Embed Size (px)
Citation preview

Hedgehog signalling and tumour-initiating cells as radioresistance
factors in esophageal adenocarcinoma
by
Jennifer Teichman
A thesis submitted in conformity with the requirements
for the degree of Master of Science
Graduate Department of Medical Biophysics
University of Toronto
© Copyright by Jennifer Teichman (2012)

ii
Hedgehog signalling and tumour-initiating cells as radioresistance factors in esophageal adenocarcinoma
Jennifer Teichman Master of Science, 2012 Medical Biophysics, University of Toronto
Abstract
Clinical management of esophageal adenocarcinoma (EAC) relies on radiation therapy, yet
radioresistance is a pervasive challenge in this disease. The mechanisms of EAC radioresistance
remain largely unknown due to a paucity of validated preclinical models. The present studies
report on the development of seven primary xenograft models established from patient
tumours. These models are used to interrogate the range of radiosensitivities and mechanisms
of radioresistance in EAC tumours. We found that radiation enriches the tumour-initiating cell
population in two xenograft lines tested. Furthermore, three tested xenograft lines respond to
irradiation by upregulating Hedgehog transcripts, a pathway involved in stem cell maintenance
and proliferation. Upregulation occurs in autocrine and paracrine patterns simultaneously,
suggesting that Hedgehog signalling may have a complex role in the radioresponse of EAC
tumours. These findings suggest that inhibiting stem cell pathways in combination with
radiotherapy may have an important role in the clinical management of EAC.

iii
Special Acknowledgements This work was co-supervised by Dr. Geoffrey Liu and Dr. Laurie Ailles. Thank you to Dr. Robert Bristow for his insight and guidance as a supervisory committee member. Thank you to Dr. Richard Hill, Dr. Helen MacKay and Dr. Naz Chaudary for their input on the Hedgehog component of this project. Thank you to Dangxiao Cheng for his help with qRT-PCR, to Joerg Schwock for his histologic evaluation of tissue sections, and to Zhuo Chen, Lorin Dodbiba, Andrew Fleet and Henry Thai for their daily assistance in the laboratory. This work would not have been possible without the collaboration of the University Health Network Tissue Bank and the generosity of the patients who donated their tissues to medical research. This work was supported by an Ontario Graduate Scholarship.

iv
Table of Contents
Ch.1: Introduction and Background............................................................................................ 1 1.1 Epidemiology of esophageal adenocarcinoma (EAC)........................................................ 1 1.2 Etiology of EAC.................................................................................................................. 2 1.3 Clinical management of EAC............................................................................................. 5 1.4 General mechanisms of radioresistance........................................................................... 7 1.5 Mechanisms of radioresistance in EAC............................................................................. 12 1.6 The primary human xenograft as a clinically-reflective model of EAC............................. 13 1.7 The tumour bed effect...................................................................................................... 16 1.8 Cancer stem cell theory.................................................................................................... 18 1.9 TICs and radioresistance................................................................................................... 20 1.10 TICs and EAC................................................................................................................... 23 1.11 Hedgehog signalling pathway......................................................................................... 25 1.12 Hedgehog pathway and EAC.......................................................................................... 29 1.13 Hedgehog pathway and radiation.................................................................................. 32 1.14 Aims and hypotheses..................................................................................................... 35 Ch.2: Primary xenografts and models of radiosensitivity and resistance (Aim 1).................. 38 2.1 Aim 1 Methods................................................................................................................. 38 2.1.1 Patient samples......................................................................................................... 38 2.1.2. Development of xenograft model............................................................................ 39 2.1.3 Precision irradiation: Identification of appropriate radiation doses......................... 40 2.1.4 Assessment of tumour bed effect............................................................................. 40 2.1.5 Xenograft growth delay............................................................................................. 42 2.1.6 Statistical analysis...................................................................................................... 42 2.2 Aim 1 Results..................................................................................................................... 45 2.2.1 A tumour bed effect, if present, is negligible at low radiation doses in our
xenograft model................................................................................................................. 45 2.2.2 Precision irradiation delays xenograft tumour growth............................................. 47 2.2.3 No passage effect on radiation growth delay was detected in our xenograft
models................................................................................................................................ 48 2.2.4 Specific growth delay cannot quantitatively distinguish between xenograft
tumour lines ....................................................................................................................... 49 2.3 Aim 1 Discussion................................................................................................................ 50 Ch. 3: Enrichment of tumourigenic and clonogenic cells through radiotherapy (Aim 2)......... 56 3.1 Methods............................................................................................................................ 56 3.1.1 Limiting dilution assay............................................................................................... 56 3.1.2 Clonogenic assay........................................................................................................ 59 3.2 Aim 2 Results..................................................................................................................... 60 3.2.1 Radiation may enrich the TIC fraction prior to repopulation in some EAC tumours. 60 3.2.2 The ability of radiation to enrich the clonogenic fraction was not demonstrated............. 65 3.3 Aim 2 Discussion.............................................................................................................. 66 Ch. 4: The Hedgehog pathway in response to irradiation......................................................... 69 4.1 Aim 3 Methods.................................................................................................................. 69 4.1.1 PCR primer design..................................................................................................... 69

v
4.1.2 Housekeeping gene selection.................................................................................... 70 4.1.3 Hedgehog gene expression........................................................................................ 71 4.1.4 5E1 validation and toxicity study............................................................................... 73 4.1.5 In vivo Hh inhibition in a xenograft model................................................................. 74 4.1.6 Statistical analysis...................................................................................................... 75 4.2 Aim 3 Results..................................................................................................................... 75 4.2.1 ACTB, HPRT1, HSP90AB1 and YWHAZ are appropriate housekeeping genes for EAC
radiation studies................................................................................................................. 75 4.2.2 Hedgehog expression in EAC xenografts displays a predominantly epithelial-to-
mesenchymal paracrine mechanism (Aim 3a)................................................................... 80 4.2.3 Radiation upregulates both autocrine and paracrine Hh signalling in some EAC
tumours (Aim 3b)............................................................................................................... 83 4.2.4 Single dose 5E1 inhibits stromal Hh activation for up to one week.......................... 89 4.2.5 The ability of 5E1 to radiosensitize EAC xenografts was not demonstrated and
warrants further study....................................................................................................... 89 4.3 Aim 3 Discussion............................................................................................................... 90 4.3.1 Hedgehog signalling follows a predominantly paracrine signalling mechanism in EAC.............................................................................................................. 90 4.3.2 Hedgehog is involved in the radiation response of EAC tumours............................. 91 Ch. 5: Limitations, alternatives and future directions............................................................... 96 Ch. 6: Conclusion......................................................................................................................... 103 Appendix A: Xenograft growth curves for all seven models........................................................ 106 Appendix B: Sample flow cytometric plots of H2K depletion for limiting dilution and
clonogenic assays............................................................................................................... 109 References................................................................................................................................... 110
List of Tables Table 1: Risk factors and level of evidence for ESCC and EAC..................................................... 4 Table 2: Comparison of experimental models............................................................................ 15 Table 3: Primary patient specimens established as xenograft lines........................................... 38 Table 4: Effect of radiation on tumour growth........................................................................... 47 Table 5: Growth delay and specific growth delay by tumour line and passage.......................... 48 Table 6: Linear regression analysis for the effect of passage on SGD......................................... 49 Table 7: Average SGD by line...................................................................................................... 49 Table 8: Clonogenic assays.......................................................................................................... 65 Table 9: PCR primers designed for Hedgehog gene expression analysis with internal controls 70 Table 10: Housekeeping gene radiation stability score............................................................... 78 Table 11: Histologic quantification of percent tumour epithelium in xenograft samples.......... 80
List of Figures Figure 1: Disease progression and molecular alterations in EAC................................................ 5 Figure 2: The Hedgehog pathway............................................................................................... 27 Figure 3: Hh signaling promotes cell survival.............................................................................. 28

vi
Figure 4: Relevant xenograft tumour volume points used for LDAs and clonogenic assays....... 36 Figure 5: Tumour bed effect schematic...................................................................................... 41 Figure 6: There is no significant TBE at low radiation doses in Line 3 passage 3........................ 45 Figure 7: Linear mixed effect model on TBE experiment............................................................ 46 Figure 8: Two representative samples of growth delay derivation using the mixed effect Model.......................................................................................................................................... 48 Figure 9: Specific growth delay by tumour line and passage...................................................... 48 Figure 10: Average SGD for each line.......................................................................................... 49 Figure 11: Spectrum of radiosensitivity among xenograft lines.................................................. 53 Figure 12: Relevant xenograft tumour volume points used for LDAs and clonogenic assays..... 57 Figure 13: Xenograft limiting dilution assay................................................................................ 59 Figure 14: Limiting dilution assay Line 3 passage 4..................................................................... 62 Figure 15: Limiting dilution assay Line 4 passage 5..................................................................... 63 Figure 16: Limiting dilution assay Line 2 passage 10................................................................... 64 Figure 17: Clonogenic assay Line 3.............................................................................................. 66 Figure 18: 5E1 validation............................................................................................................. 74 Figure 19: Housekeeping gene radiation stability in 3 xenograft lines........................................ 77 Figure 20: Selection of best housekeeping gene combination using radiation stability in three tumour lines................................................................................................................................ 79 Figure 21: Localization of Hh transcripts in epithelium versus stroma of untreated tumours from six xenograft lines........................................................................................................................ 82 Figure 22: Autocrine and paracrine Hh signaling displayed in one-colour heat maps................ 83 Figure 23: Gene expression changes in Line 8 passage 4............................................................ 86 Figure 24: Bar graphs of gene expression changes in Line 8 passage 4...................................... 86 Figure 25: Gene expression changes in Line 6 passage 4............................................................ 87 Figure 26: Bar graphs of gene expression change in Line 6 passage 4........................................ 87 Figure 27: Gene expression changes in Line 7 passage 5............................................................ 88 Figure 28: Bar graphs of gene expression changes in Line 7 passage 5...................................... 88 Figure 29: 5E1 failed to radiosensitize xenograft tumours from Line 7 passage 6..................... 90
List of Appendices:
Appendix A: Xenograft growth curves for all seven models....................................................... 106 Appendix B: Sample flow cytometric plots of H2K depletion for limiting dilution and clonogenic assays........................................................................................................................ 109

vii
Glossary of Acronyms AUP- animal use protocol BE- Barrett’s esophagus BE-3- validated esophageal adenocarcinoma cell line CDK- Cyclin-dependent kinase CRT- chemoradiotherapy CSC- cancer stem cell DDSP- DNA damage associated secretory program DHH- Desert hedgehog DISP- Dispatched DNA-PKcs- DNA protein kinase catalytic subunit DSFM- defined serum-free media EAC- esophageal adenocarcinoma ESCC- esophageal squamous cell carcinoma GD- growth delay GEJ- gastroesophageal junction GERD- gastroesophageal reflux disease GLI1-3- Glioma associated oncogene 1-3 Gy- gray HKG- housekeeping gene IHH- Indian hedgehog Lgr5- Leucine-rich-repeat-containing G-protein-coupled receptor 5 NOD/SCID- non-obese diabetic severe combined immunodeficient OE33- validated esophageal adenocarcinoma cell line PTCH1/2- Patched 1/2 qRT- PCR- quantitative reverse transcription real-time polymerase chain reaction ROS- Reactive oxygen species RCT- randomized control trial RT- radiation therapy SGD- specific growth delay SHH- Sonic hedgehog SMO- Smoothened TBE- tumour bed effect TCD50- tumour control dose 50; radiation dose required to control growth in 50% of tumours TD50- cell dose at which 50% injections give rise to tumours TIC- tumour-initiating cell

1
Chapter 1: Introduction and Background
1.1 Epidemiology of esophageal adenocarcinoma (EAC) (All aims)
Esophageal cancer is a deadly malignancy with the eighth highest incidence of all cancers and
the sixth highest mortality rate globally.1,2 The disease is comprised of two main
histopathological types with distinctly different disease mechanisms and epidemiological
patterns: esophageal squamous cell carcinoma (ESCC) and esophageal adenocarcinoma (EAC).
ESCC develops throughout the esophagus, while EAC is found predominantly in the distal third
or at the gastroesophageal junction. ESCC predominantly affects populations in the developing
world, particularly in the region from northern Iran to north central China, appropriately
termed the esophageal cancer belt. Because its risk factors include frequent consumption of
alcohol, tobacco, hot tea, low fruit and vegetable intake and malnutrition, ESCC is associated
with populations of a lower socioeconomic status. In addition, ESCC has a male to female
incidence ratio of 2-3:1. Conversely, esophageal adenocarcinoma (EAC) occurs mostly in the
developed world, particularly among Caucasian males. With risk factors including obesity and
gastroesophageal reflux disease (GERD), EAC is associated with populations of a higher
socioeconomic class, and its male to female ratio is closer to 7:1.3 A pooled analysis found a
strong correlation between smoking and EAC,2 however, unlike in ESCC studies, no significant
association between EAC and alcohol consumption has been found. The prevailing risk factors
for each histological type, and the associated levels of evidence are summarized in Table 1.
While the incidence of ESCC has been decreasing in the western world – due in part to the
declining prevalence of smoking4 – the incidence of EAC has increased by more than 600% over

2
the last three decades.4–7 In fact, the incidence of EAC has grown faster than any other tumour
type in the United States,8 outpacing the next closest cancer by almost three times.9 This
appears to be a real trend rather than overdiagnosis and reclassification of the tumour.5 The
increasing incidence of obesity and GERD in developed countries may be partly responsible for
the increasing incidence of EAC, although this hypothesis is controversial. A recent study
suggests that the increasing incidence of EAC preceded the rise in obesity prevalence by a
decade,10 and a disease simulation model found that increasing obesity may only account for a
small percentage (6.5%) of the rise in EAC incidence.11 In light of these concerning trends, the
project described here has focused exclusively on EAC.
1.2. Etiology of EAC (All aims)
It is largely accepted that EAC develops through a metaplasia-dysplasia-carcinoma sequence
that most commonly begins with reflux-induced Barrett’s esophagus (BE). Metaplastic and
dysplastic epithelia frequently present side-by-side in pathologic specimens, and endoscopically
surveyed patients have been observed to progress from metaplasia to low-grade dysplasia to
high-grade dysplasia and finally to invasive carcinoma.12 Acid reflux at the gastroesophageal
junction is associated with decreased lower esophageal sphincter pressure. Chronic exposure to
bile acids results in a change in the lining of the distal esophagus from normal stratified
squamous mucosa to a more injury-resistant mucin-secreting mucosa that may contain goblet
cells— histologically defined as specialized intestinal metaplasia13,14— and endoscopically
diagnosed as BE. It has been proposed that metaplasia results from changes in the
differentiation pattern of stem cells residing in the basal layer of the esophagus.15–18 Others

3
have proposed that differentiated squamous cells can convert directly into columnar cells
through a process termed transdifferentiation.19,20
Inflammation may play a critical role in the progression from metaplasia to dysplasia and
ultimately to adenocarcinoma, particularly through the production of reactive oxygen species
(ROS) that damage DNA, proteins and lipids. GERD can cause reflux esophagitis, and inflamed
Barrett’s metaplasia expresses the pro-inflammatory cytokines IL-1B, IL-8, and NF-κB, a
transcription factor involved in regulating pro-inflammatory genes.21,22 Animal models of reflux
esophagitis, BE and EAC have shown elevated levels of ROS,23 and biopsies of inflamed
esophageal squamous and Barrett’s mucosae show higher levels of ROS and lipid peroxidation
than uninflamed control tissues.24 Finally, in vitro studies of transformed and primary BE and
EAC cells demonstrate that exposure to low pH induces higher levels of ROS and DNA double
strand breaks.25 Figure 1 illustrates the disease progression from GERD to EAC.

4
Subtype Risk factor Reference
(PMID) Study type Level of risk (OR, RR or HR)
ESCC
Alcohol consumption
19828467 Prospective cohort study ≥ 30g ethanol/day: RR 4.61 (95% CI 2.24,
9.50)
21430021 Meta-analysis of 8 cohort and
case-control studies RR 3.36 (95% CI, 1.66–6.78)
21190191 Meta-analysis of 40 case-
control and 13 cohort studies
Light drinking: RR 1.31 (95% CI 1.10–1.57) Moderate drinking: RR 2.27 (95% CI 1.89–
2.72) Heavy drinking: RR 4.89 (95% CI 3.84–6.23)
Tobacco use 22131340 Meta-analysis of 4 cohort and
9 case-control studies
Ever vs. never smokers: RR 3.01 (95% CI 2.30-3.94). Current vs. never smokers: RR 3.73 (95%CI 2.16-6.43). Former vs. never smokers: RR 2.21 (95%CI 1.60-3.06)
Hot tea consumption
11058886 Meta-analysis of 5 hospital-based case-control studies
OR 4.14 (95%CI 2.24-7.67)
Diet (fruit and
vegetables) 18537156 Population-based case-control
No significant increase in risk after adjustment for other food groups
EAC
Obesity
16702363 Meta-analysis of 3 cohort and
case-control studies Males: OR 2.4 (95% CI 1.9-3.2)
Females: OR 2.1 (95%CI 1.4-3.2)
16061918 Meta-analysis of 7 population-
based case-control studies OR 2.78 (95% CI 1.850, 4.164)
18268119 Nested case-control study using abdominal obesity
rather than BMI
BMI-adjusted OR 4.78 (95% CI 1.14-20.11). Note: no association found with
ESCC
GERD
17461453 Population-based case-control OR 3.48 (95% CI 2.25-5.41)
20955441 Meta-analysis of 5
retrospective case-control studies
Weekly symptoms: OR 4.92 (95% CI 3.90, 6.22)
Daily symptoms: OR 7.40 (95% CI 4.94, 11.1)
10080844 Population-based case-control OR 7.7 (9% CI 5.3, 11.4)
Barrett’s esophagus
21995385
Population-based cohort
Without dysplasia: 1.0 case per 1000 person-years
With low-grade dysplasia: 5.1 cases per 1000 person-years
Tobacco use 20716718 Meta-analysis of 10
population-based case-control studies and 2 cohort studies
OR 1.96 (95% CI 1.64, 2.34)
Dietary fruit, vegetables
and antioxidants
17461453 Population-based case-control High fruit intake: OR 0.50 (95% CI 0.30-
0.86)
17581269 Meta-analysis of 1 cohort and
9 case-control Vitamin C: OR 0.49 (95%CI 0.39-0.62)
β-carotene: OR 0.46 (95%CI 0.36-0.59)
18537156 Population-based case-control
Vegetable intake: OR 0.86 (95%CI 0.75, 0.99)
Non-citrus fruit: OR 0.73 (95%CI 0.59, 0.90)
Table 1: Risk factors and level of evidence for esophageal squamous cell carcinoma (ESCC) and esophageal adenocarcinoma (EAC). RR= relative risk; OR= odds ratio; HR= hazard ratio

5
Figure 1: Disease progression and molecular alterations in EAC. Adapted from [26]. 26
1.3. Clinical management of EAC (All aims)
Since the early 1990s, treatment regimens for esophageal cancer have incorporated concurrent
chemoradiotherapy (CRT) or chemotherapy and surgical resection when possible. For locally-
advanced unresectable disease, the standard approach includes 50Gy of radiation therapy (RT)
plus 4 courses of combined cisplatin and 5-FU, with the first two courses given concurrently
with RT.27 In a phase III randomized controlled trial, patients treated with this regimen had a
median survival of 12.5 months, compared to 8.9 months in patients treated with radiation
alone. The two-year survival rate in the former group was 38% compared to 10% in the latter
group (P<0.001).28
When surgical resection is possible, treatment modalities include surgery alone, neoadjuvant
chemotherapy or CRT, and adjuvant CRT.27 With surgery alone, median survival ranges
between 13 and 19 months and five-year survival rates are between 15% and 24%.29 At least
nine randomized controlled trials (RCT) have compared neoadjuvant CRT to surgery alone, with

6
mixed results. However, two meta-analyses have shown a significant advantage to neoadjuvant
treatment. The first showed a significant reduction in three-year mortality after neoadjuvant
CRT compared with surgery alone (odds ratio 0.53, P=0.03), as well as more frequent down
staging of the tumour (odds ratio 0.43, P=0.001).30 The second meta-analysis demonstrated
improved three-year survival with neoadjuvant CRT compared to surgery alone (odds ratio 0.45,
P=0.005), but only when RCTs using concurrent rather than sequential CRT were included in the
analysis.31
Neoadjuvant CRT followed by surgery is the current standard of care for patients with locally
advanced resectable esophageal cancer, but only 20-25% of patients achieve a complete
pathologic response.32,33 Five-year survival rates remain at or below 20%, due to disease
recurrence and metastasis after therapy.34 In a phase III dose escalation study of esophageal
cancer, a higher radiation dose did not increase the two-year survival rate or local regional
control rate, but was associated with higher normal tissue toxicity and higher mortality.35 The
anatomical location of the esophagus further complicates attempts to increase radiation doses.
Major blood vessels, airways, the heart and lungs are all in close proximity to the esophagus.
Nearly all patients experience treatment related morbidities while few benefit. Thus,
radioresistance is a pervasive problem in esophageal cancer and is a major contributor to
treatment failure and patient suffering.
1.4. General mechanisms of radioresistance (All aims)

7
Many factors contribute to cellular radiosensitivity. In the context of solid tumours, some of
these factors are intrinsic to the cells themselves, such as cell proliferation rate, insensitivity to
apoptosis induction and the efficiency of DNA repair. Other radioresistance elements derive
from benign elements in the tumour microenvironment, including tissue hypoxia, cytokine
secretion and the inflammatory response to radiation. Analyses of epigenetic responses to
genotoxic stress have identified several hundred factors derived from the tumour
microenvironment, a highly-conserved secretory phenomenon termed the DNA Damage
associated Secretory Program (DDSP). The DDSP includes pro-inflammatory cytokines such as
Interleukin (IL)-6 and IL-8, extracellular matrix-altering proteases, angiogenic and growth
factors with documented roles in promoting tumour growth and invasion.36 In the 1950s,
Revesz and colleagues demonstrated that co-injection of lethally irradiated and non-irradiated
tumour cells enhanced tumour growth,37 an effect that was later attributed to factors secreted
from the irradiated cells.38 More recently, growth factors secreted by senescent prostate
fibroblasts were shown to promote prostate epithelial cell proliferation.39
Tumour cell adhesion to benign components of the microenvironment—including stromal cells,
fibronectin, collagens and laminins—may also contribute to the radiation response. Adhesion of
multiple myeloma cells to bone marrow constituents promotes therapeutic resistance through
the redistribution of anti-apoptotic proteins CASP8 and FADD-like apoptosis regulator (FLIP)
from the cytoplasm to the cell membrane, proteosomal degradation of the pro-apoptotic
protein BIM, and upregulation of the cyclin-dependent kinase inhibitor p27.40,41 Thus,
interactions between tumour cells and the microenvironment, whether through physical
contact or secreted factors, influence the radiation response.

8
Several clinical studies, particularly in head and neck cancer have demonstrated unequivocally
that tumour hypoxia has a negative impact on outcome after radiotherapy.42,43 Hypoxia can
regulate radioresistance both directly – through the deprivation of reactive oxygen species—
and indirectly by inducing gene expression changes, post-translational modifications, and by
controlling mRNA translation. Radiation-induced ionizations produce DNA radicals (DNA) that
are oxidized in aerobic conditions. Oxidized DNA (DNA-OO) results in irreversible strand
breaks. Thus, radiation will produce substantially fewer DNA strand breaks and consequently
less cell death in hypoxic compared to normoxic regions within a tumour.
A deeper understanding of indirect hypoxia-mediated radioresistance developed with the
demonstration that hypoxia stimulates angiogenesis and that hypoxia-inducible factor 1 (HIF-1)
is the major transcriptional regulator of this relationship.44 Around the same time, clinical and
preclinical evidence implicated HIF-1 in radiation resistance. Expression of HIF-1 in
oropharyngeal cancer patients was associated with failure to achieve complete remission after
radiation therapy, and HIF-1α null mouse fibroblasts were more radiosensitive than their wild-
type counterparts.45,46 The first link between HIF-1 and radioresistance was provided by Moeller
et al, who showed that irradiation-induced nuclearization of HIF-1 resulted in increased levels
of vascular endothelial growth factor (VEGF) and basic fibroblast growth factor (bFGF). These
growth factors prevent radiation-induced endothelial cell death, a critical factor in the radiation
response.47 Zhang et al subsequently demonstrated that hypoxia promotes radioresistance
among tumour cells by upregulation of mouse double minute-2 (Mdm2) and consequent
suppression of p53 in two cell lines.48

9
Instrinsic cellular factors are equally important in the radiation response. It is well established
that radiation causes both single- and double-strand DNA breaks (dsbs), although the latter is
considered the lethal event. MRE11 senses DNA-dsbs and activates ataxia telangiectasia
mutated (ATM), DNA-dependent protein kinase catalytic subunit (DNA-PKcs), and ataxia
telangiectasia and Rad3 related (ATR) kinase activity.49 These initial activations result in the
phosphorylation and activation of proteins involved in cell cycle arrest and DNA repair,
including p53, BRCA1 and RAD9. DNA repair subsequently occurs through two pathways:
homologous recombination (HR) and non-homologous end joining (NHEJ). HR requires an
undamaged DNA template, and is therefore most efficient in the S and G2 phases of the cell
cycle, when a sister chromatid is present. Repair occurs in several steps. First, the damaged
DNA is processed by 5’ to 3’ nucleolytic resection to create single-stranded 3’ overhangs. RAD51
is recruited to the single-stranded DNA and creates a nucleoprotein filament that searches for a
homologous DNA sequence. The single-stranded DNA invades the template strand, and DNA
polymerase extends the broken sequence from the 3’ end. The same process occurs on the
second 3’ overhang, creating two crossed DNA strands that are resolved to produce two
complete double-stranded molecules.50,51 Unlike HR, NHEJ does not use a complementary
strand to repair DNA, and is therefore more prone to error. Nevertheless, it is critical to cell
survival during the G1 phase of the cell cycle. NHEJ is initiated by Ku proteins that bind to
broken DNA strands. The DNA-dependent protein kinase catalytic subunit (DNA-PKcs) forms a
complex with the Ku proteins to initiate repair of the damaged DNA.52 Severe combined
immunodeficient mice harbour a DNA-PKcs mutation that renders them hypersensitive to
radiation (discussed further in section 1.6). Thus, intrinsic cellular factors such as the amount of

10
initial DNA damage, activation of cell cycle checkpoints and the efficiency of DNA damage repair
may contribute to radiosensitivity.
In a study of 19 tumour cell lines, Chavaudra et al highlighted the role of chromatin architecture
in the induction of DNA damage after radiation. Measuring both chromatin condensation—
which mediates initial DNA damage by shielding DNA strands—and residual double strand
breaks after irradiation, the authors distinguished four groups among their panel of cell lines:
the most resistant were repair-proficient and sustained chromatin condensation after
irradiation. Cell lines that exhibited either condensed chromatin and repair deficiency or
relaxed chromatin and repair proficiency displayed intermediate radiation sensitivity, while the
most radiosensitive lines were repair deficient and had relaxed chromatin.53 Thus, differential
DNA repair efficiencies can account for variations in radiosensitivity among different tumour
cell types.
Variations in DNA damage-independent apoptosis signalling may also affect radiosensitivity. For
example, cellular stress can activate acid sphingomyelinase (asmase), leading to ceramide
release and initiation of apoptosis signalling. Ceramide-induced apoptosis depends on the cell’s
ability to rearrange lipid rafts in the membrane to form macrodomains. Cells deficient in
sphingomyelinases, as well as cells with impaired lipid raft coalescence are more resistant to
radiation than their respective counterparts.54,55
Functionality of cell cycle checkpoints, mediated largely by p53 and p21, as well as the cell cycle
phase, can also influence radiosensitivity. For example, inhibition of serine/threonine-protein

11
kinase CHK1, a checkpoint regulator at the G2-M boundary radiosensitizes p53-deficient human
cells.56 Furthermore, stabilization of p21 by RNPC1 enhances EAC cell radioresistance (discussed
in Section 1.5).57 However, the contributions of p53 and p21 status to radiosensitivity are
complex; both p53 loss of function and p53 gain of function mutations have been associated
with increased radioresistance. Furthermore, some studies demonstrate increased
radioresistance upon p53 loss while others have shown either no effect or increased
radiosensitivity.58,59 Some of this variability may be cell type- and cell context-specific.
Early studies in Chinese hamster cells showed that cells respond differently to radiation
depending on their position in the cell cycle at the time of treatment. Cells are most
radiosensitive in the G2-M phase, less sensitive in the G1 phase, and least sensitive during the
latter part of the S phase due to differential degrees of chromosomal damage and repair
efficiencies in each phase.60,52 However, the relative radiosensitivities of cells in each phase of
the cell cycle varies between cell types and is dependent on the molecular profile of the cells.
For example, BRCA2 is involved in DNA repair by homologous recombination. Mice with a
truncated BRCA2 display a proliferative defect that can be restored with genetic ablation of
p53. This permits the interrogation of BRCA2-mediated cellular radiosensitivity in proliferating
cells. Reasoning that a mouse model with a truncated BRCA2 should increase cellular
radiosensitivity, Tutt et al found that the BRCA2 mutation has little effect on cells irradiated in
quiescence but radiosensitized proliferating S and G2 cells in p53-/- mice.61 Thus, the interplay of
molecular profile and cell cycle distribution is a critical determinant of intrinsic cellular
radiosensitivity, particularly for proteins with cell-cycle specific functions like BRCA2.

12
1.5. Mechanisms of radioresistance in EAC (All aims)
The mechanisms of resistance in EAC are poorly understood, due partly to a paucity of
radioresistance models in this disease. Furthermore, the recent discovery that three commonly
used esophageal adenocarcinoma cell lines are in fact derived from other cancers has
undermined a significant amount of previously reported data.62 Through chronic exposure of a
validated EAC cell line to ionizing radiation, Lynam-Lennon et al developed an isogenic model of
radioresistance, and demonstrated that radioresistant cells had an enriched capacity to repair
damaged DNA, compared to parental cells.63 However, while useful for probing the precise
molecular responses to radiation, such isogenic models deliberately discount the clinically-
observed heterogeneity in tumour radiosensitivity. Hötte et al found that RNPC1, which
stabilizes p21 and is upregulated in therapeutically-resistant patient tumours, enhanced
radioresistance in three EAC cell lines through p21 stabilization and resultant G0/G1 cell cycle
accumulation.57 However, the reported G0/G1 accumulation was modest (5%), and did not
entirely account for the observed radioresistance. In addition, the authors probed a single time
point (72 hours) after radiation; it is likely that radioresistance is a dynamic phenomenon
requiring more frequent observations. TGF-β may also be an endogenous radiation-induced
resistance factor in EAC. Kim et al found that TGF-β was upregulated by irradiated OE33 cells (a
validated EAC cell line), and could confer radioresistance to otherwise radiosensitive cells in
vitro.64 Taken together, these studies suggest that multiple mechanisms may be responsible for
the radioresistance of EAC cell lines, but further investigation using numerous primary patient
samples is necessary in order to replicate the clinically-observed heterogeneity in EAC
radioresistance. Furthermore, studies that approach radioresistance through characteristics of

13
distinct cell populations would provide valuable information missing from in vitro models that
look at bulk changes in the whole cell population.
Each aim of this project addresses these resource and knowledge gaps (see section 1.14 for
Aims and Hypotheses). Aim 1 evaluates the utility of a primary xenograft model for studying
EAC radioresistance. Aim 2 asks whether a distinct population of radioresistant stem- or
progenitor-like cells exist in these primary EAC xenografts, and Aim 3 explores the role of
Hedgehog signalling in conferring radioresistance to EAC tumours.
1.6. The primary human xenograft as a clinically-reflective model of EAC (Aim 1)
Cancer cell lines have historically been the standard for probing tumour biology. However, it is
becoming increasingly clear that long-term culturing of genetically-modified human cells
produces tissue cultures that can bear little resemblance to the original patient tumour. These
changes arise both through in vitro selection for clones that better adapt to tissue culture
plastic, and through genetic mutations acquired from the culturing conditions themselves. For
example, Lee et al demonstrated that glioblastoma stem cells isolated from cell cultures
showed marked histologic, transcriptomic, and genomic differences compared to their
counterparts derived from primary human tumours. Furthermore, these differences were most
pronounced after ten passages, suggesting that long-term culturing in serum produces cell line
models that differ significantly from the original disease.65 Other studies have shown that
mutation of p53 or silencing of the gene encoding the DNA repair protein MGMT occur
frequently in culture.66,67

14
Xenograft models are a valuable alternative to cell culture models. While significantly more
costly and labour-intensive, these in vivo models provide tumour cells with a
microenvironment—including stroma, vasculature and a support matrix—that better reflects
the true tumour environment in a patient. Xenograft models can be derived from cell lines or
from primary patient tumours, however selection pressures during in vitro culturing result in
xenograft tumours with more a homogenous, undifferentiated histology.68 Conversely,
xenografts derived from primary patient tissue appear to retain the morphological and
molecular markers of the original tumours, even after serial passaging.69 Compared to
experimental models using primary tissue directly from the patient, xenograft models permit
the expansion of minute fragments of patient samples. This allows valuable patient specimens
to be studied more extensively, by multiple investigators, and over longer periods of time than
would be possible with only the original tissue.
Xenograft assays can be further distinguished by implantation site. Implantation of tumour
fragments into the subcutaneous space is a relatively straightforward surgical technique.
Tumours established by this method are easy to monitor and appear to maintain the original
tumour biology.69 Inherent weaknesses in this model include the following: involvement of
mouse stroma derived from an organ system distinct from that of the original tumour; poor
engraftment rates due to a comparatively sparse blood supply; a lack of natural metastasis; a
lack of an immune response to tumour formation or therapeutic intervention; and an inability
to study prophylactic therapies since tumours are intentionally implanted.

15
There is evidence to suggest that orthotopic implantation (onto the same organ that gave rise
to the original tumour) provides a tumour microenvironment that more accurately reflects that
of the primary tumour, better supports spontaneous metastasis, and produces higher
engraftment rates.70 However, tumour progression (or regression) is more difficult to assess
orthotopically. Furthermore, orthotopic implantation on certain organs and tissues (such as the
esophagus) is not feasible given the size of the animal organ, among other factors. The
strengths and weaknesses of subcutaneous and orthotopic in vivo tumour models, as well as a
traditional cell culture model are summarized in Table 2.
Subcutaneous xenograft Orthotopic xenograft Cell culture
Easy surgical procedure Need surgical expertise No surgical skill required
Comparatively inexpensive More expensive Most inexpensive
Labour and time economic Labour and time intensive Labour and time economic
Easy to monitor tumour burden and progression
More difficult to monitor tumour burden and progression
Can study single cells
Gene expression is not organ-specific
Organ-specific gene-expression
Lack of tumour microenvironment
Lack of spontaneous metastasis Spontaneous metastasis Risk of contamination from other cultures
Cannot study immune response Cannot study immune response
Lack of cell heterogeneity due to selection by culture conditions
Cannot study prophylactic therapy Cannot study prophylactic therapy
Tendency for genetic mutations to arise in long term culture
Table 2: Comparison of experimental models. Columns 1 and 2 adapted from [68]. 71
To address the need for robust models of EAC, our laboratory has developed primary human
xenograft models using esophagectomy specimens from patients with histologically-confirmed
EAC. This in vivo model provides the materials to study multiple aspects of EAC tumour biology,
most notably radiation resistance and Sonic hedgehog signalling. We have chosen to use a
subcutaneous implantation procedure since orthotopic implantation would result in weight loss
and a reduced animal life span due to obstruction from the growing tumour.

16
We have used non-obese diabetic severe combined immune-deficient (NOD/SCID) mice for this
model. The Prkdcscid mutation in SCID mice is a loss of function mutation in the DNA protein
kinase catalytic subunit (DNA-PKcs). Since the DNA-PKcs repairs DNA double-strand breaks
during V(D)J recombination, SCID mice lack T and B lymphocytes.72 When this mutation is
transferred onto an inbred NOD background, the resultant NOD/SCID strain has no Pre-B or B
cells, non-functional T cells, impaired NK cells, defective macrophages, and low serum
immunoglobulins.73 In essence, NOD/SCID mice have no adaptive immunity and a reduced
innate immunity, making implanted human tumours more likely to engraft. Current work in our
laboratory has focused on characterizing this xenograft model. Preliminary data confirms that
engrafted tumours reflect the original patient tumour both histologically and genomically, even
after 13 passages (unpublished data, personal communication L. Dodbiba).
1.7. The tumour bed effect (Aim 1)
The germ line Prkdcscid mutation and consequent DNA double strand break repair deficiency
renders SCID mice substantially more sensitive to radiation-induced cell death.74 Radiation
damage to the endothelium and connective tissue composing the tumour stroma reduces the
growth rate of subsequently implanted tumours, a phenomenon known as the “tumour bed
effect” (TBE).75 The TBE has proven useful for studies seeking to either mimic tumour
recurrence and metastasis in humans, or to investigate stromal radiosensitivity.76,77 However, it
remains unclear whether a TBE can influence measurements of intrinsic tumour radiosensitivity
when tumours are irradiated in vivo.

17
The degree of influence of the TBE on tumour radioresponse is a controversial topic. In 1993,
Budach et al demonstrated that the doses required for local control in 50% of tumours (TCD50)
transplanted in SCID mice were not significantly different from the matched TCD50 values of
tumours grown in wild-type mice.78 Since then, studies using various measures of
radiosensitivity (growth delay and TCD50) and experimental animal models have challenged
these results. Several reports from Garcia-Barros et al suggest that acid spingomyelinase
(asmase)-mediated endothelial cell damage is a significant determinant of tumour
radioresponse.79,80 In contrast, the group around Leo Gerweck maintains that intrinsic tumour
cell radioresistance is the dominant factor in the overall radiation response. They demonstrated
that the ratio of the radiation-induced growth delays of DNA-PKcs-/- and DNA-PKcs+/+ tumour
cell lines grown in nude mice was equal to the ratio of their intrinsic radiosensitivities measured
by clonogenic survival.81 Taken together, the relative contributions of intrinsic tumour
radiosensitivity and stromal radiosensitivity to the overall radiation response are not clear.
These studies, as well as subsequent investigations will be discussed further in Section 2.3.
A TBE, if present in our xenograft model, would have significant implications for our study. First,
it would confound results on the intrinsic radiosensitivity of patient-derived xenografts, as
measured by specific growth delay. Second, it would undermine our ability to enrich for
radioresistant tumour-initiating cells, since a compromised tumour bed would affect cell
survival in all tumour cells, regardless of tumour-initiating capacity. We therefore sought to
determine the magnitude of the TBE in NOD/SCID mice, as a proof of principle that this strain is
an appropriate model for xenograft irradiation and TIC studies.

18
1.8. Cancer Stem Cell Theory (Aim 2)
The cancer stem cell (CSC) model offers a unique perspective for modelling EAC radioresistance.
According to the traditional clonal evolution model of cancer development, tumours arise as
cells stochastically accumulate mutations that confer growth and survival advantages over
other clones. Each mutation creates further chromosomal instability, predisposing the tissue to
malignant transformation.82,83 By this view, each cell in a tumour contributes equally to tumour
propagation through a common capacity to proliferate, metastasize and to seed a new tumour
in an immunodeficient mouse. In contrast, the CSC model proposes a hierarchical organization
of tumour cells among which only a small population is necessary and sufficient to regenerate
growth in vivo. These cells are distinguishable by phenotype, possessing properties similar to
those of stem cells, including the ability to self-renew, to proliferate extensively and to produce
progeny that differentiate into multiple lineages.84 The first evidence for the presence of CSCs
came from work by Bonnet and Dick, which demonstrated that only a small fraction of acute
myeloid leukemia cells could recapitulate the cancer in immunocompromised mice.85 Cancer
stem-like cells have since been identified in solid tumours including breast 86, brain 87, gastric 88,
hematopoietic 89, pancreatic 90, colon 91, bladder 92, head and neck 93, and lung 94 cancers.
Importantly, CSC theory views these cells as both the drivers of tumorigenesis and the
propagators after chemoradiotherapy, due to their intrinsic chemo- and radioresistance.95 It
should be noted that the clonal evolution and CSC theories are not mutually exclusive; with
accumulating evidence that non-CSCs can spontaneously convert to CSCs,96–98 a hybrid of the
two theories may more accurately reflect tumour biology.

19
Considering the controversy in terminology within the cancer stem cell field, and given the
functional tumourigenic assay used for this project, the term “tumour-initiating cell” (TIC) will
hereafter be used in lieu of “cancer stem cell.” Some exceptions will appear throughout the
text, since it remains unclear whether Hedgehog signalling—while a distinctly stem cell
pathway—is restricted to the TIC niche.
TICs are typically identified using a combination of flow cytometric analysis of cell surface
markers, in vivo tumourigenicity assays and in some cancers, in vitro or ex vivo sphere-forming
assays. While staining for cell surface markers provides information about cell phenotype, it
may over- or underestimate the fraction of cells that possess the biological function in question.
Thus, the hallmark demonstration of stemness is an enhanced ability of a cell type to grow
tumours in immune-compromised mice, and to continue doing so with serial passaging. This
would demonstrate both an ability to give rise to transit-amplifying and differentiated cells, and
to self-renew.
A limiting dilution assay is used to measure the frequency of cells possessing these properties in
a mixed population of cells. Suspensions of cells at discrete dilutions are injected
subcutaneously into mice, and the animals are monitored for tumour formation. At the limiting
dose—that is, the dose at which you have one TIC per injection volume—the probability of
injecting zero, one or greater than one TIC per mouse follows the Poisson distribution. In this
model, the probability of injecting zero cells is equal to the probability of injecting one cell, and
each of these outcomes will occur in 37% of injections at the limiting dose. However, when a
tumour forms, the observer cannot be sure whether this positive signal arose from a single TIC

20
or from more than one TIC. Thus, the fraction of injections at each dose that do not give rise to
tumours is scored, since this is probabilistically equivalent to the fraction of injections that gave
rise to a tumour from a single cell. The dose at which this negative signal occurs 37% of the time
is calculated, thus providing the TIC frequency.
In cancer biology, a clonogenic cell is defined as a neoplastic cell with the capacity to produce a
proliferating colony of descendents (generally >50 cells), and is therefore considered capable of
regrowing a tumour if left intact after treatment.52 A clonogenic assay measures the fraction of
these progenitors in a cell population. Since the stem cell compartment is likely contained
within the larger progenitor population, a clonogenic assay can be used as an indirect measure
of stem cell frequency. Dilute concentrations of cells are plated at an appropriate density such
that, after several days in culture, the observer can be sure each colony arose from a single
clonogenic cell. The clonogenic frequency is calculated as the (# of colonies per well)/(# of cell
seeded per well) x 100. Both of these assays are used to address the question stated in Aim 2—
that is, whether irradiation enriches for TICs and/or clonogenic cells in EAC tumours.
1.9. TICs and radioresistance (Aim 2)
That TICs might represent both the radioresistant and tumourigenic cell population poses
serious challenges to the clinical management of cancer, particularly since novel drug
development is usually judged by macroscopic tumour volume endpoints rather than
eradication of TICs.99 Numerous studies have demonstrated that a higher proportion of TICs
correlates with higher radioresistance.100 In a seminal demonstration of radioresistance in the
tumourigenic cell population, Hill and Milas showed that in a panel of murine tumours, TD50

21
values inversely correlated with TCD50. That is, the tumours with a higher tumourigenic
capacity required the highest doses of radiation in order to control tumour growth.101 Thus, the
number of TICs per tumour is an important determinant of tumour control after irradiation.
The advent of surface marker-based methods of cell sorting has facilitated deeper exploration
of TIC radioresistance mechanisms. Findings in the field are controversial and at times
contradictory, indicating a need for further investigation. Glioblastoma stem cells (GSCs),
characterized by expression of CD133, are less sensitive to radiation-induced apoptosis
compared to CD133- cells, and are enriched after irradiation both in culture and in mice. CD133+
cells preferentially activated checkpoint proteins in response to radiation-induced DNA
damage, and were radiosensitized by inhibition of Chk1 and Chk2 checkpoint kinases. In fact,
these cells had higher baseline levels of phosphorylated Rad17, suggesting that GSCs are
primed to respond to DNA damage. Additionally, CD133+ cells had an enriched capacity to
repair damaged DNA, measured by comet assay and phosphorylated histone 2AX nuclear
foci.102 However, the authors of this study did not report the absolute number of double strand
breaks, and did not perform clonogenic assays on the same cells used in the double strand
break analysis.
Nevertheless, in two follow-up papers the same group provided a potential mechanism for
augmented checkpoint activation in GSCs. The neuronal adhesion molecule L1CAM is
overexpressed in GSCs and increases the expression of NBS1, a critical component of the MRN
complex. Thus, L1CAM may bolster radioresistance by upregulating MRN-ATM-CHK2 signalling
in the DNA damage response of GSCs. RNA interference against L1CAM in gliomasphere

22
cultures and in vivo models suggested that this mechanism of radioresistance was restricted to
CD133+ GSCs.103,104
Building on the established principle that radiation-induced cell death is mediated by free
radicals, Diehn et al demonstrated higher levels of antioxidants and hence lower levels of
reactive oxygen species (ROS) in CD44+/CD24-/lowLin- breast TICs compared to non-TICs. In
particular, genes involved in the synthesis of the cellular reducing agent glutathione were
significantly over-expressed in a subset of TICs compared to non-TICs. Their second, albeit
controversial finding of fewer radiation-induced DNA strand breaks in breast TICs suggests that
enhanced ROS defences in TICs prevent extensive DNA damage after irradiation.105 Similar
results were obtained in a separate study by Phillips et al, which showed that while ROS levels
increased in both monolayer and mammosphere cell cultures, a smaller increase was seen in
the latter. In addition, the radiation-induced H2AX foci seen in monolayer cultures was absent
in mammosphere cultures.106 However, a recent report by Karimi-Busheri et al demonstrated
that breast TICs utilize a H2AX-independent pathway for double-strand break repair, suggesting
that the significance of this mechanism in breast TIC radioresistance had been overstated. In
addition to validating previous observations of lower ROS levels and more rapid single strand
break repair, this group also showed that downregulation of the senescence pathway through
increased telomerase activity contributed to breast TIC radioresistance.107
Taken together, these studies illustrate that TICs possess intrinsic radioresistance mechanisms
distinct from non-TICs. These mechanisms likely involve altered ROS levels and altered DNA
damage signalling with consequences for cell death pathways. However, the precise

23
mechanisms, as well as their relative contributions to overall radioresistance require further
investigations.
1.10. TICs and EAC (All aims)
To date, no study has identified bona fide TICs in esophageal cancer, and hence the
radioresistance mechanisms of this putative cell population are unknown. Once again, a lack of
validated EAC models is partly responsible, however the methods used to identify this rare cell
population may be more at fault: recent studies have produced conflicting results on whether
EAC tumours express common TIC surface markers.108,109 Thus, a surface marker approach to
identifying and isolating TICs may not be appropriate in this cancer. Aims 1 and 2 address this
methodological deficiency with a procedure for identifying tumourigenic and clonogenic cells
harvested directly from validated in vivo models of EAC.
Citing similarities in carcinogenesis among gastrointestinal cancers, particularly in the role of
chronic inflammation, several groups have used intestinal stem cell markers to track the
pathologic progression from BE to EAC. In human esophageal specimens, Musashi-1 shows
progressively increasing levels of expression from normal squamous epithelium to Barrett’s
metaplasia and dysplasia, with highest expression levels in early stage adenocarcinoma.110 In
other human specimens, the putative gastrointestinal stem cell marker DCAMKL-1 shows
increasing expression from BE to EAC, with minimal expression in normal squamous esophageal
mucosa.111 The most evidence, however, comes from studies of the intestinal stem cell marker
leucine-rich-repeat-containing G-protein-coupled receptor 5 (Lgr5).112 Lgr5 is expressed in
colon, ovarian and hepatocellular carcinoma,113,114 and is expressed in tumour spheres derived

24
from colon cancer,115 highlighting its potential as a cancer stem cell marker. Two recent
publications reported the ubiquitous expression of Lgr5 in patient samples of BE and EAC. While
the studies contradicted each other on the relative intensity of Lgr5 staining in EAC compared
to BE, both distinguished between high and low expression of Lgr5, and both correlated high
Lgr5 expression with worse patient survival.116,117 Building on these observations, Quante and
colleagues used a transgenic mouse model that over-expressed interleukin-1β to model human
esophagitis, BE and EAC. Their evidence suggested that inflammation may recruit Lgr5+ gastric
cardia progenitor cells into the squamous mouse esophagus, suggesting that BE and EAC arise
from gastric progenitor cells.118
A stem cell model of EAC development is not a recent concept. Early investigators proposed a
gastric or gastric cardia progenitor cell of origin in BE, 119,120 while other recent investigations
point to progenitors in the esophagus,15,121–123 the esophageal submucosal glands,124,125 and the
gastroesophageal junction (GEJ).126 The authors of this last report modeled Barrett’s metaplasia
using p63-deficient mice, which are unable to develop stratified squamous epithelia and quickly
develop a Barrett’s-like metaplasia. The origin of this metaplasia was traced to residual Car4-
expressing embryonic stem cells at the GEJ that opportunistically migrate into the esophagus in
the absence of squamous epithelia. Taken together, these studies provide evidence that EAC
may arise via aberrant activity in stem cell pathways, irrespective of the origins of these
progenitors. Aim 3 extends this reasoning to therapeutic resistance in EAC by asking whether
the expression of a stem cell pathway—with an established role in gastrointestinal
development—is associated with EAC radioresistance.

25
1.11 Hedgehog Signalling Pathway (Aim 3)
The hypothesis that EAC is initiated and propagated by a subset of stem-like cells is further
supported by the observation that the Hedgehog (Hh) signalling pathway, a member of the
stem cell signalling network, is aberrantly activated in BE and EAC. Hh signalling regulates
embryonic development, with key roles in pattern formation and appendage development in
insects, and neural tube differentiation in vertebrates.127 In addition to patterning, Hh signalling
controls cell proliferation and differentiation in stem cells and stem-like progenitors. 128,129 In
adult life, Hh signalling mediates tissue homeostasis 130,131 and repair after injury.132
Mammalian systems have three Hh homologues: Sonic (SHH), Indian (IHH) and Desert Hh
(DHH). Of the three, SHH is the most studied in both developmental and pathologic contexts. It
is also the predominant ligand found in gastrointestinal development and carcinogenesis. In the
absence of Hh ligand, the 12-pass transmembrane receptor Patched-1 (PTCH1) inhibits
Smoothened (SMO), a 7-pass transmembrane protein with homology to G-protein coupled
receptors (Figure 2). SMO suppression permits the assembly of a cytoplasmic inhibitory
complex including Suppressor of Fused (SUFU), which targets the glioma-associated oncogene
homologue (GLI) family of transcription factors, GLI1, GLI2 and GLI3 for proteolytic cleavage.
Vertebrates have a second isoform of the receptor, PTCH2. While PTCH1 and PTCH2 have
similar affinities for all three ligands, PTCH2 has a decreased ability to inhibit SMO.133
HH ligands are released from the signalling cell through the 12-pass transmembrane protein
Dispatched (DISP). Ligand binding to either PTCH1 or PTCH2 alleviates PTCH-mediated
suppression of SMO, allowing SMO to translocate to the primary cilium. There, it causes the

26
dissociation of the inhibitory complex, permitting the GLI family of transcription factors to
accumulate in their full length forms.134
Regulation of gene transcription by GLI proteins is better understood for the Drosophila
homolog, Cubitus interruptus (Ci). The Ci protein is a composite of positive and negative
regulatory domains. In the absence of Hh signal, Ci is processed into a repressor form, while Hh
signalling stabilizes the full length activator form. The three mammalian GLI isoforms differ
significantly in their homology to Ci and consequently in their transcriptional functions. GLI2
and GLI3 are more closely related to Ci than GLI1, and can act as both transcriptional activators
and repressors.135 However, GLI2 appears to act more potently as an activator, since GLI2 loss-
of-function diminishes SHH-induced target gene expression in mouse embryonic fibroblasts.136
Conversely, GLI3 acts primarily as a transcriptional repressor, although it may also function as a
negative regulator of the pathway by upregulating PTCH1 and Hedgehog Interacting Protein
(HIP), a transmembrane protein that binds each of the three ligands and attenuates the Hh
signal.136 GLI1 is exclusively an activator, but its role in activating the pathway appears to be
less potent than that of GLI2. In fact, GLI1 but not GLI2 is dispensable for murine
development.137 While GLI1 appears to act in concert with GLI2 to activate the pathway, it
predominantly functions as readout of activated signaling.136
Hh target genes are involved in many cell functions, including cell cycle progression,
proliferation, differentiation, stem cell maintenance, epithelial-mesenchymal transitions, cell
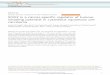
adhesion, signal transduction, angiogenesis and apoptosis138,139 ( Figure2A).
A B

27
Of the cellular processes targeted by Hh signalling, proliferation, self-renewal and survival are
most relevant to this project and warrant elaboration. Cellular proliferation requires cyclin-
dependent kinase (CDK)-mediated progression through cell cycle checkpoints (Figure 2B). CDKs
associate with Cyclins A-E at different points in the cell cycle to promote progression. Cdc25
family members activate CDKs by dephosphorylating inhibitory phosphorylation sites. CDK
inhibitors such as p16, p19 and p27 inhibit the CDK-cyclin complexes.
N-Myc promotes cell cycle progression through p27 downregulation, and FOXM1 does so
through upregulation of Cdc25B and Cyclin B1. Hedgehog signalling promotes cell cycle
progression through GLI-binding to promoter and enhancer regions of the N-Myc, Cyclin D1,
and Cyclin D2 genes. The Cyclin E and FOXM1 genes are indirect GLI targets.139 Thus, Hedgehog
signalling promotes cellular proliferation through upregulation of multiple cell-cycle mediators.
Figure 2.The Hedgehog pathway. (A) Hh signalling cascade. See text for description of pathway. (B) The role of Hh signalling in cell cycle regulation. Adapted from [139].

28
Hedgehog signalling protects cells from apoptosis through
upregulation of anti-apoptotic BCL2, CFLAR and
downregulation of pro-apoptotic BIM, p19, p53, the BH3-
only protein NOXA and FAS proteins139 (Figure 3).
Finally, Hedgehog signalling cross-talks with WNT, RTK,
NOTCH and BMP/TGFβ signalling to regulate stem cell
renewal. These interactions are complex, and will not be
outlined in detail here. Nevertheless, several interactions
are worth highlighting. Bone morphogenetic protein (BMP) signalling is involved in both self-
renewal and lineage commitment of embryonic stem (ES) cells.140 Murine ES cells utilize
autocrine BMP signalling to promote self-renewal by collaborating with LIF-STAT3 to suppress
neural lineage differentiation.141 Similarly, Notch is required for the maintenance of many self-
renewing tissues including the brain 142, blood 143 and the gut.144 Hh signals from epithelial cells
induce mesenchymal BMP4 upregulation through FOXF1 upregulation.145 Furthermore, Hh
signals both positively and negatively mediate Notch signalling. Hh upregulates the Notch
ligand JAG2. It also represses p53, which upregulates the Notch receptor NOTCH1.146
Hedgehog signalling can follow one of three mechanisms: paracrine, reverse paracrine and
autocrine. In paracrine signalling, the ligand-secreting epithelial cell signals locally to the
stroma, which expresses both the receptors and effectors of the pathway. Signal transduction
in the stroma provides a selective growth advantage for the tumour through the upregulation
Figure 3. Hh signalling promotes cell survival. Adapted from [139]

29
of growth-promoting genes. Paracrine signalling has been observed in tumours of the
gastrointestinal tract, including pancreatic, esophageal and colon cancer.147–149 In reverse
paracrine signalling, seen in B cell lymphoma,150 bone marrow- and splenic stroma-derived
ligands activate the pathway in receptor-expressing tumour cells. In autocrine signalling,
tumours synthesize and respond to their own ligands. This mechanism has been observed in
small cell lung cancer and in some cancers of the digestive tract.151,152
1.12. Hedgehog pathway and EAC (Aim 3)
Hedgehog signalling between the endoderm and mesoderm is critical for the development of
the esophagus from the endodermally-derived gut.148 Intestinal columnar epithelium such as
that lining the early esophagus is maintained by SHH signalling.153,154 Squamous epithelium
does not appear in the esophagus until Hh signalling is downregulated.154 Berman et al found
endogenous overexpression of SHH and IHH in OE33, an esophageal adenocarcinoma cell line.
Pathway activity was induced by ligand overexpression rather than mutation, suggesting that
reactivation of an embryonic pathway is involved in carcinogenesis.152 Interestingly, the results
from this study suggested that Hh signalling in EAC followed an autocrine mechanism, with
ligands, receptor and effectors all expressed in the EAC cell line used.
A subsequent study by Ma et al found elevated levels of Hh pathway transcripts and proteins in
four of four primary human esophageal adenocarcinoma specimens, compared to matched
normal esophageal epithelium. Using a combination of in situ hybridization and
immunohistochemistry, they found that SHH was restricted to the tumour cells while PTCH1
protein was detected in both the tumour and stroma.155 This suggested that a paracrine

30
signalling mechanism similar to that found in esophageal development could mediate
epithelial-mesenchymal interactions during tumorigenesis. Furthermore, epithelial PTCH1
expression indicated that autocrine and paracrine signalling could occur simultaneously. In a
separate study, Wang and colleagues looked at tissue microarrays of esophagectomy
specimens representing normal squamous epithelium, BE, BE with low- to high-grade dysplasia
and adenocarcinoma. Immunohistochemistry did not detect SHH in normal squamous
epithelium but found ubiquitous expression of SHH in BE and EAC. Using a mouse
esophagojejunostomy model, this group further demonstrated that exposure to bile reflux
resulted in marked upregulation of Hh ligands in the mouse esophagus, accompanied by
stromal expression of PTCH1 near the resulting intestinal metaplasia.156
Most recently, Yang et al compared Hh staining in 174 primary human esophageal specimens
encompassing ESCC, EAC and their respective precursor lesions, squamous dysplasia and BE.
PTCH1 was expressed in 96% of human EAC specimens, compared to 38% of ESCC specimens.
21% of dysplastic lesions were positive for PTCH1, and these positive signals were restricted to
tissues with severe dysplasia or carcinoma in situ. In contrast, PTCH1 was detected in 58% of BE
tissues, with similar frequencies in tissues with and without dysplasia. Thus, while Hh signalling
appears to promote the formation of carcinoma in situ in ESCC, pathway activation may be
among the earliest events in the pathological progression to BE and ultimately EAC.157 Taken
together, the evidence suggests that EAC carcinogenesis may be modeled as development gone
awry. That is, exposure to chronic acid reflux and inflammation may reactivate embryonic
pathways in stem cell populations, giving rise to a more resistant albeit genetically unstable
epithelium.

31
While these studies unequivocally demonstrate reactivation of Hh signalling in EAC
carcinogenesis, it remains unclear whether pathway activity occurs in an autocrine or paracrine
pattern. Furthermore, of those studies performed in primary patient tissues, most have relied
on immunostaining with Hh antibodies. This technique offers a visual representation of the
spatial distribution of Hh signalling in tissue sections, but it suffers from several limitations.
First, it has a limited ability to quantify low levels of antigen expression, a particularly potent
weakness given the often remarkably low expression levels of certain Hh genes. Second, cross-
reactivity of antibodies is a concern, given the substantial sequence homology between SHH
and IHH and between PTCH1 and PTCH2. Most antibodies against Hh ligands are designed
against the highly conserved NH2 terminal 19kDa protein.134 Since ligand and receptor isoforms
may have distinct functions,133 reliable detection of each is desirable. Aim 3a seeks to
complement the evidence provided in these studies by using quantitative real time polymerase
chain reactions (qPCR) of Hh pathway genes in primary xenograft-derived tumour tissue. In
these tumours, malignant epithelium derives from the human patient, while normal
mesenchyme and endothelium—the tumour bed—is supplied by the mouse. Thus, species-
specific qPCR primers are used in Aim 3a to determine which cell types express each Hh
transcript.
1.13. Hedgehog pathway and radiation (Aim 3)
Since the Hh pathway appears to be activated in response to tissue injury, it seems reasonable
that it may be involved in the cellular response to radiation therapy. Several clinical studies
have supported this hypothesis. qRT-PCR analysis of biopsy specimens from cervical cancer
patients undergoing chemoradiotherapy revealed a correlation between up-regulation of SMO

32
and increased risk of locoregional recurrence, supporting a role for Hh signalling in tumour
repopulation after chemoradiotherapy (CRT).158 Additionally, in a large cohort of head and
neck cancer patients treated with radiation alone, increasing GLI1 expression measured by
immunohistochemistry correlated with poorer outcomes in time to disease progression, time to
metastasis and overall survival in a multivariate analysis.159 In a similar study of esophageal
squamous cell carcinoma patients treated with CRT, the absence of nuclear Gli1 staining in pre-
treated surgically excised tumours was associated with overall patient survival, and all patients
with nuclear Gli1 staining had distant lymph node metastases.160 Thus, clinical data from
several tumour sites suggest that Hh signalling promotes cancer regrowth and metastasis after
RT and CRT. However, since access to pretreatment tumour biopsies is scarce, many of these
studies rely on surgically-resected specimens and are consequently limited in their ability to
distinguish baseline Hh expression from therapy-induced pathway activation. A particular
strength of our xenograft model is the ability to probe Hh pathway activity at multiple time
points before and after irradiation.
Other studies have directly implicated Hh signalling in the cellular response to radiation.
Recently, an in vitro study of hepatocellular carcinoma demonstrated a radioprotective effect of
autocrine Hh signalling. SHH ligand, added either as recombinant protein or as a component of
conditioned medium from irradiated and non-irradiated cells protected subsequent cultured
cells from the effects of radiation. SHH antibody neutralization partially blocked
radioprotection, and GLI1 knockdown abolished this effect.161 Exogenous SHH ligand delayed
the disappearance of γH2AX foci after irradiation, and reduced the level of phosphorylated
CHK1 after irradiation. While the mechanisms of a putative Hh-mediated response to radiation

33
are poorly understood, it appears that pathway activity may contribute to radioresistance by
overriding cell cycle checkpoints despite DNA damage.
In response to DNA damage, tumour suppressor protein p53 is covalently modified and
stabilized, resulting in cell cycle arrest and in some cases, apoptosis. Hedgehog signalling after
DNA damage in transformed mouse embryonic fibroblasts (MEFs) inhibits p53 accumulation by
inducing phosphorylation and activation of the E3 ubiquitin-protein ligase MDM2, resulting in
p53 degradation. In addition, constitutively activated SMO augments p53 binding to MDM2.
Thus Hh signalling may promote cellular proliferation after DNA damage by overriding p53-
mediated cell cycle arrest.162
Further evidence that Hh signalling abrogates cell cycle checkpoints was provided by Fernandez
et al using a murine model of medulloblastoma. Radioresistant medulloblastoma cells occupy
the perivascular niche and express Yes-associated protein (YAP), a transcriptional co-activator
and a SHH target. YAP enables cells to progress through the G2/M checkpoint with damaged
DNA by promoting insulin-like growth factor 2 (IGF2) expression and AKT activation, resulting in
ATM-CHK2 inactivation. However, the bulk of these mechanistic studies used YAP-transfected
cells rather than SHH-induced upregulation of YAP, leaving open the possibility of a SHH-
independent role for YAP in the radiation response. Nevertheless, this data supports the
hypothesis that Hh signaling might promote radioresistance through cell cycle checkpoint
abrogation.163
In spite of this evidence, none of these reports directly implicate radiation-induced SHH in

34
cellular radioresistance. Furthermore, little is known about Hh expression as a response
mechanism to radiation in EAC. In a number of experiments, Sims-Mourtada et al showed that
in cell line-derived xenografts treated with chemoradiation, increases in Hh activity
immediately preceded increases in tumour proliferation rates. Furthermore, SHH stimulation
increases cyclin D1 expression and Rb phosphorylation. Hh inhibition decreases Cyclin D1 and
CDK4 expression with a resulting accumulation of cells in the G1 phase of the cell cycle. This
suggests that pathway activity promotes G1-S phase cell cycle transitions by increasing activity
of the cyclin-Rb axis. The implications of this finding are non-trivial, given the well-documented
observation that cells in S phase are more radioresistant compared to cells in G2 and M
phases.60 Finally, Hh blockade reduced the shoulder region of the radiation survival curve,
suggesting that repair of sublethal damage was inhibited. While this last observation was made
on cells from the validated EAC cell line BE-3, it was subsequently discovered that another cell
line used to generate the bulk of data in this study, believed to be derived from an EAC tumour,
was in fact derived from another disease site. While an elegant mechanistic demonstration of
the radioresistance-conferring effect of Hh signalling, these results have yet to be replicated in
validated models of EAC. Aims 3b and 3c seek to fill in these knowledge gaps by measuring
changes in Hh expression at multiple time points after irradiation, and by evaluating the ability
of Hh inhibition to radiosensitive EAC tumours.
1.14. Aims and Hypotheses
The overall objective of this project is to apply the cancer stem cell model to interrogate
multiple contributors to radioresistance in a panel of primary human EAC xenografts. These
factors include heterogeneous TIC frequencies, clonogenic frequencies and Hedgehog pathway
activity.

35
Aim 1 overall: To evaluate our primary xenografts as a model of intrinsic tumour
radiosensitivity and of clinically-observed heterogeneity in radioresistance.
Aim 1: To determine whether the radiation-induced specific growth delay of xenograft tumours
varies more between xenograft lines (primary patient tumours) or between multiple passages
within one xenograft line. We hypothesize that specific growth delay will remain consistent
across multiple passages within a tumour line, but vary across tumour lines, thereby reflecting
both the intrinsic radiosensitivity of each patient tumour and the heterogeneity in EAC tumour
radiosensitivity.
Aim 2 overall: To determine whether radiation therapy enriches for tumourigenic and/or
clonogenic cells in primary patient xenograft tumours.
Aim 2(a): To compare the in vivo tumour TIC frequency between irradiated and non-irradiated
xenograft tumours. Figure 4 is a schematic representation of a typical xenograft growth curve
and response to irradiation. Points A, A’, B and B’ represent the volumes at which limiting
dilution and clonogenic assays are performed. We hypothesize that EAC TICs are more
radioresistant than non-TICs and thus, irradiated tumours will have a higher TIC frequency than
non-irradiated controls after some time delay post-irradiation (see Figure 4: A’ > A). However,
after tumour recovery and repopulation, the TIC frequency will have returned to baseline (non-
irradiated) levels (Figure 4: B’ = B). Importantly, we expect that the TIC frequency will not
change in control tumours (Figure 4: A = B)
Aim 2(b): To compare the ex vivo clonogenic frequency between irradiated and non-irradiated
xenograft tumours. We hypothesize that the clonogenic cell population is enriched for TICs and
is therefore more radioresistant than the non-clonogenic population. Therefore, the clonogenic

36
frequency should be higher in irradiated compared to non-irradiated tumours (Figure 4: A’ > A).
However, we predict that, like the TIC frequency, the clonogenic frequency will return to
baseline levels after tumour repopulation (Figure 4: B’ = B; A = B).
Aim 3 overall: To study the role of Hedgehog pathway activity in the radiation-response of
xenograft tumours.
Aim 3(a): To determine how Hedgehog pathway expression is distributed between the tumour
and stroma in xenograft tumours, using qRT-PCR. Given previously published data using
antibody staining methods, and acknowledging an epithelial to mesenchymal signalling
mechanism during normal esophageal development, we hypothesize that SHH and IHH
transcripts will be detected largely in the human tumour cells, while pathway activation (i.e.
PTCH1 and GLI1) will be detected in the murine stromal cells.
Aim 3(b): To track the expression changes of Hh pathway components with time after radiation.
We hypothesize that ionizing radiation will induce upregulation of Hh pathway proteins prior to
and possibly during repopulation, reflecting a survival response of remaining TICs.
Aim 3(c): To investigate the effects of Hh pathway inhibition, with and without radiation, on the
growth delay of primary EAC cells in vivo. We hypothesize that administration of 5E1, a SHH
Figure 4. Relevant xenograft tumour volume points used for LDAs and clonogenic assays. A= control LDA1; A’= radiation LDA1; B= control LDA2; B’= radiation LDA2.
A
B’ B
A’

37
inhibitor will radiosensitize xenograft tumours and significantly increase the specific growth
delay compared to radiation or inhibitor alone.

38
Chapter 2: Primary xenografts as models of radiosensitivity and resistance (Aim 1)
AIM 1: To evaluate our primary xenografts as a model of intrinsic tumour radiosensitivity and of
clinically-observed heterogeneity in radioresistance.
The background and rationale of Aim 1 are discussed in Sections 1.6 and 1.7.
2.1. Aim 1 Methods
2.1.1. Patient samples
Primary human esophageal adenocarcinoma specimens from patient esophagectomies were
obtained fresh from the University Health Network Tumour Tissue Bank. Specimens were
collected according to the institutional human ethical guidelines. Specimens were used
immediately for xenograft implantation, or else stored overnight at four degrees Celsius until
implantation the following day. Clinical data was also collected, including the patient’s age,
gender, date of diagnosis, date of recurrence, treatment regimen, tumour location and
differentiation grade, disease-free survival, overall survival, and treatment outcome. These data
are summarized in Table 3.
Tissue bank
ID
Xenograft line
Patient tumour
site
Differentiation grade
Patient Gender
Age at diagnosis
Disease stage at
diagnosis
Pre-operative
chemo
Pre-operative
rad
Disease-free
survival (days)
Overall survival (days)
Dead or alive (as
of March 2012)
59046 2 GEJ/ Gastric Cardia
Moderate F 86 2 No No 71 805 Alive
61057 3 GEJ Poor M 77 3 No No 165 312 Alive
60045 4 GEJ Moderate M 46 3 Yes No 403 786 Dead
60516 5 GEJ/Gastric Cardia
Poor F 70 3 No No 83 270 Alive
62325 6 GEJ Moderate M 57 3 No No 242 461 Alive
60745 7 Lower third/distal esophagus
Not available M 51 3 Yes No 107 193 Alive
63862 8 GEJ Moderate M 65 3 Yes Yes 43 258 Alive
Table 3. Primary patient specimens established as xenograft lines

39
2.1.2. Development of xenograft model
Xenograft experiments followed an Animal Use Protocol approved by the University Health
Network Animal Care Committee. Primary tumour samples cut into fragments roughly 2-3 mm
in each dimension were implanted subcutaneously on the abdomen or hind flank of NOD/SCID
mice that were a minimum of 3 weeks old. During surgical implantation, mice were kept under
general anesthesia with isofluorane. Tumours that engrafted were grown to a maximum of
1.5cm in the longest dimension, at which point the mice were sacrificed, the tumours were
excised and small fragments were passaged to a new cohort of mice. Mice were kept in
ventilated cages of five mice each and were given irradiated food and water ad libitum.
Engrafted primary tumours between passages three and 13 were used for xenograft irradiation
experiments. For each experiment, tumour fragments were implanted subcutaneously into the
right hind flank of 30-60 NOD/SCID mice. Once palpable, biweekly tumour measurements were
made using calipers. Tumour size was calculated using the formula for ellipsoid volume:164
Volume = length × width2 × 0.52
When tumours reached an average volume of approximately 400mm3, mice were randomly
sorted into a control (non-irradiated) or irradiated group. In general, at least ten mice were
used per treatment arm. When mice were sacrificed at different time points for a specific
experiment, it was calculated that eight to ten mice would be available for analysis at the end
of the xenograft growth curve, even after accounting for a 5- 10% premature animal death rate.

40
2.1.3. Precision irradiation: Identification of appropriate radiation doses
Radiation dose toxicity was evaluated by delivering 2, 4, 8 or 10Gy of precision radiation to the
hind flank of non-tumour bearing NOD/SCID mice. Irradiated mice were monitored for two
months for signs of radiation injury, including hair loss, erythema, dermatitis, tissue edema,
ulcers, weight loss, impaired motor skills or death. 4Gy was the highest dose that did not
produce any signs of radiation damage and was therefore selected for further xenograft
studies.
During irradiation, mice were restrained in a custom-made plastic jig with the tumour-bearing
leg extended from the abdomen and exposed to the radiation beam. 4Gy of X-rays was
delivered at a dose rate of 3.07 Gy/min using an XRAD 225 kVp precision irradiator, fitted with a
2mm thick copper filter to remove low-energy X-rays. A 2.5cm diameter columnator was used
to direct the radiation beam to the tumour bearing leg. Half of the dose was delivered from
above the tumour, and half from below. Radiation was given under ambient air breathing
conditions without anesthetic.
2.1.4. Assessment of tumour bed effect
Xenograft Line 3 was expanded into 23 NOD/SCID mice at the third passage. Tumour volume
measurements were made biweekly. When tumours reached an average volume of 400mm3,
mice were randomly sorted into three groups: control (non-irradiated), 4Gy or 8Gy of ionizing
radiation. Studies of tumour implantations into pre-irradiated tissue beds have established that
the TBE becomes apparent at a threshold dose of about 5Gy, and maintains a dose dependent
effect on tumour growth rate from 5Gy to 20Gy.165 Thus, 4Gy was chosen from the results of

41
the toxicity study, and 8Gy was chosen because it lay above the 5Gy threshold. Weekly tumour
volume measurements were continuously made until tumours reached the volume limit,
became ulcerated, or until the slopes (i.e. tumour growth rates) of the irradiated groups
stabilized.
A TBE alters the tumour growth rate by damaging normal tissue endothelium and epithelium.
Thus, if present in our xenograft model, a TBE would manifest in the growth curve as a post-
radiation slope (Figure 5, green curve) less than that of the non-irradiated controls (blue curve).
If no TBE occurs, then irradiated tumours will recapitulate the growth rate of the control
tumours (i.e. the slopes will be parallel) after some period of growth delay (a “plateau” phase).
This is graphically represented in Figure 5.
Figure 5. Tumour bed effect schematic.

42
2.1.5. Xenograft growth delay
Volume measurements were continued biweekly following irradiation until tumours reached
the volume endpoint. The specific growth delay was calculated as: (TR-TC)/TC, where TR and TC
are the times taken after irradiation for the control and irradiated tumours to triple their
volumes as measured at the time of radiation. By normalizing the growth delay to the control
tripling time, the specific growth delay can be interpreted as the number of control tumour
tripling times required for irradiated tumours to triple their own volume. In contrast to the
growth delay, the specific growth delay permits comparisons between tumour lines (or
passages) with different intrinsic growth rates.166,167
2.1.6. Statistical analysis
A linear mixed effect repeated measures model was applied to tumour volume measurements.
This model-based approach to testing the effect of radiation accounts for the repeated and
longitudinal nature of tumour measurements. Furthermore, it enables statistical inference on
the effect of irradiation through estimation of model parameters. Finally, this model can
account for outlying tumour growth profiles as well as missing data points arising from
mortality or premature sacrifice of animals. The linear mixed-effects model of longitudinal
tumour volume is formulated as:
TimeTreatmentTimeTreatmentTimeVolume TTTrteatmentTimeTreatmentTime ,2,1*0 *)log(
Here, the dichotomous Treatment covariate is 0 for control tumours and 1 for irradiated
tumours. The discrete Time covariate represents the number of days elapsed since irradiation.

43
The fixed component of this mixed-effects model is represented by the terms; 0 represents
the tumour volume of each group at the time of irradiation; Treatment represents the difference
in the average tumour volume between irradiated and control tumours over the duration of
the experiment; Time represents the rate of tumour volume growth, and
TreatmentTime*
represents the difference in the growth rate between irradiated and control tumours. The
random effects component of the model (i,1 and
i,2 ) captures the observed variation in
tumour volumes of individual mice (as opposed to the treatment group as a whole).
To determine whether a TBE is present in our model, the slopes of the curves for each
treatment (or control) group were compared using the Wald test. The model was first applied
to all data points to measure the overall effect of irradiation on tumour volume. The model was
subsequently applied only to data points in the plateau phase of the growth curve, and then to
all data points during the repopulation phase, in order to assess the effect of radiation on the
slopes during these two respective growth phases.
To determine whether radiation significantly delayed tumour growth, we tested the null
hypothesis that treatment has no effect on tumour growth rate ( 0: *0 TrteatmentTimeH ) using
SAS 9.0 statistical software.
To calculate tumour growth delay, we first obtained a model-based estimate of tumour volume
at the time of radiation, represented by 0V̂ . Tumour volume was assumed to be equivalent for
both groups at the time of irradiation. We then calculated the time taken for the average

44
volume for each treatment group to reach 30V̂ . The growth delay was calculated from the
modeled parameters as:
)ˆˆ(ˆ
)3/1log(||
*
3,3, 00
TreatmentTimeTimeTime
VVolumeControlTreatmentVVolumeRadiationTreatment TimeTimeGD
.
The specific growth delay is given by:
1ˆˆ
ˆ
|*3, 0
TreatmentTimeTime
Time
VVolumeControlTreatmentTime
GDSGD
and the standard error of the SGD is given by:
2
*
***
22
*
)ˆˆ(
)ˆ,ˆ(ˆˆ2)ˆ(ˆ)ˆ(ˆ)(
TreatmentTimeTime
TreatmentTimeTimeTreatmentTimeTimeTreatmentTimeTimeTimeTreatmentTime CovVarVarSGDSE
To test for a trend in SGD across passages (a “passage effect”), a linear regression was fitted to
the SGDs calculated for each passage within a line, and the regression slope was tested for
equivalency with zero ( 0:0 passageH ).
To test for a difference in SGD between xenograft lines, the ANOVA test was applied to the
SGDs of each experiment. The ANOVA test was also applied to the mean SGDs for each
xenograft Line.

45
2.2. Aim 1 Results
2.2.1. A tumour bed effect, if present, is negligible at low radiation doses in our xenograft
model
Figure 6 presents the growth of Line 3 passage 3 tumours that received 0Gy, 4Gy or 8Gy of
radiation. The linear mixed effect model was first applied to all data points in order to measure
the overall effect of irradiation on tumour volume (Figure 7A). A Wald test of the resultant
curves showed that the slopes of each of the treatment groups was significantly different from
that of the control, suggesting that radiation had a significant effect in both groups. However,
the difference in slopes of the 4Gy and 8Gy treatment groups was not significantly different
when tested over the whole xenograft observation period. Similar results were obtained when
the model was applied only to data points within the plateau phase of the growth curve
(between days 38 and 60), indicating that the overall effect of radiation is most pronounced
during this phase.
Figure 6. There is no significant TBE at low radiation doses in Line 3 passage 3
RT
↓
0
500
1000
1500
2000
2500
3000
0 20 40 60 80 100 120
Tum
ou
r vo
lum
e (m
m^3
)
Days since Implantation
Line 3 passage 3: Assay for TBE
Control
4Gy
8Gy
Plateau Repopulation

46
The model was subsequently applied only to data points within the repopulation phase (after
day 60 in the 4Gy and 8Gy arms), in order to evaluate a potential tumour-bed effect (Figure 7B).
Control measurements from Day 40 onwards were used for comparison. None of the pair-wise
comparisons revealed a statistically significant change in slope relative to controls. Thus, no
significant tumour bed effect was observed at either the 4Gy or 8Gy dose in xenograft line 3.
However, these experiments were performed on one xenograft line only, and further
experiments are required to confirm these results in additional lines. Furthermore, it is possible
that our experiments lacked the power to detect subtle changes in slope, i.e. small tumour bed
effects after 8Gy of radiation.
p<0.0001
p = 0.74
p<0.0001
Days since radiation Days since radiation
Figure 7. Linear mixed effect model on TBE experiment. (A) Linear regression applied on all datapoints. There is a significant effect of each radiation dose, but no significant difference between the two doses. (B) Linear regression of datapoints during repopulation phase. There is no significant difference in any of the slopes.
A B
p = 0.89
p = 0.37
p = 0.46
Tum
ou
r vo
lum
e (m
m3 )

47
2.2.2. Precision irradiation delays xenograft tumour growth
In total, 7 xenograft lines were treated with precision irradiation, 6 of which were treated on
multiple passages. The growth curves are presented in Appendix A. Radiation significantly
delayed tumour growth in all 7 lines. Only passage 4 of Line 6 showed a non-significant
response to irradiation. The results of these statistical tests are summarized in Table 4.
Line Passage P value
2 8 0.0065
2 10 0.0005
3 3a <.0001
3 3b 0.0003
3 4 0.041
4 4 <.0001
4 5 <.0001
4 6 <.0001
4 7 <.0001
5 3 0.0156
6 3 0.0448
6 4 0.0911
7 4 <.0001
7 5 <.0001
8 3 <.0001
8 4 <.0001
Table 4. Effect of radiation on tumour growth. P values <0.05 indicate a significant change in slope of irradiated versus non-irradiated tumours using all time points after treatment.

48
2.2.3. No passage effect on radiation growth delay was detected in our xenograft models
Figure 8 presents two examples of the how SGD was obtained from the linear mixed effect
model. Model parameters including slopes and intercepts were obtained from the green and
blue lines. Table 5 lists the GD and SGD for each passage. Figure 9 presents this data graphically.
Line Passage Volume at
radiation GD
(days) SGD
(fold) Standard
error SGD
2 8 386 7.5 0.47 0.19
2 10 336 18.7 1.14 0.43
3 3A 362 26.2 1.21 0.31
3 3B 342 33.5 2.05 0.72
3 4 397 34.7 1.10 0.92
4 4 413 23.7 1.08 0.32
4 5 413 25.8 0.91 0.30
4 6 372 20.8 0.75 0.18
5 3 314 16.4 0.56 0.28
6 3 458 17.6 0.49 0.29
6 4 334 9.7 0.29 0.20
7 4 516 11.1 0.56 0.13
7 5 641 13.7 0.73 0.13
8 3 770 35.5 1.98 0.77
8 4 238 9.2 0.63 0.17
Table 5. Growth delay (GD) and specific growth delay (SGD) by tumour line and passage
Figure 8. Two representative samples of growth delay derivation using the mixed effect model. (A) Tumour growth of control and treated mice from Line 7 passage 5 plotted as tumour volume over time. (1B) Tumour growth plotted as the natural logarithm of tumour volume over time. (2A) Tumour growth of Line 4 passage 6. (2B) Natural logarithm of tumour growth in Line 4 passage 6. The green and blue lines represent the overall growth of control and treated tumours, respectively. The intersection of the blue and green lines is the starting volume at the time of irradiation. The red line is the volume corresponding to 3x the starting volume. The GD is calculated from the time delay between the green and blue lines each intersecting the red line.
1A 1B 2A 2B
0
0.5
1
1.5
2
2.5
3
0 1 2 3 4 5 6 7 8
Spec
ific
gro
wth
del
ay
Passage
Line 2
Line 3
Line 4
Line 6
Line 7
Line 8
3 3a 3b 4 5 6 8 10
Figure 9. Specific growth delay by tumour line and passage

49
We speculated that the passage number of a xenograft experiment might impact the growth
delay (GD) and specific growth delay (SGD). Linear regression of the SGDs across all passages
within each line showed no significant trend with passage (Table 6).
2.2.4. Specific growth delay cannot quantitatively distinguish between xenograft tumour lines
Since there was no significant passage effect on SGD, we calculated an average SGD for each
tumour line (Table 7). These values are plotted in Figure 10. An ANOVA of the average SGD
revealed no significant difference between xenograft lines (p=0.15). Thus, our xenograft model
cannot quantitatively distinguish between patient tumours based on xenograft radiosensitivity.
Figure 10. Average SGD for each line. Line 6 had the shortest SGD, while Line 3 had the longest SGD. Lines 2, 4, 5 and 7 had intermediate SGDs. The average SGD calculated for Line 8 is questionable (discussed in section 3.3)
Table 7. Average SGD by line. The SGD is normalized to the time required for control tumours to triple their original volume. Thus, SGD can be interpreted as the number of control tumour tripling times required for irradiated tumours to grow by the same amount.
Line P value
2 0.261
3 0.402
4 0.284
5 N/A
6 0.456
7 0.373
8 0.204
Line SGD (fold) Standard error
2 0.804 0.237
3 1.454 0.402
4 0.915 0.158
5 0.558 0.282
6 0.389 0.177
7 0.643 0.091
8 1.305 0.393
Table 6. Linear regression analysis for the effect of passage on SGD. None of the tests yielded significance. Thus, H0 cannot be rejected, and it is concluded that passage number does not affect SGD.

50
2.3. Aim 1 Discussion
There has been significant debate over the role of endothelial cell damage in determining
overall tumour radioresponse. To evaluate the suitability of using NOD/SCID xenografts as a
model of radiosensitivity, it was therefore necessary to evaluate the effect of radiation on the
tumour bed.
A study of tumour growth kinetics in xenograft Line 3 suggests that there was no strong tumour
bed effect at low doses of ionizing radiation. There was an apparent reduction in tumour
growth rate during repopulation after an 8Gy dose, however these kinetics were statistically
indistinguishable from those of tumours repopulating after a 4Gy dose. The difference in slope
between these two treatment arms was slight. Nevertheless, experiments were performed on
one xenograft line only, and further investigations in additional lines are required to conclude
that a TBE is absent entirely. Furthermore, it is possible that statistically significant changes in
slope might have been found with the inclusion of more mice in each arm of the study. For
these reasons, 4Gy was used as the standard dose for all subsequent xenograft radiation
studies. No significant change in slope during tumour repopulation was seen in any subsequent
xenograft growth curve (see Appendix A). This provides further evidence that 4Gy was an
appropriate dose for our model.
These results, while performed on a single xenograft line, are supported by published literature.
Originally, Garcia-Barros et al showed that tumours grown in apoptosis-resistant acid
sphingomyelinase (asmase-/-) deficient mice were more radioresistant than the same tumours
grown in wild-type mice, suggesting that microvascular damage was indeed a significant

51
determinant of tumour radioresponse.79 Gerweck et al subsequently challenged this conclusion
by demonstrating that tumours established in nude mice using a DNA-PKcs-/- tumour cell line
were more radiosensitive than tumours established from radioresistant DNA-PKcs+/+ cells. Since
relative radiosensitivity between the isogenic tumours was proportional to their intrinsic
radiosensitivity as measured in vitro, they concluded that intrinsic tumour cell radiosensitivity
was the major determinant of tumour radiation response.81 In a follow-up paper, the same
group reported a growth delay ratio between isogenic tumours in SCID mice that was not equal
to the ratio of their respective intrinsic radiosensitivies, suggesting that stromal damage may
contribute to the radiation response in some models.168 While this appeared to contradict
earlier observations that the TCD50 was not associated with a SCID phenotype,78 it may be that
endothelial cell damage conferred by the SCID mutation increases tumour growth delay but
does not affect tumour cure as measured by TCD50. However, a more recent study by Garcia-
Barros et al failed to show enhanced tumour growth delay in SCID mice compared to
SV129/C57BL/6 wild-type mice for either MCA/129 fibrosarcomas or B16 melanomas, calling
into question a causal relationship between irradiated SCID endothelium and enhanced tumour
growth delay. Building on their earlier results,80 this group maintained that the effect of stromal
damage on tumour response was accounted for by asmase-mediated endothelial cell apoptosis,
rather than impaired DNA damage repair.
Taken together, published findings suggest that macroscopic observations of a putative tumour
bed effect depend both on context (mouse strain, tumour cell type) and on the parameter used
to measure radiation response. If TCD50 is indeed independent of a TBE, we may justifiably use
NOD/SCID mice to measure the TIC frequency, since previous studies have shown a convincing

52
correlation between TCD50 and TIC frequency.101 While the effect of tumour bed radiation
damage on tumour growth delay remains controversial, our study focuses exclusively on a
comparative analysis of primary patient tumours. Thus, if growth delay is prolonged by a TBE,
this effect would likely be similar across all our xenograft models, negating a TBE as a
confounding factor in our comparisons. That is, the magnitude of the growth delay would not
reflect the true values, but the ranking of the models in order of relative radiosensitivity would
remain the same.
We sought to evaluate (a) the in vivo radiosensitivity of our xenograft tumours and (b) the
ability of our xenograft model to distinguish between the intrinsic radiosensitivities of multiple
patient tumours. To evaluate and quantify tumour radioresponse, we first modeled tumour
growth after irradiation as linear growth in the log scale. This estimation does not distinguish
between the plateau and regrowth phases in growth curve. Rather, the control and irradiated
arms share an intercept (volume at the time of radiation) yet differ in their slopes. The
significance of this change in slope was used as an initial test of radiosensitivity. By this method,
all passages showed a response to radiation with the exception of Line 6 passage 4. Passage 3
of the same line was marginally significant (p = 0.045). We therefore concluded that Line 6 was
the most radioresistant relative to other models in our panel.
We next sought to measure the growth delay (GD) and specific growth delay (SGD) for each
passage of each line. We reasoned that SGD measurements would provide both a descriptive
assessment of heterogeneity in our tumour panel and a means to quantify this heterogeneity.
The SGDs ranged from 0.4 to 1.5 fold relative to the control tripling time, with Line 6 and Line 3

53
at the radioresistant and radiosensitive ends of this spectrum, respectively (Figure 11). This
result was in agreement with our previous observation that Line 6 is relatively radioresistant.
Importantly, we noted that the initial tumour volumes (at the time of irradiation) varied across
passages in some lines, and that significant differences in starting volume impacted the GD and
SGD (See Line 8 in Table 5). It is possible that a larger starting volume contributes to a larger GD
by increasing the tripled volume (3V0) above a particular threshold. This threshold may
represent a point at which continued tumour growth is limited by nutrient and oxygen supply.
Since irradiated tumours face more endothelial cell damage than control tumours, this putative
“volume threshold” may be more pronounced in large irradiated tumours compared to
controls. In other words, a 3V0 of approximately 1000mm3 may impose a lesser burden on
irradiated tumours than a 3V0 of 1500mm3 due to differences in levels of hypoxia and nutrients
at the two volume endpoints. Furthermore, a 3V0 above the volume endpoint outlined in our
Animal Use Protocol (AUP) requires the mixed effect model to extrapolate past real data points,
thereby introducing error into the calculated SGD. Taken together, these observations suggest
that reliable quantification of SGD can only be obtained from passages with approximately
Figure 11. Spectrum of radiosensitivity among xenograft lines
Relatively resistant Intermediate Relatively sensitive

54
equal starting volumes. For this reason, the average SGD of Line 8 should be interpreted with
caution.
Figures 9-11 (p. 48-49, 53) portray the range of radiosensitivities among the seven lines used.
However, statistical testing revealed that differences in SGD among xenograft lines were non-
significant. Thus, SGD is not quantitatively capable of distinguishing between patient-derived
tumour lines. The reduced sensitivity of our model to quantitatively distinguish patient
tumours is in part a factor of the numbers of mice used. For economical reasons, mouse
numbers were limited such that the total number of mice remaining at the end of a xenograft
experiment would be approximately ten per group. In some experiments, fewer than ten mice
per group remained due to animal morbidity, tumour burden, or because gene expression
analyses required more mice to be sacrificed than originally estimated. Such cases resulted in
truncated growth curves and missing data points that may have introduced significant error
into the linear regression. With these mouse numbers, growth delays such as those seen in Line
6 were too small to detect statistical significance. For those lines that did show statistically
significant growth delays, limited mouse numbers reduced the ability of the model to
distinguish between the average growth delays of each line.
The methods used to irradiate xenografts may also account for the reduced sensitivity of our
model to discriminate between tumours. Because our second aim was to interrogate TIC
enrichment at a distinct point after irradiation, we chose a single-dose rather than fractionated
treatment regimen, and we used a dose rate of 3.07 Gy/min. Studies have demonstrated that
the use of low-dose rate irradiation (0.01-0.05 Gy/min) in cell lines increases the spread of

55
clonogenic survival measurements, thereby enhancing the ability to detect slight differences in
radiosensitivity.169 Similarly, in a study of five human cell lines, a series of 5 and 6 fractions of
2Gy gave a more precise and reproducible measure of radiosensitivity than a single dose at
2Gy.170 These methods incorporate the influence of cellular repair of sublethal damage into the
radiosensitivity measurements, and are therefore better able to discriminate between
clonogenic survival curves. While it remains unclear whether low-dose rate or fractionated
irradiation can increase the spread of SGD measurements, future efforts may consider
incorporating these considerations into our radiosensitivity model.
Ultimately, the biological characteristics of each patient tumour may manifest in an insufficient
effect size to detect such differences, or the models may indeed all have the same degree of
radiosensitivity. Additional experiments are required to determine whether the effect size
between patient tumours is significant. Despite the resulting lack of statistical significance, our
mathematical model is robustly capable of detecting a radiation-induced growth delay within
each experiment. Thus, while our xenograft model may be less well-suited for studies of patient
heterogeneity (i.e. distinct patient subgroups classified by therapeutic response), it remains a
valuable tool for preclinical studies investigating the effects of radiation on tumour growth both
alone and in combination with other agents. This strength alone renders our model a valuable
contribution to the field of preclinical radiation studies of EAC, since few such models currently
exist. Furthermore, we utilize our model for precisely this purpose in Aim 3, and demonstrate
that Hedgehog inhibitors in combination with radiation may have an important future role in
the clinical management of EAC.

56
Chapter 3: Enrichment of tumourigenic and clonogenic cells through radiotherapy (Aim 2)
3. AIM 2: To determine whether radiation therapy enriches for tumourigenic and/or clonogenic
cells in primary patient xenograft tumours.
The background and rationale of Aim 2 are discussed in Sections 1.8 - 1.10
3.1. Aim 2 Methods
3.1.1. Limiting dilution assay
Limiting dilution assays (LDAs) were performed on control and treated mice in order to
determine changes in TIC frequency with radiation. In a typical radiation response curve, the
average volume of irradiated tumours plateaus after treatment (Figure 12). Interpreted within
the context of a TIC model, this plateau theoretically reflects an equilibrium between cell death
in the non-TIC population and the survival and/or proliferation of the much smaller TIC fraction.
Thus, we hypothesize that the point of maximum TIC enrichment occurs at the end of this
plateau phase, immediately prior to repopulation (Figure 12: point A’). This point of maximum
TIC enrichment was chosen as a target for the first LDA on the irradiated group. The second LDA
was performed after repopulation, when the tumours approached their maximum average
volume (Figure 12: B’). The clonogenic assays were performed at the same points along the
growth curve.
It is unclear whether the TIC frequency is dependent on tumour volume. Thus, to control for
this possibility, LDAs and clonogenic assays were performed on irradiated and non-irradiated
groups at the same volume (different time points). Thus, point A in Figure 12 represents the
control LDA used as a baseline for comparison with A’. Similarly, point B represents the control

57
LDA used as a baseline for comparison with B’. We hypothesized that the TIC frequency at A’
would be greater than at both A and B’.
Figure 13 is a schematic representation of the LDA methods. When average tumour volume
reached the target for each LDA, a minimum of three mice per group were sacrificed. The
tumours were excised, mechanically minced and enzymatically digested into single cell
suspension using 300 units/mL collagenase, 100 units/mL hyaluronidase (Stemcell
Technologies) and 125 units/mL deoxyribonuclease I (Worthington) in DMEM or Media 199
supplemented with penicillin and streptomycin. Cell suspensions were incubated with 1-2 mL of
red blood cell lysis buffer, rinsed with and re-suspended in phosphate buffered saline (PBS)
with 2% fetal bovine serum (FBS).
The percentage of stromal cells varied between tumour lines. It was conceivable that stromal
percentage could also vary between different treatment groups within a tumour line. Since
LDAs and clonogenic assays rely on a cell count that accurately reflects the number of epithelial
cells used, it was necessary to deplete cell suspensions of any mouse-derived stromal cells.
H2Kd is the major histocompatibility complex for NOD/SCID mice, and labels all nucleated cells
A
B’ B
A’
Figure 12. Relevant xenograft tumour volume points used for LDAs and clonogenic assays. A= control LDA1; A’= radiation LDA1; B= control LDA2; B’= radiation LDA2.

58
in the mouse. Since red blood cells were lysed in the previous step, it was reasonable to assume
that all remaining mouse cells expressed H2Kd. Thus, cell suspensions were incubated with
biotinylated rat anti-mouse H2Kd antibody (BD Biosciences) (1:2000 dilution), followed by anti-
biotin microbeads (Miltenyi Biotec) (1:10). Cells were then passed through a magnetic column
to deplete the effluent of all magnetically-labeled H2K+ cells. Viable cells were counted using
trypan blue dye exclusion.
Successful H2K depletion was confirmed by flow cytometry. Effluent cells were stained with
mouse IgG blocking antibody, followed by streptavidin-PE-Cy7, anti-EpCAM-APC, and DAPI
stain. Cells stained with all antibodies minus one (“fluorescence minus one”) were used as
gating controls. Data were collected on a BD LSRII Analytical Flow Cytometer and analyzed using
FlowJo software. The percentage of cells negative for H2K was recorded for future back-
calculations of actual number of tumour cells injected. Sample flow cytometric plots of pre-
depletion cell suspensions, column-adhering cell suspensions and effluent suspensions are
presented in Appendix B.
The remaining effluent tumour cells were re-suspended in MatrigelTM (BD Biosciences) at ten-
fold dilutions of 1x105, 1x104 and 1x103 cells/100uL. Each cell suspension was injected
subcutaneously into the right and left hind flanks of five NOD/SCID mice in 100uL injection
volumes. Thus, a total of ten injections in five mice were performed for each cell concentration.
Mice were monitored for tumour formation for up to six months. If a tumour on one flank
reached the volume limit or developed an ulcer before the other flank could be scored, the
mouse was sacrificed and the injection on the second flank was removed from the analysis. The

59
fraction of injections at each cell dose that gave rise to tumours was recorded. Extreme Limiting
Dilution Analysis (ELDA) software (available online at http://bioinf.wehi.edu.au/software/elda/)
was used to calculate the TIC fraction.
Figure 13. Xenograft limiting dilution assay. Once tumours reach the targeted volume, 2-3 mice are sacrificed, the tumours are excised and digested into single-cell suspension. Mouse-derived cells are magnetically depleted and the remaining tumour cells are injected at discrete doses into 3 cohorts of 5 mice each. Each mouse receives 2 injections for a total of 10 injections per dose.
3.1.2. Clonogenic assay Recent evidence suggests that long term culturing in serum promotes cellular differentiation.65
Furthermore, it is well documented that stem cells are highly sensitive to O2. In particular, high
oxygen tension increases the differentiation rate of certain stem cell populations.171 It is
possible that limited energy supply under hypoxia allows only strict DNA replication (otherwise
known as self-renewal) rather than activation of differentiation programs. Since the purpose of
this assay was to measure the fraction of cells possessing the stem-like property of
clonogenicity, two culturing conditions were of interest: classical DMEM supplemented with
fetal bovine serum (FBS) and cultured in ambient air, as well as a defined serum-free media
(DSFM) cultured at physiological oxygen levels (2% O2). Initially, all clonogenic assays were

60
performed in both conditions. After repeated observation that the latter better supported
colony growth (data not shown), assays were performed strictly under serum-free, low oxygen
conditions.
DSFM was prepared using the following reagents: DMEM/F12+Glutamax (Invitrogen), 1X B27
minus Vitamin A (Invitrogen), 1X non-essential amino acids (Gibco), lipids (Sigma), 4ug/mL
heparin (Sigma), 1mM N-Acetyl-cysteine (Sigma), 10mM Hepes (Sigma), 10ng/mL FGF
(Invitrogen), 10ng/mL EGF (Invitrogen), 100units/mL and 100ug/mL of penicillin and
streptomycin, respectively (Invitrogen).
Cells were prepared for clonogenic assays following the methods described above for the LDA.
After magnetic depletion of H2K+ cells and verification by FACS, cells were re-suspended in
either DSFM or DMEM in three discrete cell dilutions. Cells were typically seeded at densities of
5x104, 1x104 and 1x103 cells per well, however these densities varied with each experiment,
depending on the number of available effluent cells. Cells were plated on 24-well plates coated
with rat tail collagen (BD Biosciences) at 5ug/cm2. Media was changed 3-4 days after initial
plating, and cells remained in culture for up to four weeks, or until visible colonies formed.
Colonies containing >50 cells were counted under a microscope.
3.2. Aim 2 Results
3.2.1. Radiation may enrich the TIC fraction prior to repopulation in some EAC tumours
Limiting dilution assays were performed on Lines 2, 3 and 4 at passages 10, 4 and 5,
respectively. Lines 3 and 4 showed a >10-fold and >6-fold enrichment of the TIC fraction at

61
point A’ (day 73 and day 64 for Lines 3 and 4, respectively) (Figure 14A and Figure 15A). These
time points roughly corresponded to periods late in the plateau phase of the xenograft growth
curves, prior to repopulation of the irradiated tumours.
Results from the first LDA performed on Line 2 were not as conclusive. While the irradiated
tumours presented a TIC frequency of 1 in 17,790 cells, the control group yielded non-
interpretable results, since tumours grew more frequently at the intermediate dose than at the
highest dose (Figure 16A). Thus, radiation-induced TIC enrichment could not be evaluated in
Line 2.
In Line 3, the second LDA (performed at the volume endpoints for each group) yielded non-
interpretable results in both the control and radiation groups (Figure 14B). In Line 4, the
enriched TIC frequency was sustained even after tumour repopulation (point B’). Curiously, the
TIC frequency in the control arm increased almost 5-fold at point B (Figure 15B). In Line 2, point
B yielded a TIC frequency of 1 in 3,328 control cells (Figure 16B). This was compared to a post-
repopulation TIC frequency of 1 in 9,650 irradiated cells (point B’). The difference between
these two frequencies was insignificant. In addition, the apparent enrichment within the
irradiated tumours before and after repopulation (from 1:17,790 to 1:9,650) was insignificant.
It therefore appears that radiation may enrich the tumourigenic cell population immediately
after treatment in some EAC tumours. It remains inconclusive whether the TIC frequency
returns to baseline levels after tumour repopulation. Results from the second LDA in Lines 2
and 4 suggest that this might not be the case.

62
Control LDA time point 1
Dose Tested Response
99600 10 8
9960 10 6
996 8 2
Confidence Intervals
Lower Estimate Upper
63298 30676 14867
Radiation LDA time point 1
Dose Tested Response
95000 7 7
9500 9 8
950 10 5
Confidence Intervals
Lower Estimate Upper
6341 2928 1352
A
Control LDA time point 1
Dose Tested Response
96000 8 5
9600 10 7
960 10 2
Confidence Intervals
Lower Estimate Upper
Error Error Error
Radiation LDA time point 1
Dose Tested Response
95800 10 2
9580 8 3
958 9 2
Confidence Intervals
Lower Estimate Upper
Error Error Error
B
Control LDA1
↓ RT ↓
↑
Radiation LDA1
0
500
1000
1500
2000
0 20 40 60 80 100 120
Tum
ou
r V
olu
me
(mm
^3)
Days since Implantation
Line 3 (P4): 1st LDA
Control
Radiation
Control LDA2
↓
RT ↓
Radiation LDA2
↓
0
500
1000
1500
2000
0 20 40 60 80 100 120
Tum
ou
r V
olu
me
(mm
^3)
Days since Implantation
Line 3 (P4): 2nd LDA
Control
Radiation
Figure 14. Limiting dilution assay Line 3 passage 4. Tumours were irradiated at an average volume of 470mm3. (A) LDA1,
corresponding to points A and A’ in Figure 5. (B) LDA2, corresponding to points B and B’ in Figure 5.

63
Control LDA time point 1
Dose Tested Response
67541 9 5
13508 10 7
1351 10 3
Confidence Intervals
Lower Estimate Upper
60719 31968 16831
Radiation LDA time point 1
Dose Tested Response
88182 9 8
8818 10 10
882 10 7
Confidence Intervals
Lower Estimate Upper
9951 4871 2384
A
Control LDA time point 2
Dose Tested Response
100000 10 9
10000 10 9
1000 10 5
Confidence Intervals
Lower Estimate Upper
15915 7113 3179
Radiation LDA time point 2
Dose Tested Response
98500 9 9
9850 10 7
985 10 6
Confidence Intervals
Lower Estimate Upper
9110 4559 2282
B
Control LDA1
↓ RT ↓
↑ Radiation
LDA1
0
500
1000
1500
2000
0 20 40 60 80 100 120
Tum
ou
r vo
lum
e (m
m^3
)
Days since Implantation
Line 4 (P5): 1st LDA
Control
Radiation
Control LDA2 →
RT ↓
Radiation LDA2
←
0
500
1000
1500
2000
0 20 40 60 80 100 120
Tum
ou
r vo
lum
e (m
m^3
)
Days since Implantation
Line 4 (P5): 2nd LDA
Control
Radiation
Figure 15. Limiting dilution assay Line 4 passage 5. Tumours were irradiated at an average volume of 514mm3. (A) LDA1,
corresponding to points A and A’ in Figure 5. (B) LDA2, corresponding to points B and B’ in Figure 5.

64
Control LDA time point 1
Dose Tested Response
47440 10 5
9488 10 10
948 10 5
Confidence Intervals
Lower Estimate Upper
Error Error Error
Radiation LDA time point 1
Dose Tested Response
32016 10 7
8000 10 5
1600 10 2
Confidence Intervals
Lower Estimate Upper
32266 17790 9809
Control LDA2
↓ RT ↓
↑ Radiation
LDA1 0
500
1000
1500
2000
2500
0 20 40 60 80 100
Tum
ou
r V
olu
me
(mm
^3)
Days since Implantation
Line 2 (P10): 1st LDA
Control
Radiation
Control LDA time point 1
Dose Tested Response
31600 10 10
7900 10 8
1580 10 6
Confidence Intervals
Lower Estimate Upper
6273 3328 1766
Radiation LDA time point 1
Dose Tested Response
33400 10 10
8350 10 9
1670 10 6
Confidence Intervals
Lower Estimate Upper
17467 9650 5331
Control LDA2
↓
RT ↓
Radiation LDA2
←
0
500
1000
1500
2000
2500
0 20 40 60 80 100
Tum
ou
r V
olu
me
(mm
^3)
Days since Implantation
Line 2 (P10): 2nd LDA
Control
Radiation
A
B
Figure 16. Limiting dilution assay Line 2 passage 10. Tumours were irradiated at an average volume of 480mm3. (A) LDA1,
corresponding to points A and A’ in Figure 5. (B) LDA2, corresponding to points B and B’ in Figure 5.

65
3.2.2. The ability of radiation to enrich the clonogenic fraction was not demonstrated
Ex vivo clonogenic assays were performed on xenograft Line 2 at passage 10, Line 3 at passage 6
and Line 4 at passages 6 and 7. Results from the clonogenic assays are summarized in Table 8.
All but one passage failed to yield clonogenicity results for various reasons. The clonogenic
assays performed on Line 2 passage 10 were lost to fungal infection in the tissue culture plates.
Cells derived from Line 4 did not adapt well to tissue culture conditions. Of the small
percentage of cells that adhered, very few formed colonies and none formed colonies larger
than 50 cells. In addition, the clonogenic assay performed on Line 4 passage 7 was lost to
bacterial contamination. An in vivo streptococcus pneumoniae infection was subsequently
found in several mice harbouring tumours from Line 4. Thus, it is likely that cells from Line 4
were unable to adhere to and proliferate in tissue culture condition due to a mouse-derived
infection.
Line Passage Control frequency at Point A (%)
Radiation frequency at Point A’ (%)
Control frequency at Point B (%)
Radiation frequency at Point B’ (%)
2 10 Fungal infection Fungal infection No colonies No colonies 3 6 0.332±0.016 0.097±0.006 0.026±0.001 0.005±0.001 4 6 No colonies Bacterial infection No colonies No colonies
7 Bacterial infection Bacterial infection Bacterial infection Bacterial infection Table 8. Clonogenic assays. Points A, A’, B and B’ correspond to points on the xenograft growth curve as indicated in Figure 11.
Clonogenic frequencies were obtained in Line 3 passage 6 only (Figure 17). The clonogenic
frequency of control tumours was 0.332%±0.016 at point A and 0.026%±0.001 at point B. The
corresponding clonogenic frequencies in irradiated tumours were 0.097%±0.006 and
0.005%±0.001 at points A’ and B’ respectively. Thus, radiation did not appear to enrich the
clonogenic cell population in Line 3. Rather, the clonogenicity of irradiated tumours appeared
to be reduced relative to controls at both points on the xenograft growth curve.

66
3.3. Aim 2 Discussion
TICs have been identified in many solid tumours, but no study to date has identified this
putatively radioresistant cell population in EAC. The results reported here from LDAs on two
radiosensitive xenograft lines suggest that radiation enriches for tumourigenic cells prior to
tumour repopulation. To our knowledge, this is the first demonstration of enhanced TIC
frequencies during the radiation-induced plateau period of the growth curve. However, in
several of the LDAs performed the frequency of “negative events” was <37%, indicating that a
more accurate calculation of TIC frequency would have been obtained by extending the LDA to
lower cell doses. Thus additional studies are required to verify these results in a larger number
of samples, and using limiting dilution assays that include a lower cell dose than the minimum
dose used here.
The translational significance of radiation-induced TIC enrichment is non-trivial, particularly in
EAC. Clinical management of EAC relies heavily on chemoradiotherapy, yet EAC tumours are
notoriously resistant to cure by these methods. Residual TICs left after radiotherapy may be the
A: 0.33%
↓
B: 0.03%
↓
RT
↓
↑ A':
0.1%
B': 0.01%
↓
0
500
1000
1500
2000
0 20 40 60 80 100 120
Tum
ou
r V
olu
me
(mm
^3)
Days since Implantation
Line 3 (P4)
Control
Radiation
Figure 17. Clonogenic assay Line 3. Tumour were irradiated at an average volume of 470mm3.

67
drivers of disease progression, emphasizing the need to integrate TIC-targeted agents into
current treatment regimens. These preliminary results warrant further experimentation in
additional xenograft lines.
We hypothesized that the tumourigenic capacity of irradiated xenografts would resemble that
of non-irradiated xenografts after tumour repopulation (points B and B’ in Figure 12). We
proposed that at this point, the TICs enriched after irradiation would have undergone multiple
asymmetric cell divisions in order to repopulate the tumour with transit amplifying and
terminally-differentiated cells. However, only Line 4 yielded interpretable results at the second
LDA, and these did not indicate a return to baseline TIC levels in the irradiated tumours. Rather,
irradiated tumours sustained a high tumourigenic capacity, and control tumours developed
enhanced tumourigenicity. It is unclear what may have contributed to these observations. It is
possible that radiation selected for or generated radioresistant clones with a higher
tumourigenic capacity. If conferred with a survival advantage, these cells could contribute
disproportionately to the repopulated tumour. However, since the TIC frequency also increased
in the control arm, this is an unlikely explanation. Rather, other technical and/or biological
factors may be accountable, including the selection of poorly-representative xenograft tumours
for LDAs, inaccurate cell counts, or an intrinsic increase in TIC frequency as the tumour grows
(i.e. size-associated changes in the TIC frequency).
Of particular interest is the reversal in LDA response rates at intermediate and high doses in
Lines 2 and 3. In these cases, more tumours grew at intermediate or even low doses than at the
highest dose. Similar results have been obtained by separate work in our lab using

68
chemotherapy to enrich for TICs. We hypothesize that an inhibitory effect may be present at
the higher doses in some tumours. This inhibitory molecule could derive from residual
contaminating mouse cells, such as a secreted matrix or inflammatory factor, or from the
tumour cells themselves. In this case, the lower cell doses would have a diluted concentration
of the inhibitor and would better support tumour growth. Once again, further studies are
required to elucidate the mechanisms involved in this dose-response inversion.
We sought to determine whether the clonogenic frequency increases after irradiation. We
reasoned that the TIC population is contained within the clonogenic fraction. Thus, TIC
enrichment might yield clonogen enrichment. Of the clonogenic assays performed, only one
yielded interpretable results. These results suggest that the clonogenic frequency is reduced
both after irradiation (Figure 12: point A’) and at the tumour volume endpoints (Figure 12:
points B and B’). The reliability of these results is questionable. While TICs may be contained
within the clonogenic population, it is likely that the non-TIC clonogenic compartment,
consisting of transit amplifying and differentiated cells, is radiosensitive. In fact, the high
proliferative capacity of these cells suggests that substantial cell kill should be expected within
the clonogenic population. To truly observe TIC enrichment with an ex vivo clonogenic assay,
one would likely have to perform a re-plating assay in which colonies formed in the first assay
are seeded again in a second passage. By testing the self-renewal of clonogens, a re-plating
assay introduces an extra functional test and may therefore be more appropriate for future TIC
studies.

69
Chapter 4: Hedgehog pathway in the response to irradiation
4. AIM 3: To study the role of Hedgehog pathway activity in the radiation-response of xenograft
tumours.
4.1. Aim 3 Methods
4.1.1. PCR primer design
To accomplish the objectives of Aim 3, it was necessary that each primer be designed twice,
once for the mouse transcript and once for the human transcript. mRNA sequences were
obtained from NCBI for the following Hedgehog-related genes: SHH and IHH (ligands), PTCH1
and PTCH2 (receptors), GLI1 and SMO (effectors). In addition, primers were designed for a
panel of eight potential housekeeping genes: GAPDH, ACTB, HPRT1, GUSB, HSP90AB1, YWHAZ,
L32 and RPLP0. Mouse and human sequences for each gene were aligned using NCBI blast, and
regions of low homology were identified. The online Primer3 software was used to find short
sequences within these regions that satisfy specified requirements of primer size (20-22bp),
product size (90-150bp) melting temperature (57-63 degrees C) and %GC (40-60%). Output
sequences were then blasted against the whole genome of each species to ensure species-
specificity. Sequence synthesis was outsourced (Integrated DNA Technologies). The species
specificity of each synthesized primer was tested by RT-PCR on a sample of normal mouse
esophagus and the human EAC cell line, OE33. Primers that cross-amplified in both species, or
that produced doublet dissociation curves were redesigned. The primer sequences used are
listed in Table 9.

70
Transcript Human Mouse
SHH F: cagaggagtctctgcactacga R: cgtagtacacccagtcgaagc
F: cactatgagggtcgagcagtg R: gtggatgtgagctttggattc
IHH F: acaaagcatgggacactggt R: catgccaagctgtgaaagagt
F: gcattgctctgtcaagtctga R: tctcctggctttacagctgac
GLI1 F: acacatatggacctggctttg R: ctgccctatgtgaagccctat
F: tcctctcattccacaggacag R: ctggtatgggagttcctggtt
PTCH1 F: caccgacacacacgacaatac R: gcatggtaatctgcgtttcat
F: ctgctgggtgtactgatgctt R: agcagaaccagtccattgaga
PTCH2 F: tcttctacatggggctgacc R: gtatttgtcgtgcagccattc
F: ctccgctcaggtcattcagat R: ggaggcaaaatggtgactaca
SMO F:tgagtgggatttgttttgtgg R: ctcctcggatgaggaagtagc
F: agattgtttgccgagcagat R: ccacgaaccagactactccag
ACTB F: ttgctatccaggctgtgctat R: agggcatacccctcgtagat
F: cgttgacatccgtaaagacctc R: gtgctaggagccagagcagta
HPRT1 F: ctagttctgtggccatctgct R: gcccaaagggaactgatagtc
F: ctgtggccatctgcctagtaa R: gacaatctaccagagggtaggc
YWHAZ F: aatgcttcacaagcagagagc R: tgcttgttgtgactgatcgac
F: ctgcctacatattggtgtgtg R: tttgtgtcacagcctcacaag
HSP90AB1 F:gtcttctgctggaggttcctt R:ctttgacccgcctctcttcta
F:gaggcagacaaaaacgacaaa R:tgagaaaccagaggagagcag
GAPDH F:gacctgccgtctagaaaaacc R: accacctggtgctcagtgtag
F: aaattcaacggcacagtcaag R: tttgatgttagtggggtctcg
L32 F: atcgctcacaatgtttcctcc R: aaacagaaaacgtgcacatga
F: gccattgtagaaagagcagca R: tgcacacaagccatctactca
GUSB F: gactgcagcggtctgtacttc R: aaagacgcacttccaacttga
F: gtcaacttcaggttcccagtg R: tcccgataggaagggtgtagt
RPLP0 F: gaaactctgcattctcgcttc R: actcgtttgtacccgttgatg
F: caaagctgaagcaaaggaaga R: attaagcaggctgacttggttg
Table 9. PCR primers designed for Hedgehog gene expression analysis with internal controls
4.1.2. Housekeeping gene selection
RT-PCR requires the use of internal reference genes whose expression levels do not change
with experimental intervention, in this case, irradiation. Multiple studies have demonstrated
that expression levels of commonly-used housekeeping genes can fluctuate under cellular
stress conditions such as irradiation, hypoxia, induced differentiation and passaging.172–174 To
ensure selection of proper endogenous controls, eight housekeeping genes were evaluated for
their stable expression following irradiation in three separate xenograft tumour lines (lines 2, 3
and 4).

71
Primers were designed for the following housekeeping genes: ACTB, GAPDH, HPRT1, GUSB,
HSP90AB1, YWHAZ, RPLP0 and L32. Irradiated and control samples from Lines 2, 3 and 4, frozen
in OCT and stored at -80°C, were used to test the housekeeping genes. The results of these
experiments are discussed below.
4.1.3. Hedgehog gene expression
The time points interrogated for Hh expression varied between xenograft experiments, and
were chosen based on the number of available animals in each treatment group. Generally, the
time points of interest aligned with the volume points used for the in vivo TIC and ex vivo
clonogenic studies. Thus, at least one time point during the plateau phase of the irradiated
growth curve was chosen, with a volume-matched control. Volume endpoints (after tumour cell
repopulation) were also chosen, with volume-matched controls. In addition, several time points
immediately after irradiation (24 hours, 48 hours, 1 week) were added based on sufficient
numbers of mice. For the first three experiments (Lines 2, 3 and 4), 1-2 mice were sacrificed at
each time point. For subsequent experiments (lines 6, 7 and 8), 2-3 mice were sacrificed at each
time point.
Mice were sacrificed and the tumours were immediately excised and frozen at -80°C in
optimized cutting temperature (OCT) compound (Sakura Finetek). 10µm-thick sections were cut
from frozen specimens using a cryostat. Additional sections were cut and stained with
hematoxylin and eosin. A pathologist assessed the percentage of tumour epithelium in each
sample.

72
RNA was extracted from frozen tissue sections using the RNeasy Mini Kit (Qiagen) with on-
column DNA digestion. RNA concentration was measured using a Nanodrop® ND-1000
Spectrophotometer (Thermo Scientific). RNA quality was evaluated using the Agilent 2100
Bioanalyzer, which provides a RNA Integrity Number (RIN) as a measure of RNA degradation. As
RNA degrades, the ratio of the 28S to 18S ribosomal RNA decreases. The RIN is an integer
between 1 and 10 that accounts for both the 28S:18S ratio, as well as the entire electrophoretic
trace. RNA samples with RINs above 8 were used for qRT-PCR experiments, although most
samples had RINs above 9.
RNA was reverse-transcribed using the iScript™ cDNA Synthesis Kit (Bio Rad). Depending on the
amount of available RNA in each experiment, 2-10ng of cDNA from each tumour sample was
combined with RT2 SYBR® Green ROX™ qPCR Mastermix (Qiagen) and the primer solution for a
total volume of 10uL per well. Each sample was run in triplicate wells. PCR was performed on an
Applied Biosystems 7900HT Fast Real-Time PCR System (Life Technologies). Amplification data
was analyzed using Sequence Detection Systems (SDS) software version 2.3. For each sample,
Hh gene expression was normalized to the geometric mean of the housekeeping genes (∆Ct).
The ∆∆Ct was calculated as the difference between the ∆Ct of each irradiated sample and the
∆Ct of its volume-matched control sample. The fold difference in expression was calculated as
fold difference = 2-∆∆Ct.

73
4.1.4. 5E1 validation and toxicity study
The ability of Hedgehog inhibition to radiosensitize xenograft tumours was assessed by in vivo
administration of 5E1, an antibody for the biologically-active N-terminus of SHH.175 5E1 was
cultured from a hybridoma obtained from the laboratory of Dr. Thomas Jessell in New York.
5E1 toxicity in NOD/SCID mice was evaluated at three doses: 10mg/kg, 20mg/kg and 30mg/kg.
This range was chosen based on previous reports, as well as the quantity of antibody obtained
from the hybridoma culture. Three to five mice were each given a single intraperitoneal
injection of 5E1 antibody. While the proposed experiment required weekly dosing, there was
insufficient antibody obtained from culture to perform multiple dosing in the toxicity analyses.
Mice were monitored for six weeks. No mice died, however mice that received the highest dose
showed an average 10.7% weight loss. Thus, 20mg/kg was selected for the experiment.
The 5E1 antibody was validated by the following method: two tumour-bearing mice were
injected with 5E1 at each of the three doses listed above. In addition, two tumour-bearing mice
were injected with a PBS control. One mouse from each of the 5E1 groups and one control
mouse were sacrificed 24 hours after injection and the remaining mice were sacrificed 1 week
after 5E1 (or PBS) administration. The tumours were excised and RNA was extracted by the
same methods described in Section 4.1.3. The expression levels of SHH, IHH, GLI1, PTCH1,
PTCH2 and SMO were evaluated by qRT-PCR. ACTB was used as a single housekeeping gene.
While there was no detectable change in expression levels in the human tumour cells after 5E1
administration, mouse GLI1 and mouse PTCH1 were significantly downregulated at 24 hours
and 1 week after 5E1 administration at all three doses (Figure 18).

74
4.1.5. In vivo Hh inhibition in a xenograft model
Passage 6 of the primary human xenograft tumour line 7 was expanded into 80 NOD/SCID mice.
The implantation procedure followed that previously described. Mice were monitored for two
weeks, at which point tumours were palpable. Biweekly tumour measurements were made,
and when tumours reached an average volume of 260mm3, the mice were randomly sorted into
5 groups: radiation control (0Gy), 5E1 control (intraperitoneal (i.p.) injection of mouse
polyclonal IgG), 5E1 alone (20mg/kg), radiation alone (4Gy), and combined 5E1+radiation
(20mg/kg, 4Gy). An IgG group was included to control for the effects of off-target 5E1 antibody
binding. Mice receiving 5E1 were given a single i.p. injection of the inhibitor. Mice in the IgG
group received a single injection of IgG at the same concentration and volume as the 5E1
injections. 24 hours later, mice in the radiation and 5E1+radiation groups received 4Gy of
precision irradiation to the tumour-bearing leg. Biweekly measurements of tumour volume
0
0.05
0.1
0.15
0.2
0.25
%H
ou
seke
epin
g ge
ne
exp
ress
ion
H-SHH H-IHH H-GLI1 H-PTCH1 H-PTCH2 H-SMO
Human primers
Mouse primers
Figure 18. 5E1 validation. Human and mouse primers were used to detect expression of pathway genes in xenograft tumours of Line 7 passage 5 after 10, 20 or 30mg/kg 5E1. Mice were sacrificed at 1 and 7 days after 5E1 injection to measure sustainability of Hh inhibition.

75
were made until tumours reached the volume endpoint, or until the average tumour volume of
each group tripled in value from the time of radiation.
4.1.6. Statistical analysis
To determine whether the expression of Hedgehog transcripts fluctuated significantly after
irradiation, we took the natural logarithm of the fold difference in expression between
irradiated and control samples. The mean fold difference of the two to three samples per time
point after irradiation was calculated. A z-test was used to test the difference of this mean from
zero ( 0)ln(:0 foldH ).
4.2. Aim 3 Results
4.2.1. ACTB, HPRT1, HSP90AB1 and YWHAZ are appropriate housekeeping genes for EAC
radiation studies
Housekeeping gene (HKG) stability was evaluated on control and irradiated tumours from Lines
2, 3 and 4. The ∆Ct between each irradiated tumour sample and a baseline control (non-
irradiated) sample was calculated for all candidate genes. In addition, the ∆Ct between a
control sample at the volume endpoint and a baseline control sample was calculated in order to
determine if housekeeping gene expression was stable across volume changes within the
control arm. Gene expression stability was evaluated by plotting the ∆Ct values for each cDNA
sample across all candidate genes (Figure 19). If all candidate housekeeping genes were stably
expressed after irradiation (or after substantial tumour growth), the ∆Ct for one xenograft
sample would be constant, represented by a line with a slope of 1. This constant ∆Ct would
simply represent the difference in cDNA concentration of the irradiated and control sample.

76
Any housekeeping gene for which the ∆Ct deviated from this horizontal line would therefore be
interpreted to have unstable expression after irradiation.
Each of the eight candidate housekeeping genes was evaluated at multiple time points after
irradiation in three separate xenograft tumour lines (lines 2, 3 and 4). Most samples displayed
an approximately linear trend with a slope of zero (Figure 19). A line of best fit was plotted for
each sample, and each housekeeping gene was scored based on the number of times it fell
significantly above or below this line (Table 10). With the three highest scores of 10, 10 and 9,
GUSB, L32 and RPLP0 most frequently deviated from the estimated line and were immediately
rejected. GAPDH, with a score of 9, was retained for subsequent analysis because of its wide
use in published literature. With the remaining candidate genes, different combinations of
three were used to calculate geometric means for each sample. A previously rejected
housekeeping gene, RPLP0 was used as surrogate “gene of interest” and its expression levels in
each sample were normalized using each of the putative geometric means. These geometric
means were evaluated for their ability to “flatten” the RPLP0 expression curve (Figure 20).
Ultimately, two combinations of housekeeping genes were selected as suitable endogenous
controls: HPRT1/HSP90AB1/YWHAZ, and ACTB/HSP90AB1/YWHAZ. Interestingly, recent work
from another group within our institution has demonstrated that the former combination of
HKGs is stably expressed under hypoxia (personal communication, M. Koritzinsky). However,
while this combination is most stably expressed in the three tumour lines tested (lines 2, 3, 4),
subsequent tumour lines used for gene expression experiments (lines 6, 7, 8) showed variable
levels of HPRT1 expression. Thus, ACTB, HSP90AB1 and YWHAZ were chosen as HKGs in
subsequent experiments.

77
Figu
re
19:
Ho
use
keep
ing
gen
e (H
KG
) ra
dia
tio
n
stab
ility
in
3
xe
no
graf
t lin
es.
Eac
h l
ine
rep
rese
nts
∆C
t b
etw
een
a s
amp
le a
nd
a
con
tro
l fr
om
ear
ly i
n t
he
gro
wth
cu
rve.
Sin
ce v
aria
tio
ns
in s
amp
le
con
cen
trat
ion
are
co
nsi
sten
t ac
ross
all
gen
es w
ith
in a
sam
ple
, th
e ∆
Ct
bet
wee
n t
wo
sam
ple
s sh
ou
ld b
e co
nsi
ste
nt,
ass
um
ing
HK
G r
adia
tio
n-
stab
ility
. G
enes
th
at r
epea
ted
ly d
evia
ted
fro
m t
he
ho
rizo
nta
l tr
end
we
re r
ejec
ted
(se
e Ta
ble
10
). (
A)
4 r
adia
tio
n a
nd
1 l
arge
vo
lum
e co
ntr
ol s
amp
le f
rom
Lin
e 2
. (B
) 2
rad
iati
on
an
d 1
larg
e vo
lum
e co
ntr
ol
sam
ple
fro
m L
ine
3. (
C)
3 r
adia
tio
n a
nd
1 la
rge
volu
me
con
tro
l sam
ple
fr
om
Lin
e 4
.

78
Line Species Sample ACTB GAPDH GUSB HPRT L32 RPLP0 HSP90 YWHAZ
2
Human C/27d X X X X X
R/6hr X X
R/1d X X
R/3d X X X X
R/9d X X X
Mouse C /27d X X
R/6hr X X X X
R/1d X X X X
R/3d X X X X
3 R/9d X X X
Human C/33d X
R/26d X X X
R/67d X X
Mouse C/33d X X X
R/26d X
R/67d X X
4 Human C/36d X
R/48hr X
R/7d X X
R/15d X X X
Mouse C/36d X
R/48hr X X
R/7d X X
R/15d X X
Total score 3 9 10 8 10 9 5 5
Table 10. Housekeeping gene radiation stability score. C= control; R= Radiation; d= days after irradiation. Each housekeeping gene was scored based on the number of times it fell above or below a perceived best-fit horizontal line for each sample. Those with the highest scores were deemed unstable before and after irradiation and therefore unsuitable as housekeeping genes. ACTB, GAPDH, HPRT, HSP90 and YWHAZ were used for subsequent analysis.

79
Figure 20. Selection of best housekeeping gene combination using radiation stability in 3 tumour lines. The geometric mean of 3 housekeeping genes is used to normalize the expression of a 4
th gene, RPLP0. The blue line is the difference of the green and red
lines, and its “flatness” represents the stability of the housekeeping gene combination. (A)The geometric mean of ACTB, HSP90AB1 and YWHAZ, and (B) of HPRT, HSP90AB1 and YWHAZ were selected as suitable for endogenous controls. (C) The geometric mean of GAPDH, HSP90AB1 and YWHAZ was rejected as a normalizer because it introduced kinks into the ∆Ct line (blue).

80
4.2.2. Hedgehog expression in EAC xenografts displays a predominantly epithelial-to-
mesenchymal paracrine mechanism (Aim 3a)
Histologic examination confirmed the presence of both epithelia and stroma in all samples used
for qRT-PCR. The percent epithelia of samples used at each time point is summarized in Table
11.
Reverse-transcription followed by quantitative real-time PCR of six xenograft tumour lines
revealed that the expression of Hh ligands, Sonic and Indian Hedgehog was detected
predominantly in the patient-derived tumour epithelium. In contrast, both of the pathway’s
receptors, PTCH1 and PTCH2, as well as the co-receptor/activator SMO and transcription factor
GLI1 were predominantly expressed in the host-derived stromal cells (Figure 21). Nevertheless,
low-level expression of PTCH1/2, GLI1 and SMO was detected in human cells in five of the six
lines (Line 3 excluded), suggesting that tumour cells may be capable of receiving and
Line (passage)
Treatment Time after
radiation (days) # Samples % tumour epithelium
6(4)
Control 0 2 60-70%
Control 7 3 70-80% Control 48 3 60-90%
Radiation 1 1 70% Radiation 15 2 60% Radiation 21 2 60-80% Radiation 48 3 80%
7(5)
Control 0 2 70%
Control 7 3 70-80% Control 22 3 80%
Radiation 1 2 70% Radiation 7 3 70-80% Radiation 14 3 70-80% Radiation 38 3 80%
8(4)
Control 0 3 80-90%
Control 7 3 90% Control 28 3 90%
Radiation 1 3 80-90% Radiation 7 3 80% Radiation 14 3 80-90% Radiation 35 3 80-90%
Table 11. Histologic quantification of percent tumour epithelium in xenograft samples

81
transducing the Hh signal. Similarly, slight expression of both ligands was detected in the
stroma of each line except for Lines 2 and 4, neither of which expressed SHH.
The expression levels of Hh transcripts varied among xenograft lines and between genes within
each line. Lines 2, 3 and 4 showed the highest overall expression, with transcript levels ranging
from 1-20% those of the housekeeping genes. In contrast, Lines 6, 7 and 8 displayed ligand and
receptor/effector expression levels less than 1% and 5% of housekeeping gene levels,
respectively (see y-axes in Figure 21).

82
0
0.2
0.4
0.6
0.8
SHH IHH PTCH1 PTCH2 GLI1 SMO
Line 6 (62325) passage 4
Human
Mouse
0
4
8
12
16
20
SHH IHH PTCH1 PTCH2 GLI1 SMO
Line 4 (60045) passage 7
Human
Mouse
A B
C D
E F
Hed
geh
og
exp
ress
ion
as
per
cen
t h
ou
seke
ep
ing
gen
es
Figure 21. Localization of Hh transcripts in epithelium versus stroma of untreated tumours from 6 xenograft lines. Note that each y-axis uses a scale appropriate for the expression levels of that xenograft line. (A-C) Each bar represents the average of 8-9 control (unirradiated) xenograft tumours taken from 3 different volumes along the growth curve. For qRT-PCR, 10ng of RNA were loaded in each well. (D-F) Each bar is the average of 3-6 tumours from 3 different volumes. 2-5ng of RNA were loaded in each well for qRT-PCR. Housekeeping genes used for A-D were ACTB, YWHAZ and HSP90AB1. HPRT1 was used in lieu of ACTB for E and F.
0
1
2
3
4
5
6
7
SHH IHH PTCH1 PTCH2 GLI1 SMO
Line 3 (61057) passage 6
Human
Mouse
0
0.5
1
1.5
2
2.5
3
SHH IHH PTCH1 PTCH2 GLI1 SMO
Line 7 (60745) passage 5
Human
Mouse
0
0.4
0.8
1.2
1.6
2
SHH IHH PTCH1 PTCH2 GLI1 SMO
Line 8 (63862) passage 4
Human
Mouse
0
5
10
15
20
25
SHH IHH PTCH1 PTCH2 GLI1 SMO
Line 2 (59046) passage 10
Human
Mouse

83
4.2.3. Radiation upregulates both autocrine and paracrine Hh expression in some EAC tumours
(Aim 3b)
Since our studies interrogate transcript levels in irradiated tumours compared to a reference of
non-irradiated controls, we use the terms “upregulation” and “downregulation” to refer to
positive and negative changes in gene expression relative to non-irradiated controls, making no
inference on the subsequent levels of protein in the cell. In each of the three xenograft lines
investigated, at least five transcripts were significantly upregulated in either mouse or human
cells after irradiation. The genes themselves, the cell type demonstrating the expression
changes, and the levels and timing of fluctuation were heterogeneous across the lines. To map
out the autocrine and/or paracrine characteristics of this expression, we generated one-colour
heat maps with mouse and human data displayed separately. Two hypothetical heat maps—
one displaying autocrine signalling and the other paracrine signalling—is presented in Figure 22.
Transcript
Days since radiation
Human Mouse
1 7 14 38 1 7 14 38
SHH
IHH
PTCH1
PTCH2
GLI1
SMO
Transcript
Days since radiation
Human Mouse
1 7 14 38 1 7 14 38
SHH
IHH
PTCH1
PTCH2
GLI1
SMO
Figure 22. Autocrine and paracrine Hh signalling displayed in one-colour heat maps. In the top panel, ligands, receptors and effectors are all upregulated in the epithelium. In the bottom panel, ligands are upregulated in epithelium while pathway activation occurs in the stroma.
Autocrine
Paracrine

84
Figures 23-28 present the gene expressions as one-colour heat maps and bar graphs for each of
the three lines. Fold changes correspond to the relative expression of irradiated tumours
compared to volume-matched controls. Statistically significant upregulations of ≥1.5-fold are
coloured in red. The same data is presented as bar graphs, where the natural logarithm of the
fold change is used to visually distinguish upregulation and downregulation. Data is presented
line by line.
Line 8 Passage 4 (Figures 23-24):
IHH showed a progressive increase in expression after irradiation, reaching a 6-fold
upregulation relative to controls two weeks after irradiation. This was followed by a sharp 8-
fold downregulation at the volume endpoint (38 days after irradiation). SHH expression
mirrored this trend, with approximately 2-fold upregulation at one and two weeks, followed by
a return to baseline levels at day 38. In addition, GLI1 was approximately 2-fold upregulated in
human tumour cells at one week, two weeks and 38 days. A modest 1.5-fold upregulation was
seen in PTCH1 at one and two weeks after irradiation.
Stromal GLI1, PTCH1 and PTCH2 were all approximately 2-fold upregulated for the first two
weeks after irradiation, and PTCH1 reached a 3-fold upregulation at day 14. While SHH was
almost 2-fold upregulated in the same cells one week after radiation, no significant changes
were seen in IHH.
Line 6 Passage 4 (Figures 25-26):
Similar expression patterns were seen in Line 6. IHH was 3-fold upregulated 15 days after
irradiation. This was followed by a modest 1.6-fold upregulation in SHH at day 21. In addition,

85
GLI1 was 7-fold upregulated in human tumour cells at day 21. A 2-fold GLI1 upregulation
appeared to be maintained at the volume endpoint (day 48) with a trend towards significance
(p=0.055). No significant upregulation was seen in either PTCH1 or PTCH2 in human cells.
The stromal compartment in Line 6 also showed similar expression patterns to those of Line 8.
PTCH1 was significantly upregulated at all time-points probed. PTCH2 showed a marked 22-fold
upregulation at day 1, a 5-fold upregulation at day 15, and a return to baseline levels at day 48.
GLI1 was 2-fold upregulated at day 1, subsequently returned to baseline and was ultimately 2-
fold downregulated at day 48. IHH was 4-fold upregulated in stromal cells at day 15.
Line 7 Passage 5 (Figures 27-28):
Line 7 exhibited a unique expression profile, with a sharp increase in Hh transcripts consistent
with a pathway response in tumour cells immediately following irradiation. This was followed
by subsequent downregulation or return to baseline. GLI1 and PTCH2 were 3-fold and 6-fold
upregulated at day 1, respectively. SHH, IHH and PTCH1 were either unchanged or showed
decreased expression relative to controls in the human epithelial compartment throughout the
observation period.
In the stromal compartment, SHH was 14-fold and 9-fold upregulated one day after irradiation
and at the volume endpoint (day 38), respectively. No statistical significance was found in the
apparent fluctuations in IHH transcript levels. GLI1, PTCH1 and PTCH2 were all either
unchanged on downregulated for all time points after irradiation.

86
Transcript
Days since radiation
Human Mouse
1 7 14 38 1 7 14 38
SHH 1.03 2.02 1.89 1.00 0.17 1.81 0.11 1.78
IHH 1.42 3.99 6.20 0.12 1.04 0.87 0.78 4.86
PTCH1 0.85 1.60 1.52 0.43 1.85 2.13 2.77 0.87
PTCH2 0.62 1.16 0.74 0.62 1.70 1.30 1.88 1.10
GLI1 0.63 1.69 2.05 1.55 1.87 1.54 1.94 0.72
SMO 0.54 0.71 0.73 1.15 1.31 0.82 0.91 0.97
Figure 23. Gene expression changes in Line 8 passage 4. Statistically significant ≥1.5-fold upregulations are coloured in red.
Figure 24. Bar graphs of gene expression changes in Line 8 passage 4. Asterisks mark statistically-significant up- or downregulations.
Line 8 passage 4

87
Transcript
Days since radiation
Human Mouse
1 15 21 48 1 15 21 48
SHH 0.39 0.18 1.61 0.57 1.23 N/A * 1.39
IHH 0.59 3.38 0.29 0.22 8.33 4.16 N/A 5.18
PTCH1 1.41 0.92 0.74 0.71 2.63 6.37 3.45 1.75
PTCH2 * 0.91 2.03 0.55 22.44 4.86 0.17 1.32
GLI1 2.92 2.47 6.71 1.92 1.98 1.11 0.90 0.52
SMO 0.32 0.41 1.07 2.69 0.97 0.52 1.24 1.48
Figure 25. Gene expression changes in Line 6 passage 4. Statistically significant ≥1.5-fold upregulations are coloured in red. Asterisks represent genes that were detected in irradiated samples but not in the volume-matched controls. Thus, while a fold difference could not be calculated, upregulation was detected.
Ligands
Receptors
Activators
& targets
Figure 26. Bar graphs of gene expression changes for Line 6 passage 4. Asterisks mark statistically significant up- or downregulations.
Line 6 passage 4

88
Transcript
Days since radiation
Human Mouse
1 7 14 38 1 7 14 38
SHH 0.82 0.91 0.80 0.81 14.30 0.20 1.13 8.62
IHH 0.93 0.51 0.53 0.83 N/A 1.44 2.11 4.92
PTCH1 0.80 1.00 0.79 0.71 1.00 0.56 0.37 0.72
PTCH2 6.13 0.43 0.39 0.97 0.79 0.44 0.35 0.87
GLI1 3.30 0.49 0.53 1.49 0.92 0.57 0.56 0.84
SMO 68.05 0.23 1.50 0.55 0.92 0.75 0.76 1.22
Figure 27. Gene expression changes in Line 7 passage 5. Statistically significant ≥1.5-fold upregulations are coloured in red.
Ligands
Receptors
Activators
& targets
Figure 28. Bar graphs of gene expression changes in Line 7 passage 5. Asterisks mark statistically significant up- or downregulations
Line 7 passage 5

89
4.2.4. Single dose 5E1 inhibits stromal Hh activation for up to one week
To validate 5E1 as an inhibitor of Hh signalling in our xenograft model, we treated mice bearing
tumours from Line 7 passage 5 with a single dose of 5E1 at 20mg/kg and measured the
expression levels of Hh pathway components by RT-PCR. Two tumours from untreated mice
were used as controls. Across the three doses used, GLI1 was an average 20-fold and 13-fold
downregulated 24 hours and one 1 week after 5E1 administration, respectively. Similarly,
PTCH1 showed an average 9-fold and 7-fold downregulation at 24 hours and one week after
5E1 injection (Figure 18). In contrast, SMO expression was unchanged by 5E1 inhibition. This
result was expected given that SMO is not a target of the pathway. Thus, 5E1 inhibits Hh
signalling in our xenograft model of EAC.
4.2.5. The ability of 5E1 to radiosensitize EAC xenografts was not demonstrated and warrants
further study (Aim 3c)
Prior to selecting a xenograft line for a 5E1 inhibition study, untreated tumours from three lines
(Lines 6, 7 and 8) were tested for baseline Hh expression using qRT-PCR. All three expressed Hh
transcripts (Figure 21). Line 7 was selected for the 5E1 inhibition study. 5E1 failed to increase
the growth delay of both irradiated and non-irradiated tumours relative to radiation alone and
IgG control, respectively (Figure 29).

90
Figure 29. 5E1 failed to radiosensitize xenograft tumours from Line 7 passage 6
4.3. Aim 3 Discussion
4.3.1. Hedgehog expression in EAC xenografts is suggestive of paracrine signalling
While the activation of Hh signalling has been well characterized in EAC, studies have produced
conflicting results describing the direction of signalling in this tumour. Autocrine signalling was
reported by Berman et al using the commercially available validated EAC cell line OE33.152 In
contrast, one study using a mouse esophagojejunostomy model reported paracrine activation
of the pathway.156 Yet another study found expression patterns consistent with both autocrine
and paracrine mechanisms in primary patient samples.155
Our study uses qRT-PCR to evaluate the distribution of Hh transcripts between the epithelial
and stromal compartments of patient-derived primary xenografts. As we have not incorporated
a measure of protein readout (i.e. nuclearization of GLI), we cannot conclude that the presence
of a transcript indicates pathway activation, even if elevated relative to controls, as is the case
0
500
1000
1500
2000
2500
0 10 20 30 40 50 60 70 80 90
Tum
ou
r vo
lum
e (m
m^3
)
Days since Implantation
Line 7 passage 6 treated with 5E1 and radiation
Control
IgG
5E1
RT
5E1+RT

91
in our post-radiation gene expression experiments. Nevertheless, we found that xenograft
tumours display lower transcript levels after treatment with 5E1, suggesting that changes in
transcript levels correspond to changes in pathway activity. With this in mind, our data from
patient-derived xenografts agree with the results of the latter reports outlined above, and
support both paracrine and autocrine signalling in EAC, with the former playing the
predominant role.
The apparent contradiction between our results and those of Berman et al may be resolved
through a consideration of the experimental techniques used. Use of fresh patient samples
suggests that while both signalling mechanisms are present, a paracrine pattern predominates.
It is likely that long-term culture conditions impose selective pressures on EAC cells to become
self-sufficient in their use of the Hh pathway. Thus, lacking a stromal support network, OE33
cells may have capitalized upon and enhanced an already-present albeit low-level autocrine
mechanism to support their own growth.
4.3.2. Hedgehog is involved in the radiation response of EAC tumours
With evidence amassing in both the cancer stem cell and Hedgehog fields, carcinogenesis is
increasingly being viewed as the misuse of homeostatic mechanisms involved in tissue repair
and stem cell self-renewal. This model is particularly appealing for EAC, which develops from
chronic acid reflux-induced injury to esophageal epithelium. By extension, irradiation of EAC
tumours may provoke the same stem cell-driven responses found during normal repair.
Evidence from multiple tumour sites suggests that Hh signalling is involved in the radiation
response (see section 1.13.) However, little is known about the pathway’s role in the

92
radioresponse of EAC tumours. Our results demonstrate that Hh transcripts are significantly
modulated after irradiation of primary EAC xenografts.
Of the three xenograft lines tested, two (Line 6 and Line 8) showed a distinct increase in
expression of pathway transcripts in an epithelial-to-mesenchymal pattern suggestive of
paracrine signalling. Both SHH and IHH were significantly upregulated in Line 8 tumour cells one
week after irradiation. Despite a significant decrease in the number of transcripts
(“downregulation”) at several time points, the expression of both ligands was also upregulated
in Line 6 at two to three weeks following irradiation. In both Line 6 and Line 8 PTCH1, PTCH2
and GLI1 were upregulated in stromal cells.
Curiously, stromal receptor upregulation preceded epithelial ligand upregulation in Line 6
(Figure 25). Receptor upregulation could be interpreted as an autocrine response to stromal
IHH. However, this upregulation does not reach significance until day 15, at which point
tumour-derived IHH is also upregulated. Alternatively, stromal receptor upregulation may
reflect a response to epithelial ligand upregulation at time points in between or prior to those
tested in this experiment.
In both Line 6 and Line 8, epithelial ligand expression was accompanied by epithelial GLI1
upregulation, suggesting an underlying autocrine signalling mechanism in addition to the
paracrine activation discussed above. The use of both paracrine and autocrine Hh signalling has
been reported in other organ systems. SHH signalling occurs in an autocrine loop within the
epithelial compartment of prostate tumours, while normal prostate development utilizes the

93
pathway in a paracrine fashion between epithelia and stroma.176 Similarly, embryonic lung
epithelial cells communicate with adjacent mesenchyme using paracrine Hh signals. 177
However, epithelial Hh activation occurs in small clusters of cells later in lung development, and
in small numbers of the basal layer epithelial cells in adult bronchial epithelium. Furthermore,
this autocrine signalling mechanism is detectable in lung tissue during repair of acute airway
injury and in small cell lung cancer (SCLC). The authors of this report speculated that autocrine
Hh activation following tissue injury represents a regenerative response within the progenitor
cell population in lung tissue, and could play a role in SCLC carcinogenesis.151 Thus, while
paracrine Hh signalling in tumours may represent the misuse an embryonic development
pathway, autocrine signalling may reflect the adaptation of a cancer cell to profound Hh
dependency during inflammation, tissue repair or carcinogenesis.
Alternatively, underlying autocrine Hh signalling may represent the predominant signalling
mechanism of the TIC compartment. TICs from several disease sites have been shown to rely on
either autocrine or reverse paracrine (mesenchymal to epithelial) Hh signalling for self-renewal
and proliferation. Blockade of SMO function inhibits the self-renewal of TICs from multiple
myeloma,178 glioblastoma,179,180 colon cancer,181 gastric cancer,182 breast cancer,183 and chronic
myeloid leukaemia.184,185 Thus, TICs from multiple different tumour types rely on an active Hh
pathway through autocrine or reverse-paracrine signalling. It is possible that EAC TICs are
equally reliant on Hh pathway activation. The upregulated autocrine expression observed after
irradiation may reflect an increased burden on EAC TICs to self-renew and repopulate the
tumour.

94
As part of the rationale for investigating a putative TIC population in EAC tumours, we cited
recent findings that the intestinal stem cell marker Lgr5 is ubiquitously expressed in EAC and
marks gastric progenitors from which the EAC cell of origin may derive (see section 1.10). It is
interesting to note that Lgr5 is also a target of the Hh pathway and appears to promote cellular
proliferation and tumour formation in basal cell carcinoma.186 Furthermore, Lgr5+ hair follicle
stem cells communicate within their own population and with other follicular stem cell
populations through autocrine and paracrine Hh signalling, respectively.187 Thus, a bimodal Hh
signalling pattern has been established in the putative EAC cell of origin.
Taken together, there is substantial evidence that autocrine Hh signalling plays an important
role in stem cell and cancer stem cell self-renewal and in tissue repair. Thus, paracrine
expression patterns observed here may reflect a proliferative and/or anti-apoptotic response of
bulk or transit-amplifying EAC tumour to radiation-induced tissue injury, or it may represent
epithelial-to-mesenschymal communication between EAC TICs and the tumour
microenvironment. Since Hedgehog signalling has documented roles in both proliferation and
cell survival, this interpretation is plausible. The observed autocrine expression patterns may be
the response of a radiation-activated TIC population that must maintain self-renewing divisions
while repopulating the tumour.
Despite an initial spike in epithelial SMO expression following irradiation in Line 7, levels of the
co-receptor remain relatively unchanged across all three lines. Since SMO has not been
reported as a target of the pathway, these results are reasonable. Furthermore, stromal SMO

95
expression was unchanged after pathway inhibition with 5E1 (Figure 18). Thus, SMO expression
is pathway-independent.
The radiation-induced expression changes in Line 7 deviated from the pattern established by
Lines 6 and 8. SHH ligand in Line 7 appeared to be upregulated in the stromal compartment,
while PTCH2 and GLI1 were upregulated in tumour cells following irradiation, suggesting a
reverse-paracrine mechanism in this model. However, we cannot rule out the possibility of an
autocrine mechanism in this case. That is, epithelial expression of PTCH2, GLI1 and SMO may be
a response to undetected ligand expression in tumour cells within the first 24 hours after
irradiation (prior to the first time point interrogated). Regardless, Hh pathway expression
patterns do not appear to follow a conventionally paracrine mechanism in Line 7. Furthermore,
while Lines 6 and 8 showed a delayed albeit sustained upregulation of Hh transcripts after
irradiation, Line 7 showed acute, transient upregulation that was not sustained past the first 24
hours after irradiation. The remaining pathway components are largely downregulated or
statistically equivalent to baseline (control) expression levels, suggesting that the Hh pathway is
not active after irradiation in Line 7. That pathway activation—whether autocrine or reverse
paracrine—may not even be functional in this line is supported by results obtained from Aim
3(c). SHH inhibition with 5E1 antibody alone failed to induce a significant growth delay in Line 7
xenografts relative to control tumours. In addition, 5E1 failed to radiosensitize Line 7 xenografts
compared to radiation alone. Thus, Hh signalling may not be a critical component of the
radiation response in Line 7, and the unique expression patterns in this line should be
interpreted with caution.

96
Chapter 5: Limitations, alternatives and future directions
The limitations of the present studies shed light on alternative methods and questions we
might have pursued. Looking forward, the knowledge gained from these studies—both those
that succeeded and those that did not—will guide further exploration within each of the three
aims.
The overarching objective of the present studies was to probe multiple factors in the radiation
response of EAC tumours (radiosensitivity, TICs and Hh signalling) using clinically-reflective
experimental models. There were, however, several limitations inherent in the xenograft
models used.
First, our panel of seven xenograft lines may not represent the full spectrum of radiosensitivity
observed clinically. For instance, no single line was completely non-responsive to irradiation.
Future xenograft irradiation studies in an expanded panel of primary patient tumours will
determine whether a wider spectrum of radiosensitivity exists. This will allow us to probe the
radiation response of patient tumours in relation to parameters such as the TIC frequency and
to Hh transcript levels suggestive of pathway activation. Our panel of seven tumours currently
lacks the power to detect such correlations. In addition, our model is unable to detect SGD
differences between tumour lines. We suspect that insufficient animal numbers can account for
this. Initially, our xenograft experimental design incorporated all three aims into one
experiment. That is, one xenograft irradiation experiment would provide the materials to assess
SGD, TIC enrichment and Hh pathway activity. In retrospect, combining these aims into one

97
experimental passage may have compromised our ability to detect statistically significant
differences in tumour growth kinetics, since mice were continuously removed from the growth
delay experiment for TIC and gene expression experiments. Thus, increasing the number of
animals per experiment, as well as re-evaluating the mathematical models used to assess
tumour growth kinetics will increase the power of our xenograft model.
Second, our use of single-dose, high dose-rate precision irradiation deviates from clinical
treatment modalities. Patients receive multiple fractions of low-dose irradiation over an
extended treatment period. This permits normal tissue to repair damaged DNA in between
fractions, resulting in reduced patient toxicity. Initially, fractionated irradiation did not seem
feasible for our studies, since limiting dilution assays, clonogenic assays and gene expression
analyses were planned for two distinct points in the growth curve: during the plateau phase
and after repopulation. Fractionation would have delayed repopulation (and perhaps even
cured some mice), resulting in lengthy experiments and missing data points. Furthermore,
initial radiation studies were combined with chemotherapy and chemoradiation studies (not
reported here), both of which used single-dose chemotherapy. For these reasons, we selected a
single-dose irradiation scheme.
In retrospect, it would likely have been both feasible and desirable to incorporate fractionated
irradiation into our xenograft models. Since fractionated radiotherapy preferentially spares
benign tissue, interactions between tumour and stroma during fractionated therapy may differ
from tumour-stroma interactions after a single dose of radiation. In particular, by allowing
normal tissue to repair sublethal damage, and by allowing radioresistant tumour cells to

98
redistribute into sensitive phases of the cell cycle, fractionated irradiation may decrease the
proportion of tumour to stroma (i.e. the % tumour epithelium) compared to single-dose
therapy. Thus, measurements of Hh expression after fractionated irradiation may more closely
mirror the human radiation response. This has important implications for the translational
relevance of our work. If Hh inhibitors are to be integrated into standard clinical management
of EAC, preclinical Hh studies should aim to mimic the human treatment regimens as closely as
possible.
In addition to fractionation, future work should consider incorporating low-dose rate irradiation
into xenograft studies. As discussed in Section 2.3, these methods incorporate a measure of
sublethal cellular repair, and may therefore increase the sensitivity of our xenograft model to
slight differences in specific growth delay between patient tumours.
A third limitation of the xenograft model involves the use of NOD/SCID mice, a decision that
was made based on available resources. Chemotherapeutic studies in our laboratory resulted in
the establishment of an in-house NOD/SCID breeding colony. Our economical decision to use
the same strain for radiation studies may have undermined our efforts to recapitulate clinically-
relevant radiation responses. As discussed above, normal tissue is presumed to repair damaged
DNA more efficiently than malignant cells during fractionated radiotherapy. Being incapable of
DNA double-strand break repair, NOD/SCID mice may differ from the human system in terms of
the normal tissue response to irradiation. We showed on a macroscopic level that this genetic
defect did not compromise our ability to measure xenograft growth kinetics. However, on a
cellular and molecular level, this stromal defect may have affected our gene expression and TIC

99
studies. For example, it is widely accepted that oxygen tension fluctuates in a tumour following
irradiation. Acute hypoxia may result from endothelial cell death, but subsequent angiogenesis
permits tumour reoxygenation. This observation forms the rationale for combined
chemoradiotherapy, since radiation-induced debulking and angiogenesis can improve drug
delivery. However, it is possible that an irradiated NOD/SCID tumour bed has a reduced
capacity to stimulate angiogenesis and tumour reoxygenation relative to other model systems
or to humans. Thus, irradiated tumours in NOD/SCID mice may remain chronically hypoxic. This
microenvironmental difference between the murine and human systems would likely affect the
TIC frequency, since hypoxia is a component of the TIC niche. Furthermore, changes in oxygen
tension have profound effects on gene expression, and may result in Hh expression changes
that differ from those seen in irradiated human tissue. This genetic difference between human
and mouse stroma could be of particular concern in our models, given our observation of a
predominantly paracrine signalling mechanism.
Future xenograft irradiation studies should consider using a mouse strain with a benign tissue
radiation response that more closely resembles that of human tissue. The RAG2γc double
knockout strain, which lacks the SCID mutation, offers a suitable alternative to NOD/SCIDs.
We have chosen to model EAC radioresistance within the framework of the cancer stem cell
theory. Due to a lack of validated stem cell markers in EAC, we have used a functional assay of
tumour-initiating capability to detect TICs in this cancer. Our studies of TICs and clonogenic cells
were limited in several respects. First, our LDA results suggest but do not conclusively show that
the radioresistant cells are the same population as the TICs. It is possible that radioresistant

100
non-TICs are equally capable of driving tumour regrowth (during the repopulation phase of the
growth curve) and are therefore relevant therapeutic targets. Second, our LDA results are
limited in number. We demonstrated TIC enrichment in two xenograft Lines. Additional TIC
studies should be pursued in remaining xenograft lines in order to corroborate the findings
presented here. A final limitation of our TIC studies emerges from a potential inhibitory effect
at intermediate and high LDA doses. This suggests that factors other than the intrinsic TIC
frequency can modulate LDA outcomes. This observation has not yet been reported in
published literature, and may shed light on inherent weaknesses in the techniques used to
identify TICs.
Our data on Hh expression is limited in several ways. First, missing information about pathway
regulation at the protein level precludes us from making conclusions about pathway signalling.
The use of qRT-PCR in combination with immunohistochemistry would permit more conclusive
observations of pathway activation in response to radiotherapy. Furthermore, we have
measured the transcript levels of GLI1 only, since it is the most reliable readout of pathway
activity. Since GLI2 and GLI3 appear to be the predominant transcriptional activators of the
pathway, analysis of their expression levels and protein localization within the cell would
provide a more robust indication of pathway activation.
Second, our studies on baseline Hh expression in EAC (Section 4.2.2) do not include normal
esophageal epithelium as negative controls. This, however, would be a difficult comparison to
make, since normal squamous and malignant esophageal epithelia differ in cell phenotype (i.e.
squamous versus columnar). Ideally, matched controls from normal gastric cardia could provide

101
a reasonable measure of baseline expression prior to malignant transformation. Finally, our
data on Hh signalling after irradiation is currently restricted to two xenograft lines. The results
from these studies are promising and warrant further investigation in additional lines. If
possible, future studies should aim to demonstrate that Hh signalling promotes EAC TIC self-
renewal, either through functional assays (LDAs, re-plating clonogenic assays) or through
biomarker staining (Lgr5, Oct4, Nanog etc.). In addition, future studies should explore
alternative roles for Hedgehog in the radiation response. For example, current work in our
laboratory is focused on the ability of Hh inhibition to radiosensitize cell-line derived EAC cells
via cell cycle redistribution.
The present studies describe a set of EAC xenograft models with heterogeneous
radiosensitivities. These models are therefore valuable tools for preclinical studies of (a) the in
vivo EAC tumour radiation response, and (b) combined modality therapies incorporating novel
targeted agents. We sought to determine whether in vivo inhibition of Sonic Hh could increase
the growth delay of irradiated tumours. However, due to time constraints and the transient
availability of xenograft tumours for implantation, we unknowingly selected a xenograft line
that does not appear to upregulate Hh signalling in response to irradiation. It is therefore a
limitation of our study that experimentation was not sequential. Had gene expression data
been available at the time, a more appropriate line would have been selected for Hh inhibition.
Moving forward, our xenograft lines will continue to be used for Hh inhibition studies. Our
laboratory has obtained two clinically-used Smoothened inhibitors (LDE225, Novartis and BMS-
833923, BMS). Future work will aim to incorporate these targeted agents, as well as a research-

102
grade GLI1 antibody, into a radiation-based treatment scheme in our xenograft model. Our
immediate aim is to demonstrate that Hh inhibition in Lines 6 and 8 can increase the growth
delay and specific growth delay of irradiated tumours relative to radiation or inhibitor alone. A
demonstration of the radiosensitizing effect of Hh inhibition would have profound clinical-
translational implications.
It may be the case that Hh inhibition does not “prime” cells for irradiation, but prevents
repopulation after radiotherapy. In other words, it is unclear whether Hh inhibition would act
syngergistically or additively in combination with radiation. Future work should aim to
distinguish between these two effects in order to optimize the timing and dosing of pathway
inhibition. These initiatives will be complemented by further efforts to map out the radiation-
induced gene expression changes both in additional xenograft lines, and at additional time
points after radiotherapy. Ultimately, a more robust understanding of the role and timing of
upregulated Hh signalling after irradiation will guide clinical decision-making in a multi-modality
therapeutic setting.

103
Chapter 6: Conclusion
Despite the pervasive clinical radioresistance of EAC tumours, little is known about the
underlying mechanisms driving this resistance. A paucity of validated preclinical models has
contributed to this knowledge deficit.
We have described seven primary EAC xenograft models that, while encompassing a range of
radiosensitivities, are statistically indistinguishable from each other based on specific growth
delay. Nevertheless, a qualitative assessment of the range of GD and SGD values, both within
and between xenograft lines leaves open the possibility that subtle differences in intrinsic
radiosensitivity may be detected with increasing sample sizes and further optimization of
experimental techniques.
Using this model, we have demonstrated that ionizing radiation enriches the tumourigenic
population in two patient-derived EAC tumours. It remains inconclusive whether the TIC
frequency returns to baseline following tumour recovery after radiotherapy. Our finding that
radiation may enrich the TIC component of a solid tumour has not been reported in the
literature, and is therefore valuable not only from a clinical-translational perspective, but as a
proof-of-principle that LDAs are a valuable tool for probing the cell phenotypic factors
contributing to radioresistance.
Building on a foundation of evidence that the Hh pathway is activated in EAC, we next used our
xenograft model to interrogate the response of this pathway to irradiation. We found that in

104
the absence of radiation, Hh expression patterns in EAC suggest a predominantly paracrine
mechanism, corroborating published evidence from studies using other detection techniques.
Furthermore, we found that Hh expression is upregulated following irradiation in two primary
xenograft lines, suggesting that the pathway may be activated in response to radiotherapy.
Upregulation occurred in both autocrine (tumour cell to tumour cell) and paracrine (tumour to
stroma) patterns. A third xenograft line appeared to upregulate Hh pathway components in a
reverse paracrine direction, however pathway inhibition with 5E1 in this line did not delay
tumour growth or regrowth following radiotherapy, calling into question the functionality of
the pathway in this line.
Taken together, the results from these studies support the hypothesis that EAC radioresistance
can be modeled in parallel with EAC carcinogenesis. That is, embryonic development pathways
usurped during EAC development may take on a heightened role in the tumour response to
irradiation. In this case, pathway upregulation may reflect a widespread anti-apoptotic and/or
proliferative response following radiation-induced DNA damage. Alternatively, upregulation
following irradiation may be restricted to TICs and TIC niches, reflecting an acute signal to
promote TIC self-renewal in the face of widespread cytotoxicity. Since no EAC TIC markers have
been found to date, it remains unclear whether Hh signalling exclusively supports the EAC TIC
compartment. For this reason, efforts at measuring TIC frequencies with LDAs before and after
Hh inhibition should be pursued.
Despite the recent failure of SMO inhibitor IPI-929 in a phase II clinical trial of pancreatic
cancer, our data support findings in both EAC and other tumour sites that SMO inhibitors have

105
an important place in the clinical management of Hh-expressing cancers. We observed
upregulation of Hh transcripts in the first two to three weeks after irradiation (during the
plateau phase of the growth curve), suggesting that clinical integration of a SMO inhibitor after
radiotherapy may improve patient response. However, further efforts are needed to both
buttress our reported findings, and to determine whether Hh inhibition is most effective when
used before or after radiotherapy.

106
Appendix A: Xenograft growth curves for all seven models

107

108
\

109
Appendix B: Sample flow cytometric plots of H2K depletion for limiting dilution and clonogenic assays
EpC
AM
-AP
C
H2K-PECy7
Line 4 (P6)
Line 3(P4)
Line 2(P10)
Pre-depletion Cells remaining in column Effluent

110
References
1. Parkin, D.M., Bray, F., Ferlay, J. & Pisani, P. Global cancer statistics, 2002. CA: a cancer journal for clinicians 55, 74-108
2. Kamangar, F., Dores, G.M. & Anderson, W.F. Patterns of cancer incidence, mortality, and prevalence across five continents: defining priorities to reduce cancer disparities in different geographic regions of the world. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 24, 2137-50 (2006).
3. Weiss, E., Meneshian, A. & Brock, M. The biology of epithelial esophageal cancer. Esophageal cancer: principles and practice 27-38 (2009).
4. Cook, M.B., Chow, W.-H. & Devesa, S.S. Oesophageal cancer incidence in the United States by race, sex, and histologic type, 1977-2005. British journal of cancer 101, 855-9 (2009).
5. Pohl, H. & Welch, H.G. The role of overdiagnosis and reclassification in the marked increase of esophageal adenocarcinoma incidence. Journal of the National Cancer Institute 97, 142-6 (2005).
6. Lepage, C., Rachet, B., Jooste, V., Faivre, J. & Coleman, M.P. Continuing rapid increase in esophageal adenocarcinoma in England and Wales. The American journal of gastroenterology 103, 2694-9 (2008).
7. Bosetti, C. et al. Trends in oesophageal cancer incidence and mortality in Europe. International journal of cancer. Journal international du cancer 122, 1118-29 (2008).
8. Pera, M., Manterola, C., Vidal, O. & Grande, L. Epidemiology of esophageal adenocarcinoma. Journal of surgical oncology 92, 151-9 (2005).
9. DeMeester, S.R. Adenocarcinoma of the esophagus and cardia: a review of the disease and its treatment. Annals of surgical oncology 13, 12-30 (2006).
10. Abrams, J.A., Sharaiha, R.Z., Gonsalves, L., Lightdale, C.J. & Neugut, A.I. Dating the rise of esophageal adenocarcinoma: analysis of Connecticut Tumor Registry data, 1940-2007. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology 20, 183-6 (2011).
11. Kong, C.Y. et al. The impact of obesity on the rise in esophageal adenocarcinoma incidence: estimates from a disease simulation model. Cancer epidemiology, biomarkers & prevention : a publication of the American Association for Cancer Research, cosponsored by the American Society of Preventive Oncology 20, 2450-6 (2011).

111
12. Hameeteman, W., Tytgat, G.N., Houthoff, H.J. & van den Tweel, J.G. Barrett’s esophagus: development of dysplasia and adenocarcinoma. Gastroenterology 96, 1249-56 (1989).
13. Jankowski, J., Barr, H., Wang, K. & Delaney, B. Diagnosis and management of Barrett’s oesophagus. BMJ (Clinical research ed.) 341, c4551 (2010).
14. Zhang, H.Y., Spechler, S.J. & Souza, R.F. Esophageal adenocarcinoma arising in Barrett esophagus. Cancer letters 275, 170-7 (2009).
15. Seery, J.P. Stem cells of the oesophageal epithelium. Journal of cell science 115, 1783-9 (2002).
16. Pera, M. et al. Duodenal-content reflux esophagitis induces the development of glandular metaplasia and adenosquamous carcinoma in rats. Carcinogenesis 21, 1587-91 (2000).
17. Jankowski, J.A. & Wright, N.A. Epithelial stem cells in gastrointestinal morphogenesis, adaptation and carcinogenesis. Seminars in cell biology 3, 445-56 (1992).
18. Souza, R.F., Krishnan, K. & Spechler, S.J. Acid, bile, and CDX: the ABCs of making Barrett’s metaplasia. American journal of physiology. Gastrointestinal and liver physiology 295, G211-8 (2008).
19. Shields, H.M. et al. Scanning electron microscopy of the human esophagus: application to Barrett’s esophagus, a precancerous lesion. Microscopy research and technique 31, 248-56 (1995).
20. Wang, D.H. & Souza, R.F. Biology of Barrett’s esophagus and esophageal adenocarcinoma. Gastrointestinal endoscopy clinics of North America 21, 25-38 (2011).
21. O’Riordan, J.M. et al. Proinflammatory cytokine and nuclear factor kappa-B expression along the inflammation-metaplasia-dysplasia-adenocarcinoma sequence in the esophagus. The American journal of gastroenterology 100, 1257-64 (2005).
22. Fitzgerald, R.C. et al. Inflammatory gradient in Barrett’s oesophagus: implications for disease complications. Gut 51, 316-22 (2002).
23. Chen, X. et al. Oxidative damage in an esophageal adenocarcinoma model with rats. Carcinogenesis 21, 257-63 (2000).
24. Wetscher, G.J. et al. Reflux esophagitis in humans is mediated by oxygen-derived free radicals. American journal of surgery 170, 552-6; discussion 556-7 (1995).
25. Clemons, N.J., McColl, K.E.L. & Fitzgerald, R.C. Nitric oxide and acid induce double-strand DNA breaks in Barrett’s esophagus carcinogenesis via distinct mechanisms. Gastroenterology 133, 1198-209 (2007).

112
26. McConnell, P. & Jobe, B. Barrett’s esophagus: molecular biology. Esophageal cancer: principles and practice 53-60 (2009).
27. Sanghvi, P., Choi, M., Holland, J. & Thomas, C. Adjuvant (post-operative) therapy. Esophageal cancer: principles and practice 401-405 (2009).
28. Herskovic, A. et al. Combined chemotherapy and radiotherapy compared with radiotherapy alone in patients with cancer of the esophagus. The New England journal of medicine 326, 1593-8 (1992).
29. Rice, T.W., Adelstein, D.J., Zuccaro, G., Falk, G.W. & Goldblum, J.R. Advances in the treatment of esophageal carcinoma. The Gastroenterologist 5, 278-94 (1997).
30. Fiorica, F. et al. Preoperative chemoradiotherapy for oesophageal cancer: a systematic review and meta-analysis. Gut 53, 925-30 (2004).
31. Gebski, V. et al. Survival benefits from neoadjuvant chemoradiotherapy or chemotherapy in oesophageal carcinoma: a meta-analysis. The lancet oncology 8, 226-34 (2007).
32. Defoe, S.G. et al. Retrospective review of patients with locally advanced esophageal cancer treated at the University of Pittsburgh. American journal of clinical oncology 34, 587-92 (2011).
33. Walsh, T.N. et al. A comparison of multimodal therapy and surgery for esophageal adenocarcinoma. The New England journal of medicine 335, 462-7 (1996).
34. Polednak, A.P. Trends in survival for both histologic types of esophageal cancer in US surveillance, epidemiology and end results areas. International journal of cancer. Journal international du cancer 105, 98-100 (2003).
35. Minsky, B.D. et al. INT 0123 (Radiation Therapy Oncology Group 94-05) phase III trial of combined-modality therapy for esophageal cancer: high-dose versus standard-dose radiation therapy. Journal of clinical oncology : official journal of the American Society of Clinical Oncology 20, 1167-74 (2002).
36. Sun, Y. & Nelson, P.S. Molecular Pathways Involving Microenvironment Damage Responses in Cancer Therapy Resistance. Clinical cancer research : an official journal of the American Association for Cancer Research (2012).doi:10.1158/1078-0432.CCR-11-0768
37. Revesz, L. Effect of tumour cells killed by x-rays upon the growth of admixed viable cells. Nature 178, 1391-2 (1956).
38. Seelig, K.J. & Revesz, L. Effect of lethally damaged tumour cells upon the growth of admixed viable cells in diffusion chambers. British journal of cancer 14, 126-38 (1960).

113
39. Bavik, C. et al. The gene expression program of prostate fibroblast senescence modulates neoplastic epithelial cell proliferation through paracrine mechanisms. Cancer research 66, 794-802 (2006).
40. Hazlehurst, L.A., Damiano, J.S., Buyuksal, I., Pledger, W.J. & Dalton, W.S. Adhesion to fibronectin via beta1 integrins regulates p27kip1 levels and contributes to cell adhesion mediated drug resistance (CAM-DR). Oncogene 19, 4319-27 (2000).
41. Shain, K.H., Landowski, T.H. & Dalton, W.S. Adhesion-mediated intracellular redistribution of c-Fas-associated death domain-like IL-1-converting enzyme-like inhibitory protein-long confers resistance to CD95-induced apoptosis in hematopoietic cancer cell lines. Journal of immunology (Baltimore, Md. : 1950) 168, 2544-53 (2002).
42. Nordsmark, M., Overgaard, M. & Overgaard, J. Pretreatment oxygenation predicts radiation response in advanced squamous cell carcinoma of the head and neck. Radiotherapy and oncology : journal of the European Society for Therapeutic Radiology and Oncology 41, 31-9 (1996).
43. Brizel, D.M., Dodge, R.K., Clough, R.W. & Dewhirst, M.W. Oxygenation of head and neck cancer: changes during radiotherapy and impact on treatment outcome. Radiotherapy and oncology : journal of the European Society for Therapeutic Radiology and Oncology 53, 113-7 (1999).
44. Semenza, G.L. Targeting HIF-1 for cancer therapy. Nature reviews. Cancer 3, 721-32 (2003).
45. Aebersold, D.M. et al. Expression of hypoxia-inducible factor-1alpha: a novel predictive and prognostic parameter in the radiotherapy of oropharyngeal cancer. Cancer research 61, 2911-6 (2001).
46. Unruh, A. et al. The hypoxia-inducible factor-1 alpha is a negative factor for tumor therapy. Oncogene 22, 3213-20 (2003).
47. Moeller, B.J., Cao, Y., Li, C.Y. & Dewhirst, M.W. Radiation activates HIF-1 to regulate vascular radiosensitivity in tumors: role of reoxygenation, free radicals, and stress granules. Cancer cell 5, 429-41 (2004).
48. Zhang, L., Subarsky, P. & Hill, R.P. Hypoxia-regulated p53 and its effect on radiosensitivity in cancer cells. International journal of radiation biology 83, 443-56 (2007).
49. Kastan, M.B. DNA damage responses: mechanisms and roles in human disease: 2007 G.H.A. Clowes Memorial Award Lecture. Molecular cancer research : MCR 6, 517-24 (2008).
50. Mimitou, E.P. & Symington, L.S. Nucleases and helicases take center stage in homologous recombination. Trends in biochemical sciences 34, 264-72 (2009).

114
51. San Filippo, J., Sung, P. & Klein, H. Mechanism of eukaryotic homologous recombination. Annual review of biochemistry 77, 229-57 (2008).
52. Pawlik, T.M. & Keyomarsi, K. Role of cell cycle in mediating sensitivity to radiotherapy. International journal of radiation oncology, biology, physics 59, 928-42 (2004).
53. Chavaudra, N., Bourhis, J. & Foray, N. Quantified relationship between cellular radiosensitivity, DNA repair defects and chromatin relaxation: a study of 19 human tumour cell lines from different origin. Radiotherapy and oncology : journal of the European Society for Therapeutic Radiology and Oncology 73, 373-82 (2004).
54. Bionda, C. et al. Radioresistance of human carcinoma cells is correlated to a defect in raft membrane clustering. Free radical biology & medicine 43, 681-94 (2007).
55. Chen, M. et al. Suppression of Bcl-2 messenger RNA production may mediate apoptosis after ionizing radiation, tumor necrosis factor alpha, and ceramide. Cancer research 55, 991-4 (1995).
56. Koniaras, K., Cuddihy, A.R., Christopoulos, H., Hogg, A. & O’Connell, M.J. Inhibition of Chk1-dependent G2 DNA damage checkpoint radiosensitizes p53 mutant human cells. Oncogene 20, 7453-63 (2001).
57. Hötte, G.J., Linam-Lennon, N., Reynolds, J.V. & Maher, S.G. Radiation sensitivity of esophageal adenocarcinoma: the contribution of the RNA-binding protein RNPC1 and p21-mediated cell cycle arrest to radioresistance. Radiation research 177, 272-9 (2012).
58. Lee, J.M. & Bernstein, A. p53 mutations increase resistance to ionizing radiation. Proceedings of the National Academy of Sciences of the United States of America 90, 5742-6 (1993).
59. Bristow, R.G. et al. Radioresistant MTp53-expressing rat embryo cell transformants exhibit increased DNA-dsb rejoining during exposure to ionizing radiation. Oncogene 16, 1789-802 (1998).
60. Sinclair, W.K. & Morton, R.A. X-ray sensitivity during the cell generation cycle of cultured Chinese hamster cells. Radiation research 29, 450-74 (1966).
61. Tutt, A. et al. Cell cycle and genetic background dependence of the effect of loss of BRCA2 on ionizing radiation sensitivity. Oncogene 22, 2926-31 (2003).
62. Boonstra, J.J. et al. Verification and unmasking of widely used human esophageal adenocarcinoma cell lines. Journal of the National Cancer Institute 102, 271-4 (2010).
63. Lynam-Lennon, N. et al. Alterations in DNA repair efficiency are involved in the radioresistance of esophageal adenocarcinoma. Radiation research 174, 703-11 (2010).

115
64. Kim, A.H. et al. Transforming growth factor-beta is an endogenous radioresistance factor in the esophageal adenocarcinoma cell line OE-33. International journal of oncology 23, 1593-9 (2003).
65. Lee, J. et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer cell 9, 391-403 (2006).
66. Harris, L.C. et al. Changes in O6-methylguanine-DNA methyltransferase expression during immortalization of cloned human fibroblasts. Carcinogenesis 17, 219-24 (1996).
67. Taylor, A.C. et al. P53 mutation and MDM2 amplification frequency in pediatric rhabdomyosarcoma tumors and cell lines. Medical and pediatric oncology 35, 96-103 (2000).
68. Rubio-Viqueira, B. & Hidalgo, M. Direct in vivo xenograft tumor model for predicting chemotherapeutic drug response in cancer patients. Clinical pharmacology and therapeutics 85, 217-21 (2009).
69. Rubio-Viqueira, B. et al. An in vivo platform for translational drug development in pancreatic cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 12, 4652-61 (2006).
70. Loi, M. et al. The use of the orthotopic model to validate antivascular therapies for cancer. The International journal of developmental biology 55, 547-55 (2011).
71. Liu, M. & Hicklen, D. Tumor xenograft efficacy models. Tumor models in cancer research (2011).
72. Blunt, T. et al. Defective DNA-dependent protein kinase activity is linked to V(D)J recombination and DNA repair defects associated with the murine scid mutation. Cell 80, 813-23 (1995).
73. Shultz, L.D. et al. Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. Journal of immunology (Baltimore, Md. : 1950) 154, 180-91 (1995).
74. Biedermann, K.A., Sun, J.R., Giaccia, A.J., Tosto, L.M. & Brown, J.M. scid mutation in mice confers hypersensitivity to ionizing radiation and a deficiency in DNA double-strand break repair. Proceedings of the National Academy of Sciences of the United States of America 88, 1394-7 (1991).
75. Stenstrom, K.W., Vermund, H., Mosser, D.G. & Marvin, J.F. Effects of roentgen irradiation on the tumor bed. I. The inhibiting action of local pretransplantation roentgen irradiation (1500 r alpha) on the growth of mouse mammary carcinoma. Radiation research 2, 180-91 (1955).

116
76. Haveman, J., Rodermond, H., van Bree, C., Wondergem, J. & Franken, N.A.P. Residual late radiation damage in mouse stromal tissue assessed by the tumor bed effect. Journal of radiation research 48, 107-12 (2007).
77. Rofstad, E.K., Mathiesen, B., Henriksen, K., Kindem, K. & Galappathi, K. The tumor bed effect: increased metastatic dissemination from hypoxia-induced up-regulation of metastasis-promoting gene products. Cancer research 65, 2387-96 (2005).
78. Budach, W., Taghian, A., Freeman, J., Gioioso, D. & Suit, H.D. Impact of stromal sensitivity on radiation response of tumors. Journal of the National Cancer Institute 85, 988-93 (1993).
79. Garcia-Barros, M. et al. Tumor response to radiotherapy regulated by endothelial cell apoptosis. Science (New York, N.Y.) 300, 1155-9 (2003).
80. García-Barros, M. et al. Impact of stromal sensitivity on radiation response of tumors implanted in SCID hosts revisited. Cancer research 70, 8179-86 (2010).
81. Gerweck, L.E., Vijayappa, S., Kurimasa, A., Ogawa, K. & Chen, D.J. Tumor cell radiosensitivity is a major determinant of tumor response to radiation. Cancer research 66, 8352-5 (2006).
82. Nowell, P.C. The clonal evolution of tumor cell populations. Science (New York, N.Y.) 194, 23-8 (1976).
83. Fearon, E.R. & Vogelstein, B. A genetic model for colorectal tumorigenesis. Cell 61, 759-67 (1990).
84. Weissman, I.L., Anderson, D.J. & Gage, F. Stem and progenitor cells: origins, phenotypes, lineage commitments, and transdifferentiations. Annual review of cell and developmental biology 17, 387-403 (2001).
85. Bonnet, D. & Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nature medicine 3, 730-7 (1997).
86. Al-Hajj, M., Wicha, M.S., Benito-Hernandez, A., Morrison, S.J. & Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proceedings of the National Academy of Sciences of the United States of America 100, 3983-8 (2003).
87. Singh, S.K. et al. Identification of a cancer stem cell in human brain tumors. Cancer research 63, 5821-8 (2003).
88. Zhang, C., Li, C., He, F., Cai, Y. & Yang, H. Identification of CD44+CD24+ gastric cancer stem cells. Journal of cancer research and clinical oncology 137, 1679-86 (2011).

117
89. Lapidot, T. et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature 367, 645-8 (1994).
90. Li, C. et al. Identification of pancreatic cancer stem cells. Cancer research 67, 1030-7 (2007).
91. Ricci-Vitiani, L. et al. Identification and expansion of human colon-cancer-initiating cells. Nature 445, 111-5 (2007).
92. Chan, K.S. et al. Identification, molecular characterization, clinical prognosis, and therapeutic targeting of human bladder tumor-initiating cells. Proceedings of the National Academy of Sciences of the United States of America 106, 14016-21 (2009).
93. Prince, M.E. et al. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proceedings of the National Academy of Sciences of the United States of America 104, 973-8 (2007).
94. Eramo, A. et al. Identification and expansion of the tumorigenic lung cancer stem cell population. Cell death and differentiation 15, 504-14 (2008).
95. Zhou, B.-B.S. et al. Tumour-initiating cells: challenges and opportunities for anticancer drug discovery. Nature reviews. Drug discovery 8, 806-23 (2009).
96. Yang, G. et al. Dynamic equilibrium between cancer stem cells and non-stem cancer cells in human SW620 and MCF-7 cancer cell populations. British journal of cancer 106, 1512-9 (2012).
97. Chaffer, C.L. et al. Normal and neoplastic nonstem cells can spontaneously convert to a stem-like state. Proceedings of the National Academy of Sciences of the United States of America 108, 7950-5 (2011).
98. Liang, Y. et al. Stem-like cancer cells are inducible by increasing genomic instability in cancer cells. The Journal of biological chemistry 285, 4931-40 (2010).
99. Baumann, M., Krause, M. & Hill, R. Exploring the role of cancer stem cells in radioresistance. Nature reviews. Cancer 8, 545-54 (2008).
100. Baumann, M., Dubois, W. & Suit, H.D. Response of human squamous cell carcinoma xenografts of different sizes to irradiation: relationship of clonogenic cells, cellular radiation sensitivity in vivo, and tumor rescuing units. Radiation research 123, 325-30 (1990).
101. Hill, R.P. & Milas, L. The proportion of stem cells in murine tumors. International journal of radiation oncology, biology, physics 16, 513-8 (1989).

118
102. Bao, S. et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 444, 756-60 (2006).
103. Bao, S. et al. Targeting cancer stem cells through L1CAM suppresses glioma growth. Cancer research 68, 6043-8 (2008).
104. Cheng, L. et al. L1CAM regulates DNA damage checkpoint response of glioblastoma stem cells through NBS1. The EMBO journal 30, 800-13 (2011).
105. Diehn, M. et al. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 458, 780-3 (2009).
106. Phillips, T.M., McBride, W.H. & Pajonk, F. The response of CD24(-/low)/CD44+ breast cancer-initiating cells to radiation. Journal of the National Cancer Institute 98, 1777-85 (2006).
107. Karimi-Busheri, F., Rasouli-Nia, A., Mackey, J.R. & Weinfeld, M. Senescence evasion by MCF-7 human breast tumor-initiating cells. Breast cancer research : BCR 12, R31 (2010).
108. Grotenhuis, B.A. et al. Barrett’s oesophageal adenocarcinoma encompasses tumour-initiating cells that do not express common cancer stem cell markers. The Journal of pathology 221, 379-89 (2010).
109. Ahmad, J. et al. Re: Grotenhuis et al. Barrett’s oesophageal adenocarcinoma encompasses tumour-initiating cells that do not express common cancer stem cell markers. J Pathol 2010; 221: 379-389. The Journal of pathology 224, 143-5 (2011).
110. Bobryshev, Y.V. et al. Expression of the putative stem cell marker Musashi-1 in Barrett’s esophagus and esophageal adenocarcinoma. Diseases of the esophagus : official journal of the International Society for Diseases of the Esophagus / I.S.D.E 23, 580-9 (2010).
111. Vega, K.J. et al. Identification of the putative intestinal stem cell marker doublecortin and CaM kinase-like-1 in Barrett’s esophagus and esophageal adenocarcinoma. Journal of gastroenterology and hepatology 27, 773-80 (2012).
112. Barker, N. et al. Identification of stem cells in small intestine and colon by marker gene Lgr5. Nature 449, 1003-7 (2007).
113. McClanahan, T. et al. Identification of overexpression of orphan G protein-coupled receptor GPR49 in human colon and ovarian primary tumors. Cancer biology & therapy 5, 419-26 (2006).
114. Yamamoto, Y. et al. Overexpression of orphan G-protein-coupled receptor, Gpr49, in human hepatocellular carcinomas with beta-catenin mutations. Hepatology (Baltimore, Md.) 37, 528-33 (2003).

119
115. Vermeulen, L. et al. Single-cell cloning of colon cancer stem cells reveals a multi-lineage differentiation capacity. Proceedings of the National Academy of Sciences of the United States of America 105, 13427-32 (2008).
116. Becker, L., Huang, Q. & Mashimo, H. Lgr5, an intestinal stem cell marker, is abnormally expressed in Barrett’s esophagus and esophageal adenocarcinoma. Diseases of the esophagus : official journal of the International Society for Diseases of the Esophagus / I.S.D.E 23, 168-74 (2010).
117. von Rahden, B.H.A. et al. LgR5 expression and cancer stem cell hypothesis: clue to define the true origin of esophageal adenocarcinomas with and without Barrett’s esophagus? Journal of experimental & clinical cancer research : CR 30, 23 (2011).
118. Quante, M. et al. Bile acid and inflammation activate gastric cardia stem cells in a mouse model of Barrett-like metaplasia. Cancer cell 21, 36-51 (2012).
119. Bremner, C.G., Lynch, V.P. & Ellis, F.H. Barrett’s esophagus: congenital or acquired? An experimental study of esophageal mucosal regeneration in the dog. Surgery 68, 209-16 (1970).
120. Hamilton, S.R. & Yardley, J.H. Regnerative of cardiac type mucosa and acquisition of Barrett mucosa after esophagogastrostomy. Gastroenterology 72, 669-75 (1977).
121. Kalabis, J. et al. A subpopulation of mouse esophageal basal cells has properties of stem cells with the capacity for self-renewal and lineage specification. The Journal of clinical investigation 118, 3860-9 (2008).
122. Jankowski, J.A. et al. Molecular evolution of the metaplasia-dysplasia-adenocarcinoma sequence in the esophagus. The American journal of pathology 154, 965-73 (1999).
123. Pera, M. & Pera, M. Experimental Barrett’s esophagus and the origin of intestinal metaplasia. Chest surgery clinics of North America 12, 25-37 (2002).
124. Glickman, J.N., Chen, Y.Y., Wang, H.H., Antonioli, D.A. & Odze, R.D. Phenotypic characteristics of a distinctive multilayered epithelium suggests that it is a precursor in the development of Barrett’s esophagus. The American journal of surgical pathology 25, 569-78 (2001).
125. Leedham, S.J. et al. Individual crypt genetic heterogeneity and the origin of metaplastic glandular epithelium in human Barrett’s oesophagus. Gut 57, 1041-8 (2008).
126. Wang, X. et al. Residual embryonic cells as precursors of a Barrett’s-like metaplasia. Cell 145, 1023-35 (2011).
127. Lum, L. & Beachy, P.A. The Hedgehog response network: sensors, switches, and routers. Science (New York, N.Y.) 304, 1755-9 (2004).

120
128. Ahlgren, S.C. & Bronner-Fraser, M. Inhibition of sonic hedgehog signaling in vivo results in craniofacial neural crest cell death. Current biology : CB 9, 1304-14 (1999).
129. Rowitch, D.H. et al. Sonic hedgehog regulates proliferation and inhibits differentiation of CNS precursor cells. The Journal of neuroscience : the official journal of the Society for Neuroscience 19, 8954-65 (1999).
130. Zhang, Y. & Kalderon, D. Hedgehog acts as a somatic stem cell factor in the Drosophila ovary. Nature 410, 599-604 (2001).
131. Lai, K., Kaspar, B.K., Gage, F.H. & Schaffer, D.V. Sonic hedgehog regulates adult neural progenitor proliferation in vitro and in vivo. Nature neuroscience 6, 21-7 (2003).
132. Beachy, P.A., Karhadkar, S.S. & Berman, D.M. Tissue repair and stem cell renewal in carcinogenesis. Nature 432, 324-31 (2004).
133. Rahnama, F., Toftgård, R. & Zaphiropoulos, P.G. Distinct roles of PTCH2 splice variants in Hedgehog signalling. The Biochemical journal 378, 325-34 (2004).
134. van den Brink, G.R. Hedgehog signaling in development and homeostasis of the gastrointestinal tract. Physiological reviews 87, 1343-75 (2007).
135. Sasaki, H., Nishizaki, Y., Hui, C., Nakafuku, M. & Kondoh, H. Regulation of Gli2 and Gli3 activities by an amino-terminal repression domain: implication of Gli2 and Gli3 as primary mediators of Shh signaling. Development 126, 3915-24 (1999).
136. Lipinski, R.J., Gipp, J.J., Zhang, J., Doles, J.D. & Bushman, W. Unique and complimentary activities of the Gli transcription factors in Hedgehog signaling. Experimental cell research 312, 1925-38 (2006).
137. Bai, C.B., Auerbach, W., Lee, J.S., Stephen, D. & Joyner, A.L. Gli2, but not Gli1, is required for initial Shh signaling and ectopic activation of the Shh pathway. Development 129, 4753-4761 (2002).
138. Yoon, J.W. et al. Gene expression profiling leads to identification of GLI1-binding elements in target genes and a role for multiple downstream pathways in GLI1-induced cell transformation. The Journal of biological chemistry 277, 5548-55 (2002).
139. Katoh, Y. & Katoh, M. Hedgehog target genes: mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Current molecular medicine 9, 873-86 (2009).
140. Fei, T. et al. Genome-wide mapping of SMAD target genes reveals the role of BMP signaling in embryonic stem cell fate determination. Genome research 20, 36-44 (2010).

121
141. Ying, Q.-L., Stavridis, M., Griffiths, D., Li, M. & Smith, A. Conversion of embryonic stem cells into neuroectodermal precursors in adherent monoculture. Nature biotechnology 21, 183-6 (2003).
142. Bolós, V., Grego-Bessa, J. & de la Pompa, J.L. Notch signaling in development and cancer. Endocrine reviews 28, 339-63 (2007).
143. Duncan, A.W. et al. Integration of Notch and Wnt signaling in hematopoietic stem cell maintenance. Nature immunology 6, 314-22 (2005).
144. Radtke, F. & Clevers, H. Self-renewal and cancer of the gut: two sides of a coin. Science (New York, N.Y.) 307, 1904-9 (2005).
145. Katoh, Y. & Katoh, M. Hedgehog signaling pathway and gastrointestinal stem cell signaling network (review). International journal of molecular medicine 18, 1019-23 (2006).
146. Wei, C.-L. et al. A global map of p53 transcription-factor binding sites in the human genome. Cell 124, 207-19 (2006).
147. Tian, H. et al. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proceedings of the National Academy of Sciences of the United States of America 106, 4254-9 (2009).
148. Ramalho-Santos, M., Melton, D.A. & McMahon, A.P. Hedgehog signals regulate multiple aspects of gastrointestinal development. Development (Cambridge, England) 127, 2763-72 (2000).
149. Chen, W. et al. Canonical hedgehog signaling augments tumor angiogenesis by induction of VEGF-A in stromal perivascular cells. Proceedings of the National Academy of Sciences of the United States of America 108, 9589-94 (2011).
150. Dierks, C. et al. Essential role of stromally induced hedgehog signaling in B-cell malignancies. Nature medicine 13, 944-51 (2007).
151. Watkins, D.N. et al. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature 422, 313-7 (2003).
152. Berman, D.M. et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 425, 846-51 (2003).
153. Madison, B.B. et al. Epithelial hedgehog signals pattern the intestinal crypt-villus axis. Development (Cambridge, England) 132, 279-89 (2005).
154. Litingtung, Y., Lei, L., Westphal, H. & Chiang, C. Sonic hedgehog is essential to foregut development. Nature genetics 20, 58-61 (1998).

122
155. Ma, X. et al. Hedgehog signaling is activated in subsets of esophageal cancers. International journal of cancer. Journal international du cancer 118, 139-48 (2006).
156. Wang, D.H. et al. Aberrant epithelial-mesenchymal Hedgehog signaling characterizes Barrett’s metaplasia. Gastroenterology 138, 1810-22 (2010).
157. Yang, L. et al. Hedgehog signaling activation in the development of squamous cell carcinoma and adenocarcinoma of esophagus. International journal of biochemistry and molecular biology 3, 46-57 (2012).
158. Chaudary, N. et al. Hedgehog pathway signaling in cervical carcinoma and outcome after chemoradiation. Cancer 118, 3105-15 (2012).
159. Chung, C.H. et al. Glioma-associated oncogene family zinc finger 1 expression and metastasis in patients with head and neck squamous cell carcinoma treated with radiation therapy (RTOG 9003). Journal of clinical oncology : official journal of the American Society of Clinical Oncology 29, 1326-34 (2011).
160. Yoshikawa, R. et al. Hedgehog signal activation in oesophageal cancer patients undergoing neoadjuvant chemoradiotherapy. British journal of cancer 98, 1670-4 (2008).
161. Chen, Y.-J. et al. Sonic hedgehog signaling protects human hepatocellular carcinoma cells against ionizing radiation in an autocrine manner. International journal of radiation oncology, biology, physics 80, 851-9 (2011).
162. Abe, Y. et al. Hedgehog signaling overrides p53-mediated tumor suppression by activating Mdm2. Proceedings of the National Academy of Sciences of the United States of America 105, 4838-43 (2008).
163. Fernandez-L, A. et al. Oncogenic YAP promotes radioresistance and genomic instability in medulloblastoma through IGF2-mediated Akt activation. Oncogene 31, 1923-37 (2012).
164. Euhus, D.M., Hudd, C., LaRegina, M.C. & Johnson, F.E. Tumor measurement in the nude mouse. Journal of surgical oncology 31, 229-34 (1986).
165. Camplejohn, R.S. & Penhaligon, M. The tumour bed effect: a cell kinetic and histological investigation of tumours growing in irradiated mouse skin. The British journal of radiology 58, 443-51 (1985).
166. Rofstad, E.K. & Brustad, T. Tumour growth delay following single dose irradiation of human melanoma xenografts. Correlations with tumour growth parameters, vascular structure and cellular radiosensitivity. British journal of cancer 51, 201-10 (1985).
167. Steel, G.G., Courtenay, V.D. & Peckham, M.J. The response to chemotherapy of a variety of human tumour xenografts. British journal of cancer 47, 1-13 (1983).

123
168. Ogawa, K. et al. Influence of tumor cell and stroma sensitivity on tumor response to radiation. Cancer research 67, 4016-21 (2007).
169. Björk-Eriksson, T., West, C., Karlsson, E. & Mercke, C. Discrimination of human tumor radioresponsiveness using low-dose rate irradiation. International journal of radiation oncology, biology, physics 42, 1147-53 (1998).
170. Gerweck, L.E. & Zaidi, S.T. The reproducibility and relationship between single and fractionated dose estimates of the surviving fraction at 2 Gy. International journal of radiation oncology, biology, physics 35, 81-7 (1996).
171. Ivanovic, Z. Hypoxia or in situ normoxia: The stem cell paradigm. Journal of cellular physiology 219, 271-5 (2009).
172. Banda, M., Bommineni, A., Thomas, R.A., Luckinbill, L.S. & Tucker, J.D. Evaluation and validation of housekeeping genes in response to ionizing radiation and chemical exposure for normalizing RNA expression in real-time PCR. Mutation research 649, 126-34 (2008).
173. Caradec, J. et al. “Desperate house genes”: the dramatic example of hypoxia. British journal of cancer 102, 1037-43 (2010).
174. Fink, T. et al. Instability of standard PCR reference genes in adipose-derived stem cells during propagation, differentiation and hypoxic exposure. BMC molecular biology 9, 98 (2008).
175. Ericson, J., Morton, S., Kawakami, A., Roelink, H. & Jessell, T.M. Two critical periods of Sonic Hedgehog signaling required for the specification of motor neuron identity. Cell 87, 661-73 (1996).
176. Datta, S. & Datta, M.W. Sonic Hedgehog signaling in advanced prostate cancer. Cellular and molecular life sciences : CMLS 63, 435-48 (2006).
177. Bellusci, S. et al. Involvement of Sonic hedgehog (Shh) in mouse embryonic lung growth and morphogenesis. Development (Cambridge, England) 124, 53-63 (1997).
178. Peacock, C.D. et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proceedings of the National Academy of Sciences of the United States of America 104, 4048-53 (2007).
179. Clement, V., Sanchez, P., de Tribolet, N., Radovanovic, I. & Ruiz i Altaba, A. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal, and tumorigenicity. Current biology : CB 17, 165-72 (2007).
180. Bar, E.E. et al. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem cells (Dayton, Ohio) 25, 2524-33 (2007).

124
181. Varnat, F. et al. Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO molecular medicine 1, 338-51 (2009).
182. Song, Z. et al. Sonic hedgehog pathway is essential for maintenance of cancer stem-like cells in human gastric cancer. PloS one 6, e17687 (2011).
183. Liu, S. et al. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer research 66, 6063-71 (2006).
184. Dierks, C. et al. Expansion of Bcr-Abl-positive leukemic stem cells is dependent on Hedgehog pathway activation. Cancer cell 14, 238-49 (2008).
185. Zhao, C. et al. Hedgehog signalling is essential for maintenance of cancer stem cells in myeloid leukaemia. Nature 458, 776-9 (2009).
186. Tanese, K. et al. G-protein-coupled receptor GPR49 is up-regulated in basal cell carcinoma and promotes cell proliferation and tumor formation. The American journal of pathology 173, 835-43 (2008).
187. Jaks, V. et al. Lgr5 marks cycling, yet long-lived, hair follicle stem cells. Nature genetics 40, 1291-9 (2008).