Embed Size (px)
Citation preview

Heterologe Expression
und funktionelle Grundcharakterisierung
von Myelinproteinen aus dem ZNS
der Regenbogenforelle (Oncorrhynchus mykiss)
Dissertation
zur Erlangung des Grades eines Doktors der
Naturwissenschaften (Dr. rer. nat.)
des Fachbereiches Biologie/Chemieder Universität Osnabrück
vorgelegt von
Carsten Lanwertaus Osnabrück
Frühjahr 2000

1
1 Einleitung
Im Laufe der Evolution haben die Vertebraten ein neuronales Netzwerk entwickelt, das in
Komplexität und Leistungsfähigkeit einzigartig ist. Die besondere Leistungsfähigkeit
verdankt es dem Myelin, welches die Axone der Nervenfasern des zentralen (ZNS) und
peripheren Nervensystems (PNS) umgibt und somit gegen transmembrane Ionenströme
isoliert. Die Erregungsleitung kann sich daher nur an nicht myelinisierten Bereichen der
Nervenfaser, den Ranvierschen Schnürringen, sprunghaft ausbreiten. Diese sog. saltatorische
Erregungsleitung spart gegenüber der kontinuierlichen Erregungsleitung Zeit, Energie und
Platz und bildet somit Grundstein für die unglaubliche Rechenleistung der Vertebratengehirne
(BUNGE, 1968; MORELL UND NORTON 1980; REVIEW: HILDEBRAND ET AL., 1993). Die
Myelinhülle wird von spezialisierten Gliazellen gebildet. Im zentralen Nervensystem sind
dieses die Oligodendrozyten, im PNS übernehmen die Schwannschen Zellen die Aufgabe der
Myelinogenese. Diese myelinformenden spezialisierten Zellen wickeln die Membranen ihrer
Zellfortsätze als flache Zunge mehrfach konzentrisch um die Axone von Nervenfasern und
bilden durch Verdichtung die typische
kompakte, multilamelläre Struktur des
Myelins (WOOD UND BUNGE, 1984,
PETERS ET AL., 1976; RUMSBY UND
CRANG, 1977). Innerhalb des kompakten
Myelins werden durch die dichte
Aufeinanderlagerung der cytoplas-
matischen Seiten der Plasmamembran der
Schwannschen Zellen, bzw. des
Oligodendrozyten die sog. „major dense
line“ gebildet, und durch Apposition der
extrazellulären Seiten des Bilayers die
sog. „intraperiod line“ des Myelins, die
nach ihrer Struktur in elektronen-
mikroskopischen Aufnahmen benannt wurden.
Die Fähigkeit zur elektrischen Isolation spiegelt sich in der biochemischen Zusammensetzung
Abbildung 1: ElektronenmikroskopischeAufnahme einer typischen myelinisiertenNervenfaser aus dem ZNS (Rückenmark Hund).Der Zellkörper des Oligodendrozyten istzuerkennen (Pfeil). Man sieht deutlich diecytoplasmatische „major dense line“ als scharfeschwarze Linie und die extrazelluläre„intraperiod line“ als gräuliche Substanz(x135000) (entnommen aus: „BasicNeurochemistry“ Lippincott-Raven, 1998)

2
der Myelinmembran wieder: Sie besteht zu rund 70 % aus hydrophoben und elektrisch
isolierenden Lipiden (MORELL, 1977), unter denen das Galactocerebrosid (GalC) als
spezifischer Marker für Oligodendrozyten und Schwannschen Zellen identifiziert wurde (YU
UND IQBAL, 1979). Der Proteinanteil ist mit 30 % relativ gering, aber äußerst
charakteristisch. Eine wesentliche Funktion dieser Proteine ist es, die Adhäsion der einzelnen
Myelinlamellen im kompakten Myelin zu vermitteln. Im ZNS der höheren Vertebraten, wie
z.B. Säuger, Vögel, Reptilien und Amphibien, besteht der Proteinanteil zu 50 % aus dem
hydrophoben, nichtglykosilierten Proteolipid Protein PLP (LEES UND BROSTOFF, 1984). Die
basischen Myelinproteine (MBP’s) sind mit 30 % die zweithäufigsten Vertreter. Sie bilden
eine Klasse von Proteinen, deren Molekulargewicht von 12 bis 22 kDa reicht und von nur
einem Gen durch alternatives RNA-Splicing gebildet werden (BARBARESE ET AL., 1977;
GREENFIELD ET AL., 1982; REVIEW: KAMHOLZ UND WRABETZ, 1992). Weitere
charakteristische Proteine sind die 2’3’-cyclisches Nukleotid-3’-Phosphohydrolase (CNPase)
mit 5% und das Myelinassoziierte Glykoprotein (MAG), welches in zwei Isoformen vorliegt
und weniger als 1% der Proteine ausmacht (VOGEL UND THOMPSON, 1988; CAMPAGNONI,
1988, LAI ET AL., 1987).
Das PLP tritt ausschließlich im ZNS auf, während im PNS seine Funktion vom
transmembranen Glykoprotein P0, das mit bis zu 60 % den weitaus größten Teil der
gesamten Myelinproteinfraktion darstellt,
übernommen wird. Die Proteine PLP und P0
unterscheiden sich gravierend in der
Aminosäuresequenz, Membran-Topologie
und Genstruktur. Während das 30 kDa
große, stark acylierte PLP vier
Transmembran-Durchgänge aufweist, die
intra- und extrazellulär durch hydrophile
Schleifen verbunden sind (POPOT ET AL.,
1991), besitzt das P0-Protein nur ein
einzelnes Membran-Segment sowie eine
stark basische intrazelluläre Domäne (LEMKE UND AXEL, 1985, LAI ET AL., 1987; DING UND
BRUNDEN, 1994). Als weitere charakteristische PNS-Myelinproteine wurde das P2-Protein,
Abbildung 2: SDS-PAGE von Myelin MO: Maus; TR:Forelle; OX Rind; (Abbildung aus: Jeserich undWaehneldt, 1986b)

3
das periphere Myelinprotein 22 (PMP22) mit einem Anteil von bis zu 5 % (PAREEK ET AL.,
1993) MAG, MBP’s (bis zu 15 %) und Connexin 32 (Cx32) charakterisiert (BERGOFFEN ET
AL., 1993; REVIEW: SNIPES UND SUTER, 1995).
Das Myelin aus dem ZNS phylogenetisch älterer Vertebraten, wie den Fischen, unterscheidet
sich in seinem Proteinmuster von dem der übrigen Vertebraten (JESERICH 1983; WAEHNELDT
UND JESERICH, 1984). PLP sucht man hier vergeblich: es wird durch P0-ähnliche Proteine
ersetzt. Bei der Forelle sind dieses die intermediären, in zwei Isoformen durch alternatives
Splicing auftretenden Glykoproteine IP1 und IP2. (JESERICH UND WAEHNELDT, 1986A;
JESERICH UND WAEHNELDT, 1987). Die Sequenz von IP1 konnte bestimmt und ihre
strukturelle Verwandtschaft zu P0, auf die im folgenden noch genauer eingegangen wird,
näher charakterisiert werden (STRATMANN UND JESERICH, 1995). Interessanterweise ist die
Expression der Splice-Varianten IP1 und IP2 entwicklungsabhängig, denn die größere
Isoform wird während der Oligodendrozytenentwicklung früher exprimiert als die kleinere
(JESERICH ET AL. 1990).
Ein weiteres einzigartiges, nur im ZNS der Fische vorkommendes Protein ist ein
nichtglykosiliertes, cytoplasmatisches, membranassoziiertes Protein mit dem
Molekulargewicht von 36 kDa, genannt 36K-Protein (JESERICH, 1983; JESERICH UND
WAEHNELDT, 1986B). Die cDNA-, bzw. Proteinsequenz ist bislang nicht vollständig bekannt,
doch aufgrund von computergestützten Vorhersagen, basierend auf gDNA-Teilfragmenten,
wurde deutlich, daß sich das 36K-Protein in Sequenz und Struktur von allen von höheren
Vertebraten bekannten Myelinproteinen unterscheidet. Statt dessen konnte eine auffällige
Ähnlichkeit zu Vertretern der kurzkettigen NAD/NADP-abhängigen Oxido-Reduktasen
ausgemacht werden (STRELAU, DISSERTATION, 1997). Die Aufgabe dieser einzigartigen
Myelinkomponente ist bislang noch ein Rätsel. Im PNS der Forelle fehlt 36K. Es treten neben
zwei weiteren IP-ähnlichen Proteinen (IPb und IPc) MAG und niedermolekulare basische
Proteine auf. Zusammenfassend läßt sich demnach sagen, daß die Oligodendrozyten aus dem
ZNS der Fische bezüglich ihrer Morphologie den Säuger-Oligodendrozyten ähneln, bezüglich
ihrer biochemischen Zusammensetzung jedoch Ähnlichkeit zu Säuger-Schwannzellen
aufweisen.
Die besondere Stellung von Fisch-Oligodendrozyten zeigt sich, außer in der oben

4
beschriebenen biochemische Zusammensetzung, in einer weiteren außergewöhnlichen
Eigenschaft. Sie besitzen die Fähigkeit, Axone nach Verletzungen vollständig zu
remyelinisieren (WOLBURG, 1981), eine Fähigkeit, die sonst nur bei Schwannschen Zellen im
PNS zu finden ist. Außerdem unterstützen sie in vitro die neuronale Regeneration
(BASTMEYER ET AL. 1991). Da dem P0-Protein aus dem PNS der Säuger die Fähigkeit
nachgewiesen wurde, das Auswachsen von Neuriten zu fördern (SCHNEIDER-SCHAULIES ET
AL., 1990, YAZAKI ET AL., 1994), ist es denkbar, daß im Fisch-ZNS die P0-ähnlichen
Proteine IP1 und IP2 in den Remyelinisierungs- und Regenerationsprozessen involviert sind.
Aufgrund der auffälligen
Sekundärstruktur von IP1 und IP2
werden diese Forellenmyelinproteine als
typische Vertreter der Immunglobulin-
Superfamilie (Ig-CAM) angesehen
(STRATMANN & JESERICH, 1995).
Vertreter dieser Proteinfamilie definieren
sich über Regionen, die Ähnlichkeiten
mit der variablen Domäne der
Immunglobuline aufweisen. Diese
Immunglobulin-Domänen bestehen aus
alternierenden hydrophoben und
hydrophilen Aminosäureresten, die eine
Serie aus antiparallelen β-Strängen
bilden. Diese β-Stränge falten sich zu 2
β-Faltblättern, die durch Disulfidbrücken stabilisiert werden (WILLIAMS UND BARCLAY,
1987; EDELMAN, 1988). Zu den Ig-CAM gehören z.B. das N-CAM (neural cell adhesion
molecule), MAG und P0. Man konnte allen diesen aufgeführten Proteinen eine Funktion als
homophiles/heterophiles Adhäsionsmolekül sowohl im Nerven- als auch im Immunsystem
experimentell nachweisen (FILBIN ET AL, 1990; D’URSO ET AL., 1990; SALZER UND COLMAN,
1989; AFAR ET AL., 1991; POLTORAK ET AL., 1987).
Abbildung 3: Vertreter der Ig-CAM Superfamilie: Zuihnen gehören P0, NCAM, MAG und weitere Vertreter.Die überwältigende Mehrheit der Moleküle sindVertreter der Subklasse Typ I zu denen auch dieForellenmyelinproteine IP1/2 zählen. (entnommen aus:„Basic Neurochemistry“ Lippincott-Raven, 1998)

5
Da das IP1-Protein der Forelle eine auffällige Ähnlichkeit zu dem P0-Protein der Säuger
aufweist, wurde bereits 1995
von Stratmann und Jeserich
vorgeschlagen, daß es P0-
ähnliche Adhäsionsaufgaben
in der extrazellulären
„intraperiod line“ zur
Kompaktierung des
Forellenmyelins ausführen
könnte. Zur Kompaktierung
der cytoplasmatischen „major
dense line“ des Forellenmyelin
wäre es denkbar, daß die
cytoplasmatischen Domänen
der IP Splice-Varianten
hierbei von großer Bedeutung
sind. Zwar ist die vollständige
Sequenz der längeren IP2
Splice-Variante noch nicht
bekannt, dennoch wird deutlich, daß die cytoplasmatischen Domänen der IP-Proteine eine
positive Nettoladung aufweisen und somit über elektrostatische Wechselwirkungen an saure
d.h. negativ-geladene Phospholipide der Plasmamembran binden könnten (STRATMANN UND
JESERICH, 1995). Eine derartige Eigenschaft konnte den korrespondieren Proteindomänen
des P0-Protein und den Basischen Myelinproteinen bereits nachgewiesen werden (DING UND
BRUNDEN, 1994; CHEIFETZ ET AL., 1985, ROUX ET AL. 1994; REVIEW: STAUGAITIS ET AL.,
1995) Die Funktion des Forellen ZNS-spezifischen 36K Proteines ist noch völlig ungeklärt.
Es ist denkbar, daß es aufgrund seiner ebenfalls stark basischen d.h. positiven Ladung an
negativ-geladene saure Phospholipide der Zellmembran bindet und so die Apposition der
cytoplasmatischen „major dense line“ unterstützt.
In der vorliegenden Dissertation sollten mit Hilfe der heterologen Expression erste
experimentelle Hinweise auf die Funktion der Hauptmyelinproteine IP1 und IP2 sowie 36K
Abbildung 4: Hypothetisches Forellenmyelinmodell: Zelladhäsionan der intraperiod line wird druch die IgCAM Proteine IP1/2vermittelt. Die Apposition der cytoplasmatischen major dense lineerfolgt über die cytoplasmatischen Domänen von IP1/2 sowie überdie Basischen Myelinproteine (BP) und 36K. ElektrostatischeAnziehungskräfte sind gestrichelt angedeutet

6
gewonnen werden. Diese funktionellen Grundcharakterisierungen könnten zum besseren
Verständinis der Myelinogenese beitragen und als Basis für weiterführende Experimente
dienen.
7
2 Material und Methoden
2.1 Material
2.1.1 Bakterienstämme
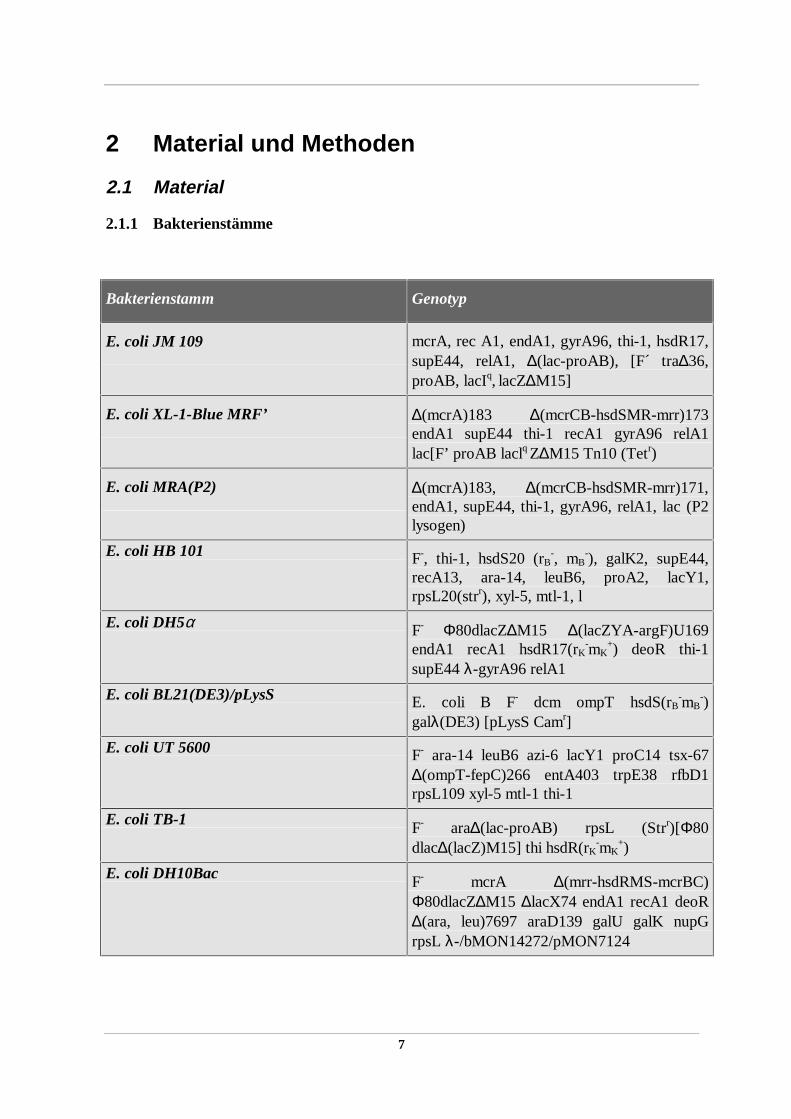
Bakterienstamm Genotyp
E. coli JM 109 mcrA, rec A1, endA1, gyrA96, thi-1, hsdR17,supE44, relA1, ∆(lac-proAB), [F´ tra∆36,proAB, lacIq, lacZ∆M15]
E. coli XL-1-Blue MRF’ ∆(mcrA)183 ∆(mcrCB-hsdSMR-mrr)173endA1 supE44 thi-1 recA1 gyrA96 relA1lac[F’ proAB laclq Z∆M15 Tn10 (Tetr)
E. coli MRA(P2) ∆(mcrA)183, ∆(mcrCB-hsdSMR-mrr)171,endA1, supE44, thi-1, gyrA96, relA1, lac (P2lysogen)
E. coli HB 101 F-, thi-1, hsdS20 (rB-, mB
-), galK2, supE44,recA13, ara-14, leuB6, proA2, lacY1,rpsL20(strr), xyl-5, mtl-1, l
E. coli DH5α F- Φ80dlacZ∆M15 ∆(lacZYA-argF)U169endA1 recA1 hsdR17(rK
-mK+) deoR thi-1
supE44 λ-gyrA96 relA1
E. coli BL21(DE3)/pLysS E. coli B F- dcm ompT hsdS(rB-mB
-)galλ(DE3) [pLysS Camr]
E. coli UT 5600 F- ara-14 leuB6 azi-6 lacY1 proC14 tsx-67∆(ompT-fepC)266 entA403 trpE38 rfbD1rpsL109 xyl-5 mtl-1 thi-1
E. coli TB-1 F- ara∆(lac-proAB) rpsL (Strr)[Φ80dlac∆(lacZ)M15] thi hsdR(rK
-mK+)
E. coli DH10Bac F- mcrA ∆(mrr-hsdRMS-mcrBC)Φ80dlacZ∆M15 ∆lacX74 endA1 recA1 deoR∆(ara, leu)7697 araD139 galU galK nupGrpsL λ-/bMON14272/pMON7124
8
2.1.2 Plasmide
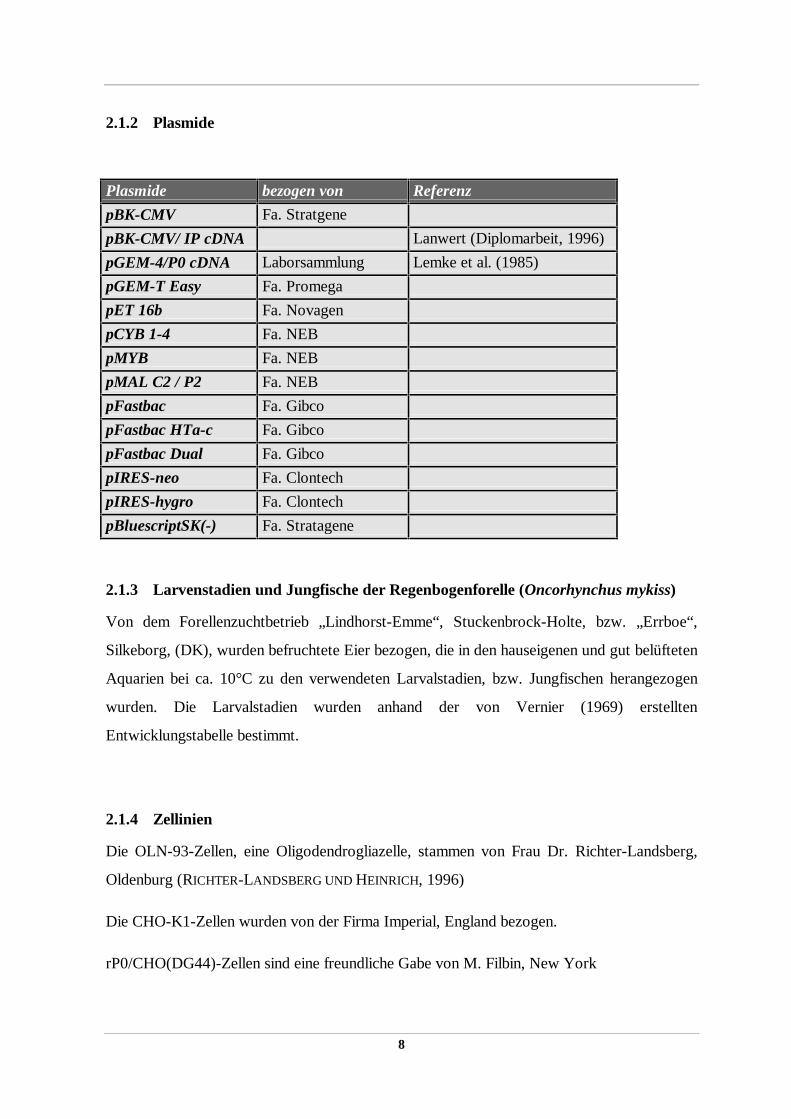
Plasmide bezogen von Referenz
pBK-CMV Fa. Stratgene
pBK-CMV/ IP cDNA Lanwert (Diplomarbeit, 1996)
pGEM-4/P0 cDNA Laborsammlung Lemke et al. (1985)
pGEM-T Easy Fa. Promega
pET 16b Fa. Novagen
pCYB 1-4 Fa. NEB
pMYB Fa. NEB
pMAL C2 / P2 Fa. NEB
pFastbac Fa. Gibco
pFastbac HTa-c Fa. Gibco
pFastbac Dual Fa. Gibco
pIRES-neo Fa. Clontech
pIRES-hygro Fa. Clontech
pBluescriptSK(-) Fa. Stratagene
2.1.3 Larvenstadien und Jungfische der Regenbogenforelle (Oncorhynchus mykiss)
Von dem Forellenzuchtbetrieb „Lindhorst-Emme“, Stuckenbrock-Holte, bzw. „Errboe“,
Silkeborg, (DK), wurden befruchtete Eier bezogen, die in den hauseigenen und gut belüfteten
Aquarien bei ca. 10°C zu den verwendeten Larvalstadien, bzw. Jungfischen herangezogen
wurden. Die Larvalstadien wurden anhand der von Vernier (1969) erstellten
Entwicklungstabelle bestimmt.
2.1.4 Zellinien
Die OLN-93-Zellen, eine Oligodendrogliazelle, stammen von Frau Dr. Richter-Landsberg,
Oldenburg (RICHTER-LANDSBERG UND HEINRICH, 1996)
Die CHO-K1-Zellen wurden von der Firma Imperial, England bezogen.
rP0/CHO(DG44)-Zellen sind eine freundliche Gabe von M. Filbin, New York

9
Sf21 und HighFive-Zellen stammen von Fa. Invitrogen und sind eine freundliche Gabe von
Herrn Dr. R.Wagner, Osnabrück
2.1.5 Chemikalien
Bacto-Agar, Bacto-Trypton, Bacto-Yeast extrakt von Difco, Fa. Nordwald, Hamburg
Tetramethylammoniumchlorid (TMAC), IPTG (Isopropyl-β-D-thio-galactopyranosid,
Tetracyclin-HCl, Kanamycin, Agarose Typ II Medium EEO, Fluoreszenz-Antikörper-
konjugate, Proteinase-Inhibitor-cocktail von der Fa. Sigma, Deisenhofen
Sterile Petrischalen und Multiwellplatten für die Zellkulturen von Nunc, Dänemark
Restriktionsenzyme und -Puffer, sowie Taq-Polymerase, T4-Ligase, Superscript RT II,
RNase H (alle samt Puffer) von Fa. Gibco/BRL, Eggenheim, bzw. Fa. NEB, Schwalbach
Transfectam von Fa. Promega, Cellfectin von Fa. Gibco
Soweit im Methodenteil (siehe 2.) nicht anders vermerkt, sind alle Reagenzien und
Chemikalien von den Firmen Riedel-de Haen, Seelze oder von der Firma Sigma, Deisenhofen
in der Qualität p.A. bezogen worden.
2.1.6 Medien zur Anzucht von Bakterien
LB-Medium 10 g Bacto tryptone; 10 g NaCl, 10 g Hefeextrakt pro 1l
LB-Bac-Medium LB mit 50µg/ml Kanamycin, 7 µg/ml Gentamycin, 10 µl/ml Tetrazyklin
LB-Agar LB-Medium mit 7,5 g Bacto-Agar pro l
LB-Bac-Agar LB-Agar mit mit 50µg/ml Kanamycin, 7 µg/ml Gentamycin, 10 µl/ml
Tetrazyklin, 100µg X-gal und 40µg/ml IPTG

10
SOB-Medium 2 % Bacto-Trypton, 0,5% Hefeextract, 10mM NaCl, 2,5 mM KCl, 10
mM MgCl2, 10 mM MgSO4
TFB-1-Medium 30 mM Kalium Acetat, 50 mM MnCl2, 100 mM RbCl2, 10 mM CaCl2,
15% Glycerin, pH 5,8
TFB-2-Medium 10 mM Na-MOPS, 75 mM CaCl2, 10 mM RbCl2, 15% Glycerin, pH 7,0
TSS-Puffer LB-Medium mit 10 % PEG 8000, 5 % DMSO 50 mM MgSO4 oder
MgCl2, pH 6,5
RM-Medium 10 g Trypton, 5g Hefeextrakt, 5 g NaCl, 2g Glucose pH 7,4 pro 1l
2xYT Medium 10g NaCl, 10g Hefeextrakt, 16g Bactotrypton pro 1l
2.1.7 PCR Primer
Die verwendeten PCR-Primer wurden von der Fa. TIB MolBiol, Berlin, Gibco BLR,
Schottland, MWG, Ebersberg, bzw. Genaxis, Spechbach synthetisiert.
Name Sequenz 5⇒́ 3´
36K-2-R TAA ACT GTT AAA GTA GTG CAA ATC
36K-CF-1 CTC GAG TAC CAC CCT GTG GAA TGG AGC AGT
36K-CL-F4 TAC AAC GCA TAT GTC TCT TTA CCG CA
36K-CL-F6 TCT GAA AGG AAT GAC TGA ATT C
36K-CL-F7 GGA CTC AAA TCC CTG GCT ACA A
36K-CL-R5 ATC GAT TTA GTT TGT AAC TTG T
36K-CL-R6 GAA TTC AGT CAT TCC TTT CAG A
36K-mal-F2 ACG GGA TCC ATG TCT CTT TAC CGC
36K-mal-R2 GTC TCT AGA TCA GTG GGG CTG GAA
36K-R2 CTC CTT GAC AAT GTC AGC CCT
HTB-F1 ATG TCG TAC TAC CAT CAC CA
IP2-C3mal-R TGA AGC TTC ATC AGC ACC CTA
IP2-C3-R GTC ATC AGC ACC CTA CCA GG

11
IP2-CIZ-F ACA GGG CCC AAC TCT CTC CAT
IP2-IZ-F GAC CCG TCC AAG CTG AAG GCC GCC
IP-4E-mal CCT AAG CTT TAG AAG AGA GAG C
IP-5F CTG GAA CCC TGC ACC CCT CTG
IP-cyto-F1 AGG GAA TTC CTG GTG GCC CGC
Po-fus3-f ATT TCT AGA TAC TGC TGG CTG CGC AGG CA
2.2 Methoden
Alle im folgenden beschriebenen Methoden wurden, wenn nicht anders vermerkt, bei RT
durchgeführt.
2.2.1 Bakterienkulturen/Kryokonservierung von Bakterienkulturen
Die Bakterienflüssigkulturen wurden in Wachstumsmedium (s.o.) mit einer Platinöse von
einer Einzelkolonie angeimpft. Am Vortage wurden die Bakterien aus einer Dauerkultur zu
einer Einzelkolonie auf der entsprechenden LB-Platte vereinzelt. Die frisch inokulierten
Flüssigkulturen wurden auf einem Roller, bzw. Schütteltisch zur besseren Durchlüftung bei
37°C bis zum nächsten Morgen, bzw. bis zur gewünschten OD650 inkubiert.
Zur Kryokonservierung von Bakterienkulturen wurden 800 µl der Flüssigkulturen mit 70 µl
DMSO vermischt und in flüssigem Stickstoff schockgefroren und bei -70°C gelagert.
2.2.2 Plasmidpräparationen
Die verwendeten Plasmidpräparations-Kits enthalten Puffer und Säulen mit einer DNA
bindenden Gelmatrix und basieren auf dem gleichen Prinzip. Nach alkalischer Lyse bindet die
Plasmid DNA in Gegenwart von chaotropen Salzen an eine Silicagelmatrix (QIAprep Spin
Plasmid Kit), bzw. Ionenaustauscher („Jet star“ Plasmid-Präparation) und kann mit Hilfe
von A. dest. von der Matrix wieder abgelöst werden.
2.2.2.1 „Spin (Mini) -Präp“-Methode
Zur schnellen Plasmid-DNA Präparation für PCR, Restriktionsanalysen oder Ligationen
wurde das QIAprep Spin Plasmid Kit (Fa. Qiagen) eingesetzt. Die Durchführung erfolgte

12
nach Protokoll des Herstellers. Nach alkalische Lyse und Zentrifugation wurde der
Überstand auf die Säulen pipettiert und für 30 sec bei 12000 rpm zentrifugiert. Nach 2-4
maligem Waschen mit Waschpuffer wurde 50 µl TE-Puffer oder A. dest. auf die Mitte der
Gelmatrix der Säule pipettiert und für 1 min inkubiert. Anschließend wurde die Plasmid-DNA
durch zweiminütiges Zentrifugieren (12000 rpm) eluiert.
2.2.2.2 „Jet star“ Plasmid-Präparation (Fa. Genomed)
Für spätere Transfektionen mußte die DNA eine besonders hohe Qualität aufweisen, da
Kontaminationen mit Proteinen, Endotoxinen, genomischer DNA oder Salzrückständen die
Transfektionseffizienz deutlich reduziert. Die „Jet Star“ Plasmid-Präparation liefert hochreine
und endotoxinfreie DNA (vergleichbar mit 2x CsCl Gradient) und wurde streng nach
Angaben des Herstellers durchgeführt. 3 ml einer Übernacht-Bakterienkultur wurden
abzentrifugiert, das Pellet in 400 µl des Resuspensionspuffers resuspendiert. Nach Zellyse
durch 400 µl des alkalischen, SDS-haltigen Lysis-Puffer (5 min bei RT) erfolgte die
Neutralisierung mit 400 µl Neutralisierungspuffer mit anschließender Zentrifugation bei RT
für 20 min (16000xg). Hierbei wurden die Proteine und Zelltrümmer pelletiert und der
Überstand auf die DNA-bindende, zuvor equilibrierte Säule gegeben. Der Puffer lief von der
Schwerkraft getrieben durch die Säule und wurde verworfen, während die Plasmid-DNA an
der Gelmatrix bindet. In nachfolgenden Waschschritten (2x 2ml Waschpuffer) wurden
Proteine, genomische DNA, RNA, Proteine und alle anderen Metabolite herausgewaschen.
Die Plasmid DNA wurde mit 0,9 ml des Elutionspuffers von der Gelmatrix eluiert und mit
630 µl Isopropanol gefällt, 30 min bei 4°C und 15000 rpm pelletiert und vorsichtig mit 200
µl 70% Ethanol gewaschen, getrocknet und in 50-100 µl A. dest. resuspendiert.
2.2.3 RNA/DNA-Quantifizierung durch Methylen-Blau-Spot-Test
Bei dem Methylen-Blau-Spot-Test wird die Fähigkeit von DNA bzw. RNA ausgenutzt, im
saurem Milieu basische Farbstoffe zu binden. Dazu wird 1 µl der zu quantifizierenden
Lösung auf eine positiv-geladene Nylonmembran (Fa. Boehringer) pipettiert und im UV-
Crosslinker (Fa. Stratagene) bei 120 mJ/cm2 mit der Membran kovalent verbunden. Die
Membran wurde in 5 % Essigsäure auf pH 4 für 15 min equilibriert. Nach anschließendem
viertelstündigem Anfärben in Methylen-Blau-Färbelösung (0,5 M Na-Acetat, 0,04%
Methylen-Blau, pH 5,2) wurde die Membran unter leichtem Schütteln in mehreren Schritten

13
in A. dest. vollständig entfärbt. Anhand einer vorher erstellten Eichreihe wird die DNA-
Menge colorimetrisch bestimmt.
2.2.4 RNA/DNA-Quantifizierung durch Photometrie
Mit Hilfe des Spectralphotometers kann die Nukleinsäurekonzentration wäßriger Lösungen
bestimmt werden. Eine OD 260 von 1,0 entspricht 50µg/ml bei DNA, bzw. 40µg/ml bei
RNA.
2.2.5 Restriktion von DNA mit Restriktionsendonukleasen
Zur Hydrolyse von DNA mit Restriktionsendonukleasen wurde ca. 1 µg DNA pro 10 µl
Ansatzvolumen mit 2-10 U des gewünschten Enzyms im Wasserbad bei 37°C (bzw.
abweichenden Temperaturoptima nach Hersteller) 1-2 h inkubiert. Als Puffersystem diente
der vom Hersteller mitgelieferte 10x Puffer.
2.2.6 Agarose-Gelelektrophorese
DNA Fragmente können mit Hilfe der Agarose-Gelelektrophorese im elektrischen Feld der
Größe nach aufgetrennt werden. Dazu wurden 0,7 bis 2 % Agarose (w/v) in 1x TAE-Puffer
(0,04 M Tris-Acetat, 0,001 M EDTA, pH 8) verwendet. Die DNA-Proben wurden mit einem
1/5 Gel-Ladepuffer (1x TAE, 0,25% Bromphenolblau, 0,25% Xylencyanol, 50 % Glycerin )
versetzt und die Elektrophorese bei 5-10 V/cm Elektrodenabstand durchgeführt
(Elektrophoresekammer Mini-Sub Electrophoresis Cell, Fa. Biorad). Als Größenstandard für
die zu analysierenden DNA-Fragmente diente EcoR I/Hind III verdaute λ-Phagen DNA (Fa.
Sigma), Smart-Ladder (Fa. Eurogentec), bzw. eine 100 bp DNA-Leiter (100-1500 bp) (Fa.
Gibco). Die DNA-Fragmente im Gel wurden anschließend für 10 min im Ethidiumbromid-
Bad (2µg/ml, Fa. Serva) gefärbt, für 10 min in dH2O gespült, auf einem UV-Transluminator
bei 302 nm sichtbar gemacht und auf einem Videoprinter (Fa. Sony) dokumentiert.
2.2.7 Gelextraktionsmethoden
2.2.7.1 DNA-Gelextraktion „Jet-sorb“ -Methode (Fa. Genomed)
Bei der Jet sorb“ Gelextraktion Methode aus TAE-Agarosegelen wird die DNA nach
Elektrophorese (2.2.6) aus einem Agarosegel herausgeschnitten und gewogen. Die Menge an
eingesetzten Puffern richtet sich nach dem Gewicht des ausgeschnittenen Fragmentes (pro
100mg Gel/ 300µl Puffer A1 und A2 mit 15 µl Silika-Beads) Der mitgelieferte Puffer A1

14
enthält einen TAE-Gel Solubilizer, der die H-Brücken zwischen den Zuckern im
Agarosepolymer zerstört. Das Gel löst sich daher bereits bei 50°C auf, so daß die DNA in
diesem Milieu an den mitgelieferten Silika-Beads binden kann. Nach diversen Waschschritten
mit Puffer A1 kann die DNA im Niedrigsalz-Puffer A2 bei 50°C eluiert werden. Die „Jet-
sorb“-Methode wurde streng nach Angaben des Herstellers durchgeführt.
2.2.7.2 DNA Gelextraktion „Qiaquick Gel Extraction Kit“ Fa. Qiagen
Die Gelextraktionmethode der Fa. Qiagen ähnelt der oben beschriebenen Jet-sorb Methode,
mit dem Unterschied, daß die Silica Matrix in Form kleiner Säulchen vorliegt und daher
einfacher in der Handhabung ist. Die Methode wurde streng nach Protokoll des Herstellers
durchgeführt.
2.2.8 Klonierungen
2.2.8.1 Dephosphorylierung von Plasmidvektoren
Zur Klonierung von DNA-Fragmenten, die nur mit einem Restriktionsenzym geschnitten
wurden und daher unter Ligationsbedingungen zur Rezirkulation neigen, müssen die 5´-
Phosphatgruppen des Vektors hydrolysiert werden. Hierzu wurde der geschnittene Vektor
vor Ligation mit alkalischer Phosphatase (CIAP calf intestinal alkaline phosphatase, Fa.
Promega) behandelt: Es wurden 5µl CIAP (0,5 U) sowie mitgelieferter 10x Reaktionspuffer
pro 2µg linearisiertem Vektor pipettiert und das Volumen des Ansatzes mit A.dest. auf 50µl
eingestellt. Die Dephosphorylierung erfolgte für 60min bei 37° C bzw. für 30 min bei 37° C
und anschließend für 30 min bei 56° C unter Zugabe von weiteren 0,5µl Enzym.
2.2.8.2 Sticky end-Ligation
Für die sog. „sticky end“-Ligation werden kohäsive, komplementäre Enden benötigt, die
durch die Restriktion der doppelsträngigen DNA entstehen können. Hierzu bedient man sich
der Eigenschaft bestimmter Restriktionsenzyme, 3’ oder 5’ überhängende Termini zu
produzieren, in dem sie den DNA-Doppelstrang „versetzt“ schneiden. Durch die
Verwendung von zwei unterschiedlichen Restriktionsenymen, kann man gezielt und gerichtet
klonieren. Für eine ungerichtete Klonierung (nur mit einem Enzym geschnitten) mußte der
geöffnete Vektor dephosphoriliert werden (2.2.8.1) Die Ligationsreaktionen wurden im 10 µl
Ansatz durchführt, wobei 0,5 µl T4-Ligase (Fa. Gibco) sowie 2 µl des vom Hersteller

15
mitgelieferten 5x Reaktionspuffer verwendet wurden. Das für die Reaktion notwendige ATP
war bereits im Reaktionpuffer enthalten. Es wurden 50 µg Vektor/Plasmid-DNA und die im
molaren Verhältnis 3-6 fachen Menge an Insert -DNA eingesetzt. Durch die größere Menge
an Insert DNA sollte die intermolekulare Ligation von Vektor/Insert gegenüber
intramolekularen Bindungen begünstigt werden. Die Reaktion erfolgte über Nacht bei 16° C
oder 1h bei RT.
2.2.8.3 blunt end-Ligation
Bei der Restriktion von DNA mit Enzymen, die nicht versetzt schneiden, entstehen sog. blunt
ends, glatte DNA-Termini. Die T4-Ligase ist in der Lage auch blunt ends zu ligieren, jedoch
ist nur eine ungerichtete Ligation möglich. Außerdem ist die Effizienz gegenüber der sticky
end-Ligation nicht so hoch. Um die Effizienz zu steigern wurde die bis zu 10-fache molare
Menge an blunt end-Insert DNA gegenüber der Vektor DNA eingesetzt.
2.2.9 Herstellung von glatten Enden mit T4 Polymerase („blunting“)
Nach einer Restriktion können 5‘ oder 3‘ Überhänge auf glatte Enden (blunt ends) aufgefüllt,
bzw. abgedaut werden. Dazu wurden 0,3 – 2 µg der verdauten und gereinigten DNA im 20
µl Ansatz mit 10 U T4-Polymerase (Gibco), 1µl 100mM DTT und dem mitgelieferten 5x
Reaktionspuffer versetzt. Im Falle einer Auffüllreaktion wurde zusätzlich 2µl 10mM dNTP-
Mix pipettiert und für 15 min bei 12° C inkubiert. Die Reaktion wurde anschließend für 10
min bei 70° C Hitzeinaktiviert.
2.2.10 Reinigung von DNA Fragment aus wäßrigen enzymatischen Lösungen
Zur Reinigung von DNA aus wäßrigen Lösungen nach enzymatischen Reaktionen, z.B.
Restriktionsverdau, PCR oder DNA-Modifikationen wurde der Ansatz mit 50 µl
Chloroform/Phenol (Fa. Roth) vermischt und 5 min bei RT (12000 rpm) zentrifugiert. Die
wäßrige Phase wurde vorsichtig abgezogen und nach Zugabe von 1/6,3 Volumen 3 M
Natriumacetat (pH 4,5) und des dreifachen Volumen eiskalten absoluten Ethanols für 30 min
bei -70° C gefällt. Anschließend wurde das Präzipitat 30 min bei 4° C und 15000 rpm
zentrifugiert, mit 70% Ethanol gewaschen und in 10 µl dH2O oder TE-Puffer (10 mM Tris, 1
mM EDTA, pH 8,0) resuspendiert.
Alternativ wurde das „Qiaquick PCR Purification Kit“ der Fa. Qiagen nach Protokoll des

16
Herstellers verwendet. Hierbei handelt es sich um kleine Säulchen mit einer DNA-bindenden
Silicamatrix, die ähnlich wie unter 2.2.2.1 beschrieben eine schnelle und phenolfreie
Aufreinigung erlaubten.
2.2.11 Klonierung von PCR-Produkten / pGEM-T easy
PCR-Produkte, die durch Taq-Polymerase amplifiziert wurden, konnten mit Hilfe des
pGEM-T Easy System (Fa. Promega) einfach kloniert werden. Hierbei wird die terminale
Transferase-Aktivität (hohe Affinität für dATP) der Taq-Polymerase ausgenutzt, die am 3‘
Ende eines Taq-Amplifikates ein d(A)-Überhang entsteht läßt. Über eine A/T sticky end
Ligation mit dem vom Hersteller mit einem d(T)-Überhang modifizierten, linearisierten
pGEM-T easy Vektor konnte sehr effektiv kloniert werden. Der 10µl Ligationsansatz
bestand aus 0,5µl pGEM-T easy, 1µl (3U) Ligase, 5µl Ligasepuffer (alles Bestandteile des
Kits) sowie 4 µl des gereinigten PCR-Produktes. Die Ligation wurde entweder für 1h bei RT
oder ÜN bei 4°C inkubiert.
2.2.12 Plasmid Transformation
2.2.12.1 TFB-Methode und Blau/Weiß-Selektion
Zur Herstellung kompetenter E. coli-Zellen wurde die TFB-Methode (buffers for
transformation and frozen storage of competent cells), nach dem modifizierten Protokoll von
Hanahan (1983) durchgeführt. Von einer Einzelkolonie wurde eine 5 ml Übernachtkultur
angeimpft. Von 1 ml dieser Übernachtkultur wurden 200 ml SOB-Medium angeimpft und bei
37°C auf dem Schüttler bis zu einer OD650 von 0,4 bis 0,5 inkubiert. Anschließend wurden
sie bei 4°C und 500 g für 10 min abzentrifugiert, in 60 ml TFB-1-Medium resuspendiert und
für 10 min auf Eis gekühlt. Die in diesem speziellen Transformationsmedium enthaltenen
Salze sollen die Bakterienmembran „löcherig“ und somit für die Aufnahme der Plasmid-DNA
fähig, d.h. kompetent machen. Nach erneutem Pelletieren und Resuspendieren in 8 ml TFB-2,
wurden die erhaltenen kompetenten Zellen zu 200 µl aliquotiert, in flüssigen Stickstoff
schockgefroren und bei -70° C gelagert.
Pro Transformationsansatz wurden 100 µl der eingefrorenen kompetenten Zellen auf
Eiswasser aufgetaut und vorsichtig gemischt. Nach Zugabe von 50 ng klonierter DNA (bis zu
10 µl des Ligationsansatzes) und 10 minütiger Inkubation auf Eis erfolgte der Hitzeschock

17
für 75 sec bei genau 42° C mit anschließender Abkühlung auf Eis für 2 min. Während dieser
Hitzeschock-Phase nehmen die Bakterien die DNA auf. Zur phänotypische Expression des
Resistenzgenes wurde der Ansatz mit 900µl LB versetzt und für 1 h bei 37°C im Roller
inkubiert, 30 sec bei 12000xg abzentrifugiert, der Überstand verworfen, das Pellet in 100 µl
LB-Medium resuspendiert, mit einem Drigalski-Spatel auf einer entsprechenden
Selektionsplatte ausplattiert und bei 37° C ÜN inkubiert. Im Falle einer Blau-Weiß-Selektion
(αKomplementationstest nach SAMBROCK ET AL., 1989) wurde zusätzlich 100 µl 100 mM
IPTG und 100 µl X-Gal (2% in Dimethylformamid) vor dem Plattieren der Bakterien
eingespatelt.
2.2.12.2 TSS Methode
Die TSS- (Transformation and storage solution) Methode wurde nach dem Protokoll von
Chung et al. (1989) durchgeführt. Eine mitlogarhythmische Bakterienkultur wurde bei 2000
g, 4°C, abzentrifugiert und die Pellets in jeweils 100 µl TSS-Medium resuspendiert. Nach 10
min Inkubation der Zellen auf Eis wurden 10 µl des Ligationsansatzes dazugegeben und nach
weiteren 20 min auf Eis 900 µl TSS-Puffer dazupipettiert. Nach phänotypischer Expression
wurden die Zellen auf Selektionsplatten ausplattiert und ÜN inkubiert.
2.2.13 Polymerase-chain-reaction (PCR)-Techniken
2.2.13.1 PCR Primer Design und PCR-gerichtetete Mutagenese
Beim Primerdesign wurde auf einen ausgewogenen GC-Gehalt (zwischen 40 und 60 %)
geachtet. Bei einer Länge von 20-26 Nukleotiden resultierte daraus eine Schmelztemperatur
von 48-68°C. Um Primer-Dimere auszuschließen, besaßen die Primerpaare möglichst wenig
Komplementarität zueinander, sowie möglichst wenig palindrome Sequenzen, um die
Ausbildung von Sekundärstrukturen zu vermeiden.
Zur PCR-gerichteten Mutagenese wurde die gewünschte Mutation, z.B. Basenaustausche
zum Einführen von Restriktionsschnittstellen, in den sequenzspezifischen Primer im Bereich
des 5‘-Endes eingefügt. Im Falle von Restriktionsschnittstellen wurden zusätzlich 4-6
Nukleotide an das 5‘ Ende angefügt.
2.2.13.2 Polymerase-Ketten-Reaktion (PCR)
Die PCR zur Amplifizierung von DNA-Templates (SAIKI ET AL., 1988) wurde in einem 50µl-

18
Reaktionsansatz durchgeführt. Die Menge an eingesetztem Template betrug 0,1-30ng. Pro
Reaktion wurden je 50pMol der beiden PCR-Primer eingesetzt, die dNTP-Konzentration war
200µM. Die MgCl2 Konzentrationen variierten je nach Anwendung von 10mM (Standard)
bis zu 17,5 mM bei Amplifikationen größer als 4 kb. Eingesetzt wurden die Taq DNA
Polymerasen/Reaktionspuffer der Firmen Gibco, bzw. Boehringer (Hifi-Taq)
Die PCR-Reaktionen bestanden aus 30-40 Temperaturzyklen. Dabei wurde die DNA zur
Denaturierung auf eine Temperatur von 95° C (1 min) erhitzt. Da die Taq-Polymerase
(Temperaturoptimum 72° C) bereits bei 30-50°C eine partielle Aktivität besitzt, wurde
generell die sog „Hot Start-PCR“ durchgeführt, um unspezifische Reaktionen zu vermeiden
(ERLICH ET AL., 1991). Bei der Hot Start-PCR wird das Enzym erst nach einer
Hitzeinkubation von 1-5 min bei 95° C und einem anschließenden Abkühlen auf 72° C
dazugegeben. Für die nachfolgende Bindung der PCR-Primer an das DNA-Template wurde
die sog. Annealing-Temperatur der Schmelztemperatur der Primer angepaßt. Sie betrug
zwischen 50-60° C (30 sec). Anschließend folgte die 5´→3´ Kettenverlängerung (sog.
Extension) des Primer-Template-Komplexes bei einer Temperatur von 72°C (45 sec für
Amplifikationen bis 1kb, Extension bis zu 6min +5 sec /Zyklus bei longdistance PCR). Um
unspezifische Primer-Template-Bindungen zu unterbinden, wurden wahlweise 2,5 µl
Tetramethylammoniumchlorid (100mM TMAC) oder bis zu 2µl DMSO in den
Reaktionsansatz gegeben. Anschließend wurde das Reaktionsgemisch mit zwei bis drei
Tropfen DNase-freiem Mineralöl (Fa. Sigma) überschichtet, um eine Evaporation der Lösung
während der Reaktion zu verhindern.
Alle PCR-Reaktionen wurden in einem programmierbaren Thermocycler (Nr. IHB101) der
Fa. Biometra bzw. Primus Thermocycler der Fa. MWG-Biotech durchgeführt.
2.2.13.3 Invers-PCR
Die inverse PCR wurde erstmal von Ochman et al. (1988) beschrieben. Die inverse PCR
wurde zur Identifizierung unbekannter gDNA Fragmente im Anschluß an ein Southern-
Blotting (2.2.41) durchgeführt. Der Größenbereich, der dem positiven Signal des Southern-
Blottes entspricht, wurde in einem Parallelansatz aus dem Agarosegel herausgeschnitten und
aufgereinigt (2.2.7). Da beide Enden des mit nur einem Enzym geschnittenen DNA-
Fragmentes mit sich selbst kompatibel sind, wurde in einem Ligationsansatz für 1h bei RT in

19
einer sticky-end Ligation (2.2.8.2) das DNA-Fragment zirkularisiert. Dieses kovalent
geschlossenen DNA-Fragment diente als Template für die subsidiäre Invers-PCR. Mit Hilfe
der Invers-PCR kann ein zirkuläres, kovalent geschlossenes DNA Fragment vollständig
amplifiziert werden. Dazu werden PCR-Primer so generiert, daß sie invers zur
konventionellen PCR orientiert ist. So erfolgt während der PCR die Kettenverlängerung
„run-around“. Die Invers PCR wurde ausschließlich mit „Expand HiFi Taq“ (Fa. Boehringer)
durchgeführt.
2.2.13.4 rapid PCR /Transformanden Screening
Mit einem sterilen Zahnstocher wurde etwas Zellmaterial einer Bakterien-
Transformandenkolonie von der Selektionsplatte gepickt und direkt in ein PCR-Gefäß
überführt, welches den vollständigen PCR-Mix enthält. Anschließend wurde mit diesem
Zahnstocher eine Duplikatsplatte ausgestrichen und ÜN bei 37°C inkubiert. Die PCR-Primer
wurden nach Möglichkeit so gewählt, daß einer komplementär zu Sequenzen im Insert, der
andere komplementär zu Sequenzen im Vektors ist („Vektor/Insert-PCR“). So ist nur bei
gewünschter Klonierungsrichtung eine Bande zu erwarten.
2.2.13.5 RT-PCR
Eine PCR im Anschluß an eine Reverse Transkription wird Reverse Transkription-PCR (RT-
PCR) genannt. Es wurden zur RT-PCR die gleichen Mengen an Puffern, Lösungen und
Primern, sowie dieselben Zyklen verwendet wie unter 2.2.13.2. beschrieben. Als Template
dienten hier 1-50 ng der nach der Reversen Transkription erhaltenen Erststrang-cDNA (siehe
2.2.53).
2.2.13.6 5‘ bzw. 3‘ RACE (random amplification of cDNA ends) –PCR und „nested“PCR
Die Methode der RACE-PCR dient der Amplifikation von vollständigen cDNA Sequenzen
ausgehend von einer bekannten „internen“ Sequenz und den 5‘ bzw. 3‘ Endes der cDNA. Die
RACES wurden mit dem 5‘/3‘ RACE-Kit der Firma Boehringer, Mannheim streng nach
Protokoll des Herstellers durchgeführt. Hierzu wurde poly(A)+-mRNA (2.2.52) isoliert und
mit der im Kit enthaltenen AMV-Reversen Transkriptase bzw. der Superscript II (Gibco) in
cDNA umgeschrieben (2.2.53) Für das 3‘ RACE erfolgte die RT mit dem mitgelieferten
oligo d(T) Ankerprimer 1, der außer der oligo d(T) Sequenz eine zusätzliche Sequenz trägt,

20
die vom Ankerprimer 2 erkannt werden kann. Mit Hilfe einer anschließenden PCR mit einem
„gene of interest“-spezifischen vorwärts Primer im Kombination mit Ankerprimer 1 wird das
3‘Ende der gesuchten cDNA komplettiert. Durch eine zweite „nested“ PCR mit einem weiter
innenliegenden cDNA-spezifischem Primer in Kombination mit dem Ankerprimer 2 kann die
Spezifität des erhaltenen Amplifikates überprüft werden.
Im Falle eines 5‘ RACES wurde die mRNA mit einem reversen genspezifischen Primer
umgeschrieben und somit vorselektiert. Mit Hilfe der im Kit enthaltenen Terminalen
Transferase wurde an das 5‘ Ende der erhaltenen cDNAs ein poly(A)-Schwanz angehängt. In
zwei PCR Reaktionen im Anschluß wurden als reverse Primer zwei genspezifische Primer
zusammen mit den Ankerprimern 1 und 2 verwendet.
2.2.13.7 Herstellung von nicht-radioaktiven DIG-Sonden per PCR
Für Southern-Blots (2.2.41) und Plaque-Hybridisierungen (2.2.18) wurden nicht-radioaktive
Digoxygenin (DIG) markierte Gensonden hergestellt. Digoxygenin ist ein aus Digitalis
purpurea gewonnenes Cardenolid-Steroid und kann nach Hybridisierung durch einen
Antikörper immunologisch detektiert werden (2.2.35). Die Markierung der DNA-Sonde
erfolgte mit Hilfe der PCR (2.2.13). Anstelle des „normalen“ dNTP Mixes wurde ein DIG-
dNTP-labelling-Mix (Fa. Boehringer) verwendet, der einen Teil der dTTP’s durch DIG-11-
dUTP substituiert hat. Durch den Einbau dieses Nukleotides in das Amplifikat wird die
Sonde mit DIG markiert.
2.2.14 Chemolumineszenzdetektion/ ECL-Kit (Fa. Amersham)
Diese Methode wurde zum Nachweis von Proteinen im Westernblot (2.2.35) mit sekundären
Peroxidase-gekoppelten Antikörpern angewandt. Das ECL Kit basiert auf einer Peroxidase-
gekoppelten Reaktion, wodurch ein Luminol (Bestandteil des Kits) oxidiert wird. Bei dieser
Oxidation wird Licht mit einer Wellenlänge von 428nm mit einer Halbwertzeit von ca. 60
min emittiert und kann nach Exposition auf einem Röntgenfilm (Hyperfilm, Fa. Amersham)
nachgewiesen werden. Die Durchführung der Detektion erfolgte streng nach den Angaben
des Herstellers.
2.2.15 Alkalische Phosphatase-NBT/BCIP-Farbreaktion
Die Alkalische Phosphatase-NBT/BCIP-Farbreaktion basiert auf der Oxidation von 5-

21
Bromo-4-chloro-3-indolyl-Phosphat (BCIP) zu Indigo nach Freisetzung der Phosphatgruppe
durch die alkalische Phosphatase. In einer simultanen gekoppelten Reaktion wird Nitroblau
Tetrazoliumchlorid (NBT) zu Diformazan reduziert. Beide Prozesse führen zur Bildung eines
bläulichen unlöslichen Farbpräzipitates. Die NBT/BCIP Farbreaktion wurde zur
immunologische Identifizierung mit Alkalischer Phosphatase gekoppeltem anti-DIG-Anti-
körper von DIG-Sonden hybridisierten Phagenplaques wie unter 2.2.18 beschrieben. Dazu
wurde die NC-Membran in AP-Puffer (100 mM Tris-HCl pH9,5, 100 mM NaCl, 50 mM
MgCl2) equilibriert und mit 99 µl BCIP (50 mg/ml in DMF) und 132 µl NBT (75µg/ml in
70% (v/v) DMF) pro 15 ml AP-Puffer bis zum Farbumschlag inkubiert. Die Reaktion wurde
durch Waschen mit Wasser oder TE gestoppt.
2.2.16 Alkalische Phophatasenachweis mit CSPD (Fa. Boehringer)
Der Nachweis mit alkalischer Phophatase gekoppelter Antikörper wurde streng nach
Vorschrift des Herstellers mit dem CSPD-Kit (Fa. Boehringer) durchgeführt. Es handelt sich
hierbei um eine Chemolumineszenzreaktion, die auf Röntgenfilm dokumentiert werden kann
und ist in Sensitivität und Reaktionsmechanismus vergleichbar mit dem ECL-Kit (2.2.14)
2.2.17 Peroxidasenachweis mit 4-Chloro-1-Naphtol (4CN) (nach Sambrock)
Meerrettich-Peroxidase gekoppelte Antikörper wurden mit Hilfe von 4 Chloro-1-Naphtol
(4CN) nachgewiesen. 4CN ist farblos und wird unter Zugabe von H2O2 und Peroxidase
oxidiert, was durch einen blauen unlöslichen Niederschlag sichtbar wird. Dazu wurden 15mg
4CN in 5ml Methanol gelöst und mit 25 ml TBS (vorgewärmt auf 37°C) vermischt. Die
Reaktion wurde mit 40µl H2O2 gestartet und mit A. dest. nach Farbumschlag gestoppt.
2.2.18 Screening einer λ-UNI ZAP-cDNA Libary mit DIG markierten DNA-Sonden
Eine cDNA Phagen-libary aus mRNA des Forellengehirns (Stadium 37) lag vor Beginn
dieser Arbeit vor (Dissertation Astrid Stratmann, 1995). Die Durchführung dieses
Hybridisierungsscreenings erfolgte im wesentlichen nach der von Sambrock et al. (1989)
beschriebenen Methode und ist bereits in mehreren Dissertationen (Stratmann, 1995, Strelau,
1997, Nguyen, 1998) detailliert beschrieben.
Eine lamB –induzierte ÜK von E. coli XL-1Blue (Wachstum in LB Medium mit 0,2%
Maltose, 10 mM MgSO4) wurde mit 3-4 x 105 pfu der Phagenlibary infiziert und in

22
verflüssigtem Topagar auf NZY-Platten ausplattiert. Nach Inkubation ÜN bei 37°C sind
klare einzelne Phagenplaques im Bakterienrasen zuerkennen. Nach Abkühlung der Agarplatte
auf 4°C wurde ein Nitrocellulose-Filter aufgelegt, markiert und so ein Abdruck der Plaques
erstellt. Der NC-Filter wurde für 5 min in Denaturierungslösung (1,5M NaCl, 0,5M NaOH)
inkubiert und anschließend mit 1,5M NaCl, 0,5M TrisHCl pH8,0 neutralisiert. Die Filter
wurden kurz in 2xSSC gewaschen und die DNA auf dem Filter durch 20 min 120°C
immobilisiert.
Zur Hybridisierung wurden die Filter in Hybridisierungsbeutel (Fa. Gibco) transferiert und
mit 10 ml Hybridisierungslösung (50% Formamid, 5xSSC, 2% Blockierungsreagenz Fa.
Boehringer, 0,1% N-Laurylsarcosin, 0,02% SDS) für 1h bei 42°C prähybridisiert. Die
anschließende Hybridisierung mit der DIG-Sonde (50-75 ng/ml Hybridisierungslösung, frisch
denaturiert durch 10 min 95°C, anschließend 5 min auf Eis) erfolgte ÜN bei 42°C.
Der Filter wurde zum stringenten Waschen aus dem Beutel genommen und 2 mal mit
Stringenz A (2xSSC, 0,1% SDS) bei RT und zweimal mit Stringenz B (0,1x SSC, 0,1%
SDS) bei 68°C für je 15 min gewaschen.
Der subsequente Nachweis von hybridisierter DIG-DNA-Sonden erfolgte mit NBT/BCIP
Farbnachweis (siehe 2.2.15). Positive blaugefärbte Signale auf den Filtern konnten eindeutig
Plaques auf den Agarplatten zugewiesen werden. Diese Plaques wurden mit einer
abgeschnittenen Pasteurpipette ausgestochen und in 1 ml SM-Puffer Puffer (5,8g NaCl, 2g
MgSO4 x 7H2O, 50ml 1M TrisHCl pH 7,5, 5ml 2% Gelatine pro Liter) gevortext und ÜN
bei 4°C leicht geschüttelt. Die Phagen diffundieren während dieser Zeit in den SM-Puffer.
Mit Hilfe einer PCR konnte die Spezifität der Hybridisierung überprüft und die
rekombinanten Plaques von falsch positiven diskriminiert werden. Durch zwei
„rescreenings“, in denen der oben beschriebene Ablauf noch zweimal durchgeführt wurde,
wurde ein positiver Plaque auf nahezu Homogenität aufgereinigt.
2.2.19 In vivo excision positiver cDNA/Phagen-Klone
Die in vivo Excision positiver Phagenklone erfolgte mit Hilfe von R408 Helferphagen (Fa.
Stratagene). Bei der in-vivo Excision nutzt man in trans wirkende Proteine des
Helferphagen, die zwei Domänen (Initiator und Terminator des f1 origin) erkennen, welche

23
flankierend zu den cDNA-Inserts auf dem λ-UNI-ZAP Phagenarmen positioniert sind. Die
dazwischenliegenden DNA-Sequenzen (pBluescript mit cDNA-Inserts) wird als
zirkularisierter Einzelstrang in filamentöse Phagmide verpackt und sekretiert. Diese
ssPhagmide können neue Bakterien infizieren, so daß die cDNAs als dsPlasmid-DNA
vorliegen. Die vivo Excision erfolgte streng nach Vorschrift des Herstellers.
Zur in vivo Excision wurden lamB-induzierte E. coli XL1-Blue Zellen (200 µl einer OD600
von 1,0 in 10 mM MgSO4)mit ca. 5x105 pfu des positiven λ-UNI-ZAP Klones und 1x104
R408 Helferphagen für 15 min bei 37° C coinfiziert. Nach Inkubation mit 2xYT Medium für
3h bei 37°C sind mit Hilfe der Helferphagen pBluescript-Phagmide entstanden, die die
gesuchte cDNA als Insert in der multi cloning-site tragen, sekretiert worden. Die Bakterien
werden durch 20 minütiger Hitzeinkubation bei 70° C abgetötet. Die Hitzeresistenten
pBluescriptphagmide liegen im Überstand vor. Durch eine anschließende Infektion frischer
XL1-Blue-Zellen mit diesen Phagmiden und subsequentes Ausplattieren auf einer Amp-
Selektionplatte (ÜN 37° C) wurde auf die pBluescript-Plasmid tragenden Bakterien
selektioniert. Die Plasmid-DNA, die das gesuchte cDNA Insert trägt, konnte mit unter 2.2.2
beschriebenen Methoden präpariert werden.
2.2.20 Heterologe Expression in Insektenzellen mit Baculoviren / Bac-to-Bac-System(Fa.Gibco)
Der Autographa californica nuclear polyhedrosis virus (AcNPV), kurz auch Baculovirus
genannt, infiziert Insektenzellen, schleust sein Genom in die Wirtszelle ein und bedient sich
des Proteinbiosyntheseapparates des Wirtes zur eigenen Vermehrung. Die Wirtszelle lysiert
nach Virusvermehrung und setzt so neue Viren frei. Da das Virusgenom bei einer Größe von
ca. 130 kb auch nicht-essentielle Bereiche enthält, ist der Baculovirus ein idealer Transporter
für Fremd-cDNA und daher geeignet für heterologe Proteinexpression. Aufgrund der Größe
des AcNPV-Genomes ist eine direkte Klonierung mit Fremd-DNA ähnlich der Konstruktion
rekombinanter Plasmide nicht möglich. Das verwendete Bac-to-Bac-System löst das
Klonierungsproblem durch gerichtete Transposition. Die Ziel-cDNA wird zunächst in
pFastbac unter der Kontrolle des für Insektenzellen konstitutiven Polyhedrin-Promoters
(bzw. für pFastbacDual P10-Promoters) subkloniert. Der Vektor pFastbac trägt flankierend
zum Polylinker (Multicloningsite) Konsensussequenzen für das bakterielle Tn7 Transposon

24
und dient einzig als Klonierungs-Vektor. Das AcNPV-Genom ist ebenfalls rekombinant
verändert worden und liegt als Shuttle-Vektor (Bacmid genannt) in einem speziellen
DH10Bac-E. coli-Stamm vor. Dieses Bacmid (bMON14272) trägt die essentiellen AcNPV-
Gene, darüber hinaus aber u.a. Resistenzmarker, mini-F-Replikon und eine lacZ-Kassette zur
α-Komplementation (Blau/Weiß-Selektion, 2.2.12.1), die es möglich machen, das AcNPV
Genom in E. coli zu replizieren. Das Bacmid trägt außerdem Tn7-Transposon attachment
sites. Die Tn7 Transpositionsfunktionen werden in trans durch ein weiteres Helferplasmid
(pMON7124) in DH10Bac zur Verfügung gestellt. Transformiert man nun sein Zielgen mit
Hilfe des pFastbac in kompetente DH10Bac, so springt es samt Polyhedrinpromoter durch
gerichtete Transposition in das Bacmid. Durch Blau/Weiß-Selektion kann man auf
rekombinante Bacmid-Kolonien diskriminieren. Nach Präparation der rBacmid-DNA wird sie
in Insektenzellen transfiziert, so daß nach 4-6 Tagen rekombinante AcNPV geerntet werden
können.
2.2.20.1 Herstellung rekombinanter AcNPV / Lagerung
Das Bac-to-Bac System wurde im wesentlichen nach dem Herstellerprotokoll durchgeführt:
Nach Subklonierung des Zielgenes in pFastbac erfolgte die Transformation in DH10Bac nach
der TFB-Methode (2.2.12), wobei mehrere Verdünnungsstufen auf LB-Bac Agarplatte
ausplattiert wurden. Nach 2 d Inkubation bei 37° C zur Transposition/Blau-Weiß-Selektion
wurden weiße Kolonien erneut auf einer Selektionsplatte ausgestrichen, um falsch positive
Kolonien auszuschließen. Die positiven Bacmidklone wurden ÜN bei 37°C in LB Bac-
Medium (2.1.6) kultiviert. Abweichend zum Herstellerprotokoll erfolgte die Bacmid-
Präparation mit Hilfe der Jet-Star Präparation (2.2.2.2), wobei der Elutionspuffer auf 65°C
erhitzt wurde, um die rBacmidDNA (>100kb) besser zu eluieren. Die initiale Transfektion
von Insektenzellen erfolgte mit Cellfectin in 35 mm Zellkulturschälchen (2.2.50). Wurde das
Reportergen GFP verwendet, so waren bereits nach 2 Tagen Inkubation bei 27°C die ersten
positiven Zellen detektierbar. Nach weiteren 3-6 Tagen waren nahezu alle Zellen infiziert,
einige bereits lysiert. Das rAcNPV enthaltende Medium wurde abpipettiert und sterilfiltriert.
So wurden pro 35 mm Transfektion 1 ml Primärviren gewonnen. Die rAcNPV wurden nun in
Nunc-Kryoröhrchen dunkel bei 4°C gelagert und waren länger als 1 Jahr infektiös.
Zur Virusvermehrung wurden 1 ml der Primärviren auf eine 50ml HighFive, bzw. Sf21

25
Kultur (2.2.45) gegeben und für 6-7 Tage bis annähernd zur vollständigen Zellyse bei 27°C
inkubiert, der Virusüberstand abgenommen, sterilfiltriert und bei 4°C gelagert. Zur
Kryokonservierung wurde dem Medium DMSO (Endkonzentration 10%) und FCS
(Endkonz. 10%) hinzugefügt und wie eine Bakteriendauerkultur bei -70°C gelagert. Zum
Auftauen werden sie bei 37°C im Wasserbad zügig erwärmt und für 1-2 h auf eine
vorbereitete Flasche konfluenter Insektenzellen gegeben. Anschließend kann das Medium
durch frisches Kulturmedium ersetzt werden.
2.2.20.2 Virustiterbestimmung und Klonierung durch limitierende Verdünnung
Der Titer der rAcNPV wurde durch limitierende Verdünnung bestimmt (Nur bei GFP oder
anderen Reportergen-tragenden rAcNPV möglich). Dazu wurde in einer 96 Napf-(well)
Mikrotiterplatte Sf21 bzw. HighFive Zellen in einem Volumen von je 100 µl auf ca. 50 %
Konfluenz angezogen. In Napf A1 wurden 100 µl der zu untersuchenden Viruslösung
gegeben, gut durchmischt und 100 µl des Überstandes in B1 pipettiert. Aus diesem Napf
wurden nach dem gleichen Schema 100 µl in den jeweilig nächsten Napf pipettiert bis H1
erreicht wird. Von den dort anfallenden 200 µl wurden 100 µl nach Durchmischung
verworfen. Nach gleichen Schema wurden die senkrechten wells reihenweise verdünnt. Die
so pipettierte Mikrotiterplatte enthält in der Waagerechten und Senkrechten subsequente
50% Verdünnungen, wobei die aufsteigenden Diagonalen jeweils die gleichen
Verdünnungsstufen aufweisen. Nach anschließender Inkubation der Platte bei 27°C wurde
die GFP Expression 4-6 Tage später kontrolliert. Mit Hilfe der Verdünnungsstufen konnte
der Virustiter errechnet werden. Er lag bei 2-4x 106 pfu/ml. Der Virenüberstand der letzten
positiven Verdünnungsstufe wurde abgenommen und als Virenklon amplifiziert. Aufgrund
der großen Zeitspanne des Experimentes wurden 2 –3 verschiedene Virusvorverdünnungen
parallel getestet.
2.2.20.3 Infektion und Expression/Aufreinigung im Baculovirus-System
Der Primärvirusüberstand eines Virenklones wurde zur heterologen Expression eingesetzt.
Dazu wurde eine konfluente 5 ml Kultur mit HighFive-Zellen auf 100 ml Kulturmedium
verdünnt und in einer 600 ml Kulturflasche bei 27° C bis zum Anheften der Zellen inkubiert.
Die Infektion erfolgte mit 200 µl des Primärvirusüberstand eines Virenklones (d.h. ca. 8x 105
pfu). 5 Tage nach Infektion wurden die Zellen mit einem Rubberpoliceman geerntet, 5 min

26
800xg pelletiert und der Virusüberstand für weitere Expressionen aufbewahrt. Die weitere
Aufreinigung erfolgte mit dem His-Bind-Kit (2.2.24 f) mit folgenden Veränderungen: Das
Zellpellet wurde in 1 ml 1x His-Bindingpuffer, supplementiert mit ca. 20 µg/ml DNase I (Fa.
Boehringer) und 200 µl Proteinase-Inhibitor-Cocktail, der keine Metall-Chelat-
Affinitätschromatographie inhibierenden Agenzien enthielt (Fa. Sigma), resuspendiert. Die
Zellyse erfolgte durch 3x Gefrier/Auftau-Zyklen mit subsidiärem Abbau der freigewordenen
und hochviskosen gDNA durch DNase I (15 min Inkubation bei 37°C). Diese
Proteinsuspension wurde auf 10 ml 1x His-Bindepuffer aufgefüllt. Diesem Bindepuffer wurde
wahlweise 1% NP-40, 2% Triton X-100 oder 6 M Guanidin-HCl (Endkonzentrationen)
zugesetzt. Nach zusätzlichem Scheren der DNA durch mehrmaliges Passieren einer 21 gauge
Kanüle wurden die verbleibenden Zelltrümmer bei 21000xg bei 4°C für 20 min pelletiert. Im
Falle der denaturierenden Aufreinigung in Gegenwart von 6 M Gu-HCl wurde die
Zellsuspension vor der Zentrifugation für 1 h auf Eiswasser inkubiert, um die an den
Zellmembrantrümmern gebundenen Proteine abzulösen. Die weitere Aufreinigung erfolgte
wie unter 2.2.24, mit dem Unterschied, daß Detergenzien bzw. 6 M Gu-HCl allen
verwendeten Puffern zugesetzt wurden. Proteinproben, die in Gegenwart von 6 M Gu-HCl
erstellt worden sind, mußten vor einer SDS-PAGE erst mit Hilfe von Slide-A-Lyzern
Dialysemembranrähmchen (Fa. Pierce) gegen entsprechenden Puffer ohne das chaotrope Salz
dialysiert werden.
2.2.21 Heterologe Expression in E. coli /pMAL-System (Fa. NEB)
Der pMAL-C2 Vektor trägt unter der Kontrolle des starken induzierbaren bakteriellen tac
Promotors das malE Gen, welches für das Maltose-Binde-Protein aus E. coli codiert. Durch
Insertion des Zielproteingenes in den dahinterliegenden Polylinker entsteht unter Beachtung
des Leserahmens bei Expression eine c-terminale Fusion des Maltose-Binde-Proteins mit dem
Zielprotein. Dieses Fusionsprotein kann aufgrund der Affinität des Maltose-Binde-Proteins
zu Amylose durch eine Affinitätschromatographie aufgereinigt werden.
Durch PCR-gerichteter Mutagenese konnten die entsprechenden Zielproteine in den pMAL-
C2 Vektor kloniert werden. Das entstandene Konstrukt wurde durch PCR und
Restriktionsverdau, sowie durch Sequenzierung auf Richtigkeit kontrolliert. Nach
Transformation in den lon und omp Protease-defizienten E. coli-Stamm TB-1 wurden 500 ml

27
RM-Medium mit 5 ml dieser ÜK angeimpft und bis zu einer OD600 von 0,8-1,0 im
Schikanekolben bei 37°C 150 rpm inkubiert, mit IPTG (Endkonzentration 0,3 mM) induziert
und für weitere 3h bei 37°C inkubiert. Zur Zellernte wurden die Zellen bei 4°C für 5 min und
5000g abzentrifugiert und in 10 ml Bindepuffer (siehe 2.2.22) resuspendiert und bei –20°C
ÜN gelagert.
2.2.22 Affinitätschromatographie mit Amylose-Beads (pMAL-System)
Die Affinitätschromatographie mit Amylose-Beads erfolgte nach Protokoll des Herstellers.
Die Zellen wurden in Bindepuffer (20 mM TrisHCl, 500 mM NaCl, 1 mM EDTA, pH 7,4)
im Eisbad aufgetaut und mit dem Sonifier (Fa. Branson) für ca. 20 min im Eisbad mit
Pulslängen von ca. 1 Sekunde mit Ultraschall lysiert. Die lysierten Zellen wurden für 30 min
bei 9000xg 4° C zentrifugiert und der Überstand auf die zuvor mit Bindepuffer equilibrierte
Amylosebead-Säule (Fa. NEB) gegeben. Die Säule wurde subsidiär mit 10-fachem
Säulenvolumen Bindepuffer gespült. Die an der Säule gebundene Maltosebindeproteinfusion
wurde mit Elutionspuffer (Bindepuffer mit 20 mM Maltose) von der Säule abgelöst, in 1 ml
Fraktionen aufgefangen, die Proteinkonzentrationen bestimmt und die proteinhaltigen
Fraktionen durch eine anschließende SDS-PAGE analysiert. Die so aufgereinigten
Proteinfusionen wurden im Elutionspuffer bei 4°C gelagert bzw. lyophilisiert.
2.2.23 Heterologe Expression in E. coli/pET-System (Fa. Novagen)
Die heterologe Expression in E. coli/pET-System erfolgte im wesentlichen nach Protokoll
des Herstellers (Fa. Novagen).
Der pET-16b Vektor trägt unter der Kontrolle des induzierbaren Bakteriophagenpromotors
T7 eine Nukleotidsequenz, die für 10 aufeinanderfolgende Histidine codiert. Durch
Klonierung des Zielproteins unter Beachtung des Leserahmens in den distalen Polylinker
entsteht bei Expression eine c-terminale „His(10)-Tag“-Zielprotein-Fusion, die durch Ni2+-
chelatierende Sepharose durch Affinitätschromatographie (2.2.24) aufgereinigt werden kann.
Die codierende Sequenz des 36K Proteins wurde unter Berücksichtigung des Leserahmens
mit Hilfe der PCR-Mutagenese in pET 16b kloniert. Das Konstrukt wurde sequenziert und in
den für dieses System optimierten E. coli Expressionsstamm BL21 DE3 (pLysS )
transformiert. Dieser Bakterienstamm trägt auf einem Prophagen das Gen für die T7-
Polymerase, welche nach Induktion mit IPTG die Expression des His-Tag-Fusionsproteines

28
erlaubt. Zur Expression wurden 500 ml LB-Amp Medium mit 5 ml einer ÜK angeimpft bei
37° C bei 150 rpm im Schikanekolben inkubiert und bei einer OD600 von 0,8 bis 1,0 mit
IPTG (Endkonz. 1 mM) für 3 h induziert.
2.2.24 Affinitätschromatographie mit Ni 2+-chelatierender Sepharose
Die Affinitätschromatographie mit Ni2+-chelatierender Sepharose kann unter nativen und
unter denaturierenden Bedingungen in Gegenwart von chaotropen Salzen durchgeführt
werden. Sie erfolgte nach dem Protokoll des Herstellers (His-Bind Kit, Fa. Novagen).
Native Aufreinigung: Nach Zellernte wurde das Zellpellet in His-Binde Puffer (5 mM
Imidazol, 500 mM NaCl, 20 mM TrisHCl, pH 7,9) resuspendiert und bis zur Zellyse
sonifiziert. Nach Pelletierung der unlöslichen Bestandteile und Zelltrümmer für 15 min bei
21000xg wurde der lösliche Überstand auf eine zuvor mit NiS04 geladene (Ladepuffer 50
mM NiS04) und mit His-Bindepuffer equilibrierte His-Bind Sepharose (Fa. Novagen)
gegeben. Anschließendes Säulenwaschen erfolgte durch His-Bindepuffer (10 faches
Säulenvolumen ) und 5x Säulenvolumen His-Waschpuffer (His-Bindepuffer mit 60 mM
Imidazol). Nach Elution mit His-Elutionspuffer (His-Bindepuffer mit 1M Imidazol) erfolgte
die Fraktionierung und Proteinanalyse wie unter 2.2.22 beschrieben.
Zur denaturierenden Aufreinigung (2.2.25) wurde den Puffermedien (außer dem Ladepuffer)
6M Harnstoff bzw. 6M Guanidin-Hydrochlorid zugefügt. Unter denaturierenden
Bedingungen ist die Bindung von His-Tag-Proteine an die Säule nicht mehr so stark. Um
Proteinverluste zu vermeiden, erfolgten die Waschschritte mit reduzierten
Imidazolkonzentrationen (His-Bindepuffer mit 15 mM Imidazol).
2.2.25 Aufschluß von Einschlußkörpern (inclusion bodies)
Nach Induktion liegen viele hochexprimierten Proteine in E. coli als Einschlußkörper
(inclusion bodies) vor. Diese sind nicht löslich und werden nach Zellyse wie oben beschrieben
mit den Zelltrümmern sedimentiert. Um Proteine aus diesem unlöslichen Bestandteilen
herauslösen zu können, wurden chaotrope Salze wie 6 M Harnstoff oder 6 M Guanidin-
Hydrochlorid verwendet. Die His-Tag Fusionsproteine binden unter diesen denaturierenden
Bedingungen immer noch selektiv an die Säulenmatrix. Zur Aufreinigung von Zielprotein aus

29
inclusion bodies wurde das Zellpellet nach Zellyse (siehe 2.2.24) in Bindepuffer mit 6 M
Harnstoff aufgenommen, durch eine Kanüle resuspendiert und für 1h auf Eis inkubiert. Durch
anschließende Zentrifugation bei 20000xg für 45 min wurde der Überstand mit einem 0,45
µm Sterilfilter filtriert, um letzte unlösliche Bestandteile und DNA-Reste von der
niedrigviskosen und klaren Proteinlösung abzutrennen. Dieser Proteinüberstand wurde wie
oben beschrieben in Gegenwart von 6 M Harnstoff auf die His-Bind Säule gegeben und
weiter aufgereinigt.
2.2.26 Affinitätschromatographie mit Farbsepharosen (reactive-red, Fa. Merck)
Farbsepharosen wie „reactive-red“ (Fa. Merck) ähneln auf ihrer Oberfläche den
Dinukleotiden NAD/NADP. Es ist daher möglich mit „reactive-red“ NAD/NADP-bindende
Proteine in einer Affinitätschromatographie aufzureinigen. Die Affinitätschromatographie mit
Farbsepharosen wurde daher als alternative native Aufreinigungsmethode für das 36K
Protein eingesetzt. Die Farbsepharose wurde mit 50 mM TrisHCl pH 7,4 equilibriert, die
induzierten Bakterienzellen geerntet, in 50 mM TrisHCl pH 7,4 resuspendiert und lysiert. Der
lösliche Überstand wurde auf die Säule gegeben. Nach Waschen der Säule mit 10-fachen
Säulenvolumen mit 50 mM TrisHCl wurde das gebundene Protein mit Hilfe von TrisHCl
supplementiert mit 25 mM NAD bzw. NADP eluiert. Die eluierten Proteinfraktionen wurden
gefällt (2.2.30 bzw.2.2.31) und im Westernblot (2.2.34 f) detektiert.
2.2.27 Heterologe Expression in E. coli /IMPACT-System (Fa. NEB)
Bei dem IMPACT-System wird das Zielprotein als N-Terminale Fusion unter Beachtung des
Leserahmens exklusive des Stop-Codons vor ein Gen kloniert, welches für eine
Inteinsequenz codiert, gefolgt von einer Chitinbindedomäne. So entsteht nach Induktion des
pTac-Promotors eines Proteinfusion, die über eine Affinitätschromatographie mit Chitin-
Kügelchen aufgereinigt werden kann. Durch die selbstspleißende Aktivität innerhalb der
Inteinsequenz zwischen Zielprotein und Chitinbindedomäne wird die Fusion zielgerichtet
unter Verwendung von reduzierenden Agenzien wie DTT hydrolysiert und kann daher ohne
Erweiterung von der Chitinsäule eluiert werden. Die Durchführung dieser Methode erfolgte
streng nach Protokoll des Herstellers.
2.2.28 Proteinbestimmung (nach Bradford, 1979)

30
Es wurde die zu bestimmende Proteinlösung auf 500 µl A. dest. verdünnt und mit 500 µl
Bradford-Farbreagenz (50 mg Coomassie Brilliant Blue G 250 gelöst in 10 ml absolutem
Ethanol p.A., 175 ml ortho-Phosphosäure (85%) auf 1000 ml A.dest.) in einer 1 ml
Mikroküvette 15 min bei RT inkubiert. Die Absorption bei 595 nm wurde im
Spektralphotometer bestimmt. Durch eine Referenzkurve (1 bis 10 µg BSA) konnte die
Proteinkonzentration errechnet werden.
2.2.29 Proteinbestimmung mit Amidoschwarz-Tüpfel Test
Es wurde 1 µl der zu bestimmenden Proteinlösung auf einen Nitroacetatstreifen aufgespottet
und für 3 min in Amidoschwarz (0,5% (w/v) Amidoschwarz in 9:1 Methanol/Eisessig)
gefärbt. Durch anschließendes Entfärben in 9:1 Methanol/Eisessig kann mit Hilfe einer
parallel durchgeführten Standard Eichreihe die Proteinkonzentration bestimmt werden.
2.2.30 Proteinfällung mit Aceton
Zur Proteinfällung mit Aceton wurde die wäßrige Proteinlösung mit 3x Volumen –20°C
kaltem Aceton durchmischt und für 30 min bei –20° C inkubiert. Die präzipitierten Proteine
wurden bei 4° C bei 20.000xg für 20 min pelletiert und zweimal mit kaltem Aceton
gewaschen. Nach Absaugen des Acetonüberstandes und Evaporation von Acetonresten in
der Speedvac (1 min, Schalterstellung low), wurde das Präzipitat in 1x Laemmlipuffer (siehe
2.2.32), bzw. Puffer nach Wahl resuspendiert.
2.2.31 Proteinfällung mit TCA
Die Proteinlösung wurde mit Trichloressigsäure (TCA) versetzt (Endkonzentration 10%), 10
min auf Eis inkubiert und bei 4°C, 20.000xg für 10 min abzentrifugiert. Das Pellet wurde
zweimal mit eiskaltem Aceton gewaschen und schließlich wie unter 2.2.30 beschrieben
resuspendiert.
2.2.32 SDS-Polyacrylamid-Gelelektrophorese (SDS-PAGE) (nach Laemmli)
Zur analytischen Proteingelelektrophorese nach Laemmli wurden diskontinuierliche Puffer-
Systeme gewählt. Zunächst wurden die Trenngele (12-16 % Endkonz. bei einer AA/BAA
Konzentration von 37,5:1 (Fertiglösung Fa. Roth)), und 1/1000 Volumen TEMED, 1/500
Volumen Ammoniumpersulfatlsg. (10% w/v), 0,375 M TrisHCl pH 8,8, 1% SDS im Hoefer
Mighty Small Gelcaster gegossen und mit n-Butanol überschichtet. Nach Polymerisation

31
wurde das Butanol abgegossen, kurz mit Wasser gespült, das Sammelgel (5% wie oben mit
0,125 M TrisHCl, pH 6,8) eingefüllt und der Teflonkamm eingeführt. Proteinproben wurden
vor dem Lauf mit 2x Lämmlipuffer (Präzipitate mit 1x Lämmlipuffer) für 3 min aufgekocht.
Die Elektrophorese erfolgte bei 15 mA im Sammelgel und 30 mA im Trenngel.
2.2.33 Native Polyacrylamid-Gelelektrophorese (Native-PAGE)
Die Native PAGE (nicht-denaturierend ohne SDS) wurde mit fertigen 4-20% „NuPAGE“
Tris-Glycin Gelen (Fa. Novex) nach Vorschrift des Herstellers in Nativ-Laufpuffer bei
konstant 125V gefahren.
2.2.34 Proteintransfer auf Nitrozellulosemembran (Westernblot)
Es wurden drei Lagen Blotpapier (Fa. Schleicher & Schuell) in Größe des zu blottenden Gels
in Transferpuffer (25 mM Tris, 20 mM Glycin, 20% (v/v) Methanol) getränkt und auf die
Anode der Semi-Dry-Blotting Apparatur (Fa. Phase) gelegt. Darüber wurde ein gleich
großes mit Transferpuffer getränktes Stück Nitrozellulose (Fa. Schleicher & Schuell)
luftblasenfrei aufgelegt. Das darüber aufgelegte SDS-Gel bzw. native Gel wurde vorher für 5
min in Transferpuffer equilibriert und mit weiteren drei Lagen getränktem Blotting-Papier
blasenfrei bedeckt. Die Kathode der Blotkammer wurde auf das Sandwich aufgelegt und mit
ca. 1 kg beschwert. Der Transfer erfolgte bei 150 mA für 1,5 h. Die auf der Nitrozellulose
transferierten Proteine wurden durch 5 min Inkubation in Ponceau-S-Lösung (0,1% (w/v)
Ponceau-S, 5% (v/v) Essigsäure) und anschließendes Spülen mit TBS (10 mM Tris HCl pH
8,0, 150 mM NaCl) reversibel angefärbt, wobei der Größenstandard vor Entfärbung mit dem
Skalpell abgetrennt wurde.
2.2.35 Immunologischer Nachweis von Proteinen auf Nitrozellulosemembran
Die proteintragende Nitrozellulosemembran wurde für 30 min in TBST/Gelatine (10 mM
TrisHCl pH 8,0, 150 mM NaCl, 0,05% Tween 20, 0,3% Gelatine) geschwenkt, um
unspezifische Antikörperbindung zu vermeiden. Nach 30 min Inkubation mit dem
entsprechenden Antikörper (verdünnt in TSBT/Gelatine) und 3x5 minütigem Waschen der
Membran mit TBST/Gelatine wurde der sekundäre Peroxidase oder Alkalische Phosphatase
gekoppelte Antikörper (ebenfalls verdünnt in TBST/Gelatine) für 30 min dazugegeben.

32
Anschließend wurde der überschüssige Antikörper durch 3x5 min Waschen der Membran in
TBS entfernt. Die Bindung des Antikörpers konnte wie unter 2.2.14 - 2.2.17 beschrieben
detektiert werden.
2.2.36 Zellaggregations/Adhäsionstest
Im Zellaggregationstest konnte die Adhäsivität von heterolog exprimierten
Oberflächenproteinen in stabil transfizierten rekombinanten Zellinien (2.2.51) detektiert
werden. Der Assay basiert auf der Tatsache, daß Einzelzellen in Suspension unter leichtem
Schwenken in charakteristischer Weise zu Zellaggregaten aus vielen Einzelzellen aggregieren
können, was zur Abnahme der absoluten Partikelzahl in der Suspension führt. Die Abnahme
der absoluten Partikelzahl mit der Zeit kann mit Hilfe eines Partikelzählgerätes (Coulter
Counter Mod. ZM) quantifiziert werden und ergibt ein Maß für die adhäsiven Eigenschaften
der Oberflächenproteine der Zelle, ausgedrückt durch den Quotient der absoluten
Partikelanzahl zum gewählten Zeitpunkt / absoluten Partikelanzahl zum Startzeitpunkt t=0
(N(t) /N(0)) und kann visuell unter dem Mikroskop verifiziert werden.
Dazu wurden je drei 100 mm-Zellkulturschälchen stabiler Expressoren sowie eine nicht
transfizierte Kontrolle der gleichen Zellinie bis zu einer Konfluenz von 80-90% inkubiert,
zweimal mit serumfreien DMEM gewaschen und mild trypsiniert (0,0025% (w/v) Trypsin in
DMEM). Die Zellen wurden im Mikroskop beobachtet und nach ca. 10 min die leicht
angelösten Zellen mit einer 10 ml Pipette vom Zellkulturschälchen abgespült, dreimal mit 4°
C kaltem Kulturmedium gewaschen (Zentrifugation für 5 min bei 800xg 4°C) und in 1 ml
4°C kalten DMEM (supplementiert mit 10% FCS, 20 mM Glutamin) durch viermaliges
Pipettieren durch eine 18 gauge Kanüle zu Einzelzellen resuspendiert. Unter dem Mikroskop
wurde die Suspension auf das Vorhandensein von mindestens 99% Einzelzellen kontrolliert
und die absolute Partikelanzahl mit Hilfe eines Coulter Counters bestimmt. Es wurden jeweils
2 x106 Zellen/ml für den Aggregationsassay eingesetzt. Die Zellen wurden bis zum Start des
Assays auf 4° C gehalten. Der Assay erfolgte in Doppelproben auf einer Wippe (Fa.
Heidolph) bei 37° C und 5-10 rpm im 2 ml-Eppendorfcup. Nach 5 min Inkubation in 37° C
hatte die Zellsuspension die Assaytemperatur von 37° C erreicht. Es wurde die Zellzahl
bestimmt und als Referenzwert zum Zeitpunkt 0 definiert. Die weitere Probenentnahme nach
definierten Zeitabständen erfolgte mit einer abgeschnittenen Pipettenspitze mit einer so

33
vergrößerten Öffnung, um mögliche Zellaggregate durch auftretende Scherkräfte während
des Pipettierens nicht zu zerstören. Vor der Probenentnahme wurde das Reaktionsgefäß
einmal vorsichtig invertiert. Die absolute Partikelanzahl wurde mit dem Coulter Counter
bestimmt. Es wurden pro Doppel-Probe je drei unabhängige Experimente durchgeführt und
gemittelt.
2.2.37 Trypan-Blau-Ausschlußtest
Durch einen Trypan-Blau-Auschlußtest konnten lebende von toten Zellen unterschieden
werden. 0,1 ml der Färbelösung (0,4 % Trypan-Blau in PBS, pH 7,2) wurde pro ml
Zellkulturmedium auf die adhärenten Zellen gegeben und unter dem Mikroskop betrachtet.
Tote Zellen färben sich blau, während lebende Zellen nicht angefärbt werden.
2.2.38 ELISA zur Quantifikation extrazellulärer IP-Proteindomänen
Um das Expressionsniveau der IP-Proteine bzw. ihrer Konstrukte auf der Zellmembran stabil
transfizierter Expressoren zu bestimmen, wurde ein indirekter ELISA (weitestgehend nach
DOHERTY ET AL., 1990) durchgeführt. Dazu wurden ca. 2000-3000 Zellen (quantifiziert mit
dem Coulter Counter) pro Napf in einer Nunc-96-well Mikrotiterplatte gegeben und ÜN im
Brutschrank inkubiert. Am nächsten Morgen wurde eine Reihe Zellen (8 Näpfe) trypsiniert,
abgelöst und die mittlere Zellzahl/Napf erneut bestimmt. Die verbleibenden Zellen wurden
zweimal mit kaltem PBS gespült, bei RT mit 4% Paraformaldehyd/PBS für 30 min fixiert und
wiederum mit PBS gespült. Die fixierten Zellen wurden für 30 min in 1%BSA/PBS zur
Blockierung unspezifischer Antikörperbindungen bei RT inkubiert. Nach Zugabe des
primären Antikörpers (αIP1 bzw. αIP2 beide 1:100 in PBS/BSA verdünnt) für 30 min
wurden die Zellen erneut dreimal für 5 min mit PBS/BSA gespült. Der sekundäre
Peroxidase-gekoppelte Antikörper (Fa. Biorad, 1:3000) wurde nach 30 min Inkubation
fünfmal mit PBS abgespült und anschließend mit 100 µl dH2O überschichtet. Die
subsequente Farbreaktion erfolgte durch Zugabe von 50 µl 0,2% (w/v) o-phenylenediamine
(OPD, Fa. Sigma) in 0,05 M Citratpuffer pH 5,5 mit 0,02% H2O2 pro Napf. Die
enzymatische Reaktion wurde nach 15 – 45 min durch Zugabe von 50% H2SO4 gestoppt und
die optische Dichte im Multiwellreader (Fa. Molecular Devices) bei 490 nm bestimmt. Als
Kontrolle dienten nicht transfizierte CHO-Zellen. Es wurden pro Zelltyp mindestens 28

34
Näpfe je dreimal bestimmt und so die OD 490/Zellanzahl gemittelt. Der Quotient (Verhältnis)
aus mittlerer OD490/Zellzahl transfizierter Zellen und mittlerer OD490/Zahl nichtransfizierter
Zellen ergibt eine dimensionslose Einheit für das Maß der Expression der extrazellulären
Epitope der IP-Proteine bzw. IP-Konstrukte auf der Oberfläche der CHO-Zellmembran.
2.2.39 Protein-Phospholipid-Bindungstudien mit Transil-Kügelchen (Fa. Nimbus)
Transil-Kügelchen sind auf ihrer Oberfläche mit Phospholipiden beschichtet, die in der
Zusammensetzung definiert variiert werden können. Sie stellen daher ein ideales System zur
Untersuchung von Protein-Phospholipid-Wechselwirkungen dar. Hierzu wurde das zu
untersuchende Zielprotein (gelöst in 20 mM HEPES pH 7 und 50 mM NaCl) für 15 min bei
RT mit Transilen unterschiedlicher Phospholipidzusammensetzung gegeben und unter
leichtem Schwenken inkubiert. Die Transilkügelchen wurden pelletiert, das gebundene
Protein durch Zugabe von 2 % SDS abgelöst, die gebundene Proteinmenge nach Bradford
(2.2.28) bestimmt und auf ein SDS-PAGE aufgetragen werden. Die Transil-Bindungsstudien
wurden als Auftragsarbeit von der Firma Nimbus durchgeführt.
2.2.40 Protein-Protein-Bindungsstudien („overlay-assay“)
Für Protein-Protein-Bindungsstudien wurde ein ca. 1 µg Protein als „Köder“ auf eine
Nitrozellulosemembran aufgespottet und so immobilisiert. Die Membran wurde zur
Absättigung unspezifischer Proteinbindungen für 30 min in TSB/ 1% BSA inkubiert.
Anschließend wurde die Membran in der zu detektierenden Proteinlösung (ca. 1 mg/ml in 20
mM TrisHCl pH 7,4, 200 mM NaCl, 10 mM Maltose) für 30 min geschwenkt. Die Detektion
der Bindung des Zielproteins an das Köderprotein erfolgte immunologisch wie unter 2.2.35
beschrieben.
2.2.41 DNA-Transfer auf positivierter Nylonmembran (Southern-Blot)
Nach elektrophoretischer Auftrennung von DNA-Fragmenten (ca. 5 µg gDNA pro
Restriktionsverdau) wurde das 0,8 % ige Gel für 10 min in Denaturierungspuffer (1,5 M
NaCl, 0,5 N NaOH) denaturiert, kurz in A. dest. gespült und anschließend für 2 x 15 min im
Neutralisierungspuffer (1 M TrisHCl, 1,5 M NaCl, pH 7,4) unter leichtem Schütteln
neutralisiert. Danach wurde überschüssiges Gelmaterial abgetrennt und das Gel vor Beginn

35
des Transfers für 30 min in 20 x SSC inkubiert und der Blot-Sandwich wie unter Sambrock
et al. (1989) beschrieben installiert. Als Blot-Membran diente positivierte Nylonmembran
(Fa. Boehringer). Der Kapillartransfer erfolgte über Nacht. Die Hybridisierung mit der DIG-
markierten Sonde erfolgte wie unter unter 2.2.18 beschrieben. Der Farbnachweis erfolgte mit
Hilfe des CSPD-Kits (2.2.16) nach Vorschrift des Herstellers.
2.2.42 Radioaktive DNA-Kettenabbruch-Sequenzierungen (nach Sanger)
Die Sequenzierung von DNA wurde nach der Didesoxy-Kettenabbruch-Methode (SANGER
ET AL., 1977) mit Sequenase Version 2.0 (Fa. USB) durchgeführt. Als Template für die
Sequenzierung diente Plasmid-DNA, die durch Jet-Star-Präp (2.2.2.2) gewonnen wurde.
Als Sequenzgele wurden 5%, 6% oder 8% Long Ranger (chemisch modifiziertes Acrylamid-
monomer mit neuartigem Cross-Linker, Fa. AT Biochem) und 7 M Harnstoff (in 1,2-fach
TBE-Puffer) enthaltende denaturierende Gele gegossen. Die Gele waren 0,23 mm dünn und
die Probentaschen wurden mit einem Sägezahnkamm geformt. Als Laufpuffer diente 0,6 x
TBE (1xTBE: 45 mM Tris, 45 mM Borsäure, 1 mM EDTA, pH 8). Für die Elektrophorese
der Sequenzgele wurde die Sequenzgelapparatur DS 91 (Fa. Biometra) eingesetzt. Die
Elektrophorese wurde mittels einer temperierbaren Laminarstromplatte bei einer konstanten
Temperatur von 50°C und einer Spannung von 2500 V, bei max. 96 W durchgeführt. Nach
der Elektrophorese wurde das Gel in 10% Essigsäure für 20 min fixiert. Nach Auswaschen
des Harnstoffs mit Leitungswasser wurde das Gel bei 80° C für 2 hrs in einem Sterilofen
getrocknet. Anschließend wurde das Gel in einer Exponierkassette 12-48 hrs auf X-OMAT,
XAR 351 (Fa. Kodak) exponiert. Die Entwicklung des Films erfolgte mit den entsprechenden
Fotochemikalien ( GBX Developer, GBX Fixer, Fa. Kodak).
2.2.43 Auftragssequenzierungen
Auftragssequenzierungen wurden von der Firma Seqlab GmbH, Göttingen auf ABI Prism
377 Sequenzautomaten durchgeführt.
2.2.44 Computergestützte Sequenzanalysen
Die computergestützten Sequenzanalysen wurden mit dem Softwarepaket DNasis for
Windows 3.5 (Fa. Hitachi), bzw. im Internet mit Hilfe der biocomputing Programme am

36
NCBI oder EMBL durchgeführt. Datenbankenvergleich wurden mit dem BLAST-Programm
in den Datenbanken GenBank, Swissprot durchgeführt.
2.2.45 Eukaryotische Zellkulturlinien
Die Zellkulturlinie OLN-93 wurde in „Dulbecco’s modifiziertes essentielles Eagles Medium“
(DMEM) der Fa. ICN mit 10 % fötalem Rinderserum (FCS) gehalten. Das Medium wurde
zusätzlich mit 20 mM L-Glutamin, 7,5 % Bicarbonat und 1 % Gentamycin (alles Fa. Gibco)
supplementiert.
Die CHO-K-1-Zellen wurden in einer 1:1 Mischung aus DMEM /Ham’s F-12 Medium (Fa.
Gibco) mit 10 % FCS, 1 % Gentamycin, 7,5% Bicarbonat, 20 mM Glutamin gehalten.
rP0/CHO(DG44)-Zellen wurden in DMEM mit 10 % FCS, 0,5 µm MTX, 20 mM Glutamin,
7,5 % Bicarbonat, supplementiert mit 40 mg/l Prolin, 0,73 mg/l Thymidin, 7,5 mg/l Glycin
gehalten.
Die Zellkulturen wuchsen als adhärente Monolayer-Kulturen auf unbeschichteten Nuncolon
Delta-Dishes (Fa. Nunc, Dänemark) bei 37°C in einer Wasserdampf-gesättigten Atmosphäre
mit 5 % CO2 im begasbaren Zellkulturschrank (Fa. Kendro/Heraeus).
Sf21 und HighFive-Zellen wuchsen in 50 ml Zellkulturflaschen der Fa. Greiner. Sie wurden
pro 50 ml Flasche in 5 ml Insect-Xpress (BioWhittaker, Belgien) mit 2% FCS und 100 µg/ml
Gentamycin bei 27 °C ohne CO2 Begasung gehalten.
2.2.46 Subkultivieren („passagieren“) von Säuger-Zellkulturen
Bei einer Konfluenz > 90 % (d.h. fast Monolayer) wurden die Zellen passagiert. Dazu
wurden die Zellen mit 0,25% Trypsin in DMEM (Fa. Sigma) unter leichtem Schwenken für
ca. 10 min trypsiniert. Anschließend wurde 500 µl Trypsininhibitor (0,025 % mit 0,004 %
DNase I Fa. Sigma in DMEM) dazugegeben. Durch vorsichtiges Spülen mit Medium mit der
Pipettenspitze wurden die restlichen Zellen vom Untergrund abgelöst und für 5 min bei ca.
800 g, 18°C zentrifugiert. Nach zweimaligem Waschen mit DMEM wurde das Zellpellet mit
in Kulturmedium (siehe oben) resuspendiert und auf die gewünschte Zelldichte ausplattiert.
2.2.47 Subkultivieren („passagieren“) von Insektenzellen
Insektenzellen wurden bei einer Konfluenz von ca. 90% durch einfaches Abschlagen (leichtes

37
Schlagen der Zellkulturflasche an den Handballen) vom Untergrund abgelöst. Noch
anheftende Zellen wurden durch Spülen mit Medium mit der Pipette vorsichtig abgewaschen.
Je nach gewünschter Zelldichte wurden 0,5 bis 2 ml dieser Zellsuspension in eine neue
Zellkulturflasche mit 5 ml frischen InsectXpress (siehe oben) überimpft.
2.2.48 Kryokonservierung von eukaryotischen Zellinien
Eukaryotische Zellinien können ähnlich einer Bakterienkultur (siehe 2.2.1) als DMSO-
Dauerkultur gehalten werden. Dazu wurden die Zellen von ihren Kulturschalen, bzw. Kultur–
flaschen, abgelöst und mit nichtsupplementiertem Medium gewaschen (siehe 2.2.46). Die
Zellen wurden anschließend mit einem DMSO-haltigen Gefriermedium (DMEM, 20% FCS,
10% DMSO) in einem Kyro-Röhrchen resuspendiert. Die Zellen wurden über Nacht auf -70°
C gekühlt und danach in flüssigem Stickstoff gelagert.
Zum Auftauen wurden die Zellen schnell im Wasserbad bei 37° C aufgetaut und pro ml
Gefriermedium mit 10 ml frischem Kulturmedium verdünnt und ÜN bei 37° C ,bzw. 27° C
kultiviert. Am nächsten Morgen wurde das Medium durch frisches Kulturmedium ersetzt.
2.2.49 Transfektionen von Säugerzellen /Transfectam Methode
Die Transfektion von Säugerzellen erfolgte nach der Transfectam Methode (Fa. Promega),
wie bereits im Detail beschrieben (LANWERT, DIPLOMARBEIT 1996). Sie wurde über Nacht
(für stabile Transfektionen), bzw. für 5 h für transiente Transfektionen durchgeführt. Es
wurden pro 35 mm-Zellkulturschale 2 µg DNA und 4 µl Transfectam verwendet.
2.2.50 Transfektion von Insektenzellen/ Cellfectin (Gibco)
Zur Transfektion von Sf21 und HighFive-Zellen wurden 50% konfluente 35 mm-Kulturen
verwendet. Die Zellen wurde einmal mit nichtsupplementiertem InsectXpress gespült. Pro
Kultur wurden 2-6 µg DNA gelöst in 200 µl Medium mit 15 µl Cellfectin (ebenfalls in 200 µl
verdünnt), gemischt und für 15 min bei RT inkubiert. Dieses Transfektionsgemisch wurde auf
1 ml mit InsectXpress aufgefüllt, auf die Zellkulturen gegeben und für 6 h bei 27° C
inkubiert. Anschließend wurde das Transfektionsmedium entfernt und durch Kulturmedium
ersetzt.
2.2.51 Hygromycin und G-418 („Geneticin“) Selektion /Stabile Transfektion mitpIRES-neo

38
Hygromycin wie G-418 sind Antibiotika, welche für die meisten eukaryotischen Zellen, wie
Hefen, Pflanzen und Säugerzellen, aber auch für Bakterien toxisch sind. Hygromycin bzw. G-
418 wirkt auf die ribosomale Proteinbiosynthese der Zellen. Beide Antibiotika eignen sich in
Verbindung zur Selektion auf stabil transfizierte Zellkulturen. Als Expressionsvektor zur
stabilen Transfektion diente pIRES-neo bzw. pIRES-hygro. Diese bicistronischen Vektoren
tragen hinter der Multicloningsite, in der das Zielprotein ohne poly(A)-Signale kloniert
werden kann, eine IRES-Erkennungssequenz gefolgt von der codierenden Region des G-
418/Hygromycin Genes. Die virale IRES-Sequenz (intra ribosomal entry site ) erlaubt es den
Ribosomen (ähnlich der polycistronische Genstruktur der Prokaryoten) an der mRNA
festzumachen und die Translation des zweiten (Resistenz-)Proteines von einem Transkript zu
initiieren. Da Zielprotein und Resistenzgen somit auf dieselbe mRNA zurückgehen, ist die
Wahrscheinlichkeit sehr hoch, daß resistente Zellen auch das Zielprotein exprimieren.
2 Tage nach Übernacht-Transfektion von CHO-K1-Zellen (siehe 2.2.49) wurden die Zellen
passagiert (siehe 2.2.46) und auf das mindestens 200 faches Volumen in mit 1000µg G-
418/ml (bzw. 200 µg Hygromycin/ml) supplementierten Kulturmedium gebracht und auf
100-mm-Dishes verteilt. Ca. 14-20 Tage nach Transfektion entstanden Zellkolonien, die aus
einer bzw. wenigen Zellen hervorgegangen sind. Es wurden nach dem Zufallsprinzip ca. 20
Zellkolonien mit einem Klonierungszylinder (Fa. Sigma) abgegrenzt und selektiv von der
Kulturschale mit Trypsin abgelöst. Ein Teil dieser Zellsuspension wurde zur
immunologischen Kontrolle der stabilen Expression (siehe 2.2.54) erneut ausplattiert, die
andere Hälfte in 96 well-Platten zu Einzelzellen ausverdünnt. So wurde gewährleistet, daß es
sich bei den verwendeten stabilen Expressoren um reine Klone handelt. Nach weiteren 8-14
Tagen hatten sich die Zellen eines Klones soweit vermehrt, daß wiederum Duplikatkulturen
erstellt und so die Reinheit des Klones anhand der Expressionsrate immunologisch
kontrolliert werden konnten. Stabile Expressoren wurden zweimal rekloniert und auch nach
erfolgreicher Selektion und Klonierung stets weiterhin in Selektionsmedium gehalten.
2.2.52 Dynabead oligo d(T)-mRNA-Isolierungen aus Hirngewebe
Die Dynabead oligo d(T)-mRNA-Isolierung basiert auf der Hybridisation zwischen den 3’
poly(A)-Enden der eukaryotischen mRNAs und den oligod(T)-Resten der supramagnetischen
Dynabeads. Die mRNA-bindenden supramagnetischen Beads können während der

39
Wachsschritte durch einen Magneten fixiert und dadurch separiert und gereinigt werden. Das
Gewebe (0,1g) wurde in einem Glas-Homogenisator in 1 ml Lysis/Binde-Puffer (100 mM
TrisHCl, 500 mM LiCl, 10 mM EDTA, 1% SDS, 5 mM DTT, pH 8 ) für 1-2 min manuell
homogenisiert. Das Lysat wurde anschließend in ein Eppendorf-Reaktionsgefäß dekantiert
und 30 sec in einer Eppendorf Zentrifuge zentrifugiert. Danach wurde der Überstand in ein
neues Eppendorf-Reaktionsgefäß überführt und 1,5 mg oligo d(T)25-Dynabeads zugegeben.
Nach vorsichtigem Mischen und einer Inkubationszeit von 5 min wurde das Gefäß für 1 min
in die Magnethalterung gestellt. Die paramagnetischen Kügelchen mit der gebundenen
mRNA wanderten während dieser Zeit zu der dem Magneten zugewandten Gefäßwand.
Anschließend konnte der Überstand vorsichtig abpipettiert und verworfen werden. Die an der
Gefäßwand fixierten Kügelchen wurden nun mit 0,5 ml Waschpuffer (10 mM TrisHCl, 0,15
M LiCl, 1 mM EDTA, 0,1-0,3 % SDS, pH 7,5) resuspendiert und der Separationsvorgang
wurde mehrmals wiederholt. Nach dem letzten Waschschritt wurde die an den „Beads“
gebundene mRNA durch Zugabe von 10 µl Elutionspuffer (2 mM EDTA, pH 7,5) und
Erhitzen (2 min, 65° C) von den „Beads“ getrennt. Danach wurde das Eluat im
Magnetpartikelkonzentrator von den Kügelchen befreit und der mRNA-enthaltende
Überstand in ein neues Gefäß überführt. Die so gewonnene mRNA (400-800 ng/µl) wurde
sofort zur cDNA Erststrangsynthese eingesetzt.
2.2.53 cDNA Erststrangsynthese (Reverse Transkription)
Zur cDNA Erststrangsynthese wurden pro 20 µl Gesamtreaktionsansatz ca. 600 ng mRNA
eingesetzt. Als Primer für die Reverse Transkriptase wurde 1 µl des oligo dT-Primers (Fa.
Gibco) verwendet. Das Gemisch wurde für 10 min auf 70°C erhitzt, um Sekundärstrukturen
der RNA zu unterbinden und das Anlagern des Primers zu ermöglichen. Anschließend
wurden pro Ansatz 4 µl des 5x Erststrangpuffers, 2 µl 10 mM dNTP-Mix , 2 µl 0,1 M DTT
und 1 µl (200 U) Superscript RT II (alles Fa. Gibco) dazupipettiert. Die Reverse
Transkription wurde zunächst für 10 min bei RT, anschließend für weitere 50 min bei 42°C
durchgeführt. Nach anschließendem Stoppen durch Erhitzen auf 95°C für 5 min wurde 1 µl
RNase H (Fa. Gibco) dazugegeben und für 20 min bei 37°C inkubiert, um die RNA-
Komponente in den entstandenen RNA-DNA-Hybriden abzubauen. Die so erhaltene

40
Erststrang cDNA wurde bei -20°C gelagert oder sofort für die PCR verwandt.
2.2.54 Immunocytochemie/indirekte Immunfluoreszenz
Gelvatol:
Lösung A 6 g Glyzerin (Fa. Fluka), 6 ml dH2O, 2,4 g Gelvatol, 2ml TrisHCl
(pH 8,5)
Lösung B 0,5 g N-Propyl-Gallat (Fa. Sigma), 12,6 g Glyzerin, 0,176 g
NaCl, 10 ml 0,2 M TrisHCl (pH 8,5)
Die Lösungen A und B wurden im Verhältnis 1:3 gemischt und unter Alufolie bei 4° C
gelagert.
Zur indirekten Immunfluoreszenz wurden mono- bzw. polyklonale Antikörper verwendet, die
ein oder mehrere spezifische Epitope des speziellen Proteins erkennen. Die verwendeten
primären Antikörper sind bereits in früheren Studien hinreichend charakterisiert worden. Die
Zellkulturen wurden zweimal mit MEM („Minimal essential Medium“ nach Eagles, 10 mM
Hepes, 0,076 % Bicarbonat; pH 7,4) gewaschen und wahlweise mit Methanol für 20 min bei
-20° C oder mit 4 % Paraformaldehyd für 10 min bei RT fixiert. Die Methanolfixierung
permeabilisiert die Zellmembranen und bietet daher die Möglichkeit auch cytoplasmatische
Epitope zu erkennen. Die Paraformaldehydfixierung wurde für Oberflächenmarkierungen
eingesetzt. Die Zellen wurden vor der Inkubation mit einem Antikörper jeweils fünfmal mit
MEM gewaschen. Zur Absättigung unspezifischer Bindungen wurden die Zellen jeweils 45
min bei RT mit MEM/1% BSA inkubiert. Die verwendeten monoklonalen Antikörper
wurden unverdünnt, die polyklonalen ersten Antikörper wurden 1:50-1:100 verdünnt in
MEM/BSA auf die Zellen gegeben und für 30 min bis 24 h bei RT inkubiert. Als zweite
Antikörper dienten FITC oder Cy3-markierte Fluoreszenzkonjugate, die ebenfalls in
MEM/BSA 1:50 (bzw. 1:400 bei Cy3) verdünnt wurden und für 30 min bis 2 h inkubiert
wurden. Die Präparate wurden abschließend fünfmal mit MEM gewaschen und anschließend
mit wenigen Tropfen Gelvatol mit einem Deckgläschen eingedeckelt, bzw. ohne
Deckgläschen in TBS gehalten. Sie wurden unter einem Olympus Fluoreszenz Mikroskop
Typ ITM-2. mit geeigneten Fluoreszenzfiltern betrachtet und ggf. auf einem Foto
dokumentiert.

41

42
3 Ergebnisse
3.1 Isolierung der vollständigen cDNA Sequenz für das IP2-Protein
Vor Beginn dieser Arbeit lag eine vollständige
exprimierbare cDNA für das in der cytoplasmatischen
Domäne kürzeren IP1 Splicevariante vor, während von
der längeren Isoform (IP2) lediglich ein Teilfragment
existierte (STRATMANN & JESERICH, 1995). Mit Hilfe
der IP2-Teilfragment-Sequenzinformation wurde ein
spezifischer PCR-Primer generiert und für eine 3‘
RACE PCR (2.2.13.6) eingesetzt. So konnte die
fehlende codierende Region für die cytoplasmatische
Domäne inkl. 3‘ UTR gefunden werden. Durch eine
unabhängige RT-PCR (2.2.13.5) wurde die vollständige
codierende Region der IP2 Splice-Variante verifiziert
und kloniert (Abbildung 5). Die Nukleotidsequenz der
cDNA der IP2 Isoform in Abbildung 8 ist ab den in
beiden Varianten unterschiedlichen cytoplasmatischen Domänen dargestellt. Die codierende
Region der cytoplasmatischen Domäne der IP2 Variante ist im Vergleich zu IP1 um 90 bp
(inkl. Stop-Codon) auf 180 bp verlängert. Im Anschluß an das Stop-Codon erstreckt sich
eine IP2-spezifische 3‘ UTR von 133 bp Länge, welche in Position 940 in eine Sequenz
mündet, die mit der 3‘ UTR der bereits bekannten kürzeren Splice-Variante identisch ist.
Eine schematische Darstellung der cDNA Sequenzen der beiden Splice-Varianten IP1 und
IP2 ist in Abbildung 6 dargestellt. Aufgrund der neugewonnenen Sequenzdaten und der aus
der cDNA abgeleiteten Membran-Topologie konnte ein hypothetisches Modell für die
zugrundeliegende Struktur des IP-Genes vorgeschlagen werden (Abbildung 6).
1 2 St
1000 bp
Abbildung 5: Agarosegel Spur 1:RT-PCR mit IP-5-F/ IP2 C3-R; Spur2: 3‘RACE-PCR mit IP2-CIZ-F/Ankerprimer 2; St: Standard (smartladder, Fa. Eurogentec)

43
Abbildung 6: Hypothetische Genstruktur des IP-Genes, schwarze Winkel: konstitutive Splice-Vorgänge, roteWinkel: alternative Splicevorgänge
Abbildung 7: cDNA Schema der IP Splice-Varianten

44
Abbildung 8: IP2 cDNA Sequenz dargestellt ab Beginn der cytoplasmatischen Domäne; codierende Regionist grau unterlegt, IP2 spezifische 3‘ UTR ist rot unterstrichen, Rest 3‘ UTR ist identisch mit IP1 3‘UTR
3.1.1 Aminosäuresequenz der vollständigen cytoplasmatischen Domäne von IP2
625 ATG AGG TTC CTG GTG GCC CGC AGA GTC TTC AAC CTC AGT GTC172 Met Arg Phe Leu Val Ala Arg Arg Val Phe Asn Leu Ser Val
667 AGC AAA CAT GGA AAG AAA GGG AAG GGT AAA GAG GGA TCA CAG186 Ser Lys His Gly Lys Lys Gly Lys Gly Lys Glu Gly Ser Gln
709 CAG AGA CAG GGC CCA ACT CTC TCC ATC GGA GAC CCG TCC AAG200 Gln Arg Gln Gly Pro Thr Leu Ser Ile Gly Asp Pro Ser Arg
751 CTG AAG GCC GCC GCC TCA GAA AAG AAG AAG CAG GAG TCG CGC214 Leu Arg Ala Ala Ala Ser Glu Lys Lys Lys Gln Glu Ser Arg
793 AAG GAT AAG AAA TAG228 Lys Asp Lys Lys ---
Abbildung 9: Translatierte AS-Sequenz der cytoplasmatischen Domäne von IP2
Die cytoplasmatische Domäne der IP2 Splice-Variante (IP2cyto) ist im Vergleich zu IP1 um

45
29 AS länger und somit insgesamt 59 Aminosäuren lang, woraus sich ein vollständiges IP2-
Protein von 231 AS und ein Molekulargewicht des reifen Proteines von 23 kDa ergibt.
Dieses paßt gut zu früheren Untersuchungen von Jeserich & Waehneldt (1987), in denen dem
reifen deglykosiliertem IP2 Protein ein apparentes Molekulargewicht von 23 kDa zugewiesen
werden konnte. Ein Sequenzvergleich des verlängerten C-Terminus des IP2 Proteins mit der
intrazellulären Domäne des P0-Proteins verschiedener Vertebratenspezies (Abbildung 10
nächste Seite) ließ mehrere konservierte, meist basische, positiv geladene Aminosäuren in
diesem Bereich erkennen. Das gehäufte Auftreten von Lysinen und Argininen am C-
Terminus der cytoplasmatischen Domäne steht im Einklang mit der ansonsten stark positiven
Ladung dieser Proteinregion. Während die cytoplasmatischen Domänen der aufgeführten
Vertebraten Spezies zwischen 69 AS (Mensch und Ratte) bzw. 68 AS (Hai) aufweisen, ist
der IP2 C-Terminus der Forelle mit einer Länge von 59 AS um 9 bzw. 10 AS kürzer. Säuger
und Hai besitzen im Vergleich zu IP2 einen „Einschub“ von 9 AS im Anschluß an das
Transmembransegment zu Beginn der cytoplasmatischen Domäne. In diesem Bereich ist die
Aminosäureübereinstimmung zwischen IP2 und den aufgeführten Spezies daher mit 34 %
relativ gering, während der C-Terminus von IP2cyto ca. 71% Sequenzübereinstimmung mit
den P0-Homologen aufweist. Die letzten 7 AS sind in allen aufgeführten Vertebratenspezies
identisch. In IP2cyto konnten zwei putative Phosphorylierungs-Stellen für Casein-Kinase II
(Position 207-210: SIGD bzw. Position 226-229: SRKD) ausgemacht werden, wobei die
letztere ebenfalls als putative Phosphorylierungsstelle für die Protein-Kinase C (Position 226-
228: SRK) angesehen werden kann. Darüber hinaus wurde eine zweite potentielle PKC-
Phosphorylierungsstelle in Position 219-221: (SEK) ausgemacht.
3.2 Untersuchungen zur Funktion von IP1/2 alsAdhäsionsmolekül
In der vorliegenden Dissertation sollten experimentelle Hinweise auf die Funktion von IP1/2
als homophiles Adhäsionsmolekül gesammelt werden. Hierzu wurde der Ansatz der
heterologen Expression von IP in eukaryotischen Zellinien gewählt. Transfizierte Zellen
sollten sich aufgrund ihres differierenden Membranproteinrepertoires in Suspension unter
definierten Bedingungen im Adhäsionsverhalten von nicht-transfizierten Kontrollzellen

46
unterscheiden.
Human P0 178 VRYCWLRRQAA.....LQRRLSAMEKGKLHKPGKDA..SKRatte P0 178 IRYCWLRRQAA.....LQRRLSAMEKGKFHKSSKDS..SKHai P0 177 FRYI.VRRRARSETSFLQRRRSAAERGKV..SGKAGTVSKIP1/2 172 MRF............LVARRVFNLSVSKHGKKGKGKEGSQ
+ ++ - + ++ + +- -
Human P0 211 RGRQTPVLYAMLDHSRSTKAVSEKKAKGLGESRKDKKRatte P0 211 RGRQTPVLYAMLDHSRSTKAASEKKSKGLGESRKDKKHai P0 214 .G...PVLYATLDQSKSGKGASEKKSK.LSESKRDKKIP1/2 200 Q.RQGPTL.SIGDPSKLKAAASEKK.KQ..ESRKDKK
- +- - + + -++ +- - ++-++ ↑Abbildung 10: Aminosäuresequenzvergleich zwischen den cytoplasmatischen Domänen von P0verschiedener Vertebratenspezies mit IP1 bzw. IP2. Identische AS-Reste sind schwarz unterlegt, ähnlichegrau. +/- repräsentieren geladene AS. Das Ende der kürzeren IP1 Isoform ist mit einem Pfeil markiert.Auffällig ist die Häufung konservierter positiv-geladener AS am C-Terminus der verglichenen Proteine.
3.2.1 Aufbau eines Adhäsionsassays
Mit Hilfe der Zellinien OLN-93, einer Oligodendrogliazelle, (RICHTER-LANDSBERG UND
HEINRICH, 1996) , CHO-K1, einer Fibroblastenzelle und stabil exprimierenden rP0/CHO-
DG44-Zellen (freundliche Gabe von M. Filbin, New York) konnte ein Adhäsionsassay
etabliert werden, der es möglich machte, die Abnahme der absoluten Partikelzahl mit der Zeit
einhergehend mit der Zunahme von Zellaggregaten zu quantifizieren. Im Rahmen dieser
Dissertation wurden verschiedene Protokolle mit unterschiedlichen Inkubationsbedingungen,
Medien und Temperaturen erarbeitet und verglichen. Durch Trypan-Blau-Ausschlußtest
(2.2.37) wurde die Vitalität der Zellen überprüft und somit sichergestellt, daß eine Adhäsion
nicht auf die unspezifische Verklumpung toter Zellen zurückzuführen ist. Desweiteren wurde
deutlich, daß die Intensität der Zellablösung durch Trypsin entscheidenden Einfluß auf das
Adhäsionsverhalten hatte. Wurden die Zellen zu lange trypsiniert, war absolut keine
Adhäsion mehr detektierbar, was belegt, daß die gemessenen Adhäsionen auf
Proteininteraktionen zurückzuführen sind. Es zeigte sich, daß das unter 2.2.36 detailliert
beschriebene Protokoll geeignet war und reproduzierbare Ergebnisse lieferte.
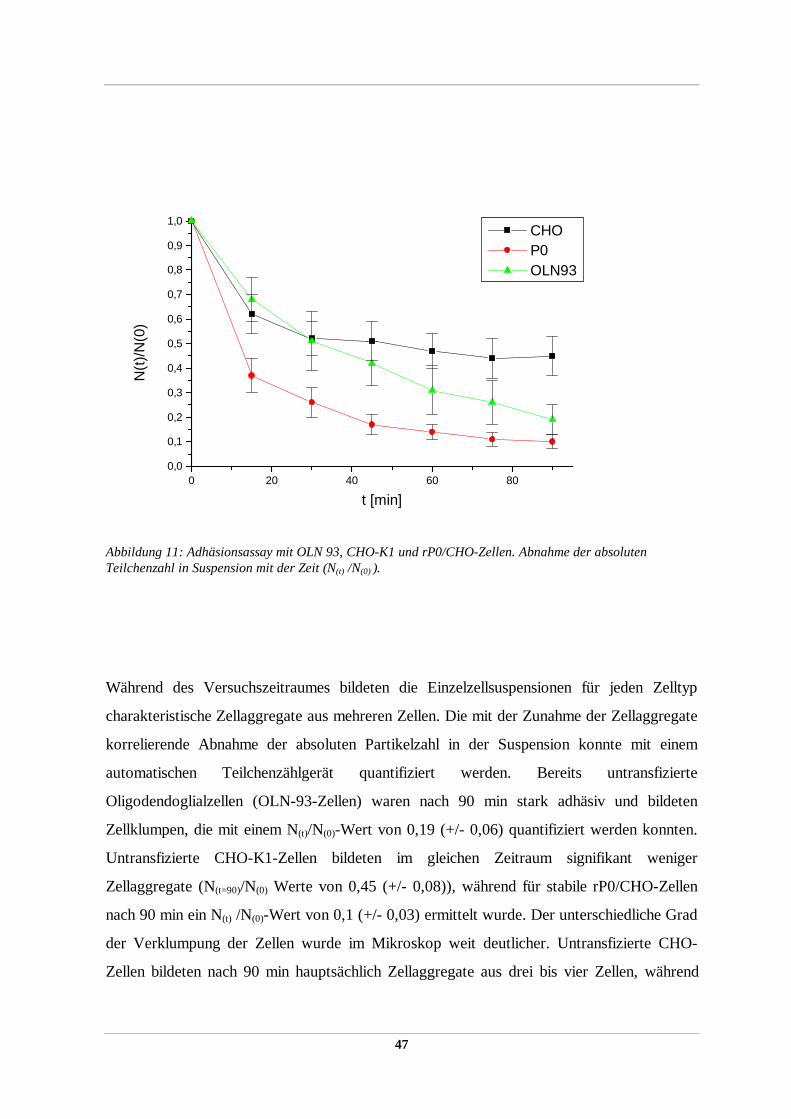
47
Abbildung 11: Adhäsionsassay mit OLN 93, CHO-K1 und rP0/CHO-Zellen. Abnahme der absolutenTeilchenzahl in Suspension mit der Zeit (N(t) /N(0) ).
Während des Versuchszeitraumes bildeten die Einzelzellsuspensionen für jeden Zelltyp
charakteristische Zellaggregate aus mehreren Zellen. Die mit der Zunahme der Zellaggregate
korrelierende Abnahme der absoluten Partikelzahl in der Suspension konnte mit einem
automatischen Teilchenzählgerät quantifiziert werden. Bereits untransfizierte
Oligodendoglialzellen (OLN-93-Zellen) waren nach 90 min stark adhäsiv und bildeten
Zellklumpen, die mit einem N(t)/N(0)-Wert von 0,19 (+/- 0,06) quantifiziert werden konnten.
Untransfizierte CHO-K1-Zellen bildeten im gleichen Zeitraum signifikant weniger
Zellaggregate (N(t=90)/N(0) Werte von 0,45 (+/- 0,08)), während für stabile rP0/CHO-Zellen
nach 90 min ein N(t) /N(0)-Wert von 0,1 (+/- 0,03) ermittelt wurde. Der unterschiedliche Grad
der Verklumpung der Zellen wurde im Mikroskop weit deutlicher. Untransfizierte CHO-
Zellen bildeten nach 90 min hauptsächlich Zellaggregate aus drei bis vier Zellen, während
0 20 40 60 800,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1,0 CHO P0 OLN93
N(t
)/N
(0)
t [min]

48
OLN-93 Zellklumpen aus bis zu 30 Zellen und rP0/CHO-DG44 Aggregate aus mehreren
hundert Zellen bildeten. Das hier ermittelte Aggregationsverhalten von rP0/CHO-
Kontrollzellen paßt gut zu früheren Studien (WONG UND FILBIN, 1996) und belegt somit die
Funktionalität und Reproduzierbarkeit des etablierten Adhäsionsassays.
Da die intrinsischen Adhäsionseigenschaften der myelinbildenden Oligodendrogliazelle OLN-
93 Zellen wie erwartet bereits zu starker Aggregation führten, wurden für die folgenden
Untersuchungen die Fibroblastenzelline CHO-K1 als Wirtszelle für die Adhäsionsassays
verwendet.
3.2.2 Heterologe transiente Expression von IP1 und IP2 in CHO-K1-Zellen
Zur heterologen Expression in CHO-K1-Zellen wurden die vollständigen codierenden
Regionen der cDNAs von IP1 bzw. IP2 in den bicistronischen Expressionsvektor pIRES-neo
kloniert (7.1 und 7.2). Mit Hilfe der Transfectam-Methode (2.2.49) konnten ca. 5 %. einer
CHO-K1-Zellkultur erfolgreich transfiziert werden. 48 h nach Transfektion zeigten
transfizierte CHO-K1-Zellen nach Methanolfixierung eine starke granuläre Immunfärbung im
Cytoplasma der Zelle. Mit Hilfe der Paraformaldehydfixierung / Oberflächenmarkierung
(2.2.54) konnten mit den polyklonalen Antikörpern αIP1 und αIP2 eine Fluoreszenzfärbung
der extrazellulären Domäne von rIP1-, bzw. rIP2-CHO-Zellen detektiert werden, die die
Integration der Forellenmyelinproteine in die Zellmembran der Säugerwirtszelle belegte
(Abbildung 12). Oberflächenfluoreszenzmarkierungen an rIP1/CHO-Zellen mit dem
monoklonalen Antikörper 7C4, spezifisch für die intrazelluläre Domäne von IP1
(STROTMANN, DIPLOMARBEIT, 1988), blieben negativ und stützten obiges Resultat.
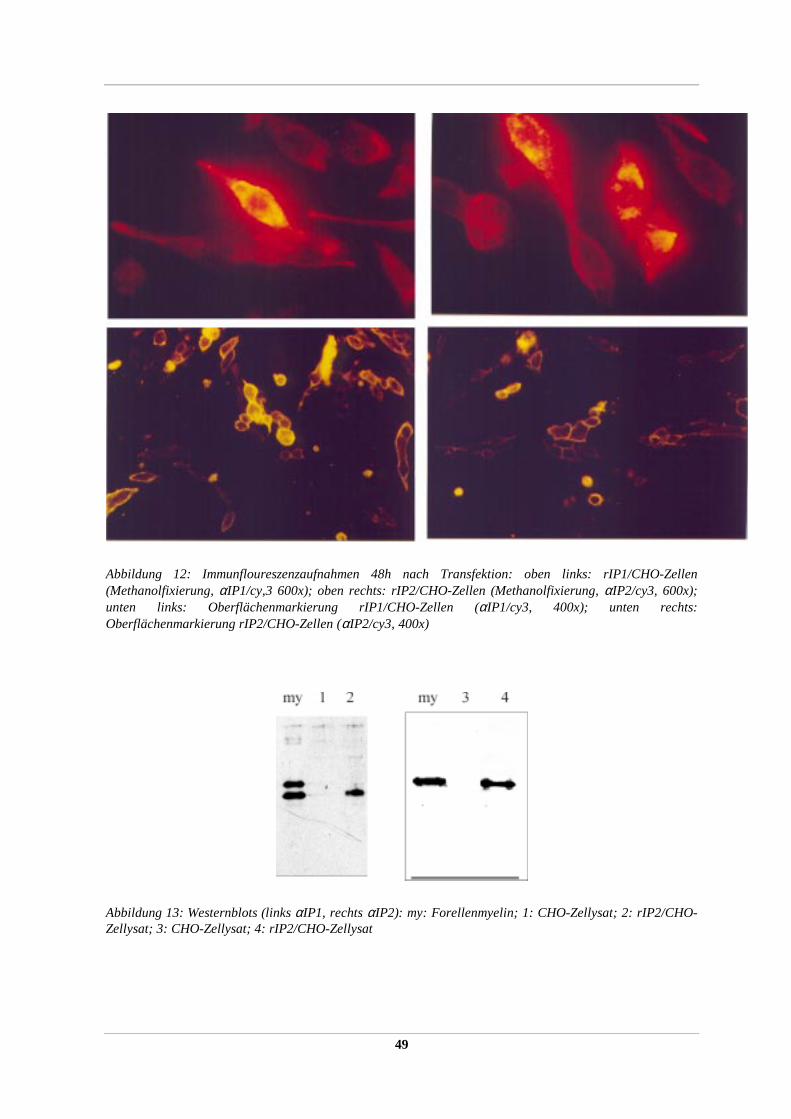
49
Abbildung 12: Immunfloureszenzaufnahmen 48h nach Transfektion: oben links: rIP1/CHO-Zellen(Methanolfixierung, αIP1/cy,3 600x); oben rechts: rIP2/CHO-Zellen (Methanolfixierung, αIP2/cy3, 600x);unten links: Oberflächenmarkierung rIP1/CHO-Zellen (αIP1/cy3, 400x); unten rechts:Oberflächenmarkierung rIP2/CHO-Zellen (αIP2/cy3, 400x)
Abbildung 13: Westernblots (links αIP1, rechts αIP2): my: Forellenmyelin; 1: CHO-Zellysat; 2: rIP2/CHO-Zellysat; 3: CHO-Zellysat; 4: rIP2/CHO-Zellysat

50
Abbildung 13 zeigt die korrespondierenden Westernblots. Aus dem elektrophoretischen
Laufverhalten von rIP1 und rIP2 wird deutlich, daß beide Isoformen auch in CHO-K1 in der
glykosilierten Form ähnlich der nichtrekombinanten Variante aus Forellenmyelin vorliegen
muß.
3.2.3 Klonierung von stabil-transfizierten rIP1/CHO-K1-Zellen
Durch die Zugabe des Antibiotikums G-418 nach Transfektion, Selektion und zweimaligem
Reklonieren konnte nach ca. 12-16 Wochen ein stabil-exprimierender rIP1/CHO-Zellklon
separiert werden (2.2.51). Nahezu 100% der Zellen konnten durch Immunfluoreszenz als
Expressoren eingestuft und die Expression außerdem durch Westernblot-Analyse verifiziert
werden. Die Klonierung stabiler rIP2/CHO-Expressoren ist trotz mehrmaliger Versuche nicht
gelungen.
3.2.4 Adhäsion von rIP1/CHO-Zellen unter definierten Bedingungen
Abbildung 14 zeigt das charakteristische Adhäsionsverhalten des rIP1/CHO-Klones. Im
Partikelzählgerät konnte nach 90 min ein leichter Unterschied in der Zellaggregation
zwischen rIP1/CHO-, und CHO-Zellen festgestellt werden. rIP1/CHO-Zellen fielen im Laufe
des Experimentes auf einen N(t=90) /N(0) Wert von 0,36 (+/-0,09) zurück, während nicht
transfizierte CHO-Zellen einen N(t=90) /N(0) Wert von 0,45 (+-0,08) aufwiesen. Deutlicher
wurden diese Unterschiede in der visuellen mikroskopischen Analyse: CHO-Zellen fanden
sich über den Versuchszeitraum nur zu Duplett-, oder Triplett-Aggregaten zusammen,
während in der rIP1/CHO-Population überwiegend Aggregate aus 6 bis 10 Zellen beobachtet
werden konnten (Abbildung 15).
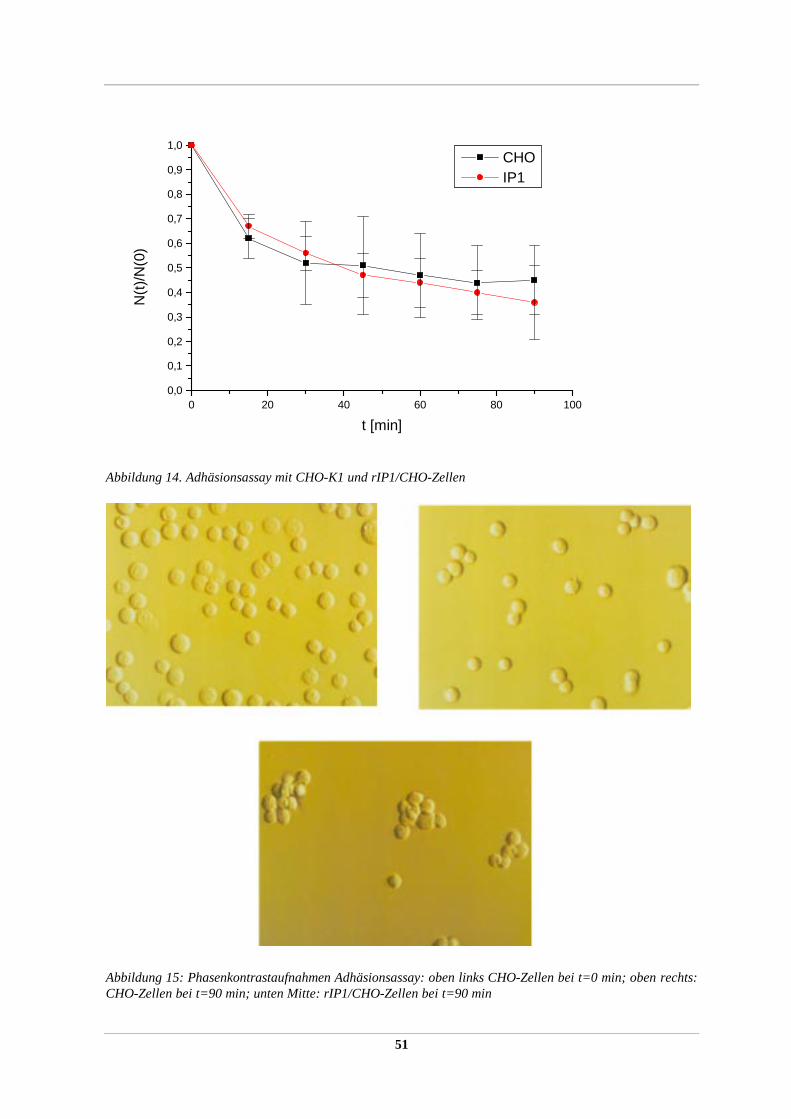
51
Abbildung 14. Adhäsionsassay mit CHO-K1 und rIP1/CHO-Zellen
Abbildung 15: Phasenkontrastaufnahmen Adhäsionsassay: oben links CHO-Zellen bei t=0 min; oben rechts:CHO-Zellen bei t=90 min; unten Mitte: rIP1/CHO-Zellen bei t=90 min
0 20 40 60 80 1000,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1,0 CHO IP1
N(t
)/N
(0)
t [min]

52
3.2.5 Konstruktion einer IP-P0-Chimäre
Für viele Adhäsionsproteine, wie z.B. P0 und den Cadherinen, gibt es gibt es Hinweise, daß
die intakte vollständige cytoplasmatische Domäne eine für die Adhäsionfunktion des
Proteines essentielle Aufgabe als Membran-, bzw. Cytoskelettanker ausübt (NAGAFUCHI UND
TAKEICHI, 1988; NAGAFUCHI ET AL., 1991; HIRANO ET AL., 1992). So waren in der
cytoplasmatischen Domäne trunkierte P0-Expressoren trotz der Expression der Adhäsion-
vermittelnden extrazellulären Domäne im Aggregationsverhalten nicht von untransfizierten
Kontrollzellen zu unterscheiden (WONG & FILBIN, 1994 UND 1996). Bei dem IP1-Protein der
Forelle handelt es sich um eine natürlich vorkommende, im Vergleich zu IP2 in der
cytoplasmatischen Domäne kürzeren Splice-Variante. Um also Hinweise auf einen möglichen
Einfluß der cytoplasmatischen Domäne von IP1 auf die Zelladhäsion zu erhalten, wurde ein
chimäres Fusionsprotein konstruiert (7.3). Es bestand aus der extrazellulären Domäne sowie
der Transmembrandomäne des IP-Proteins. Die gesamte intrazelluläre Domäne stammt von
dem P0-Protein der Ratte (Abbildung 16).
Abbildung 16: Schematische Darstellung der IP-P0-Chimäre
3.2.6 Stabile Expression und Adhäsionsverhalten von rIP-P0/CHO-Zellen
Nach Transfektion und G418-Selektion konnte wie oben beschrieben ein stabiler rIP-
P0/CHO-Klon selektiert werden. Die Oberflächenexpression der extrazellulären IP-Domäne

53
konnte durch Immunfluoreszenz mit dem Antikörper αIP1 nachgewiesen werden (Abbildung
18). Im korrespondierenden Westernblot (Abbildung 17) wurde eine immunopositive Bande
detektiert, die im elektrophoretischen Laufverhalten zwischen IP1 und IP2 liegt.
Abbildung 17: Westernblot. Spur My: Forellenmyelin; CHO: untransfizierte CHO-Zellen; CHO/IP-P0: stabiltransfizierte rIP-P0/CHO-Zellen; IP0: Proteoloyse-Produkt von IP1/2; Nachweis mit monoklonalem 4C5Antikörper (spezifisch gegen IPex)
Abbildung 18: Immunfloureszenz (Oberflächenmarkierung) 48h nach Transfektion an rIP-P0/CHO-Zellen(αIP1/cy3, 400x)
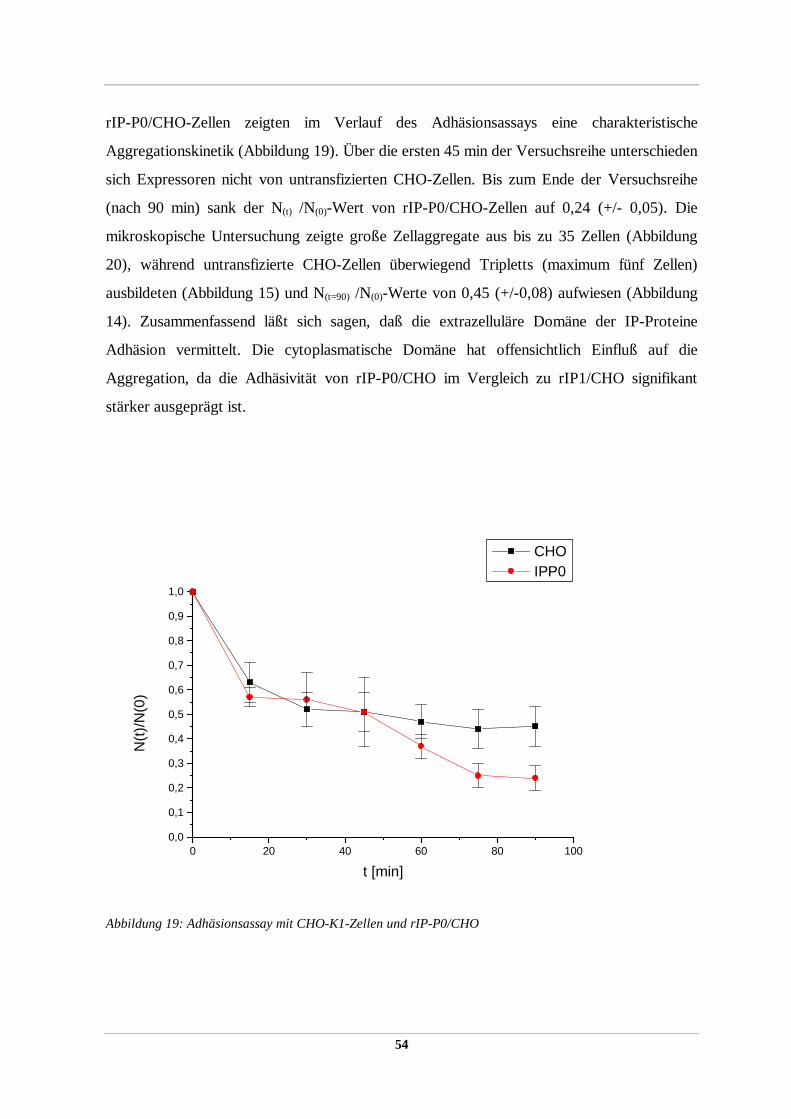
54
rIP-P0/CHO-Zellen zeigten im Verlauf des Adhäsionsassays eine charakteristische
Aggregationskinetik (Abbildung 19). Über die ersten 45 min der Versuchsreihe unterschieden
sich Expressoren nicht von untransfizierten CHO-Zellen. Bis zum Ende der Versuchsreihe
(nach 90 min) sank der N(t) /N(0)-Wert von rIP-P0/CHO-Zellen auf 0,24 (+/- 0,05). Die
mikroskopische Untersuchung zeigte große Zellaggregate aus bis zu 35 Zellen (Abbildung
20), während untransfizierte CHO-Zellen überwiegend Tripletts (maximum fünf Zellen)
ausbildeten (Abbildung 15) und N(t=90) /N(0)-Werte von 0,45 (+/-0,08) aufwiesen (Abbildung
14). Zusammenfassend läßt sich sagen, daß die extrazelluläre Domäne der IP-Proteine
Adhäsion vermittelt. Die cytoplasmatische Domäne hat offensichtlich Einfluß auf die
Aggregation, da die Adhäsivität von rIP-P0/CHO im Vergleich zu rIP1/CHO signifikant
stärker ausgeprägt ist.
Abbildung 19: Adhäsionsassay mit CHO-K1-Zellen und rIP-P0/CHO
0 20 40 60 80 1000,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1,0
CHO IPP0
N(t
)/N
(0)
t [min]

55
Abbildung 20: Phasenkontrastaufnahme Adhäsionsassay rIP-P0/CHO-Zellen bei t=90 min
3.2.7 Vergleichende Quantifizierung der Oberflächenexpression
Um auszuschließen, daß das unterschiedliche Aggregationsverhalten von rIP1/CHO und rIP-
P0/CHO auf ein unterschiedliches Oberflächenexpressionsniveau der extrazellulären Domäne
von IP (IPex) zurückzuführen ist, wurde ein Zell-ELISA an fixierten, nicht-permeabilisierten
Expressoren mit den Antikörpern αIP1 und αIP2 durchgeführt. Als Kontrolle dienten
untransfizierte Zellen (2.2.38). Die relative Oberflächenexpression ist in Abbildung 21
dargestellt. Es waren geringe, nicht signifikante Unterschiede zwischen rIP1/CHO und rIP-
P0/CHO auszumachen. Das rel. Oberflächenexpressionsniveau von rIP-P0/CHO wurde mit
5,12 (+/- 1,6), von rIP1/CHO mit 4,08 (+/- 1,59) quantifiziert. rIP-P0/CHO-Zellen
exprimierten demnach das 1,25 fache (d. h. 25% mehr) IPex auf ihrer Zelloberfläche im
Vergleich zu rIP1/CHO-Zellen. Bei Verwendung des αIP1-Antikörpers lagen die Werte der
relativen Expressionseinheiten insgesamt höher als beim vergleichbaren αIP2-Antikörper.
Wurde dieser Antikörper verwendet, konnte für rIP-P0/CHO-Zellen (2,27 (+/- 0,85) 1,41-
fach mehr Antigen auf der Zelloberfläche detektiert werden (rIP1/CHO: 1,6 (+/- 1,43)). Das
geringfügig höhere Expressionsniveau von rIP-P0 ist jedoch nicht signifikant und kann daher
die unterschiedlichen Aggregationseigenschaften der beiden Zellpopulationen von rIP1/CHO
und rIP-P0/CHO nicht erkären.
56
Abbildung 21: rel. Oberflächenexpressionsniveau von rIP-P0/CHO und rIP1/CHO-Zellen; links detektiertmit αIP1; rechts mit αIP2
3.2.8 Inhibierung der Zell/Zell-Adhäsion von rIP-P0/CHO durch Antikörper
Um zu überprüfen, ob die Aggregation von rIP-P0/CHO auf die Oberflächenexpression von
IPex zurückzuführen ist, wurde vor dem Start des Adhäsionsassays der für das
Oberflächenepitop von IP spezifische Antikörper αIP1 (3.3) der Zellsuspension hinzugefügt.
Es zeigte sich, daß die adhäsiven Eigenschaften und die damit einhergehende Zellaggregation
vollständig und selektiv inhibiert werden konnte (Abbildung 22). In Gegenwart von αIP1-
Antikörpern bildeten rIP-P0/CHO-Zellen nach 90 min Zellaggregate bestehend aus bis zu
fünf Zellen mit korrespondierenden N(t) /N(0) Werten von 0,57 (+/- 0,1) im Gegensatz zu 0,24
(bis zu 35 Zellen) ohne Antikörper (3.2.4). Die Zugabe des Antikörpers hatte keinen Einfluß
auf das Aggregationsverhalten untransfizierter CHO-Kontrollzellen (Abbildung 23). Durch
mikroskopische Analyse der Zellaggregate war kein Unterschied zwischen beiden
Kontrollversuchen erkennbar. Diese Ergebnisse geben weitere Hinweise darauf, daß die
Zell/Zell-Adhäsion auf die Expression der IPex-Domäne auf der Zelloberfläche transfizierter
Zellen zurückzuführen ist, da sie durch einen spezifischen Antikörper vollständig inhibiert
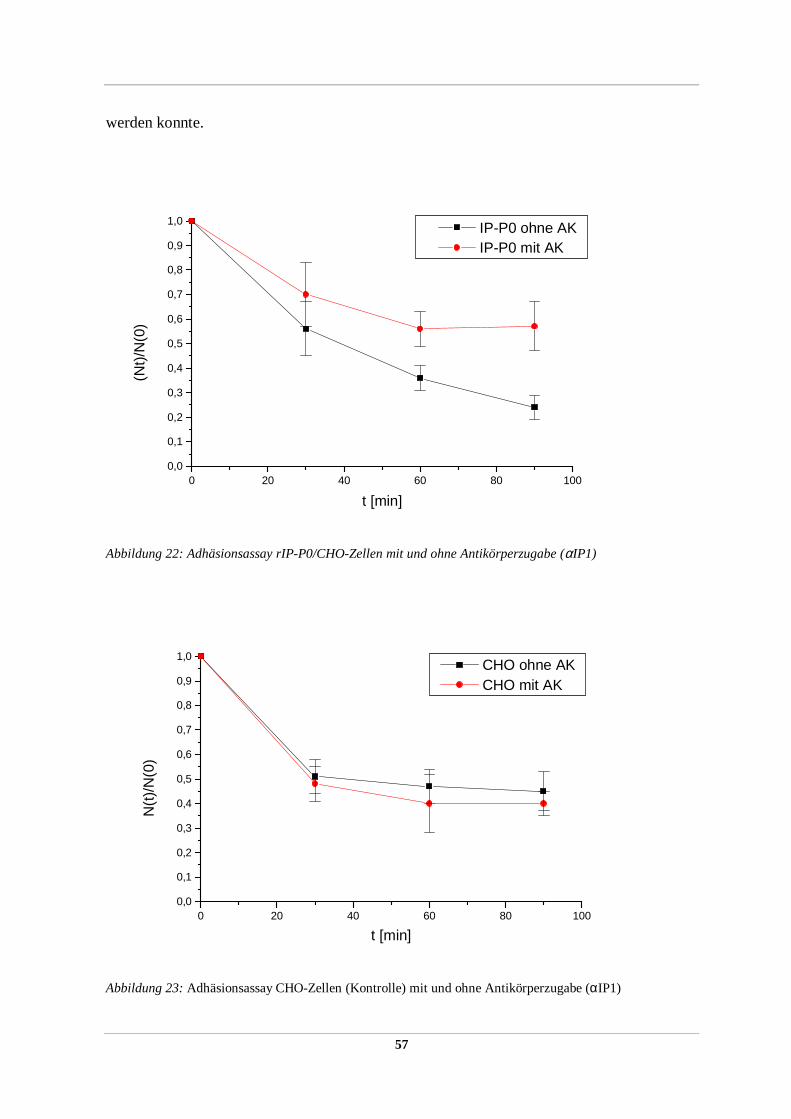
57
werden konnte.
Abbildung 22: Adhäsionsassay rIP-P0/CHO-Zellen mit und ohne Antikörperzugabe (αIP1)
Abbildung 23: Adhäsionsassay CHO-Zellen (Kontrolle) mit und ohne Antikörperzugabe (αIP1)
0 20 40 60 80 1000,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1,0 IP-P0 ohne AK IP-P0 mit AK
(Nt)
/N(0
)
t [min]
0 20 40 60 80 1000,0
0,1
0,2
0,3
0,4
0,5
0,6
0,7
0,8
0,9
1,0 CHO ohne AK CHO mit AK
N(t
)/N
(0)
t [min]

58
3.2.9 Heterologe Expression und Adhäsion von IP1 in Insektenzellen
Wie unter 7.4 beschrieben, wurde die vollständige codierende Region der IP1cDNA in
pFastbac kloniert und so rIP1AcNPV isoliert, die zur Infektion von Sf21-Zellen eingesetzt
wurden. Ca. 48 h nach Infektion zeigten ca. 80% der Zellen eine deutliche Immunfärbung.
Diese Zellen wurden in einem Adhäsionsassay eingesetzt. Als Kontrolle der
Aggregationskinetik dienten mit den Reportergen Grünfluoreszierendes Protein (GFP)
infizierte Zellen (rekombinante GFP-Viren waren freundliche Gabe von R. Wagner,
Osnabrück) sowie nicht transfizierte Sf21-Zellen. Es zeigte sich, daß IP1-Expressoren mit
N(t) /N(0) Werten von 0,68 (+/- 0,28) im Vergleich zu GFP infizierten und nicht transfizierten
Sf21-Zellen (N(t) /N(0) Wert von 0,85 bzw. 0,94) geringfügig adhäsiver waren. Der Nachteil
des hier beschriebenen Baculovirus-Systems zur Detektion von Adhäsionen liegt allerdings
eindeutig darin, daß infizierte Zellen nach einer gewissen Expressionsdauer lysierten. Es muß
daher an dieser Stelle erwähnt werden, daß der Zeitpunkt der Zellernte sowie die zur
Infektion eingesetzte Virusmenge offensichtlichen Einfluß auf das Aggregationsverhalten der
Wirtszelle hatte. Die Standardisierung dieser Parameter erwies sich als äußerst schwierig und
wenig konsistent (siehe Standardfehler (+/- SE) in Abbildung 24). Hilfreich wäre
diesbezüglich die Konstruktion von IP1/2-GFP-Koexpressoren, die eine einfache visuelle
Kontrolle über das Infektions-, bzw. Expressionsniveau der Zellkultur erlauben würde (siehe
3.8).
N(t=90) /N(0) -Wert +/- SE
Sf21 nicht inf. 0,94 +/- 0,09
IP1/Sf21 0,68 +/- 0,28
GFP/Sf21 0,85 +/- 0,29
Abbildung 24: N(t) /N(0) -Werte (nach 90 min) im Adhäsionsassay mit Baculovirus-infizierten Sf21-Zellen3 Tage nach Infektion

59
3.3 Heterologe Expression der cytoplasmatischen Domänen vonIP1 und IP2 in E. coli
Im Rahmen dieser Dissertation wurden dazu drei verschiedene E. coli Expressionssysteme
vergleichend eingesetzt. Es zeigte sich, daß weder das pET- noch das IMPACT-System
(2.2.27 und 2.2.23) eine Expression und Aufreinigung von kurzkettigen Peptiden, wie sie
IP1cyto mit ca. 3,5 kDa und IP2cyto mit ca. 6,5 kDa darstellen, geeignet waren und wurden
daher verworfen (Daten nicht gezeigt). Einzig das pMAL-System erlaubte eine Aufreinigung
von rIP1/2cyto in ausreichenden Ausbeuten.
Zur pMAL-Expression wurden die korrespondierenden cDNAs der vollständigen
cytoplasmatischen Domänen von IP1 und IP2, wie unter 7.8 beschrieben, in den E. coli
Expressionsvektor pMAL-C2 kloniert. Auf diese Weise entstanden Fusionen der Zielpeptide
mit dem Maltose-Binde-Protein. Aufgrund der hohen Affinität des Maltose-Binde-Proteins
zu Amylose-Sepharose, konnten die nativen Fusionsproteine präparativ aufgereinigt werden
(2.2.21 und 2.2.22).
Abbildung 26 zeigt eine typische Expression und Aufreinigung von MBP/IP1cyto und
MBP/IP2cyto im SDS-PAGE. Nach Induktion war in beiden Fällen eine prominente Bande in
der erwarteten Höhe von 46 kDa (MBP/IP1cyto) bzw. 49 kDa (MBP/IP2cyto) sichtbar. In
den eluierten Proteinfraktionen befanden sich in beiden Fällen außer dem Zielprotein eine
weitere ca. 42 kDa große Proteinbande. Die korrespondierenden Westernblots (Abbildung
25) zeigen ein stark immunopositives Signal jeweils in Höhe der oberen Bande, die dem
erwarteten molekularen Gewicht der Zielproteine entsprachen. Hierbei zeigte sich, daß der
monoklonale 7C4-Antikörper im Westernblot spezifisch IP1cyto erkennt, während die
längere IP2-Variante nicht markiert wurde. Der polyklonale αIP2-Antikörper hingegen
erkannte im Westernblot ausschließlich IP2cyto, während IP1cyto nicht detektiert wurde.
Interessanterweise konnten weder IP1cyto noch IP2cyto mit dem polyklonalen αIP1-
Antikörper markiert werden. Hierdurch konnte gezeigt werden, das der polyklonale αIP1-
Antikörper ausschließlich extrazelluläre Epitope erkennt (3.2.2 und 3.2.8).

60
Bei dem co-eluierten 42kDa-Protein handelte es sich offensichtlich um ein Abbauprodukt
bzw. eine nichtfusionierte Form des Maltose-Binde-Proteins. Eine typische Aufreinigung von
MBP/IP1cyto ergab Ausbeuten von ca. 1,5 mg Zielprotein/ 1l Kultur, eine Aufreinigung von
MBP/IP2cyto ergab ca. 2,5 mg Zielprotein/ 1l Kultur.
3.3.1 Bindestudien von MBP/IP2cyto an phospholipidbeschichteten Trägermatrices(Transil)
Da sich beide MBP/IPcyto-Fusionen als resistent gegen einen Faktor Xa-Verdau erwiesen,
der spezifisch die IP1/2cyto-Peptide von ihrem „Ankerprotein“ MBP trennen sollte, wurde
MBP/IP2cyto-Fusion für Protein/Phospholipid- Bindungsstudien mit der Transil-Methode
(2.2.39) eingesetzt. Als Kontrolle diente das in der eluierten Proteinfraktion enthaltene
nichtfusionierte MBP. Eine Analyse der gebundenen Proteine ist in Abbildung 27 zu sehen.
Spur 6 zeigt die eingesetzte Probe vor Transilbindung in einem annähernd equimolaren
Verhältnis beider Proteinbanden. Es zeigte sich, daß im Falle von Transil PS (Transil
Oberfläche mit 20 % Phophatidylserin), Transil PC (Transil Oberfläche mit 20 %
Phophatidylcholin) und Transil Duo (Oberfläche mit positiven und negativen Ladungen) eine
Verschiebung dieses Verhältnisses auf ca. 80:20 zugunsten des MBP/IP2-Fusionsproteines
verschoben wurde. Durch die Zugabe von 150 mM Maltose konnte dieses Verhältnis nicht
Abbildung 25: Westernblot vonIP1/2cyto MBP-Fusionen nachElution, 1: IP2 cyto mit 7C4, 2:IP1 cyto mit 7C4; 3: IP2cyto mitαIP2; 4: IP1cyto mit αIP2
Abbildung 26: SDS-PAGE (CBB) 1:Zellysat 3h nach InduktionMBP/IP2cyto; 2: Eluat MBP/IP2cyto; 3: Zellysat 3h nachInduktion MBP/IP1cyto; 4: EluatMBP/IP1cyto/

61
variiert werden (Daten nicht gezeigt). Der Einfluß des MBP-Fusionsanteiles ist unter diesen
Bedingungen nicht genau bestimmbar, so daß quantitative Messungen zur Membranaffinität
des IP2cyto mit der MBP/IP2cyto-Fusion nicht möglich waren.
Abbildung 27: SDS-PAGE nach Elution von Transilkügelchen; 1: Transil neutral, 2: Transil positiv geladen,3: Transil neg. und pos. geladen, 4: Transil mit Phosphatidylserin; 5: Transil mit Phophatidylcholin; 6:Probe ohne Transil
3.3.2 Untersuchung zu Protein-Protein-Wechselwirkungen von IP1/2cyto
Die eluierten Proteinfraktionen MBP/IP1cyto und MBP/IP2cyto wurden auf ein natives-
PAGE (2.2.33) in Abwesenheit von denaturierenden Detergenzien wie SDS aufgetragen. Im
apparenten Laufverhalten der nativen MBP/IP1cyto und MBP/IP2cyto ist im Vergleich zur
denaturierenden SDS-PAGE kein Unterschied erkennbar (Abbildung 28). Dieses ist als
Hinweis zu deuten, daß die cytoplasmatischen Domänen von IP1/2 nicht zur
Oligomerisierung neigen und daher als Monomer vorliegen.
Abbildung 28: Natives NuPAGE (4-20%, Fa. Novex). 1: MBP/IP1cyto; 2: MBP/IP2cyto
Um Hinweise auf eine denkbare Interaktion von IP1cyto und IP2cyto zu erhalten, wurde eine
Protein-Protein-Bindungsstudie mit dem Overlay-Assay (2.2.40) durchgeführt. Hierzu wurde

62
sowohl MBP/IP1cyto auf einer Nitrozellulosemembran als „Köder“ immobilisiert und in einer
Proteinlösung mit MBP/IP2cyto inkubiert, als auch vice versa. Mit Hilfe der für die jeweilige
Isoform spezifischen Antikörperkombination (7C4/anti-Maus-IgG -Peroxidase für Ip1cyto
und αIP2/anti-Hase-IgG-Peroxidase für IP2cyto) konnte gezeigt werden, daß
MBP/IP1/2cyto offensichtlich nicht die Tendenz haben, spezifische intermolekulare Protein-
Protein-Interaktionen einzugehen (Abbildung 29). Es konnte in keinem Fall am Köderprotein
gebundenes Zielprotein detektiert werden.
Abbildung 29: Overlayblot: 1: immobil. IP1cyto mit IP2cyto inkubiert (Nachweis mit α IP2); 2: immobil.IP2cyto mit IP1cyto inkubiert (Nachweis mit 7C4); 3: Positiv-Kontrolle immobil. IP1cyto (Nachweis mit7C4); 4: Positiv-Kontrolle immobil. IP2cyto (Nachweis αIP2)

63
3.4 Isolierung der cDNA Sequenz für das 36K- Protein
Das zweite Hauptmyelinprotein der Forelle ist das cytoplasmatische und membranassoziierte
36K. Wie bereits in der Einleitung erwähnt, ist es denkbar, daß 36K eine Rolle in der
Apposition der cytoplasmatischen „major dense line“ ausübt, ähnlich wie oben für IP1/2cyto
vermutet. Um erste Hinweise auf die Funktion dieses zweiten Hauptmyelinproteines zu
erhalten, wurde im Rahmen dieser Dissertation wiederum die Methode der heterologen
Expression gewählt.
Vor Beginn dieser Arbeit existierte keine exprimierbare
cDNA des 36K-Proteins. Es lag lediglich ein
Teilfragment der 36K cDNA Sequenz sowie eine gDNA
Teilsequenz für das 36K-Protein vor (STRELAU,
DISSERTATION 1997), so daß zunächst eine vollständige
cDNA isoliert werden mußte. Mit Hilfe der bereits
bekannten Teilsequenzen war es möglich, per PCR eine
DIG-markierte Sonde herzustellen, mit der eine Forellen
cDNA λ-Phagenbank durchsucht werden konnte. Wie
unter 2.2.18 f beschrieben, konnte ein positiver
Phagenklon isoliert, durch dreimaliges Rescreening
gereinigt und amplifiziert werden. Nach in vivo Excision
lag das cDNA-Fragment als Insert in pBluescript vor
und konnte sequenziert werden. In einer unabhängigen RT-PCR an Forellen konnte die
Existenz der isolierten cDNA-Sequenz verifiziert werden. Abbildung 30 zeigt das PCR-
Amplifikat in der erwarteten Höhe von 950 bp. Das Amplifikat wurde kloniert und
sequenziert und stimmte mit der in Abbildung 31 gezeigten durch Genbank-Screening
gefundenen cDNA Sequenz zu 100% überein.
950 bp
Abbildung 30: Agarosegelelek-trophorese, Spur 1: RT-PCR mit36K-CL-F4/36K2R an ForellenHirn; Spur 2: Standard (smartladder, Fa. Eurogentec); Spur 3:H2O-PCR (neg. Kontrolle)

64
Abbildung 31: Sequenz der 36K cDNA nach λ-Phagenbank Screening, ATG und TGA sind durch Fettdruckhervorgehoben; die codierende Region in Großbuchstaben ist grau unterlegt, die 5‘ UTR und die 3‘ UTR istdurch Kleinbuchstaben dargestellt; poly(a) Signale fett

65
Die vollständige 36K cDNA ist 2120 bp lang. Der größte offene Leserahmen ist 963 bp lang,
befindet sich zwischen Position 10 (Start ATG) und Stop-Codon (TGA) in Position 973 und
codiert für ein 320 Aminosäuren großes Protein. Die untranslatierte 5‘Region ist mit 10 bp
auffällig kurz, während die untranslatierte 3‘ Region 1167 bp lang ist. Sie trägt in Position
1838, 2034 und überlappend in Position 2087 und 2089 am 3‘ Ende 3 poly (A) Signale (nach
PROUDFOOT UND BROWNLEE, 1976).
3.4.1 Aminosäuresequenz des 36K-Proteins
Die aus den 320 Triplett des größten offenen Leserahmens abgeleitete Aminosäuresequenz
ist in Abbildung 32 dargestellt.
1 ATG TCT CTT TAC CGC AAC TCT GCG TGG TTT CTG AAA GGA ATG1 Met Ser Leu Tyr Arg Asn Ser Ala Trp Phe Leu Lys Gly Met
42 ACT GAA TTC ACC AGG AGT GCG TTC CTC TCT GCC GCC AAA CAC14 Thr Glu Phe Thr Arg Ser Ala Phe Leu Ser Ala Ala Lys His
84 TTT GTG GAA AAG GAC CTG GAG GTG TCC ATG GCA GGC AGG GTC28 Phe Val Glu Lys Asp Leu Glu Val Ser Met Ala Gly Arg Val
126 TTC ATG ATC ACC GGA GCC AAC AGT GGC ATA GGG AGA GCT ACA42 Phe Met Ile Thr Gly Ala Asn Ser Gly Ile Gly Arg Ala Thr
168 GCC ATG GCC ATC GCC AAG AGA GGT GGT ACA GTC CAC ATG GTG56 Ala Met Ala Ile Ala Lys Arg Gly Gly Thr Val His Met Val
210 TGC AGG AAC AAG GAC AAA GCA GAA GAG GCC AGG GCT GAC ATT70 Cys Arg Asn Lys Asp Lys Ala Glu Glu Ala Arg Ala Asp Ile
252 GTC AAG GAG TCA GGA AAT AAA GAA ATA TAT GTC CAC ATT CTG84 Val Lys Glu Ser Gly Asn Lys Glu Ile Tyr Val His Ile Leu
294 GAC CTG TCT GAG ACA CGG AAG GTT TGG GAG TTT GCA GAG GCC98 Asp Leu Ser Glu Thr Arg Lys Val Trp Glu Phe Ala Glu Ala
336 TTC AAG AGG AAG TAC AAA GCC TTG AAT GTA CTG ATA AAT AAT112 Phe Lys Arg Lys Tyr Lys Ala Leu Asn Val Leu Ile Asn Asn
378 GCA GGC TGC ATC ATG AGT GAG AGG GAT GTG AAC GCT GAG GGG126 Ala Gly Cys Ile Met Ser Glu Arg Asp Val Asn Ala Glu Gly

66
420 CTG GAG AAG AGC TTT GCC AGT AAC GTC ATG GGT GTG TAT ATC140 Leu Glu Lys Ser Phe Ala Ser Asn Val Met Gly Val Tyr Ile
462 CTG ACC AAG GGC CTG ATT CCT TTA CTG GAG AAG AGT GCA GAG154 Leu Thr Lys Gly Leu Ile Pro Leu Leu Glu Lys Ser Ala Glu
504 CCA AGA GTG ATC ACA GTT TCA TCT GGA GGG ATG CTG GTC CAG168 Pro Arg Val Ile Thr Val Ser Ser Gly Gly Met Leu Val Gln
546 AAG CTG AGG ACA GGG AAC CTG CAG ACA GAA ATA GGT CGC TAT182 Lys Leu Arg Thr Gly Asn Leu Gln Thr Glu Ile Gly Arg Tyr
588 GAC GGC ACC ATG GTC TAC GCA CAG CAC AAG AGG CAA CAG GTG196 Asp Gly Thr Met Val Tyr Ala Gln His Lys Arg Gln Gln Val
630 GTG ATG ACA GAG CAG TGG GCC AAG ACC CAT AGC AAC GTC CAC210 Val Met Thr Glu Gln Trp Ala Lys Thr His Ser Asn Val His
672 TTC TCA GTG ATG CAC CCT GGC TGG GTG GAC ACG CCG GCG GTG224 Phe Ser Val Met His Pro Gly Trp Val Asp Thr Pro Ala Val
714 GCC AAT GCC ATG CCT GAC TTC CAC CAG TCT ATG AAG GAC AGT238 Ala Asn Ala Met Pro Asp Phe His Gln Ser Met Lys Asp Ser
756 ATG AGG ACC CCG GAG CAG GGG GCT GAC ACC ACG GTG TGG TTG252 Met Arg Thr Pro Glu Gln Gly Ala Asp Thr Thr Val Trp Leu
798 GCC ATC TCA GAG GCC GCC GCC ACC AAG CCC AGC GGA AGC TTC266 Ala Ile Ser Glu Ala Ala Ala Thr Lys Pro Ser Gly Ser Phe
840 TTC CAG GAT CGG AGG ATG GTG TCG GCC CAC CTG CCC CTG GCC280 Phe Gln Asp Arg Arg Met Val Ser Ala His Leu Pro Leu Ala
882 TGG ACC CAC AGT TCC CAG TTG GAG CAG CAG AAG TTC ATG AGT294 Trp Thr His Ser Ser Gln Leu Glu Gln Gln Lys Phe Met Ser
924 GTG ATG GAG GAT CTG GCC AAG ACC TTC CAG CCC CAC TGA308 Val Met Glu Asp Leu Ala Lys Thr Phe Gln Pro His ***
Abbildung 32: Translatierte Aminosäuresequenz der isolierten 36K cDNA
Es konnte eine eindeutige Codonpräferenz festgestellt werden; dieses gilt vor allem für die
Tripletts der Aminosäuren Ala (20 von 33: GCC), Phe (10 von 14: TTC), Leu (15 von 17:
CTG), His (10 von 11: CAC), Gln (12 von 13: CAG), Lys (17 von 21: AAG), Arg (10 von
17: AGG), Val (14 von 25: GTG). Diese Codonnutzung entspricht der von WADA (1991) für

67
Knochenfische festgestellten Präferenz.
Anhand der Aminosäuresequenz wurde ein Molekulargewicht errechnet, welches mit 35,7
kDa annähernd dem apparenten Molekulargewicht des 36K-Proteins in der SDS-
Gelelektrophorese entspricht. Der kalkulierte isoelektrische Punkt liegt bei pH 9,54.
Computergestützte Sequenzanalysen sagen kein sekretorisches Signalpeptid voraus.
Ebenfalls fehlt ein Transmembransegment, so daß es sich um ein cytoplasmatisches Protein
handeln dürfte. Es können 4 potentielle Phosphorylierungsstellen für Protein-Kinase-C
ausgemacht werden (Position 103-105: TRK; Position 132-134: SER; Position 248-250:
SMK; Position 252-254: SMR). Außerdem sind 4 putative Phosphorylierungsstellen für
Casein Kinase II auszumachen (Position 132-135: SERD; Position 248-251: SMKD;
Position 299-302: SQLE; Position 308-311: SVME). Wie schon in der Einleitung erwähnt,
konnte vor Beginn dieser Arbeit bereits anhand von gDNA-Fragmenten ein putatives 36K-
Protein vorausgesagt werden. Vergleicht man diese Voraussage mit der hier isolierten
translatierten cDNA Sequenz, fällt auf, daß sich beide Proteine im N-Terminus
unterscheiden, was aber keine Auswirkungen auf die vorhergesagte Sekundärstruktur des
Proteines hat (näheres unter 3.5.1).
Sekundärstrukturanalysen mit der hier isolierten Sequenz nach dem Chou/Fasman
Algorithmus (1978) ergaben eine charakteristische Abfolge von 6 β-Faltblätter und 6 α-
Helices. Innerhalb dieser 6 β-Faltblätter wurden außerdem Bereiche mit hohem β-Turn-
Potential identifiziert. Diese β6/α6-Struktur ist bereits 1974 von Rossman et al. beschrieben
worden. Gestützt durch Proteindatenbank-Sequenzvergleiche wurde deutlich, daß 36K daher
eine signifikante Ähnlichkeit zu klassischen Vertretern dieser charakteristisch strukturierten
Proteine, den NAD/NADP-abhängigen Oxido-Reduktasen aufweist. Diese bestehen zumeist
aus einer Dinukleotid- und einer substratbindenden Domäne. Das aktive Zentrum der
Enzyme liegt dabei zwischen beiden Abschnitten. Die im Rahmen der vorliegenden Arbeit
durchgeführten Sekundärstrukturvorhersagen stützten die bereits von Strelau postulierte
Struktur des vorhergesagten putativen 36K-Proteins, so daß an dieser Stelle auf diese Arbeit
verwiesen werden kann. (STRELAU, DISSERTATION 1997).

68
Abbildung 33: Hypothetisches Sekundärstrukturmodell von 36K, gestützt durch Protein-Datenbankvergleiche; β-Faltblätter sind durch blaue Pfeile dargestellt, die alternierenden α-Helices durchrote Tonnen; eine putative NAD/NADP-Bindestelle ist durch die rote Strukturformel angedeutet. DieseSekundärstruktur ähnelt den Rossman-gefalteten Oxido-Reduktasen.
3.5 Untersuchungen zur 36K-Genstruktur durch PCR
Um Aufschluß über die zugrundeliegenden Genstruktur zu
erhalten und somit die Intron/Exon-Grenzen festlegen zu
können, wurde gDNA aus der Forellenleber isoliert und
als Template für eine PCR eingesetzt. Es ergab sich ein
heterologes Amplifikat aus Fragmenten unterschiedlicher
Länge. Es konnten jedoch fünf prominente Banden
ausgemacht werden (Abbildung 34), die zur Verifikation
kloniert und sequenziert wurden. Durch
Sequenzvergleiche mit der in dieser Arbeit isolierten
cDNA konnten die erhaltenen Amplifikate auf ihre
Spezifität verifiziert und das größte Amplifikat von ca.
2000 bp Länge eindeutig als das zugrundeliegende
Genfragment identifiziert werden. So war es möglich, die Intron/Exon-Strukturen des 36K-
Abbildung 34: AgarosegelelektrophoreseSpur 1: gDNA-PCR mit 36K-CL-F4/36KR2; 1968 bp Amplifikat ist 36KGenfragment, Spur 2: Smart Ladder(Standard); Spur 3: gleiche PCR mit H20(neg. Kontrolle)

69
Genes in diesem Bereich festzulegen (Abbildung 36). Das 5‘ Ende des gDNA-Amplifikates
entsprach exakt dem 5‘ Ende des codierenden Bereiches der in dieser Arbeit isolierten
cDNA-Sequenz. Der als Exon I bezeichnete Bereich endet nach 56 bp in einen kanonischen
5‘ Splice Donor (GT). Das 1551 bp lange Intron I endet bei Nukleotid 1607 in einen
kanonischen 3‘ Splice Akzeptor (AG). Strangabwärts folgen 138 bp Exon II, die wiederum
in einen kanonischen GT 5‘ Splice Donor münden (Position 1745). Das Intron II umfaßt die
folgenden 172 bp und endet mit AG als 3‘ Splice-Akzeptor, an dem sich das Exon III
anschließt, welches bis zur Primersequenz dargestellt ist. Alle gefundenen Splice-Punkte des
36K-Genes unterliegen demnach der GT/AG Regel von Breathnach et al. (1978). Eine
Intron/Exon-Karte des 36K-Genes ist in Abbildung 35 dargestellt.
Abbildung 35: Karte der Intron/Exon-Struktur des 36K-Genes (modifiziert nach Strelau); für die Rossman-Faltung der Proteinstruktur wichtige Domänen (βn αn) sind in keinem Falle durch Introns getrennt.

70

71
Abbildung 36 (vorherige Seite): Sequenz des zugrundeliegenden 36K-Genes (gDNA-PCR): verwendetePrimer sind fett gedruckt, Start-ATG ist unterstrichen; codierende Regionen (Exons) in Großbuchstaben sindgrau unterlegt; Splice-Punkte sind rot dargestellt. Von Strelau (Dissertation,1997) ermittelterTranskriptionsstart ist fett/kursiv, die postulierten alternativen Start-ATG’s sind unterstrichen(Erläuterungen hierzu siehe 3.5.1)
3.5.1 Diskrepanz zwischen isolierter und vorhergesagter 36K-Sequenz
Vergleicht man die Aminosäuresequenz bzw. die zugrundeliegende Nukleotidsequenz der
hier isolierten 36k cDNA mit der von Strelau (DISSERTATION, 1997) anhand von gDNA-
Fragmenten vorhergesagten cDNA-Sequenz, fällt auf, daß sie sich im N-Terminus der
codierenden Region unterscheiden. Diese Diskrepanz erklärt sich durch eine abweichende
Nukleotidfolge (TTCCA statt TTCA) in der gDNA-Sequenz, die als Grundlage für die
Vorhersage der putativen 36K-Sequenz diente. Interessanterweise ergibt sich durch die
abweichende Sequenz eine translatierte Proteinsequenz, die bis auf 40 AS im N-Terminus mit
der hier isolierten Sequenz identisch ist und keinen Einfluß auf die vorhergesagte
Sekundärstruktur des Proteines hat (siehe auch 3.4.1, bzw. STRELAU, DISSERTATION, 1997).
ATGGAGCAGTGGATGGATAGCCTCCACCTGACTGTTTATCTTTGTGTTG-ATGTCTCTTTACCGCAACTCTGCGTGGTTTCTGAAAGGAATGACTGAATT
---CAGGAGTGCGTTCCTGTCTGCAGCCAAGCACTTTGTGGAAAAGGACCCACCAGGAGTGCGTTCCTCTCTGCCGCCAAACACTTTGTGGAAAAGGACC
TGGAGGTGTCCATGGCAGGCAGGGTCTTCCATGATCACCGGAGCCAAC..TGGAGGTGTCCATGGCAGGCAGGGTCTTC-ATGATCACCGGAGCCAAC..
Abbildung 37: Nukleotidvergleich zwischen N-Terminus der vorhergesagten cDNA-Sequenz (Strelau,Dissertation, 1997) in rot und hier isolierter cDNA Sequenz in schwarz; Start-ATG sind unterstrichen;Sequenzunterschiede (Cytosin-Insertion) ist eingerahmt
Die von Strelau zur cDNA-Vorhersage verwendetet gDNA-Sequenz ist mit der im Rahmen
dieser Dissertation ermittelten und oben beschriebenen Gensequenz in Abbildung 36 bis zur
Erkennungsstelle für das Restriktionsenzym ClaI identisch (exklusive der Cytosin-Insertion;
Abbildung). Durch RT-PCR konnte in dieser Arbeit das von Strelau durch Primerextension
gefundene Transkript (Transkriptionsstart und putative Start-ATGs sind in Abbildung 36
verzeichnet) verifiziert werden. Die ermittelte Sequenz entsprach wiederum der TTCA-
Nukleotidfolge, wie sie im Rahmen dieser Arbeit stets gefunden wurde. Eine Translation

72
dieses Transkriptes, ausgehend von den von Strelau vorgeschlagenen Start-ATGs, ergibt ein
Protein von 78 AS Länge.
Abbildung 38: Agarosegelelektrophorese Verifikation eines weiteren nicht 36K-Transkriptes Spur 1: RT-PCR mit 36K CF1/36KR2; Spur 2: H2O-PCR; St: 100bp Ladder (Fa. Gibco); Spur 3: RT-PCR 36KCL-F4/36KR2; Spur 4: H2O-PCR
3.5.2 Klonierung einer putativen Promotorregion des 36K-Genes durch „inversePCR“
Nachdem die oben beschriebene Intron/Exon-Struktur des 36K-Genes nunmehr vollständig
aufgeklärt war (Abbildung 35), ergab sich die Frage nach der Sequenz und Struktur des
zugrundeliegenden Promotors. Damit einher ging die Frage nach dem Transkriptionsstart des
36K codierenden Transkriptes. Der bisher unbekannte Bereich strangaufwärts des Start-
ATGs sollte mit Hilfe der inversen PCR (2.2.13.3), die im Rahmen dieser Dissertation
etabliert wurde, kloniert werden.
Per PCR wurde eine ca. 300 bp lange DIG-markierte
Sonde spezifisch gegen den Bereich strangaufwärts
der Erkennungsstelle für Cla I inkl. des Exon I,
codierend für den N-Terminus von 36K, generiert.
Nach southernbotting von Cla I verdauter Forellen
gDNA war in Höhe von 1,2 kb sowie in Höhe von ca.
3,5 kb ein schwach positives Signal erkennbar. Aus
einem Parallelgel wurde Cla I verdaute gDNA diesesAbbildung 39: Agarosegel-elektrophorese, inverse PCR anzirkularisierter Cla I verdautergDNA mit 36K-CL-R6/ 36K-CL-F71: Template 3,5 kb gDNA; 2: 1,2 kbgDNA

73
Größenbereiches herausgeschnitten, aufgereinigt und zur Selbstligation/Zirkularisierung über
die Cla I kompatiblen Enden eingesetzt (2.2.13.3). Die inverse PCR an zirkularisierter 1,2 kb
DNA als Matrize ergab eine prominente Bande in der erwarteten Höhe, an zirklarisierter 3 kb
DNA war kein PCR-Amplifikat detektierbar (Abbildung 39). Nach Klonierung,
Sequenzierung und Sequenzvergleich mit der bislang bekannten 36K-Gensequenz konnte die
ermittelte Sequenz der 1,2 kb Bande eindeutig als distaler gDNA Bereich stromaufwärts des
Start-ATGs eingestuft werden (Abbildung 40). Die Existenz und Orientierung dieser
neugewonnenen Nukleotidsequenz konnte in einer unabhängigen PCR an unverdauter gDNA
verifiziert werden.
1 ATCGATAAAC CGCTTCTTAT GCAAGACAGA CGGCACAACT GTTCAAGTGG
50 GAGATGTGGT TTCAGTGGTG CCAGCCAGCA ACGCTTCTTT AAGCTACTGT
100 AGTGTCCGGG CAAACAGGTG GAGACAACCT GCCAAATGAA CTGGGTGTGG
150 CATTGACAAC ACCGTTACTG TAGCCTACAC ACCTTTTGGA CAGGTGCATG
200 TTCGATAGAG AGAATGTACT TGCCCTGTAA AATGTCAAAA CATGGAACCA
250 AAAGCAGACA TTTTACTCAC CATCGTGAAG CTGCAATGTG ACAGTAAGGA
300 CAACAGTTGA GGGAAAATAT GTTAACTACA TTCTTGCGTA AAACCGGTAC
350 ATAGGGACTG GCAACGTCAA ATTCCCACAA TGCTGTGGGT TAAGTTGCGA
400 AACCATATCT TCTCTTGTCT TGACTGTGCC GCGCCCCGCN TTTTGCTTTG
450 CTTGGTCAGA AAAGGTTCAC ACCTTTCACA GGAGGTTCCT GGGCAAGGTG
500 CATTGTAGTA CTGTACTAAC TGGTTTATGA TCAAATATAG ATATTATCCT
550 GTTTGTCAAG CCAAGTTATG TGCGTCTATT ATTTCATTTC CAGCGACAGA
600 CATATTGCGT GTTCTTTTGG ATTTTTCTAT TGTAGCCTAA AGGCCTATAA
650 AAACCGACAA TTAGTCTCAG TTCCCCCAGA ATTATTATCA ATAATTCCCT
700 GATTTAATTT TATTCTGTTA CTNAAATATG CGGTCGTTGA AATGTAATTT
750 GAAATGCTGT GCATGCAACT CCTGAATTTG CCATGCAAAC CTATTGGAGA
800 TTCAGTAGGA TTGTTTTTTA ACATTAATTC AAGCCTATAG ACATCATGGA
850 CAGTGTCGAG CTGACACCAG TTGACACCGA GTTTGATTGT GCCTCAGTTG
900 TTGATCCCAA CTCGCCGCTG GGTGGCCGAA GGTGTCATCC AATGTAAATT
950 CACACACGTG GGTTTGGTCC AAGTTAAAAC CGGAGGAGAG AGTGCAATAA
1000 CGGACGACGG GACGAGAGGA TTCCATGGGT AATTCAACGA TGTCTCTTTA
1050 CCGCAACTCC GCGTGGTTTC TGAAAGGAAT GACTGAATTC
Abbildung 40: Durch inverse PCR erhaltene gDNA-Sequenz (putative Promotorregion); codierende Regionist grau unterlegt; Erkennungsstelle für Cla I ist umrahmt; Start-ATG der isolierten 36K cDNA ist rot und

74
unterstrichen; putatives Start-ATG in Position 1024 rot; strangauwärts „in-frame“ Stop-codon rot; putativeTATA-Boxen kursiv
Die neugewonnene putative Promotor-, bzw. 5‘ UTR-Sequenz ist in Abbildung 40 in der
korrespondierenden Genstruktur dargestellt. In Position 1039 befindet sich das Start-ATG
der isolierten cDNA. Die N-terminale codierende Region erstreckt sich bis zum Ende der
dargestellten Sequenz. Der Bereich zwischen Position 1029-1039 ist bis auf ein
Nukleotidaustausch (Position 1034 T statt A) mit der als 5‘ UTR interpretierten Sequenz der
isolierten 36K cDNA (Abbildung 31) identisch. In Position 1024 befindet sich im gleichen
Leserahmen wie das isolierte 36K-Transkript als erstes ATG hinter einem Stop-Codon
(Position 998) ein weiteres putatives Start-ATG. Innerhalb der putativen Promotorregion
konnten mehrere potentielle TATA-Boxen gefunden werden (Position 589, Position 681;
Position 703; Position 823). Die potentiellen TATA-Boxen in Position 823 (Sequenz
TTAATTCAA), in Position 589 (Sequenz TTATTTCAT) und in Position 681 (Sequenz
TTATTATCAA) weisen auffällige Ähnlichkeiten zu der für das IP-Gen gefundenen TATA-
Box (JESERICH, STRELAU UND LANWERT, 1997), dargestellt in Abbildung 41 auf.
Abbildung 41: Sequenzvergleich zwischen putativen TATA-Boxen des 36K-Genes mit der TATA-Box des IP-Genes; identische Nukleotide sind grau hinterlegt
3.6 Heterologe Expression der isolierten 36K cDNA in CHO-K1
Die codierende Region der isolierten 36K cDNA wurde wie unter 7.5 beschrieben, in den
pIRES-hygro Expressionsvektor kloniert und diente zur Transfektion von CHO-K1-Zellen
(2.2.49). 48h nach Transfektion zeigten ca. 5% der CHO-Zellen eine deutliche granuläre
Immunfärbung im Bereich des Cytoplasmas (Abbildung 43). Der korrespondierende
Westernblot (Abbildung 42) zeigt eine immunopositve Bande, die leicht unterhalb der
nichtrekombinanten Forellenmyelin 36K-Probe läuft. Eine Klonierung von stabilen
Expressoren mit Hilfe der Hygromycin/G418-Selektion (2.2.51) ist leider trotz mehrerer

75
Ansätze nicht gelungen.
Abbildung 42. Westernblot mit 36K transfizierten CHO-Zellen (36K/CHO); daneben Forellenmyelin (My)bzw. untransfizierte CHOZellen (CHO); (Nachweis mit α36K)
Abbildung 43: Immunfloureszenz 48 h nach Transfektion: r36K/CHO-Zellen (Methanolfixierung α36K/cy3,600x)
3.7 Expression in E. coli (pET-System)Im Rahmen dieser Dissertation wurden drei verschiedene präparative E. coli
Expressionssysteme etabliert und vergleichend eingesetzt. Hierbei handelte es sich um das
IMPACT-System, pMAL-System (2.2.27 und 2.2.21) sowie das pET-System (2.2.23).
Es zeigte sich, daß weder das IMPACT-System noch das pMAL-System für die Expression

76
von r36K geeignet war. Nach Induktion und Aufreinigung (2.2.27 und 2.2.21 f ) waren nur
marginale Proteinausbeuten von ≤ 550µg/ 1l Kultur detektierbar, die außerdem stark mit
Fremdprotein kontaminiert waren. Das Zielprotein machte somit nur ca. 15 % aus (Daten
nicht gezeigt). Diese geringen Ausbeuten waren durch Veränderung der
Induktionsbedingungen (Zeitpunkt und -dauer und/oder Temperatur nach Induktion) nicht zu
verbessern. Daher wurden diese Ansätze verworfen. Das pET-System lieferte jedoch
ansprechende Resultate.
In dieser Dissertation ist es gelungen, 36K mit Hilfe des pET-System (Klonierungsschema
siehe 7.7) unter denaturierenden Bedingungen in Gegenwart von 6M Harnstoff in großen
Mengen aufzureinigen (2.2.23 ff). Nach
dreistündiger Induktion bei 37° C war im
Vergleich zur nicht induzierten Spur auf dem
SDS-PAGE in Abbildung 44 eine prominente
Bande in der erwarteten Größe sichtbar.
Nach Ni2+-Chelat-Affinitätschromatographie
in Gegenwart von 6M Harnstoff konnte das
rHis-Tag-36K-Protein nahezu ohne
Fremdproteinkontaminationen aufgereinigt
werden. Eine vergleichende Aufreinigung
unter nativen Bedingungen ohne Harnstoff
brachte nur unbefriedigende Ausbeuten und
starke Kontaminationen durch Fremdprotein
(Abbildung 44) Das rekombinante His-Tag-
36K liegt in der Bakterienwirtszelle nach
Induktion offensichtlich zum größten Teil in
Einschlußkörpern („inclusion bodies“) vor. Der korrespondierende Westernblot zeigt ein
immunopositives Signal auf der Höhe der eluierten Bande. Das elektrophoretische
Laufverhalten des rHis-Tag-36K Proteins weicht von dem des „nativen“ 36K aus dem Myelin
der Forelle aufgrund der „His-Tag“-Erweiterung durch zusätzliche 21 Aminosäuren, davon
10 geladene Histidine, in der erwarteten Weise ab (Abbildung 44). Die denaturierende
Aufreinigung des rHis Tag-36K ergab Ausbeuten von ≥ 7,5 mg Zielprotein / 1l Kultur.
Abbildung 44: SDS-PAGE/Westernblot einertypischen Expression von 36KHT in E.coli BL 21(DE3)/pLysS-36K/pET16b und Aufreinigung inGegenwart von 6M Harnstoff; Spur 1: nicht induzierteZellen; Spur 2: 3h nach Induktion bei 37°C; Spur 3:denaturiernd aufgereinigtes 36KHT in Gegenwart von6M Harnstoff; Spur4: nativ aufgereinigtes Eluat; Spur5: Westernblot vom 36KHT Eluat denat.

77
3.7.1 Alternative Aufreinigung von His-Tag-36K durch Affinitätschromatographiemit Farbsepharosen
Um erste Hinweise auf eine denkbare NAD- bzw. NADP-bindende Funktion des 36K
Proteines zu erhalten wurde eine Affinitätschromatographie mit Farbsepharosen (2.2.26)
durchgeführt. Wie aus Abbildung 45 deutlich wird, konnte jedoch natives HisTag-36K mit
dieser Methode unter den gewählten Bedingungen nicht aufgereinigt werden. Durch
Veränderung der Pufferbedingungen und/oder der Farbsepharose könnten weiterführende
Studien mit renaturiertem, gereinigtem 36KHT jedoch Hinweise auf eine NAD/NADP-
Abhängigkeit des HisTag-36K-Proteines bringen.
Abbildung 45: Westernblot alternative 36K Aufreinigung mit „reactive red“ Sepharose; my: ZNS Myelin;neg.: E coli Zellysat nicht induziert; 1: nativ aufgereinigtes 36KHT Eluat nach Umpuffern; Spur 2: nicht anSäule gebundener Durchlauf; Spur 3: Waschdurchlauf; Spur 4 und 5: Elution mit 20 mM NAD bzw. NADP
3.8 Expression in Insektenzellen / Baculovirus-SystemVergleichend zu den E. coli Systemen wurde in der vorliegenden Dissertation die Methode
der Baculovirus-Expression „Bac-to-Bac“ (siehe 2.2.20 f.) als weitere heterologe
eukaryotische Proteinexpressionsmethode etabliert. Als Wirtszellen dienten HighFive-Zellen
(H5) bzw. Sf21-Zellen. Da die Baculovirus-Expression zur Lyse der infizierten Wirtszelle
führt, ist es günstig, den Erfolg und den zeitlichen Verlauf der Infektion überprüfen zu
können (3.2.9). Hierzu diente in der vorliegenden Arbeit das Grünfluoreszierende Protein
(GFP) als Reportergen. Das komplexe Klonierungsschema des Konstruktes, bestehend aus

78
Reportergen und Zielproteingen, ist unter 7.6 ausführlich dargestellt. Wie unter 2.2.20.1 f
beschrieben, konnten r36KHT/GFP-DualAcNPV kloniert werden, die für die
Proteinexpression (2.2.20.3) eingesetzt wurden.
Abbildung 46 zeigt zeigt r36KHT/GFP-DualAcNPV infizierte HighFive-Zellen im
Fluoreszenzmikroskop 72h nach Infektion. Zu diesem Zeitpunkt waren bereits ca. 50 % aller
Zellen infiziert und zeigten eine deutliche Immunfärbung.
Abbildung 46: links: Immunfloureszenz 72h nach Infektion 36KHT/GFP-Dual/High Five (α36K/cy3, 400x),GFP-Markierung der 36KHT/GFP-Dual-Expressoren
Abbildung 47: SDS-PAGE; 1: HighFive Zellysat; 2: HighFive 3 Tage nach Infektion mit r36KHT/AcNPV; 3:r36KHT Eluat nach Aufreinigung; 4: Westernblot (α36K) an r36KHT-Eluat
Die SDS-PAGE in Abbildung 47 zeigt eine typische Expression und His-Tag -Aufreinigung
(2.2.24) des rHis-Tag 36K aus HighFive-Zellen in Gegenwart von 6M Guanidin-HCl,

79
daneben der korrespondierende Westernblot mit einer immunopositiven Bande in der
erwarteten Höhe. Das Laufverhalten des rHis-Tag-36K weicht aufgrund der His-Tag-
Erweiterung um insgesamt 28 Aminosäuren wie schon unter 3.7 beobachtet ab. Die
Ausbeuten der Baculovirus-Expression fielen mit ca. 500µg Zielprotein /1l Kultur um eine
10er Potenz geringer aus als das vergleichbare E. coli System (3.7).

80
4 Diskussion
Im Rahmen dieser Dissertation konnten die cDNAs der Hauptmyelinproteine IP2 und 36K
aus dem ZNS der Forelle vervollständigt und in unterschiedlichen Systemen heterolog
exprimiert werden, so daß eine erste Grundcharakterisierung dieser Proteine möglich wurde.
Es konnten erste experimentelle Hinweise auf die Funktionen dieser putativen
Adhäsionsmyelinproteine in mimetischen Systemen gewonnen und darüberhinaus weitere
Einsichten in die Sequenz und Struktur der zugrundeliegenden Myelinproteingene erhalten
werden.
Da die Sequenz der IP2 cDNA vor Beginn der Arbeit nicht vollständig bekannt war, wurde
sie zunächst durch PCR-gestützte Techniken komplettiert. Die IP2 cDNA Nukleotidsequenz
ist in dem codierenden Bereich mit der cDNA-Sequenz der bereits isolierten IP1-Isoform
identisch, am 3‘ Ende aber um 90 bp verlängert. Die 3‘ UTR der IP2 cDNA ist mit ca. 1000
bp auffällig lang und besitzt charakteristische Merkmale. Im Anschluß an die verlängerte
codierende Region erstreckt sich eine kurze IP2 spezifische 3‘UTR, die in eine für beide IP-
Isoformen identische 3‘UTR inkl. poly (A). mündet. Die cDNAs der beiden Splice-Varianten
IP1 und IP2 unterscheiden sich also außer in der verlängerten cytoplasmatischen Domäne nur
durch kurze Isoform-spezifische 3‘ UTR-Abschnitte. Es ist zur Zeit noch nicht geklärt, ob
die gemeinsame 3‘ UTR-Sequenz zweimal auf der gemeinsamen prä-mRNA vertreten ist
oder aber als konstitutives Exon innerhalb der 3‘ UTR entsprechend für beide Varianten
prozessiert wird. Über die physiologische Bedeutung von 3‘ UTRs ist allgemein wenig
bekannt, doch dürften sie eine wichtige Rolle bei der Prozessierung von Transkripten und
somit bei der Regulation des alternativen Splicings spielen. Beiden IP-Isoformen liegt der
gleiche Promotor zugrunde (JESERICH, STRELAU UND LANWERT, 1997), die Expression der
Proteinvarianten wird jedoch entwicklungsabhängig gesteuert. Hierbei wird zunächst die
längere IP2 Isoform exprimiert, erst in späteren Entwicklungsstadien folgt die Expression
von IP1 (JESERICH ET AL., 1990A). Es ist daher anzunehmen, daß sich auf der beiden
Varianten zugrundeliegenden gemeinsamen prä-mRNA Sequenzmotive befinden, die die
Auswahl der alternativen Splice-Punkte beeinflussen und somit die differentielle
Proteinexpression regulieren. Die Regulation von alternativen Splice-Vorgängen ist bisher
nur wenig verstanden, doch konnten hierfür bereits Protein-Protein-, Protein-RNA-, und

81
RNA-RNA-Interaktionen verantwortlich gemacht werden, die ihrerseits
entwicklungsabhängig reguliert werden. (STOJDL UND BELL, 1999; ZHAO ET AL, 1999;
LOPEZ, 1998; VARANI UND NAGAI, 1998). Innerhalb der gemeinsamen reifen 3‘ UTR von
IP1 und IP2 konnten außerdem AT-reiche Sequenzmotive gefunden werden. Diese Motive
könnten Determinanten für die Instabilität einer mRNA darstellen und ihre Degradation
beeinflussen (SHAW UND KAMEN, 1986). Weiterhin gibt es Hinweise, daß die 3‘ UTR-
Sequenzen von Myelinproteinen die Translationseffizienz beeinflussen und so zur Regulation
der Proteinexpression beitragen können. So sind z. B. für das Basische Myelinprotein durch
progressive Deletionen der 3‘ UTR bis zu zehnfache Unterschiede in der Translations-
effizienz beobachtet worden (UENO ET AL., 1994). Auch für das Periphere Myelinprotein 22
(PMP22) ist seit kurzem bekannt, daß die 3‘ UTR cis-inhibierenden Motive enthält, welche
die Translationseffizienz beeinflussen (BOSSE ET AL., 1999) Der molekulare Mechanismus
hinter dieser post-transkriptionellen Regulation sowie eventuelle konservierte Konsensus-
Sequenzmotive sind bislang jedoch noch nicht bekannt. Ein weiterer denkbarer Aspekt für
die Funktion der IP1/2 spezifischen 3‘ UTR ist die Regulation der Proteinlokalisation des
translatierten Proteines. Erst kürzlich konnte in Drosophila-Oozyten gezeigt werden, daß die
subzelluläre Lokalisation dreier durch alternatives Splicing entstandener Genprodukte durch
unterschiedliche 3‘ UTRs reguliert wird. (WHITTAKER ET AL. 1999). Ähnliche mRNA-
abhängige Proteintranslokation ist ebenfalls für das Basische Myelinprotein beschrieben,
wobei die regulatorischen Sequenzmotive jedoch nicht ausschließlich auf die 3‘ UTR
beschränkt, sondern auch innerhalb der codierenden Region auszumachen sind (BOCCACIO ET
AL., 1999; WOODRUFF UND FRANKLIN,1998; COLMAN ET AL., 1982; GOTTLIEB, 1992;
REVIEW: CARSON ET AL.,1998). Weiterführende Arbeiten, in denen die 3‘UTR von IP1/2
hinter die codierende Region eines Reportergenes wie das Grünfluoreszierende Protein
(GFP) oder Chloramphenicol-Acetly-Transferase (CAT) kloniert werden würde sowie die
Isolierung der zugrundeliegenden Gen-, bzw. prä-mRNA-Sequenz, könnten interessante
physiologische Aufgaben der proteinspezifischen bzw. gemeinsamen 3‘ UTR liefern.
Anhand der isolierten cDNA-Sequenz wurde die Aminosäuresequenz des IP2-Proteines
abgeleitet. Für viele der in der IP2 cDNA-Sequenz codierten Aminosäuren konnte eine
auffällige Codonpräferenz ausgemacht werden. Die bevorzugten Codons stehen in
Übereinstimmung mit der von Wada et al. (1991) veröffentlichten Codonpräferenz von

82
Knochenfischen. Das IP2-Protein ist 231 Aminosäuren lang und in der cytoplasmatischen
Domäne im Vergleich zur IP1-Splice-Variante um 29 Aminosäurereste verlängert. Das
kalkulierte Molekulargewicht des reifen Proteines von 23 kDa steht in guter
Übereinstimmung mit der elektrophoretischen Mobilität des reifen deglykosilierten IP2-
Moleküls (JESERICH UND WAEHNELDT, 1987). Außerdem stimmt der vorhergesagte
isoelektrische Punkt des Proteines (pI: 10,53) mit früher gewonnenen Resultaten aus 2 D-
Gelektrophorese-Analysen überein (JESERICH UND WAEHNELDT, 1986).
Sequenzvergleiche der cytoplasmatischen Domäne des IP2-Porteins aus dem ZNS der Forelle
mit den cytoplasmatischen Domänen des obligatorischen PNS-Adhäsionsprotein P0 von
Ratte (LEMKE UND AXEL, 1985), Mensch (HAYASAKA ET AL., 1991) und Hai (SAAVEDRA ET
AL., 1989) zeigten eine charakteristische Verteilung von identischen Aminosäuren. Diese
Aminosäureübereinstimmung ist im verlängerten C-terminalen Proteinbereich, der nur in der
IP2 Splice-Variante zu finden ist, mit 71% besonders hoch. Die letzten sieben Aminosäuren
mit einer positiven Nettoladung sind in allen verglichenen Spezies identisch. In dem Teil der
cytoplasmatischen Domäne, der von IP1 und IP2 geteilt wird, beträgt die Aminosäure-
übereinstimmung lediglich 34%. Phylogenetisch konservierte Aminosäuren sind meist
basisch, d.h. positiv-geladen. Zu den konservierten Bereichen gehören auch zwei Protein-
Kinase C-Phosphorylierungsstellen. Aufgrund der auffälligen Verteilung von konservierten
Aminosäuren ist eine wichtige funktionelle Rolle der cytoplasmatischen Domäne des IP2-
Proteines zu vermuten. Es ist denkbar, daß die stark positiv-geladene, basische Domäne
aufgrund von elektrostatischen Wechselwirkungen an saure negativ-geladene
Membranphospholipide bindet, also als „Membrananker“ fungiert und somit die Apposition
der cytoplasmatischen „major dense line“ unterstützt. Eine derartige Membranassoziation
wurde bereits 1985 von Lemke und Axel für das P0-Protein postuliert und konnte
experimentell mit Hilfe von artifiziellen negativ-geladenen Liposomen und die durch
chemische Spaltung als Peptidfragmente isolierte cytoplasmatische Domäne des P0-Proteins
nachgewiesen werden (DING UND BRUNDEN, 1994). In diesem Zusammenhang ist
erwähnenswert, daß die verwendeten cytoplasmatischen P0-Domänen eine
Mikroheterogenität in der Nettoladung aufwiesen, die auf einen unterschiedlichen
Phosphorylierungsgrad zurückzuführen ist. Die Zelle ist so in der Lage, aufgrund von
Phosphorylierungen die Membranbindung der cytoplasmatischen Domäne zu modulieren. Die

83
phylogenetische Konservierung der Position der Protein-Kinase C-Phosphorylierungstellen
zwischen dem evolutiv weit entfernten P0-Protein der Säuger und dem IP2-Protein der
Forelle läßt eine vergleichbare Funktion nicht abwegig erscheinen. Eine ebenfalls für P0
beschriebene Membranverankerung durch Palmytilierung der cytoplasmatischen Domäne
(GAO UND FILBIN, 1999), die z.B. auch in dem neuralen Zelladhäsionsmolekül N-CAM
(LITTLE ET AL., 1998) und PLP (POPOT ET AL., 1991) gefunden werden konnte, scheidet für
das IP2-Protein aus, da es keine cytoplasmatischen acylierbaren Cysteinreste besitzt.
Zusammenfassend läßt sich sagen, daß durch die hier gefundenen auffälligen Sequenzüberein-
stimmungen zwischen IP2 und P0 innerhalb der cytoplasmatischen Domäne die bereits von
Stratmann und Jeserich (1995) beschriebene strukturelle und sequentielle Ähnlichkeit
zwischen IP und dem obligatorischen Adhäsionsprotein P0 gestützt werden konnte.
Im Rahmen dieser Arbeit ist es gelungen, die adhäsiven Eigenschaften der in beiden
Isoformen identischen extrazellulären Domänen von IP in einem mimetischen Zellsystems zu
untersuchen. Dazu wurde ein Adhäsionstest etabliert, der es ermöglichte, das
Aggregationsverhalten von transfizierten und nicht transfizierten Einzelzell-Suspensionen zu
vergleichen. Da sich eine transfizierte Zelle nur durch die Expression des jeweiligen
Zielproteins von einer nicht-transfizierten Zelle unterscheidet, konnten Unterschiede im
Aggregationsverhalten auf adhäsive Eigenschaften des heterolog exprimierten Zielproteins
zurückgeführt werden. Durch Trypanblau-Ausschluß-Test wurde überprüft, daß die
aggregierten Zellen vital sind, so daß eine unspezifische Verklumpung von toten Zellen
auszuschließen ist. Die Zunahme der gebildeten Zellaggregate in der Zellsuspension wurde
mit einem automatischen Teilchenzählgerät quantifiziert und zusätzlich mikroskopisch
analysiert. Wie erwartet aggregierten bereits untransfizierte Zellen der myelinbildenden
oligodrendroglialen OLN-93-Zellinie aufgrund starker intrinsischer Adhäsionseigenschaften
zu großen Zellaggregaten und kamen daher als Wirtszelle für die Adhäsionsassays nicht in
Frage. Untransfizierte CHO-K1-Zellen waren deutlich weniger adhäsiv, so daß sie als
Wirtszelle für alle folgenden Experimente eingesetzt wurde. Es wurde deutlich, daß bei
alleiniger Betrachtung der durch die automatische Quantifizierung der absoluten Teilchenzahl
in der Zellsuspension ermittelten N(t)/N(0)-Werte die durch mikroskopische Analyse ermittelte
Aggregation unterschätzt werden kann. Es ist denkbar, daß durch auftretenden Scherkräfte
während der Partikelzählung Zellaggregate partiell dissoziieren können, so daß nur sehr

84
starke Adhäsionen quantifizierbar sind. Durch die mikroskopische Analyse ist daher
zusätzlich eine Detektion von schwächeren Bindungen bzw. Interaktionen möglich. Die
Funktionalität und Reproduzierbarkeit des etablierten Adhäsionsassays konnte mit Hilfe der
rP0/CHO-Kontrollzellen überprüft werden, da die im Rahmen dieser Dissertation
gemessenen und beobachteten Adhäsionen frühere Untersuchungen mit diesen Zellen
stützten (FILBIN ET AL, 1990; WONG UND FILBIN, 1996; GAO UND FILBIN, 1999).
IP1 und IP2 konnten transient in CHO-Zellen exprimiert werden. 48 h nach Transfektion
zeigten ca. 5% der transfizierten Zellen eine deutliche Immunfärbung. Durch eine
Oberflächenmarkierung von nicht permeabilisierten transfizierten Wirtszellen wurde deutlich,
daß IP1 und IP2 auch in CHO-Zellen in die Zellmembran integriert wurden. Im
korrespondierenden Westernblot zeigte sich, daß IP1/2 auch in der CHO-Wirtszelle
glykosiliert werden, da das apparente Laufverhalten der heterolog exprimierten IP-Splice-
Varianten exakt mit dem Laufverhalten der aus Forellenmyelin isolierten IP-Proteine
übereinstimmte. Diese Glykosilierung könnte eine wichtige funktionelle Bedeutung für IP1/2
besitzen. Zwar ist die Kohlenhydratseitenkette von IP noch nicht im Detail analysiert, doch
konnte in gereinigtem PNS-Myelin der Forelle sowie im ZNS-Myelin des Karpfens durch
Immunreaktion das Kohlenhydrat-Epitop HNK-1 nachgewiesen werden (HAMMER ET AL.,
1993; TAKEI ET AL., 1993), welches außer im Immunsystem und einer Anzahl von neuralen
Adhäsionsproteinen auch im P0-Protein auftritt (BOLLENSEN UND SCHACHNER, 1987). Die
Kohlenhydratseitenkette wurde für P0 durch Kristallstrukturanalysen an der Basis der
extrazellulären Domäne in der Nähe der Zellmembran positioniert (SHAPIRO ET AL., 1996).
Die Funktion ist bislang umstritten. Während Filbin und Tennekoon 1991 dem
Kohlenhydratrest eine für die homophile Adhäsion essentielle Lectin-ähnliche Funktion
zugeschrieben haben, fanden Schachner und Mitarbeiter, daß die bakteriell heterolog
exprimierte extrazelluläre P0-Domäne, die demnach nicht-glykosiliert vorliegt, ebenfalls
homophile Bindekapazitäten besitzt (GRIFFITH ET AL., 1992). Kürzlich konnte die Struktur
des HNK-1 Kohlenhydratkette des P0-Proteines aus Rind durch NMR-Spektroskopie
bestimmt werden, die genaue Funktion bleibt aber weiterhin unbekannt (VOSHOL ET AL.,
1996). Durch Untersuchungen mit Endoglykosidase F konnten neben dem gemeinsamen
HNK-1-Epitop signifikante Unterschiede im Kohlenhydratrest der IP-Proteine der Forelle
und dem P0-Protein aufgedeckt werden (JESERICH UND WAEHNELDT, 1987), so daß auch

85
über die Funktion der IP-Glykosilierung bislang nur spekuliert werden kann.
Es konnte ein stabiler IP1/CHO-Zellklon durch Antibiotikaselektion und limitierender
Verdünnung selektiert, expandiert und für die Adhäsionstests eingesetzt werden. Nahezu
100% der Zellen wurden per Immunfärbung als positive Expressoren identifiziert. Aufgrund
der im Vergleich zur transienten Expression schwächeren Fluoreszenzfärbung von stabilen
Transfektanden, kann davon ausgegangen werden, daß das Expressionsniveau stabiler
Expressoren pro Zelle unter dem von transient exprimierenden Zellen lag. Während bei der
transienten Expression offensichtlich mehrere Kopien des Expressionsvektor-Konstruktes in
der Zelle vorliegen, integriert bei der stabilen Expression durch das seltene Ereignis der
nicht-homologen Rekombination nur eine oder wenige Kopien des Expressionsvektors in das
Genom der Zelle, so daß das Expressionsniveau sinkt. Es ist an dieser Stelle wichtig zu
erwähnen, daß die als Positivkontrolle verwendeten rP0/CHO-Zellen ein deutlich höheres
Expressionsniveau aufweisen: Bei den verwendeten P0-Zellen handelt es sich nämlich um
eine spezielle Dihydrofolat-Reduktase (dhfr) defiziente CHO-DG44-Zelle. Dieser Gendefekt
des essentielle Enzymes wurde in trans mit dem Expressionsvektor, der auch das P0-
Zielprotein codiert, komplementiert. So war es möglich, durch Zellkultivierung in
ansteigenden Methotrexat (MTX)-Dosen (dhfr-Antagonist), aufgrund des bislang nicht
detailliert beschriebenen Phänomens der Genamplifikation, das Expressionsniveau von P0 zu
erhöhen. Im Vergleich zu nicht geboosteten transfizierten Zellen liegen mehrere Kopien
integriert im Genom der Wirtszelle vor, was mit einer Steigerung der P0 Expression um
590% einhergeht und somit 8-16 fach über dem Expressionsniveau von kultivierten
Sekundären Schwannzellen liegt (FILBIN UND TENNEKOON, 1990). Das Phänomen der
Genamplifikation ist offensichtlich auf einige spezielle Gene, darunter die beschrieben dhfr-
Amplifikation, beschränkt (SINGER UND BERG, 1992; KAUFMAN ET AL., 1979; MONTOYA-
ZAVALA ET AL., 1985), da es in dieser Arbeit nicht gelungen ist, durch ansteigende
Antibiotikakonzentrationen das Expressionsniveau von IP in CHO-Zellen zu steigern. Eine
vergleichende Quantifizierung des Oberflächenexpressionsniveaus der P0-Zellen mit den in
dieser Arbeit isolierten stabilen IP-Expressoren, z.B. durch einen Zell-ELISA, ist nicht
möglich, da hierfür unterschiedliche Antikörper verwendet werden müßten, zwischen denen
nicht verglichen werden kann. Folglich ist auch ein direkter quantitativer Vergleich zwischen
allen im Rahmen dieser Arbeit gemessenen Adhäsionswerten der IP-Expressoren und den P0-

86
Kontrollzellen nicht möglich.
In den im Rahmen dieser Dissertation erstmals durchgeführten Adhäsionstests konnte gezeigt
werden, daß das IP-Protein der Forelle eine Funktion als Adhäsionsmolekül ausübt. Stabil
transfizierte rIP1/CHO-Zellen waren im Vergleich zu nicht transfizierten CHO-Zellen
schwach adhäsiv. Die absolute Partikelzahl ging in der rIP1/CHO-Zellpopulation auf ca. 36%
zurück, während in nicht-transfizierten CHO-Kontrollzellen aufgrund ihrer intrinsischen
Adhäsionseigenschaften die Teilchenzahl auf ca. 45% zurückging. Noch deutlicher wurde die
gemessene Adhäsion in der mikroskopischen Anlayse: rIP1/CHO-Zellen bildeten
hauptsächlich Zellaggregate aus bis zu 10 Zellen, während sich untransfizierte CHO-Zellen
überwiegend zu Tripletts zusammenfanden. Die gebildeten rIP1/CHO-Zellaggregate konnten
allerdings durch mehrmaliges Invertieren wieder dissoziiert werden. Es dürfte sich daher bei
der IP1-vermittelten Adhäsion um recht schwache Interaktionen handeln. Dieses könnte den
Unterschied zwischen der durch automatische Teilchenzählung (Scherkräfte) gemessenen
und der mikroskopisch beobachteten Adhäsion erklären.
rIP-P0/CHO-Zellen, die ein chimäres Fusionsprotein bestehend aus der extrazellulären und
transmembranellen Domäne des IP und der vollständigen cytoplasmatischen Domäne des P0
Proteins exprimierten, waren deutlich adhäsiver als rIP1/CHO-Zellen. Die absolute
Partikelzahl von rIP-P0/CHO ging im Adhäsionsassay auf ca. 24% zurück, im Gegensatz zu
ca. 45% bei den nicht-transfizierten Kontrollzellen und 36% bei rIP1/CHO. Auch in diesem
Fall wurde die Adhäsion von rIP-P0/CHO-Zellen in der mikroskopischen Kontrolle
deutlicher: rIP-P0/CHO-Zellen bildeten während des Adhäsionsassays Zellaggregate von bis
zu 35 Zellen, während sich IP1/CHO-Zellen zu kleineren Aggregaten von 6-10 Zellen
zusammenfanden. Diese Unterschiede im Aggregationsverhalten lassen sich nicht durch die in
einem vergleichenden Zell-ELISA detektierten geringen, aber nicht-signifikanten
Unterschiede im Oberflächenexpressionsniveau (ca. 1,3 fach mehr IP-P0 als IP1) erklären.
Der Unterschied im Adhäsionsverhalten zwischen rIP-P0/CHO und rIP1/CHO ist daher nicht
auf einen Gendosiseffekt, sondern auf die unterschiedlichen cytoplasmatischen Domänen der
Adhäsionsproteine zurückzuführen. Die cytoplasmatische Domäne wirkt offenbar als eine Art
„Membran/Cytoskelettanker“ was somit eine stärkere Adhäsion ermöglichen würde. Der hier
gefundene Einfluß der cytoplasmatischen Domäne auf die Adhäsionseigenschaften des IP-

87
Proteins steht in Übereinstimmung mit Untersuchungen beim P0-Protein. Mutierte bzw.
trunkierte P0 Varianten (Deletion der cytoplasmatischen Domäne) waren im Adhäsionsassay
nicht von untransfizierten Zellen zu unterscheiden (GAO UND FILBIN 1999; WONG UND
FILBIN, 1994). Nicht nur für P0, sondern auch für die Cadherine, eine Gruppe von
Adhäsionsmolekülen, die nicht wie IP und P0 zur Immunglobulin-Superfamilie gezählt
werden, ist bekannt, daß die intakte cytoplasmatische Domäne mit Bestandteilen des
Cytoskeletts interagiert und so essentiell für die Funktion als Adhäsionsmolekül ist
(NAGAFUCHI UND TAKEICHI, 1988; NAGAFUCHI ET AL., 1991; HIRANO ET AL., 1992).
Die adhäsiven Eigenschaften der rIP-P0/CHO-Zellen waren durch Zugabe des αIP1-
Antikörpers, der in dieser Arbeit als spezifischer Antikörper gegen die extrazelluläre Domäne
von IP charakterisiert wurde, vollständig und selektiv zu blockieren. Durch Antikörper
blockierte rIP-P0/CHO-Zellen waren praktisch nicht von untransfizierten CHO-
Kontrollzellen zu unterscheiden, auf die der Antikörper keinen Effekt hatte. Beide
Zellpopulationen bildeten Zellaggregate aus bis zu 5 Zellen, während sich rIP-P0/CHO-
Zellen ohne Antikörperzugabe zu Zellaggregaten aus bis zu 35 Zellen zusammenfanden.
Diese selektive Inhibierung der Adhäsion durch einen spezifischen Antikörper gegen ein
Oberflächenepitop der IP-Proteine unterstreicht die Korrelation zwischen IP-Expression und
Zellaggregation und paßt gut zu vergleichbaren Studien mit dem P0-Protein (ZHANG ET AL.,
1996). Der Antikörper bindet an der extrazellulären Domäne und maskiert sie gegen
Proteininteraktionen. Zusammenfassend zeigen die vorliegenden Untersuchungen, daß die
extrazelluläre Immunglobulin-ähnliche Domäne der IP-Splice-Varianten Zell-Zell-Adhäsion
vermittelt. Die cytoplasmatische Domäne fungiert wahrscheinlich als Membran-, bzw.
Cytoskelettanker und festigt somit die Zell-Zell-Kontakte. IP1/2 ist demnach als Vertreter
der Ig-CAM-Superfamilie ebenfalls als typisches Adhäsionsprotein einzustufen.
Weiterführende Studien mit mehreren verschiedenen Zellklonen stabiler IP2-Expressoren
könnten wichtige neue Einsichten zur Funktion der cytoplasmatischen Domänen der IP-
Splicevarianten ermöglichen. Aufgrund der hohen Sequenzübereinstimmung innerhalb der
cytoplasmatischen Domänen zwischen P0 und IP2, könnte für IP2 ein dem chimären IP-P0-
Fusionsprotein ähnliches Aggregationsverhalten vermutet werden. Besonders interessant
wären auch Untersuchungen zur Adhäsivität von IP1/IP2 Co-Expressoren. Es ist nämlich für

88
das P0-Protein bekannt, daß die Fähigkeit zur Adhäsion durch Co-Expression einer in der
cytoplasmatischen Domäne trunkierten Proteinvariante zusammen mit einem vollständigen
P0-Protein inhibiert wurde. Die trunkierte P0-Variante übt also einen inhibierenden
dominant-negativen Einfluß auf die Adhäsion aus (WONG UND FILBIN 1996). Im
Forellenmyelin hingegen liegen eine „verkürzte“ und eine „vollständige“ IP-Splicevariante in
vivo gemeinsam im kompakten Forellenmyelin vor. Es wäre daher denkbar, daß durch die
Expression zweier putativ unterschiedlich adhäsiver Proteine die Kompaktierung des
Myelinmembran moduliert werden kann. Weiterführende Arbeiten mit entsprechenden
IP1/IP2 Co-Expressoren könnten Einblick in diesen interessanten Aspekt der Myelinogenese
ermöglichen.
Es ist an dieser Stelle interessant zu erwähnen, daß das IP-P0-Fusionsprotein im Westernblot
nur durch die Antikörper αIP1 bzw. 4C5 detektiert werden konnte. Diese Antikörper sind
demnach spezifisch für die extrazelluläre Domäne des IP-Proteins. Überaschenderweise
weicht das apparente Laufverhalten von IP-P0 von seinem kalkulierten Molekulargewicht ab.
Es wurde bereits mehrfach beschrieben, daß P0 äußerst empfindlich auf Proteolyse reagiert
(CAMMER ET AL., 1981; WONG UND FILBIN, 1996). Da die Zellyse von rIP-P0/CHO-Zellen
zur Probenvorbereitung ohne Proteaseinhibitoren durchgeführt wurde, ist es wahrscheinlich,
daß es sich hierbei um ein Proteolyseprodukt handelt. Da diese Proteolyse nur bei lysierten
Zellen auftritt, dürfte sie die Zelladhäsion der vitalen Zelle nicht beeinflussen.
Um weitere Hinweise auf die Funktion der unterschiedlichen cytoplasmatischen IP-Domänen
zu erhalten, wurden sie als Peptidfusionen in E. coli heterolog exprimiert. Hierzu wurden
vergleichend drei verschiedene Expressionssysteme eingesetzt. Es zeigte sich, daß nur das
pMAL-System geeignet war, um ausreichende Mengen der Peptidfragmente präparativ
aufzureinigen. Es konnte ein Protokoll entwickelt werden, welches die Aufreinigung von ca.
2 mg/l Kultur ermöglichte. Neben den IP1/2cyto-Maltosebindeproteinfusionen (MBP) wurde
stets ein ca. 42 kDa Protein co-präpariert, welches in subsidiären Westernblot-Analysen
immuno-negativ blieb, so daß es sich hierbei offensichtlich um degradiertes, nicht-fusioniertes
Maltosebindeprotein handelt. Durch die Westernblot-Analysen wurde auch deutlich, daß der
monoklonale Antikörper 7C4 spezifisch für die cytoplasmatische Domäne der IP1-Splice-

89
Variante ist, während sich der polyklonale Antikörper αIP2 im Westernblot spezifisch für die
cytoplasmatische Domäne der IP2-Isoform erwies. Es waren keine Kreuzreaktionen
detektierbar. Der monoklonale Antikörper 7C4 erkennt also ein Epitop, welches
offensichtlich mit der letzten freien Aminosäure (Glutamin) der IP1-Sequenz korreliert,
während der αIP2-Antikörper Epitope im IP2-spezifischen cytoplasmatischen Proteinbereich
erkennt.
Da sich das Laufverhalten der IP-MBP-Fusionen in einem nativen PAGE nicht von dem
Laufverhalten im SDS-PAGE unterscheidet, ist offensichtlich keine Tendenz zur
Oligomerisierung der cytoplasmatischen Domänen auszumachen. Ein inhibierender Einfluß
des Maltosebindeproteines hierbei ist eher unwahrscheinlich, da mit einem ähnlichen Ansatz
bereits Untersuchungen zur Dimerisierung des Blauzungen-Virus Nucleocapsid Protein VP4
ohne störende Einflüsse des Fusionsanteils durchgeführt werden konnten (RAMADEVI ET AL.,
1998). Für das P0-Protein ist bekannt, daß das Protein in großen Clustern in der
Zellmembran auftritt (D’URSO ET AL., 1990, FILBIN ET AL., 1990). Dieses Clustering ist
essentiell für die Funktion des P0 als Adhäsionsmolekül. In der cytoplasmatischen Domäne
trunkierte P0-Proteine clustern nicht und sind nicht adhäsiv. (WONG UND FILBIN, 1996). Es
ist bislang jedoch nicht bekannt, ob das Clustering von P0, wie von Shapiro et al. (1996)
aufgrund von Kristallstrukturuntersuchungen vorgeschlagen, auf Protein-Protein-
Wechselwirkungen zwischen den extrazellulären Domänen beruht, so daß eine Inhibierung
des Clustering durch eine trunkierte cytoplasmatische Domäne auf sterische Veränderungen
innerhalb des Proteinkomplexes zurückzuführen wäre, oder aber, ob die cytoplasmatische
Domäne selbst Sequenzmotive bzw. Proteinbereiche besitzt, die für die Interaktionen
verantwortlich sind. Für einen Vergleich der Funktionen der cytoplasmatischen Domänen von
IP1/2 und P0 wäre in der Zukunft daher ein oben beschriebenes Experiment mit P0-MBP-
Fusionen äußerst interessant.
Desweiteren wurden erste Experimente zur Detektion von putativen Protein-Protein-
Interaktionen zwischen den cytoplasmatischen Domänen von IP1 und IP2 durchgeführt. Es
war nicht möglich, durch Immobilisierung einer Splice-Variante als Köderprotein die andere
Splice-Variante zu binden und immunologisch zu detektieren. Zwar müssen weiterführende
Studien mit sensitiveren Detektionsmethoden und anderen Puffersystemen, sowie Studien mit

90
nicht-fusionierten IP-Peptidfragmenten abgewartet werden, doch ist vom derzeitigen
Standpunkt aus eine IP1 /IP2-Interaktion auf Seiten der cytoplasmatischen Domäne eher
unwahrscheinlich.
Wahrscheinlicher ist eine Interaktion der cytoplasmatischen Domäne von IP2 mit sauren
Phospholipiden der Zellmembran. Erste experimentelle Untersuchungen mit artifiziellen
Zellmembranen definierter Phospholipidzusammensetzung (Transil), ergaben eine Bindung
von IP2cyto/MBP an eine Oberfläche mit negativ-geladenem Phosphatidylserin, und in
verminderter Form auch an Phophatidylcholin. Aufgrund des stark basischen isoelektrischen
Punktes der IP2-Domäne dürften diese Wechselwirkungen elektrostatischer Natur sein. Der
Einfluß des Maltosebindeproteinanteils ist zur Zeit nicht genau quantifizierbar, doch stehen
die gefundenen Ergebnisse für IP2 in Einklang mit der für P0cyto gefundenen
elektrostatischen Wechselwirkung mit sauren Phospholipiden (DING UND BRUNDEN, 1994).
Es ist daher denkbar, daß auch die cytoplasmatische Domäne von IP2 an der Apposition der
cytoplasmatischen „major dense line beteiligt“ ist. Weiterführende Untersuchung mit
nichtfusionierten IP1 bzw. IP2 Peptidfragmenten wären diesbezüglich von großem Nutzen.
Hierfür könnte ein erst kürzlich speziell zur Expression von kurzen Peptidfragmenten unter
10 kDa entwickeltes heterologes Expressionsystem (Fa. Novagen) besonders hilfreich sein.
Aufgrund der im Rahmen dieser Dissertation neu gewonnenen Erkenntnisse wird folgendes
Modell zur Kompaktierung des Forellenmyelin in Anlehnung an das von Wong und Filbin
1996 für P0 entwickelte Adhäsionsmodell vorgeschlagen: IP1/2 sind typische Vertreter der
Ig-CAM Superfamilie und fungieren als Adhäsionsproteine innerhalb der extrazellulären
„intraperiod line“. Die cytoplasmatische Domäne dient als Membran-, bzw.
Cytoskelettverankerung und beeinflußt so die Adhäsion. Die in der cytoplasmatischen
Domäne längere IP2-Variante, welche in der Embryonalentwicklung der Forelle zuerst
exprimiert wird (JESERICH ET AL., 1990), ermöglicht die initiale starke Adhäsion der
Myelinmembranen. In diesem Entwicklungsstadium ist die Myelinmembran noch recht locker
um das Axon gewunden und das Cytoskelett der Zelle für die Membranproteine zugänglich.
Durch Interaktionen der cytoplasmatischen Domäne von IP2 mit sauren negativ-geladenen
Phospholipiden der Zellmembran und/oder mit dem Cytoskelett wird die Adhäsion gefestigt.
Im Zuge der weiteren Entwicklung kompaktiert das Myelin zunehmend, welches mit der

91
Expression der Myelinproteine 36K und IP1 korreliert. Da im kompakten Myelin kein
Cytoskelett mehr detektierbar ist, wird die gefundene Membranadhäsion durch eine
Cytoskelett unabhängige Adhäsion durch die kürzere IP1 Splice-Variante vermittelt.
In diesem Zusammenhang stellt sich die spannende Frage nach putativen Protein-
Bindungspartnern von IP. So konnte für das P0-Protein durch Co-
Immunpräzipitationsuntersuchungen eine Interaktion mit PMP22 nachgewiesen werden
(D’URSO ET AL., 1999). Für das Forellenmyelin wäre es daher denkbar, daß IP1/2 während
der Myelinogenese mit dem Basischen Myelinprotein und/oder dem 36K-Protein interagieren
und somit die Adhäsion der Myelinmembranen festigen. Darüber hinaus sind im kompakten
adulten Forellenmyelin Interaktionen von IP1/2 mit dem DM20 Protein denkbar. 1996
konnte nämlich von Yoshida und Colman durch immunologische Analysen eine Co-
Lokalisation von DM20 und IP1/2, im kompakten Goldfischmyelin nachgewiesen werden.
Tang et al. (1996) fanden DM20 auch im Hirn der Forelle. Die Funktion des DM20 im
Forellenmyelin ist bislang noch unbekannt, so daß über die Funktion einer denkbaren
IP/DM20 Interaktion nur spekuliert werden kann. Das Expressionsniveau von DM20 ist
während der Myelinogenese jedoch äußerst gering. Erst im jungadulten Zustand fünf Wochen
nach Eischlupf ist eine erhöhte DM20-Expression durch Northernblot-Analysen detektiert
worden. Eine mögliche Funktion der IP/DM20-Interaktion könnte weniger zur Festigung der
Adhäsion während der Myelinogenese, sondern vielmehr zur Modulation der Adhäsion im
kompakten Myelin der Forelle genutzt werden. Co-Immunpräziptationsstudien, Detektion
von Proteininteraktion mittels des Hefe-, oder Säuger-Two-Hybrid-Systems, sowie
Adhäsionsassays von entsprechenden Co-Expressoren wären diesbezüglich sehr hilfreich und
von großem Interesse in der Zukunft.
Im Rahmen der vorliegenden Dissertation konnte die cDNA des zweiten
Hauptmyelinproteins aus dem ZNS der Forelle, dem 36K-Protein kloniert werden. Der
größte offene Leserahmen codiert für ein 320 Aminosäuren großes Protein. Die
untranslatierte 5‘ Region ist auffällig kurz, während die untranslatierte 3‘ Region mit über
1000 bp eine ähnliche Länge wie die 3‘UTR der IP1/2 Splice-Varianten aufweist. Wie bereits
oben für das IP-Protein ausführlich diskutiert, ist die physiologische Funktion der 3‘UTR des
36K-Proteins bisher noch unklar.

92
Vor Beginn der vorliegenden Arbeit waren bereits Teile der zugrundeliegenden 36K-
Genstruktur bekannt (STRELAU, DISSERTATION, 1997). Durch PCR an genomischer Forellen-
DNA konnten die Intron/Exon-Grenzen der zugrundeliegenden Genstruktur im bisher
fehlenden 5‘-Bereich identifiziert werden, so daß nunmehr die vollständige Genstruktur
aufgeklärt ist. Das 5‘ Ende des gDNA-Amplifikates entsprach exakt dem 5‘ Ende des
codierenden Bereiches der in dieser Arbeit isolierten cDNA-Sequenz. Es folgt ein ca. 1500
bp langes Intron I, welches in einen kanonischen 3‘ Splice Akzeptor endet. Strangabwärts
konnten ebenfalls die Intron/Exon-Grenzen exakt festgelegt werden. Alle gefundenen Splice-
Punkte des 36K-Genes unterliegen der GT/AG Regel von Breathnach et al. (1978).
Da die Expression des 36K Proteins nicht nur gewebespezifisch, sondern auch
entwicklungsabhängig reguliert wird (JESERICH ET AL. 1990), könnte die Entschlüsselung der
zugrundeliegenden Promotorregion interessante neue Einblicke in die differentielle
Genexpression ermöglichen. Daher wurde mit Hilfe der inversen PCR ein Genabschnitt
kloniert, der durch Sequenzvergleich mit der isolierten 36K cDNA als putativer Promotor
bzw. 5‘ UTR einzustufen ist. Die Existenz dieses Genabschnittes konnte durch unabhängige
gDNA-PCRs verifiziert werden. Es wurde deutlich, daß sich 15bp strangaufwärts neben dem
durch cDNA-Isolierung identifizierten Start-ATG ein weiteres potentielles Start-ATG im
gleichen Leserahmen befindet. Innerhalb des putativen 36K Promotors konnten TATA-Box-
ähnliche Sequenzmotive gefunden werden. Diese besaßen eine auffällige
Sequenzübereinstimmung mit der für das IP-Gen identifizierten TATA-Box (JESERICH,
STRELAU UND LANWERT, 1997). Die für viele Promotoren typischen TATA-Sequenzen
(Konsensus: TATAA/TAA/T) befinden sich in einem Abstand von annähernd 30 bp vom
Transkriptionsstart entfernt und stellen Bindesequenzen für das TATA-Bindeprotein TBP
bzw. TFIID dar (LATCHMAN, 1995). Alle gefundenen putativen TATA-Boxen sind im
Vergleich zur Konsensussequenz degeneriert, was für Myelinproteine typisch ist (FORS ET
AL.,1993; TAMURA ET AL., 1990; NAVE ET AL., 1991). Da der Transkriptionsstart, der die
Promotorregion und daher auch putative TATA-Boxen und alle weiteren putativen
regulatorischen Sequenzmotive definiert, für die 36K codierende mRNA bislang aber noch
nicht bekannt ist, ist eine weiterführende Interpretation dieser Sequenzmotive zur Zeit nicht
möglich. In weiterführenden Studien nach exakter Identifikation des Transkriptionsstart des
36K-Transkriptes durch Primerextension könnten Sequenzvergleiche mit verschiedenen

93
Myelinproteinpromotoren Aufschluß über etwaige gemeinsame putativ-regulatorische
Sequenzbereiche liefern. Die Transkriptionsaktivität des 36K-Promotors könnte weiterhin
durch progressive Deletionen in Reportergenassays quantifiziert und charakterisiert und so
eventuelle funktionelle und regulatorische Ähnlichkeiten mit dem bereits charakterisierten IP-
Promotor deutlich werden (JESERICH, STRELAU UND LANWERT, 1997).
Anhand der isolierten 36K cDNA konnte ein 320 Aminosäuren großes Protein abgeleitet
werden. Die gefundene Codonpräferenz entsprach der Präferenz von Knochenfischen.
(WADA ET AL., 1991) Das korrespondierende Molekulargewicht entspricht mit 35,7 kDa
annähernd dem apparenten Molekulargewicht des 36K Proteins in der SDS-
Gelelektrophorese (JESERICH, 1983). Der kalkulierte isoelektrische Punkt von pH 9,54 steht
in Übereinstimmung mit früher gewonnenen Resultaten aus 2-D-Gelelektrophorese-Analysen
(JESERICH UND WAEHNELDT, 1986). Es ist an dieser Stelle erwähnenswert, daß Strelau mit
Hilfe von gDNA-Fragmenten eine cDNA-Sequenz vorausgesagt hat, die sich von der in
dieser Arbeit isolierten Sequenz im N-Terminus unterscheidet (STRELAU, DISSERTATION,
1997). Diese unterschiedlichen N-Termini sind auf einen Sequenzierfehler innerhalb der für
die Vorhersage genutzten gDNA-Sequenzen zurückzuführen und haben offensichtlich keine
Auswirkung auf die vorhergesagte Sekundärstruktur des Proteins. Sekundärstrukturanalysen
mit der hier isolierten Sequenz nach dem Chou/Fasman Algorithmus, ergaben eine
charakteristische Abfolge von 6 β-Faltblätter und 6 α-Helices. Innerhalb dieser 6 β-
Faltblätter wurden außerdem Bereiche mit hohem β-Turn-Potential identifiziert. Diese
β6/α6-Struktur ist bereits 1974 von Rossman et al. beschrieben worden. Gestützt durch
Proteindatenbank-Sequenzvergleiche wurde deutlich, daß 36K eine signifikante Ähnlichkeit
zu klassischen Vertretern dieser charakteristisch strukturierten Proteine, den NAD/NADP-
abhängigen Oxido-Reduktasen aufweist. Charakteristisches Merkmal dieser Proteinfamilie ist
eine Konzentrierung der phylogenetisch konservierten Sequenzmotive auf die Dinukleotid-
und der substratbindenden Domäne. Das aktive Zentrum der Enzyme liegt dabei zwischen
beiden Abschnitten und ist äußerst variabel (SCRUTTON ET AL., 1990; PERSON ET AL., 1991).
Das 36K-Protein unterscheidet sich von allen bislang gefundenen Myelinproteinen, was gut
zu früheren Untersuchungen paßt, in denen der 36K-Antikörper keinerlei Kreuzreaktionen
gegen Myelinproteine der Säuger, Vögel, Reptilien und Amphibien zeigte (WAEHNELDT ET
AL., 1986B). Überraschenderweise ähnelt das 36K-Protein der Protochlorophyllid-Reduktase

94
aus der Thylakoidmembran der Erbse (STRELAU, DISSERTATION 1997). Die Gruppe der
Oxido-Reduktasen ist in Bezug auf ihre Funktionen äußerst heterogen (LABESSE ET AL.,
1994). Sie reichen von der Reduktion verschiedenster Carbonylverbindungen über die
Inaktivierung von Steroiden bis hin zu wichtigen Funktionen bei der Synthese von
Antibiotika (ALBALAT ET AL., 1992; CHEN ET AL., 1993; BAKER, 1994). Dieses weite
Aktivitätsspektrum und die großen Sequenzunterschiede im katalytischen Bereich der
Enzyme machen eine funktionelle Interpretation äußerst schwierig. Über eine mögliche
enzymatische Funktion der einzigartigen Myelinkomponente 36K kann daher bislang nur
spekuliert werden. Eine für das Myelinprotein 36K sehr naheliegende putative Funktion ist
jedoch, daß es aufgrund seines kalkulierten isoelektrischen Punktes von pH 9,54 wie oben
bereits detailliert für die cytoplasmatischen Domänen von IP beschrieben, an der Apposition
der „major dense line“ durch elektrostatische Wechselwirkung mit sauren
Membrankomponenten beteiligt ist. Durch eine NAD-, bzw. NADP-Abhängigkeit wäre eine
regulatorische Modulation dieser Apposition denkbar.
Um erste Hinweise auf die Funktion des 36K-Proteines zu erhalten wurde im Rahmen dieser
Dissertation verschiedene Ansätze zur heterologen Expression in Eukaryoten wie in
Prokaryoten vergleichend eingesetzt. Permeabilisierte CHO-Zellen zeigten 48h nach
Transfektion eine starke granuläre Immunfärbung im Bereich des Cytoplasmas. Durch
Oberflächenmakierung konnte keine 36K-Expression detektiert werden, was den
cytoplasmatischen Charakter des 36K-Proteines unterstreicht. Im Westernblot wies das
heterolog exprimierte 36K-Protein im Vergleich zur native Form aus dem Forellenmyelin ein
etwa 0,5 bis 1 kDa kleineres apparentes Molekulargewicht auf. Bei der im Rahmen dieser
Arbeit isolierten 36K-cDNA handelt es sich offenbar um eine nicht ganz vollständige
Sequenz. Wie bereits oben im Zuge der Promotorklonierung beschrieben, befindet sich auf
der zugrundeliegenden Genstruktur strangaufwärts vom 5‘ Ende der klonierten cDNA-
Sequenz ein weiteres putatives Start-ATG im gleichen Leserahmen. Es ist daher sehr
wahrscheinlich, daß die vollständige codierende Region der 36K-cDNA am N-Terminus um
5 Aminosäuren länger ist als der isolierte Klon, was die Diskrepanz im Laufverhalten erklären
würde. Die Existenz eines entsprechenden Transkriptes konnte durch unabhängige RT-PCR-
Analysen bereits verifiziert werden. Diese N-terminale „Verlängerung“ hat keinen Einfluß auf
die vorhergesagte Sekundärstruktur des 36K-Proteines. Leider sind alle Versuche stabile

95
r36K/CHO-Expressoren zu klonieren bislang erfolglos geblieben, so daß ein möglicher
Einfluß auf das Aggregationsverhalten der Zellen im Adhäsionsassay nicht untersucht werden
konnte. Weiterführende Studien, vorallem Co-Expressoren mit IP1 bzw. IP2 könnten
interessante neue Hinweise auf die putative Funktion des Proteines ermöglichen.
Weiterhin wurde das 36K Protein als His-Tag-Fusion (36KHT) in einem E. coli und einem
Baculovirus-System exprimiert und präparativ aufgereinigt. Es zeigte sich, daß das
bakterielle System weitaus höhere Proteinausbeuten ermöglichte als das Baculovirus-System,
und ist daher zur Produktion von rekombinantem Protein im großen Maßstab gut geeignet.
36K wird in E. coli hauptsächlich in Einschlußkörpern gebildet, so daß eine Aufreinigung nur
unter denaturierenden Bedingungen in Gegenwart von chaotropen Salzen wie 6M Harnstoff
bzw. Guanidin-Hydrochlorid möglich war. Das elektrophoretische Laufverhalten des
präparierten 36KHT-Proteins weicht von dem des „nativen“ 36K aus dem Myelin der Forelle
aufgrund der „His-Tag“ Erweiterung durch zusätzliche Aminosäuren, davon 10 geladene
Histidine, in der erwarteten Weise ab. Interessanterweise war auch in dem Baculovirus-
System eine Aufreinigung nur im Gegenwart von chaotropen Salzen möglich. Da 36KHT
unter nativen Bedingungen stets zusammen mit unlöslichen Bestandteilen der Wirtszelle
durch Zentrifugation pelletiert wurde, könnte dieses ein erster Hinweis auf eine Membran-,
bzw. Cytoskelettassoziation sein, was jedoch in weiterführenden Untersuchungen geklärt
werden müßte. Im Rahmen der vorliegenden Dissertation ist erstmals eine Methode etabliert
worden, die die Überexpression und präparative Aufreinigung eines Forellenmyelinproteins
ermöglichte. Diese erarbeitete methodische Basis dürfte vielfältige weiterführende Studien
zur funktionellen Bedeutung des Proteines ermöglichen. Es wären z.B. denkbar mit
renaturiertem 36KHT-Protein Bindestudien mit artifiziellen Lipidmembranen, wie oben für
die cytoplasmatische Domäne von IP2 beschrieben, durchzuführen. Darüberhinaus könnten
mit Hilfe der Circulardichroismus (CD)-Spektroskopie Aussagen über die Sekundärstruktur
des renaturierten Proteins möglich sein. Hierbei wäre es von besonderem Interesse, ob eine
putative Konformationsänderung des 36K-Protein aufgrund von Interaktionen mit
Lipidvesikeln, ähnlich wie für das Basische Myelinprotein beschrieben, mit Hilfe der CD-
Spektren detektierbar wäre (KENIRY UND SMITH, 1978, CHEIFETZ ET AL. 1985). Die erzielten
Ausbeuten der heterologen Expression von 36KHT in E. coli würden es eventuell erlauben,
das Protein zu kristallisieren und seine Struktur durch Röntgenstrukturanalyse aufzuklären.

96

97
5 Zusammenfassung
Im Rahmen der vorliegenden Dissertation konnte die cDNA der IP2 Splice-Variante
vervollständigt werden. Es zeigte sich, daß der codierende Bereich exakt mit der IP1-
Splicevariante übereinstimmte, die cytoplasmatische Domäne ist jedoch um 90 bp verlängert.
Anschließend folgt eine IP2-spezifische 3‘ UTR von 133 bp Länge, die in eine beiden IP-
Splicevariante identische 3‘UTR mündet. Da den beiden IP-Isoformen der gleiche Promotor
zugrundeliegt, beide Proteine während der Myelinogenese jedoch unterschiedlich d.h.
entwicklungsabhängig exprimiert werden, ist ein Einfluß der 3‘ UTR auf die Regulation des
alternativen Splicings denkbar.
Die translatierte Aminosäuresequenz der verlängerten cytoplasmatischen Domäne der IP2-
Isoform wies, im Gegensatz zur IP1cyto-Domäne, eine auffällige Sequenzübereinstimmung
mit der cytoplasmatischen Domäne des P0-Proteins auf. Phylogenetisch konservierte
Aminosäuren sind meist basisch. Auch zwei PKC-Kinase-Stellen sind zwischen allen Spezies
konserviert, so daß man in diesem Bereich wichtige funktionelle Bereiche vermuten kann.
Beide IP Splice-Varianten konnten in CHO-Zellen exprimiert und durch Immunfluoreszenz,
sowie durch Westernblot detektiert werden. IP1/2 werden auch in der heterologen Wirtszelle
glykosiliert und in die Zellmembran integriert.
Es konnten ein Adhäsionsassay in einem mimetischen Zellsystem etabliert werden, mit dem
die adhäsive Funktion der extrazellulären Domäne der IP-Proteine charakterisiert werden
konnte. Nach Klonierung eines stabil-exprimierenden rIP1/CHO-Zellklones zeigte sich, daß
IP1 eine Adhäsion vermittelt, die zur Aggregation von bis zu zehn rIP1/CHO-Zellen führte.
Um zu untersuchen, ob die cytoplasmatische Domäne Einfluß auf die Adhäsion ausübt,
wurde eine Chimäre aus der extrazellulären und transmembranellen Domäne des IP-Proteins
und der P0cyto Domäne konstruiert und exprimiert. Stabile rIP-P0/CHO-Zellen waren im
Adhäsionsassay deutlich quantifizierbar adhäsiver. Sie bildeten Zellaggregate aus bis zu 35
Zellen. Die cytoplasmatische Domäne hat offensichtlich einen Einfluß auf die Adhäsion. Die
geringen, aber nicht-signifikanten Unterschiede im Oberflächenexpressionsniveau zwischen
rIP/CHO und rIP-P0/CHO-Zellen können die Unterschiede im Adhäsionsverhalten nicht
erklären. Die cytoplasmatischen Domänen von IP1 und vorallem von IP2 sind stark basisch,

98
d.h. positiv geladen und könnten somit durch Bindung an saure, d.h. negativ geladene
Phospholipide der Zellmembran die Apposition der cytoplasmatischen „major dense line“
unterstützen. Durch die Zugabe eines spezifischen Antikörpers gegen die extrazelluläre
Domäne von IP konnte die Adhäsion von rIP-P0/CHO-Zellen vollständig inhibiert werden.
IP ist daher als typisches Adhäsionsprotein der Immunglobulin-Superfamilie einzustufen. Die
cytoplasmatische Domäne hat Einfluß auf die Adhäsion des Proteins.
Die cytoplasmatischen Domänen von IP1/2 konnten in E. coli als Peptidfragmente heterolog
exprimiert und aufgereinigt werden. Es zeigte sich, daß hierzu nur ein von drei getesteten
Expressionssytemen geeignet war. Mit der aufgereinigten IP2cyto/MBP-Fusion konnten
Bindestudien mit artifiziellen Lipidoberflächen durchgeführt werden. Zwar ist der Einfluß des
zur Aufreinigung benötigtem MBP-Fusionsanteils nicht genau quantifizierbar, doch konnten
erste experimentelle Hinweise auf eine Bindung von IP2cyto an negativ geladene
Phophatidylserin-haltige Lipidoberflächen gewonnen werden, die die obige Theorie stützen.
Eine Protein-Protein Interaktion zwischen den IP1cyto-, und IP2cyto-Domänen ist zum
derzeitigen Standpunkt eher unwahrscheinlich.
Darüber hinaus konnte die cDNA des 36K-Proteines isoliert, der korrespondierende putative
Promotor bzw. 5‘ UTR durch invers PCR kloniert, sowie die Intron/Exon-Grenzen eindeutig
kartiert werden. Somit ist die vollständige Struktur des 36K-Genes aufgeklärt. Innerhalb des
putativen 36K-Promotors konnten putative TATA-Boxen identifiziert werden, die auffällige
Ähnlichkeit zur TATA-Box des IP-Genes aufwiesen.
Das 36K-Protein besitzt keine Ähnlichkeit mit Myelinproteinen, sondern ähnelt den
NAD/NADP-abhängigen Oxido-Reduktasen. Seine Funktion ist bislang noch unklar, doch
aufgrund seines basisch-positiven Charakters könnte es wie oben beschrieben als
Membrananker bei der Apposition der cytoplasmatischen „major dense line“ beteiligt sein.
36K konnte als His-Tag-Fusion sowohl mit Hilfe eines Baculoviren-Systems als auch mit
einem von drei getesteten E. coli Systemen exprimiert und präparativ aufgereinigt werden.
Weiterführende Studien mit artifiziellen Lipidoberflächen, sowie spektroskopische Ansätze
könnten neue Einblicke in die Funktion und Struktur des Proteins ermöglichen.

99
6 Literatur
• Afar DE, Marius RM, Salzer JL, Stanners CP, Braun PE, Bell JC (1991): „Celladhesion properties of myelin-associated gylcoprotein in L cell fibroblasts“, JNeurosci Res 29:429-436
• Albalat R, Gonzales-Duarte, Atrian S (1992): „Protein engineering of Drosophilaalcohol dehydrogenase. The hydoxyl group of Tyr 152 is involved in the active site ofthe enzyme“ FEBS 308(3):235-239
• Altschul, Stephen F., Thomas L. Madden, Alejandro A. Schäffer, Jinghui Zhang,Zheng Zhang, Webb Miller, and David J. Lipman (1997), „Gapped BLAST andPSI-BLAST: a new generation of protein database search programs“, NucleicAcids Res. 25:3389-3402.
• Baker ME (1994): „Protochlorophyllide reductase is homologous to humancarbonyl and pig 20β-hydroxysteroid dehydrogenase“ J Biochem 300:605-607
• Barbarese E, Braun PE, Carson JH (1977): „Identification of prelarge and presmallbasic proteins in mouse myelin and their structural relationship to small and largebasic proteins.“ Proc Natl Acad Sci USA 74: 3360-3364
• Bastmeyer M, Beckmann M, Schwab ME und Stuermer CAO (1991): „ Growth ofregenerating goldfish axons is inhibited by rat oligodendrocytes and CNS myelin butnot goldfish optic nerve trct oligodendrocyte-like cells and fish CNS myelin“ JNeurosc 11: 626-640
• Behr JP, Demeneix B, Loeffler JP, Perez-Mutul J (1989): „ Efficent gene transferinto mammalian primary endocrine cells with lipopolyamine-coated DNA.“ ProcNatl Acad Sci USA 86:6982-6986
• Bergoffen J, Scherer SS, Wang S, Oronzi Scott M, Bone LJ, Paul DL et al. (1993):„Connexin mutations in X-linked Charcot-Marie-Tooth disease.“ Science 262:2039-2042
• Boccaccio GL, Carminatti H, Colman DR (1999): „Subcellular fractionation andassociation with the cytoskeleton of messengers encoding myelin proteins“ JNeurosci Res 58(4):480-91
• Bollensen E, Schachner M (1987): „The peripheral myelin glycoprotein expressesthe L2/HNK-1 and L3 carbohydrate structurs shared by neural cell adhesionmolecules“, Neurosci Lett 82:77-82
• Bosse F, Brodbeck J, Müller HW (1999). „Nach-transcriptional regulation of theperipheral myelin protein gene PMP22/Gas3“ J Neursci Res 55:164-177

100
• Bradford, M.M. (1976): „A rapid and sensitive method for the quantification ofmicrogram quantities of protein utilizing the principle of protein-dye binding.“ Anal.Biochem. 72: 248-254.
• Braun PE (1984) „Molecular organization of myelin“ in Myelin (ed P. Morell)Plenum, New York, pp. 97-116
• Breathnach R, Benoist C, O’Hare K, Gannon F, Chambon P (1978): Ovalbumingene: evidence for a leader sequence in mRNA and DNA sequences at the exon-intron boundaries“ Proc Natl Acad Sci USA 75:4853-4857
• Bunge RP (1968): „Glial cells and the central myelin sheath“ Phyiol Rev 48: 197-251
• Cammer W, Brosnan CF, Bloom BR, Norton WT (1981): „Degradation of the P0,P1 and Pr proteins in the peripheral nervous system myelin by plasmin: implicationsregrding the role of macrophages in demyelination disease“ J Neurochem 36:1506-1516
• Campagnoni AT (1988): „Molecular Biology of myelin proteins from the CNS.“ JNeurochem 51:2129-2138
• Carson JH, Kwon S, Barbarese E (1998): „RNA trafficking in myelinating cells“Curr Op Neurobiol 8:607-612
• Cheifetz S, Boggs JM, Moscarello MA (1985): „Increase in vesicle permeabilitymediated by myelin basic protein: Effect of phosphorylation of basic protein“Biochemistry 24:5170-75
• Chen C, Okayama H (1987): „High efficency transformation of mamalian cell byplasmid DNA“ Mol Cell Biol 7:2745-2752
• Chou PY, Fasman GD (1978): „Rediction of the secondary structure of proteinsfrom their amino acid sequence“ Adv. Enzymol 47:45-148
• Chung CT, Niemela SL, Miller RH (1989). „ One-step preparation of competent E.coli : Transformation and storage of bacterial cells in the same solution“ Proc NatlAcad Sci USA 86:2172-2175
• Colman DR, Kreibich G, Frey AB, Sabatini DD (1982): „Synthesis andincorporation of myelin polypeptides into CNS myelin“ J Cell Biol 95:598-608
• D’Urso D, Brophy PJ, Staugiatis SM, Gillespie CS, Frey AB, Stempak JG, ColmanDR (1990): „Protein Zero of peripheral nerve myelin: biosythesis, membraneinsertion and evidence for homotypic interaction.“ Neuron 2: 449-460
• D’Urso D, Ehrhardt P, Müller HW (1999): „Peripheral Myelin Protein 22 andProtein Zero: a novel association in peripheral nervous system myelin“ J Neurosci19(9):3396-3403

101
• Ding Y, Brunden KR (1994): „The cytoplasmatic domain of myelin glycoprotein P0interacts with negative charged phospholipid bilayers.“ J Biol Chem 269: 10764-10770
• Doherty P, Fruns M, Seaton P, Dickson G, Barton CH, Sears TA, Walsh FS(1990): „N-CAM and neurite outgrowth: evidence for a threshold effect and theefficacy of the major isoforms“ Nature 343:464-466
• Edelman GM (1987): „CAMs and Igs: Cell adhesionand the evolutionary origins ofimmunity“ Immunol Rev 100:11-45
• Erlich HA, Gelfand O, Sninsky JJ (1991): „Recent advances in the polymerase chainreaction“ Science 252: 1643-1651
• Filbin MT, Tennekoon GI (1991): „The role of complex carbohydrates in adhesionof the myelin protein P0“ Neuron 7:845-855
• Filbin MT, Tennekoon GI (1993): „Both P0-Proteins must be glycosylated forhomophilic adhesion.“ J Cell Biol 122:451-459
• Filbin MT, Walsh FS, Trapp BD, Pizzey JA, Tennekoon GI (1990): „Role of myelinP0-protein as a homophilic adhesion molecule.“ Nature 344: 871-872
• Foecking MK, Hofstetter (1986): „Powerful and versatile enhancer-promotor unitfor mammalian expression vectors.“ Gene 45:101-105
• Fors L, Hood L, Saavedra RA, (1993): „Sequence similarities of Myelin BasicProtein promotors from mouse and shark: Implications for the control of geneexpression in myelinating cells“ J Neurochem 60(2):513-521
• Gao Y, Filbin MT (1999): „Characterization of the palmitoylation of myelin P0protein“ Nacher Presentaion on ISN Satelltie-Meeting „Myelination and MyelinDisorders: NewVistas“ 1999
• Gorman CM, Howard BH, (1983): „Expression of recombinant plasmids isenhanced by sodium butyrate“ Nucl Acid Res 11: 7631-7648
• Gottlieb E (1992): „The 3‘ UTR of localized maternal messages contains aconserved motif involved in mRNA localization.“ Proc Natl Acad Sci USA89:7164-7168
• Greenfield S, Brostoff S, Eylar EH, Morell P (1973): „Protein composition ofmyelin of the peripheral nervous system.“ J Neuro Chem 20: 1207-1216
• Greenfield S, Weise MJ, Gantt G, Hogan EL, Brostoff SW (1982): „Basic Proteinsof rodent peripheral nerve myelin: immunochemical identification of the 21,5K,18,5K, 17K, 14K and P2 proteins.“ J Neuro Chem 39:1278-1282

102
• Griffiths LS, Schmitz B, Schachner M (1992): „L2/HNK-1 carbohydrate andprotein-protein interactions mediate the homophilic binding of the neural adhesionmolecule P0“ J Neurosci Res 33:639-648
• Hames BD, Glover DM (eds.)(1988): in „Transcription & splicing“ pp: 111-113IRL Press Oxford-Washington
• Hammer JA, Oshannessy DJ, Deleon M, Gould R, Zand D, Daune G, Quarles RH(1993): „Immunoreactivity of PMP-22, P0 and other 19 to 28 kDa glycoproteins inperipheral nerve myelin of mammals and fish with HNK-1 and related antibodies“ JNeurosci Res 35:546-558
• Hanahan, D. (1983): „Studies of transformation of Escherichia coli with plasmids“.J. Mol. Biol. 166: 557-580.
• Hayasaka K, Nanao K, Tahara M, Sato W, Takada G, Miura M, Uyemura K(1991): „Isolationa nd sequence determination of cDNA encoding the majorstructural protein of human peripheral myelin“ Biochem Biophys Res Commun180(2):515-518
• Herdandez M-C, Erkmann L, Matter-Sadzinski L., Roztocil T; Ballivet M, Matter J-M (1995): „Characterization of the nicotinic acetylcholine receptor β3 gene: istregulation within the avian nervous system is affected by a promotor 143 bp inlength.“ J Biol Chem 270:3224-3233
• Hildebrand C, Remahl S, Persson H, Bjartmar C (1993): „Myelinatednerve fibers inthe CNS“ Prog. Neurobiol.40: 319-384
• Hirano S, Kimoto N, Shimoyama Y, Hirohashi S, Takeishi M (1992): „Identificationod a neural α-catenin as a key regulator of cadherin function and multicellularorganization“ Cell 70:293-301
• Holmes TC, Fadool DA, Levitan IB (1996): „ Tyrosine phosporylation of the Kv1.3potassium channel.“ J Neurosci 16(5):1581-1590
• Jeserich G (1983): „Protein analysis of myelin isolated from the CNS of fish:developmental and species comparisons“ Neurochem Res 8:957-969
• Jeserich G, Müller A, Jacque C (1990): „Developmental expression of myelinproteins by oligodendrocytes in the CNS of trout.“ Dev Brain Res 51: 27-34
• Jeserich G, Stratmann A (1995): „Molecular clonig and tissue expression of acDNA encoding IP1 - a P0-like glycoprotein of trout CNS myelin.“ J Neurochem64:2427-2436
• Jeserich G, Strelau J, Lanwert C (1997): „Partial characterization of the 5‘-flankingregion of trout IP: a P0-like gene containing a PLP-like promotor“ J Neurosci Res50:1-10

103
• Jeserich G, Waehneldt TV (1986a): „Bony fish myelin: evidence for common majorstructural glycoproteins in central and peripheral myelin of trout.“ J Neurochem46:525-533
• Jeserich G, Waehneldt TV (1986b): „Characterization of antibodies against majorfish CNS myelin proteins: immunoblot analysis immunohistochemical loccalizationof 36K and IP2 proteins in trout nerve tissue.“ J Neurosci Res 15:147-158
• Jeserich G, Waehneldt TV (1987): „Antigenic sites common to major fish myelinglycoproteins (IP) and to major tetrapod PNS myelin glycoprotein P0 reside in theamino acid chains.“ Neurochem Res 12:821-825
• Kamholz J., Wrabetz L (1992): „Molecular genetics of myelin basic protein“ In:Biology and Chemistry (ed. R.E. Martenson) pp. 367-385, Boca Raton: CRC Press
• Kaufman RJ, Brown PC, Schimke RT (1979): „Amplified Dihydrofolate ReductaseGenes in unstably methotrexate-resistent cells are associated with double minutechromosomes“ Proc Natl Acad Sci 76:5669-5673
• Keniry MA, Smith R (1978): „Circular dichroic analysis of the secondary structureof myelin basic protein and derived peptides bound to detergents and to lipidvesicles“ BBA 578:381-391
• Kozak M (1984): „Compilation and analysis of sequenzes upstream from thetranslational start site in eukaryotic mRNA’s.“ Nucl Acids Res 12: 857-872
• Labesse G, Vidal-Cros A, Chomilier J, Gaudry M, Mornon JP (1994): structuralcomparisons lead to the definition of a new superfamily of NAD(P)(H)-acceptingoxidoreductases: the single-domain reductases/epimerases/dehydrogenases (the„RED“ family) Biochem J 304:95-99
• Laemmli, K. (1970): „Cleavage of structural proteins during the assembly of thehead of bacteriophage T4“ Nature 227, 680-685
• Lai C, Brow MA, Nave KA, Noronha AB, Quarles RH, Bloom FE et al. (1987):„Two forms of 1B236/myelin-associated glycoprotein, a cell adhesion molecules fornachnatal neural development, are produced by alternative splicing.“ Proc Nat AcSci 84:4337-4341
• Lai C, Watson JB, Bloom FE, Sutcliff JG, Milner RJ (1987): „Neural protein1B236/myelin associates glycoprotein (MAG) defines a subgroup of theimmunoglobulin superfamily.“ Immunol. Rev. 100: 129
• Lanwert C (1996): „Untersuchungen zur Expression von Myelinproteingenen derRegenbogenforelle in OLN 93 Zellen der Ratte“ Diplomarbeit UniversitätOsnabrück
• Latchman DS (1995): „Gene Regulation. A eukaryotic perspektive „ (Secondedition) publ. Chapman & Hall, 2-6 Boundary Row, London SE1 8HN, UK

104
• Laursen RA, Samiullah A, Lees MB (1984): „The structure of bovine brain myelinproteolipid and ist organization in myelin“ Proc Natl Acad Sci USA 81:2912-2916
• Lees MB, Brostoff SW (1984): „Proteins of myelin“ in: Myelin 2 nd ed. (ed. P.Morell), pp. 197-224. New York: Plenum Press
• Lemke G (1988): „Unwrapping the genes of myelin“ Neuron 1: 535-543
• Lemke G, Axel R (1985): „Isolation and sequence of a cDNA encoding the majorstructural protein of peripheral myelin“ Cell 40: 501-508
• Lemke G, Axel R (1985): „Isolation and sequence of a cDNA encoding the majorstructural protein of peripheral myelin.“ Cell 40: 501-508
• Little EB, Edelman GM, cunningham BA (1998): „Palmitoylation of thecytoplasmic domain of the Neural Cell Adhesion Moelcule N-CAM serves as ananchor to cellular membranes“ Cell Adhesion Comm 6(5):415-430
• Loeffler JP, Barthel F, Feltz P, Behr JP, Sassone-Corsi P, Feltz A (1990):„Lipopolyamine-mediated transfection allows gene expression studies in primaryneuronal cells.“ J Neurochem 54:1812-1815
• Lubon et al (1989): „Cell specific activity of the modulator region in the humancytomegalovirus immediately early gene.“ Mol Cell Biol 9:1342-1345
• McCutchan JH, Pagano JS (1968): „Enhancement of the infectivity of simian virus40 deoxyribonucleic acid with diethylaminoethyl-dextran“ J Nat Cancer Inst 41:351
• Milner RJ, Sutcliff JG (1983): „ Gene expression in rat brain.“ Nucl Acids Res11:5497-5520
• Montoya-Zavala M, Hamlin JL (1985): „Similar 150 kb DNA sequences areamplified in independently derived methotrexate resistent Chinese Hamster cells“Mol Cell Biol 5:619-627
• Morell P (1977): in „Myelin“, Plenum Press, New York
• Morell P, Norton WT (1980): „Myelin“ Sci Am 242: 88-118
• Mukhopadhay A (1997), Adv Biochem Eng Biotechnol 56:61-109
• Nagafuchi A und Takeichi M (1988): „ Cell binding function of E-cadherin isregulated by the cytoplasmic domain“ EMBO J 7:3679-3684
• Nagafuchi A, Takeichi M, Tsukita S (1991): „ The 102 kDa cadherin-associatedprotein: similarity to vinculin and nachtranscriptional regulation of expression“ Cell65:849-857

105
• Nave KA, Lemke G (1991): „Induction of the myelin proteolipid protein (PLP) genein C6 Glioblastoma cells: Functional analysis of the PLP-promotor“ J Neurosci11(10):3060-3069
• O’Mahoney JV, Adams TE (1994): „Opimization of experimental variablesinfluencing reporter gene expressoin in heptoma cells following calcium phosphatetransfection.“ DNA and Cell Biol 13 (12):1227-1232
• Ochman H, Gerber AS, Hartl DL (1988): „Genetic applications of an inversepolymerase chain reaction“ Genetics 120(3):621-623
• Pareek S, Suter U, Snipes GJ, Welcher AA, Shooter EM, Murphy RA (1993):„Detection aund processing of peripheral myelin protein PMP22 in culturedSchwann cells.“ J Biol Chem 268: 10372-10379
• Persson B, Krook M, Jörnvall H (1991): „Characteristics of short-chain alcoholdehydrodenases andrelated enzymes“ Eur J Biochem 200:537-543
• Peters A, Palay S, Webster HdeF (1976): „The fine structure of the nervoussystem.“ WB Saunders, Philadelphia
• Poltorak M, Sadoul R, Keilhauer G Landa C Fahrig T, Schachner M (1987):„Myelin-associated glyoprotein, a member of the L2/HNK-1 family of neural celladhesion molecules, is involved in neuron-oligodendrocyte interaction“ J Cell Biol105:1893-1899
• Popot JL, Pham-Dinh D, Dautigny A (1991): „Major myelin proteolipid protein: the4-alpha-helix-topology.“ J Mem Biol 120: 233-246
• Proudfoot NJ, Brownlee GG (1976): „3‘ noncoding region sequences in eukaryoticmessenger RNA“ Nature 263:211-214
• Ramadevi N, Rodriguez J, Roy P (1998): „A leucine zipper-like domain is essentialfor dimerization and encapsidation of Bluetongue Virus Nucleocapsid Protein VP4“J Virol 72(4):2983-90
• Richter-Landsberg C, Heinrich M (1996): „OLN-93: A new permanentoligodendroglia cell line derived from primary rat brain gilal cultures“ J NeurosciRes 45:161-173
• Rossman MG, Moras D, Olsen KW (1974): „Chemical and biological evolution of anucleotide-binding protein“ Nature 250: 194-199
• Roux M, Nezil FA, Monck M, Bloom M (1994): „Fragmentation of phospholipidbilayers by myelin basic protein“ Biochem 33:307-311
• Rumsby MG, Crang AJ (1977): „The myelin sheath: a structural examination in thesynthesis, assembly and turnover oc Cel surface components.“ (Nache G, Nicolson

106
GL eds.), North-Holland, Amsterdam, pp 247-362
• Saavedra RA, Fors L, Aebersold RH, Arden B, Horvath S, Sanders J, Hood L(1989): „The myelin proteins of the shark brain are similar to the myelin proteins ofthe mammalian peripheral nervous system“ J Mol Evol 29:149-156
• Sabbioni S, Negrini M, Rimessi P, Manservigi R, Barbanti-Brodano G (1995): „ABK virus episomal vector for constitutive high expression of exogenous cDNA’s inhuman cells“ Arch. Virol. 140:335-339
• Saiki RK, Gelfand DH, Stoffel S, Scharf SJ, Higuchi R, Horn GT, Mullis KB ErlichHA (1988): „Primer-directed enzymatic amplification of DNA with a thermostableDNA Polymerase“ Science 239:487-491
• Salzer JL, Colman DR (1989): „Mechanisms of cell adhesion in the nervous system:role of the immunoglobulin gene superfamily“ Dev Neurosci 11:377-390
• Salzer JL, Holmes WP, Colman DR (1987): „The amino acid sequences of theMyelin-associated Glycoproteins: Homology to the Immunoglobulin GeneSuperfamily“ J Cell Biol 104:957-965
• Sambrook, J., Fritsch, E.F. & Maniatis, T. (1989) Molecular cloning: a laboratorymanual, Cold Spring Harbor Laboratory, Cold Spring Harbor, New York.
• Sanger, F., Nicklen, S. & Coulsen, A.R. (1977): „DNA sequencing with chainterminating inhibitors.“ Proc. Natl. Acad. Sci. USA 74: 5463-5467.
• Schneider-Schaulies J, von Brunn A, Schachner M (1990): „Recombinant peripheralmyelin protein P0 confers both adhesion and neurite outgrowth-promotingproperties.“ J Neurosci Res 27:286-297
• Scrutton NS, Berry A, Perham RN (1990): „Redesign of the coenzyme specificity ofa dehydrogenase by protein engineering“ Nature 343:38-43
• Shapiro L, Doyle JP, Hensley P, Colman DR, Hendrickson WA (1996): „ Crystalstructure of the extracellular domain fron P0, the major structural protein ofperipheral nerve myelin“ Neuron 17:435-449
• Shaw G, Kamen R (1986): „A conserved AU sequence from the 3‘ untranslatedregion of GM-CSF mRNA mediates selektive mRNA degradation“ Cell 46:659
• Singer M und Berg P (1992): „Gene und Genome“ Spektrum Akademischer Verlag;Heidelberg, Berlin, New York
• Snipes GJ. und Suter U (1995): „Molecular anatomy and genetics of myelin proteinsin the peripheral nervous system (review).“ J. Anat. 186:483-494
• Staugaitis SM, Colman DR, Pedraza L (1995): „Membrane adhesion and otherfunctions for the myelin basic proteins) BioEssays 18(1):13-18

107
• Stoffel W, Hillen H, Giersiefen R (1984): „Structure and molecular arrangement ofproteolipd protein of central nervous system myelin“ Proc Natl Acad Sci USA81:5012-5016
• Stojdl DF, Bell JC (1999): „SR protein kinases: the splice of life“ Biochem Cell Biol77(4):293-298
• Stratmann (1995): „ Molekulare Charakterisierung von Myelinproteinen aus demZNS der Regenbogenforelle Oncorhynchus mykiss.“ Dissertation Uni Osnabrück
• Stratmann A, Jeserich G (1995): „Molecular clonig and tissue expression of acDNA encoding IP1 - a P0-like glycoprotein of trout CNS myelin.“ J Neurochem64:2427-2436
• Strelau J (1997): „Molekulare Charakterisierung von Promotoren Myelinprotein-kodierender Gene aus dem ZNS der Regenbogenforelle (Oncorhynchus mykiss),Dissertation, Universität Osnabrück
• Strotmann: (1988): „ Produktion und Charakterisierung monoklonaler Antikörpergegen die Hauptmyelinglykoproteine IP1 und IP2 aus dem ZNS derRegenbogenforelle Salmo gairdneri, Rich.“ Diplomarbeit Uni Osnabrück
• Takamura T, Sumita K, Hirose S, Mikoshiba K (1990): „Core promotor of themouse myelin basic protein gene governs brain-specific transcription in vitro“ EmboJ 9(10):3101-3108
• Takei K, Kitamura K, Banno K, Uyemura K (1993): „Major glycoproteins in carpCNS myelin: Homology to P0 protein with HNK-1/L2 carbohydrate epitope“Neurochem Int 23:239-248
• Tang S, Panno JP, McKeown BA (1996): „Cloning and expression of theproteolipid DM20 cDNA from the brain of the rainbow trout, oncorhynchusmykiss“ Mol Brain Res 41:134-139
• Thompson SH, Cass KH, Stellwagen E (1975): „ Blue dextran-sepharose: anaffinity column for the dinucleotide fold in proteins“ Proc Natl Acad Sci USA72(2):669-672
• Ueno S, Handley VW, Byravan S, Campagnoni AT (1994): „ Structural features ofmyelin basic protein messenger RNAs influence their translational efficiencies.“ JNeurochem 62:1254-1259
• Vernier JM (1969): „Table chronologique du developpement embryonaire de latruite arc-en-ciel Salmo gairdneri, Rich. 1836“ Ann. Embryol Morphogent 2:495-520
• Vogel US, Thompson RJ (1988): „Molecular structure, localisation and posiiblefunctions of the myelin-associated enzyme 2’3’-cyclic nucleotide 3’-phosphodiesterase.“ J Neurochem 50:1667-1677

108
• Von Heijne G (1986): „A new method for predicting signal sequence cleavagesites.“ Nucl Acids Res 14: 4683-4690
• Voshol H, van Zuylen CWEM, Orberger G, Vliegenthart JFG, Schachner MT(1996): „Structure of the HNK-1-Epitope on Bovine Peripheral MyelinGlycoprotein P0“ J Biol Chem 271(38):22957-60
• Wada K, Wada Y, Doi H Ishibashi F, Gojobori T, Ikemura T (1991): „Codon usagetabulated from GenBank genetic sequence data“ Nucl Acids Res. 19:1981-1986
• Waehneldt TV, Jeserich G (1984): „Biochemical characterization of the centralnervous system myelin proteins of the rainbow trout, Salmo gaidneri.“ Brain Res309:127-134
• Waehneldt TV, Stocklas S, Jeserich G, Matthieu JM (1986b): „Central nervoussystem myelin of teleosts: comparative electrophoretic analysis of ist proteins bystaining and immunoblotting.“ Comp Biochem Physiol 84B:273-278
• Whitelaw E , Proudfoot N (1986): „α-thalassaemia caused by a poly(A) sitemutation reveals that transcriptional termination is linked to 3’ end processing in thehuman α2 globin gene“ Embo J 5:2915-2922
• Whittaker KL, Ding D, Fisher WW, Lipshitz HD (1999): „Different 3‘ UTR targetalternatively processed hu-li tai shao (hts) transcripts to distinct cytoplasmiclocations during drosophila oogenesis.“ J Cell Sci 112 (pt 19): 3385-98
• Williams AF, Barclay AN (1988): „The immunoglobulin superfamily - Domains forcell surface recognition“ Annu Rev Immunol 6:381-405
• Wolburg H (1981): Myelination and remyelination in the regenerating visual systemof the goldfish“ Exp Brain Res 43: 199-206
• Wong MH, Filbin MT (1994): „The cytoplasmatic domain of the myelin P0 proteininfluences tha adhesive interactions of ist extracellular domain.“ J Cell Biol 126:1089-1097
• Wong MH, Filbin MT (1996): „Dominant-negative effect on adhesion by myelin P0protein truncated in its cytoplasmic domain“ J Cell Biol 134:1531-1541
• Wood P, Bunge RP (1984): „The biology of the oligodendrocyte“, in „Advances inNeurochemistry, vol 5: Oligodendroglia“(Norton WT, ed.), Plenum Press, NewYork, pp. 1-46
• Woodruff RH, Franklin RJ (1998): „The expression of myelin basic protein exon 1and exon 2 containing transcripts during myelination of the neonatal rat spinal cord -- an in situ hybridization study“ J Neurocytol 27(9):683-93
• Yazaki T, Miura M, Asou H, Toya S, Uyemura K (1994): „Peripheral myelin P0protein mediates neurite outgrowth of cortical neurons in vitro und axonal

109
regeneration in vivo.“ Neurosci Lett 176: 13-16
• Yoshida M, Colman DR (1996): „Parallel evolution and coexpression of theproteolipid proteins and P0 in vertebrate myelin“ Neuron 16:1115-1126
• Yu RK, Iqbal K (1979): „Sialosylgalactosylceramide as a specific marker for humanmyelin and oligodendroglia.“ J Neurochem 32: 293-300
• Zhang K, Filbin MT (1994): „Formation of a disulfide bond in the immunoglobulindomain of the myelin P0 protein is essentiell for its adhesion“ J Neurochem623:367-370
• Zhang K, Merazga Y, Filbin MT (1996): „Mapping the adhesive domains of themyelin P0 protein“ J Neursci Res 45:525-533

110
7 Anhang
7.1 Klonierung von IP1/pIRES-neo
Die Klonierung der codierenden Region der IP1 cDNA ohne poly(A)-Signale erfolgte wie
folgt: IP1-cDNA wurde HindIII verdaut, anschließend gebluntet und mit EcoRI
nachgeschnitten. Der pIRES-neo Vektor wurde mit BamHI geöffnet, anschließend gebluntet
und mit EcoRI nachgeschnitten. So konnte IP1 EcoRI/blunt gerichtet kloniert werden.
7.2 Klonierung von IP2/pIRES-neo
Da die vollständige codierende Region der IP2 cDNA durch PCR mit den Primern IP-5F und
IP2 C3-R gewonnen und in pGEM-T easy subkloniert wurde, erfolgte eine ungerichtete
Ligation in pIRES-neo über die flankierenden Not I Schnittstellen.
7.3 Klonierung von IP-P0/pIRES-neo
Die cytoplasmatische Domäne von P0 wurde mit Hilfe der PCR mit den Primern P0-fus3 ,
der eine Xba I Schnittstelle trägt und T7 an pBK-CMV mod/P0 ohne 3‘ UTR amplifiziert
Das Amplifikat trägt am 5‘ Ende eine XbaI Schnittstelle von Primer P0-fus3 und am 3‘ Ende
kurz vor der T7 Sequenz ebenfalls eine XbaI Schnittstelle, die aus der Multicloningsite des
pBK-CMV mod/P0 ohne 3‘ UTR stammt. Es wurde XbaI verdaut und in den gleichermaßen
geöffneten pBK-CMV ligiert
Es befindet sich innerhalb der codierenden Region der IP cDNA (direkt an der Grenze
zwischen Transmembransegment und cytoplasmatischer Domäne) eine BspHI Schnittstelle.
Die cDNA wurde BspHI geschnitten und mit T4-Polymerase zu blunt-ends aufgefüllt.
Anschließend wurde der Extrazellulär- und Transmembrandomäne umfassende cDNA Teil
durch EcoRI Verdau abgetrennt. Der pBK-CMV/P0cyto Vektor, der bereits wie oben
beschrieben die P0cyto Domäne trägt, wurde durch EcoRI/ScaI geöffnet. Das EcoRI/ blunt
IP-Eluat konnte so in den pBK-CMV/P0cyto ligiert werden. Da sich der IP-Teil und P0-Teil
im gleichen Leserahmen befinden, entsteht eine Proteinfusion aus IP und P0 mit einer
Linkersequenz von nur 9 bp (3 AS). Die entstandene Fusionsstelle wurde durch
Sequenzierung verifiziert. Das vollständige IP-P0 Fusionskonstrukt wurde EcoRI/SmaI
verdaut und konnte so in den oben unter 7.1 beschriebenen pIRES-neo EcoRI/blunt ligiert.

111
7.4 Klonierung von IP1 in pFastbac
Die komplette IP1-cDNA wurde über seine flankierenden EcoRI/XhoI Schnittstellen aus
pBluescript in den gleichermaßen geöffneten pFastbac-Vektor ligiert.

112
7.5 Klonierung von 36K/pIRES-hygro
Die Klonierung der codierenden Region der 36K cDNA ohne poly(A)-Signale erfolgte über
seine flankierenden BamHI/ScaI Schnittstellen aus pBluescript. Der pIRES-hygro Vektor
wurde BstXI verdaut, anschließend gebluntet und mit BamHI nachgeschnitten. So konnte
36K BamHI/blunt gerichtet kloniert werden.
7.6 Klonierung von 36KHT/Dual Fastbac
Zur Expression wurde durch PCR Mutagenese mit den Primern 36K-mal F2/36K-mal R2 an
der isolierten 36KcDNA eine BamHI vor dem Start ATG und eine Xba I Schnittstelle hinter
dem Stop-TGA eingefügt. Das PCR-Amplifikat wurde mit diesen beiden
Restriktionsenzymen geschnitten und pFastbac-HTb-Vektors mit den gleichen Enzymen
geöffnet. Auf diese Weise konnte eine His(6)-Tag-36K Fusion kloniert werden. Parallel dazu
wurde die codierende Region des Grünfluoreszierenden Proteins (GFP) aus dem
pGreenlantern Vektor (Fa. Gibco) über seine flankierenden Not I Sequenzen
herausgeschnitten, gebluntet und in die Multicloningsite II (p10 Promotor) des mit Sma I
geöffneten pFastbac Dual Vektors kloniert. Das oben beschriebene His-Tag-36K-Konstrukt
aus pFastbac HTb wurde mit Hilfe einer weiteren PCR-Mutagenese mit den Primern HTb-
F1/ 36K-mal R2 in pGEM-T easy (2.2.11) subkloniert, anschließend über seine flankierenden
Not I Schnittstellen in die Multicloningsite I des auf gleiche Weise geöffneten pFastbac-
Dual/GFP kloniert und subsidiär durch Sequenzierung auf Richtigkeit und eventuelle
Nukleotidaustausche überprüft. Das entstandene Konstrukt erlaubt die Expression des His-
Tag-36K Fusionsproteines unter der Kontrolle des konstitutiven Polyhedrin-Promotors,
sowie zeitgleich die Expression des GFP-Reportergenes unter der Kontrolle des ebenfalls
konstitutiven p10-Promotors.

113
7.7 Klonierung von 36K/pET16b
Durch PCR-gerichtete Mutagenese mit dem Vorwärts Primer 36K-CL-F4, der eine NdeI
Schnittstelle trägt und direkt am Start ATG bindet und dem 36K2R Primer entstanden PCR-
Amplifikate, die nach blunten mit T4 Polymerase mit Nde I verdaut wurde. Der pET 16b
Vektor wurde mit BamHI geöffnet, anschließend gebluntet und mit Nde I nachgeschnitten.
So konnte Nde/blunt gerichtet kloniert werden. Es entstand eine His -Tag 36K Fusion, die
durch Sequenzieren auf Richtigkeit kontrolliert wurde.
7.8 Klonierung von IP1/2cyto/pMal-C2
Durch PCR-gerichtete Mutagenese mit dem vorwärts Primer IP-Cyto F1, der am Beginn der
cytoplasmatischen Domäne beider Isoformen bindet, in Kombination mit den reversen
Primern IP-4E-mal (für IP1) bzw. IP2-C3-malR (für IP2), entstanden PCR-Fragmente, die

114
am 5‘ Ende eine eingefügte EcoRI, am 3‘ Ende eine Hind III Schnittstelle besaßen. Diese
Amplifikate wurden EcoRI/Hind III verdaut, der Restriktionsansatz über Qiagen PCR
Purification Kit aufgereinigt und in den gleichermaßen vorbereiten pMAL-C2 Vektor
kloniert. Das entstandene Konstrukt wurde durch PCR und Restriktionsverdau, sowie durch
Sequenzierung auf Richtigkeit kontrolliert.
7.9 Abkürzungsverzeichnis% v/v Volumenprozente% w/v Gewichtsprozenteµ mikroADP AdenosindiphosphatAmp Ampicillinbp BasenpaarBSA RinderserumalbumincDNA komplementäre DesoxyribonukleinsäureDNA DesoxyribonukleinsäuredNTP 2’-DesoxyribonukleosidtriphosphatE. coli Escherichia coliEDTA Etylendiamintetraessigsäureg 1x ErdanziehungskraftHis-tag Histidin-„tag“ (engl. für Fortsatz, Anhang)IPcyto (bzw. IP2, P0) cytoplasmatische Domäne des IP1, bzw. IP2, P0-ProteinesIPTG Isopropylthiogalaktosidkb Kilobase(n)kDa Kilo-Daltonm milliM molarMBP Maltose-Binde-ProteinMCS „Multiple Cloning Site“ (multiple Klonierungsstelle)mRNA „Messenger“-RibonukleinsäureNAD(P) Nicotinamid-adenin-dinukleotid(-phosphat)OD600 optische Dichte bei einer Wellenlänge von 600 nmPAGE Polyacrylamid-GelelektrophoresePCR „Polymerase Chain Reaction“ (Polymerase-Kettenreaktion)PEG PolyethlenglykolPLP ProteolipidproteinPNS peripheres Nervensystemr ResistenzgenRif RifampicinrIP1/CHO bzw. IP-P0 rekombinant exprimiertes IP1 bzw. IP-P0 in CHO-ZellenRNA Ribonukleinsäurerpm Umdrehung pro MinuteSDS Natrium-(„Sodium“-)dodecylsulfatTCA TrichloressigsäureTet TetrazyklinTris Tris-HydroxymethylaminomethanUTR untranslatierte RegionZNS zentrales Nervensystem

115
7.10 Publikationen
Jeserich G, Strelau J, Lanwert C (1997): „Partial characterization of the 5‘-flanking region of
trout IP: a P0-like gene containing a PLP-like promotor“ J Neurosci Res 50:1-10
C. Lanwert und G. Jeserich: „Heterologous expression of fish myelin genes in mammalian
cell lines“ nacher presentation at Gordon Research Conference – „Myelin“ Ventura
California, (1998)
C. Lanwert und G. Jeserich: „Recombinantly expressed fish myelin proteins as tools to
mimick protein interactions at the major dense line“ nacher presentation at ISN Satellite
Meeting „Myelination and Myelin Disorders: New Vistas“ Rügen, Deutschland (1999)

116
Inhaltsverzeichnis
1 Einleitung.................................................................................................................... 1
2 Material und Methoden............................................................................................... 7
2.1 Material............................................................................................................................ 72.1.1 Bakterienstämme....................................................................................................................72.1.2 Plasmide.................................................................................................................................82.1.3 Larvenstadien und Jungfische der Regenbogenforelle (Oncorhynchus mykiss).........................82.1.4 Zellinien .................................................................................................................................82.1.5 Chemikalien ...........................................................................................................................92.1.6 Medien zur Anzucht von Bakterien.........................................................................................92.1.7 PCR Primer .......................................................................................................................... 10
2.2 Methoden ....................................................................................................................... 112.2.1 Bakterienkulturen/Kryokonservierung von Bakterienkulturen ............................................... 112.2.2 Plasmidpräparationen ........................................................................................................... 112.2.3 RNA/DNA-Quantifizierung durch Methylen-Blau-Spot-Test ................................................ 122.2.4 RNA/DNA-Quantifizierung durch Photometrie..................................................................... 132.2.5 Restriktion von DNA mit Restriktionsendonukleasen............................................................ 132.2.6 Agarose-Gelelektrophorese ................................................................................................... 132.2.7 Gelextraktionsmethoden ....................................................................................................... 132.2.8 Klonierungen........................................................................................................................ 142.2.9 Herstellung von glatten Enden mit T4 Polymerase („blunting“) ............................................ 152.2.10 Reinigung von DNA Fragment aus wäßrigen enzymatischen Lösungen ................................ 152.2.11 Klonierung von PCR-Produkten / pGEM-T easy................................................................... 162.2.12 Plasmid Transformation........................................................................................................ 162.2.13 Polymerase-chain-reaction (PCR)-Techniken........................................................................ 172.2.14 Chemolumineszenzdetektion/ ECL-Kit (Fa. Amersham)....................................................... 202.2.15 Alkalische Phosphatase-NBT/BCIP-Farbreaktion ................................................................. 202.2.16 Alkalische Phophatasenachweis mit CSPD (Fa. Boehringer)................................................. 212.2.17 Peroxidasenachweis mit 4-Chloro-1-Naphtol (4CN) (nach Sambrock).................................. 212.2.18 Screening einer λ-UNI ZAP-cDNA Libary mit DIG markierten DNA-Sonden...................... 212.2.19 In vivo excision positiver cDNA/Phagen-Klone.................................................................... 222.2.20 Heterologe Expression in Insektenzellen mit Baculoviren / Bac-to-Bac-System (Fa.Gibco) ... 232.2.21 Heterologe Expression in E. coli /pMAL-System (Fa. NEB) ................................................. 262.2.22 Affinitätschromatographie mit Amylose-Beads (pMAL-System) ........................................... 272.2.23 Heterologe Expression in E. coli/pET-System (Fa. Novagen) ................................................ 272.2.24 Affinitätschromatographie mit Ni2+-chelatierender Sepharose............................................... 282.2.25 Aufschluß von Einschlußkörpern (inclusion bodies).............................................................. 282.2.26 Affinitätschromatographie mit Farbsepharosen (reactive-red, Fa. Merck).............................. 292.2.27 Heterologe Expression in E. coli /IMPACT-System (Fa. NEB)............................................. 292.2.28 Proteinbestimmung (nach Bradford, 1979)............................................................................ 292.2.29 Proteinbestimmung mit Amidoschwarz-Tüpfel Test.............................................................. 302.2.30 Proteinfällung mit Aceton..................................................................................................... 302.2.31 Proteinfällung mit TCA........................................................................................................ 302.2.32 SDS-Polyacrylamid-Gelelektrophorese (SDS-PAGE) (nach Laemmli) .................................. 302.2.33 Native Polyacrylamid-Gelelektrophorese (Native-PAGE)...................................................... 312.2.34 Proteintransfer auf Nitrozellulosemembran (Westernblot) ..................................................... 312.2.35 Immunologischer Nachweis von Proteinen auf Nitrozellulosemembran................................. 312.2.36 Zellaggregations/Adhäsionstest............................................................................................. 322.2.37 Trypan-Blau-Ausschlußtest................................................................................................... 332.2.38 ELISA zur Quantifikation extrazellulärer IP-Proteindomänen............................................... 33

117
2.2.39 Protein-Phospholipid-Bindungstudien mit Transil-Kügelchen (Fa. Nimbus) ......................... 342.2.40 Protein-Protein-Bindungsstudien („overlay-assay“) ............................................................... 342.2.41 DNA-Transfer auf positivierter Nylonmembran (Southern-Blot) ........................................... 342.2.42 Radioaktive DNA-Kettenabbruch-Sequenzierungen (nach Sanger)....................................... 352.2.43 Auftragssequenzierungen...................................................................................................... 352.2.44 Computergestützte Sequenzanalysen..................................................................................... 352.2.45 Eukaryotische Zellkulturlinien .............................................................................................. 362.2.46 Subkultivieren („passagieren“) von Säuger-Zellkulturen ....................................................... 362.2.47 Subkultivieren („passagieren“) von Insektenzellen ................................................................ 362.2.48 Kryokonservierung von eukaryotischen Zellinien.................................................................. 372.2.49 Transfektionen von Säugerzellen /Transfectam Methode ...................................................... 372.2.50 Transfektion von Insektenzellen/ Cellfectin (Gibco).............................................................. 372.2.51 Hygromycin und G-418 („Geneticin“) Selektion /Stabile Transfektion mit pIRES-neo.......... 372.2.52 Dynabead oligo d(T)-mRNA-Isolierungen aus Hirngewebe................................................. 382.2.53 cDNA Erststrangsynthese (Reverse Transkription)................................................................ 392.2.54 Immunocytochemie/indirekte Immunfluoreszenz .................................................................. 40
3 Ergebnisse ..................................................................................................................42
3.1 Isolierung der vollständigen cDNA Sequenz für das IP2-Protein.................................. 423.1.1 Aminosäuresequenz der vollständigen cytoplasmatischen Domäne von IP2 .......................... 44
3.2 Untersuchungen zur Funktion von IP1/2 als Adhäsionsmolekül.................................... 453.2.1 Aufbau eines Adhäsionsassays .............................................................................................. 463.2.2 Heterologe transiente Expression von IP1 und IP2 in CHO-K1-Zellen .................................. 483.2.3 Klonierung von stabil-transfizierten rIP1/CHO-K1-Zellen .................................................... 503.2.4 Adhäsion von rIP1/CHO-Zellen unter definierten Bedingungen............................................ 503.2.5 Konstruktion einer IP-P0-Chimäre........................................................................................ 523.2.6 Stabile Expression und Adhäsionsverhalten von rIP-P0/CHO-Zellen .................................... 523.2.7 Vergleichende Quantifizierung der Oberflächenexpression ................................................... 553.2.8 Inhibierung der Zell/Zell-Adhäsion von rIP-P0/CHO durch Antikörper ................................ 563.2.9 Heterologe Expression und Adhäsion von IP1 in Insektenzellen............................................ 58
3.3 Heterologe Expression der cytoplasmatischen Domänen von IP1 und IP2 in E. coli .... 593.3.1 Bindestudien von MBP/IP2cyto an phospholipidbeschichteten Trägermatrices (Transil) ....... 603.3.2 Untersuchung zu Protein-Protein-Wechselwirkungen von IP1/2cyto..................................... 61
3.4 Isolierung der cDNA Sequenz für das 36K- Protein...................................................... 633.4.1 Aminosäuresequenz des 36K-Proteins................................................................................... 65
3.5 Untersuchungen zur 36K-Genstruktur durch PCR....................................................... 683.5.1 Diskrepanz zwischen isolierter und vorhergesagter 36K-Sequenz ......................................... 713.5.2 Klonierung einer putativen Promotorregion des 36K-Genes durch „inverse PCR“................. 72
3.6 Heterologe Expression der isolierten 36K cDNA in CHO-K1 ...................................... 74
3.7 Expression in E. coli (pET-System) ............................................................................... 753.7.1 Alternative Aufreinigung von His-Tag-36K durch Affinitätschromatographie mitFarbsepharosen .................................................................................................................................... 77
3.8 Expression in Insektenzellen / Baculovirus-System ....................................................... 77
4 Diskussion ..................................................................................................................80
5 Zusammenfassung .....................................................................................................97
6 Literatur .....................................................................................................................99
7 Anhang.....................................................................................................................110
7.1 Klonierung von IP1/pIRES-neo................................................................................... 110

118
7.2 Klonierung von IP2/pIRES-neo................................................................................... 110
7.3 Klonierung von IP-P0/pIRES-neo ............................................................................... 110
7.4 Klonierung von IP1 in pFastbac .................................................................................. 111
7.5 Klonierung von 36K/pIRES-hygro.............................................................................. 112
7.6 Klonierung von 36KHT/Dual Fastbac ......................................................................... 112
7.7 Klonierung von 36K/pET16b....................................................................................... 113
7.8 Klonierung von IP1/2cyto/pMal-C2............................................................................. 113
7.9 Abkürzungsverzeichnis ................................................................................................ 114
7.10 Publikationen................................................................................................................ 115

119
&DUVWHQ�/DQZHUW
geboren am 30.6.70 in Osnabrückverheiratet
Schule und Grundwehrdienst
1976 - 1980 Grundschule Wallenhorst
1980 - 1982 Thomas-Morus-Schule Osnabrück
1982 - 1989 Angelaschule Osnabrück, Abschluß mit Abitur
1989 - 1990 Grundwehrdienst im Jägerbataillon 522 in Fürstenau
Studium
10/90 – 4/96 Immatrikulation an der Universität Osnabrück Fach: Biologie/Diplom
Diplomarbeit mit dem Thema: „Heterologe Expression vonMyelinproteingenen aus dem ZNS der Forelle in OLN- 93 Zellen derRatte“
23.4.96 Erfolgreicher Abschluß und Verleihung des akademischen Grades desDiplom Biologen
Promotion
5/96 bis 12/99 wissenschaftlicher Angestellter an der Universität Osnabrück imSonderforschungsbereich 171
5/96 bis 3/00 Doktorand in der Arbeitsgruppe Zoophysiologie, Anfertigung derDissertation unter der Leitung von Prof. Dr. G. Jeserich mit dem Thema:„Heterolge Expression und funktionelle Grundcharakterisierung vonMyelinproteinen aus dem ZNS der Regenbogenforelle (Oncorhynchusmykiss)“

120
Eidesstattliche Versicherung
Ich versichere, daß ich die vorliegende Dissertation entsprechend § 3 Absatz 2 der
Promotionsordnung selbständig und ohne unerlaubte Hilfe verfaßt und die benutzten
Hilfsmittel vollständig angegeben habe.
Ich versichere, daß ich weder an der Universität Osnabrück noch anderweitig versucht habe,
eine Dissertation einzureichen oder mich einer Doktorprüfung zu unterziehen.
Osnabrück, den 6.3.2000
Carsten Lanwert





























