Embed Size (px)
Citation preview

Journal of Neuroimmunology 276 (2014) 71–79
Contents lists available at ScienceDirect
Journal of Neuroimmunology
j ourna l homepage: www.e lsev ie r .com/ locate / jneuro im
Gold drug auranofin could reduce neuroinflammation by inhibitingmicroglia cytotoxic secretions and primed respiratory burst
Jocelyn M. Madeira a, Ekta Bajwa a, Maegan J. Stuart a, Sadayuki Hashioka b,c, Andis Klegeris a,⁎a Department of Biology, University of British Columbia Okanagan Campus, Kelowna, BC, Canadab Kinsmen Laboratory of Neurological Research, Department of Psychiatry, University of British Columbia, Vancouver, BC, Canadac Department of Psychiatry, Faculty of Medicine, Shimane University, Izumo, Shimane, Japan
⁎ Corresponding author at: Department of Biology, UOkanagan Campus, 3187 University Way, Kelowna, BC V807 9557; fax: +1 250 807 8830.
E-mail address: [email protected] (A. Klegeris).
http://dx.doi.org/10.1016/j.jneuroim.2014.08.6150165-5728/© 2014 Elsevier B.V. All rights reserved.
a b s t r a c t
a r t i c l e i n f oArticle history:Received 30 September 2013Received in revised form 6 August 2014Accepted 11 August 2014
Keywords:Alzheimer's diseaseParkinson's diseaseNeuroprotectionAurothiomalateAurothiosulfateRespiratory burst priming
Neuroinflammation contributes to the pathogenesis of neurological disorders. Anti-inflammatory treatmentscould potentially be used to slow down the progression of these diseases. We studied the anti-neuroinflammatory activity of gold compounds which have been used to treat rheumatoid arthritis. Non-toxicconcentrations of auranofin (0.1–1 μM) significantly reduced the cytotoxic secretions by primary humanmicrog-lia andmicroglia-like THP-1 promonocytic cells. Auranofin inhibited primed NADPH-oxidase dependent respira-tory burst and secretion of tumor necrosis factor (TNF)-α and nitric oxide by monocytic cells. It had a directneuroprotective effect on SH-SY5Y neuronal cells. Auranofin could have a novel application in the treatment ofneurodegenerative diseases.
© 2014 Elsevier B.V. All rights reserved.
1. Introduction
In recent years, accumulating data have indicated that inflammationin the central nervous system (CNS) contributes to several neurologicalimpairments including Alzheimer's and Parkinson's diseases (Zhanget al., 2005; Frohman et al., 2006; Lee et al., 2010). Currently, there areno effective clinical treatments to prevent or stop this inflammation;therefore, research into novel therapeutic approaches is warranted.Inflammation in the CNS is driven by twomain glial cell types:microgliaand astrocytes (Li et al., 2011; Zilka et al., 2012). Both these cell typescould be targeted by therapies aimed at reducing neuroinflammation(Block et al., 2007). In their activated pro-inflammatory state, microgliaand astrocytes increase the secretion of toxins and inflammatorymediators. This could lead to reduced viability of the healthy neuronssurrounding activated glia (Cameron and Landreth, 2010). Pharmaco-logical means could potentially be used to reduce the neuronal losscaused by neuroinflammation through either suppressing the releaseof neurotoxins from glia or inducing their secretion of neurotrophicfactors. Substances that act on neurons directly protecting them fromglial neurotoxins could also be beneficial in AD and other neuropathol-ogies that are partially driven by neuroinflammation.
niversity of British Columbia1V 1V7, Canada. Tel.: +1 250
Rheumatoid arthritis is a chronic autoimmune disorder character-ized by inflammation in the joints (Kean, 1990). Gold compoundshave been used in the treatment of rheumatoid arthritis. In NorthAmerica, clinically available gold-containing therapeutics include intra-muscular injections of aurothiomalate (ATM) and the oral gold com-pound 2,3,4,6-tetra-o-acetyl-L-thio-β-D-glucopyrano-sato-S-(triethyl-phosphine) gold manufactured as auranofin (AF) (Champion et al.,1990; Kean, 1990; Kean et al., 1997). The exact mechanisms of AF'santi-inflammatory activity have not been established; though a rangeof different effects of AF on peripheral immune cells have beendocumented (Yamashita et al., 1997; Stern et al., 2005; Kim et al.,2007, 2010; Nakaya et al., 2011). AF affects cytokine levels by increasingsecretion of interleukin (IL)-8 and reducing IL-6 secretion from lipo-polysaccharide (LPS) stimulated human monocytic cells (Stern et al.,2005; Kim et al., 2007). AF also induces the anti-inflammatory enzymeheme oxygenase (HOX)-1 in monocytic THP-1 cells (Kim et al., 2010).The variousmechanisms of AF action have been summarized in a recentreview (Madeira et al., 2012a). The peripheral anti-inflammatory activ-ity of gold compounds has been characterized extensively, whereas theeffects of these compounds on neuroimmune reactions are less studied,even though AF has been shown to reach the CNS after its oral adminis-tration (Walz et al., 1983; Madeira et al., 2012a, 2013).
In this study we used three different monocytic cell lines to modelmicroglia as well as cultured primary human cells. Microglia representthe mononuclear phagocyte system in the CNS and therefore play acentral role in neuroimmune reactions (Ransohoff and Brown, 2012).

72 J.M. Madeira et al. / Journal of Neuroimmunology 276 (2014) 71–79
We investigated two of the clinically available drugs: ATM and AF, alongwith aurothiosulfate (ATS), which is a structurally similar monovalentgold thiol compound shown to be ineffective as an anti-inflammatorydrug (Bruze et al., 1995). Compounds were tested for their ability toreduce the secretion of pro-inflammatory cytokines and cytotoxins pro-duced by activated humanmonocytic cells. In addition, we investigatedthe effects of gold compounds on phagocyte respiratory burst and in-vestigated the ability of the gold compounds to protect neuronal cellsfrom toxicity induced by supernatants from stimulated microglia-likecells. Of the three gold compounds studied, only AF exhibited potential-ly anti-neurotoxic and neuroprotective activity.
2. Materials and methods
2.1. Reagents
The following substances were obtained from Sigma-Aldrich(Oakville, ON, Canada): ATM, LPS (from Escherichia coli 055-B5), diapho-rase (EC 1.8.1.3, from Clostridium kluyveri, 5.8 U/mg solid), dimethylsulfoxide (DMSO), Triton X-100, luminol sodium salt, N-formyl-met-leu-phe (fMLP), p-iodonitrotetrazolium violet, NAD+, 3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT), sulfanilamideand N-1-naphthylethylenediamine. AF was supplied by CedarlaneCanada (Burlington, ON, Canada). ATS was from VWR International(Mississauga, ON, Canada). Human and murine recombinant interferon(IFN)-γ, monocyte chemoattractant protein (MCP)-1 and tumor necro-sis factor (TNF)-α enzyme linked immunosorbent assay (ELISA) kitswere purchased fromPeprotech (RockyHill, NJ, USA). All other reagentswere obtained from Thermo Fisher Scientific (Nepean, ON, Canada) un-less stated otherwise.
2.2. Cell culture
The human monocytic THP-1 and promyelocytic HL-60 cell lineswere obtained from the American Type Culture Collection (ATCC,Manassas, VA, USA). The human neuroblastoma SH-SY5Y cell line wasa gift from Dr. R. Ross, Fordham University, NY. Murine BV-2 microglialcell line was a gift from Dr. G. Garden, University of Washington, WA.Human primarymicroglia were obtained from epileptic patients under-going temporal lobe surgery. The specimens were from normal tissueoverlying the epileptic foci. The use of human brain materials wasapproved by the Clinical Screening Committee for Human Subjects ofthe University of British Columbia. Microglia were isolated followingprotocols described by Hashioka et al. (2009). All cells were grown inDulbecco's modified Eagle's medium-nutrient mixture F12 ham(DMEM-F12) supplemented with 10% fetal bovine serum (FBS) andantibiotics (100 U/ml penicillin, 100 μg/ml streptomycin). The THP-1and SH-SY5Y cell lines were used without differentiation; HL-60 cellswere differentiated prior to use in experiments as described below.
2.3. Effects of compounds on cell viability and cytokine secretion
Human monocytic THP-1 cells were seeded into 24-well plates at aconcentration of 5 × 105 cells/ml in 0.9 ml of DMEM-F12 containing5% FBS. Human microglia were plated at 7.5 × 104 cells per well inDMEM-F12 containing 5% FBS. Following 48 h incubation, media wereremoved and replaced with fresh medium. Cells were incubated in thepresence of gold compounds or their vehicle solution (DMSO) for15 min prior to the addition of the activating stimulus (0.5 μg/ml LPSplus 150 U/ml human recombinant IFN-γ). The final concentration ofDMSO in cell culture medium did not exceed 0.13%; at this concentra-tion, DMSO did not have any detectable effects on cell viability or func-tion. After 24–48 h incubation, 0.1 ml of cell culturemedia was sampledfor lactate dehydrogenase (LDH) enzymatic activity to determine thepercentage of dead cells, while the evaluation of the surviving cellswas performed by the MTT assay. The concentration of MCP-1 and
TNF-α (ng/ml) in THP-1 cell supernatants was measured in 0.1 ml ofcell-free supernatants by ELISA according to the protocol provided bythe supplier of antibodies (Peprotech).
2.4. Cytotoxicity of THP-1 cell and human microglia supernatants towardsSH-SY5Y neuronal cells
The experiments were performed as previously described (Klegeriset al., 2003). Briefly, THP-1 cells and primary human microglia wereseeded into 24-well plates and stimulated in the presence or absenceof various compounds as described above. After 24 h or 48 h incubation,0.4 ml of cell-free supernatant was transferred to each well containingSH-SY5Y cells that had been plated 24 h earlier at a concentration of2 × 105 cells/ml in 0.4 ml of DMEM-F12 medium containing 5% FBS.After 72 h incubation, neuronal cell death was assessed by the LDHassay and neuronal cell viability was assessed by the MTT assay. SH-SY5Y cells in culturewere also observedwith an inverted phase contrastmicroscope (Motic, Richmond, BC, Canada) and photographed using aMotic 3000 digital camera.
2.5. Protective activity of AF on SH-SY5Y neuronal cells exposed tosupernatants from stimulated THP-1 cells
THP-1 cells were plated in 10 cm tissue culture plates at2 × 105 cells/ml in 15 ml of DMEM-F12 containing 5% FBS. Following15 min incubation, THP-1 cells were stimulated with a combinationof 0.5 μg/ml LPS and 150 U/ml IFN-γ. After 24 h, 0.4 ml of cell-freesupernatant was transferred to each well containing SH-SY5Y cells.Immediately after transfer of supernatants, SH-SY5Y cells were treatedwith AF or its vehicle solution (DMSO). Following 72 h incubation, thesurvival of neuronal cells was measured by the MTT assay. Controlexperimentswere performed to evaluate the direct effects of AF on neu-ronal cell viability by treating SH-SY5Y cells grown in DMEM-F12containing 5% FBS with AF or its vehicle solution (DMSO) for 72 h andevaluating neuronal cell viability by the MTT assay.
2.6. Cell viability assays: LDH release
Cell death was evaluated by measuring LDH enzymatic activity incell culture supernatants as described by Decker and Lohmann-Matthes (1988). In this assay, formation of the formazan product ofiodonitrotetrazolium dye was followed colorimetrically. Briefly, 0.1 mlof cell culture supernatants was pipetted into thewells of 96-well plates,followed by an addition of 15 μl lactate (36 mg/ml) and 15 μl p-iodonitrotetrazolium violet (2 mg/ml) solutions. The enzymatic reactionwas started by the addition of NAD+/diaphorase solution (3 mg/mlNAD+, 2.3 mg solid/ml diaphorase). After a 15–30 min incubationperiod, optical densities at 490 nm were measured by a FLUOstarOmega microplate reader (BMG Labtech, Offenburg, Germany). Theamount of LDH which had been released was expressed as a fraction ofthe value obtained in comparative wells where the remaining cellswere completely lysed by 1% Triton X-100.
2.7. Cell viability assay: reduction of formazan dye (MTT)
The MTT assay was performed as previously described (Mosmann,1983; Hansen et al., 1989). This assay is based on the ability of viable,but not dead, cells to convert the tetrazolium salt MTT to coloredformazan. The viability of cultured cells was determined by addingMTT to reach a final concentration of 0.5 mg/ml. Following a 1–2 h incu-bation at 37 °C, the dark crystals which had formed were dissolved byadding an equal volume of SDS/DMF (20% sodium dodecyl sulfate, 50%N,N-dimethyl formamide, pH 4.7) to eachwell. The plates were then in-cubated at 37 °C for 3 h. Optical densities were measured at 570 nmusing a microplate reader after transferring 0.1 ml aliquots to 96-well

Fig. 1.AF reduces cytotoxicity of humanTHP-1monocytic cells towardshumanneuroblas-toma SH-SY5Y cells. THP-1 cells were pre-treated with various concentrations of the goldcompounds or their vehicle solution (DMSO) for 15min before stimulation with a combi-nation of LPS (0.5 μg/ml) and IFN-γ (150 U/ml). After 24 h incubation, THP-1 cell viabilitywas assessed by the MTT (B) and LDH (C) assays. The viability of SH-SY5Y neuronal cellsexposed for 72 h to cell-free supernatants from stimulated THP-1 cells was assessed bythe MTT assay (A). The horizontal dashed line (A) indicates viability of SH-SY5Y cellsexposed to supernatants from unstimulated THP-1 cells. Data from 4 independent exper-iments are presented. The concentration-dependent effects of the compounds wereassessed by the randomized block design ANOVA, followed by Fisher's LSD post-hoc test.*P b 0.05 significantly different from samples treated with the vehicle solution only.
73J.M. Madeira et al. / Journal of Neuroimmunology 276 (2014) 71–79
plates. The viable cell value was calculated as a percent of the valueobtained from cells incubated with fresh medium only.
2.8. Nitric oxide (NO) production by murine BV-2 microglial cells
NO production was determined indirectly by measuring nitrite(NO2
−) accumulation using the Griess reagent (Ding et al., 1988). Mu-rine microglial BV-2 cells were plated at 2 × 105 cells/ml in 0.5 mlDMEM-F12 medium containing 5% FBS and incubated for 24 h toallow for adherence of cells. The media was refreshed and cells werepre-treated with vehicle (0.1% DMSO) or varying AF concentrations(0.05–0.5 μM) for 15 min before stimulation with 0.5 μg/ml LPS and150 U/ml murine IFN-γ. Cell-free culture media were collected 24 hlater and nitrite measurements were performed by mixing equal vol-umes (50 μl) of culture medium and Griess reagent (1% sulfanilamide,0.1% N-1-naphthylethylenediamine, 2.5% phosphoric acid) followed byabsorbance measurement at 570 nm. Standard sodium nitrite solutionswere used to calibrate absorbance readings.
2.9. Measuring unprimed respiratory burst by luminol-dependentchemiluminescence
DMSO-differentiated human promyelocytic HL-60 cellswere used tostudy the effects of AF on the phagocyte respiratory burst. These cellshave been shown to express all subunits of phagocyte NADPH oxidase(Muranaka et al., 2005). Real-time chemiluminescence measurementswere performed as described by Muranaka et al. (2005). First, HL-60cellswere differentiated by plating them into 10 cm tissue culture platesat a density of 2 × 105 cells/ml in DMEM-F12 medium containing 10%FBS and 1.3% DMSO. After a 5–7 day incubation period, which was con-firmed in preliminary studies to enhance the respiratory burst activitysignificantly, HL-60 cells were washed and transferred into DMEM-F12 medium without phenol red containing 5% FBS. Cells were seededinto a 96-well plate at a concentration of 1 × 106 cells/ml in 80 μl of me-dium and the plate was inserted into the FLUOstar Omega plate-reader.The respiratory burst responsewasmeasured after injecting luminol so-lution first, followed by a 7.5 min baseline recording and subsequentfMLP injection directly into the wells. The final reaction volume in thewell was 0.1 ml, the final luminol concentration was 5 mM and fMLPwas at 1 μM. Chemiluminescence response of HL-60 cells was recordedfor 30 min and expressed as the area under the curve in Relative LightUnits. Plates were maintained at 37 °C during all experiments. The ef-fect of AF on respiratory burst of unprimed cellswas studied by injectingluminol, followed by fMLP, 15 min after addition of AF to HL-60 cells.Concentration of AF vehicle solvent (DMSO) in control wells did not ex-ceed 0.5%; at this concentration DMSO did not affect the chemilumines-cent response of cells.
2.10. Measuring LPS-primed respiratory burst by luminol-dependentchemiluminescence
In some experiments, the respiratory burst response of DMSO-differentiated HL-60 cells was enhanced (primed) by adding 0.5 μg/mlLPS to HL-60 cells 24 h before the injection of luminol and fMLP.HL-60 cells were plated and chemiluminescence measurements wereperformed exactly as described above. The effect of AF on respiratoryburst priming was studied after its addition to cells 15 min before thepriming agent LPS (24 h treatment), or 30 min before injecting fMLP(30min treatment). Concentration of AF vehicle solvent (DMSO) in con-trol wells did not exceed 0.5%; at this concentration DMSO did not affectthe chemiluminescent response of cells.
2.11. Statistical analyses
Due to considerable variability in the absolute values obtained fromindependent experiments performed on different days, randomized
block design Analysis of Variance (ANOVA) was used, followed byFisher's least significant difference (LSD) post-hoc test. A P value lessthan 0.05 was considered statistically significant.
3. Results
3.1. Effects of gold compounds on human THP-1 promonocytic cell viabilityand cytotoxic secretions
Gold compounds (0.1–2.5 μM)were tested for their ability to inhibithuman THP-1 promonocytic cell toxicity towards human neuronal SH-SY5Y cells and results were compared to those obtained from samplestreated with DMSO vehicle solution only. At the concentration used(b0.13%, v/v), DMSO alone had no detectable effects in the assaysused (data not shown). THP-1 cells were treated with the compoundsfor 15min prior to stimulation with LPS plus IFN-γ. Following a 24 h in-cubation period, viability of THP-1 cells was assessed using theMTT cellviability (Fig. 1B) and LDH cell death (Fig. 1C) assays, which showed no

74 J.M. Madeira et al. / Journal of Neuroimmunology 276 (2014) 71–79
toxicity of AF, ATMor ATS towards stimulated THP-1 cells at the concen-trations tested.
After the 24 h stimulation period, the cell-free supernatants fromTHP-1 cell cultures were transferred to SH-SY5Y neuroblastoma cellsto assess their cytotoxic effects. Following a 72 h incubation periodwith THP-1 supernatants, the viability of neuronal cells was assessedusing the MTT assay (Fig. 1A). Supernatants from unstimulated THP-1cells did not significantly affect the viability of SH-SY5Y cells (seedashed line in Fig. 1A). Transfer of cell-free supernatants from stimulat-ed THP-1 cells to SH-SY5Y cells resulted in significantly reduced neuro-nal cell viability (Fig. 1A). A combination of LPS and IFN-γ was used toachieve maximal stimulation of cells (Klegeris et al., 2005). Fig. 1Ashows that ATM and ATS did not exhibit anti-neurotoxic activity whileAF (0.5–2 μM) inhibited secretion of toxins by stimulated THP-1 cells.This anti-neurotoxic activity of AF was not due to its toxicity towardsTHP-1 cells, but most likely due to specific inhibition of the cytotoxicsecretions of THP-1 cells.
3.2. Effects of AF on primary human microglia cell viability and cytotoxicsecretions
The anti-neurotoxic activity of AF was also confirmed by using pri-mary microglia cultures prepared from human surgical tissue samples.AF was tested at 0.1 μM and cell viability values were compared tothose obtained from samples treated with DMSO vehicle solution only.Following 48 h incubation, the viability of primary human microgliawas assessed using the MTT assay (Fig. 2B); at 0.1 μM, AF was nottoxic to microglia. Cell-free supernatants from microglia cultures weretransferred to SH-SY5Y neuroblastoma cells in order to assess possiblecytotoxic effects. Following a 72 h incubation period with these super-natants, the viability of neuronal cells was measured using the MTTassay (Fig. 2A), which demonstrated that at 0.1 μM AF inhibited the
Fig. 2.AF reduces cytotoxicity of humanmicroglia towards humanneuroblastoma SH-SY5Y cellsfor 15min before stimulationwith a combination of LPS (0.5 μg/ml) and IFN-γ (150 U/ml). AfteSH-SY5Y neuronal cells exposed for 72h to supernatants from stimulatedmicrogliawas assesseddifferent surgical cases are presented. The effects of AFwere assessed by the randomized block dstimulated samples treated with the vehicle solution only. Phase contrast microscopy of SH-unstimulated (C) or stimulated with LPS plus IFN-γ in the absence (D) or presence of 0.1 μ(=50 μm) in C is representative for all three panels.
toxicity of stimulated human microglia towards neuronal cells(Fig. 2A), confirming results observed with the THP-1 cells whichwere used as microglia model. The morphology of SH-SY5Y cells incu-bated with supernatants of human microglia was also analyzed(Fig. 2C–E). The supernatants of activated microglia caused significantchanges of cellular morphology with many cells showing bright andcircularly shrunk cytoplasm (Fig. 2D). This was different from controlsamples exposed to vehicle solution only, which showed mostlypyramidal cell bodies (Fig. 2C). The adverse changes in SH-SY5Y cellmorphology induced by stimulated microglia supernatants wereconsiderably inhibited by pre-treatment with 0.1 μM AF (Fig. 2E).These observations were consistent with the results obtained by theMTT assay (Fig. 2A).
3.3. Direct neuroprotective effects of AF
The direct neuroprotective effect of AF on cultured SH-SY5Y cellswas assessed by adding AF (0.1–1 μM) to supernatants from stimulatedTHP-1 cells at the time of their transfer to SH-SY5Y cells (Fig. 3A). AF at0.5 and 1 μM significantly increased viability of SH-SY5Y cells. Fig. 3Bdemonstrates that at the concentrations studied, AF had no direct effecton SH-SY5Y cell viability in the absence of THP-derived toxins.
3.4. Effects of gold compounds on the respiratory burst
The three gold compounds; AF, ATM and ATS, were tested for theirability to inhibit the NADPH oxidase-dependent respiratory burst(data not shown for ATM and ATS). DMSO-differentiated human HL-60 cells were used as a model of the microglia/phagocyte respiratoryburst as THP-1 cells do not express the NADPH enzyme at levels highenough to generate detectable levels of reactive oxygen species (ROS)in our experiments (data not shown). Gold compounds were tested at
. Primary humanmicrogliawere pre-treatedwith 0.1 μMAFor its vehicle solution (DMSO)r 24h incubation,microglial cell viabilitywas assessed by theMTT assay (B). The viability ofby theMTT assay (A). Data from3 independent experimentswith cells obtained from twoesign ANOVA, followed by Fisher's LSD post-hoc test. *P b 0.05, significantly different fromSY5Y cell cultures after incubation with supernatants from human microglia that wereM AF (E). Photos are representative of 3 independent experiments. Magnification bar
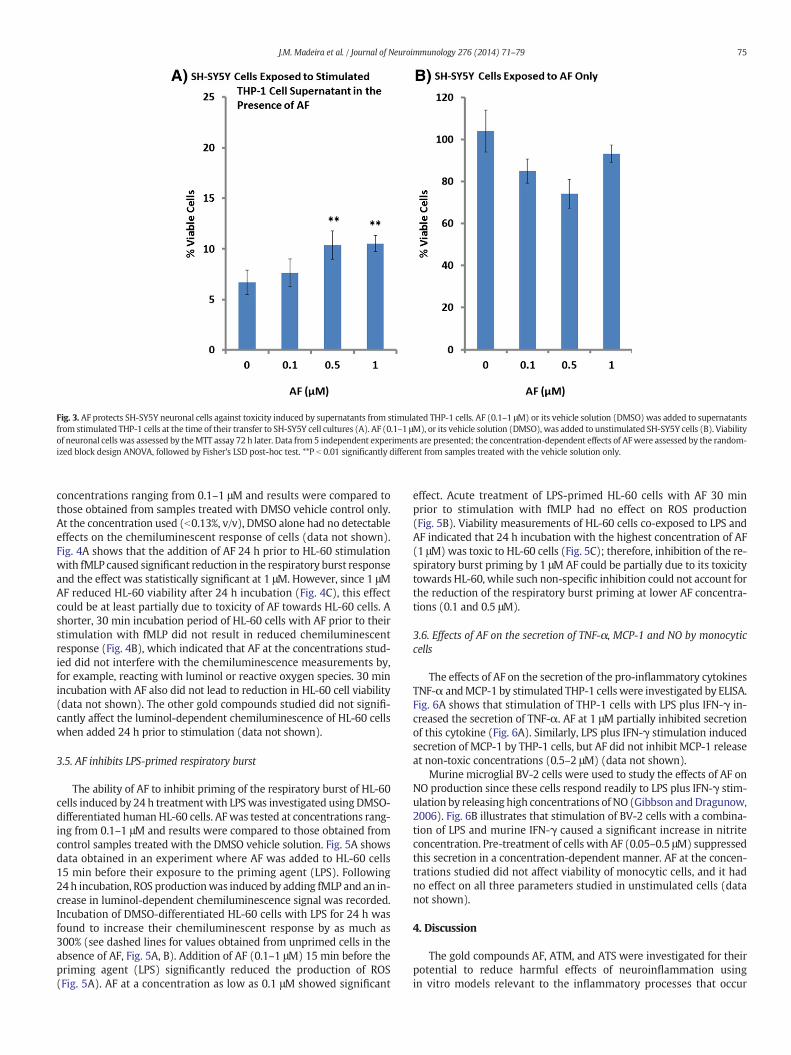
Fig. 3. AF protects SH-SY5Y neuronal cells against toxicity induced by supernatants from stimulated THP-1 cells. AF (0.1–1 μM) or its vehicle solution (DMSO) was added to supernatantsfrom stimulated THP-1 cells at the time of their transfer to SH-SY5Y cell cultures (A). AF (0.1–1 μM), or its vehicle solution (DMSO), was added to unstimulated SH-SY5Y cells (B). Viabilityof neuronal cells was assessed by theMTT assay 72 h later. Data from 5 independent experiments are presented; the concentration-dependent effects of AFwere assessed by the random-ized block design ANOVA, followed by Fisher's LSD post-hoc test. **P b 0.01 significantly different from samples treated with the vehicle solution only.
75J.M. Madeira et al. / Journal of Neuroimmunology 276 (2014) 71–79
concentrations ranging from 0.1–1 μM and results were compared tothose obtained from samples treated with DMSO vehicle control only.At the concentration used (b0.13%, v/v), DMSO alone had no detectableeffects on the chemiluminescent response of cells (data not shown).Fig. 4A shows that the addition of AF 24 h prior to HL-60 stimulationwith fMLP caused significant reduction in the respiratory burst responseand the effect was statistically significant at 1 μM. However, since 1 μMAF reduced HL-60 viability after 24 h incubation (Fig. 4C), this effectcould be at least partially due to toxicity of AF towards HL-60 cells. Ashorter, 30 min incubation period of HL-60 cells with AF prior to theirstimulation with fMLP did not result in reduced chemiluminescentresponse (Fig. 4B), which indicated that AF at the concentrations stud-ied did not interfere with the chemiluminescence measurements by,for example, reacting with luminol or reactive oxygen species. 30 minincubation with AF also did not lead to reduction in HL-60 cell viability(data not shown). The other gold compounds studied did not signifi-cantly affect the luminol-dependent chemiluminescence of HL-60 cellswhen added 24 h prior to stimulation (data not shown).
3.5. AF inhibits LPS-primed respiratory burst
The ability of AF to inhibit priming of the respiratory burst of HL-60cells induced by 24 h treatmentwith LPSwas investigated using DMSO-differentiated human HL-60 cells. AF was tested at concentrations rang-ing from 0.1–1 μM and results were compared to those obtained fromcontrol samples treated with the DMSO vehicle solution. Fig. 5A showsdata obtained in an experiment where AF was added to HL-60 cells15 min before their exposure to the priming agent (LPS). Following24h incubation, ROS productionwas induced by adding fMLP and an in-crease in luminol-dependent chemiluminescence signal was recorded.Incubation of DMSO-differentiated HL-60 cells with LPS for 24 h wasfound to increase their chemiluminescent response by as much as300% (see dashed lines for values obtained from unprimed cells in theabsence of AF, Fig. 5A, B). Addition of AF (0.1–1 μM) 15 min before thepriming agent (LPS) significantly reduced the production of ROS(Fig. 5A). AF at a concentration as low as 0.1 μM showed significant
effect. Acute treatment of LPS-primed HL-60 cells with AF 30 minprior to stimulation with fMLP had no effect on ROS production(Fig. 5B). Viability measurements of HL-60 cells co-exposed to LPS andAF indicated that 24 h incubation with the highest concentration of AF(1 μM)was toxic to HL-60 cells (Fig. 5C); therefore, inhibition of the re-spiratory burst priming by 1 μM AF could be partially due to its toxicitytowards HL-60, while such non-specific inhibition could not account forthe reduction of the respiratory burst priming at lower AF concentra-tions (0.1 and 0.5 μM).
3.6. Effects of AF on the secretion of TNF-α, MCP-1 and NO by monocyticcells
The effects of AF on the secretion of the pro-inflammatory cytokinesTNF-α andMCP-1 by stimulated THP-1 cells were investigated by ELISA.Fig. 6A shows that stimulation of THP-1 cells with LPS plus IFN-γ in-creased the secretion of TNF-α. AF at 1 μM partially inhibited secretionof this cytokine (Fig. 6A). Similarly, LPS plus IFN-γ stimulation inducedsecretion of MCP-1 by THP-1 cells, but AF did not inhibit MCP-1 releaseat non-toxic concentrations (0.5–2 μM) (data not shown).
Murine microglial BV-2 cells were used to study the effects of AF onNO production since these cells respond readily to LPS plus IFN-γ stim-ulation by releasing high concentrations of NO (Gibbson andDragunow,2006). Fig. 6B illustrates that stimulation of BV-2 cells with a combina-tion of LPS and murine IFN-γ caused a significant increase in nitriteconcentration. Pre-treatment of cells with AF (0.05–0.5 μM) suppressedthis secretion in a concentration-dependent manner. AF at the concen-trations studied did not affect viability of monocytic cells, and it hadno effect on all three parameters studied in unstimulated cells (datanot shown).
4. Discussion
The gold compounds AF, ATM, and ATS were investigated for theirpotential to reduce harmful effects of neuroinflammation usingin vitro models relevant to the inflammatory processes that occur

Fig. 4. Only high concentrations of AF inhibit the chemiluminescent response of HL-60cells to fMLP stimulation. DMSO-differentiated HL-60 cells were seeded into 96-wellplates and pre-treated with various concentrations of AF or its vehicle solution (DMSO)for 24 h (A) or 30 min (B). Luminol-dependent chemiluminescence response of HL-60cells was recorded for 30 min after injection of 1 μM fMLP. Viability of HL-60 cells at theend of the experiment (conditions shown in A) was assessed by the MTT assay (C). Datafrom 4 independent experiments are presented; the concentration-dependent effects ofthe compounds were assessed by the randomized block design ANOVA, followed byFisher's LSD post-hoc test. *P b 0.05 significantly different from samples treated with thevehicle solution only.
Fig. 5. AF inhibits LPS-primed respiratory burst of DMSO-differentiated HL-60 cells. Cellswere seeded into 96-well plates and their respiratory burst response primed by 24 hincubation with 0.5 μg/ml LPS, followed by injection of 1 μM fMLP which is a trigger ofthe respiratory burst. The concentration-dependent effects of AF were studied by addingthis drug 15 min before HL-60 exposure to LPS (A) or 30 min before fMLP injection(B). Luminol-dependent chemiluminescence response of HL-60 cells in the presence orabsence of AFwas recorded for 30min after injection of fMLP (A, B). The horizontal dashedlines indicate chemiluminescence values obtained from unprimed cells in the absence ofAF (A, B). Viability of HL-60 cells at the end of the experiment (conditions shown inA) was assessed by the MTT assay (C). Data from 4-5 independent experiments are pre-sented; the concentration-dependent effects of AF were assessed by the randomizedblock design ANOVA, followed by Fisher's LSD post-hoc test. *P b 0.05, **P b 0.01 signifi-cantly different from samples treated with the vehicle solution only.
76 J.M. Madeira et al. / Journal of Neuroimmunology 276 (2014) 71–79
during neurodegeneration. We studied the effects of gold-containingcompounds at low micromolar concentrations, which in the case of AFhave been shown to be attainable after its oral administration (Walzet al., 1983;Madeira et al., 2013). It was found that at non-toxic concen-trations (0.1–2 μM) AF reduced cytotoxicity of both stimulated humanTHP-1 promonocytic cells (0.5–2 μM) and primary human microglia(0.1 μM) towards neuronal SH-SY5Y cells, indicating a potential anti-neurotoxic effect of AF. ATM and ATS, the other two gold compoundsstudied, were inactive. Previous research using similar assays hasalready shown that primary human cells could be more sensitive toanti-inflammatory drug treatment than cell lines; therefore, the lowereffective concentration of AF in primary cells was not unexpected(Yamada et al., 1999; Polanski et al., 2011). Similarly, 0.1 μM AFinhibited toxicity of primary human astrocytes, while 1–5 μM concen-trations of AF were needed to observe such inhibitory activity by usingan astrocytoma cell line (Madeira et al., 2013).
To determine whether the observed anti-neurotoxic activity of AFwas due to its interaction with monocytic THP-1 and microglial cells,or whether this effect was due to the transfer of some of the compoundwith the monocytic supernatants and its subsequent direct action onneuronal cells, experiments were performed where AF was applieddirectly to neuronal cells at the time of their exposure to cytotoxic su-pernatants from stimulated THP-1 cells. Treating SH-SY5Y neuronalcells with 0.5–1 μM AF protected them from toxicity induced by super-natants from stimulated THP-1 cells. A direct protective effect of AF(0.5–1 μM) on neuronal cells that had been exposed to toxic concentra-tions of hydrogen peroxide or astrocytic toxinswas reported previously(Madeira et al., 2013). These observations indicate that AF may conferneuroprotection in addition to its better-known and accepted anti-inflammatory properties. The direct protective activity of AF on neuro-nal cells was unlikely the sole mechanism responsible for the anti-neurotoxic activity of AF observed in our study since AF effectivelyinhibited primary human microglial toxic secretions at 0.1 μM, a con-centration that was ineffective at rescuing neuronal cells from severaldifferent types of toxins (Fig. 3;Madeira et al., 2013). It is also importantto note that the inhibitory effect of AF onmonocytic cells was not due toits non-specific toxicity since both of the cell viability assays employed

Fig. 6. AF inhibits TNF-α and NO secretion by monocytic cells. Human monocytic THP-1cells (A) and mouse microglial BV-2 (B) cells were pre-treated with various concentra-tions of AF or the vehicle solution (DMSO) for 15min before stimulation with a combina-tion of LPS and either human (A) or murine (B) IFN-γ. Following a 48 h (A) or 24 h(B) incubation period, TNF-α concentration in culture supernatants was measured byELISA (A), and nitrite concentration was measured by Griess reagent (B). Data from 4(A) and 7 (B) independent experiments are presented. The horizontal dashed lines corre-spond to values obtained in supernatants from unstimulated cells. The concentration-dependent effect of AF was assessed by the randomized block design ANOVA, followedby Fisher's LSD post-hoc test. *P b 0.05, **P b 0.01 significantly different from samplestreated with the vehicle solution only.
77J.M. Madeira et al. / Journal of Neuroimmunology 276 (2014) 71–79
to monitor monocytic cell viability showed no toxic effects of AF at lowmicromolar concentrations. In addition, AF at the concentrationsstudied did not inhibit secretion of MCP-1 by THP-1 monocytic cells.MCP-1 is a chemotactic cytokine, which is sometimes used as an indica-tor of inflammatory activation of mononuclear phagocytes (Abramsonand Gallin, 1990).
A wide range of substances have been shown to contribute to theneurotoxic and cytotoxic activity of microglia; most likely a mixture ofsubstances is responsible for such effects, the composition of which de-pends on the cell type and stimuli used (for review see Block et al., 2007;Madeira et al., 2012b). Our data indicate that AF at non-toxic lowmicro-molar concentrations inhibits the secretion of two known microglialtoxins, TNF-α and NO (Gibbson and Dragunow, 2006; Brown, 2010;Lull and Block, 2010), which could be the basis for the anti-neurotoxicactivity of AF observed in this study.
One of the proposed strategies for the treatment of neurodegenera-tive diseases is to enhance neuronal viability by increasing tolerance ofneurons to oxidative damage and inflammatory toxins released byactivated glial cells (Pocernich and Butterfield, 2012; Cornelius et al.,2013). Glial cells themselves release several endogenous neuroprotec-tive molecules, including insulin and pro-insulin, which have receivedattention as potential therapeutic agents for preventing the neuronaldeath in Alzheimer's and Parkinson's diseases (Ellingsen et al., 2001;Block et al., 2007). In addition to endogenous molecules, syntheticdrugs and purified compounds have been developed in an attempt toincrease neuronal viability. For example, flavonoids, such as quercetin,have been recognized as neuroprotective molecules; Dajas et al.(2003) found that 25 μM quercetin protected neuronal cells againsthydrogen peroxide toxicity. This protective effect was thought to bedue to the anti-oxidant activity of the flavonoid (Dajas et al., 2003). Incontrast, AF has been shown to activate caspase-3 and disrupt redoxbalance within cells; therefore, the protective mechanisms of AF arelikely different from those of flavonoids (Mirabelli et al., 1985; Jeonet al., 2000; Ellingsen et al., 2001).
Ineffectiveness of AF as anti-oxidant has been reported before(Krause et al., 2011). It was confirmed in this study by the lack of the ef-fect of AF on the luminol-dependent chemiluminescence response ofHL-60 cells. This assay is dependent on production of ROS by stimulatedcells and therefore drugs which are anti-oxidants and can scavenge ROSoften inhibit the chemiluminescence signal (Lu et al., 2012; Lee et al.,2013). HL-60 cells were used as surrogates of mononuclear phagocytesthat possess enzymatically active NADPH oxidase complexes capable ofgenerating strong respiratory burst response. It is known that mamma-lianmicroglia possess this enzymatic activity (Gao et al., 2012) but somecell lines, including humanmonocytic THP-1 cells, express low levels ofNADPH oxidase, which does not allow respiratory burst measurementin these cells (Makino et al., 2012). Low yield of primary humanmicrog-lia precluded respiratory burst experiments with this cell type.
Since a 30min treatment of HL-60 cells with AF did not inhibit theirfMLP-induced luminol-dependent chemiluminescence response, effectsof AF on the priming of respiratory burst were studied. Priming ofNADPH oxidase is a good target for pharmacological interventionbecause its inhibition would reduce the pathological increase in ROSproduction while maintaining the normal physiological NADPHoxidase-mediated immune responses (Muranaka et al., 2005). HL-60cells were primed with LPS, a known priming agent, and were treatedwith AF either before the addition of the priming agent or 30 minprior to stimulation with fMLP. The former treatment with AF at thenon-toxic 0.1–0.5 μM range inhibited LPS priming of the respiratoryburst. Treating HL-60 cells with AF for 30 min did not inhibit the respi-ratory burst response,whichwas similar to the lack of the effect of AF onun-primed cells. The extended incubation time needed for the inhibito-ry effect of AF to appear indicates that AF does not inhibit NADPH oxi-dase directly; therefore, AF may regulate expression of genes encodingNADPH oxidase components or affect slow-acting cellular pathways(Anderson et al., 1991).
Previous research indicates that the majority of AF is taken up bycells within 20min (Grahamet al., 1994); therefore, a 30min treatmentis sufficient for AF to enter cells and interact with NADPH-oxidase sub-units if this were the mechanism of AF action (Wong et al., 1990).Agents that inhibit the priming of NADPH-oxidase, but not the function-ing of this enzymatic complex, are not common. IL-4 and lipid A ana-logues have both been shown to inhibit the priming of respiratoryburst in neutrophils (Means and Dedman, 1980; Brodie et al., 1998;Chomarat and Banchereau, 1998). IL-4 inhibits NADPH-oxidase assem-bly by decreasing the expression of its gp91-phox subunit, thus effec-tively reducing the number of active enzyme complexes (Means andDedman, 1980; Brodie et al., 1998; Chomarat and Banchereau, 1998).Lipid A analogues are structurally similar to LPS and they are thoughtto inhibit NADPH-oxidase priming by antagonizing the LPS-binding re-gions on cell surfaces (Van Dervort et al., 1992). It is possible that AFinhibits priming of the respiratory burst through either of thesemechanisms. Future investigations into the exact mechanism bywhich AF inhibits the LPS-primed respiratory burst should focus onexpression of NADPH-oxidase subunits and potential antagonism ofLPS receptors.
The direct neuroprotective activity of AF has already been reported,and it was shown that up-regulation of HOX-1 could contribute to thisprotective activity in neuronal SH-SY5Y cells (Madeira et al., 2013).Previous studies by Kim et al. (2010) have shown that AF upregulatesHOX-1 in THP-1 cells, therefore upregulation of HOX-1 could be respon-sible for the anti-inflammatory effects of AF in different types of mono-nuclear phagocytes, including microglia. HOX-1 has been previouslysuggested as a protective molecule in neuroinflammatory conditions.Yamamoto et al. (2010) demonstrated that induction of HOX-1protected dopaminergic neurons in an in vitromodel of Parkinson's dis-ease. By up-regulating protective enzymes like HOX-1, AF could directlyincrease the ability of neurons to withstand toxic insults.
Our study identifies potential anti-neurotoxic and neuroprotectiveeffects of AF in microglia-like cells. Previous studies of the in vivo

78 J.M. Madeira et al. / Journal of Neuroimmunology 276 (2014) 71–79
distribution of AF after its oral administration in rodents (1–2 mg/kgper day for 5–7 days) have shown that the CNS concentration of AFcould reach 0.2–5 μM range (Walz et al., 1983; Madeira et al., 2013).This indicates that AF can cross the blood–brain barrier and may reachanti-neurotoxic and neuroprotective concentrations in the CNS.Furthermore, the blood–brain barrier becomes compromised in neuro-degenerative diseases, such as Alzheimer's and Parkinson's diseases(Kortekaas et al., 2005; Persidsky et al., 2006), which could lead toeven higher brain concentrations of AF after its oral administration.Therefore, further testing of AF in animal models of neuroinflammationis warranted.
Extensive clinical use of AF has shown that this drug has a relativelysafe pharmacological profile in humans including elderly patients(Glennas et al., 1997; Kean et al., 1997; Kim et al., 2010). This, in combi-nation with its anti-inflammatory and cytoprotective activity demon-strated by this study may make AF a good treatment option forinflammatory conditions other than rheumatoid arthritis. Anti-inflammatory and neuroprotective treatment strategies have the poten-tial to slow the progression of neurological disorders includingAlzheimer's and Parkinson's diseases and should be explored in clinicalstudies since currently there is no effective treatment for these diseases(Klegeris et al., 2007; Heneka et al., 2010).
Acknowledgments
This work was supported by grants from the Natural Sciences andEngineering Research Council of Canada and the Jack Brown and FamilyAlzheimer's Disease Research Foundation. We would like to thank Mr.C.J. Renschler for his technical assistance.
References
Abramson, S.L., Gallin, J.I., 1990. IL-4 inhibits superoxide production by human mononu-clear phagocytes. J. Immunol. 144, 625–630.
Anderson, R., Van Rensburg, C.E., Joone, G.K., Lessing, A., 1991. Auranofin inactivatesphosphofructokinase in human neutrophils, leading to depletion of intracellularATP and inhibition of superoxide generation and locomotion. Mol. Pharmacol. 40,427–434.
Block, M.L., Zecca, L., Hong, J.S., 2007. Microglia-mediated neurotoxicity: uncovering themolecular mechanisms. Nat. Rev. Immunol. 8, 57–69.
Brodie, C., Goldreich, N., Haiman, T., Kazimirsky, G., 1998. Functional IL-4 receptors onmouse astrocytes: IL-4 inhibits astrocyte activation and induces NGF secretion. J.Neuroimmunol. 81, 20–30.
Brown, G., 2010. Nitric oxide and neuronal death. Nitric Oxide 23, 153–165.Bruze, M., Björkner, B., Möller, H., 1995. Skin testing with gold sodium thiomalate and
gold sodium thiosulfate. Contact Dermatitis 32, 5–8.Cameron, B., Landreth, G.E., 2010. Inflammation, microglia, and Alzheimer's disease.
Neurobiol. Dis. 37, 503–509.Champion, G.D., Graham, G.G., Ziegler, J.B., 1990. The gold complexes. Baillieres Clin.
Rheumatol. 4, 491–534.Chomarat, P., Banchereau, J., 1998. Interleukin-4 and interleukin-13: their similarities and
discrepancies. Int. Rev. Immunol. 17, 1–52.Cornelius, C., Crupi, R., Calabrese, V., Graziano, A., Milone, P., Pennisi, G., Radak, Z.,
Calabrese, E.J., Cuzzocrea, S., 2013. Traumatic brain injury: oxidative stress andneuroprotection. Antioxid. Redox Signal. 19, 836–853.
Dajas, F., Rivera, F., Blasina, F., Arredondo, F., Echeverry, C., Lafon, L., Morquio, A., Heinzen,H., 2003. Cell culture protection and in vivo neuroprotective capacity of flavonoids.Neurotox. Res. 5, 425–432.
Decker, T., Lohmann-Matthes, M.L., 1988. A quick and simple method for the quantitationof lactate dehydrogenase release in measurements of cellular cytotoxicity and tumornecrosis factor (TNF) activity. J. Immunol. Methods 115, 61–69.
Ding, A.H., Nathan, C.F., Stuehr, D.J., 1988. Release of reactive nitrogen intermediates andreactive oxygen intermediates from mouse peritoneal macrophages. Comparison ofactivating cytokines and evidence for independent production. J. Immunol. 141,2407–2412.
Ellingsen, T., Buus, A., Stengaard-Pedersen, K., 2001. Plasma monocyte chemoattractantprotein 1 is a marker for joint inflammation in rheumatoid arthritis. J. Rheumatol.28, 41–46.
Frohman, E.M., Racke, M.K., Raine, C.S., 2006. Multiple sclerosis — the plaque and itspathogenesis. N. Engl. J. Med. 354, 942–955.
Gao, H.M., Zhou, H., Hong, J.S., 2012. NADPH oxidases: novel therapeutic targets forneurodegenerative diseases. Trends Pharmacol. Sci. 33, 295–303.
Gibbons, H., Dragunow, M., 2006. Microglia induce neural cell death via a proximity-dependent mechanism involving nitric oxide. Brain Res. 1084, 1–15.
Glennas, A., Kvien, T.K., Andrup, O., Clarke-Jenssen, O., Karstensen, B., Brodin, U., 1997.Auranofin is safe and superior to placebo in elderly-onset rheumatoid arthritis. Br.J. Rheumatol. 36, 870–877.
Graham, G.G., Champion, G.D., Ziegler, J.B., 1994. The cellular metabolism and effects ofgold complexes. Metal-Based Drugs 1, 395–404.
Hansen, M.B., Nielsen, S.E., Berg, K., 1989. Re-examination and further development of aprecise and rapid dye method for measuring cell growth/cell kill. J. Immunol.Methods 119, 203–210.
Hashioka, S., Klegeris, A., McGeer, P.L., 2009. Proton pump inhibitors exert anti-inflammatory effects and decrease human microglial and monocytic THP-1 cellneurotoxicity. Exp. Neurol. 217, 177–183.
Heneka, M.T., O'Banion, M.K., Terwel, D., Kummer, M.P., 2010. Neuroinflammatoryprocesses in Alzheimer's disease. J. Neural Transm. 117, 919–947.
Jeon, K.I., Jeong, J.Y., Jue, D.M., 2000. Thiol-reactive metal compounds inhibit NF-kappa Bactivation by blocking I kappa B kinase. J. Immunol. 164, 5981–5989.
Kean, W.F., 1990. Intramuscular versus oral gold therapy. Baillieres Clin. Rheumatol. 4,219–246.
Kean, W.F., Hart, L., Buchanan, W.W., 1997. Auranofin. Br. J. Rheumatol. 36, 560–572.Kim, N.H., Lee, M.Y., Park, S.J., Choi, J.S., Oh, M.K., Kim, I.S., 2007. Auranofin blocks
interleukin-6 signalling by inhibiting phosphorylation of JAK1 and STAT3. Immunol-ogy 122, 607–614.
Kim, N.H., Oh, M.K., Park, H.J., Kim, I.S., 2010. Auranofin, a gold (I)-containingantirheumatic compound, activates Keap1/Nrf2 signaling via Rac1/iNOS signal andmitogen-activated protein kinase activation. J. Pharmacol. Sci. 113, 246–254.
Klegeris, A., Bissonnette, C.J., McGeer, P.L., 2003. Reduction of human monocytic cellneurotoxicity and cytokine secretion by ligands of the cannabinoid-type CB2 recep-tor. Br. J. Pharmacol. 139, 775–786.
Klegeris, A., Bissonnette, C.J., McGeer, P.L., 2005. Modulation of human microglia andTHP-1 cell toxicity by cytokines endogenous to the nervous system. Neurobiol.Aging 26, 673–682.
Klegeris, A., McGeer, E.G., McGeer, P.L., 2007. Therapeutic approaches to inflammation inneurodegenerative disease. Curr. Opin. Neurol. 20, 351–357.
Kortekaas, R., Leenders, K.L., van Oostrom, J.C., Vaalburg, W., Bart, J., Willemsen, A.T.,Hendrikse, N.H., 2005. Blood–brain barrier dysfunction in Parkinsonian midbrainin vivo. Ann. Neurol. 57, 176–179.
Krause, D., Suh, H.S., Tarassishin, L., Cui, Q.L., Durafourt, B.A., Choi, N., Bauman, A., Cosenza-Nashat, M., Antel, J.P., Zhao, M.L., Lee, S.C., 2011. The tryptophan metabolite3-hydroxyanthranilic acid plays anti-inflammatory and neuroprotective roles duringinflammation: role of hemeoxygenase-1. Am. J. Pathol. 179, 1360–1372.
Lee, Y.J., Han, S.B., Nam, S.Y., Oh, K.W., Hong, J.T., 2010. Inflammation and Alzheimer'sdisease. Arch. Pharm. Res. 33, 1539–1556.
Lee, C.W., Ko, H.H., Lin, C.C., Chai, C.Y., Chen, W.T., Yen, F.L., 2013. Artocarpin attenuatesultraviolet B-induced skin damage in hairless mice by antioxidant and anti-inflammatory effect. Food Chem. Toxicol. 60, 123–129.
Li, C., Zhao, R., Gao, K., Wei, Z., Yin, M.Y., Lau, L.T., Chui, D., Hoi Yu, A.C., 2011. Astrocytes:implications for neuroinflammatory pathogenesis of Alzheimer's disease. Curr.Alzheimer Res. 8, 67–80.
Lu, D., Shao, H.T., Ge, W.P., Liu, N., Zhang, X., Ma, C.M., Qin, C., Zhang, L.F., 2012.Ginsenoside-RB1 and tetramethylpyrazine phosphate act synergistically to preventdilated cardiomyopathy in cTnTR141W transgenic mice. J. Cardiovasc. Pharmacol.59, 426–433.
Lull, M.E., Block, M.L., 2010. Microglial activation and chronic neurodegeneration.Neurotherapeutics 7, 354–365.
Madeira, J.M., Gibson, D.L., Kean, W.F., Klegeris, A., 2012a. The biological activity ofauranofin: implications for novel treatment of diseases. Inflammopharmacology 20,297–306.
Madeira, J.M., Little, J.P., Klegeris, A., 2012b. Microglia secretome: from neurotoxins toneurotrophins. In: Kaur, C., Ling, E.A. (Eds.), Microglia: Biology, Functions and Rolesin Disease. Nova Publishers, New York, pp. 73–92.
Madeira, J.M., Renschler, C.J., Mueller, B., Hashioka, S., Gibson, D.L., Klegeris, A., 2013.Novel protective properties of auranofin: inhibition of human astrocyte cytotoxicsecretions and direct neuroprotection. Life Sci. 92, 1072–1080.
Makino, J., Kamiya, T., Hara, H., Adachi, T., 2012. TPA induces the expression of EC-SOD inhumanmonocytic THP-1 cells: involvement of PKC, MEK/ERK and NOX-derived ROS.Free Radic. Res. 46, 637–644.
Means, A.R., Dedman, J.R., 1980. Calmodulin—an intracellular calcium receptor. Nature285, 73–77.
Mirabelli, C.K., Johnson, R.K., Sung, C.M., Faucette, L., Muirhead, K., Crooke, S.T., 1985. Eval-uation of the in vivo antitumor activity and in vitro cytotoxic properties of auranofin,a coordinated gold compound, in murine tumor models. Cancer Res. 45, 32–39.
Mosmann, T., 1983. Rapid colorimetric assay for cellular growth and survival: applicationto proliferation and cytotoxicity assays. J. Immunol. Methods 65, 55–63.
Muranaka, S., Fujita, H., Fujiwara, T., Ogino, T., Sato, E.F., Akiyama, J., Imada, I., Inoue, M.,Utsumi, K., 2005. Mechanism and characteristics of stimuli-dependent ROS genera-tion in undifferentiated HL-60 cells. Antioxid. Redox Signal. 7, 1367–1376.
Nakaya, A., Sagawa, M., Muto, A., Uchida, H., Ikeda, Y., Kizaki, M., 2011. The gold compoundauranofin induces apoptosis of human multiple myeloma cells through both down-regulation of STAT3 and inhibition of NF-kappaB activity. Leuk. Res. 35, 243–249.
Persidsky, Y., Ramirez, S.H., Haorah, J., Kanmogne, G.D., 2006. Blood–brain barrier: struc-tural components and function under physiologic and pathologic conditions. J.Neuroimmune Pharmacol. 1, 223–236.
Pocernich, C.B., Butterfield, D.A., 2012. Elevation of glutathione as a therapeutic strategy inAlzheimer disease. Biochim. Biophys. Acta 1822, 625–630.
Polanski, W., Reichmann, H., Gille, G., 2011. Stimulation, protection and regeneration ofdopaminergic neurons by 9-methyl-β-carboline: a new anti-Parkinson drug? Expert.Rev. Neurother. 11, 845–860.

79J.M. Madeira et al. / Journal of Neuroimmunology 276 (2014) 71–79
Ransohoff, R.M., Brown,M.A., 2012. Innate immunity in the central nervous system. J. Clin.Invest. 122, 1164–1171.
Stern, I., Wataha, J.C., Lewis, J.B., Messer, R.L., Lockwood, P.E., Tseng, W.Y., 2005. Anti-rheumatic gold compounds as sublethal modulators of monocytic LPS-inducedcytokine secretion. Toxicol. in Vitro 19, 365–371.
Van Dervort, A.L., Doerfler, M.E., Stuetz, P., Danner, R.L., 1992. Antagonism oflipopolysaccharide-induced priming of human neutrophils by lipid A analogs. J.Immunol. 149, 359–366.
Walz, D.T., DiMartino, M.J., Griswold, D.E., Intoccia, A.P., Flanagan, T.L., 1983. Biologicactions and pharmacokinetic studies of auranofin. Am. J. Med. 75, 90–108.
Wong, K., Parente, J., Prasad, K.V., Ng, D., 1990. Auranofin modulated cytoplasmic freecalcium in neutrophils by mobilizing intracellular calcium and inhibiting proteinkinase. J. Biol. Chem. 265, 21454–21461.
Yamada, R., Sano, H., Hla, T., Hashiramoto, A., Fukui, W., Miyazaki, S., Kohno, M.,Tsubouchi, Y., Kusaka, Y., Kondo, M., 1999. Auranofin inhibits interleukin-1beta-induced transcript of cyclooxygenase-2 on cultured human synoviocytes. Eur. J.Pharmacol. 385, 71–79.
Yamamoto, N., Izumi, Y., Matsuo, T., Wakita, S., Kume, T., Takada-Takatori, Y., Sawada, H.,Akaike, A., 2010. Elevation of heme oxygenase-1 by proteasome inhibition affords do-paminergic neuroprotection. J. Neurosci. Res. 88, 1934–1942.
Yamashita, M., Niki, H., Yamada, M., Watanabe-Kobayashi, M., Mue, S., Ohuchi, K., 1997.Dual effects of auranofin on prostaglandin E2 production by rat peritoneal macro-phages. Eur. J. Pharmacol. 325, 221–227.
Zhang, W., Wang, T., Pei, Z., Miller, D.S., Wu, X., Block, M.L., Wilson, B., Zhang,W., Zhou, Y.,Hong, J.S., Zhang, J., 2005. Aggregated alpha-synuclein activates microglia: a processleading to disease progression in Parkinson's disease. FASEB J. 19, 533–542.
Zilka, N., Kazmerova, Z., Jadhav, S., Neradil, P., Madari, A., Obetkova, D., Bugos, O., Novak,M., 2012. Who fans the flames of Alzheimer's disease brains? Misfolded tau on thecrossroad of neurodegenerative and inflammatory pathways. J. Neuroinflammation9, 47.





























