Embed Size (px)
Citation preview

Review article
n engl j med 364;20 nejm.org may 19, 20111932
From the Departments of Pediatrics (V.C.S.) and Ophthalmology and Visual Sciences (V.C.S., E.M.S.), Howard Hughes Medical Institute, University of Iowa Carver College of Medicine, Iowa City. Address reprint re-quests to Dr. Stone at the University of Iowa Institute for Vision Research, 375 Newton Rd., Iowa City, IA 52242, or at edwin-stone@ uiowa.edu.
This article (10.1056/NEJMra1012354) was updated on May 19, 2011, at NEJM.org.
N Engl J Med 2011;364:1932-42.Copyright © 2011 Massachusetts Medical Society.
Genomic MedicineW. Gregory Feero, M.D., Ph.D., and Alan E. Guttmacher, M.D., Editors
Genomics and the EyeVal C. Sheffield, M.D., Ph.D., and Edwin M. Stone, M.D., Ph.D.
The eye has had a pivotal role in the evolution of human genom-ics. At least 90% of the genes in the human genome are expressed in one or more of the eye’s many tissues and cell types at some point during a person’s
life. Consistent with this impressive genomic footprint is the observation that about a third of entries in the Online Mendelian Inheritance in Man database for which a clinical synopsis is provided include a term that refers to the structure or function of the eye.1 Moreover, the phenotypic effects of even small genetic variations are made readily apparent by the many layers of amplification in the human visual system. For example, a single-nucleotide change in PAX6 can cause an anatomic abnormality of the macula less than a millimeter in diameter that results in noticeably reduced vi-sual acuity and nystagmus.2
The heritable inability to correctly perceive the color green, known as Daltonism (after the English chemist John Dalton, who himself was affected), was the first hu-man trait mapped to the X chromosome.3 (See Fig. 1 for a timeline of historic dis-coveries.) The Coppock cataract was the first human trait mapped to an autosome,4 and Leber’s hereditary optic neuropathy was the first human disease shown to be caused by a mutation in mitochondrial DNA.5 More recently, age-related macular degeneration (AMD) and glaucoma6,7 — two common causes of human blindness — have been shown to be largely genetic, as has Fuchs’ endothelial dystrophy,8 the most common cause of corneal transplantation in developed countries. Here, we review discoveries in mendelian and complex ophthalmic disorders and their impli-cations for genetic testing and therapeutic intervention.
Gene tic Tes ting
The modern era of molecular ophthalmology began in 1985, with the discovery of the retinoblastoma gene.9 Since then, hundreds of other genes that are responsible for a wide variety of important diseases have been discovered, including those as-sociated with AMD, glaucoma, congenital cataract, syndromic and nonsyndromic forms of photoreceptor degeneration, and multiple macular dystrophies, corneal dystrophies, vitreoretinopathies, and optic neuropathies. The discovery of each new genetic cause of disease affords the possibility of using molecular investigation of DNA samples collected from individual patients as an adjunct to clinical diagnosis, prognosis, and counseling. In addition, persons who are found to carry known disease-causing mutations can be enrolled in clinical trials of new therapies or carefully studied with a variety of clinical instruments to fully explore the behavior of their disease over time.10,11 Samples from patients who lack mutations in known disease-causing genes can also serve as a valuable resource for scientists who seek to find additional disease-causing genes.
A major challenge in using this emerging genetic information in the clinical
The New England Journal of Medicine Downloaded from nejm.org on February 23, 2013. For personal use only. No other uses without permission.
Copyright © 2011 Massachusetts Medical Society. All rights reserved.

genomic medicine
n engl j med 364;20 nejm.org may 19, 2011 1933
domain is the gap that exists between the amount of information that is needed to convincingly demonstrate a pathogenic role of a given gene in a group of research subjects and the amount of information that is needed to reliably assert that a given genetic variation is responsible for a disease in an individual patient. Some of the factors that are responsible for this gap include the large number of non–phenotype-altering vari-ations scattered throughout many genes, the wide variety of types of true disease-causing mutations
(e.g., missense, nonsense, splice-site, promoter-inactivation, and copy-number variation) (see Glos-sary), the genetic differences among different ethnic groups, the genetic heterogeneity of many phenotypes (e.g., retinitis pigmentosa can be caused by a mutation in any one of more than 40 genes), and the clinical variation among patients with similar genotypes.
Because all genetic variations are not equally likely to cause disease, some investigators have suggested methods for taking this uncertainty
1869Leber describescongenitalamaurosis
1917Ophthalmology is the first medical specialty to create itsown assessment board
1953Structure of DNA is deduced
1963Coppock cataractis the first human trait mapped to an autosome
1971 Folkman recognizestherapeutic implications oftumor angiogenesis factor
1997First human glaucoma gene, MYOC,is identified
Stargardt’s diseasegene, ABCA4, is identified
1983 Polymerase chain reaction is introduced
460–322 B.C.E.Hippocrates and Aristotle studyfamilial transmission of ocular traits
1847Charles Babbageinvents theophthalmoscope
1865Mendelexperimentswith plant hybridization
1911Thomas Morgan identifies the chromosomeas the physical repository of genetic material
Color blindness is the first human traitmapped to the X chromosome
1900s 1910s 1920s 1930s 1940s 1950s 1960s 1970s 1980s 1990s 2000s 2010s1800s
1985First human cancer gene, retinoblastoma, is cloned
1987Rhodopsin is the first gene associated with a retinal degenerative disease
1989Leber’s hereditary optic neuropathy is the first human disease
shown to be caused by a mutation of mitochondrial DNA
2000Gene therapy for canine blindness caused by Leber’s congenital amaurosis is successful
2004Bevacizumab is approved for use in colon cancer
2005AMD is the first eye disease to yield a positive genomewide association
2006Off-label bevacizumab is shown to be effective in neovascular AMD
Ranibizumab is approved for use in neovascular AMD
2007 First gene-replacement therapy for a human eye disease, Leber’s congenital amaurosis, is successful
2011Ranibizumab and bevacizumab are shown to be essentially equivalent in neovascular AMD
1
Phimister
5/02/11
AUTHOR PLEASE NOTE:Figure has been redrawn and type has been reset
Please check carefully
Author
Fig #
Title
ME
DEArtist
Issue date
COLOR FIGURE
Draft 5Stone
Knoper
5/19/11
Figure 1. Timeline of Landmarks in Ophthalmic Genetics.
AMD denotes age-related macular degeneration.
The New England Journal of Medicine Downloaded from nejm.org on February 23, 2013. For personal use only. No other uses without permission.
Copyright © 2011 Massachusetts Medical Society. All rights reserved.

T h e n e w e ngl a nd j o u r na l o f m e dic i n e
n engl j med 364;20 nejm.org may 19, 20111934
into account in a standardized fashion when in-terpreting the results of genetic testing.12 The advent of whole-exome sequencing as a diagnos-tic tool accentuates the need for this type of probabilistic interpretation, because every person carries several recessive disease-causing mutations that would be incidental and medically irrelevant to any disease that they might have in their life-times. For example, approximately 1 in 30 Euro-peans is heterozygous for the deletion of codon 508 in the gene that is associated with cystic fi-brosis.13 The ability to distinguish between newly encountered benign variants and those that might confer risk is central to the task of interpreting genetic data, especially those generated by large data sets, such as the whole genome of an indi-vidual. As in all of medicine, a genetic test result is more likely to be meaningful when it is accom-
panied by a robust pretest hypothesis. Thus, the growth of molecular ophthalmology has increased the need for experienced clinicians who can place the observed genetic variations in the correct clini-cal context.
Mendeli a n Disor der s
According to the World Health Organization, the most common causes of blindness across the globe are cataracts, glaucoma, AMD, corneal opacity, diabetic retinopathy, infections, and parasitic dis-eases.14 Genetic factors play a role in many of these conditions, sometimes in the form of rela-tively rare, high-penetrance monogenic diseases and sometimes in the form of more common conditions caused by the complex interplay of multiple genes and the environment. From the
Glossary
Allele: One of two or more versions of a genetic sequence at a particular location in the genome.
Autosome: All the chromosomes except for the sex chromosomes and the mitochondrial chromosome.
Chaperone complex: An oligomeric protein that assists in the folding, unfolding, assembly, or disassembly of other macromolecular structures without being permanently incorporated into the assisted structures.
Codon: A three-nucleotide sequence of DNA or RNA that specifies a single amino acid.
Copy-number variation: Variation from one person to the next in the number of copies of a particular gene or DNA se-quence. The full extent to which copy-number variation contributes to human disease is not yet known.
De novo mutation: Any DNA sequence change that occurs during replication, such as a heritable gene alteration occur-ring in a family for the first time as a result of a DNA sequence change in a germ cell or fertilized egg.
Genomewide association study: An approach used in genetics research to look for associations between typically hun-dreds of thousands of specific genetic variations (most commonly single-nucleotide polymorphisms) and particu-lar diseases.
Linkage analysis: An approach to the discovery of the genetic basis of a disease that correlates the pattern of disease inheritance within families with specific alleles of genetic markers of known location.
Locus: The specific chromosomal location of a gene or other DNA sequence of interest.
Loss-of-function mutation: A mutation that decreases the production or function of a protein (or does both).
Missense mutation: The alteration of a single DNA nucleotide so that the resulting codon specifies a different amino acid.
Nonsense mutation: The alteration of a single DNA nucleotide so that the resulting codon signals a termination of translation, thus leading to truncation of the encoded protein.
Penetrance: The likelihood that a person carrying a particular genetic variant will have a detectably altered phenotype.
Population attributable risk: The difference in the rate of disease between a population that is exposed to a given factor and one that is not. The population attributable risks of individual factors that contribute to a single clinical entity, such as age-related macular degeneration, often total more than 100% because the disease in a specific patient may be caused by a combination of factors that are counted more than once when individual population attribut-able risks are summed.
Promoter-inactivation mutation: A genetic variation in the promoter of an otherwise normal gene that results in a dra-matic reduction in gene expression.
Single-nucleotide polymorphism: A single-nucleotide variation in a genetic sequence, a common form of variation in the human genome.
Splice-site mutation: A sequence variation at or near an intron–exon boundary that perturbs normal splicing of the ad-jacent intron.
The New England Journal of Medicine Downloaded from nejm.org on February 23, 2013. For personal use only. No other uses without permission.
Copyright © 2011 Massachusetts Medical Society. All rights reserved.

genomic medicine
n engl j med 364;20 nejm.org may 19, 2011 1935
many known monogenic eye disorders, we have selected three to illustrate the wide variety of pathophysiological mechanisms involved in hu-man blindness.
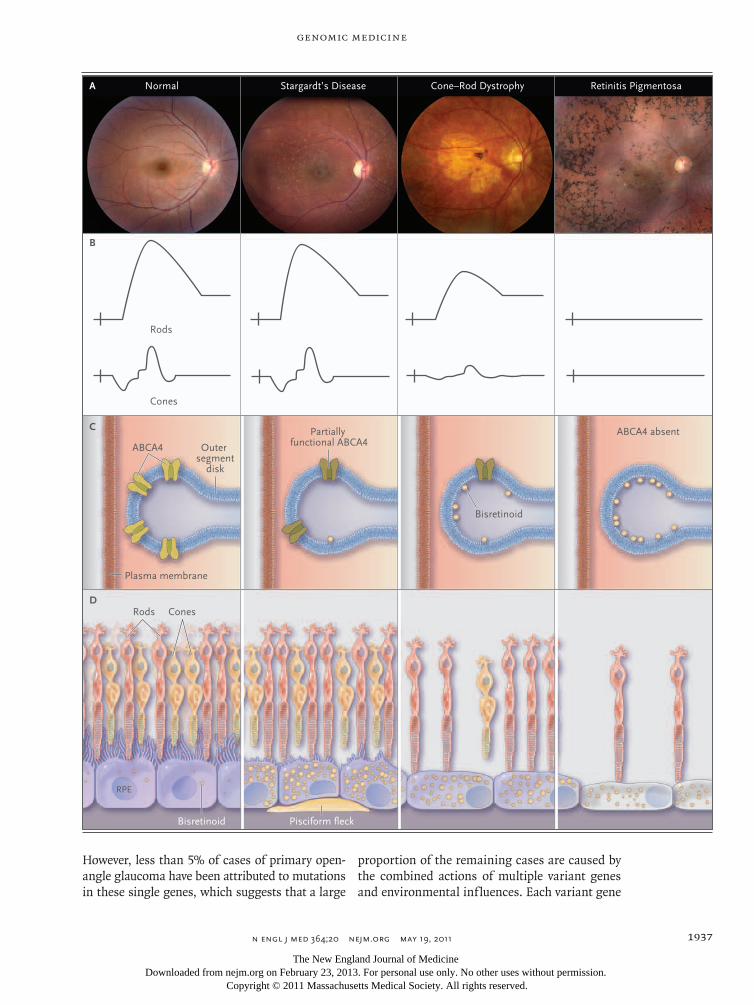
Mutation in ABCA4 — identified by Allikmets in 1997 as a cause of Stargardt’s disease15 — is one of the most important causes of monogenic retinal disease in humans. ABCA4 is an enzyme that flips a retinoid intermediate of the visual cycle known as N-retinylidene-phosphatidyletha-nolamine (N-retPE) from the inner leaflet to the outer leaflet of the photoreceptor outer segment disk membrane16 (Fig. 2, and interactive graphic, available with the full text of this article at NEJM .org). Mutations in ABCA4 result in an intradiscal accumulation of N-retPE, which in turn leads to the formation of a toxic, insoluble bisretinoid known as A2E. Variations in ABCA4 are responsible for more than 95% of cases of Stargardt’s disease, 30% of cases of cone–rod dystrophy, and 8% of cases of autosomal recessive retinitis pigmento-sa.17 This range of phenotypes results from the interplay of at least three factors: the degree of residual enzymatic function associated with a given genotype, the fact that cones are more read-ily harmed than rods by the accumulation of A2E, and the fact that injury to the retinal pigment epithelium results in secondary injury to both rods and cones.10 ABCA4 mutations with a rela-tively mild effect result in the accumulation of A2E within and beneath the retinal pigment epi-thelium, those with an intermediate effect result in a direct injury to photoreceptors that is some-what cone-selective, and those with the most se-vere effect result in injury to both cone and rod photoreceptors.17
A second example, mutation in MYOC in autoso-mal dominant juvenile-onset primary open-angle glaucoma, involves mistrafficking of a normally secreted trabecular meshwork protein to the per-oxisome. Linkage analysis of several large fami-lies mapped the causal mutations to the long arm of chromosome 1,18 and further genetic dissec-tion of this locus revealed mutations in MYOC as the cause of the disease.19 Certain missense mu-tations are associated with very high intraocular pressures and early onset of vision loss, whereas a nonsense mutation at codon 368 is associated with milder disease and a later onset,20 an unex-pected finding because nonsense mutations typi-cally have a more severe effect on protein integrity than do missense mutations. It was later discov-
ered that the missense mutations in MYOC cause the myocilin protein to misfold, with consequent unmasking of an otherwise cryptic signal that targets myocilin to the peroxisome.21 The result-ing intracellular retention of myocilin causes in-jury to the cells that make up the trabecular meshwork, which in turn reduces the outflow of aqueous humor. The elevated intraocular pressure resulting from this reduced outflow causes inju-ry to the optic nerve. MYOC mutations have been shown to be involved in approximately 4% of all cases of primary open-angle glaucoma, including adult-onset disease.19
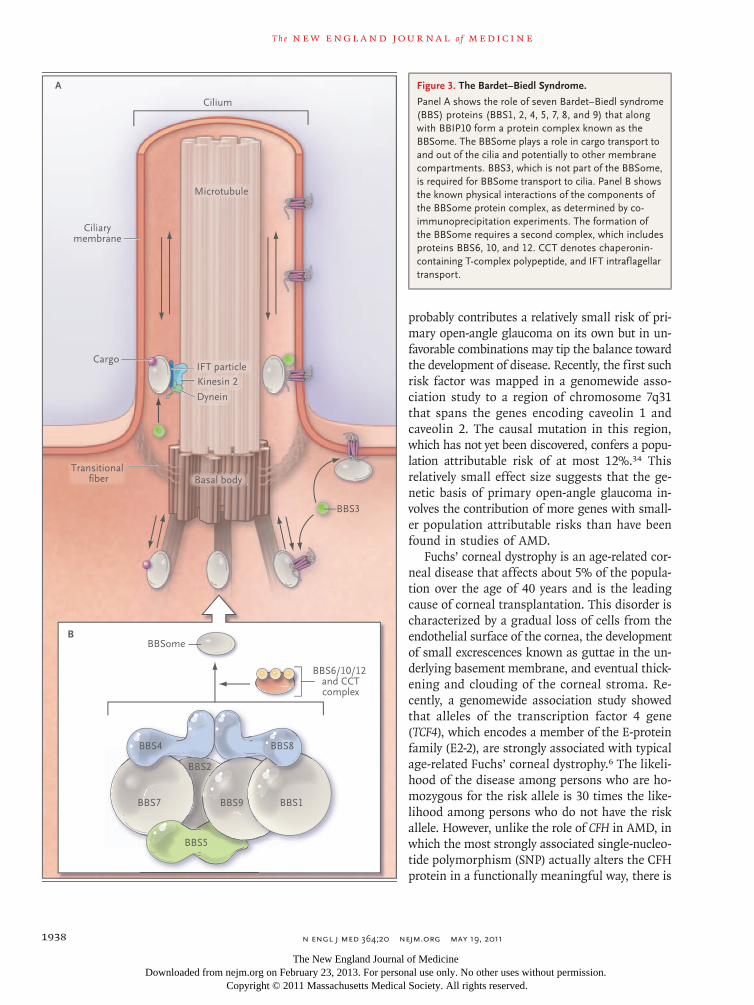
A third example illustrates the phenomenon of genetic heterogeneity: mutations in at least 14 genes cause a clinical syndrome known as the Bardet–Biedl syndrome (BBS). BBS is a pleiotropic autosomal recessive disorder that is characterized by the combination of retinitis pigmentosa, obe-sity, polydactyly, congenital heart defects, renal abnormalities, hypogenitalism, cognitive impair-ment, and an increased incidence of hypertension and diabetes mellitus.22 Patients with BBS pres-ent with progressive photoreceptor degeneration and are usually blind by the third decade of life. Studies of animal models have shown that pro-teins that are affected by mutations causing BBS are components of cilia or serve in intraflagellar or intracellular transport.23 The fact that the mu-tation of multiple different genes can be associ-ated with a single pleiotropic phenotype has now been largely explained by the discovery of two BBS protein complexes: the BBSome (consisting of seven BBS proteins), which plays a role in intra-flagellar transport; and a chaperone complex (con-sisting of three BBS proteins), which is required for BBSome assembly (Fig. 3).
Comple x Disor der s
Disorders with complex inheritance result from the interaction of multiple genetic loci and envi-ronmental factors such that a mendelian inheri-tance pattern is not observed. As a result, an allele contributing to a complex disease has a much lower penetrance than an allele involved in a sin-gle-gene disorder. This fact affects both the man-ner in which investigators identify such alleles and the way in which the presence or absence of such alleles has an effect on the care and counseling of patients and their families. As a general rule, linkage analysis of affected families has been
An interactive graphic regarding ABCA4 mutations is available at NEJM.org
The New England Journal of Medicine Downloaded from nejm.org on February 23, 2013. For personal use only. No other uses without permission.
Copyright © 2011 Massachusetts Medical Society. All rights reserved.

T h e n e w e ngl a nd j o u r na l o f m e dic i n e
n engl j med 364;20 nejm.org may 19, 20111936
more successful in identifying disease-causing al-leles in single-gene disorders (e.g., those described in the previous section), whereas genomewide or candidate association studies have been more suc-cessful in identifying factors involved in complex diseases, such as AMD, glaucoma, and Fuchs’ en-dothelial dystrophy. In a counseling context, al-leles of single-gene disorders can often be reason-ably said to cause disease, whereas alleles that are involved in complex diseases are more commonly said to increase the risk of disease.
Three of the most common causes of blindness — AMD, glaucoma, and Fuchs’ endothelial cor-neal dystrophy — have both genetic heterogeneity and genetic complexity, and genomewide asso-ciation studies have recently revealed clues to the pathogenicity of all three disorders.
AMD is the leading cause of blindness in de-veloped countries. As the name implies, the disorder usually affects persons over the age of 60 years and results in loss of macular (central) vision (Fig. 4). The prevalence of AMD increases with age, and more than 30% of persons over the age of 75 years will have some manifestation of the disease.6 Initial attempts to identify AMD loci involved screening genes that were known to cause monogenic macular disease in patients with AMD and in ethnically matched control sub-jects.24 Later, the development of cost-effective high-throughput genotyping made genomewide association studies possible. Of all diseases stud-ied in this manner, AMD has been one of the most successful in that it has yielded loci that contribute a high relative risk. For example, per-sons who carry a certain variant of the gene en-coding complement factor H (CFH)25-28 have a rela-tive risk of AMD that is more than 2.7 times that of persons without this variant.25,26 A variant at chromosome 10q26, in the vicinity of three genes (ARMS2, HTRA1, and PLEKHA1), is also strongly associated with AMD.29-31 In all, more than a dozen genes have been linked to AMD.6 Although these studies are important to the further under-standing of the pathophysiological mechanisms of AMD and may aid in the development of new therapies, clinical testing for AMD-associated poly-morphisms is of little value in the clinical man-agement of AMD at this time. AMD will develop in only about a third of persons with the highest-risk CFH genotype by the age of 70 years. Thus, unless and until a safe and effective treatment specific for CFH-associated AMD is developed,
there will be little clinical benefit in a genetic test that is less sensitive and specific for the detection of AMD than a routine eye exam.
Glaucoma is the second leading cause of blind-ness in the United States and the leading cause of blindness among blacks. As many as 60 million people worldwide currently have glaucoma.32 The most common form of glaucoma in the United States is primary open-angle glaucoma, which is characterized by optic-nerve damage and loss of peripheral visual field. Studies of mendelian (monogenic) forms of the disorder have implicat-ed two genes (MYOC and OPTN) and mapped the chromosomal location of an additional 13 genes.33
Figure 2 (facing page). Retinal Disease Associated with ABCA4 Mutations.
Panel A shows a series of photographs of the retinas of patients with progressively decreasing amounts of ABCA4 function (from left to right), ranging from a nor-mal retina to those of patients with Stargardt’s disease, cone–rod dystrophy, and retinitis pigmentosa. Panel B shows the effects of reduced ABCA4 function on full-field electroretinograms. The relatively mild reduction in ABCA4 activity in patients with Stargardt’s disease has little effect on global photoreceptor function. Moderate loss of ABCA4 function in patients with cone–rod dystro-phy has a greater effect on cone photoreceptors than it does on rods. Complete loss of ABCA4 function in some patients with retinitis pigmentosa is associated with ex-tensive loss of both cones and rods and a nonrecordable electroretinogram. Panel C shows the effects of reduced ABCA4 function on the accumulation of bisretinoid (yellow symbols) on the inner leaflet of the photoreceptor outer segment disk membranes. Mild reduction in ABCA4 ac-tivity in Stargardt’s disease is associated with some bis-retinoid formation; moderate loss of function in cone–rod dystrophy is associated with intermediate amounts of ac-cumulation; and complete loss of function in retinitis pigmentosa results in maximal accumulation. Panel D shows the histopathological effects of reduced ABCA4 activity. In patients with Stargardt’s disease, the rate of bisretinoid formation in the outer segments is relatively slow and the photoreceptors are not directly injured. Bis-retinoids are delivered to the secondary lysosomes of the retinal pigment epithelium (RPE) during the normal phagocytosis of photoreceptor outer segments. Some of this material accumulates beneath the RPE as accumula-tions known as pisciform flecks that are visible on oph-thalmoscopy. In patients with cone–rod dystrophy, mod-erate loss of ABCA4 function results in sufficient accumulation of bisretinoids in photoreceptor outer seg-ments to cause some apoptosis of photoreceptors (in cones more than rods). In patients with retinitis pigmen-tosa, complete loss of ABCA4 function causes extensive accumulation of bisretinoids in photoreceptor outer seg-ments, apoptosis of both rod and cone photoreceptors, and associated RPE thinning.
The New England Journal of Medicine Downloaded from nejm.org on February 23, 2013. For personal use only. No other uses without permission.
Copyright © 2011 Massachusetts Medical Society. All rights reserved.
genomic medicine
n engl j med 364;20 nejm.org may 19, 2011 1937
However, less than 5% of cases of primary open-angle glaucoma have been attributed to mutations in these single genes, which suggests that a large
proportion of the remaining cases are caused by the combined actions of multiple variant genes and environmental influences. Each variant gene
B
D
Rods
Rods
Cones
Cones
RPE
Pisciform fleckBisretinoid
ABCA4
C
Plasma membrane
Outer segment
disk
Partially functional ABCA4
ABCA4 absent
Bisretinoid
A Normal Stargardt’s Disease Cone–Rod Dystrophy Retinitis Pigmentosa
2
Phimister
5/02/11
AUTHOR PLEASE NOTE:Figure has been redrawn and type has been reset
Please check carefully
Author
Fig #
Title
ME
DEArtist
Issue date
COLOR FIGURE
Draft 13Stone
Knoper
5/19/11
The New England Journal of Medicine Downloaded from nejm.org on February 23, 2013. For personal use only. No other uses without permission.
Copyright © 2011 Massachusetts Medical Society. All rights reserved.
T h e n e w e ngl a nd j o u r na l o f m e dic i n e
n engl j med 364;20 nejm.org may 19, 20111938
probably contributes a relatively small risk of pri-mary open-angle glaucoma on its own but in un-favorable combinations may tip the balance toward the development of disease. Recently, the first such risk factor was mapped in a genomewide asso-ciation study to a region of chromosome 7q31 that spans the genes encoding caveolin 1 and caveolin 2. The causal mutation in this region, which has not yet been discovered, confers a popu-lation attributable risk of at most 12%.34 This relatively small effect size suggests that the ge-netic basis of primary open-angle glaucoma in-volves the contribution of more genes with small-er population attributable risks than have been found in studies of AMD.
Fuchs’ corneal dystrophy is an age-related cor-neal disease that affects about 5% of the popula-tion over the age of 40 years and is the leading cause of corneal transplantation. This disorder is characterized by a gradual loss of cells from the endothelial surface of the cornea, the development of small excrescences known as guttae in the un-derlying basement membrane, and eventual thick-ening and clouding of the corneal stroma. Re-cently, a genomewide association study showed that alleles of the transcription factor 4 gene (TCF4), which encodes a member of the E-protein family (E2-2), are strongly associated with typical age-related Fuchs’ corneal dystrophy.6 The likeli-hood of the disease among persons who are ho-mozygous for the risk allele is 30 times the like-lihood among persons who do not have the risk allele. However, unlike the role of CFH in AMD, in which the most strongly associated single-nucleo-tide polymorphism (SNP) actually alters the CFH protein in a functionally meaningful way, there is
Cilium
Microtubule
Basal body
Ciliarymembrane
Transitional fiber
Cargo
Dynein
Kinesin 2IFT particle
BBS6/10/12 and CCTcomplex
A
B
BBS1BBS9
BBS5
BBS7
BBSome
BBS4 BBS8
BBS2
BBS3
3
Phimister
5/02/11
AUTHOR PLEASE NOTE:Figure has been redrawn and type has been reset
Please check carefully
Author
Fig #
Title
ME
DEArtist
Issue date
COLOR FIGURE
Draft 4Stone
Knoper
5/19/11
Figure 3. The Bardet–Biedl Syndrome.
Panel A shows the role of seven Bardet–Biedl syndrome (BBS) proteins (BBS1, 2, 4, 5, 7, 8, and 9) that along with BBIP10 form a protein complex known as the BBSome. The BBSome plays a role in cargo transport to and out of the cilia and potentially to other membrane compartments. BBS3, which is not part of the BBSome, is required for BBSome transport to cilia. Panel B shows the known physical interactions of the components of the BBSome protein complex, as determined by co-immunoprecipitation experiments. The formation of the BBSome requires a second complex, which includes proteins BBS6, 10, and 12. CCT denotes chaperonin-containing T-complex polypeptide, and IFT intraflagellar transport.
The New England Journal of Medicine Downloaded from nejm.org on February 23, 2013. For personal use only. No other uses without permission.
Copyright © 2011 Massachusetts Medical Society. All rights reserved.

genomic medicine
n engl j med 364;20 nejm.org may 19, 2011 1939
currently little corroborating biologic evidence to support the involvement of TCF4 in Fuchs’ corneal dystrophy. For example, the linked SNP lies within an intron of TCF4 and is unlikely to alter TCF4 expression. Also, persons with de novo loss-of-function mutations in TCF4 have a severe neuro-logic disease that does not have corneal endothelial dysfunction as a feature.8,35 Thus, as with many findings obtained through a genomewide asso-ciation study, more work is needed to unravel the mechanism through which the statistically asso-ciated locus is linked to the corneal phenotype.
Ther a pies for Gene tic E y e Dise a se
Physicians have sought to treat inherited eye dis-eases at every level of the disease process — ranging from a very specific inhibition of a single mutant allele with a small inhibitory RNA mole-cule36 to a broad alteration of the metabolic mi-lieu with the use of a cocktail of vitamins with an uncertain mechanism of action.37 Gene discov-ery experiments have aided in this effort by pro-viding an improved understanding of specific biologic pathways that when perturbed lead to disease or susceptibility to disease. Such path-ways can become important targets for therapeu-tic agents, and scientists have been very creative in devising treatments aimed at those targets.
For example, the discovery that ABCA4 is in-volved in transporting vitamin A derivatives out of outer segment disks16 led to the discovery that vitamin A inhibitors such as fenretinide inhibit the accumulation of lipofuscin in animal models of Stargardt’s disease.38 The identification of the role of vascular endothelial growth factor in cho-roidal neovascularization led to the development of therapeutic antibodies (e.g., ranibizumab and bevacizumab) to combat the major blinding com-plication of AMD (Fig. 4). (An article describing a test of noninferiority of these drugs in the treatment of AMD appears in this issue of the Journal.39) Growth factors40 and neuroprotective agents41 have also been used to reduce the rate of an apoptotic response to inherited cellular abnormalities. In recent years, gene-replacement therapy, therapeutic stem cells, and retinal pros-theses have also moved to the threshold of clini-cal use for the treatment of genetic eye disease. A potential advantage of the latter two approach-
es is that a complete knowledge of the specific molecular cause of a patient’s disease may not be necessary for the treatment to be successful.
A
B
C
200 µm
200 µm
Figure 4. Treatment of Age-Related Macular Degeneration (AMD) with Bevacizumab.
A retinal photograph of a 67-year-old patient with a subretinal neovascular membrane (Panel A) shows yellow deposits (drusen) beneath the retinal pigment epithelium (arrows), which are the clinical hallmarks of AMD. Cloudy subretinal fluid and small hemorrhages in the center of the macula are suggestive of subretinal neovascularization. The horizontal black line indicates the center of the macula. The visual acuity in this eye is 20/80. A spectral-domain optical coherence tomogram (SDOCT) taken through the center of the macula reveals subretinal neovascular tissue and fluid-filled spaces within the retina (Panel B). A repeat SDOCT taken after three intravitreal injections of bevacizumab during a 3-month period reveals a dramatic reduction of neovascular tissue and intraretinal fluid (Panel C). The visual acuity has improved to 20/50.
The New England Journal of Medicine Downloaded from nejm.org on February 23, 2013. For personal use only. No other uses without permission.
Copyright © 2011 Massachusetts Medical Society. All rights reserved.

T h e n e w e ngl a nd j o u r na l o f m e dic i n e
n engl j med 364;20 nejm.org may 19, 20111940
Gene Ther a py
Most human photoreceptor diseases are inherited in an autosomal recessive manner, and in these cases the mechanism of disease usually involves a profound loss of function of the gene product. More than a dozen recessive retinal diseases have been successfully treated with viral- or nanoparti-cle-based gene transfer in animal models.42 For example, one molecular form of Leber’s congenital amaurosis is caused by the lack of the retinoid isomerase encoded by RPE65. A decade ago, Acland and colleagues43 successfully restored vision in a naturally occurring canine model of this disease using an adeno-associated viral vector to transfer a normal version of RPE65 to the retinal pigment epithelium. More recently, three different groups have extended this work to humans.44-46 For ex-ample, 3 years ago, Maguire and colleagues44 re-ported results in 12 patients who were treated with gene-replacement therapy. They observed vi-sual improvement in all 12 patients, with the great-est gains among younger patients.
Tr a nspl a n tation of S tem Cell s
Several important cell types in the eye have little if any capacity for endogenous regeneration, and as a result the only viable treatment option for pa-tients with hereditary disorders that involve the loss of such cells is some type of cell-replacement therapy. Although the replacement of highly dif-ferentiated cells, such as photoreceptors, poses challenges, a number of recent experiments sug-gest that the use of stem cells to achieve such a goal is now feasible.
In 2004, Klassen and colleagues47 found that transplanted retinal progenitor cells could devel-op into functional photoreceptors and give rise to enhanced visual function in mice with retinal degeneration. Since these original reports, an as-sortment of different cell types, ranging from the fate-restricted photoreceptor precursor to the pluripotent embryonic stem cell,48-51 have been used to replace photoreceptors in animals with inherited retinal disease. Embryonic stem cells have been of particular interest because of their ability to undergo unlimited self-renewal and tissue-specific cell production. For instance, Eiraku and colleagues52 recently found that by using a three-dimensional cell-culture system, they could recapitulate development and reliably gen-
erate functional photoreceptor cells in vitro. These properties permit the generation of cells in suffi-cient numbers to perform clinical transplantation from a single isolation rather than the multiple donations that are required when more develop-mentally restricted cell types are used.
Regardless of the theoretical utility of these cell types in humans, the isolation of cells from human embryos is rife with ethical concerns and immuno-logic limitations. As a result, freshly isolated em-bryonic stem cells seem unlikely to be widely used in the treatment of degenerative eye disease. A cell type that overcomes the majority of these limita-tions is the induced pluripotent stem cell (iPSC). Initially produced by Takahashi and Yamanaka 5 years ago,53 iPSCs can be generated by genetic re programming of dermal fibroblasts to pluripo-tency through retroviral transduction of only four transcription factors.53 Several groups of investi-gators have been able to show that iPSCs have the capacity to generate a variety of retinal-cell types, including photoreceptors,54,55 and that after trans-plantation these photoreceptors will integrate with in the dystrophic retinal architecture,56,57 which results in partial recovery of the electro-retinographic response.57 Although methodologic barriers, such as the use of retroviruses, preclude the immediate clinical translation of this tech-nology, and recent studies suggest that the pro-cess of somatic-cell reprogramming may result in the introduction of pathway-specific genetic de-fects,58-60 this field is evolving rapidly, and it is possible that these cells will eventually make their way into clinical use.
R e tina l Pros theses
In normal vision, decreased glutamate at the photoreceptor axon terminals stimulates bipolar and amacrine cells, which in turn release gluta-mate to stimulate the ganglion cells that com-municate with the brain. In an attempt to bypass photoreceptors and other neuronal elements that have been damaged by degenerative retinal dis-ease, investigators have explored the possibility of stimulating the ganglion cells directly with the use of electrical impulses delivered from a planar array of microelectrodes. Several different designs for retinal prostheses have had promis-ing results in both animals and humans,61 and one of these designs has recently been approved for clinical use in Europe.
The New England Journal of Medicine Downloaded from nejm.org on February 23, 2013. For personal use only. No other uses without permission.
Copyright © 2011 Massachusetts Medical Society. All rights reserved.

genomic medicine
n engl j med 364;20 nejm.org may 19, 2011 1941
Conclusion
The eye has figured prominently in the develop-ment of genetic and genomic approaches to human disease. Vision is critically important to most ac-tivities of daily living, and cures for blindness will remain an important goal for medicine for many years to come. Physicians and scientists will be aided in the pursuit of this goal by the optical and anatomic accessibility of the organ, as well as by the large amount of visual cortex devoted to the interpretation of the neural information originating in the retina. That is, a patient with a disease that injures only a few thousand neurons in the fovea
can describe this injury to his physician in great detail, and the physician can in turn view these neurons in the living patient at microscopic resolu-tion by taking advantage of the near perfect optics of the anterior portion of the eye. These natural optics also contribute to a surgical accessibility that is unmatched by any other part of the central ner-vous system. This latter attribute will be a tremen-dous advantage for clinician scientists seeking to translate all the recent progress in gene-transfer and stem-cell biology into effective therapies for their patients with genetic eye diseases.
Disclosure forms provided by the authors are available with the full text of this article at NEJM.org.
References
1. Senju S, Haruta M, Matsunaga Y, et al. Characterization of dendritic cells and macrophages generated by directed dif-ferentiation from mouse induced pluripo-tent stem cells. Stem Cells 2009;27:1021-31.2. Azuma N, Nishina S, Yanagisawa H, Okuyama T, Yamada M. PAX6 missense mutation in isolated foveal hypoplasia. Nat Genet 1996;13:141-2.3. Wilson EB. The sex chromosomes. Arch Mikrosk Anat Enwicklungsmech 1911;77:249-71.4. Renwick JH, Lawler SD. Probable linkage between a congenital cataract lo-cus and the Duffy blood group locus. Ann Hum Genet 1963;27:67-84.5. Newman NJ, Lott MT, Wallace DC. The clinical characteristics of pedigrees of Leber’s hereditary optic neuropathy with the 11778 mutation. Am J Ophthal-mol 1991;111:750-62.6. Swaroop A, Chew EY, Rickman CB, Abecasis GR. Unraveling a multifactorial late-onset disease: from genetic susceptibil-ity to disease mechanisms for age-related macular degeneration. Annu Rev Genom-ics Hum Genet 2009;10:19-43.7. Kwon YH, Fingert JH, Kuehn MH, Al-ward WL. Primary open-angle glaucoma. N Engl J Med 2009;360:1113-24.8. Baratz KH, Tosakulwong N, Ryu E, et al. E2-2 protein and Fuchs’s corneal dys-trophy. N Engl J Med 2010;363:1016-24.9. Cavenee WK, Hansen MF, Nordensk-jold M, et al. Genetic origin of mutations predisposing to retinoblastoma. Science 1985;228:501-3.10. Schindler EI, Nylen EL, Ko AC, et al. Deducing the pathogenic contribution of recessive ABCA4 alleles in an outbred population. Hum Mol Genet 2010;19:3693-701.11. Cideciyan AV, Swider M, Aleman TS, et al. ABCA4 disease progression and a proposed strategy for gene therapy. Hum Mol Genet 2009;18:931-41.
12. Stone EM. Finding and interpreting genetic variations that are important to ophthalmologists. Trans Am Ophthalmol Soc 2003;101:437-84.13. Hamosh A, FitzSimmons SC, Macek M Jr, Knowles MR, Rosenstein BJ, Cutting GR. Comparison of the clinical manifes-tations of cystic fibrosis in black and white patients. J Pediatr 1998;132:255-9.14. Resnikoff S, Pascolini D, Etya’ale D, et al. Global data on visual impairment in the year 2002. Bull World Health Organ 2004;82:844-51.15. Allikmets R. A photoreceptor cell-specific ATP-binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nat Genet 1997;17: 122.16. Weng J, Mata NL, Azarian SM, Tzekov RT, Birch DG, Travis GH. Insights into the function of Rim protein in photore-ceptors and etiology of Stargardt’s dis-ease from the phenotype in abcr knockout mice. Cell 1999;98:13-23.17. Maugeri A, Klevering BJ, Rohrsch-neider K, et al. Mutations in the ABCA4 (ABCR) gene are the major cause of auto-somal recessive cone-rod dystrophy. Am J Hum Genet 2000;67:960-6.18. Sunden SL, Alward WL, Nichols BE, et al. Fine mapping of the autosomal domi-nant juvenile open angle glaucoma (GLC1A) region and evaluation of candidate genes. Genome Res 1996;6:862-9.19. Stone EM, Fingert JH, Alward WLM, et al. Identification of a gene that causes primary open angle glaucoma. Science 1997;275:668-70.20. Alward WL, Fingert JH, Coote MA, et al. Clinical features associated with muta-tions in the chromosome 1 open-angle glaucoma gene (GLC1A). N Engl J Med 1998;338:1022-7.21. Shepard AR, Jacobson N, Millar JC, et al. Glaucoma-causing myocilin mutants require the peroxisomal targeting sig-nal-1 receptor (PTS1R) to elevate intraoc-
ular pressure. Hum Mol Genet 2007;16: 609-17.22. Sheffield VC. The blind leading the obese: the molecular pathophysiology of a human obesity syndrome. Trans Am Clin Climatol Assoc 2010;121:172-81.23. Shah AS, Farmen SL, Moninger TO, et al. Loss of Bardet-Biedl syndrome pro-teins alters the morphology and function of motile cilia in airway epithelia. Proc Natl Acad Sci U S A 2008;105:3380-5.24. Stone EM, Webster AR, Vandenburgh K, et al. Allelic variation in ABCR associ-ated with Stargardt disease but not age-related macular degeneration. Nat Genet 1998;20:328-9.25. Haines JL, Hauser MA, Schmidt S, et al. Complement factor H variant increases the risk of age-related macular degenera-tion. Science 2005;308:419-21.26. Edwards AO, Ritter R III, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age-related macular degeneration. Sci-ence 2005;308:421-4.27. Hageman GS, Anderson DH, Johnson LV, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A 2005;102:7227-32.28. Klein RJ, Zeiss C, Chew EY, et al. Complement factor H polymorphism in age-related macular degeneration. Sci-ence 2005;308:385-9.29. Jakobsdottir J, Conley YP, Weeks DE, Mah TS, Ferrell RE, Gorin MB. Suscepti-bility genes for age-related maculopathy on chromosome 10q26. Am J Hum Genet 2005;77:389-407.30. Rivera A, Fisher SA, Fritsche LG, et al. Hypothetical LOC387715 is a second ma-jor susceptibility gene for age-related macular degeneration, contributing inde-pendently of complement factor H to dis-ease risk. Hum Mol Genet 2005;14:3227-36.
The New England Journal of Medicine Downloaded from nejm.org on February 23, 2013. For personal use only. No other uses without permission.
Copyright © 2011 Massachusetts Medical Society. All rights reserved.

n engl j med 364;20 nejm.org may 19, 20111942
genomic medicine
31. Dewan A, Liu M, Hartman S, et al. HTRA1 promoter polymorphism in wet age-related macular degeneration. Sci-ence 2006;314:989-92.32. Quigley HA, Broman AT. The number of people with glaucoma worldwide in 2010 and 2020. Br J Ophthalmol 2006;90: 262-7.33. Fan BJ, Wiggs JL. Glaucoma: genes, phenotypes, and new directions for therapy. J Clin Invest 2010;120:3064-72.34. Thorleifsson G, Walters GB, Hewitt AW, et al. Common variants near CAV1 and CAV2 are associated with primary open-angle glaucoma. Nat Genet 2010;42: 906-9.35. Zweier C, Peippo MM, Hoyer J, et al. Haploinsufficiency of TCF4 causes syn-dromal mental retardation with intermit-tent hyperventilation (Pitt-Hopkins syn-drome). Am J Hum Genet 2007;80:994- 1001.36. Gorbatyuk M, Justilien V, Liu J, Haus-wirth WW, Lewin AS. Preservation of photoreceptor morphology and function in P23H rats using an allele independent ribozyme. Exp Eye Res 2007;84:44-52.37. Age-Related Eye Disease Study Research Group. A randomized, placebo-controlled, clinical trial of high-dose supplementa-tion with vitamins C and E and beta caro-tene for age-related macular degeneration and vision loss: AREDS report no. 9. Arch Ophthalmol 2001;119:1439-52.38. Radu RA, Han Y, Bui TV, et al. Reduc-tions in serum vitamin A arrest accumu-lation of toxic retinal f luorophores: a potential therapy for treatment of lipo-fuscin-based retinal diseases. Invest Oph-thalmol Vis Sci 2005;46:4393-401. [Erratum, Invest Ophthalmol Vis Sci 2006;47:3735.]39. The CATT Research Group. Ranibi-zumab and bevacizumab for neovascular age-related macular degeneration. N Engl J Med 2011;364:1897-908.40. Sieving PA, Caruso RC, Tao W, et al. Ciliary neurotrophic factor (CNTF) for human retinal degeneration: phase I trial of CNTF delivered by encapsulated cell intraocular implants. Proc Natl Acad Sci U S A 2006;103:3896-901.
41. Boatright JH, Moring AG, McElroy C, et al. Tool from ancient pharmacopoeia prevents vision loss. Mol Vis 2006;12: 1706-14.42. den Hollander AI, Black A, Bennett J, Cremers FP. Lighting a candle in the dark: advances in genetics and gene therapy of recessive retinal dystrophies. J Clin Invest 2010;120:3042-53. [Erratum, J Clin Invest 2011;121:456-7.]43. Acland GM, Aguirre GD, Ray J, et al. Gene therapy restores vision in a canine model of childhood blindness. Nat Genet 2001;28:92-5.44. Maguire AM, Simonelli F, Pierce EA, et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med 2008;358:2240-8.45. Bainbridge JW, Smith AJ, Barker SS, et al. Effect of gene therapy on visual func-tion in Leber’s congenital amaurosis. N Engl J Med 2008;358:2231-9.46. Hauswirth WW, Aleman TS, Kaushal S, et al. Treatment of Leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-asso-ciated virus gene vector: short-term re-sults of a phase I trial. Hum Gene Ther 2008;19:979-90.47. Klassen HJ, Ng TF, Kurimoto Y, et al. Multipotent retinal progenitors express developmental markers, differentiate into retinal neurons, and preserve light-medi-ated behavior. Invest Ophthalmol Vis Sci 2004;45:4167-73.48. Osakada F, Ikeda H, Mandai M, et al. Toward the generation of rod and cone photoreceptors from mouse, monkey and human embryonic stem cells. Nat Bio-technol 2008;26:215-24.49. MacLaren RE, Pearson RA, MacNeil A, et al. Retinal repair by transplantation of photoreceptor precursors. Nature 2006;444:203-7.50. Lamba DA, Karl MO, Ware CB, Reh TA. Efficient generation of retinal pro-genitor cells from human embryonic stem cells. Proc Natl Acad Sci U S A 2006; 103:12769-74.51. Ikeda H, Osakada F, Watanabe K, et al. Generation of Rx+/Pax6+ neural reti-
nal precursors from embryonic stem cells. Proc Natl Acad Sci U S A 2005;102: 11331-6.52. Eiraku M, Takata N, Ishibashi H, et al. Self-organizing optic-cup morphogenesis in three-dimensional culture. Nature 2011; 472:51-6.53. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse em-bryonic and adult fibroblast cultures by defined factors. Cell 2006;126:663-76.54. Meyer JS, Shearer RL, Capowski EE, et al. Modeling early retinal development with human embryonic and induced pluri-potent stem cells. Proc Natl Acad Sci U S A 2009;106:16698-703.55. Osakada F, Jin ZB, Hirami Y, et al. In vitro differentiation of retinal cells from human pluripotent stem cells by small-molecule induction. J Cell Sci 2009;122: 3169-79.56. Lamba DA, McUsic A, Hirata RK, Wang PR, Russell D, Reh TA. Generation, purification and transplantation of pho-toreceptors derived from human induced pluripotent stem cells. PLoS One 2010;5(1): e8763.57. Tucker B, Park I-H, Qi SD, et al. Trans-plantation of adult mouse iPS cell-derived photoreceptor precursors restores retinal structure and function in retinal degen-erative mice. PLoS One (in press).58. Gore A, Li Z, Fung HL, et al. Somatic coding mutations in human induced plu-ripotent stem cells. Nature 2011;471:63-7.59. Hussein SM, Batada NN, Vuoristo S, et al. Copy number variation and selection during reprogramming to pluripotency. Nature 2011;471:58-62.60. Lister R, Pelizzola M, Kida YS, et al. Hotspots of aberrant epigenomic repro-gramming in human induced pluripotent stem cells. Nature 2011;471:68-73.61. Dowling J. Current and future pros-pects for optoelectronic retinal prosthe-ses. Eye (Lond) 2009;23:1999-2005.Copyright © 2011 Massachusetts Medical Society.
The New England Journal of Medicine Downloaded from nejm.org on February 23, 2013. For personal use only. No other uses without permission.
Copyright © 2011 Massachusetts Medical Society. All rights reserved.





























