Embed Size (px)
Citation preview

Herstellung und Charakterisierung von
Antikörpern gegen das Prion-Protein mit
inhibitorischer Wirkung auf die Prionreplikation
in Zellkultur
Dissertation zur Erlangung des
naturwissenschaftlichen Doktorgrades
der Bayerischen Julius-Maximilians-Universität Würzburg
vorgelegt von
Tanja Hoffmann
aus Datteln
Würzburg, Juli 2008

Eingereicht am: …………………………………………..…………..
Mitglieder der Promotionskommission:
Vorsitzender: ………………………………………………………….
Gutachter: Prof. Dr. M. A. Klein
Gutachter: Prof. Dr. J. Hacker
Tag des Promotionskolloquiums: ……………………………..……
Doktorurkunde ausgehändigt am: ………………………………….

„Magna pars est profectus velle proficere.“
„Ein großer Teil des Fortschreitens besteht darin, dass wir fortschreiten wollen.“
(Lucius Annaeus Seneca, 4 v. Chr./65 n. Chr.)

INHALTSVERZEICHNIS
i
1 Einführung und Problemstellung 5
1.1 Prionen und Prionenerkrankungen 5
1.1.1 Übertragbare spongiforme Enzephalopathien 5
1.1.2 Prionenerkrankungen bei Tieren 6
1.1.3 Prionenerkrankungen beim Menschen 6
1.1.4 Das Prion-Konzept 9
1.1.5 Das Prion-Protein 11
1.1.6 Funktionen von PrPC 12
1.1.7 Strukturelle und biochemische Eigenschaften von PrPC und PrPSc 13
1.1.8 Replikationsmechanismen und Prionenstämme 15
1.1.9 Therapeutische Ansätze für Prionenerkrankungen 18
1.2 Immunsystem und Antikörper 22
1.2.1 Das Immunsystem 22
1.2.2 Struktur und Isotypen von Maus-Immunglobulinen 23
1.2.3 Antikörpervielfalt durch somatische Rekombination 25
1.2.4 Antigen-Antikörper-Interaktionen 27
1.2.5 Produktion von Antikörpern 28
1.2.6 Therapeutische Anwendung von monoklonalen Antikörpern 30
1.3 Ziel der Arbeit 34
2 Material und Methoden 35
2.1 Material 35
2.1.1 Geräte 35
2.1.2 Chemikalien 36
2.1.3 Zelllinien 36
2.1.4 Lösungen zur Analyse und Klonierung von DNA 36
2.1.5 Standardlösungen 37
2.1.6 Antikörper und Farbstoffe 38
2.1.7 Scrapie-Prionenstamm 38
2.1.8 Plasmide 38
2.1.9 Primer 39
2.1.10 Proteine 40
2.1.11 Versuchstiere 40
2.2 Methoden 40
2.2.1 Herstellung von monoklonalen Antikörpern 40

INHALTSVERZEICHNIS
ii
2.2.1.1 Immunisierung und Isolierung von Milzzellen 40
2.2.1.2 Kultivierung und Vorbereitung von Myelomzellen 41
2.2.1.3 Fusion der Milzzellen mit Myelomzellen 42
2.2.1.4 Screening der Hybridomzellen 42
2.2.1.5 Klonierung der Hybridomzelllinien und Nomenklatur der Klone 43
2.2.1.6 Produktion der monoklonalen Antikörper 43
2.2.1.7 Aufreinigung der monoklonalen Antikörper mittels Protein G 44
2.2.1.8 Bestimmung der Antikörperklasse und der Isotypen 45
2.2.2 Zellbiologische Methoden 46
2.2.2.1 Allgemeine Kulturbedingungen 46
2.2.2.2 Bestimmung der Zellzahl mit dem Hämazytometer 46
2.2.2.3 Einfrieren und Auftauen von Zellen 47
2.2.2.4 Behandlung von Zellen mit Anti-PrP-Antikörpern 47
2.2.2.4.1 Inhibition der Prionreplikation in ScN2a-Zellen 47
2.2.2.4.2 Analyse der Retention auf ScN2a-Zellen 48
2.2.2.5 MTT-Test 49
2.2.2.6 Transfektion von 293T-Zellen mittels Kalziumphosphat-Präzipitation 49
2.2.3 Proteinbiochemische Methoden 50
2.2.3.1 Herstellung von Zelllysaten 50
2.2.3.2 Herstellung von Gehirnhomogenaten 51
2.2.3.3 Proteinbestimmung nach Bradford 51
2.2.3.4 Proteinase K-Behandlung von Homogenaten 51
2.2.3.5 Immunpräzipitation 52
2.2.3.6 SDS-Polyacrylamidgelelektrophorese (SDS-PAGE) 53
2.2.3.7 Western Blot 53
2.2.3.7.1 Blocken und Antikörperinkubation 54
2.2.3.7.2 Chemolumineszenz-Detektion 55
2.2.4 Immunologische Methoden 55
2.2.4.1 Enzym Linked Immunsorbent Assay (ELISA) 55
2.2.4.2 Bestimmung der Avidität von Antikörper 56
2.2.4.3 Bestimmung der Dissoziationskonstanten (KD) mittels ELISA 57
2.2.4.4 Durchflusszytometrische Analyse (FACS) 57
2.2.4.5 Bestimmung der Dissoziationskonstanten (KD) mittels FACS 58
2.2.4.6 Immunfluoreszenzfärbung von Zellen 58
2.2.5 Tierexperimentelle Methoden 59
2.2.5.1 Betäubung der Mäuse mit Ether 59
2.2.5.2 Blutentnahme und Serumgewinnung 60

INHALTSVERZEICHNIS
iii
2.2.5.3 Herzpunktion und Organentnahme 60
2.2.5.4 Bestimmung der Pharmakokinetik der Anti-PrP-Antikörper 60
2.2.5.5 Bioassay 61
2.2.6 Molekularbiologische Methoden 61
2.2.6.1 Klonierung 61
2.2.6.1.1 Präparative Polymerasekettenreaktion (PCR) 61
2.2.6.1.2 Analytische Polymerasekettenreaktion (PCR) 63
2.2.6.1.3 Restriktionsverdau 64
2.2.6.1.4 Agarose-Gelelektrophorese 65
2.2.6.1.5 Gelextraktion 65
2.2.6.1.6 Dephosphorylierung der 5'-DNA-Enden 66
2.2.6.1.7 Phosphorylierung von DNA-Fragmenten 66
2.2.6.1.8 Ligation 67
2.2.6.1.9 Transformation von E.coli mit Plasmiden 67
2.2.6.1.10 Plasmidpräparation aus E.coli 68
2.2.6.1.11 Quantifizierung von doppelsträngiger DNA 68
2.2.6.1.12 Sequenzierung von DNA 69
2.2.6.2 Ortsgerichtete Mutagenese 70
3 Ergebnisse 71
3.1 Herstellung und Reinigung von monoklonalen Anti-PrP-Antikörper 71
3.1.1 Antikörpertiter der immunisierten Prnp0/0-Mäuse und Fusionsausbeute 71
3.1.2 Isolierung von monoklonalen Anti-PrP-Antikörpern 73
3.1.3 Antikörperproduktion und -konzentrierung in hoher Reinheit 76
3.2 Charakterisierung der Anti-PrP-Antikörper 77
3.2.1 Klassen und Subklassen der Anti-PrP-Antikörper 77
3.2.2 Epitop-Bestimmung der Anti-PrP-Antikörper 77
3.2.2.1 PrP-Plasmide mit einer N-terminalen und einer internen Deletion 77
3.2.2.2 Bestimmung des Epitops mittels FACS 80
3.2.2.3 Bestimmung des Minimalepitops mittels Alanin-Scanning 82
3.2.3 Avidität der Anti-PrP-Antikörper 84
3.2.4 Dissoziationskonstanten (KD) der Anti-PrP-Antikörper 86
3.3 Hemmung der Prionreplikation durch Anti-PrP-Antikörper in vitro 87
3.3.1 Einfluss der Anti-PrP-Antikörper auf die PrPSc-Akkumulation in
ScN2a-Zellen 87
3.3.2 Untersuchung zur Zytotoxizität der Anti-PrP-Antikörper 88

INHALTSVERZEICHNIS
iv
3.3.3 Bindung der Anti-PrP-Antikörper an PrPSc von ScN2a-Zellen 89
3.3.4 Untersuchungen zur dosisabhängigen Wirkung der Anti-PrP-Antikörper 91
3.3.5 Untersuchungen zur zeitabhängigen Wirkung der Anti-PrP-Antikörper 92
3.3.6 Bestimmung des PrPSc-Gehalts von ScN2a-Zellen nach Beendigung
der Antikörperbehandlung 93
3.3.7 Analyse der Infektiosität von Antikörper-behandelten ScN2a-Zellen 95
3.4 Untersuchungen zum Wirkungsmechanismus der Anti-PrP-Antikörper 97
3.4.1 Untersuchungen zur inhibitorischen Wirkung eines PrP/Ak-Komplexes 97
3.4.2 Untersuchung des Einflusses der Anti-PrP-Antikörper auf die Retention 99
3.5 Pharmakokinetik der Anti-PrP-Antikörper im in vivo Modell 102
4 Diskussion 106
4.1 Die Herstellung von PrPC- und PrPSc-spezifischen Antikörpern 107
4.2 Inhibition der PrPSc-Akkumulation durch Helix 1-bindende Antikörper 109
4.3 Unterschiede zwischen den Helix 1-bindenden Antikörpern 113
4.4 Hypothesen über den Wirkungsmechanismus der Anti-PrP-Antikörper 115
4.5 Ausblick 118
5 Zusammenfassung 120
5.1 Deutsch 120
5.2 Englisch 122
6 Literaturverzeichnis 124
7 Anhang 143
7.1 Abkürzungsverzeichnis 143
7.2 Tabellen- und Abbildungsverzeichnis 146
7.3 Lebenslauf 148
7.4 Veröffentlichungen und Kongressbeiträge 149
7.5 Erklärung 150
7.6 Danksagung 151

EINLEITUNG
5
1 Einführung und Problemstellung
1.1 Prionen und Prionenerkrankungen
1.1.1 Übertragbare spongiforme Enzephalopathien
Prionenerkrankungen oder übertragbare spongiforme Enzephalopathien (engl.: transmissible
spongiform encephalopathies, TSE) sind letal verlaufende, neurodegenerative Erkrankungen,
die sowohl bei Tieren als auch beim Menschen auftreten können (Collinge, 2001). Unter Tieren
sind die Traberkrankheit (Scrapie) bei Schafen und Ziegen (McGowan, 1922), die Bovine
Spongiforme Enzephalopathie (BSE) bei Rindern (Wells et al., 1987) und die Chronic Wasting
Disease (CWD) bei bestimmten Hirscharten in Nordamerika (Williams und Young, 1980)
bekannt (1.1.2). Die Creutzfeldt-Jakob-Krankheit (CJD) (Creutzfeldt, 1920), das Gerstmann-
Sträussler-Scheinker-Syndrom (GSS) (Gerstmann et al., 1936), Kuru (Gajdusek und Zigas,
1957) sowie die fatale familiäre Schlaflosigkeit (FFI) (Lugaresi et al., 1986) zählen zu den
humanen Prionenerkrankungen. Die Krankheiten werden durch fortschreitende motorische
Störungen (Myoklonien), Gangunsicherheit (Ataxie) sowie den Verlust kognitiver Fähigkeiten
(Demenz) im terminalen Stadium charakterisiert. Zu den typischen neuropathologischen
Veränderungen gehören schwammartige Veränderungen im Gehirn (Spongiose), die
Aktivierung von Astrozyten und Mikrogliazellen (Astrozytose und Gliose), Neuronenverlust und
extrazelluläre Ablagerungen von amyloiden Aggregaten. Im Gegensatz zu anderen
neurodegenerativen Krankheiten können Prionenerkrankungen nicht nur sporadisch und
genetisch auftreten, sondern auch durch Infektionen erworben werden (Collinge, 1997;
Collinge, 2001; Prusiner, 1998). Während die Anzahl der sporadischen Fälle weltweit mit nur
einem Fall pro einer Million relativ gering ist, führte das Auftreten der BSE-Epidemie in
Großbritannien und die Übertragung der Erkrankung auf den Menschen durch BSE-
kontaminierte Produkte zu einem starken öffentlichen Interesse (1.1.3).
Das Prion-Konzept besagt, dass der infektiöse Erreger aus einem körpereigenen Protein mit
veränderter Gestalt besteht. Gelangt diese Form in einen Organismus, repliziert sie sich, indem
ein endogenes Protein in eine fehlgefaltete Form umgewandelt wird (1.1.4). Hierbei spielt das
zelluläre Prion-Protein (PrPC) eine zentrale Rolle. Seine physiologische Funktion ist noch nicht
genau geklärt (1.1.5-6). PrPC unterscheidet sich strukturell und biochemisch von seiner
fehlgefalteten Form, dem pathologischen Prion-Protein, PrPSc (1.1.7). Der Mechanismus der
Konversion ist noch hypothetisch (1.1.8). Vom Zeitpunkt der Infektion oder der spontanen
Konversion bis zum Auftreten der klinischen Krankheit können mehrere Jahre oder Jahrzehnte
vergehen (Collinge et al., 2006). Gegenwärtig gibt es noch keine Therapie für die tödlich
verlaufenden Krankheiten, jedoch wurden in den letzen Jahrzehnten systematisch verschiedene
therapeutische Ansätze untersucht (1.1.9).

EINLEITUNG
6
1.1.2 Prionenerkrankungen bei Tieren
Die Traberkrankheit (Scrapie) bei Schafen und Ziegen wurde bereits vor 250 Jahren
beschrieben (Parry, 1962). Der Name „Scrapie“ bezieht sich auf die Symptome erkrankter Tiere,
die ihr Fell an Zäunen oder Bäumen abscheuern (engl.: to scrape = abkratzen, schaben). 1936
gelang erstmals die experimentelle Übertragung von Scrapie und somit der Beweis, dass es
sich um eine Infektionskrankheit handelt (Cuillé und Chelle, 1936). Die Bovine Spongiforme
Enzephalopathie (BSE) erlangte besonders hohe Aufmerksamkeit durch ihr epidemieartiges
Auftreten bei Rindern. 1986 traten die ersten BSE-Fälle in Großbritannien auf (Wells et al.,
1987). Seitdem sind mehr als 200.000 Rinder in ganz Europa und Japan erkrankt und
Schätzungen gehen weltweit von bis zu einer Million infizierter Tiere aus (Brown et al., 2001).
Ausgelöst wurde diese Epidemie wahrscheinlich durch die Verfütterung von Prion-
kontaminiertem Tiermehl. Ende der siebziger Jahre erfolgte aus wirtschaftlichen Gründen eine
Verminderung der Inaktivierungstemperatur bei der Herstellung von Tiermehl, so dass eine
vollständige Inaktivierung des Erregers nicht mehr gewährleistet war (Ford, 1996). Nach
Aufkommen der ersten Verdachtsmomente wurde 1988 in Großbritannien die
Tiermehlverfütterung an Wiederkäuer verboten, so dass die Anzahl infizierter Tiere wieder
rückläufig ist. Mittlerweile beweisen zahlreiche Studien, dass in diesem Zeitraum die
Übertragung von BSE auf andere Tiere und sogar den Menschen erfolgte (1.1.3).
Andere bekannte Prionenerkrankungen bei Tieren sind die übertragbare Enzephalopathie bei
Nerzen (engl.: Transmissible Mink Encephalopathy, TME) und die übertragbare Katzen-
Enzephalopathie (engl.: Feline Spongiforme Encephalopathy, FSE). Auch hier gilt die
Verfütterung von infektiösem Futter als Ursache der Übertragung (Hanson et al., 1971; Pearson
et al., 1991). Die Chronic Wasting Disease (CWD) ist die einzige bekannte
Prionenerkrankung, die auch bei frei lebenden Tieren, wie bestimmten Hirscharten der Familie
Cervidae, in Nordamerika und Kanada auftritt (Miller et al., 2000).
1.1.3 Prionenerkrankungen beim Menschen
Gemäß ihrer Ätiologie können Prionenerkrankungen spontan (sporadisch), vererbbar (hereditär)
oder durch Infektion (infektiös) auftreten (Tabelle 1). Das Gen für das humane Prion-Protein
(PRNP) ist auf Chromosom 20 lokalisiert (Sparkes et al., 1986).

EINLEITUNG
7
Tabelle 1: Zusammenfassung humaner Prionenerkrankun gen
Manifestation Erkrankung Übertragungsmechanismus
sporadisch sCJD unbekannt
hereditär fCJD
GSS
FFI
autosomal dominante Mutationen im PRNP-Gen
infektiös Kuru
iCJD
vCJD
Endokannibalismus
iatrogene Übertragung
Konsum von BSE-kontaminierten Fleischprodukten
Die bekannteste und häufigste humane TSE-Erkrankung ist die Creutzfeldt-Jakob-Krankheit
(CJD). Etwa 85% aller CJD-Fälle sind sporadischen Ursprungs (sporadische Creutzfeldt-
Jakob-Krankheit, sCJD ), welche weltweit mit einer konstanten Inzidenz von ~1:1.000.000 pro
Einwohner auftreten. Die Krankheit ist nicht mit einer Mutation im PRNP-Gen assoziiert, so dass
der genaue Ursprung bis heute unbekannt ist. Hypothetische Erklärungen für die Ätiologie von
sCJD gehen davon aus, dass die Konversion von PrPC spontan stattfindet oder durch das
zufällige Auftreten von altersbedingten somatischen Mutationen erleichtert wird (Prusiner et al.,
1998). Die Infektiosität von sCJD konnte 1968 durch die experimentelle Übertragung auf
Schimpansen gezeigt werden (Gibbs et al., 1968).
Zu den hereditär auftretenden Prionenerkrankungen gehören die familiäre Creutzfeldt-Jakob-
Krankheit (fCJD) , das Gerstmann-Sträussler-Scheinker-Syndrom (GSS) und die fatale
familiäre Schlaflosigkeit (engl.: Fatal Familial Insomnia, FFI). Sie werden durch Mutationen im
PRNP-Gen verursacht, welche autosomal-dominant vererbt werden. Dabei handelt es sich
entweder um Punktmutationen, die zur Substitution einer Aminosäure oder zur Entstehung
eines Stop-Kodons führen, oder um Insertionen im Bereich der N-terminalen Oktarepeat-
Region. Mittlerweile wurden über dreißig verschiedenen Mutationen beschrieben (Abbildung 1).
Bei der fCJD, die ca. 10-15% aller hereditären CJD-Fälle ausmacht, sind Mutationen an Kodon
200 (E200K) oder Kodon 178 (D178N) am häufigsten (Goldfarb et al., 1990a; Goldfarb et al.,
1990b; Kovanen, 1993).
Das Auftreten von GSS kann mit sieben verschiedenen Mutationen assoziiert sein, jedoch tritt in
den meisten betroffenen Familien eine Mutation von Prolin zu Leucin an Kodon 102 auf (Hsiao
et al., 1989). Die FFI ist ebenfalls mit der Mutation an Kodon 178 (D178N) assoziiert, deren
Phänotyp aber nur entsteht, wenn dasselbe Allel an Kodon 129 für Methionin kodiert. Wird
dagegen Valin kodiert, entsteht das Krankheitsbild der fCJD (Goldfarb et al., 1992).
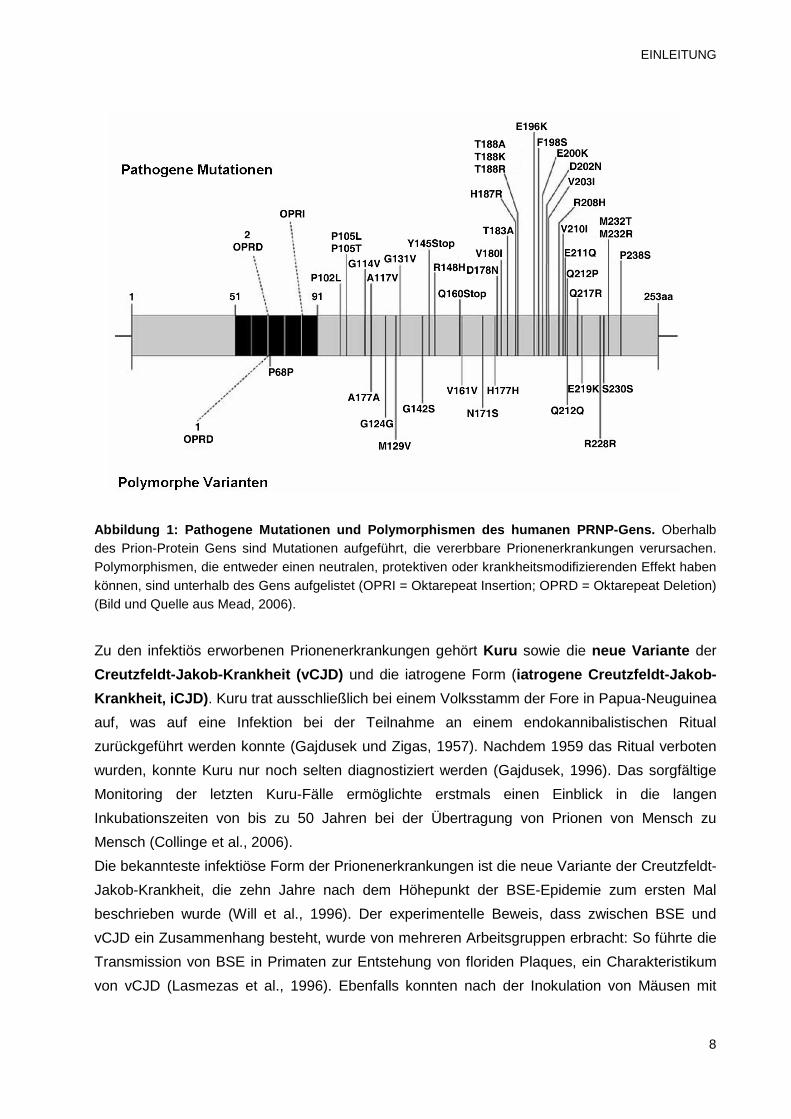
EINLEITUNG
8
Abbildung 1: Pathogene Mutationen und Polymorphisme n des humanen PRNP-Gens. Oberhalb des Prion-Protein Gens sind Mutationen aufgeführt, die vererbbare Prionenerkrankungen verursachen. Polymorphismen, die entweder einen neutralen, protektiven oder krankheitsmodifizierenden Effekt haben können, sind unterhalb des Gens aufgelistet (OPRI = Oktarepeat Insertion; OPRD = Oktarepeat Deletion) (Bild und Quelle aus Mead, 2006).
Zu den infektiös erworbenen Prionenerkrankungen gehört Kuru sowie die neue Variante der
Creutzfeldt-Jakob-Krankheit (vCJD) und die iatrogene Form (iatrogene Creutzfeldt-Jakob-
Krankheit, iCJD) . Kuru trat ausschließlich bei einem Volksstamm der Fore in Papua-Neuguinea
auf, was auf eine Infektion bei der Teilnahme an einem endokannibalistischen Ritual
zurückgeführt werden konnte (Gajdusek und Zigas, 1957). Nachdem 1959 das Ritual verboten
wurden, konnte Kuru nur noch selten diagnostiziert werden (Gajdusek, 1996). Das sorgfältige
Monitoring der letzten Kuru-Fälle ermöglichte erstmals einen Einblick in die langen
Inkubationszeiten von bis zu 50 Jahren bei der Übertragung von Prionen von Mensch zu
Mensch (Collinge et al., 2006).
Die bekannteste infektiöse Form der Prionenerkrankungen ist die neue Variante der Creutzfeldt-
Jakob-Krankheit, die zehn Jahre nach dem Höhepunkt der BSE-Epidemie zum ersten Mal
beschrieben wurde (Will et al., 1996). Der experimentelle Beweis, dass zwischen BSE und
vCJD ein Zusammenhang besteht, wurde von mehreren Arbeitsgruppen erbracht: So führte die
Transmission von BSE in Primaten zur Entstehung von floriden Plaques, ein Charakteristikum
von vCJD (Lasmezas et al., 1996). Ebenfalls konnten nach der Inokulation von Mäusen mit

EINLEITUNG
9
BSE- und vCJD-Prionen gleiche Läsionsprofile und Inkubationszeiten bestimmt werden (Bruce
et al., 1997), und das Glykosylierungsmuster von PrPSc ist bei BSE- und vCJD-Prionen
identisch (Collinge et al., 1996b). Die Anfälligkeit gegenüber der neuen Variante von CJD
scheint an einen Polymorphismus von Kodon 129 des PRNP-Gens gekoppelt zu sein. 37% der
Bevölkerung sind an dieser Stelle homozygot für Methionin (Met), 12% sind homozygot für Valin
(Val) und 51% sind Met/Val heterozygot. Alle bisher aufgetretenen vCJD-Fälle sind an diesem
Kodon homozygot für Methionin (Collinge et al., 1996a; Ironside und Head, 2004).
Die iatrogene Form von CJD umfasst etwa 1% aller CJD-Fälle und tritt durch eine
unbeabsichtigte Übertragung von sCJD durch medizinische Eingriffe beim Menschen auf. So
wurden Fälle beschrieben, die nach Hirnhauttransplantationen oder
Augenhornhautverpflanzungen auftraten (Duffy et al., 1974), deren Spender offenbar
asymptomatische Träger von sCJD waren. Zahlreiche Empfänger von humanen
Wachstumshormonen, die aus Leichenhypophysen gewonnen wurden, erkrankten. Die
Übertragung durch CJD-kontaminierte chirurgische Instrumente, wie z.B. Gehirn-Elektroden, ist
ebenfalls dokumentiert (Brown et al., 2000). Auch eine Übertragung von vCJD durch
kontaminierte Bluttransfusionen ist möglich, da deren Empfänger kurze Zeit später an vCJD
erkrankten und starben (Llewelyn et al., 2004; Peden et al., 2004).
1.1.4 Das Prion-Konzept
Die Natur des krankheitserregenden Agens von Prionenerkrankungen konnte aufgrund seiner
außergewöhnlichen Eigenschaften lange Zeit nicht genau aufgeklärt werden. 1936 gelang die
experimentelle Übertragung von Scrapie auf gesunde Schafe und Ziegen durch die Inokulation
von Gehirnhomogenat nach einer sehr langen Inkubationszeit (Cuillé und Chelle, 1936). Infolge
dieser Beobachtungen nahm man zunächst an, dass es sich bei dem Krankheitserreger um
einen „Slow-Virus“ handelte, d.h. um ein sehr langsam replizierendes Virus (Sigurdsson, 1954;
Thormar, 1971). Gegen diese Virus-Hypothese sprach jedoch die Beobachtung, dass die
Infektiosität des Erregers weder durch die Behandlung mit DNA- oder RNA-schädigender
Strahlung noch mit Nukleasen reduziert werden konnte (Alper et al., 1967). Stattdessen führten
Verfahren, die Proteine modifizierten oder hydrolysierten, wie z.B. die Behandlung mit Harnstoff
oder Natriumhydroxid (Alper et al., 1967; Alper et al., 1966; Prusiner, 1982; Prusiner et al.,
1981), zur Inaktivierung des Erregers. Diese Untersuchungen zeigten, dass der Erreger aus
einem nukleinsäurefreien Pathogen bestehen könnte. Griffith postulierte 1967 erstmals die
Hypothese, dass das infektiösen Agens ein Protein ist (Griffith, 1967). Diese „Nur Eiweiss“-
Hypothese konnte 1982 durch die Isolation eines Protease-resistenten Glykoproteins, welches
in hoch aufgereinigten Gehirnproben von infizierten Hamstern mit der Infektiosität assoziiert
war, bestätigt werden. In vitro bildete dieses aufgereinigte Protein ebenfalls schwer lösliche

EINLEITUNG
10
Aggregate, die denen in den Ablagerungen von Prion-infizierten Gehirnen ähnlich waren (Bolton
et al., 1982; McKinley et al., 1983; Prusiner, 1982).
Um diese neue Erregerklasse von konventionellen Pathogenen wie Viren oder Bakterien
abzugrenzen, prägte Stanley Prusiner den Begriff Prion (proteinaceous infectious particle) als
Bezeichnung für infektiöse proteinartige Partikel ohne Nukleinsäuren (Bolton et al., 1982;
Prusiner, 1982).
Nach Bestimmung der N-terminalen Sequenz des infektiösen Prion-Proteins (Prusiner et al.,
1984) gelang mit Hilfe von Gensonden die Klonierung des kodierenden Gens.
Überraschenderweise handelte es sich um ein zelluläres Gen, das sowohl in gesunden als auch
in infizierten Hamstern in gleichen Maßen exprimiert wird (Chesebro et al., 1985; Oesch und
Tempst, 1985). Da in der Primärstruktur des zellulären und pathologischen Proteins keine
Unterschiede festgestellt werden konnten (Basler et al., 1986), wurde angenommen, dass sich
die beiden Formen nur in ihrer Konformation unterscheiden (Pan et al., 1993). Um zwischen
diesen beiden Isoformen zu differenzieren, wurde das normale zelluläre Protein PrPC (cellular
PrP) und die infektiöse pathogene Form PrPSc (PrP scrapie) genannt. Die „Nur Eiweiss“-
Hypothese besagt, dass das infektiöse Agens von Prionenerkrankungen nur aus der
pathogenen Isoform besteht (Prusiner, 1982; Prusiner, 1997; Prusiner, 1998; Prusiner et al.,
1984). Die Replikation erfolgt vermutlich dadurch, dass PrPSc dem endogenen PrPC seine
eigene Konformation aufzwingt und sich somit vermehrt (1.1.8) (Cohen et al., 1994).
Ein bedeutender Hinweis für die Richtigkeit der „Nur Eiweiss“-Hypothese lieferte der Befund,
dass Mäuse ohne PrPC (Prnp0/0-Mäuse) nach Inokulation mit Prionen vollständig resistent
waren und nicht erkrankten (Bueler et al., 1993; Prusiner et al., 1993), die Empfänglichkeit der
Tiere jedoch durch die Einführung des PrP-kodierenden Transgens wieder hergestellt werden
konnte (Whittington et al., 1995).
Obwohl diese Hypothese mittlerweile weitestgehend akzeptiert ist, erklärt sie nicht, wie ein
einzelnes pathogenes Protein stammspezifische Unterschiede kodieren kann, die zu
Variationen in der Inkubationszeit oder der Neuropathologie bei Mäusen führen (Bruce et al.,
1991) (1.1.8).
Eine weitere Unterstützung für die „Nur Eiweiss“-Hypothese wurde durch die de novo
Generation von Proteinase K-resistenten PrPSc erbracht (Mestel, 1996). So gelang die in vitro
Konversion von PrPC in Proteinase K-resistentes PrPSc durch die Koinkubation beider Isoformen
(Kocisko et al., 1994). Ob dabei auch Infektiosität erzeugt wurde, konnte nicht bestimmt werden.
Kürzlich gelang die Herstellung von amyloiden Fibrillen aus rekombinantem PrP, welche nach
Inokulation auf eine transgene Mauslinie nach langer Inkubationszeit zu „Scrapie“-typischen
Symptomen führten (Legname et al., 2004). Da sich jedoch nach Transmission dieser Fibrillen
auf wildtypische Mäuse keine Krankheit entwickelte, ist die Interpretation der Daten, ob wirklich
de novo Infektiosität erzeugt wurde, noch unklar.

EINLEITUNG
11
Signalsequenz 5 Oktarepeats
STE H1 H2 H3NH2- -COOHTM
51 91 95 112 126 144 151
S-S
179 193 200 217
ß1 ß2
N-Glykosylierung180 196
231 254
SignalsequenzSignalsequenz 5 Oktarepeats
STE H1 H2 H3NH2- -COOHTM
51 91 95 112 126 144 151
S-S
179 193 200 217
ß1 ß2
N-Glykosylierung180 196
231 254231 254
Signalsequenz
1.1.5 Das Prion-Protein
Das Prion-Protein ist innerhalb der Säugetiere hoch konserviert (Schatzl et al., 1995; Wopfner
et al., 1999). In Vögeln (Harris et al., 1991), Fischen (Gibbs und Bolis, 1997) und Beuteltieren
(Windl et al., 1995) konnte ebenfalls die Expression eines PrPC-Homologs bestätigt werden.
PrPC wird hauptsächlich in Neuronen (Kretzschmar et al., 1986) und in geringen Mengen auch
in Astrozyten und Oligodendrozyten exprimiert (Moser et al., 1995). Außerhalb des Gehirns
wurde PrPC in der Skelett-Muskulatur (Brown et al., 1998), lymphoiden Geweben (Brown et al.,
1999b; Burthem et al., 2001; Liu et al., 2001) sowie im Darmgewebe nachgewiesen (Morel et
al., 2004).
Das murine Prnp-Gen besteht aus drei Exons und ist auf Chromosom 2 lokalisiert (Sparkes et
al., 1986). Der offene Leserahmen liegt komplett in Exon 3 (Basler et al., 1986). Das
vollständige Translationsprodukt besteht bei der Maus aus 254 Aminosäuren (Abbildung 2).
Abbildung 2: Schematische Darstellung des murinen P rion-Proteins. Das zelluläre Prion-Protein besteht aus einer flexiblen N-terminalen Domäne (Reste 23-121) und einer globulären C-terminalen Domäne. N-terminal sind fünf hochkonservierte Oktarepeat-Regionen (PQGGTWGQ), eine Stopp-Transfer-Effektor-Sequenz (STE) und eine transmembrane Region (TM), die auch hydrophobe Domäne genannt wird, angeordnet. Der globuläre Teil besteht aus drei α-Helices und zwei β-Faltblättern. Die Signalpeptide für den Import ins rER (Reste 1-22) und den GPI-Anker (Reste 231-254) werden abgespalten, so dass das reife PrPC-Molekül aus 209 Aminosäuren besteht.
PrPC wird im rauen endoplasmatischen Retikulum (rER) synthetisiert und gelangt durch den
Golgi-Apparat über den sekretorischen Weg zur Plasmamembran, mit der es über einen
Glykosylphosphatidylinositol (GPI)-Anker verknüpft ist (Borchelt et al., 1990; Caughey, 1991;
Taraboulos et al., 1990). Während der Biosynthese von PrPC erfolgen mehrere post-
translationale Modifikationen: Abspaltung der N-terminalen Signalsequenz für den Import in das
rER (Basler et al., 1986; Oesch und Tempst, 1985), N-Glykosylierung der beiden Asparagin-
Reste 180 und 196 (Haraguchi et al., 1989), Ausbildung einer Disulfidbrücke zwischen den
Cystein-Resten 179 und 214 (Hope et al., 1986; Safar et al., 1998) und Abspaltung der
C-terminalen hydrophoben Signalsequenz für die Anheftung des GPI-Ankers (Stahl et al.,
1987).

EINLEITUNG
12
PrPC gelangt im Verlauf seiner Reifung vom ER über den Golgi-Apparat auf die Zelloberfläche
und kann dort entweder mit spezialisierten Mikrodomänen (lipid rafts) (Naslavsky et al., 1997;
Vey et al., 1996), die sich durch einen besonders hohen Gehalt an Cholesterol und
Sphingolipiden auszeichnen (Simons und Ikonen, 1997), oder mit „Clathrin-coated pits“
assoziiert sein (Laine et al., 2001; Madore et al., 1999; Shyng et al., 1994; Sunyach et al.,
2003). Von der Zelloberfläche werden die PrPC-Moleküle kontinuierlich über Endosome
internalisiert. Ein Teil gelangt zurück an die Oberfläche, während ein anderer Teil abgebaut wird
(Shyng et al., 1993). Der Mechanismus der PrPC-Internalisierung wird gegenwärtig kontrovers
diskutiert. Eine Beteiligung von sowohl lipid rafts bzw. raft-ähnlichen Strukturen wie zum
Beispiel Caveolin-1 enthaltende Caveolaes als auch Clathrin-abhängige Mechanismen sind
möglich.
Für eine Clathrin-abhängige Endozytose könnte die Lokalisation von PrPC in Clathrin-Vesikeln
sprechen, jedoch fehlt PrPC eine zytoplasmatischen Domäne, die direkt mit einem
Adapterprotein oder mit Clathrin interagieren könnte. Deswegen wurde ein mutmaßlicher PrP-
Rezeptor postuliert, der die Internalisierung über Clathrin-abhängige Mechanismen vermittelt
(Harris et al., 1996; Shyng et al., 1995). Ein möglicher Kandidat wäre der 37kDa/67kDa
Laminin-Rezeptor, welcher in der Zellkultur für die PrPC-Internalisierung notwendig ist
(Gauczynski et al., 2001). Weitere Daten zeigen, dass die Internalisierung von PrPC offenbar
über mehrere alternative Routen möglich ist (Prado et al., 2004).
1.1.6 Funktionen von PrP C
Die physiologische Funktion von PrPC ist heute noch nicht genau geklärt. Die Generierung von
Prnp0/0-Mäusen sollte erste Hinweise auf die Funktion liefern, jedoch zeigten die Tiere
überraschenderweise keinen ausgeprägten Phänotyp und entwickelten sich normal (Bueler et
al., 1992). Lediglich eine leichte Beeinträchtigung der GABAA-Rezeptor-vermittelten Inhibition
und der Langzeit-Potenzierung (Collinge et al., 1994) sowie geringe Veränderungen im
zirkadianen Rhythmus wurden beobachtet. Diese phänotypischen Veränderungen lassen sich
durch die Expression von PrP-Transgenen wieder aufheben (Tobler et al., 1997; Whittington et
al., 1995).
Mittlerweile werden verschiedene Funktionen für PrPC diskutiert. Einige Untersuchungen deuten
auf eine funktionelle Rolle im Kupfer-Metabolismus hin. Dies basiert auf der Fähigkeit von
PrPC, Cu2+-Ionen zu binden. Diese Bindung kann sowohl über die Histidine in den Oktarepeats
als auch über Histidin 96 und 111 erfolgen (Jackson et al., 2001). Da die Bindung von
Cu2+-Ionen die Clathrin-abhängige Endozytose stimuliert, könnte PrPC eine regulierende
Funktion beim Kupfer-Transport besitzen (Pauly und Harris, 1998). Eine protektive Wirkung von
PrPC als Cu/Zn Superoxiddismutase-1 (SOD-1) wurde ebenfalls beschrieben (Brown et al.,

EINLEITUNG
13
1999a). Diese Funktion, die von der Aufnahme von Kupfer als essentiellem Kofaktor abhängt,
führt zu einem Schutz gegen oxidativen Stress. Da aber keine direkte Korrelation zwischen der
PrPC-Expression und der Kupfer-Konzentration in verschiedenen Mausmodellen beobachtet
wurde und auch die SOD-1-Funktion von anderen Arbeitsgruppen nicht bestätigt wurde, bleibt
die genaue Beteiligung von PrPC an der Kupferhomöostase spekulativ (Hutter et al., 2003;
Waggoner et al., 2000).
Da PrPC als GPI-verankertes Protein auf der Zelloberfläche exprimiert wird, liegt eine weitere
Funktion in der Signaltransduktion nahe. So wurde z.B. nach Antikörper-vermittelter
Vernetzung von PrPC eine erhöhte Aktivität der Tyrosin-Kinase Fyn beobachtet (Mouillet-
Richard et al., 2000). Die physiologische Bedeutung dieses Befundes ist noch unklar. Ebenfalls
führte die Interaktion von PrPC mit dem neuralen Zelladhäsionsmolekül (NCAM) zu einer
Aktivierung der Fyn-Kinase, die, wie auch die Interaktion mit dem 37kDa/67kDa Laminin-
Rezeptor, das Neuritenwachstum stimulierte (Graner et al., 2000; Rieger et al., 1997;
Santuccione et al., 2005). Ein neuroprotektiver Effekt wurde nach Interaktionsstudien mit dem
Stress-induzierbaren Protein 1 (Zanata et al., 2002) oder dem anti-apoptotischen Faktor Bcl-2
(Kurschner und Morgan, 1995) postuliert. Gegenwärtig sind mehr als 20 Interaktionspartner von
PrPC bekannt, jedoch muss die physiologische Relevanz der Interaktion noch weiter untersucht
werden (Caughey und Baron, 2006).
1.1.7 Strukturelle und biochemische Eigenschaften v on PrP C und PrP Sc
Während PrPC und PrPSc identische Aminosäuresequenzen und gleiche post-translationale
Modifikationen besitzen, unterscheiden sie sich in ihren biochemischen und strukturellen
Eigenschaften (Tabelle 2).
Tabelle 2: Vergleich der Eigenschaften von PrP C, PrPSc und PrP27-30
PrP Isoform PrP C PrPSc PrP27-30
Infektiosität nicht-infektiös infektiös infektiös
Proteasesensitivität sensitiv partiell resistent resistent
Löslichkeit löslich unlöslich unlöslich
Halbwertszeit kurz (2-4h) lang (> 24h) lang (> 24h)
Molekulargewicht 33-35 kDa 27-35 kDa 27-30 kDa
Aggregationsstatus Monomere,
Dimere, Oligomere
Monomere,
Dimere, Aggregate
amyloide Fibrillen
Sekundärstruktur α-Helices (42%)
β-Faltblätter (3%)
α-Helices (30%)
β-Faltblätter (43%)
α-Helices (21%)
β-Faltblätter (54%)

EINLEITUNG
14
Im Gegensatz zu PrPC besitzt PrPSc eine partielle Resistenz gegenüber Proteinase K, ist in
nicht-ionischen Detergenzien unlöslich und tendiert zur Aggregatbildung (Cohen und Prusiner,
1998). Die Inkubation von murinem PrPSc mit Proteinase K führt zur Abspaltung von ca. 90
Aminosäureresten am N-Terminus (Oesch und Tempst, 1985). Das resultierende Fragment wird
entweder nach seiner Größe PrP27-30 oder nach seiner Protease-resistenten Eigenschaft als
PrPres bezeichnet (Bolton et al., 1982). Im Verlauf der Krankheit kann die Akkumulation von
PrPSc zu Ablagerungen in Form von Amyloid-Plaques führen. Eine biochemische Anreicherung
aus Prion-infizierten Hamstergehirnen führt zur Entstehung von amyloiden Fibrillen („prion
rods”), die ebenfalls in wässrigen und organischen Lösungsmitteln sowie in nicht-ionischen
Detergenzien unlöslich sind (McKinley et al., 1991).
Diese Eigenschaften erschweren bis heute die genaue Bestimmung der Struktur von PrPSc.
Nachdem eine partielle Aufreinigung von nativem PrPC und PrPSc gelang, wurden verschiedene
Verfahren zur Strukturanalysen, wie z.B. die Fourier-Transformations-Infrarotspektroskopie und
die Zirkulardichroismus-Spektroskopie, eingesetzt. Bei PrPC beträgt der Anteil an α-helikalen
Bereichen 42%, während nur 3% in einer β-Faltblatt-Struktur vorliegen. Dieses Verhältnis
verschiebt sich bei der Konversion zu PrPSc, wobei mit 45% die β-Faltblätter dominieren und der
α-helikale Anteil nur noch 30% beträgt (Pan et al., 1993).
Abbildung 3: NMR-Struktur von rekombinantem PrP (Re ste 121-231). Die Struktur von PrPC besteht aus drei α-Helices (rot) und zwei anti-parallelen β-Faltbättern (blau) (Bild und Quelle aus Eghiaian, 2005).
Durch die Herstellung von rekombinantem Prion-Protein der Maus (Reste 23-231) in E.coli
wurde die Struktur von PrPC mittels Kernresonanz-Spektroskopie (NMR) aufgeklärt. Die
C-terminale, globuläre Domäne besteht aus drei α-Helices sowie aus zwei antiparallelen
β-Faltblättern, welche die α-Helix 1 flankieren. Für den N-Terminus (Reste 23-120) konnte keine

EINLEITUNG
15
A B CA B C
Struktur bestimmt werden. Man geht davon aus, dass PrPC in diesem Bereich flexibel ist
(Hornemann et al., 1997; Riek et al., 1997) (Abbildung 3). Die Bindung von Cu2+-Ionen im
N-Terminus führt vermutlich zu einer Stabilisierung der flexiblen Struktur (Jones et al., 2004;
Leclerc et al., 2006).
Huang und Kollegen entwickelten für PrPSc ein theoretisches Struktur-Modell, in dem Helix 2
und Helix 3 erhalten bleiben und Helix 1 durch vier β-Faltblätter ersetzt wird (Huang et al.,
1996). Neuere Untersuchungen von 2D-Kristallen, die aus PrP27-30 gewonnen wurden (Wille
et al., 2002), weisen jedoch darauf hin, dass der Bereich der Helix 1 inklusive der beiden
angrenzenden β-Faltblätter (Reste 98-175) in eine linksgängige β-Helix umgewandelt wird
(Abbildung 4). Helix 2 und Helix 3, die durch eine Disulfidbrücke verbunden sind, sowie die
flexible N-terminale Domäne bleiben erhalten. Drei dieser Moleküle lagern sich zu Trimeren
zusammen und bilden eine Einheit der 2D-Kristalle. Dabei liegen die β-Helices der Monomere
im Zentrum und die beiden C-terminalen α-Helices ragen nach außen. Lagern sich mehrere
dieser Trimere übereinander zusammen, bilden sich so genannte „prion rods“ (Govaerts et al.,
2004; Wille et al., 2002).
Abbildung 4: Schematisches Modell der PrP Sc-Konformation und Aggregation. (A) In einem PrPSc-Monomer bildet die Sequenz der Aminosäuren 90 bis 176 eine linksgängige β-Helix (gelb) aus, während die Struktur von Helix 2 und Helix 3, wie bei PrPC, erhalten ist. (B) Trimeres Modell bei der Assoziation von PrPSc-Monomeren. (C) Modell für die Aggregation von PrPSc-Trimeren zu einer PrPSc-Fibrille („prion rod“). Die Zuckerreste sind nicht dargestellt (Govaerts et al., 2004).
1.1.8 Replikationsmechanismen und Prionenstämme
Die Replikation der pathogenen Form spielt die zentrale Rolle in der Prion-Pathogenese. Da bis
heute keine Prion-spezifischen Nukleinsäuren oder Nukleinsäuren, die groß genug für die

EINLEITUNG
16
Kodierung eines Proteins sind, identifiziert wurden (Kellings et al., 1992; Safar et al., 2005),
nimmt man an, dass PrPSc repliziert, indem es an PrPC bindet und es in seine eigene
Konformation umwandelt (Prusiner, 1991). Die genaue zelluläre Lokalisation der Replikation ist
noch nicht geklärt, aber Untersuchungen deuten darauf hin, dass die Konversion vermutlich
innerhalb des endosomalen/lysosomalen Kompartiments oder sogar an der Zelloberfläche in
lipid rafts stattfindet (Arnold et al., 1995; Baron und Caughey, 2003; Borchelt et al., 1992; Botto
et al., 2004; Caughey und Raymond, 1991; Mayer et al., 1992).
Für den Replikationsmechanismus von PrPSc werden gegenwärtig zwei Modelle diskutiert: Das
Faltungs- oder Heterodimermodell und das Keimbildungsmodell (Abbildung 5).
Das Faltungs- oder Heterodimermodell geht davon aus, dass die spontane Konversion von
PrPSc zu PrPC durch eine hohe Energiebarriere kontrolliert wird. Exogen eingeführtes PrPSc
kann die Entstehung der stabilen PrPSc-Form favorisieren und somit PrPC in PrPSc umwandeln.
Dabei bindet ein PrPSc-Monomer an ein partiell entfaltetes PrPC-Molekül, wodurch ein
Heterodimer entsteht und dadurch PrPC konvertiert wird. Dieser Komplex zerfällt wiederum in
zwei PrPSc-Monomere, die in einem autokatalytischen Prozess weitere PrPC-Moleküle in PrPSc
konvertieren können (Cohen et al., 1994; Prusiner et al., 1990).
Beim Keimbildungsmodell besteht zwischen PrPC und PrPSc ein thermodynamisch
kontrolliertes Gleichgewicht, d.h. die Umfaltung ist reversibel, jedoch ist die PrPC-Konformation
begünstigt. PrPSc wird nur stabilisiert, wenn sich mehrere PrPSc-Monomere zu einem
kristallähnlichen Keim zusammenlagern. Entsteht so ein „Kristallisationskeim“, lagern sich sehr
schnell weitere PrPC-Monomere an, die konvertieren und Aggregate bilden. Um den
exponentiellen Anstieg von PrPSc im Krankheitsverlauf erklären zu können, müssen diese
Aggregate kontinuierlich fragmentiert werden, um so weitere Anlagerungsflächen liefern zu
können (Jarrett und Lansbury, 1993; Orgel, 1996).
Das Keimbildungsmodell liefert eine hypothetische Erklärung für die Existenz von
verschiedenen Prionenstämmen. Solche Stämme unterscheiden sich sowohl in ihren
biologischen als auch ihren biochemischen Eigenschaften. So wurden nach experimenteller
Inokulation von Prionenstämmen in gleiche Mauslinien Unterschiede in der Inkubationszeit und
der Neuropathologie beobachtet. PrPSc-Moleküle von einzelnen Prionenstämmen können sich
in der Sensitivität gegenüber Proteinase K und in der Anzahl der proteolytisch entfernten
Aminosäuren im N-Terminus unterscheiden, wodurch stammspezifische Profile nach der
SDS-Elektrophorese entstehen können. Die Unterschiede betreffen sowohl die Fragmentgröße
als auch das Verhältnis der di-, mono- und unglykosylierten PrPSc-Formen untereinander
(Bessen und Marsh, 1992; Bruce, 2003; Collinge et al., 1996b; Telling et al., 1996). Die
stammspezifischen Charakteristika bleiben auch nach Passage des Inokulums, unabhängig von
der primären PrPC-Sequenz des Wirts, erhalten (Telling et al., 1996).

EINLEITUNG
17
Im Rahmen des Keimbildungsmodells würde das bedeuten, dass bei der Fragmentierung
unterschiedliche Kristallisationskeime entstehen, welche die Information für die
Stammeigenschaften in ihrer Struktur kodieren. Dies würde für die Existenz mehrerer stabiler
PrPSc-Konformationen sprechen.
Beide Modelle konnten in vivo noch nicht bestätigt werden, aber das Keimbildungsmodell wird
von experimentellen Daten gut unterstützt. Einerseits besteht die Möglichkeit, rekombinantes
PrP in vitro zu konvertieren und dabei die stammspezifischen biochemischen Charakteristika
verschiedener Prionenstämme zu erhalten (Bessen et al., 1995; Kocisko et al., 1995; Vanik et
al., 2004). Andererseits existiert mittlerweile eine Methode (protein misfolding cyclic
amplification, PMCA), mit der es möglich ist, PrPSc-Moleküle zu amplifizieren (Saborio et al.,
2001). Während der ersten Phase der PMCA wird eine geringe Menge von PrPSc in Form von
infektiösem Gehirnhomogenat mit einem großen Überschuss von gesundem Gehirnhomogenat
inkubiert, um das Wachstum von PrPSc-Polymeren zu induzieren. In der zweiten Phase werden
diese Polymere durch die Behandlung mit Ultraschall fragmentiert, wodurch die Anzahl an
konvertierenden Einheiten erhöht wird. Durch das mehrmalige Wiederholen dieser Phasen
können PrPSc-Moleküle exponentiell amplifiziert werden (Saborio et al., 2001).

EINLEITUNG
18
Abbildung 5: Modelle für die Konversion von PrP C zu PrPSc. A Faltungsmodell . Ein Konformationswechsel von PrPC zu PrPSc wird durch eine hohe Energiebarriere verhindert. Interagiert PrPC jedoch mit exogen eingeführtem PrPSc, entsteht ein Heterodimer, welches PrPC in PrPSc konvertiert. Das entstandene PrPSc-Homodimer dissoziiert und beide Monomere können weitere Konversionswechsel katalysieren. B Keimbildungsmodell . Zwischen PrPC und dem unstabilen PrPSc besteht ein thermodynamisch kontrolliertes Gleichgewicht zu Gunsten von PrPC. Zu einer Stabilisierung und Akkumulation von PrPSc kommt es nur nach Bildung eines Kristallisationskeims, der durch die Anlagerung weiterer PrPSc-Moleküle schnell wächst. Durch die Fragmentierung dieses Keims kommt es zu einer exponentiellen Konversionsrate (Bild und Quelle aus Aguzzi und Sigurdson, 2004).
Es gibt Hinweise darauf, dass neben der direkten Interaktion zwischen den beiden Isoformen
ein weiterer Faktor für die Konversion von PrPC zu PrPSc notwendig ist. So wurde gezeigt, dass
transgene Mäuse, die humanes PrPC exprimieren, nicht mit CJD infiziert werden können,
während Mäuse, die ein chimäres Mensch/Maus-PrP exprimieren, die Krankheit entwickeln
(Telling et al., 1994). Daraus folgerte man, dass ein endogener Faktor, der als Faktor X
bezeichnet wurde, für die Konversion notwendig ist. Indem Faktor X mit einer höheren Affinität
an PrP der eigenen Spezies bindet, könnte er die Konversion des transgenen PrPC einer
anderen Spezies inhibieren (Telling et al., 1995). Ob es sich hierbei um ein molekulares
Chaperon handelt, ist noch unklar. Mittlerweile wurde eine mögliche Bindestelle des Faktor X in
der C-terminalen Domäne des Prion-Proteins identifiziert (Kaneko et al., 1997).
1.1.9 Therapeutische Ansätze für Prionenerkrankunge n
Das Interesse an Prionenerkrankungen nahm durch den Ausbruch der BSE-Epidemie und dem
damit verbundenen Auftreten von vCJD stark zu. Seit 1995 sind weltweit über 180 Menschen
an vCJD verstorben (Hilton, 2006). Aufgrund der langen Inkubationszeiten wird in den nächsten
Jahren mit weiteren Krankheitsfällen gerechnet. Eine genaue Vorhersage über die Anzahl der
erwarteten Fälle wird durch fehlende Angaben über Faktoren, welche die Inkubationszeit
beeinflussen, wie z.B. die Infektionsroute, die Inokulum-Menge oder die genetische
Prädisposition, erschwert (Wisniewski und Sigurdsson, 2007). Zudem besteht das Risiko einer
iatrogenen Infektion, da bereits Fälle einer Übertragung von vCJD durch Bluttransfusionen von
Spendern bekannt sind, welche bereits mit vCJD infiziert, aber noch nicht erkrankt waren
(Llewelyn et al., 2004; Peden et al., 2004) (1.1.3). Diese Beobachtungen sprechen für die
Notwendigkeit, eine effektive Therapie für Prionenerkrankungen zu entwickeln.

EINLEITUNG
19
amyloid
PrPC
PrPScSYNTHESE
ZELLOBERFLÄCHE
ABBAU6
FAKTOR X ?
1
25
3
4
amyloid
PrPC
PrPScPrPScSYNTHESE
ZELLOBERFLÄCHE
ABBAU6
FAKTOR X ?
1
25
3
4
Sowohl die Synthese von PrPC als auch der Konversionsprozess liefern mehrere Ansatzpunkte,
an denen Therapeutika effektiv eingreifen könnten (Abbildung 6):
(1) Hemmung der PrPC-Synthese
(2) Blockierung des PrPC-Transports zur Zelloberfläche
(3) Stabilisierung der PrPC-Konformation
(4) Hemmung der Interaktion von PrPC und PrPSc
(5) Hemmung der Bindung von Faktor X
(6) Stimulierung der Degradation von PrPSc bzw. des Abbaus der PrPSc-Aggregate
Abbildung 6: Potentielle Angriffspunkte für therape utische Interventionen bei Prionen-erkrankungen am Beispiel des Faltungsmodells. Die möglichen Ansatzpunkte sind durch Zahlen gekennzeichnet und werden im Text erklärt. Diese Ansätze können auch auf das Keimbildungsmodell übertragen werden.
Durch die Generierung von Prnp0/0-Mäusen wurde bereits früh gezeigt, dass das endogene
PrPC für eine Infektion essentiell ist. Nach Inokulation replizierten diese Mäuse keine Prionen
und entwickelten keine Pathologie (Bueler et al., 1993; Bueler et al., 1992). Somit bietet sich die
Reduktion bzw. Beseitigung von PrPC als therapeutischer Interventionspunkt an (Abbildung 6
(1)). Dies kann entweder durch RNA-Interferenzen (RNAi) auf Ebene der mRNA von PrPC
(Daude et al., 2003; Tilly et al., 2003) oder auf Ebene des Gens erfolgen. Seit der BSE-Krise
wurde verstärkt an der Generierung von PrP-defizienten Rindern gearbeitet. Da Prnp0/0-Mäuse
nicht mit Prionen infiziert werden können, geht man davon aus, dass dies auch bei PrP-
defizienten Rindern nicht möglich ist, wodurch die Gefahr, die von BSE-kontaminierten

EINLEITUNG
20
Produkten ausgeht, vollständig eliminiert wäre. Kürzlich gelang die erfolgreiche Herstellung von
PrPC-defizienten Rindern (Richt et al., 2007). Bisher wurden in den 20 Monaten
Beobachtungszeitraum keine phänotypischen Auffälligkeiten beobachtet (Richt et al., 2007).
Auch die Hemmung der Genexpression nach erfolgter Infektion erscheint vielversprechend.
Durch einen Knock-out der neuralen PrP-Expression in acht Wochen alten Mäusen, die bereits
infiziert waren und erste neurodegenerative Veränderungen zeigten, konnte sogar die
spongiforme Pathologie im Gehirn wieder aufgehoben werden, und die Tiere blieben trotz
fortschreitender Gliose und extraneuraler PrPSc-Akkumulation asymptomatisch (Mallucci et al.,
2003).
Eine Blockade des Transports von PrPC zu den Kompartimenten, in denen die Konversion
stattfindet (Abbildung 6 (2)), konnte nach Behandlung mit Suramin und den beiden Statinen
Lovastatin und Squalestatin beobachtet werden. Während Suramin zu einer PrPC-Aggregation
im Trans-Golgi-Netzwerk mit anschließender lysosomaler Degradation führt, hemmen die
Statine die Cholesterol-Biosynthese und beeinflussen so die Organisation der lipidhaltigen
Raftmikrodomänen (1.1.5) (Bate et al., 2004; Gilch et al., 2001; Taraboulos et al., 1995). Zu
einer Stabilisierung der PrPC-Konformation können molekulare Chaperone, wie z.B. DMSO oder
Kongo Rot beitragen (Head und Ironside, 2000; Tatzelt et al., 1996) (Abbildung 6 (3)).
Ein anderer Ansatzpunkt ist die Inhibition des Kontakts zwischen PrPC und PrPSc, der für die
Konversion notwendig ist (Abbildung 6 (4)). Durch die Bindung von PrP-Aptameren (Proske et
al., 2002), „Beta-sheet breaker“-Peptiden (Chabry et al., 1999; Soto et al., 2000) oder Anti-PrP-
Antikörpern könnte die Bindestelle maskiert werden (Enari et al., 2001; Peretz et al., 2001). In
transgenen Mäusen, die ein Fusionsprotein bestehend aus zwei murinen PrP-Molekülen
exprimierten, welche über einen Fc-Teil (1.2.2) verbunden waren (PrP-Fc2), wurde eine
Hemmung der Prionreplikation beobachtet. Diese PrP-Fc2-Moleküle waren nicht in PrPSc
konvertierbar, so dass vermutet wurde, dass die gebundenen PrPSc-Moleküle dem
Konversionsprozess entzogen werden (Meier et al., 2003). Ein weiterer Ansatzpunkt zur
Hemmung der Konversion ist die Blockade des postulierten Faktor X (Prusiner, 1998; Telling et
al., 1995) (Abbildung 6 (5)). Die vermutete Bindestelle dieses Faktors beinhaltet im murinen PrP
die Reste Q167, Q171, T214 und Q218 (Kaneko et al., 1997). In Prion-infizierten Zellen führte
die Expression solcher PrP-Mutanten zu einer Reduktion des PrPSc-Gehalts und zu der
Annahme, dass diese Mutanten einen dominant-negativen Effekt auf die Prionreplikation
besitzen. Indem sie an den Faktor X binden, machen sie ihn unzugänglich für den
Konversionprozess (Crozet et al., 2004; Kaneko et al., 1997; Kishida et al., 2004).
Substanzen, welche den Abbau von PrPSc fördern, können ebenfalls für therapeutische Zwecke
eingesetzt werden (Abbildung 6 (6)). Ein Beispiel für so eine Substanz ist der Tyrosin-Kinase-
Inhibitor STI571 (Gleevec®), der zwar nicht die de novo Synthese von PrPSc verhindert, aber
den Abbau von bereits vorhandenem PrPSc verstärkt (Ertmer et al., 2004). Dieser Effekt konnte

EINLEITUNG
21
ebenfalls mit verzweigten Polyaminen beobachtet werden (Supattapone et al., 1999;
Supattapone et al., 2001; Winklhofer und Tatzelt, 2000).
Nachteile vieler der vorgestellten Substanzen sind ihre Toxizität in hohen Dosen und ihre
Unfähigkeit, die Blut-Hirn-Schranke zu passieren, womit meistens nur eine Hemmung der
Prionreplikation in der Peripherie möglich ist und kein therapeutischer Effekt im Gehirn erzielt
werden kann. Zurzeit werden drei Substanzen in klinischen Studien getestet: das Anti-Malaria-
Mittel Quinacrine (Barret et al., 2003; Korth et al., 2001; Nakajima et al., 2004),
Pentosanpolysulfat (PPS) (Todd et al., 2005; Whittle et al., 2006) und Flupirtine (Otto et al.,
2004). Keines der Medikamente führte bisher zu einer signifikanten Beeinflussung des
Krankheitsverlaufs von CJD-Patienten (Haik et al., 2004; Whittle et al., 2006). Nach der
Behandlung mit Flupirtinen wurde lediglich eine leichte Verbesserung der kognitiven
Fähigkeiten beobachtet (Otto et al., 2004). In einem Einzelfall von vCJD führte die
intraventrikuläre Applikation von PPS zu einer Verlängerung der Inkubationszeit (Parry et al.,
2007). Der Einsatz dieser Medikamente wird zusätzlich durch den Beginn der Behandlung
eingeschränkt, der erst nach der Diagnose erfolgen kann und somit zu einem Zeitpunkt, an dem
bereits neuropathologische Veränderungen vorliegen. Die Entwicklung einer
Krankheitsprophylaxe im Sinne einer Impfung wäre somit wünschenswert.
Zahlreiche Studien demonstrierten bereits die erfolgreiche Hemmung der Prionreplikation durch
Anti-PrP-Antikörper in in vitro Zellsystemen (Beringue et al., 2004; Enari et al., 2001; Feraudet
et al., 2005b; Kim et al., 2004; Miyamoto et al., 2005; Pankiewicz et al., 2006; Peretz et al.,
2001). Da PrPC jedoch ubiquitär exprimiert wird, besitzt der Organismus gegenüber PrPC eine
Autotoleranz (1.2.1) und baut keine Immunantwort auf. Somit führen aktive Immunisierungen
mit rekombinantem murinem PrP oder PrP-Peptiden nur zu schwachen Antikörpertitern und
lediglich zu einer moderaten Verlängerung der Inkubationszeit (Magri et al., 2005; Schwarz et
al., 2003; Sigurdsson et al., 2002). Um dieses Problem zu umgehen, wurde in einem
Mausmodell ein definierter Anti-PrP-Antikörper als Transgen exprimiert. Es wurden hohe Titer
des transgenen Antikörpers nachgewiesen, der eine Erkrankung nach peripherer Infektion mit
Prionen verhinderte (Heppner et al., 2001). Auch passive Immunisierungsversuche wurden
durchgeführt, die nach intraperitonealer Inokulation von wildtypischen Mäusen zu einem
deutlich reduzierten PrPSc-Level in der Milz sowie zu einer signifikanten Verlängerung der
Inkubationszeit führten (White et al., 2003).
Eine andere Möglichkeit zur Überwindung der Toleranz ist der Einsatz von
immunstimulatorischen Komponenten wie z.B. CpGs. CpGs sind unmethylierte Cytosin-
Guanosin-Nukleotid-Motive aus Bakterien, welche die angeborene Immunantwort direkt
aktivieren (Krieg et al., 1995; Lipford et al., 1998). In einer Pilotstudie führte die Applikation von
CpGs zwar zu einer vollständigen Hemmung der Prionreplikation, jedoch wurde dadurch

EINLEITUNG
22
gleichzeitig die Milz-Architektur pathologisch verändert (Heikenwalder et al., 2004; Sethi et al.,
2002).
Die Evaluierung solcher therapeutischen Ansätze sollte gleichzeitig mit der Entwicklung von
präklinischen Diagnose-Tests einhergehen, da bis dato Prionenerkrankungen nur post mortem
eindeutig diagnostiziert werden können. Die Chancen einer erfolgreichen Therapie könnten
durch einen möglichst frühzeitigen Beginn der Behandlung, am besten noch in der präklinischen
Phase, gesteigert werden.
1.2 Immunsystem und Antikörper
1.2.1 Das Immunsystem
Das Immunsystem vermittelt dem Organismus einen Schutz gegenüber Pathogenen wie
Bakterien, Parasiten oder Viren. Hierzu ist die Unterscheidung zwischen körperfremden und
körpereigenen Antigenen notwendig, um nur bei körperfremden Antigenen mit der
entsprechenden Immunantwort zu reagieren und bei körpereigenen Antigenen tolerant zu
reagieren (Toleranz).
Die Immunantwort von Vertebraten ist in zwei Phasen unterteilt: Pathogene werden zunächst
unspezifisch erkannt, wobei kein Schutz gegen eine erneute Infektion mit dem gleichen Erreger
entwickelt wird. Diese sogenannte angeborene Immunantwort besteht sowohl aus einfachen
physikalischen Barrieren als auch aus humoralen Faktoren und verschiedenen Zelltypen.
Hierbei interagieren Zellen wie Makrophagen, Granulozyten, dendritische Zellen (DC) und
natürliche Killerzellen (NK-Zellen) mit zahlreichen humoralen Faktoren wie Komponenten des
Komplementsystems, verschiedenen Chemokinen, Zytokinen oder anderen chemischen
Mediatoren. Erst wenn die unspezifische Immunantwort eine Infektion nicht mehr kontrollieren
kann, beginnt die zweite Phase, in der Makrophagen und DCs als antigenpräsentierende Zellen
(APC) das spezifische oder auch adaptive Immunsystem aktivieren. Es besteht aus zellulären
und humoralen Komponenten, welche hauptsächlich über Lymphozyten wie T-Zellen oder
B-Zellen vermittelt werden. In dieser Phase werden Antigene spezifisch erkannt und ein
immunologisches Gedächtnis aufgebaut, so dass bei erneutem Kontakt die Immunantwort
schneller und effizienter ausfällt. T-Zellen erkennen über ihren T-Zell-Rezeptoren die
zellgebundenen Antigene auf der Oberfläche von APCs und differenzieren entweder in
zytotoxische T-Zellen, die dann in APCs Apoptose induzieren, oder in T-Helferzellen, die
B-Zellen aktivieren können. Nach der Antigen-spezifischen Aktivierung von B-Zellen entstehen
Plasma- und Gedächniszellen. Plasmazellen sezernieren in großen Mengen Antikörper
(Immunglobuline, Ig), welche dann an spezifische Antigene des Pathogens binden und somit
deren Inaktivierung vermitteln können (1.2.2).

EINLEITUNG
23
Eine wichige Voraussetzung für die effektive Erkennung von körperfremden Antigenen durch
ausgereifte T-Zellen besteht darin, dass während der Zellreifung alle T-Zellen entfernt werden
können, deren T-Zell-Rezeptoren körpereigene Antigene binden können. Dazu finden
verschiedene Selektionen statt: Einerseits werden im Thymus, dem Reifungsort der T-Zellen,
alle T-Vorläuferzellen, welche körpereigene Antigene erkennen können, durch Induktion von
Apoptose entfernt. Somit wird gewährleistet, dass nur T-Zellen, die nicht gegen die im Thymus
exprimierten körpereigenen Antigene gerichtet sind, ausreifen und als funktionsfähige T-Zellen
in die Peripherie auswandern (zentrale Toleranz). Andererseits werden aber nicht alle
Selbstantigene im Thymus exprimiert, so dass ein Teil der autoreaktiven T-Zellen in die
Peripherie gelangt. Bei fehlender Kostimulation gehen diese T-Zellen in einen anergen Zustand
über, d.h. sie werden funktionell inaktiviert. Die wiederholte Begegnung einer autoreaktiven
T-Zelle mit peripheren Antigenen führt ebenfalls zu Apoptose. Darüber hinaus halten
regulatorische T-Zellen die periphere Toleranz aufrecht (Referenzen in Janeway et al., 2005).
1.2.2 Struktur und Isotypen von Maus-Immunglobuline n
Antikörper spielen eine wichtige Rolle beim Schutz des Organismus gegen eindringende
Krankheitserreger. Zum einen können sie Pathogene durch ihre Bindung neutralisieren, zum
anderen durch Opsonierung die Antikörper- bzw. Komplement-abhängige zellvermittelte
Zytotoxizität (ADCC bzw. CDCC) vermitteln.
Unabhängig von ihrem Isotypen besitzen alle Immunglobuline eine gemeinsame Struktur. Sie
bestehen aus vier Polypeptidketten, die nach ihrem Molekulargewicht in je zwei identische
leichte (je 25 kDa) und zwei schwere Ketten (je 50-70 kDa) unterschieden werden. Die vier
Ketten sind über mehrere Disulfidbrücken miteinander verbunden und bilden so eine
charakteristische Y-Struktur. Die Gelenkregion, d.h. der Bereich, in dem die beiden schweren
Ketten durch Disulfidbrücken verbunden sind, vermittelt dem Antikörper die notwendige
Flexibilität. Die leichten Ketten bestehen aus jeweils einer variablen und einer konstanten
Domäne (VL und CL), während die schweren Ketten aus einer variablen Domäne (VH) und drei
konstanten Domänen (CH1, CH2, CH3) gebildet werden. Das Antigen-bindende Fragment (Fab-
Fragment), das oberhalb der Gelenkregion liegt, enthält die beiden identischen Antigen-
bindenden Domänen, welche aus den beiden variablen Domänen der leichten und schweren
Ketten gebildet werden. Unterhalb der Gelenkregion befindet sich das FC-Fragment (engl.:
fragment crystallizable) (Abbildung 7A), welches die wichtigen Effektorfunktionen (ADCC,
CDCC) vermittelt.
Die Primärstruktur der konstanten Domäne eines Isotyps ist weitgehend konserviert, während
die variablen Regionen zur Bildung der Antigenbindungsstelle große Sequenzunterschiede
aufweisen müssen. Diese Sequenzvariationen konzentrieren sich auf drei hypervariable

EINLEITUNG
24
A B
L1
L3L2 H3 H1 H2
variable Region derleichtenKette
variable Region derschwerenKette
Antigenbindungs-regionFab
Fc
A B
L1
L3L2 H3 H1 H2
variable Region derleichtenKette
variable Region derschwerenKette
Antigenbindungs-regionFab
Fc
Regionen (CDR), die von weniger variablen Gerüstregionen (FR) umgeben sind. Beim
gefalteten Antikörpermolekül ragen die jeweils drei CDRs der leichten und schweren Kette als
Schleifen aus den β-Faltblättern der restlichen Antikörperstruktur heraus und bilden so die
Antigenbindungsstelle (Abbildung 7B). Durch sie wird die Spezifität des Antikörpers bestimmt.
Abbildung 7: Modell eines IgG-Antikörpers und Struk tur der Antigenbindungsstelle. A) Der IgG-Antikörper besteht aus zwei schweren Ketten (blau) und zwei leichten Ketten (lila), die über Disulfidbrücken (-) miteinander verbunden sind. B) Die Struktur der Immunglobulin-Domänen wird von den β-Faltblatt-Bereichen (grün) der FR-Regionen bestimmt, während die Antigenbindungsstelle aus sechs Schleifen der CDRs (H1-H3, L1-L3) gebildet wird (Bild und Quelle aus Schmiedl und Dübel, 2004).
Es existieren fünf Typen von schweren Ketten (γ, α, µ, δ und ε), welche die Zugehörigkeit zu
einer der Hauptklassen bzw. Isotypen der sezernierten Antikörper definieren (IgG, IgA, IgM, IgD
und IgE). Bei der Maus wird der Isotyp IgG aufgrund von leichten Sequenzunterschieden in vier
weitere Unterklassen (IgG1, IgG2a, IgG2b, IgG3) unterteilt. Diese Isotypen werden zu
verschiedenen Zeitpunkten der Immunantwort gebildet und vermitteln unterschiedliche
Effektorfunktionen (Tabelle 3).
Die beiden Isotypen der leichten Kette, κ und λ, weisen keine funktionellen Unterschiede auf.
Eine Antikörper-produzierende Zelle exprimiert Antikörper entweder mit κ- oder λ-Leichtketten.
Das Verhältnis von κ- zu λ-Ketten beträgt bei der Maus 20:1 (Janeway et al., 2005).

EINLEITUNG
25
Tabelle 3: Eigenschaften muriner Immunglobuline
IgG IgM IgA IgD IgE
schwere Kette γ µ α δ ε
leichte Kette κ oder λ κ oder λ κ oder λ κ oder λ κ oder λ
Valenz 2 10 2, 4 oder 6 2 2
Struktur
Molekular- gewicht (kDa)
150 900 160-320 180 200
Funktion sekundäre
Immunantwort
primäre
Immunantwort
Schutz von
mukosen
Membranen
unbekannt Schutz
gegen
Parasiten
Innerhalb der B-Zell-Zellentwicklung können verschiedene schwere Ketten exprimiert werden.
Am Ende der Reifung steht die Bildung eines funktionierenden B-Zell-Rezeptors (BCR), der von
einem membrangebundenen Antikörper gebildet wird. Beim Übergang von primärer zu
sekundärer Immunantwort erfolgt ein Isotypenwechsel von IgM- zu IgG-Immunglobulinen mit
gleicher Spezifität („class switch“).
1.2.3 Antikörpervielfalt durch somatische Rekombina tion
Das große Antikörperrepertoire wird durch somatische Rekombination innerhalb einer kleinen
Gruppe von Gensegmenten ermöglicht, welche die V-Domänen kodieren (Immunglobulin-
Rearrangement).
Die V-Domäne der leichten Kette wird von V- und J (engl.: joining)-Gensegmenten kodiert,
während die V-Domäne der schweren Kette zusätzlich ein drittes D (engl.: diversity)-
Gensegment zur Steigerung der kombinatorischen Vielfalt enthält. Bei der somatischen
Rekombination der leichten Kette erfolgt zuerst eine Verknüpfung zwischen einem V- und
einem J-Segment. Bei der schweren Kette wird zunächst ein DJ-Segment generiert, das dann
mit dem V-Segment verknüpft wird. Beide Ketten werden abschließend mit einem C-Segment,
das die konstante Domäne kodiert, kombiniert. Diese Reihenfolge des Rearrangements wird
durch die 12/23-Regel gewährleistet. Die kodierenden Sequenzen der einzelnen Segmente
werden durch direkt angrenzende nicht-kodierende Signalsequenzen markiert (RSS), die aus
einer konservierten Reihenfolge von sieben (Heptamer) oder neun (Nonamer) Nukleotiden

EINLEITUNG
26
L L
V V
D
H1 H135
H1
Cµ
JH4
DH11
DH13
JH1
L L
V
D
H1
H1
Cµ
JH4
L L
VH1 Cµ
JH4
L
Cµ
JH4
AAAAA
L
VH135 Cµ
JH2
DH11
AAAAA
VH135
VH135
JH2
DH11
VH135
JH2
DH11
JH2
DH11
primäre RNA
mRNA
gDNA
umgelagerte DNA
umgelagerte DNA V-J-D-Rekombination
Transkription
RNA-Prozessierung
Translation
J-D-Rekombination
bestehen. Zwischen diesen Regionen liegt eine unkonservierte Region, die immer aus 12 oder
23 Basenpaaren besteht (Spacer). Durch die Bindung der beiden Rekombinasen RAG-1 und
RAG-2 an die RSS werden die Gensegmente so verknüpft, dass ein Gensegment, das von
einem 12-bp-Spacer flankiert wird, nur mit einem Gensegment verknüpft werden kann, das
durch eine RSS mit einem 23-bp-Spacer markiert ist (12/23-Regel) (Abbildung 8).
Abbildung 8: Schematische Darstellung der somatisch en Rekombination am Beispiel der schweren Kette. Die Umlagerung der genomischen DNA findet zunächst auf Keimbahnebene statt. Bei der schweren Kette erfolgt zuerst eine DJ-Verknüpfung und anschließend eine Kombination des DJ-Segments mit dem V-Segment. Anschließend wird die DNA in primäre RNA transkribiert, Introns und überzählige Gensegmente werden durch RNA-Prozessierung entfernt und die mRNA wird in ein Protein translatiert. Da Immunglobuline extrazelluläre Proteine sind, befindet sich vor jedem V-Segment eine kurze Signalsequenz (L), die den Antikörper in den sekretorischen Weg dirigiert (modifiziert nach Janeway et al., 2005). Die Gencluster der einzelnen Ketten, die bei der Maus auf Chromosom 2 (κ), 22 (λ) und 14 (H)
liegen, unterscheiden sich in ihrem Aufbau und in der Anzahl der einzelnen V-, J-, D- und

EINLEITUNG
27
C-Segmente. Während der Lokus der κ-Kette nur ein einzelnes Cκ-Segment beinhaltet,
existieren im Gencluster der λ-Kette vier Cλ-Segmente, von denen jedes mit einem der vier
Jλ-Segmente verbunden ist. Entsprechend der fünf Isotypen und vier Subtypen existieren für die
murine schwere Kette acht CH-Gensegmente. Die Anzahl der V-Segmente reicht von nur zwei
Segmenten bei der κ-Kette über 85 Vλ-Gensegmenten bis zu einer Anzahl von 135 bei der
schweren Kette. Dadurch entstehen bis zu 1,8 x 107 Kombinationsmöglichkeiten. Da aber nicht
jede dieser Kombination zu einem stabilen Antikörpermolekül führt und auch nicht jedes
Gensegment mit der gleichen Häufigkeit kombiniert wird, wird die Quantität des
Antikörperrepertoires zusätzlich durch Nukleotid-Addition sowie somatische Hypermutation
erhöht (Referenzen in Janeway et al., 2005).
1.2.4 Antigen-Antikörper-Interaktionen
Die Erkennung und Bindung eines Antigens durch einen Antikörper muss nicht immer eine
Immunantwort auslösen. Haptene, d.h. Substanzen mit einer relativ kleinen Molekülmasse
(< 4000 Da), binden zwar an Antikörper, führen jedoch nur zu einer immunologischen Reaktion,
wenn sie an größere Carrier-Moleküle (z.B. Keyhole Limpet Hemocyanin (KLH)) gekoppelt sind.
Dagegen führen als Antigen bezeichnete Substanzen durch die Bindung an einen Antikörper
zur Aktivierung des Immunsystems. Die Antigenität dieser Substanzen wird unter anderem von
ihrer chemischen Struktur und der Molekülgröße beeinflusst. Proteine, die einen hohen Anteil
an aromatischen Aminosäuren (z.B. Tyrosin) besitzen, sind häufig besonders immunogen.
Antikörper binden nur an einen definierten Bereich des Antigens, der als Epitop bezeichnet
wird. Epitope, die aus hintereinander liegenden Aminosäuren bestehen, bezeichnet man als
linear oder kontinuierlich, während ein Konformationsepitop (diskontinuierliches Epitop) durch
die Faltung des Proteins entsteht und unabhängig von der Sequenz der Primärstruktur ist
(Atassi und Smith, 1978).
Antikörper erkennen Antigene über ihren Fab-Molekülteil, dessen Spezifität durch die insgesamt
sechs hypervariablen CDRs bestimmt wird (Abbildung 7B). Innerhalb der CDRs sind bestimmte
Aminosäuren wie Asparagin, Histidin, Tryptophan und Tyrosin (Kabat et al., 1977; Mian et al.,
1991; Padlan, 1990) besonders häufig. Die Bindung zwischen Antigen und Antikörper ist
reversibel und basiert auf nicht-kovalenten Bindungen wie Wasserstoffbrückenbindungen,
Van-der-Waals-Kräften sowie hydrophoben und elektrostatischen Wechselwirkungen zwischen
den Aminosäuren des Antikörpers und dem Epitop. Die Komplementarität zwischen Epitop und
Antikörper ermöglicht eine räumliche Nähe, so dass diese relativ schwachen Kräfte zu einer
starken Bindung führen können. Dabei wird die Bindungsstärke zwischen einem Antikörper und
einem einzelnen Epitop als Affinität bezeichnet. Niedrig-affine Antikörper binden nur sehr
schwach an das Antigen, und der gebildete Komplex dissoziiert sehr schnell, während hoch-

EINLEITUNG
28
affine Antikörper zu einer lang anhaltenden starken Bindung führen. Ein Maß für die Affinität
eines Antikörpers ist die Dissoziationskonstante (KD-Konstante). Die Affinität reflektiert aber
nicht immer die wirkliche Stärke einer Antigen-Antikörper-Interaktion, da sowohl Antikörper als
auch Antigen multivalent sein können. Die Gesamtstärke aller Bindungen zwischen Antikörper
und Antigen wird als Avidität bezeichnet und ist abhängig von der Affinität des Antikörpers
sowie von der Multivalenz und der geometrischen Struktur von Antikörper und Antigen. Hohe
Avidität kann die niedrige Affinität eines Antikörpers kompensieren und zu einer Stabilisierung
des Antigen-Antikörper-Komplexes führen. Ein Beispiel hierfür ist die Tatsache, dass während
der primären Immunantwort hauptsächlich niedrig-affine, dafür aber hoch-avide IgM-Moleküle
sezerniert werden. Erst nach erneutem Antigenkontakt bei der sekundären Immunantwort
werden überwiegend IgG-Antikörper mit einer hohen Affinität gebildet (1.2.2). Diese
Affinitätsreifung basiert hauptsächlich auf somatischen Hypermutationen innerhalb der CDRs
und klonaler Deletion von weniger affinen Antikörpern.
1.2.5 Produktion von Antikörpern
Bereits 1890 zeigten Emil von Behring und Shibasabo Kitasato, dass das Serum von Tieren,
denen zuvor das Diphterietoxin injiziert wurde, Antitoxine enthält, die beim Menschen zu einer
passiven Immunität führen (Behring und Kitasato, 1890). Dies schuf die Grundlage der
„Serumtherapie“, die auf der Anwendung von polyklonalen Antikörperseren, die aus vielen
Antikörpern mit unterschiedlichen Spezifitäten und Affinitäten für ein bestimmtes Antigen
bestehen, basierte.
Polyklonale Antikörperseren werden durch die wiederholte Immunisierung von Versuchstieren
(Kaninchen, Maus, Ratte, Hamster, Meerschweinchen, Ziege, Schaf, Huhn) mit einem
bestimmten Antigen hergestellt (Hanly et al., 1995). Zur Verstärkung der Antigenität wird
gleichzeitig ein immunstimulatorisches Adjuvans injiziert. Das bekannteste Adjuvans ist das
komplette Freund-Adjuvans (CFA), das aus einer Emulsion aus hitzeinaktiviertem
Mycobacterium tuberculosis und Paraffinöl besteht. Durch seine entzündungsfördernde
Eigenschaft führt es zu starken Nebeneffekten (Abszesse, Granulome, Peritonitis), so dass es
nur bei der ersten Immunisierung verwendet werden kann. Bei den anschließenden Booster-
Injektionen wird inkomplettes Freund-Adjuvans (IFA) eingesetzt, das auf den Zusatz der
Mykobakterien verzichtet. Alternative Adjuvantien sind Mineralgele, wie z.B. Aluminiumhydroxid
oder bakterielle CpGs. Obwohl polyklonale Antikörperseren relativ schnell produziert werden
können (4 bis 8 Wochen) und erfolgreich für diagnostische und therapeutische Zwecke
eingesetzt werden, sind sie mit einigen Nachteilen behaftet. Zum einen ist die Produktion
abhängig von der Größe und Lebensspanne des verwendeten Tiers und zum anderen
entstehen immer unterschiedliche Antiseren, auch, wenn gleiche Immunisierungsschemata bei

EINLEITUNG
29
genetisch identischen Tieren durchgeführt werden. Zudem kommt es häufig zu
Kreuzreaktivitäten, da die Seren auch nach Aufreinigung durch Affinitätschromatografie noch
mit minimalen Mengen unerwünschter Antikörper verunreinigt sein können.
Abbildung 9: Schematische Darstellung der Herstellu ng von monoklonalen Antikörpern. Nach der Immunisierung einer Maus werden die B-Zellen aus der Milz isoliert und mit immortalisierten Myelomzellen zu Hybridomzellen fusioniert. Aufgrund eines Stoffwechseldefekts können nur fusionierte Hybridomzellen im HAT (Hypoxanthin, Aminopterin, Thymidin)-Medium überleben. Positive Klone, die den gewünschten monoklonalen Antikörper sezernieren, werden dann durch spezielle Selektionsverfahren identifiziert. 1975 führten Georg Köhler und César Milstein die „Hybridom“-Technik ein (Kohler und Milstein,
1975). Diese Methode ermöglichte es, eine B-Zelle aus der Milz einer immunisierten Maus mit
einer Myelomzelle zu fusionieren und dadurch zu immortalisieren (Hybridomzelle). Somit
konnten monoklonale Antikörper mit einer bestimmten Spezifität in beliebigen Mengen
produziert werden (Abbildung 9). Da die Myelomzellen keine eigenen Immunglobulinketten
exprimieren, ist gewährleistet, dass der fusionierte Hybridomklon nur den Antikörper der B-Zelle

EINLEITUNG
30
sezernieren kann. Um den gewünschten Hybridomklon zu selektionieren, erfolgt nach der
Fusion die Kultivierung in einem Selektionsmedium, das Hypoxanthin, Aminopterin und
Thymidin (HAT-Medium) enthält (Littlefield, 1964). Aminopterin hemmt die de novo-Biosynthese
von Nukleotiden. Da B-Zellen über alternative Stoffwechselwege verfügen, die entsprechende
Nukleotide aus Hypoxanthin und Thymidin produzieren können, wachsen fusionierte B-Zellen
weiter. Nicht-fusionierte Myelomzellen, welche diesen Stoffwechselweg nicht besitzen, sterben
ab und die Lebensdauer von nicht-fusionierten B-Zellen ist limitiert. Somit können nur die
entstandenen Hybridomzellen im HAT-Medium weiter kultiviert werden. Durch immunologische
Nachweisverfahren kann dann der Klon ermittelt werden, der den monoklonalen Antikörper mit
der gewünschten Spezifität sezerniert.
1.2.6 Therapeutische Anwendung von monoklonalen Ant ikörpern
Die unbegrenzte Verfügbarkeit von Hybridomzellen zur Produktion von monoklonalen
Antikörpern mit einer gewünschten Spezifität eröffnet ein breites Spektrum neuer
Einsatzmöglichkeiten für Therapie und Diagnostik. Therapeutische Anwendungen von murinen
monoklonalen Antikörpern beim Menschen sind jedoch begrenzt, da sie aufgrund ihrer hohen
Immunogenität zur Bildung von humanen Anti-Maus-Antikörpern (HAMA) führen,
Effektorfunktionen nur ineffizient vermitteln können und eine verkürzte Halbwertszeit im
Vergleich mit humanen Antikörpern besitzen. Mittels gentechnischer Methoden ist es
mittlerweile möglich, diese Nachteile durch die Generierung von chimären oder humanisierten
Antikörpern zu reduzieren. Bei der Chimärisierung eines murinen Antikörpers wird die konstante
Domäne durch die äquivalente humane Antikörperdomäne ersetzt (Boulianne et al., 1984). Bei
humanisierten Antikörpern werden zusätzlich auch die FR-Domänen substituiert (Jones et al.,
1986). Somit wird der murine Anteil bei chimären Antikörpern auf ~25% und bei humanisierten
Antikörpern auf ~5% reduziert (Stern und Herrmann, 2005), was zu einer deutlichen
Herabsetzung der Immunogenität bei gleichzeitiger Erhaltung der Spezifität führt. Doch auch
diese Antikörper lösen eine geringe Immunantwort aus, so dass die Herstellung von vollständig
humanen Antikörpern die beste Lösung bedeuten würde. Eine Strategie verwendete dazu das
Phagen-Display-System (McCafferty et al., 1990; Smith, 1985), in dem Bibliotheken von
humanen cDNAs, die für die variablen Regionen der Antikörper kodieren, exprimiert werden.
Jeder Phage dieser Bibliothek exprimiert auf seiner Oberfläche eine Kombination aus VH und VL
in Form eines single-chain-Fragments (scFv) (Abbildung 10). Nach Isolierung eines
gewünschten Phagenklons können E.coli-Zellen infiziert werden, welche das humane scFv-
Fragment dann sezernieren (McCafferty et al., 1990; Smith, 1985).
Eine andere Strategie beinhaltet die Generierung von transgenen Mäusen, die ausschließlich
Immunglobulin Gen-Cluster vom Menschen in ihrer Keimbahn tragen. Durch die Verwendung

EINLEITUNG
31
CH1
CH2
CH3
IgG F(ab´) 2 Minibody Fab scFv DiabodyVL
VH
CH1
CH2
CH3
IgG F(ab´) 2 Minibody Fab scFv DiabodyVL
VH
von Yeast artificial chromosome (YAC)-Vektoren gelang die Herstellung der XenoMouse®, die
einen Großteil des humanen VLκ- und VHκ-Repertoires trägt. Nach Immunisierung dieser Mäuse
können durch die Fusion mit humanen Myelomzellen monoklonale Antikörper mittels
„Hybridom“-Technik hergestellt werden. Ein Beispiel für die erfolgreiche Nutzung dieser
Strategie ist der IgG2κ-Antikörper Vectibix®, der zur Behandlung des Kolonkarzinoms eingesetzt
werden soll und derzeit in Phase III Studien getestet wird (Green et al., 1994; Yang et al.,
2001).
Je nach therapeutischer Anwendung bietet sich auch die Verwendung von kleineren Antikörper-
fragmenten an (Abbildung 10). Durch eine proteolytische Behandlung von Antikörpern mit der
Thiolprotease Papain oder der Carboxypeptidase Pepsin können entweder zwei Fab-
Fragmente oder ein F(ab`)2-Fragment hergestellt werden. Rekombinante Antikörper-abgeleitete
Moleküle wie z.B. scFvs oder Minibodies welche nur die Antigenbindungsstelle des parentalen
Antikörpers enthalten, können durch die Expression in Bakterien oder eukaryontischen Zellen
generiert werden. Die Größe des Antikörperformats beeinflusst verschiedene Eigenschaften wie
Avidität, Pharmakokinetik, die Fähigkeit zur Gewebepenetration sowie die Ausschleusung durch
die Niere (Clearance). Fab-Fragmente, scFv, Diabodies und Minibodies eignen sich aufgrund
ihrer besseren Clearance und Gewebepenetration z.B. eher für radiodiagnostische Methoden.
Dagegen haben vollständige Antikörpermoleküle eine längere Halbwertszeit
(~ 3 Wochen) und können Effektorfunktionen über das FC-Fragment vermitteln.
Abbildung 10: Darstellung von verschiedenen Antikör performaten. Die abgebildeten Formate besitzen ein Molekulargewicht von 27 bis 150 kDa. Die konstanten Domänen sind in lila dargestellt, die variable Domäne der leichten Kette in orange und der schweren Kette in grün. Diabodies bestehen aus zwei scFv, die über ein kurzes Peptid verbunden sind. Minibodies bestehen ebenfalls aus zwei scFv, werden aber über die Gelenkregion und die CH3-Domäne verbunden (Fab = Fragment antigen binding, scFv = single chain fragment variable) (Bild und Quelle aus Carter, 2006).
EINLEITUNG
32
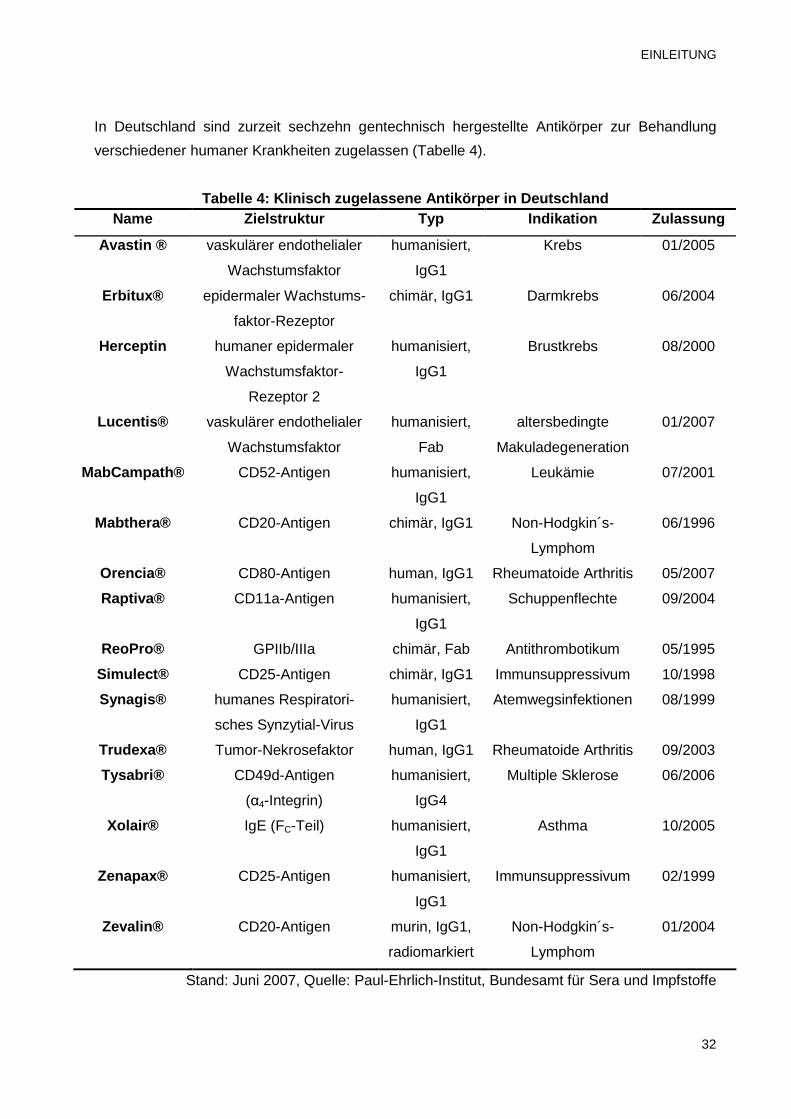
In Deutschland sind zurzeit sechzehn gentechnisch hergestellte Antikörper zur Behandlung
verschiedener humaner Krankheiten zugelassen (Tabelle 4).
Tabelle 4: Klinisch zugelassene Antikörper in Deuts chland Name Zielstruktur Typ Indikation Zulassung
Avastin ® vaskulärer endothelialer
Wachstumsfaktor
humanisiert,
IgG1
Krebs 01/2005
Erbitux® epidermaler Wachstums-
faktor-Rezeptor
chimär, IgG1 Darmkrebs 06/2004
Herceptin humaner epidermaler
Wachstumsfaktor-
Rezeptor 2
humanisiert,
IgG1
Brustkrebs 08/2000
Lucentis® vaskulärer endothelialer
Wachstumsfaktor
humanisiert,
Fab
altersbedingte
Makuladegeneration
01/2007
MabCampath® CD52-Antigen humanisiert,
IgG1
Leukämie 07/2001
Mabthera® CD20-Antigen chimär, IgG1 Non-Hodgkin´s-
Lymphom
06/1996
Orencia® CD80-Antigen human, IgG1 Rheumatoide Arthritis 05/2007
Raptiva® CD11a-Antigen humanisiert,
IgG1
Schuppenflechte 09/2004
ReoPro® GPIIb/IIIa chimär, Fab Antithrombotikum 05/1995
Simulect® CD25-Antigen chimär, IgG1 Immunsuppressivum 10/1998
Synagis® humanes Respiratori-
sches Synzytial-Virus
humanisiert,
IgG1
Atemwegsinfektionen 08/1999
Trudexa® Tumor-Nekrosefaktor human, IgG1 Rheumatoide Arthritis 09/2003
Tysabri® CD49d-Antigen
(α4-Integrin)
humanisiert,
IgG4
Multiple Sklerose 06/2006
Xolair® IgE (FC-Teil) humanisiert,
IgG1
Asthma 10/2005
Zenapax® CD25-Antigen humanisiert,
IgG1
Immunsuppressivum 02/1999
Zevalin® CD20-Antigen murin, IgG1,
radiomarkiert
Non-Hodgkin´s-
Lymphom
01/2004
Stand: Juni 2007, Quelle: Paul-Ehrlich-Institut, Bundesamt für Sera und Impfstoffe

EINLEITUNG
33
Einer der zugelassenen Antikörper ist Natalizumab (Tysabri®), der bei der Behandlung der
neurodegenerativen Krankheit Multiple Sklerose eingesetzt wird. Indem der Antikörper durch
die Bindung an α4-Integrin auf aktivierten T-Zellen und Monozyten den Eintritt dieser Zellen über
VCAM-1 (engl.: vascular cell adhesion molecul-1) ins Gehirn verhindert, kann die
inflammatorische Symptomatik reduziert werden (Polman et al., 2006). Auch wenn im Vorfeld
potentielle Risiken der monoklonalen Antikörper im Tiermodell evaluiert und minimiert werden,
können unerwartete Nebenwirkungen bei der Übertragung auf den Menschen nicht vollständig
ausgeschlossen werden, wie die kürzlich durchgeführte Phase I Studie mit dem Anti-CD28-
Antikörper TGN1412 zeigte. Dieser Antikörper, der als Immunsuppressivum bei
Autoimmunkrankheiten eingesetzt werden sollte, führte bei den sechs Probanden der Studie zu
multiplem Organversagen (Suntharalingam et al., 2006; Weerasekera und Hudson, 2007).
Bei der Alzheimerschen Erkrankung (AD), die wie Prionenerkrankungen durch die Akkumulation
eines falsch gefalteten Proteins ausgelöst wird, werden ebenfalls monoklonale Antikörper für
therapeutische Zwecke eingesetzt. Diese Krankheit zeichnet sich durch Ablagerungen eines
amyloiden Peptids (Aβ40/42) aus, das durch die proteolytische Spaltung von einer β- und
γ-Sekretase aus dem endogenen Amyloid-Precursor-Protein (APP) entsteht. Passive
Immunisierungen mit Antikörpern gegen das Amyloid-β-Peptid führten in einem transgenen AD-
Mausmodell zu einer deutlichen Reduktion der Aβ-Plaques im Gehirn (Bard et al., 2000;
DeMattos et al., 2001; Lee et al., 2006; McLaurin et al., 2002). Man nimmt an, dass die
Antikörper entweder direkt an das Aβ-Peptid binden und so zur Auflösung der Plaques führen,
welche über eine FCR-vermittelte Phagozytose durch Mikrogliazellen möglich wäre, oder, dass
die monoklonalen Antikörper im Blut eine Reduktion des freien Aβ-Gehalts bewirken und so
einen Rückfluss von Aβ aus dem Gehirn ins Plasma induzieren („peripheral sink hypothesis“)
(DeMattos et al., 2001; Schenk et al., 1999). Zurzeit wird die passive Immunisierung mit einem
humanisiertem Anti-Aβ-Antikörper (AAB-001) in einer Phase II Studie untersucht. Auch aktive
Immunisierungen mit einem fibrillärem Aβ1-42-Peptid (AN1792) wurden bereits in einer Phase II
Studie durchgeführt. Da jedoch 6% der Probanden eine Meningoenzephalitis entwickelten,
musste die Studie vorzeitig abgebrochen werden. Trotzdem konnten bei einem Teil der
Patienten verbesserte kognitive Leistungen beobachtet werden, und post mortem Sektionen
zeigten eine deutlich reduzierte Ablagerung von amyloiden Plaques im Gehirn (Gilman et al.,
2005; Hock et al., 2003; Masliah et al., 2005). Mittlerweile nimmt man an, dass die
Meningoenzephalitis durch ein bisher unbekanntes T-Zell-Epitop innerhalb des Aβ-Peptids
verursacht wurde und eine Immunisierung mit ausgewählten B-Zell-Epitopen des Aβ-Peptids die
Nebenwirkungen minimieren könnte (Monsonego und Weiner, 2003; Monsonego et al., 2003).
Der erfolgreiche Einsatz von monoklonalen Antikörpern bei der Behandlung der
Alzheimerschen Erkrankung führte zu der Annahme, dass humanisierte Antikörper gegen das
Prion-Protein bei humanen Prionenerkrankungen zu ähnlichen Erfolgen führen können.

EINLEITUNG
34
1.3 Ziel der Arbeit
Prionenerkrankungen wie die Creutzfeldt-Jakob Krankheit beim Menschen oder die Bovine
Spongiforme Enzephalopathie beim Rind sind letal verlaufende, neurodegenerative
Erkrankungen. Der Erreger dieser Krankheiten ist das infektiöse Prion-Protein PrPSc, bei dem
es sich um ein pathogenes Konformationsisomer des zellulären Prion-Proteins PrPC handelt
und welches PrPC seine Konformation aufzwingen kann.
Zurzeit existieren keine Medikamente, welche den Krankheitsverlauf beim Menschen signifikant
beeinflussen können (Prusiner, 1998). Bei einer anderen neurodegenerativen Erkrankung, der
Alzheimerschen Krankheit, wurden im Tiermodell gute Erfolge mit aktiven und passiven
Immunisierungsstrategien erzielt (Janus et al., 2000; Schenk et al., 1999; Sigurdsson et al.,
2001). Der Erfolg von aktiven Immunisierungsstrategien bei Prionenerkrankungen wird jedoch
durch die körpereigene Toleranz gegenüber dem Prion-Protein erschwert. Nach passiver
Immunisierung von wildtypischen Mäusen wurden hingegen signifikante Erfolge beobachtet
(White et al., 2003). Das Ziel der vorliegenden Arbeit war somit die Herstellung von neuen Anti-
PrP-Antikörpern mittels „Hybridom“-Technik, welche für diagnostische und therapeutische
Zwecke genutzt werden können. Insgesamt sollten folgende Fragestellungen untersucht
werden:
• Wie unterscheiden sich die Anti-PrP-Antikörper bezüglich des
Epitops, der Avidität und Affinität?
• Hemmen die Anti-PrP-Antikörper die PrPSc-Akkumulation in Prion-
infizierten Zellen und wenn ja, ist dieser Effekt dosis- und
zeitabhängig?
• Beeinflussen die Anti-PrP-Antikörper auch die Infektiosität von Prion-
infizierten Zellen?
• Welchen Wirkungsmechanismus nutzen die Anti-PrP-Antikörper, um
die PrPSc-Akkumulation in vitro zu inhibieren?

MATERIAL UND METHODEN
35
2 Material und Methoden
2.1 Material
2.1.1 Geräte
Autoklav Münchner Medizin GmbH
Bakterienkulturschüttler Braun AG (Certomat)
Bio-Imager Fuji GmbH (FLA 3000-2R, Image Reader V1.8E),
Digitale Kamera Diagnostic Instruments, Inc.
Eismaschine Scotsman
Elektrophoresekammern Hauswerkstatt
ELISA-Reader Molecular Devices, Inc. (Vmax® kinetic)
FACS Becton Dickinson GmbH (FACS Calibur)
Fein- und Laborwaage Sartorius AG
Fluoreszenzmikroskop Leica GmbH
Gefrierschränke Bosch GmbH
Großzentrifuge Sorvall GmbH(SLC-6000, SLC-3000, SLC-1500)
Heizblock Eppendorf AG
Image Reader Fuji GmbH (LAS-3000 Version 2.0)
Inkubatoren Heraeus GmbH
Kapillarsequenzierer Perkin Elmer, Inc. (ABI Prism 310)
PE Applied Biosystems GmbH (ABI Prism 3100)
Kühlschränke Bosch GmbH
Küvetten Eppendorf AG
Magnetrührer Ika® Werke GmbH (Ikamag®RH)
Mikrowellenherd Severin GmbH
PC Mac OS X; Windows XP
pH-Meter Denver Instrument GmbH
Pipetten Gilson, Inc., Eppendorf AG
Pipettierhilfe Gilson, Inc.
Sonifier (Ultraschall) Branson, Inc.
Spektralphotometer Pharmacia Corp. (Ultrospec III)
Sterilbank Nuaire, Inc.
Thermocycler Eppendorf AG
Tisch-Wippe Heidolph GmbH & Co. KG
Tischzentrifuge Eppendorf AG
UV-Handlampe Vilber Lourmat, Inc.

MATERIAL UND METHODEN
36
UV-Leuchttisch/Videodrucker MWG AG, Mitsubishi Corp.
Vortexer Heidolph GmbH & Co. KG
Wasserbäder GFL GmbH
Zellkulturzentrifuge Hettich GmbH & Co. KG
Zentrifugen Kendro Corp.
2.1.2 Chemikalien
Die verwendeten Chemikalien wurden bei den Firmen AppliChem GmbH, USB Europe GmbH,
Sigma-Aldrich Chemie GmbH, Serva GmbH, Carl Roth GmbH & Co. KG und Merck Chemicals
Ltd. bestellt. Verbrauchsmaterialien wie Reaktionsgefäße, Handschuhe oder Pipettenspitzen
wurden bei der Firma Hartenstein Laborbedarf GmbH erworben. Materialien für die Zellkultur
stammten von der Firma Nunc GmbH & Co. KG.
2.1.3 Zelllinien
HEK 293T humane embryonale Nierenzelllinie,
immortalisiert mit dem Ad5-E1A/B-Protein
und dem SV-40-T-Antigen
(Graham et al., 1977)
N2a murine Neuroblastoma-Zelllinie (Olmsted et al., 1970)
ScN2a Scrapie-infizierte N2a-Zelllinie (Butler et al., 1988)
cuScN2a ScN2a-Zellen, die nach Behandlung mit
Pentosanpolysulfat (Caughey und Raymond,
1993) kein PrPSc tragen
J. Springer (AG Klein)
SP2/0-AG14 Hybrid aus Milz- und Myelomzellen der Maus (Shulman et al., 1978)
2.1.4 Lösungen zur Analyse und Klonierung von DNA
50x TAE Tris pH 8,0
Konz. Essigsäure
EDTA-Na2 pH 8,0
2 M
5,7% (v/v)
0,05 M
6x DNA-Probenpuffer Bromphenolblau
Xylencyanol
Glycerin
0,25% (w/v)
0,25% (w/v)
30%
3 M Natriumacetat, pH 5,2 Natriumacetat
pH mit konz. Essigsäure einstellen
3 M
MATERIAL UND METHODEN
37
Plasmidpräparation
Qiagen-Puffer P1
(Resuspensionspuffer)
Tris-HCl pH 8,0
EDTA
RNase A
0,05 M
0,01 M
100 µg/ml
Plasmidpräparation
Qiagen-Puffer P2
(Lyse-Puffer)
NaOH
SDS
0,2 M
1% (w/v)
Plasmidpräparation
Qiagen-Puffer P3
(Neutralisationspuffer)
Kaliumacetat pH 5,5 3 M
Plasmidpräparation
Qiagen-Puffer QBT
(Äquilibrierungspuffer)
NaCl
MOPS pH 7,0
Isopropanol
Triton X 100
0,75 M
0,05 M
15% (v/v)
0,15% (v/v)
Plasmidpräparation
Qiagen-Puffer QC
(Waschpuffer)
NaCl
MOPS pH 7,0
Isopropanol
1 M
0,05 M
15% (v/v)
Plasmidpräparation
Qiagen-Puffer QF
(Elutionspuffer)
NaCl
Tris-HCl pH 8,5
Isopropanol
1,25 M
0,05 M
15% (v/v)
1 M KOH KOH 1 M
1 M Tris-HCl pH 7,5 Tris
pH mit konzentrierter HCl einstellen
1 M
2.1.5 Standardlösungen
PBS NaCl
KCl
Na2HPO4
KH2PO2
CaCl2
MgCl2
0,137 M
0,0027 M
0,0043 M
0,0014 M
0,0015 M
0,001 M
PBS- NaCl
KCl
Na2HPO4
KH2PO2
0,137 M
0,0027 M
0,0043 M
0,0014 M
MATERIAL UND METHODEN
38
2.1.6 Antikörper und Farbstoffe
Antikörper Spezies Isotyp Konjugat Hersteller
N-CAM (CD 56) Maus IgG2a / BD GmbH
Mouse IgG1, к Maus Ascites / Sigma GmbH
Mouse IgG2a, к Maus Ascites / Sigma GmbH
α-mouse-IgG (H+L) Kaninchen / ALEXA
Fluor® 488
Molecular Probes, Inc.
α-mouse-IgG1 Kaninchen / HRP Zymed GmbH
α-mouse-IgG2a Kaninchen / HRP Zymed GmbH
α-mouse-Ig Ziege pAk HRP Dako GmbH
α-rabbit-IgG (H+L) Ziege / HRP Zymed GmbH
α-mouse-IgG (H+L) Ziege / FITC Dianova GmbH
α-mouse-IgM Ziege / HRP Zymed GmbH
α-PrP (SA2124) Kaninchen pAk / AG Klein
α-PrP (ICSM18) Maus IgG1 / D-Gen Ltd.
α-PrP (ICSM35) Maus IgG2b / D-Gen Ltd.
α-PrP (6H4) Maus IgG1 / Prionics AG
Farbstoff Konjugat Hersteller
Cholera Toxin Subunit B ALEXA Fluor® 549 Molecular Probes, Inc.
2.1.7 Scrapie-Prionenstamm
Für die Inokulationsstudien wurde der RML (Rocky Mountain Laboratories)-Prionenstamm
eingesetzt. Hierbei handelt es sich um ein Maus-adaptiertes Scrapie-Isolat (Chandler, 1961),
das in CD-1 Mäusen passagiert wurde. Das isolierte Gehirn wurde gewogen und in neun
Volumen 0,32 M Sucrose aufgenommen. Um das Zellmaterial zu homogenisieren, wurde es
durch unterschiedlich große Kanülen (G18-G21) gezogen. Nach Zentrifugation (2000 rpm,
20 min) erfolgte die Aufbewahrung des Überstandes als 10%iges-Homogenat bei -20°C.
2.1.8 Plasmide
Plasmid Herkunft
pBluescript II SK(+) Stratagene GmbH
pAd-CMV-PrP ∆32-93 E.Flechsig
pAd-CMV-PrP ∆32-106 E.Flechsig

MATERIAL UND METHODEN
39
pAd-CMV-PrP ∆32-93 & ∆141-176 E.Flechsig
Während dieser Arbeit hergestellte Konstrukte:
pAd-CMV-PrP ∆32-93 & ∆142-157
pAd-CMV-PrP ∆32-93 & ∆158-171
pAd-CMV-PrP ∆32-93 & Y148A
pAd-CMV-PrP ∆32-93 & E151A
pAd-CMV-PrP ∆32-93 & H176A
2.1.9 Primer
Primer Sequenz (5´ →→→→ 3´)
Klonierung
BS_MCS-NotI for GTG GCG GCC GCT CTA GAA CTA GTG G
SAP_PrP_MfeI rev TAG CCT ATG GGG GAC ACA GAG AAG C
PrP ∆157for TGT ACC GCT CTT CTA ACC AAG TGT ACT ACA GGC CAG TGG
PrP ∆142rev CGG TCC GCT CTT CCG TTG CCA AAA TTG ATC ATG GGC CTG C
PrP ∆171for AGT ACA GCT CTT CGA ACA ACT TCG TGC ACG ACT GC
PrP ∆158rev CTG TAG GCT CTT CGG TTA GGG TAG CGG TAC ATG TTT TCA CG
Ortsgerichtete Mutagenese
E151A for CCG CTA CTA CCG TGC CAA CAT GTA CCG CTA CC
E151A rev GGT AGC GGT AGA TGT TGG CAC GGT AGT AGC GG
Y148A for GGA GGA CCG CGC CTA CCG TGC TAA CAT GTA CC
Y148A rev GGT ACA TGT TAG CAC GGT AGG CGC GGT CCT CC
M153A for GCC TAC CGT GCT AAC GCC TAC CGC TAC CCT AAC C
M153A rev GGT TAG GGT AGC GGT AGG CGT TAG CAC GGT AGG C
H176A for CAG AAC AAC TTC GTG GCC GAC TGC GTC AAT ATC ACC
H176A rev GGT GAT ATT CAC GCA GTC GGC GAC GAA GTT GTT CTG
Screening und Sequenzierung
M13 for GGA AAC AGC TAT GAC CAT G
M13 rev GTA AAA CGA CGG CCA GT
Sig1 for TAT GTG GAC TGA TGT CGG CC
3-NC rev CCC TCC CCC AGC CTA GAC CAC GA
Mut217 rev CCT GGG ACT CCT TCT GGT ACC GGG TGA CGC

MATERIAL UND METHODEN
40
2.1.10 Proteine
rekombinantes Maus-PrP (23-231) Thorsten Lührs (Zahn et al., 1997)
rekombinantes Maus-PrP (23-231), fibrillär Thorsten Lührs (Luhrs et al., 2006)
2.1.11 Versuchstiere
Tiere Bezeichnung Quelle
C57BL/6 Prnp+/+ (Wildtyp PrPC) Jackson Laboratories
Prnp0/0 Knockout des Prnp Gens (kein PrPC) (Bueler et al., 1992)
Tga20 Prnp0/0 mit PrPC-Transgen (Fischer et al., 1996)
2.2 Methoden
2.2.1 Herstellung von monoklonalen Antikörpern
2.2.1.1 Immunisierung und Isolierung von Milzzellen
rekombinantes Maus-PrP (23-231) Thorsten Lührs (Zahn et al., 1997)
rekombinantes Maus-PrP (23-231),
fibrillär
Thorsten Lührs (Luhrs et al., 2006)
CpG 1668 MWG AG
komplettes Freund-Adjuvans Sigma GmbH
inkomplettes Freund-Adjuvans Sigma GmbH
serumfreies Medium RPMI 1640mit Glutamin(Gibco Ltd.)
100x Pyruvat (Sigma GmbH) 1% (v/v)
100x Aminosäuren (Sigma GmbH) 1% (v/v)
NH4Cl/HEPES 0,83% (w/v)
Im Abstand von vier Wochen wurden die Prnp0/0-Mäuse immunisiert. Jeweils eine Woche nach
der Immunisierung wurden die Antikörpertiter der einzelnen Tiere mittels ELISA (2.2.4.1)
bestimmt. Als Negativkontrolle wurde das Serum einer nicht-immunisierten Prnp0/0-Maus
verwendet. Die Immunisierung für die erste Fusion erfolgte mit rekombinantem Maus-PrP,
während für die zweite Fusion eine fibrilläre Form des rekombinanten Maus-PrP (Luhrs et al.,
2006) verwendet wurde. Für beide Fusionen wurden jeweils 50 µg PrP in einem
Volumenverhältnis von 1:1 mit komplettem Freund-Adjuvans gemischt und intraperitoneal
injiziert. Für die anschließenden Booster-Injektionen wurde jeweils die gleiche Proteinmenge im
gleichen Verhältnis mit inkomplettem Freund-Adjuvans gemischt und ebenfalls intraperitoneal
appliziert. Die erste bzw. zweite Fusion erfolgte nach der vierten bzw. dritten Boosterung. Für
die dritte Fusion erfolgte eine subkutane Immunisierung mit 10 µg rekombinantem Maus-PrP im

MATERIAL UND METHODEN
41
Gemisch mit 10 µg CpG 1668 als Adjuvans. Nach zweimaliger Booster-Injektion mit gleichen
Antigenmengen und gleicher Applikationsroute erfolgte die dritte Fusion.
Drei Tage nach der letzen Immunisierung wurde die Maus getötet und die entnommene Milz für
die Fusion verwendet. Hierzu wurde die Bauchseite mit 70% Ethanol desinfiziert und das Fell
entfernt. Nach Öffnung der Bauchdecke wurde die Milz vorsichtig mit einer Pinzette angehoben
und mit einer Schere vom Bindegewebe abgeschnitten. Abschließend wurde die Milz in eine mit
10 ml vorgewärmtem und serumfreiem Medium befüllte Petrischale überführt.
Zur Herstellung einer Einzelzellsuspension wurde die Milz in kleine Stücke geschnitten und
durch ein steriles Zellsieb gedrückt bis die Milzzellen einzeln im Medium vorlagen. Nach
mehrmaligem Spülen des Zellsiebs wurden alle Fraktionen gesammelt, in ein
Zentrifugenröhrchen überführt und ca. zwei Minuten stehen gelassen, damit sich gröbere
Partikel absetzen konnten. Der Überstand wurde in ein neues Zentrifugenröhrchen transferiert
und die enthaltenen Zellen dreimal in serumfreiem Medium gewaschen (6 min, 1100 rpm, 4°C).
Zur Bestimmung der Zellzahl (2.2.2.2) wurde die Suspension zuvor mit 0,83% NH4Cl (1:10)
verdünnt, um die Erythrozyten zu lysieren. Die so präparierten Milzzellen wurden für die Fusion
eingesetzt.
2.2.1.2 Kultivierung und Vorbereitung von Myelomzel len
RPMI 1640 + Glutamax FCS (PAA GmbH)
Pyruvat (Gibco Ltd.)
50x 8-Azaguanin (Sigma GmbH)
10% (v/v)
1% (v/v)
2% (w/v)
Für alle Fusionen wurde die murine Myelomzelllinie SP2/0 verwendet (Shulman et al., 1978).
Dieser Myelomzelllinie fehlt das Enzym Hypoxanthin-Guanin-Phosphoribosyl-Transferase
(HGPRT) für die alternative Biosynthese von Nukleotiden, wodurch die Zellen nur nach einer
Fusion in Gegenwart von Hypoxanthin, Aminopterin und Thymidin im Medium (HAT-Medium)
überleben können. Sie sezerniert keine Immunglobulinketten. Die Kultivierung der SP2/0-Zellen
erfolgte in 500 ml Zellkulturflaschen unter Standardbedingungen bei 37°C in Gegenwart von
5% CO2. Um eine genetische Reversion des Selektionsmarkers zu verhindern, wurden die
Myelomzellen etwa vier Wochen vor der geplanten Fusion für acht Tage in 8-Azaguanin
kultiviert, so dass alle Zellen mit einer intakten alternativen Biosynthese für Nukleotide eliminiert
wurden. Einen Tag vor der Fusion wurden die SP2/0-Zellen in neue Kulturflaschen überführt
und die Zellzahl auf 5 x 105 Zellen pro ml eingestellt.
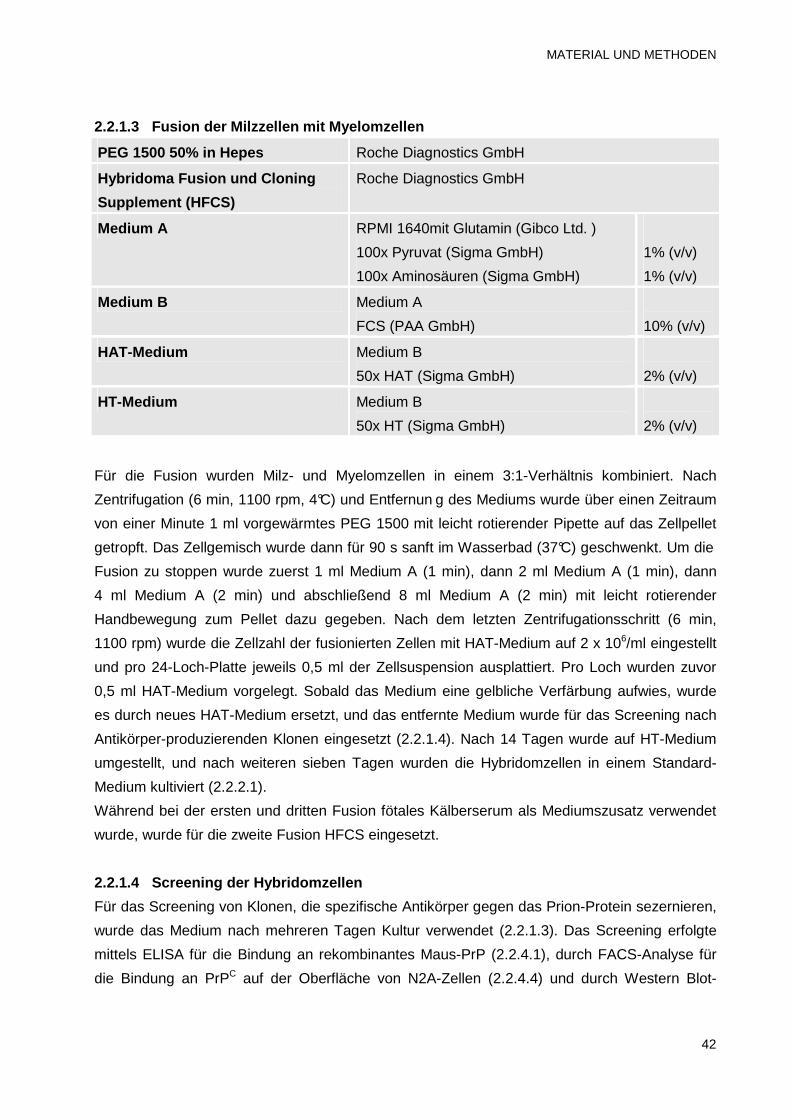
MATERIAL UND METHODEN
42
2.2.1.3 Fusion der Milzzellen mit Myelomzellen
PEG 1500 50% in Hepes Roche Diagnostics GmbH
Hybridoma Fusion und Cloning
Supplement (HFCS)
Roche Diagnostics GmbH
Medium A RPMI 1640mit Glutamin (Gibco Ltd. )
100x Pyruvat (Sigma GmbH)
100x Aminosäuren (Sigma GmbH)
1% (v/v)
1% (v/v)
Medium B Medium A
FCS (PAA GmbH)
10% (v/v)
HAT-Medium Medium B
50x HAT (Sigma GmbH)
2% (v/v)
HT-Medium Medium B
50x HT (Sigma GmbH)
2% (v/v)
Für die Fusion wurden Milz- und Myelomzellen in einem 3:1-Verhältnis kombiniert. Nach
Zentrifugation (6 min, 1100 rpm, 4°C) und Entfernun g des Mediums wurde über einen Zeitraum
von einer Minute 1 ml vorgewärmtes PEG 1500 mit leicht rotierender Pipette auf das Zellpellet
getropft. Das Zellgemisch wurde dann für 90 s sanft im Wasserbad (37°C) geschwenkt. Um die
Fusion zu stoppen wurde zuerst 1 ml Medium A (1 min), dann 2 ml Medium A (1 min), dann
4 ml Medium A (2 min) und abschließend 8 ml Medium A (2 min) mit leicht rotierender
Handbewegung zum Pellet dazu gegeben. Nach dem letzten Zentrifugationsschritt (6 min,
1100 rpm) wurde die Zellzahl der fusionierten Zellen mit HAT-Medium auf 2 x 106/ml eingestellt
und pro 24-Loch-Platte jeweils 0,5 ml der Zellsuspension ausplattiert. Pro Loch wurden zuvor
0,5 ml HAT-Medium vorgelegt. Sobald das Medium eine gelbliche Verfärbung aufwies, wurde
es durch neues HAT-Medium ersetzt, und das entfernte Medium wurde für das Screening nach
Antikörper-produzierenden Klonen eingesetzt (2.2.1.4). Nach 14 Tagen wurde auf HT-Medium
umgestellt, und nach weiteren sieben Tagen wurden die Hybridomzellen in einem Standard-
Medium kultiviert (2.2.2.1).
Während bei der ersten und dritten Fusion fötales Kälberserum als Mediumszusatz verwendet
wurde, wurde für die zweite Fusion HFCS eingesetzt.
2.2.1.4 Screening der Hybridomzellen
Für das Screening von Klonen, die spezifische Antikörper gegen das Prion-Protein sezernieren,
wurde das Medium nach mehreren Tagen Kultur verwendet (2.2.1.3). Das Screening erfolgte
mittels ELISA für die Bindung an rekombinantes Maus-PrP (2.2.4.1), durch FACS-Analyse für
die Bindung an PrPC auf der Oberfläche von N2A-Zellen (2.2.4.4) und durch Western Blot-

MATERIAL UND METHODEN
43
Analyse für die Erkennung von PrPC und PrPSc in Gehirnhomogenaten der Maus (2.2.3). Nur
Hybridomzellen, die in allen drei Methoden ein positives Signal lieferten, wurden für
Zellklonierungen weiter verwendet.
2.2.1.5 Klonierung der Hybridomzelllinien und Nomen klatur der Klone
Interleukin-6 Sigma GmbH
Nach dem Screening lagen die Antikörper-produzierenden Hybridome in einem polyklonalen
Zustand vor. Die Klonierung dieser Hybridomzellen sollte schnell erfolgen, da die Gefahr
besteht, dass der produzierende Klon von anderen Zellen überwachsen wird. Dazu wurden die
Hybridomzellen so verdünnt, dass sich nach der Aussaat maximal eine, zwei oder fünf Zellen
pro Loch befanden (Coller und Coller, 1983). Von jeder Zellkonzentration wurde eine 96-Loch-
Platte angelegt. Um die Wachstumsbedingungen zu verbessern, wurden zusätzlich 5 x 105
Prnp0/0-Milzzellen pro Loch ausgesät, die zuvor 20 min mit 35 Gy bestrahlt wurden. Die
mikroskopische Kontrolle der Monoklonalität fand nach einer Woche statt. Die
Zellkulturüberstände von vereinzelten Klonen wurden einem zweiten Screening unterzogen, um
zu gewährleisten, dass auch der Antikörper-produzierende Klon weiter kultiviert wurde (2.2.1.4).
Die Zellklonierung musste ein- bis zweimal wiederholt werden. Über die Häufigkeit dieser
Subklonierungen gibt die Nomenklatur der Antikörper Auskunft. Beispielsweise wurde der Klon
„7-12-5“ zweimal subkloniert, wobei sich der selektierte Klon direkt nach der Fusion in Loch Nr.
7 befand und nach der ersten bzw. der zweiten Subklonierung in Loch Nr. 12 bzw. Loch Nr. 5.
Die positiven Klone wurden schrittweise in 24-Loch-Platten, 6-Loch-Platten und abschließend in
Zellkulturflaschen umgesetzt. Klone, die schlecht wuchsen, wurden zusätzlich mit Interleukin-6
behandelt (0,05 ng/ml Medium).
2.2.1.6 Produktion der monoklonalen Antikörper
Integra CELLine CL350/ CL 1000 INTEGRA Biosciences AG, Schweitz
Hybridomed DIF 1000 Biochrom AG
Zur Produktion von großen Mengen monoklonaler Antikörper wurden Zwei-Kompartiment-
Bioreaktoren eingesetzt. Diese Bioreaktoren zeichnen sich durch die Trennung in ein
Versorgungs- und ein Kultivierungskompartiment aus. Separiert werden die beiden
Kompartimente durch eine 10 kDa semipermeable Zellulose-Acetat Membran, welche den
Nährstoffaustausch garantiert, jedoch die Hybridomzellen sowie die sezernierten Antikörper im
Kultivierungskompartiment zurückhält. Dadurch entsteht in diesem Kompartiment eine hohe
Anreicherung von Hybridomzellen und Antikörpern.
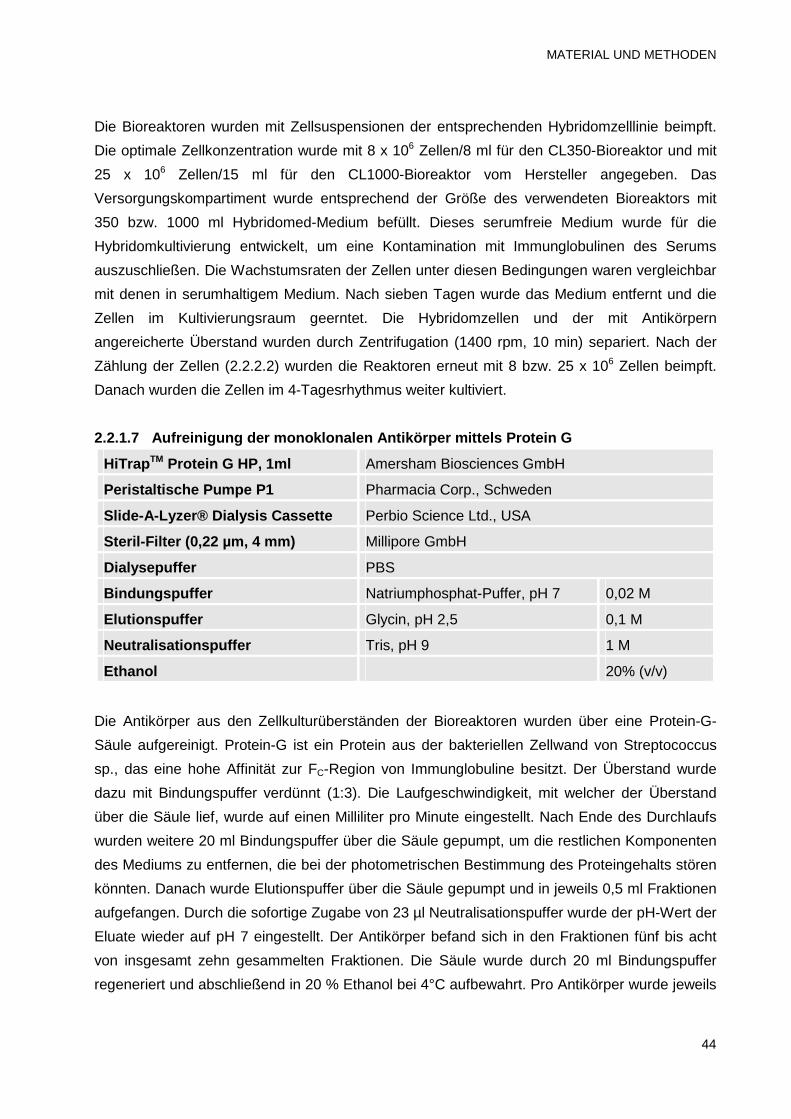
MATERIAL UND METHODEN
44
Die Bioreaktoren wurden mit Zellsuspensionen der entsprechenden Hybridomzelllinie beimpft.
Die optimale Zellkonzentration wurde mit 8 x 106 Zellen/8 ml für den CL350-Bioreaktor und mit
25 x 106 Zellen/15 ml für den CL1000-Bioreaktor vom Hersteller angegeben. Das
Versorgungskompartiment wurde entsprechend der Größe des verwendeten Bioreaktors mit
350 bzw. 1000 ml Hybridomed-Medium befüllt. Dieses serumfreie Medium wurde für die
Hybridomkultivierung entwickelt, um eine Kontamination mit Immunglobulinen des Serums
auszuschließen. Die Wachstumsraten der Zellen unter diesen Bedingungen waren vergleichbar
mit denen in serumhaltigem Medium. Nach sieben Tagen wurde das Medium entfernt und die
Zellen im Kultivierungsraum geerntet. Die Hybridomzellen und der mit Antikörpern
angereicherte Überstand wurden durch Zentrifugation (1400 rpm, 10 min) separiert. Nach der
Zählung der Zellen (2.2.2.2) wurden die Reaktoren erneut mit 8 bzw. 25 x 106 Zellen beimpft.
Danach wurden die Zellen im 4-Tagesrhythmus weiter kultiviert.
2.2.1.7 Aufreinigung der monoklonalen Antikörper mi ttels Protein G
HiTrap TM Protein G HP, 1ml Amersham Biosciences GmbH
Peristaltische Pumpe P1 Pharmacia Corp., Schweden
Slide-A-Lyzer® Dialysis Cassette Perbio Science Ltd., USA
Steril-Filter (0,22 µm, 4 mm) Millipore GmbH
Dialysepuffer PBS
Bindungspuffer Natriumphosphat-Puffer, pH 7 0,02 M
Elutionspuffer Glycin, pH 2,5 0,1 M
Neutralisationspuffer Tris, pH 9 1 M
Ethanol 20% (v/v)
Die Antikörper aus den Zellkulturüberständen der Bioreaktoren wurden über eine Protein-G-
Säule aufgereinigt. Protein-G ist ein Protein aus der bakteriellen Zellwand von Streptococcus
sp., das eine hohe Affinität zur FC-Region von Immunglobuline besitzt. Der Überstand wurde
dazu mit Bindungspuffer verdünnt (1:3). Die Laufgeschwindigkeit, mit welcher der Überstand
über die Säule lief, wurde auf einen Milliliter pro Minute eingestellt. Nach Ende des Durchlaufs
wurden weitere 20 ml Bindungspuffer über die Säule gepumpt, um die restlichen Komponenten
des Mediums zu entfernen, die bei der photometrischen Bestimmung des Proteingehalts stören
könnten. Danach wurde Elutionspuffer über die Säule gepumpt und in jeweils 0,5 ml Fraktionen
aufgefangen. Durch die sofortige Zugabe von 23 µl Neutralisationspuffer wurde der pH-Wert der
Eluate wieder auf pH 7 eingestellt. Der Antikörper befand sich in den Fraktionen fünf bis acht
von insgesamt zehn gesammelten Fraktionen. Die Säule wurde durch 20 ml Bindungspuffer
regeneriert und abschließend in 20 % Ethanol bei 4°C aufbewahrt. Pro Antikörper wurde jeweils

MATERIAL UND METHODEN
45
eine Protein-G-Säule verwendet, um Kontaminationen der Antkörper untereinander zu
vermeiden.
Die Antikörper-enthaltenden Eluate wurden in Dialyse-Kassetten überführt und in einem
1 l Becherglas dialysiert. Der Wechsel des Dialysepuffers, von dem das 300fache Volumen der
Probe eingesetzt wurde, erfolgte jeweils zweimal nach zwei Stunden bei RT und ein drittes Mal
für einen letzten Dialyseschritt, der über Nacht bei 4°C stattfand. Der aufgereinigte Antikörper
wurde abschließend steril filtriert, und die Konzentration wurde photometrisch nach folgender
Formel bestimmt:
Protein 1,35 OD280nm = 1 mg IgG/ml
Die Aufbewahrung erfolgte bei 4°C.
2.2.1.8 Bestimmung der Antikörperklasse und der Iso typen
Mouse Monoclonal Antibody Isotyping Test Kit Serotec GmbH
Clonotyping TM System/HRP Southern Biotech. Associates, USA
Die Identifizierung der Klasse und des Isotyps der monoklonalen Antikörper erfolgte mit Hilfe
des Mouse Monoclonal Antibody Isotyping Test Kits. Dafür wurde Antikörper-angereichertes
Medium 1:100 mit PBS verdünnt. Von dieser Verdünnung wurden 0,15 ml in das
Entwicklerröhrchen pipettiert und für 30 s inkubiert. Anschließend wurde der Inhalt des
Röhrchens durchmischt, um die mit Antikörper beschichteten farbigen Latexkugeln zu
suspendieren. In diese Suspension wurde der Entwicklerstreifen eingetaucht und das Ergebnis
nach 5 min abgelesen. Mit dem ClonotypingTM System/HRP wurden die Isotypen der
aufgereinigten Antikörper bestätigt. Die HRP-gekoppelten Antikörper aus diesem Kit wurden als
sekundäre Antikörper (1:5000) für einen ELISA gegen rekombinantes Maus-PrP eingesetzt
(2.2.4.1). Als primäre Antikörper wurden Verdünnungsreihen der Anti-PrP-Antikörper verwendet.
Bei beiden Tests wurde der monoklonale Anti-PrP-Antikörper 6H4 (2.1.6) als Positivkontrolle
eingesetzt.
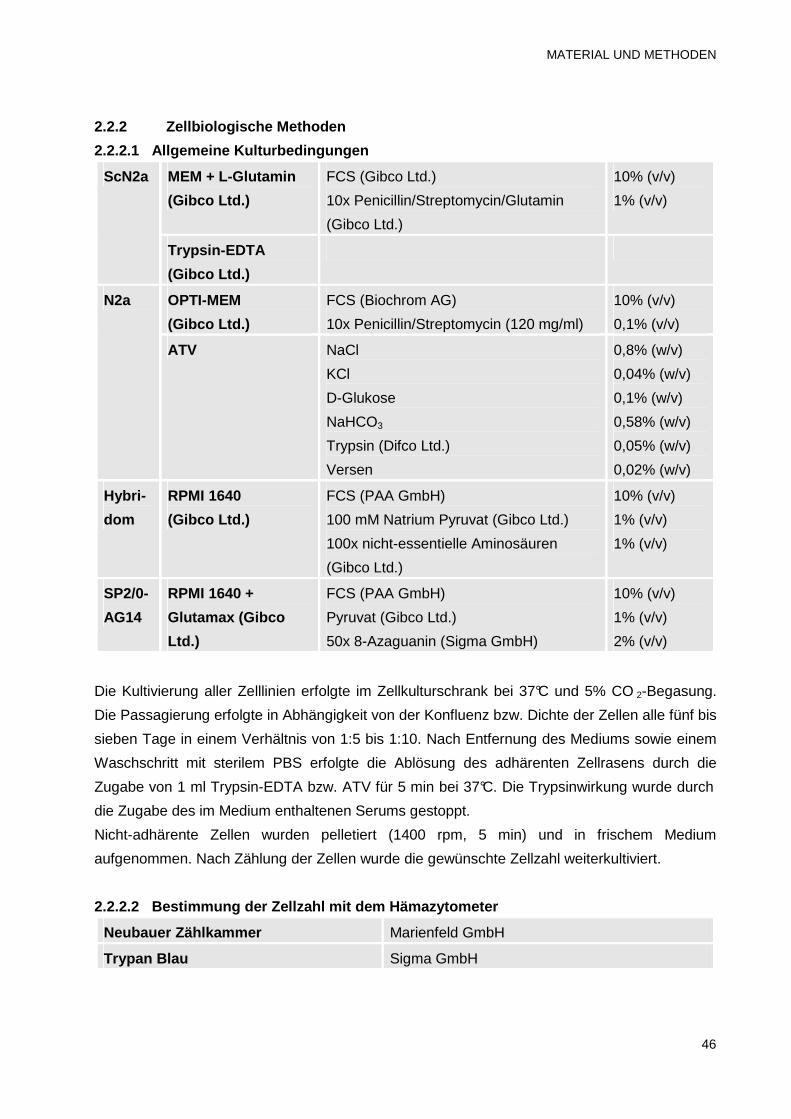
MATERIAL UND METHODEN
46
2.2.2 Zellbiologische Methoden
2.2.2.1 Allgemeine Kulturbedingungen
MEM + L-Glutamin
(Gibco Ltd.)
FCS (Gibco Ltd.)
10x Penicillin/Streptomycin/Glutamin
(Gibco Ltd.)
10% (v/v)
1% (v/v)
ScN2a
Trypsin-EDTA
(Gibco Ltd.)
OPTI-MEM
(Gibco Ltd.)
FCS (Biochrom AG)
10x Penicillin/Streptomycin (120 mg/ml)
10% (v/v)
0,1% (v/v)
N2a
ATV NaCl
KCl
D-Glukose
NaHCO3
Trypsin (Difco Ltd.)
Versen
0,8% (w/v)
0,04% (w/v)
0,1% (w/v)
0,58% (w/v)
0,05% (w/v)
0,02% (w/v)
Hybri-
dom
RPMI 1640
(Gibco Ltd.)
FCS (PAA GmbH)
100 mM Natrium Pyruvat (Gibco Ltd.)
100x nicht-essentielle Aminosäuren
(Gibco Ltd.)
10% (v/v)
1% (v/v)
1% (v/v)
SP2/0-
AG14
RPMI 1640 +
Glutamax (Gibco
Ltd.)
FCS (PAA GmbH)
Pyruvat (Gibco Ltd.)
50x 8-Azaguanin (Sigma GmbH)
10% (v/v)
1% (v/v)
2% (v/v)
Die Kultivierung aller Zelllinien erfolgte im Zellkulturschrank bei 37°C und 5% CO 2-Begasung.
Die Passagierung erfolgte in Abhängigkeit von der Konfluenz bzw. Dichte der Zellen alle fünf bis
sieben Tage in einem Verhältnis von 1:5 bis 1:10. Nach Entfernung des Mediums sowie einem
Waschschritt mit sterilem PBS erfolgte die Ablösung des adhärenten Zellrasens durch die
Zugabe von 1 ml Trypsin-EDTA bzw. ATV für 5 min bei 37°C. Die Trypsinwirkung wurde durch
die Zugabe des im Medium enthaltenen Serums gestoppt.
Nicht-adhärente Zellen wurden pelletiert (1400 rpm, 5 min) und in frischem Medium
aufgenommen. Nach Zählung der Zellen wurde die gewünschte Zellzahl weiterkultiviert.
2.2.2.2 Bestimmung der Zellzahl mit dem Hämazytomet er
Neubauer Zählkammer Marienfeld GmbH
Trypan Blau Sigma GmbH

MATERIAL UND METHODEN
47
Für die Zählung der Zellen wurde eine Neubauer Zählkammer eingesetzt. Um nur die lebenden
Zellen zu zählen, wurde die Zellsuspension mit dem sauren Farbstoff Trypanblau verdünnt
(1:2). Das Trypanblau dringt durch defekte Zellmembranen toter Zellen in das Cytosol ein und
färbt diese Zellen tiefblau. Lebende Zellen erscheinen unter dem Mikroskop dagegen leuchtend
grün. Die Zellen wurden in allen vier Großquadraten des Hämazytometers ausgezählt. Zur
Berechnung der Zellzahl pro ml Suspension wurde folgende Formel verwendet:
Zellzahl pro ml = Zellzahl/4 x Verdünnungsfaktor x 104
2.2.2.3 Einfrieren und Auftauen von Zellen
Einfriermedium DMSO (Sigma GmbH)
FCS (PAA GmbH)
10% (v/v)
90% (v/v)
Einfrierboxen Cryo 1°C Freezing Container, Nalgene TM, UK
Kryoröhrchen Greiner AG
Von allen Antikörper-produzierenden Hybridomzelllinien wurden Aliquots in flüssigem Stickstoff
eingefroren. Dazu wurde die gewünschte Zellzahl 10 min bei 1300 rpm und 4°C sedimentiert
und anschließend in kaltem Einfriermedium aufgenommen (2 x 106 Zellen/ml). Dieses
Einfriermedium enthält Dimethylsulfoxid (DMSO), das beim Gefriervorgang die Bildung von
Eiskristallen verhindert. In jedes Kryoröhrchen wurde 1 ml pipettiert und direkt in eine
vorgekühlte Einfrierbox (-20°C) überführt. Die Box wurde zunächst für zwei Stunden bei -20°C
aufbewahrt und anschließend für mindestens vier Stunden bei -80°C. Die Langzeitlagerung
erfolgte in der Gasphase über flüssigem Stickstoff (-196°C). Durch diesen Prozess findet die
Abnahme der Temperatur in einer optimalen Geschwindigkeit statt (1°C pro Stunde) und
gewährleistet somit das langsame Eindringen des DMSO in die Zellen.
Um die Zellen aufzutauen, wurden die Kryoröhrchen so lange im Wasserbad (37°C)
geschwenkt, bis nur noch ein kleiner Eiskristall sichtbar war. Die aufgetauten Zellen wurden in
20 ml Kultivierungsmedium überführt und zweimal mit Medium gewaschen (1400 rpm, 5 min).
Da DMSO zytotoxisch ist, müssen beide Methoden zügig durchgeführt werden. Diese Methoden
wurden für alle Zelllinien angewendet.
2.2.2.4 Behandlung von Zellen mit Anti-PrP-Antikörp ern
2.2.2.4.1 Inhibition der Prionreplikation in ScN2a- Zellen
Um den Effekt der aufgereinigten Anti-PrP-Antikörper auf die Prionreplikation in Zellkultur zu
bestimmen, wurden ScN2a-Zellen verwendet. Für den Standardassay wurden jeweils 105 Zellen
in 6-Loch-Platten eingesät, das Medium (1 ml) am nächsten Tag erneuert und mit 10 µg

MATERIAL UND METHODEN
48
Antikörper versetzt. Nach fünf Tagen wurde der Gehalt von PrPSc in den behandelten Zellen
mittels Western Blot bestimmt (2.2.3). Zur Bestimmung des IC50-Werts eines Antikörpers
erfolgte die Behandlung mit abnehmenden Antikörperkonzentrationen von 10, 5, 2,5 bis
0,0006 µg/ml. Zur Bestimmung eines möglichen Langzeiteffekts der Antikörper wurden
105 ScN2a-Zellen mit 10 µg Ak/ml behandelt und die Behandlung im 6-Tagesrhythmus
fortgeführt. Nach den angegebenen Behandlungsintervallen erfolgte dann eine Bestimmung von
PrPSc mittels Western Blot (2.2.3) oder der Infektiosität mittels Bioassay (2.2.5.5). Um zu
bestimmen, ob der Antikörpereffekt auf die Prionreplikation transient oder dauerhaft ist, wurden
die Zellen nach Beendigung der Langzeitbehandlung für weitere 36 Tage in Abwesenheit der
Antikörper kultiviert. Dazu wurde der PrPSc-Gehalt ebenfalls im 6-Tagesrhythmus mittels
Western Blot (2.2.3) überprüft.
Als Kontrollen für die Behandlungen dienten entweder Antikörper gegen PrP (6H4 und ICSM18)
oder Antikörper gegen nicht-PrP Epitope (NCAM, Anti-IgG1) (2.1.6).
Um zu bestimmen, ob der freie Anti-PrP-Antikörper oder ein PrP/Anti-PrP-Antikörper-Komplex
einen inhibitorischen Effekt auf die Prionreplikation hat, wurde der Standardassay modifiziert.
Die Behandlung mit freiem Antikörper erfolgte mit abnehmenden Konzentrationen (1,5, 0,75,
0,37, …, 0 µg/ml). Für die Komplexbildung wurden direkt vor der Behandlung die angegebenen
Antikörperkonzentrationen mit rekombinantem PrP (1 µg) 30 min bei RT inkubiert und dann zu
den Zellen gegeben. Als Negativkontrolle wurden weitere Zellen nur mit rekombinantem PrP
(1 µg) kultiviert. Nach Beendigung der Behandlung wurde der PrPSc-Gehalt der Zellen mittels
Western Blot (2.2.3) bestimmt und der PrP/Antikörper-Komplex im Medium mittels
Immunpräzipitation (2.2.3.5) nachgewiesen.
2.2.2.4.2 Analyse der Retention auf ScN2a-Zellen
Um zu bestimmen, wie lange PrP auf der Zelloberfläche von ScN2a-Zellen durch die Bindung
der Anti-PrP-Antikörper gehalten wird (Retention), wurde eine FACS-Analyse durchgeführt.
Dazu wurden am ersten Tag 2 x 105 ScN2a-Zellen in eine 6-Loch-Platte eingesät. Am nächsten
Tag erfolgte eine einstündige Inkubation bei 37°C m it den Anti-PrP-Antikörpern. Damit die
Fluoreszenzsignale nicht im gesättigten Bereich liegen, wurden dazu Antikörperkonzentrationen
eingesetzt, die den KD-Werten entsprachen (2.2.4.5). Nach Entfernung des Antikörpers erfolgte
die weitere Inkubation für unterschiedlich lange Zeitintervalle (0, 1, 2, 3, und 4 Stunden) bei
37°C. Danach wurden die Zellen mit 0,5 ml 50 mM EDT A/PBS abgelöst (5 min, 37°C), in 1,5 ml
PBS aufgenommen und im Durchflusszytometer analysiert (2.2.4.4).
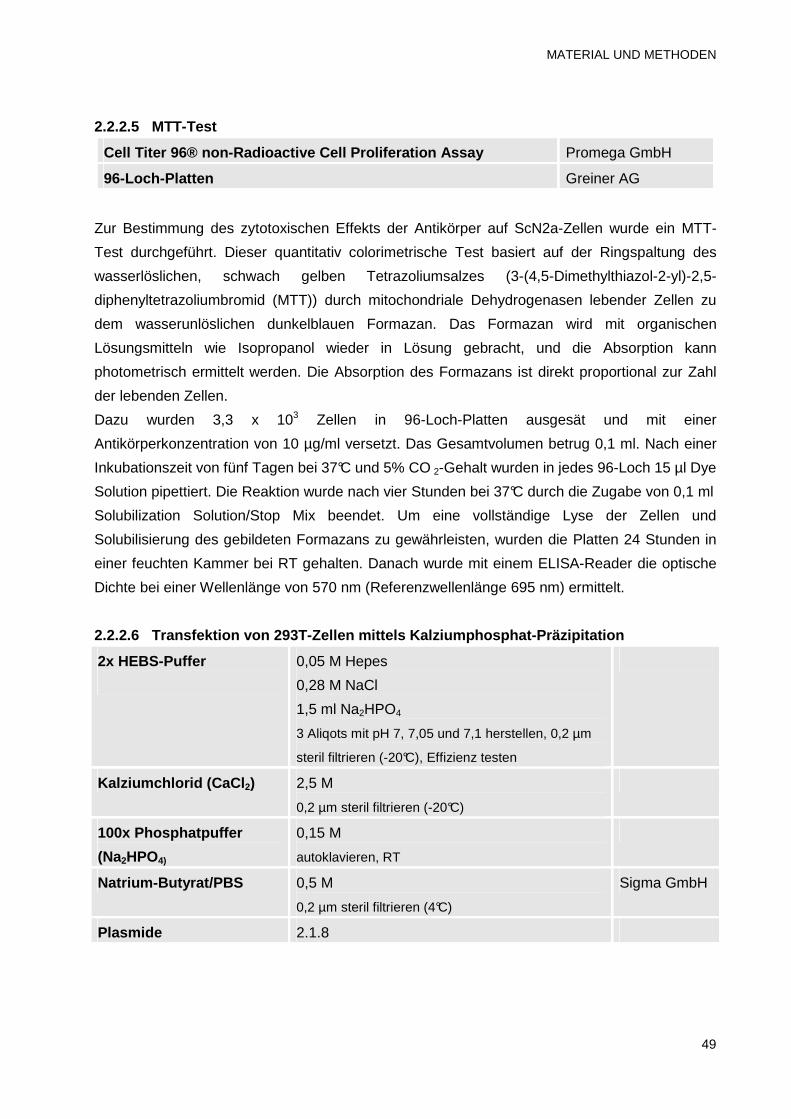
MATERIAL UND METHODEN
49
2.2.2.5 MTT-Test
Cell Titer 96® non-Radioactive Cell Proliferation A ssay Promega GmbH
96-Loch-Platten Greiner AG
Zur Bestimmung des zytotoxischen Effekts der Antikörper auf ScN2a-Zellen wurde ein MTT-
Test durchgeführt. Dieser quantitativ colorimetrische Test basiert auf der Ringspaltung des
wasserlöslichen, schwach gelben Tetrazoliumsalzes (3-(4,5-Dimethylthiazol-2-yl)-2,5-
diphenyltetrazoliumbromid (MTT)) durch mitochondriale Dehydrogenasen lebender Zellen zu
dem wasserunlöslichen dunkelblauen Formazan. Das Formazan wird mit organischen
Lösungsmitteln wie Isopropanol wieder in Lösung gebracht, und die Absorption kann
photometrisch ermittelt werden. Die Absorption des Formazans ist direkt proportional zur Zahl
der lebenden Zellen.
Dazu wurden 3,3 x 103 Zellen in 96-Loch-Platten ausgesät und mit einer
Antikörperkonzentration von 10 µg/ml versetzt. Das Gesamtvolumen betrug 0,1 ml. Nach einer
Inkubationszeit von fünf Tagen bei 37°C und 5% CO 2-Gehalt wurden in jedes 96-Loch 15 µl Dye
Solution pipettiert. Die Reaktion wurde nach vier Stunden bei 37°C durch die Zugabe von 0,1 ml
Solubilization Solution/Stop Mix beendet. Um eine vollständige Lyse der Zellen und
Solubilisierung des gebildeten Formazans zu gewährleisten, wurden die Platten 24 Stunden in
einer feuchten Kammer bei RT gehalten. Danach wurde mit einem ELISA-Reader die optische
Dichte bei einer Wellenlänge von 570 nm (Referenzwellenlänge 695 nm) ermittelt.
2.2.2.6 Transfektion von 293T-Zellen mittels Kalziu mphosphat-Präzipitation
2x HEBS-Puffer
0,05 M Hepes
0,28 M NaCl
1,5 ml Na2HPO4
3 Aliqots mit pH 7, 7,05 und 7,1 herstellen, 0,2 µm
steril filtrieren (-20°C), Effizienz testen
Kalziumchlorid (CaCl 2) 2,5 M
0,2 µm steril filtrieren (-20°C)
100x Phosphatpuffer
(Na2HPO4)
0,15 M
autoklavieren, RT
Natrium-Butyrat/PBS 0,5 M
0,2 µm steril filtrieren (4°C)
Sigma GmbH
Plasmide 2.1.8

MATERIAL UND METHODEN
50
Für die transiente Transfektion von 293T-Zellen wurde die klassische Methode der
Kalziumphosphat-Präzipitation verwendet (Wigler et al., 1978).
Einen Tag vor der Transfektion wurden 5 x 106 Zellen in 10 cm Kulturschalen ausgesät und
über Nacht bei 37°C und 5% CO 2-Gehalt inkubiert. Eine halbe bis zwei Stunden vor Beginn der
Transfektion erfolgte ein Mediumwechsel mit equilibriertem Medium. Danach wurde das
Kalziumphosphat-DNA-Präzipitat hergestellt. Dazu wurden 10 µg des jeweiligen Plasmids mit
Fresenius-Wasser auf ein Endvolumen von 0,45 ml eingestellt und mit 50 µl CaCl2 versetzt.
Dieses Gemisch wurde unter kontinuierlichem Einblasen von steriler Luft mit 0,5 ml 2x HEBS-
Puffer vermischt und für 30 min bei RT inkubiert. In dieser Zeit bildete sich ein feines Präzipitat
aus DNA und Kalziumphosphat, das tropfenweise zur Zellkultur zugegeben wurde. Während
der anschließenden fünfstündigen Inkubationszeit bildeten sich Kristalle, welche die Aufnahme
der DNA in die Zelle über einen endozytotischen Prozess erleichtern und somit die
Transfektionsrate erhöhen. Danach erfolgte für acht bis zwölf Stunden eine Induktion durch
0,2 ml Natriumbutyrat, um den CMV-Promoter zu stimulieren und so die Expressionsrate des
Proteins zu erhöhen. Abschließend wurden die Zellen gezählt, jeweils 1 x 105 Zellen pro FACS-
Röhrchen ausgesät und die Bindung der Anti-PrP-Antikörper an die transfizierten Zellen im
Durchflusszytometer analysiert (2.2.4.4).
2.2.3 Proteinbiochemische Methoden
2.2.3.1 Herstellung von Zelllysaten
Lyse-Puffer
0,05 M Tris, pH 7,5
0,1 M NaCl
0,5% (v/v) Igepal (NP-40)
0,5% (w/v) Natrium Deoxycholat Monohydrat
Zellschaber, 13mm Hartenstein Laborbedarf GmbH
Zur Herstellung von Zelllysaten wurde das Medium entfernt und die Zellen mit eiskaltem PBS
gewaschen. Der Zellrasen wurde mit 60 bis 70 µl Lyse-Puffer bedeckt und für mindestens 5 min
auf Eis inkubiert. Danach wurden die Zellen mit einem Zellschaber abgelöst, in ein
Reaktionsgefäß überführt und 3 min bei 5000 rpm zentrifugiert. Der Überstand wurde bis zur
weiteren Bearbeitung bei -20°C gelagert.

MATERIAL UND METHODEN
51
2.2.3.2 Herstellung von Gehirnhomogenaten
Lyse-Puffer
0,5% (v/v) Igepal (NP-40)
0,5% (w/v) Natrium Deoxycholat Monohydrat
PBS
Kanülen (G18-G21) Braun AG
Die gefrorenen Maus-Gehirne wurden gewogen und mit neun Volumen des Lyse-Puffers
versetzt (10% Gehirnhomogenat). Indem das Zellmaterial mehrmals durch unterschiedlich
große Kanülen (G18-G21) gezogen wurde, wurde die Zellmasse homogenisiert. Nach
Zentrifugation (1000 rpm, 5 min) erfolgte die Lagerung der Überstände bei -20°C. Zuvor wurde
die Proteinkonzentration dieser Homogenate bestimmt, die zwischen 8 bis 10 mg/ml lag
(2.2.3.3).
2.2.3.3 Proteinbestimmung nach Bradford
Bradfordreagenz Bio-Rad GmbH
bovines Serumalbumin (BSA)
Die quantitative Bestimmung der gelösten Proteine in den Zelllysaten wurde photometrisch
mittels Bradford-Test vorgenommen. Hierbei bildet der Farbstoff Coomassie-Brilliant-Blau einen
Komplex mit den gelösten Proteinen, durch den es zu einer Verschiebung des
Absorptionsmaximums von 470 nm zu 595 nm kommt. Die Zunahme der Absorption bei 595 nm
kann somit als Maß für die Proteinkonzentration einer Lösung genommen werden (Bradford,
1976). Die jeweiligen Zelllysate wurden in drei unterschiedlichen Verdünnungen gemessen. Mit
Hilfe einer Eichgeraden, die mit verschiedenen BSA-Konzentrationen erstellt wurde, konnte die
Proteinkonzentration genau bestimmt werden.
2.2.3.4 Proteinase K-Behandlung von Homogenaten
Proteinase K, rekombinant, PCR Grade Roche Diagnostics GmbH
ββββ-Mercapto-Ethanol 9,5% (v/v)
Phenylmethylsulfonylfluorid (PMSF) 0,075 M (AppliChem GmbH)
5x SDS Probenpuffer SDS
Glycerol
Tris-HCl, pH 6,8
Bromphenolblau
2% (w/v)
25% (v/v)
0,06 M
0,05% (w/v)
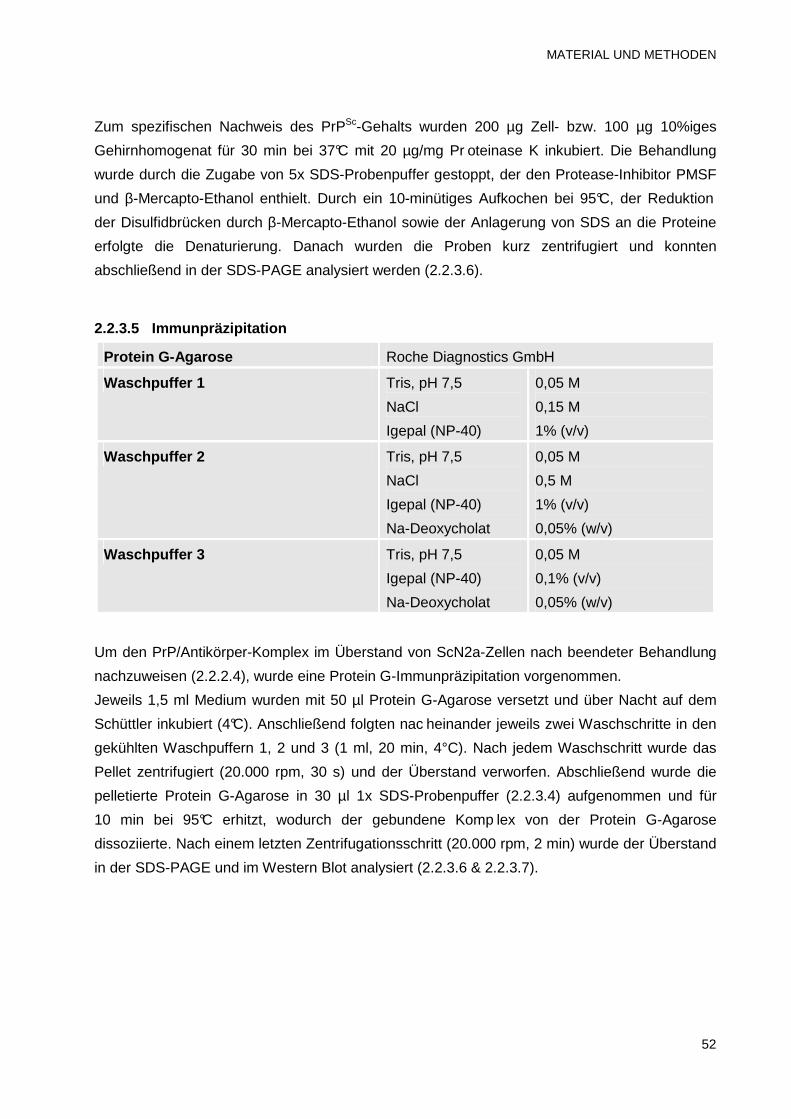
MATERIAL UND METHODEN
52
Zum spezifischen Nachweis des PrPSc-Gehalts wurden 200 µg Zell- bzw. 100 µg 10%iges
Gehirnhomogenat für 30 min bei 37°C mit 20 µg/mg Pr oteinase K inkubiert. Die Behandlung
wurde durch die Zugabe von 5x SDS-Probenpuffer gestoppt, der den Protease-Inhibitor PMSF
und β-Mercapto-Ethanol enthielt. Durch ein 10-minütiges Aufkochen bei 95°C, der Reduktion
der Disulfidbrücken durch β-Mercapto-Ethanol sowie der Anlagerung von SDS an die Proteine
erfolgte die Denaturierung. Danach wurden die Proben kurz zentrifugiert und konnten
abschließend in der SDS-PAGE analysiert werden (2.2.3.6).
2.2.3.5 Immunpräzipitation
Protein G-Agarose Roche Diagnostics GmbH
Waschpuffer 1 Tris, pH 7,5
NaCl
Igepal (NP-40)
0,05 M
0,15 M
1% (v/v)
Waschpuffer 2 Tris, pH 7,5
NaCl
Igepal (NP-40)
Na-Deoxycholat
0,05 M
0,5 M
1% (v/v)
0,05% (w/v)
Waschpuffer 3 Tris, pH 7,5
Igepal (NP-40)
Na-Deoxycholat
0,05 M
0,1% (v/v)
0,05% (w/v)
Um den PrP/Antikörper-Komplex im Überstand von ScN2a-Zellen nach beendeter Behandlung
nachzuweisen (2.2.2.4), wurde eine Protein G-Immunpräzipitation vorgenommen.
Jeweils 1,5 ml Medium wurden mit 50 µl Protein G-Agarose versetzt und über Nacht auf dem
Schüttler inkubiert (4°C). Anschließend folgten nac heinander jeweils zwei Waschschritte in den
gekühlten Waschpuffern 1, 2 und 3 (1 ml, 20 min, 4°C). Nach jedem Waschschritt wurde das
Pellet zentrifugiert (20.000 rpm, 30 s) und der Überstand verworfen. Abschließend wurde die
pelletierte Protein G-Agarose in 30 µl 1x SDS-Probenpuffer (2.2.3.4) aufgenommen und für
10 min bei 95°C erhitzt, wodurch der gebundene Komp lex von der Protein G-Agarose
dissoziierte. Nach einem letzten Zentrifugationsschritt (20.000 rpm, 2 min) wurde der Überstand
in der SDS-PAGE und im Western Blot analysiert (2.2.3.6 & 2.2.3.7).

MATERIAL UND METHODEN
53
2.2.3.6 SDS-Polyacrylamidgelelektrophorese (SDS-PAG E)
Tris-Glycin Gel (12%) Biozym GmbH
Western Blot-Gelkammeraparatur Biozym GmbH
1x SDS-Laufpuffer (1l) SDS
Tris
Glycin
1% (w/v)
0,25 M
2 M
MagicMark TM Standard Invitrogen Ltd.
Die SDS-PAGE erlaubt die Auftrennung von Proteinen abhängig von ihrer relativen
Molekülmasse unter denaturierenden Bedingungen (Laemmli, 1970). Diese Methode basiert auf
der durch hydrophobe Wechselwirkungen bedingten Bindung des negativ geladenen Detergens
Natriumdodecylsulfat (SDS) an denaturierte Proteine.
Auf das Gel wurden 5 µl des Größenstandards sowie 150 bis 200 µg der Proteinproben
aufgetragen. Die Auftrennung erfolgte bei 110 V im Sammelgel (ca. 10 min) und 150 V im
Trenngel (ca. 1 Stunde). Die Gelelektrophorese wurde beendet, sobald die Lauffront des
Bromphenolblaus das untere Ende des Gels erreicht hatte.
MagicMarkTM Größenstandard (kDa)
220, 120, 100, 80, 60, 50, 40, 30, 20
2.2.3.7 Western Blot
Polyvinyldifluorid (PVDF)-Membran Roth GmbH & Co. KG
Whatman Schleicher & Schuell ®-Filter Sigma GmbH
Methanol
1 x Transferpuffer (1l) SDS
Tris
Glycin
Methanol
0,75% (w/v)
0,25 M
2 M
20% (v/v)
Nach Auftrennung durch die SDS-PAGE wurden die Proteine mittels Tank-Blot-Technik
elektrophoretisch auf eine PVDF-Membran transferiert (Towbin et al., 1979). Zuvor wurde die
PVDF-Membran in Methanol aktiviert (2 min) und in Transferpuffer equilibriert (10 min). Danach
wurde das Gel luftblasenfrei auf die Membran gelegt. Auf beiden Seiten wurden jeweils ein
Whatman-Filter und eine Schwammstoffmatte platziert, die vorab in Transferpuffer getränkt
wurden. Die PVDF-Membran zeigte bei dieser Anordnung in Richtung Anode. Der Transfer
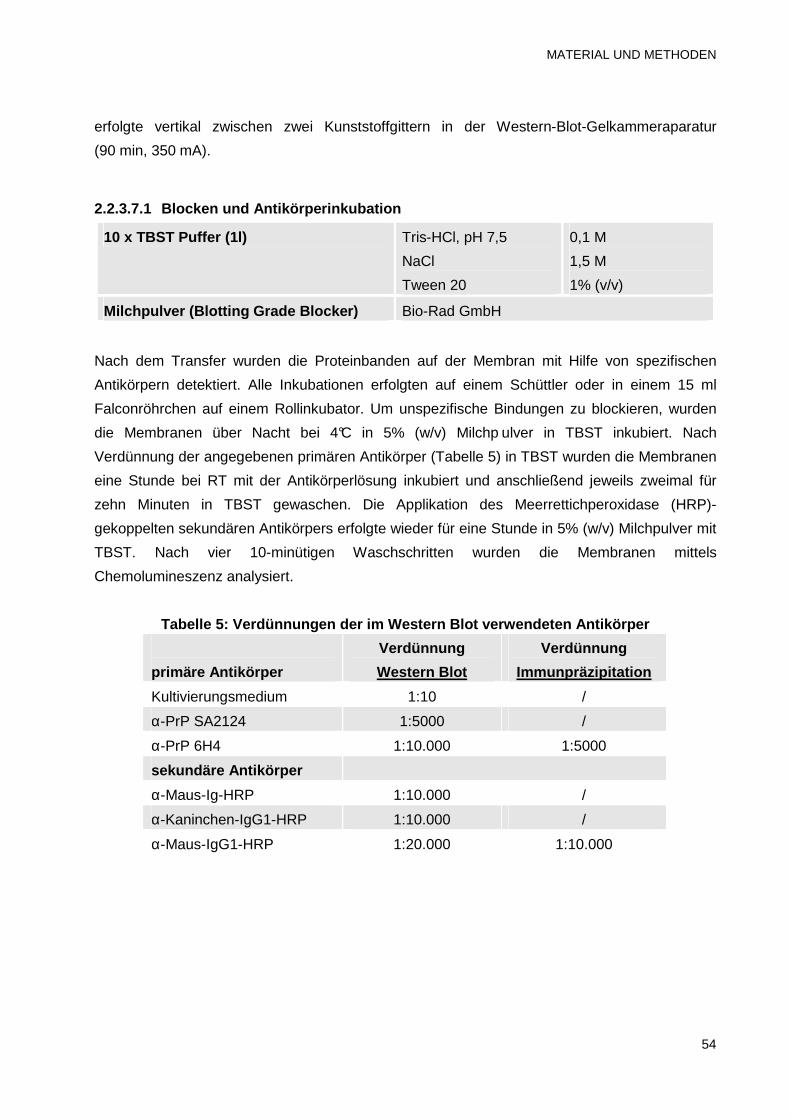
MATERIAL UND METHODEN
54
erfolgte vertikal zwischen zwei Kunststoffgittern in der Western-Blot-Gelkammeraparatur
(90 min, 350 mA).
2.2.3.7.1 Blocken und Antikörperinkubation
10 x TBST Puffer (1l) Tris-HCl, pH 7,5
NaCl
Tween 20
0,1 M
1,5 M
1% (v/v)
Milchpulver (Blotting Grade Blocker) Bio-Rad GmbH
Nach dem Transfer wurden die Proteinbanden auf der Membran mit Hilfe von spezifischen
Antikörpern detektiert. Alle Inkubationen erfolgten auf einem Schüttler oder in einem 15 ml
Falconröhrchen auf einem Rollinkubator. Um unspezifische Bindungen zu blockieren, wurden
die Membranen über Nacht bei 4°C in 5% (w/v) Milchp ulver in TBST inkubiert. Nach
Verdünnung der angegebenen primären Antikörper (Tabelle 5) in TBST wurden die Membranen
eine Stunde bei RT mit der Antikörperlösung inkubiert und anschließend jeweils zweimal für
zehn Minuten in TBST gewaschen. Die Applikation des Meerrettichperoxidase (HRP)-
gekoppelten sekundären Antikörpers erfolgte wieder für eine Stunde in 5% (w/v) Milchpulver mit
TBST. Nach vier 10-minütigen Waschschritten wurden die Membranen mittels
Chemolumineszenz analysiert.
Tabelle 5: Verdünnungen der im Western Blot verwend eten Antikörper
primäre Antikörper
Verdünnung
Western Blot
Verdünnung
Immunpräzipitation
Kultivierungsmedium 1:10 /
α-PrP SA2124 1:5000 /
α-PrP 6H4 1:10.000 1:5000
sekundäre Antikörper
α-Maus-Ig-HRP 1:10.000 /
α-Kaninchen-IgG1-HRP 1:10.000 /
α-Maus-IgG1-HRP 1:20.000 1:10.000

MATERIAL UND METHODEN
55
2.2.3.7.2 Chemolumineszenz-Detektion
ECL-Detektion-Kit (SuperSignal ® West Pico
Chemoluminescent Substrate)
Pierce Ltd., Rockford, USA
Röntgenfilme
Amersham Biosciences GmbH
Autoradiographiekassette/Hyperkassette TM Amersham Biosciences GmbH
Die Visualisierung der markierten Proteine erfolgte mittels ECL-Kit. Hierbei katalysiert die an
den sekundären Antikörper gebundene Peroxidase eine Luminol-Wasserstoffperoxid-Reaktion.
Luminol wird oxidiert und emittiert dadurch Lichtquanten, die auf einem Röntgenfilm zur
Schwärzung führen.
Die gewaschenen PVDF-Membranen wurden in einer 1:1-Mischung der Detektionsreagenzien
für eine Minute inkubiert. Nachdem die überschüssige ECL-Lösung mit einem Whatman-Filter
entfernt wurde, erfolgte direkt die Exponierung der Röntgenfilme in einer
Autoradiographiekassette (30 s bis 15 min). Die Entwicklung und Fixierung der Röntgenfilme
wurde in einer Dunkelkammer manuell durchgeführt. Abschließend wurden die Filme unter
fließendem Wasser gewaschen und luftgetrocknet.
Zur Quantifizierung von Signalintensitäten wurden die Membranen in einem Image Reader
entwickelt und anschließend mit der AIDA Software, Version 3.20.116 (raytest
Isotopenmessgeräte GmbH) analysiert. Für die Auswertung des IC50-Werts wurde eine
sigmoidale Dosis-Wirkungs-Kurve erstellt.
2.2.4 Immunologische Methoden
2.2.4.1 Enzyme Linked Immunosorbent Assay (ELISA)
Waschpuffer (PBST) Tween 20
PBS
0,1% (v/v)
Blockpuffer Milchpulver
PBST
5% (w/v)
Assay-Lösung BSA
PBST
1% (w/v)
96-Loch-Maxisorb-Flachbodenplatten Nunc GmbH & Co. KG
TMB-Substrat BD Biosciences GmbH
H2SO4 1 M
rekombinantes Maus-PrP (23-231) Thorsten Lührs (Zahn et al., 1997)

MATERIAL UND METHODEN
56
Beim ELISA-Verfahren werden Antigen-Antikörper-Wechselwirkungen durch eine enzymatische
Farbreaktion nachgewiesen. In dieser Arbeit wurde ein indirekter ELISA eingesetzt.
Zur Immobilisierung des Antigens wurden 96-Loch-Platten mit rekombinantem PrP
(50 ng/0,1 ml PBS/Loch) beschichtet, über Nacht bei 4°C inkubiert und anschließend dreimal
mit 0,3 ml PBST gewaschen. Die unbesetzten Bindestellen wurden durch die Zugabe von
0,1 ml Blockpuffer für zwei Stunden bei 37°C abgesä ttigt, und danach wurden die Platten erneut
dreimal mit PBST gewaschen. Die Lagerung der Platten erfolgte für maximal einen Monat bei -
20°C.
Vor Beginn des ELISAs wurden die Platten 30 min bei RT gelagert. Alle anschließenden
Schritte wurden ebenfalls bei RT durchgeführt. Die Primärantikörper wurden verdünnt
(2.2.4.2-3), pro Loch wurden jeweils 0,1 ml Antikörperlösung pipettiert und für zwei Stunden
inkubiert. Nach jeweils sechs Waschschritten mit 0,3 ml PBST wurde der zweite Peroxidase-
gekoppelte Antikörper (1:10.000) für zwei Stunden aufgetragen. Sowohl Primär- als auch
Sekundärantikörper wurden in Assay-Lösung verdünnt. Nach sechs weiteren Waschschritten
erfolgte die Visualisierung des Antigen-Antikörper-Komplexes durch TMB als Peroxidase-
Substrat. Diese Farbreaktion wurde nach zehn Minuten durch die Zugabe von 50 µl H2SO4
gestoppt und abschließend bei einer Wellenlänge von 450 nm (Referenzwellenlänge 560 nm)
photometrisch gemessen. Die Messungen wurden immer in Duplikaten durchgeführt.
2.2.4.2 Bestimmung der Avidität von Antikörpern
Ammoniumthiozyanat (NH 4SCN) Sigma GmbH
Die Bestimmung der Avidität der Anti-PrP-Antikörper erfolgte mittels ELISA (2.2.4.1). Zur
Standardisierung der optischen Dichte (OD) wurden zuvor Verdünnungen der primären
Antikörper bestimmt, bei denen die OD zwischen 0,9 und 1,3 lag. Nach zweistündiger
Inkubation der Platten mit der Primärantikörperlösung erfolgten zwei Waschschritte.
Anschließend wurden die einzelnen Löcher 15 min bei RT mit verschiedenen NH4SCN-
Molaritäten (0 / 0,5 / 1 / 1,5 / 2 / 2,5 / 3 / 3,5 M) inkubiert. Dieses chaotrophe Salz hebt die
Wasserstoffbrückenbindungen zwischen Antigen und Antikörper auf. Für die Lösung hoch
avider Bindungen ist eine höher-molare NH4SCN-Lösung notwendig als für Bindungen mit
niedriger Avidität (Pullen et al., 1986). Die Avidität wird hier als Aviditätsindex (AI) angegeben,
welcher der Molarität von NH4SCN entspricht, bei der 50% des Antikörpers eluiert wurden.
Nach erneutem sechsmaligem Waschen wurde das ELISA-Standardprotokoll durchgeführt
(2.2.4.1).

MATERIAL UND METHODEN
57
2.2.4.3 Bestimmung der Dissoziationskonstanten (K D) mittels ELISA
Die KD-Konstante wurde auf rekombinantem Maus-PrP mittels ELISA bestimmt (2.2.4.1). Die
Anti-PrP-Antikörper wurden in verschiedenen Molaritäten (0,0016 bis 0,06 pmol) eingesetzt.
Danach erfolgte die Bestimmung der KD-Konstante mittels Sättigungskurve und Scatchard-
Diagramm (PRISM®, Version 4.0, GraphPad Software, Inc.). Es wurden nur Sättigungskurven
zur Auswertung verwendet, deren Korrelationskoeffizient R2 größer als 0,98 war.
2.2.4.4 Durchflusszytometrische Analyse (FACS)
FACS-Puffer
2% (v/v) FCS (Biochrom AG)
0,01 M EDTA, pH 8
0,1% (w/v) Natriumazid
PBS, 1l
EDTA/PBS 0,02 M
FACS-Röhrchen
Mit Hilfe der Durchflusszytometrie (Fluorescence-Activated-Cell-Sorting; FACS) können
Fluoreszenz-markierte Antigene auf der Oberfläche von Zellen nachgewiesen werden. Hierzu
wurde das Durchflusszytometer FACS-Calibur (Becton Dickinson GmbH) eingesetzt und die
erhaltenen Daten mit der Software Cell Quest (Becton Dickinson GmbH) ausgewertet.
Diese Methode wurde für das Screening nach Antikörper-sezernierenden Hybridomklonen
(2.2.1.4), die Bindung der Anti-PrP-Antikörper an transfizierte 293T-Zellen (2.2.2.6), sowie
Bindungs- und Retentionsanalysen der Anti-PrP-Antikörper auf ScN2a-Zellen (2.2.4.5,
2.2.2.4.2) eingesetzt.
Je nach Anwendung wurden entweder alle behandelten Zellen aus einem 6-Loch in ein FACS-
Röhrchen überführt oder die Zellen wurden nach einem PBS-Waschschritt durch Inkubation mit
EDTA/PBS (5 min, 37°C) abgelöst und gezählt. Danach wurden jeweils 1 x 105 Zellen in ein
FACS-Röhrchen transferiert. Die Antikörper wurden in FACS-Puffer verdünnt (Tabelle 6) und
die Inkubation von sowohl Primär- als auch Sekundärantikörper fand für eine Stunde auf Eis
statt. Die Inkubation mit dem zweiten Antikörper erfolgte in Dunkelheit. Zellen aus der
Retentions- und Bindungsanalyse wurden zuvor mit dem Primärantikörper behandelt, so dass
diese Zellen nur noch mit dem Sekundärantikörper inkubiert wurden. Bei allen Anwendungen
erfolgten vor und nach der Inkubation mit dem Sekundärantikörper jeweils zwei Waschschritte
mit 3 ml FACS-Puffer (1250 rpm, 5 min). Abschließend wurden die Proben im
Durchflusszytometer analysiert. Für die Einstellungen am FACS-Gerät wurden ungefärbte
Zellen verwendet, und als Negativkontrolle dienten Zellen, die nur mit dem sekundären
Antikörper inkubiert wurden.

MATERIAL UND METHODEN
58
Tabelle 6: Verdünnungen der im Durchflusszytomter v erwendeten Antikörper.
Anwendung primärer Antikörper Verdünnung
Screening Kultivierungsmedium 1:3
Transfektion Anti-PrP-Antikörper 1 µg/Röhrchen
Bindungsanalyse Anti-PrP-Antikörper 2.2.4.5
Retentionsanalyse Anti-PrP-Antikörper 2.2.2.4.2
Für alle Experimente wurde ein Ziege-α-Maus-IgG (H+L) mit FITC-
Konjugat als sekundärer Antikörper verwendet, der 1:100 in FACS-
Puffer verdünnt wurde.
2.2.4.5 Bestimmung der Dissoziationskonstanten (K D) mittels FACS
Zur Bestimmung der KD-Konstante wurden 2 x 105 ScN2a-Zellen in 6-Loch-Platten eingesät. Am
nächsten Tag erfolgte eine einstündige Inkubation mit verschiedenen Konzentrationen von Anti-
PrP-Antikörpern bei 37°C (5 / 2,5 / 1,25, …, 0,0006 µg/ml). Anschließend wurden die Zellen mit
0,5 ml 20 mM EDTA/PBS (5 min, 37°C) abgelöst und de m Standardprotokoll entsprechend im
FACS analysiert (2.2.4.4). Zur Ermittlung des KD-Werts wurden eine Sättigungskurve und ein
Scatchard-Diagramm erstellt (PRISM®, Version 4.0, GraphPad Software, Inc.). Nur Analysen,
bei denen der Korrelationskoeffizient R2 größer als 0,98 war, wurden verwendet.
2.2.4.6 Immunfluoreszenzfärbung von Zellen
Paraformaldehyd (PFA) 4% (w/v) in 4% Sucrose (v/v)
NH4Cl 0,05 M
Triton-X/PBS 0,1% (v/v)
Guanidiniumthiozyanat
(GnSCN)
3 M
Kaninchenserum 10% (v/v)
30 min bei 56°C inaktiviert
Eindeckmedium Cyto Fluor Mounting Solution (Dako GmbH)
PBS-
MEM + L-Glutamin
(Gibco Ltd.)
FCS (Gibco Ltd.) 10% (v/v)
10x Penicillin/Streptomycin/Glutamin (Gibco Ltd.) 1% (v/v)
Objektträger Lab-Tek™ Chamber Slide™ (Nunc GmbH & Co. KG)
Deckgläschen 10 mm, rund

MATERIAL UND METHODEN
59
Mit Hilfe von Immunfluoreszenzanalysen wurde ermittelt, ob die Anti-PrP-Antikörper an PrPSc
von ScN2a-Zellen binden.
Dazu wurden 4 x 104 ScN2a-Zellen auf speziellen Objektträgern in Kammern ausgesät und
zwei Tage in MEM-Medium bei 37°C und 5% CO 2 kultiviert. Am dritten Tag wurden die Zellen
mit 4% PFA fixiert und anschließend mit NH4Cl und Triton-X permeabilisiert. Die Denaturierung
von PrPC erfolgte durch die Behandlung mit Guanidiniumthiozyanat. Durch die Inkubation mit
Kaninchenserum wurden die unspezifischen Bindestellen abgesättigt. Danach wurden die
Zellen mit einer Lösung, bestehend aus Anti-PrP-Antikörper (1,5 µg/ml) und ALEXA Fluor® 549
gekoppelter Cholera-Toxin Untereinheit B (1:800), versetzt. Als Sekundärantikörper wurde ein
ALEXA Fluor® 488-gekoppelter IgG-Antikörper (1:1.000) verwendet. Die Antikörper wurden in
jeweils 1%igem Kaninchenserum verdünnt. Alle Schritte wurden bei RT durchgeführt. Der
Zellrasen wurde abschließend mit Eindeckmedium überschichtet, mit einem Deckgläschen
bedeckt und für zwei Stunden bei 4°C getrocknet. Al s Kontrolle wurden ScN2a-Zellen
verwendet, die nach der Behandlung mit Pentosanpolysulfat (Caughey und Raymond, 1993)
kein PrPSc mehr trugen (cuScN2a-Zellen). Die Präparate wurden mittels konfokaler
Laserscanning-Mikroskopie (Axiovert 200M, Zeiss AG) analysiert.
Das genaue Protokoll ist nachfolgend aufgeführt:
1) Fixierung PFA, 5 min Lösung entfernen PFA, 15 min 3 x mit PBS waschen 2) Permeabilisierung NH4Cl, 15 min 3 x mit PBS waschen Triton X, 5 min 2 x mit PBS waschen 3) Denaturierung GnSCN, 5 min 2 x mit PBS waschen 4) Blockierung Kaninchen-Serum, 30 min 5) Färbung 1. Antikörper, 1 h 3 x mit PBS waschen 2. Antikörper, 1 h, Dunkelheit 3 x mit PBS waschen 2 x mit Aqua bidest. waschen
2.2.5 Tierexperimentelle Methoden
2.2.5.1 Betäubung der Mäuse mit Ether
Diethylether Roth GmbH & Co. KG

MATERIAL UND METHODEN
60
Für Immunisierungen, Inokulationen und Blutentnahmen wurden die Mäuse mit Ether
narkotisiert. Sobald die Mäuse eine flache Atmung aufwiesen und keinen „Pinch-Reflex“ mehr
zeigten, erfolgte der Eingriff.
2.2.5.2 Blutentnahme und Serumgewinnung
Mikro-Hämatokritkapillaren, heparinisiert Assistent ®
Serum separator tubes (Mikrotainer) Becton Dickinson GmbH
Bei narkotisierten Tieren erfolgte die Blutentnahme durch Punktierung des retroorbitalen
Venenplexus. Dazu wurde eine Kapillare über den medialen Augenwinkel am Augapfel entlang
zum Augenhintergrund eingeführt. Mit einer drehenden Bewegung wurde der retrobulbuläre
Plexus eröffnet (Herbert und Kristensen, 1986) und das entnommene Blut direkt in spezielle
Sammelröhrchen überführt. Durch Zentrifugation der Sammelröhrchen (5 min, 6000 rpm)
erfolgte eine Separation in Blutplasma und -serum. Die Lagerung des gewonnenen Blutserums
fand bei -20°C statt.
2.2.5.3 Herzpunktion und Organentnahme
Insulinspritze Becton Dickinson GmbH
Tissue-Tek® Sakura Finetek, Niederlande
4% Paraformaldehyd (PFA), pH 7,2
Die terminale Herzblutentnahme erfolgte an euthanisierten Mäusen. Nach Eröffnung des
Thorax wurde eine Insulinspritze (G26) in den rechten Herzventrikel eingeführt und das Blut
entnommen. Abschließend wurden Milz und Gehirn entfernt und entweder in 4% PFA oder
Tissue-Tek eingebettet. Die Aufbewahrung der PFA-Proben erfolgte bei RT, während die
Tissue-Tek-Proben bei -20°C gelagert wurden.
2.2.5.4 Bestimmung der Pharmakokinetik der Anti-PrP -Antikörper
Für die Bestimmung der Pharmakokinetik der Anti-PrP-Antikörper wurden fünf Gruppen,
bestehend aus jeweils sechs wildtypischen Mäusen, mit 0,1 ml 1% RML-Inokulum
intraperitoneal (i.p.) inokuliert. Nach 63 Tagen erfolgte die erste i.p. Injektion von 1 mg
Antikörper. Eine und vier Stunden später wurde den Mäusen Blut entnommen. Im
3-Tagesrhythmus wurden zwei weitere Injektionen der Anti-PrP-Antikörper vorgenommen. Nach
drei weiteren Tagen wurde die Untersuchung beendet und die Tiere mittels zervikaler
Dislokation getötet. Vor jeder Applikation sowie bei Beendigung des Experiments wurde den
MATERIAL UND METHODEN
61
Tieren erneut Blut abgenommen (2.2.5.2-3). Nach Bestimmung der Antikörpertiter im Serum der
Mäuse mittels ELISA (2.2.4.1) wurde die Pharmakokinetik der Anti-PrP-Antikörper berechnet.
2.2.5.5 Bioassay
Kanülen Braun AG
EDTA/PBS 0,02 M, pH 7,3
PBS-
Zur Bestimmung der Infektiosität von unbehandelten und Antikörper-behandelten ScN2a-Zellen
(2.2.2.4.1) wurden Zellhomogenate hergestellt und intrazerebral in Tga20-Indikatormäuse
injiziert. Dazu wurden die Zellen einmal mit PBS gewaschen und danach durch die Inkubation
mit 0,5 ml EDTA/PBS (5 min, 37°C) abgelöst. Anschli eßend wurden die Zellen in 10 ml PBS
aufgenommen und für 10 min bei 2000 rpm zentrifugiert. Die Zellzahl wurde bestimmt (2.2.2.2)
und auf 104 bzw. 106 Zellen pro 30 µl PBS eingestellt. Die Lagerung der Zelllysate fand bei
-80°C statt. Am Tag der Inokulation wurden die Prob en aufgetaut und durch das Ziehen durch
unterschiedlich große Kanülen (G18, G21, G23) homogenisiert. Jeweils 30 µl des
Zellhomogenats wurden in die Indikatormäuse injiziert. Die Prüfung der Mäuse auf
Krankheitssymptome wie Ataxie, Kyphose und Gewichtsverlust begann 40 Tage nach der
Inokulation und wurde dreimal pro Woche vorgenommen. Im terminalen Stadium der
Erkrankung erfolgte der Abbruch des Experiments. Die Mäuse wurden durch zervikale
Dislokation getötet und Hirn und Milz wurden entnommen (2.2.5.3).
2.2.6 Molekularbiologische Methoden
2.2.6.1 Klonierung
2.2.6.1.1 Präparative Polymerasekettenreaktion (PCR )
MgCl 2 (1,5 mM) Promega GmbH
ddNTPs (0,25 mM) Promega GmbH
reverse und forward Primer (50 pmol/µl) 2.1.9
10x Polymerase-Puffer Promega GmbH/ Stratagene GmbH
Pfu-DNA-Polymerase Stratagene GmbH
ddH 2O
DNA-Matrize
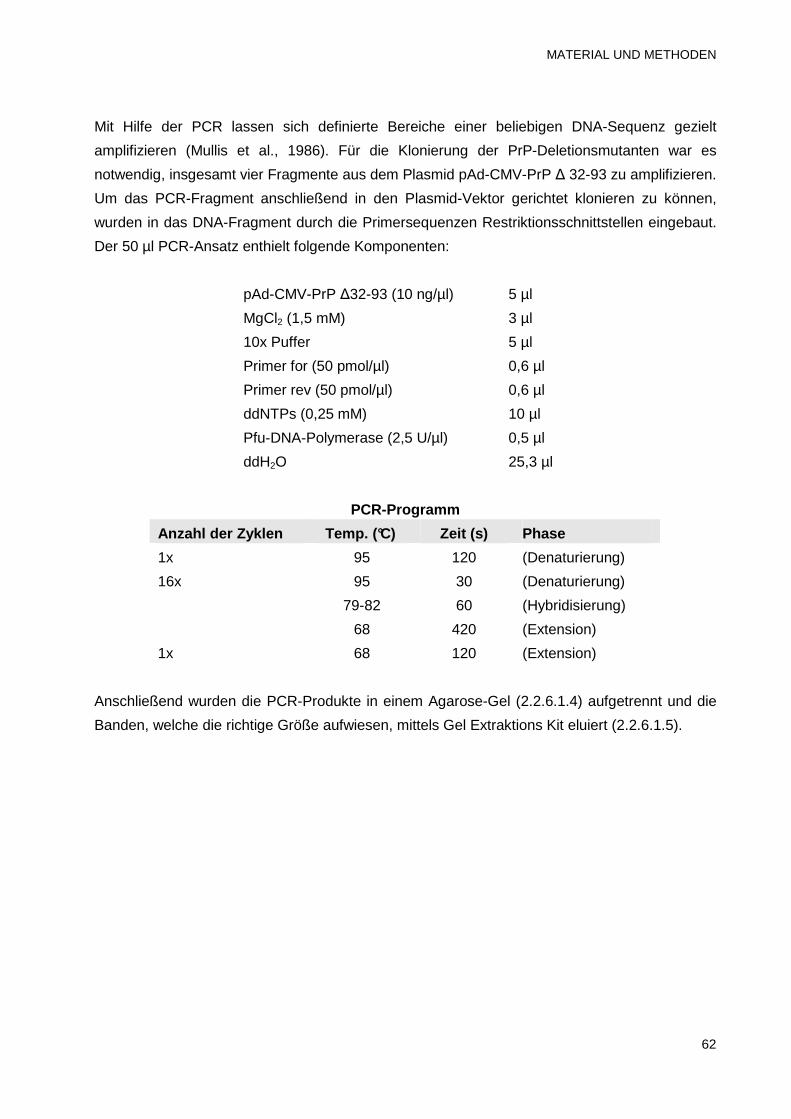
MATERIAL UND METHODEN
62
Mit Hilfe der PCR lassen sich definierte Bereiche einer beliebigen DNA-Sequenz gezielt
amplifizieren (Mullis et al., 1986). Für die Klonierung der PrP-Deletionsmutanten war es
notwendig, insgesamt vier Fragmente aus dem Plasmid pAd-CMV-PrP ∆ 32-93 zu amplifizieren.
Um das PCR-Fragment anschließend in den Plasmid-Vektor gerichtet klonieren zu können,
wurden in das DNA-Fragment durch die Primersequenzen Restriktionsschnittstellen eingebaut.
Der 50 µl PCR-Ansatz enthielt folgende Komponenten:
pAd-CMV-PrP ∆32-93 (10 ng/µl) 5 µl
MgCl2 (1,5 mM) 3 µl
10x Puffer 5 µl
Primer for (50 pmol/µl) 0,6 µl
Primer rev (50 pmol/µl) 0,6 µl
ddNTPs (0,25 mM) 10 µl
Pfu-DNA-Polymerase (2,5 U/µl) 0,5 µl
ddH2O 25,3 µl
PCR-Programm
Anzahl der Zyklen Temp. (°C) Zeit (s) Phase
1x 95 120 (Denaturierung)
16x 95
79-82
68
30
60
420
(Denaturierung)
(Hybridisierung)
(Extension)
1x 68 120 (Extension)
Anschließend wurden die PCR-Produkte in einem Agarose-Gel (2.2.6.1.4) aufgetrennt und die
Banden, welche die richtige Größe aufwiesen, mittels Gel Extraktions Kit eluiert (2.2.6.1.5).
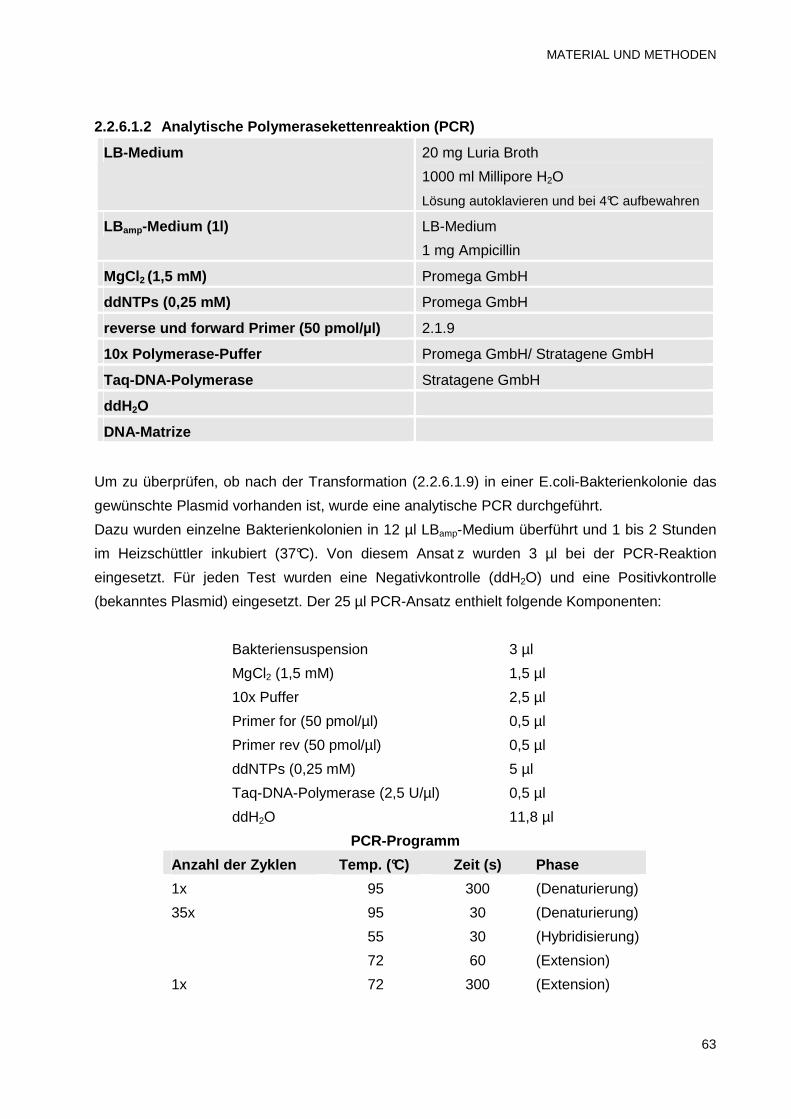
MATERIAL UND METHODEN
63
2.2.6.1.2 Analytische Polymerasekettenreaktion (PCR )
LB-Medium 20 mg Luria Broth
1000 ml Millipore H2O
Lösung autoklavieren und bei 4°C aufbewahren
LB amp-Medium (1l) LB-Medium
1 mg Ampicillin
MgCl 2 (1,5 mM) Promega GmbH
ddNTPs (0,25 mM) Promega GmbH
reverse und forward Primer (50 pmol/µl) 2.1.9
10x Polymerase-Puffer Promega GmbH/ Stratagene GmbH
Taq-DNA-Polymerase Stratagene GmbH
ddH 2O
DNA-Matrize
Um zu überprüfen, ob nach der Transformation (2.2.6.1.9) in einer E.coli-Bakterienkolonie das
gewünschte Plasmid vorhanden ist, wurde eine analytische PCR durchgeführt.
Dazu wurden einzelne Bakterienkolonien in 12 µl LBamp-Medium überführt und 1 bis 2 Stunden
im Heizschüttler inkubiert (37°C). Von diesem Ansat z wurden 3 µl bei der PCR-Reaktion
eingesetzt. Für jeden Test wurden eine Negativkontrolle (ddH2O) und eine Positivkontrolle
(bekanntes Plasmid) eingesetzt. Der 25 µl PCR-Ansatz enthielt folgende Komponenten:
Bakteriensuspension 3 µl
MgCl2 (1,5 mM) 1,5 µl
10x Puffer 2,5 µl
Primer for (50 pmol/µl) 0,5 µl
Primer rev (50 pmol/µl) 0,5 µl
ddNTPs (0,25 mM) 5 µl
Taq-DNA-Polymerase (2,5 U/µl) 0,5 µl
ddH2O 11,8 µl
PCR-Programm
Anzahl der Zyklen Temp. (°C) Zeit (s) Phase
1x 95 300 (Denaturierung)
35x 95
55
72
30
30
60
(Denaturierung)
(Hybridisierung)
(Extension)
1x 72 300 (Extension)

MATERIAL UND METHODEN
64
Abschließend wurde die amplifizierte DNA in einem Agarosegel aufgetrennt (2.2.6.1.4) und
ausgewertet.
2.2.6.1.3 Restriktionsverdau
Restriktionsenzyme New England Biolabs GmbH
MBI Fermentas GmbH
Restriktionsenzym-spezifische Puffer New England Biolabs GmbH
MBI Fermentas GmbH
QIAquick PCR Purification Kit Qiagen GmbH
Plasmid-DNA/PCR-Produkte
ddH 2O
Um sowohl die Richtigkeit der amplifizierten PCR-Fragmente zu überprüfen als auch zur
Herstellung der passenden Ligationsstellen für die Klonierung der PrP-Deletionsmutanten
wurden Restriktionsverdaue nach den Angaben der Firmen New England Biolabs GmbH oder
MBI Fermentas GmbH durchgeführt. Es wurden 3 Units pro µg DNA eingesetzt, wobei das
Volumen des Enzyms 10% des Endvolumens nicht überschreiten durfte. Der Restriktionsansatz
wurde für 1 bis 2 Stunden oder über Nacht bei 37°C inkubiert. Anschließend wurde die verdaute
DNA mit dem QIAquick PCR Purification Kit aufgereinigt und in 30 µl Elutionspuffer
aufgenommen. Beim Verdau mit zwei Enzymen, die unterschiedliche Puffer erforderten, wurden
die Enzyme nacheinander eingesetzt und ein Aufreinigungsschritt zwischengeschaltet. Der
25 µl Ansatz enthielt folgende Komponenten:
DNA 5 µl
Mastermix: 20 µl
2,5 µl 10x Puffer
17,5 µl ddH2O – x µl Restriktionsenzym
Abschließend erfolgte die elektrophoretische Auftrennung der verschiedenen DNA-Fragmente
(2.2.6.1.4). Als Kontrollen wurden unbehandelte DNA bzw. DNA, die nur mit einem Enzym
behandelt wurde, mitgeführt.

MATERIAL UND METHODEN
65
2.2.6.1.4 Agarose-Gelelektrophorese
Agarose Standard AppliChem GmbH
1 kb DNA-Marker MBI Fermentas GmbH
Ethidiumbromid Roth GmbH & Co. KG
6x Probenpuffer MBI Fermentas GmbH
Gelelektrophoresekammer Hauswerkstatt
Gelschlitten Hauswerkstatt
1xTAE (Tris-Acetat-EDTA )Puffer, pH 8,0
Die Auftrennung und der Nachweis von DNA-Fragmenten zu analytischen und präparativen
Zwecken erfolgte mit Hilfe der Elektrophorese in 1x TAE-Puffer. Je nach Größe der
aufzutrennenden DNA wurden Gele von 0,8% bis 1,0% (w/v) Agarose verwendet.
Die entsprechende Menge Agarose wurde abgewogen, in TAE gelöst und in der Mikrowelle
aufgekocht. Nach Abkühlung wurde die lauwarme Agarose in einen Gelschlitten mit
Probentaschenkamm zum Polymerisieren gegossen. Das ausgehärtete Agarosegel wurde in
die mit TAE gefüllte Elektrophoresekammer gelegt, der Kamm wurde entfernt, und
anschließend wurden die Taschen mit 6x Probenpuffer-versetzten Proben und dem 1 kb DNA-
Marker (6 µl) befüllt. Die elektrophoretische Auftrennung erfolgte für analytische Zwecke bei
180 V und für präparative Analysen bei 80 V. Nach Trennung der Fragmente wurde das Gel 15
min in einem TAE-Ethidiumbromid-Bad (0,0001% Ethidiumbromid (10 mg/ml Stammlösung))
gefärbt, zehn Minuten in TAE gewaschen und unter der UV-Lampe (366 nm) analysiert. Danach
wurde das Ergebnis entweder fotografisch dokumentiert oder die DNA wurde aus dem Agarose-
Gel eluiert (2.2.6.1.5).
2.2.6.1.5 Gelextraktion
UV-Lampe Vilber Lourmat, Inc.
Gel Extraction Kit Qiagen GmbH
Für die Aufreinigung von DNA aus Agarose-Gelen wurden die PCR-Fragmente (2.2.6.1.1) oder
Verdauansätze (2.2.6.1.3) in einem 1%igem Agarose-Gel elektrophoretisch aufgetrennt.
Anschließend wurde die gesuchte Bande unter UV-Licht bestimmt, aus dem Gel ausgeschnitten
und in ein Reaktionsgefäß überführt. Diese Proben wurden entsprechend den Angaben des
Herstellers mittels Gel Extraktions Kit eluiert.

MATERIAL UND METHODEN
66
2.2.6.1.6 Dephosphorylierung der 5'-DNA-Enden
Shrimp alkaline phosphatase (SAP) New England Biolabs GmbH
SAP-Puffer New England Biolabs GmbH
Plasmid-DNA
ddH2O
Um eine Religation der linearisierten Plasmidvektoren zu verhindern, wurden die 5'-Enden der
DNA durch alkalische Phosphatase (Shrimp Alkaline Phosphatase (SAP)) dephosphoryliert. Der
50 µl umfassende Ansatz enthielt folgende Komponenten:
Vektor 23 µl
SAP 1 µl
SAP-Puffer 5 µl
ddH2O 21 µl
Dieser Ansatz wurde für 30 min bei 37°C inkubiert. Die Reaktion wurde durch eine
Hitzeinaktivierung (10 min, 75°C) gestoppt.
2.2.6.1.7 Phosphorylierung von DNA-Fragmenten
ATP New England Biolabs GmbH
PEG 4000 New England Biolabs GmbH
Puffer A New England Biolabs GmbH
T4-Polynukleotidkinase (T4-PNK) New England Biolabs GmbH
DNA-Fragmente
ddH 2O
Um die Effizienz der Ligation zu erhöhen, wurde mit Hilfe der T4-Polynukleotidkinase eine
Phosphatgruppe von ATP auf die 5'-terminale Hydroxylgruppe der PCR-Produkte übertragen.
Der 20 µl umfassende Ansatz enthielt folgende Komponenten:
DNA 10 µl
T4-PNK 1 µl
ATP 2 µl
PEG 4000 2 µl
Puffer A 2 µl
ddH2O 3 µl

MATERIAL UND METHODEN
67
Dieser Ansatz wurde für 30 min bei 37°C inkubiert. Abschließend erfolgte ein Pufferwechsel
mittels QIAquick PCR Purification Kit.
2.2.6.1.8 Ligation
T4-DNA-Ligase MBI Fermentas GmbH oder NEB GmbH
10x T4-Ligationspuffer MBI Fermentas GmbH oder NEB GmbH
Plasmid-DNA
PCR-Produkte
ddH 2O
Bei der Ligation werden linearisierte Plasmid-DNA und das geschnittene PCR-Produkt mit Hilfe
der T4-DNA-Ligase miteinander ligiert. Für die 3-Weg-Ligation wurde ein Molekülverhältnis
Plasmid-DNA:PCR-Fragment von 1:4:4 gewählt. Als Negativkontrolle wurde die Religation der
Plasmid-DNA verglichen. Der 20 µl umfassende Ligationsansatz enthielt folgende
Komponenten:
Vektor 2 µl
PCR-Produkt 1 8 µl
PCR-Produkt 2 8 µl
10x Puffer 2 µl
T4-DNA-Ligase 1 µl
ddH2O 1 µl
Dieser Ansatz wurde über Nacht bei 16°C inkubiert. Abschließend erfolgte eine
Hitzeinaktivierung für 10 min bei 65°C.
2.2.6.1.9 Transformation von E.coli mit Plasmiden
XL 10-Gold Ultrakompetente Zellen Stratagene GmbH
LB-Medium 20 mg Luria Broth
1000 ml Millipore H2O
Lösung autoklavieren und bei 4°C aufbewahren
LB amp-Agarplatten 35 mg Luria Broth Agar
1 ml Ampicillin (100µg/ml)
1000 ml Millipore H2O
Lösung autoklavieren, bei einer Temperatur von ca.
60°C das Antibiotikum zugeben und auf Petri-
schalen verteilen

MATERIAL UND METHODEN
68
Die Transformation von E.coli erfolgte mittels Hitzeschock. Dazu wurden 0,1 ml kompetente
Bakterien zu 10 µl des inaktivierten Ligationsansatzes hinzugefügt, vorsichtig gemischt und für
30 min auf Eis inkubiert. Der Hitzeschock erfolgte für 90 s bei 42°C, gefolgt von einer
zweiminütigen Inkubation auf Eis. Nach der Zugabe von 0,9 ml LB-Medium wurde die
Suspension für eine Stunde bei 37°C auf dem Schüttl er inkubiert. Abschließend erfolgte eine
Zentrifugation (10 min, 6000 rpm). Das Pellet wurde in 0,1 ml Medium resuspendiert, auf
Agarplatten, welche das entsprechende Antibiotikum enthielten, ausplattiert und über Nacht mit
der Agarschicht nach oben bei 37°C im Brutschrank i nkubiert. Danach erfolgte die
Aufbewahrung der Agarplatten bei 4°C.
2.2.6.1.10 Plasmidpräparation aus E.coli
LB amp-Medium 2.2.2.1.8
Plasmid Mini Kit Qiagen GmbH
Plasmid Midi Kit Qiagen GmbH
Bakterien-Übernachtkulturen
Zur Erzeugung größerer Mengen an Plasmid-DNA wurden E.coli-Flüssigkulturen angelegt.
Dazu wurde LBamp-Medium mit einem Bakterienklon beimpft und über Nacht bei 37°C im
Inkubator geschüttelt. Für das Screening nach positiven Klonen wurde eine 2 ml
Übernachtkultur abzentrifugiert (6000 rpm, 8 min) und die DNA abschließend aus den Bakterien
mittels Plasmid Mini Kit extrahiert. Für die DNA-Extraktion aus 100 ml Übernachtkulturen wurde
das Plasmid Midi Kit verwendet. Diese Kulturen wurden für Plasmide verwendet, die in einer
Transfektion eingesetzt werden sollten. Die Durchführung der DNA-Extraktion fand
entsprechend der Angaben des Herstellers statt. Abschließend wurde die gewonnene DNA in
25 bis 200 µl sterilem Wasser aufgenommen und die Konzentration photometrisch bestimmt
(2.2.6.1.11). Je nach verwendetem Kit lag die Ausbeute zwischen 20 µg und 100 µg DNA.
2.2.6.1.11 Quantifizierung von doppelsträngiger DNA
UVette® Eppendorf AG
Spektralphotometer Eppendorf AG
Die Konzentrationsbestimmung von doppelsträngiger DNA wurde photometrisch in sterilem
Wasser durchgeführt. Die Messung der Absorption erfolgte zwischen 260 und 280 nm.
Nukleinsäuren besitzen ein Absorptionsmaximum bei 260 nm, da jedoch auch Phenol und
Aminosäuren in der Probe vorliegen, wird ebenfalls bei 280 nm gemessen. Die OD260 gibt

MATERIAL UND METHODEN
69
Auskunft über die DNA-Konzentration und der Quotient OD260/OD280 über den Reinheitsgrad der
Präparation. Liegt dieser Quotient zwischen 1,7 und 2,0 beträgt die Reinheit der DNA 70-95%.
doppelsträngige DNA 1 OD260nm = 50 µg DNA/ml
2.2.6.1.12 Sequenzierung von DNA
Primer (5 pmol/µl) 2.1.9
BDT-Puffer (BigDye) V.1.1 Applied Biosystems GmbH
2,5x Puffer 200 mM Tris-HCl, pH 9,0
500 mM MgCl2
Plasmid-DNA (50 ng/µl)
steriles ddH 2O
Die Sequenzierung der DNA wurde in modifizierter Form nach der Kettenabbruchmethode
(Sanger et al., 1977) durchgeführt. Die Reaktion wird durch die AmpliTaq FS-Polymerase
katalysiert. Der 5 µl umfassende Sequenzierungsansatz enthielt folgende Komponenten:
DNA 2 µl
2,5x Puffer 1 µl
Primer (5 pmol/µl) 1 µl
BDT-Puffer 1 µl
Der Ansatz wurde in einen Thermozykler gestellt, und das folgende Programm wurde gestartet:
Denaturierung 96°C 30 s
Annealing 50°C 15 s
Elongation 60°C 4 min → 25 Zyklen
Nach der PCR-Reaktion wurden die Proben mit dem Qiagen Dye Ex Kit über eine
chromatographische Säule aufgereinigt, mit 2 Volumen Hi-Di-Formamid versetzt und bis zur
Sequenzanalyse bei 4°C aufbewahrt.
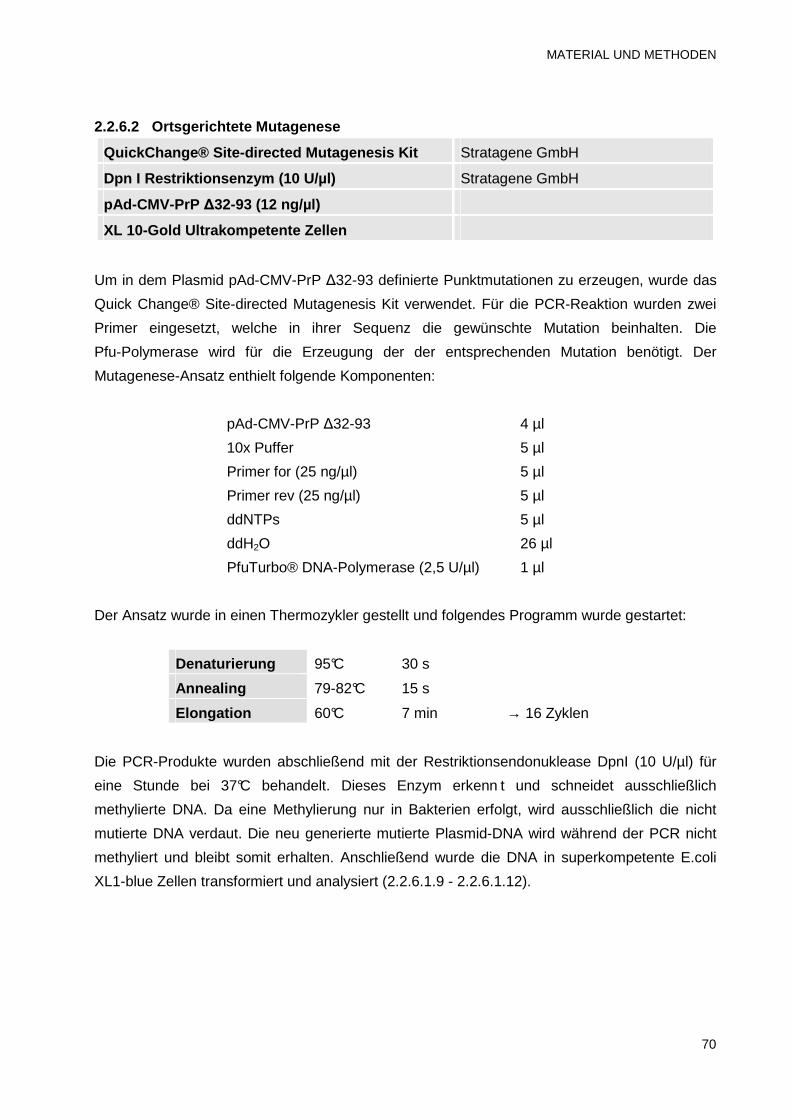
MATERIAL UND METHODEN
70
2.2.6.2 Ortsgerichtete Mutagenese
QuickChange® Site-directed Mutagenesis Kit Stratagene GmbH
Dpn I Restriktionsenzym (10 U/µl) Stratagene GmbH
pAd-CMV-PrP ∆32-93 (12 ng/µl)
XL 10-Gold Ultrakompetente Zellen
Um in dem Plasmid pAd-CMV-PrP ∆32-93 definierte Punktmutationen zu erzeugen, wurde das
Quick Change® Site-directed Mutagenesis Kit verwendet. Für die PCR-Reaktion wurden zwei
Primer eingesetzt, welche in ihrer Sequenz die gewünschte Mutation beinhalten. Die
Pfu-Polymerase wird für die Erzeugung der der entsprechenden Mutation benötigt. Der
Mutagenese-Ansatz enthielt folgende Komponenten:
pAd-CMV-PrP ∆32-93 4 µl
10x Puffer 5 µl
Primer for (25 ng/µl) 5 µl
Primer rev (25 ng/µl) 5 µl
ddNTPs 5 µl
ddH2O 26 µl
PfuTurbo® DNA-Polymerase (2,5 U/µl) 1 µl
Der Ansatz wurde in einen Thermozykler gestellt und folgendes Programm wurde gestartet:
Denaturierung 95°C 30 s
Annealing 79-82°C 15 s
Elongation 60°C 7 min → 16 Zyklen
Die PCR-Produkte wurden abschließend mit der Restriktionsendonuklease DpnI (10 U/µl) für
eine Stunde bei 37°C behandelt. Dieses Enzym erkenn t und schneidet ausschließlich
methylierte DNA. Da eine Methylierung nur in Bakterien erfolgt, wird ausschließlich die nicht
mutierte DNA verdaut. Die neu generierte mutierte Plasmid-DNA wird während der PCR nicht
methyliert und bleibt somit erhalten. Anschließend wurde die DNA in superkompetente E.coli
XL1-blue Zellen transformiert und analysiert (2.2.6.1.9 - 2.2.6.1.12).

ERGEBNISSE
71
3 Ergebnisse
Monoklonale Antikörper gegen PrP haben in den letzten Jahren eine zunehmende Bedeutung
für diagnostische und therapeutische Anwendungen in der Prionenforschung bekommen. Ziel
der vorliegenden Arbeit war die Herstellung von neuen monoklonalen Antikörpern gegen
murines PrP und ihre Charakterisierung für diagnostische und therapeutische Fragestellungen.
Durch die Immunisierung von Prnp0/0-Mäusen mit rekombinantem PrP der Maus wurden sieben
neue monoklonale Antikörper isoliert. Drei verschiedene Immunisierungsprotokolle führten
jeweils zu hohen Antikörpertitern, so dass pro Set die Milz einer Maus entnommen wurde und
die B-Zellen jeweils für eine Fusion verwendet wurden. Die Antikörpertiter der immunisierten
Mäuse und die Anzahl der potentiellen Klone pro Fusion sind in Abschnitt 3.1 beschrieben. Ein
dreistufiges Screeningverfahren ermöglichte die Isolierung der sieben Klone. Die
Antikörpermenge in den Überständen wurde nach Kultivierung in Zwei-Kompartiment-
Bioreaktoren bestimmt und die monoklonalen Antikörper durch Säulenchromatographie in hoher
Reinheit gewonnen. Die Charakterisierung der aufgereinigten Anti-PrP-Antikörper ist im zweiten
Abschnitt (3.2) beschrieben. Dabei wurden die Epitope, die Aviditäten und
Dissoziationskonstanten der monoklonalen Antikörper bestimmt. Anschließend wurden die Anti-
PrP-Antikörper auf ihr therapeutisches Potential, die Prionreplikation in Zellkultur zu hemmen,
untersucht (3.3). Vier der sieben Antikörper konnten die Prionreplikation hemmen. Eine
vollständige Hemmung der PrPSc-Akkumulation und komplette Inaktivierung der Prionreplikation
in den Zellen wurde jedoch nur durch eine Behandlung mit dem Antikörper B2-43-133 erreicht.
Um den Wirkungsmechanismus der Antikörper-vermittelten Hemmung der Prionreplikation zu
untersuchen, wurden zwei Hypothesen experimentell getestet (3.4). Im Hinblick auf eine
therapeutische Anwendung des Antikörpers B2-43-133 wurde abschließend die
Pharmakokinetik in Prionen-infizierten Mäusen untersucht (3.5).
3.1 Herstellung und Reinigung von monoklonalen Anti -PrP-Antikörpern
3.1.1 Antikörpertiter der immunisierten Prnp0/0-Mäuse und Fusionsausbeute
Zur Herstellung von monoklonalen Antikörpern gegen murines PrP wurden drei Gruppen von
Prnp0/0-Mäusen mit verschiedenen Protokollen immunisiert (Abbildung 11). Als Antigen diente
jeweils rekombinantes PrP der Maus, welches entweder in seiner nativ gefalteten Form in Set I
und III (Zahn et al., 1997) oder in einer fibrillären Form (Luhrs et al., 2006) in Set II appliziert
wurde. Die fibrilläre Form besitzt einen hohen β-Faltblattanteil und kann leicht in amyloide
Fibrillen aggregieren, so dass seine physiko-chemischen Eigenschaften denen von PrPSc
ähnlich sind, ohne aber selbst infektiös zu sein (Luhrs et al., 2006). Für Set I und II wurde das
Antigen mit Freund-Adjuvans gemischt und die Antikörpertiter im Serum der Mäuse mittels

ERGEBNISSE
72
1. Boost 2. Boost 3. Bo ost 4. BoostMonate
Immunisierung
50 µg rek.PrP+ CFA (1:1)
50 µg fib.PrP+ CFA (1:1)
10 µg fib.PrP10 µg CpG 1668
+
50 µg rek.PrP+ IFA (1:1)
50 µg rek.PrP+ IFA (1:1) X X X FUSION
X X FUSION
X X FUSION
Set I (i.p.)
Set III (i.c.)
Set II (i.p.)
1. Boost 2. Boost 3. Bo ost 4. BoostMonate
Immunisierung
1. Boost 2. Boost 3. Bo ost 4. BoostMonate
Immunisierung
50 µg rek.PrP+ CFA (1:1)
50 µg fib.PrP+ CFA (1:1)
10 µg fib.PrP10 µg CpG 1668
+
50 µg rek.PrP+ IFA (1:1)
50 µg rek.PrP+ IFA (1:1) X X X FUSION
X X FUSION
X X FUSION
Set I (i.p.)
Set III (i.c.)
Set II (i.p.)
ELISA bei einer Verdünnung von 1:20.000 bestimmt. Während in Set I bereits drei Tage nach
der ersten Immunisierung alle vier Tiere hohe Antikörpertiter (OD um 1,5) zeigten, konnte in Set
II nur in zwei von vier Tieren vergleichbar hohe Titer bestimmt werden. Nach der zweiten
Immunisierung zeigten jedoch alle Tiere in beiden Gruppen ähnlich hohe Titer (OD um 2), die
auch nach weiteren Immunisierungen auf einem konstanten Wert blieben. Ein Unterschied in
der Antigenität zwischen der nativ gefalteten und der fibrillären Form von PrP konnte aus dieser
Beobachtung nicht geschlossen werden. In Set III wurde die Immunisierung ebenfalls mit der
nativ gefalteten Form durchgeführt, wobei jedoch eine subkutane Antigengabe in Gegenwart
von CpGs 1668 als Adjuvans erfolgte (Abbildung 11).
Pro Immunisierungsset wurde jeweils das Tier mit dem höchsten Titer zu dem angegebenen
Zeitpunkt für die Entnahme der Milz ausgewählt und für die Fusion verwendet (Abbildung 11).
Während die Titer der ausgewählten Tiere aus Set I und II eine OD von über zwei zeigten, war
der Titer des Tieres aus Set III bei gleicher Verdünnung deutlich tiefer (Tabelle 7). Da Set III für
ein anderes Projekt etabliert wurde, stand für die Titerbestimmung und Fusion nur eine Maus
zur Verfügung. Ob der Titer in diesem Tier im Vergleich zu Set I und II wirklich tiefer war, konnte
nicht geklärt werden, da die Bestimmung auf einer ELISA-Platte erfolgte, auf der ebenfalls der
interne Standard niedrigere Werte zeigte (Tabelle 7).
Abbildung 11: Immunisierungsschema der Prnp0/0-Mäuse und Zeitpunkte der Fusion. Die Tiere wurden in 4-Wochenabständen entweder intraperitoneal (i.p.) oder subkutan (s.c.) mit rekombinantem PrP (rek. PrP) oder seiner fibrillären Form (fib. PrP) immunisiert. Als Adjuvans wurde entweder komplettes (CFA) und inkomplettes (IFA) Freund-Adjuvans oder CpGs 1668 verwendet. Drei Tage nach den Immunisierungen wurden die Antikörpertiter im Serum der Mäuse mittels ELISA bestimmt. Zu den angegebenen Zeitpunkten wurde in jeder Gruppe die Milz der Maus mit dem höchsten Titer entnommen und für die Fusion verwendet.
Die Milzen aller drei Mäuse, die für die Fusionen ausgewählt wurden, waren von normaler
Größe und wiesen keine besonderen Auffälligkeiten auf. Aus den drei Fusionen resultierten
durchschnittlich 135 potentielle Hybridomklone pro 108 Milzzellen (Tabelle 7). Von allen Klonen,
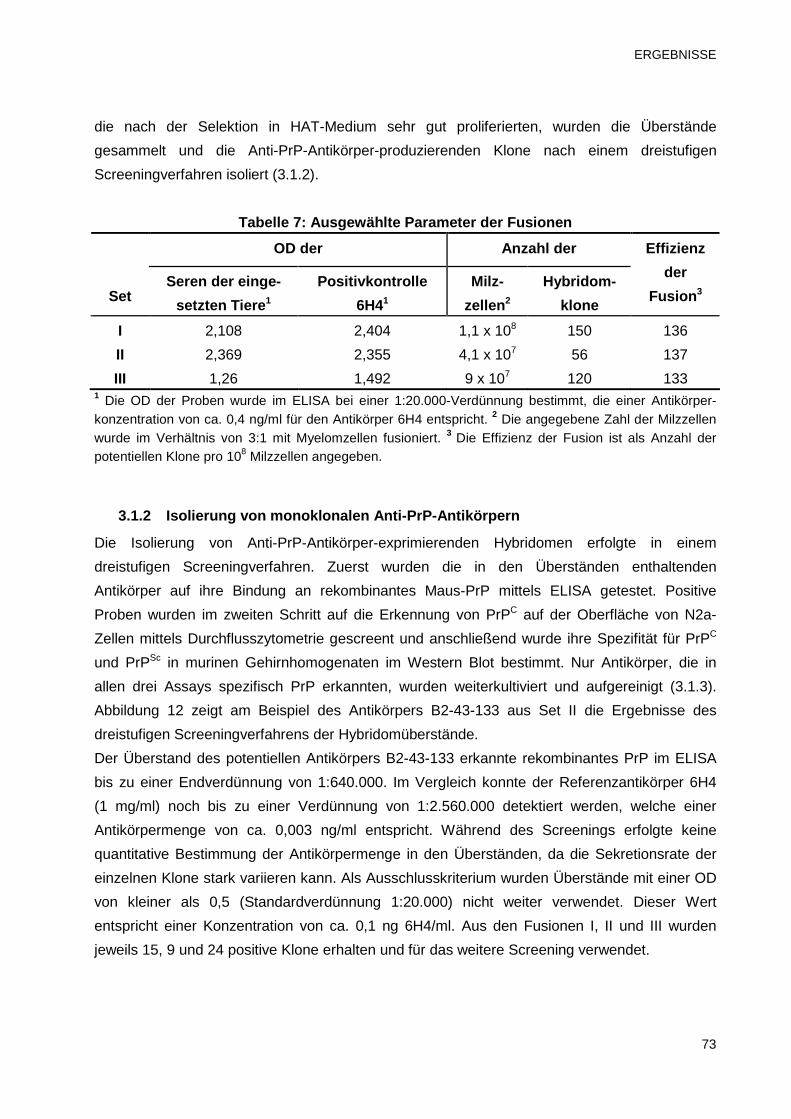
ERGEBNISSE
73
die nach der Selektion in HAT-Medium sehr gut proliferierten, wurden die Überstände
gesammelt und die Anti-PrP-Antikörper-produzierenden Klone nach einem dreistufigen
Screeningverfahren isoliert (3.1.2).
Tabelle 7: Ausgewählte Parameter der Fusionen
OD der Anzahl der
Set Seren der einge-
setzten Tiere 1
Positivkontrolle
6H41
Milz-
zellen 2
Hybridom-
klone
Effizienz
der
Fusion 3
I 2,108 2,404 1,1 x 108 150 136
II 2,369 2,355 4,1 x 107 56 137
III 1,26 1,492 9 x 107 120 133 1 Die OD der Proben wurde im ELISA bei einer 1:20.000-Verdünnung bestimmt, die einer Antikörper- konzentration von ca. 0,4 ng/ml für den Antikörper 6H4 entspricht. 2 Die angegebene Zahl der Milzzellen wurde im Verhältnis von 3:1 mit Myelomzellen fusioniert. 3 Die Effizienz der Fusion ist als Anzahl der potentiellen Klone pro 108 Milzzellen angegeben.
3.1.2 Isolierung von monoklonalen Anti-PrP-Antikörp ern
Die Isolierung von Anti-PrP-Antikörper-exprimierenden Hybridomen erfolgte in einem
dreistufigen Screeningverfahren. Zuerst wurden die in den Überständen enthaltenden
Antikörper auf ihre Bindung an rekombinantes Maus-PrP mittels ELISA getestet. Positive
Proben wurden im zweiten Schritt auf die Erkennung von PrPC auf der Oberfläche von N2a-
Zellen mittels Durchflusszytometrie gescreent und anschließend wurde ihre Spezifität für PrPC
und PrPSc in murinen Gehirnhomogenaten im Western Blot bestimmt. Nur Antikörper, die in
allen drei Assays spezifisch PrP erkannten, wurden weiterkultiviert und aufgereinigt (3.1.3).
Abbildung 12 zeigt am Beispiel des Antikörpers B2-43-133 aus Set II die Ergebnisse des
dreistufigen Screeningverfahrens der Hybridomüberstände.
Der Überstand des potentiellen Antikörpers B2-43-133 erkannte rekombinantes PrP im ELISA
bis zu einer Endverdünnung von 1:640.000. Im Vergleich konnte der Referenzantikörper 6H4
(1 mg/ml) noch bis zu einer Verdünnung von 1:2.560.000 detektiert werden, welche einer
Antikörpermenge von ca. 0,003 ng/ml entspricht. Während des Screenings erfolgte keine
quantitative Bestimmung der Antikörpermenge in den Überständen, da die Sekretionsrate der
einzelnen Klone stark variieren kann. Als Ausschlusskriterium wurden Überstände mit einer OD
von kleiner als 0,5 (Standardverdünnung 1:20.000) nicht weiter verwendet. Dieser Wert
entspricht einer Konzentration von ca. 0,1 ng 6H4/ml. Aus den Fusionen I, II und III wurden
jeweils 15, 9 und 24 positive Klone erhalten und für das weitere Screening verwendet.
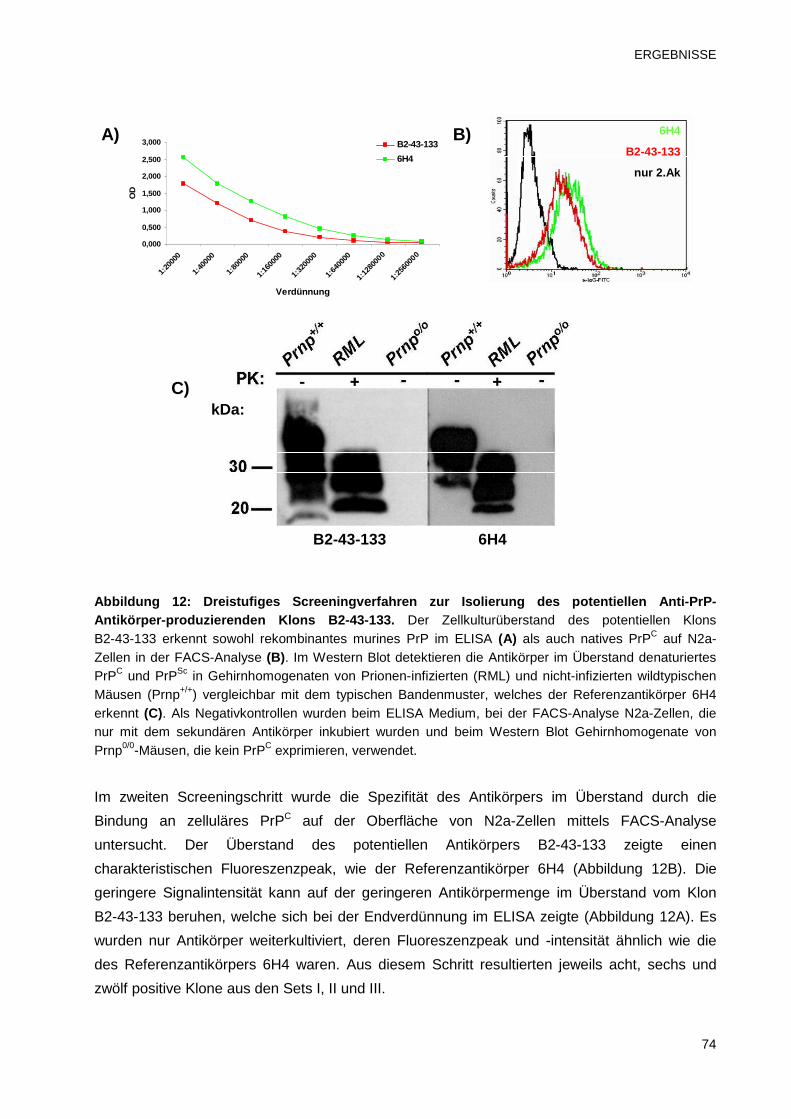
ERGEBNISSE
74
C)
B)A) 6H4
B2-43-133
nur 2.Ak
kDa:
0,000
0,500
1,000
1,500
2,000
2,500
3,000
1:20
000
1:40
000
1:80
000
1:160
000
1:32
0000
1:640
000
1:12
8000
0
1:256
0000
Verdünnung
OD
B2-43-133
6H4
B2-43-133 6H4
C)
B)A) 6H4
B2-43-133
nur 2.Ak
kDa:
0,000
0,500
1,000
1,500
2,000
2,500
3,000
1:20
000
1:40
000
1:80
000
1:160
000
1:32
0000
1:640
000
1:12
8000
0
1:256
0000
Verdünnung
OD
B2-43-133
6H4
B2-43-133 6H4
C)
B)A) 6H4
B2-43-133
nur 2.Ak
kDa:
0,000
0,500
1,000
1,500
2,000
2,500
3,000
1:20
000
1:40
000
1:80
000
1:160
000
1:32
0000
1:640
000
1:12
8000
0
1:256
0000
Verdünnung
OD
B2-43-133
6H4
B2-43-133 6H4
Abbildung 12: Dreistufiges Screeningverfahren zur I solierung des potentiellen Anti-PrP-Antikörper-produzierenden Klons B2-43-133. Der Zellkulturüberstand des potentiellen Klons B2-43-133 erkennt sowohl rekombinantes murines PrP im ELISA (A) als auch natives PrPC auf N2a-Zellen in der FACS-Analyse (B). Im Western Blot detektieren die Antikörper im Überstand denaturiertes PrPC und PrPSc in Gehirnhomogenaten von Prionen-infizierten (RML) und nicht-infizierten wildtypischen Mäusen (Prnp+/+) vergleichbar mit dem typischen Bandenmuster, welches der Referenzantikörper 6H4 erkennt (C). Als Negativkontrollen wurden beim ELISA Medium, bei der FACS-Analyse N2a-Zellen, die nur mit dem sekundären Antikörper inkubiert wurden und beim Western Blot Gehirnhomogenate von Prnp0/0-Mäusen, die kein PrPC exprimieren, verwendet.
Im zweiten Screeningschritt wurde die Spezifität des Antikörpers im Überstand durch die
Bindung an zelluläres PrPC auf der Oberfläche von N2a-Zellen mittels FACS-Analyse
untersucht. Der Überstand des potentiellen Antikörpers B2-43-133 zeigte einen
charakteristischen Fluoreszenzpeak, wie der Referenzantikörper 6H4 (Abbildung 12B). Die
geringere Signalintensität kann auf der geringeren Antikörpermenge im Überstand vom Klon
B2-43-133 beruhen, welche sich bei der Endverdünnung im ELISA zeigte (Abbildung 12A). Es
wurden nur Antikörper weiterkultiviert, deren Fluoreszenzpeak und -intensität ähnlich wie die
des Referenzantikörpers 6H4 waren. Aus diesem Schritt resultierten jeweils acht, sechs und
zwölf positive Klone aus den Sets I, II und III.
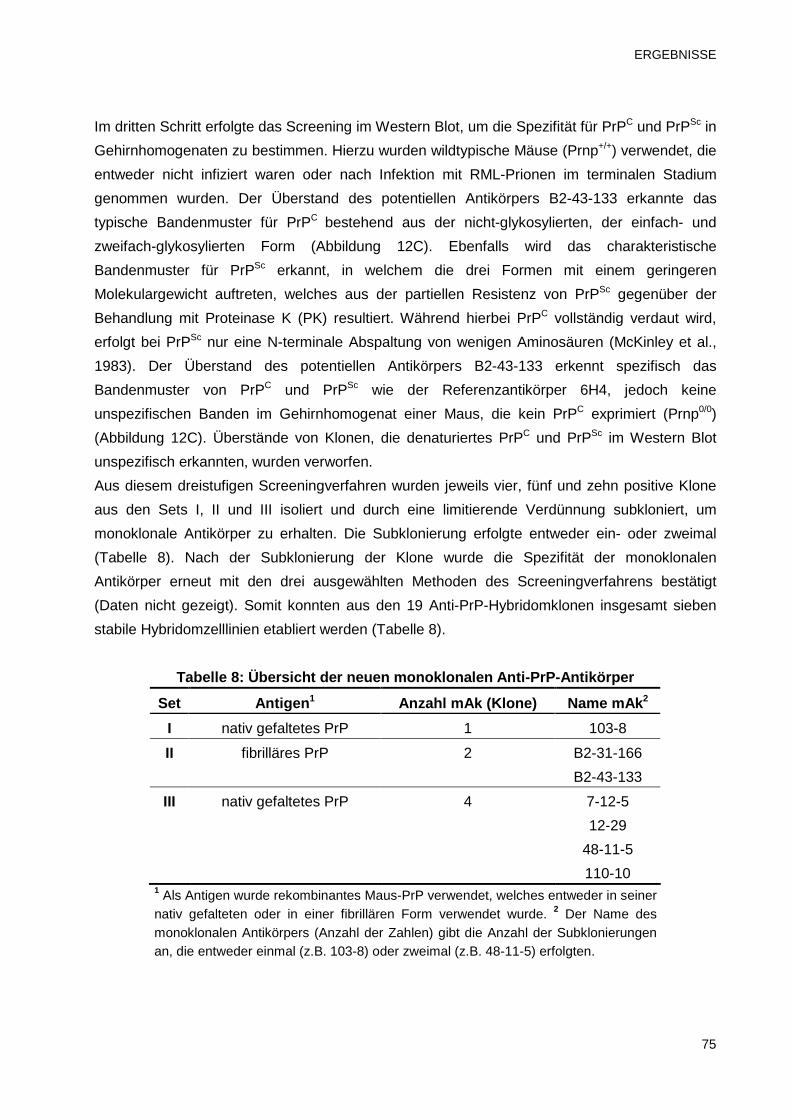
ERGEBNISSE
75
Im dritten Schritt erfolgte das Screening im Western Blot, um die Spezifität für PrPC und PrPSc in
Gehirnhomogenaten zu bestimmen. Hierzu wurden wildtypische Mäuse (Prnp+/+) verwendet, die
entweder nicht infiziert waren oder nach Infektion mit RML-Prionen im terminalen Stadium
genommen wurden. Der Überstand des potentiellen Antikörpers B2-43-133 erkannte das
typische Bandenmuster für PrPC bestehend aus der nicht-glykosylierten, der einfach- und
zweifach-glykosylierten Form (Abbildung 12C). Ebenfalls wird das charakteristische
Bandenmuster für PrPSc erkannt, in welchem die drei Formen mit einem geringeren
Molekulargewicht auftreten, welches aus der partiellen Resistenz von PrPSc gegenüber der
Behandlung mit Proteinase K (PK) resultiert. Während hierbei PrPC vollständig verdaut wird,
erfolgt bei PrPSc nur eine N-terminale Abspaltung von wenigen Aminosäuren (McKinley et al.,
1983). Der Überstand des potentiellen Antikörpers B2-43-133 erkennt spezifisch das
Bandenmuster von PrPC und PrPSc wie der Referenzantikörper 6H4, jedoch keine
unspezifischen Banden im Gehirnhomogenat einer Maus, die kein PrPC exprimiert (Prnp0/0)
(Abbildung 12C). Überstände von Klonen, die denaturiertes PrPC und PrPSc im Western Blot
unspezifisch erkannten, wurden verworfen.
Aus diesem dreistufigen Screeningverfahren wurden jeweils vier, fünf und zehn positive Klone
aus den Sets I, II und III isoliert und durch eine limitierende Verdünnung subkloniert, um
monoklonale Antikörper zu erhalten. Die Subklonierung erfolgte entweder ein- oder zweimal
(Tabelle 8). Nach der Subklonierung der Klone wurde die Spezifität der monoklonalen
Antikörper erneut mit den drei ausgewählten Methoden des Screeningverfahrens bestätigt
(Daten nicht gezeigt). Somit konnten aus den 19 Anti-PrP-Hybridomklonen insgesamt sieben
stabile Hybridomzelllinien etabliert werden (Tabelle 8).
Tabelle 8: Übersicht der neuen monoklonalen Anti-Pr P-Antikörper
Set Antigen 1 Anzahl mAk (Klone) Name mAk 2
I nativ gefaltetes PrP 1 103-8
II fibrilläres PrP 2 B2-31-166
B2-43-133
III nativ gefaltetes PrP 4 7-12-5
12-29
48-11-5
110-10 1 Als Antigen wurde rekombinantes Maus-PrP verwendet, welches entweder in seiner nativ gefalteten oder in einer fibrillären Form verwendet wurde. 2 Der Name des monoklonalen Antikörpers (Anzahl der Zahlen) gibt die Anzahl der Subklonierungen an, die entweder einmal (z.B. 103-8) oder zweimal (z.B. 48-11-5) erfolgten.

ERGEBNISSE
76
Nach Abschluss der Subklonierung proliferierte der Klon B2-31-166 nur schlecht. Um die
Wachstumsbedingungen zu verbessern, wurden der Klon bis zu zwei Monate mit Interleukin-6
(IL-6) behandelt. IL-6 ist ein proinflammatorisches Interleukin, das als B-Zell-stimulierender
Faktor bereits aktivierte B-Zellen zur Ausdifferenzierung und Antikörpersekretion anregt
(Hansson, 1990). Nach der Behandlung wuchs auch dieser Klon in einem regelmäßigen
Rhythmus. Die anderen Hybridomzelllinien wurden problemlos kultiviert.
3.1.3 Antikörperproduktion und -konzentrierung in h oher Reinheit
Um für weitere Studien aufgereinigte Antikörper unter standardisierten Bedingungen verwenden
zu können, wurden die Antikörper aus den Überständen nach Kultivierung der Klone in Zwei-
Kompartiment-Bioreaktoren mittels Protein G-Säulen in hoher Reinheit isoliert, konzentriert und
deren Konzentration bestimmt. Hierbei zeigten die Klone vergleichbare Wachstumsraten,
jedoch unterschieden sich die Mengen der sezernierten Antikörper erheblich und lagen
zwischen 0,004 und 1,4 mg pro ml Überstand (Tabelle 9). Da sowohl einzelne Klone aus Set II
(12-29) und Set III (B2-31-166, B2-43-133) nur geringe Antikörpermengen sezernierten
(< 0,06 mg/ml), konnte kein spezifischer Effekt des Antigens festgestellt werden.
Die Reinheit der aufgereinigten Antikörper wurde mittels Silbergel und isolelektrischer
Fokussierung überprüft und war mit der des kommerziellen Antikörpers 6H4 vergleichbar (Daten
nicht gezeigt). Von allen sieben etablierten Hybridomzelllinien konnten ausreichende Mengen
an aufgereinigten Antikörpern für die Charakterisierung (3.2) gewonnen werden.
Im Gegensatz zu den anderen Klonen stoppte die Hybridomzellinie B2-31-166 trotz normaler
Proliferation in einem synthetischen Medium bereits nach kurzer Zeit die Sezernierung von
Antikörpern, so dass diese Zellen für die Produktion nach wenigen Passagen durch neue
Hybridomzellen ersetzt werden mussten. Dieses Phänomen wurde bereits in einer früheren
Studie beobachtet, in der als mögliche Ursache für die kurzen Sezernierungszeiträume eine
Interaktion des Antikörpers mit intra- oder extrazellulärem PrPC angenommen wurde
(Williamson et al., 1996).
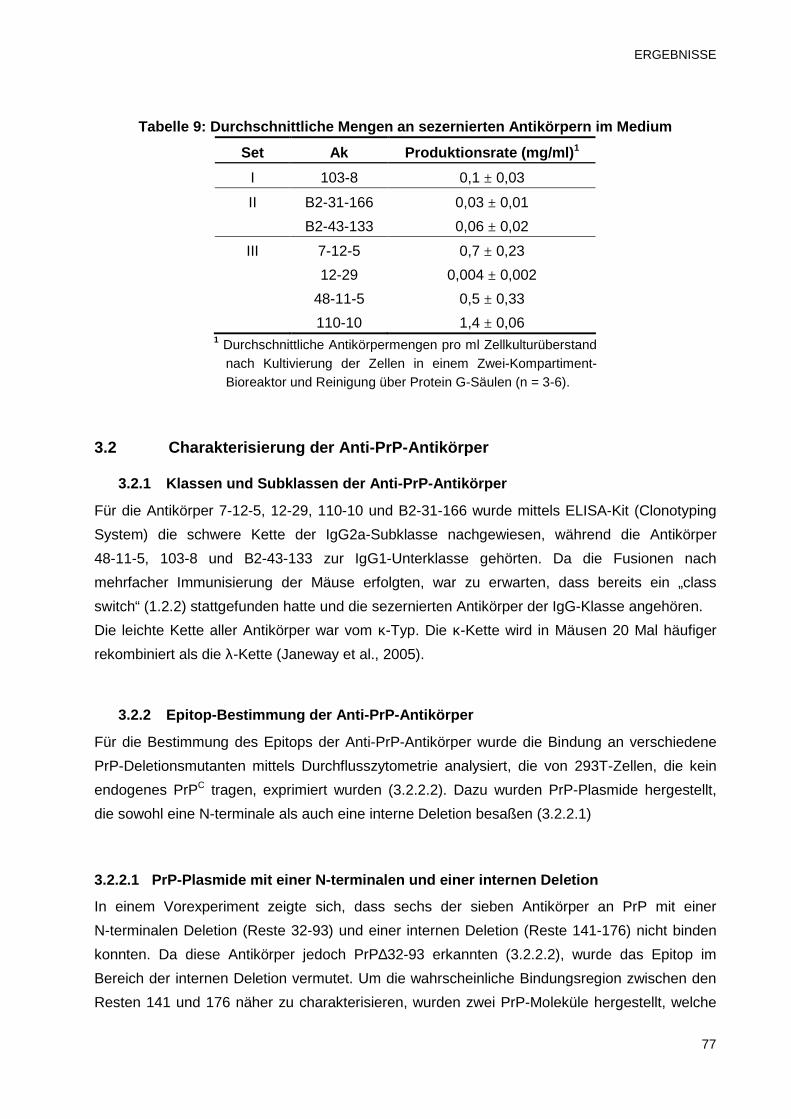
ERGEBNISSE
77
Tabelle 9: Durchschnittliche Mengen an sezernierten Antikörpern im Medium
Set Ak Produktionsrate (mg/ml) 1
I 103-8 0,1 ± 0,03
II B2-31-166 0,03 ± 0,01
B2-43-133 0,06 ± 0,02
III 7-12-5 0,7 ± 0,23
12-29 0,004 ± 0,002
48-11-5 0,5 ± 0,33
110-10 1,4 ± 0,06 1 Durchschnittliche Antikörpermengen pro ml Zellkulturüberstand
nach Kultivierung der Zellen in einem Zwei-Kompartiment-Bioreaktor und Reinigung über Protein G-Säulen (n = 3-6).
3.2 Charakterisierung der Anti-PrP-Antikörper
3.2.1 Klassen und Subklassen der Anti-PrP-Antikörpe r
Für die Antikörper 7-12-5, 12-29, 110-10 und B2-31-166 wurde mittels ELISA-Kit (Clonotyping
System) die schwere Kette der IgG2a-Subklasse nachgewiesen, während die Antikörper
48-11-5, 103-8 und B2-43-133 zur IgG1-Unterklasse gehörten. Da die Fusionen nach
mehrfacher Immunisierung der Mäuse erfolgten, war zu erwarten, dass bereits ein „class
switch“ (1.2.2) stattgefunden hatte und die sezernierten Antikörper der IgG-Klasse angehören.
Die leichte Kette aller Antikörper war vom κ-Typ. Die κ-Kette wird in Mäusen 20 Mal häufiger
rekombiniert als die λ-Kette (Janeway et al., 2005).
3.2.2 Epitop-Bestimmung der Anti-PrP-Antikörper
Für die Bestimmung des Epitops der Anti-PrP-Antikörper wurde die Bindung an verschiedene
PrP-Deletionsmutanten mittels Durchflusszytometrie analysiert, die von 293T-Zellen, die kein
endogenes PrPC tragen, exprimiert wurden (3.2.2.2). Dazu wurden PrP-Plasmide hergestellt,
die sowohl eine N-terminale als auch eine interne Deletion besaßen (3.2.2.1)
3.2.2.1 PrP-Plasmide mit einer N-terminalen und ein er internen Deletion
In einem Vorexperiment zeigte sich, dass sechs der sieben Antikörper an PrP mit einer
N-terminalen Deletion (Reste 32-93) und einer internen Deletion (Reste 141-176) nicht binden
konnten. Da diese Antikörper jedoch PrP∆32-93 erkannten (3.2.2.2), wurde das Epitop im
Bereich der internen Deletion vermutet. Um die wahrscheinliche Bindungsregion zwischen den
Resten 141 und 176 näher zu charakterisieren, wurden zwei PrP-Moleküle hergestellt, welche

ERGEBNISSE
78
171
15832 93PrP ∆32-93
∆158-171
157PrP ∆32-93
∆142-157
sSPrP
32 93 142
STE TM H1 H3H2
OKTAREPEATS ß2ß1
innerhalb dieser Sequenz kleinere Deletionen enthielten. Dazu wurden mittels 3-Weg-Ligation
die Aminosäuren 141 und 158 sowie 157 und 172 aneinander fusioniert (Abbildung 13).
Abbildung 13: Schematische Darstellung der Struktur von Maus-PrP und der klonierten Mutanten mit N-terminaler ( ∆∆∆∆32-93) sowie interner Deletion ( ∆∆∆∆142-157 oder ∆∆∆∆158-171). Die drei α-Helices (H1-3), die beiden β-Faltblattstrukturen (β1, β2) sowie die Stopp-Transfer-Effektor-Sequenz (STE), die putative Transmembrandomäne (TM) und der GPI-Anker sind dargestellt.
Als Ausgangsplasmid für die Klonierung wurde das adenovirale Transferplasmid
pAd-CMV-PrP∆32-93 benutzt, welches bereits die N-terminale Deletion der Aminosäuren 32 bis
93 im offenen Leserahmen der murinen PrP-Sequenz unter der Kontrolle eines CMV-Promoters
enthält. Für die Klonierung wurde die Sequenz des mutierten PrP (ca. 830 bp) durch einen
Verdau mit den beiden Enzymen BamHI und MfeI aus dem pAd-CMV-PrP∆32-93-Plasmid
entfernt. Danach wurden für die beiden gewünschten Deletionen jeweils zwei PCR-Produkte
erzeugt und durch eine 3-Weg-Ligation mittels BamHI und MfeI in den verbleibenden Vektor
(6,1 kb) kloniert (Abbildung 14).
Die nahtlose Fusion der beiden PCR-Fragmente wurde durch den Einbau einer
Restriktionsschnittstelle an die zu fusionierenden PCR-Enden mittels PCR-Primern ermöglicht.
Hierbei wurde das Restriktionsenzym der Klasse II SapI verwendet, bei dem die spezifische
Erkennungssequenz außerhalb der Schnittstelle liegt (Padgett und Sorge, 1996). Da die
Kapazität von SapI direkt an den Enden der PCR-Produkte zu schneiden limitiert ist, wurden die
Fragmente zunächst ohne Verdau in den pBluescript II SK(+) Vektor über eine EcoRV-
Schnittstelle subkloniert. Alle subklonierten Fragmente wurden dann mittels M13-Primerset
herausamplifiziert, wodurch an die ursprünglichen PCR-Fragmente zusätzlich 72 bp an das
5'-Ende und 113 bp an das 3'-Ende angehängt wurden. Die zusätzlichen Basenpaare
ermöglichten einen analytischen Restriktionsverdau, um die Orientierung der Fragmente und
das Schneiden der eingeführten Schnittstellen kontrollieren zu können. Die verifizierten DNA-
Fragmente wurden isoliert und für die 3-Weg-Ligation verwendet, aus der die beiden
gewünschten Deletionen (PrP∆32-93 & ∆142-157 und PrP∆32-93 & ∆158-171) resultierten
(Abbildung 14).
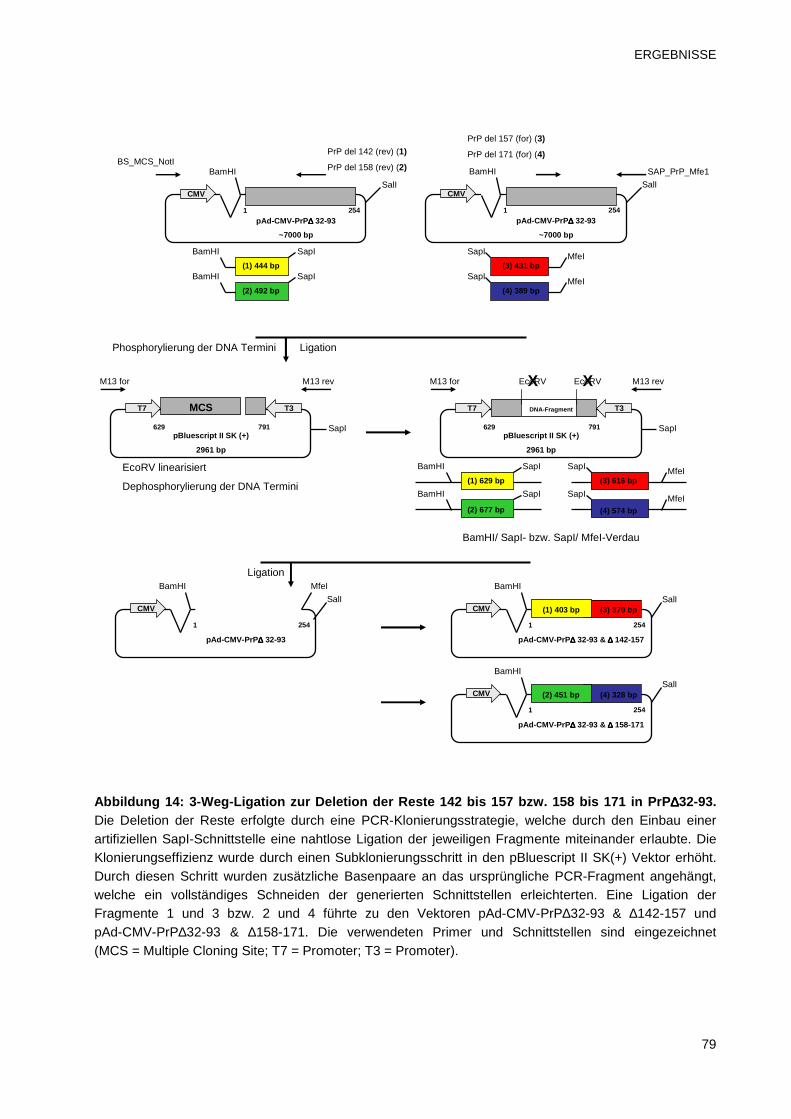
ERGEBNISSE
79
SalI
1
pAd-CMV-PrP ∆∆∆∆ 32-93
~7000 bp
CMV
254
BS_MCS_NotIBamHI
PrP del 142 (rev) (1)
PrP del 158 (rev) (2)
SalI
1
pAd-CMV-PrP ∆∆∆∆ 32-93
~7000 bp
CMV
254
SAP_PrP_Mfe1BamHI
PrP del 157 (for) (3)
PrP del 171 (for) (4)
BamHI SapI SapI MfeIEcoRV linearisiert
Dephosphorylierung der DNA Termini
SapI629
M13 rev
pBluescript II SK (+)
2961 bp
T7 T3
791
M13 for
Phosphorylierung der DNA Termini Ligation
SapI629
M13 rev
pBluescript II SK (+)
2961 bp
T7 T3
791
M13 for
DNA-Fragment
BamHI SapI
(1) 444 bp
BamHI/ SapI- bzw. SapI/ MfeI-Verdau
Ligation
SalI
1
pAd-CMV-PrP ∆∆∆∆ 32-93 & ∆∆∆∆ 142-157
CMV
254
BamHIMfeI
1
pAd-CMV-PrP ∆∆∆∆ 32-93
CMV
254
BamHI
SalI
SapI MfeI(3) 431 bp
(1) 629 bp (3) 616 bp
(3) 370 bp
BamHI SapI
(2) 492 bp
SapIMfeI
(4) 389 bp
MCS
EcoRV EcoRVX X
BamHI SapI SapI MfeI(2) 677 bp (4) 574 bp
SalI
1
pAd-CMV-PrP ∆∆∆∆ 32-93 & ∆∆∆∆ 158-171
CMV
254
BamHI
(4) 328 bp
(1) 403 bp
(2) 451 bp
SalI
1
pAd-CMV-PrP ∆∆∆∆ 32-93
~7000 bp
CMV
254
BS_MCS_NotIBamHI
PrP del 142 (rev) (1)
PrP del 158 (rev) (2)
SalI
1
pAd-CMV-PrP ∆∆∆∆ 32-93
~7000 bp
CMV
254
SAP_PrP_Mfe1BamHI
PrP del 157 (for) (3)
PrP del 171 (for) (4)
BamHI SapI SapI MfeIEcoRV linearisiert
Dephosphorylierung der DNA Termini
SapI629
M13 rev
pBluescript II SK (+)
2961 bp
T7 T3
791
M13 for
Phosphorylierung der DNA Termini Ligation
SapI629
M13 rev
pBluescript II SK (+)
2961 bp
T7 T3
791
M13 for
DNA-Fragment
BamHI SapI
(1) 444 bp
BamHI/ SapI- bzw. SapI/ MfeI-Verdau
Ligation
SalI
1
pAd-CMV-PrP ∆∆∆∆ 32-93 & ∆∆∆∆ 142-157
CMV
254
BamHIMfeI
1
pAd-CMV-PrP ∆∆∆∆ 32-93
CMV
254
BamHI
SalI
SapI MfeI(3) 431 bp
(1) 629 bp (3) 616 bp
(3) 370 bp
BamHI SapI
(2) 492 bp
SapIMfeI
(4) 389 bp
MCS
EcoRV EcoRVX X
BamHI SapI SapI MfeI(2) 677 bp (4) 574 bp
SalI
1
pAd-CMV-PrP ∆∆∆∆ 32-93 & ∆∆∆∆ 158-171
CMV
254
BamHI
(4) 328 bp
(1) 403 bp
(2) 451 bp
Abbildung 14: 3-Weg-Ligation zur Deletion der Reste 142 bis 157 bzw. 158 bis 171 in PrP ∆∆∆∆32-93. Die Deletion der Reste erfolgte durch eine PCR-Klonierungsstrategie, welche durch den Einbau einer artifiziellen SapI-Schnittstelle eine nahtlose Ligation der jeweiligen Fragmente miteinander erlaubte. Die Klonierungseffizienz wurde durch einen Subklonierungsschritt in den pBluescript II SK(+) Vektor erhöht. Durch diesen Schritt wurden zusätzliche Basenpaare an das ursprüngliche PCR-Fragment angehängt, welche ein vollständiges Schneiden der generierten Schnittstellen erleichterten. Eine Ligation der Fragmente 1 und 3 bzw. 2 und 4 führte zu den Vektoren pAd-CMV-PrP∆32-93 & ∆142-157 und pAd-CMV-PrP∆32-93 & ∆158-171. Die verwendeten Primer und Schnittstellen sind eingezeichnet (MCS = Multiple Cloning Site; T7 = Promoter; T3 = Promoter).

ERGEBNISSE
80
Die Sequenzanalyse des Plasmids pAd-CMV-PrP∆32-93 & ∆142-157 zeigte einen
unerwünschten Aminosäureaustausch des Rests 139 (Histidin→ Asparaginsäure). Da diese
PrP-Mutante nach transienter Expression korrekt auf der Oberfläche von 293T-Zellen exprimiert
wurde (Abbildung 15), wurde dieses Plasmid für die Epitop-Bestimmung verwendet (3.2.2.2).
Obwohl die Sequenzanalyse des zweiten Konstrukts pAd-CMV-PrP∆32-93 & ∆158-171 keine
Mutation zeigte, konnte PrP nicht auf der Oberfläche von 293T-Zellen nachgewiesen werden.
Ob dies aus einer Instabilität der PrP-Mutante nach Entfernung des zweiten β-Faltblatts
resultierte, wurde nicht weiter untersucht.
3.2.2.2 Bestimmung des Epitops mittels FACS
Für die Bestimmung des Epitops der aufgereinigten Anti-PrP-Antikörper wurden humane
293T-Zellen mit verschiedenen PrP-Deletionsmutanten (Abbildung 15B) transient transfiziert.
Die Expression aller PrP-Deletionsmutanten wurde mit einem polyklonalen Kaninchenserum
bestätigt (Daten nicht gezeigt). Da diese Zellen kein PrPC exprimieren, kann die Bindung des
Antikörpers an das entsprechende PrP-Molekül mittels FACS-Analyse direkt bestimmt werden
(Abbildung 15A).
Die N-terminale Deletionsmutante PrP∆32-93 enthält keine Oktarepeatsequenz und wird von
allen sieben Antikörpern mit vergleichbarer Fluoreszenzintensität erkannt, die auch der
Referenzantikörper 6H4 zeigt, dessen Epitop zwischen den Aminosäuren 144 bis 151 liegt
(Korth et al., 1997). Die nächstgrößere Deletion PrP∆32-106, in der neben der
Oktarepeatsequenz auch die Stopp-Transfer-Effektor-Sequenz (STE) deletiert ist, wurde mit
Ausnahme des Antikörpers B2-31-166 von allen sechs Antikörpern erkannt. Somit liegt das
Epitop des Antikörpers B2-31-166 innerhalb der Sequenz der Reste 93 bis 106 von Maus-PrP
(Abbildung 16). Da die PrP-Mutante mit einer N-terminalen (PrP∆32-93) und einer internen
Deletion (PrP∆141-176) nur von dem Antikörper B2-31-166 erkannt wird, wurde das Epitop der
verbleibenden Antikörper im Bereich der Reste 141 und 176 lokalisiert. Wird die interne
Deletion auf die Reste 142 bis 157 (Helix 1) reduziert (3.2.2.1), können die Antikörper 7-12-5,
12-29 und 48-11-5 wieder binden (Abbildung 15A). Somit liegt das Epitop dieser drei Antikörper
vermutlich in der Region um das β-Faltblatt 2 (Reste 158 bis 176) (Abbildung 16). Da die drei
Antikörper 103-8, 110-10 und B2-43-133 ebenso wie der Referenzantikörper 6H4 an diese
Mutante nicht binden können, sind deren Epitope im Bereich der Helix 1 (Reste 142-157)
lokalisiert. Dieses Ergebnis stimmt mit dem bekannten Epitop des Antikörpers 6H4 überein
(Korth et al., 1997). Abbildung 16 zeigt eine Zusammenfassung der Epitope der sieben
Antikörper.
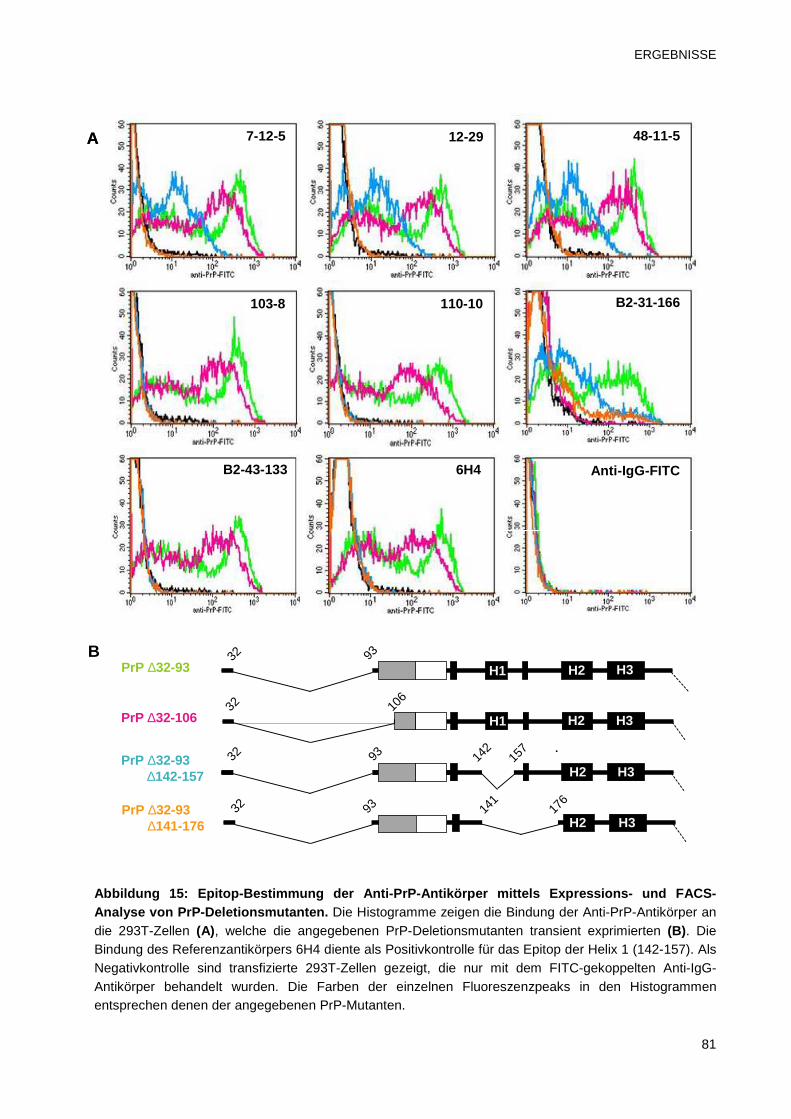
ERGEBNISSE
81
B
103-8 110-10 B2-31-166
B2-43-133 6H4 Anti-IgG-FITC
7-12-5 12-29 48-11-5A
32 106
PrP ∆32-106 H1 H2 H3
PrP ∆32-93 H1 H232 93
H3
157
PrP ∆32-93∆142-157
PrP ∆32-93∆141-176
32 141
176
32 93 142
93
H2
H2
H3
H3
B
103-8 110-10 B2-31-166103-8103-8 110-10 B2-31-166
B2-43-133 6H4 Anti-IgG-FITC
7-12-5 12-29 48-11-5A 7-12-5 12-29 48-11-5A
32 106
PrP ∆32-106 H1 H2 H3
PrP ∆32-93 H1 H232 93
H3
157
PrP ∆32-93∆142-157
PrP ∆32-93∆141-176
32 141
176
32 93 142
93
H2
H2
H3
H3
Abbildung 15: Epitop-Bestimmung der Anti-PrP-Antikö rper mittels Expressions- und FACS-Analyse von PrP-Deletionsmutanten. Die Histogramme zeigen die Bindung der Anti-PrP-Antikörper an die 293T-Zellen (A), welche die angegebenen PrP-Deletionsmutanten transient exprimierten (B). Die Bindung des Referenzantikörpers 6H4 diente als Positivkontrolle für das Epitop der Helix 1 (142-157). Als Negativkontrolle sind transfizierte 293T-Zellen gezeigt, die nur mit dem FITC-gekoppelten Anti-IgG-Antikörper behandelt wurden. Die Farben der einzelnen Fluoreszenzpeaks in den Histogrammen entsprechen denen der angegebenen PrP-Mutanten.

ERGEBNISSE
82
7-12-5
N C
GPI
93-106 142-157 158-176
S-S
STE TM H1 H2 H3ββββ1 ββββ2Oktarepeats
23 95-112
112-126
128-131
161-164
144-151
179-193
200-217
23151-91
12-29
103-8
B2-31-166
48-11-5
B2-43-133
110-10
7-12-5
N C
GPI
93-106 142-157 158-176
S-S
STE TM H1 H2 H3ββββ1 ββββ2Oktarepeats
23 95-112
112-126
128-131
161-164
144-151
179-193
200-217
23151-91
12-29
103-8
B2-31-166
48-11-5
B2-43-133
110-10
Eine weitere Einteilung der sieben Antikörper ist entsprechend der lokalisierten Epitope
möglich, so dass Helix 1-bindendende Antikörper (103-8, 110-10 und B2-43-133),
β-Faltblatt 2-bindendende Antikörper (7-12-5, 12-29 und 48-11-5) und ein Stopp-Transfer-
Effektor-Sequenz (STE)-bindender Antikörper (B2-31-166) unterschieden werden können.
Abbildung 16: Schematische Darstellung der Struktur von Maus-PrP und Lokalisierung der Epitope der Anti-PrP-Antikörper. Als Orientierung wurden sowohl die Aminosäurereste als auch die drei α-Helices (H1-3), die beiden β-Faltblattstrukturen (β1, β2), die Stopp-Transfer-Effektor-Sequenz (STE), die putative Transmembrandomäne (TM) und der GPI-Anker dargestellt (S-S = Disulfidbrücke).
3.2.2.3 Bestimmung des Minimalepitops mittels Alani n-Scanning
Da einige Antikörper mit inhibitorischem Effekt auf die PrPSc-Akkumulation in Zellkultur PrPC im
Bereich der Helix 1 (Reste 143-153) binden (Tabelle 18), sollte im nächsten Schritt das Epitop
der drei Helix 1-bindenden Antikörper (103-8, 110-10 und B2-43-133) genauer charakterisiert
werden. Dazu wurde mittels ortsgerichteter Mutagenese ein Alanin-Scanning von einzelnen
Resten im Bereich der Helix 1 vorgenommen. Um die Helix 1 dabei nicht zu destabilisieren,
wurden die stabilisierenden Salzbrücken erhalten (Abbildung 17) (Norstrom und Mastrianni,
2006). Innerhalb der Helix 1 (Reste 143-153) wurden die beiden Aminosäuren Tyrosin 148 und
Glutaminsäure 151 ausgewählt. Zusätzlich wurde die Aminosäure Histidin 176 in der Nähe der
Helix 2 (Reste 179-192) gegen ein Alanin ausgetauscht (Abbildung 18).
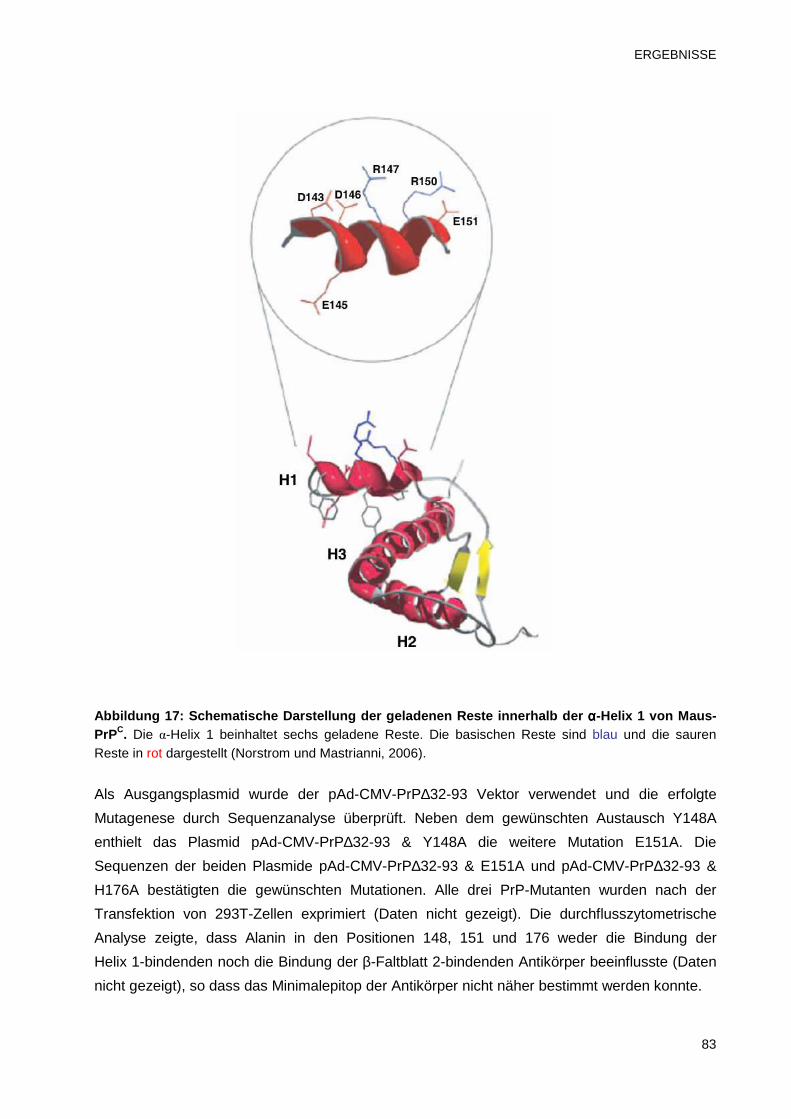
ERGEBNISSE
83
Abbildung 17: Schematische Darstellung der geladene n Reste innerhalb der αααα-Helix 1 von Maus-PrPC. Die α-Helix 1 beinhaltet sechs geladene Reste. Die basischen Reste sind blau und die sauren Reste in rot dargestellt (Norstrom und Mastrianni, 2006).
Als Ausgangsplasmid wurde der pAd-CMV-PrP∆32-93 Vektor verwendet und die erfolgte
Mutagenese durch Sequenzanalyse überprüft. Neben dem gewünschten Austausch Y148A
enthielt das Plasmid pAd-CMV-PrP∆32-93 & Y148A die weitere Mutation E151A. Die
Sequenzen der beiden Plasmide pAd-CMV-PrP∆32-93 & E151A und pAd-CMV-PrP∆32-93 &
H176A bestätigten die gewünschten Mutationen. Alle drei PrP-Mutanten wurden nach der
Transfektion von 293T-Zellen exprimiert (Daten nicht gezeigt). Die durchflusszytometrische
Analyse zeigte, dass Alanin in den Positionen 148, 151 und 176 weder die Bindung der
Helix 1-bindenden noch die Bindung der β-Faltblatt 2-bindenden Antikörper beeinflusste (Daten
nicht gezeigt), so dass das Minimalepitop der Antikörper nicht näher bestimmt werden konnte.

ERGEBNISSE
84
oktarepeats GPIß1 ß2
N C
GPIS-S
STE TM H1 H2 H3ββββ1 ββββ2Oktarepeats
23 231
Y148A H176A
E151A
oktarepeats GPIß1 ß2
N C
GPIS-S
STE TM H1 H2 H3ββββ1 ββββ2Oktarepeats
23 231
Y148A H176A
E151A
Abbildung 18: Schematische Darstellung von Maus-PrP C und der drei Substitutionen für die Bestimmung des Minimalepitops. Bei den ersten beiden Konstrukten wurde jeweils eine Aminosäure im Bereich der Helix 1 (Reste 143-153) durch ein Alanin ausgetauscht (pAd-CMV-PrP∆32-93 & Y148A und pAd-CMV-PrP∆32-93 & E151A). Eine weitere Aminosäure an Position 176, kurz vor der Helix 2 (Reste 179-193), wurde ebenfalls in ein Alanin mutiert (pAd-CMV-PrP∆32-93 & H176A) (H1-3 = α-Helix 1-3, β1-2 = β-Faltblatt 1-2, STE = Stopp-Transfer-Effektor-Sequenz, TM = Transmembrandomäne, S-S = Disulfidbrücke, GPI = Glycosylphosphatidylinositol-Anker).
3.2.3 Avidität der Anti-PrP-Antikörper
Die Avidität ist ein Maß für die Gesamtstärke aller Antigen-Antikörper-Bindungen und ist
abhängig von der Valenz des Antikörpers und des Antigens. Zur Bestimmung der Avidität wurde
ein indirekter ELISA gegen rekombinantes PrP durchgeführt und ein Ammoniumthiocyanat
(NH4SCN)-Elutionsschritt zwischengeschaltet. Je avider ein Antikörper ist, desto höhere
Molaritäten von NH4SCN waren notwendig, um die Antigen-Antikörper-Interaktionen zu lösen
(Abbildung 19). Der Aviditätsindex (AI) wurde als Molarität von NH4SCN angegeben, bei der
das Signal um 50% reduziert wurde (Tabelle 10).
Die höchsten Aviditätsindizes wurden für die Gruppe der Helix 1-bindenden Antikörper
bestimmt. Beim Antikörper 103-8 war für eine 50%ige Reduktion der Absorption eine
1,37 molare NH4SCN-Lösung notwendig und bei den Antikörpern 110-10 und B2-43-133 eine
Molarität von 0,67 und 0,57. Der Referenzantikörper ICSM18 (Beringue et al., 2004) zeigte mit
2,45 M den höchsten Aviditätsindex, während bei einer NH4SCN-Molarität von 0,94 50% der
Interaktionen mit dem Referenzantikörper 6H4 aufgehoben wurden. Die Bindungen der
β-Faltblatt 2- und STE-bindenden Antikörper waren dagegen weniger avid. Bei diesen
Antikörpern, zu denen auch der Referenzantikörper ICSM35 (Beringue et al., 2004) gehörte,
waren bereits Molaritäten kleiner als 0,5 M ausreichend, um 50% der Interaktionen aufzuheben.
Die Helix 1-bindenden Antikörper besitzen somit in der vorgenommenen Untersuchung die
höchste Avidität.
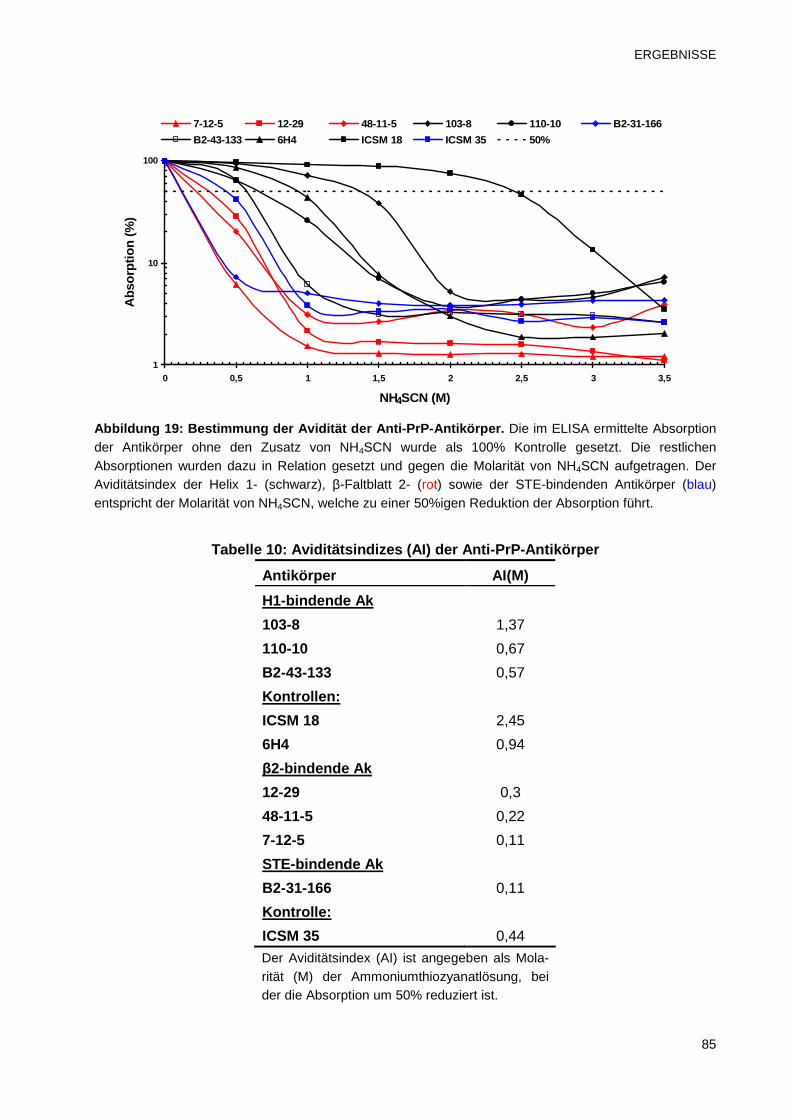
ERGEBNISSE
85
1
10
100
0 0,5 1 1,5 2 2,5 3 3,5
NH SCN (M)
Abs
orpt
ion
(%)
7-12-5 12-29 48-11-5 103-8 110-10 B2-31-166
B2-43-133 6H4 ICSM 18 ICSM 35 50%
4
1
10
100
0 0,5 1 1,5 2 2,5 3 3,5
NH SCN (M)
Abs
orpt
ion
(%)
7-12-5 12-29 48-11-5 103-8 110-10 B2-31-166
B2-43-133 6H4 ICSM 18 ICSM 35 50%
4
Abbildung 19: Bestimmung der Avidität der Anti-PrP- Antikörper. Die im ELISA ermittelte Absorption der Antikörper ohne den Zusatz von NH4SCN wurde als 100% Kontrolle gesetzt. Die restlichen Absorptionen wurden dazu in Relation gesetzt und gegen die Molarität von NH4SCN aufgetragen. Der Aviditätsindex der Helix 1- (schwarz), β-Faltblatt 2- (rot) sowie der STE-bindenden Antikörper (blau) entspricht der Molarität von NH4SCN, welche zu einer 50%igen Reduktion der Absorption führt.
Tabelle 10: Aviditätsindizes (AI) der Anti-PrP-Anti körper
Antikörper AI(M)
H1-bindende Ak
103-8 1,37
110-10 0,67
B2-43-133 0,57
Kontrollen:
ICSM 18 2,45
6H4 0,94
β2-bindende Ak
12-29 0,3
48-11-5 0,22
7-12-5 0,11
STE-bindende Ak
B2-31-166 0,11
Kontrolle:
ICSM 35 0,44
Der Aviditätsindex (AI) ist angegeben als Mola-rität (M) der Ammoniumthiozyanatlösung, bei der die Absorption um 50% reduziert ist.

ERGEBNISSE
86
3.2.4 Dissoziationskonstanten (K D) der Anti-PrP-Antikörper
Die Affinität ist im Gegensatz zur Avidität ein Maß für die Stärke einer monovalenten Bindung
zwischen Antikörper und Antigen. Über die Affinität eines Antikörpers gibt die
Dissoziationskonstante KD Auskunft. Dabei gilt: Je höher die Affinität eines Antikörpers zu
seinem Antigen ist, desto niedriger ist die KD-Konstante. In der vorliegenden Arbeit wurden die
KD-Konstanten auf zwei unterschiedlichen Substraten mit zwei verschiedenen Methoden
bestimmt. Für beide Bestimmungen wurde die KD-Konstante aus einer Sättigungskurve und
einem Scatchard-Diagramm mittels PRISM®- Software ermittelt. In Tabelle 11 sind die
KD-Konstanten als Mittelwerte mit Standardabweichungen von drei Versuchen
zusammengefasst.
Alle Anti-PrP-Antikörper zeigen eine ähnliche Affinität zu rekombinantem PrP, die mit den
beiden Referenzantikörpern 6H4 und ICSM18 vergleichbar ist. Es wurde kein signifikanter
Unterschied zwischen Helix 1- und β-Faltblatt 2-bindenden Antikörpern festgestellt. Im Vergleich
zum Antikörper 6H4, welcher die niedrigste KD-Konstante besitzt (0,001 pmol), ist die Bindung
des Antikörpers B2-43-133 an rekombinantes PrP 16-fach weniger affin (0,0158 pmol), jedoch
liegen beide Dissoziationskonstanten im pikomolaren Bereich.
Tabelle 11: K D-Konstanten der Anti-PrP-Antikörper
KD (pmol)
Methode ELISA FACS
Substrat rek. PrP natives PrP Sc
H1-bindende Ak
103-8 0,0067 ± 0,0006 4,9 ± 0,125
110-10 0,0032 ± 0,0002 /
B2-43-133 0,0158 ± 0,0018 0,49 ± 0,055
Kontrollen
6H4 0,001 ± 0,0006 0,049 ± 0,004
ICSM 18 0,0053 ± 0,0005 0,7 ± 0,14
ββββ2-bindende Ak
7-12-5 0,0035 ± 0,0004 /
48-11-5 0,0039 ± 0,0003 1,7 ± 0,27
Die KD-Konstanten, die auf nativem PrP von ScN2a-Zellen mittels FACS-Analyse ermittelt
wurden, sind dagegen 50- bis 300-fach höher. Dieser Unterschied kann auf methodische
Ursachen oder ein verändertes Bindeverhalten der Antikörper an PrPC auf der Oberfläche von
Zellen zurückgeführt werden. Auch bei dieser Bestimmung besitzt der Referenzantikörper 6H4

ERGEBNISSE
87
die kleinste KD-Konstante (0,049 pmol). Im Vergleich dazu ist die Affinität der Antikörper
B2-43-133 und 103-8 10-bzw. 100-fach geringer. Mit einer KD-Konstante von 1,7 pmol ist die
Bindung des β-Faltblatt 2-bindenden Antikörpers 48-11-5 deutlich affiner als die des Antikörpers
103-8, jedoch auch weniger affin als die des Antikörpers B2-43-133.
Die Affinität aller Anti-PrP-Antikörper zu rekombinantem PrP ist somit relativ einheitlich,
während sich auf ScN2a-Zellen bis zu 100-fache Unterschiede zeigen.
3.3 Hemmung der Prionreplikation durch Anti-PrP-Ant ikörper in vitro
Ein wichtiges Ziel dieser Arbeit war die Herstellung von Anti-PrP-Antikörpern mit
therapeutischem Potential, die Prionreplikation in Zellkultur zu hemmen.
3.3.1 Einfluss der Anti-PrP-Antikörper auf die PrP Sc-Akkumulation in ScN2a-Zellen
Um den Einfluss der Anti-PrP-Antikörper auf die PrPSc-Akkumulation in ScN2a-Zellen zu
untersuchen, wurde nach einer fünftägigen Behandlung der Zellen mit den monoklonalen
Antikörpern (10 µg/ml) der PrPSc-Gehalt bestimmt. Die in Abbildung 20 dargestellten Resultate
wurden mehrfach reproduziert. Nach Behandlung mit den Helix 1-bindenden Antikörpern 103-8,
110-10 und B2-43-133 erfolgte eine vollständige Reduktion des PrPSc-Gehalts, die ebenfalls für
den Referenzantikörper 6H4 detektiert wurde. Der β-Faltblatt 2-bindende Antikörper 48-11-5
zeigte nur einen unvollständigen inhibitorischen Effekt, während die beiden β-Faltblatt 2-
bindenden Antikörper 7-12-5 und 12-29 sowie der STE-bindende Antikörper B2-31-166 die
PrPSc-Akkumulation in ScN2a-Zellen nicht beeinflussten. Eine Behandlung mit dem Anti-NCAM
Antikörper wirkte sich ebenfalls nicht auf den PrPSc-Gehalt der ScN2a-Zellen aus, da dieser
Antikörper nicht an PrPC bindet, sondern an das neurale Zelladhäsionsmolekül (NCAM), das
hauptsächlich im zentralen und peripheren Nervensystem exprimiert wird (Abbildung 20).
Um den Effekt zu quantifizieren, wurde die Intensität der PrPSc-Signale densitometrisch
bestimmt. Die Signalintensität der unbehandelten Zellen (MOCK) wurde als 100%-Wert gesetzt.
Nach der Behandlung mit den Antikörpern 103-8, 110-10 und B2-43-133 wurde der PrPSc-
Gehalt der Zellen um 95%, 93% und 99% reduziert. Diese Werte sind mit dem
Referenzantikörper 6H4 vergleichbar, dessen inhibitorische Wirkung bereits demonstriert wurde
(Enari et al., 2001; Heppner et al., 2001) und der in der vorliegenden Arbeit den PrPSc-Gehalt
um 97% reduzierte (Abbildung 20). Die unvollständig inhibitorische Wirkung des Antikörpers 48-
11-5 führte zu einer Reduktion von 73% des PrPSc-Gehalts. In ScN2a-Zellen, die mit den
Antikörpern 7-12-5, 12-29 und B2-31-166 behandelt wurden, konnte noch ein PrPSc-Gehalt von
74%, 74% und 56% detektiert werden.
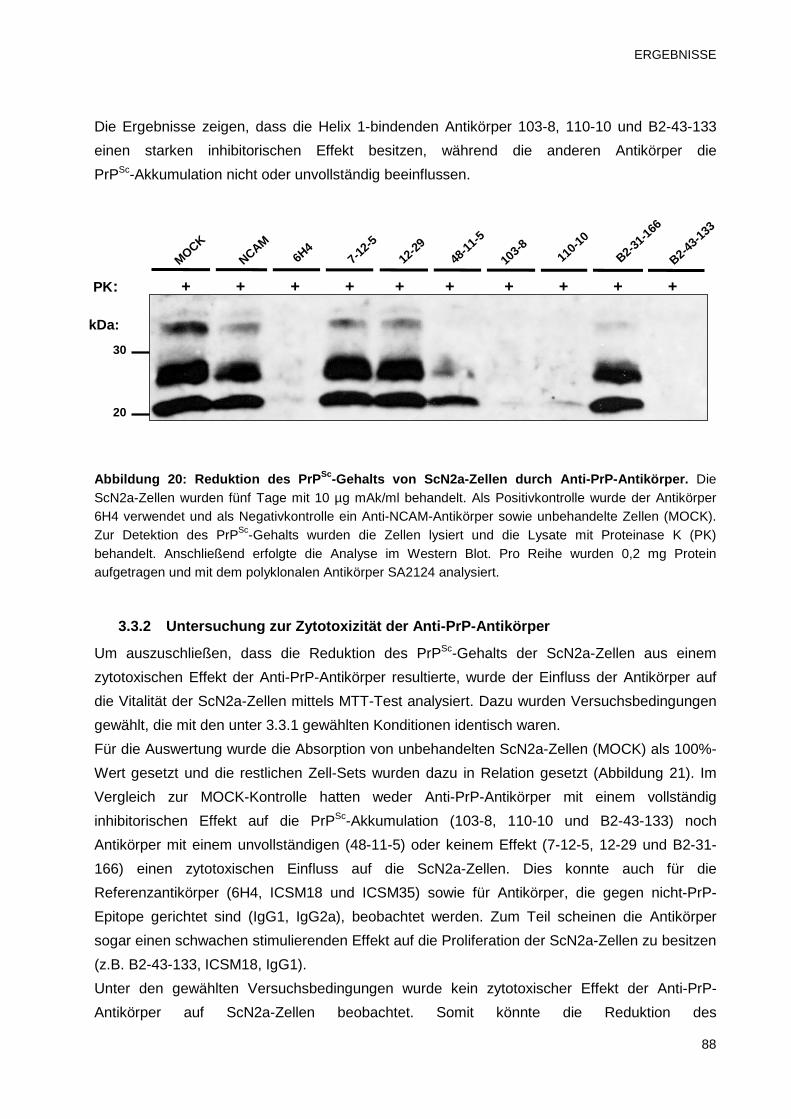
ERGEBNISSE
88
20
30
PK: + + + + + + + + + +
48-1
1-5
103-
811
0-10
B2-43
-133
7-12
-56H
4MOCK
12-2
9NCAM
B2-31
-166
kDa:
20
30
PK: + + + + + + + + + +
48-1
1-5
103-
811
0-10
B2-43
-133
7-12
-56H
4MOCK
12-2
9NCAM
B2-31
-166
kDa:
Die Ergebnisse zeigen, dass die Helix 1-bindenden Antikörper 103-8, 110-10 und B2-43-133
einen starken inhibitorischen Effekt besitzen, während die anderen Antikörper die
PrPSc-Akkumulation nicht oder unvollständig beeinflussen.
Abbildung 20: Reduktion des PrP Sc-Gehalts von ScN2a-Zellen durch Anti-PrP-Antikörper . Die ScN2a-Zellen wurden fünf Tage mit 10 µg mAk/ml behandelt. Als Positivkontrolle wurde der Antikörper 6H4 verwendet und als Negativkontrolle ein Anti-NCAM-Antikörper sowie unbehandelte Zellen (MOCK). Zur Detektion des PrPSc-Gehalts wurden die Zellen lysiert und die Lysate mit Proteinase K (PK) behandelt. Anschließend erfolgte die Analyse im Western Blot. Pro Reihe wurden 0,2 mg Protein aufgetragen und mit dem polyklonalen Antikörper SA2124 analysiert.
3.3.2 Untersuchung zur Zytotoxizität der Anti-PrP-A ntikörper
Um auszuschließen, dass die Reduktion des PrPSc-Gehalts der ScN2a-Zellen aus einem
zytotoxischen Effekt der Anti-PrP-Antikörper resultierte, wurde der Einfluss der Antikörper auf
die Vitalität der ScN2a-Zellen mittels MTT-Test analysiert. Dazu wurden Versuchsbedingungen
gewählt, die mit den unter 3.3.1 gewählten Konditionen identisch waren.
Für die Auswertung wurde die Absorption von unbehandelten ScN2a-Zellen (MOCK) als 100%-
Wert gesetzt und die restlichen Zell-Sets wurden dazu in Relation gesetzt (Abbildung 21). Im
Vergleich zur MOCK-Kontrolle hatten weder Anti-PrP-Antikörper mit einem vollständig
inhibitorischen Effekt auf die PrPSc-Akkumulation (103-8, 110-10 und B2-43-133) noch
Antikörper mit einem unvollständigen (48-11-5) oder keinem Effekt (7-12-5, 12-29 und B2-31-
166) einen zytotoxischen Einfluss auf die ScN2a-Zellen. Dies konnte auch für die
Referenzantikörper (6H4, ICSM18 und ICSM35) sowie für Antikörper, die gegen nicht-PrP-
Epitope gerichtet sind (IgG1, IgG2a), beobachtet werden. Zum Teil scheinen die Antikörper
sogar einen schwachen stimulierenden Effekt auf die Proliferation der ScN2a-Zellen zu besitzen
(z.B. B2-43-133, ICSM18, IgG1).
Unter den gewählten Versuchsbedingungen wurde kein zytotoxischer Effekt der Anti-PrP-
Antikörper auf ScN2a-Zellen beobachtet. Somit könnte die Reduktion des
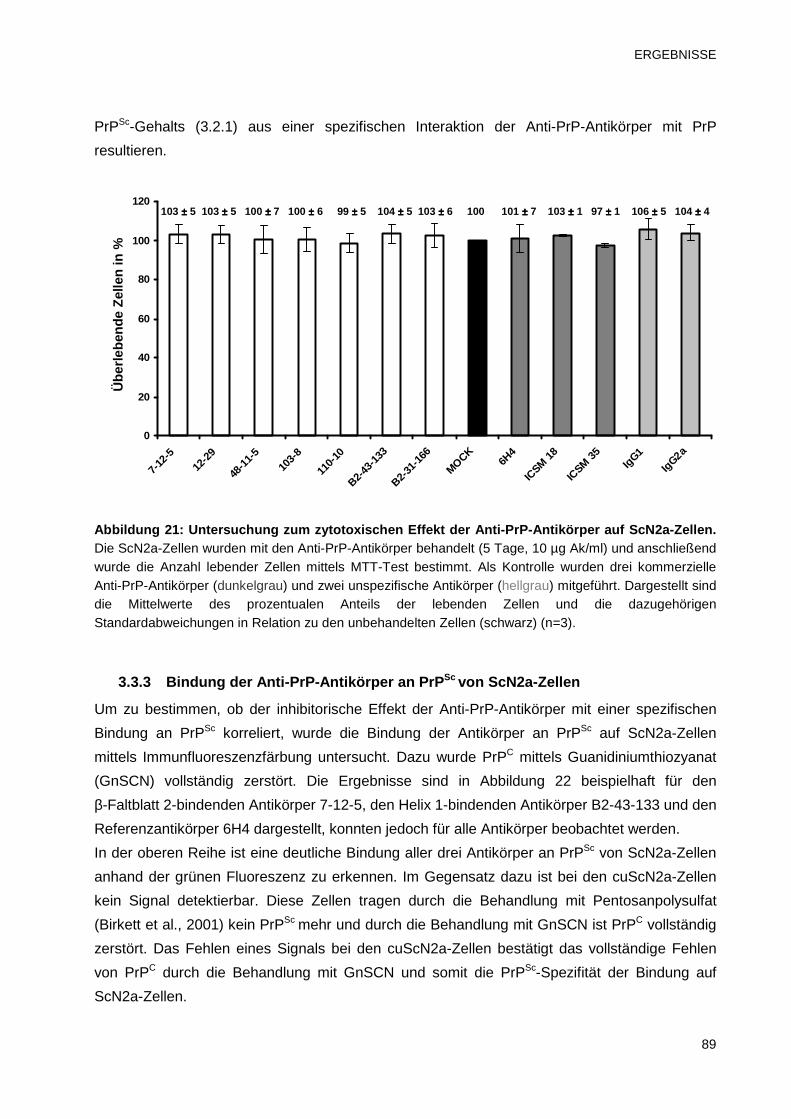
ERGEBNISSE
89
0
20
40
60
80
100
120
7-12
-512
-29
48-1
1-5
103-
8
110-
10
B2-43
-133
B2-31
-166
MOCK6H
4
ICSM
18
ICSM
35Ig
G1Ig
G2a
Übe
rlebe
nde
Zelle
n in
%
103 ±±±± 5 103 ±±±± 5 100 ±±±± 7 100 ±±±± 6 99 ±±±± 5 104 ±±±± 5 103 ±±±± 6 100 101 ±±±± 7 103 ±±±± 1 97 ±±±± 1 106 ±±±± 5 104 ±±±± 4
Übe
rlebe
nde
Zel
len
in %
0
20
40
60
80
100
120
7-12
-512
-29
48-1
1-5
103-
8
110-
10
B2-43
-133
B2-31
-166
MOCK6H
4
ICSM
18
ICSM
35Ig
G1Ig
G2a
Übe
rlebe
nde
Zelle
n in
%
103 ±±±± 5 103 ±±±± 5 100 ±±±± 7 100 ±±±± 6 99 ±±±± 5 104 ±±±± 5 103 ±±±± 6 100 101 ±±±± 7 103 ±±±± 1 97 ±±±± 1 106 ±±±± 5 104 ±±±± 4
Übe
rlebe
nde
Zel
len
in %
PrPSc-Gehalts (3.2.1) aus einer spezifischen Interaktion der Anti-PrP-Antikörper mit PrP
resultieren.
Abbildung 21: Untersuchung zum zytotoxischen Effekt der Anti-PrP-Antikörper auf ScN2a-Zellen. Die ScN2a-Zellen wurden mit den Anti-PrP-Antikörper behandelt (5 Tage, 10 µg Ak/ml) und anschließend wurde die Anzahl lebender Zellen mittels MTT-Test bestimmt. Als Kontrolle wurden drei kommerzielle Anti-PrP-Antikörper (dunkelgrau) und zwei unspezifische Antikörper (hellgrau) mitgeführt. Dargestellt sind die Mittelwerte des prozentualen Anteils der lebenden Zellen und die dazugehörigen Standardabweichungen in Relation zu den unbehandelten Zellen (schwarz) (n=3).
3.3.3 Bindung der Anti-PrP-Antikörper an PrP Sc von ScN2a-Zellen
Um zu bestimmen, ob der inhibitorische Effekt der Anti-PrP-Antikörper mit einer spezifischen
Bindung an PrPSc korreliert, wurde die Bindung der Antikörper an PrPSc auf ScN2a-Zellen
mittels Immunfluoreszenzfärbung untersucht. Dazu wurde PrPC mittels Guanidiniumthiozyanat
(GnSCN) vollständig zerstört. Die Ergebnisse sind in Abbildung 22 beispielhaft für den
β-Faltblatt 2-bindenden Antikörper 7-12-5, den Helix 1-bindenden Antikörper B2-43-133 und den
Referenzantikörper 6H4 dargestellt, konnten jedoch für alle Antikörper beobachtet werden.
In der oberen Reihe ist eine deutliche Bindung aller drei Antikörper an PrPSc von ScN2a-Zellen
anhand der grünen Fluoreszenz zu erkennen. Im Gegensatz dazu ist bei den cuScN2a-Zellen
kein Signal detektierbar. Diese Zellen tragen durch die Behandlung mit Pentosanpolysulfat
(Birkett et al., 2001) kein PrPSc mehr und durch die Behandlung mit GnSCN ist PrPC vollständig
zerstört. Das Fehlen eines Signals bei den cuScN2a-Zellen bestätigt das vollständige Fehlen
von PrPC durch die Behandlung mit GnSCN und somit die PrPSc-Spezifität der Bindung auf
ScN2a-Zellen.
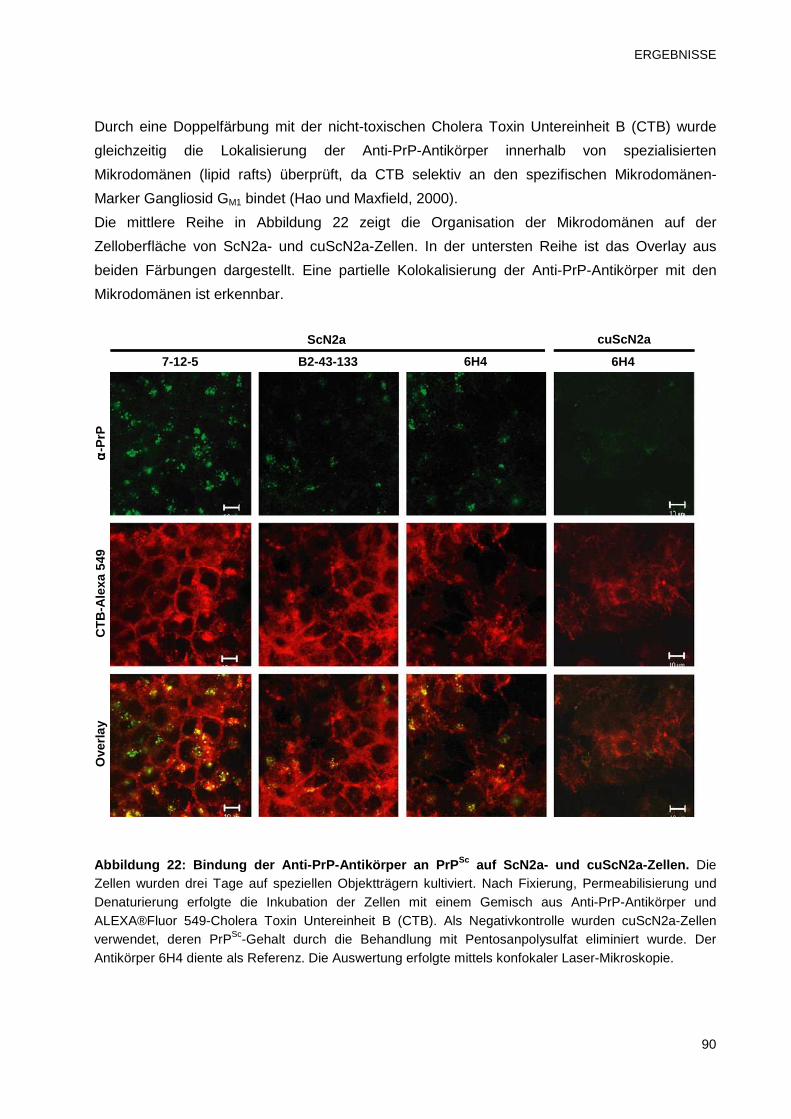
ERGEBNISSE
90
7-12-5 B2-43-133 6H4 6H4
cuScN2aScN2a
αα αα-P
rPC
TB
-Ale
xa54
9O
verla
y
7-12-5 B2-43-133 6H4 6H4
cuScN2aScN2a
αα αα-P
rPC
TB
-Ale
xa54
9O
verla
y
Durch eine Doppelfärbung mit der nicht-toxischen Cholera Toxin Untereinheit B (CTB) wurde
gleichzeitig die Lokalisierung der Anti-PrP-Antikörper innerhalb von spezialisierten
Mikrodomänen (lipid rafts) überprüft, da CTB selektiv an den spezifischen Mikrodomänen-
Marker Gangliosid GM1 bindet (Hao und Maxfield, 2000).
Die mittlere Reihe in Abbildung 22 zeigt die Organisation der Mikrodomänen auf der
Zelloberfläche von ScN2a- und cuScN2a-Zellen. In der untersten Reihe ist das Overlay aus
beiden Färbungen dargestellt. Eine partielle Kolokalisierung der Anti-PrP-Antikörper mit den
Mikrodomänen ist erkennbar.
Abbildung 22: Bindung der Anti-PrP-Antikörper an Pr PSc auf ScN2a- und cuScN2a-Zellen. Die Zellen wurden drei Tage auf speziellen Objektträgern kultiviert. Nach Fixierung, Permeabilisierung und Denaturierung erfolgte die Inkubation der Zellen mit einem Gemisch aus Anti-PrP-Antikörper und ALEXA®Fluor 549-Cholera Toxin Untereinheit B (CTB). Als Negativkontrolle wurden cuScN2a-Zellen verwendet, deren PrPSc-Gehalt durch die Behandlung mit Pentosanpolysulfat eliminiert wurde. Der Antikörper 6H4 diente als Referenz. Die Auswertung erfolgte mittels konfokaler Laser-Mikroskopie.
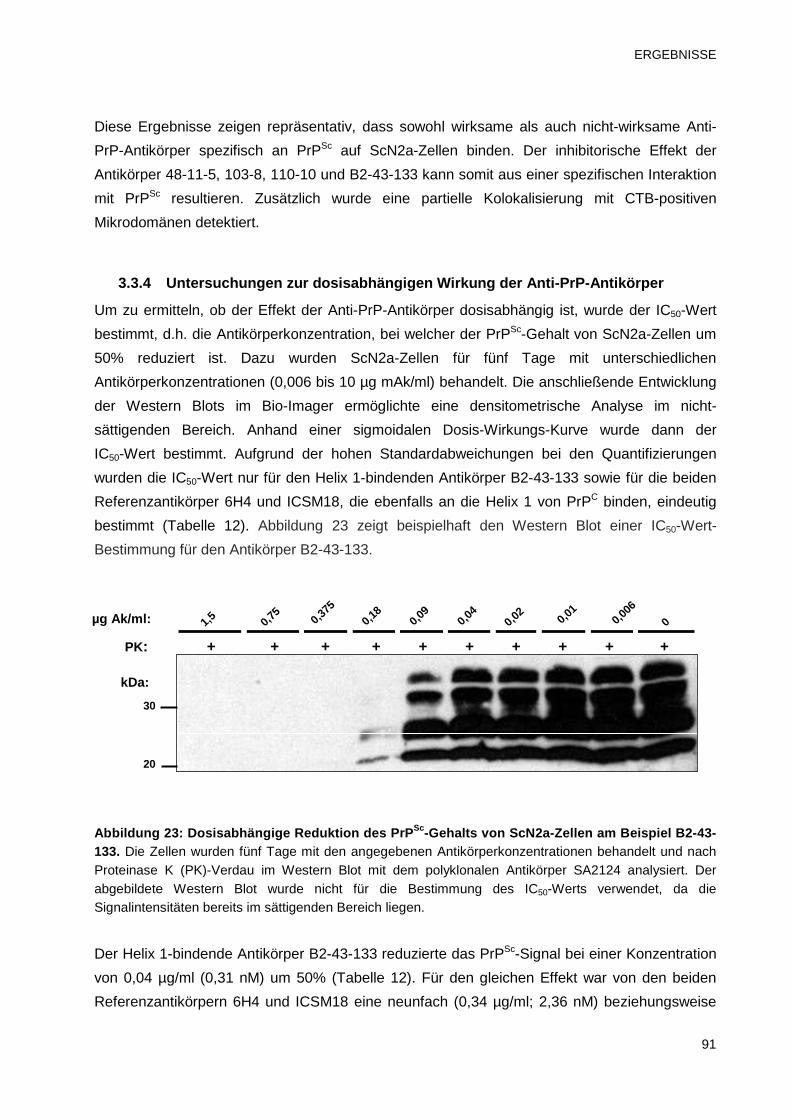
ERGEBNISSE
91
20
30
PK: + + + + + + + + + +
0,04
0,02
0,01
00,18
0,375
1,5 0,09
0,75
0,006
kDa:
µg Ak/ml:
20
30
PK: + + + + + + + + + +
0,04
0,02
0,01
00,18
0,375
1,5 0,09
0,75
0,006
kDa:
µg Ak/ml:
Diese Ergebnisse zeigen repräsentativ, dass sowohl wirksame als auch nicht-wirksame Anti-
PrP-Antikörper spezifisch an PrPSc auf ScN2a-Zellen binden. Der inhibitorische Effekt der
Antikörper 48-11-5, 103-8, 110-10 und B2-43-133 kann somit aus einer spezifischen Interaktion
mit PrPSc resultieren. Zusätzlich wurde eine partielle Kolokalisierung mit CTB-positiven
Mikrodomänen detektiert.
3.3.4 Untersuchungen zur dosisabhängigen Wirkung de r Anti-PrP-Antikörper
Um zu ermitteln, ob der Effekt der Anti-PrP-Antikörper dosisabhängig ist, wurde der IC50-Wert
bestimmt, d.h. die Antikörperkonzentration, bei welcher der PrPSc-Gehalt von ScN2a-Zellen um
50% reduziert ist. Dazu wurden ScN2a-Zellen für fünf Tage mit unterschiedlichen
Antikörperkonzentrationen (0,006 bis 10 µg mAk/ml) behandelt. Die anschließende Entwicklung
der Western Blots im Bio-Imager ermöglichte eine densitometrische Analyse im nicht-
sättigenden Bereich. Anhand einer sigmoidalen Dosis-Wirkungs-Kurve wurde dann der
IC50-Wert bestimmt. Aufgrund der hohen Standardabweichungen bei den Quantifizierungen
wurden die IC50-Wert nur für den Helix 1-bindenden Antikörper B2-43-133 sowie für die beiden
Referenzantikörper 6H4 und ICSM18, die ebenfalls an die Helix 1 von PrPC binden, eindeutig
bestimmt (Tabelle 12). Abbildung 23 zeigt beispielhaft den Western Blot einer IC50-Wert-
Bestimmung für den Antikörper B2-43-133.
Abbildung 23: Dosisabhängige Reduktion des PrP Sc-Gehalts von ScN2a-Zellen am Beispiel B2-43-133. Die Zellen wurden fünf Tage mit den angegebenen Antikörperkonzentrationen behandelt und nach Proteinase K (PK)-Verdau im Western Blot mit dem polyklonalen Antikörper SA2124 analysiert. Der abgebildete Western Blot wurde nicht für die Bestimmung des IC50-Werts verwendet, da die Signalintensitäten bereits im sättigenden Bereich liegen.
Der Helix 1-bindende Antikörper B2-43-133 reduzierte das PrPSc-Signal bei einer Konzentration
von 0,04 µg/ml (0,31 nM) um 50% (Tabelle 12). Für den gleichen Effekt war von den beiden
Referenzantikörpern 6H4 und ICSM18 eine neunfach (0,34 µg/ml; 2,36 nM) beziehungsweise

ERGEBNISSE
92
zweifach (0,09 µg/ml; 0,625 nM) höhere Konzentration notwendig. Obwohl keine quantitative
Bestimmung der IC50-Werte der Antikörper 103-8 und 110-10 erfolgte, lässt sich aus den
erhaltenen Ergebnissen ableiten, dass für eine 50%ige Reduktion des PrPSc-Signals eine
höhere Konzentration als 0,34 µg/ml notwendig war (Daten nicht gezeigt).
Tabelle 12: IC 50-Werte der Anti-PrP-Antikörper
mAk IC 50-Wert (µg/ml) 1 IC50-Wert (nM)
B2-43-133 0,04±0,01 0,31±0,07
6H4
ICSM 18
0,34±0,14
0,09±0,02
2,36±0,97
0,63±0,14 1 Die IC50-Werte wurden aus drei unabhängig voneinander durchge- führten Experimenten ermittelt.
Somit ist in dieser Untersuchung für eine 50%ige Reduktion des PrPSc-Gehalts von dem Helix
1-bindenden Antikörper B2-43-133 im Vergleich zu den anderen untersuchten Antikörpern die
geringste Konzentration notwendig.
3.3.5 Untersuchungen zur zeitabhängigen Wirkung der Anti-PrP-Antikörper
Da in den zuvor beschriebenen Experimenten die Behandlungsdauer auf fünf Tage beschränkt
war, sollte im nächsten Schritt untersucht werden, ob die Verlängerung des
Behandlungszeitraums auf 30 Tage den Effekt der Anti-PrP-Antikörper verstärkt. Diese
Untersuchung ist besonders interessant im Hinblick auf den Antikörper 48-11-5, der nach fünf
Tagen Applikation die PrPSc-Akkumulation nur unvollständig inhibierte.
Der PrPSc-Gehalt der unbehandelten ScN2a-Zellen (MOCK) und der Zellen, die mit einem
Kontrollantikörper behandelt wurden (IgG1), blieb über den gesamten Behandlungszeitraum
konstant bei 100% (Abbildung 24 und 25). Im Vergleich dazu reduzierte der Anti-PrP-Antikörper
48-11-5 nach fünf Tagen den PrPSc-Gehalt um ca. 80% (Abbildung 24). Diese Reduktion wurde
auch durch 24 weitere Behandlungstage nicht verstärkt, so dass nach 30 Tagen immer noch
20% des PrPSc-Gehalts detektiert wurden. Somit ist die Wirkung des Antikörpers 48-11-5 bei
einer konstanten Antikörperkonzentration nicht zeitabhängig.
Bei Abschluss der Behandlung der ScN2a-Zellen mit den Helix 1-bindenden Antikörpern wurde
kein PrPSc detektiert (Abbildung 25).
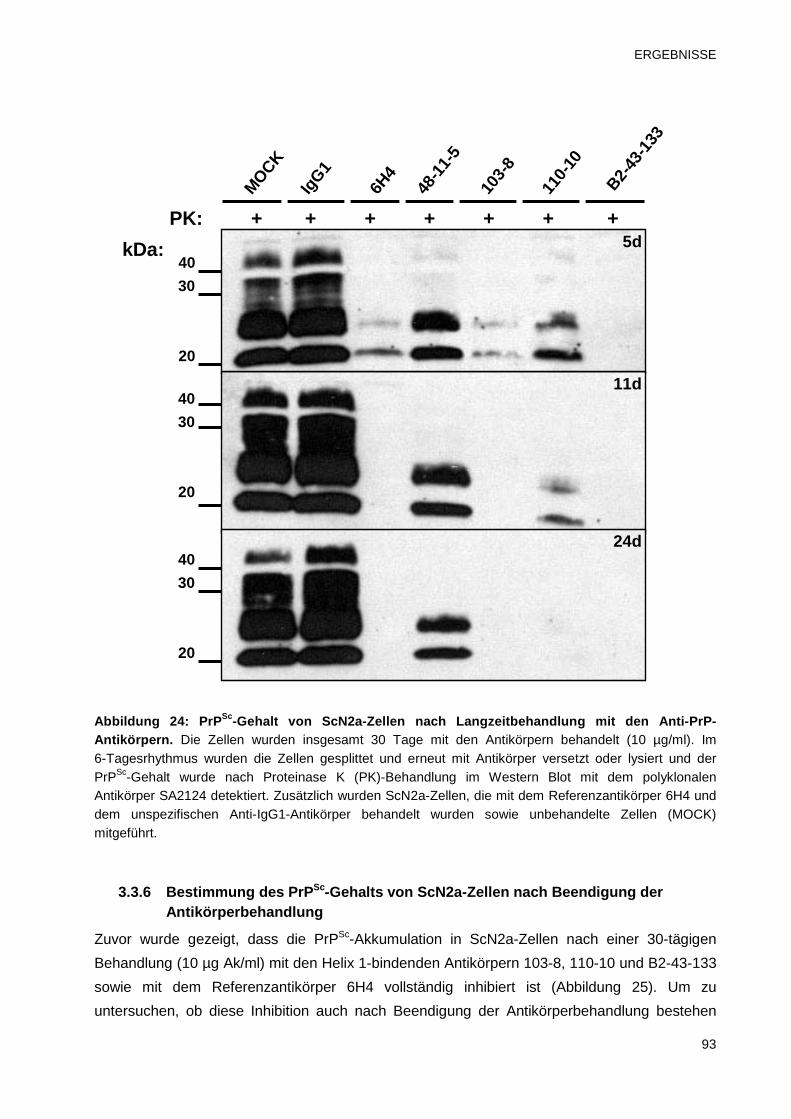
ERGEBNISSE
93
PK: + + + + + + +M
OCK
IgG
1
6H4
48-1
1-5
103-
8
110-
10
B2-43
-133
40
20
30
40
20
30
40
20
30
kDa: 5d
11d
24d
PK: + + + + + + +M
OCK
IgG
1
6H4
48-1
1-5
103-
8
110-
10
B2-43
-133
40
20
30
40
20
30
40
20
30
kDa: 5d
11d
24d
Abbildung 24: PrP Sc-Gehalt von ScN2a-Zellen nach Langzeitbehandlung mi t den Anti-PrP-Antikörpern. Die Zellen wurden insgesamt 30 Tage mit den Antikörpern behandelt (10 µg/ml). Im 6-Tagesrhythmus wurden die Zellen gesplittet und erneut mit Antikörper versetzt oder lysiert und der PrPSc-Gehalt wurde nach Proteinase K (PK)-Behandlung im Western Blot mit dem polyklonalen Antikörper SA2124 detektiert. Zusätzlich wurden ScN2a-Zellen, die mit dem Referenzantikörper 6H4 und dem unspezifischen Anti-IgG1-Antikörper behandelt wurden sowie unbehandelte Zellen (MOCK) mitgeführt.
3.3.6 Bestimmung des PrP Sc-Gehalts von ScN2a-Zellen nach Beendigung der Antikörperbehandlung
Zuvor wurde gezeigt, dass die PrPSc-Akkumulation in ScN2a-Zellen nach einer 30-tägigen
Behandlung (10 µg Ak/ml) mit den Helix 1-bindenden Antikörpern 103-8, 110-10 und B2-43-133
sowie mit dem Referenzantikörper 6H4 vollständig inhibiert ist (Abbildung 25). Um zu
untersuchen, ob diese Inhibition auch nach Beendigung der Antikörperbehandlung bestehen
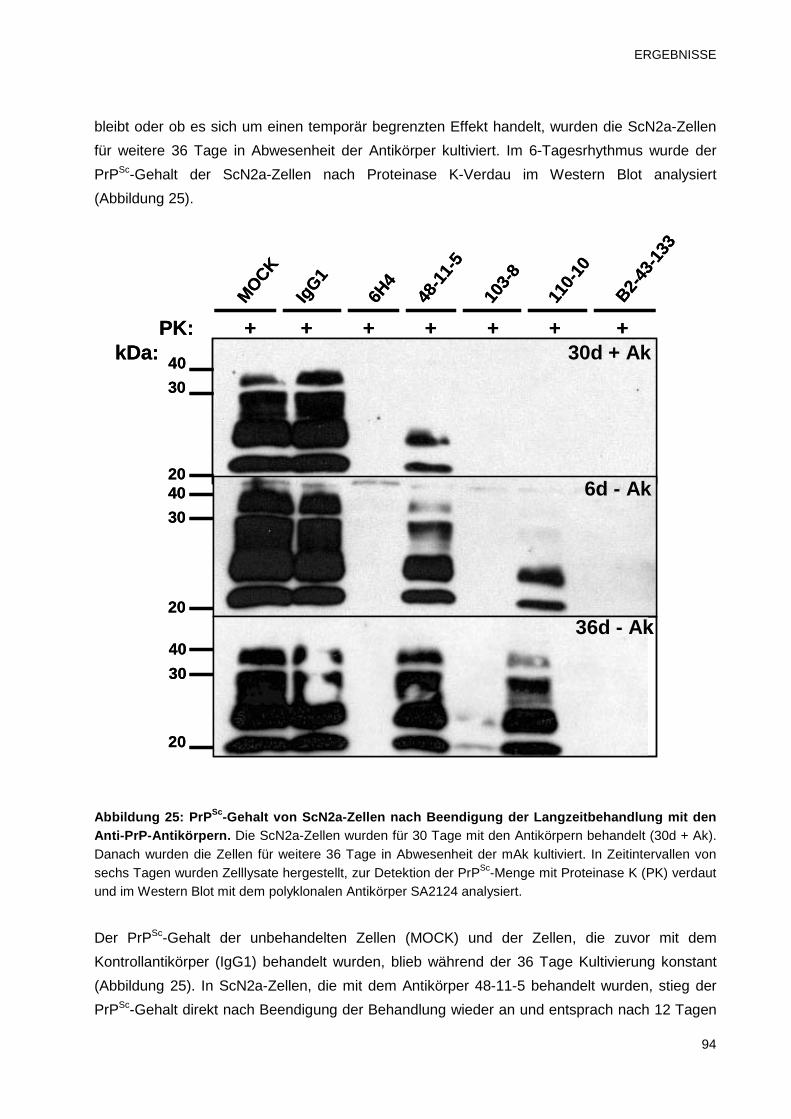
ERGEBNISSE
94
30d + AkPK: + + + + + + +
MOCK
IgG1
6H4
48-1
1-5
103-
8
110-
10
B2-43
-133
40
20
30
kDa:
5d
40
20
30
6d - Ak
36d - Ak
40
20
30
30d + AkPK: + + + + + + +
MOCK
IgG1
6H4
48-1
1-5
103-
8
110-
10
B2-43
-133
40
20
30
kDa:
5d
40
20
30
6d - Ak
36d - Ak
40
20
30
30d + AkPK: + + + + + + +
MOCK
IgG1
6H4
48-1
1-5
103-
8
110-
10
B2-43
-133
40
20
30
kDa:
5d
40
20
30
6d - Ak
36d - Ak
40
20
30
bleibt oder ob es sich um einen temporär begrenzten Effekt handelt, wurden die ScN2a-Zellen
für weitere 36 Tage in Abwesenheit der Antikörper kultiviert. Im 6-Tagesrhythmus wurde der
PrPSc-Gehalt der ScN2a-Zellen nach Proteinase K-Verdau im Western Blot analysiert
(Abbildung 25).
Abbildung 25: PrP Sc-Gehalt von ScN2a-Zellen nach Beendigung der Langze itbehandlung mit den Anti-PrP-Antikörpern. Die ScN2a-Zellen wurden für 30 Tage mit den Antikörpern behandelt (30d + Ak). Danach wurden die Zellen für weitere 36 Tage in Abwesenheit der mAk kultiviert. In Zeitintervallen von sechs Tagen wurden Zelllysate hergestellt, zur Detektion der PrPSc-Menge mit Proteinase K (PK) verdaut und im Western Blot mit dem polyklonalen Antikörper SA2124 analysiert.
Der PrPSc-Gehalt der unbehandelten Zellen (MOCK) und der Zellen, die zuvor mit dem
Kontrollantikörper (IgG1) behandelt wurden, blieb während der 36 Tage Kultivierung konstant
(Abbildung 25). In ScN2a-Zellen, die mit dem Antikörper 48-11-5 behandelt wurden, stieg der
PrPSc-Gehalt direkt nach Beendigung der Behandlung wieder an und entsprach nach 12 Tagen

ERGEBNISSE
95
der PrPSc-Menge in unbehandelten ScN2a-Zellen (Daten nicht gezeigt). In ScN2a-Zellen, in
denen zuvor die PrPSc-Akkumulation durch die Behandlung mit dem Helix 1-bindenden
Antikörper 110-10 vollständig inhibiert wurde, konnte nach sechs Tagen eine Zunahme des
PrPSc-Gehalts von ca. 20% detektiert werden, der nach weiteren 30 Tagen auf fast 100%
anstieg. Im Gegensatz dazu wurde in ScN2a-Zellen, die mit dem Antikörper 103-8 behandelt
wurden, erst nach 36 Tagen eine schwache Zunahme des PrPSc-Gehalts von 6% detektiert. Zu
diesem Zeitpunkt wurde in ScN2a-Zellen, die mit dem Antikörper B2-43-133 behandelt wurden,
weiterhin kein PrPSc detektiert. Dies wurde ebenfalls für den Referenzantikörper 6H4
beobachtet (Abbildung 25).
Diese Ergebnisse zeigen, dass innerhalb der Gruppe der Helix 1-bindenden Antikörper die
Antikörper 103-8 und 110-10 zwar die PrPSc-Akkumulation in ScN2a-Zellen scheinbar
vollständig inhibieren, jedoch beginnt die Akkumulation in Abwesenheit der Antikörper erneut,
so dass diese Antikörper nur einen temporär begrenzten inhibitorischen Effekt besitzen. Im
Gegensatz dazu führt eine Behandlung mit dem Antikörper B2-43-133 oder dem
Referenzantikörper 6H4 zu einer vollständigen Hemmung der Prionreplikation, da auch nach
36 Tagen ohne Behandlung kein PrPSc in den Zellen nachweisbar ist.
3.3.7 Analyse der Infektiosität von Antikörper-beha ndelten ScN2a-Zellen
Da es sich bei PrPSc nur um einen Surrogat-Marker handelt, besteht die Möglichkeit, dass
Antikörper-behandelte ScN2a-Zellen trotz nicht detektierbarem PrPSc-Gehalts weiterhin infektiös
sind. Um die Infektiosität von ScN2a-Zellen, die zuvor 30 Tage mit den Anti-PrP-Antikörpern
behandelt wurden (3.3.4), zu überprüfen, wurden Lysate aus 106 ScN2a-Zellen intrazerebral in
Tga20-Indikatormäuse (Fischer et al., 1996) inokuliert (Bioassay). In Abbildung 26 ist die
prozentuale Überlebensrate der verschiedenen Versuchsgruppen als Kaplan-Meyer-Kurve
dargestellt.
Bei ScN2a-Zellen handelt es sich um RML-infizierte N2a-Zellen. Um zellspezifische Einflüsse
auszuschließen, wurde einer Gruppe ein Lysat aus 106 uninfizierten N2a-Zellen injiziert. Von
diesen fünf Mäusen waren bei Versuchende noch vier Tiere gesund, während eine Maus nach
292 Tagen an einer unspezifischen Ursache verstarb. Somit können zellspezifische Ursachen
für die Mortalität der Mäuse ausgeschlossen werden.
Die durchschnittliche Inkubationszeit von Mäusen, denen 106 unbehandelten ScN2a-Zellen
injiziert wurden, lag bei 81±10 Tagen. Im Vergleich dazu beeinflusste die vorhergegangene
Behandlung der ScN2a-Zellen mit dem unvollständig wirksamen Antikörper 48-11-5 und dem
wirksamen Antikörper 110-10 die Inkubationszeiten der Tga20-Mäuse nicht (73±13 Tage und
84±7 Tage, Tabelle 13) und zeigte somit keinen signifikanten Einfluss auf die Infektiosität.
Dagegen verlängerte sich die Inkubationszeit der Mäuse nach Inokulation von nur
104 unbehandelten ScN2a-Zellen um ca. 30 Tage (110±7 Tage, P=0,0284; Log-Rank-Test)
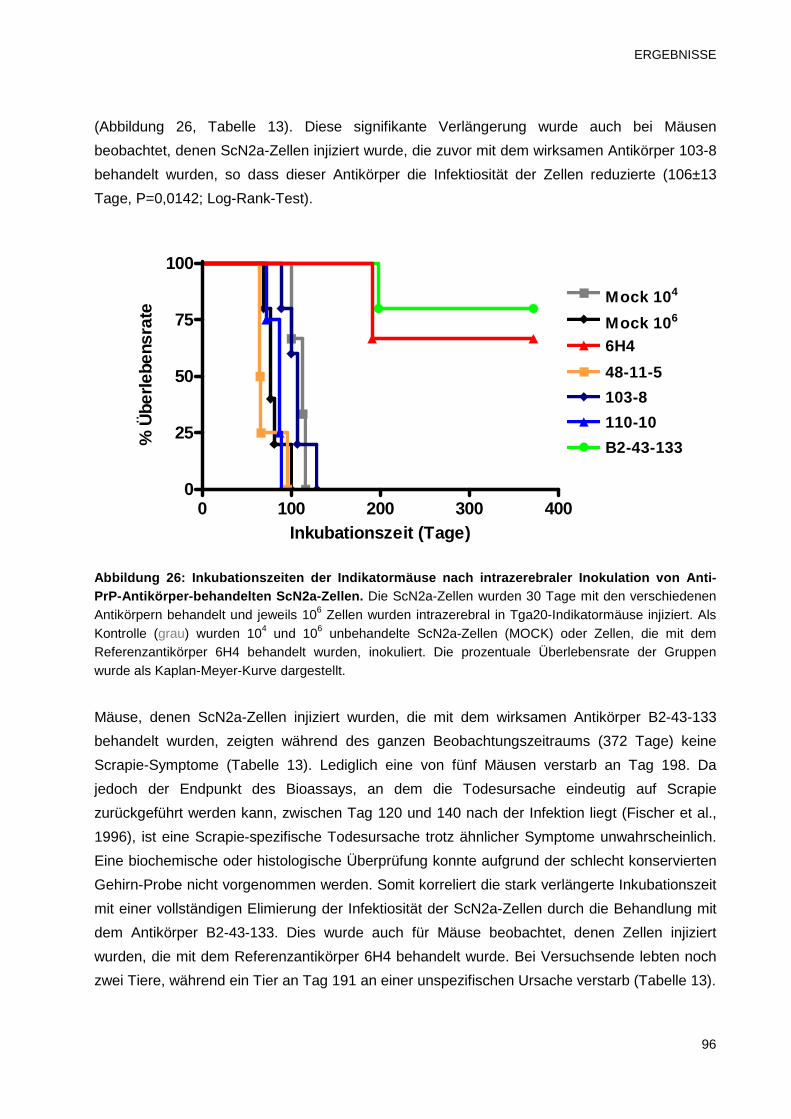
ERGEBNISSE
96
0 100 200 300 4000
25
50
75
100
Mock 10 4
Mock 10 6
48-11-5
103-8
110-10
B2-43-133
6H4
Inkubationszeit (Tage)
% Ü
berle
bens
rate
(Abbildung 26, Tabelle 13). Diese signifikante Verlängerung wurde auch bei Mäusen
beobachtet, denen ScN2a-Zellen injiziert wurde, die zuvor mit dem wirksamen Antikörper 103-8
behandelt wurden, so dass dieser Antikörper die Infektiosität der Zellen reduzierte (106±13
Tage, P=0,0142; Log-Rank-Test).
Abbildung 26: Inkubationszeiten der Indikatormäuse nach intrazerebraler Inokulation von Anti-PrP-Antikörper-behandelten ScN2a-Zellen. Die ScN2a-Zellen wurden 30 Tage mit den verschiedenen Antikörpern behandelt und jeweils 106 Zellen wurden intrazerebral in Tga20-Indikatormäuse injiziert. Als Kontrolle (grau) wurden 104 und 106 unbehandelte ScN2a-Zellen (MOCK) oder Zellen, die mit dem Referenzantikörper 6H4 behandelt wurden, inokuliert. Die prozentuale Überlebensrate der Gruppen wurde als Kaplan-Meyer-Kurve dargestellt.
Mäuse, denen ScN2a-Zellen injiziert wurden, die mit dem wirksamen Antikörper B2-43-133
behandelt wurden, zeigten während des ganzen Beobachtungszeitraums (372 Tage) keine
Scrapie-Symptome (Tabelle 13). Lediglich eine von fünf Mäusen verstarb an Tag 198. Da
jedoch der Endpunkt des Bioassays, an dem die Todesursache eindeutig auf Scrapie
zurückgeführt werden kann, zwischen Tag 120 und 140 nach der Infektion liegt (Fischer et al.,
1996), ist eine Scrapie-spezifische Todesursache trotz ähnlicher Symptome unwahrscheinlich.
Eine biochemische oder histologische Überprüfung konnte aufgrund der schlecht konservierten
Gehirn-Probe nicht vorgenommen werden. Somit korreliert die stark verlängerte Inkubationszeit
mit einer vollständigen Elimierung der Infektiosität der ScN2a-Zellen durch die Behandlung mit
dem Antikörper B2-43-133. Dies wurde auch für Mäuse beobachtet, denen Zellen injiziert
wurden, die mit dem Referenzantikörper 6H4 behandelt wurde. Bei Versuchsende lebten noch
zwei Tiere, während ein Tier an Tag 191 an einer unspezifischen Ursache verstarb (Tabelle 13).

ERGEBNISSE
97
Tabelle 13: Bioassay für die Bestimmung der Infekti osität von ScN2a-Zellen nach der
Behandlung mit Anti-PrP-Antikörpern
Inokulum
Zellzahl mAk
Mortalität der
Mäuse #
durchschnittliche
Inkubationszeit
(Tage post
Inokulation)
Signifikanz
(Log-Rank-Test)
Kontrollen
3/3 110 ± 7 P = 0,0284
5/5 81 ± 10 /
1*1/3 > 372 P = 0,0018§
104 ScN2a
106 ScN2a
106 ScN2a
106 N2a
/
/
6H4
/ 1*2/5 > 372 /
Helix 1-bindende Ak
5/5 106 ± 13 P = 0,0142
4/4 84 ± 7 nicht signifikant
106 ScN2a
106 ScN2a
106 ScN2a
103-8
110-10
B2-43-133 1*3/5 > 372 P = 0,0046§
ββββ-Faltblatt 2-bindender Ak
106 ScN2a 48-11-5 4/4 73 ± 13 nicht signifikant # n/n0 = Anzahl der an Scrapie verstorbenen Mäuse/Anzahl der inokulierten Mäuse. * = Mäuse, die an einer Scrapie-unabhängigen Ursache an Tag 191 (1), Tag 292 (2) und Tag 198 (3) verstarben. § = Der Bioassay wurde nach 372 Tage nach der Inokulation beendet. Diese Ergebnisse zeigen, dass die Behandlung von ScN2a-Zellen mit dem wirksamen
Antikörper 110-10 die Infektiosität der Zellen nicht beeinflusste, während der ebenfalls
wirksame Antikörper 103-8 die Infektiosität reduzierte. Nur der Antikörper B2-43-133 sowie der
der Referenzantikörper 6H4 hemmten die PrPSc-Akkumulation vollständig und eliminierten die
Infektiosität von ScN2a-Zellen komplett.
3.4 Untersuchungen zum Wirkungsmechanismus der Anti -PrP-Antikörper
Da bis heute unbekannt ist, welchen Mechanismus Anti-PrP-Antikörper für die Hemmung der
PrPSc-Akkumulation nutzen, wurden zwei Hypothesen bezüglich des Wirkungsmechanismusses
der Antikörper untersucht.
3.4.1 Untersuchungen zur inhibitorischen Wirkung ei nes PrP/Ak-Komplexes
Um zu untersuchen, ob der eigentliche inhibitorische Effekt der Anti-PrP-Antikörper durch eine
Komplexbildung mit freiem PrP vermittelt wird (Feraudet et al., 2005a), wurden ScN2a-Zellen
fünf Tage lang mit einem Komplex aus Antikörper und rekombinantem PrP (1 µg) behandelt.
Für die Untersuchung wurde der Helix 1-bindende Antikörper B2-43-133 ausgewählt, da er die
stärkste inhibitorische Wirkung zeigt (3.3) und den niedrigsten IC50-Wert besitzt (Tabelle 12).

ERGEBNISSE
98
Indem der Komplex mit verschiedenen Konzentrationen des Antikörpers im Bereich des
IC50-Werts gebildet wurde, konnte gleichzeitig eine mögliche dosisabhängige Wirkung des
Komplexes untersucht werden. Zum Vergleich wurden ScN2a-Zellen mit den identischen
Konzentrationen an freien Antikörpern behandelt. Die Bestimmung des PrPSc-Gehalts erfolgte
nach Proteinase K-Verdau der Lysate im Western Blot. Zusätzlich wurde der PrP/Antikörper-
Komplex nach Abschluss der Behandlung mittels Immunpräzipitation aus den Überständen
isoliert.
Ein direkter Einfluss von rekombinantem PrP auf den PrPSc-Gehalt von ScN2a-Zellen wurde
ausgeschlossen, da das PrPSc-Signal dieser Zellen der Intensität von unbehandelten Zellen
entsprach (Daten nicht gezeigt). Die Behandlung mit 0,18 µg B2-43-133/ml führte, wie bereits in
Abbildung 20 gezeigt, zu einer vollständigen Löschung des PrPSc-Signals (Abbildung 27A). Bei
ScN2a-Zellen, die mit dem PrP/Antikörper-Komplex behandelt wurden, konnte dieser Effekt erst
bei einer etwa achtfach höheren Antikörperkonzentration beobachtet werden (Abbildung 27A).
Erfolgte die Komplexbildung mit einer geringeren Menge von rekombinantem PrP (0,1 µg), war
für die vollständige Löschung des PrPSc-Signals eine Antikörperkonzentration von 0,37 µg/ml
ausreichend (Daten nicht gezeigt).
Nach Abschluss der Behandlung wurde der PrP/Antikörper-Komplex aus den Überständen der
ScN2a-Zellen isoliert, die mit den drei höchsten Konzentrationen des PrP/Antikörper-Komplexes
behandelt wurden (Abbildung 27B). Die 50 kDa-Bande repräsentiert die schwere Kette des
Antikörpers und die 20 kDa-Bande das rekombinante PrP, dessen Signalintensität proportional
zur verwendeten Antikörpermenge abnimmt. Die leichte Kette des Antikörpers (~25 kDa) konnte
aufgrund des verwendeten sekundären Antikörpers nicht detektiert werden. Da rekombinantes
PrP ohne vorhergegangene Komplexbildung nicht präzipitiert werden konnte (Abbildung 27B),
lag das rekombinante PrP in den untersuchten Überständen auch noch nach fünf Tagen als
stabiler Komplex vor.
Diese Ergebnisse zeigen, dass die Komplexbildung des Antikörpers B2-43-133 mit
rekombinantem PrP zu einer Reduktion der inhibitorischen Wirkung führt. Die Stabilität des
Komplexes über fünf Tage wurde mittels Immunpräzipitation bestätigt. Unter den gewählten
Versuchsbedigungen ist auszuschließen, dass die inhibitorische Wirkung der Anti-PrP-
Antikörper durch eine Komplexbildung vermittelt wird. Die Wirkung eines Komplexes aus
Antikörper und endogenem PrPC wurde nicht untersucht.
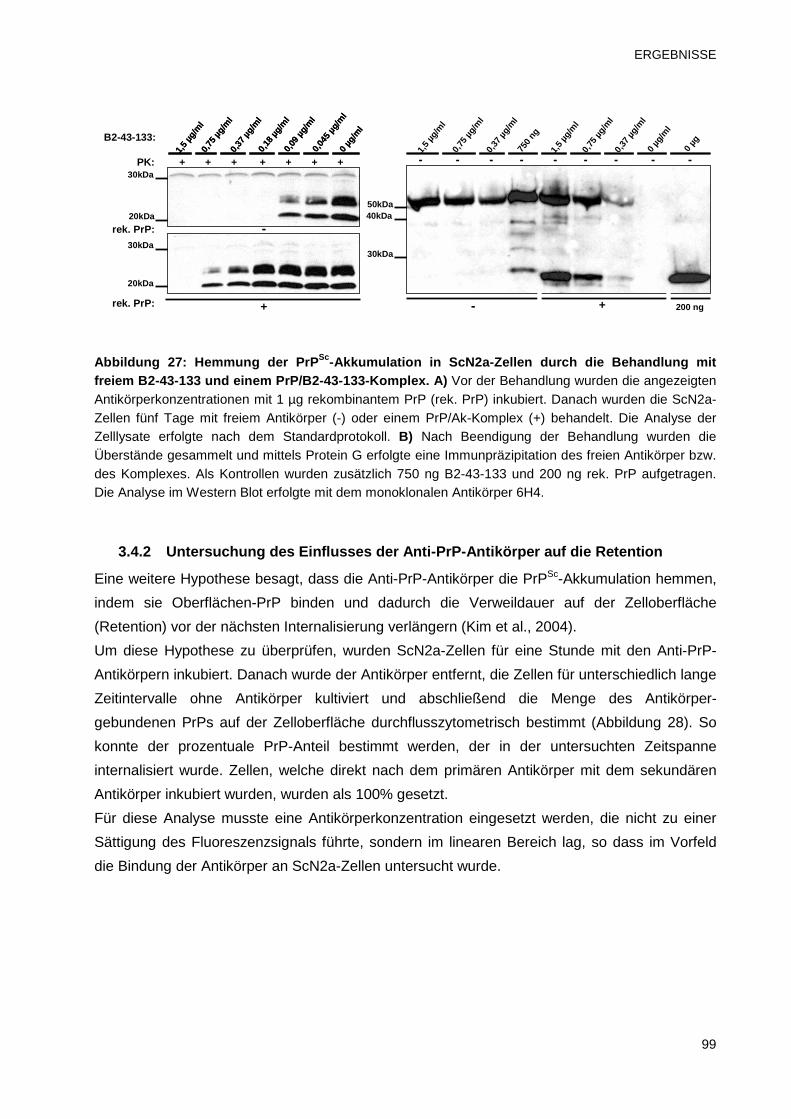
ERGEBNISSE
99
1,5
µg/m
l0,
75 µ
g/m
l0,
37 µ
g/m
l0,
18 µ
g/m
l0,
09 µ
g/m
l0,
045
µg/m
l0
µg/m
l
-
+
+ + + + + + +
rek. PrP:
rek. PrP:
PK:30kDa
20kDa
30kDa
20kDa
B2-43-133:
1,5
µg/m
l0,
75 µ
g/m
l0,
37 µ
g/m
l75
0 ng
0 µg
/ml
0 µg
1,5
µg/m
l0,
75 µ
g/m
l0,
37 µ
g/m
l
- + 200 ng
- - - - - - - - -
30kDa
50kDa40kDa
1,5
µg/m
l0,
75 µ
g/m
l0,
37 µ
g/m
l0,
18 µ
g/m
l0,
09 µ
g/m
l0,
045
µg/m
l0
µg/m
l
1,5
µg/m
l0,
75 µ
g/m
l0,
37 µ
g/m
l0,
18 µ
g/m
l0,
09 µ
g/m
l0,
045
µg/m
l0
µg/m
l
-
+
+ + + + + + +
rek. PrP:
rek. PrP:
PK:30kDa
20kDa
30kDa
20kDa
B2-43-133:
1,5
µg/m
l0,
75 µ
g/m
l0,
37 µ
g/m
l75
0 ng
0 µg
/ml
0 µg
1,5
µg/m
l0,
75 µ
g/m
l0,
37 µ
g/m
l
- + 200 ng
- - - - - - - - -
30kDa
50kDa40kDa
Abbildung 27: Hemmung der PrP Sc-Akkumulation in ScN2a-Zellen durch die Behandlung mit freiem B2-43-133 und einem PrP/B2-43-133-Komplex. A ) Vor der Behandlung wurden die angezeigten Antikörperkonzentrationen mit 1 µg rekombinantem PrP (rek. PrP) inkubiert. Danach wurden die ScN2a-Zellen fünf Tage mit freiem Antikörper (-) oder einem PrP/Ak-Komplex (+) behandelt. Die Analyse der Zelllysate erfolgte nach dem Standardprotokoll. B) Nach Beendigung der Behandlung wurden die Überstände gesammelt und mittels Protein G erfolgte eine Immunpräzipitation des freien Antikörper bzw. des Komplexes. Als Kontrollen wurden zusätzlich 750 ng B2-43-133 und 200 ng rek. PrP aufgetragen. Die Analyse im Western Blot erfolgte mit dem monoklonalen Antikörper 6H4.
3.4.2 Untersuchung des Einflusses der Anti-PrP-Anti körper auf die Retention
Eine weitere Hypothese besagt, dass die Anti-PrP-Antikörper die PrPSc-Akkumulation hemmen,
indem sie Oberflächen-PrP binden und dadurch die Verweildauer auf der Zelloberfläche
(Retention) vor der nächsten Internalisierung verlängern (Kim et al., 2004).
Um diese Hypothese zu überprüfen, wurden ScN2a-Zellen für eine Stunde mit den Anti-PrP-
Antikörpern inkubiert. Danach wurde der Antikörper entfernt, die Zellen für unterschiedlich lange
Zeitintervalle ohne Antikörper kultiviert und abschließend die Menge des Antikörper-
gebundenen PrPs auf der Zelloberfläche durchflusszytometrisch bestimmt (Abbildung 28). So
konnte der prozentuale PrP-Anteil bestimmt werden, der in der untersuchten Zeitspanne
internalisiert wurde. Zellen, welche direkt nach dem primären Antikörper mit dem sekundären
Antikörper inkubiert wurden, wurden als 100% gesetzt.
Für diese Analyse musste eine Antikörperkonzentration eingesetzt werden, die nicht zu einer
Sättigung des Fluoreszenzsignals führte, sondern im linearen Bereich lag, so dass im Vorfeld
die Bindung der Antikörper an ScN2a-Zellen untersucht wurde.
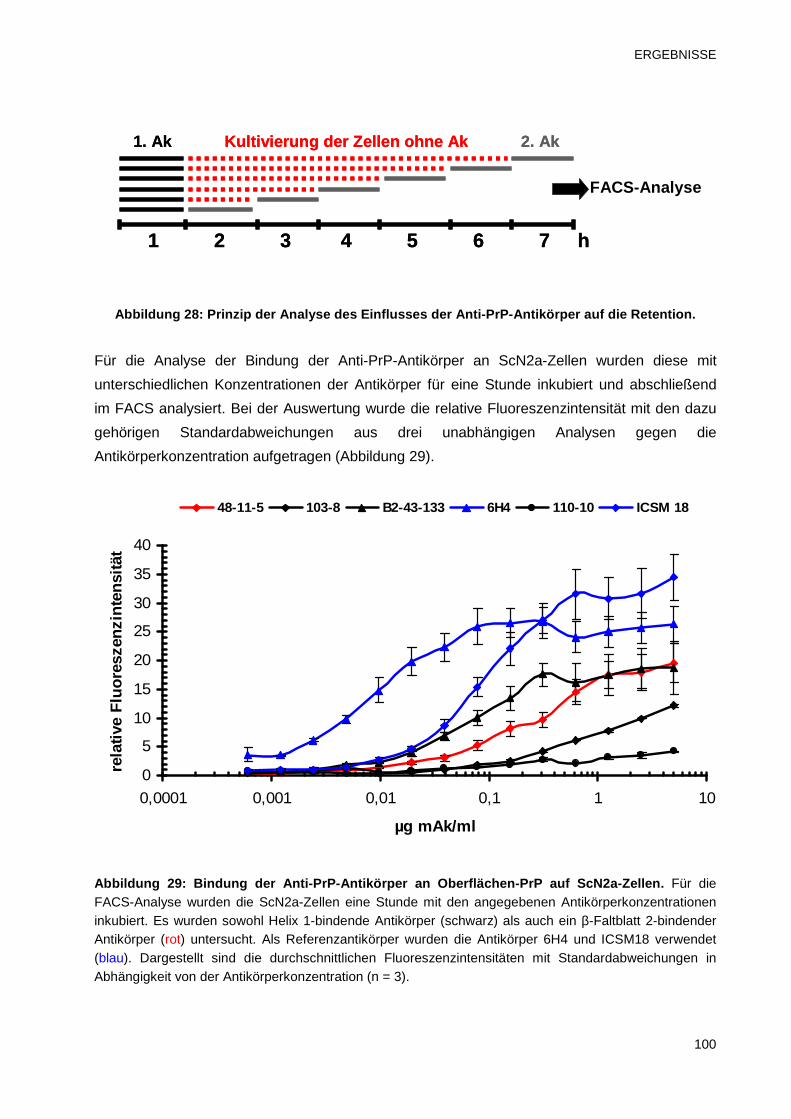
ERGEBNISSE
100
0
5
10
15
20
25
30
35
40
0,0001 0,001 0,01 0,1 1 10
µg mAk/ml
rela
tive
Flu
ores
zenz
inte
nsitä
t
48-11-5 103-8 B2-43-133 6H4 110-10 ICSM 18
0
5
10
15
20
25
30
35
40
0,0001 0,001 0,01 0,1 1 10
µg mAk/ml
rela
tive
Flu
ores
zenz
inte
nsitä
t
48-11-5 103-8 B2-43-133 6H4 110-10 ICSM 18
1 2 3 4 5 6 7 h
1. Ak Kultivierung der Zellen ohne Ak 2. Ak
FACS-Analyse
1 2 3 4 5 6 7 h
1. Ak Kultivierung der Zellen ohne Ak 2. Ak
1 2 3 4 5 6 7 h
1. Ak Kultivierung der Zellen ohne Ak 2. Ak
FACS-Analyse
Abbildung 28: Prinzip der Analyse des Einflusses de r Anti-PrP-Antikörper auf die Retention.
Für die Analyse der Bindung der Anti-PrP-Antikörper an ScN2a-Zellen wurden diese mit
unterschiedlichen Konzentrationen der Antikörper für eine Stunde inkubiert und abschließend
im FACS analysiert. Bei der Auswertung wurde die relative Fluoreszenzintensität mit den dazu
gehörigen Standardabweichungen aus drei unabhängigen Analysen gegen die
Antikörperkonzentration aufgetragen (Abbildung 29).
Abbildung 29: Bindung der Anti-PrP-Antikörper an Ob erflächen-PrP auf ScN2a-Zellen. Für die FACS-Analyse wurden die ScN2a-Zellen eine Stunde mit den angegebenen Antikörperkonzentrationen inkubiert. Es wurden sowohl Helix 1-bindende Antikörper (schwarz) als auch ein β-Faltblatt 2-bindender Antikörper (rot) untersucht. Als Referenzantikörper wurden die Antikörper 6H4 und ICSM18 verwendet (blau). Dargestellt sind die durchschnittlichen Fluoreszenzintensitäten mit Standardabweichungen in Abhängigkeit von der Antikörperkonzentration (n = 3).
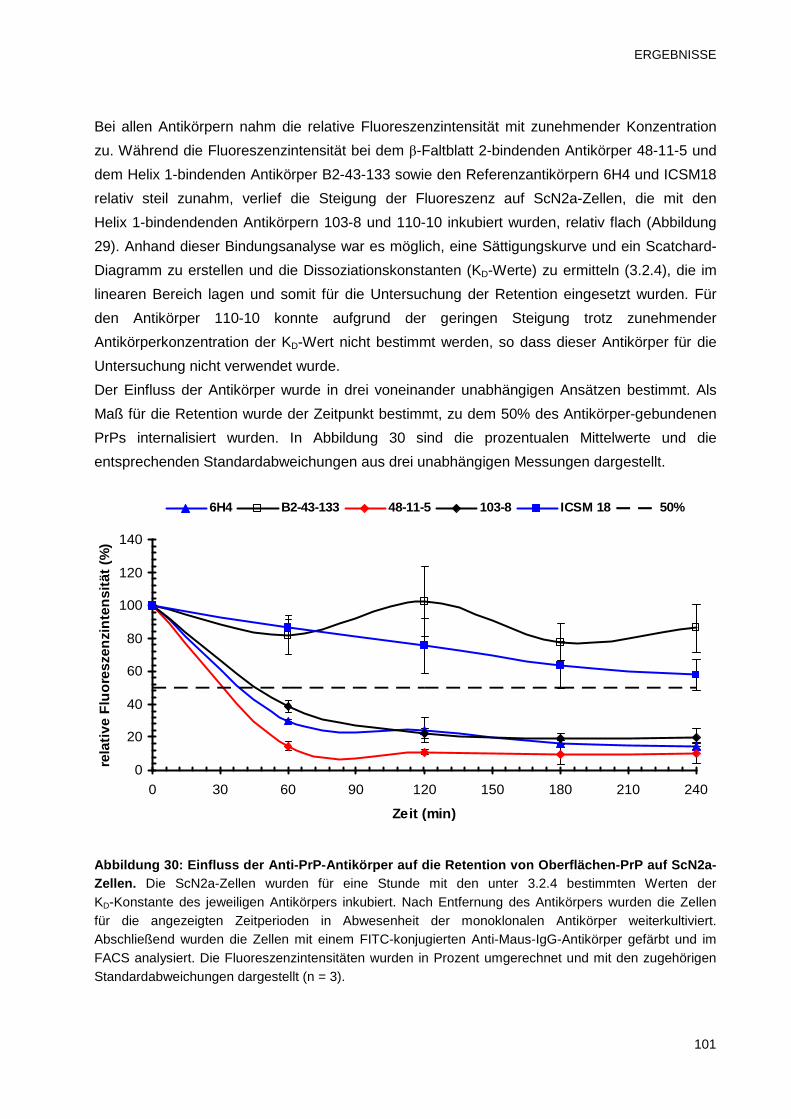
ERGEBNISSE
101
0
20
40
60
80
100
120
140
0 30 60 90 120 150 180 210 240
Zeit (min)
rela
tive
Flu
ores
zenz
inte
nsitä
t (%
)
6H4 B2-43-133 48-11-5 103-8 ICSM 18 50%
0
20
40
60
80
100
120
140
0 30 60 90 120 150 180 210 240
Zeit (min)
rela
tive
Flu
ores
zenz
inte
nsitä
t (%
)
6H4 B2-43-133 48-11-5 103-8 ICSM 18 50%
Bei allen Antikörpern nahm die relative Fluoreszenzintensität mit zunehmender Konzentration
zu. Während die Fluoreszenzintensität bei dem β-Faltblatt 2-bindenden Antikörper 48-11-5 und
dem Helix 1-bindenden Antikörper B2-43-133 sowie den Referenzantikörpern 6H4 und ICSM18
relativ steil zunahm, verlief die Steigung der Fluoreszenz auf ScN2a-Zellen, die mit den
Helix 1-bindendenden Antikörpern 103-8 und 110-10 inkubiert wurden, relativ flach (Abbildung
29). Anhand dieser Bindungsanalyse war es möglich, eine Sättigungskurve und ein Scatchard-
Diagramm zu erstellen und die Dissoziationskonstanten (KD-Werte) zu ermitteln (3.2.4), die im
linearen Bereich lagen und somit für die Untersuchung der Retention eingesetzt wurden. Für
den Antikörper 110-10 konnte aufgrund der geringen Steigung trotz zunehmender
Antikörperkonzentration der KD-Wert nicht bestimmt werden, so dass dieser Antikörper für die
Untersuchung nicht verwendet wurde.
Der Einfluss der Antikörper wurde in drei voneinander unabhängigen Ansätzen bestimmt. Als
Maß für die Retention wurde der Zeitpunkt bestimmt, zu dem 50% des Antikörper-gebundenen
PrPs internalisiert wurden. In Abbildung 30 sind die prozentualen Mittelwerte und die
entsprechenden Standardabweichungen aus drei unabhängigen Messungen dargestellt.
Abbildung 30: Einfluss der Anti-PrP-Antikörper auf die Retention von Oberflächen-PrP auf ScN2a-Zellen. Die ScN2a-Zellen wurden für eine Stunde mit den unter 3.2.4 bestimmten Werten der KD-Konstante des jeweiligen Antikörpers inkubiert. Nach Entfernung des Antikörpers wurden die Zellen für die angezeigten Zeitperioden in Abwesenheit der monoklonalen Antikörper weiterkultiviert. Abschließend wurden die Zellen mit einem FITC-konjugierten Anti-Maus-IgG-Antikörper gefärbt und im FACS analysiert. Die Fluoreszenzintensitäten wurden in Prozent umgerechnet und mit den zugehörigen Standardabweichungen dargestellt (n = 3).

ERGEBNISSE
102
Nach Inkubation mit dem β-Faltblatt 2-bindenden Antikörper 48-11-5, dem Helix 1-bindenden
Antikörper 103-8 sowie mit dem Referenzantikörper 6H4 wurden 50% des Antikörper-
gebundenen PrPs bereits innerhalb der ersten Stunde nach Entfernung des Antikörpers
internalisiert. Im Vergleich dazu verlängerte die Bindung des Antikörpers B2-43-133 die
Retention von PrP deutlich, da nach vier Stunden immer noch mehr als 50% des Oberflächen-
PrPs detektiert wurden. Dies wurde ebenfalls für ScN2a-Zellen beobachtet, die mit dem
Referenzantikörper ICSM18 inkubiert wurden und auf denen nach vier Stunden noch ca. 55%
Antikörper-gebundenes PrP nachgewiesen wurde (Abbildung 30).
Diese Ergebnisse zeigen, dass von den untersuchten Antikörpern lediglich der Helix 1-bindende
Antikörper B2-43-133 sowie der Referenzantikörper ICSM18 fähig sind, die Internalisierung von
PrP zu verzögern und so die Retention von PrP auf der Zelloberfläche zu verlängern. Da aber
zu den Antikörpern, welche die Retention von PrP nicht verlängern können, auch der
Referenzantikörper 6H4 gehört, der mit 0,34 µg/ml ebenfalls einen sehr niedrigen IC50-Wert
besitzt (Tabelle 12), ist es unwahrscheinlich, dass die Verlängerung der Retention von PrP der
Wirkungsmechanismus ist. Jedoch kann die Möglichkeit, dass die Anti-PrP-Antikörper
verschiedene Mechanismen nutzen, durch diese Untersuchung nicht ausgeschlossen werden.
3.5 Pharmakokinetik der Anti-PrP-Antikörper im in vivo Modell
Da der Antikörper B2-43-133 sowohl die PrPSc-Akkumulation in ScN2a-Zellen inhibiert als auch
deren Infektiosität eliminiert (3.3), handelt es sich bei diesem Antikörper um einen potentiellen
Kandidaten für passive Immunisierungen von RML-infizierten wildtypischen Mäusen. Im Hinblick
auf solche Immunisierungsexperimente, für die ein Antikörper mit langer Halbwertszeit
wünschenswert ist, wurde die Pharmakokinetik untersucht, welche den Einfluss des
Organismusses auf einen applizierten Antikörper bezüglich seiner Resorption, Distribution und
Elimination beschreibt. Dazu wurden wildtypische Mäuse intraperitoneal mit RML-Prionen
inokuliert. 63 Tage nach der Inokulation erhielten diese Mäuse im Abstand von drei Tagen drei
intraperitoneale Antikörperinjektionen (1 mg). Für die Untersuchung der Pharmakokinetik wurde
den Mäusen eine Stunde und vier Stunden nach der ersten Antikörperapplikation sowie kurz vor
jeder weiteren Injektion und am Ende des Versuches Blut abgenommen. Die Antikörpermengen
wurden dann im Serum mittels ELISA bestimmt (Abbildung 31).

ERGEBNISSE
103
1 63 66 69 72 dpi 1 mg mAk 1mg mAk 1 mg mAk VE
0,1 ml RML 5 (i.p)(1% Gehirnhomogenat)
Blut 1h Blut Blut BlutBlut 4h
1 63 66 69 72 dpi 1 mg mAk 1mg mAk 1 mg mAk VE
0,1 ml RML 5 (i.p)(1% Gehirnhomogenat)
Blut 1h Blut Blut BlutBlut 4h
Abbildung 31: Immunisierungsschema von RML-infizier ten C57BL/6-Mäusen für die Analyse der Pharmakokinetik. Fünf Gruppen, bestehend aus jeweils sechs Tieren, wurden mit den monoklonalen Antikörpern (mAk) 7-12-5, 48-11-5, 103-8, 110-10 und B2-43-133 nach dem abgebildeten Schema intraperitoneal (i.p.) immunisiert (dpi = days post inoculation). Zu den angegebenen Zeitpunkten wurde den Mäusen Blut entnommen und die Antikörpermenge im Serum mittels ELISA zur Analyse der Pharmakokinetik bestimmt (VE = Versuchsende).
Da die Wirkung der Antikörper in Zellkultur nicht mit der Wirkung in vivo korrelieren muss, wurde
zusätzlich die Pharmakokinetik des nicht wirksamen Antikörpers 7-12-5, des unvollständig
wirksamen Antikörpers 48-11-5 sowie der beiden wirksamen Antikörper 103-8 und 110-10
untersucht. In Abbildung 32 sind die durchschnittlich ermittelten Antikörpermengen mit
Standardabweichungen pro Gruppe (n = 6) zu den untersuchten Zeitpunkten dargestellt.
Eine Stunde nach der ersten Antikörperapplikation wurden 36 ± 11% der ursprünglich
applizierten Menge des Antikörpers B2-43-133 im Serum der Mäuse nachgewiesen (Tabelle
14). Im Vergleich dazu wurde von den beiden Antikörpern 7-12-5 und 48-11-5 eine geringere
Menge detektiert. Drei Stunden später stieg die Menge des Antikörpers B2-43-133 auf 46 ± 8%
an. Ein ähnlicher Anstieg ist in allen Versuchsgruppen zu beobachten und resultiert daraus,
dass die Antikörper nach der intraperitonealen Applikation vom Blut aufgenommen werden
müssen (Resorption). Da dieser Prozess zeitabhängig ist, kommt es vier Stunden nach der
Applikation zu einer Zunahme der detektierbaren Antikörpermenge im Vergleich zu einer
Stunde nach der Injektion. Vier Stunden nach der ersten Antikörperapplikation wurden in allen
Gruppen die höchsten Antikörpermengen nachgewiesen (Tabelle 14).
Drei Tage nach der ersten Antikörperapplikation (Tag 3) wurden vom Antikörper B2-43-133 nur
noch 11 ± 22% im Serum nachgewiesen. Auch die nachweisbare Menge der anderen
Antikörper war zu diesem Zeitpunkt geringer, jedoch wurden von diesen Antikörpern im
Vergleich zum Antikörper B2-43-133 zwischen 17% und 26% detektiert (Tabelle 14). Die
Abnahme der Antikörpermengen zu diesem Zeitpunkt ist darauf zurückzuführen, dass die
Antikörper innerhalb der drei Tage nach Antikörperinjektion ins Gewebe diffundiert sind und
zum Teil degradiert wurden.
Drei Tage nach der zweiten Antikörperapplikation (Tag 6) wurden mit 23 bis 57% insgesamt
wieder höhere Antikörpermengen im Serum aller Gruppen detektiert als an Tag 3, während an
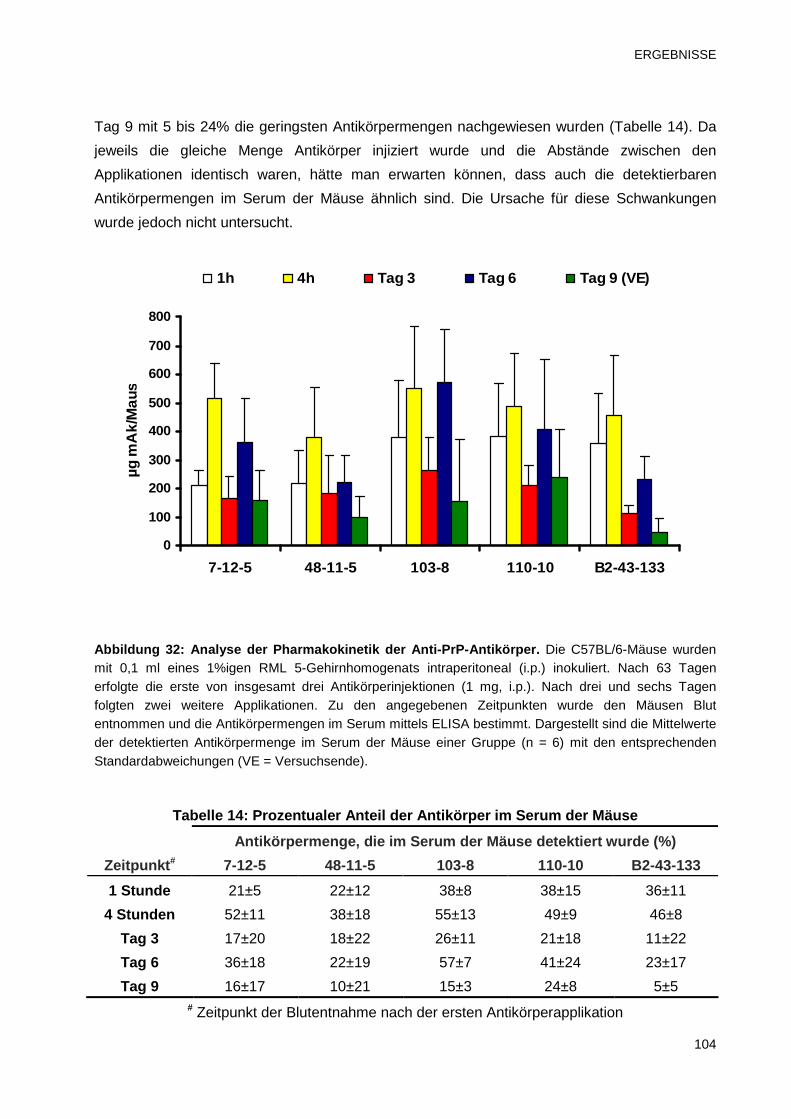
ERGEBNISSE
104
0
100
200
300
400
500
600
700
800
7-12-5 48-11-5 103-8 110-10 B2-43-133
µg m
Ak/
Mau
s
1h 4h Tag 3 Tag 6 Tag 9 (VE)
Tag 9 mit 5 bis 24% die geringsten Antikörpermengen nachgewiesen wurden (Tabelle 14). Da
jeweils die gleiche Menge Antikörper injiziert wurde und die Abstände zwischen den
Applikationen identisch waren, hätte man erwarten können, dass auch die detektierbaren
Antikörpermengen im Serum der Mäuse ähnlich sind. Die Ursache für diese Schwankungen
wurde jedoch nicht untersucht.
Abbildung 32: Analyse der Pharmakokinetik der Anti- PrP-Antikörper. Die C57BL/6-Mäuse wurden mit 0,1 ml eines 1%igen RML 5-Gehirnhomogenats intraperitoneal (i.p.) inokuliert. Nach 63 Tagen erfolgte die erste von insgesamt drei Antikörperinjektionen (1 mg, i.p.). Nach drei und sechs Tagen folgten zwei weitere Applikationen. Zu den angegebenen Zeitpunkten wurde den Mäusen Blut entnommen und die Antikörpermengen im Serum mittels ELISA bestimmt. Dargestellt sind die Mittelwerte der detektierten Antikörpermenge im Serum der Mäuse einer Gruppe (n = 6) mit den entsprechenden Standardabweichungen (VE = Versuchsende).
Tabelle 14: Prozentualer Anteil der Antikörper im S erum der Mäuse
Antikörpermenge, die im Serum der Mäuse detektiert wurde (%)
Zeitpunkt # 7-12-5 48-11-5 103-8 110-10 B2-43-133
1 Stunde 21±5 22±12 38±8 38±15 36±11
4 Stunden 52±11 38±18 55±13 49±9 46±8
Tag 3 17±20 18±22 26±11 21±18 11±22
Tag 6 36±18 22±19 57±7 41±24 23±17
Tag 9 16±17 10±21 15±3 24±8 5±5 # Zeitpunkt der Blutentnahme nach der ersten Antikörperapplikation

ERGEBNISSE
105
Der Verlauf der Pharmakokinetik aller untersuchten Anti-PrP-Antikörper war ähnlich, jedoch
wurden vom Antikörper B2-43-133 insgesamt geringere Mengen im Serum der Mäuse
detektiert. Dies deutet auf eine geringe Halbwertszeit des Antikörpers hin, die jedoch durch
weitere Untersuchungen genau bestimmt werden müsste.

DISKUSSION
106
4 Diskussion
Für die Therapie von humanen Prionenerkrankungen existieren zurzeit keine Substanzen,
welche die Inkubationszeit signifikant beeinflussen (1.1.9).
Bereits Ende der achtziger Jahre konnte gezeigt werden, dass die Inkubation von infektiösem
Hamster-Gehirnhomogenat mit Anti-PrP-Antiseren vor der Inokulation in Indikatortiere zu einer
deutlichen Reduktion der Infektiosität führte (Gabizon et al., 1988). Zusammen mit der
Beobachtung, dass Anti-PrP-Antikörper die Konversion von PrPC zu PrPSc in einem zellfreien
System inhibierten (Horiuchi und Caughey, 1999), deuteten diese beide Experimente auf das
therapeutische Potential von Anti-PrP-Seren bzw. -Antikörpern hin.
Der Erfolg von aktiven Immunisierungsstrategien ist bei Prionenerkrankungen durch die
körpereigene Toleranz gegenüber dem Prion-Protein limitiert. Obwohl Untersuchungen darauf
hindeuten, dass zumindest die B-Zell-Toleranz nur partiell ist (Heppner et al., 2001), induzierten
Immunisierungen von wildtypischen Mäusen mit verschiedenen PrP-spezifischen Peptiden
(Arbel et al., 2003; Polymenidou et al., 2004; Rosset et al., 2004; Schwarz et al., 2003), PrP-
Dimeren (Gilch et al., 2003) sowie mit rekombinantem PrP (Arbel et al., 2003; Polymenidou et
al., 2004; Rosset et al., 2004; Schwarz et al., 2003; Sigurdsson et al., 2002; Wisniewski und
Sigurdsson, 2002) nur moderate Antikörpertiter, welche nicht zu einer signifikanten
Beeinflussung der Inkubationszeit führten. Auch die Erhöhung der Immunogenität von PrP
durch die Verwendung von unterschiedlichen Adjuvantien (Freund-Adjuvans, Titermax®,
filamentöse Phagen, CpG-ODN) oder die Kopplung an immunstimulatorische Proteine
(Heat-shock Proteine, Keyhole limpet Hämocyanin) verstärkte den therapeutischen Effekt nicht.
Aktive DNA-Immunisierungsversuche mit retroviralen PrP-exprimierenden Partikeln (Nikles et
al., 2005) oder PrP-kodierenden Plasmiden, die zusätzlich ein toleranzbrechendes Tetanus
Toxin-Epitop exprimierten (Nitschke et al., 2007), führten ebenfalls nur zu marginalen Erfolgen.
Lediglich nach einer mukosalen Immunisierung mit einer abgeschwächte Maus-PrP-
exprimierenden Salmonella Vakzine, die vor der Prioninfektion erfolgte, verlängerte sich die
Inkubationszeit von wildtypischen Mäusen signifikant (Goni et al., 2005).
Der Einsatz von monoklonalen Anti-PrP-Antikörpern erscheint dagegen erfolgversprechender,
da zum einen die PrPSc-Akkumulation in verschiedenen Prion-infizierten Zelllinien durch die
Behandlung mit Anti-PrP-Antikörpern gehemmt wurde (Enari et al., 2001; Feraudet et al.,
2005b; Kim et al., 2004; Miyamoto et al., 2005; Pankiewicz et al., 2006), und zum anderen die
passive Immunisierung von wildtypischen Mäusen mit bereits fortgeschrittener Prioninfektion zu
einer signifikanten Verlängerung der Inkubationszeit führte (White et al., 2003). Jedoch
scheinen nur bestimmte Anti-PrP-Antikörper auch eine inhibitorische Wirkung auf die
Prionreplikation zu besitzen.

DISKUSSION
107
Das Ziel der vorliegenden Arbeit war somit monoklonale Antikörper gegen das Prion-Protein
herzustellen und diese bezüglich ihrer Eigenschaften und ihrer inhibitorischen Wirkung in vitro
zu analysieren. Zusätzlich sollte der Mechanismus, den Anti-PrP-Antikörper bei der Hemmung
der Prionreplikation nutzen, näher untersucht werden.
4.1 Die Herstellung von PrP C- und PrP Sc-spezifischen Antikörpern
Die ersten polyklonalen Antikörper gegen das Prion-Protein wurden durch Immunisierungen von
Kaninchen mit Spezies-fremdem PrP hergestellt. Als Antigene wurden entweder filamentöse
amyloide PrPSc-Ablagerungen (scrapie-associated fibrils (SAF)) oder PrP27-30 aus
Gehirnhomogenaten von Prion-infizierten Hamstern verwendet. Die geringen Abweichungen in
der Primärsequenz des Antigens führten zur Erhöhung der Antigenität in der heterologen
Spezies, so dass die Herstellung von polyklonalen Antiseren gelang. 1987 wurde nach der
Immunisierung einer wildtypischen Maus mit Hamster-SAFs der PrP-spezifische Antikörper 3F4
isoliert, der Hamster- und Mensch-PrP detektierte, jedoch nicht Maus-PrP (Kascsak et al.,
1987). Diese Spezifität resultierte aus zwei abweichenden Aminosäuren an den Positionen 109
und 112 im Epitop von Maus-PrP (Bolton et al., 1991; Lund et al., 2007). Da das Mausmodell
jedoch für die Untersuchung von Prionenerkrankungen besonders geeignet ist, war es
notwendig, monoklonale Antikörper gegen Maus-PrP zu generieren.
Erst die Herstellung von PrPC-defizienten Mäusen, die auf die Immunisierung mit Maus-PrP mit
einer starken humoralen Antwort reagierten (Bueler et al., 1992; Prusiner et al., 1993),
ermöglichte die Generierung von murinen Anti-PrP-Antikörpern mittels Hybridom-Technik.
Mittlerweile existieren zahlreiche monoklonale PrPC-spezifische Antikörper (Tabelle 15), die
sowohl die zelluläre als auch die pathogene Form des Prion-Proteins detektieren. Antikörper,
die nur PrPSc detektieren, sind hingegen sehr selten (Moroncini et al., 2004). PrPSc-spezifische
Antikörper können von großem diagnostischem Nutzen sein, da sie zum einen das
Detektionslimit von präzipitiertem PrPSc in peripheren Geweben, in denen der PrPSc-Gehalt nur
gering ist, herabsetzen, und zum anderen PrPSc ohne Proteinase K-Verdau detektiert werden
kann. Zusätzlich könnten die Antikörper helfen, die pathogene Isoform näher zu
charakterisieren.
In der vorliegenden Arbeit wurden Prnp0/0-Mäuse mit einer fibrillären Form von rekombinantem
PrP immunisiert, welche durch die Behandlung mit bizellulären Lösungen einen höheren
β-Faltblatt-Gehalt besitzt und somit der Konformation von PrPSc ähnelt (Luhrs et al., 2006).
Durch die Verwendung von diesem Antigen sollte die Wahrscheinlichkeit erhöht werden, einen
PrPSc-spezifischen Antikörper zu generieren. Die beiden Antikörper, B2-31-166 und B2-43-133,
die nach der Fusion isoliert wurden, detektierten jedoch sowohl PrPC als auch PrPSc (Abbildung
12A). Auch die fünf Antikörper, 7-12-5, 12-29, 48-11-5, 103-8 und 110-10, die nach der Fusion

DISKUSSION
108
mit rekombinantem PrP-immunisierten Milzzellen selektiert wurden, diskriminierten nicht
zwischen den beiden Isoformen.
Der erste beschriebene PrPSc-spezifische Antikörper 15B3 entstand zufällig durch die
Immunisierung von Prnp0/0-Mäusen mit bovinem rekombinanten PrP. Dieser Antikörper, ein
IgM-Immunglobulin, präzipitierte selektiv bovines, humanes und murines PrPSc aus Prion-
infizierten Gehirnhomogenaten (Korth et al., 1997). Die gezielte Herstellung solcher Antikörper
ist erschwert, da man davon ausgeht, dass PrPSc-spezifische Antikörper an ein
diskontinuierliches Epitop binden, wie es zum Beispiel für den Antikörper 15B3 bestätigt wurde
(Korth et al., 1997). Somit mussten für die gezielte Herstellung von PrPSc-spezifischen
Antikörpern spezielle Strategien angewendet werden.
Durch den Austausch der CDR3-Region der schweren Kette eines Gerüst-Antikörpers durch
Epitop-Sequenzen von PrPC-spezifischen Antikörpern, die in einer in vitro Studie einen hohen
inhibitorischen Effekt auf die Prionreplikation zeigten, gelang die Herstellung eines Antikörpers,
der in biochemischen und immunhistologischen Experimenten spezifisch PrPSc erkannte
(Moroncini et al., 2004; Moroncini et al., 2006). 2003 führte die Arbeitsgruppe um Neil
Cashmann Immunisierungen mit KLH-gekoppelten PrP-Peptiden durch, welche die Sequenz
YYRRYYRYY repräsentierten (Paramithiotis et al., 2003). Dieses Peptid-Motiv wurde
ausgewählt, weil die Behandlung von rekombinantem PrP mit niedrigen pH-Werten dazu führte,
dass Tyrosin (Y)-Seitenketten, die im zellulären PrP verdeckt sind, exponiert werden. Da die
pH-Behandlung mit einer Erhöhung des β-Faltblatt-Gehalts und einer erhöhten Tendenz zur
Aggregation einhergeht, wurde angenommen, dass dieser Prozess den Konformationswechsel
von PrPC zu PrPSc unter physiologischen Bedingungen reflektieren könnte. Somit sollten die
Tyrosin-Motive PrPSc-spezifische Epitope repräsentieren. Die generierten polyklonalen
Kaninchenseren sowie die murinen monoklonalen Antikörper bestätigten diese Hypothese, da
sie PrPSc mehrerer Spezies selektiv präzipitierten (Paramithiotis et al., 2003).
Eine eindeutige Demonstration der inhibitorischen Wirkung von PrPSc-spezifischen Antikörpern
auf die Prionreplikation steht noch aus. Die transgene Expression der schweren Kette des
15B3-Antikörpers in einem Mausmodell führte weder zu einem detektierbaren Antikörpertiter
noch zur Beeinflussung der Inkubationszeit. Als möglicher Grund hierfür wurde eine veränderte
Spezifität des Antikörpers angenommen, ausgelöst durch die Verwendung einer anderen
Leichtkette (Heppner et al., 2001). Nach Behandlung von ScN2a-Zellen mit den
YYR-spezifischen Antikörpern wurde lediglich eine unvollständige Reduktion des PrPSc-Gehalts
detektiert, so dass die Autoren vermuteten, dass die Konversion zum Teil in Kompartimenten
stattfinden muss, welche für die Antikörper unzugänglich sind (Lehto et al., 2004).

DISKUSSION
109
4.2 Inhibition der PrP Sc-Akkumulation durch Helix 1-bindende Antikörper
Mittlerweile wurde in zahlreichen in vitro-Studien die inhibitorische Wirkung von Anti-PrP-
Antikörpern auf die PrPSc-Akkumulation bestätigt. Eine Auswahl von inhibitorisch wirksamen
Antikörpern ist in Tabelle 15 zusammengefasst.
In der vorliegenden Arbeit wurden drei Anti-PrP-Antikörper, 7-12-5, 12-29 und B2-31-166,
identifiziert, welche die PrPSc-Akkumulation in ScN2a-Zellen nicht hemmten und ein Antikörper,
48-11-5, der zu einer unvollständigen Hemmung führte (Abbildung 20). Auch die Verlängerung
der Behandlungsdauer mit dem Antikörper 48-11-5 verstärkte diesen Effekt nicht (Abbildung
24). Dagegen reduzierten die drei Helix 1-bindenden Antikörper, 103-8, 110-10 und B2-43-133,
ähnlich wie der Referenzantikörper 6H4, den PrPSc-Gehalt von ScN2a-Zellen vollständig
(Abbildung 20). Es konnte ausgeschlossen werden, dass diese PrPSc-Reduktion auf einen
zytotoxischen Effekt der Anti-PrP-Antikörper zurückzuführen ist (Abbildung 21). Der Effekt
basiert wahrscheinlich auf einer spezifischen Interaktion der Antikörper mit PrP (Abbildung 22).
Der inhibitorische Effekt aller bisher untersuchten Anti-PrP-Antikörper zeigte eine
Dosisabhängigkeit. In der Studie von Feraudet lagen die Konzentrationen, bei denen die
37 inhibitorisch wirksamen Antikörper 50% des PrPSc-Signals reduzierten (IC50-Wert), zwischen
0,1 und 7 µg/ml (Feraudet et al., 2005a). In anderen Veröffentlichungen, in denen eine kleinere
Anzahl von Antikörpern untersucht wurde, waren Konzentrationen kleiner als 1 µg/ml
ausreichend (Kim et al., 2004; Miyamoto et al., 2005; Pankiewicz et al., 2006; Peretz et al.,
2001; Perrier et al., 2004). In der vorliegenden Arbeit wurde für den wirksamen Antikörper
B2-43-133 ein IC50-Wert von 0,044 ± 0,01 µg/ml bestimmt. Für die ebenfalls inhibitorisch
wirksamen Antikörper 103-8 und 110-10 konnte der IC50-Wert aufgrund der hohen
Standardabweichungen nicht präzise bestimmt werden. Jedoch kann von den erhaltenen
Analysen abgeleitet werden, dass deutlich höhere Konzentrationen für eine 50%ige Reduktion
des PrPSc-Gehalts notwendig waren als von dem Antikörper B2-43-133 (Daten nicht gezeigt).
Im Vergleich mit bisher publizierten IC50-Werten besitzt der Antikörper B2-43-133 einen sehr
niedrigen IC50-Wert (Tabelle 15). Für die beiden Referenzantikörper 6H4 und ICSM18 wurden
die IC50-Werte 0,34 ± 0,14 µg/ml und 0,09 ± 0,02 µg/ml bestimmt. In einer anderen Studie, die
auf einer epithelialen Kaninchen-Zelllinie (ScRov-Zellen) durchgeführt wurde, lag der IC50-Wert
des Antikörpers ICSM18 dagegen bei 5 µg/ml. Für den Antikörper ICSM35 wurde in der
gleichen Studie ein IC50-Wert von 0,004 µg/ml bestimmt (Beringue et al., 2004). Auch dieser
Wert konnte auf ScN2a-Zellen nicht reproduziert werden, da selbst eine Konzentration von 5 µg
ICSM35/ml nur zu einer schwachen Verringerung des PrPSc-Gehalts von ScN2a-Zellen führte
(Daten nicht gezeigt). Die Ursachen für diese kontroverse Beobachtung liegen wahrscheinlich in
den unterschiedlichen Behandlungszeiträumen, den gewählten Antikörperkonzentrationen oder
den verwendeten Zelllinien. Bei ScN2a-Zellen handelt es sich um die murine
Neuroblastomazelllinie N2a, die durch die Kokultivierung mit infektiösem Maus-

DISKUSSION
110
Gehirnhomogenat infiziert wurde und deren empfänglichster Zellklon abschließend mittels
Subklonierung isoliert und kultiviert wurde (Butler et al., 1988; Enari et al., 2001). Im Gegensatz
dazu kann in ScRov-Zellen die Überexpression von ovinem PrPC durch Doxyzyklin induziert
werden und die Zellen sind mit Schaf-Prionen infizierbar (Vilette et al., 2001). Die verwendeten
Zelllinien haben somit nicht nur einen unterschiedlichen Spezies-Ursprung, sondern
exprimieren PrPC auch in unterschiedlichen Mengen.
Miyamoto und Kollegen berichteten, dass nach einer 8-tägigen Behandlung von N2a/22L-Zellen
mit dem Antikörper 3S9 (10 µg/ml) selbst nach einer einjährigen Weiterkultivierung ohne
Antikörper kein erneuter PrPSc-Gehalt detektiert wurde (Miyamoto et al., 2005). In der
vorliegenden Arbeit konnte nach einer 30-tägigen Behandlung mit den Antikörpern 110-10 und
103-8 lediglich ein transienter Effekt beobachtet werden, da nach Beendigung der Behandlung
PrPSc nach 6 bzw. 36 Tagen in den Zellen wieder nachgewiesen wurde (Abbildung 25).
Dagegen wurde in ScN2a-Zellen, die mit den beiden Helix 1-bindenden Antikörpern 6H4 und
B2-43-133 behandelt wurden, auch nach 36 Tagen Kultivierung ohne Antikörper kein PrPSc
detektiert.
Um zu überprüfen, ob die Antikörper, neben dem PrPSc-Gehalt, auch die Infektiosität der
ScN2a-Zellen beeinflussen, wurde ein Bioassay durchgeführt (Abbildung 26, Tabelle 13). Die
Inkubationszeiten von Mäusen, denen ScN2a-Zellen inokuliert wurden, die mit dem Antikörper
110-10 behandelt wurden, zeigten keinen signifikanten Unterschied zu der gleichen Zellzahl von
unbehandelten Zellen (81 ± 10 Tage) und verstarben 84 ± 7 Tage nach der Inokulation an
Scrapie. Dies belegt, dass der Antikörper 110-10 zwar den PrPSc-Gehalt nach einer 30-tägigen
Behandlung vollständig reduzierte, jedoch die Infektiosität der Zellen nicht beeinflusste. Diese
Schlussfolgerung wird durch die Beobachtung gestützt, dass nach Beendigung der Behandlung
das PrPSc-Signal sofort wieder detektierbar war (Abbildung 25). Dagegen führte die Behandlung
mit dem Antikörper 103-8 zu einer signifikanten Reduktion der Infektiosität der Zellen. Die
Inkubationszeit dieser Tiere ist vergleichbar mit der Gruppe, denen nur 104 unbehandelte
ScN2a-Zellen inokuliert wurden (108 ± 13 dpi, P=0,0142; Log-Rank-Test). Da bei diesen
ScN2a-Zellen erst nach 36 Tagen ein schwaches PrPSc-Signal detektiert wurde (Abbildung 25),
spricht dies dafür, dass eine verzögerte Rückkehr des PrPSc-Signals nach Beendigung der
Antikörperbehandlung mit einer Reduktion der Infektiosität einhergeht. Diese Beobachtung
wurde durch die beiden Antikörper 6H4 und B2-43-133 unterstützt. Bei beiden Antikörpern
wurde 36 Tage nach Beendigung der Behandlung kein PrPSc in ScN2a-Zellen detektiert
(Abbildung 25). Zudem lebten nach Abschluss des Bioassays (372 Tage) aus beiden Gruppen
noch Tiere. Der Endpunkt des Bioassays in Tga20-Mäusen liegt bei 120 bis maximal 140 Tagen
(Fischer et al., 1996). Um eine Verschiebung der Dosis-Wirkungs-Kurve zu beobachten, wurden
die Tiere dieser Gruppen jedoch über einen längeren Zeitraum beobachtet. Bei der verlängerten
Beobachtungsperiode verstarb jeweils ein Tier aus beiden Gruppen (B2-43-133 = 198dpi;

DISKUSSION
111
6H4 = 191dpi) an einer unspezifischen Ursache. Scrapie konnte weder biochemisch noch
histologisch bei beiden Tieren beobachtet werden. Somit reduziert der Helix 1-bindende
Antikörper B2-43-133, ähnlich wie der Referenzantikörper 6H4, sowohl den PrPSc-Gehalt als
auch die Infektiosität von ScN2a-Zellen vollständig.
Zusammenfassend zeigen diese Ergebnisse, dass von den sieben neu generierten Anti-PrP-
Antikörpern der Helix 1-bindende Antikörper B2-43-133 den stärksten inhibitorischen Effekt
besitzt. Diese Einschätzung basiert auf dem sehr niedrigen IC50-Wert, der dauerhaften
Hemmung der PrPSc-Akkumulation in ScN2a-Zellen und der Löschung der Infektiosität von
ScN2a-Zellen nach einer Langzeitbehandlung.
White und Kollegen konnten 2003 zeigen, dass Antikörper, welche die PrPSc-Akkumulation in
vitro inhibieren, auch in vivo eine inhibitorische Wirkung besitzen. Die Behandlung von
intraperitoneal (i.p.) RML-infizierten wildtypischen Mäusen mit den beiden Antikörpern ICSM18
und ICSM35 reduzierte das PrPSc-Level in der Milz und verlängerte die Inkubationszeit um
150%. Dies wurde ebenfalls beobachtet, wenn die Antikörperbehandlung erst dreißig Tage
nach der Inokulation begann, also zu einem Zeitpunkt, an dem die PrPSc-Akkumulation in der
Milz nach einer i.p.-Infektion bereits eine Plateau-Phase erreicht hat (White et al., 2003). Auch
die einmalige Injektion der Antikörper 8H4 und 8B4 direkt nach einer i.p. Inokulation führte zu
einer signifikanten Verlängerung der Inkubationszeit (Sigurdsson et al., 2003). Dieser Effekt ist
jedoch wahrscheinlich auf eine Neutralisierung des Inokulums zurückzuführen, da zwischen der
Inokulation und Antikörperapplikation nur ein geringer zeitlicher Abstand lag.
Beide Studien untersuchten die Pharmakokinetik der applizierten Antikörper, da für
therapeutische Anwendungen von Antikörpern eine lange Halbwertszeit wünschenswert ist. Vier
Tage nach der letzten Injektion der beiden Antikörper ICSM18 und ICSM35 (2 mg pro Injektion)
zirkulierten noch ca. 20% im Serum (White et al., 2003). Die Antikörper 8H4 und 8B4
(0,05 mg Ak) konnten bereits vier Stunden nach der Applikation nicht mehr nachgewiesen
werden (Sigurdsson et al., 2003). In einer anderen Untersuchung wurden drei Tage nach einer
einzelnen Injektion des Antikörpers BAR236 (1 mg Ak), der zu einer signifikanten Verlängerung
der Inkubationszeit führte (P<0,001), ca. 15% detektiert, während von einem nicht-wirksamen
Antikörper noch 30% im Serum nachgewiesen wurden (Feraudet et al., 2005a). In der
vorliegenden Arbeit wurde von dem wirksamen Antikörper B2-43-133 zu diesem Zeitpunkt
zwischen 5% und 23% der ursprünglich applizierten Antikörpermenge (1 mg) im Serum
nachgewiesen (Tabelle 14). Im Vergleich dazu wurden von den anderen untersuchten
Antikörpern (7-12-5, 48-11-5, 103-8 und 110-10) höhere Mengen detektiert. Ein direkter
Vergleich mit den beiden ersten Studien ist aufgrund der unterschiedlichen Antikörpermengen
und Zeiträume nicht möglich.
Die Ergebnisse deuten auf eine geringe Halbwertszeit des Antikörpers B2-43-133 hin, so dass
dieser Antikörper für passive Immunisierungen wahrscheinlich nicht optimal geeignet ist.
DISKUSSION
112
Antikörper Antigen Epitop Effekt K IC Referenz(µg/ml(nM))
6H4 rek.boPrP 144-152 + Korth, 1997(µg/ml(nM))
D13 HaPrP27-30 95-104 0,16 (3,3) 0,6 (12)D18 HaPrP27-30 132-156 + 0,07 (1,6) 0,45 (9)R1 HaPrP27-30 200-231 + 0,09 (1,7) 2,5 (50)
R2 HaPrP27-30 225-231 +/- 0,11 (2,2) 2 (40)
8B4 rek.PrP 35-45 + (0,09/6,5/0,05)
8F9 rek.PrP 220-231 (0,7/43/2,1)
8H4 rek.PrP 175-185 + (0,07/0,09/0,04)
ICSM18 α-rek.huPrP 146-159 + 5 (45,5)ICSM19 β-rek.huPrP K + 0,36 (3,3)
ICSM35 β-rek.huPrP 91-110 + 0,004 (0,03)Sha31 Ha SAF* 145-152 + 0,1 (0,7)
BAR236 rek.ovPrP ? + 0,1 (0,7)
SAF83 Ha SAF* 126-141 + 0,25 (1,7) & 161-164
BAR214 rek.ovPrP ? + 0,25 (1,7)
BAR221 rek.ovPrP 141-151 + 0, 6 (4)
SAF34 HaSAF** 59-89(4x) + 1 (6,7)
SAF61 HaSAF** 142-153 + 2,5 (16,7)BAR224 rek.ovPrP 141-151
βH3 β-rek.PrP ?
31C6 rek.PrP 143-149 + 0,1 (0,7) Kim, 2004110 Maus-PrP 59-65 + 0,2 (1,2)
& 83-8944B1 rek.PrP 89-231 + 0,3 (1,7)72 Maus-PrP + 0,6 (4,1)
3S9 rek.huPrP 141-161 + 0,08 (0,6)2H9 rek.huPrP 151-221 + 1,2 (8,4)
6D11 ? 93-109 + (0,085/0,15) 0,07(0,47) Pankiewitcz, 2006
9H7 rek.PrP 143-231 (0,103)7D9 ? ? (0,82)
7H6 ? 130-140 + (0,22) 0,16 (1,07)
8H4 rek.PrP 175-185 + (0,38)
8B4 rek.PrP 35-45 + (0,24)
8F9 rek.PrP 220-231 (10,6)
SAF34 HaSAF** 59-89(4x) + 1,3 (8,7) Perrier, 2004
SAF61 HaSAF** 142-153 + 0,8 (5,3)
D 50
Sc
Sc
-
-
-
-
-
-
KK
+ Peretz, 2001
Sigurdsson, 2003
Beringue, 2004
Feraudet, 2004
Miyamoto, 2005
Tabelle 15: Auswahl von Anti-PrP-Antikörpern
Bei den Antikörpern 8B4, 8F9 und 8H4 beziehen sich die KD-Werte auf rekombinantes PrP (rek. PrP), Maus-PrPC und Maus-PrPSc. Der KD-Wert des Antikörpers 6D11 wurde auf rek. PrP und auf PrP93-122 bestimmt. * Proteinase K (PK)-behandelte, nicht-denaturierte SAF vom Hamster, ** PK-behandelte, Ameisensäure-denaturierte SAF vom Hamster, K Konformationsepitop

DISKUSSION
113
4.3 Unterschiede zwischen den Helix 1-bindenden Ant i-PrP-Antikörpern
Von den sieben neu generierten Anti-PrP-Antikörpern führten nur die drei Helix 1-bindenden
Antikörper 103-8, 110-10 und B2-43-133 zu einer vollständigen Inhibition der PrPSc-
Akkumulation in ScN2a-Zellen (Abbildung 20). Der Einfluss auf die Infektiosität der behandelten
ScN2a-Zellen war jedoch unterschiedlich, da der Antikörper 110-10 keinen Effekt zeigte, der
Antikörper 103-8 die Infektiosität reduzierte und der Antikörper B2-43-133 die Infektiosität der
behandelten ScN2a-Zellen vollständig löschte (Tabelle 13). Um Unterschiede in dieser Gruppe
von Helix 1-bindenden Antikörpern zu identifizieren, wurde ihre Charakterisierung im Hinblick
auf die inhibitorische Effizienz analysiert.
Obwohl das Epitop von allen drei Antikörpern innerhalb der Aminosäuresequenz 142-157 von
PrPC lokalisiert wurde, deutet die Analyse der Speziesreaktivität darauf hin, dass zumindest der
Antikörper 103-8 innerhalb dieser Sequenz ein anders definiertes Epitop als die Antikörper
110-10 und B2-43-133 erkennt (Daten nicht gezeigt).
Bei der Bindung der Antikörper an ScN2a-Zellen wurden ebenfalls Unterschiede zwischen den
Helix 1-bindenden Antikörper bestimmt (Abbildung 29). Im Vergleich zum Antikörper 110-10 und
103-8 erkannte der Antikörper B2-43-133 PrPC und PrPSc mit einer höheren
Fluoreszenzintensität. Da jedoch auch nach der Bindung des unvollständig wirksamen
Antikörpers 48-11-5 an native ScN2a-Zellen eine stärkere Fluoreszenzintensität als mit den
wirksamen Antikörpern 110-10 und 103-8 detektiert wurde, ist es unwahrscheinlich, dass diese
Fähigkeit ausschlaggebend für den inhibitorischen Effekt der Antikörper ist.
Die Affinität der Antikörper zu rekombinantem PrP war hingegen sehr ähnlich (Tabelle 11). Trotz
der minimalen Unterschiede innerhalb der Gruppe der Helix 1-bindenden Antikörper und im
Vergleich zu den β-Faltblatt 2- und STE-bindenden Antikörpern lagen alle bestimmten
KD-Konstanten im pikomolaren Bereich und waren niedriger als viele der zuvor bestimmten
KD-Konstanten (Tabelle 15). Somit kann über die KD-Konstante kein Rückschluß auf das
inhibitorische Potential eines Antikörpers gezogen werden.
Zusätzlich wurde die Avidität der Antigen-Antikörper-Interaktion analysiert. Im Vergleich zu den
β-Faltblatt 2- und STE-bindenden Antikörpern waren die Aviditätsindizes der Helix 1-bindenden
Antikörper größer als 0,5 M. Die stärkste Bindung innerhalb dieser Gruppe zeigte der Antikörper
103-8 (1,37 M), während die beiden Antikörper 110-10 und B2-43-133 relativ ähnliche
Aviditätsindizes zeigten (~0,6 M). Würde jedoch die Avidität die Eigenschaft der Antikörper sein,
welche den inhibitorischen Effekt bestimmt, so sollte der Antikörper B2-43-133, und ebenso der
wirksame Referenzantikörper 6H4 (0,94 M), einen höheren Aviditätsindex als der Antikörper
103-8 besitzen.
In einer anderen Studie wurde die Hypothese aufgestellt, dass Antikörper, die ausschließlich
PrPC binden weniger effizient sind als Antikörper, die an beide PrP-Isoformen binden (Beringue
et al., 2004). In der vorliegenden Arbeit waren alle Antikörper fähig, neben PrPC, spezifisch

DISKUSSION
114
N C
GPI
93-106 142-157 158-171
S-S
STE TM H1 H2 H3ββββ1 ββββ2Oktarepeats
23 95-112
112-126
128-131
161-164
144-151
179-193
200-217
23151-91
Antigen: rek. PrP
Antigen: fib. PrP
7-12-5
12-29
48-11-5
103-8
110-10
B2-43-133
B2-31-166
Inhibition von PrP Sc
(10 µg/ml)Avidität
(M)KD (M)rek. PrP
KD (M)natives PrP
--
+
+
+
+/-
+/-
0,11
0,3
0,22
1,37
0,57
0,67
0,11
3,5x10-14
1,7x10-12
4,9x10-12
6,7x10-14
3,2x10-14
1,6x10-13 4,9x10-13
/
/ /
/
/
/
3,9x10-14
N C
GPI
93-106 142-157 158-171
S-S
STE TM H1 H2 H3ββββ1 ββββ2Oktarepeats
23 95-112
112-126
128-131
161-164
144-151
179-193
200-217
23151-91
N C
GPI
93-106 142-157 158-171
S-S
STE TM H1 H2 H3ββββ1 ββββ2Oktarepeats
23 95-112
112-126
128-131
161-164
144-151
179-193
200-217
23151-91
Antigen: rek. PrP
Antigen: fib. PrP
7-12-5
12-29
48-11-5
103-8
110-10
B2-43-133
B2-31-166
Inhibition von PrP Sc
(10 µg/ml)Avidität
(M)KD (M)rek. PrP
KD (M)natives PrP
--
+
+
+
+/-
+/-
0,11
0,3
0,22
1,37
0,57
0,67
0,11
3,5x10-14
1,7x10-12
4,9x10-12
6,7x10-14
3,2x10-14
1,6x10-13 4,9x10-13
/
/ /
/
/
/
3,9x10-14
PrPSc von ScN2a-Zellen zu detektieren (Abbildung 22). Die Bindungsanalyse der Antikörper an
PrP von ScN2a-Zellen zeigte jedoch, dass die Affinitäten der einzelnen Antikörper
unterschiedlich waren (Abbildung 29). Interessanterweise war die Affinität des Antikörpers
110-10, der Antikörper aus der Helix 1-bindenden Gruppe mit dem schwächsten inhibitorischen
Effekt, so gering, dass die Dissoziationskonstante auf diesem Substrat nicht bestimmt werden
konnte. Hingegen wurden für die drei Antikörper 6H4, ICSM18 und B2-43-133, diejenigen
Antikörper mit dem stärksten inhibitorischen Effekt, die kleinsten KD-Konstanten ermittelt und
somit die höchste Affinitäten (Tabelle 11). Da jedoch die KD-Konstante des β-Faltblatt
2-bindenden Antikörpers 48-11-5 auf ScN2a-Zellen, welcher nur unvollständig inhibitorisch
wirkt, deutlich geringer ist, als die des Antikörpers 103-8, spricht dies ebenfalls gegen eine
direkte Korrelation von Affinität und inhibitorischer Wirkung. Auch andere Studien konnten keine
Korrelation zwischen der Immunreaktivität gegenüber PrPSc und dem inhibitorischen Effekt der
Antikörper finden (Feraudet et al., 2005b).
Tabelle 16: Charakterisierung der neuen Anti-PrP-An tikörper
H1-3 = α-Helix 1-3, ββββ1-2 = β-Faltblatt 1-2, STE = Stopp-Transfer-Effektor-Sequenz, TM = Transmembran-domäne, S-S = Disulfidbrücke, GPI = Glycosylphosphatidylinositol-Anker, M = Molarität, fib. PrP = fibrilläres rekombinantes PrP, rek. PrP = rekombinantes PrP
Zusammenfassend lässt sich sagen, dass innerhalb der Gruppe der Helix 1-bindenden
Antikörper sowohl Unterschiede im Epitop als auch in den Bindungen an PrPC und PrPSc

DISKUSSION
115
bestehen (Tabelle 16). Bezieht man jedoch die Ergebnisse der β-Faltblatt 2-bindenden
Antikörper in die Analyse mit ein, kann kein direkter Zusammenhang zwischen den
untersuchten Eigenschaften und dem inhibitorischen Effekt der Antikörper bestimmt werden.
4.4 Hypothesen über den Wirkungsmechanismus der Ant i-PrP-Antikörper
Die inhibitorische Wirkung von Anti-PrP-Antikörpern auf die PrPSc-Akkumulation in
verschiedenen Zelllinien wurde bereits mehrfach demonstriert (Beringue et al., 2004; Enari et
al., 2001; Feraudet et al., 2005b; Kim et al., 2004; Miyamoto et al., 2005; Pankiewicz et al.,
2006). Der Wirkungsmechanismus, den die Antikörper nutzen, um den Konformationswechsel
von PrPC zu PrPSc zu verhindern, ist jedoch bis heute unbekannt. Ausgehend von dem Wissen,
dass der Konformationswechsel entweder auf der Zelloberfläche oder in endozytotischen
Kompartimenten stattfindet (Arnold et al., 1995; Borchelt et al., 1992; Caughey und Raymond,
1991; Mayer et al., 1992) und eine direkte Interaktion zwischen PrPC und PrPSc notwendig ist,
an der eventuell Hilfsmoleküle beteiligt sind (Deleault et al., 2003; Telling et al., 1995), wurden
verschiedene Hypothesen über den Wirkungsmechanismus formuliert (Abbildung 33).
Viele der wirksamen Anti-PrP-Antikörper (Tabelle 15) binden PrPC innerhalb der Helix 1 (Reste
143-151) (Beringue et al., 2004; Enari et al., 2001; Miyamoto et al., 2005; Peretz et al., 2001;
Perrier et al., 2004; White et al., 2003). Auch das Epitop der drei inhibitorisch wirksamen
Antikörper 103-8, 110-10 und B2-43-133 wurde in dieser Region lokalisiert (Reste 142-157)
(Abbildung 16). Beim Konformationswechsel wird dieser Bereich und die N-terminale Domäne
in eine β-helikale Struktur umgewandelt (Eghiaian et al., 2004; Govaerts et al., 2004). Dies legt
die Vermutung nahe, dass die Antikörperbindung an PrPC zu einer sterischen Blockade des
Konformationswechsels führt.
Die Bindung könnte aber auch die Interaktion von PrPC mit PrPSc inhibieren. PrPSc bindet mit
einer hohen Affinität an die Aminosäuresequenzen 23-33, 98-110 und 136-158 von PrPC
(Solforosi et al., 2007). Die Epitope aller Antikörper, die einen inhibitorischen Effekt zeigten,
beschränken sich auf vier Regionen (Reste 59-89, 90-109, 144-156 und 225-231) (Tabelle 15),
von denen zwei mit den PrPSc-affinen Epitopen übereinstimmen. Somit könnten die Antikörper,
welche an diese Epitope binden, direkt die Interaktion zwischen PrPC und PrPSc inhibieren
(Abbildung 33B). Die Studie zeigte jedoch auch, dass die Sequenzen 29-49, 110-136 und
158-231 keine intrinsische Affinität für PrPSc besitzen (Solforosi et al., 2007). Trotzdem
existieren Antikörper, wie zum Beispiel die Antikörper 8H4 und 8F9 oder die Fab-Fragmente R1
und R2, deren Epitope zwischen den Aminosäuren 175 bis 231 lokalisiert sind und welche die
PrPSc-Akkumulation inhibieren (Tabelle 15). Auch in der vorliegenden Studie wurde ein
Antikörper generiert, 48-11-5, welcher an die Sequenz 158 bis 176 bindet und zu einer
unvollständigen Inhibition der Prionreplikation führt. Dies lässt vermuten, dass, neben der oben
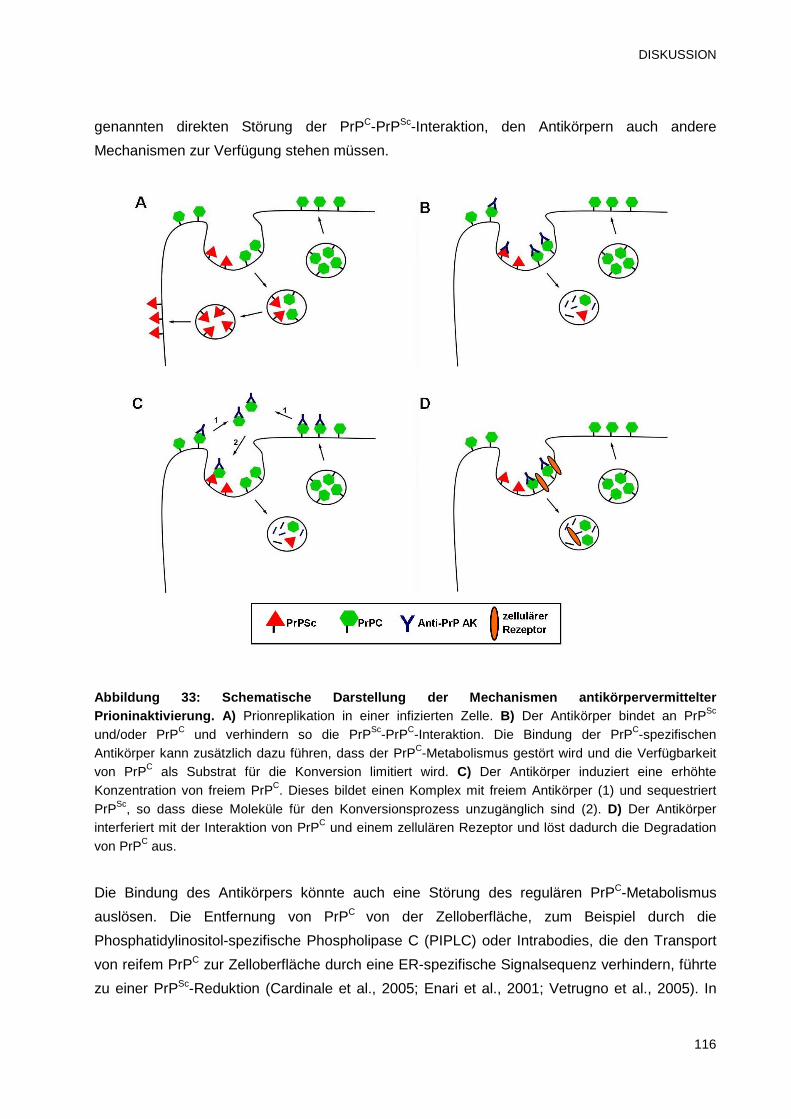
DISKUSSION
116
genannten direkten Störung der PrPC-PrPSc-Interaktion, den Antikörpern auch andere
Mechanismen zur Verfügung stehen müssen.
Abbildung 33: Schematische Darstellung der Mechanis men antikörpervermittelter Prioninaktivierung. A) Prionreplikation in einer infizierten Zelle. B) Der Antikörper bindet an PrPSc und/oder PrPC und verhindern so die PrPSc-PrPC-Interaktion. Die Bindung der PrPC-spezifischen Antikörper kann zusätzlich dazu führen, dass der PrPC-Metabolismus gestört wird und die Verfügbarkeit von PrPC als Substrat für die Konversion limitiert wird. C) Der Antikörper induziert eine erhöhte Konzentration von freiem PrPC. Dieses bildet einen Komplex mit freiem Antikörper (1) und sequestriert PrPSc, so dass diese Moleküle für den Konversionsprozess unzugänglich sind (2). D) Der Antikörper interferiert mit der Interaktion von PrPC und einem zellulären Rezeptor und löst dadurch die Degradation von PrPC aus.
Die Bindung des Antikörpers könnte auch eine Störung des regulären PrPC-Metabolismus
auslösen. Die Entfernung von PrPC von der Zelloberfläche, zum Beispiel durch die
Phosphatidylinositol-spezifische Phospholipase C (PIPLC) oder Intrabodies, die den Transport
von reifem PrPC zur Zelloberfläche durch eine ER-spezifische Signalsequenz verhindern, führte
zu einer PrPSc-Reduktion (Cardinale et al., 2005; Enari et al., 2001; Vetrugno et al., 2005). In

DISKUSSION
117
einer in vitro Studie wurde beobachtet, dass durch die Antikörperbehandlung die Halbwertszeit
von PrPC auf der Zelloberfläche reduziert wird, indem die PrPC-Degradation verstärkt wird (Gilch
et al., 2003; Perrier et al., 2004). Es wurde vermutet, dass die Degradation durch eine Störung
der Interaktion von PrP mit dem Laminin-Rezeptor (LRP/LR) ausgelöst wurde (Perrier et al.,
2004), der für die Internalisierung von PrP notwendig ist (Gauczynski et al., 2001; Morel et al.,
2005) (Abbildung 33D).
Gleichzeitig könnten Anti-PrP-Antikörper aber auch einen stabilisierenden Effekt auf PrPC
besitzen, indem sie die Internalisierung von PrPC verhindern und somit auch die Konversion in
den subzellulären Kompartimenten (Abbildung 33B). Einige inhibitorische Antikörper, wie zum
Beispiel SAF34 und 110 (Tabelle 15), binden an die Oktarepeats der N-terminalen Domäne.
Diese Domäne ist zwar für die Replikation nicht essentiell (Flechsig et al., 2000; Rogers et al.,
1993), jedoch für den subzellulären Transport und für die Endozytose von PrPC (Nunziante et
al., 2003; Shyng et al., 1995), so dass diese Antikörper zu einer verringerten Endozytoserate
führen könnten.
Ebenfalls konnte gezeigt werden, dass Antikörper den PrPC-Gehalt auf der Zelloberfläche
Epitop-unabhängig stabilisieren. Die Bindung des Helix 1-bindenden Antikörpers 31C6
verlängerte die Halbwertszeit von PrPC auf der Zelloberfläche (Retention) und verhinderte so
die PrPC-Internalisierung und die Konversion zu PrPSc (Kim et al., 2004). Dies wurde in der
vorliegenden Arbeit auf ScN2a-Zellen für den Antikörper B2-43-133 und den Referenzantikörper
ICSM18 beobachtet (Abbildung 30). Bei den wirksamen Antikörpern 103-8 und 110-10 sowie
bei dem Referenzantikörper 6H4 verlängerte die Bindung jedoch nicht die Retention von PrPC.
Das gebundene PrPC wurde innerhalb eines ähnlich kurzen Zeitraums wie bei dem
unvollständig wirksamen Antikörpern 48-11-5 internalisiert.
Eine andere Hypothese geht davon aus, dass die Inhibition der Prionreplikation nicht durch
einen freien Antikörper, sondern durch einen freien PrP/Antikörper-Komplex vermittelt wird
(Feraudet et al., 2005a). Dies wurde von den Beobachtungen abgeleitet, dass eine einzelne
Anti-PrP-Antikörperinjektion in wildtypische Mäuse zu einem Anstieg von freiem PrPC im Blut
führte und gleichzeitig ein PrPC/Ak-Komplex mit einer langen Halbwertszeit nachgewiesen
wurde, während der freie Antikörper nur eine relativ kurze Halbwertszeit im Blut besaß. Bindet
dieser PrP/Ak-Komplex an PrPC oder PrPSc, kann er entweder die Interaktion zwischen den
beiden Isoformen verhindern oder er neutralisiert bereits existierendes PrPSc (Feraudet et al.,
2005a) (Abbildung 33C). In der vorliegenden Arbeit reduzierte die Komplexbildung mit
rekombinantem PrP den inhibitorischen Effekt des Antikörpers B2-43-133, obwohl ein stabiler
Komplex auch noch nach fünf Tagen im Medium nachgewiesen wurde (Abbildung 27). Zwar
wurde auch eine vollständige Reduktion des PrPSc-Signals nach der Behandlung mit dem
Komplex beobachtet, jedoch war eine ca. achtfach höhere Antikörperkonzentration im Vergleich
zu freiem Antikörper notwendig. Da der IC50-Wert des Antikörpers B2-43-133 sehr niedrig ist

DISKUSSION
118
(0,044 µg/ml), legt dies die Vermutung nah, dass der Effekt auf freien Antikörper
zurückzuführen ist, der durch die unspezifische Spaltung eines kleinen Teils des Komplexes
entstanden ist.
Ob diese Modellvorstellungen auch auf die in vivo-Situation übertragbar sind, ist noch unklar.
Der Antikörper 7D9, der in Zellkulturversuchen keinen inhibitorischen Effekt zeigte, reduzierte
die Infektiosität eines Inokulums durch Präinkubation und verlängerte nach einer einmaligen
Injektion die Inkubationszeit von infizierten Mäusen signifikant (Pankiewicz et al., 2006).
Die Überprüfung mehrere Hypothesen bezüglich des Wirkungsmechanismusses von Anti-PrP-
Antikörpern lieferte kein eindeutiges Ergebnis. Alle wirksamen Antikörper (103-8, 110-10,
B2-43-133) können theoretisch aufgrund ihres Epitopes mit der PrPC-PrPSc-Interaktion
interferieren. Zusätzlich verlängerte der Antikörper B2-43-133 die Retention von PrP auf der
Zelloberfläche. Da dies aber für den Referenzantikörper 6H4, der einen ähnlich starken
inhibitorschen Effekt wie der Antikörper B2-43-133 zeigte, nicht beobachtet wurde, ist es
unwahrscheinlich, dass die Retention von PrP der Wirkungsmechanismus ist. Jedoch muss in
Betracht gezogen werden, dass verschiedene Antikörper verschiedene Mechanismen nutzen
könnten und dass sich einzelne Mechanismen nicht gegenseitig ausschließen, sondern auch
durch ihre Kombination die inhibitorische Effizienz erhöhen könnten.
4.5 Ausblick
Die vorgestellten Ergebnisse zeigen, dass der Antikörper B2-43-133 ein idealer Kandidat für
weiterführende Untersuchungen bezüglich möglicher therapeutischer Implikationen ist. Die
kontinuierliche Applikation eines Antikörpers, der gegen ein körpereigenes Protein gerichtet ist,
birgt jedoch mehrere Risiken. Zum einen besteht die Gefahr, dass die Toleranz gegen das
Protein überwunden wird und es zur Entwicklung von Autoimmunkrankheiten kommt, zum
anderen könnten die Antikörper die Funktion von PrPC beeinflussen. Bei wildtypischen Mäusen,
die für 53 Wochen zweimal wöchentlich mit hohen Dosen Anti-PrP-Antikörpern behandelt
wurden, wurden keine unerwünschten Nebeneffekte beobachtet (White et al., 2003).
Ein weiteres Problem bei der Behandlung von Prionenerkrankungen ist, dass es nach einer
peripheren Infektion mit Prionen, die in der Regel bei Scrapie, BSE und vCJD vorliegt, nach
einer Replikationsphase in den sekundären lymphoiden Organen zu einer Prioneninvasion ins
ZNS kommt (Neuroinvasion). Obwohl die Antikörper in den vorgestellten Studien die
Prionpathogenese nach intraperitonealer Infektion signifikant beeinflussten, konnte nach
intrazerebraler Infektion weder die Applikation von Antikörpern noch die kontinuierliche
Expression einer Anti-PrP-µ-Kette die Entwicklung einer vollständigen Scrapie-Pathogenese
stoppen (Heppner et al., 2001; White et al., 2003). Da die Antikörper die Blut-Hirn-Schranke
nicht effizient passieren können, ist ihre Verfügbarkeit im Gehirn limitiert. Wird jedoch die

DISKUSSION
119
Prionenerkrankung erst nach der Neuroinvasion diagnostiziert, ist es notwendig, dass effektive
Immuntherapeutika auch ins ZNS eindringen können.
Eine Möglichkeit, um die Bioverfügbarkeit der Antikörper im Gehirn zu maximieren, ist die
direkte intrazerebrale Applikation. In einer Pilotstudie führte jedoch die direkte Injektion von
Anti-PrP-Antikörpern, die an die Sequenz 95-105 binden, in den Hippocampus wildtypischer
Mäuse zu neuronaler Apoptose. Da die Applikation der korrespondierenden Fab-Fragmente
keine Apoptose induzierte, wurde ein Komplement-vermittelter Effekt über den FC-Teil
ausgeschlossen. Die Autoren folgerten vielmehr, dass durch die Dimerisierung von
extrazellulärem PrPC über ein bestimmtes Epitop eine Signalkaskade ausgelöst wurde
(Solforosi et al., 2004).
Diese Beobachtungen sprechen dafür, dass passive Immunisierungsstrategien mit
humanisierten Antikörpern eher zur Postexpositionsprophylaxe geeignet sind, d.h. wenn PrP für
die im Blut zirkulierenden Antikörpern noch zugänglich ist. Im Falle von asymptomatischen
Trägern pathogener PRNP-Mutationen oder bestimmten Risikogruppen wie z.B. Empfänger von
vCJD-kontaminiertem Blut könnte eine postprophylaktische Behandlung mit Anti-PrP-
Antikörpern zu therapeutischen Erfolgen führen.
Gentherapeutische Strategien bieten eine Möglichkeit, um die oben genannten Limitationen zu
umgehen. Kürzlich wurde gezeigt, dass eine humane Zelllinie, die kontinuierlich scFv des
monoklonalen Antikörpers 6H4 sezernierte, nach Kokultivierung mit ScN2a-Zellen zu einer
parakrinen Hemmung der Prionenpropagation führte (Donofrio et al., 2005). Solche genetisch
veränderten Zellen bieten den Vorteil, dass sie in die Nähe der Zielstruktur implantiert werden
können und dort über einen längeren Zeitraum kontinuierlich Antikörper bzw.
Antikörperfragmente sezernieren.
Um diese Zellen vor dem Immunsystem des Empfängers zu schützen, können sie zusätzlich in
polykationische Kapseln eingeschlossen werden. Eine semipermeable Membran gewährleistet
dann, dass die Zellen mit Nährstoffen versorgt werden und die sezernierten Antikörper in den
Metabolismus gelangen (Orive et al., 2003). Diese Technologie wurde bereits für die Krankheit
Chorea Huntington in einer Phase I Studie untersucht (Bachoud-Levi et al., 2000; Bloch et al.,
2004).
Im Hinblick auf diese Untersuchungen soll in zukünftigen Experimenten der Anti-PrP-Antikörper
B2-43-133 kloniert werden und eine Zelllinie etabliert werden, welche diesen Antikörper
kontinuierlich sezerniert und für therapeutische Zwecke einsetzbar ist.

ZUSAMMENFASSUNG
120
5 Zusammenfassung
5.1 Deutsch
Der Verlauf von Prionenerkrankungen wird durch die Akkumulation einer abnormal gefalteten
Isoform des zellulären Prion-Proteins PrPC bestimmt. Diese infektiöse Isoform, die PrPSc
genannt wird, entsteht, indem sie mit PrPC interagiert und dieses die Konformation von PrPSc
übernimmt. Die Konversion des zellulären Prion-Proteins PrPC in seine pathogene Isoform
PrPSc und die damit verbundene PrPSc-Akkumulation sind demnach die wesentlichen Merkmale
von Prionenerkrankungen und bieten mögliche therapeutische Ansatzpunkte. Studien aus den
letzten Jahren haben gezeigt, dass Antikörper, die gegen PrPC und/oder PrPSc gerichtet sind, in
vitro und in vivo in den Konversionsprozess eingreifen können und so die PrPSc-Akkumulation
inhibieren (Enari et al., 2001; Feraudet et al., 2005b; Kim et al., 2004; Miyamoto et al., 2005;
Pankiewicz et al., 2006; White et al., 2003). Das Ziel der vorliegenden Arbeit war somit die
Generation neuer Anti-PrP-Antikörper, welche die PrPSc-Konversion effizient hemmen können
und somit die Auswahl an Anti-PrP-Antikörpern für therapeutische und diagnostische Zwecke zu
erweitern.
Insgesamt wurden sieben Anti-PrP-Antikörper mittels „Hybridom“-Technik hergestellt. Zwei der
Antikörper (B2-31-166 und B2-43-133) resultierten aus einer Fusion mit Milzzellen von Prnp0/0-
Mäusen, die zuvor mit fibrillärem PrP immunisiert wurden. Bei fünf Antikörpern (7-12-5, 12-29,
48-11-5, 103-8 und 110-10) wurde für die Immunisierung rekombinantes PrP verwendet.
Alle Antikörper detektierten rekombinantes PrP sowie natives und denaturiertes PrPC und PrPSc.
Die bestimmten Aviditäten und die relativ einheitlichen Dissoziationskonstanten (KD-Konstante)
waren mit kommerziellen Referenzantikörpern vergleichbar. Das Epitop des Antikörpers
B2-31-166 wurde im unstrukturierten N-Terminus von PrP lokalisiert (Reste 96-110), während
die Antikörper 7-12-5, 12-29 und 48-11-5 PrP im globulären C-Terminus binden (Reste 158-
176). Das Epitop der Antikörper 103-8, 110-10 und B2-43-133 wurde ebenfalls im C-Terminus
bestimmt (Reste 142-157). Diese Antikörper binden PrP im Bereich der Helix 1 (Reste 144-
152), einem Epitop, das bereits häufiger für inhibitorisch wirksame Antikörper bestimmt wurde.
Drei der sieben Anti-PrP-Antikörper (7-12-5, 12-29 und B2-31-166) hemmten die
PrPSc-Akkumulation in Prion-infizierten N2a-Zellen (ScN2a-Zellen) nicht. Der Antikörper 48-11-5
führte zu einer unvollständigen Reduktion des PrPSc-Gehalts, die sich auch durch die
Verlängerung des Applikationszeitraums nicht verstärken ließ. Dagegen inhibierten die
Antikörper 103-8, 110-10 und B2-43-133 die PrPSc-Akkumulation vollständig. Dieser Effekt
wurde auf eine spezifische Interaktion der Antikörper mit PrP zurückgeführt, da ein
zytotoxischer Effekt, der den PrPSc-Gehalt unspezifisch hätte verringern können, nicht
beobachtet wurde.

ZUSAMMENFASSUNG
121
Bei der Analyse der Infektiosität von ScN2a-Zellen zeigte sich jedoch, dass der Antikörper
110-10 zwar die PrPSc-Akkumulation inhibierte, aber die Infektiosität der Zellen nicht reduzieren
konnte. Die Behandlung mit dem Antikörper 103-8 reduzierte die Infektiosität der ScN2a-Zellen
zwar, jedoch setzte 30 Tage nach Abschluss der Behandlung eine erneute PrPSc-Akkumulation
in ScN2a-Zellen ein. Dagegen reduzierte die Behandlung mit dem Antikörper B2-43-133 die
Infektiosität der ScN2a-Zellen vollständig. Des Weiteren wurde bei diesen Zellen auch nach
36 Tagen Kultivierung ohne Antikörper keine erneute PrPSc-Akkumulation detektiert. Die starke
inhibitorische Wirkung dieses Antikörpers wird zusätzlich durch einen sehr niedrigen IC50-Wert
(0,31 nM) unterstützt, der mit kommerziellen Referenzantikörpern vergleichbar ist.
Zusammenfassend sprechen diese Ergebnisse dafür, dass von den sieben neu generierten
Anti-PrP-Antikörpern der Antikörper B2-43-133 ein Kandidat für weiterführende Experimente mit
therapeutischem Hintergrund ist.
Welchen Mechanismus die Anti-PrP-Antikörper für die Inhibition nutzen, konnte nicht geklärt
werden. Obwohl der Antikörper B2-43-133 die Verweildauer von PrPC auf der Zelloberfläche
(Retention) verlängerte, wodurch hypothetisch die gebundenen PrPC-Moleküle der
PrPSc-Konversion entzogen werden können, wurde dies für einen Referenzantikörper, welcher
einen ähnlichen inhibitorischen Effekt besitzt, nicht beobachtet. Dies spricht entweder dafür,
dass die Verlängerung der Retention nur ein weiteres Charakteristikum der Antikörper ist und
ein anderer Mechanismus genutzt wird, oder dafür, dass verschiedene Antikörper verschiedene
Mechanismen nutzen können. Auch die Hypothese, dass die inhibitorische Wirkung des
Antikörpers durch eine Komplexbildung mit PrP vermittelt wird, konnte nicht bestätigt werden,
da eine stabile Komplexbildung des Antikörpers B2-43-133 mit rekombinantem PrP zu einer
Erhöhung des IC50-Werts im Vergleich zu freiem Antikörper führte.

ZUSAMMENFASSUNG
122
5.2 Englisch
The development of prion diseases is characterized by accumulation of an abnormal folded
isoform of the cellular prion protein (PrPC). This infectious isoform, called PrPSc, emerges
through interaction with PrPC adopting the conformation of PrPSc. Thus, conversion of PrPC to its
pathogenic isoform and the resulting PrPSc accumulation are substantial hallmarks of prion
diseases that provide possible therapeutical intervention points. Recent studies have shown
that antibodies recognizing PrPC and/or PrPSc interfere with the conversion process in vitro and
in vivo and inhibit PrPSc accumulation (Enari et al., 2001; Feraudet et al., 2005b; Kim et al.,
2004; Miyamoto et al., 2005; Pankiewicz et al., 2006; White et al., 2003).
This study was initiated to generate new anti-PrP antibodies that could efficiently inhibit the
PrPSc conversion and thereby extend the repertoire of monoclonal antibodies (mAbs) for
therapeutic and diagnostic use. Altogether, seven antibodies could be established by
hybridoma-technique: two mAbs from immunisation of Prnp0/0 mice with fibrillar PrP (B2-31-166
and B2-43-133) and the remainder (7-12-5, 12-29, 48-11-5, 103-8 und 110-10) by using
recombinant PrP as immunogen.
All antibodies were able to detect recombinant PrP as well as native and denaturated PrPC and
PrPSc. The avidities and similar dissociation constants were comparable to commercial
reference antibodies. The epitope of mAb B2-31-166 was determined in the unstructured N-
terminal domain (residues 96-110), whereas mAbs 7-12-5, 12-29 and 48-115 detected an
epitope in the C-terminal globular domain (residues 158-176). The epitopes of mAbs 103-8,
110-10 and B2-43-133 are located in the C-terminus (residues 142-157) as well and correspond
to the α-helix 1 of PrPC (residues 144-152). This region was already determined as a potential
epitope for other inhibitory effective antibodies.
Three antibodies (7-12-5, 12-29 and B2-31-166) did not inhibit PrPSc accumulation in prion-
infected cells (ScN2a cells). MAb 48-11-5 only led to an incomplete reduction of the PrPSc
content. This effect could not be strengthened through extending the treatment period. In
contrast, mAbs 103-8, 110-10 and B2-43-133 were able to completely inhibit the PrPSc
accumulation in ScN2a cells. This effect was attributed to a specific interaction between
antibodies and PrP as no cytotoxic effect was detectable that could lead to an unspecific
reduction of the PrPSc content.
Interestingly, analysis of the infectivity of antibody-treated ScN2a cells showed that mAb 110-10
was indeed able to inhibit PrPSc accumulation, although it did not affect the infectivity of ScN2a
cells. MAb 103-8 reduced the infectivity level, but new PrPSc accumulation could be detected in
ScN2a cells 30 days after the treatment had been finished. Only treatment with mAb B2-43-133
completely eliminated the infectivity of ScN2a cells and no PrPSc could be detected in ScN2a
cells even 36 days after treatment. The strong inhibitory effect of this antibody is supported by a
very low IC50 value (0,31 nM) which is also comparable with commercial anti-PrP antibodies. In

ZUSAMMENFASSUNG
123
summary, these results indicate that anti-PrP antibody B2-43-133 represents a good candidate
for future immunotherapeutic approaches.
Furthermore, it was impossible to clearly identify the underlying mechanisms of the antibodies
for the inhibition of PrPSc accumulation. Although mAb B2-43-133 prolonged the retention of
PrPC on the cell surface, whereby the bound PrPC molecules could hypothetical withdraw the
PrPSc conversion, this could not be observed for commercial antibodies with similar inhibitory
effect. These data imply that the prolongation of the PrP retention is only one feature of mAb
B2-43-133. However, it is possible that different antibodies use different mechanisms to inhibit
PrPSc conversion and that the retention is one of them. In addition, the inhibition of PrPSc
accumulation is probably not mediated by the formation of an antibody-protein complex since
the treatment of ScN2a cells with a stable complex consisting of B2-43-133 and recombinant
PrP complex decreased the inhibitory effect compared to free antibody.

LITERATURVERZEICHNIS
124
6 Literaturverzeichnis Aguzzi, A., und Sigurdson, C. J. (2004). Antiprion immunotherapy: to suppress or to stimulate? Nat Rev Immunol 4, 725-736.
Alper, T., Cramp, W. A., Haig, D. A., und Clarke, M. C. (1967). Does the agent of scrapie replicate without nucleic acid? Nature 214, 764-766.
Alper, T., Haig, D. A., und Clarke, M. C. (1966). The exceptionally small size of the scrapie agent. Biochem Biophys Res Commun 22, 278-284.
Arbel, M., Lavie, V., und Solomon, B. (2003). Generation of antibodies against prion protein in wild-type mice via helix 1 peptide immunization. J Neuroimmunol 144, 38-45.
Arnold, J. E., Tipler, C., Laszlo, L., Hope, J., Landon, M., und Mayer, R. J. (1995). The abnormal isoform of the prion protein accumulates in late-endosome-like organelles in scrapie-infected mouse brain. J Pathol 176, 403-411.
Atassi, M. Z., und Smith, J. A. (1978). A proposal for the nomenclature of antigenic sites in peptides and proteins. Immunochemistry 15, 609-610.
Bachoud-Levi, A. C., Deglon, N., Nguyen, J. P., Bloch, J., Bourdet, C., Winkel, L., Remy, P., Goddard, M., Lefaucheur, J. P., Brugieres, P., et al. (2000). Neuroprotective gene therapy for Huntington's disease using a polymer encapsulated BHK cell line engineered to secrete human CNTF. Hum Gene Ther 11, 1723-1729.
Bard, F., Cannon, C., Barbour, R., Burke, R. L., Games, D., Grajeda, H., Guido, T., Hu, K., Huang, J., Johnson-Wood, K., et al. (2000). Peripherally administered antibodies against amyloid beta-peptide enter the central nervous system and reduce pathology in a mouse model of Alzheimer disease. Nat Med 6, 916-919.
Baron, G. S., und Caughey, B. (2003). Effect of glycosylphosphatidylinositol anchor-dependent and -independent prion protein association with model raft membranes on conversion to the protease-resistant isoform. J Biol Chem 278, 14883-14892.
Barret, A., Tagliavini, F., Forloni, G., Bate, C., Salmona, M., Colombo, L., De Luigi, A., Limido, L., Suardi, S., Rossi, G., et al. (2003). Evaluation of quinacrine treatment for prion diseases. J Virol 77, 8462-8469.
Basler, K., Oesch, B., Scott, M., Westaway, D., Walchli, M., Groth, D. F., McKinley, M. P., Prusiner, S. B., und Weissmann, C. (1986). Scrapie and cellular PrP isoforms are encoded by the same chromosomal gene. Cell 46, 417-428.
Bate, C., Salmona, M., Diomede, L., und Williams, A. (2004). Squalestatin cures prion-infected neurons and protects against prion neurotoxicity. J Biol Chem 279, 14983-14990.
Behring, E. A., und Kitasato, S. (1890). Ueber das zustandekommen der diptherie-immunität und der tetanus-immunität bei thieren. DeutchMedWoch 49, 1113-1114.
Beringue, V., Vilette, D., Mallinson, G., Archer, F., Kaisar, M., Tayebi, M., Jackson, G. S., Clarke, A. R., Laude, H., Collinge, J., und Hawke, S. (2004). PrPSc binding antibodies are potent inhibitors of prion replication in cell lines. J Biol Chem 279, 39671-39676.

LITERATURVERZEICHNIS
125
Bessen, R. A., Kocisko, D. A., Raymond, G. J., Nandan, S., Lansbury, P. T., und Caughey, B. (1995). Non-genetic propagation of strain-specific properties of scrapie prion protein. Nature 375, 698-700.
Bessen, R. A., und Marsh, R. F. (1992). Biochemical and physical properties of the prion protein from two strains of the transmissible mink encephalopathy agent. J Virol 66, 2096-2101.
Birkett, C. R., Hennion, R. M., Bembridge, D. A., Clarke, M. C., Chree, A., Bruce, M. E., und Bostock, C. J. (2001). Scrapie strains maintain biological phenotypes on propagation in a cell line in culture. Embo J 20, 3351-3358.
Bloch, J., Bachoud-Levi, A. C., Deglon, N., Lefaucheur, J. P., Winkel, L., Palfi, S., Nguyen, J. P., Bourdet, C., Gaura, V., Remy, P., et al. (2004). Neuroprotective gene therapy for Huntington's disease, using polymer-encapsulated cells engineered to secrete human ciliary neurotrophic factor: results of a phase I study. Hum Gene Ther 15, 968-975.
Bolton, D. C., McKinley, M. P., und Prusiner, S. B. (1982). Identification of a protein that purifies with the scrapie prion. Science 218, 1309-1311.
Bolton, D. C., Seligman, S. J., Bablanian, G., Windsor, D., Scala, L. J., Kim, K. S., Chen, C. M., Kascsak, R. J., und Bendheim, P. E. (1991). Molecular location of a species-specific epitope on the hamster scrapie agent protein. J Virol 65, 3667-3675.
Borchelt, D. R., Scott, M., Taraboulos, A., Stahl, N., und Prusiner, S. B. (1990). Scrapie and cellular prion proteins differ in their kinetics of synthesis and topology in cultured cells. J Cell Biol 110, 743-752.
Borchelt, D. R., Taraboulos, A., und Prusiner, S. B. (1992). Evidence for synthesis of scrapie prion proteins in the endocytic pathway. J Biol Chem 267, 16188-16199.
Botto, L., Masserini, M., Cassetti, A., und Palestini, P. (2004). Immunoseparation of Prion protein-enriched domains from other detergent-resistant membrane fractions, isolated from neuronal cells. FEBS Lett 557, 143-147.
Boulianne, G. L., Hozumi, N., und Shulman, M. J. (1984). Production of functional chimaeric mouse/human antibody. Nature 312, 643-646.
Bradford, M. M. (1976). A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72, 248-254.
Brown, D. R., Schmidt, B., Groschup, M. H., und Kretzschmar, H. A. (1998). Prion protein expression in muscle cells and toxicity of a prion protein fragment. Eur J Cell Biol 75, 29-37.
Brown, D. R., Wong, B. S., Hafiz, F., Clive, C., Haswell, S. J., und Jones, I. M. (1999a). Normal prion protein has an activity like that of superoxide dismutase. Biochem J 344 Pt 1, 1-5.
Brown, K. L., Stewart, K., Ritchie, D. L., Mabbott, N. A., Williams, A., Fraser, H., Morrison, W. I., und Bruce, M. E. (1999b). Scrapie replication in lymphoid tissues depends on prion protein-expressing follicular dendritic cells. Nat Med 5, 1308-1312.
Brown, P., Preece, M., Brandel, J. P., Sato, T., McShane, L., Zerr, I., Fletcher, A., Will, R. G., Pocchiari, M., Cashman, N. R., et al. (2000). Iatrogenic Creutzfeldt-Jakob disease at the millennium. Neurology 55, 1075-1081.

LITERATURVERZEICHNIS
126
Brown, P., Will, R. G., Bradley, R., Asher, D. M., und Detwiler, L. (2001). Bovine spongiform encephalopathy and variant Creutzfeldt-Jakob disease: background, evolution, and current concerns. Emerg Infect Dis 7, 6-16.
Bruce, M. E. (2003). TSE strain variation. Br Med Bull 66, 99-108.
Bruce, M. E., McConnell, I., Fraser, H., und Dickinson, A. G. (1991). The disease characteristics of different strains of scrapie in Sinc congenic mouse lines: implications for the nature of the agent and host control of pathogenesis. J Gen Virol 72 ( Pt 3), 595-603.
Bruce, M. E., Will, R. G., Ironside, J. W., McConnell, I., Drummond, D., Suttie, A., McCardle, L., Chree, A., Hope, J., Birkett, C., et al. (1997). Transmissions to mice indicate that 'new variant' CJD is caused by the BSE agent. Nature 389, 498-501.
Bueler, H., Aguzzi, A., Sailer, A., Greiner, R. A., Autenried, P., Aguet, M., und Weissmann, C. (1993). Mice devoid of PrP are resistant to scrapie. Cell 73, 1339-1347.
Bueler, H., Fischer, M., Lang, Y., Bluethmann, H., Lipp, H. P., DeArmond, S. J., Prusiner, S. B., Aguet, M., und Weissmann, C. (1992). Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature 356, 577-582.
Burthem, J., Urban, B., Pain, A., und Roberts, D. J. (2001). The normal cellular prion protein is strongly expressed by myeloid dendritic cells. Blood 98, 3733-3738.
Butler, D. A., Scott, M. R., Bockman, J. M., Borchelt, D. R., Taraboulos, A., Hsiao, K. K., Kingsbury, D. T., und Prusiner, S. B. (1988). Scrapie-infected murine neuroblastoma cells produce protease-resistant prion proteins. J Virol 62, 1558-1564.
Cardinale, A., Filesi, I., Vetrugno, V., Pocchiari, M., Sy, M. S., und Biocca, S. (2005). Trapping prion protein in the endoplasmic reticulum impairs PrPC maturation and prevents PrPSc accumulation. J Biol Chem 280, 685-694.
Carter, P. J. (2006). Potent antibody therapeutics by design. Nat Rev Immunol 6, 343-357.
Caughey, B. (1991). In vitro expression and biosynthesis of prion protein. Curr Top Microbiol Immunol 172, 93-107.
Caughey, B., und Baron, G. S. (2006). Prions and their partners in crime. Nature 443, 803-810.
Caughey, B., und Raymond, G. J. (1991). The scrapie-associated form of PrP is made from a cell surface precursor that is both protease- and phospholipase-sensitive. J Biol Chem 266, 18217-18223.
Caughey, B., und Raymond, G. J. (1993). Sulfated polyanion inhibition of scrapie-associated PrP accumulation in cultured cells. J Virol 67, 643-650.
Chabry, J., Priola, S. A., Wehrly, K., Nishio, J., Hope, J., und Chesebro, B. (1999). Species-independent inhibition of abnormal prion protein (PrP) formation by a peptide containing a conserved PrP sequence. J Virol 73, 6245-6250.
Chandler, R. L. (1961). Encephalopathy in mice produced by inoculation with scrapie brain material. Lancet 1, 1378-1379.

LITERATURVERZEICHNIS
127
Chesebro, B., Race, R., Wehrly, K., Nishio, J., Bloom, M., Lechner, D., Bergstrom, S., Robbins, K., Mayer, L., Keith, J. M., und et al. (1985). Identification of scrapie prion protein-specific mRNA in scrapie-infected and uninfected brain. Nature 315, 331-333.
Cohen, F. E., Pan, K. M., Huang, Z., Baldwin, M., Fletterick, R. J., und Prusiner, S. B. (1994). Structural clues to prion replication. Science 264, 530-531.
Cohen, F. E., und Prusiner, S. B. (1998). Pathologic conformations of prion proteins. Annu Rev Biochem 67, 793-819.
Coller, H. A., und Coller, B. S. (1983). Statistical analysis of repetitive subcloning by the limiting dilution technique with a view toward ensuring hybridoma monoclonality. Hybridoma 2, 91-96.
Collinge, J. (1997). Human prion diseases and bovine spongiform encephalopathy (BSE). Hum Mol Genet 6, 1699-1705.
Collinge, J. (2001). Prion diseases of humans and animals: their causes and molecular basis. Annu Rev Neurosci 24, 519-550.
Collinge, J., Beck, J., Campbell, T., Estibeiro, K., und Will, R. G. (1996a). Prion protein gene analysis in new variant cases of Creutzfeldt-Jakob disease. Lancet 348, 56.
Collinge, J., Sidle, K. C., Meads, J., Ironside, J., und Hill, A. F. (1996b). Molecular analysis of prion strain variation and the aetiology of 'new variant' CJD. Nature 383, 685-690.
Collinge, J., Whitfield, J., McKintosh, E., Beck, J., Mead, S., Thomas, D. J., und Alpers, M. P. (2006). Kuru in the 21st century--an acquired human prion disease with very long incubation periods. Lancet 367, 2068-2074.
Collinge, J., Whittington, M. A., Sidle, K. C., Smith, C. J., Palmer, M. S., Clarke, A. R., und Jefferys, J. G. (1994). Prion protein is necessary for normal synaptic function. Nature 370, 295-297.
Creutzfeldt, H. G. (1920). Über eine eigenartige Erkrankung des Zentralnervensystems. Vorläufige Mitteilung Z f d ges Neurol und Psych, 1-18.
Crozet, C., Lin, Y. L., Mettling, C., Mourton-Gilles, C., Corbeau, P., Lehmann, S., und Perrier, V. (2004). Inhibition of PrPSc formation by lentiviral gene transfer of PrP containing dominant negative mutations. J Cell Sci 117, 5591-5597.
Cuillé, J., und Chelle, P.-L. (1936). La maladie dite "tremblante" du mouton: est-elle inoculable? Compte Rend Acad Sci 203, 1552.
Daude, N., Marella, M., und Chabry, J. (2003). Specific inhibition of pathological prion protein accumulation by small interfering RNAs. J Cell Sci 116, 2775-2779.
Deleault, N. R., Lucassen, R. W., und Supattapone, S. (2003). RNA molecules stimulate prion protein conversion. Nature 425, 717-720.
DeMattos, R. B., Bales, K. R., Cummins, D. J., Dodart, J. C., Paul, S. M., und Holtzman, D. M. (2001). Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A 98, 8850-8855.

LITERATURVERZEICHNIS
128
Donofrio, G., Heppner, F. L., Polymenidou, M., Musahl, C., und Aguzzi, A. (2005). Paracrine inhibition of prion propagation by anti-PrP single-chain Fv miniantibodies. J Virol 79, 8330-8338.
Duffy, P., Wolf, J., Collins, G., DeVoe, A. G., Streeten, B., und Cowen, D. (1974). Letter: Possible person-to-person transmission of Creutzfeldt-Jakob disease. N Engl J Med 290, 692-693.
Eghiaian, F. (2005). Structuring the puzzle of prion propagation. Curr Opin Struct Biol 15, 724-730.
Eghiaian, F., Grosclaude, J., Lesceu, S., Debey, P., Doublet, B., Treguer, E., Rezaei, H., und Knossow, M. (2004). Insight into the PrPC-->PrPSc conversion from the structures of antibody-bound ovine prion scrapie-susceptibility variants. Proc Natl Acad Sci U S A 101, 10254-10259.
Enari, M., Flechsig, E., und Weissmann, C. (2001). Scrapie prion protein accumulation by scrapie-infected neuroblastoma cells abrogated by exposure to a prion protein antibody. Proc Natl Acad Sci U S A 98, 9295-9299.
Ertmer, A., Gilch, S., Yun, S. W., Flechsig, E., Klebl, B., Stein-Gerlach, M., Klein, M. A., und Schatzl, H. M. (2004). The tyrosine kinase inhibitor STI571 induces cellular clearance of PrPSc in prion-infected cells. J Biol Chem 279, 41918-41927.
Feraudet, C., Lacroux, C., Morel, N., Simon, S., Volland, H., Frobert, Y., Créminon, C., Andréoletti, O., und Grassi, J. (2005a). In vivo immunotherapeutic effect of anti-PrP monoclonal antibodies on mouse models of TSE infection: pharmacodynamic and pharmacokinetic analysis, Paper presented at: Prion 2005 Between fundamentals and society´s need (Düsseldorf).
Feraudet, C., Morel, N., Simon, S., Volland, H., Frobert, Y., Creminon, C., Vilette, D., Lehmann, S., und Grassi, J. (2005b). Screening of 145 anti-PrP monoclonal antibodies for their capacity to inhibit PrPSc replication in infected cells. J Biol Chem 280, 11247-11258.
Fischer, M., Rulicke, T., Raeber, A., Sailer, A., Moser, M., Oesch, B., Brandner, S., Aguzzi, A., und Weissmann, C. (1996). Prion protein (PrP) with amino-proximal deletions restoring susceptibility of PrP knockout mice to scrapie. Embo J 15, 1255-1264.
Flechsig, E., Shmerling, D., Hegyi, I., Raeber, A. J., Fischer, M., Cozzio, A., von Mering, C., Aguzzi, A., und Weissmann, C. (2000). Prion protein devoid of the octapeptide repeat region restores susceptibility to scrapie in PrP knockout mice. Neuron 27, 399-408.
Ford, B. J. (1996). BSE - the facts (mad cow disease and the risk of mankind). (London: Gorgi Books).
Gabizon, R., McKinley, M. P., Groth, D., und Prusiner, S. B. (1988). Immunoaffinity purification and neutralization of scrapie prion infectivity. Proc Natl Acad Sci U S A 85, 6617-6621.
Gajdusek, D. C. (1996). Infectious amyloids: subacute spongiform encephalopathies as transmissible cerebral amyloidoses (Philadelphia Pennsylvania: Lippencott-Raven Publishers). Gajdusek, D. C., und Zigas, V. (1957). Degenerative disease of the central nervous system in New Guinea; the endemic occurrence of kuru in the native population. N Engl J Med 257, 974-978.

LITERATURVERZEICHNIS
129
Gauczynski, S., Peyrin, J. M., Haik, S., Leucht, C., Hundt, C., Rieger, R., Krasemann, S., Deslys, J. P., Dormont, D., Lasmezas, C. I., und Weiss, S. (2001). The 37-kDa/67-kDa laminin receptor acts as the cell-surface receptor for the cellular prion protein. Embo J 20, 5863-5875.
Gerstmann, J., Sträussler, E., und Scheinker, I. (1936). Über eine eigenartige hereditär-familiäre Erkrankung des Zentralnervensystems. Z Neurol Psych 154, 736-762.
Gibbs, C. J., Jr., und Bolis, C. L. (1997). Normal isoform of amyloid protein (PrP) in brains of spawning salmon. Mol Psychiatry 2, 146-147.
Gibbs, C. J., Jr., Gajdusek, D. C., Asher, D. M., Alpers, M. P., Beck, E., Daniel, P. M., und Matthews, W. B. (1968). Creutzfeldt-Jakob disease (spongiform encephalopathy): transmission to the chimpanzee. Science 161, 388-389.
Gilch, S., Winklhofer, K. F., Groschup, M. H., Nunziante, M., Lucassen, R., Spielhaupter, C., Muranyi, W., Riesner, D., Tatzelt, J., und Schatzl, H. M. (2001). Intracellular re-routing of prion protein prevents propagation of PrP(Sc) and delays onset of prion disease. Embo J 20, 3957-3966.
Gilch, S., Wopfner, F., Renner-Muller, I., Kremmer, E., Bauer, C., Wolf, E., Brem, G., Groschup, M. H., und Schatzl, H. M. (2003). Polyclonal anti-PrP auto-antibodies induced with dimeric PrP interfere efficiently with PrPSc propagation in prion-infected cells. J Biol Chem 278, 18524-18531.
Gilman, S., Koller, M., Black, R. S., Jenkins, L., Griffith, S. G., Fox, N. C., Eisner, L., Kirby, L., Rovira, M. B., Forette, F., und Orgogozo, J. M. (2005). Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology 64, 1553-1562.
Goldfarb, L. G., Brown, P., Haltia, M., Cathala, F., McCombie, W. R., Kovanen, J., Cervenakova, L., Goldin, L., Nieto, A., Godec, M. S., und et al. (1992). Creutzfeldt-Jakob disease cosegregates with the codon 178Asn PRNP mutation in families of European origin. Ann Neurol 31, 274-281.
Goldfarb, L. G., Korczyn, A. D., Brown, P., Chapman, J., und Gajdusek, D. C. (1990a). Mutation in codon 200 of scrapie amyloid precursor gene linked to Creutzfeldt-Jakob disease in Sephardic Jews of Libyan and non-Libyan origin. Lancet 336, 637-638.
Goldfarb, L. G., Mitrova, E., Brown, P., Toh, B. K., und Gajdusek, D. C. (1990b). Mutation in codon 200 of scrapie amyloid protein gene in two clusters of Creutzfeldt-Jakob disease in Slovakia. Lancet 336, 514-515.
Goni, F., Knudsen, E., Schreiber, F., Scholtzova, H., Pankiewicz, J., Carp, R., Meeker, H. C., Rubenstein, R., Brown, D. R., Sy, M. S., et al. (2005). Mucosal vaccination delays or prevents prion infection via an oral route. Neuroscience 133, 413-421.
Govaerts, C., Wille, H., Prusiner, S. B., und Cohen, F. E. (2004). Evidence for assembly of prions with left-handed beta-helices into trimers. Proc Natl Acad Sci U S A 101, 8342-8347.
Graham, F. L., Smiley, J., Russell, W. C., und Nairn, R. (1977). Characteristics of a human cell line transformed by DNA from human adenovirus type 5. J Gen Virol 36, 59-74.
Graner, E., Mercadante, A. F., Zanata, S. M., Forlenza, O. V., Cabral, A. L., Veiga, S. S., Juliano, M. A., Roesler, R., Walz, R., Minetti, A., et al. (2000). Cellular prion protein binds laminin and mediates neuritogenesis. Brain Res Mol Brain Res 76, 85-92.

LITERATURVERZEICHNIS
130
Green, L. L., Hardy, M. C., Maynard-Currie, C. E., Tsuda, H., Louie, D. M., Mendez, M. J., Abderrahim, H., Noguchi, M., Smith, D. H., Zeng, Y., und et al. (1994). Antigen-specific human monoclonal antibodies from mice engineered with human Ig heavy and light chain YACs. Nat Genet 7, 13-21.
Griffith, J. S. (1967). Self-replication und scrapie. Nature 215, 1043-1044.
Haik, S., Brandel, J. P., Salomon, D., Sazdovitch, V., Delasnerie-Laupretre, N., Laplanche, J. L., Faucheux, B. A., Soubrie, C., Boher, E., Belorgey, C., et al. (2004). Compassionate use of quinacrine in Creutzfeldt-Jakob disease fails to show significant effects. Neurology 63, 2413-2415.
Hanly, W. C., Artwohl, J. E., und Bennett, B. T. (1995). Review of Polyclonal Antibody Production Procedures in Mammals and Poultry. Ilar J 37, 93-118.
Hanson, R. P., Eckroade, R. J., Marsh, R. F., Zu Rhein, G. M., Kanitz, C. L., und Gustafson, D. P. (1971). Susceptibility of mink to sheep scrapie. Science 172, 859-861.
Hansson, M. (1990). Growth and differentiation factors for B and T cells. Leuk Res 14, 705-710.
Hao, M., und Maxfield, F. R. (2000). Characterization of rapid membrane internalization and recycling. J Biol Chem 275, 15279-15286.
Haraguchi, T., Fisher, S., Olofsson, S., Endo, T., Groth, D., Tarentino, A., Borchelt, D. R., Teplow, D., Hood, L., Burlingame, A., und et al. (1989). Asparagine-linked glycosylation of the scrapie and cellular prion proteins. Arch Biochem Biophys 274, 1-13.
Harris, D. A., Falls, D. L., Johnson, F. A., und Fischbach, G. D. (1991). A prion-like protein from chicken brain copurifies with an acetylcholine receptor-inducing activity. Proc Natl Acad Sci U S A 88, 7664-7668.
Harris, D. A., Gorodinsky, A., Lehmann, S., Moulder, K., und Shyng, S. L. (1996). Cell biology of the prion protein. Curr Top Microbiol Immunol 207, 77-93.
Head, M. W., und Ironside, J. W. (2000). Inhibition of prion-protein conversion: a therapeutic tool? Trends Microbiol 8, 6-8.
Heikenwalder, M., Polymenidou, M., Junt, T., Sigurdson, C., Wagner, H., Akira, S., Zinkernagel, R., und Aguzzi, A. (2004). Lymphoid follicle destruction and immunosuppression after repeated CpG oligodeoxynucleotide administration. Nat Med 10, 187-192.
Heppner, F. L., Musahl, C., Arrighi, I., Klein, M. A., Rulicke, T., Oesch, B., Zinkernagel, R. M., Kalinke, U., und Aguzzi, A. (2001). Prevention of scrapie pathogenesis by transgenic expression of anti-prion protein antibodies. Science 294, 178-182.
Herbert, W. J., und Kristensen, F. (1986). Laboratory animal techniques for immunology, Vol 4, 4 edn (Oxford: Blackwell Scientific ).
Hilton, D. A. (2006). Pathogenesis and prevalence of variant Creutzfeldt-Jakob disease. J Pathol 208, 134-141.
Hock, C., Konietzko, U., Streffer, J. R., Tracy, J., Signorell, A., Muller-Tillmanns, B., Lemke, U., Henke, K., Moritz, E., Garcia, E., et al. (2003). Antibodies against beta-amyloid slow cognitive decline in Alzheimer's disease. Neuron 38, 547-554.

LITERATURVERZEICHNIS
131
Hope, J., Morton, L. J., Farquhar, C. F., Multhaup, G., Beyreuther, K., und Kimberlin, R. H. (1986). The major polypeptide of scrapie-associated fibrils (SAF) has the same size, charge distribution and N-terminal protein sequence as predicted for the normal brain protein (PrP). Embo J 5, 2591-2597.
Horiuchi, M., und Caughey, B. (1999). Specific binding of normal prion protein to the scrapie form via a localized domain initiates its conversion to the protease-resistant state. Embo J 18, 3193-3203.
Hornemann, S., Korth, C., Oesch, B., Riek, R., Wider, G., Wuthrich, K., und Glockshuber, R. (1997). Recombinant full-length murine prion protein, mPrP(23-231): purification and spectroscopic characterization. FEBS Lett 413, 277-281.
Hsiao, K., Baker, H. F., Crow, T. J., Poulter, M., Owen, F., Terwilliger, J. D., Westaway, D., Ott, J., und Prusiner, S. B. (1989). Linkage of a prion protein missense variant to Gerstmann-Straussler syndrome. Nature 338, 342-345.
Huang, Z., Prusiner, S. B., und Cohen, F. E. (1996). Scrapie prions: a three-dimensional model of an infectious fragment. Fold Des 1, 13-19.
Hutter, G., Heppner, F. L., und Aguzzi, A. (2003). No superoxide dismutase activity of cellular prion protein in vivo. Biol Chem 384, 1279-1285.
Ironside, J. W., und Head, M. W. (2004). Neuropathology and molecular biology of variant Creutzfeldt-Jakob disease. Curr Top Microbiol Immunol 284, 133-159.
Jackson, G. S., Murray, I., Hosszu, L. L., Gibbs, N., Waltho, J. P., Clarke, A. R., und Collinge, J. (2001). Location and properties of metal-binding sites on the human prion protein. Proc Natl Acad Sci U S A 98, 8531-8535.
Janeway, C. A., Travers, P., Walport, M., und Shlomchik, M. (2005). Immunobiology: the immune system in health and disease, 6 edn (Oxford: Garland Science Publishing).
Janus, C., Pearson, J., McLaurin, J., Mathews, P. M., Jiang, Y., Schmidt, S. D., Chishti, M. A., Horne, P., Heslin, D., French, J., et al. (2000). A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer's disease. Nature 408, 979-982.
Jarrett, J. T., und Lansbury, P. T., Jr. (1993). Seeding "one-dimensional crystallization" of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie? Cell 73, 1055-1058.
Jones, C. E., Abdelraheim, S. R., Brown, D. R., und Viles, J. H. (2004). Preferential Cu2+ coordination by His96 and His111 induces beta-sheet formation in the unstructured amyloidogenic region of the prion protein. J Biol Chem 279, 32018-32027.
Jones, P. T., Dear, P. H., Foote, J., Neuberger, M. S., und Winter, G. (1986). Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature 321, 522-525.
Kabat, E. A., Wu, T. T., und Bilofsky, H. (1977). Unusual distributions of amino acids in complementarity-determining (hypervariable) segments of heavy and light chains of immunoglobulins and their possible roles in specificity of antibody-combining sites. J Biol Chem 252, 6609-6616.

LITERATURVERZEICHNIS
132
Kaneko, K., Zulianello, L., Scott, M., Cooper, C. M., Wallace, A. C., James, T. L., Cohen, F. E., und Prusiner, S. B. (1997). Evidence for protein X binding to a discontinuous epitope on the cellular prion protein during scrapie prion propagation. Proc Natl Acad Sci U S A 94, 10069-10074.
Kascsak, R. J., Rubenstein, R., Merz, P. A., Tonna-DeMasi, M., Fersko, R., Carp, R. I., Wisniewski, H. M., und Diringer, H. (1987). Mouse polyclonal and monoclonal antibody to scrapie-associated fibril proteins. J Virol 61, 3688-3693.
Kellings, K., Meyer, N., Mirenda, C., Prusiner, S. B., und Riesner, D. (1992). Further analysis of nucleic acids in purified scrapie prion preparations by improved return refocusing gel electrophoresis. J Gen Virol 73 ( Pt 4), 1025-1029.
Kim, C. L., Karino, A., Ishiguro, N., Shinagawa, M., Sato, M., und Horiuchi, M. (2004). Cell-surface retention of PrPC by anti-PrP antibody prevents protease-resistant PrP formation. J Gen Virol 85, 3473-3482.
Kishida, H., Sakasegawa, Y., Watanabe, K., Yamakawa, Y., Nishijima, M., Kuroiwa, Y., Hachiya, N. S., und Kaneko, K. (2004). Non-glycosylphosphatidylinositol (GPI)-anchored recombinant prion protein with dominant-negative mutation inhibits PrPSc replication in vitro. Amyloid 11, 14-20.
Kocisko, D. A., Come, J. H., Priola, S. A., Chesebro, B., Raymond, G. J., Lansbury, P. T., und Caughey, B. (1994). Cell-free formation of protease-resistant prion protein. Nature 370, 471-474.
Kocisko, D. A., Priola, S. A., Raymond, G. J., Chesebro, B., Lansbury, P. T., Jr., und Caughey, B. (1995). Species specificity in the cell-free conversion of prion protein to protease-resistant forms: a model for the scrapie species barrier. Proc Natl Acad Sci U S A 92, 3923-3927.
Kohler, G., und Milstein, C. (1975). Continuous cultures of fused cells secreting antibody of predefined specificity. Nature 256, 495-497.
Korth, C., May, B. C., Cohen, F. E., und Prusiner, S. B. (2001). Acridine and phenothiazine derivatives as pharmacotherapeutics for prion disease. Proc Natl Acad Sci U S A 98, 9836-9841.
Korth, C., Stierli, B., Streit, P., Moser, M., Schaller, O., Fischer, R., Schulz-Schaeffer, W., Kretzschmar, H., Raeber, A., Braun, U., et al. (1997). Prion (PrPSc)-specific epitope defined by a monoclonal antibody. Nature 390, 74-77.
Kovanen, J. (1993). Clinical characteristics of familial and sporadic Creutzfeldt-Jakob disease in Finland. Acta Neurol Scand 87, 469-474.
Kretzschmar, H. A., Prusiner, S. B., Stowring, L. E., und DeArmond, S. J. (1986). Scrapie prion proteins are synthesized in neurons. Am J Pathol 122, 1-5.
Krieg, A. M., Yi, A. K., Matson, S., Waldschmidt, T. J., Bishop, G. A., Teasdale, R., Koretzky, G. A., und Klinman, D. M. (1995). CpG motifs in bacterial DNA trigger direct B-cell activation. Nature 374, 546-549.
Kurschner, C., und Morgan, J. I. (1995). The cellular prion protein (PrP) selectively binds to Bcl-2 in the yeast two-hybrid system. Brain Res Mol Brain Res 30, 165-168.

LITERATURVERZEICHNIS
133
Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680-685.
Laine, J., Marc, M. E., Sy, M. S., und Axelrad, H. (2001). Cellular and subcellular morphological localization of normal prion protein in rodent cerebellum. Eur J Neurosci 14, 47-56.
Lasmezas, C. I., Deslys, J. P., Demaimay, R., Adjou, K. T., Lamoury, F., Dormont, D., Robain, O., Ironside, J., und Hauw, J. J. (1996). BSE transmission to macaques. Nature 381, 743-744.
Leclerc, E., Serban, H., Prusiner, S. B., Burton, D. R., und Williamson, R. A. (2006). Copper induces conformational changes in the N-terminal part of cell-surface PrPC. Arch Virol 151, 2103-2109.
Lee, E. B., Leng, L. Z., Zhang, B., Kwong, L., Trojanowski, J. Q., Abel, T., und Lee, V. M. (2006). Targeting amyloid-beta peptide (Abeta) oligomers by passive immunization with a conformation-selective monoclonal antibody improves learning and memory in Abeta precursor protein (APP) transgenic mice. J Biol Chem 281, 4292-4299.
Legname, G., Baskakov, I. V., Nguyen, H. O., Riesner, D., Cohen, F. E., DeArmond, S. J., und Prusiner, S. B. (2004). Synthetic mammalian prions. Science 305, 673-676.
Lehto, M., Ashman, D. A., und Cashman, N. R. (2004). Treatment of ScN2a cells with prionspecific YYR antibodies, Paper presented at: Proceedings of the First International Conference of the European Network of Excellence Neuroprion (Paris, Frankreich).
Lipford, G. B., Heeg, K., und Wagner, H. (1998). Bacterial DNA as immune cell activator. Trends Microbiol 6, 496-500.
Littlefield, J. W. (1964). Selection of Hybrids from Matings of Fibroblasts in Vitro and Their Presumed Recombinants. Science 145, 709-710.
Liu, T., Li, R., Wong, B. S., Liu, D., Pan, T., Petersen, R. B., Gambetti, P., und Sy, M. S. (2001). Normal cellular prion protein is preferentially expressed on subpopulations of murine hemopoietic cells. J Immunol 166, 3733-3742.
Llewelyn, C. A., Hewitt, P. E., Knight, R. S., Amar, K., Cousens, S., Mackenzie, J., und Will, R. G. (2004). Possible transmission of variant Creutzfeldt-Jakob disease by blood transfusion. Lancet 363, 417-421.
Lugaresi, E., Montagna, P., Baruzzi, A., Cortelli, P., Tinuper, P., Zucconi, M., Gambetti, P. L., und Medori, R. (1986). [Familial insomnia with a malignant course: a new thalamic disease]. Rev Neurol (Paris) 142, 791-792.
Luhrs, T., Zahn, R., und Wuthrich, K. (2006). Amyloid formation by recombinant full-length prion proteins in phospholipid bicelle solutions. J Mol Biol 357, 833-841.
Lund, C., Olsen, C. M., Tveit, H., und Tranulis, M. A. (2007). Characterization of the prion protein 3F4 epitope and its use as a molecular tag. J Neurosci Methods 165, 183-190.
Madore, N., Smith, K. L., Graham, C. H., Jen, A., Brady, K., Hall, S., und Morris, R. (1999). Functionally different GPI proteins are organized in different domains on the neuronal surface. Embo J 18, 6917-6926.

LITERATURVERZEICHNIS
134
Magri, G., Clerici, M., Dall'Ara, P., Biasin, M., Caramelli, M., Casalone, C., Giannino, M. L., Longhi, R., Piacentini, L., Della Bella, S., et al. (2005). Decrease in pathology and progression of scrapie after immunisation with synthetic prion protein peptides in hamsters. Vaccine 23, 2862-2868.
Mallucci, G., Dickinson, A., Linehan, J., Klohn, P. C., Brandner, S., und Collinge, J. (2003). Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science 302, 871-874.
Masliah, E., Hansen, L., Adame, A., Crews, L., Bard, F., Lee, C., Seubert, P., Games, D., Kirby, L., und Schenk, D. (2005). Abeta vaccination effects on plaque pathology in the absence of encephalitis in Alzheimer disease. Neurology 64, 129-131.
Mayer, R. J., Landon, M., Laszlo, L., Lennox, G., und Lowe, J. (1992). Protein processing in lysosomes: the new therapeutic target in neurodegenerative disease. Lancet 340, 156-159.
McCafferty, J., Griffiths, A. D., Winter, G., und Chiswell, D. J. (1990). Phage antibodies: filamentous phage displaying antibody variable domains. Nature 348, 552-554.
McGowan, J. P. (1922). Scrapie in sheep. ScottJAgric 5, 365-375.
McKinley, M. P., Bolton, D. C., und Prusiner, S. B. (1983). A protease-resistant protein is a structural component of the scrapie prion. Cell 35, 57-62.
McKinley, M. P., Meyer, R. K., Kenaga, L., Rahbar, F., Cotter, R., Serban, A., und Prusiner, S. B. (1991). Scrapie prion rod formation in vitro requires both detergent extraction and limited proteolysis. J Virol 65, 1340-1351.
McLaurin, J., Cecal, R., Kierstead, M. E., Tian, X., Phinney, A. L., Manea, M., French, J. E., Lambermon, M. H., Darabie, A. A., Brown, M. E., et al. (2002). Therapeutically effective antibodies against amyloid-beta peptide target amyloid-beta residues 4-10 and inhibit cytotoxicity and fibrillogenesis. Nat Med 8, 1263-1269.
Mead, S. (2006). Prion disease genetics. Eur J Hum Genet 14, 273-281.
Meier, P., Genoud, N., Prinz, M., Maissen, M., Rulicke, T., Zurbriggen, A., Raeber, A. J., und Aguzzi, A. (2003). Soluble dimeric prion protein binds PrP(Sc) in vivo and antagonizes prion disease. Cell 113, 49-60.
Mestel, R. (1996). Putting prions to the test. Science 273, 184-189.
Mian, I. S., Bradwell, A. R., und Olson, A. J. (1991). Structure, function and properties of antibody binding sites. J Mol Biol 217, 133-151.
Miller, M. W., Williams, E. S., McCarty, C. W., Spraker, T. R., Kreeger, T. J., Larsen, C. T., und Thorne, E. T. (2000). Epizootiology of chronic wasting disease in free-ranging cervids in Colorado and Wyoming. J Wildl Dis 36, 676-690.
Miyamoto, K., Nakamura, N., Aosasa, M., Nishida, N., Yokoyama, T., Horiuchi, H., Furusawa, S., und Matsuda, H. (2005). Inhibition of prion propagation in scrapie-infected mouse neuroblastoma cell lines using mouse monoclonal antibodies against prion protein. Biochem Biophys Res Commun 335, 197-204.

LITERATURVERZEICHNIS
135
Monsonego, A., und Weiner, H. L. (2003). Immunotherapeutic approaches to Alzheimer's disease. Science 302, 834-838.
Monsonego, A., Zota, V., Karni, A., Krieger, J. I., Bar-Or, A., Bitan, G., Budson, A. E., Sperling, R., Selkoe, D. J., und Weiner, H. L. (2003). Increased T cell reactivity to amyloid beta protein in older humans and patients with Alzheimer disease. J Clin Invest 112, 415-422.
Morel, E., Andrieu, T., Casagrande, F., Gauczynski, S., Weiss, S., Grassi, J., Rousset, M., Dormont, D., und Chambaz, J. (2005). Bovine prion is endocytosed by human enterocytes via the 37 kDa/67 kDa laminin receptor. Am J Pathol 167, 1033-1042.
Morel, E., Fouquet, S., Chateau, D., Yvernault, L., Frobert, Y., Pincon-Raymond, M., Chambaz, J., Pillot, T., und Rousset, M. (2004). The cellular prion protein PrPc is expressed in human enterocytes in cell-cell junctional domains. J Biol Chem 279, 1499-1505.
Moroncini, G., Kanu, N., Solforosi, L., Abalos, G., Telling, G. C., Head, M., Ironside, J., Brockes, J. P., Burton, D. R., und Williamson, R. A. (2004). Motif-grafted antibodies containing the replicative interface of cellular PrP are specific for PrPSc. Proc Natl Acad Sci U S A 101, 10404-10409.
Moroncini, G., Mangieri, M., Morbin, M., Mazzoleni, G., Ghetti, B., Gabrielli, A., Williamson, R. A., Giaccone, G., und Tagliavini, F. (2006). Pathologic prion protein is specifically recognized in situ by a novel PrP conformational antibody. Neurobiol Dis 23, 717-724.
Moser, M., Colello, R. J., Pott, U., und Oesch, B. (1995). Developmental expression of the prion protein gene in glial cells. Neuron 14, 509-517.
Mouillet-Richard, S., Ermonval, M., Chebassier, C., Laplanche, J. L., Lehmann, S., Launay, J. M., und Kellermann, O. (2000). Signal transduction through prion protein. Science 289, 1925-1928.
Mullis, K., Faloona, F., Scharf, S., Saiki, R., Horn, G., und Erlich, H. (1986). Specific enzymatic amplification of DNA in vitro: the polymerase chain reaction. Cold Spring Harb Symp Quant Biol 51 Pt 1, 263-273.
Nakajima, M., Yamada, T., Kusuhara, T., Furukawa, H., Takahashi, M., Yamauchi, A., und Kataoka, Y. (2004). Results of quinacrine administration to patients with Creutzfeldt-Jakob disease. Dement Geriatr Cogn Disord 17, 158-163.
Naslavsky, N., Stein, R., Yanai, A., Friedlander, G., und Taraboulos, A. (1997). Characterization of detergent-insoluble complexes containing the cellular prion protein and its scrapie isoform. J Biol Chem 272, 6324-6331.
Nikles, D., Bach, P., Boller, K., Merten, C. A., Montrasio, F., Heppner, F. L., Aguzzi, A., Cichutek, K., Kalinke, U., und Buchholz, C. J. (2005). Circumventing tolerance to the prion protein (PrP): vaccination with PrP-displaying retrovirus particles induces humoral immune responses against the native form of cellular PrP. J Virol 79, 4033-4042.
Nitschke, C., Flechsig, E., van den Brandt, J., Lindner, N., Luhrs, T., Dittmer, U., und Klein, M. A. (2007). Immunisation strategies against prion diseases: Prime-boost immunisation with a PrP DNA vaccine containing foreign helper T-cell epitopes does not prevent mouse scrapie. Vet Microbiol 123, 367-376.

LITERATURVERZEICHNIS
136
Norstrom, E. M., und Mastrianni, J. A. (2006). The charge structure of helix 1 in the prion protein regulates conversion to pathogenic PrPSc. J Virol 80, 8521-8529.
Nunziante, M., Gilch, S., und Schatzl, H. M. (2003). Essential role of the prion protein N terminus in subcellular trafficking and half-life of cellular prion protein. J Biol Chem 278, 3726-3734.
Oesch, B., Westaway,D., Walchli,M., McKinley,M.P., Kent,S.B., Aebersold,R., Barry,R.A.,, und Tempst, P., Teplow,D.B., Hood,L.E. (1985). A cellular gene encodes scrapie PrP 27-30 protein. Cell 40, 735-746.
Olmsted, J. B., Carlson, K., Klebe, R., Ruddle, F., und Rosenbaum, J. (1970). Isolation of microtubule protein from cultured mouse neuroblastoma cells. Proc Natl Acad Sci U S A 65, 129-136.
Orgel, L. E. (1996). Prion replication and secondary nucleation. Chem Biol 3, 413-414.
Orive, G., Hernandez, R. M., Gascon, A. R., Calafiore, R., Chang, T. M., De Vos, P., Hortelano, G., Hunkeler, D., Lacik, I., Shapiro, A. M., und Pedraz, J. L. (2003). Cell encapsulation: promise and progress. Nat Med 9, 104-107.
Otto, M., Cepek, L., Ratzka, P., Doehlinger, S., Boekhoff, I., Wiltfang, J., Irle, E., Pergande, G., Ellers-Lenz, B., Windl, O., et al. (2004). Efficacy of flupirtine on cognitive function in patients with CJD: A double-blind study. Neurology 62, 714-718.
Padgett, K. A., und Sorge, J. A. (1996). Creating seamless junctions independent of restriction sites in PCR cloning. Gene 168, 31-35.
Padlan, E. A. (1990). On the nature of antibody combining sites: unusual structural features that may confer on these sites an enhanced capacity for binding ligands. Proteins 7, 112-124.
Pan, K. M., Baldwin, M., Nguyen, J., Gasset, M., Serban, A., Groth, D., Mehlhorn, I., Huang, Z., Fletterick, R. J., Cohen, F. E., und et al. (1993). Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc Natl Acad Sci U S A 90, 10962-10966.
Pankiewicz, J., Prelli, F., Sy, M. S., Kascsak, R. J., Kascsak, R. B., Spinner, D. S., Carp, R. I., Meeker, H. C., Sadowski, M., und Wisniewski, T. (2006). Clearance and prevention of prion infection in cell culture by anti-PrP antibodies. Eur J Neurosci 23, 2635-2647.
Paramithiotis, E., Pinard, M., Lawton, T., LaBoissiere, S., Leathers, V. L., Zou, W. Q., Estey, L. A., Lamontagne, J., Lehto, M. T., Kondejewski, L. H., et al. (2003). A prion protein epitope selective for the pathologically misfolded conformation. Nat Med 9, 893-899.
Parry, A., Baker, I., Stacey, R., und Wimalaratna, S. (2007). Long term survival in a patient with variant Creutzfeldt-Jakob disease treated with intraventricular pentosan polysulphate. J Neurol Neurosurg Psychiatry 78, 733-734.
Parry, H. B. (1962). Scrapie: a transmissible and hereditary disease of sheep. Heredity 17, 75-105.
Pauly, P. C., und Harris, D. A. (1998). Copper stimulates endocytosis of the prion protein. J Biol Chem 273, 33107-33110.

LITERATURVERZEICHNIS
137
Pearson, G. R., Gruffydd-Jones, T. J., Wyatt, J. M., Hope, J., Chong, A., Scott, A. C., Dawson, M., und Wells, G. A. (1991). Feline spongiform encephalopathy. Vet Rec 128, 532.
Peden, A. H., Head, M. W., Ritchie, D. L., Bell, J. E., und Ironside, J. W. (2004). Preclinical vCJD after blood transfusion in a PRNP codon 129 heterozygous patient. Lancet 364, 527-529.
Peretz, D., Williamson, R. A., Kaneko, K., Vergara, J., Leclerc, E., Schmitt-Ulms, G., Mehlhorn, I. R., Legname, G., Wormald, M. R., Rudd, P. M., et al. (2001). Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature 412, 739-743.
Perrier, V., Solassol, J., Crozet, C., Frobert, Y., Mourton-Gilles, C., Grassi, J., und Lehmann, S. (2004). Anti-PrP antibodies block PrPSc replication in prion-infected cell cultures by accelerating PrPC degradation. J Neurochem 89, 454-463.
Polman, C. H., O'Connor, P. W., Havrdova, E., Hutchinson, M., Kappos, L., Miller, D. H., Phillips, J. T., Lublin, F. D., Giovannoni, G., Wajgt, A., et al. (2006). A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med 354, 899-910.
Polymenidou, M., Heppner, F. L., Pellicioli, E. C., Urich, E., Miele, G., Braun, N., Wopfner, F., Schatzl, H. M., Becher, B., und Aguzzi, A. (2004). Humoral immune response to native eukaryotic prion protein correlates with anti-prion protection. Proc Natl Acad Sci U S A 101 Suppl 2, 14670-14676.
Prado, M. A., Alves-Silva, J., Magalhaes, A. C., Prado, V. F., Linden, R., Martins, V. R., und Brentani, R. R. (2004). PrPc on the road: trafficking of the cellular prion protein. J Neurochem 88, 769-781.
Proske, D., Gilch, S., Wopfner, F., Schatzl, H. M., Winnacker, E. L., und Famulok, M. (2002). Prion-protein-specific aptamer reduces PrPSc formation. Chembiochem 3, 717-725.
Prusiner, S. B. (1982). Novel proteinaceous infectious particles cause scrapie. Science 216, 136-144.
Prusiner, S. B. (1991). Molecular biology of prion diseases. Science 252, 1515-1522.
Prusiner, S. B. (1997). Prion diseases and the BSE crisis. Science 278, 245-251.
Prusiner, S. B. (1998). Prions. Proc Natl Acad Sci U S A 95, 13363-13383.
Prusiner, S. B., Groth, D., Serban, A., Koehler, R., Foster, D., Torchia, M., Burton, D., Yang, S. L., und DeArmond, S. J. (1993). Ablation of the prion protein (PrP) gene in mice prevents scrapie and facilitates production of anti-PrP antibodies. Proc Natl Acad Sci U S A 90, 10608-10612.
Prusiner, S. B., Groth, D. F., Bolton, D. C., Kent, S. B., und Hood, L. E. (1984). Purification and structural studies of a major scrapie prion protein. Cell 38, 127-134.
Prusiner, S. B., Groth, D. F., McKinley, M. P., Cochran, S. P., Bowman, K. A., und Kasper, K. C. (1981). Thiocyanate and hydroxyl ions inactivate the scrapie agent. Proc Natl Acad Sci U S A 78, 4606-4610.
Prusiner, S. B., Scott, M., Foster, D., Pan, K. M., Groth, D., Mirenda, C., Torchia, M., Yang, S. L., Serban, D., Carlson, G. A., und et al. (1990). Transgenetic studies implicate interactions between homologous PrP isoforms in scrapie prion replication. Cell 63, 673-686.

LITERATURVERZEICHNIS
138
Prusiner, S. B., Scott, M. R., DeArmond, S. J., und Cohen, F. E. (1998). Prion protein biology. Cell 93, 337-348.
Pullen, G. R., Fitzgerald, M. G., und Hosking, C. S. (1986). Antibody avidity determination by ELISA using thiocyanate elution. J Immunol Methods 86, 83-87.
Richt, J. A., Kasinathan, P., Hamir, A. N., Castilla, J., Sathiyaseelan, T., Vargas, F., Sathiyaseelan, J., Wu, H., Matsushita, H., Koster, J., et al. (2007). Production of cattle lacking prion protein. Nat Biotechnol 25, 132-138.
Rieger, R., Edenhofer, F., Lasmezas, C. I., und Weiss, S. (1997). The human 37-kDa laminin receptor precursor interacts with the prion protein in eukaryotic cells. Nat Med 3, 1383-1388.
Riek, R., Hornemann, S., Wider, G., Glockshuber, R., und Wuthrich, K. (1997). NMR characterization of the full-length recombinant murine prion protein, mPrP(23-231). FEBS Lett 413, 282-288.
Rogers, M., Yehiely, F., Scott, M., und Prusiner, S. B. (1993). Conversion of truncated and elongated prion proteins into the scrapie isoform in cultured cells. Proc Natl Acad Sci U S A 90, 3182-3186.
Rosset, M. B., Ballerini, C., Gregoire, S., Metharom, P., Carnaud, C., und Aucouturier, P. (2004). Breaking immune tolerance to the prion protein using prion protein peptides plus oligodeoxynucleotide-CpG in mice. J Immunol 172, 5168-5174.
Saborio, G. P., Permanne, B., und Soto, C. (2001). Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 411, 810-813.
Safar, J., Wille, H., Itri, V., Groth, D., Serban, H., Torchia, M., Cohen, F. E., und Prusiner, S. B. (1998). Eight prion strains have PrP(Sc) molecules with different conformations. Nat Med 4, 1157-1165.
Safar, J. G., Kellings, K., Serban, A., Groth, D., Cleaver, J. E., Prusiner, S. B., und Riesner, D. (2005). Search for a prion-specific nucleic acid. J Virol 79, 10796-10806.
Sanger, F., Nicklen, S., und Coulson, A. R. (1977). DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A 74, 5463-5467.
Santuccione, A., Sytnyk, V., Leshchyns'ka, I., und Schachner, M. (2005). Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J Cell Biol 169, 341-354.
Schatzl, H. M., Da Costa, M., Taylor, L., Cohen, F. E., und Prusiner, S. B. (1995). Prion protein gene variation among primates. J Mol Biol 245, 362-374.
Schenk, D., Barbour, R., Dunn, W., Gordon, G., Grajeda, H., Guido, T., Hu, K., Huang, J., Johnson-Wood, K., Khan, K., et al. (1999). Immunization with amyloid-beta attenuates Alzheimer-disease-like pathology in the PDAPP mouse. Nature 400, 173-177.
Schmiedl, A., und Dübel, S. (2004). Rekombinante Antikörper und Phagen-Display. Molekulare Biotechnologie.

LITERATURVERZEICHNIS
139
Schwarz, A., Kratke, O., Burwinkel, M., Riemer, C., Schultz, J., Henklein, P., Bamme, T., und Baier, M. (2003). Immunisation with a synthetic prion protein-derived peptide prolongs survival times of mice orally exposed to the scrapie agent. Neurosci Lett 350, 187-189.
Sethi, S., Lipford, G., Wagner, H., und Kretzschmar, H. (2002). Postexposure prophylaxis against prion disease with a stimulator of innate immunity. Lancet 360, 229-230.
Shulman, M., Wilde, C. D., und Kohler, G. (1978). A better cell line for making hybridomas secreting specific antibodies. Nature 276, 269-270.
Shyng, S. L., Heuser, J. E., und Harris, D. A. (1994). A glycolipid-anchored prion protein is endocytosed via clathrin-coated pits. J Cell Biol 125, 1239-1250.
Shyng, S. L., Huber, M. T., und Harris, D. A. (1993). A prion protein cycles between the cell surface and an endocytic compartment in cultured neuroblastoma cells. J Biol Chem 268, 15922-15928.
Shyng, S. L., Moulder, K. L., Lesko, A., und Harris, D. A. (1995). The N-terminal domain of a glycolipid-anchored prion protein is essential for its endocytosis via clathrin-coated pits. J Biol Chem 270, 14793-14800.
Sigurdsson, B. (1954). Rida, a chronic encephalitis of sheep: With general remarks on infections which develop slowly and some of their special characteristics. Br Vet J 110, 341–354.
Sigurdsson, E. M., Brown, D. R., Daniels, M., Kascsak, R. J., Kascsak, R., Carp, R., Meeker, H. C., Frangione, B., und Wisniewski, T. (2002). Immunization delays the onset of prion disease in mice. Am J Pathol 161, 13-17.
Sigurdsson, E. M., Scholtzova, H., Mehta, P. D., Frangione, B., und Wisniewski, T. (2001). Immunization with a nontoxic/nonfibrillar amyloid-beta homologous peptide reduces Alzheimer's disease-associated pathology in transgenic mice. Am J Pathol 159, 439-447.
Sigurdsson, E. M., Sy, M. S., Li, R., Scholtzova, H., Kascsak, R. J., Kascsak, R., Carp, R., Meeker, H. C., Frangione, B., und Wisniewski, T. (2003). Anti-prion antibodies for prophylaxis following prion exposure in mice. Neurosci Lett 336, 185-187.
Simons, K., und Ikonen, E. (1997). Functional rafts in cell membranes. Nature 387, 569-572.
Smith, G. P. (1985). Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science 228, 1315-1317.
Solforosi, L., Bellon, A., Schaller, M., Cruite, J. T., Abalos, G. C., und Williamson, R. A. (2007). Toward molecular dissection of PrPC-PrPSc interactions. J Biol Chem 282, 7465-7471.
Solforosi, L., Criado, J. R., McGavern, D. B., Wirz, S., Sanchez-Alavez, M., Sugama, S., DeGiorgio, L. A., Volpe, B. T., Wiseman, E., Abalos, G., et al. (2004). Cross-linking cellular prion protein triggers neuronal apoptosis in vivo. Science 303, 1514-1516.
Soto, C., Kascsak, R. J., Saborio, G. P., Aucouturier, P., Wisniewski, T., Prelli, F., Kascsak, R., Mendez, E., Harris, D. A., Ironside, J., et al. (2000). Reversion of prion protein conformational changes by synthetic beta-sheet breaker peptides. Lancet 355, 192-197.

LITERATURVERZEICHNIS
140
Sparkes, R. S., Simon, M., Cohn, V. H., Fournier, R. E., Lem, J., Klisak, I., Heinzmann, C., Blatt, C., Lucero, M., Mohandas, T., und et al. (1986). Assignment of the human and mouse prion protein genes to homologous chromosomes. Proc Natl Acad Sci U S A 83, 7358-7362.
Stahl, N., Borchelt, D. R., Hsiao, K., und Prusiner, S. B. (1987). Scrapie prion protein contains a phosphatidylinositol glycolipid. Cell 51, 229-240.
Stern, M., und Herrmann, R. (2005). Overview of monoclonal antibodies in cancer therapy: present and promise. Crit Rev Oncol Hematol 54, 11-29.
Suntharalingam, G., Perry, M. R., Ward, S., Brett, S. J., Castello-Cortes, A., Brunner, M. D., und Panoskaltsis, N. (2006). Cytokine storm in a phase 1 trial of the anti-CD28 monoclonal antibody TGN1412. N Engl J Med 355, 1018-1028.
Sunyach, C., Jen, A., Deng, J., Fitzgerald, K. T., Frobert, Y., Grassi, J., McCaffrey, M. W., und Morris, R. (2003). The mechanism of internalization of glycosylphosphatidylinositol-anchored prion protein. Embo J 22, 3591-3601.
Supattapone, S., Nguyen, H. O., Cohen, F. E., Prusiner, S. B., und Scott, M. R. (1999). Elimination of prions by branched polyamines and implications for therapeutics. Proc Natl Acad Sci U S A 96, 14529-14534.
Supattapone, S., Wille, H., Uyechi, L., Safar, J., Tremblay, P., Szoka, F. C., Cohen, F. E., Prusiner, S. B., und Scott, M. R. (2001). Branched polyamines cure prion-infected neuroblastoma cells. J Virol 75, 3453-3461.
Taraboulos, A., Scott, M., Semenov, A., Avrahami, D., Laszlo, L., und Prusiner, S. B. (1995). Cholesterol depletion and modification of COOH-terminal targeting sequence of the prion protein inhibit formation of the scrapie isoform. J Cell Biol 129, 121-132.
Taraboulos, A., Serban, D., und Prusiner, S. B. (1990). Scrapie prion proteins accumulate in the cytoplasm of persistently infected cultured cells. J Cell Biol 110, 2117-2132.
Tatzelt, J., Prusiner, S. B., und Welch, W. J. (1996). Chemical chaperones interfere with the formation of scrapie prion protein. Embo J 15, 6363-6373.
Telling, G. C., Parchi, P., DeArmond, S. J., Cortelli, P., Montagna, P., Gabizon, R., Mastrianni, J., Lugaresi, E., Gambetti, P., und Prusiner, S. B. (1996). Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science 274, 2079-2082.
Telling, G. C., Scott, M., Hsiao, K. K., Foster, D., Yang, S. L., Torchia, M., Sidle, K. C., Collinge, J., DeArmond, S. J., und Prusiner, S. B. (1994). Transmission of Creutzfeldt-Jakob disease from humans to transgenic mice expressing chimeric human-mouse prion protein. Proc Natl Acad Sci U S A 91, 9936-9940.
Telling, G. C., Scott, M., Mastrianni, J., Gabizon, R., Torchia, M., Cohen, F. E., DeArmond, S. J., und Prusiner, S. B. (1995). Prion propagation in mice expressing human and chimeric PrP transgenes implicates the interaction of cellular PrP with another protein. Cell 83, 79-90.
Thormar, H. (1971). Slow infections of the central nervous system. II. Z Neurol 199, 151-166.
Tilly, G., Chapuis, J., Vilette, D., Laude, H., und Vilotte, J. L. (2003). Efficient and specific down-regulation of prion protein expression by RNAi. Biochem Biophys Res Commun 305, 548-551.

LITERATURVERZEICHNIS
141
Tobler, I., Deboer, T., und Fischer, M. (1997). Sleep and sleep regulation in normal and prion protein-deficient mice. J Neurosci 17, 1869-1879.
Todd, N. V., Morrow, J., Doh-ura, K., Dealler, S., O'Hare, S., Farling, P., Duddy, M., und Rainov, N. G. (2005). Cerebroventricular infusion of pentosan polysulphate in human variant Creutzfeldt-Jakob disease. J Infect 50, 394-396.
Towbin, H., Staehelin, T., und Gordon, J. (1979). Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A 76, 4350-4354.
Vanik, D. L., Surewicz, K. A., und Surewicz, W. K. (2004). Molecular basis of barriers for interspecies transmissibility of mammalian prions. Mol Cell 14, 139-145.
Vetrugno, V., Cardinale, A., Filesi, I., Mattei, S., Sy, M. S., Pocchiari, M., und Biocca, S. (2005). KDEL-tagged anti-prion intrabodies impair PrP lysosomal degradation and inhibit scrapie infectivity. Biochem Biophys Res Commun 338, 1791-1797.
Vey, M., Pilkuhn, S., Wille, H., Nixon, R., DeArmond, S. J., Smart, E. J., Anderson, R. G., Taraboulos, A., und Prusiner, S. B. (1996). Subcellular colocalization of the cellular and scrapie prion proteins in caveolae-like membranous domains. Proc Natl Acad Sci U S A 93, 14945-14949.
Vilette, D., Andreoletti, O., Archer, F., Madelaine, M. F., Vilotte, J. L., Lehmann, S., und Laude, H. (2001). Ex vivo propagation of infectious sheep scrapie agent in heterologous epithelial cells expressing ovine prion protein. Proc Natl Acad Sci U S A 98, 4055-4059.
Waggoner, D. J., Drisaldi, B., Bartnikas, T. B., Casareno, R. L., Prohaska, J. R., Gitlin, J. D., und Harris, D. A. (2000). Brain copper content and cuproenzyme activity do not vary with prion protein expression level. J Biol Chem 275, 7455-7458.
Weerasekera, N., und Hudson, I. (2007). Lessons from TGN-1412 - the impact on phase I clinical trials. IDrugs 10, 230-232.
Wells, G. A., Scott, A. C., Johnson, C. T., Gunning, R. F., Hancock, R. D., Jeffrey, M., Dawson, M., und Bradley, R. (1987). A novel progressive spongiform encephalopathy in cattle. Vet Rec 121, 419-420.
White, A. R., Enever, P., Tayebi, M., Mushens, R., Linehan, J., Brandner, S., Anstee, D., Collinge, J., und Hawke, S. (2003). Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature 422, 80-83.
Whittington, M. A., Sidle, K. C., Gowland, I., Meads, J., Hill, A. F., Palmer, M. S., Jefferys, J. G., und Collinge, J. (1995). Rescue of neurophysiological phenotype seen in PrP null mice by transgene encoding human prion protein. Nat Genet 9, 197-201.
Whittle, I. R., Knight, R. S., und Will, R. G. (2006). Unsuccessful intraventricular pentosan polysulphate treatment of variant Creutzfeldt-Jakob disease. Acta Neurochir (Wien) 148, 677-679; discussion 679.
Wigler, M., Pellicer, A., Silverstein, S., und Axel, R. (1978). Biochemical transfer of single-copy eucaryotic genes using total cellular DNA as donor. Cell 14, 725-731.

LITERATURVERZEICHNIS
142
Will, R. G., Ironside, J. W., Zeidler, M., Cousens, S. N., Estibeiro, K., Alperovitch, A., Poser, S., Pocchiari, M., Hofman, A., und Smith, P. G. (1996). A new variant of Creutzfeldt-Jakob disease in the UK. Lancet 347, 921-925.
Wille, H., Michelitsch, M. D., Guenebaut, V., Supattapone, S., Serban, A., Cohen, F. E., Agard, D. A., und Prusiner, S. B. (2002). Structural studies of the scrapie prion protein by electron crystallography. Proc Natl Acad Sci U S A 99, 3563-3568.
Williams, E. S., und Young, S. (1980). Chronic wasting disease of captive mule deer: a spongiform encephalopathy. J Wildl Dis 16, 89-98.
Williamson, R. A., Peretz, D., Smorodinsky, N., Bastidas, R., Serban, H., Mehlhorn, I., DeArmond, S. J., Prusiner, S. B., und Burton, D. R. (1996). Circumventing tolerance to generate autologous monoclonal antibodies to the prion protein. Proc Natl Acad Sci U S A 93, 7279-7282.
Windl, O., Dempster, M., Estibeiro, P., und Lathe, R. (1995). A candidate marsupial PrP gene reveals two domains conserved in mammalian PrP proteins. Gene 159, 181-186.
Winklhofer, K. F., und Tatzelt, J. (2000). Cationic lipopolyamines induce degradation of PrPSc in scrapie-infected mouse neuroblastoma cells. Biol Chem 381, 463-469.
Wisniewski, T., und Sigurdsson, E. M. (2002). Immunization treatment approaches in Alzheimer's and prion diseases. Curr Neurol Neurosci Rep 2, 400-404.
Wisniewski, T., und Sigurdsson, E. M. (2007). Therapeutic approaches for prion and Alzheimer's diseases. Febs J 274, 3784-3798.
Wopfner, F., Weidenhofer, G., Schneider, R., von Brunn, A., Gilch, S., Schwarz, T. F., Werner, T., und Schatzl, H. M. (1999). Analysis of 27 mammalian and 9 avian PrPs reveals high conservation of flexible regions of the prion protein. J Mol Biol 289, 1163-1178.
Yang, X. D., Jia, X. C., Corvalan, J. R., Wang, P., und Davis, C. G. (2001). Development of ABX-EGF, a fully human anti-EGF receptor monoclonal antibody, for cancer therapy. Crit Rev Oncol Hematol 38, 17-23.
Zahn, R., von Schroetter, C., und Wuthrich, K. (1997). Human prion proteins expressed in Escherichia coli and purified by high-affinity column refolding. FEBS Lett 417, 400-404.
Zanata, S. M., Lopes, M. H., Mercadante, A. F., Hajj, G. N., Chiarini, L. B., Nomizo, R., Freitas, A. R., Cabral, A. L., Lee, K. S., Juliano, M. A., et al. (2002). Stress-inducible protein 1 is a cell surface ligand for cellular prion that triggers neuroprotection. Embo J 21, 3307-3316.

ABKÜRZUNGSVERZEICHNIS
143
7 Anhang
7.1 Abkürzungsverzeichnis
Über die gebräuchlichen SI- und IUPAC-Einheiten hinausgehend wurden die folgenden Abkürzungen verwendet: A Alanin Aβ Amyloid-β A.dest. destilliertes Wasser AD Alzheimer-Krankheit AI Aviditätsindex Ak Antikörper amp Ampicillin APC Antigen-präsentierende Zelle APP Amyloid-Precursor-Protein AS Aminosäure boPrP bovines PrP bp Basenpaare BSA Rinderserumalbumin BSE Bovine Spongiforme Enzephalopathie bzw. beziehungsweise CD Lymphozyten-Differenzierungsmarker cDNA complementary DNA CDR complementary determining region CFA Komplettes Freund-Adjuvans CH konstante Domäne der schweren Kette CJD Creutzfeldt-Jakob-Krankheit CL konstante Domäne der leichten Kette CMV Cytomegalievirus CO2 Kohlendioxid CpG Cytosin-Guanosin-Dinukleotide CTB Cholera Toxin Untereinheit B Cu Kupfer CWD Chronic Wasting Disease D Asparaginsäure DC dendritische Zellen ddH2O doppelt destilliertes Wasser ddNTP Didesoxyribonukleotid-Triphosphat D-Gensement Diversity-Gensegement DMSO Dimethylsulfoxid DNA Desoxyribonukleinsäure dpi days post inoculation E Glutaminsäure E.coli Escherichia coli ECL Enhanced Chemiluminescense EDTA Ethylendiamintetraacetat ELISA Enzyme-linked-immunosorbent-assay et al. et altera Fab Fragment antigen binding FACS Fluorescence-activated-cell-sorting

ABKÜRZUNGSVERZEICHNIS
144
FC Fragment crystallizable fCJD familiäre Creutzfeldt-Jakob-Krankheit FCS fötales Kälberserum FDC follikulär dendritische Zellen FFI Fatal Familiäre Insomnie fib.PrP fibrilläres PrP FITC Fluoresceinisothiocyanat FR framework region FSE Feline Spongiforme Enzephalopathie GABA γ-Aminobuttersäure GPI Glykosyl-Phosphatidyl-Inositol-Gruppe GSS Gerstmann-Sträussler-Scheinker-Syndrom H Histidin HAMA humaner Anti-Maus-Antikörper HaPrP Hamster-PrP HAT-Medium Hypoxanthin-Aminopterin-Thymidin-Medium H-Kette heavy chain HRP Horseraddish Peroxidase i.c. intrazerebral i.p. intraperitoneal iCJD iatrogene Creutzfeldt-Jakob-Krankheit IFA inkomplettes Freund-Adjuvants Ig Immunglobulin IL Interleukin in vitro in Zellkultur/Reagenzglas in vivo am Lebenden J-Gensegment Joining-Gensegement K Lysin kb Kilobasenpaar KD Dissoziationskonstante kDa Kilodalton KLH Keyhole limpet Hämocyanin LB Luria Broth (Medium) L-Kette light chain mAk monoklonaler Antikörper Met Methionin MHC Haupthistokompatibilitätskomplex MOCK unbehandelt N Asparagin NCAM neurales Zellenadhäsionsmolekül NH4SCN Ammoniumthiocyanat NMR Kernresonanz-Spektroskopie OD Optische Dichte ODN Oligodinukleotid(e) ovPrP ovines PrP PAGE Polyacrylamidgelelektrophorese pAK polyklonaler Antikörper PBS Phosphat-gepufferte Salzlösung PCR Polymerasekettenreaktion PEG Polyethylenglykol pH negativer dekadischer Logarithmus der Hydroniumionenkonzentration PK Proteinase K PMSF Phenylmethylsulfonylfluorid

ABKÜRZUNGSVERZEICHNIS
145
PPS Pentosanpolyphosphat PRNP Genlocus des humanen Prion-Proteins Prnp0/0 PrP-defiziente Mäuse (Knockout) PrPC zelluläres Prion-Protein PrPSc pathologische Form des Prion-Proteins PVDF Polyvinylidene Fluoride Q Glutamin R Arginin RAG Rekombination-aktivierendes Gen rek. PrP rekombinantes Maus-PrP rER raues endoplasmatisches Retikulum RML Rocky-Mountain-Laboratory RNA Ribonukleinsäure rpm Runden pro Minute RSS Rekombination-Signal-Sequenz RT Raumtemperatur SAF Scrapie-assoziierte Fibrillen scFv single chain fragment variable sCJD sporadische Creutzfeldt-Jakob-Krankheit SDS Natriumdodecylsulfat SOD-1 Superoxiddismutase-1 STE Stop-Transfer-Effektor-Seite T Threonin TAE Tris-Essigsäure-EDTA-Puffer TBST Tris-gepufferte Salzlösung mit Tween 20 Temp. Temperatur TM transmembrane Region TME Transmissible Mink Encephalopathy TSE transmissible spongiforme Enzephalopathien U unit (Enzymaktivitätseinheit) V Volt Val Valin vCJD variante Form der Creutzfeldt-Jakob-Disease VH variable Domäne der schweren Kette
VL variable Domäne der leichten Kette Y Tyrosin z.B. zum Beispiel z.T. zum Teil Zn Zink ZNS zentrales Nervensystem

TABELLEN- UND ABBILDUNGSVERZEICHNIS
146
7.2 Tabellen- und Abbildungsverzeichnis
Tabelle 1: Zusammenfassung humaner Prionenerkrankungen. 7
Tabelle 2: Vergleich der Eigenschaften von PrPC, PrPSc und PrP27-30. 13
Tabelle 3: Eigenschaften muriner Immunglobuline. 25
Tabelle 4: Klinisch zugelassene Antikörper in Deutschland. . 32
Tabelle 5: Verdünnungen der im Western Blot verwendeten Antikörper. 54
Tabelle 6: Verdünnungen der im Durchflusszytometer verwendeten Antikörper. 58
Tabelle 7: Ausgewählte Parameter der Fusionen. 73
Tabelle 8: Übersicht der neuen monoklonalen Anti-PrP-Antikörper. 75
Tabelle 9: Durchschnittliche Mengen an sezernierten Antikörpern im Medium. 77
Tabelle 10: Aviditätsindizes (AI) der Anti-PrP-Antikörper. 85
Tabelle 11: KD-Konstanten der Anti-PrP-Antikörper. 86
Tabelle 12: IC50-Werte der Anti-PrP-Antikörper. 92
Tabelle 13: Bioassay für die Bestimmung der Infektiosität von ScN2a-Zellen nach
der Behandlung mit Anti-PrP-Antikörpern. 97
Tabelle 14: Prozentualer Anteil der Antikörper im Serum der Mäuse. 104
Tabelle 15: Auswahl von Anti-PrP-Antikörpern. 112
Tabelle 16: Charakterisierung der neuen Anti-PrP-Antikörper. 114
Abb. 1: Pathogene Mutationen und Polymorphismen des humanen PRNP-Gens. 8
Abb. 2: Schematische Darstellung des murinen Prion-Proteins. 11
Abb. 3: NMR-Struktur von rekombinantem PrP (Reste 121-231). 14
Abb. 4: Schematisches Modell der PrPSc-Konformation und Aggregation. 15
Abb. 5: Modelle für die Konversion von PrPC zu PrPSc. 17
Abb. 6: Potentielle Angriffspunkte für therapeutische Interventionen bei Prionen-
erkrankungen am Beispiel des Faltungsmodells. 19
Abb. 7: Modell eines IgG-Antikörpers und Struktur der Antigenbindungsstelle. 24
Abb. 8: Schematische Darstellung der somatischen Rekombination am Beispiel der
schweren Kette. 26
Abb. 9: Schematische Darstellung der Herstellung von monoklonalen Antikörpern. 29
Abb. 10: Darstellung von verschiedenen Antikörperformaten. 31
Abb. 11: Immunisierungsschema der Prnp0/0-Mäuse und Zeitpunkte der Fusion. 72
Abb. 12: Dreistufiges Screeningverfahren zur Isolierung des potentiellen Anti-PrP-
Antikörper-produzierenden Klons B2-43-133. 74
Abb. 13: Schematische Darstellung der Struktur von Maus-PrP und der klonierten

TABELLEN- UND ABBILDUNGSVERZEICHNIS
147
Mutanten mit N-terminaler (∆32-93) sowie interner Deletion (∆142-157
oder ∆158-171). 78
Abb. 14: 3-Weg-Ligation zur Deletion der Reste 142 bis 157 bzw. 158 bis 171
in PrP ∆32-93. 79
Abb. 15: Epitop-Bestimmung der Anti-PrP-Antikörper mittels Expressions- und
FACS-Analyse von PrP-Deletionsmutanten. 81
Abb. 16: Schematische Darstellung der Struktur von Maus-PrP und Lokalisierung der
Epitope der Anti-PrP-Antikörper. 82
Abb. 17:Schematische Darstellung der geladenen Reste innerhalb der α-Helix 1
von Maus-PrPC. 83
Abb. 18: Schematische Darstellung von Maus-PrPC und der drei Substitutionen für
die Bestimmung des Minimalepitops. 84
Abb. 19: Bestimmung der Avidität der Anti-PrP-Antikörper. 85
Abb. 20: Reduktion des PrPSc-Gehalts von ScN2a-Zellen durch Anti-PrP-Antikörper. 88
Abb. 21: Untersuchung zum zytotoxischen Effekt der Anti-PrP-Antikörper auf
ScN2a-Zellen. 89
Abb. 22: Bindung der Anti-PrP-Antikörper an PrPSc auf ScN2a- und cuScN2a-Zellen. 90
Abb. 23: Dosisabhängige Reduktion des PrPSc-Gehalts von ScN2a-Zellen am
Beispiel B2-43-133. 91
Abb. 24: PrPSc-Gehalt von ScN2a-Zellen nach Langzeitbehandlung mit den
Anti-PrP-Antikörpern. 93
Abb. 25: PrPSc-Gehalt von ScN2a-Zellen nach Beendigung der Langzeitbehandlung
mit den Anti-PrP-Antikörpern. 94
Abb. 26: Inkubationszeiten der Indikatormäuse nach intrazerebraler Inokulation von
Anti-PrP-Antikörper-behandelten ScN2a-Zellen. 96
Abb. 27: Hemmung der PrPSc-Akkumulation in ScN2a-Zellen durch die Behandlung
mit freiem B2-43-133 und einem PrP/B2-43-133-Komplex. 99
Abb. 28: Prinzip der Analyse des Einflusses der Anti-PrP-Antikörper auf die Retention. 100
Abb. 29: Bindung der Anti-PrP-Antikörper an Oberflächen-PrP auf ScN2a-Zellen. 100
Abb. 30: Einfluss der Anti-PrP-Antikörper auf die Retention von Oberflächen-PrP
auf ScN2a-Zellen. 101
Abb. 31: Immunisierungsschema von RML-infizierten C57BL/6-Mäusen für die Analyse
der Pharmakokinetik. 103
Abb. 32: Analyse der Pharmakokinetik der Anti-PrP-Antikörper. 104
Abb. 33: Schematische Darstellung der Mechanismen antikörpervermittelter
Prioninaktivierung. 116

LEBENSLAUF
148
7.3 Lebenslauf
Persönliche Daten
Name Tanja Hoffmann
Geburtsdatum, -ort 26.11.1976, Datteln
Familienstand ledig, keine Kinder
Schulabschluss
1996 Abitur, Theodor Heuss-Gymnasium, Waltrop (Note: gut)
Studium
1996 – 2002 Diplom-Biologiestudium, Ruhr-Universität Bochum (Note: sehr gut)
2001 – 2002
Thema:
Diplomarbeit in der AG Spezielle Zoologie, Ruhr-Universität Bochum
(Prof. Dr. G. A. Schaub)
„Untersuchungen zur bakteriellen Darmflora von Panstrongylus
megistus, Triatoma klugi und Triatoma brasiliensis und Identifizierung
symbiontischer Aktinomyceten“ (Note: sehr gut)
Promotion
seit 04/2003
Thema:
Promotionsstudium, Bayerischen Julius-Maximilians-Universität
Würzburg, Institut für Virologie und Immunbiologie
(Prof. Dr. M. A. Klein)
„Herstellung und Charakterisierung von Antikörpern gegen das Prion-
Protein mit inhibitorischer Wirkung auf die Prionreplikation in
Zellkultur“
Würzburg, 01.07.2008
Tanja Hoffmann

VERÖFFENTLICHUNGEN UND KONGRESSBEITRÄGE
149
7.4 Veröffentlichungen und Kongressbeiträge
Veröffentlichungen
Christine von Poser-Klein, Eckhard Flechsig, Tanja Hoffmann, Petra Schwarz, Harry Harms,
Raymond Bujdoso, Adriano Aguzzi and Michael A. Klein „Alteration of B cell subsets enhance
neuroinvasion in mouse scrapie.“
J Virol. 2008 Jan 16
Tanja Hoffmann, Julia Merk, Nele Lindner, Thorsten Lührs, Michael A. Klein and Eckhard
Flechsig „Characterization of novel monoclonal antibodies against PrP.”
in progress
Kongressbeiträge in Form einer Posterpräsentation
07/2004
10/2004
03/2005
07/2005
10/2005
05/2006
05/2007
Tanja Hoffmann, Nele Ruoff, Julia Günter, Michael A. Klein, Eckhard
Flechsig “Characterization of novel monoclonal antibodies against PrP for
diagnostic and therapeutic approaches.”
• Treffen des Bayrischen Forschungsverbundes Prionen (ForPrion),
Würzburg
• Nationale TSE-Forschungsplattform, Düsseldorf
Tanja Hoffmann, Nele Ruoff, Julia Günter, Michael A. Klein, Eckhard
Flechsig “Characterization of novel monoclonal antibodies against PrP.”
• Jahrestagung der Gesellschaft für Virologie, Hannover
Tanja Hoffmann, Nele Lindner, Julia Günter, Michael A. Klein, Eckhard
Flechsig “Cell-surface retention of PrPC by novel anti-PrP antibodies
correlates with inhibition of PrPSc accumulation in cultured cells.”
• Treffen des Bayrischen Forschungsverbundes Prionen (ForPrion),
Hohenkammer
• International Conference, Prion 2005: Between fundamentals and
society's needs, Düsseldorf
• Treffen des Bayrischen Forschungsverbundes Prionen (ForPrion),
München
• Treffen des Bayrischen Forschungsverbundes Prionen (ForPrion),
München

ERKLÄRUNG
150
7.5 Erklärung
Hiermit erkläre ich ehrenwörtlich, dass ich diese Dissertation selbständig angefertigt und keine
anderen als die von mir angegebenen Hilfsmittel und Quellen benutzt habe.
Zudem erkläre ich, dass diese Dissertation weder in gleicher noch in anderer Form bereits in
einem Prüfungsverfahren vorgelegen hat.
Ich habe früher, außer den mit dem Zulassungsgesuch urkundlichen Graden, keine weiteren
akademischen Grade erworben oder zu erwerben versucht.
Würzburg, 01.07.2008
Tanja Hoffmann

DANKSAGUNG
151
7.6 Danksagung
Mein Dank gilt Prof. Dr. Michael Klein für die Vergabe des interessanten Themas, die
hervorragende Betreuung sowie für die Unterstützung und Förderung während der gesamten
Arbeit.
Ich danke Prof. Dr. Dr. Jörg Hacker für die Bereitschaft, die Zweitkorrektur zu übernehmen und
für sein Interesse am Fortschritt der Arbeit.
Mein besonderer Dank gilt Julia Günter und Nele Lindner. Die Bewältigung von so vielen
„Mamas“ und „Babys“ war nur mit drei Paar tatkräftigen Händen möglich (…und bald gibt es
echte Babys, juchuh!).
Ganz herzlich möchte ich mich auch bei den restlichen Mitgliedern der AG Klein/Flechsig –
Angela Bahlo, Vladimir Ermolayev, Katja Hochgräfe, Patrick Porps, Jan Springer – und allen
ehemaligen Mitgliedern – besonders bei Cindy Nitschke – bedanken. Ohne sie wäre diese
Arbeit nur mit halb so viel Spaß verbunden gewesen. Danke für die vielen lustigen Keks-
Pausen, die zahlreichen Gespräche über Gott und die Welt und den Spaß, den wir vor allen
Dingen auch außerhalb des Labors hatten.
Bedanken möchte ich mich noch herzlicher bei Familie Porps – Patrick, Corinna und Daria –,
die mir während meiner Zeit in Würzburg sehr ans Herz gewachsen sind.
Nicht zuletzt danke ich meinen Waltroper Mädels – Gesche, Ilka, Jessi, Moni, Rebecca und
Wibke (Dir gilt ein zusätzliches Danke für das Einfügen vieler englischer Artikel, Bindestriche
und Zeilenumbrüche) –, die auch aus der Ferne immer ein offenes Ohr für meine Probleme
hatten und mich in jeglicher Hinsicht uneingeschränkt unterstützt und motiviert haben.
Zu guter Letzt bedanke ich mich von ganzem Herzen bei meinen Eltern und meiner Tante dafür,
dass sie immer an mich geglaubt haben.
DANKE ☺





























