Embed Size (px)
Citation preview

Good Documentation
Practice
Conference Document| 22nd February 2018

Significant share of work on the shop floor is
actually documentation1
Good documentation is critical!
SOURCE: McKinsey warning letter database
• As part of GDP, ALCOA++
principles to be followed:
• Attributable
• Legible
• Contemporaneous
• Original
• Accurate
• Complete
• Consistent
• Enduring
• Available
Documentation accounts for a large share of
observations
72%
28%
Others
Documentation relatedPercentage of
observations in
2016, and 2017
1 Based on observations from 2-3 leading Indian pharmacos
200+
25+
Number of entries in typical
Batch Manufacturing Record
(BMR)
Total number of templates with
Quality Assurance
Number of document changes
or change controls per 100
SOPs
30

Recent observations span the document lifecycle
SOURCE: McKinsey Warning letter database
QA provided analysts blank controlled document forms that are already approved and signed
... not all pages of the batch record used in the production area are stamped and dated when issued.
Failure to control issuance, revision and superseding of documents by maintaining revision histories
Employees completed batch production records days after operations had ended, and failed to maintain original manufacturing data for critical steps
Failure to prevent unauthorized access and provide adequate controls to prevent manipulation and omission of data
Appropriate controls are not exercised over computers or related systems to assure changes in master production and control
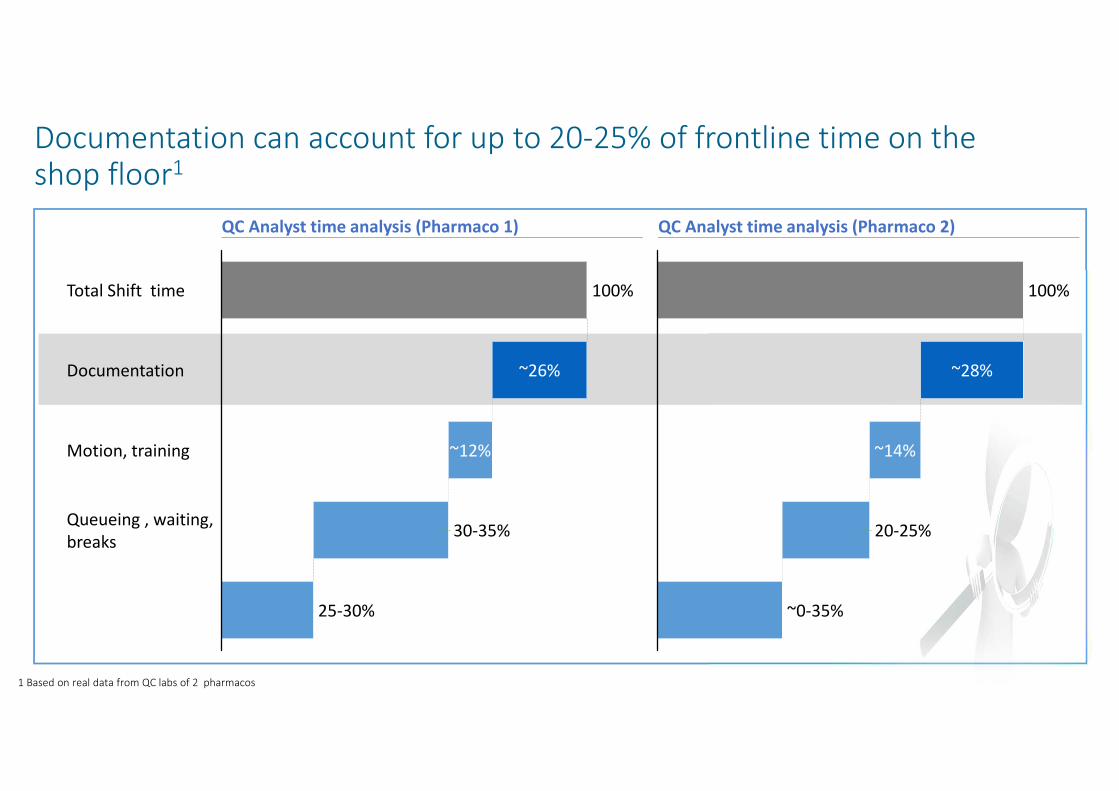
Documentation can account for up to 20-25% of frontline time on the shop floor1
~12%Motion, training
Documentation
Total Shift time 100%
~26%
Queueing , waiting,
breaks 30-35%
25-30%
100%
~14%
~0-35%
~28%
20-25%
QC Analyst time analysis (Pharmaco 1) QC Analyst time analysis (Pharmaco 2)
1 Based on real data from QC labs of 2 pharmacos

Good Documentation can include various aspects…
Error proofing templates
Documentation for Non Oral solid dosage forms
Efficient and effective test record design
Efficient and effective BMR
design
Deep dive on document lifecycle

… However, focus on few common elements is crucial
Set up strong processes across document lifecycle
Simplify and “right size” documentation
Continuous improvement driven by users
Manage the transition to electronic systems effectively
Campaigns to build shop floor awareness an capability
Set up metrics and track improvement
1
2
3
4
5
6
Detailed later

Key design balance to "right size" documentation
Simple
documentation
minimizing chance of
errorsCoverage of critical
quality parameters
2
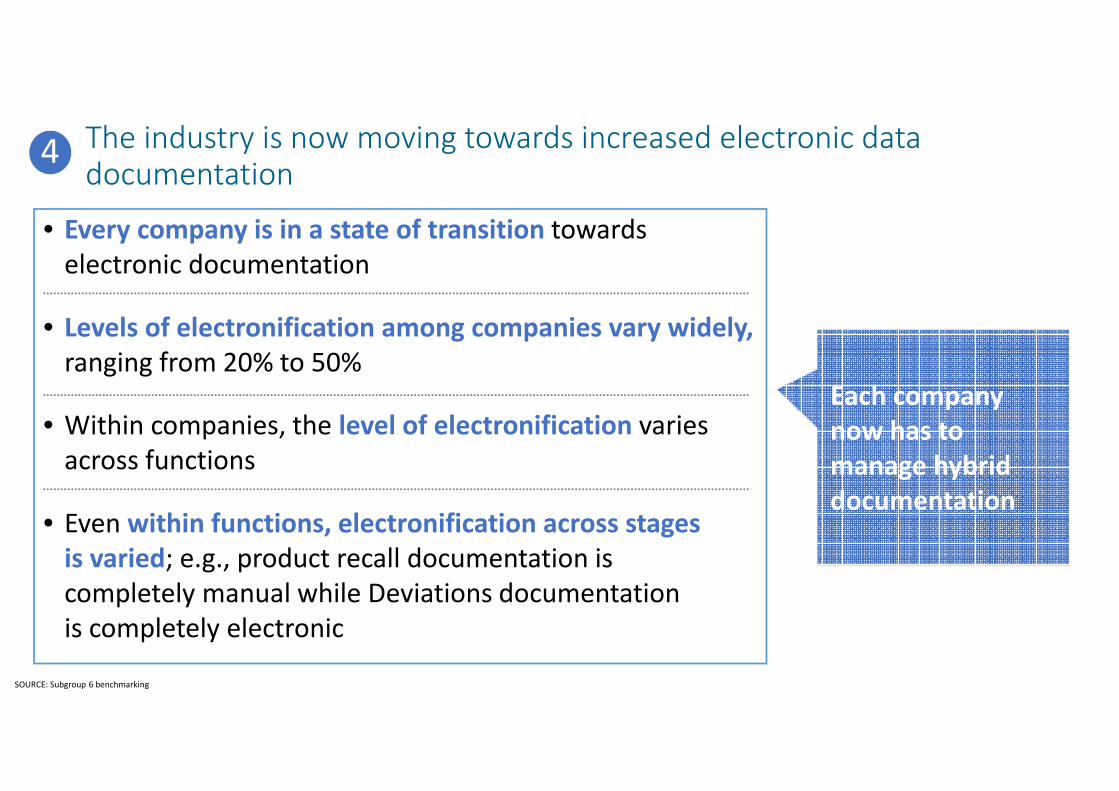
The industry is now moving towards increased electronic data documentation
• Every company is in a state of transition towards
electronic documentation
• Levels of electronification among companies vary widely,
ranging from 20% to 50%
• Within companies, the level of electronification varies
across functions
• Even within functions, electronification across stages
is varied; e.g., product recall documentation is
completely manual while Deviations documentation
is completely electronic
Each company
now has to
manage hybrid
documentation
SOURCE: Subgroup 6 benchmarking
4

Increased electronification can have its benefits… ILLUSTRATIVE
4

But mitigating risks during transition is critical !
Operating system
People system
• Multiple handover points between manual and electronic
• Duplication of process in hybrid state
• No standard approach for migration
• Adoption challenges
• Investment required in terms of resources (new systems/
instruments, etc.), and effort (tweaking an operational SOP)
• Need for capabilities in both, operations, and IT
Management system
• Identification of requirements for the IT system/tool
• Need for effective project management to ensure
on-time migration
4

Campaigns can improve awareness and capability at the shop floor level
Bottom-up document simplification drive
Documentation-themed Gemba walks
Performance management and error tracking
Weekly quizzes on documentation
Daily dialogue discussions structured in a role-playing format
Multi channel reinforcement communication
1
2
3
4
5
6
‘Dos & Don’ts’ on the shop-floor
"Train the trainer" program
7
8
5

Broad questions for the subgroup
• How can we ensure good documentation practices
across different stages of the lifecycle
• How do you manage a hybrid documentation system
• How can we ensure smooth transition to electronic
documentation with minimal risk

Good Documentation Practice guidelinesSub-group 6

IPA QF sub-group 6 focused on Good Documentation practice
Sanjay Shetgar
Vice President, Head Quality Global Generics
Vice President, Formulation Technology Transfer
Rakesh Sheth PanjatcharamM
Head, CMC Quality Operations
Executive Vice President, Global Quality Head
Rajiv Desai
Vice President, Quality
Sanjeev Asgekar
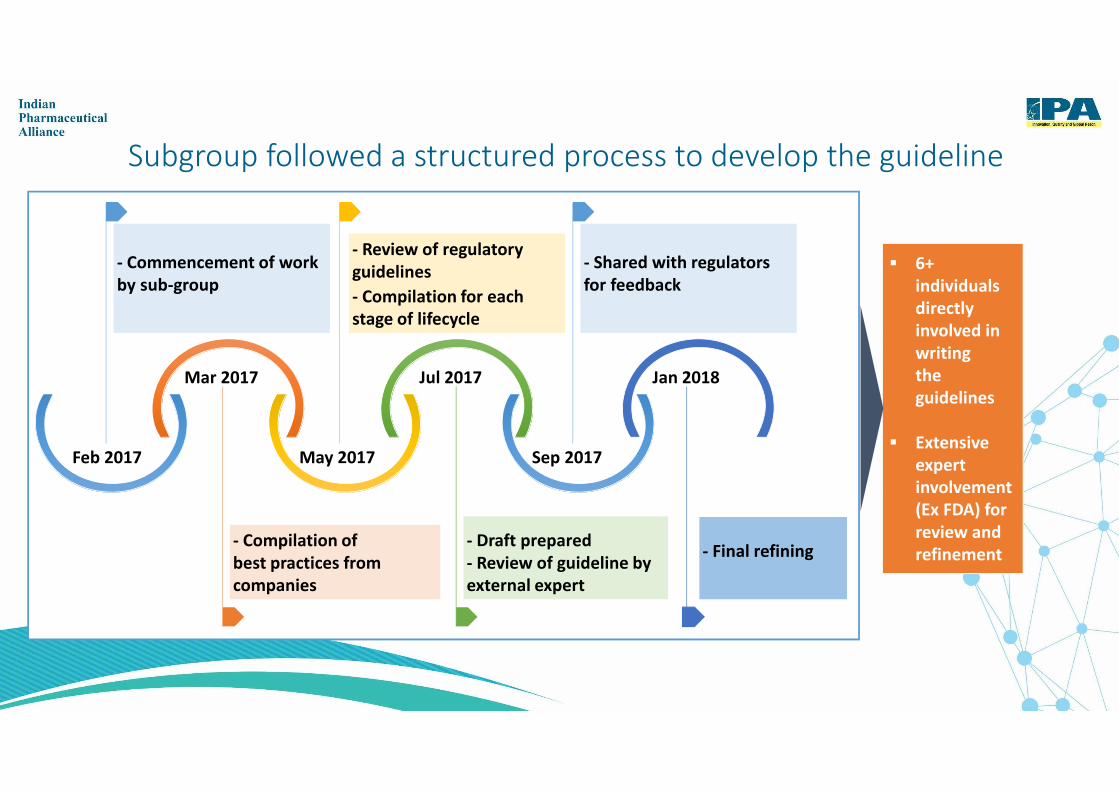
Subgroup followed a structured process to develop the guideline
� 6+
individuals
directly
involved in
writing
the
guidelines
� Extensive
expert
involvement
(Ex FDA) for
review and
refinement
- Commencement of work
by sub-group
- Shared with regulators
for feedback
- Compilation of
best practices from
companies
- Draft prepared
- Review of guideline by
external expert
Sep 2017
Jan 2018
Feb 2017
Mar 2017
May 2017
Jul 2017
- Final refining
- Review of regulatory
guidelines
- Compilation for each
stage of lifecycle

A comprehensive guideline is designed covering each stage of the document lifecycle
SOURCE: Team analysis
Define controls at each stage from generation to destruction
Document issue
Review and ApprovalDesign/generation
Change
Mgmt.
Storage Review and reconciliation
Record-ing / data capture
Destruction
Guidance for Good Documentation Practice
▪ Clarifies application of data management procedures throughout the lifecycle of the document – includes preparation, recording and correction of data and maintenance of records
▪ Provides guidance for best practices for both manual and electronic documentation
▪ Provides guidance on shifting from manual to electronic documentation
1 2 3 4
5678
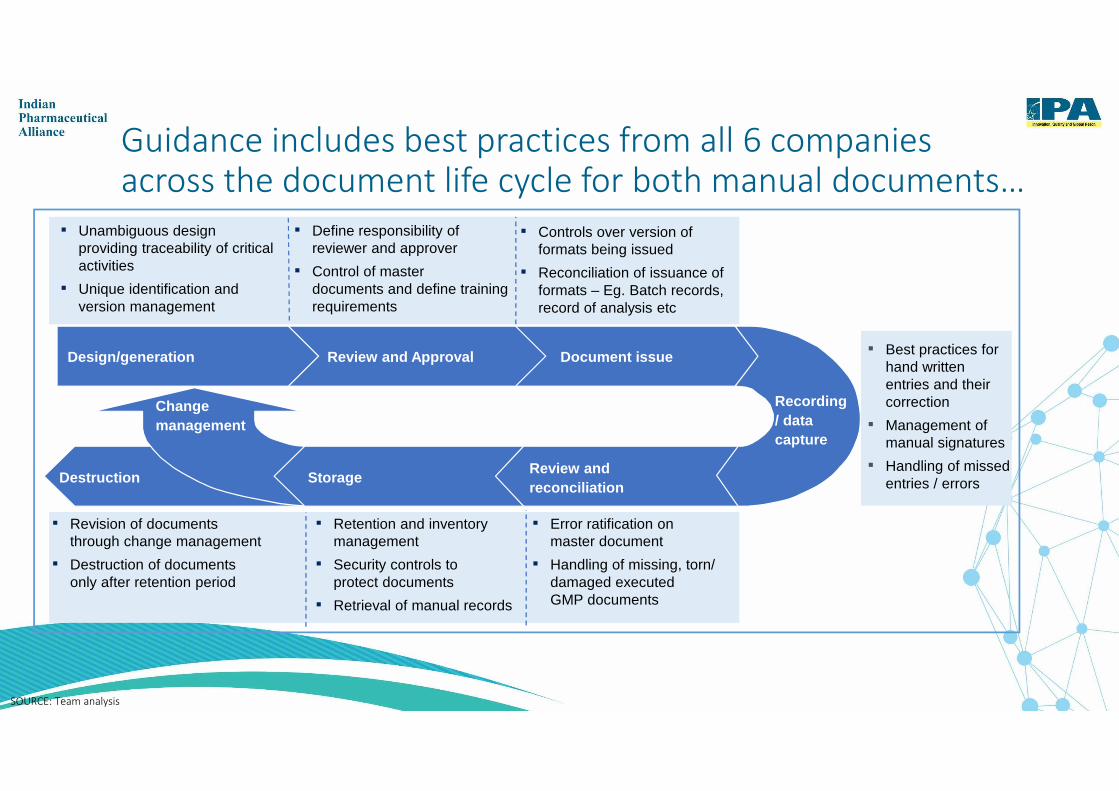
Guidance includes best practices from all 6 companies across the document life cycle for both manual documents…
SOURCE: Team analysis
▪ Best practices for hand written entries and their correction
▪ Management of manual signatures
▪ Handling of missed entries / errors
Document issueReview and ApprovalDesign/generation
Change management
StorageReview and reconciliation
Recording / data capture
Destruction
▪ Unambiguous design providing traceability of critical activities
▪ Unique identification and version management
▪ Define responsibility of reviewer and approver
▪ Control of master documents and define training requirements
▪ Controls over version of formats being issued
▪ Reconciliation of issuance of formats – Eg. Batch records, record of analysis etc
▪ Revision of documents through change management
▪ Destruction of documents only after retention period
▪ Retention and inventory management
▪ Security controls to protect documents
▪ Retrieval of manual records
▪ Error ratification on master document
▪ Handling of missing, torn/ damaged executed GMP documents
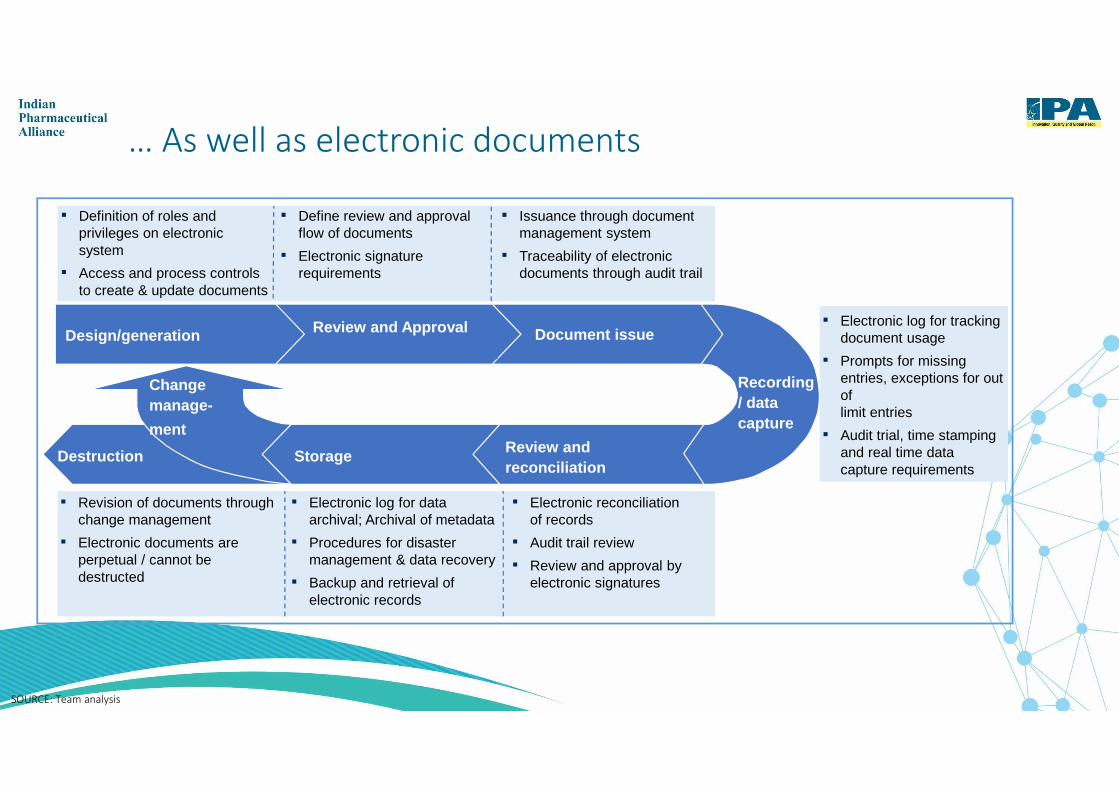
… As well as electronic documents
SOURCE: Team analysis
Document issueReview and ApprovalDesign/generation
Change manage-
ment
Storage Review and reconciliation
Recording / data capture
Destruction
▪ Definition of roles and privileges on electronic system
▪ Access and process controls to create & update documents
▪ Define review and approval flow of documents
▪ Electronic signature requirements
▪ Issuance through document management system
▪ Traceability of electronic documents through audit trail
▪ Electronic log for tracking document usage
▪ Prompts for missing entries, exceptions for out of limit entries
▪ Audit trial, time stamping and real time data capture requirements
▪ Revision of documents through change management
▪ Electronic documents are perpetual / cannot be destructed
▪ Electronic log for data archival; Archival of metadata
▪ Procedures for disaster management & data recovery
▪ Backup and retrieval of electronic records
▪ Electronic reconciliation of records
▪ Audit trail review
▪ Review and approval by electronic signatures

Design / Generation: Illustrative points1
Manual Documentation
1.1 All documents must be accurate and written in a manner that prevents errors and ensures consistency
1.2 Documents shall have unambiguous contents; title, nature and purpose shall be clearly stated
…
1.8 All documents shall have the signature and date of the person who prepared, reviewed and approved the document
1.9 All documents shall have an effective date and review period if applicable
…
1.15 The use of temporary recording practices, Eg. Scraps of paper, shall be prohibited
Electronic Documentation
1.1 Appropriate roles shall be defined with relevant privileges on the electronic system to ensure there is no overlap of roles
1.2 Access to the master template shall be controlled
1.3 Access control shall be provided either through biometric or two level controls (unique username and password)
…
1.9 For activities where an electronic document is generated in addition to a paper document, it shall be determined in advance whether the electronic record or paper record is used in any decision making process
SOURCE: Good Documentation Practice guideline

Review and Approval: Illustrative points
SOURCE: Good Documentation Practice guideline
2
Manual Documentation
2.1 Review of GMP documents
2.1.1 Documents within Quality Management System shall be regularly reviewed and kept up to date
…
2.1.5 Unsigned or incomplete documents or records shall not be used to perform any task or considered as evidence of a completed task
2.2 Approval of GMP documents
2.2.1 Document shall be approved, signed and dated by the appropriate responsible persons. No document shall be changed without authorization and approval
…
2.2.4 All GMP documents shall be approved by QA
Electronic Documentation
2.1 Review of GMP electronic documents
2.1.1 For data generated from a computerized system, regular review of audit trails shall be conducted to identify incorrect processing of data and prevent incorrect results from being reported. It shall be ensured that both administrative audit trail and business workflow related audit trail are reviewed for each system/application.
2.2 Approval of GMP electronic documents
…
2.2.4 Approval with electronic signatures should have appropriate date/time stamps
2.3 Signing of electronic GMP documents
…
2.3.11 A hybrid approach may be used to sign electronic records when the system lacks features for electronic signatures

Issuance: Illustrative points
SOURCE: Good Documentation Practice guideline
3
Manual Documentation
3.1 The formats or records associated with the activity shall be part of respective SOPs.
3.2 Master copies of controlled documents (paper-based and electronic) must be stored in a secure manner and accessible only to authorized individuals.
…
3.8 Reconciliation of issued documents shall be performed and recorded in the respective issuance log.
Electronic Documentation
3.1 Issuance of GMP electronic documents
3.1.1 Automated mail communication should reach the users upon document version changes
…
3.1.7 Periodic audit trail/system audit trail reviews must be conducted
3.2 Controls on GMP electronic documents
3.2.1 Master documents should be stored in a manner which prevents unauthorized changes
…
3.2.11 The ability to restore and read the back-up electronic data should be verified as per a predefined schedule.

Recording / data capture: Illustrative points
SOURCE: Good Documentation Practice guideline
4
Manual Documentation
4.1 Recording of data on GMP documents
….
4.1.27 In the case of modifications to a document before final approval, an additional page can be attached to the original document as attachment /annexure with the changes made, which shall be cross-referred to the original/mother document. The concerned department involved in the approval process shall be informed and involved in the signatory cycle.
4.2 Signing of GMP documents
….
4.2.6 (Use of a personal seal to sign documents requires storage of seal in a secure location with access limited only to the assigned individual, or other means of preventing potential misuse
4.3 Handling of missing entries and corrections
4.3.7 The nature of corrections made shall be reviewed to analyze if they can be incorporated on a permanent basis.
Electronic Documentation
4.1 Electronic log must be maintained for document usage. In case of electronic data, the computer systems shall have adequate controls in order to prevent alteration of time/date stamps
4.2 Data shall be entered electronically
4.3 The system shall prompt for error messages for missing data.
4.4 Exceptions shall be generated for out-of-limit results
…
4.15 File and project naming conventions are defined by procedure and should be followed accordingly

Review and Reconciliation: Illustrative points
SOURCE: Good Documentation Practice guideline
5
Manual Documentation
5.1 Error ratification
5.1.1 The errors identified in documents shall be notified to the head of concerned department, who owns/is responsible for the document.
….
5.1.13 Any change in input quantity, numerical value/formula, change in calculation, yield, and addition/deletion of processing step should be routed ONLY through change control procedure.
5.2 Cancellation of GMP documents
….
5.2.2 The data shall be transcribed with proper justification. The voided data shall be cross referenced in the new document by attaching the original document for traceability.
5.3 Missing documents
5.3.1 If any executed record has been lost or not traceable then it shall be handled though a deviation procedure.
Electronic Documentation
5.1 Cancellation of GMP documents
5.1.1 Cancellation of electronic records shall be allowed only in rare cases supported by suitable justification with the approval of Quality Assurance.

Storage and retrieval: Illustrative points
SOURCE: Good Documentation Practice guideline
6
Manual Documentation
6.1 Retention of document
6.1.1 There shall be pre-defined retention periods for different sets of documents
6.1.2 An inventory of documents within the quality management system should be maintained.
6.1.3 It shall be clearly defined as to which record is related to each manufacturing activity and where this record is located.
….
6.1.8 Justification for this shall be documented and shall take into account the requirements for retention of batch documentation; for example, in the case of process validation data, the accompanying raw data shall be retained for a period at least as long as the records for all batches whose release has been supported on the basis of that validation exercise
Electronic Documentation
6.1 Storage of electronic documents
6.1.1 Privileged personnel should have access to the document archival system
…
6.1.8 Reconciliation log shall be maintained for archived data and shall have date/time stamp
6.2 Retention of electronic documents
6.2.1 If an electronic record is copied to another system, the metadata including the audit trail and electronic signature is not required to be copied, but must be retained in the original system. If original system is no longer maintained, the metadata and audit trail and electronic signature must be migrated along with the electronic record. .

Revision: Illustrative points
SOURCE: Good Documentation Practice guideline
7
Manual Documentation Electronic Documentation
7.1 All revised documents shall mention its revision history.
7.2 Periodic reviews of controlled documents and forms shall be done as per approved procedure and shall be handled through a change approval process.
7.3 Only authorized personnel shall revise the documents
7.4 Document revisions shall always be version controlled and only the latest version shall be used at any point in time
…
7.7 All revisions of GMP documents shall be handled through change approval process
7.1 Revision of electronic documents shall have revision history on the document.
7.2 Revision history shall be periodically reviewed and any changes shall be handled through change control procedure with assessment of impact on validation.
7.3 Privileged persons shall revise the documents.
7.4 Document revisions shall be version controlled.
7.5 Obsoleted versions shall be automatically removed after uploading the latest versions in distribution system.

Destruction: Illustrative points
SOURCE: Good Documentation Practice guideline
8
Manual Documentation Electronic Documentation
8.1 Record shall not be destroyed before its stated retention period or validity without appropriate justification and consultation with Quality Assurance.
8.2 Any approved or under approval GMP documents shall not be discarded or destroyed without the appropriate stamp authorisingcancellation/obsolescence.
…
8.4 Labels used at different process levels shall be destroyed by defacing with a cross “X” mark on it (e.g., status label of equipment, cleaning labels, visual inspection status labels, leak testing status label, product quarantine and release labels, etc.).
8.1 Electronic data shall not be destroyed. All electronic data shall be perpetual

This GDP guideline combines inputs from regulatory guidelines and best practices from the 6 companies
SOURCE: Team analysis
▪ Single document which captures controls required across document lifecycle serving as the single source of truth which is usually covered across multiple documents in the industry
▪ The document provides processes and controls for both manual and electronic documentation
▪ It incorporates the best practices from all 6 companies and hence bringing out best of the best from the current industry practices
▪ Provides a forward looking view that documents will be increasingly electronic. Vision for transition from manual to electronic documents is being built into the guideline

The final guideline adds to, and standardizes the existing best practices across individual companies
SOURCE: Team analysis

We also carried out a benchmarking exercise on level of automation in documentation across 6 pharma companies
SOURCE: Team analysis
In the spirit to move towards completely electronic documentation, following documents can be addressed:� QC: OOS, OOT, analysis records (analysis protocols, weighing records etc.)� QA: Customer complaints, Internal audits
From a long term lens, following documents can be converted to fully electronic format:▪ QC: Lab notebook, calibration management and maintenance management▪ QA: Product recall, APQR
Creation Issuance Recording Archival
C1 C2 C3 C4 C5 C1 C2 C3 C4 C5 C1 C2 C3 C4 C5 C1 C2 C3 C4 C5
Quality Control Standard documents (COA, specs, MOA etc.) C C H M M C C M M M M C C M M M C H M M
Analysis records (analysis protocols, weighing records etc.) C C M C M H H M C H H H H C M H M M C H
Lab equipment qualification M M M H M M M M H M M M M H M M M M H M
Lab notebook H M H H M H M H H M H M H H M M M H H M
OOS H C M H C H C M H C H C M H C M C M H C
OOT H C M H C H C M H C H C M H C M C M H C
Sample management C C C M M C C C M M C C C M M C C C M M
Reserve sample management C C C M C C H H M C C M M M C C M M M C
Stability management C C C C H C C M C C C C M C C C C M C C
Calibration management C M M H C C M M H C C C C H C C C C H C
Maintenance management M M M H C M M M H C M M M H C M M M H H
Chromatographic documentation system C C C C C C NA NA C C C C C C C C H H C C
Networking of standalone systems C C C H C C NA NA H C C C C H C C C H H C
Quality Assurance SOPs C H M M C H M M M H C M C H H M
Change control C C C C C C C C C C M C C C C C C C C C
Deviations C C C C C C C C C C H C C C C C C C C C
Investigations C C C H M C C C H M H C C H C C C C H C
Master batch records C M M H H H M M H C M M M H M C M M H M
Master formulation records C M M H H H M M H C M M M H M C M M H M
CAPA C C C H C C C NA H C H C C H C C C H H C
Product recall M M M M M M M M M M M M M M M M M M M M
Customer complaints C C M H C C C M H C H C M H C C C M H C
Internal audit M M M H C M M M H C M M M H C M M M H C
APQR M M M H H M M M H M M M M H H M M M H H
Process validatiion protocols / reports M M M H M M M M H M M M M H M M M M H M
Cleaning validation protocol / Report M M M H M M M M H M M M M H M M M M H M
Department Type of documentationLevel of automation

Going forward, the benchmarking exercise could be utilized for transitioning from manual to electronic documentation
SOURCE: Team analysis
▪ Align internally on which document types should be prioritized for moving to completely electronic systemacross the 6 companies
▪ Conduct gap analysis to identify specific resources and infrastructure required to move to electronic documentation for the immediate and long term documents
▪ Create a roadmap for transition to fully electronic document and an implementation plan





























