Embed Size (px)
Citation preview

GO Annotation Fall 2005
Introduction: Proteins & Function
Guest Lecturer: Bindu Nanduri [email protected]

Post genomic world
The Genome Covers
2001 IHGSC and Celera corporation


From Sequence to Function
•The genomic sequence identifies the 'parts'the next trick is understanding gene function
•Post genomic era = functional genomics
•Critical concept: genes of similar sequence may have similar functions
•Inferring function for a new gene begins with searching for it’s nearest neighbor (or homolog) of known function

Evolution Allows us to Infer Function
• The most powerful method for inferring function of a gene or protein is by similarity searching a sequence database.
• Our ability to characterize biological properties of a protein using sequence data alone stems from properties conserved through evolutionary time.
• Homologous (evolutionarily related) proteins always share a common 3-dimensional folding structure.
• They often contain common active sites or binding domains.
• They frequently share common functions.

Orthologs• Homologs = genes that are evolutionarily related
• There are two kinds of homologs:
• Orthologs = genes in different species that have diverged from a common gene in an ancestral species.
• Paralogs = genes that have diverged due to gene duplication.
• Orthologs are more likely than paralogs to have conserved function.
• Orthologs cannot be identified using BLAST or FASTA sequence comparison alone.
• Reliable ortholog identification requires phylogenetic methods.

Rice-2b
Rice-2a
Maize-2
Wheat-2
Sorghum-2
Barley-1
Wheat-1
Maize-1
Sorghum-1
Arabidopsis
Example Gene Tree (with plant genes)
The outgroup, Arabidopsis is a dicot. The cereals are monocots. Dicots and monocots diverged ~230 million years ago. These monocots diverged from each other ~60 mya.
orthologs
paralogs

Why shouldn’t we depend on inferences based on paralogs?
• Paralogs emerge after a gene duplication.
• Possible fates of duplicated genes:
– Loss of function for one of the duplicates - lack of selective pressure allows gene to mutate beyond recognition
– Emergence of new functional paralogs - one duplicate aquires a new function, so selection favors its maintenance in the genome
– Sub-functionalization - both duplicates are required to maintain the function of the original

“Central Dogma” (Francis Crick):
1 gene gives 1 mRNA gives 1 protein (predicted hundreds of thousands of genes in humans)
Information flow
DNA----- >>Proteins---->> Function

Finding Genes in Genomic DNA• Translate (in all 6 reading frames) and look for similarity to known protein
sequences• Look for long Open Reading Frames (ORFs) between start and stop
codons(start=ATG, stop=TAA, TAG, TGA)
• Look for known gene markers• TAATAA box, intron splice sites, etc.
• Statistical methods (codon preference)
Intron/exon boundaries• Gene finding programs work well in bacteria• None of the gene prediction programs do an adequate job predicting
intron/exon boundaries• The only reasonable gene models are based on alignment of cDNAs to
genome sequence• Some human genes still do not have a correct coding sequence defined
(transcription start, intron splice sites)

Proteins As Modules• Proteins are derived from a limited number of
basic building blocks (Domains)
• Evolution has shuffled these modules giving rise to a diverse repertoire of protein sequences
• As a result, proteins can share a global or local relationship
(Higgins, 2000)

Protein Domains
Motifs describe the domain
Janus Kinase 2 Modular Sequence Architecture
SH2Motif

Protein Families• Protein Family - a group of proteins that share a common function
and/or structure, that are potentially derived from a common ancestor (set of homologous proteins)
• Characterizing a Family - Compare the sequence and structure patterns of the family members to reveal shared characteristics that potentially describe common biological properties
• Motif/Domain - sequence and/or structure patterns common to protein family members (a trait)

Protein Families
Family A
Family B
Family D Family E
Family C
Separate Families canBe Interrelated

Creating Protein Families• Use domains to identify family members
– Use a sequence to search a database and characterize a pattern/profile
– Use a specific pattern/profile to identify homologous sequences (family members)
Find Domains Find Family Members
Pattern/Profile Searches
BLAST and Alignments

Motifs are built from Multiple Alignments

Protein Motif Databases• Known protein motifs have been collected in databases• One such database is PROSITE
Each domain is defined by a simple pattern– Patterns can have alternate amino acids in each position and defined
spaces, but no gaps– Pattern searching is by exact matching, so any new variant will not
be found (can allow mismatches, but this weakens the algorithm)

Profiles• Profiles are tables of amino acid frequencies at each position in a motif• They are built from multiple alignments• PROSITE entries also contain profiles built from an alignment of proteins
that match the pattern• Profile searching is more sensitive than pattern searching - uses an
alignment algorithm, allows gaps

Hidden Markov Models
• Hidden Markov Models (HMMs) are a more sophisticated form of profile analysis.
• Rather than build a table of amino acid frequencies at each position, they model the transition from one amino acid to the next.
• Pfam is built with HMMs.

So far……..
Given a gene/protein sequence we have learned ways to determine the function
With this wealth of information, there arises a need for ANNOTATION
an·no·ta·tion (ăn'ō-tā'shən)
The act or process of furnishing critical commentary or explanatory notes
Genome sequence annotation can be
---structural demarcate functional nucleotide sequences in the genome (ORFs)
---functional assign functions to the identified functional element (gene products)
ONTOLOGY controlled structured vocabulary

Gene Ontology (GO)
•Molecular function: tasks performed by gene product
•Biological process: broad biological goals accomplished by ordered assemblies of molecular functions
•Cellular component: subcellular structures, locations and macromolecular complexes

Example, Bioworks output from a MudPIT experiment (Bovine)



Gene association file

Guilt-by-Association
Compare T with seqs of known function in a db
Assign to T same function as homologs
Confirm with suitable wet experiments
Discard this functionas a candidate

Basic Local Alignment Search ToolBLAST
Altschul et al., JMB, 215:403--410, 1990
find from db seqswith short perfectmatches to queryseq
find seqs withgood flanking alignment

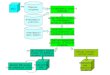
Annotating the Chicken GenomeCHICKEN GENOME
known gene products
(UniProt)
‘predicted’gene products(NCBI/UniParc)
unique gene products
(Ensembl?)
no annotationannotation
IEA annotation
non-IEA annotation
requiresmanual annotation
further experimentation
35% 35%30%
(0.91%)

Gene Ontology Annotation Training: Introduction
Fiona McCarthy November 2005

GO Annotation Training
• Primarily focus on training to be a biocurator, ie. training to assign GO terms to gene products
• GO Users: introduction to GO

Overview1. Bio-Ontologies and the Gene Ontology2. The Gene Ontology Consortium and
AgBase3. The 3 Gene Ontologies4. GO Terms5. DAGs6. Annotating to GO7. Nomenclature8. GO Term Rules and Relationships

1. Bio-Ontologies and the Gene Ontology (GO)

What is an Ontology?
Gruber, 1993:• “ontologies provide controlled, consistent
vocabularies to describe concepts and relationships, thereby enabling knowledge sharing”

OBO: Open Biomedical Ontologies

Why is Gene Ontology Needed?• The post-genome ‘problem’!
- a huge body of information with an extremely large vocabulary to describe it
• Vocabulary used is poorly defined- one word can have different meanings- different names for the same concept
• Biological systems are complex and our knowledge of such systems is incomplete
• Results in large databases which are difficult to mine computationally
Lewis SE, Genome Biology 2004. PMID:15642104

What is the Gene Ontology?Emily Dimmer, GOA EBI, 2004:GO: “a controlled vocabulary that can be applied to all organisms even as knowledge of gene and protein roles in cells is accumulating and changing”
• a collaborative effort to address the need for consistent descriptions of gene products in different databases• facilitates uniform queries across them • structured to be queried at different levels, eg. you can use GO to find all the maize gene products in the genome that are involved in signal transduction or you can zoom in on all the receptor tyrosine kinases• annotators can assign properties to gene products at different levels, depending on how much is known about a gene product• provides a standard, species-neutral way of representing biological function• GO covers ‘normal’ functions and processes
- no pathology, no experimental conditions

What GO is NOT
• GO is not a database of gene sequences or gene products
• GO is not a unified database; rather it is a shared vocabulary
• GO does not attempt to describe every aspect of biology
• GO is not a dictated standard

2. The GO Consortium and AgBase

The GO Consortium• development of ontologies that describe gene products
in terms of their associated biological processes, cellular components and molecular functions in a species-independent manner
• There are three separate aspects to this effort: 1. Writing and maintaining the ontologies; 2. Making associations between the ontologies and the
genes and gene products in the collaborating databases; 3. Developing tools that facilitate the creation, maintenance
and use of ontologies.

© Emily Dimmer, GOA

© Emily Dimmer, GOA

AgBase Data Submission• new associations are made by biocurators• gene associations are prepared as a gene
association file• the gene association file is submitted to AgBase
for QC checking• AgBase collates and submits gene associations
to the GO Consortium via GOA• AgBase submissions are QC checked by GOA
and MGI• AgBase gene associations are available through
the AgBase homepage, GOA and UniProt

3. The Gene Ontologies
http://www.geneontology.org/

The Gene OntologiesThe three organizing principles of GO are:1. Molecular Function (mf or F)
- describes activities, such as catalytic or binding activities, at the molecular level
- represent activities rather than the entities (molecules or complexes) that perform the actions
- eg. catalytic activity, transporter activity 2. Biological Process (bp or P)
- series of events accomplished by one or more ordered assemblies of molecular functions
- a bp term does not represent a pathway- eg.signal transduction or pyrimidine metabolism
3. Cellular Component (cc or C)- a component of a cell which may be a subcellular structure or a gene
product group- eg. nucleus or proteasome

Gene products can be described by more that one term in each ontology.1. Molecular Function (mf or F)cytochrome c can be described by the mf term electron transporter activity2. Biological Process (bp or P)cytochrome c is used in several bp: oxidative phosphorylation and induction of cell death3. Cellular Component (cc or C)the gene product cytochrome c has 2 cc: mitochondrial matrix and mitochondrial inner membrane.

Ontology Guides• Cellular Component ontology guide:http://www.geneontology.org/GO.component.guid
elines.shtml• Molecular Function ontology guide:http://www.geneontology.org/GO.function.guidelin
es.shtml• Biological Process ontology guide:http://www.geneontology.org/GO.process.guidelin
es.shtml

• Many gene products associate into entities that function as complexes, or 'gene product groups', – eg. hemoglobin contains the gene products alpha-
globin and beta-globin, and the small molecule heme
– eg. the ribosome is a complex assemblies of numerous different gene products
• To avoid confusion between gene products and molecular function, mf terms are described as an activity– eg. alcohol dehydrogenase can be annotated to the
term alcohol dehydrogenase activity


4. GO Terms

What is a GO Term?• The purpose of GO is to define particular
attributes of gene products• A GO Term is simply the text string used to
describe an entry in GO– eg. signal transduction
• GO Terms do not include – mutant or disease specific terms, eg. oncogenesis– protein domains or structural features, eg. the death
domain• A GO Node is a GO Term and all its children

The Anatomy of a GO Term
GO Terms are composed of:• term name• unique 7-digit GO ID• term definition (93% of GO terms are
defined)• synonyms (optional)• database cross-references (optional)• relationships to other GO terms

The Anatomy of a GO Term
relationship to other GO terms
record of term update
term nameunique GO ID
ontology that the GO term fits intosynonyms for the term name (to aid searching)
term definition

General Conventions for GO Terms
• Use “U.S. English”• Avoid abbreviations• Use full element names, not symbols, eg. “zinc” not Zn• Spell Greek symbols out in full, eg. alpha• Use lower case, except where demanded by context,
eg,. DNA• Use singular form where possible• Do not use anatomical qualifiers, eg. mitochondrial DNA
polymerase• Avoid gene product names where possible (may be used
as a synonym)• Cross-reference other databases where possible

GO ID Numbers
• A GO ID is a 7-digit number, padded with zeros, if necessary– numbers are assigned as they are required
• a GO ID is NEVER deleted: GO IDs should be conserved at all times so that, even if a term is defunct or has a new GO ID, someone searching using the old GO ID can find it.
• A GO ID is associated with a definition rather than with the term name, eg. – if the wording but not the meaning of a term is changed, the GO ID
stays the same, – a new meaning requires a new GO ID, even if the text string doesn't
change.

Changing GO ID Numbers
• Assume that we have a term 'mouse', GO ID GO:0000123, in an ontology; it is defined as a small furry mammal.
• We decide to change the wording to 'Mus musculus', keeping the definition the same. In this case we merely update the text; the GO ID stays the same because the meaning stays the same. We may choose to keep 'mouse' as a synonym, but there would still only be one ID associated with the term.
• We decide that the term 'mouse' should instead mean a piece of computer equipment. In this case, the old term and ID are moved to the 'obsolete' category, and 'mouse' as newly defined gets a new GO ID, GO:0000456. The old GO ID and definitions are saved for posterity in case we ever need to know what happened to them.
mouseAccession: GO:0000123Aspect: biological_processSynonyms: MickeyDefinition: small, furry rodent

GO Term Definition
• Always define new terms– If you create a new term, or refine a term, you should add a definition for
it, and note the references used in composing the definition– Refer to Oxford Dictionary of Chemistry and Molecular Biology– Wherever a 'standard' definition exists for a group of related terms, it
should be used (standard definitions available from the ontology guides)• Write definitions carefully in full sentences• Document where your definition came from• Always define which ontology the term belongs to• Show where new terms fit into the ontology
– show all possible paths– all possible paths must be true

GO Term Synonyms
• Synonyms are important as they facilitate database searching
• Don’t add synonyms where the only difference is case• Synonyms may not always be strictly 'synonymous',
eg. – may be broader or narrower than the term definition– may be a related phrase– may be alternative wording, spelling or use a different
system of nomenclature

GO Term SynonymsThe synonym relationship types are:
• the term is an exact synonym:ornithine cycle is an exact synonym of urea cycle
• the terms are related:cytochrome bc1 complex is a related synonym of ubiquinol-cytochrome-c reductase activity
• the synonym is broader than the term name:cell division is a broad synonym of cytokinesis
• the synonym is narrower or more precise than the term name:pyrimidine-dimer repair by photolyase is a narrow synonym of photoreactive repair
• the synonym is related to the term, but is not exact, broader or narrower:virulence has a synonym type of other related to the term pathogenesis

Cross-referencing Other Databases General database cross references (general dbxrefs) should be used whenever a GO term has an identical meaning to an object in another database.

5. Fitting GO Terms together: DAGs

Directed Acyclic Graphs
• The ontologies are organized as directed acyclic graphs (DAGs)
• DAGs differ from hierarchies in that a “child”, or more specialized, term can have many “parents”, or less specialized, terms
• Terms are linked by two types of relationships:- is_a & part_of

AmiGO Browser http://www.godatabase.org/cgi-bin/amigo/go.cgi?search_constraint=terms&action=replace_tree

MGI GO Browser http://www.informatics.jax.org/searches/GO_form.shtml

Browsing the GO DAGsThere are three GO Browsers online:• AmiGO browserhttp://www.godatabase.org/cgi-bin/amigo/go.cgi
• QuickGO browserhttp://www.ebi.ac.uk/ego/
• MGI browserhttp://www.informatics.jax.org/searches/GO_form.
shtml

6. Annotating to GO: attributing function

Annotating to GO
• Using GO terms to represent the activities and localizations of a gene product
• Annotations contributed by members of the GO Consortium– model organism databases– cross-species databases, eg. UniProt
• Annotations made freely available from GO website under ‘downloads’

Annotating to GO
Information required:1. database object (eg. a protein or gene
identifier)2. reference ID (eg. PubMed ID)3. GO Term ID (eg. GO:0004674)4. evidence code (eg. IDA)

Two Types of Annotation:1. electronic annotation2. manual annotation
All annotations must:• Every annotation must be attributed to a source,
which may be a literature reference, another database or a computational analysis.
• The annotation must indicate what kind of evidence is found in the cited source to support the association between the gene product and the GO term.

Making Gene Associations
• GO terms should be associated with database objects representing gene products rather than genes – a single gene may encode very different products with
very different attributes – if the database object is a gene, it is associated with all
GO terms applicable to any of its products • GO annotations should be attributed to a source • Each annotation should indicate the evidence on
which it is based

Evidence CodesIMP inferred from mutant phenotype IGI inferred from genetic interaction
[with <database:gene_symbol[allele_symbol]>] IPI inferred from physical interaction
[with <database:protein_name>] ISS inferred from sequence similarity
[with <database:sequence_id>] IDA inferred from direct assay IEP inferred from expression pattern IEA inferred from electronic annotation
[to <database:id>] TAS traceable author statement NAS non-traceable author statement ND no biological data available RCA inferred from reviewed computational analysis IC inferred by curator

• Because a single gene may encode very different products with very different attributes, GO recommends associating GO terms with database objects representing gene products rather than genes. • At present, however, many participating databases are unable to associate GO terms to gene products, and therefore use genes instead. • If the database object is a gene, it is associated with all GO terms applicable to any of its products.

The Gene Association File• collaborating databases export to GO a tab
delimited file, the "gene association file" of links between database objects and GO terms
• the database object may represent a gene or a gene product (transcript or protein)
• gene association files are available from both collaborating databases and the GO Consortium
• the gene association file is the mechanism by which gene products functions are shared/released

The Gene Association FileTo make gene associations we have to fill in up to 15 fields; 11 are compulsory


7. Nomenclature

Why Do We Care?
DB_Object_Symbol
To make gene associations we have to fill in 11 compulsory fields

Why Do We Care?• The 3rd gene association field is
“DB_Object_Symbol”• The entry in the DB_Object_Symbol field should be
a symbol that means something to a biologist, wherever possible (gene symbol, for example).
• It is not an ID or an accession number (the second column, DB_Object_ID, provides the unique identifier), although IDs can be used in DB_Object_Symbol if there is no more biologically meaningful symbol available (e.g., when an unnamed gene is annotated).

Nomenclature Guidelines• chicken & cow will follow human guidelines
– controlled by HUGO Gene Nomenclature Committee (HGNC)
– http://www.gene.ucl.ac.uk/nomenclature/• Arabidopsis:
– http://www.arabidopsis.org/jsp/processor/genesymbol/symbol_main.jsp
• Rice nomeclature committee: Gramene, MaizeGDB
• catfish: “Gene Nomenclature for Protein-Coding Loci in Fish” Transactions of the American Fisheries Society: Vol. 119, No. 1, pp. 2–15.

Guidelines for HGNC
• Hester M. Wain, Elspeth A. Bruford, Ruth C. Lovering, Michael J. Lush, Mathew W. Wright and Sue PoveyGenomics 79(4):464-470 (2002)
• Available online (last updated 2004):http://www.gene.ucl.ac.uk/nomenclature/gui
delines.html

Guidelines Summary1. Each approved gene symbol must be unique. 2. Symbols are short-form representations (or abbreviations) of the descriptive gene name. 3. Symbols should only contain Latin letters and Arabic numerals. 4. Symbols should not contain punctuation. 5. Symbols should not end in "G" for gene. 6. Symbols do not contain any reference to species, for example "H/h" for human.

Designating Gene symbols• The initial character of the symbol should always be a letter.
Subsequent characters may be other letters, or if necessary, Arabic numerals.
• No superscripts or subscripts or Roman numerals may be used. • All Greek letters should be changed to letters in the Latin alphabet
and placed at the end of the gene symbol, eg. GLA "galactosidase, alpha“
• No punctuation may be used, with the exception of the HLA, immunoglobulin and T cell receptor gene symbols (which may be hyphenated).
• Gene symbols will not usually be assigned to alternative transcripts.• Tissue specificity or molecular weight should be avoided.• Some letters or combination of letters are used as prefixes or
suffixes in a symbol to give a specific meaning and their use for other meanings should be avoided (Section 10).
• Oncogenes are given symbols corresponding to the homologous retroviral oncogene, but without the "v-" or "c-" prefices.

Gene Symbols can be assigned to: • Genes - a DNA segment that contributes to phenotype/function
- includes pseudogenes, genes coded by the opposite (antisense) strand that overlap a known gene • Locus - a point in the genome, identified by a marker, which can be mapped by some means. It does not necessarily correspond to a gene• Chromosome Region - a genomic region which has been associated with a particular syndrome or phenotype, particularly when there is a possibility that several genes within it may be involved in the phenotype. • Transcribed but untranslated functional DNA segments e.g. XIST "X (inactive)-specific transcript". • EST clusters which suggest a putative gene e.g. C1orf1 "chromosome 1 open reading frame 1". • Predicted genes (in silico) which show a high degree of sequence homology to well characterized genes will be assigned the same symbol with an "L" for like• Gene symbols will not usually be assigned to alternative transcripts or to genes predicted solely from in silico data (with no other supporting evidence e.g. significant homology to a characterized gene).

Homologies With Other Species
• ORTHOLOGS: to determine orthologs, use NCBI Entrez Gene/RefSeq; PHiGs– recognizable orthologs should be named the same as
human• HOMOLOGS: (across species) can be named “-
like”, “-homolog” or “-related”– gene symbols for homologs can be designated with
an “L”• PARALOGS: (within the same species) can be
named “-related sequence:

Genes Identified from Sequence Information
•Antisense - A gene of unknown function, encoded at the same genomic locus (with overlapping exons) as another gene should have its own symbol.•Opposite Strand - Genes of unknown function on the opposite strand should be assigned the suffix OS for "opposite strand".•Untranslated Functional RNAs - These may be assigned symbols, however the approved name should contain "untranslated RNA" •Related (-like) sequences - The designation of the suffix "L" is used where no other functional information is available and there is some sequence similarity with a known gene •Genes of unknown function - Genes predicted with EST evidence, but showing no structural or functional homology, are regarded as putative. These are designated by the chromosome of origin, the letters "orf" for open reading frame and a number in a series e.g. C2orf1, "chromosome 2 open reading frame 1".•Pseudogenes - pseudogenes will usually be assigned the next number in the relevant symbol series, suffixed by a "P" for pseudogene (or "PS" in the specific cases) if requested, however, the designation "pseudogene" will remain in the gene name.


Enzymes and Proteins • The rules described in the sections on gene names and
symbols apply, but in addition the names of genes coding for enzymes should be based on those recommended by the Nomenclature Committee of the International Union of Biochemistry and Molecular Biology
• These can be found at http://ca.expasy.org/enzyme/• Names of genes encoding plasma proteins,
hemoglobins, and specialized proteins are based on standard names and those recommended by their respective committees e.g. HBA1 "hemoglobin, alpha 1".

8. GO term rules and relationships
For the most up to date information, refer to the GO Annotation Guide:
http://www.geneontology.org/GO.annotation.shtml

The True Path Rule
"the pathway from a child term all the way up to its top-level parent(s) must
always be true"

GO Term Relationships• A GO Term can have multiple parents
– Need to keep in mind relationships between ‘child’and ‘parent’ terms
– A child term can have one of two different relationships to its parent(s): is_a or part_of.
– The same term can have different relationships to different parents:
is_a
is_a
is_a
part_of


The is_a relationship • an is_a relationship means that the term is a
subclass of its parent • eg. mitotic cell cycle is_a cell cycle• The is_a relationship is transitive, which
means that if 'GO term A' is a subclass of 'GO term B', and 'GO term B' is an subclass of 'GO term C', 'GO term A' is also a subclass of 'GO term C:
is_a
is_a
is_a

The part_of relationship There are four basic levels of restriction for a part_of relationship:

The True Path Rule
the part_of relationship is restricted to those types where a child term must always be part_of its parent

The part_of relationship
2. 'necessarily is_part', means that wherever the child exists, it is as part of the parent, eg. replication fork is part_of chromosome, so whenever replication fork occurs, it is as part_of chromosome, but chromosome does not necessarily have part replication fork.

The nucleus always has_partchromosome, but chromosome isn't necessarily part_of nucleus

The part_of relationship
4. The final type is a combination of both two and three, 'has_part' and 'is_part‘,eg. nuclear membrane is part_of nucleus. So nucleus always has_part nuclear membrane, and nuclear membrane is always part_of nucleus.

• The part_of relationship used in GO is usually type two, 'necessarily is_part'.
• Like ‘is_a’, ‘part_of’ is transitive, so that if 'GO term A' is part_of 'GO term B', and 'GO term B' is part_of 'GO term C', 'GO term A' is part_of'GO term C':
part_of
part_of
part_of
For example: laminin-1 is part_of basal lamina. basal lamina is part_of basement membrane. laminin-1 is part_of basement membrane.

is_a & part_of Designations
Designations shown by GO Browsers
is_a %part_of < Plain Text Designations



Species-specific terms • GO nodes should aggressively avoid using species-
specific definitions. • Nevertheless, many functions, processes and
components are not common to all life forms. • The current convention is to include any term that can
apply to more than one taxonomic class of organism. • The are cases where a word or phrase has different
meanings when applied to different organisms, eg. embryonic development
• Such terms are distinguished from one another by their definitions and by the sensu designation (sensu means 'in the sense of'), as in the term embryonic development (sensu Insecta)

To find taxonomic groups used with sensu:http://www.geneontology.org/GO.usage.shtml#sensu

Fiona’s Homework

1. Maize NomenclatureWith thanks to Bela:http://www.maizegdb.org/maize_nomenclature.php

“Unknown” & Unannotated“Unknown vs Unannotated: "Unknown" means that someone has tried annotating the gene, but didn't find any information. Absence of annotation implies that no one has looked.
AgBase: ‘unknown’ will be used when a curator has looked at all the current literature for the product AND tried to assign function using sequence similarity (ISS).

Evidence Codes & Gene Association Files
For the most up to date information, refer to the GO Annotation Guide:
http://www.geneontology.org/GO.annotation.shtml
Fiona McCarthy July 7 2005

1. Evidence Codes
For the most up to date information, refer to the GO Annotation Guide:
http://www.geneontology.org/GO.annotation.shtml

Annotating to GO
Information required:1. database object (eg. a protein or gene
identifier)2. reference ID (eg. PubMed ID)3. GO Term ID (eg. GO:0004674)4. evidence code (eg. IDA)

Evidence CodesIMP inferred from mutant phenotype IGI inferred from genetic interaction
[with <database:gene_symbol[allele_symbol]>] IPI inferred from physical interaction
[with <database:protein_name>] ISS inferred from sequence similarity
[with <database:sequence_id>] IDA inferred from direct assay IEP inferred from expression pattern IEA inferred from electronic annotation
[to <database:id>] TAS traceable author statement NAS non-traceable author statement ND no biological data available RCA inferred from reviewed computational analysis IC inferred by curator

IMP: inferred from mutant phenotype • Any gene mutation/knockout • Overexpression/ectopic expression of wild-type or mutant genes • Anti-sense experiments • RNAi experiments • Specific protein inhibitors • Comment: anything that is concluded from looking at mutations or abnormal
levels of the product(s) only of the gene of interest is IMP (compare IGIs). Use this code for experiments that use antibodies or other specific inhibitors of RNA or protein activity, even though no gene may be mutated (the rationale is that IMP is used where an abnormal situation prevails in a cell or organism).
• In addition to such 'abnormal' situations, IMP may also be used for experiments (e.g. QTL analyses) in which an inference is based on the observed effects of naturally occurring variations in a gene.
• IMP also covers phenotypic similarity: a phenotype that is informative because it is similar to that of another independent phenotype (which may have been described earlier or documented more fully) is IMP (not IGI).

IGI: inferred from genetic interaction • "Traditional" genetic interactions such as suppressors, synthetic lethals, etc. • Functional complementation, eg. when a gene from one organism complements a deletion or other mutation in another species. (See notes for WITH field.)• Rescue experiments • Inference about one gene drawn from the phenotype of a mutation in a different gene • Includes any combination of alterations in the sequence (mutation) or expression of more than one gene/gene product. (Covers any of the IMP experiments that are done in a non-wild-type background)• When redundant copies of a gene must all be mutated to see an informative phenotype, that's IGI. • Use IMP for "phenotypic similarity," as described below. • We have also decided to use this category for situations where a mutation in one gene (gene A) provides information about the function, process, or component of another gene (gene B; i.e. annotate gene B using IGI).

IPI: inferred from physical interaction • 2-hybrid interactions • Co-purification • Co-immunoprecipitation• Ion/protein binding experiments • Covers physical interactions between the gene product of
interest and another molecule (or ion, or complex).
For functions such as protein binding or nucleic acid binding, abinding assay is simultaneously IPI and IDA; IDA is preferred because the assay directly detects the binding. For both IPI andIGI, it would be good practice to qualify them with the gene/protein/ion. We thought that antibody binding experiments were not suitable as evidence for function or process.

ISS: inferred from sequence or structural similarity
This code can be used for any analysis based on sequence alignment, structure comparison, or evaluation of sequence features such as composition, including:• Sequence similarity (homologue of/most closely related to) • Recognized domains • Structural similarity • Protein features, predicted or observed (e.g. hydrophobicity, sequence composition) • Southern blotting • Use this code for BLAST (or other sequence similarity detectionmethod) results that have been reviewed for accuracy by a curator. If the result has not been reviewed, use IEA. • ISS can also be used for sequence similarities reported in published papers, if the curator thinks the result is reliable enough.

Usage of the ISS code within GOAhttp://www.ebi.ac.uk/GOA/goaHelp.html#6
There are three ways in which a curator can use the ISS evidence code:1. If a curator reads a paper that provides functional information and states an orthologybetween two proteins, a curator can transfer the annotation to the corresponding orthologs by changing the evidence code to 'ISS' in the target protein's entry. The original literature identifier is still shown. Any information that was previously in the 'with' column of the original entry is changed in the target entry to contain the original entry's accession number. This allows the source of the annotation to be traced.
2. If a curator is confident that a protein shows high similarity to another (e.g. from using BLAST or UniRef90), and it seemed reasonable to infer that the two proteins have a common function, then annotation can be transferred from one protein to another using the 'ISS' code. The evidence code in the target annotation will change to 'ISS'. The target entry's accession number will replace the journal identifier and any information that was previously contained in the 'with' column of the original entry is changed in the target entry to contain the original entry's accession number. This allows the source of the annotation to be traced.
3. If sequence similarity and evidence for human annotation was reported in two different papers, the annotation can be transferred using the 'ISS' evidence code. In the target entry, the evidence code changes to 'ISS' and the curator will add a new journal identifier. Any information that was previously contained in the 'with' column of the original entry is changed in the target entry to the original entry's accession number. This allows the source of the annotation to be traced.

IDA: Inferred from Direct Assay• Enzyme assays • In vitro reconstitution (e.g. transcription) • Immunofluorescence (for cellular component) • Cell fractionation (for cellular component) • Physical interaction/binding assay (sometimes appropriate for cellular
component or molecular function) • Important: this code is used to indicate a direct assay for the function,
process, or component indicated by the GO term. Curators therefore need to be careful, because an experiment considered as direct assay for a term from one ontology may be a different kind of evidence for the other ontologies. In particular, we thought of more kinds of direct assays for cellular component than for function or process. For example, a fractionation experiment might provide "direct assay" evidence that a gene product is in the nucleus, but "protein interaction" evidence for its function or process. Binding assays can provide direct assay evidence for ... binding molecular function terms.

IEP: inferred from expression pattern • Transcript levels (e.g. Northerns, microarray data) • If a GO term is inferred from the results of a microarray
experiment, this code will usually be used. There are cases, however, where RCA may be appropriate, such as studies that combine results of microarray and other types of experiments. Protein levels (e.g. Western blots)
• Covers cases where the annotation is inferred from the timing or location of expression of a gene. Expression data will be most useful for process annotation rather than function. For example, several of the heat shock proteins are thought to be involved in the process of stress response because they are upregulated during stress conditions. Use this category with caution!
• Note: The "database identifier" column in the gene_associationfile should be filled in whenever possible, to help avoid circular annotations between GO and other databases.

Notes on IEP •Notes: Addition of the IEP category generated a lot of discussion via email. One theme that emerged is that curators and users will have to be careful when interpreting expression results, especially if there's no other kind of evidence linking a gene product with a process. For instance, we certainly don't want to look at a cluster of genes, and, based on previous knowledge of one of them being involved in protein folding, annotate the rest of the genes in that cluster to the same process. This is certainly a dangerous thing to do. But having the IEP code allows curators to include expression data when they deem it appropriate, and allows researchers to make their own decisions/judgments about the reliability of the annotation. (Midori Harris, 2000-03-08; updated 2000-03-09) •Another important theme, indeed one of the reasons we opted to add the category, is that systematic analysis will prove to be very informative. It was especially well stated by Richard Baldarelli of MGI, so I've included his message here: It seems that expression data will be very useful for process and cellular component mapping, but caution should be used for function mapping (as Allan and Kara point out [in email messages]). While conventional expression assays will provide useful evidence in several cases, the real benefit will come from expression profiling. The rationale behind expression profiling from chip data is that genes that are coordinately regulated over a range of environments are likely to be involved in the same biological processes, and thus may have interrelated functions. As expression technology evolves to consider other aspects of gene expression (e.g. transcription and post-transcription chips, Mass-spec on 2D protein data), profiling will become an even more valuable tool for process implication. With the genome sequences here or on the way, the most significant information we may have for many genes will be expression profiling data (at least for a while anyway). Accuracy levels for process implication aside, this type of evidence is necessarily indirect. Having an evidence type "expression" takes this into account and remains fairly non-specific.

IEA: inferred from electronic annotation • Annotations based on "hits" in sequence similarity searches, if they have not
been reviewed by curators (curator-reviewed hits would get ISS) • Annotations transferred from database records, if not reviewed by curators
(curator-reviewed items may use NAS, or the reviewing process may lead toprint references for the annotation)
• Comment: Used for annotations that depend directly on computation or automated transfer of annotations from a database. The key feature that distinguishes this evidence code from others is what a curator has done IEA is used when no curator has checked the annotation to verify its accuracy. The actual method used (BLAST search, SwissProt keyword mapping, etc.) doesn't matter. An entry may be made in the 'with' column if relevant (e.g. for sequence comparisons).

TAS: traceable author statement• Anything in a review article where the original experiments are traceable
through that article (material from introductions to non-review papers will sometimes meet this standard; discussion sections should usually be regarded with greater skepticism)
• Anything found in a text book or dictionary; usually text book material has become common knowledge (e.g. "everybody" knows that enolase is a glycolytic enzyme).
• TAS and NAS are both used for cases where the publication that acurator uses to support an annotation doesn't show the evidence (experimental results, sequence comparison, etc.). With a few exceptions, TAS should be used only if references to the original work are available. TAS is meant for the more reliable cases, such as reviews (presumably written by experts) or material sufficiently well established to appear in a text book, but there isn't really a sharp cutoff between TAS and NAS. Curator discretion is advised!
• Comment: Formerly ASS ("author said so").

NAS: non-traceable author statement
• Database entries that don't cite a paper (e.g. UniProt Knowledgebase records,)
• Statements in papers (abstract, introduction, or discussion) that a curator cannot trace to another publication
• Comment: Formerly NA (not available).

ND: no biological data available • Used for annotations to "unknown" molecular function, biological process, or cellular
component. • Curators at the contributing database found no information supporting an annotation
to any term from the ontology in question (molecular function, biological process, or cellular component) as of the date indicated.
• This reference documents the fact that a database curator has reviewed the literature describing a gene product but found no biologically useful information*; it should only be cited for annotations to "unknown," i.e. molecular function unknown ; GO:0005554, biological process unknown ; GO:0000004 or cellular component unknown ; GO:0008372.
• This code is used only for annotations to "unknown," and it is the only evidence code recommended for annotations to unknown (except in cases where a cited source explicitly says that something is unknown). It should be accompanied by a reference that explains that curators looked but found no information. The GO Reference collection includes a generic reference that can be used with ND; to use it insert "GO_REF:0000015" in the reference column of a gene_association file.
• Note that ND can be used with any one (or two) of the 'unknown' terms, even if there is data available to support annotation to a term from one or both of the other ontologies (e.g. ND can be used with cellular component unknown ; GO:0008372if the function and process are known but component is not).
* under review: GOA to decide whether or not this code can be used if there is ISS data.

RCA: inferred from reviewed computational analysis
• Predictions based on large-scale experiments (e.g. genome-wide two-hybrid, genome-wide synthetic interactions)
• Predictions based on integration of large-scale datasets of several types • Text-based computation (e.g. text mining) • This code is used for annotations based on a non-sequence-based
computational method, where the results have been reviewed by an author or a curator.
• IEA should be used for any computational annotations that are not checked for accuracy by a curator (or by the authors of a paper describing such analysis), and sequence comparisons that have been reviewed use ISS. For microarray results alone, IEP is preferred, but RCA is used when microarray results are combined with results of other types of large-scale experiments.
• Cellular component using DDF-MudPIT?

IC: Inferred by Curator • To be used for those cases where an annotation is not supported by any evidence, but can be reasonably inferred by a curator from other GO annotations, for which evidence is available. • An example would be when there is evidence (be it direct assay,sequence similarity or even from electronic annotation) that a particular gene product has the function transcription factor activity. There is no evidence whatsoever that this gene product has the cellular location nucleus, but this would be a perfectly reasonable inference for a curator to make (if the curator is annotating a eukaryotic gene product, of course). • Note that the With/From field should always be filled in with a GO id when using this evidence code. • Example:gene_product: jubireference: Ashburner et al. 2006 J. irreprod. data 107:11989-11990 molecular_function: general RNA polymerase II transcription factor ; GO:0016251 | inferred from sequence similarity cellular_location: nucleus ; GO:0005634 | inferred by curator from GO:0016251

Gene Association Files

Two Types of Annotation:
1. electronic annotation – IEA- see July 19 lecture
1. manual annotation – sequence data (ISS)- papers (all other codes)

Making Gene Associations
• GO terms should be associated with database objects representing gene products rather than genes – a single gene may encode very different products with
very different attributes – if the database object is a gene, it is associated with all
GO terms applicable to any of its products • GO annotations should be attributed to a source • Each annotation should indicate the evidence on
which it is based

The Gene Association File
• the gene association file is the mechanism by which gene products functions are shared/released
• collaborating databases export to GO a tab delimited file, the "gene association file" of links between database objects and GO terms
• the database object may represent a gene or a gene product (transcript or protein)

The Gene Association FileTo make gene associations we have to fill in up to 15 fields; 11 are compulsory

Attributing a Source
• Every annotation must be attributed to a source, which may be a literature reference, another database or a computational analysis.
• The annotation must indicate what kind of evidence is found in the cited source to support the association between the gene product and the GO term.

General Recommendations (1)• A gene product can be annotated to zero or more
nodes of each ontology. • Annotation of a gene product to one ontology is
independent of its annotation to other ontologies. • Annotate gene products in each species database to
the most detailed level in the ontology that correctly describes the biology of the gene product.
• Keep to the True Path Rule: annotating to a term implies annotation to all parents via any path, so check the parentage of a term before annotating.

General Recommendations (2) • “Unknown vs Unannotated: "Unknown" means that someone
has tried annotating the gene, but didn't find any information. Absence of annotation implies that no one has looked.
• Annotate to terms from all three ontologies, using "unknown" if necessary, citing a reference within their database that explains that they found no relevant biological information in the literature (or any other sources they may have considered).
• Uncertain knowledge of where a gene product operates should be denoted by annotating it to two nodes, one of which can be a parent of the other, eg. a gene product known to be in the nucleolus, but also experimentally observed in the nucleus generally, can be annotated to both nucleolus and nucleus in the cell component ontology. The two annotations may have the same or different supporting evidence.
• Annotate to multiple nodes that conflict with each other if there are conflicting claims in the literature.

General Recommendations (3)• If the database object is a gene, it is associated with all GO
terms applicable to any of its products. • “Normal" depends on the point of view taken by the annotator,
eg. many viruses use host proteins to carry out viral processes. The host protein is then doing something abnormal from the perspective of the host, but completely normal from the perspective of the virus.
• In this case, use two taxon IDs in the "Taxon" column of the gene association file, the first being the organism that encodesthe gene product, and the second that of the organism that uses the gene product, and whose perspective is considered "normal" for that annotation.

The Gene Association File

1. DB
• refers to the database contributing the gene_association file• this field is mandatory• currently, we use ‘UniProt’• ‘UniParc’ and ‘Genbank’ are also acceptable

2. DB_Object_ID
• refers to a unique identifier in the database for the gene product being annotated • may or may not correspond exactly to what is described in a paper, eg. a paper describing a protein may support annotations to the gene encoding the protein (gene ID in DB_Object_ID field) or annotations to a protein object (protein ID in DB_Object_ID field)• this field is mandatory• we use the UniProt accession

3. DB_Object_Symbol
• refers to a unique (and valid) symbol to which the DB_Object_ID is matched• this should be a gene symbol, wherever possible• it is not an ID or an accession number (the DB_Object_IDprovides the unique identifier), although IDs can be used if there is no more biologically meaningful symbol available (e.g.,when an unnamed gene is annotated)• can use ORF name for otherwise unnamed gene or protein this field is mandatory

4. Qualifier
• flags that modify the interpretation of an annotation• one (or more) of NOT, contributes_to, colocalizes_with (pipes delimited)• this field is not mandatory
Allowable values:1. ‘NOT’
Use NOT when a gene product is not associated with the GO term to document conflicting claims in the literature.Not is used when there is some reason to expect an association, but experimental evidence proves the expection wrong.2. ‘Contributes to’ (used with GO Function Ontology)distinguishes between individual subunits functions and whole complex functions3. ‘Colocalizes with’ (used with GO Component Ontology)Transiently or peripherally associated with an organelle or complex where the resolution of an assay is not accurate.

5. GO ID
• refers to the GO ID number for the term attributed to the DB_Object_ID• this field is mandatory
Note that the GO term name is NOT used in the gene association file.

6. DB:Reference
• refers to the unique identifier that gives authority for the attribution of the GOid to the DB_Object_ID• for manual curation, this will be a PubMed ID number• may also be a database record, eg. electronic annotations & annotations to unknown function (ND) will require that AgBase have a protocol code• only one reference can be cited on a single line • when database references also refer to PMID, entries are pipes delimited (separated by a “|”)• this field is mandatory

7. Evidence
• either IMP, IGI, IPI, ISS, IDA, IEP, IEA, TAS, NAS, ND, IC or RCA• after filling in the evidence code, check to see if you need tofill in ‘With (or) From’ field• this field is mandatory • evidence codes are changing & updated: check definitions regularly

8. With (or) From
• This field is used to hold an additional identifier for annotations using certain evidence codes (IC, IEA, IGI, IPI, ISS). • IC: if you are inferring an association based on another, populate the From field with the associated GOID number. Mandatory for the IC.
•Example: if a particular gene product has the function transcription factor activity. There is no evidence whatsoever that this gene product has the cellular location nucleus, but this would be a perfectly reasonable inference for a curator to make (if the curator is annotating a eukaryotic gene product, of course). Both associations would have the same reference. •molecular_function: general RNA polymerase II transcription factor ; GO:0016251 | inferred from sequence similarity •cellular_location: nucleus ; GO:0005634 | inferred by curator from GO:0016251
• IEA and ISS: use “with” to populate the field with a gene identifier (DB:gene_id) unless ISS is used to denote predicted sequence features (such as hydrophobicity, alpha-helices, etc.
• IGI and IPI: use ‘with’ to include an identifier for the "other" gene involved in the interaction. The entry in the "with" column does not have to refer to the same species that is being annotated.

IGI, IPI and ‘WITH’For IGI & IPI codes, we recommend making an entry in the "with"
column (i.e. include an identifier for the "other" gene involved in the interaction). If more than one independent genetic interaction supports the association, use separate lines for each. In cases where the gene of interest interacts simultaneously with more than one other gene, put both/all of the interacting genes on the same line (separate identifiers by pipes in the "with" column).
To help clarify: GOterm IGI FB:gene1|FB:gene2 means that the GO term is supported by evidence from its interaction
with both of these genes; i.e. neither of these statements are true: GOterm IGI FB:gene1
GOterm IGI FB:gene2

9. Aspect
• either P (biological process), F (molecular function) or C (cellular component) • this field is mandatory

10. DB_Object_Name
• name of gene or gene product• white space allowed• this field is not mandatory

11. DB_Object_Synonym
• any alternative names (pipes delimited)• aids searching so be thorough• generally, names that differ only by case are not recorded, butnames that differ by punctuation are, eg, CAP22|CAP-22• white space allowed• this field is not mandatory

12. DB_Object_Type
• refers to the database entry, ie. does it match a gene, transcript, protein, protein_structure or complex • we will enter ‘protein’• MUST match the database entry identified by DB_Object_ID• this column does not reflect anything about the GO term or the evidence on which the annotation is based. eg. if your database entry represents a gene, then 'gene' goes in the DB_Object_ID column, even if the annotation is relevant to the localization of a protein product of the gene• several alternative transcripts from one gene may be annotated separately, each with a unique transcript DB_Object_ID, but list the same gene symbol in the DB_Object_Symbol column. • this field is mandatory

13. taxon
• refers to the taxon identifier(s)• usually use the ID of the species encoding the gene product• can also have 2 IDs: the first ID is that of the species encoding the gene product; the second ID is that of the species using the gene product (pipes delimited)
Chicken: 9031Corn: 4577Cow: 9913 (Bos taurus)Channel catfish: 7998 (Ictalurus punctatus)

14. Date
• date on which the association was made• YYYYMMDD format

15. Assigned_by
• refers to the database that made the association (AgBase; ‘AB’)• used for tracking the source of the annotation

The Gene Association File

HOMEWORK!
1. Curate these papers:
PMID: 15356338 7490283 1512296 12097608 11119244
2. Annotate this gene product:UniProt: Q9IAK1Try to get comprehensive GO annotation

Gene Ontology Annotation Training: Electronic Annotation
and GO Tools
Fiona McCarthy November 2005

Overview• Electronic Annotation Strategies
– Interpro2GO– spkw2GO– ec2GO
• Blast Strategies– SeqHound– UniRef
• GO Slims• GO Tools
- GO slims- GO tools available from the GO Homepage - Other sources for GO tools - GO Tools available from AgBase

IEA: Electronic Annotation
November 14, 2005

Mappings of External Classification Systems to GO
http://www.geneontology.org/GO.indices.shtml

IEA Mappings at AgBase• IEA mappings provide “higher order” or more
general GO terms• allow large scale assignment of function• ALL IEA mappings need to be updated monthly• currently can take the IEA mappings from
UniProt/GOA• Process needs to be updateable (relies on
protocol references)• Need to be able to look for obsolete terms and
to change mappings as GO terms & their mappings change.

ISS: Blast Strategies
November 14, 2005

SeqHound

SeqHound
• uses gi numbers• processes multiple entries• can process whole taxon entries• uses ‘ontoglyphs’ – may lose granularity• shows sequence neighbors (but not their
alignments)

SeqHound

UniRef Database

UniRef Database

GO Tools

1. GO Slims

What is a GOSlim?• GO slims are cut-down versions of the GO ontologies
containing a subset of the terms in the whole GO. They give a broad overview of the ontology content without the detail of thespecific fine grained terms.
• GO slims are particularly useful for giving a summary of the results of GO annotation.
• GO slims are created by users according to their needs, and may be specific to species or to particular areas of the ontologies.
• GO provides a generic GO slim which should be suitable for most purposes. Alternatively, users can create their own GO slims; TAIR (plant), SGD (yeast) and GOA (generic) have submitted GO slims which have been integrated into the GO flat file.
http://www.geneontology.org/GO.slims.shtml

map2slim.pl
•The map2slim.pl script, distributed as part of the go-perl package, maps a set of annotations up to their parent GO slim terms. •Further documentation, installation help and instructions on running the script are available from: http://www.godatabase.org/dev/pod/scripts/map2slim.htmlhttp://search.cpan.org/~cmungall/go-perl-0.01/

Figure 3. GO Slim Viewer and its Output
A. The GO Slim Viewer Page.
B. GO Slim Viewer Output and a Chart plotted in Excel using this output.

Membrane Proteins: Biological Process
B-cells Stroma
ion/proton transportcell migration
cell adhesioncell growthapoptosisimmune response
cell cycle/cell proliferation cell-cell signalingfunction unknowndevelopmentendocytosisproteolysis and peptidolysis
protein modificationsignal transduction

Nuclear Proteins: Biological Process
B-cells Stroma
chromosome organization and biogenesisDNA metabolismchromatin assembly or disassembly
cell differentiationcell growthchromosome segregationchromatin modification
cell cycle/cell proliferation function unknownnuclear organization and biogenesisprotein catabolismprotein modificationRNA processingnuclear transport
regulation of transcription, DNA-dependentsignal transduction

2. GO tools available from the GO Homepage



3. Other sources for GO tools


Using GO Tools
• new GO tools may not be listed on the GO Consortium webpage
• check PubMED literature for new GO Tools
• always check when the latest DAG version and GO annotations were loaded into a GO tool

4. GO Tools available from AgBase


Online Demonstration
Nan Wang Computer Science & Engineering, MSU