Embed Size (px)
Citation preview

R
G
SBK
a
ARRAA
samacsattwnvBwtpacffmrha
i
h1
Pharmacological Research 121 (2017) 219–229
Contents lists available at ScienceDirect
Pharmacological Research
j ourna l h om epage: w ww.elsev ier .com/ locate /yphrs
eview
enomics of hypertension
andosh Padmanabhan, Alisha Aman, Anna F. Dominiczak ∗
HF Glasgow Cardiovascular Research Centre, Institute of Cardiovascular and Medical Sciences, 126 University Place, University of Glasgow, Unitedingdom
r t i c l e i n f o
rticle history:eceived 18 April 2017eceived in revised form 28 April 2017ccepted 29 April 2017
a b s t r a c t
A complex network of interacting pathways involving renal, neural, endocrine, vascular and other mecha-nisms controls the main determinants of blood pressure – cardiac output and total peripheral resistance.Multiple genes within each of these systems contribute to the specialized functions regulating blood
vailable online 8 May 2017pressure. The monogenic forms of blood pressure dysregulation have provided valuable insights intoblood pressure regulation and expanded our understanding of both the mechanisms and the treatmentof hypertension. Genome wide association studies have identified over 100 single nucleotide polymor-phisms associated with blood pressure phenotypes and have identified plausible novel pathways of BPregulation and putative drug targets.
© 2017 Published by Elsevier Ltd.
From an epidemiological and clinical perspective, blood pres-ure (BP) at the higher end of the normal population distribution isssociated with an increased risk of cardiovascular mortality andorbidity. Traditionally cardiovascular risk assessment is based on
predefined threshold at which the quantitative BP phenotype isonverted into a binary trait (hypertension). Modern managementtrategies are directed towards BP reduction below this thresholdt which the risk of excess cardiovascular events is reduced. Thus,he BP or hypertension (HTN) phenotype is a measurable charac-eristic of clinical risk, and discovering their genetic determinantsill allow early risk prediction and targeted treatment. A complexetwork of interacting pathways involving renal, neural, endocrine,ascular, and other mechanisms controls the main determinants ofP – cardiac output and total peripheral resistance. Multiple genesithin each of these systems contribute to the specialized func-
ions regulating BP, and hence it is likely that many genes willarticipate in the development of HTN. Thus, by definition, BP is
complex trait that refers to any phenotype that does not exhibitlassic Mendelian inheritance attributed to a single gene and resultsrom the interactions between multiple genes and environmentalactors. The distribution of BP in the population is a normal uni-
odal distribution supporting a complex multifactorial basis of BP
egulation [1,2]. However, there are monogenic forms of HTN orypotension (“Simple trait”) which are very rare in the populationnd have little to no impact on public health, in contrast to essen-∗ Corresponding author at: University of Glasgow, Wolfson Medical School Build-ng, University Avenue, Glasgow G12 8QQ, United Kingdom.
E-mail address: [email protected] (A.F. Dominiczak).
ttp://dx.doi.org/10.1016/j.phrs.2017.04.031043-6618/© 2017 Published by Elsevier Ltd.
tial HTN with a prevalence of ∼27% amongst the adults worldwide.The monogenic forms of BP dysregulation have provided valuableinsights into BP regulation and expanded our understanding of boththe mechanisms and the treatment of HTN. The monogenic formshave also been the most successful examples of gene mapping withmutations in over 25 genes now linked to perturbed gene functionand the BP dysregulation. The goal is to extend these successfulexamples to essential HTN.
1. Evidence for a genetic basis of essential hypertension
Whilst monogenic syndromes of HTN provide evidence that dis-ruption of gene function can have a major impact on BP, to ventureinto formal genetic dissection of BP or essential HTN requires evi-dence for a genetic contribution to these traits. Strong indicationthat BP and essential HTN may have a genetic component camefrom family studies demonstrating correlation of BP among sib-lings and between parents and children with part of this correlationattributable to genetic factors. Sir George Pickering noted a corre-lation coefficient of about 0.2 among BP of hypertensive propositi[3] and the relationship was similar to those of height. The Mon-treal adoption study [4] demonstrated correlation coefficients of0.38 and 0.16 between biological and adoptive sibs, respectively,while the Victorian Family Heart Study [5] estimated correlationcoefficients of 0.44 for non-twin siblings, 0.78 for monozygoustwins, 0.50 for dizygous twins and 0.12 for spouse-spouse pairs.
All these data indicate presence of a genetic component if the envi-ronmental influence is assumed to be similar between comparisongroups. Hunt et al. [6] studied life table data for 94,292 personsand found the relative risks of developing HTN were 4.1 in men
2 ologic
atuwgopcBBtabmenawHawmhg
2
sgaataweHbhrmfaievqaIeoaltisdweimbmei
20 S. Padmanabhan et al. / Pharmac
nd 5 in women aged 20–39 who had at least two first degree rela-ives affected by HTN. Two additional measures that are commonlysed to assess the genetic component of a trait are heritability (h2),hich is the fraction of variation in disease susceptibility due to
enetic factors, and sibling recurrent risk (�s) which is the degreef elevated risk of disease for a sibling of an affected individual com-ared with a member of the general population. The heritability oflinic systolic BP is around 15–40% and 15–30% for clinic diastolicP [7,8], whereas for ambulatory night-time systolic and diastolicP the heritabilities are 32–70% and 32–50% [7–11]. It is pertinento point out that though the heritability estimates are consider-ble, this does not equate to magnitude of genetic effect. This isecause the denominator in the estimate of heritability compriseseasurement error and variances attributable to genes, shared
nvironment, non-shared environment and unmeasured determi-ants. Heritability is also a property of the population studiednd low heritability estimates would suggest that genetic mappingould be difficult for that phenotype. The sibling recurrent risk ofTN is around 1.2–1.5 [12] and taking this along with heritabilitynd correlation estimates, the HTN and BP can be considered a traitith relatively modest genetic effect. As noted above, minimizingeasurement error by using ambulatory recordings provide higher
eritability estimates and using this phenotype can maximize theenetic signal [9–11].
. Genetic architecture of blood pressure and hypertension
To successfully map the genetic basis of a trait, a good under-tanding of the genetic architecture of the trait is required. Theenetic architecture of a trait refers to the number of disease vari-nts that exist, their allele frequencies, the risks that they confer,nd the interactions between multiple genetic and environmen-al factors. Mutations that account for Mendelian forms of HTNre highly penetrant and are usually under very strong selection,hich keeps them at low frequencies with high levels of allelic het-
rogeneity. In contrast, susceptibility variants involved in essentialTN are likely to have low or medium penetrance and are proba-ly not subject to such strong selection resulting in lower alleliceterogeneity. There is an ongoing debate whether common orare variants contribute to essential HTN. Single nucleotide poly-orphisms (SNPs) with minor allele frequency (MAF) > 1% account
or more than 90% of the genetic differences between individu-ls and thus are likely to contribute to the population variationn BP rather than rare variants. This is the basis of the common dis-ase/common variant (CDCV) hypothesis which states that geneticariants underlying complex traits occur with a relatively high fre-uency, have undergone little or no selection in earlier populationsnd are likely to date back to more than 100,000 years ago [13].ndeed from an evolutionary perspective, essential HTN is a dis-ase of civilization and may be an undesirable pleiotropic effectf a preserved genotype that could have optimized fitness in thencient environment [14]. It is well recognized that HTN occurs ear-ier and with more severity in people of African ancestry comparedo those of European ancestry [15]. Differing predispositions to HTNn different populations may simply reflect different evolutionaryelection pressures (“bottlenecks”) and the fact that populationso not share the same ancestral histories. In addition, an alleleith no effect on reproductive fitness is expected to achieve high
quilibrium frequency and this is likely to be the case for genesnfluencing HTN. The other competing model for HTN is the com-
on disease rare variant hypothesis, with an inverse relationship
etween the magnitude of genetic effect and allele frequency. Thisodel argues that diseases are common because of highly prevalentnvironmental influences, not because of common disease allelesn the population. Recently support for this has come from stud-
al Research 121 (2017) 219–229
ies of rare variants of three genes namely Solute Carrier Family12 Members 1 and 3 (SLC12A1, SLC12A3), and Potassium Voltage-Gated Channel Subfamily J Member 1 (KCNJ1) (major mutationsof which cause Gitelman syndrome, and Bartter syndrome type 1and 2 respectively) in the general population producing clinicallysignificant BP reduction and protection from development of HTN[16]. The most likely scenario would be that the allelic spectrumof the disease variants is the same as the general spectrum of alldisease variants. Under this neutral model, although most suscep-tibility variants are rare with minor allele frequencies (MAF) <1%,SNPs with MAF > 1% would account for more than 90% of the geneticdifferences between individuals. It is plausible that these commonvariants might contribute significantly to those common diseasesin which susceptibility alleles might not be under intense negativeselection. For the genome as a whole, it has been predicted thatof the expected 10–15 million SNPs with MAF > 1%, approximatelyhalf have MAF > 10%. Given that the number of disease variants con-ferring mild to moderate risks might be large, there are likely to behundreds of common and rare variants contributing to the familialclustering of HTN.
3. Monogenic forms of hypertension and hypotension
Table 1 and Fig. 1 summarises the rare monogenic forms of HTNthat have contributed to our understanding BP regulation and tar-geted treatment [17,18]. We briefly describe each of the syndromesbelow.
3.1. Glucocorticoid-remediable aldosteronism or familialhyperaldosteronism type 1
This rare autosomal dominant disorder is caused by the forma-tion of a chimeric gene due to unequal crossing during meiosis.This results in aldosterone secretion stimulated by ACTH insteadof angiotensin II resulting in HTN in childhood and early adult-hood due to the secretion of ACTH as a fusion product of 5′ endof Cytochrome P450 Family 11 Subfamily B Member 1 (CYP11B1)and the distal coding sequence of member 2 of the same family(CYP11B2) [19]. Affected individuals are treated with a low-doseof glucocorticoids to suppress ACTH secretion, amiloride to blockENaC, or spironolactone to prevent aldosterone binding to the min-eralocorticoid receptor.
3.2. Apparent mineralocorticoid excess
Normally cortisol has mineralocorticoid activity which doesnot manifest as cortisol is metabolised promptly into cortisoneby 11-beta-Hydroxysteroid Dehydrogenase Type 2 (HSD11B2). InApparent Mineralocorticoid excess (AME), absence or reducedactivity of HSD11B2 results in cortisol acting as a potent mineralo-corticoid. HSD11B2 activity is reduced by loss of function mutationsand from excess ingestion of licorice whose active constituent is theglycyrrhizic acid. Treatment is with spironolactone which blocksthe binding of cortisol to mineralocorticoid receptor and a lowsodium diet [20].
3.3. Pseudohypoaldosteronism type II (Gordon’s syndrome)
This syndrome is caused by mutations in the WNK Lysine Defi-cient Protein Kinase 1 and 4 (WNK1, WNK4), Kelch Like FamilyMember 3 (KLHL3), and Cullin 3 (CUL3). Mutations in the WNKgenes cause a disruption in the signalling pathway that regulates
the BP by controlling the ion cotransporters NCC and NKCC2 inthe kidney. CUL3 and KLH3 mutations, on the other hand, inhibitthe ubiquitylation of WNK4 and other WNK isoforms leading to anover-activation of NCC/NKCC2 and thereby increased salt retention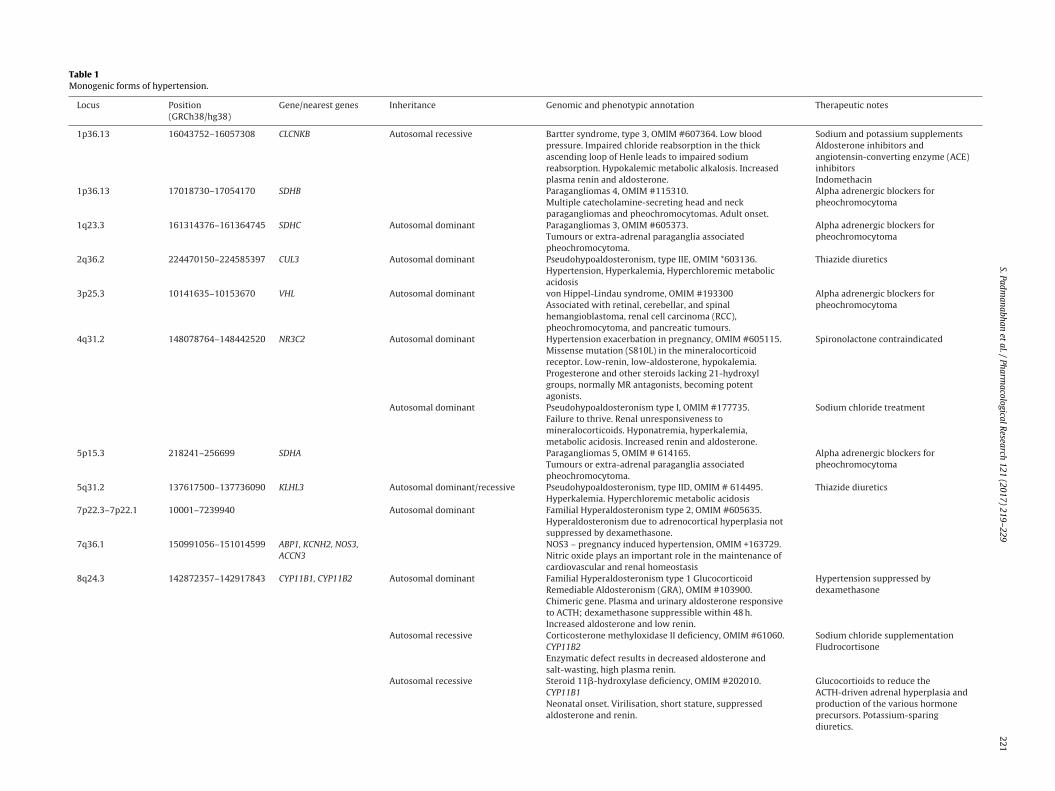
S. Padm
anabhan et
al. /
Pharmacological
Research
121 (2017)
219–229
221
Table 1Monogenic forms of hypertension.
Locus Position(GRCh38/hg38)
Gene/nearest genes Inheritance Genomic and phenotypic annotation Therapeutic notes
1p36.13 16043752–16057308 CLCNKB Autosomal recessive Bartter syndrome, type 3, OMIM #607364. Low bloodpressure. Impaired chloride reabsorption in the thickascending loop of Henle leads to impaired sodiumreabsorption. Hypokalemic metabolic alkalosis. Increasedplasma renin and aldosterone.
Sodium and potassium supplementsAldosterone inhibitors andangiotensin-converting enzyme (ACE)inhibitorsIndomethacin
1p36.13 17018730–17054170 SDHB Paragangliomas 4, OMIM #115310.Multiple catecholamine-secreting head and neckparagangliomas and pheochromocytomas. Adult onset.
Alpha adrenergic blockers forpheochromocytoma
1q23.3 161314376–161364745 SDHC Autosomal dominant Paragangliomas 3, OMIM #605373.Tumours or extra-adrenal paraganglia associatedpheochromocytoma.
Alpha adrenergic blockers forpheochromocytoma
2q36.2 224470150–224585397 CUL3 Autosomal dominant Pseudohypoaldosteronism, type IIE, OMIM *603136.Hypertension, Hyperkalemia, Hyperchloremic metabolicacidosis
Thiazide diuretics
3p25.3 10141635–10153670 VHL Autosomal dominant von Hippel-Lindau syndrome, OMIM #193300Associated with retinal, cerebellar, and spinalhemangioblastoma, renal cell carcinoma (RCC),pheochromocytoma, and pancreatic tumours.
Alpha adrenergic blockers forpheochromocytoma
4q31.2 148078764–148442520 NR3C2 Autosomal dominant Hypertension exacerbation in pregnancy, OMIM #605115.Missense mutation (S810L) in the mineralocorticoidreceptor. Low-renin, low-aldosterone, hypokalemia.Progesterone and other steroids lacking 21-hydroxylgroups, normally MR antagonists, becoming potentagonists.
Spironolactone contraindicated
Autosomal dominant Pseudohypoaldosteronism type I, OMIM #177735.Failure to thrive. Renal unresponsiveness tomineralocorticoids. Hyponatremia, hyperkalemia,metabolic acidosis. Increased renin and aldosterone.
Sodium chloride treatment
5p15.3 218241–256699 SDHA Paragangliomas 5, OMIM # 614165.Tumours or extra-adrenal paraganglia associatedpheochromocytoma.
Alpha adrenergic blockers forpheochromocytoma
5q31.2 137617500–137736090 KLHL3 Autosomal dominant/recessive Pseudohypoaldosteronism, type IID, OMIM # 614495.Hyperkalemia. Hyperchloremic metabolic acidosis
Thiazide diuretics
7p22.3–7p22.1 10001–7239940 Autosomal dominant Familial Hyperaldosteronism type 2, OMIM #605635.Hyperaldosteronism due to adrenocortical hyperplasia notsuppressed by dexamethasone.
7q36.1 150991056–151014599 ABP1, KCNH2, NOS3,ACCN3
NOS3 – pregnancy induced hypertension, OMIM +163729.Nitric oxide plays an important role in the maintenance ofcardiovascular and renal homeostasis
8q24.3 142872357–142917843 CYP11B1, CYP11B2 Autosomal dominant Familial Hyperaldosteronism type 1 GlucocorticoidRemediable Aldosteronism (GRA), OMIM #103900.Chimeric gene. Plasma and urinary aldosterone responsiveto ACTH; dexamethasone suppressible within 48 h.Increased aldosterone and low renin.
Hypertension suppressed bydexamethasone
Autosomal recessive Corticosterone methyloxidase II deficiency, OMIM #61060.CYP11B2Enzymatic defect results in decreased aldosterone andsalt-wasting, high plasma renin.
Sodium chloride supplementationFludrocortisone
Autosomal recessive Steroid 11�-hydroxylase deficiency, OMIM #202010.CYP11B1Neonatal onset. Virilisation, short stature, suppressedaldosterone and renin.
Glucocortioids to reduce theACTH-driven adrenal hyperplasia andproduction of the various hormoneprecursors. Potassium-sparingdiuretics.
222
S. Padm
anabhan et
al. /
Pharmacological
Research
121 (2017)
219–229
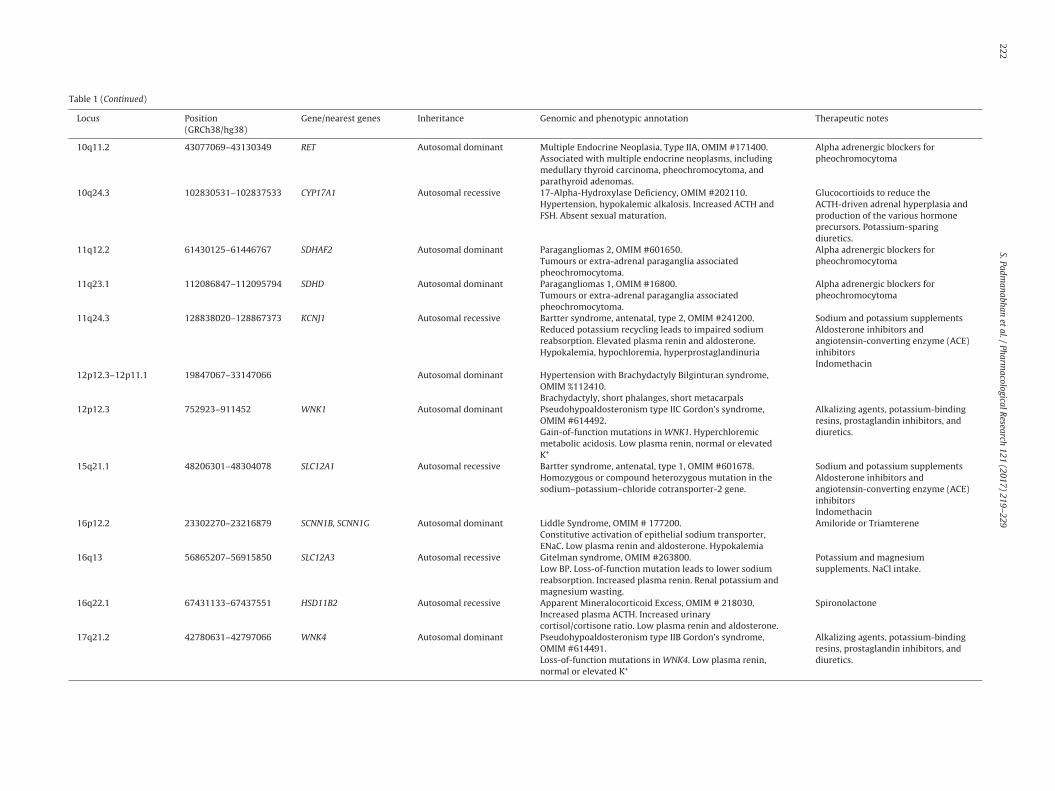
Table 1 (Continued)
Locus Position(GRCh38/hg38)
Gene/nearest genes Inheritance Genomic and phenotypic annotation Therapeutic notes
10q11.2 43077069–43130349 RET Autosomal dominant Multiple Endocrine Neoplasia, Type IIA, OMIM #171400.Associated with multiple endocrine neoplasms, includingmedullary thyroid carcinoma, pheochromocytoma, andparathyroid adenomas.
Alpha adrenergic blockers forpheochromocytoma
10q24.3 102830531–102837533 CYP17A1 Autosomal recessive 17-Alpha-Hydroxylase Deficiency, OMIM #202110.Hypertension, hypokalemic alkalosis. Increased ACTH andFSH. Absent sexual maturation.
Glucocortioids to reduce theACTH-driven adrenal hyperplasia andproduction of the various hormoneprecursors. Potassium-sparingdiuretics.
11q12.2 61430125–61446767 SDHAF2 Autosomal dominant Paragangliomas 2, OMIM #601650.Tumours or extra-adrenal paraganglia associatedpheochromocytoma.
Alpha adrenergic blockers forpheochromocytoma
11q23.1 112086847–112095794 SDHD Autosomal dominant Paragangliomas 1, OMIM #16800.Tumours or extra-adrenal paraganglia associatedpheochromocytoma.
Alpha adrenergic blockers forpheochromocytoma
11q24.3 128838020–128867373 KCNJ1 Autosomal recessive Bartter syndrome, antenatal, type 2, OMIM #241200.Reduced potassium recycling leads to impaired sodiumreabsorption. Elevated plasma renin and aldosterone.Hypokalemia, hypochloremia, hyperprostaglandinuria
Sodium and potassium supplementsAldosterone inhibitors andangiotensin-converting enzyme (ACE)inhibitorsIndomethacin
12p12.3–12p11.1 19847067–33147066 Autosomal dominant Hypertension with Brachydactyly Bilginturan syndrome,OMIM %112410.Brachydactyly, short phalanges, short metacarpals
12p12.3 752923–911452 WNK1 Autosomal dominant Pseudohypoaldosteronism type IIC Gordon’s syndrome,OMIM #614492.Gain-of-function mutations in WNK1. Hyperchloremicmetabolic acidosis. Low plasma renin, normal or elevatedK+
Alkalizing agents, potassium-bindingresins, prostaglandin inhibitors, anddiuretics.
15q21.1 48206301–48304078 SLC12A1 Autosomal recessive Bartter syndrome, antenatal, type 1, OMIM #601678.Homozygous or compound heterozygous mutation in thesodium–potassium–chloride cotransporter-2 gene.
Sodium and potassium supplementsAldosterone inhibitors andangiotensin-converting enzyme (ACE)inhibitorsIndomethacin
16p12.2 23302270–23216879 SCNN1B, SCNN1G Autosomal dominant Liddle Syndrome, OMIM # 177200.Constitutive activation of epithelial sodium transporter,ENaC. Low plasma renin and aldosterone. Hypokalemia
Amiloride or Triamterene
16q13 56865207–56915850 SLC12A3 Autosomal recessive Gitelman syndrome, OMIM #263800.Low BP. Loss-of-function mutation leads to lower sodiumreabsorption. Increased plasma renin. Renal potassium andmagnesium wasting.
Potassium and magnesiumsupplements. NaCl intake.
16q22.1 67431133–67437551 HSD11B2 Autosomal recessive Apparent Mineralocorticoid Excess, OMIM # 218030.Increased plasma ACTH. Increased urinarycortisol/cortisone ratio. Low plasma renin and aldosterone.
Spironolactone
17q21.2 42780631–42797066 WNK4 Autosomal dominant Pseudohypoaldosteronism type IIB Gordon’s syndrome,OMIM #614491.Loss-of-function mutations in WNK4. Low plasma renin,normal or elevated K+
Alkalizing agents, potassium-bindingresins, prostaglandin inhibitors, anddiuretics.

S. Padmanabhan et al. / Pharmacological Research 121 (2017) 219–229 223
F low bs ital adr
ae
3
ectb[SEtiT
3
aloCS
ig. 1. Genes and BP regulation. Genes linked to monogenic syndromes of high andtudies are in bold green. APA – aldosterone-producing adenoma; CAH – congenemediable aldosteronism, PHA-I – pseudohypoaldosteronism type I.
nd HTN [21,22]. Affected individuals or subjects are treated withither a low-salt diet or thiazide diuretics which block NCC.
.4. Liddle syndrome
This is an autosomal dominant condition with low lev-ls of aldosterone and renin. Normally, a regulatory repressoralled Nedd4-2: Neural Precursor Cell Expressed, Developmen-ally Down-Regulated 4-Like, E3 Ubiquitin Protein Ligase (NEDD4L)inds to the sodium channel ENaC and promotes its degradation23]. Mutations in Sodium Channel Epithelial 1 Beta and Gammaubunits (SCNN1 and SCNN1G), coding for the � and � subunits ofNaC respectively, disrupts this binding leading to the constitu-ive expression and prolongation of the half-life of ENaC resultingn increased sodium reabsorption, volume expansion, and HTN.reatment is with amiloride or triamterene.
.5. Bartter’s syndrome
This syndrome leads to loss of salt with hypokalemic metaboliclkalosis, normal to low BP and increased renin and aldosterone
evels. It is caused by the direct loss of function of NKCC2 or a sec-ndary cause disrupting the functions of NKCC2-ROMK channel,hloride Voltage-Gated Channel Kb (CLCNKB), Bartin and Calciumensing Receptor (CaSR) in the TAL cells [24]. Individuals withlood pressure are shown in bold black and genes that are putative loci from GWASrenal hyperplasia, HTB – hypertension with brachydactyly, GRA – glucocorticoid
this syndrome exhibit an increased level of urine prostaglandin E2and reduced susceptibility to angiotensin II and norepinephrine.Treatment is with a potassium supplementation, and use ofcyclooxygenase inhibitors, ACE-inhibitors and potassium sparingdiuretics.
3.6. Gitelman’s syndrome
Loss of function mutations in the SLC12A3, producing NCC inthe DCT cells, lead to Gitelman’s syndrome, which has a prevalenceof 1:40,000. Treatment options include oral potassium and magne-sium supplementations with adequate salt and water consumption,and prescription of indomethacin, amiloride and eplerenone [25].
3.7. Primary aldosteronism
This syndrome is characterised by the constitutive productionof aldosterone leading to HTN with hypokalemia and suppressedcirculating renin. Gain of function mutations in the gene PotassiumVoltage-Gated Channel Subfamily J Member 5 (KCNJ5) resulting inmembrane depolarisation and enhanced aldosterone production
has been associated with over 40% of these aldosterone-producingadenomas (APAs). Other implicated genes include ATPase Na+/K+Transporting Subunit Alpha 1 (ATP1A1), encoding the �1 sub-unit of Na+/K+-ATPase, in 7% of APAs; ATPase Plasma Membrane
2 ologic
CaefA
3
amonVM1(dSgpgTca
4
BGoamowpcatTpPGGtfiienttaelgdiivtmb
w
24 S. Padmanabhan et al. / Pharmac
a2+ Transporting 3 (ATP2B3), encoding Ca2+-ATPases (SERCA);nd Calcium Voltage-Gated Channel Subunit Alpha1 D (CACNA1D),ncoding an L-type Ca2+ channel, CaV1.3 [26]; and other gain ofunction mutations in genes regulating Na+, Ca2+, CACNA1D andTP1A1.
.8. Phaeochromocytomas and paragangliomas
These are rare neuroendocrine tumours in the adrenal glands,nd sympathetic and parasympathetic paraganglia. Phaeochro-ocytomas, inherited as an autosomal dominant trait, are a result
f mutations in the Ret Proto-Oncogene (RET), responsible for theormal development of neurons. Other implicated genes includeon Hippel-Lindau Tumor Suppressor (VHL), Kinesin Familyember 1B (KIF1Bbeta), Egl-9 Family Hypoxia Inducible Factor
(PHD2), Succinate Dehydrogenase Complex Assembly Factor 2SDHAF2), and the genes encoding the four subunits of succinateehydrogenase: Succinate Dehydrogenase Complex Iron Sulfurubunit A, B, C, and D (SDHA, SDHB, SDHC and SDHD). Heterozy-ous germline mutations in SDHC and SDHD are associated witharaganglioma type 1, 3 and 4 [27]. Germline mutation in the SDHBene is the only reliable predictor of malignant Paragangliomas.ype 2 multiple endocrine neoplasia (MEN 2) is a rare familialancer syndrome caused by mutations in the RET gene and isssociated with phaeochromocytoma.
. Polygenic hypertension
Major advances in identifying common variants associated withP and HTN arose from genome wide association studies (GWAS).WAS are large scale association mapping making no assumptionsf the genomic location or function of the causal variant and provide
comprehensive approach to testing the hypothesis that com-on alleles contribute to heritable phenotype variation. GWAS rely
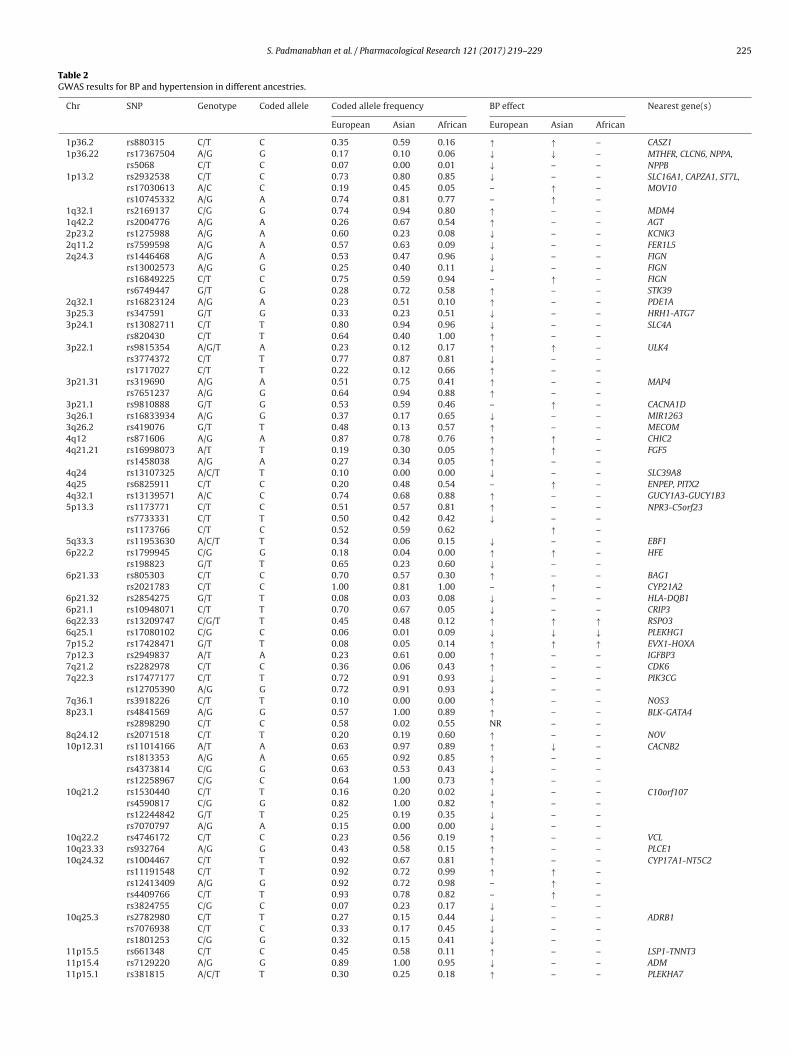
n the linkage disequilibrium (LD) or correlation patterns of SNPsith functional variants and, therefore, identified SNPs are usuallyroxies of untyped functional variants. A typical GWAS experimentonsists of genotyping 500,000 to 1 million SNPs across the genome,s depending on the population this number of SNPs is adequateo interrogate 80% of common SNPs with MAF greater than 5%.o adjust for multiple testing and to decrease type I error (false-ositive rates), the statistical burden of proof relies on stringent-values usually P < 5 × 10−8. Table 2 and Fig. 1 summarises theWAS results for BP and HTN in different ancestries. One successfulWAS for HTN used an extreme case-control design representing
he top 2% and bottom 9% of the BP distribution. Combined withollow-up validation analyses in 19,845 cases and 16,541 controls,t confirmed a locus near the Uromodulin (UMOD) gene [28]. UMODs exclusively expressed in the kidney, suggesting that the discov-red variant may influence sodium homeostasis. Additionally, rareonsynonymous variants in salt-handling genes have been showno have comparatively large effects on BP in the general popula-ion [16]. It is striking that the signals from all the GWAS for HTNnd BP do not contain genes from highly plausible pathways, forxample, the renin–angiotensin–aldosterone pathway or epithe-ial sodium channels. An important limitation of GWASs is thatenome-wide significant SNPs often merely tag but do not provideirect information on the causal variants. To translate those signals
nto biological function, follow-up studies are necessary. Anotherssue arising from GWAS studies is the small fraction of populationariance of BP (<5%) and BP heritability (∼2%) that are explained byhe collective effect of all the GWAS loci identified so far [29,30]. The
issing heritability [30] conundrum is not unique to BP genetics,ut is observed in most of the common phenotypes.
Despite the increasing pace of discovery of variants associatedith BP and HTN, the limited predictive utility of these variants
al Research 121 (2017) 219–229
either singly or as part of a composite risk score is striking [29].The population distribution of the number of BP increasing alle-les with nearly similar allele frequencies is normally distributed, aseach SNP is inherited independently and hence the number of indi-viduals in the population expected to carry all harmful risk alleleswould be vanishingly small. One way of maximizing informationabout the genetic signals is to create a composite genetic risk scorecoding for the presence or absence of risk alleles and their numbersfor all the BP GWAS SNPs [29]. In the ICBPGWAS [31], between thetop and the bottom quintiles of the risk score a 4.6 mmHg systolicand 3.0 mmHg diastolic BP difference was detected, and the preva-lence of HT was 29% compared to 16% in the top and bottom deciles.The score was also associated with early and advanced target organdamage including left ventricular hypertrophy, stroke and coronaryartery disease but not chronic kidney disease or markers of renalfunction [31]. The lack of association between BP risk score and kid-ney function may reflect just an association with GFR but not withother renal phenotypes such as albuminuria, or may indicate thathigh BP and renal disease do not necessarily have the same molec-ular origin. Using panels of genetic markers to predict risk has verypoor discrimination and that the utility of GWAS approaches areprimarily in identification of novel pathways. Below we describesome examples of plausible pathways identified by GWAS studies.
5. Functional characterisation of GWAS loci – novelpathways
5.1. Uromodulin
Uromodulin is a protein expressed exclusively in the thickascending limb of the loop of Henle (TAL) and encoded by the geneUMOD. Although the specific function is unknown, knockout micedemonstrate an increased localisation of NKCC2 in the subapicalvesicles of TAL cells with reduced phosphorylation [32]. This resultsin reduced co-transporter activity and greater sodium excretion.Alternatively, overexpression of Umod causes a dose dependentincrease in the excretion of the protein, associated with an eleva-tion of BP. A GWAS of BP extremes linked an UMOD SNP rs13333226to HTN, with the minor G allele conferring a lower risk of HTN andreduced urinary UMOD excretion [28]. Furosemide, a loop diureticand specific blocker of NKCC2, has been shown to enhance natri-uresis and reduced BP in both transgenic mice and hypertensiveindividuals homozygous for the A allele of this SNP [33].
5.2. Natriuretic peptide
Polymorphisms in the ANP and BNP propeptides encodinggenes, Natriuretic Peptide A and B (NPPA, NPPB), have been associ-ated with natriuretic peptides and BP [34,35]. Healthy individualswho are homozygous for the risk allele of SNP rs5068, presentin the 3′-UTR of NPPA, were found to have lower expression ofANP propeptide, NT-proANP, possibly mediated by miR-425. Thiseffect on circulating NT-pro-ANP is comparable to the environmen-tal change induced by switching from an extremely low salt dietof 230 mg/dl to 4600 mg/dl [36]. Natriuretic Peptide Receptor 3(NPR3) is another plausible candidate identified by GWAS. This geneencodes for the natriuretic peptide receptor C/guanylate cyclase C[31,37]; KO studies have found the mutant mice to have lower BPdue to the reduced clearance of the natriuretic peptides [38]. In aGWAS conducted for HTN, SNPs nearby this gene-rs1173771 hasbeen associated with BP in Europeans [31] while rs1173766 in eastAsians [37].
5.2.1. SH2B adaptor protein 3 (SH2B3)SH2B adaptor protein 3 is an intracellular adaptor protein
expressed in the hematopoietic and endothelial cells. GWAS stud-
S. Padmanabhan et al. / Pharmacological Research 121 (2017) 219–229 225
Table 2GWAS results for BP and hypertension in different ancestries.
Chr SNP Genotype Coded allele Coded allele frequency BP effect Nearest gene(s)
European Asian African European Asian African
1p36.2 rs880315 C/T C 0.35 0.59 0.16 ↑ ↑ – CASZ11p36.22 rs17367504 A/G G 0.17 0.10 0.06 ↓ ↓ – MTHFR, CLCN6, NPPA,
NPPBrs5068 C/T C 0.07 0.00 0.01 ↓ – –1p13.2 rs2932538 C/T C 0.73 0.80 0.85 ↓ – – SLC16A1, CAPZA1, ST7L,
MOV10rs17030613 A/C C 0.19 0.45 0.05 – ↑ –rs10745332 A/G A 0.74 0.81 0.77 – ↑ –
1q32.1 rs2169137 C/G G 0.74 0.94 0.80 ↑ – – MDM41q42.2 rs2004776 A/G A 0.26 0.67 0.54 ↑ – – AGT2p23.2 rs1275988 A/G A 0.60 0.23 0.08 ↓ – – KCNK32q11.2 rs7599598 A/G A 0.57 0.63 0.09 ↓ – – FER1L52q24.3 rs1446468 A/G A 0.53 0.47 0.96 ↓ – – FIGN
rs13002573 A/G G 0.25 0.40 0.11 ↓ – – FIGNrs16849225 C/T C 0.75 0.59 0.94 – ↑ – FIGNrs6749447 G/T G 0.28 0.72 0.58 ↑ – – STK39
2q32.1 rs16823124 A/G A 0.23 0.51 0.10 ↑ – – PDE1A3p25.3 rs347591 G/T G 0.33 0.23 0.51 ↓ – – HRH1-ATG73p24.1 rs13082711 C/T T 0.80 0.94 0.96 ↓ – – SLC4A
rs820430 C/T T 0.64 0.40 1.00 ↑ – –3p22.1 rs9815354 A/G/T A 0.23 0.12 0.17 ↑ ↑ – ULK4
rs3774372 C/T T 0.77 0.87 0.81 ↓ – –rs1717027 C/T T 0.22 0.12 0.66 ↑ – –
3p21.31 rs319690 A/G A 0.51 0.75 0.41 ↑ – – MAP4rs7651237 A/G G 0.64 0.94 0.88 ↑ – –
3p21.1 rs9810888 G/T G 0.53 0.59 0.46 – ↑ – CACNA1D3q26.1 rs16833934 A/G G 0.37 0.17 0.65 ↓ – – MIR12633q26.2 rs419076 G/T T 0.48 0.13 0.57 ↑ – – MECOM4q12 rs871606 A/G A 0.87 0.78 0.76 ↑ ↑ – CHIC24q21.21 rs16998073 A/T T 0.19 0.30 0.05 ↑ ↑ – FGF5
rs1458038 A/G A 0.27 0.34 0.05 ↑ – –4q24 rs13107325 A/C/T T 0.10 0.00 0.00 ↓ – – SLC39A84q25 rs6825911 C/T C 0.20 0.48 0.54 – ↑ – ENPEP, PITX24q32.1 rs13139571 A/C C 0.74 0.68 0.88 ↑ – – GUCY1A3-GUCY1B35p13.3 rs1173771 C/T C 0.51 0.57 0.81 ↑ – – NPR3-C5orf23
rs7733331 C/T T 0.50 0.42 0.42 ↓ – –rs1173766 C/T C 0.52 0.59 0.62 ↑ –
5q33.3 rs11953630 A/C/T T 0.34 0.06 0.15 ↓ – – EBF16p22.2 rs1799945 C/G G 0.18 0.04 0.00 ↑ ↑ – HFE
rs198823 G/T T 0.65 0.23 0.60 ↓ – –6p21.33 rs805303 C/T C 0.70 0.57 0.30 ↑ – – BAG1
rs2021783 C/T C 1.00 0.81 1.00 – ↑ – CYP21A26p21.32 rs2854275 G/T T 0.08 0.03 0.08 ↓ – – HLA-DQB16p21.1 rs10948071 C/T T 0.70 0.67 0.05 ↓ – – CRIP36q22.33 rs13209747 C/G/T T 0.45 0.48 0.12 ↑ ↑ ↑ RSPO36q25.1 rs17080102 C/G C 0.06 0.01 0.09 ↓ ↓ ↓ PLEKHG17p15.2 rs17428471 G/T T 0.08 0.05 0.14 ↑ ↑ ↑ EVX1-HOXA7p12.3 rs2949837 A/T A 0.23 0.61 0.00 ↑ – – IGFBP37q21.2 rs2282978 C/T C 0.36 0.06 0.43 ↑ – – CDK67q22.3 rs17477177 C/T T 0.72 0.91 0.93 ↓ – – PIK3CG
rs12705390 A/G G 0.72 0.91 0.93 ↓ – –7q36.1 rs3918226 C/T T 0.10 0.00 0.00 ↑ – – NOS38p23.1 rs4841569 A/G G 0.57 1.00 0.89 ↑ – – BLK-GATA4
rs2898290 C/T C 0.58 0.02 0.55 NR – –8q24.12 rs2071518 C/T T 0.20 0.19 0.60 ↑ – – NOV10p12.31 rs11014166 A/T A 0.63 0.97 0.89 ↑ ↓ – CACNB2
rs1813353 A/G A 0.65 0.92 0.85 ↑ – –rs4373814 C/G G 0.63 0.53 0.43 ↓ – –rs12258967 C/G C 0.64 1.00 0.73 ↑ – –
10q21.2 rs1530440 C/T T 0.16 0.20 0.02 ↓ – – C10orf107rs4590817 C/G G 0.82 1.00 0.82 ↑ – –rs12244842 G/T T 0.25 0.19 0.35 ↓ – –rs7070797 A/G A 0.15 0.00 0.00 ↓ – –
10q22.2 rs4746172 C/T C 0.23 0.56 0.19 ↑ – – VCL10q23.33 rs932764 A/G G 0.43 0.58 0.15 ↑ – – PLCE110q24.32 rs1004467 C/T T 0.92 0.67 0.81 ↑ – – CYP17A1-NT5C2
rs11191548 C/T T 0.92 0.72 0.99 ↑ ↑ –rs12413409 A/G G 0.92 0.72 0.98 – ↑ –rs4409766 C/T T 0.93 0.78 0.82 – ↑ –rs3824755 C/G C 0.07 0.23 0.17 ↓ – –
10q25.3 rs2782980 C/T T 0.27 0.15 0.44 ↓ – – ADRB1rs7076938 C/T C 0.33 0.17 0.45 ↓ – –rs1801253 C/G G 0.32 0.15 0.41 ↓ – –
11p15.5 rs661348 C/T C 0.45 0.58 0.11 ↑ – – LSP1-TNNT311p15.4 rs7129220 A/G G 0.89 1.00 0.95 ↓ – – ADM11p15.1 rs381815 A/C/T T 0.30 0.25 0.18 ↑ – – PLEKHA7

226 S. Padmanabhan et al. / Pharmacological Research 121 (2017) 219–229
Table 2 (Continued)
Chr SNP Genotype Coded allele Coded allele frequency BP effect Nearest gene(s)
European Asian African European Asian African
rs757081 C/G G 0.63 0.66 1.00 ↑ – – PIK3C2A, NUCB2,NCR3LG1
11p15.2 rs2014408 C/T T 0.20 0.21 0.03 ↑ ↑ ↑ SOX6rs4757391 C/T C 0.18 0.17 0.23 – ↑ –
11q13.1 rs4601790 A/G G 0.25 0.42 0.07 ↑ – – EHBP1L1rs3741378 A/G A 0.15 0.38 0.36 ↓ – – RELA
11q22.1 rs633185 C/G G 0.68 0.55 0.82 ↓ – – FLJ32810-TMEM13311q24.3 rs11222084 A/T T 0.40 0.07 0.26 ↑ – – ADAMTS812q13.13 rs7297416 A/C C 0.25 0.62 0.38 ↓ – – HOXC412q21.33 rs11105354 A/G G 0.12 0.42 0.11 ↓ – – ATP2B1
rs2681492 A/G A 0.88 0.58 0.84 ↑ – –rs2681472 C/T T 0.88 0.58 0.89 ↑ ↑ –rs17249754 A/G G 0.88 0.59 0.84 ↑ ↑ –
12q24.12 rs3184504 C/T T 0.45 0.00 0.00 ↑ – – SH2B3rs653178 A/G A 0.56 1.00 1.00 ↓ – –
12q24.13 rs11066280 A/T T 1.00 0.75 1.00 – ↑ – RPL6-ALDH212q24.21 rs35444 C/T T 0.59 0.73 0.55 ↑ – – TBX5-
TBX3rs2384550 A/G A 0.37 0.09 0.34 ↓ – –rs10850411 C/T T 0.72 0.46 0.66 ↑ – –rs1991391 C/T C 0.64 0.91 0.58 ↑ – –rs11067763 A/G A 0.89 0.61 0.65 – ↑ – MED13L
15q21.1 rs1036477 A/G G 0.10 0.42 0.61 ↓ – – FBN115q24.1 rs6495122 A/C A 0.38 0.78 0.78 ↑ – – CYP1A1-ULK3
rs1378942 G/T G 0.32 0.79 1.00 ↑ – –15q24.2 rs11072518 C/T T 0.34 0.42 0.44 ↑ – – COX5A
rs1133323 A/G A 0.59 0.17 0.00 ↓ – –15q26.1 rs2521501 A/T T 0.63 0.93 0.80 ↑ – – FURIN-FES16p12.3 rs13333226 A/G G 0.18 0.05 0.38 ↑ – – UMOD16q22.1 rs33063 A/G A 0.19 0.15 0.00 ↑ – – NFAT517q21.31 rs12946454 C/T T 0.71 0.62 0.80 ↑ – – PLCD317q21.32 rs17608766 C/T T 0.91 1.00 1.00 ↓ – – GOSR217q21.33 rs12940887 C/T T 0.41 0.09 0.03 ↑ – – ZNF652
rs16948048 A/G G 0.42 0.09 0.42 ↑ – –20p12.2 rs1327235 A/G G 0.52 0.48 0.53 ↑ – – JAG1
0.43
0.00
0.00
i[tatTrtSr[o
5(
actiaoc
5(
gs3
rs1887320 A/G A 0.58
20q13.32 rs6015450 A/G G 0.07
rs6092743 A/G A 0.06
es have found it to be a candidate gene for HTN and renal disease39–42]. Studies using mutant Dahl SS/MCW rats with a 6-bp dele-ion wherein the native proline-leucine-glutamate is replaced with
single glutamine, Sh2b3em1Mcwi, resulted in the modification ofhe phosphotyrosine peptide binding pocket of the SH2 domain.his significantly reduced the BP of the mutant rat, along witheduced renal damage and blunted infiltration of the immune cellso the kidneys. Again, transplantation of the bone marrow fromh2b3em1Mcwi mutants to Dahl SS/MCW rats fed at a 2% NaCl saltich diet led to a significant decrease in the MAP and kidney injury43]. Further studies in KO Sh2b3 mice also have implicated the rolef this gene in the development of HTN [44].
.2.2. Cytochrome P450 family 17 subfamily A member 1CYP17A1)
This gene encodes for cytochrome P450 which mediates thectivity of 17a-hydroxylase in the biosynthesis of mineralocorti-oids and glucocorticoids. It also mediates 17,20-lyase activity inhe sex-steroid biosynthesis [34]. Missense mutations in this gene ismplicated in congenital adrenal hyperplasia with apparent miner-locorticoid excess, salt retention and HTN caused by the deficiencyf 17a-hydroxylase. GWAS on HTN have found this gene to be asso-iated with systolic BP [42].
.2.3. Guanylate cyclase 1 soluble subunit alpha and betaGUCY1A3, GUCY1B3)
These genes encode the alpha and beta subunits of solubleuanylate cyclase (GC) which form a heterodimer to propagate NOignalling [45,46]. Activation of GCs also leads to the production of′,5′ cyclic GMP that blocks calcium influx and dephosphorylates
0.54 – ↑ –0.22 ↑ – – GNAS-EDN30.06 ↑ – – C20orf174
myosin light chains, resulting in smooth muscle relaxation [47,48].KO studies in mice have proved to cause HTN [49]. GWAS on Euro-pean ancestry have identified SNP rs13139571 on GUCY1A3-1B3 tobe associated with BP regulation [31].
5.2.4. Glutamyl aminopeptidase (ENPEP)This gene encodes for a glutamyl aminopeptidase converts
angiotensin II to angiotensin III, which then activates the AT2 recep-tor promoting vasodilation. A nonsense variant, rs33966350, in ahighly conserved region of this gene, is predicted to result in a trun-cated protein with either a reduced enzymatic activity or a targetto nonsense-mediated decay. This truncated protein is predictedto lead to a predominant angiotensin II signalling, which activatesthe AT1 receptor causing vasoconstriction, and ultimately resultin elevated BP [50]. This could be used as a plausible alternativetherapeutic target to ACE inhibitors [31]. ENPEP KO mice have beenfound to develop HTN [51]. GWAS has identified SNP rs6825911 tobe associated with BP in east Asians [37].
Other genes that are plausible candidates from GWAS includePleckstrin Homology Domain Containing A7 (PLEKHA7) [42,52–55];ATPase Plasma Membrane Ca2+ Transporting 1 (ATP2B1) [31,56]which encodes for a plasma membrane calcium/calmodulin-dependent ATPase, which pumps calcium from the cytosol into theECM; Solute Carrier Family 4 Member 7 (SLC4A7) which encodes foran electro-neutral sodium bicarbonate co-transporter [31]; NADPHOxidase 4 (NOX4) encodes for NADPH oxidase 4, which enhances
vasodilation [57]; phosphodiesterase 5A (PDE5A) encodes for phos-phodiesterase which is responsible for the hydrolysis of cGMP andis a target of hypertensive drug sildenafil [57]. Solute Carrier Fam-ily 14 Member 2 (SLC14A2), a target of the calcium channel blocker
ologic
deiAfhm
6
itibpagoottio
6
tEmUsmAcpgawmSo
gtcB
6
sbdtdbasaatsTo
S. Padmanabhan et al. / Pharmac
rug nifedipine [57]; Solute Carrier Family 8 Member A1 (SLC8A1)ncodes for a Na+–Ca2+ exchanger that alters cardiac contractil-ty and hypertrophy and is expressed in the cardiomyocytes [57];ldehyde Dehydrogenase 2 Family (ALDH2, mitochondrial) encodes
or the mitochondrial enzyme alcohol dehydrogenase 2 for alco-ol metabolism and has been associated with HTN through theodification of alcohol consumption [37].
. Future directions
Whilst high throughput genomic methods have resulted in thedentification of robust signals for blood pressure, the transla-ion into clinical applications have been limited. In this context,t is worthwhile recognizing that GWAS studies are population-ased studies focussing on associations at a population level. Otheroints to note are GWAS studies SNPs and phenotypes in isolationnd has so far ignored pleiotropy, environmental interactions andene–gene interactions. All these are crucial in the complex biol-gy of blood pressure and the era of GWAS has laid the foundationf robust association study methods which now needs to progressowards studies of pathways and networks and biological functionhat would allow population level discoveries to be relevant on anndividual level. Alongside this, efforts are ongoing on integratingther omics methods such as epigenomics and metabolomics.
.1. Epigenomics and hypertension
Epigenetics refers to heritable changes in gene expressionhat does not involve changes to the underlying DNA sequence.pigenetic mechanisms involve modification of DNA throughethylation, histone modification or effect of non-coding RNA.nlike genomic variants, epigenetic alterations can be difficult to
tudy as they are often tissue or cell-type specific. A genome wideethylation study of leucocyte DNA in a small cohort of 8 African-merican hypertensive subjects and 8 normotensive age-matchedontrols showed no significant associations, but there was oneutative association between methylation of Sulfatase 1 (SULF1)ene and HTN [58]. In another study of 60 hypertensive patientsnd controls, lower levels of 5mC were observed in DNA of patientsith essential HTN [59]. In animal models, a study of renal outeredullary tissue from the Dahl S hypertensive rat model and the
S.13BN26 congenic strain showed differential methylation in 80%f the CpG islands in response to salt [60].
Whilst these studies offer exploratory support for the role of epi-enetics in HTN, the evidence that epigenetic changes can explainhe missing heritability is lacking [61–66] and further studies arelearly needed to assess the causal implications of epigenetics inP regulation.
.2. Metabolomics
Metabolomics is the systematic study of metabolites, which aremall molecules generated by the process of metabolism, and haseen important in elucidating the pathways underlying metabolicisorders. Metabolomic profiling of over 3000 adult twins iden-ified a putative novel pathway for BP regulation involving aicarboxylic acid (hexadecanedioate) with a causal role supportedy in vivo studies in rats [67]. The role of hexadecanedioate in
vascular mechanism for HTN is supported by evidence from atudy of pulmonary HTN, indicating a disruption of �-oxidationnd an increase of �-oxidation in this condition and pointing to
putative role in elevating pressure in both the systemic and
he pulmonary circulations [68]. The strongest genetic associationeen with hexadecanedioate maps to Solute Carrier Organic Anionransporter Family Member 1B1 (SLCO1B1), an association previ-usly reported in a metabolome-wide genetic study in Caucasiansal Research 121 (2017) 219–229 227
[69]. Targeted metabolomics profiling in the European Prospec-tive Investigation Into Cancer and Nutrition (EPIC)-Potsdam studyshowed higher concentrations of serine, glycine, and acyl-alkyl-phosphatidylcholines C42:4 and C44:3 tended to be associatedwith higher and diacyl-phosphatidylcholines C38:4 and C38:3 withlower predicted 10-year HTN-free survival [70]. Other metaboliteassociations with incident HTN and BP come from two US studieswhich found 4-hydroxyhippurate, a metabolic sex steroids pat-tern and 2 diacylglycerols 16:0/22:5 and 16:0/22:6 to be associatedwith BP and incident HTN [71,72]. Finally, Menni et al. showed 12metabolites to be strongly associated with pulse wave velocity withuridine, phenylacetylglutamine, and serine appearing to stronglycorrelate with PWV in women [73].
Some important factors to be addressed in future studies of BPas a quantitative trait would be to more accurately model BP in sub-jects on antihypertensive treatment by considering the number ofdrugs, drug dosage and adherence as well as the use of longitudinalBP data. Novel strategies are needed to efficiently discover causaland clinically useful genetic markers. The next level of discoverywill be more challenging as the molecular and functional dissec-tion of the novel variants require more detailed functional studiesin contrast to the high-throughput screening methods applied sofar. The identification of a genomic approach to discover novelpathways and/or predict response (or lack of response) to existinganti-hypertensive drugs will be of benefit in developing the novelclasses of drugs and may ultimately define an indication for themat an early stage in the treatment of HTN.
Appendix A. Glossary of terms
Phenotype
Phenotype or trait is the observable or measurable characteristicthat is the target of genetic dissection.
Genetic architecture
The genetic architecture of a trait refers to the number of dis-tinct alleles that impact on the trait variation, their frequencies andpenetrance.
Penetrance
Penetrance is the likelihood, or probability, that a particulargenotype will be expressed in the phenotype. A penetrance of 100%means that the associated phenotype always occurs when the cor-responding genotype is present, while a penetrance of 30% indicatesthat only 30% of those carrying a particular allele exhibit a pheno-type.
Heritability
Heritability is the proportion of phenotypic variation due tothe genetic differences between individuals within a population.Estimates of heritability can be used to describe the relative compo-nents of variance attributable to genetic factors and environmentalfactors. The heritability of a continuous trait is defined as the pro-portion of its total variance that is attributable to genetic factors ina particular population.
Linkage disequilibrium (LD)
LD can be simply defined as a non-random association betweenalleles at adjacent loci. That is, the presence of combination ofalleles or markers in a population more often or less often than

2 ologic
eumoteaplauratp
H
stc
T
huo
H
tge
L
mepbwedscsed
A
u(acfmctmp
[
[
[
[
[
[
[
[
[
28 S. Padmanabhan et al. / Pharmac
xpected if the loci were segregating independently in the pop-lation. When a variant is first introduced into a population byutation, it will be perfectly correlated with nearby variants, but
ver successive generations meiotic recombination will break uphe correlations, and LD will decay. The LD between two mark-rs within the same genomic region is commonly measured by thebsolute value of D′ and r2. The higher the value of D′, the lower theossibility that a recombination event occurred between these two
oci (D′ = 1 means that the two markers have not been separated by recombination event). The absolute value of r2 is more commonlysed to quantify and compare LD in the context of mapping. When
2 = 1, the two markers have not been separated by recombinationnd have the same allele frequency. In this case of perfect LD, thewo markers are completely linked and observation at one markerrovides complete prediction about the other.
aplotype
Haplotype is a linear arrangement of closely linked alleles on theame chromosome that is inherited as a unit. Individual’s geno-ypes at multiple tightly linked SNPs have two haplotypes, eachontaining alleles from one parent.
he International HapMap project
The international HapMap project (http://www.hapmap.org),as constructed genome-wide maps of LD patterns in multiple pop-lations. One of the main objectives of the project is to identify setf SNPs that are in LD blocks to allow more efficient genotyping.
ardy Weinberg equilibrium (HWE)
The Hardy–Weinberg equilibrium states that allele and geno-ype frequencies in a population will remain constant fromeneration to generation in the absence of other evolutionary influ-nces.
inkage studies
The concept of linkage analysis is to search for alleles or chro-osomal segments that are shared by affected relatives more than
xpected by random Mendelian segregation. These segments areassed entirely from the parents to the offspring without recom-ination at meiosis. Linkage analysis is only carried out in familiesith affected relatives, and involves genotyping of several mark-
rs that spread over the entire genome. Markers that flank theisease gene or mutation tend to highly segregate with diseasetatus in families. Identifying markers within such a segment thatonsistently accompanies the disease may indicate presence ofusceptibility genetic factors nearby these markers. However, pres-nces of such factors are neither necessary nor sufficient for theisease to develop.
ssociation studies
Association mapping is based on the idea that genetic variantsnderlying complex traits occur with a relatively high frequency>1%), have undergone little or no selection in earlier populationsnd are likely to date back to >100,000 years ago (common diseaseommon variant hypothesis). Association analysis has potentiallyar greater power than linkage analysis for detecting variants with
odest effect on disease risk, provided that the genetic marker is
lose enough to exhibit strong linkage disequilibrium (LD) withhe functional variant. Unless targeting a specific, known poly-orphism, all genetic association studies utilize one importantopulation-genomic feature in their design: linkage disequilibrium
[
al Research 121 (2017) 219–229
(LD). LD is an extremely useful feature, as it means investigatorsdo not need to genotype all polymorphisms in a region of inter-est. Instead, they can select a subset of SNPs that are proxies forthe majority of all common genetic variation nearby (so-called“tagSNPs”).
Genome-wide association studies (GWAS)
Genome-wide association studies (GWAS) offer a hypothesis-free approach that systematically tests hundreds of thousands ormore variants in the genome without prior knowledge of the loca-tion of the causal variants. GWA studies were made possible afterassembling human genetic variants in large human genome refer-ence projects such as the International HapMap Project, the HumanGenome Project, and the 1000 Genome Project. For GWAS stud-ies, large sample sizes are required to generate sufficient statisticalpower to overcome the multiple hypotheses that are tested. Thehigh number of false-positive results are addressed by stringentmultiple testing correction and seeking evidence from multiplereplication and validation studies of the top signals. GWAS are alsoblind to rare and structural variants.
References
[1] A.C. Guyton, T.G. Coleman, J.C. Fourcade, L.G. Navar, Physiologic control ofarterial pressure, Bull. N. Y. Acad. Med. 45 (9) (1969) 811–830.
[2] A.C. Guyton, T.G. Coleman, A.W. Cowley, J.-F. Liard, R.A. Norman, R.D.Manning, Systems analysis of arterial pressure regulation and hypertension,Ann. Biomed. Eng. 1 (2) (1972) 254–281.
[3] G.W. Pickering, The genetic factor in essential hypertension, Ann. Intern. Med.43 (3) (1955) 457–464.
[4] J.-G. Mongeau, P. Biron, C.F. Sing, The influence of genetics and householdenvironment upon the variability of normal blood pressure: the MontrealAdoption Survey, Clin. Exp. Hypertens. A 8 (4–5) (1986) 653–660.
[5] S.B. Harrap, M. Stebbing, J.L. Hopper, H.N. Hoang, G.G. Giles, Familial patternsof covariation for cardiovascular risk factors in adults: the Victorian FamilyHeart Study, Am. J. Epidemiol. 152 (8) (2000) 704–715.
[6] S.C. Hunt, R.R. Williams, G.K. Barlow, A comparison of positive family historydefinitions for defining risk of future disease, J. Chronic Dis. 39 (10) (1986)809–821.
[7] T.A. Kotchen, J.M. Kotchen, C.E. Grim, V. George, M.L. Kaldunski, A.W. Cowley,et al., Genetic determinants of hypertension: identification of candidatephenotypes, Hypertension 36 (1) (2000) 7–13.
[8] D. Levy, A.L. DeStefano, M.G. Larson, C.J. O’Donnell, R.P. Lifton, H. Gavras, et al.,Evidence for a gene influencing blood pressure on chromosome 17. Genomescan linkage results for longitudinal blood pressure phenotypes in subjectsfrom the Framingham heart study, Hypertension 36 (4) (2000) 477–483.
[9] R. Fagard, J. Brguljan, J. Staessen, L. Thijs, C. Derom, M. Thomis, et al.,Heritability of conventional and ambulatory blood pressures. A study intwins, Hypertension 26 (6 Pt 1) (1995) 919–924.
10] C. Fava, P. Burri, P. Almgren, L. Groop, U.L. Hulthen, O. Melander, Heritabilityof ambulatory and office blood pressure phenotypes in Swedish families, J.Hypertens. 22 (9) (2004) 1717–1721.
11] N. Kupper, G. Willemsen, H. Riese, D. Posthuma, D.I. Boomsma, E.J. de Geus,Heritability of daytime ambulatory blood pressure in an extended twindesign, Hypertension 45 (1) (2005) 80–85.
12] M. Caulfield, P. Munroe, J. Pembroke, N. Samani, A. Dominiczak, M. Brown,et al., Genome-wide mapping of human loci for essential hypertension, Lancet361 (9375) (2003) 2118–2123.
13] E.S. Lander, The new genomics: global views of biology, Science 274 (5287)(1996) 536–539.
14] J.V. Neel, Diabetes mellitus: a “thrifty” genotype rendered detrimental by“progress”? Am. J. Hum. Genet. 14 (4) (1962) 353–362.
15] V.L. Burt, P. Whelton, E.J. Roccella, C. Brown, J.A. Cutler, M. Higgins, et al.,Prevalence of hypertension in the US adult population. Results from the ThirdNational Health and Nutrition Examination Survey, 1988–1991, Hypertension25 (3) (1995) 305–313.
16] W. Ji, J.N. Foo, B.J. O’Roak, H. Zhao, M.G. Larson, D.B. Simon, et al., Rareindependent mutations in renal salt handling genes contribute to bloodpressure variation, Nat. Genet. 40 (5) (2008) 592–599.
17] S. Padmanabhan, M. Caulfield, A.F. Dominiczak, Genetic and molecularaspects of hypertension, Circ. Res. 116 (6) (2015) 937–959.
18] R.P. Lifton, A.G. Gharavi, D.S. Geller, Molecular mechanisms of human
hypertension, Cell 104 (4) (2001) 545–556.19] R.P. Lifton, R.G. Dluhy, M. Powers, G.M. Rich, S. Cook, S. Ulick, et al., Achimaeric 11 beta-hydroxylase/aldosterone synthase gene causesglucocorticoid-remediable aldosteronism and human hypertension, Nature355 (6357) (1992) 262–265.

ologic
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[Metabolomics and incident hypertension among blacks: the atherosclerosisrisk in communities study, Hypertension 62 (2) (2013) 398–403.
S. Padmanabhan et al. / Pharmac
20] B.I. Cerame, M.I. New, Hormonal hypertension in children:11beta-hydroxylase deficiency and apparent mineralocorticoid excess, J.Pediatr. Endocrinol. Metab. 13 (9) (2000) 1537–1547.
21] L.M. Boyden, M. Choi, K.A. Choate, C.J. Nelson-Williams, A. Farhi, H.R. Toka,et al., Mutations in kelch-like 3 and cullin 3 cause hypertension andelectrolyte abnormalities, Nature 482 (7383) (2012) 98–102.
22] F.H. Wilson, S. Disse-Nicodeme, K.A. Choate, K. Ishikawa, C. Nelson-Williams,I. Desitter, et al., Human hypertension caused by mutations in WNK kinases,Science 293 (5532) (2001) 1107–1112.
23] R.A. Shimkets, D.G. Warnock, C.M. Bositis, C. Nelson-Williams, J.H. Hansson,M. Schambelan, et al., Liddle’s syndrome: heritable human hypertensioncaused by mutations in the beta subunit of the epithelial sodium channel, Cell79 (3) (1994) 407–414.
24] D.B. Simon, F.E. Karet, J.M. Hamdan, A. DiPietro, S.A. Sanjad, R.P. Lifton,Bartter’s syndrome, hypokalaemic alkalosis with hypercalciuria, is caused bymutations in the Na-K-2Cl cotransporter NKCC2, Nat. Genet. 13 (2) (1996)183–188.
25] D.B. Simon, C. Nelson-Williams, M.J. Bia, D. Ellison, F.E. Karet, A.M. Molina,et al., Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemicalkalosis, is caused by mutations in the thiazide-sensitive Na-Clcotransporter, Nat. Genet. 12 (1) (1996) 24–30.
26] M.C. Zennaro, S. Boulkroun, F. Fernandes-Rosa, An update on novelmechanisms of primary aldosteronism, J. Endocrinol. 224 (2) (2015) R63–R77.
27] L. Fishbein, Pheochromocytoma and paraganglioma: genetics, diagnosis, andtreatment, Hematol. Oncol. Clin. North Am. 30 (1) (2016) 135–150.
28] S. Padmanabhan, O. Melander, T. Johnson, A.M. Di Blasio, W.K. Lee, D.Gentilini, et al., Genome-wide association study of blood pressure extremesidentifies variant near UMOD associated with hypertension, PLoS Genet. 6(10) (2010) e1001177.
29] S. Padmanabhan, Prospects for genetic risk prediction in hypertension,Hypertension 61 (5) (2013) 961.
30] T.A. Manolio, F.S. Collins, N.J. Cox, D.B. Goldstein, L.A. Hindorff, D.J. Hunter,et al., Finding the missing heritability of complex diseases, Nature 461 (7265)(2009) 747–753.
31] G.B. Ehret, P.B. Munroe, K.M. Rice, M. Bochud, A.D. Johnson, D.I. Chasman,et al., Genetic variants in novel pathways influence blood pressure andcardiovascular disease risk, Nature 478 (7367) (2011) 103–109.
32] K. Mutig, T. Kahl, T. Saritas, M. Godes, P. Persson, J. Bates, et al., Activation ofthe bumetanide-sensitive Na+,K+,2Cl− cotransporter (NKCC2) is facilitated byTamm-Horsfall protein in a chloride-sensitive manner, J. Biol. Chem. 286 (34)(2011) 30200–30210.
33] M. Trudu, S. Janas, C. Lanzani, H. Debaix, C. Schaeffer, M. Ikehata, et al.,Common noncoding UMOD gene variants induce salt-sensitive hypertensionand kidney damage by increasing uromodulin expression, Nat. Med. 19 (12)(2013) 1655–1660.
34] C. Newton-Cheh, T. Johnson, V. Gateva, M.D. Tobin, M. Bochud, L. Coin, et al.,Genome-wide association study identifies eight loci associated with bloodpressure, Nat. Genet. 41 (6) (2009) 666–676.
35] C. Newton-Cheh, M.G. Larson, R.S. Vasan, D. Levy, K.D. Bloch, A. Surti, et al.,Association of common variants in NPPA and NPPB with circulatingnatriuretic peptides and blood pressure, Nat. Genet. 41 (3) (2009) 348–353.
36] P. Arora, C. Wu, A.M. Khan, D.B. Bloch, B.N. Davis-Dusenbery, A. Ghorbani,et al., Atrial natriuretic peptide is negatively regulated by microRNA-425, J.Clin. Invest. 123 (8) (2013) 3378–3382.
37] N. Kato, F. Takeuchi, Y. Tabara, T.N. Kelly, M.J. Go, X. Sim, et al., Meta-analysisof genome-wide association studies identifies common variants associatedwith blood pressure variation in east Asians, Nat. Genet. 43 (6) (2011)531–538.
38] N. Matsukawa, W.J. Grzesik, N. Takahashi, K.N. Pandey, S. Pang, M. Yamauchi,et al., The natriuretic peptide clearance receptor locally modulates thephysiological effects of the natriuretic peptide system, Proc. Natl. Acad. Sci. U.S. A. 96 (13) (1999) 7403–7408.
39] C.A. Böger, I.M. Heid, Chronic kidney disease: novel insights fromgenome-wide association studies, Kidney Blood Press. Res. 34 (4) (2011)225–234.
40] T. Huan, T. Esko, M.J. Peters, L.C. Pilling, K. Schramm, C. Schurmann, et al., Ameta-analysis of gene expression signatures of blood pressure andhypertension, PLoS Genet. 11 (3) (2015) e1005035.
41] T. Huan, Q. Meng, M.A. Saleh, A.E. Norlander, R. Joehanes, J. Zhu, et al.,Integrative network analysis reveals molecular mechanisms of blood pressureregulation, Mol. Syst. Biol. 11 (4) (2015) 799.
42] D. Levy, G.B. Ehret, K. Rice, G.C. Verwoert, L.J. Launer, A. Dehghan, et al.,Genome-wide association study of blood pressure and hypertension, Nat.Genet. 41 (6) (2009) 677–687.
43] N.P. Rudemiller, H. Lund, J.R.C. Priestley, B.T. Endres, J.W. Prokop, H.J. Jacob,et al., Mutation of SH2B3 (LNK), a GWAS candidate for hypertension,attenuates Dahl SS hypertension via inflammatory modulation, Hypertension65 (5) (2015) 1111–1117.
44] B.L. Dale, M.S. Madhur, Linking Inflammation and Hypertension viaLNK/SH2B3, Curr. Opin. Nephrol. Hypertens. 25 (2) (2016) 87–93.
45] D.A. Baker, J.M. Kelly, Structure, function and evolution of microbial adenylyl
and guanylyl cyclases, Mol. Microbiol. 52 (5) (2004) 1229–1242.46] S. Yamagami, N. Suzuki, Diverse forms of guanylyl cyclases in medaka fish –their genomic structure and phylogenetic relationships to those invertebrates and invertebrates, Zoolog. Sci. 22 (8) (2005) 819–835.
[
al Research 121 (2017) 219–229 229
47] L.J. Ignarro, R.G. Harbison, K.S. Wood, P.J. Kadowitz, Activation of purifiedsoluble guanylate cyclase by endothelium-derived relaxing factor fromintrapulmonary artery and vein: stimulation by acetylcholine, bradykinin andarachidonic acid, J. Pharmacol. Exp. Ther. 237 (3) (1986) 893.
48] F. Murad, Cyclic guanosine monophosphate as a mediator of vasodilation, J.Clin. Invest. 78 (1) (1986) 1–5.
49] A. Friebe, E. Mergia, O. Dangel, A. Lange, D. Koesling, Fatal gastrointestinalobstruction and hypertension in mice lacking nitric oxide-sensitive guanylylcyclase, PNAS 104 (18) (2007) 7699–7704.
50] L. te Riet, J.H.M. van Esch, A.J.M. Roks, A.H. van den Meiracker, A.H.J. Danser,Hypertension, Circ. Res. 116 (6) (2015) 960.
51] M. Ishii, A. Hattori, Y. Numaguchi, X. Ma, T. Nagasaka, M. Tsujimoto, et al., Theeffect of recombinant aminopeptidase A (APA) on hypertension in pregnantspontaneously hypertensive rats (SHRs), Early Hum. Dev. 85 (9) (2009)589–594.
52] E.R. Fox, J.H. Young, Y. Li, A.W. Dreisbach, B.J. Keating, S.K. Musani, et al.,Association of genetic variation with systolic and diastolic blood pressureamong African Americans: the Candidate Gene Association Resource study,Hum. Mol. Genet. 20 (11) (2011) 2273–2284.
53] J.E. Ho, D. Levy, L. Rose, A.D. Johnson, P.M. Ridker, D.I. Chasman, Discovery andreplication of novel blood pressure genetic loci in the Women’s GenomeHealth Study, J. Hypertens. 29 (1) (2011) 62–69.
54] K.-W. Hong, H.-S. Jin, J.-E. Lim, S. Kim, M.J. Go, B. Oh, Recapitulation of twogenomewide association studies on blood pressure and essentialhypertension in the Korean population, J. Hum. Genet. 55 (6) (2010) 336–341.
55] Y. Lin, X. Lai, B. Chen, Y. Xu, B. Huang, Z. Chen, et al., Genetic variations inCYP17A1, CACNB2 and PLEKHA7 are associated with blood pressure and/orhypertension in She ethnic minority of China, Atherosclerosis 219 (2) (2011)709–714.
56] G.R. Monteith, E.P.W. Kable, T.H. Kuo, B.D. Roufogalis, Elevated plasmamembrane and sarcoplasmic reticulum Ca2+ pump mRNA levels in culturedaortic smooth muscle cells from spontaneously hypertensive rats, Biochem.Biophys. Res. Commun. 230 (2) (1997) 344–346.
57] H.R. Warren, E. Evangelou, C.P. Cabrera, H. Gao, M. Ren, B. Mifsud, et al.,Genome-wide association analysis identifies novel blood pressure loci andoffers biological insights into cardiovascular risk, Nat. Genet. 49 (3) (2017)403–415.
58] X. Wang, B. Falkner, H. Zhu, H. Shi, S. Su, X. Xu, et al., A genome-widemethylation study on essential hypertension in young African Americanmales, PLOS ONE 8 (1) (2013) e53938.
59] I. Smolarek, E. Wyszko, A.M. Barciszewska, S. Nowak, I. Gawronska, A.Jablecka, et al., Global DNA methylation changes in blood of patients withessential hypertension, Med. Sci. Monit. 16 (3) (2010) CR149–CR155.
60] Y. Liu, P. Liu, C. Yang, A.W. Cowley Jr., M. Liang, Base-resolution maps of5-methylcytosine and 5-hydroxymethylcytosine in Dahl S rats: effect of saltand genomic sequence, Hypertension 63 (4) (2014) 827–838.
61] X. Wang, B.P. Prins, S. Sober, M. Laan, H. Snieder, Beyond genome-wideassociation studies: new strategies for identifying genetic determinants ofhypertension, Curr. Hypertens. Rep. 13 (6) (2011) 442–451.
62] X. Wang, H. Snieder, Genome-wide association studies and beyond: what’snext in blood pressure genetics? Hypertension 56 (6) (2010) 1035–1037.
63] S. Friso, C.A. Carvajal, C.E. Fardella, O. Olivieri, Epigenetics and arterialhypertension: the challenge of emerging evidence, Transl. Res. 165 (1) (2015)154–165.
64] T.A. Kotchen, A.W. Cowley Jr., M. Liang, Ushering hypertension into a new eraof precision medicine, JAMA 315 (4) (2016) 343–344.
65] M. Liang, A.W. Cowley Jr., D.L. Mattson, T.A. Kotchen, Y. Liu, Epigenomics ofhypertension, Semin. Nephrol. 33 (4) (2013) 392–399.
66] A.W. Cowley Jr., J.H. Nadeau, A. Baccarelli, K. Berecek, M. Fornage, G.H.Gibbons, et al., Report of the National Heart, Lung, and Blood InstituteWorking Group on epigenetics and hypertension, Hypertension 59 (5) (2012)899–905.
67] C. Menni, D. Graham, G. Kastenmuller, N.H. Alharbi, S.M. Alsanosi, M. McBride,et al., Metabolomic identification of a novel pathway of blood pressureregulation involving hexadecanedioate, Hypertension 66 (2) (2015) 422–429.
68] Y. Zhao, J. Peng, C. Lu, M. Hsin, M. Mura, L. Wu, et al., Metabolomicheterogeneity of pulmonary arterial hypertension, PLOS ONE 9 (2) (2014)e88727.
69] K. Suhre, S.Y. Shin, A.K. Petersen, R.P. Mohney, D. Meredith, B. Wagele, et al.,Human metabolic individuality in biomedical and pharmaceutical research,Nature 477 (7362) (2011) 54–60.
70] S. Dietrich, A. Floegel, C. Weikert, T. Pischon, H. Boeing, D. Drogan,Identification of serum metabolites associated with incident hypertension inthe European prospective investigation into cancer and nutrition-potsdamstudy, Hypertension 68 (2) (2016) 471–477.
71] H. Kulkarni, P.J. Meikle, M. Mamtani, J.M. Weir, C.K. Barlow, J.B. Jowett, et al.,Plasma lipidomic profile signature of hypertension in Mexican Americanfamilies: specific role of diacylglycerols, Hypertension 62 (3) (2013) 621–626.
72] Y. Zheng, B. Yu, D. Alexander, T.H. Mosley, G. Heiss, J.A. Nettleton, et al.,
73] C. Menni, M. Mangino, M. Cecelja, M. Psatha, M.J. Brosnan, J. Trimmer, et al.,Metabolomic study of carotid-femoral pulse-wave velocity in women, J.Hypertens. 33 (4) (2015) 791–796, discussion 6.

本文献由“学霸图书馆-文献云下载”收集自网络,仅供学习交流使用。
学霸图书馆(www.xuebalib.com)是一个“整合众多图书馆数据库资源,
提供一站式文献检索和下载服务”的24 小时在线不限IP
图书馆。
图书馆致力于便利、促进学习与科研,提供最强文献下载服务。
图书馆导航:
图书馆首页 文献云下载 图书馆入口 外文数据库大全 疑难文献辅助工具





























