Embed Size (px)
Citation preview

Aus dem Department für Pathobiologie
Der Veterinärmedizinischen Universität Wien
Institut für Mikrobiologie
(Leiterin: Univ.-Prof. Dr. rer. nat. Dipl. Ing. agr. Monika Ehling-Schulz)
Genotypic analysis of selected antibiotic resistance patterns in Coxiella burnetii
Bachelorarbeit
Veterinärmedizinische Universität Wien
vorgelegt von
Maximilian F. Mayerhofer
Wien, im Juni 2018

Betreuung
Univ.-Prof. Dr. rer. nat. Dipl. Ing. agr. Monika Ehling-Schulz
Institut für Mikrobiologie, Veterinärmedizinische Universität Wien
Oberfeldarzt Priv.-Doz. Dr. med. Dimitrios Frangoulidis
Institut für Mikrobiologie der Bundeswehr, München

Acknowledgements
First of all, I would like to thank Oberfeldarzt Priv.-Doz. Dr. med. Dimitrios Frangoulidis for
the possibility to work in his group and the Fachgruppe Metagenomik for their oversight and
technical tutelage in the fields of bioinformatics.
In addition, I would like to express my gratitude to Prof Monika Ehling-Schulz for her
supervision and sparking my interest in the fields of microbiology and infectiology.
I owe my sincere gratitude to Brigadier General Michael Janisch and DI Helga Plicka for their
personal commitment and guidance in my scientific career.

Table of content
1. Summary of the bachelor´s thesis ...................................................................................................... 1
2. Introduction ........................................................................................................................................ 2
2.1. Q fever ............................................................................................................................................ 2
2.2. Military personnel – an underappreciated factor? ........................................................................ 4
2.3. Genomic Classification .................................................................................................................... 5
2.4. Antibiotic Resistance Mechanisms ................................................................................................. 8
2.5. Phenotypic antimicrobial Susceptibility-testing of C. burnetii ..................................................... 15
2.6. Aim of this study ........................................................................................................................... 17
3. Material and Methods ...................................................................................................................... 18
3.1. In NCBI published Genomes of strains used for sequence alignments ........................................ 18
3.2. Bioinformatic “Pipeline” ............................................................................................................... 20
3.3. ABRICATE with CARD, ARG-ANNOT, ResFinder ............................................................................ 26
3.4. Genotyping via DNA Simpleprobe™ probe assay ......................................................................... 26
3.5. Structural modelling of predicted beta lactamase ampC of C. burnetii ....................................... 29
4. Results ............................................................................................................................................... 30
4.1. ABRICATE – screening for resistance genes ................................................................................. 30
4.2. Alignment of candidate genes ...................................................................................................... 30
4.3. Genotyping of selected strains ..................................................................................................... 33
4.3.1. parE-PCR for specifity of SimpleProbe-assay ........................................................................... 33
4.3.2. SimpleProbe-analysis of parE of selected strains ..................................................................... 34
4.3.3. Beta lactamase ampC of C. burnetii ......................................................................................... 35
5. Discussion ......................................................................................................................................... 39
5.1. Absence of known resistance genes ............................................................................................. 39
5.2. parE polymorphism A1411G ......................................................................................................... 40
5.3. Beta lactamase ampC ................................................................................................................... 41
5.4. Outlook ......................................................................................................................................... 42
5.5. Deutsche Zusammenfassung der Bachelorarbeit ......................................................................... 43
5.6. Reference List ............................................................................................................................... 45
5.7. Appendix ....................................................................................................................................... 55

1
1. Summary of the bachelor´s thesis
The obligate intracellular organism C. burnetii is the causative agent of zoonotic Query (Q)
fever and poses challenges to the development of novel drugs due to its intracellular life cycle.
The infectious dose of C. burnetii can be as low as 1 to 10 single organisms and can be
distributed by wind as highly environmental stable spore-like particles. Human infection
usually occurs after contact with infected animals. Most infections occur due to contact with
highly contagious maternal blood during birth of farm animals. After an incubation period of
two to three weeks, flu-like symptoms with fever, loss of appetite, headache and atypical
pneumonia and hepatitis can develop. About 2 % of all cases develop chronic Q fever with the
potential of fatal endocarditis if untreated. Chronic Q fever therapy has a mortality of around
10 %. Most Q fever infections are not detected, since a stable immune system is able to fight
the infection without clinical symptoms. Therapy usually consists of tetracyclines, macrolides
and gyrase inhibitors.
Reported resistances against antimicrobial substances in C. burnetii have been scarce and
antibiotic susceptibility testing has primarily been done in a pre-sequencing era and so far, this
work is the first to examine various strains of C. burnetii for antibiotic resistance genes.
A pipeline was written using the command language Bash and the interpreted high-level
programming language Python. Via the stand-alone version of blast+, possible antibiotic
resistance determining genes were located in whole genome data of C. burnetii. These genes
were then aligned using the MAFFT algorithm. Additionally, ABRICATE was used to screen
for known antibiotic resistance genes using the databases CARD, ARGANNOT and Resfinder.
After analysis of genetic markers, a PCR-based single probe DNA assay was designed for a
reliable genotypic identification of potential antibiotic resistances in various strains

2
2. Introduction
The intracellular, Gram-negative bacterium Coxiella burnetii is the causative agent of the
zoonotic disease Query (Q) -fever. First described in 1937, it is the sole member of the family
Coxiellaceae in the order of Legionellales in the class of Gammaproteobacteria. Due to low
infectivity, high morbidity, difficult diagnosis and long environmental stability combined with
aerially transmission, C.burnetii is listed as a category B potential bioterrorism agent1 by the
Centers for Disease Control and Prevention (CDC) (Madariaga et al. 2003).
Human infection usually occurs through contact with cattle, sheep and goats. Its strict obligate
intracellular life cycle leads to various predicaments in the study of this organism, namely,
cultivation and testing of antibiotic. Since antibiotics have to be specifically designed for
intracellular uptake, antibiotic resistance of Coxiella burnetii poses a major threat in therapeutic
regimens. Reported resistances are scarce and most of the identified resistances lack the
knowledge of the exact molecular pathway and mechanisms. The following review gives an
overview on current used antibiotics, known resistances-mechanisms and potential outlooks on
future research and the impact on clinical therapies.
2.1. Q fever
Human infection with C. burnetii usually occurs by inhalation of spore-like small cell variants
(SCV) through contaminated animal products or direct contact. Coxiella burnetii is able to
infect a wide spectrum of vertebrate and invertebrate hosts and is nearly found all over the
world, except New Zealand. Beside the predominate hosts like mammals, cattle, sheep and
goats as farming animals, various wildlife has been observed contributing to the transmission
including kangaroos and wallabies (Stevenson et al. 2015) in Australia and three-toed sloths
(Edouard 2014) in Cayenne.
Coxiella is able to infect an abundance of arthropod species, including 40 hard ticks, at least
14 soft ticks, as well as flies, bed bugs and mites. C. burnetii does not seem to have any vector
specificity, not all of them were able to infect animal models. Alas, the exact part of arthropods
in Coxiella life cycle is still unknown and needs further investigation. Nonetheless the
epidemiological impact of ticks in Coxiella transmission remains controversial (Duron et al.
2015) with the recent discovery of coxiella-like endosymbionts in ticks suspected of causing
potential false-positive reports. Since previous detection was based on morphological
1 https://www.selectagents.gov/SelectAgentsandToxinsList.html

3
observations, staining and immunological methods, recent PCR-based and modern next
generation sequencing (NGS) techniques provide a more precise tool for diagnostics
(Frangoulidis et al. 2012). Acute Coxiella infections present themselves with unspecific clinical
symptoms or mild infection very often self-limiting, but in rare cases, can also result in severe
chronic illness, which needs a long lasting antibiotic treatment with poor outcome.
The role of foodborne Q fever in humans via the digestive route through consumption of dairy
products originating from Coxiella-infected animals is contentious at best. Although, C.
burnetii DNA has been regularly detected in up to 64 % of French dairy products (Eldin et al.
2013) no viable bacteria could be isolated. Fairly old studies concluded that pasteurization is
sufficient in elimination of C. burnetii (HUEBNER et al. 1949) and experiments of
consumption with infected milk did not lead to a development of infection or any detectable
antibody-mediated response (Krumbiegel and Wisniewski 1970) despite consumption of
pasteurized goat cheese being handled as an independent risk factor during an epidemic in
Newfoundland in Canada (Hatchette et al. 2001). In conclusion, consumption of infected milk
products may not play a direct role in human infection, but may contribute to the transmission
of C. burnetii from cattle to cattle (Guatteo et al. 2006, Rodolakis et al. 2007).
After inhalation, C. burnetii usually targets alveolar macrophages through passive actin-
dependent phagocytosis. αβ-3 integrin is the main receptor for cellular uptake and is usually
involved in phagocytosis of apoptotic cells and associated with inhibition of inflammation. C.
burnetii may exploit this as avoidance of induction of the inflammatory response machinery.
Phase variants seem to have an impact on uptake kinetics, whereas nearly avirulent phase II
exhibits faster internalization than the virulent phase I counterpart. The Coxiella containing
vacuole (CCV) promotes a series of fusion and fission events with various effectors being
secreted through T4 secretion system. The acquisition of RAB GTPases leads to acidification
and the maturation of the vacuole to the phagolysosome.
Previous cultivation was usually done in Vero cells, but recent developments on axenic or cell
free culture models changed research and lead to less labour-intensive cultivation protocols.
The newest axenic media allows direct cultivation of infected animal tissue homogenates
(Omsland et al. 2011).
Potential infectious doses range from 1-10 organisms in the lungs and acute Q fever manifests
after 2 to 6 weeks after initial exposure. Although over 50 % of cases linger asymptomatic,
symptomatic disease is defined by severe retro-orbital headache, high fever, myalgia, malaise,
pneumonia and hepatitis lasting 1 to 2 weeks (Fournier and Marrie 1998).

4
Some studies suggest that 1 to 2 % of acute cases manifest in chronic Q fever with endocarditis,
hepatitis or an imprecise defined post-Q fever fatigue syndrome (Keijmel et al. 2013, Morroy
et al. 2016, Marmion et al. 2018). Until now, the most accurate definition has been phase-
dependent antibody titers, with high levels of Phase I antibody usually corresponding to chronic
Q fever. The classification of acute versus chronic Q fever was recently changed by Million
and Raoult 2017 to acute vs persistent C. burnetii infection. But up until now, no complete
consent in this issue has been made.
The last major outbreak of Q fever was displayed in infections in the Netherlands from 2007-
2010 with over 4000 reported cases. Noord-Brabant province was the centre of the outbreak
with the highest rates of infection, possibly due to extensive growth of dairy goat farming in
recent years and proximity to urban areas (Dijkstra et al. 2011).
Abortion rates of up to 90 % (Brom et al. 2015) in an abundance of farm animals combined
with long lasting shedding numbers (Rodolakis 2009) of Coxiella in vaginal blood and milk
(Rodolakis et al. 2007) may have had a major impact in this outbreak.
2.2. Military personnel – an underappreciated factor?
Since military organizations often operate in remote areas without direct availability of the
complete spectrum of medical support, they are at an extraordinary risk of acquiring infectious
diseases. “Undifferentiated fever”, “unexplained febrile illness”, “acute fever of unknown
origin” or related terms and combinations are usually used to describe these short-duration (less
than 2 weeks) and often clinical indistinct illnesses and therefore, a primarily symptom-
descriptive diagnosis is insufficient for therapy decision. Another problem arises, due to the
fact that not all military field hospitals have the capability of full spectrum laboratory
diagnostics at their disposal (Bailey et al. 2011). But Q fever outbreaks in a military context are
nothing new. During World War 2, allied forces in Italy experienced ‘primarily atypical
pneumonia’ which is now thought to be cases of Q fever2. With recent missions on Iraq and
Afghanistan, Q fever-related pneumonia had a revival among infectious illnesses: In 2011 a Q
fever and brucellosis outbreak in Bamyan province in Afghanistan highlighted the inherent risk
of contracting rickettsial-like diseases once more. To tackle risk assessment, retrospective
studies by the Naval Medical Research Unit 2 found 117 positive cases with detectable
antibodies against either phase I or phase II antigens of C. burnetii of a total of 879 samples in
Marines deployed to Afghanistan from 2001 to 2010 (Farris et al. 2016). The rate of
2 http://nzetc.victoria.ac.nz/tm/scholarly/tei-WH2Surg-pt2-c15.html , Accessed 8.3.2018

5
seroconversions in US Marines (3.4 %) were similar to findings for the UK military (3.1%)
during deployment to Helmand Province from 2008-2011. The thus-termed ‘Helmand Fever’
summarizes infections with Rickettsia ssp., Coxiella burnetii, sandfly fever virus, hantavirus
and Crimean-Congo hemorrhagic fever virus. In 2008, six of a reported 26 cases of Helmand
fever were identified to be Q fever (Bailey et al. 2011). It should however be noted that some
caution is required when dealing with figures that largely rely on self-reporting: previous
research has shown that values associated with military culture often leads soldiers to
underreport health issues. Correspondingly, only just 90 of the 467 UK service members
reported flu-like or unwell feeling to their chain of command (Bailey et al. 2011).
With these combined factors of uncertainty in Q fever diagnostics and treatment during military
operations, chronic Q fever developments pose a threat after deployment.
2.3. Genomic Classification
The first complete genome of C. burnetii was created from strain RSA493 (Nine Mile) in 2003
(Seshadri et al. 2003). Due to the intracellular life cycle of C. burnetii and the therefore
dependency on organic nutrients of the host cell, the genome contains a high number of
transporters. Additional there are many ionic exchangers present, possible to detoxify the
phagolysosome-like vacuole containing C. burnetii. A recent pangenomic (Amato et al. 2015)
analysis showed that strains of C. burnetii exhibit strong genomic similarity with a core
genome/pangenome ratio of 96%. Nonetheless, a few distinct properties have been observed:
C. burnetii strain Cb175 from Cayenne, French Guiana, isolated from a patient with severe
endocarditis featured a 6,105 bp deletion compared to the reference strain Nine Mile. This
genome reduction is not present in any of the other 298 C. burnetii isolates. A rather speculative
explanation was proposed by Fournier et al. 2009 due to deletion of nonvirulence genes in
hyperpathogenic strains, rather than acquisition of virulence factors.
The gold standard for genotyping of C. burnetii consists of three distinct methods: multispacer
sequence typing (MST), single nucleotide polymorphism (SNP) genotyping and Multiple-locus
variable-number tandem repeat (VNTR) analysis (MLVA). MLVA, being based on number of
repeats units being present in eukaryote or prokaryote genomes (Van Belkum 2007), has already
been used for identification of potential agents of bioterrorism in the past (Klevytska et al. 2001,
Lista et al. 2006) with new systems being developed for faster and more accurate genotyping
and identification (Farlow et al. 2001, Johansson et al. 2001). Beside the conventional agarose-
gel-based method, a chip-based assay has been reported (Prigent et al. 2015). Since its

6
development, a dutch-based MLVA database for C. burnetii typing has been released3 that
enable quick ‘online’ comparison. However, the abundant presence of the mobile element
IS1111 in the genome of C. burnetii may negatively impact the transferability between
laboritories and therefor overall translatability in the epidemiological context. Partial genotypes
(Ceglie et al. 2015), ranging from 22% to 78% of analyzed samples, amplification failures or
unexpected amplicon sizes (Prigent et al. 2015) and IS1111-based genome heterogeneity call
for further standardization and harmonization constraining C. burnetii MLVA genotyping
schemes (Sidi-Boumedine et al. 2015) . A four-year surveillance report following the Dutch
outbreak by the Belgian Federal Agency for the Safety of the Food Chain (FASFC) covering
2009 to 2013 identified CbNL01 as the responsible clone for human Q fever cluster cases, as
well as identified an emerging CbNL01-like genotype circulating among goat farms and
assessed the mandatory vaccination on these positive farms as a contribution in the decrease of
shedding (Boarbi et al. 2014).
MST genotyping classifies 30 different genotypes and three monophyletic groups among 173
isolates through discrimination of 10 highly variable spacers between open reading frames
(ORF) (Glazunova et al. 2005). Plasmid types were also partially considered. Though still
incomplete, MST genotyping was used in seventeen studies so far. MST genotyping determined
the source of the Netherland outbreak as MST 33 in goats rather than cows, which harboured
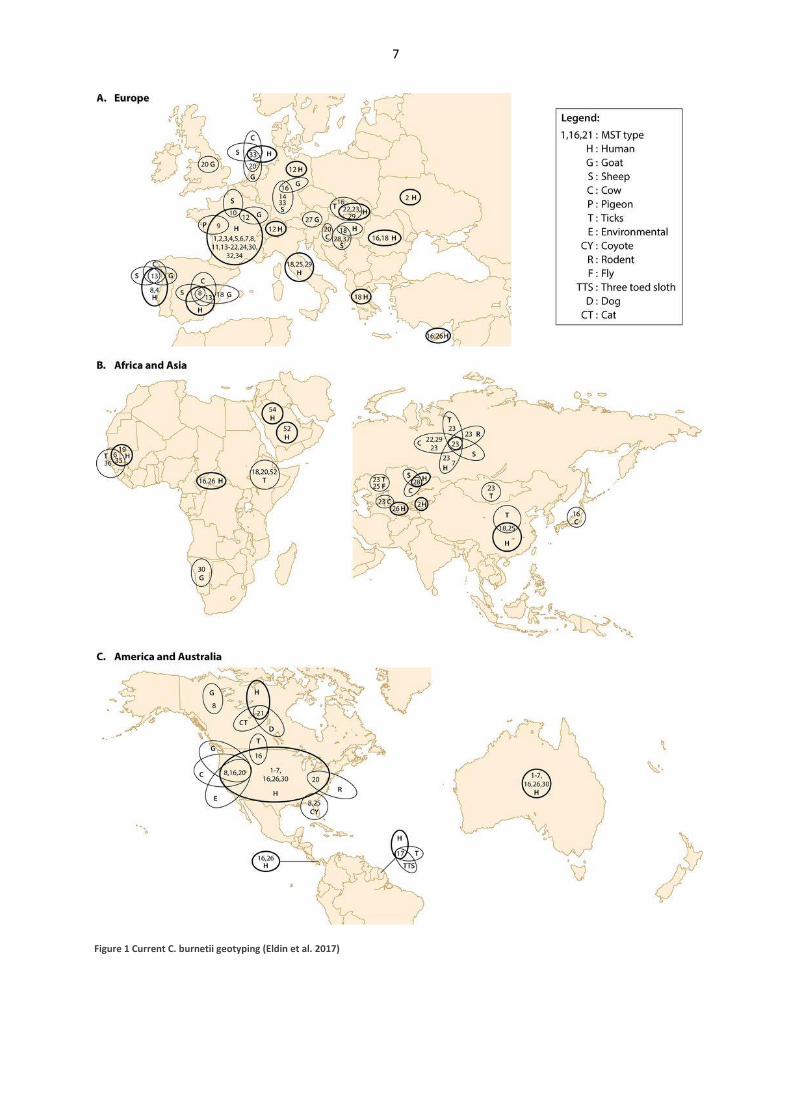
MST 20 (Fig 1, Eldin et al. 2017) and reinforced the Dutch results. The first SNP genotyping
was used during the Netherland outbreak as a fast and direct applicable method for samples
without the need for time-consuming enrichment by culture (Huijsmans et al. 2011). Hornstra
et al. (2011) reported a SNP method developed on MST genotyping by extracting SNP
signatures of MST loci resulting in a 14 SNP-base assays.
3http://mlva.u-psud.fr/mlvav4/genotyping/view.php
7
Figure 1 Current C. burnetii geotyping (Eldin et al. 2017)

8
2.4. Antibiotic Resistance Mechanisms
“There may be a danger, though, in underdosage. It is not difficult to make microbes resistant
to penicillin in the laboratory by exposing them to concentrations not sufficient to kill them,
and the same thing has occasionally happened in the body.”
- Alexander Fleming, Nobel lecture, 1945
Through evolution and natural exposure and in recent years, over usage, inappropriate
description, extensive agricultural usage and reduced availability of novel therapeutics due to
regulatory barriers in development, resulting in declining broad-level research (Ventola 2015),
bacterial organisms are able to gain various forms of antimicrobial escape mechanisms. Four
different mechanisms can be described (Table 1): Modifications of antimicrobial molecules
including chemical alterations or enzymatic degradation, decreased antibiotic penetration
and efflux including decreased permeability through cytoplasmic membrane and alterations or
active removal with efflux pumps, changes in target site including target protection with
specific proteins or modifications of target site like amino acid switch or methylation and
resistance due to global cell adaptions, which are not yet fully understood (Munita et al.
2016).
The intracellular life cycle of C. burnetii proposes a considerable challenge in the design of
novel therapeutics and the natural or intentional occurrence of resistant strains poses a threat to
public health Adjacent to the intracellular uptake, drugs have to be constructed for the high
acidic environment of all phases of phagocytosis, ranging from early phagosomes (pH 5,4) to
mature phagolysosomes (pH 4,5).

9
Table 1 Modes of resistance and candidate genes
Antibiotic agents Target Mode of Resistance Candidate Genes
Tetracyclines
Ribosome Enzymatic Degradation
Ribosomal Protection
Efflux
Tet genes
Gyrase inhibitors Bacterial DNA Gyrase Target alteration gyrA, gyrB, parC, parE
Beta lactam Peptidoglycan synthetase Target alteration
enzymatic degradation
Overexpression
PBP1, PBP2, PBP3
amp genes
Macrolides 50S ribosomal subunit Target alteration
Efflux
23SrRNA
erm, mef,
Rifampicin Bacterial polymerase Target alteration
Efflux
rpoB
Cotrimoxazole Folate pathway Target alteration sul1, sul2, sul3
dfr

10
2.4.1. Tetracyclines
The group of tetracyclines include a series of polycyclic antibiotics with a broad spectrum of
activity against both Gram-positive and Gram-negative organisms. Its name is derived from the
linear fused nucleus of four rings. Being one of the most widely used antibiotics in the last
century due to wide spectrum of activity, low toxicity and low production cost, it is now
discontinued as a first-line therapy in general bacterial infection since the rise of widespread
resistance. Tetracyclines target both mammalian (80s) and bacterial (70S) ribosomes. The
chemical derivates of Glycylcyclines are binding with higher affinity to the ribosome and are
able to evade tetracycline-specific Efflux Tet(A-E) proteins and ribosomal protection proteins
Tet(M) (Allgaier et al. 2013) but are still transported by chromosomally-encoded multidrug
efflux pumps of Pseudomonas aeruginosa (Dean et al. 2003) and degraded by mono-
oxygenase Tet(X) (Yang et al. 2004).
Doxycycline has been the primary choice of treatment for both acute and chronic Q fever with
MIC ranging from 0.05 to 2 μg/ml (Kersh 2013). Due to structural differences compared to
tetracycline, Doxycycline has a higher lipophilicity and tissue penetration, resulting in lower
oral doses and GI side effects (Sloan and Scheinfeld 2008). Although Doxycycline has been the
treatment of choice, it is not feasible for pregnant women or children and rarely anaphylactic
and immunogenic reactions have been reported (Fernando and Hudson 2013). A Q fever patient
in Israel has been successfully treated by desensitization of Doxycycline after immunogenic
reactions and initial unsuccessful treatment with azithromycin (Caplunik-Pratsch et al. 2018).
Tigecycline is a Tetracycline-derived semisynthetic antibiotic glycylcycline as a response to
the rise of antibiotic resistance. It is inter alia used as treatment for Legionella pneumophilia, a
close relative to C. burnetii as well as a last resort in the therapy of Methillin-resistant
Staphylococcus aureus.
Spyridaki et al. (2009) reported the use of Tigecycline against six isolates of C. burnetii of
Greek origin and two reference strains, Q212 and Nine Mile with MIC of 0.25 μg/ml.
Tetracycline resistance in human pathogens is almostalways acquired via horizontal gene
transfer. The broad use in clinical and agricultural environments for the past 60 years lead to
selection of various tetracycline resistance genes. In 2015, Forsberg et al. 2015 proposed the
finding of a novel family of tetracycline-inactivating enzymes: tetracycline destructases.
As a special form of resistances genes, there is a variety of resistances against tetracyclines
conferred through transposable elements (TE). They belong to the Tn916 transposon family
and carry the tet(M) gene.

11
Until today, there have been 59 different tetracycline genes (tet genes, tet(M), tet(O), tet(Q),
tet(L), tet(X)) reported. The major modes of resistance are ribosomal protection, efflux and
enzymatic inactivation (Marosevic et al. 2017).
Strains with acquired resistance against doxycycline have been described in three different
cases. The first reported doxycycline-resistant strain (Cb109) has been isolated from a patient
that suffered fatal C. burnetii endocarditis. No underlying sequence for resistance could be
annotated to this genome (Rouli et al. 2012). Two additional isolates have been described as
doxycycline-resistant, one goat isolate and one human patient with acute Q fever (Rolain et al.
2005) with no published genome sequence up to day.
2.4.2. Gyrase inhibitors
Fluorquinolones are based on 4-quinolone and have excellent anti-Gram-negative bacterial
activity. Due to their uptake in the cerebrospinal fluid, they have been recommended also for
treatment of meningitis. They include second-generation quinolones like Ciprofloxacin and the
third-generation quinolone Levofloxacin and fourth-generation quinolone Moxifloxacin.
Fluorquinolones inhibit DNA replication by targeting DNA Topoisomerase IV and DNA
gyrase. The current model explains interaction as binding of quinolones to the enzyme-DNA
complex at the unpaired bases of the unwound strand with hydrogen bonds. Thus preventing
DNA gyrase from relegating the cleaved DNA and accumulating DNA double-strand breaks.
Fluorquinolones are inhibited by acidic settings (pH<6) (Allgaier et al. 2013) Beside standard
mode of delivery Liposome-encapsulated Ciprofloxacin has been used in animal models as an
alternative therapy of Q fever. Intra nasal liposome-encapsulated antibiotics results in high
concentration in the lungs, since unencapsulated Ciprofloxacin is quickly reabsorbed and
cleared. In comparison to Doxycycline, mice showed minimal effects on body weight after
seven days (Norville et al. 2014). Although Ciprofloxacin has been successfully used to treat Q
fever endocarditis (Stunkard et al. 1990, Zekanovic et al. 2010) it is not used as mono therapy.
The common mechanism of high fluoroquinolone resistance occurs through mutations in the
target region, typically in genes that encode for topoisomerase IV (parC and parE) or gyrase
(gyrA and gyrB), This conserved region of resistance-leading mutations is called quinolone
resistance-determining region (QRDR) and is based on previously reported resistance
associated mutations. Most often, this leads to mutations at serine 83 (E. coli numbering) within
gyrA for Gram-negative bacteria (Fu et al. 2013) and reduce the binding efficiency of
fluoroquinolones. Vranikis et al. reported amino acid substituation in gyrA mutations leading

12
to Asp87Gly, gyrB mutations leading to Ser431Pro and Met518Ile, and parC mutations leading
to Asp69Asn, Thr80Ile and Gly104Ser (Spyridaki et al. 2000a, Vranakis 2010).
Resistance to Perfloxacin in Coxiella was reported as a point mutation in gyrA gene at codon
87 (E.coli numbering) and allowed PCR-restriction fragment length polymorphism (PCR-
RFLP) detection (Spyridaki et al. 2000b, 2002). Data suggests, that the causative basis of
resistance is not an energy-dependent efflux pump, since no differences in the presence or
absence of carbonyl cyanide m-chlorophenylhydrazone (CCMC), a metabolic stressor, have
been found as this metabolic stress would have had an impact on efflux capabilities.
Additionally, no difference in outer membrane proteins (OMPs) could be detected by SDS-
Page (Spyridaki et al. 2002).
Resistance against Ciprofloxacin in E. coli has been connected to amino acid changes at codons
416 (Leu → Phe), 444 (Ile → Phe), 445 (Leu → His), 458 (Ser → Ala), 460 (Glu → Asp), 464
(Ile → Phe) and 476 (Asp → Asn). and 529 (Ile → Leu) of the parE gene, but the relation to
fluoroquinolone resistance of gyrA mutations is not well understood, since these regions of
parE. lay outside of QRDRs. If mutations of parE solely or partly are the reason for quinolone
resistance or if their co-occurance to gyrA mutations play a role in increased fluoroquinolone
resistance (64 µM to 128 µM) is still a pending question (Bansal and Tandon 2011, Nawaz et
al. 2015).
Transmissible quinolone-resistances are often encoded as mobile elements and known as
plasmid-mediated quinolone resistance (PMQR) genes (Redgrave et al. 2014) but have not been
found in C. burnetii yet.

13
2.4.3. Beta lactam
Beta Lactam antibiotics and their derivates contain a beta-lactam ring and inhibit bacterial
peptidoglycan synthetase by irreversibly binding to penicillin binding proteins (PBPs) and
therefore halting final crosslinking of peptidoglycan layers during cell wall synthesis (Fisher et
al. 2005).
Resistance to beta-lactam antibiotics is mostly conferred by either acquisition of beta
lactamases, changes in structure of bacterial cell wall or in PBPs. The beta-lactamase ampC has
been predicted by homology in C. burnetii Dugway 5J108-111 (Beare et al. 2009). In 1999 a
group successfully transformed C. burnetii to Ampicillin resistance using a C. burnetii-E.coli
shuttle vector by inserting the beta lactamase blaM-gene (Suhan and Thompson 2000).
Although, no genetic base of beta lactam resistance could be determined up to date, beta lactam
antibiotics have not been useful in clinical therapy and in in-vitro susceptibility testing (Raoult
et al. 1991).
Ampillicin has been shown to inhibit growth of C. burnetii (Brennan and Samuel 2003) due to
good intracellular uptake, although clinical relevant efficiency in Q fever is lacking. Both low
oral uptake and occurrence of gastrointestinal disorder lead to the replacement with newer
derivates which have not been tested for suitability for C. burnetii (Allgaier et al. 2013).
2.4.4. Macrolide
Macrolides are targeting50S subunit of 70S ribosomes, inhibiting protein synthesis by blocking
the translocation of peptidyl-tRNA and in the end, resulting in bacteriostasis. Telithromycin is
distinguished from Erythromycin in its different chemical group on the Lacton-Ring (Rolain et
al. 2005, Allgaier et al. 2013).
Macrolide resistance genes are usually summarized in the group of MLS antibiotics
(Macrolides, Lincosamide, Streptogranine) and contain a total of 92 genes. There is a rough
classification into three different groups: methylation of rRNA as a target modification, active
efflux and inactivation of antibiotics. These resistance genes include the following gene
variants: erm (erythromycin rRNA methylase/MLSb Resistance), mef (macrolide efflux), msr
(macrolide and streptogramin B efflux), lsa (efflux for lincosamide and streptogramin A) and
the Tn916 transposon family associated erm(B) and MEGA (macrolide efflux genetic assembly)
gene (mef(E)-msr(D)operon) (Marosevic et al. 2017).
Macrolide resistance is also conferred through mutations in domain V of the 23S ribosomal
RNA (23SrRNA) at position 2057, 2058, 2059 and 2611 and through amino acid changes in

14
rplD (L4) gene at codon 62 – 66 and in rplV (L22) at codon 87 - 91 in L. pneumophilia
(Descours et al. 2017).
Eldin et al. 2015 were the first to describe susceptibility for Telithromycin in a strain of Coxiella
MST 17 from Cayenne, French Guiana.
Furthermore resistance against Erythromycin was described in 13 clinical isolates of C. burnetii
of French Guiana origin (Raoult et al. 1991, Rolain et al. 2005).
2.4.5. Rifampicin
Rifampicin mainly targets DNA-dependent RNA polymerase (Allgaier et al. 2013) and data
suggests Rifampicin binds to the pocket of RNA polymerase (RNAP) beta subunit (Campbell
et al. 2001). Resistance to rifampicin is either through changes in RNAP beta subunit or
plasmid-mediated efflux mechanisms or permeability changes (Louw et al. 2009, Goldstein
2014). Rifampicin-resistance conferring mutations in the rpoB gene usually occur in the RNA-
binding domain 3, which is highly conserved across species (Nielsen et al. 2000).
Rifampicin is used as a treatment for Legionnaires disease and tuberculosis. Since its strong
ability to induce the hepatic cytochrome p450 enzyme system it is increasing the metabolism
of other drugs. No clinical or in vitro resistance against Rifampicin has been reported in
Coxiella. It has been used in a failed Q fever endocarditis therapy of a 38 year old worker after
tetracycline withdrawal and did not supress C. burnetii (Subramanya et al. 1982) . This seemed
to have been a desperate deed, since Rifampicin has not played a major role in Q fever therapy
ever since.
2.4.6. Trimethroprim/Sulfamethoxazole (“Co-trimoxazole”)
Co-trimoxazole inhibits the folate synthesis pathway. Sulfamethoxazole interferes with p-
amonibenzoic acid (PABA), an intermediate in the de novo synthesis of dihydrofolate.
Trimetroprim inhibits dihydrofolate reductase (DFHR), thereby halting the production of
tetrahydrofolate. This combination shuts down the de novo synthesis of folate in Gram-negative
and Gram-positive bacteria. Its good toleration, cheap production costs and abundant
availability lead to extensive use in poor countries, which lead to decreased susceptibility of
Gram-negative bacteria. Reports from Tanzania showed up to 96% of Gram-negative bacteria
in bloodstream infections, urinary tract infections and surgical site infections being resistant to
Cotrimoxazole (Kayange et al. 2010, Manyahi et al. 2014).

15
Resistance against Cotrimoxazol is primarly mediated through plasmids. Sul1, sul2, sul3 genes
encode for variants of enzymes that are not affected by sulfamethoxazole. Over 30 variants of
dfr gene have been reported, conferring to an additional drug-resistant DFHR (Manyahi et al.
2017).
No genetic resistance against Cotrimoxazol of C. burnetii has been reported to date. But the use
of Cotrimoxazole as a standalone therapy for Q fever endocarditis has not been proficient
(Raoult 1993), in combination with doxycycline it has been deemed an apparent success (Didier
Raoult, et al. 1999).
2.5. Phenotypic antimicrobial Susceptibility-testing of C. burnetii
The shell-vial assay was the most common used method for determination of antibiotic
susceptibility, but is now replaced by a more precise PCR-based approach (Brennan and Samuel
2003). The understanding of antibiotic susceptibility of C. burnetii is fragmentary at best, with
different antibiotic susceptibility methods, namely qPCR-based and shell-vial assays,
concluding to diverging results of minimal inhibitory concentrations (Table 2).
Additionally, not all isolates have been tested against all antibiotics in question for treatment
(Table 2) and deviating underlying correlations of occurring mutations to phenotypic
susceptibility is deemed impossible.
Since most isolates that have been tested for antibiotic susceptibility, have not been sequenced,
accession numbers of these strains are not available and a sequenced-based phenotype to
genotype correlation for antibiotic susceptibility cannot be established.

16
Table 2 Published Antibiotic Susceptibility of C. burnetii
MIC [mg/l]
Beta lactams Tetracycline Macrolide Fluoroquinolone
Strain AMPI DOXY MINO TIGE CL LIN ERY AZI CLA TELI RIF CO/TRI CIPR TROVA LEVO MOXI PERF OFL
RSA 493 4 0.25- 1 0.25 8 2 8 2 1 4 4 1 0.5 0.5 1 1
Q212 2 0.5 4 8 4 1 8 2 1 2 4
2
Greek CP1 2 0.5 4 8 4 8 2 4 2
CP2,3,5,6,7,8 1 0.25 2 8 2 4 1 1 1
CP4 2 0.25 2 8 2 4 1 2 1
Priscilla 1 8 1 0.5 1 1
Cb109 8 8 2
Goat 8 8 0.5
Acute Q fever (blood)
8 8 0.5
French Guyana
1 0.25 0.25 8 8 2 0.25 8/1.6 0.5
MST 17 2 0.25 0.5 8 8 2 0.25 8/1.6 0.5
3 0.25 0.25 8 8 4 0.25 8/1.6 0.5
4 0.25 0.25 8 8 4 0.5 8/1.6 0.5
5 0.25 0.25 8 8 2 0.25 8/1.6 0.1
6 0.25 0.25 8 8 8 0.25 8/1.6 0.5
Japanese 307 0.5-1 4-8 0.5 0.5-1
605 0.25-0.5 8 0.5 1
Note: DOXY, Doxycycline; MINO, Minocycline; TIGE, Tigecycline; LIN, Linezolid; ERY, Erythromycin; AZI, Azithromycin; CLA, Clathrimycin; TELI, Telithromycin; CO/TRI, Cotrimoxazole; CIPR, Ciprofloxacin; TROVA, Trovafloxacin; LEVO, Levofloxacin; MOXI, Moxifloxacin; PERF, Perfloxacin; OFL, Ofloxacin; RIF, Rifampicin; CL, Chloramphenicol; AMPI, Ampicillin Background: White: tested via qPCR-based assays; Grey: tested via shell-vial assays Source: Raoult et al. 1991, Gikas et al. 1998, 2001, Rolain et al. 2001, 2005, Brennan and Samuel 2003, Andoh et al. 2004, Spyridaki et al. 2009, Eldin et al. 2015

17
2.6. Aim of this study
The objective of this thesis was to analyse the occurrence of antibiotic resistance genes and
resistance conferring mutations in published whole-genome sequences of Coxiella burnetii
against beta lactams, tetracyclines, macrolides and gyrase inhibitors antibiotics using a
bioinformatical approach and to develop a PCR-based method for detection of genetic antibiotic
resistance markers.
18
3. Material and Methods
3.1. In NCBI published Genomes of strains used for sequence alignments
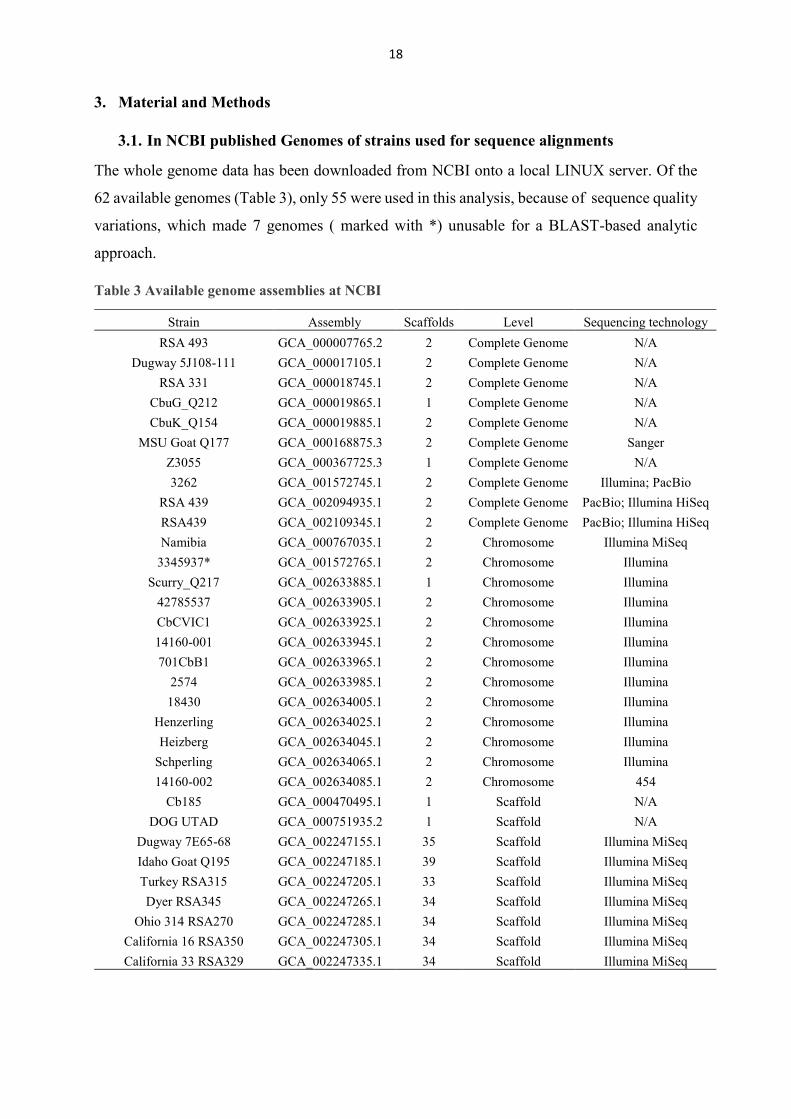
The whole genome data has been downloaded from NCBI onto a local LINUX server. Of the
62 available genomes (Table 3), only 55 were used in this analysis, because of sequence quality
variations, which made 7 genomes ( marked with *) unusable for a BLAST-based analytic
approach.
Table 3 Available genome assemblies at NCBI
Strain Assembly Scaffolds Level Sequencing technology RSA 493 GCA_000007765.2 2 Complete Genome N/A
Dugway 5J108-111 GCA_000017105.1 2 Complete Genome N/A RSA 331 GCA_000018745.1 2 Complete Genome N/A
CbuG_Q212 GCA_000019865.1 1 Complete Genome N/A CbuK_Q154 GCA_000019885.1 2 Complete Genome N/A
MSU Goat Q177 GCA_000168875.3 2 Complete Genome Sanger Z3055 GCA_000367725.3 1 Complete Genome N/A 3262 GCA_001572745.1 2 Complete Genome Illumina; PacBio
RSA 439 GCA_002094935.1 2 Complete Genome PacBio; Illumina HiSeq RSA439 GCA_002109345.1 2 Complete Genome PacBio; Illumina HiSeq Namibia GCA_000767035.1 2 Chromosome Illumina MiSeq
3345937* GCA_001572765.1 2 Chromosome Illumina Scurry_Q217 GCA_002633885.1 1 Chromosome Illumina
42785537 GCA_002633905.1 2 Chromosome Illumina CbCVIC1 GCA_002633925.1 2 Chromosome Illumina 14160-001 GCA_002633945.1 2 Chromosome Illumina 701CbB1 GCA_002633965.1 2 Chromosome Illumina
2574 GCA_002633985.1 2 Chromosome Illumina 18430 GCA_002634005.1 2 Chromosome Illumina
Henzerling GCA_002634025.1 2 Chromosome Illumina Heizberg GCA_002634045.1 2 Chromosome Illumina
Schperling GCA_002634065.1 2 Chromosome Illumina 14160-002 GCA_002634085.1 2 Chromosome 454
Cb185 GCA_000470495.1 1 Scaffold N/A DOG UTAD GCA_000751935.2 1 Scaffold N/A
Dugway 7E65-68 GCA_002247155.1 35 Scaffold Illumina MiSeq Idaho Goat Q195 GCA_002247185.1 39 Scaffold Illumina MiSeq Turkey RSA315 GCA_002247205.1 33 Scaffold Illumina MiSeq Dyer RSA345 GCA_002247265.1 34 Scaffold Illumina MiSeq
Ohio 314 RSA270 GCA_002247285.1 34 Scaffold Illumina MiSeq California 16 RSA350 GCA_002247305.1 34 Scaffold Illumina MiSeq California 33 RSA329 GCA_002247335.1 34 Scaffold Illumina MiSeq

19
Strain Assembly Scaffolds Level Sequencing technology Ohio 314 RSA338 GCA_002249845.1 34 Scaffold Illumina MiSeq
Leningrad-2 GCA_002591355.1 93 Scaffold Illumina MiSeq Q321* GCA_000169495.2 98 Contig N/A
Cb_C2* GCA_000612785.1 73 Contig Illumina Cb_B1 GCA_000613025.1 40 Contig Illumina
EV-Cb_BK10 GCA_000723245.1 37 Contig Illumina Cb_O184* GCA_000723265.1 268 Contig Illumina
EV-Cb_C13* GCA_000723285.1 109 Contig Illumina Cb_B18 GCA_000723305.1 41 Contig Illumina AuQ01 GCA_000756325.1 67 Contig Illumina HiSeq
Cb196_Saudi_Arabia GCA_000820465.1 3 Contig N/A DOG UTAD* GCA_000820825.1 67 Contig N/A
Cb171_QLYMPHOMA* GCA_000826165.2 1 Contig N/A NL-Limburg GCA_000967075.1 5 Contig PacBio
Ko Q229 GCA_002247225.1 40 Contig Illumina MiSeq Q532 GCA_002896735.1 38 Contig Illumina MiSeq Q545 GCA_002896755.1 37 Contig Illumina MiSeq Q556 GCA_002896775.1 42 Contig Illumina MiSeq
Cb3506 GCA_002896795.1 60 Contig Illumina MiSeq Q540 GCA_002896815.1 111 Contig Illumina MiSeq Q559 GCA_002896835.1 39 Contig Illumina MiSeq
Cb175_Guyana GCA_000359545.5 2 Complete Genome N/A cb109 GCA_000300315.1 257 Contig 454
Australia RSA297 GCA_002924305.1 34 Scaffold Illumina MiSeq California 16 RSA350 clone 2 GCA_002924325.1 34 Scaffold Illumina MiSeq
Nine Mile RSA363 GCA_002924345.1 35 Scaffold Illumina MiSeq M44 RSA461 GCA_002924385.1 66 Scaffold Illumina MiSeq
Nine Mile RSA514 GCA_002924395.1 33 Scaffold Illumina HiSeq RSA425 GCA_002924425.1 34 Scaffold Illumina MiSeq
Note: All available genome assemblies of NCBI (56 in total); 7 (marked with *) were not used in this analysis due to poor sequence quality. 1 is an earlier release of DOG UTAD genome.

20
3.2. Bioinformatic “Pipeline”
A combination of bash-commands and python3´s library biopython was used to automatize
multiple sequence alignments. First, the sequences of resistance associated genes (Table 4) were
downloaded. Biopython was used to create a local blast database. The standalone blast 2.6+
was used with default parameters. The raw output file was parsed using the XML parser
provided by biopython and using bash-commands. After obtaining a FASTA file for each gene
of interest, reference sequences of the correlating genes of C. burnetii str. RSA 493, L.
pneumophilia str. Philadelphia 1 and E. coli str. K-12 substr. MG1655 were added to compare
any differences between species after alignment. MAFFT (Kuraku et al. 2013, Katoh et al.
2017) was then used to align all multiFASTA files.
Figure 2 File structure of bioinformatics pipeline
All executable scripts are in the ‘scripts’ folder. ‘database’ contains ‘references’ with all
reference sequences as well as the local custom blast database in the folder ‘blastdb’. ‘genome’
contains all genomes of C. burnetii as FASTA files and the output is stored as alignment files
in the ‘output’ folder (Figure 2).
The pipeline consists of a master script (Code 1) with six subscripts. This file tree (Figure 2)
allows easy and clean addition of genes and genomes.

21
Code 1 (master script of pipeline)
Master script #!/bin/bash echo 'Starting Gene Extraction of genomes in dir' ./blast.sh echo 'Finished extracting. Parsing results.' python3 parsing.py echo 'Creating Subsets of Data' ./parse2.sh echo 'Your datasets have been created.' rm ../output/*.xml rm ../output/presults.fasta echo 'Spiking Results.' ./spike.sh echo 'Aligning parsed multifasta.' ./align.sh echo 'Done.'
For use of the standalone blast 2.6.0+ function (Camacho et al. 2009) a local blast database was
created (Code 2). The Master script runs the blast.sh file. After the local blast, parsing.py and
parse.sh are used to parse the blast results to a usable output. Spike.sh adds the reference
sequences of E. coli and L. pneumophilia of each gene of interest for comparison. Align.sh
aligns all multiFASTA files.
Code 2
Blast database #!/bin/bash cat ../database/blastdb/*.fasta>../database/blastdb/GOI.fasta makeblastdb -in ../database/blastdb/GOI.fasta -out ../database/blastdb/dbGOI.fasta -dbtype nucl
The local database is created from all fasta files by using the cat function and concatenating
into one multifasta file. Makeblastdb creates a local nucleotide database.

22
The blast algorithm (Code 3) runs with default settings and extracts all target sequences into a
raw data sheet by using all .fna files as input in the genomes directory and creating an .xml file
as output.
Code 3
Blast script #!/bin/bash for F in ../genomes/*.fna do blastn -query $F -db ../database/blastdb/dbGOI.fasta -outfmt 5 >> ../output/results.xml done echo -e "Blast has finished. "
Script 4 (Code 4) extracts the title of the alignment (gene of interest), the length of the query
sequence, the length of the subject and the subject sequence for comparison out of the .xml file
and writes it in a .fasta file
Code 4
Parsing of raw data #!/usr/bin/env python3 from Bio.Blast import NCBIXML results=open('../output/results.xml', 'r' ) records=NCBIXML.parse(results) save_file = open("../output/presults.fasta", "w") for blast_record in records: for alignment in blast_record.alignments: for hsp in alignment.hsps: save_file.write('\n>%s;%s;%s,%s\n%s ' % (blast_record.query, alignment.hit_def, hsp.identities, alignment.length, hsp.query)) save_file.close()

23
Script 5 (Code 5) divides the processed output (fasta files) in separate multifasta-files for each
gene of interest using the bash command “grep”. The argument “-A1” specifies for the line after
the query words in brackets.
Code 5
Restructuring of sequences #!/bin/bash grep -A1 "23S" ../output/presults.fasta>../output/23S.fasta grep -A1 "rpoB" ../output/presults.fasta>../output/rpoB.fasta grep -A1 "gyrA" ../output/presults.fasta>../output/gyrA.fasta grep -A1 "gyrB" ../output/presults.fasta>../output/gyrB.fasta grep -A1 "parC" ../output/presults.fasta>../output/parC.fasta grep -A1 "parE" ../output/presults.fasta>../output/parE.fasta grep -A1 "rplD" ../output/presults.fasta>../output/rplD.fasta grep -A1 "rpsG" ../output/presults.fasta>../output/rpsG.fasta grep -A1 "folA" ../output/presults.fasta>../output/folA.fasta grep -A1 "rpsL" ../output/presults.fasta>../output/rpsL.fasta grep -A1 "rplV" ../output/presults.fasta>../output/rplV.fasta grep -A1 "folP" ../output/presults.fasta>../output/folP.fasta grep -A1 "ampC" ../output/presults.fasta>../output/ampC.fasta echo "your scripts are done"

24
Script 6 (Code 6) concatenates the reference sequences (Table 5) of E. coli str. K-12 substr. MG1655 and L. pneumophilia str. Philadelphia 1 and C.
burnetii Q212Levo (Table 4) to each multifasta-file by the using cat function for comparison of sequences using the sequence alignment software
MAFFT.
Code 6
Spiking files with reference sequences #!/bin/bash echo 'Spiking Fasta Files with RefSeq' cat ../database/references/23SEC.rrlB.fasta ../database/references/23SLP.fasta ../output/23S.fasta>../output/23SS.fasta cat ../database/references/folAEC.fasta ../database/references/folALP.fasta ../output/folA.fasta>../output/folAS.fasta cat ../database/references/folPEC.fasta ../database/references/folPLP.fasta ../output/folP.fasta>../output/folPS.fasta cat ../database/references/gyrAEC.fasta ../database/references/gyrALP.fasta ../database/references/CBQ212Levo.gyrA_parcds.fasta ../output/gyrA.fasta>../output/gyrAS.fasta cat ../database/references/gyrBEC.fasta ../database/references/gyrBLP.fasta ../database/references/CBQ212Levo.gyrB_parcds.fasta ../output/gyrB.fasta>../output/gyrBS.fasta cat ../database/references/parCEC.fasta ../database/references/parCLP.fasta ../database/references/CBQ212Levo.parC_parcds.fasta ../output/parC.fasta>../output/parCS.fasta cat ../database/references/parEEC.fasta ../database/references/parELP.fasta ../output/parE.fasta>../output/parES.fasta cat ../database/references/rplDEC.fasta ../database/references/rplDLP.fasta ../output/rplD.fasta>../output/rplDS.fasta cat ../database/references/rplVEC.fasta ../database/references/rplVLP.fasta ../output/rplV.fasta>../output/rplVS.fasta cat ../database/references/rpsGEC.fasta ../database/references/rpsGLP.fasta ../output/rpsG.fasta>../output/rpsGS.fasta cat ../database/references/rpoBEC.fasta ../database/references/rpoBLP.fasta ../output/rpoB.fasta>../output/rpoBS.fasta cat ../database/references/rpsLEC.fasta ../database/references/rpsLLP.fasta ../output/rpsL.fasta>../output/rpsLS.fasta echo 'Done with spiking'
25
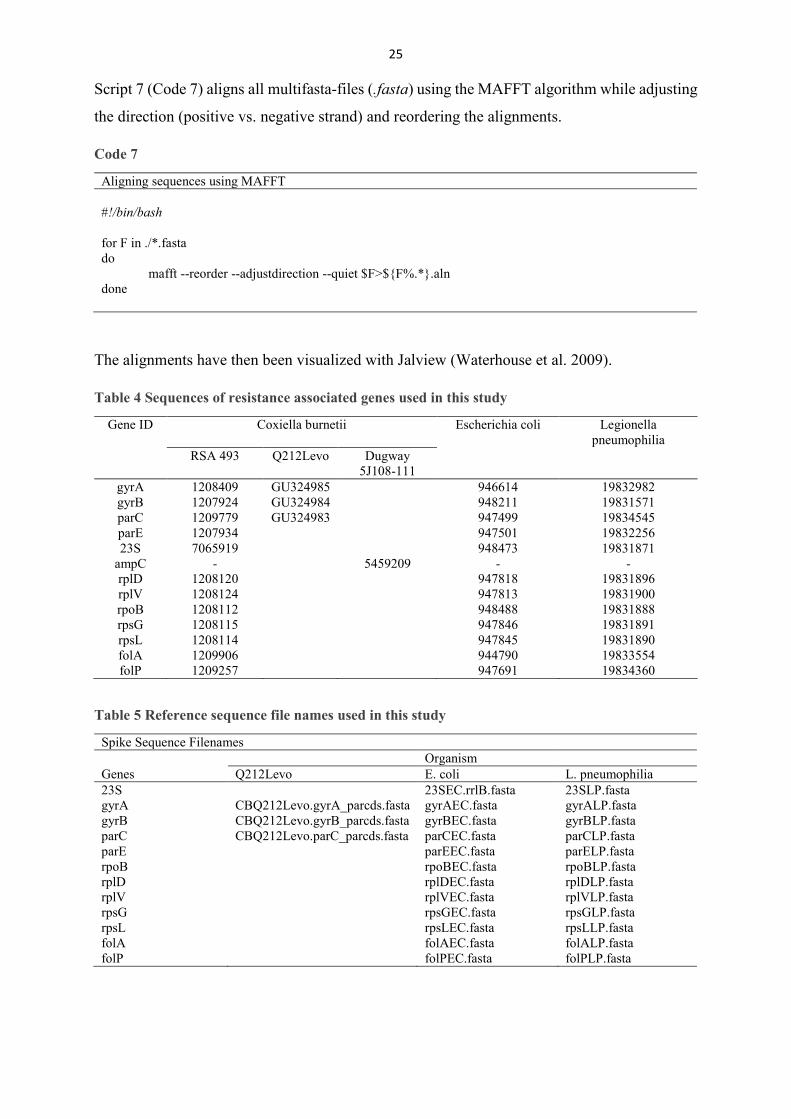
Script 7 (Code 7) aligns all multifasta-files (.fasta) using the MAFFT algorithm while adjusting
the direction (positive vs. negative strand) and reordering the alignments.
Code 7
Aligning sequences using MAFFT #!/bin/bash for F in ./*.fasta do mafft --reorder --adjustdirection --quiet $F>${F%.*}.aln done
The alignments have then been visualized with Jalview (Waterhouse et al. 2009).
Table 4 Sequences of resistance associated genes used in this study
Gene ID Coxiella burnetii Escherichia coli Legionella pneumophilia
RSA 493 Q212Levo Dugway 5J108-111
gyrA 1208409 GU324985 946614 19832982 gyrB 1207924 GU324984 948211 19831571 parC 1209779 GU324983 947499 19834545 parE 1207934 947501 19832256 23S 7065919 948473 19831871
ampC - 5459209 - - rplD 1208120 947818 19831896 rplV 1208124 947813 19831900 rpoB 1208112 948488 19831888 rpsG 1208115 947846 19831891 rpsL 1208114 947845 19831890 folA 1209906 944790 19833554 folP 1209257 947691 19834360
Table 5 Reference sequence file names used in this study
Spike Sequence Filenames Organism Genes Q212Levo E. coli L. pneumophilia 23S 23SEC.rrlB.fasta 23SLP.fasta gyrA CBQ212Levo.gyrA_parcds.fasta gyrAEC.fasta gyrALP.fasta gyrB CBQ212Levo.gyrB_parcds.fasta gyrBEC.fasta gyrBLP.fasta parC CBQ212Levo.parC_parcds.fasta parCEC.fasta parCLP.fasta parE parEEC.fasta parELP.fasta rpoB rpoBEC.fasta rpoBLP.fasta rplD rplDEC.fasta rplDLP.fasta rplV rplVEC.fasta rplVLP.fasta rpsG rpsGEC.fasta rpsGLP.fasta rpsL rpsLEC.fasta rpsLLP.fasta folA folAEC.fasta folALP.fasta folP folPEC.fasta folPLP.fasta

26
3.3. ABRICATE with CARD, ARG-ANNOT, ResFinder
The perl-written ABRICATE 4 tool was used to screen the genomes for resistance genes (Code
8). This blast-based bioinformatical tool can implement various antibiotic resistance databases
as references. Since the Comprehensive Antibiotic Resistance Database (CARD; (Jia et al.
2017)) and ResFinder (Zankari et al. 2012) do not account for point mutations, a third database,
ARG-ANNOT was used to cover point mutations (Gupta et al. 2014). ABRICATE was run
with a minimal coverage of 0% and minimal identity of 75%.
Code 8
Code for ABRICATE #!/bin/bash abricate --db argannot ../genomes/*.fna > ../output/argres.tab abricate --db card ../genomes/*.fna > ../output/cardres.tab abricate --db resfinder ../genomes/*.fna > ../output/resfres.tab echo 'Abricate done.'
3.4. Genotyping via DNA Simpleprobe™ probe assay
SimpleProbes™ were designed complimentary to the reference strain RSA 493 (Nine Mile) to
span across the regions in genes where sequence data confirmed polymorphisms in the parE
gene (Table 6). ParE harbors a A→G polymorphism at position 1411 of C. burnetii (1378 in
E. coli MK-13).
Primer-BLAST software (Ye et al. 2012) was used to design primers to amplify DNA templates
within the determined regions. The probe was designed by TIBMOLBIO to the target amplicon.
Table 6 Primer and probe sequences
Strain
Target gene
Mutation position
Primer (5´→3´) Simpleprobe™ Lot #
RSA 493
parE 1411/ 1378
F: ATACATgggAAgTTgATACCgAA
gCACggCTTgAgAXIAgCTAATACTTg--NH2
44031801
R: gCATCCgCTAAgATgCAA
4 https://github.com/tseemann/abricate, accessed 9.4.2018
27
Table 7 SimpleProbe protocol #1
LightCycler FastStart DNA
Master Hybridization Probes Kit
Company Lot
number
Concentration Concentration
/ 20 µl
1x
[µl]
10x
[µl]
H2O LightCycler
FastStart DNA
Master
Hybridization
Probes Kit Cat#
12239272001
Roche
Diagnostics,
Mannheim,
Germany
10559100 13,4 134
MgCl2 25 mM 3 mM 1,6 16
LC FastStart
– Reaction
Mix
10x 1x 2 20
Primer/probe
Mix
TIB
MOLBIOL,
Berlin,
Germany
1 10
Target DNA 2
To test for specifity and suitability, the probe was first used to discriminate between known
genotypes of C. burnetii str. RSA 493 (A1411), RT-Schperling, Priscilla, Arandale and
Namibia (1411G;, Table 10) with 103 and 104 copies of DNA per µl.
Table 8 SimpleProbe protocol #2
LightCycler FastStart DNA
Master Hybridization Probes Kit
Company Lot
number
Concentration Concentration
/ 20 µl
1x
[µl]
29x
[µl]
H2O LightCycler
FastStart DNA
Master
Hybridization
Probes Kit Cat#
12239272001
Roche
Diagnostics,
Mannheim,
Germany
10559100 13,4 388,6
MgCl2 25 mM 3 mM 1,6 46,4
LC FastStart
– Reaction
Mix
10x 1x 2 58
Primer/probe
Mix
TIB
MOLBIO
1 29
Target DNA 2
28
Table 9 LightCycler 480 protocol
Parameter Temperature (°C) Time Ramp Rate (°C/s) Acquisition mode
Activation of FastStart Taq
DNA polymerase
95 10:00 min 4.4 None
Amplification (45 cycles) 95
60
72
10 sec
10 sec
15 sec
4.4
2.2
4.4
None
Single
None
Melting curve analysis 95
40
75
30 sec
2:00 min
0
4.4
1.5
-
None
None
Continuous 3
Cooling 40 30 sec 1.5 None
SimpleProbes™ enable discrimination between wild and mutant genotypes via one probe
(Gossé et al. 2016).
The Lightcycler 4.0 software modules Tm calling was used to analyze the SimpleProbe™
melting curve data.
DNA of selected strains (Table 10) for further analysis and genotyping was provided by the
Bundeswehr Institute for Microbiology of the German Armed Forces. The DNA was isolated
using the MagnaPureCompact kit of Roche. For each reaction, a DNA copy number of 104 DNA
molecules per µl was used (Table 8).

29
Table 10 Strains analyzed in this study for parE polymorphism
strain Host Clinical Info origin
CS-Florian Human, Acute Q fever Slovakia
Andelfingen Cow N/A Switzerland
Soyta Cow N/A Switzerland
Geier Human Pneumonia Romania
Balaceanu Human Pneumonia Romania
F1 Human Endocarditis Paris, France
F2 Human Actute hepatitis Paris, France
Z-3574 Sheep N/A Berlin, Germany
Z349-36/94 Sheep N/A Sontra, Germany
J1 Cow N/A Japan
J3 Cow N/A Japan
J27 Cow N/A Japan
Frankfurt Cow N/A Frankfurt, Germany
Herzberg Human N/A Greece
Z11 Tick N/A Germany
Z5A Tick N/A Germany
CB3468 Sheep N/A Germany
OSH1 Cow N/A Germany
S-1 Sheep N/A Sweden
S-4 Sheep N/A Sweden
C5-Polsko Ixodes ricinus N/A Poland
Ohio Milk, cow N/A USA
RT-III Ixodes ricinus N/A Luga, Russia
RT-Schperling Human Fever Kyrgyzstan
RSA 493 (A1411) Tick N/A USA
Arandale (1411G) Human Acute Q fever Australia
Priscilla Q177 (1411G) Goat Abortion USA
Namibia (1411G) Goat N/A Namibia
Sources: Jiménez, Beare et al. 2017 Database of the Institute for Microbiology of the Bundeswehr
3.5. Structural modelling of predicted beta lactamase ampC of C. burnetii
Phyre2 algorithm was used to model the protein sequence of the predicted beta lactamase ampC
of C. burnetii (Kelly et al. 2015). The ampC sequence was taken from Uniprot (Entry:
A9KDR4). Due to unavailability of X-ray crystallography data, the ligand binding site was
predicted using the 3DLigandSite software (Wass et al. 2010).

30
4. Results
4.1. ABRICATE – screening for resistance genes
Screening for resistance genes with ABRICATE could not detect any resistance genes with the
databases ARGANNOT and ResFinder.
With the CARD database, a macrolide resistant rpoB variant was found in all genomes of C.
burnetii with a coverage of 6% and identity of 75%.
The CARD5 database currently holds 1435 entries for determinant of beta-lactam resistance,
132 entries for determinant of fluoroquinolone resistance, 18 entries for determinant of
rifamycin resistance, 81 entries for determinant of macrolide resistance and 22 entries for
determinant of tetracycline resistant. Determinant is defined as a mutation, single nucleotide
polymorphism, gene, or gene product that confers antibiotic resistance.
ResFinder6 database currently holds 1184 entries for beta lactam resistance genes, 95 entries
for fluoroquinolone resistance genes, 73 entries for MLS resistance genes, 6 entries for
rifampicin resistance genes, 42 entries for tetracycline resistance genes and 42 entries for
trimethoprim resistance genes.
ARG-ANNOT7 database currently holds 2615 entries for beta lactam (bla) resistance genes, 74
entries for fluoroquinolone (Flq) resistance genes, 103 entries for MLS (MLS) resistance genes,
113 entries for tetracycline (Tet) resistance genes, 47 entries for trimethoprim (Tmt) resistance
genes and 9 entries for rifampicin (Rif) resistance genes
4.2. Alignment of candidate genes
The complete list of all 13 candidate genes was present in all selected genomes. The sequence
identity and overall length varied between Coxiella, Legionella and Escherichia, but was
overall consistent within species, apart from single nucleotide deletions in Coxiella genes. If
these deletions are artificial sequencing errors due to repetitive nucleotides or are correct could
not be determined. The alignment of the candidate genes (Table 4) did not show any known
resistance conferring mutations previously described. The topoisomerase II coding genes gyrA
and gyrB, as well as topoisomerase IV coding parC did not have any resistance-linked
mutations as described above. No macrolide resistance conferring 23SrRNA mutations could
5 Accessed on 31.5.2018; https://card.mcmaster.ca/ 6 Accessed on 31.5.2018; https://cge.cbs.dtu.dk/services/ResFinder/database.php 7 Accessed on 31.5.2018; http://en.mediterranee-infection.com/article.php?laref=283%26titre=arg-annot

31
be detected. The described ribosomal proteins rplD, rplV, rpsG and rpsL did not show any
previously described mutations linked to antibiotic resistance. The amino acid sequence of
RNA-interacting domain III of rpoB was completely identical between Coxiella strains and
exhibited differences between amino acid sequences of Legionella, Escherichia and Coxiella
could not be connected to increased resistance. Also, since the pipeline would detect possible
gene duplications as double entries in the alignment files, no possible resistant gene variants
have been missed.
The topoisomerase IV subunit B coding gene parE harbours a polymorphism at nucleotide
A1411G (Figure 3, 1378 in E. coli str. K-12 substr. MG1655) which leads to amino acid change
at codon 471 (Figure 4, codon 460 in E. coli str. K-12 substr. MG1655).
Figure 3 Alignment of nucleotide sequence of parE

32
Figure 4 Alignment of amino acid sequence of parE
A SimpleProbe-assay was designed on basis of the A1411G polymorphism of parE to analyse
isolates of C. burnetii.
33
4.3. Genotyping of selected strains
4.3.1. parE-PCR for specifity of SimpleProbe-assay
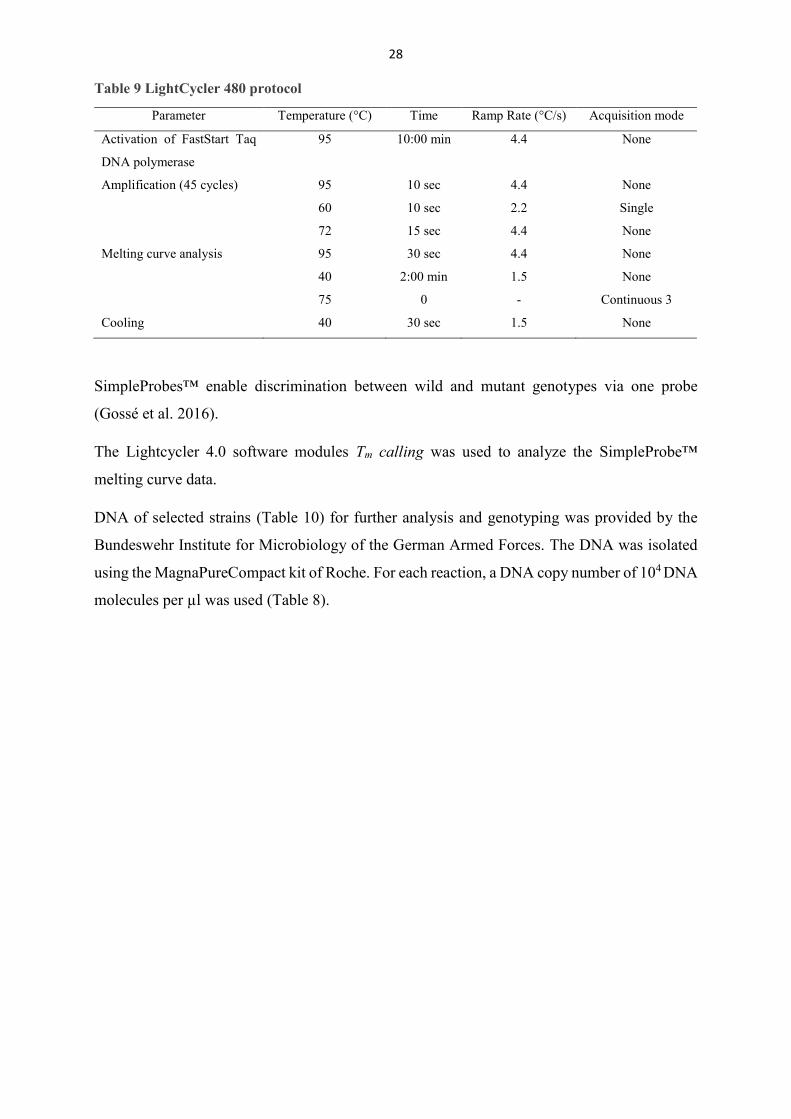
Figure 5 Melting peaks
Tm calling was not able to retrieve a temperature for each reaction (Figure 5), but by visual
discrimination Arandale 10^3, Arandale 10^4, RT-Schperling 10^4 harboured the 1411G
genotype and RSA 493 10^3 had the A1411 genotype. All sequences got amplified as can be
seen in the equal rise of all curves in the left. It seems as if probes did not bind equally in all
reactions.
strain DNA copy number Tm [°C] genotype RSA 493 103 A
104 62,33 A Arandale 103 G
104 68,82 G Priscilla 103
104 Namibia 103
104 RT-Schperling 103
104 G
34
4.3.2. SimpleProbe-analysis of parE of selected strains
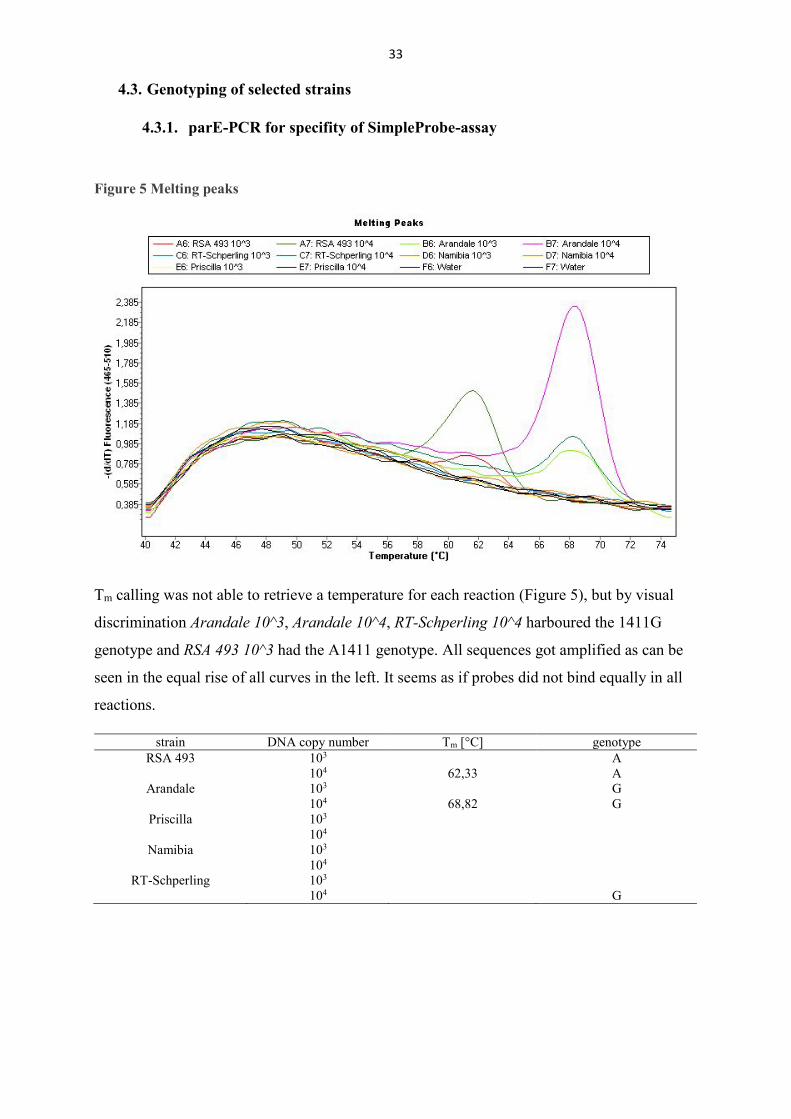
Figure 6 Melting peaks of selected strains
Tm calling did not show results for all used isolates (Table 11) but by visual discrimination all isolates could be interpreted. Of the tested 25 isolates, Coxiella burnetii str. F1, F2 and Z349-36/94 harboured the parE polymorphism A1411G (Figure 6).
35
Table 11 parE polymorphisms of selected isolates
strain Tm genotype CS-Florian A Andelfingen 59,24 A Soyta 59,30 A Geier A Balaceanu 59,46 A F1 G F2 G Z-3574 A Z349-36/94 G J1 59,67 A J3 A J27 A Frankfurt-Mumlan A Herzberg A Z11 59,32 A Z5A A CB3468 59,60 A OSH1 A S-1 59,43 A S-4 59,61 A C5-Polsko 59,12 A Ohio 59,65 A RT-III A RT-Sperling A RSA 493 (A1411) A Arandale (1411G) G
4.3.3. Beta lactamase ampC of C. burnetii
The ampC gene of C. burnetii was first predicted and annoted in C. burnetii str. Dugway by
Beare et al. 2009 using ERGO™ genome analysis suite. Up to date, no additional information
on this predicted beta lactamase gene has been published. The predicted sequence has been
found in all used genomes with no frameshift mutations using a blast-based approach. This
could conclude the inactivity of beta lactams in Q fever therapy. Genetic activity of ampC or if
it is just a pseudogene could not be concluded from sequence data alone. Variations in sequence
data between strains of Coxiella could either be sequencing artifacts or due to genome plasticity
due to the abundance of genetic mobile elements.
36
Figure 7 Sequence alignment of ampC of C. burnetii
37
Figure 8 Predicted secondary structure of ampC of C. burnetii str. Dugway

38
Figure 9 Predicted structure with binding site
3dLigandSite predicted the binding site on amino acids Val146, Ser148, Gln149, Leu152,
Val153, Leu156, Leu178, Met181, Asp233, Ser234 and Thr235 (Unique Job identifier:
56d67ae68213c426) ampC of C. burnetii str. Dugway. The blue-colored ribbons visualize the
binding site to the pink antibiotic molecule. 3dLigandSite compares known models to Phyre2
database entries to predict possible binding structures.

39
5. Discussion
5.1. Absence of known resistance genes
All genes (Table 4) submitted to the pipeline have been found in all used genomes of C. burnetii
using the programmed pipeline. Resistance-associated regions did not show differences
between strains of C. burnetii, differences between species of Coxiella, Legionella and
Escherichia could not be connected to differences in antibiotic susceptibility, apart from the
parE polymorphism.
The pipeline enables to discriminate between genetic differences of genes of interest, but does
not enable to assess for sequence quality of used genomes (namely, repetitive regions of poly
nucleotides resulting in sequencing errors). If these poly nucleotides are artificial and if they
play a role in genetic activity cannot be concluded from sequence data alone.
As tetracyclines account for first line treatments of Q fever, any potential rise of resistant
variants is of utmost importance for adaption in therapy.
Tetracycline resistance genes are usually encoded on plasmids as being transferred via
horizontal gene transfer. All isolates of C. burnetii do harbor an autonomous replicating plasmid
or chromosomally integrated plasmid-like sequences (IPS) which has not been associated with
resistance genes but with the Dot/Icm type IV secretion system (T4SS) and its related effector
proteins (Voth et al. 2011) and plasmid-derived antibiotic resistant gene variants have not been
associated with C. burnetii.
As previously reported (Rouli et al. 2012), the genetic basis of the clinically doxycycline-
resistant isolate Cb109 could not be determined from antibiotic resistance databases. Another
problem arises with the used sequence data Cb109 as it was sequenced with 454 pyrosequencing
(Rothberg and Leamon 2008) that generates long-read lengths of ~500 base pairs. C. burnetii
str. Nine Mile contains about 20 copies of a mobile element IS1111 with an average length of
~600 base pairs (Denison et al. 2007) which is usually used in various detection methods (Chen
and Ching 2016). This discrepancy of read length versus abundance of mobile elements and the
non-availability of raw data for possible reassembly proposes major difficulties in the
investigation of this rare case of antibiotic resistance in C. burnetii. Therefore, re-sequencing
of C. burnetii isolate Cb109 or at last, release of raw data is the only suitable possibility for
further investigation and may uncover a previously missed mechanism of antibiotic resistance.
Macrolide resistance could not be detected by occurrence of acquired resistance genes (e.g. erm
or mef) and no differences in domain V of 23S ribosomal RNA associated with macrolide

40
resistance could be identified and occurring mutations could not be connected to previously
reported reduced antibiotic susceptibility.
Fluoroquinolone resistance through acquisition of resistant gene variants or through resistance
conferring mutations of gyrA, gyrB and parC as previously reported (Musso et al. 1996,
Spyridaki et al. 2000a, Vranakis 2010) could not be identified either via ABRICATE algorithm
or the bioinformatical pipeline. Variation in susceptibility to Ciprofloxacin between strains like
Nine Mile (MIC: 4µg/l) versus Q212 (MIC: 8µg/l) could not be connected to variations in
topoisomerase II and IV subunit B genes.
Although, Rifampicin does not play a role in the clinical setting. Resistance through mutations
of rpoB domain III responsible for DNA binding and association with reduced susceptibility in
the phenotype could not be identified and domain III was throughout all genomes highly
conserved. The resistant rpoB variant found in all genomes via ABRICATE is probable a false
positive hit since an identity of 75% with a coverage of 6% does not represent a functional
resistant variant of the rpoB gene. The low coverage resulted in a short sequence of about 200
base pairs with high identity and therefore detection in all used genomes. Although Rifampicin
has not been successfully used in Q fever therapy, rpoB could not be identified as the culprit of
said failure.
Beta lactam resistance could not be connected to mutations in penicillin-binding proteins since
gene variants of C. burnetii (fstI, pbpA) differ greatly in identity (41%) of proteins variants from
known resistant variants of E. coli and comparison is not possible.
5.2. parE polymorphism A1411G
Literature research uncovered a previously unknown polymorphism of parE, lying outside of
regions that are usually associated with increased fluoroquinolone resistance. This
polymorphism could lead to a possible explanation of difference in susceptibility against
Ciprofloxacin of Coxiella strains.
Both E. coli and L. pneumophilia harbour the codon Glu460 (E. coli numbering) when
susceptible to Fluoroquinolones, while C. burnetii harbours a Thr471Ala polymorphism (codon
460 in E. coli) in the parE gene. The parE mutation at codon 471 (in Coxiella) has been
observed in combination with gyrA mutations in Fluoroquinolone-resistant isolates of E. coli
increasing antibiotic resistance from 64 to 128 mg/l and its role in sole contribution to antibiotic
resistance of Coxiella burnetii needs further investigation. Due to this recent discovery of non-

41
QRDR mutations of parE in fluoroquinolone-resistant strains of E. coli, available data on the
occurance of Glu460 mutations of parE is scarce (Shaheen et al. 2013, Nawaz et al. 2015). If
soley mutations of parE or their combinatory effect with gyrA mutations play a role in reduced
susceptibility is still unknown and needs to be addressed.
The polymorphism carrying isolates F1, F2 and Z3574 do not seem to share an underlying
connection, F1 resulted in a chronic Q fever endocarditis in 1992, F2 resulted in chronic Q fever
hepatitis probable in 1991 and Z3574 was isolated from a sheep near Berlin in 1992. Since
gyrase inhibitors are frequently used in veterinary settings, a possible adaption of the chronic
isolates due to previous exposure could have occurred.
5.3. Beta lactamase ampC
The bioinformatical finding of the predicted beta lactamase ampC serves as an indicator for the
clinical resistance against beta lactam antibiotics of C. burnetii. This work is the first to find it
in previously published genomes. It serves as a first hint for the in-vitro and clinical resistance
of C. burnetii against Amoxicillin (Raoult et al. 1991). Although C. burnetii was classified as
‘susceptible’ for Ampicillin in a study from Brennan and Samuel 2003 with a MIC of 4µg/ml
(Table 2) it may explain this elevated value in comparison to E. coli with a MIC of 0.5µg/ml
(HUANG et al. 2014). However, this bioinformatical detection is still in need of wet-lab based
confirmation due to the genetic flexibility of C. burnetii and different genetic activity of certain
genes being based on the high number of genetic elements like insertion sequences (IS) (Beare
et al. 2009), thereby casting a shadow of doubt on the in vivo activity of ampC or its simple
appearance as a pseudogene like in the Ank gene family (Beare et al. 2009). Using blastp to
explore any relation to known ampC variants uncovered a 41% identity to class C
betalactamase variants of Hafnia alvei and Providencia alcalifaciens both being associated
with gastrointestinal infections. Therefor, a direct comparison cannot be made.
The predicted structure of ampC of C. burnetii str. Dugway and potential binding site of ampC
serve as a first hint for spatial arrangement. Next to genetic activity and being more than a
pseudogene, X-ray data is still needed for further investigation, since most software is still far
away from being perfectly usable as a prediction tool for protein structure.

42
5.4. Outlook
This work was the first assess the genetic base of antibiotic resistance of all sequenced genomes
of C. burnetii by using previously reported antibiotic resistance databases as well as a novel
programmed pipeline. Apart from the reference strain Nine Mile and Dugway, not many
isolates have been tested for susceptibility for all suitable antibiotics. Current antibiotic
resistance gene prediction has to rely on reported resistances of well-studied organisms like
Escherichia coli or related species like Legionella spp.. Databases like CARD or ARG-ANNOT
present a proper foundation and source in resistance prediction and annotation. Novel NGS
approaches with reduced cost, effort and increased quality of reads facilitate metagenomics
studies of C. burnetii, exempli gratia ampC detection and comparison to known beta lactamases
The lack of standardization of antibiotic susceptibility testing is still a problem that needs to be
addressed, since time-consuming cultivation and shell-vial assays perform equal or less against
newer cPCR-based (Brennan and Samuel 2003) or cell-free culture methods (Clay et al. 2017).
Alas, historic data needs to be re-investigated to tackle broad fluctuations in minimal inhibitory
concentrations between laboratories and novel antibiotics need to be evaluated for usability for
clinical therapy. Also, cheap sequence services like Illumina may offer less financial effort but
result in poor sequence quality that torpedize bioinformatical based antibiotic resistance
research. The absence of acquired resistance genes is possible due to the strict intracellular
lifestyle of C. burnetii in comparison to L. pneumophilia, which exhibits an extracellular phase
and therefore could acquire foreign genetic elements outside of host cells and the rather slow
metabolic activity inside the host.
Genetic elements would need to be taken up by the host cell, localized to vacuoles, resist the
high acidic environment, taken up by C. burnetii and be integrated into the genome. All these
events seem very unlikely and seem to be a possible explanation.

43
5.5. Deutsche Zusammenfassung der Bachelorarbeit
Das Ziel dieser Bachelorarbeit war die Analyse von publizierten Gesamtgenom Daten von
Coxiella burnetii auf das Vorkommen von Veränderungen in Hinblick auf
Antibiotikaresistenzen. Neben den, in der Therapie des Q-Fiebers, klinisch relevanten
Substanzklassen, Tetrazyklinen, Gyrasehemmern, Makroliden und Folsäureantagonisten
wurden auch noch exemplarisch Rifampicin sowie Betalaktame untersucht. Ergänzend sollte
aufgrund der ermittelten Daten ein PCR-basiertes Nachweissystem entwickelt und dieses an
ausgewählten C. burnetii – Isolaten überprüft werden. Das obligat intrazelluläre Bakterium C.
burnetti ist der ursächliche Erreger des Query (Q) – Fiebers mit einer Größe von 0,2-0,4 μm x
0,4-1,0 μm. Bei dieser weltweit vorkommenden Zoonose, mit Ausnahme von Neuseeland und
Antarktis, dienen fast alle Tierarten - vor allem Paarhufer und Nutztiere des Menschen - als
Reservoir. Umweltstabile Dauerformen können als Aerosol über weite Strecken verteilt
werden. Aufgrund der geringen Infektionsdosis (1-10 Erreger) und leichten inhalativen
Aufnahme, ist die Infektionsgefahr bei Umgang mit kontaminiertem Erregermaterial äußerst
hoch. Die häufigste Ursache der Infektion ist direkter Kontakt mit Geburtsmaterial von
Nutztieren, da hier die Erregerlast bis zu 109 Erreger pro ml betragen kann. Nach Inhalation
kommt es nach 2-3 Wochen zu grippeähnlichen Symptomen mit hohem Fieber,
Appetitlosigkeit sowie Kopfschmerzen. Infektionen bleiben meistens unerkannt, da ein intaktes
menschliches Immunsystem den Erreger in etwa 50% der Fälle symptomlos bekämpft. Kommt
es zum Ausbruch von akutem Q-Fieber kann in etwa 2% der Fälle die Entwicklung von
chronischem Q-Fieber beobachtet werden, welche eine langanhaltende Antibiotika-Therapie
erfordert.
Bisherige publizierte klinisch aufgetretene Antibiotikaresistenzen bei C. burnetii beschränkten
sich auf Tetrazykline. Insgesamt liegen bisher sehr wenige Informationen zum
Resistenzverhalten von C. burnetii vor. Diese Arbeit ist die erste, die alle 66 seit 2003
publizierten Genome von C. burnetii berücksichtigt. Zur vereinfachten, standardisierten
Analayse wurde eine bioinformatische Befehls-Pipeline zum automatischen Sequenzabgleich
von antibiotikaresistenz-relevanten Genen programmiert.
Von den 12 untersuchten resistenz-assoziierten Genen bzw. Polymorphismen konnten zwar alle
Gene identifiziert werden, jedoch wurde keine bekannte resistenz-vermittelnde Mutation
gefunden. Ein erst kürzlich bei E.coli beschriebener Polymorphismus im Gen parE konnte
ebenfalls in Coxiella nachgewiesen werden.

44
In silico konnte ein Polymorphismus im Topoisomerase IV Gen jedoch in zehn Genomen
ermittelt werden und mittels eines selbstentwickelten SimpleProbe™ Sonden-Assays an
weiteren Coxiella-Isolaten untersucht werden. Von 25 untersuchten Proben wiesen drei Isolate
den veränderten Genotyp im Gen parE auf. Zusätzlich gelang es eine bisher nur im Stamm
Dugway vorhergesagte Betalaktamase ampC bioinformatisch auch in allen anderen
untersuchten Coxiellen-Genomen nachzuweisen und damit einen Erklärungsansatz für die
mangelhafte therapeutische Wirksamkeit dieser Substanzklasse beim Q-Fieber zu geben.

45
5.6. Reference List
Allgaier C, Aktories K, Barth H, Bechthold A, Biel M, Dekant W, Dilger K, Eichelbaum M,
Engelhard K, Eschenhagen T, et al. 2013. Allgemeine und spezielle Pharmakologie und
Toxikologie
Amato FD, Eldin C, Georgiades K, Edouard S, Delerce J, Labas N, Raoult D. 2015.
Comparative Immunology , Microbiology and Infectious Diseases Loss of TSS1 in
hypervirulent Coxiella burnetii 175 , the causative agent of Q fever in French Guiana.
‘Comparative Immunology, Microbiology and Infectious Diseases’ 41:35–41.
Andoh M, Naganawa T, Yamaguchi T, Fukushi H, Hirai K. 2004. In vitro susceptibility to
tetracycline and fluoroquinolones of Japanese isolates of Coxiella burnetii. Microbiology
and Immunology 48(9):661–664.
Bailey MS, Trinick TR, Dunbar JA, Hatch R, Osborne JC, Brooks TJ, Green AD. 2011.
Undifferentiated Febrile Illnesses Amongst British Troops in Helmand , Afghanistan.
7(2):150–155.
Bansal S, Tandon V. 2011. Contribution of mutations in DNA gyrase and topoisomerase IV
genes to ciprofloxacin resistance in Escherichia coli clinical isolates. International Journal
of Antimicrobial Agents 37(3):253–255.
Beare PA, Unsworth N, Andoh M, Voth DE, Omsland A, Gilk SD, Williams KP, Sobral BW,
Iii JJK, Porcella SF, et al. 2009. Comparative Genomics Reveal Extensive Transposon-
Mediated Genomic Plasticity and Diversity among Potential Effector Proteins within the
Genus Coxiella ᰔ †. 77(2):642–656.
Beare PA, Jeffrey BM, Martens CA, Pearson T, Heinzen RA. 2017. Draft Genome Sequences
of Historical Strains of Coxiella burnetii Isolated from Cow’s Milk and a Goat Placenta.
Genome Announcements 5(39):e00985-17.
Van Belkum A. 2007. Tracing isolates of bacterial species by multilocus variable number of
tandem repeat analysis (MLVA). FEMS Immunology and Medical Microbiology
49(1):22–27.
Boarbi S, Mori M, Rousset E, Sidi-Boumedine K, Van Esbroeck M, Fretin D. 2014. Prevalence
and molecular typing of Coxiella burnetii in bulk tank milk in Belgian dairy goats, 2009-
2013. Veterinary Microbiology 170(1–2):117–124.
Brennan RE, Samuel JE. 2003. Evaluation of Coxiella burnetii Antibiotic Susceptibilities by

46
Real-Time PCR Assay. 41(5):1869–1874.
Brom R Van Den, Engelen E Van, Roest HIJ, Hoek W Van Der, Vellema P. 2015. Coxiella
burnetii infections in sheep or goats : an opinionated review. Elsevier B.V.
Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, Madden TL. 2009.
BLAST+: Architecture and applications. BMC Bioinformatics 10:1–9.
Campbell EA, Korzheva N, Mustaev A, Murakami K, Nair S, Goldfarb A, Darst SA. 2001.
Structural Mechanism for Rifampicin Inhibition of Bacterial RNA Polymerase. 104:901–
912.
Caplunik-Pratsch AL, Potasman I, Kessel A, Paz A. 2018. Doxycycline desensitization in
chronic Q fever—A critical tool for the clinician. IDCases 11(January):70–72.
Ceglie L, Guerrini E, Rampazzo E, Barberio A, Tilburg JJHC, Hagen F, Lucchese L, Zuliani
F, Marangon S, Natale A. 2015. Molecular characterization by MLVA of Coxiella burnetii
strains infecting dairy cows and goats of north-eastern Italy. Microbes and Infection
17(11–12):776–781.
Chen H, Ching W. 2016. The Development of Lyophilized Loop-mediated Isothermal
Amplification Reagents for the Detection of Coxiella burnetii 6 . Perform LAMP Reaction
with Reconstitution Buffer Containing HNB or Fluorescent. (April):1–6.
Clay KA, Hartley MG, Russell P, Norville IH. 2017. Use of axenic media to determine
antibiotic efficacy against Coxiella burnetii. International Journal of Antimicrobial
Agents.
Dean CR, Visalli M a, Projan SJ, Sum P, Bradford P a. 2003. Efflux-Mediated Resistance to
Tigecycline ( GAR-936 ) in Pseudomonas aeruginosa Efflux-Mediated Resistance to
Tigecycline ( GAR-936 ) in Pseudomonas aeruginosa PAO1. Antimicrobial agents and
chemoterapy 47(3):972–78.
Denison AM, Thompson HA, Massung RF. 2007. IS1111 insertion sequences of Coxiella
burnetii: Characterization and use for repetitive element PCR-based differentiation of
Coxiella burnetii isolates. BMC Microbiology 7:1–8.
Descours G, Ginevra C, Jacotin N, Forey F, Chastang J, Kay E, Etienne J, Lina G, Doublet P,
Jarraud S. 2017. Ribosomal Mutations Conferring Macrolide Resistance in Legionella
pneumophila. 61(3):1–12.

47
Dijkstra F, Hoek W Van Der, Wijers N, Schimmer B, Rietveld A. 2011. The 2007–2010 Q
fever epidemic in the Netherlands: characteristics of notified acute Q fever patients and
the association with dairy goat farming.
Duron O, Sidi-boumedine K, Rousset E, Moutailler S, Jourdain E. 2015. The Importance of
Ticks in Q Fever Transmission : What Has ( and Has Not ) Been Demonstrated ? Trends
in Parasitology xx.
Edouard S. 2014. Three-Toed Sloth as Putative Reservoir of Coxiella burnetii , Cayenne ,
French Guiana Marburgvirus Resurgence in Kitaka Mine Bat Population after
Extermination Attempts , Uganda. 20(10):1–3.
Eldin C, Angelakis E, Renvoisé A, Raoult D. 2013. Coxiella burnetii DNA, but not viable
bacteria, in dairy products in France. American Journal of Tropical Medicine and Hygiene
88(4):765–769.
Eldin C, Perreal C, Mahamat A, Djossou F, Edouard S, Raoult D. 2015. Antibiotic susceptibility
determination for six strains of Coxiella burnetii MST 17 from Cayenne, French Guiana.
International Journal of Antimicrobial Agents:17–18.
Eldin C, Mélenotte C, Mediannikov O, Ghigo E, Million M, Edouard S, Mege J, Maurin M.
2017. From Q Fever to Coxiella burnetii Infection : a Paradigm Change. Clinical
Microbiology Reviews 30(1):115–190.
Farlow J, Smith KL, Wong J, Abrams M, Lytle M, Keim P. 2001. Francisella tularensis Strain
Typing Using Multiple-Locus , Variable-Number Tandem Repeat Analysis. Society
39(9):3186–3192.
Farris CM, Pho N, Myers TE, Richards AL. 2016. Seroconversions for. 22(8):8–10.
Fernando SL, Hudson BJ. 2013. Rapid desensitization to doxycycline. Annals of Allergy,
Asthma and Immunology 111(1):73–74.
Fisher JF, Meroueh SO, Mobashery S. 2005. Bacterial resistance to β-lactam antibiotics:
Compelling opportunism, compelling opportunity. Chemical Reviews 105(2):395–424.
Forsberg KJ, Patel S, Wencewicz TA, Dantas G. 2015. The Tetracycline Destructases: A Novel
Family of Tetracycline-Inactivating Enzymes. Chemistry and Biology 22(7):888–897.
Fournier P, Marrie TJ. 1998. MINIREVIEW Diagnosis of Q Fever. 36(7):1823–1834.

48
Fournier P, Karkouri K El, Leroy Q, Robert C, Giumelli B, Renesto P, Socolovschi C, Parola
P, Audic S, Raoult D. 2009. Analysis of the Rickettsia africae genome reveals that
virulence acquisition in Rickettsia species may be explained by genome reduction. 15:1–
15.
Frangoulidis D, Meyer H, Kahlhofer C, Splettstoesser WD. 2012. ‘Real-time’ PCR-based
detection of Coxiella burnetii using conventional techniques. FEMS Immunology and
Medical Microbiology 64(1):134–136.
Fu Y, Zhang W, Wang H, Zhao S, Chen Y, Meng F, Zhang Y, Xu H, Chen X, Zhang F. 2013.
Specific patterns of gyrA mutations determine the resistance difference to ciprofloxacin
and levofloxacin in Klebsiella pneumoniae and Escherichia coli. BMC Infectious Diseases
13(1):1.
Gikas A, Spyridaki I, Psaroulaki A. 1998. In Vitro Susceptibility of Coxiella burnetii to
Trovafloxacin in Comparison with Susceptibilities to Pefloxacin ,. 42(10):2747–2748.
Gikas A, Kofteridis DP, Manios A, Pediaditis J, Tselentis Y. 2001. Newer Macrolides as
Empiric Treatment for Acute Q Fever Infection. 45(12):3644–3646.
Glazunova O, Roux V, Freylikman O, Sekeyova Z, Fournous G, Tyczka J, Tokarevich N, Kov
E, Marrie TJ, Raoult D. 2005. Coxiella burnetii Genotyping. 11(8):1211–1217.
Goldstein BP. 2014. Resistance to rifampicin : a review. The Journal of Antibiotics 67(9):625–
630.
Gossé M, Lysvand H, Pukstad B, Nordbø SA. 2016. A novel SimpleProbe PCR for detection
of mutations in the 23S rRNA gene associated with macrolide resistance in Mycoplasma
genitalium in clinical samples. Journal of Clinical Microbiology 54(1):JCM.01233-16.
Guatteo R, Beaudeau F, Berri M, Rodolakis A, Jolyc A, Seegers H. 2006. Shedding routes of
Coxiella burnetii in dairy cows: Implications for detection and control. Veterinary
Research 37(6):827–833.
Gupta SK, Padmanabhan R, Diene SM, Lopez-rojas R, Kempf M, Landraud L. 2014. ARG-
ANNOT , a New Bioinformatic Tool To Discover Antibiotic. 58(1):212–220.
Hatchette TF, Hudson RC, Schlech WF, Campbell NA, Hatchette JE, Ratnam S, Raoult D,
Donovan C, Marrie TJ. 2001. Goat-associated Q fever: a new disease in Newfoundland.
Emerging infectious diseases 7(3):413–419.

49
Hornstra HM, Priestley RA, Georgia SM, Kachur S, Birdsell DN, Hilsabeck R, Gates LT,
Samuel JE, Heinzen RA, Kersh GJ, et al. 2011. Rapid Typing of Coxiella burnetii. 6(11).
HUANG K, XU C-W, ZENG B, XIA Q-Q, ZHANG A-Y, LEI C-W, GUAN Z-B, CHENG H,
WANG H-N. 2014. Dynamics of Quinolone Resistance in Fecal Escherichia coli of
Finishing Pigs after Ciprofloxacin Administration. Journal of Veterinary Medical Science
76(9):1213–1218.
HUEBNER RJ, JELLISON WL, BECK D, WILCOX FP. 1949. Sage Publications, Inc.,
Association of Schools of Public Health. 64(16):499–511.
Huijsmans CJJ, Schellekens JJA, Wever PC, Toman R, Savelkoul PHM, Janse I, Hermans
MHA. 2011. Single-nucleotide-polymorphism genotyping of Coxiella burnetii during a Q
fever outbreak in the Netherlands. Applied and Environmental Microbiology 77(6):2051–
2057.
Jia B, Raphenya AR, Alcock B, Waglechner N, Guo P, Tsang KK, Lago BA, Dave BM, Pereira
S, Sharma N, et al. 2017. CARD 2017 : expansion and model-centric curation of the
comprehensive antibiotic resistance database. 45(October 2016):566–573.
Jiménez PH. Genetische Unterschiede zwischen Coxiella burnetii -Isolaten und ihre
Korrelation.
Johansson A, Göransson I, Larsson P, Sjöstedt A. 2001. Extensive allelic variation among
Francisella tularensis strains in a short-sequence tandem repeat region. Journal of Clinical
Microbiology 39(9):3140–3146.
Katoh K, Rozewicki J, Yamada KD. 2017. MAFFT online service: multiple sequence
alignment, interactive sequence choice and visualization. Briefings in
Bioinformatics(April):1–7.
Kayange N, Kamugisha E, Mwizamholya DL, Jeremiah S, Mshana SE. 2010. Predictors of
positive blood culture and death among neonates with suspected neonatal sepsis in a
tertiary hospital, Mwanza-Tanzania. BMC Pediatr 10.
Keijmel SP, Delsing CE, Sprong T, Bleijenberg G, Meer JWM Van Der, Knoop H, Bleeker-
rovers CP. 2013. The Qure study : Q fever fatigue syndrome – response to treatment ; a
randomized placebo-controlled trial. BMC Infectious Diseases 13(1):1.
Kelly LA, Mezulis S, Yates C, Wass M, Sternberg M. 2015. The Phyre2 web portal for protein

50
modelling, prediction, and analysis. Nature Protocols 10(6):845–858.
Kersh GJ. 2013. Antimicrobial therapies for Q fever. 8:1–8.
Klevytska AM, Schupp JM, Worsham PL, Wong J, Keim P. 2001. Identi cation and
Characterization of Variable-Number Tandem Repeats in the. Society 39(9):3179–3185.
Krumbiegel ER, Wisniewski HJ. 1970. II consumption of infected raw milk by human
volunteers. Archives of Environmental Health 21(1):63–65.
Kuraku S, Zmasek CM, Nishimura O, Katoh K. 2013. aLeaves facilitates on-demand
exploration of metazoan gene family trees on MAFFT sequence alignment server with
enhanced interactivity. Nucleic acids research 41(Web Server issue):22–28.
Lista F, Faggioni G, Valjevac S, Ciammaruconi A, Vaissaire J, Le Doujet C, Gorgé O, De
Santis R, Carattoli A, Ciervo A, et al. 2006. Genotyping of Bacillus anthracis strains based
on automated capillary 25-loci Multiple Locus Variable-Number Tandem Repeats
Analysis. BMC Microbiology 6:1–14.
Louw GE, Warren RM, Gey Van Pittius NC, McEvoy CRE, Van Helden PD, Victor TC. 2009.
A balancing act: Efflux/influx in mycobacterial drug resistance. Antimicrobial Agents and
Chemotherapy 53(8):3181–3189.
Madariaga MG, Rezai K, Trenholme GM, Weinstein RA. 2003. Review Q fever : a biological
weapon in your backyard. :709–721.
Manyahi J, Matee MI, Majigo M, Moyo S, Mshana SE, Lyamuya EF. 2014. Predominance of
multi-drug resistant bacterial pathogens causing surgical site infections in Muhimbili
national hospital, Tanzania. BMC Research Notes 7(1):1–7.
Manyahi J, Tellevik MG, Ndugulile F, Moyo SJ, Langeland N, Blomberg B. 2017. Molecular
Characterization of Cotrimoxazole Resistance Genes and Their Associated Integrons in
Clinical Isolates of Gram-Negative Bacteria from Tanzania. Microbial Drug Resistance
23(1):37–43.
Marmion BP, Storm PA, Ayres JG, Semendric L, Mathews L. 2018. Original papers Long-term
persistence of Coxiella burnetii after acute primary Q fever. 98(1):7–20.
Marosevic D, Kaevska M, Jaglic Z. 2017. Resistance to the tetracyclines and macrolide-
lincosamide-streptogramin group of antibiotics and its genetic linkage – A review. Annals
of Agricultural and Environmental Medicine 24(2):338–344.

51
Million M, Raoult D. 2017. No Such Thing as Chronic Q Fever. 23(5):9–10.
Morroy G, Keijmel SP, Delsing CE, Bleijenberg G. 2016. Fatigue following Acute Q-Fever :
A Systematic Literature Review. :1–21.
Munita JM, Arias CA, Unit AR, Santiago A De. 2016. Mechanisms of Antibiotic Resistance.
Microbiol Spectr. 4(2):1–37.
Musso D, Drancourt M, Osscini S, Raoult D. 1996. Sequence of Quinolone Resistance-
Determining Region of gyrA Gene for Clinical Isolates and for an In Vitro-Selected
Quinolone-Resistant Strain of Coxiella burnetii. 40(4):870–873.
Nawaz M, Sung K, Kweon O, Khan S. 2015. Characterisation of novel mutations involved in
quinolone resistance in Escherichia coli isolated from imported shrimp. International
Journal of Antimicrobial Agents 45(5):471–476.
Nielsen K, Hindersson P, Høiby N. 2000. Sequencing of the rpoB Gene in Legionella
pneumophila and Characterization of Mutations Associated with Rifampin Resistance in
the Legionellaceae. 44(10):2679–2683.
Norville IH, Hatch GJ, Bewley KR, Atkinson DJ, Hamblin KA, Blanchard JD, Armstrong SJ,
Pitman JK, Rayner E. 2014. Efficacy of Liposome-Encapsulated Ciprofloxacin in a
Murine Model. 58(9):5510–5518.
Omsland A, Beare PA, Hill J, Cockrell DC, Howe D, Hansen B, Samuel JE, Heinzen RA. 2011.
Isolation from animal tissue and genetic transformation of Coxiella burnetii are facilitated
by an improved axenic growth medium. Applied and Environmental Microbiology
77(11):3720–3725.
Prigent M, Rousset E, Yang E, Thiéry R, Sidi-Boumedine K. 2015. Validation study for using
lab-on-chip technology for Coxiella burnetii multi-locus-VNTR-analysis (MLVA) typing:
Application for studying genotypic diversity of strains from domestic ruminants in France.
Microbes and Infection 17(11–12):782–788.
Raoult D. 1993. Minireview: Treatment of Q Fever. Antimicrobial Agents and Chemotherapy
37(9):1733–1736.
Raoult D, Torres H, Reference CN De, Peacock MG, Williams JC, Anacker RL, Vestris G,
Enea M. 1991. Antibiotic Susceptibility, Tested in 13 Isolates of Coxiella burnetii. :2070–
2077.

52
Raoult D, Pierre Houpikian M, Herve´ Tissot Dupont M, Jean Marc Riss M, J. Arditi-Djiane
M, Philippe Brouqui M. 1999. Treatment of Q Fever Endocarditis. 159:167–173.
Redgrave LS, Sutton SB, Webber MA, Piddock LJ V. 2014. Fluoroquinolone resistance :
mechanisms , impact on bacteria , and role in evolutionary success. Trends in
Microbiology 22(8):438–445.
Rodolakis A. 2009. Q Fever in Dairy Animals. In RICKETTSIOLOGY AND RICKETTSIAL
DISEASES-FIFTH INTERNATIONAL CONFERENCE 90–93.
Rodolakis A, Berri M, He C, Camuset P, Devillechaise P, Natorp JC, Vadet JP. 2007.
Comparison of Coxiella burnetii Shedding in Milk of Dairy Bovine , Caprine , and Ovine
Herds. :5352–5360.
Rolain J, Maurin MAX, Raoult D. 2001. Bacteriostatic and Bactericidal Activities of
Moxifloxacin against Coxiella burnetii. 45(1):301–302.
Rolain J, Lambert F, Raoult D. 2005. Activity of Telithromycin against Thirteen New Isolates
of C . burnetii Including Three Resistant to Doxycycline. 256:252–256.
Rothberg JM, Leamon JH. 2008. The development and impact of 454 sequencing. Nature
Biotechnology 26(10):1117–1124.
Rouli L, Rolain J-M, Filali A El, Robert C, Raoult D. 2012. Genome Sequence of Coxiella
burnetii 109, a Doxycycline-Resistant Clinical Isolate. 194(24):7278.
Seshadri R, Paulsen IT, Eisen JA, Read TD, Nelson KE, Nelson WC, Davidsen TM, Beanan
MJ, Deboy RT, Daugherty SC, et al. 2003. Complete genome sequence of the Q-fever
pathogen Coxiella burnetii. (20).
Shaheen BW, Nayak R, Foley SL, Boothe DM. 2013. Chromosomal and plasmid-mediated
fluoroquinolone resistance mechanisms among broad-spectrum-cephalosporin-resistant
Escherichia coli isolates recovered from companion animals in the USA. Journal of
Antimicrobial Chemotherapy 68(5):1019–1024.
Sidi-Boumedine K, Duquesne V, Prigent M, Yang E, Joulié A, Thiéry R, Rousset E. 2015.
Impact of IS1111 insertion on the MLVA genotyping of Coxiella burnetii. Microbes and
Infection 17(11–12):789–794.
Sloan B, Scheinfeld N. 2008. The use and safety of doxycycline hyclate and other second-
generation tetracyclines. Expert Opin Drug Saf 7(5):571–577.

53
Spyridaki I, Psaroulaki A, Aransay A, Scoulica E. 2000a. Diagnosis of Quinolone-Resistant
Coxiella burnetii Strains by PCR-RFLP. 63(August 1999):59–63.
Spyridaki I, Psaroulaki A, Aransay A, Scoulica E. 2000b. Diagnosis of Quinolone-Resistant
Coxiella burnetii Strains by PCR-RFLP. 63(October 1999):59–63.
Spyridaki I, Psaroulaki A, Kokkinakis E, Gikas A, Tselentis Y. 2002. JAC Mechanisms of
resistance to fluoroquinolones in Coxiella burnetii. :379–382.
Spyridaki I, Psaroulaki A, Vranakis I, Tselentis Y, Gikas A. 2009. Bacteriostatic and
Bactericidal Activities of Tigecycline against Coxiella burnetii and Comparison with
Those of Six Other Antibiotics ᰔ. 53(6):2690–2692.
Stevenson S, Gowardman J, Tozer S, Woods M. 2015. Life-threatening Q fever infection
following exposure to kangaroos and wallabies. :1–3.
Stunkard A, Harris J, Pederson N. 1990. The New England Journal of Medicine Downloaded
from nejm.org on October 11, 2014. For personal use only. No other uses without
permission. Copyright © 1990 Massachusetts Medical Society. All rights reserved. The
New England Journal of Medicine 322(21):1483–1486.
Subramanya NI, Wright JS, Khan MA. 1982. Failure of rifampicin and co-trimoxazole in Q
fever endocarditis. Bmj 285(6338):343–344.
Suhan ML, Thompson HA. 2000. Expression of L -lactamase in Coxiella burnetii
transformants. 184:304–307.
Ventola CL. 2015. The antibiotic resistance crisis: part 1: causes and threats. P & T : A peer-
reviewed journal for formulary management (2015) 40(4):277–83.
Voth DE, Beare PA, Howe D, Sharma UM, Samoilis G, Cockrell DC, Omsland A, Heinzen
RA. 2011. The Coxiella burnetii cryptic plasmid is enriched in genes encoding type IV
secretion system substrates. Journal of Bacteriology 193(7):1493–1503.
Vranakis I. 2010. DNA Gyrase and Topoisomerase IV Mutations in an In Vitro
Fluoroquinolone-Resistant Coxiella burnetii Strain. Microbial Drug Resistance 16(2).
Wass MN, Kelley LA, Sternberg MJE. 2010. 3DLigandSite: Predicting ligand-binding sites
using similar structures. Nucleic Acids Research 38(SUPPL. 2):469–473.
Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. 2009. Jalview Version 2-A

54
multiple sequence alignment editor and analysis workbench. Bioinformatics 25(9):1189–
1191.
Yang W, Moore IF, Koteva KP, Bareich DC, Hughes DW, Wright GD. 2004. TetX is a flavin-
dependent monooxygenase conferring resistance to tetracycline antibiotics. Journal of
Biological Chemistry 279(50):52346–52352.
Ye J, Coulouris G, Zaretskaya I, Cutcutache I, Rozen S, Madden TL. 2012. Primer-BLAST: a
tool to design target-specific primers for polymerase chain reaction. BMC bioinformatics
13:134.
Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O, Aarestrup FM,
Larsen MV. 2012. Identification of acquired antimicrobial resistance genes. Journal of
Antimicrobial Chemotherapy 67(11):2640–2644.
Zekanovic D, Morovic M, Borcilo MN, Rode OD. 2010. First case of Q fever endocarditis in
Croatia and a short review. Collegium antropologicum 34(3):1135–1137.

55
5.7. Appendix
List of Figures Figure 1 Current C. burnetii geotyping (Eldin et al. 2017) ............................................................................. 7
Figure 2 File structure of bioinformatics pipeline ......................................................................................... 20
Figure 3 Alignment of nucleotide sequence of parE ..................................................................................... 31
Figure 4 Alignment of amino acid sequence of parE .................................................................................... 32
Figure 5 Melting peaks .................................................................................................................................. 33
Figure 6 Melting peaks of selected strains .................................................................................................... 34
Figure 7 Sequence alignment of ampC of C. burnetii ................................................................................... 36
Figure 8 Predicted secondary structure of ampC of C. burnetii str. Dugway................................................ 37
Figure 9 Predicted structure with binding site ............................................................................................... 38
List of Tables Table 1 Modes of resistance and candidate genes ........................................................................................... 9
Table 2 Published Antibiotic Susceptibility of C. burnetii ........................................................................... 16
Table 3 Available genome assemblies at NCBI ............................................................................................ 18
Table 4 Sequences of resistance associated genes used in this study ............................................................ 25
Table 5 Reference sequence file names used in this study ............................................................................ 25
Table 6 Primer and probe sequences ............................................................................................................. 26
Table 7 SimpleProbe protocol #1 .................................................................................................................. 27
Table 8 SimpleProbe protocol #2 .................................................................................................................. 27
Table 9 LightCycler 480 protocol ................................................................................................................. 28
Table 10 Strains analyzed in this study for parE polymorphism .................................................................. 29
Table 11 parE polymorphisms of selected isolates ....................................................................................... 35
Chemicals
Name Company Product number Lot number
LightCycler FastStart DNA
Master Hybridization Probes Kit
Roche 12239272001 11158300

56
Abbreviations
QRDR quinolone determing resistance region PMQR plasmid-mediated quinolone resistance MLVA multiple locus variable-number tandem repeat VNTR variable number tandem repeat SNP single nucleotide polymorphism MST multispacer typing TE transposable element IS insertion sequence













![White Paper Antibiotic Use and Resistance: Moving Forward ... - Symp... · White Paper: Antibiotic Use & Resistance [3] BACKGROUND The symposium Antibiotic Use and Resistance: Moving](https://img.pdfslide.net/doc/110x75/5f0aa9c27e708231d42cb9b0/white-paper-antibiotic-use-and-resistance-moving-forward-symp-white.jpg)















