are shown in Table 6.
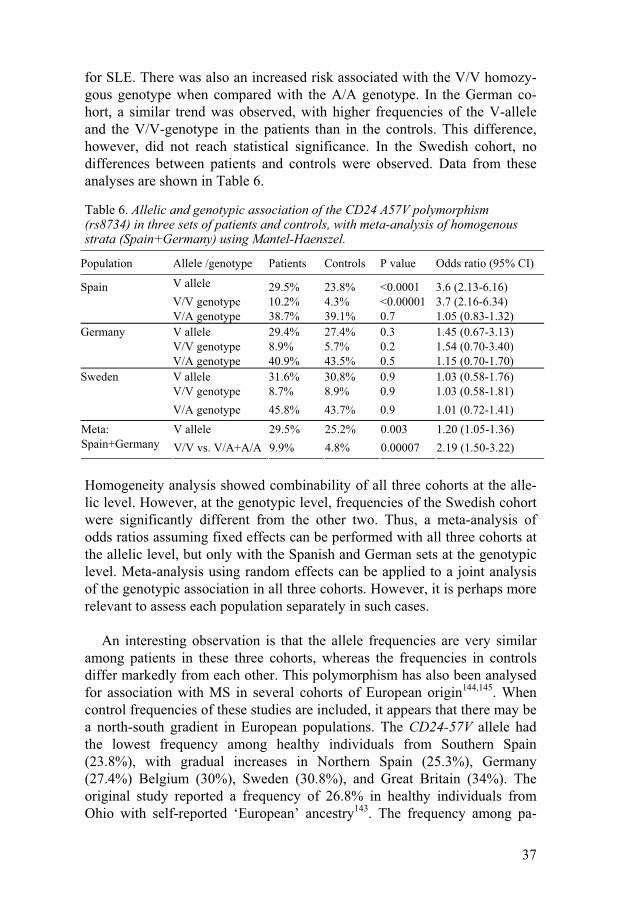
Table 6. Allelic and genotypic association of the CD24 A57V polymorphism (rs8734) in three sets of patients and controls, with meta-analysis of homogenous strata (Spain+Germany) using Mantel-Haenszel.
Population Allele /genotype Patients Controls P value Odds ratio (95% CI)
Spain V allele 29.5% 23.8% <0.0001 3.6 (2.13-6.16) V/V genotype 10.2% 4.3% <0.00001 3.7 (2.16-6.34) V/A genotype 38.7% 39.1% 0.7 1.05 (0.83-1.32)
Germany V allele 29.4% 27.4% 0.3 1.45 (0.67-3.13) V/V genotype 8.9% 5.7% 0.2 1.54 (0.70-3.40) V/A genotype 40.9% 43.5% 0.5 1.15 (0.70-1.70) Sweden V allele 31.6% 30.8% 0.9 1.03 (0.58-1.76) V/V genotype 8.7% 8.9% 0.9 1.03 (0.58-1.81)
V/A genotype 45.8% 43.7% 0.9 1.01 (0.72-1.41)
V allele 29.5% 25.2% 0.003 1.20 (1.05-1.36) Meta: Spain+Germany V/V vs. V/A+A/A 9.9% 4.8% 0.00007 2.19 (1.50-3.22)
Homogeneity analysis showed combinability of all three cohorts at the alle-lic level. However, at the genotypic level, frequencies of the Swedish cohort were significantly different from the other two. Thus, a meta-analysis of odds ratios assuming fixed effects can be performed with all three cohorts at the allelic level, but only with the Spanish and German sets at the genotypic level. Meta-analysis using random effects can be applied to a joint analysis of the genotypic association in all three cohorts. However, it is perhaps more relevant to assess each population separately in such cases.
An interesting observation is that the allele frequencies are very similar
among patients in these three cohorts, whereas the frequencies in controls differ markedly from each other. This polymorphism has also been analysed for association with MS in several cohorts of European origin144,145. When control frequencies of these studies are included, it appears that there may be a north-south gradient in European populations. The CD24-57V allele had the lowest frequency among healthy individuals from Southern Spain (23.8%), with gradual increases in Northern Spain (25.3%), Germany (27.4%) Belgium (30%), Sweden (30.8%), and Great Britain (34%). The original study reported a frequency of 26.8% in healthy individuals from Ohio with self-reported ‘European’ ancestry143. The frequency among pa-

38
tients has been relatively constant, regarding both MS (30.1-33.2%) and SLE (29.4-31.6%). This population heterogeneity correlates with the inconsistent results. Association with MS has been detected in cohorts with Southern European145 and admixed European143 ancestry, and lack of association was reported for Belgian and British cohorts144. A similar observation has been made regarding association of PDCD1 with SLE146.
The lack of replication could also have other explanations. There may be differences in LD patterns between the populations, and the CD24-57V allele could be a marker for another causative allele, which is in stronger LD in Southern European populations. There could also be epistatic effects with a risk allele that is more frequent in those populations. Alternatively, failure to replicate in different populations may be reflective of a false negative result, although this is less probable if association is detected in multiple independ-ent studies.
In conclusion, we find that the CD24-57V allele is associated with in-
creased risk of SLE in a Spanish set of patients and controls. This associa-tion is supported by a trend for association with the same allele in a German cohort, whereas a Swedish cohort shows no differences between patients and controls.
The associated allele encodes an amino acid substitution, which has been shown to correlate with cell-surface expression of the CD24 antigen143. Re-cently, protective association for both MS and SLE was also found with a dinucleotide deletion in the 3’UTR of CD24 mRNA147. This deletion was reported to decrease the stability of the CD24 mRNA. LD between this dele-tion and the A57V polymorphism was low, which indicates that they repre-sent independent effects.
Paper III: Functional variants in the B-cell gene BANK1 are associated with systemic lupus erythematosus
Background
The B-cell scaffold protein with ankyrin repeats (BANK1) is an adaptor protein involved in B-cell responses to antigen stimulation. BANK1 has been shown to associate with the tyrosine kinase Lyn and the inositol 1,4,5-triphosphate receptor (IP3R). It is believed to regulate signals from the B-cell receptor by connecting protein tyrosine kinases to the IP3R, which subse-quently leads to calcium mobilisation and B-cell activation148.
B cells are generally considered to have a key role in SLE pathogenesis by autoantibody production and regulation of innate and adaptive immune responses by antigen presentation and cytokine signalling. The importance

39
of this cell type is confirmed by the success of novel therapies for SLE, which are aimed at depleting the B-cell population149.
Results and discussion
In a GWA study of Swedish SLE patients and controls, we identified asso-ciation with the SNP rs10516487, encoding a non-synonymous substitution (R61H) in exon 2 of the BANK1 gene. This association was replicated in SLE cases and controls from Germany, Italy and Spain, and a trend for asso-ciation was observed in samples from Argentina. A small set of patients from Denmark was also included, and was analysed together with the Swed-ish samples in a joint Scandinavian cohort. Association with SLE was found in all five cohorts, except the borderline-significant p value in Argentina (Table 7).
Fine-mapping of the genetic region in the Swedish cohort, using 30 tag SNPs in the BANK1 genetic region, revealed association with nine SNPs located between introns 1 and 7 of the gene.
The B-cell specificity of BANK1 was confirmed by expression analysis, where only very weak expression in other cell types was detected, possibly due to contamination with other cell types. Surprisingly, when full-length BANK1 cDNA was amplified, and PCR products were verified by gel elec-trophoresis, two main bands were detected. Sequencing of these bands re-vealed that the larger size PCR product represented a full-length cDNA, and the smaller was a novel isoform with an in-frame deletion of exon 2 (�2 isoform). This �2 isoform lacks the putative IP3R-binding domain, and one may therefore speculate that the encoded protein has different functions than does the full-length isoform. When cDNA from 83 healthy individuals and 30 SLE patients were analysed, the �2 isoform was present in every sample. Moreover, cDNA from chimpanzee and mouse spleen also contained this isoform, suggesting conservation across species.
Evaluation of known polymorphisms in the gene identified a SNP located in a potential branch-point site, which could affect the splicing of exon 2. This SNP, rs17266594, was in strong (although not complete) LD with the previously associated rs10516487. Association between rs17266594 and SLE was detected in all five cohorts (Table 7).
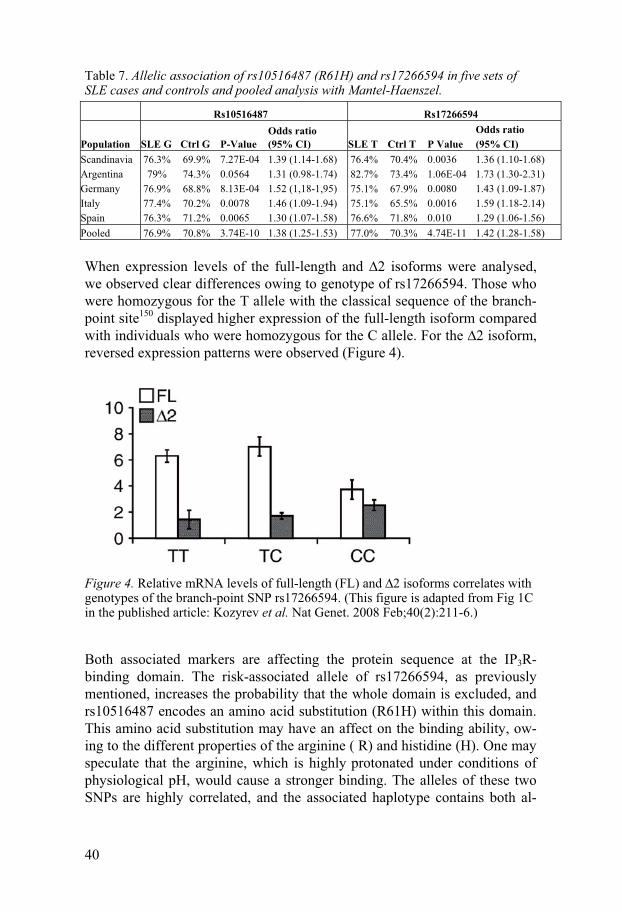
40
Table 7. Allelic association of rs10516487 (R61H) and rs17266594 in five sets of SLE cases and controls and pooled analysis with Mantel-Haenszel.
Rs10516487 Rs17266594
Population SLE G Ctrl G P-Value Odds ratio (95% CI) SLE T Ctrl T P Value
Odds ratio (95% CI)
Scandinavia 76.3% 69.9% 7.27E-04 1.39 (1.14-1.68) 76.4% 70.4% 0.0036 1.36 (1.10-1.68) Argentina 79% 74.3% 0.0564 1.31 (0.98-1.74) 82.7% 73.4% 1.06E-04 1.73 (1.30-2.31) Germany 76.9% 68.8% 8.13E-04 1.52 (1,18-1,95) 75.1% 67.9% 0.0080 1.43 (1.09-1.87) Italy 77.4% 70.2% 0.0078 1.46 (1.09-1.94) 75.1% 65.5% 0.0016 1.59 (1.18-2.14) Spain 76.3% 71.2% 0.0065 1.30 (1.07-1.58) 76.6% 71.8% 0.010 1.29 (1.06-1.56) Pooled 76.9% 70.8% 3.74E-10 1.38 (1.25-1.53) 77.0% 70.3% 4.74E-11 1.42 (1.28-1.58)
When expression levels of the full-length and �2 isoforms were analysed, we observed clear differences owing to genotype of rs17266594. Those who were homozygous for the T allele with the classical sequence of the branch-point site150 displayed higher expression of the full-length isoform compared with individuals who were homozygous for the C allele. For the �2 isoform, reversed expression patterns were observed (Figure 4).
Figure 4. Relative mRNA levels of full-length (FL) and �2 isoforms correlates with genotypes of the branch-point SNP rs17266594. (This figure is adapted from Fig 1C in the published article: Kozyrev et al. Nat Genet. 2008 Feb;40(2):211-6.)
Both associated markers are affecting the protein sequence at the IP3R-binding domain. The risk-associated allele of rs17266594, as previously mentioned, increases the probability that the whole domain is excluded, and rs10516487 encodes an amino acid substitution (R61H) within this domain. This amino acid substitution may have an affect on the binding ability, ow-ing to the different properties of the arginine ( R) and histidine (H). One may speculate that the arginine, which is highly protonated under conditions of physiological pH, would cause a stronger binding. The alleles of these two SNPs are highly correlated, and the associated haplotype contains both al-

41
leles that are hypothesised to generate a BANK1 protein with higher capac-ity to interact with the IP3R.
Association with the SNP rs3733197, causing an alanine to threonine sub-stitution (A383T) in the ankyrin domain, was also detected in our datasets. The potential effect of alterations in ankyrin repeats were recently high-lighted151, which makes this an additional potentially functional variant. However, the association with this SNP was distinctively weaker than with the other two in our datasets, which indicates that such an effect would be a minor contributor to the association of this gene.
In conclusion, these findings implicate BANK1 as a susceptibility gene for
SLE, with variants affecting functional domains. The disease-associated variants could contribute to hyperactive B cells with sustained reactions, which is characteristic in SLE. Previously published studies have reported that over-expression of BANK1 increases IP3R signalling148. A seemingly disparate observation was that a knock-out mouse for the BANK1 gene showed increased antibody secretion and germinal centre formation152. The contradictions of these results may be owing to the differences in experimen-tal design and model systems used. Furthermore and importantly, the BANK1 gene was knocked-out by interruption of exon 2, which leaves the possibility that the Δ2-isoform was still expressed. If the full-length and �2 isoforms have distinct functions, this could explain the apparent contradic-tions in the previous studies. Unpublished data indicates that the two iso-forms may indeed have different roles in B cell activation153.
Paper IV: STAT4 associates with SLE through two independent effects that correlate with gene expression and act additively with IRF5 to increase risk
Background
The Signal Transducer and Activator of Transcription 4 (STAT4) is a cyto-plasmic transcription factor involved in cytokine signal transduction. It is primarily activated by interleukin-12 (IL-12), and is an important factor in both innate and adaptive immune reactions. In the initial stages of an im-mune response, STAT4 causes natural killer (NK) cells and antigen-presenting cells to produce interferon-γ (IFN-γ), cytokines and chemokines that are critical in generating an inflammatory reaction108,154. It has been demonstrated that Stat4 knock-out mice have impaired IL-12 mediated func-tions, including Th1 response, IFN-γ production, cellular proliferation and NK cell cytotoxic activity155,156. Furthermore, in mouse models for several autoimmune diseases, mice that are deficient for STAT4 exhibit less in-

42
flammation and milder disease symptoms than wild-type mice157-160. Other studies observe that increased expression of IL-12 and IFN- γ in the kidneys of MRL-lpr/lpr mice precede the development of glomerulonephritis161,162.
We found association to SLE with SNPs in this gene when analysing a GWA scan of Argentinean patients and controls, and considered it a strong candidate due to its function in regulation of immune functions. This gene appears to have been discovered in parallel by several independent research groups, and yielded strong signals in all three GWAS for SLE published thus far, which speaks for a significant role in the risk of SLE28-30,106,107. The asso-ciation of STAT4 has also been found to be particularly strong with severe manifestations of SLE, especially nephritis163.
Results and discussion
Fine-mapping and replication After detecting association with STAT4 in our Argentinean cohort, and veri-fying this association in other populations, we proceeded with fine-mapping of the genetic region in a set of Spanish SLE cases and controls. This cohort is considerably larger than the Argentinean and therefore has a higher power to detect association. Since the STAT4 gene is located proximal to the STAT1 gene, which also has important functions in the immune system, we decided to include both genes as well as the intergenic region in the fine-mapping. We selected 29 tag SNPs in the STAT1-STAT4 region, and during the analy-sis of these SNPs, the article by Remmers et al was published, announcing strong evidence for association with rs7574865 in their datasets. We then chose to include this SNP in our fine-mapping.
The strongest association in our Spanish cohort was observed with SNPs rs3821236 and rs3024866, with rs7574865 in third place, followed by rs1467199 (Table 8). These four SNPs are spread across the major part of the STAT4 gene, with rs1467199 in the intergenic region between STAT1 and STAT4, rs7574865 in intron 3, and rs3024866 and rs3821236 (both in the same LD block) in introns 12 and 16, respectively. Of the six LD blocks in the STAT1-STAT4 region, we observe association in the four blocks covering STAT4 and the intergenic region, but find no association in STAT1 blocks. These results differ from the original study, where the association mainly was restricted to intron 3106. A second, recently published fine-mapping study, also detected association throughout most of the STAT4 gene, al-though it was concluded that the association could be explained by the intron 3 SNPs alone107.
The four strongest SNPs from our fine-mapping study were then geno-typed in independent sets of cases and controls from Germany, Italy, Argen-tina and two sets from Mexico (adult and paediatric). In the German cohort, only a weak borderline association with rs7574865 was observed (Table 8).

43
In the Italian and Mexican adult cohorts, we observed strong association with rs7574865, as well as association with rs3821236 and rs3024866. In the Argentinean set, the strongest association was observed with rs3821236, but there was also strong association with rs7574865 and rs3024866, which is similar to the results from the Spanish cohort (Table 8). These results indi-cate that the STAT4 gene may contain other effects independent of the previ-ously reported rs7574865.
Two independent effects Rs3821236 and rs3024866 are located in the same LD block, although not
in complete LD (r2=0.64). They are both poorly correlated with rs7574865 (r2 is 0.42 and 0.29, respectively). The correlation of rs1467199 with rs7574865 is also low (r2=0.30). Conditional logistic regression analysis indicated that there are two independent risk effects in the STAT4 gene, rep-resented by rs7574865 and rs3821236: when conditioning on rs7574865, rs3821236 remains significant, and vice versa. Analysis by PLINK164 also showed that rs7574865 and the haplotype tagged by rs3821236 represent two independent associations. A joint conclusion of these analyses would be that there are at least two independent risk variants in the STAT4 gene, repre-sented by rs7574865 and rs3821236 (or rs3024866). Association with rs1467199, which was found in the Spanish and the Mexican paediatric co-horts, could possibly represent a third risk factor, however, less convincing than the other two.
Population differences and meta-analysis The results from our replication studies indicate that there may be popula-
tion differences in susceptibility alleles for SLE in the STAT4 gene. It is not unlikely that a certain degree of allelic heterogeneity exists, where various risk alleles have different effects in different populations. Two other studies of STAT4 and SLE have found association with SNPs in intron 3 but no strong signals from other genetic regions in individuals of Northern Euro-pean descent106,107. This is similar to what we observe in our German cohort. However, the results from our study indicate that additional risk factors may be involved in populations from Southern Europe and Latin America.
Homogeneity analysis revealed borderline heterogeneity between the strata (p values ranging between 0.013 and 0.138). Thus, for a meta-analysis, it is uncertain if a fixed effects model (for homogeneous strata) or random effects (for heterogeneous strata) should be used. We therefore apply both methods for meta-analysis for all for markers (Table 8). The Mexican paedi-atric set only showed association for rs1467199, and was significantly het-erogeneous from the other cohorts for all four SNPs. We therefore excluded this set from the analysis. It is not unlikely that child-onset SLE has, at least partly, different mechanisms than adult-onset SLE.
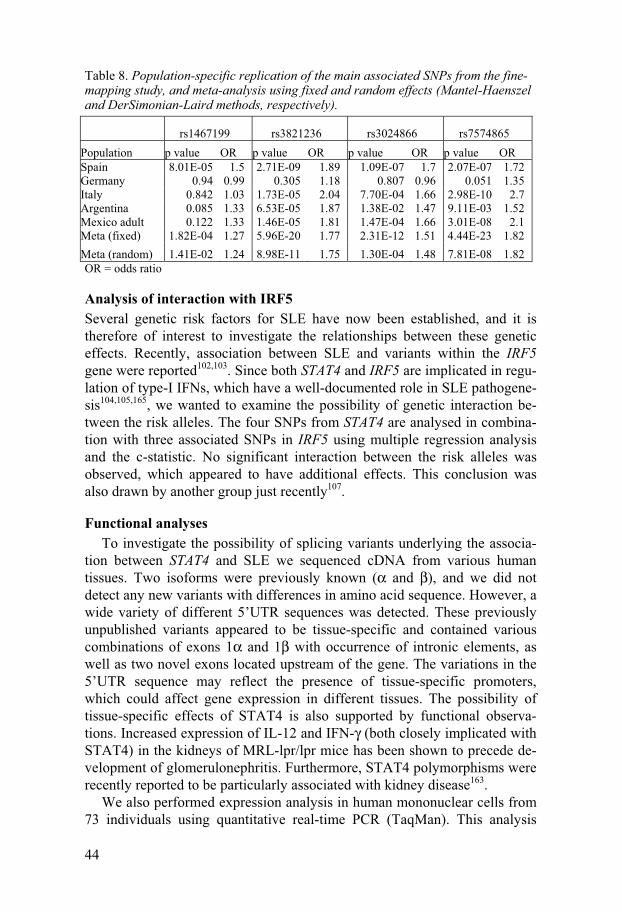
44
Table 8. Population-specific replication of the main associated SNPs from the fine-mapping study, and meta-analysis using fixed and random effects (Mantel-Haenszel and DerSimonian-Laird methods, respectively).
rs1467199 rs3821236 rs3024866 rs7574865
Population p value OR p value OR p value OR p value OR Spain 8.01E-05 1.5 2.71E-09 1.89 1.09E-07 1.7 2.07E-07 1.72 Germany 0.94 0.99 0.305 1.18 0.807 0.96 0.051 1.35 Italy 0.842 1.03 1.73E-05 2.04 7.70E-04 1.66 2.98E-10 2.7 Argentina 0.085 1.33 6.53E-05 1.87 1.38E-02 1.47 9.11E-03 1.52 Mexico adult 0.122 1.33 1.46E-05 1.81 1.47E-04 1.66 3.01E-08 2.1 Meta (fixed) 1.82E-04 1.27 5.96E-20 1.77 2.31E-12 1.51 4.44E-23 1.82
Meta (random) 1.41E-02 1.24 8.98E-11 1.75 1.30E-04 1.48 7.81E-08 1.82 OR = odds ratio
Analysis of interaction with IRF5 Several genetic risk factors for SLE have now been established, and it is therefore of interest to investigate the relationships between these genetic effects. Recently, association between SLE and variants within the IRF5 gene were reported102,103. Since both STAT4 and IRF5 are implicated in regu-lation of type-I IFNs, which have a well-documented role in SLE pathogene-sis104,105,165, we wanted to examine the possibility of genetic interaction be-tween the risk alleles. The four SNPs from STAT4 are analysed in combina-tion with three associated SNPs in IRF5 using multiple regression analysis and the c-statistic. No significant interaction between the risk alleles was observed, which appeared to have additional effects. This conclusion was also drawn by another group just recently107.
Functional analyses To investigate the possibility of splicing variants underlying the associa-
tion between STAT4 and SLE we sequenced cDNA from various human tissues. Two isoforms were previously known (α and β), and we did not detect any new variants with differences in amino acid sequence. However, a wide variety of different 5’UTR sequences was detected. These previously unpublished variants appeared to be tissue-specific and contained various combinations of exons 1α and 1β with occurrence of intronic elements, as well as two novel exons located upstream of the gene. The variations in the 5’UTR sequence may reflect the presence of tissue-specific promoters, which could affect gene expression in different tissues. The possibility of tissue-specific effects of STAT4 is also supported by functional observa-tions. Increased expression of IL-12 and IFN-γ (both closely implicated with STAT4) in the kidneys of MRL-lpr/lpr mice has been shown to precede de-velopment of glomerulonephritis. Furthermore, STAT4 polymorphisms were recently reported to be particularly associated with kidney disease163.
We also performed expression analysis in human mononuclear cells from 73 individuals using quantitative real-time PCR (TaqMan). This analysis

45
indicated that homozygosity for the risk allele of rs3024866 was associated with a modestly elevated increase in expression levels of the α isoform. Similar correlations were also observed for rs3821236 and rs7574865, but not for rs1467199. The β isoform followed the same pattern as α isoform, although with much lower levels.
Conclusions This study confirms the association between STAT4 and SLE. In addition
to the previously reported association of rs7574865 in intron 3106, we iden-tify at least one independent risk factor represented by SNPs rs3024866 and rs3821236 located in introns 12 and 16, respectively. This effect appears to be the strongest in populations of southern European or Latin American ori-gin. Correlation is also observed between moderately elevated expression of STAT4 and risk alleles of rs3024866, rs3821236 and rs7574865. However, the best correlation is not found with the most strongly associated SNP, which indicates that the matter will need further investigations. Furthermore, if two independent genetic effects are observed, these are likely to reflect the presence of more than one functional variant.
Identifying the functional variants responsible for the association of STAT4 may prove difficult considering the size of the gene and the potential presence of several effects. However, the overwhelming evidence for a role in SLE from several large association studies indicates that it may be worth the extra effort.

46
Concluding remarks
SLE is a complex disease with substantial heterogeneity regarding both symptoms and aetiology. Until quite recently, there were relatively few breakthroughs in the genetics of SLE and other complex diseases. However, the last few years have brought highly convincing evidence for a number of risk genes, much thanks to the combination of technical advancements and large collaborations, enabling comprehensive association studies of large cohorts. If the launch of GWA studies brought a sudden boost, the speed of detecting new disease variants will most probably decelerate in the future. On the other hand, the future may bring new technical inventions, which create opportunities to detect similar amounts of information regarding the underlying mechanisms of SLE.
In this thesis, five candidate genes for SLE are analysed. Two of these genes, PD-L1 and PD-L2, appear not to contain any major risk factors for SLE in the analysed European and South American populations. In two other genes, CD24 and STAT4, there appears to be population-specific effects. The A57V amino acid substitution in the CD24 antigen, previously implicated with MS, was associated in a Spanish cohort, with a weak trend in German samples, and no association in Swedish. The previously reported and highly convincing association of the STAT4 transcription factor was confirmed in all our cohorts. Interestingly, the results indicate the presence of at least two independent risk variants: the first, represented by a previously reported SNP, was the strongest in individuals of Northern European ancestry, and the second was more pronounced in individuals from Southern Europe and Latin America. We also report the identification of a novel susceptibility gene, BANK1. This gene encodes a scaffold protein involved in B-cell acti-vation, and contains functional variants affecting important domains, which are associated in all investigated cohorts from Europe and Latin America. The results of these studies confirm the existence of replicable associations between genetic variants and SLE, which are common and present in many populations. The results also illustrate a certain degree of heterogeneity, where some risk factors could have variable effect in different populations.
The identification of genetic variants associated with SLE is a first step in obtaining knowledge regarding the causes of the disease. It will now be of high interest to search for functional explanations to the increase in suscepti-bility. For several associated genes, some clues have been identified to how the genetic risk variants affect protein function and expression. However,

47
much remains to be unravelled regarding these and other associated risk variants and the effect in their pathways. The identification of causative variants and their downstream consequences will be a major challenge in the field of complex disease genetics. Nevertheless, such knowledge may be of great value for developing new tools for diagnosis and treatment.

48
Acknowledgements
I would like to express my sincere gratitude to the following people, without whom this thesis would not have seen the light of day. I would especially like to thank:
My supervisor, Marta Alarcón-Riquelme, for convincing me to start my
graduate studies, for your admirable level of enthusiasm and energy, and for always being available for discussions, local or trans-Atlantic, and regarding small or big issues.
My co-supervisor, Ulf Gyllensten, for valuable discussions and for creat-
ing a stimulating research environment in the ‘PCR group’ and the Rudbeck laboratory.
All the past and present members of the SLE group: Cissi, who almost
deserves an acknowledgement page of her own for all her valuable input, especially regarding the exhausting story of the ligands, and for being a great desk neighbour and fika friend. Veronica, for introducing me to the field of SLE genetics back in my undergraduate days. Bobo and Ludmila, our NIH stars, for your inspiring passion for science. Sergey, for excellent work on ‘functional stuff’ on both paper III and IV. Casimiro, for asking all those tricky questions and for entertaining the office with exotic Spanish expres-sions. Hong, for all the work with DNA samples, and for keeping me com-pany during odd hours at work and at the gym. Prasad, for giving me the BANK1 project when you were too busy with IRF5, for excellent Indian cooking, and for being such a nice guy and true gentleman. Angélica, my fellow TaqWoman, for great work on paper IV, for good comments on this thesis, and for all the discussions regarding statistical methods, Colombian practical jokes etc. Ammar, for discovering the �2 isoform, and for all the nice talks at lunch and fika. Juan, for keeping an eye on the lab during the night shift and for your great smile. Sara, for organising the weekly group fika. Helga, Eduardo and Bryndis, for founding a strong Icelandic branch of the group. Serena, for taking good care of my desk when I was away, and for your great smile and Italian determination. And finally, all the other nice and enthusiastic students through the years, especially Madhu, Sandra, Susanna, Jonas and David.

49
The rest of the ‘PCR group’, for making meetings and lunch breaks fun and/or interesting: Marie Allen’s forensic girls Hanna x2, Martina (CSI Stockholm), Anna-Maria, and all their nice students. Ulf G’s group, includ-ing Emma, Malin, Anna W, Jessica, Inger G, Max, Åsa, Anna B and Veronika V. The former members Lucia and Marie S, and all the new members like Tobias et al.
Genomcentret, especially Inger J, Jenny, Charlotte, Ann-Sofi and
Katarina for help with sequencing, for letting me borrow your machines etc and for fun lunch and coffee breaks.
All the people that make things work at work. Especially Gunilla Å,
Frida, Ulla, Lena, and the other administrators, and the computer guys Per-Ivan (PIW) and Viktor.
All other people on the 2nd and 3rd floor of the Rudbeck Laboratory for making it such a nice working place. Ulf P’s group: Lioudmila, Sara, Mårten and the rest. The Lannfelt group, especially Anna, Hille, and Char-lotte. Niklas Dahl’s group, especially Anne-Sophie, Miriam, Malin, Larry and Johanna, and all the other nice people with whom I’ve shared many ‘close’ moments in the not-so-spacious kitchen… Maja, Mehdi, my låtsast-villing Jenny (thanks for help with spiking-issues), my teaching colleagues Caroline and Chatarina…
Also, my sincere thanks to all collaborators at research centres and clin-
ics, and all participating patients. Jag vill också rikta ett stort och hjärtligt tack till alla för mig viktiga per-
soner i världen utanför Rudbeck: Världens bästa mamma och pappa, tack för att ni trott på mig och stöttat
mig i allt! Världens bästa och bussigaste syster Stina, som till och med kom och städade mina badrum när jag inte hann med! Och alla trevliga och roliga släktingar och ingifta familjemedlemmar, som sällan missar ett stort kalas, även om det är på andra sidan Sverige.
Stort tack också till alla de vänner som bidragit till att göra studietiden så
rolig. Världens bästa Sofie & Liv som hängt med hela livet, känns det som. Alla trevliga kursare på biologprogrammet, särskilt Liv J. Världens kanske

50
näst bästa studentorkester Glasblåsarna, med Lotten, Petter, Sanna, Lise… och alla andra roliga och svårt musikaliska personer. Äsch, ni vet vilka ni är allihop. Och tack till Jenny, Linda och Karin i Barnvagnsmaffi-an för att ni sett till att jag fått både schysst motion och en jämn koffeinnivå hela våren. Sist och såklart störst tack till min familj. Världens bästa Klas, som varit världens bästa hemmafru hemmaman under den här intensiva avhandlings-skrivar-, husköpar- och hussäljarhösten. Tack för allt ditt stöd, för alla ca-shewnötter och vaknätter, och för allt kul vi haft. Och tack mina älskade Alva och Stig för att ni alltid påminner mig om vad som är viktigt i livet. Om jag utelämnat någon viktigt person ber jag allra ödmjukast om ursäkt, och ber denne fylla i nödvändig kompletterande information här nedan.
I especially wish to thank ……………………………………………… (your name here)
for (please select) � all your help
� giving me inspiration � being such a nice person � all the fun times we’ve had � ………………………….. � all of the above

51
References
1. Druery, C.T. & Bateson, W. Experiments in plant hybridization.
Journal of the Royal Horticultural Society 26, 1-32 (1901). 2. Mendel, J.G. Versuche über Pflanzenhybriden. Verhandlungen des
naturforschenden Vereines in Brünn, Bd. IV für das Jahr 1865 (1866).
3. Watson, J.D. & Crick, F.H. Molecular structure of nucleic acids; a structure for deoxyribose nucleic acid. Nature 171, 737-8 (1953).
4. Lander, E.S. et al. Initial sequencing and analysis of the human ge-nome. Nature 409, 860-921 (2001).
5. Venter, J.C. et al. The sequence of the human genome. Science 291, 1304-51 (2001).
6. The International HapMap Project. Nature 426, 789-96 (2003). 7. Kruglyak, L. & Nickerson, D.A. Variation is the spice of life. Nat
Genet 27, 234-6 (2001). 8. Mills, R.E. et al. An initial map of insertion and deletion (INDEL)
variation in the human genome. Genome Res 16, 1182-90 (2006). 9. Feuk, L., Carson, A.R. & Scherer, S.W. Structural variation in the
human genome. Nat Rev Genet 7, 85-97 (2006). 10. Redon, R. et al. Global variation in copy number in the human ge-
nome. Nature 444, 444-54 (2006). 11. Iafrate, A.J. et al. Detection of large-scale variation in the human
genome. Nat Genet 36, 949-51 (2004). 12. Sebat, J. et al. Large-scale copy number polymorphism in the human
genome. Science 305, 525-8 (2004). 13. Iafrate, A. et al. Database of Genomic Variants,
http://projects.tcag.ca/variation/. (2008). 14. Aitman, T.J. et al. Copy number polymorphism in Fcgr3 predisposes
to glomerulonephritis in rats and humans. Nature 439, 851-5 (2006). 15. Fanciulli, M. et al. FCGR3B copy number variation is associated
with susceptibility to systemic, but not organ-specific, autoimmu-nity. Nat Genet 39, 721-3 (2007).
16. Gonzalez, E. et al. The influence of CCL3L1 gene-containing seg-mental duplications on HIV-1/AIDS susceptibility. Science 307, 1434-40 (2005).

52
17. McKinney, C. et al. Evidence for an influence of chemokine ligand 3-like 1 (CCL3L1) gene copy number on susceptibility to rheuma-toid arthritis. Ann Rheum Dis 67, 409-13 (2008).
18. Yang, Y. et al. Gene copy-number variation and associated poly-morphisms of complement component C4 in human systemic lupus erythematosus (SLE): low copy number is a risk factor for and high copy number is a protective factor against SLE susceptibility in Eu-ropean Americans. Am J Hum Genet 80, 1037-54 (2007).
19. Taylor, R.W. & Turnbull, D.M. Mitochondrial DNA mutations in human disease. Nat Rev Genet 6, 389-402 (2005).
20. Lander, E. & Kruglyak, L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet 11, 241-7 (1995).
21. Risch, N. & Merikangas, K. The future of genetic studies of com-plex human diseases. Science 273, 1516-7 (1996).
22. Altmuller, J., Palmer, L.J., Fischer, G., Scherb, H. & Wjst, M. Ge-nomewide scans of complex human diseases: true linkage is hard to find. Am J Hum Genet 69, 936-50 (2001).
23. Mueller, J.C. Linkage disequilibrium for different scales and appli-cations. Brief Bioinform 5, 355-64 (2004).
24. Gabriel, S.B. et al. The structure of haplotype blocks in the human genome. Science 296, 2225-9 (2002).
25. Terwilliger, J.D. & Hiekkalinna, T. An utter refutation of the "Fun-damental Theorem of the HapMap". Eur J Hum Genet 14, 426-37 (2006).
26. Lander, E.S. The new genomics: global views of biology. Science 274, 536-9 (1996).
27. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447, 661-78 (2007).
28. Graham, R.R. et al. Genetic variants near TNFAIP3 on 6q23 are associated with systemic lupus erythematosus. Nat Genet (2008).
29. Harley, J.B. et al. Genome-wide association scan in women with systemic lupus erythematosus identifies susceptibility variants in ITGAM, PXK, KIAA1542 and other loci. Nat Genet 40, 204-10 (2008).
30. Hom, G. et al. Association of systemic lupus erythematosus with C8orf13-BLK and ITGAM-ITGAX. N Engl J Med 358, 900-9 (2008).
31. Frazer, K.A. et al. A second generation human haplotype map of over 3.1 million SNPs. Nature 449, 851-61 (2007).
32. Hirschhorn, J.N. & Daly, M.J. Genome-wide association studies for common diseases and complex traits. Nat Rev Genet 6, 95-108 (2005).
33. Wang, W.Y., Barratt, B.J., Clayton, D.G. & Todd, J.A. Genome-wide association studies: theoretical and practical concerns. Nat Rev Genet 6, 109-18 (2005).

53
34. Ge, D. et al. WGAViewer: software for genomic annotation of whole genome association studies. Genome Res 18, 640-3 (2008).
35. Sherer, Y., Gorstein, A., Fritzler, M.J. & Shoenfeld, Y. Autoanti-body explosion in systemic lupus erythematosus: more than 100 different antibodies found in SLE patients. Semin Arthritis Rheum 34, 501-37 (2004).
36. Hochberg, M.C. Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythemato-sus. Arthritis Rheum 40, 1725 (1997).
37. Tan, E.M. et al. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum 25, 1271-7 (1982).
38. Cervera, R. et al. Systemic lupus erythematosus in Europe at the change of the millennium: lessons from the "Euro-Lupus Project". Autoimmun Rev 5, 180-6 (2006).
39. Danchenko, N., Satia, J.A. & Anthony, M.S. Epidemiology of sys-temic lupus erythematosus: a comparison of worldwide disease bur-den. Lupus 15, 308-18 (2006).
40. Jimenez, S., Cervera, R., Font, J. & Ingelmo, M. The epidemiology of systemic lupus erythematosus. Clin Rev Allergy Immunol 25, 3-12 (2003).
41. Petri, M. Epidemiology of systemic lupus erythematosus. Best Pract Res Clin Rheumatol 16, 847-58 (2002).
42. Morgan, B.P. & Walport, M.J. Complement deficiency and disease. Immunol Today 12, 301-6 (1991).
43. Lee-Kirsch, M.A. et al. Mutations in the gene encoding the 3'-5' DNA exonuclease TREX1 are associated with systemic lupus ery-thematosus. Nat Genet 39, 1065-7 (2007).
44. Alarcon-Segovia, D. et al. Familial aggregation of systemic lupus erythematosus, rheumatoid arthritis, and other autoimmune diseases in 1,177 lupus patients from the GLADEL cohort. Arthritis Rheum 52, 1138-47 (2005).
45. Hochberg, M.C. The application of genetic epidemiology to sys-temic lupus erythematosus. J Rheumatol 14, 867-9 (1987).
46. Block, S.R. Twin studies: genetic factors are important. Arthritis Rheum 36, 135-6 (1993).
47. Deapen, D. et al. A revised estimate of twin concordance in systemic lupus erythematosus. Arthritis Rheum 35, 311-8 (1992).
48. Reichlin, M., Harley, J.B. & Lockshin, M.D. Serologic studies of monozygotic twins with systemic lupus erythematosus. Arthritis Rheum 35, 457-64 (1992).
49. Zhou, Y. & Lu, Q. DNA methylation in T cells from idiopathic lu-pus and drug-induced lupus patients. Autoimmun Rev 7, 376-83 (2008).
50. Kaufman, K.M., Kirby, M.Y., Harley, J.B. & James, J.A. Peptide mimics of a major lupus epitope of SmB/B'. Ann N Y Acad Sci 987, 215-29 (2003).

54
51. McClain, M.T. et al. Early events in lupus humoral autoimmunity suggest initiation through molecular mimicry. Nat Med 11, 85-9 (2005).
52. Pender, M.P. Infection of autoreactive B lymphocytes with EBV, causing chronic autoimmune diseases. Trends Immunol 24, 584-8 (2003).
53. Harley, J.B., Harley, I.T., Guthridge, J.M. & James, J.A. The curi-ously suspicious: a role for Epstein-Barr virus in lupus. Lupus 15, 768-77 (2006).
54. Sarzi-Puttini, P., Atzeni, F., Iaccarino, L. & Doria, A. Environment and systemic lupus erythematosus: an overview. Autoimmunity 38, 465-72 (2005).
55. Borchers, A.T., Keen, C.L. & Gershwin, M.E. Drug-induced lupus. Ann N Y Acad Sci 1108, 166-82 (2007).
56. Parks, C.G. & Cooper, G.S. Occupational exposures and risk of sys-temic lupus erythematosus: a review of the evidence and exposure assessment methods in population- and clinic-based studies. Lupus 15, 728-36 (2006).
57. Prete, P.E. The mechanism of action of L-canavanine in inducing autoimmune phenomena. Arthritis Rheum 28, 1198-200 (1985).
58. Costenbader, K.H. & Karlson, E.W. Cigarette smoking and systemic lupus erythematosus: a smoking gun? Autoimmunity 38, 541-7 (2005).
59. Bynoe, M.S., Grimaldi, C.M. & Diamond, B. Estrogen up-regulates Bcl-2 and blocks tolerance induction of naive B cells. Proc Natl Acad Sci U S A 97, 2703-8 (2000).
60. Grimaldi, C.M. Sex and systemic lupus erythematosus: the role of the sex hormones estrogen and prolactin on the regulation of autore-active B cells. Curr Opin Rheumatol 18, 456-61 (2006).
61. Grimaldi, C.M., Cleary, J., Dagtas, A.S., Moussai, D. & Diamond, B. Estrogen alters thresholds for B cell apoptosis and activation. J Clin Invest 109, 1625-33 (2002).
62. Grimaldi, C.M., Jeganathan, V. & Diamond, B. Hormonal regulation of B cell development: 17 beta-estradiol impairs negative selection of high-affinity DNA-reactive B cells at more than one developmen-tal checkpoint. J Immunol 176, 2703-10 (2006).
63. Peeva, E., Venkatesh, J. & Diamond, B. Tamoxifen blocks estrogen-induced B cell maturation but not survival. J Immunol 175, 1415-23 (2005).
64. Cooper, G.S., Dooley, M.A., Treadwell, E.L., St Clair, E.W. & Gil-keson, G.S. Hormonal and reproductive risk factors for development of systemic lupus erythematosus: results of a population-based, case-control study. Arthritis Rheum 46, 1830-9 (2002).
65. Costenbader, K.H., Feskanich, D., Stampfer, M.J. & Karlson, E.W. Reproductive and menopausal factors and risk of systemic lupus ery-thematosus in women. Arthritis Rheum 56, 1251-62 (2007).

55
66. Sanchez-Guerrero, J. et al. Past use of oral contraceptives and the risk of developing systemic lupus erythematosus. Arthritis Rheum 40, 804-8 (1997).
67. Clowse, M.E., Magder, L.S., Witter, F. & Petri, M. The impact of increased lupus activity on obstetric outcomes. Arthritis Rheum 52, 514-21 (2005).
68. Petri, M., Howard, D. & Repke, J. Frequency of lupus flare in preg-nancy. The Hopkins Lupus Pregnancy Center experience. Arthritis Rheum 34, 1538-45 (1991).
69. Lockshin, M.D. Pregnancy does not cause systemic lupus erythema-tosus to worsen. Arthritis Rheum 32, 665-70 (1989).
70. Meehan, R.T. & Dorsey, J.K. Pregnancy among patients with sys-temic lupus erythematosus receiving immunosuppressive therapy. J Rheumatol 14, 252-8 (1987).
71. Lahita, R.G., Bradlow, H.L., Kunkel, H.G. & Fishman, J. Altera-tions of estrogen metabolism in systemic lupus erythematosus. Ar-thritis Rheum 22, 1195-8 (1979).
72. Scofield, R.H. et al. Klinefelter's syndrome (47,XXY) in male sys-temic lupus erythematosus patients: support for the notion of a gene-dose effect from the X chromosome. Arthritis Rheum 58, 2511-7 (2008).
73. James, J.A., Gross, T., Scofield, R.H. & Harley, J.B. Immunoglobu-lin epitope spreading and autoimmune disease after peptide immuni-zation: Sm B/B'-derived PPPGMRPP and PPPGIRGP induce spli-ceosome autoimmunity. J Exp Med 181, 453-61 (1995).
74. Manderson, A.P., Botto, M. & Walport, M.J. The role of comple-ment in the development of systemic lupus erythematosus. Annu Rev Immunol 22, 431-56 (2004).
75. Cantor, R.M. et al. Systemic lupus erythematosus genome scan: support for linkage at 1q23, 2q33, 16q12-13, and 17q21-23 and nov-el evidence at 3p24, 10q23-24, 13q32, and 18q22-23. Arthritis Rheum 50, 3203-10 (2004).
76. Gaffney, P.M. et al. A genome-wide search for susceptibility genes in human systemic lupus erythematosus sib-pair families. Proc Natl Acad Sci U S A 95, 14875-9 (1998).
77. Gaffney, P.M. et al. Genome screening in human systemic lupus erythematosus: results from a second Minnesota cohort and com-bined analyses of 187 sib-pair families. Am J Hum Genet 66, 547-56 (2000).
78. Gray-McGuire, C. et al. Genome scan of human systemic lupus ery-thematosus by regression modeling: evidence of linkage and epista-sis at 4p16-15.2. Am J Hum Genet 67, 1460-9 (2000).
79. Johansson, C.M. et al. Chromosome 17p12-q11 harbors susceptibil-ity loci for systemic lupus erythematosus. Hum Genet 115, 230-8 (2004).

56
80. Koskenmies, S. et al. Linkage mapping of systemic lupus erythema-tosus (SLE) in Finnish families multiply affected by SLE. J Med Genet 41, e2-5 (2004).
81. Lindqvist, A.K. et al. A susceptibility locus for human systemic lupus erythematosus (hSLE1) on chromosome 2q. J Autoimmun 14, 169-78 (2000).
82. Moser, K.L. et al. Genome scan of human systemic lupus erythema-tosus: evidence for linkage on chromosome 1q in African-American pedigrees. Proc Natl Acad Sci U S A 95, 14869-74 (1998).
83. Nath, S.K. et al. Linkage at 12q24 with systemic lupus erythemato-sus (SLE) is established and confirmed in Hispanic and European American families. Am J Hum Genet 74, 73-82 (2004).
84. Olson, J.M. et al. A genome screen of systemic lupus erythematosus using affected-relative-pair linkage analysis with covariates demon-strates genetic heterogeneity. Genes Immun 3 Suppl 1, S5-S12 (2002).
85. Shai, R. et al. Genome-wide screen for systemic lupus erythemato-sus susceptibility genes in multiplex families. Hum Mol Genet 8, 639-44 (1999).
86. Lee, Y.H. & Nath, S.K. Systemic lupus erythematosus susceptibility loci defined by genome scan meta-analysis. Hum Genet 118, 434-43 (2005).
87. Forabosco, P. et al. Meta-analysis of genome-wide linkage studies of systemic lupus erythematosus. Genes Immun 7, 609-14 (2006).
88. Vyse, T.J. & Kotzin, B.L. Genetic susceptibility to systemic lupus erythematosus. Annu Rev Immunol 16, 261-92 (1998).
89. Morel, L. et al. Genetic reconstitution of systemic lupus erythemato-sus immunopathology with polycongenic murine strains. Proc Natl Acad Sci U S A 97, 6670-5 (2000).
90. Brown, E.E., Edberg, J.C. & Kimberly, R.P. Fc receptor genes and the systemic lupus erythematosus diathesis. Autoimmunity 40, 567-81 (2007).
91. Prokunina, L. et al. A regulatory polymorphism in PDCD1 is associ-ated with susceptibility to systemic lupus erythematosus in humans. Nat Genet 32, 666-9 (2002).
92. Nath, S.K. et al. A nonsynonymous functional variant in integrin-alpha(M) (encoded by ITGAM) is associated with systemic lupus erythematosus. Nat Genet 40, 152-4 (2008).
93. Horton, R. et al. Gene map of the extended human MHC. Nat Rev Genet 5, 889-99 (2004).
94. Goldberg, M.A., Arnett, F.C., Bias, W.B. & Shulman, L.E. Histo-compatibility antigens in systemic lupus erythematosus. Arthritis Rheum 19, 129-32 (1976).
95. Fernando, M.M. et al. Defining the role of the MHC in autoimmu-nity: a review and pooled analysis. PLoS Genet 4, e1000024 (2008).

57
96. Graham, R.R. et al. Visualizing human leukocyte antigen class II risk haplotypes in human systemic lupus erythematosus. Am J Hum Genet 71, 543-53 (2002).
97. Tsao, B.P. Update on human systemic lupus erythematosus genetics. Curr Opin Rheumatol 16, 513-21 (2004).
98. Kim, T.G. et al. Systemic lupus erythematosus with nephritis is strongly associated with the TNFB*2 homozygote in the Korean population. Hum Immunol 46, 10-7 (1996).
99. Wilson, A.G. et al. A genetic association between systemic lupus erythematosus and tumor necrosis factor alpha. Eur J Immunol 24, 191-5 (1994).
100. Sjoholm, A.G., Jonsson, G., Braconier, J.H., Sturfelt, G. & Trueds-son, L. Complement deficiency and disease: an update. Mol Immu-nol 43, 78-85 (2006).
101. Yang, Y. et al. The intricate role of complement component C4 in human systemic lupus erythematosus. Curr Dir Autoimmun 7, 98-132 (2004).
102. Sigurdsson, S. et al. Polymorphisms in the tyrosine kinase 2 and interferon regulatory factor 5 genes are associated with systemic lu-pus erythematosus. Am J Hum Genet 76, 528-37 (2005).
103. Graham, R.R. et al. A common haplotype of interferon regulatory factor 5 (IRF5) regulates splicing and expression and is associated with increased risk of systemic lupus erythematosus. Nat Genet 38, 550-5 (2006).
104. Baechler, E.C. et al. Interferon-inducible gene expression signature in peripheral blood cells of patients with severe lupus. Proc Natl Acad Sci U S A 100, 2610-5 (2003).
105. Ronnblom, L.E., Alm, G.V. & Oberg, K. Autoimmune phenomena in patients with malignant carcinoid tumors during interferon-alpha treatment. Acta Oncol 30, 537-40 (1991).
106. Remmers, E.F. et al. STAT4 and the risk of rheumatoid arthritis and systemic lupus erythematosus. N Engl J Med 357, 977-86 (2007).
107. Sigurdsson, S. et al. A risk haplotype of STAT4 for systemic lupus erythematosus is over-expressed, correlates with anti-dsDNA and shows additive effects with two risk alleles of IRF5. Hum Mol Genet 17, 2868-76 (2008).
108. Kaplan, M.H. STAT4: a critical regulator of inflammation in vivo. Immunol Res 31, 231-42 (2005).
109. Fagerholm, S.C., Varis, M., Stefanidakis, M., Hilden, T.J. & Gahm-berg, C.G. alpha-Chain phosphorylation of the human leukocyte CD11b/CD18 (Mac-1) integrin is pivotal for integrin activation to bind ICAMs and leukocyte extravasation. Blood 108, 3379-86 (2006).
110. Dymecki, S.M., Zwollo, P., Zeller, K., Kuhajda, F.P. & Desiderio, S.V. Structure and developmental regulation of the B-lymphoid ty-rosine kinase gene blk. J Biol Chem 267, 4815-23 (1992).

58
111. Bottini, N. et al. A functional variant of lymphoid tyrosine phos-phatase is associated with type I diabetes. Nat Genet 36, 337-8 (2004).
112. Begovich, A.B. et al. A missense single-nucleotide polymorphism in a gene encoding a protein tyrosine phosphatase (PTPN22) is associ-ated with rheumatoid arthritis. Am J Hum Genet 75, 330-7 (2004).
113. Smyth, D. et al. Replication of an association between the lymphoid tyrosine phosphatase locus (LYP/PTPN22) with type 1 diabetes, and evidence for its role as a general autoimmunity locus. Diabetes 53, 3020-3 (2004).
114. Kyogoku, C. et al. Genetic association of the R620W polymorphism of protein tyrosine phosphatase PTPN22 with human SLE. Am J Hum Genet 75, 504-7 (2004).
115. Lee, Y.H. et al. The PTPN22 C1858T functional polymorphism and autoimmune diseases--a meta-analysis. Rheumatology (Oxford) 46, 49-56 (2007).
116. Graham, D.S. et al. Polymorphism at the TNF superfamily gene TNFSF4 confers susceptibility to systemic lupus erythematosus. Nat Genet 40, 83-9 (2008).
117. Gramaglia, I. et al. The OX40 costimulatory receptor determines the development of CD4 memory by regulating primary clonal expan-sion. J Immunol 165, 3043-50 (2000).
118. Ahmed, S. et al. Association of CTLA-4 but not CD28 gene poly-morphisms with systemic lupus erythematosus in the Japanese popu-lation. Rheumatology (Oxford) 40, 662-7 (2001).
119. Flores-Borja, F., Kabouridis, P.S., Jury, E.C., Isenberg, D.A. & Ma-geed, R.A. Decreased Lyn expression and translocation to lipid raft signaling domains in B lymphocytes from patients with systemic lu-pus erythematosus. Arthritis Rheum 52, 3955-65 (2005).
120. Monticielo, O.A., Mucenic, T., Xavier, R.M., Brenol, J.C. & Chies, J.A. The role of mannose-binding lectin in systemic lupus erythema-tosus. Clin Rheumatol 27, 413-9 (2008).
121. Beebe, A.M., Cua, D.J. & de Waal Malefyt, R. The role of inter-leukin-10 in autoimmune disease: systemic lupus erythematosus (SLE) and multiple sclerosis (MS). Cytokine Growth Factor Rev 13, 403-12 (2002).
122. Mehrian, R. et al. Synergistic effect between IL-10 and bcl-2 geno-types in determining susceptibility to systemic lupus erythematosus. Arthritis Rheum 41, 596-602 (1998).
123. Livak, K.J., Marmaro, J. & Todd, J.A. Towards fully automated genome-wide polymorphism screening. Nat Genet 9, 341-2 (1995).
124. Spielman, R.S., McGinnis, R.E. & Ewens, W.J. Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM). Am J Hum Genet 52, 506-16 (1993).

59
125. Schulze, T.G. & McMahon, F.J. Genetic association mapping at the crossroads: which test and why? Overview and practical guidelines. Am J Med Genet 114, 1-11 (2002).
126. Terwilliger, J.D. & Ott, J. A haplotype-based 'haplotype relative risk' approach to detecting allelic associations. Hum Hered 42, 337-46 (1992).
127. Laird, N.M., Horvath, S. & Xu, X. Implementing a unified approach to family-based tests of association. Genet Epidemiol 19 Suppl 1, S36-42 (2000).
128. Dudbridge, F. Pedigree disequilibrium tests for multilocus haplo-types. Genet Epidemiol 25, 115-21 (2003).
129. Balding, D.J. A tutorial on statistical methods for population asso-ciation studies. Nat Rev Genet 7, 781-91 (2006).
130. McCarthy, M.I. et al. Genome-wide association studies for complex traits: consensus, uncertainty and challenges. Nat Rev Genet 9, 356-69 (2008).
131. Keir, M.E., Butte, M.J., Freeman, G.J. & Sharpe, A.H. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol 26, 677-704 (2008).
132. Khlat, M., Cazes, M.H., Genin, E. & Guiguet, M. Robustness of case-control studies of genetic factors to population stratification: magnitude of bias and type I error. Cancer Epidemiol Biomarkers Prev 13, 1660-4 (2004).
133. Seldin, M.F. et al. Argentine population genetic structure: large vari-ance in Amerindian contribution. Am J Phys Anthropol 132, 455-62 (2007).
134. Wang, S.C. et al. Ligands for programmed cell death 1 gene in pa-tients with systemic lupus erythematosus. J Rheumatol 34, 721-5 (2007).
135. De Bruijn, M.L., Peterson, P.A. & Jackson, M.R. Induction of heat-stable antigen expression by phagocytosis is involved in in vitro ac-tivation of unprimed CTL by macrophages. J Immunol 156, 2686-92 (1996).
136. Hubbe, M. & Altevogt, P. Heat-stable antigen/CD24 on mouse T lymphocytes: evidence for a costimulatory function. Eur J Immunol 24, 731-7 (1994).
137. Liu, Y. et al. Heat-stable antigen is a costimulatory molecule for CD4 T cell growth. J Exp Med 175, 437-45 (1992).
138. Zhou, Q., Wu, Y., Nielsen, P.J. & Liu, Y. Homotypic interaction of the heat-stable antigen is not responsible for its co-stimulatory activ-ity for T cell clonal expansion. Eur J Immunol 27, 2524-8 (1997).
139. Lim, S.C. CD24 and human carcinoma: tumor biological aspects. Biomed Pharmacother 59 Suppl 2, S351-4 (2005).
140. Wu, Y., Zhou, Q., Zheng, P. & Liu, Y. CD28-independent induction of T helper cells and immunoglobulin class switches requires costi-mulation by the heat-stable antigen. J Exp Med 187, 1151-6 (1998).

60
141. Hahne, M., Wenger, R.H., Vestweber, D. & Nielsen, P.J. The heat-stable antigen can alter very late antigen 4-mediated adhesion. J Exp Med 179, 1391-5 (1994).
142. McMurray, R.W. Adhesion molecules in autoimmune disease. Se-min Arthritis Rheum 25, 215-33 (1996).
143. Zhou, Q. et al. CD24 is a genetic modifier for risk and progression of multiple sclerosis. Proc Natl Acad Sci U S A 100, 15041-6 (2003).
144. Goris, A. et al. CD24 Ala/Val polymorphism and multiple sclerosis. J Neuroimmunol 175, 200-2 (2006).
145. Otaegui, D. et al. CD24 V/V is an allele associated with the risk of developing multiple sclerosis in the Spanish population. Mult Scler 12, 511-4 (2006).
146. Ferreiros-Vidal, I. et al. Association of PDCD1 with susceptibility to systemic lupus erythematosus: evidence of population-specific ef-fects. Arthritis Rheum 50, 2590-7 (2004).
147. Wang, L. et al. A dinucleotide deletion in CD24 confers protection against autoimmune diseases. PLoS Genet 3, e49 (2007).
148. Yokoyama, K. et al. BANK regulates BCR-induced calcium mobili-zation by promoting tyrosine phosphorylation of IP(3) receptor. Em-bo J 21, 83-92 (2002).
149. Anolik, J., Sanz, I. & Looney, R.J. B cell depletion therapy in sys-temic lupus erythematosus. Curr Rheumatol Rep 5, 350-6 (2003).
150. Burge, C.B., Tuschl, T. & Sharp, P.A. in The RNA World II (eds. Gesteland, R.F., Cech, T.R. & Atkins, J.F.) 525–560 (Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, 1999).
151. Mohler, P.J. et al. Ankyrin-B mutation causes type 4 long-QT car-diac arrhythmia and sudden cardiac death. Nature 421, 634-9 (2003).
152. Aiba, Y. et al. BANK negatively regulates Akt activation and subse-quent B cell responses. Immunity 24, 259-68 (2006).
153. Castillejo-Lopez, C. Personal communication. (2008). 154. Wurster, A.L., Tanaka, T. & Grusby, M.J. The biology of Stat4 and
Stat6. Oncogene 19, 2577-84 (2000). 155. Kaplan, M.H., Sun, Y.L., Hoey, T. & Grusby, M.J. Impaired IL-12
responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature 382, 174-7 (1996).
156. Thierfelder, W.E. et al. Requirement for Stat4 in interleukin-12-mediated responses of natural killer and T cells. Nature 382, 171-4 (1996).
157. Chitnis, T. et al. Effect of targeted disruption of STAT4 and STAT6 on the induction of experimental autoimmune encephalomyelitis. J Clin Invest 108, 739-47 (2001).
158. Finnegan, A. et al. IL-4 and IL-12 regulate proteoglycan-induced arthritis through Stat-dependent mechanisms. J Immunol 169, 3345-52 (2002).
159. Simpson, S.J. et al. T cell-mediated pathology in two models of ex-perimental colitis depends predominantly on the interleukin

61
12/Signal transducer and activator of transcription (Stat)-4 pathway, but is not conditional on interferon gamma expression by T cells. J Exp Med 187, 1225-34 (1998).
160. Yang, Z. et al. Autoimmune diabetes is blocked in Stat4-deficient mice. J Autoimmun 22, 191-200 (2004).
161. Fan, X., Oertli, B. & Wuthrich, R.P. Up-regulation of tubular epithe-lial interleukin-12 in autoimmune MRL-Fas(lpr) mice with renal in-jury. Kidney Int 51, 79-86 (1997).
162. Schwarting, A. et al. IL-12 drives IFN-gamma-dependent autoim-mune kidney disease in MRL-Fas(lpr) mice. J Immunol 163, 6884-91 (1999).
163. Taylor, K.E. et al. Specificity of the STAT4 genetic association for severe disease manifestations of systemic lupus erythematosus. PLoS Genet 4, e1000084 (2008).
164. Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81, 559-75 (2007).
165. Ronnblom, L., Eloranta, M.L. & Alm, G.V. The type I interferon system in systemic lupus erythematosus. Arthritis Rheum 54, 408-20 (2006).

Acta Universitatis UpsaliensisDigital Comprehensive Summaries of Uppsala Dissertationsfrom the Faculty of Medicine 395
Editor: The Dean of the Faculty of Medicine
A doctoral dissertation from the Faculty of Medicine, UppsalaUniversity, is usually a summary of a number of papers. A fewcopies of the complete dissertation are kept at major Swedishresearch libraries, while the summary alone is distributedinternationally through the series Digital ComprehensiveSummaries of Uppsala Dissertations from the Faculty ofMedicine. (Prior to January, 2005, the series was publishedunder the title “Comprehensive Summaries of UppsalaDissertations from the Faculty of Medicine”.)
Distribution: publications.uu.seurn:nbn:se:uu:diva-9367
ACTAUNIVERSITATIS
UPSALIENSISUPPSALA
2008













