Embed Size (px)
Citation preview

High-throughput sequencing with R MappingBiostrings and ShortRead
Kasper Daniel HansenMargaret Taub
based on slides developed byJim Bullard
University of CopenhagenAugust 17-21 2009
1 38
Introduction
These slides will discuss mapping of sequence data as well as theBiostrings and ShortRead packages
I Alignment and tools (mostly external to R)
I Biostrings overview
I Alignment tools in Biostrings
I Mapping data and reading it into R
2 38
A comment
Analysis of high-throughput sequencing data and especially RNA-Seq isstill in its infancy
It is unclear what is the best way to think about and analyze these dataIt is also unclear are the right entities to compute on
We focus on some tools and some computations we have found useful
3 38
Alignment input FASTQ files
FASTQ files represent a common ldquoend-pointrdquo from the various sequencingplatforms (ie NCBI short read archivehttpwwwncbinlmnihgovTracessrasracgi) Qualities areencoded in ASCII and depending on the platform have slightly differentmeaningsranges and encoding More details can be found athttpenwikipediaorgwikiFASTQ_format
GA-EAS46_2_209DG61890752TTCTCTTAAGTCTTCTAGTTCTCTTCTTTCTCCACT+GA-EAS46_2_209DG61890752hhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhGA-EAS46_2_209DG61905558TCTGGCTTAACTTCTTCTTTTTTTTCTTCTTCTTCT+GA-EAS46_2_209DG61905558hhhfhhhhhhhhhhhhhhhhhhhhhhhhehhGhhJh
4 38
Mapping reads to the transcriptome
Mapping reads to the transcriptome
Transcriptome
2^Genome
Reads
Genome
Illustration from Lior Patcher
Well established
5 38
Mapping reads to the transcriptome 2
Mapping transcripts
Genome
Transcript
Length in genome space
paired-end reads
6 38
Mapping reads to the transcriptome 3
Junction reads
chrX
Conservation
d_simulansd_sechelliad_yakubad_erectad_ananassaed_pseudoobscurad_persimilisd_willistonid_virilisd_mojavensisd_grimshawia_gambiaea_melliferat_castaneum
1067300010673500106740001067450010675000106755001067600010676500106770001067750010678000106785001067900010679500106800001068050010681000
CG8144_6lane_0MM
S2_DRSC_6_lanes
FlyBase Protein-Coding Genes
RefSeq Genes
12 Flies Mosquito Honeybee Beetle Multiz Alignments amp phastCons Scores
Repeating Elements by RepeatMasker
CG15211CG15211CG15211CG15211
Ant2Ant2
sesBsesBsesBsesB
CG15211CG15211CG15211CG15211
Ant2Ant2
sesBsesBsesBsesB
_ 1000
_ 0
_ 1000
_ 0
ps RNAi
S2 Untreated
Splice JunctionReads
GenomicReads
Splice JunctionReads
GenomicReads
Image from Brenton Gravely
Image courtesy of Brenton Graveley
7 38
Mapping reads to the transcriptome 4
Junction reads zoom
chrX
Conservation
d_simulansd_sechelliad_yakubad_erectad_ananassaed_pseudoobscurad_persimilisd_willistonid_virilisd_mojavensisd_grimshawia_gambiaea_melliferat_castaneum
1067300010673500106740001067450010675000106755001067600010676500106770001067750010678000106785001067900010679500106800001068050010681000
CG8144_6lane_0MM
S2_DRSC_6_lanes
FlyBase Protein-Coding Genes
RefSeq Genes
12 Flies Mosquito Honeybee Beetle Multiz Alignments amp phastCons Scores
Repeating Elements by RepeatMasker
CG15211CG15211CG15211CG15211
Ant2Ant2
sesBsesBsesBsesB
CG15211CG15211CG15211CG15211
Ant2Ant2
sesBsesBsesBsesB
_ 1000
_ 0
_ 1000
_ 0
ps RNAi
S2 Untreated
Splice JunctionReads
GenomicReads
Splice JunctionReads
GenomicReads
Image from Brenton Gravely
Image courtesy of Brenton Graveley
8 38
Sequencing errors Genome differences
Sample Genome
Ref Genome
True read
read with sequence error
Sample different from reference
Sequence errorMapping
Evidence suggests that Illumina sequencing does not introduce indels
9 38
Alignment Tools
The number of short read aligners have exploded but a couple tools haveemerged as the de facto standards
I Eland Illuminarsquos aligner quality aware fast paired end capable
I MAQ Good SNP caller quality aware paired end capable
I Bowtie Super fast offers different alignment strategies paired endcapable
I BWA Fast indel support paired ends qualities
I NovoAlign MAQ like speed many features
A superior overview of the different aligners is available athttpwwwsangeracukUserslh3NGSalignshtml Additionallya comparison of two of the best aligners can be found here httpwwwmassgenomicsorg200907maq-bwa-and-bowtie-comparedhtml
10 38
Common Alignment Strategies
I Use qualities (default for Bowtie and MAQ)
I Perfect match no repeats (Strict Lenient)
I Mismatches no repeats
I Paired end data (PET) (harder for RNA-Seq)
The standard Illumina protocol yields unstranded reads
Most aligners are evaluated in terms of ldquohow many reads are mappedrdquo Isthis the right objective
Watch out for the output of the program there are many differentconventions (0-based what happens to read hitting the reverse strandetc)
11 38
SAMBAM Formats
SAM (BAM) is a new general format for storing mapped reads It isdeveloped as part of the 1000 Genomes project and is quickly becoming akind of standard Some alignment tools output this format directlyotherwise there are scripts in samtools for doing it for most popularaligners Details at httpsamtoolssourceforgenetindexshtml
12 38
A comment
There are no great comprehensive tools for analyzing deep-sequence dataIt will involve a fair amount of coding and gluing together various tools
We will introduce some tools from Bioconductor that can be useful
I use a lot of shell scripting
13 38
Biostrings overview
I A package for working with large (biological) strings
I Two main types of objects A really long single string (thinkchromosome) or a set of shorter strings (think reads or genes)BString vs BStringSet
I These classes are implemented efficiently minimizing copy andmemory loadingunloading
I The BSgenome contains some infrastructure for whole genomes
I Methods for dealing with biological data including basicmanipulation (complementation translation etc) string searching(exactinexact matching Smith-Waterman PWMs)
I Fairly complicated class structure
Irsquom sorry to say that at least for me this has becomehopelessly confusing and I imagine that many other usersfeel the same ndash Simon Anders on bioc-sig-sequencing
Computing on genomes is not trivial The approach to a givencomputation is important
14 38
Strings in Biostrings
I BString (general) DNAString RNAString AAString (Amino Acid)all examples of XString
I complement reverse reverseComplement translate for the classeswhere ldquoit makes senserdquo
I Convert to and from a standard R character string
I Constructor DNAString(ACGGGGG)
I Support for IUPAC alphabet
I Subsetting subseq using the SEW format (two out of the threestartendwidth) Efficient
I StringSets are collections of Strings like DNAStringSet
15 38
BSgenomes
As examples we will use whole genomes Use availablegenomes to getavailable genomes Long package names but always a shorter objectname
gt library(BSgenomeScerevisiaeUCSCsacCer1)
gt Scerevisiae
gt Scerevisiae[[1]]
gt Scerevisiae[[chr1]]
A BSgenome may also have masks We will ignore this for now
16 38
Biostrings more
I alphabetFrequency oligonucleotideFrequency and others
gt alphabetFrequency(Scerevisiae[[chr1]])
gt oligonucleotideFrequency(Scerevisiae[[chr1]]
+ width = 3)
I chartr for character translation (ldquomake all As into Csrdquo)
I Various IO functions (also ShortRead)
17 38
Views
A view is a set of substrings of an XString stored and manipulated veryefficiently Example to store exon sequences one can just store thegenomic location of the exons
gt Views(Scerevisiae[[chr1]] start = c(300
+ 400 500) width = 50)
Views on a 230208-letter DNAString subjectsubject CCACACCACACCCAGTGGTGTGTGTGGGviews
start end width[1] 300 349 50 [CTGTTCTTAAATAAC][2] 400 449 50 [CCCTCACTAGTATAT][3] 500 549 50 [TCTCTCACCGGCACT]
A view is associated with a subject Internally they are essentiallyIRanges so they are very fast to compute with Views can in many casesbe treated exactly as other strings There are methods such as narrowtrim gaps restrict
18 38
Matching in Biostrings
There are various ways of matching or aligning strings to each other Weuse matching to denote searching for an exact match or possibly a matchwith a certain number of mismatches Alignment denotes a more generalstrategy eg Smith-Waterman
I matchPattern countPattern match 1 sequence to 1 sequence
I vmatchPattern vcountPattern match 1 sequence to manysequences
I pairwiseAlignment align many sequences to 1 sequence
I matchPDict countPDict match many sequences to 1 sequence(ldquodictrdquo indicates that the many sequences are preprocessed into adictionary)
I matchPWM trimLRpattern
The different functions are optimized for different situations They alsouse different algorithms which has a big impact EspeciallypairwiseAlignment is flexible and therefore has a complicated syntax
19 38
Example mapping probes to a genome
Get a list of Scerevisiae probes from the ldquoyeast2rdquo Affymetrix array
gt library(yeast2probe)
gt ids lt- scan(s_pombemsk skip = 2
+ what = list(probeset = character(0)
+ junk = character(0)))$probeset
gt probes lt- yeast2probe$sequence[yeast2probe$ProbeSetName in
+ ids]
gt probes lt- DNAStringSet(probes)
Mapping
gt require(BSgenomeScerevisiaeUCSCsacCer1)
gt dict0 lt- PDict(probes)
gt dict0r lt- PDict(reverseComplement(probes))
gt hits lt- matchPDict(dict0 Scerevisiae[[1]])
gt table(countIndex(hits))
gt table(countPDict(dict0 Scerevisiae[[1]]))
20 38
Example contrsquod
Over all chromosomes
gt allhits lt- lapply(116 function(i)
+ countPDict(dict0 Scerevisiae[[i]]) +
+ countPDict(dict0r Scerevisiae[[i]])
+ )
gt table(rowSums(docall(cbind allhits)))
21 38
ShortRead
ShortRead contains tools for
I Work with GERALDBUSTARD output
I Generate QA report on BUSTARD files
I Read a variety of short read data formats
22 38
Data
We will use data from (Lee Hansen Bullard Dudoit and Sherlock(2008) PLoS Genetics)
We are considering a wild-type and a mutant strain of yeast both grownin rich media The two strains were sequenced using an Illumina GenomeAnalyzer
We are using a subset of the data 1M reads from each of two lanes ofthe two strains
23 38
Reading the unaligned data
gt require(ShortRead)
gt fq lt- readFastq(seqdata mut_1_ffastq$)
gt fq
class ShortReadQlength 1000000 reads width 36 cycles
There are various simple accessor functions for this object especiallysread and quality
24 38
A brief look at the unaligned data
Using alphabetByCycle and as( rdquomatrixrdquo) (which creates a big read timescycle matrix) we can do
gt alp lt- alphabetByCycle(sread(fq))
gt matplot(t(proptable(alp[DNA_BASES
+ ] margin = 2)) type = l)
gt qaMatraw lt- as(quality(fq) matrix)
gt plot(colMeans(qaMatraw))
25 38
Aligning the data
We now align the data using Bowtie
binbash
BOWTIE_OPTS=-m 1 -v 2 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-m 1 -v 0 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 2 -k 2 -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 0 -k 1 -p 2 -q --quiet -3 10
for f in `ls seqdatafastq`dobowtie s_cerevisiae $BOWTIE_OPTS $f gt $fbowtiedone
How many hits do we get
26 38
Reading in the aligned data
We read the data into R as a 4-component list (one lane per component)
gt files lt- listfiles(seqdata
+ pattern = bowtie)
gt aligned lt- sapply(files function(f)
+ readAligned(seqdata pattern = f
+ type = Bowtie)
+ )
gt names(aligned) lt- gsub(_ffastqbowtie
+ names(aligned))
gt aligned[[1]]
gt save(aligned file = alignedrda)
27 38
Some exercises
1 Determine the number of times the motif ldquoTATAArdquo occurs in theyeast genome How often does it occur with one mismatch
2 The seqdata directory has an object called ste12 which is aposition weight matrix for the transcription factor ldquoSte12rdquo (obtainedfrom SCPD) Where does it occur in the genome
3 Compute the average quality for each cycle for the aligned reads andcompare to the qualities for the unaligned reads What is thedifference
28 38
Solution 1
gt sum(sapply(116 function(i)
+ motif lt- DNAString(TATAA)
+ countPattern(motif Scerevisiae[[i]]) +
+ countPattern(reverseComplement(motif)
+ Scerevisiae[[i]])
+ ))
29 38
Solution 2
30 38
Solution 3
31 38
SessionInfo
gt toLatex(sessionInfo())
I R version 292 RC (2009-08-17 r49312) i386-apple-darwin980
I Localeen_USUTF-8en_USUTF-8CCen_USUTF-8en_USUTF-8
I Base packages base datasets graphics grDevices grid methodsstats utils
I Other packages Biostrings 2128 BSgenome 1123BSgenomeScerevisiaeUCSCsacCer1 1313 classGraph 07-2graph 1222 IRanges 123 lattice 017-25 Rgraphviz 1234ShortRead 121
I Loaded via a namespace (and not attached) Biobase 241hwriter 11 tools 292
32 38
Some ShortRead classes
ShortRead
ShortReadQ
AlignedRead
33 38
Some Biostrings classes (single strings)
XString
BString DNAString RNAString AAString
34 38
Some Biostrings classes (sets of strings)
XStringSet
BStringSet DNAStringSet RNAStringSet AAStringSet PhredQualitySolexaQualityQualityScaledXStringSet
QualityScaledBStringSetQualityScaledDNAStringSetQualityScaledRNAStringSetQualityScaledAAStringSet
35 38

Introduction
These slides will discuss mapping of sequence data as well as theBiostrings and ShortRead packages
I Alignment and tools (mostly external to R)
I Biostrings overview
I Alignment tools in Biostrings
I Mapping data and reading it into R
2 38
A comment
Analysis of high-throughput sequencing data and especially RNA-Seq isstill in its infancy
It is unclear what is the best way to think about and analyze these dataIt is also unclear are the right entities to compute on
We focus on some tools and some computations we have found useful
3 38
Alignment input FASTQ files
FASTQ files represent a common ldquoend-pointrdquo from the various sequencingplatforms (ie NCBI short read archivehttpwwwncbinlmnihgovTracessrasracgi) Qualities areencoded in ASCII and depending on the platform have slightly differentmeaningsranges and encoding More details can be found athttpenwikipediaorgwikiFASTQ_format
GA-EAS46_2_209DG61890752TTCTCTTAAGTCTTCTAGTTCTCTTCTTTCTCCACT+GA-EAS46_2_209DG61890752hhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhGA-EAS46_2_209DG61905558TCTGGCTTAACTTCTTCTTTTTTTTCTTCTTCTTCT+GA-EAS46_2_209DG61905558hhhfhhhhhhhhhhhhhhhhhhhhhhhhehhGhhJh
4 38
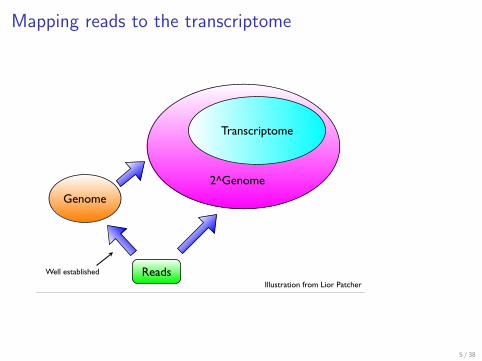
Mapping reads to the transcriptome
Mapping reads to the transcriptome
Transcriptome
2^Genome
Reads
Genome
Illustration from Lior Patcher
Well established
5 38
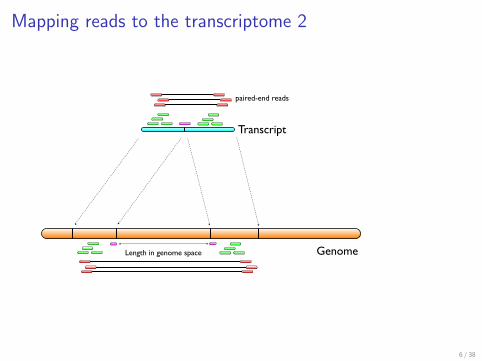
Mapping reads to the transcriptome 2
Mapping transcripts
Genome
Transcript
Length in genome space
paired-end reads
6 38
Mapping reads to the transcriptome 3
Junction reads
chrX
Conservation
d_simulansd_sechelliad_yakubad_erectad_ananassaed_pseudoobscurad_persimilisd_willistonid_virilisd_mojavensisd_grimshawia_gambiaea_melliferat_castaneum
1067300010673500106740001067450010675000106755001067600010676500106770001067750010678000106785001067900010679500106800001068050010681000
CG8144_6lane_0MM
S2_DRSC_6_lanes
FlyBase Protein-Coding Genes
RefSeq Genes
12 Flies Mosquito Honeybee Beetle Multiz Alignments amp phastCons Scores
Repeating Elements by RepeatMasker
CG15211CG15211CG15211CG15211
Ant2Ant2
sesBsesBsesBsesB
CG15211CG15211CG15211CG15211
Ant2Ant2
sesBsesBsesBsesB
_ 1000
_ 0
_ 1000
_ 0
ps RNAi
S2 Untreated
Splice JunctionReads
GenomicReads
Splice JunctionReads
GenomicReads
Image from Brenton Gravely
Image courtesy of Brenton Graveley
7 38
Mapping reads to the transcriptome 4
Junction reads zoom
chrX
Conservation
d_simulansd_sechelliad_yakubad_erectad_ananassaed_pseudoobscurad_persimilisd_willistonid_virilisd_mojavensisd_grimshawia_gambiaea_melliferat_castaneum
1067300010673500106740001067450010675000106755001067600010676500106770001067750010678000106785001067900010679500106800001068050010681000
CG8144_6lane_0MM
S2_DRSC_6_lanes
FlyBase Protein-Coding Genes
RefSeq Genes
12 Flies Mosquito Honeybee Beetle Multiz Alignments amp phastCons Scores
Repeating Elements by RepeatMasker
CG15211CG15211CG15211CG15211
Ant2Ant2
sesBsesBsesBsesB
CG15211CG15211CG15211CG15211
Ant2Ant2
sesBsesBsesBsesB
_ 1000
_ 0
_ 1000
_ 0
ps RNAi
S2 Untreated
Splice JunctionReads
GenomicReads
Splice JunctionReads
GenomicReads
Image from Brenton Gravely
Image courtesy of Brenton Graveley
8 38
Sequencing errors Genome differences
Sample Genome
Ref Genome
True read
read with sequence error
Sample different from reference
Sequence errorMapping
Evidence suggests that Illumina sequencing does not introduce indels
9 38
Alignment Tools
The number of short read aligners have exploded but a couple tools haveemerged as the de facto standards
I Eland Illuminarsquos aligner quality aware fast paired end capable
I MAQ Good SNP caller quality aware paired end capable
I Bowtie Super fast offers different alignment strategies paired endcapable
I BWA Fast indel support paired ends qualities
I NovoAlign MAQ like speed many features
A superior overview of the different aligners is available athttpwwwsangeracukUserslh3NGSalignshtml Additionallya comparison of two of the best aligners can be found here httpwwwmassgenomicsorg200907maq-bwa-and-bowtie-comparedhtml
10 38
Common Alignment Strategies
I Use qualities (default for Bowtie and MAQ)
I Perfect match no repeats (Strict Lenient)
I Mismatches no repeats
I Paired end data (PET) (harder for RNA-Seq)
The standard Illumina protocol yields unstranded reads
Most aligners are evaluated in terms of ldquohow many reads are mappedrdquo Isthis the right objective
Watch out for the output of the program there are many differentconventions (0-based what happens to read hitting the reverse strandetc)
11 38
SAMBAM Formats
SAM (BAM) is a new general format for storing mapped reads It isdeveloped as part of the 1000 Genomes project and is quickly becoming akind of standard Some alignment tools output this format directlyotherwise there are scripts in samtools for doing it for most popularaligners Details at httpsamtoolssourceforgenetindexshtml
12 38
A comment
There are no great comprehensive tools for analyzing deep-sequence dataIt will involve a fair amount of coding and gluing together various tools
We will introduce some tools from Bioconductor that can be useful
I use a lot of shell scripting
13 38
Biostrings overview
I A package for working with large (biological) strings
I Two main types of objects A really long single string (thinkchromosome) or a set of shorter strings (think reads or genes)BString vs BStringSet
I These classes are implemented efficiently minimizing copy andmemory loadingunloading
I The BSgenome contains some infrastructure for whole genomes
I Methods for dealing with biological data including basicmanipulation (complementation translation etc) string searching(exactinexact matching Smith-Waterman PWMs)
I Fairly complicated class structure
Irsquom sorry to say that at least for me this has becomehopelessly confusing and I imagine that many other usersfeel the same ndash Simon Anders on bioc-sig-sequencing
Computing on genomes is not trivial The approach to a givencomputation is important
14 38
Strings in Biostrings
I BString (general) DNAString RNAString AAString (Amino Acid)all examples of XString
I complement reverse reverseComplement translate for the classeswhere ldquoit makes senserdquo
I Convert to and from a standard R character string
I Constructor DNAString(ACGGGGG)
I Support for IUPAC alphabet
I Subsetting subseq using the SEW format (two out of the threestartendwidth) Efficient
I StringSets are collections of Strings like DNAStringSet
15 38
BSgenomes
As examples we will use whole genomes Use availablegenomes to getavailable genomes Long package names but always a shorter objectname
gt library(BSgenomeScerevisiaeUCSCsacCer1)
gt Scerevisiae
gt Scerevisiae[[1]]
gt Scerevisiae[[chr1]]
A BSgenome may also have masks We will ignore this for now
16 38
Biostrings more
I alphabetFrequency oligonucleotideFrequency and others
gt alphabetFrequency(Scerevisiae[[chr1]])
gt oligonucleotideFrequency(Scerevisiae[[chr1]]
+ width = 3)
I chartr for character translation (ldquomake all As into Csrdquo)
I Various IO functions (also ShortRead)
17 38
Views
A view is a set of substrings of an XString stored and manipulated veryefficiently Example to store exon sequences one can just store thegenomic location of the exons
gt Views(Scerevisiae[[chr1]] start = c(300
+ 400 500) width = 50)
Views on a 230208-letter DNAString subjectsubject CCACACCACACCCAGTGGTGTGTGTGGGviews
start end width[1] 300 349 50 [CTGTTCTTAAATAAC][2] 400 449 50 [CCCTCACTAGTATAT][3] 500 549 50 [TCTCTCACCGGCACT]
A view is associated with a subject Internally they are essentiallyIRanges so they are very fast to compute with Views can in many casesbe treated exactly as other strings There are methods such as narrowtrim gaps restrict
18 38
Matching in Biostrings
There are various ways of matching or aligning strings to each other Weuse matching to denote searching for an exact match or possibly a matchwith a certain number of mismatches Alignment denotes a more generalstrategy eg Smith-Waterman
I matchPattern countPattern match 1 sequence to 1 sequence
I vmatchPattern vcountPattern match 1 sequence to manysequences
I pairwiseAlignment align many sequences to 1 sequence
I matchPDict countPDict match many sequences to 1 sequence(ldquodictrdquo indicates that the many sequences are preprocessed into adictionary)
I matchPWM trimLRpattern
The different functions are optimized for different situations They alsouse different algorithms which has a big impact EspeciallypairwiseAlignment is flexible and therefore has a complicated syntax
19 38
Example mapping probes to a genome
Get a list of Scerevisiae probes from the ldquoyeast2rdquo Affymetrix array
gt library(yeast2probe)
gt ids lt- scan(s_pombemsk skip = 2
+ what = list(probeset = character(0)
+ junk = character(0)))$probeset
gt probes lt- yeast2probe$sequence[yeast2probe$ProbeSetName in
+ ids]
gt probes lt- DNAStringSet(probes)
Mapping
gt require(BSgenomeScerevisiaeUCSCsacCer1)
gt dict0 lt- PDict(probes)
gt dict0r lt- PDict(reverseComplement(probes))
gt hits lt- matchPDict(dict0 Scerevisiae[[1]])
gt table(countIndex(hits))
gt table(countPDict(dict0 Scerevisiae[[1]]))
20 38
Example contrsquod
Over all chromosomes
gt allhits lt- lapply(116 function(i)
+ countPDict(dict0 Scerevisiae[[i]]) +
+ countPDict(dict0r Scerevisiae[[i]])
+ )
gt table(rowSums(docall(cbind allhits)))
21 38
ShortRead
ShortRead contains tools for
I Work with GERALDBUSTARD output
I Generate QA report on BUSTARD files
I Read a variety of short read data formats
22 38
Data
We will use data from (Lee Hansen Bullard Dudoit and Sherlock(2008) PLoS Genetics)
We are considering a wild-type and a mutant strain of yeast both grownin rich media The two strains were sequenced using an Illumina GenomeAnalyzer
We are using a subset of the data 1M reads from each of two lanes ofthe two strains
23 38
Reading the unaligned data
gt require(ShortRead)
gt fq lt- readFastq(seqdata mut_1_ffastq$)
gt fq
class ShortReadQlength 1000000 reads width 36 cycles
There are various simple accessor functions for this object especiallysread and quality
24 38
A brief look at the unaligned data
Using alphabetByCycle and as( rdquomatrixrdquo) (which creates a big read timescycle matrix) we can do
gt alp lt- alphabetByCycle(sread(fq))
gt matplot(t(proptable(alp[DNA_BASES
+ ] margin = 2)) type = l)
gt qaMatraw lt- as(quality(fq) matrix)
gt plot(colMeans(qaMatraw))
25 38
Aligning the data
We now align the data using Bowtie
binbash
BOWTIE_OPTS=-m 1 -v 2 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-m 1 -v 0 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 2 -k 2 -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 0 -k 1 -p 2 -q --quiet -3 10
for f in `ls seqdatafastq`dobowtie s_cerevisiae $BOWTIE_OPTS $f gt $fbowtiedone
How many hits do we get
26 38
Reading in the aligned data
We read the data into R as a 4-component list (one lane per component)
gt files lt- listfiles(seqdata
+ pattern = bowtie)
gt aligned lt- sapply(files function(f)
+ readAligned(seqdata pattern = f
+ type = Bowtie)
+ )
gt names(aligned) lt- gsub(_ffastqbowtie
+ names(aligned))
gt aligned[[1]]
gt save(aligned file = alignedrda)
27 38
Some exercises
1 Determine the number of times the motif ldquoTATAArdquo occurs in theyeast genome How often does it occur with one mismatch
2 The seqdata directory has an object called ste12 which is aposition weight matrix for the transcription factor ldquoSte12rdquo (obtainedfrom SCPD) Where does it occur in the genome
3 Compute the average quality for each cycle for the aligned reads andcompare to the qualities for the unaligned reads What is thedifference
28 38
Solution 1
gt sum(sapply(116 function(i)
+ motif lt- DNAString(TATAA)
+ countPattern(motif Scerevisiae[[i]]) +
+ countPattern(reverseComplement(motif)
+ Scerevisiae[[i]])
+ ))
29 38
Solution 2
30 38
Solution 3
31 38
SessionInfo
gt toLatex(sessionInfo())
I R version 292 RC (2009-08-17 r49312) i386-apple-darwin980
I Localeen_USUTF-8en_USUTF-8CCen_USUTF-8en_USUTF-8
I Base packages base datasets graphics grDevices grid methodsstats utils
I Other packages Biostrings 2128 BSgenome 1123BSgenomeScerevisiaeUCSCsacCer1 1313 classGraph 07-2graph 1222 IRanges 123 lattice 017-25 Rgraphviz 1234ShortRead 121
I Loaded via a namespace (and not attached) Biobase 241hwriter 11 tools 292
32 38
Some ShortRead classes
ShortRead
ShortReadQ
AlignedRead
33 38
Some Biostrings classes (single strings)
XString
BString DNAString RNAString AAString
34 38
Some Biostrings classes (sets of strings)
XStringSet
BStringSet DNAStringSet RNAStringSet AAStringSet PhredQualitySolexaQualityQualityScaledXStringSet
QualityScaledBStringSetQualityScaledDNAStringSetQualityScaledRNAStringSetQualityScaledAAStringSet
35 38

A comment
Analysis of high-throughput sequencing data and especially RNA-Seq isstill in its infancy
It is unclear what is the best way to think about and analyze these dataIt is also unclear are the right entities to compute on
We focus on some tools and some computations we have found useful
3 38
Alignment input FASTQ files
FASTQ files represent a common ldquoend-pointrdquo from the various sequencingplatforms (ie NCBI short read archivehttpwwwncbinlmnihgovTracessrasracgi) Qualities areencoded in ASCII and depending on the platform have slightly differentmeaningsranges and encoding More details can be found athttpenwikipediaorgwikiFASTQ_format
GA-EAS46_2_209DG61890752TTCTCTTAAGTCTTCTAGTTCTCTTCTTTCTCCACT+GA-EAS46_2_209DG61890752hhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhGA-EAS46_2_209DG61905558TCTGGCTTAACTTCTTCTTTTTTTTCTTCTTCTTCT+GA-EAS46_2_209DG61905558hhhfhhhhhhhhhhhhhhhhhhhhhhhhehhGhhJh
4 38
Mapping reads to the transcriptome
Mapping reads to the transcriptome
Transcriptome
2^Genome
Reads
Genome
Illustration from Lior Patcher
Well established
5 38
Mapping reads to the transcriptome 2
Mapping transcripts
Genome
Transcript
Length in genome space
paired-end reads
6 38
Mapping reads to the transcriptome 3
Junction reads
chrX
Conservation
d_simulansd_sechelliad_yakubad_erectad_ananassaed_pseudoobscurad_persimilisd_willistonid_virilisd_mojavensisd_grimshawia_gambiaea_melliferat_castaneum
1067300010673500106740001067450010675000106755001067600010676500106770001067750010678000106785001067900010679500106800001068050010681000
CG8144_6lane_0MM
S2_DRSC_6_lanes
FlyBase Protein-Coding Genes
RefSeq Genes
12 Flies Mosquito Honeybee Beetle Multiz Alignments amp phastCons Scores
Repeating Elements by RepeatMasker
CG15211CG15211CG15211CG15211
Ant2Ant2
sesBsesBsesBsesB
CG15211CG15211CG15211CG15211
Ant2Ant2
sesBsesBsesBsesB
_ 1000
_ 0
_ 1000
_ 0
ps RNAi
S2 Untreated
Splice JunctionReads
GenomicReads
Splice JunctionReads
GenomicReads
Image from Brenton Gravely
Image courtesy of Brenton Graveley
7 38
Mapping reads to the transcriptome 4
Junction reads zoom
chrX
Conservation
d_simulansd_sechelliad_yakubad_erectad_ananassaed_pseudoobscurad_persimilisd_willistonid_virilisd_mojavensisd_grimshawia_gambiaea_melliferat_castaneum
1067300010673500106740001067450010675000106755001067600010676500106770001067750010678000106785001067900010679500106800001068050010681000
CG8144_6lane_0MM
S2_DRSC_6_lanes
FlyBase Protein-Coding Genes
RefSeq Genes
12 Flies Mosquito Honeybee Beetle Multiz Alignments amp phastCons Scores
Repeating Elements by RepeatMasker
CG15211CG15211CG15211CG15211
Ant2Ant2
sesBsesBsesBsesB
CG15211CG15211CG15211CG15211
Ant2Ant2
sesBsesBsesBsesB
_ 1000
_ 0
_ 1000
_ 0
ps RNAi
S2 Untreated
Splice JunctionReads
GenomicReads
Splice JunctionReads
GenomicReads
Image from Brenton Gravely
Image courtesy of Brenton Graveley
8 38
Sequencing errors Genome differences
Sample Genome
Ref Genome
True read
read with sequence error
Sample different from reference
Sequence errorMapping
Evidence suggests that Illumina sequencing does not introduce indels
9 38
Alignment Tools
The number of short read aligners have exploded but a couple tools haveemerged as the de facto standards
I Eland Illuminarsquos aligner quality aware fast paired end capable
I MAQ Good SNP caller quality aware paired end capable
I Bowtie Super fast offers different alignment strategies paired endcapable
I BWA Fast indel support paired ends qualities
I NovoAlign MAQ like speed many features
A superior overview of the different aligners is available athttpwwwsangeracukUserslh3NGSalignshtml Additionallya comparison of two of the best aligners can be found here httpwwwmassgenomicsorg200907maq-bwa-and-bowtie-comparedhtml
10 38
Common Alignment Strategies
I Use qualities (default for Bowtie and MAQ)
I Perfect match no repeats (Strict Lenient)
I Mismatches no repeats
I Paired end data (PET) (harder for RNA-Seq)
The standard Illumina protocol yields unstranded reads
Most aligners are evaluated in terms of ldquohow many reads are mappedrdquo Isthis the right objective
Watch out for the output of the program there are many differentconventions (0-based what happens to read hitting the reverse strandetc)
11 38
SAMBAM Formats
SAM (BAM) is a new general format for storing mapped reads It isdeveloped as part of the 1000 Genomes project and is quickly becoming akind of standard Some alignment tools output this format directlyotherwise there are scripts in samtools for doing it for most popularaligners Details at httpsamtoolssourceforgenetindexshtml
12 38
A comment
There are no great comprehensive tools for analyzing deep-sequence dataIt will involve a fair amount of coding and gluing together various tools
We will introduce some tools from Bioconductor that can be useful
I use a lot of shell scripting
13 38
Biostrings overview
I A package for working with large (biological) strings
I Two main types of objects A really long single string (thinkchromosome) or a set of shorter strings (think reads or genes)BString vs BStringSet
I These classes are implemented efficiently minimizing copy andmemory loadingunloading
I The BSgenome contains some infrastructure for whole genomes
I Methods for dealing with biological data including basicmanipulation (complementation translation etc) string searching(exactinexact matching Smith-Waterman PWMs)
I Fairly complicated class structure
Irsquom sorry to say that at least for me this has becomehopelessly confusing and I imagine that many other usersfeel the same ndash Simon Anders on bioc-sig-sequencing
Computing on genomes is not trivial The approach to a givencomputation is important
14 38
Strings in Biostrings
I BString (general) DNAString RNAString AAString (Amino Acid)all examples of XString
I complement reverse reverseComplement translate for the classeswhere ldquoit makes senserdquo
I Convert to and from a standard R character string
I Constructor DNAString(ACGGGGG)
I Support for IUPAC alphabet
I Subsetting subseq using the SEW format (two out of the threestartendwidth) Efficient
I StringSets are collections of Strings like DNAStringSet
15 38
BSgenomes
As examples we will use whole genomes Use availablegenomes to getavailable genomes Long package names but always a shorter objectname
gt library(BSgenomeScerevisiaeUCSCsacCer1)
gt Scerevisiae
gt Scerevisiae[[1]]
gt Scerevisiae[[chr1]]
A BSgenome may also have masks We will ignore this for now
16 38
Biostrings more
I alphabetFrequency oligonucleotideFrequency and others
gt alphabetFrequency(Scerevisiae[[chr1]])
gt oligonucleotideFrequency(Scerevisiae[[chr1]]
+ width = 3)
I chartr for character translation (ldquomake all As into Csrdquo)
I Various IO functions (also ShortRead)
17 38
Views
A view is a set of substrings of an XString stored and manipulated veryefficiently Example to store exon sequences one can just store thegenomic location of the exons
gt Views(Scerevisiae[[chr1]] start = c(300
+ 400 500) width = 50)
Views on a 230208-letter DNAString subjectsubject CCACACCACACCCAGTGGTGTGTGTGGGviews
start end width[1] 300 349 50 [CTGTTCTTAAATAAC][2] 400 449 50 [CCCTCACTAGTATAT][3] 500 549 50 [TCTCTCACCGGCACT]
A view is associated with a subject Internally they are essentiallyIRanges so they are very fast to compute with Views can in many casesbe treated exactly as other strings There are methods such as narrowtrim gaps restrict
18 38
Matching in Biostrings
There are various ways of matching or aligning strings to each other Weuse matching to denote searching for an exact match or possibly a matchwith a certain number of mismatches Alignment denotes a more generalstrategy eg Smith-Waterman
I matchPattern countPattern match 1 sequence to 1 sequence
I vmatchPattern vcountPattern match 1 sequence to manysequences
I pairwiseAlignment align many sequences to 1 sequence
I matchPDict countPDict match many sequences to 1 sequence(ldquodictrdquo indicates that the many sequences are preprocessed into adictionary)
I matchPWM trimLRpattern
The different functions are optimized for different situations They alsouse different algorithms which has a big impact EspeciallypairwiseAlignment is flexible and therefore has a complicated syntax
19 38
Example mapping probes to a genome
Get a list of Scerevisiae probes from the ldquoyeast2rdquo Affymetrix array
gt library(yeast2probe)
gt ids lt- scan(s_pombemsk skip = 2
+ what = list(probeset = character(0)
+ junk = character(0)))$probeset
gt probes lt- yeast2probe$sequence[yeast2probe$ProbeSetName in
+ ids]
gt probes lt- DNAStringSet(probes)
Mapping
gt require(BSgenomeScerevisiaeUCSCsacCer1)
gt dict0 lt- PDict(probes)
gt dict0r lt- PDict(reverseComplement(probes))
gt hits lt- matchPDict(dict0 Scerevisiae[[1]])
gt table(countIndex(hits))
gt table(countPDict(dict0 Scerevisiae[[1]]))
20 38
Example contrsquod
Over all chromosomes
gt allhits lt- lapply(116 function(i)
+ countPDict(dict0 Scerevisiae[[i]]) +
+ countPDict(dict0r Scerevisiae[[i]])
+ )
gt table(rowSums(docall(cbind allhits)))
21 38
ShortRead
ShortRead contains tools for
I Work with GERALDBUSTARD output
I Generate QA report on BUSTARD files
I Read a variety of short read data formats
22 38
Data
We will use data from (Lee Hansen Bullard Dudoit and Sherlock(2008) PLoS Genetics)
We are considering a wild-type and a mutant strain of yeast both grownin rich media The two strains were sequenced using an Illumina GenomeAnalyzer
We are using a subset of the data 1M reads from each of two lanes ofthe two strains
23 38
Reading the unaligned data
gt require(ShortRead)
gt fq lt- readFastq(seqdata mut_1_ffastq$)
gt fq
class ShortReadQlength 1000000 reads width 36 cycles
There are various simple accessor functions for this object especiallysread and quality
24 38
A brief look at the unaligned data
Using alphabetByCycle and as( rdquomatrixrdquo) (which creates a big read timescycle matrix) we can do
gt alp lt- alphabetByCycle(sread(fq))
gt matplot(t(proptable(alp[DNA_BASES
+ ] margin = 2)) type = l)
gt qaMatraw lt- as(quality(fq) matrix)
gt plot(colMeans(qaMatraw))
25 38
Aligning the data
We now align the data using Bowtie
binbash
BOWTIE_OPTS=-m 1 -v 2 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-m 1 -v 0 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 2 -k 2 -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 0 -k 1 -p 2 -q --quiet -3 10
for f in `ls seqdatafastq`dobowtie s_cerevisiae $BOWTIE_OPTS $f gt $fbowtiedone
How many hits do we get
26 38
Reading in the aligned data
We read the data into R as a 4-component list (one lane per component)
gt files lt- listfiles(seqdata
+ pattern = bowtie)
gt aligned lt- sapply(files function(f)
+ readAligned(seqdata pattern = f
+ type = Bowtie)
+ )
gt names(aligned) lt- gsub(_ffastqbowtie
+ names(aligned))
gt aligned[[1]]
gt save(aligned file = alignedrda)
27 38
Some exercises
1 Determine the number of times the motif ldquoTATAArdquo occurs in theyeast genome How often does it occur with one mismatch
2 The seqdata directory has an object called ste12 which is aposition weight matrix for the transcription factor ldquoSte12rdquo (obtainedfrom SCPD) Where does it occur in the genome
3 Compute the average quality for each cycle for the aligned reads andcompare to the qualities for the unaligned reads What is thedifference
28 38
Solution 1
gt sum(sapply(116 function(i)
+ motif lt- DNAString(TATAA)
+ countPattern(motif Scerevisiae[[i]]) +
+ countPattern(reverseComplement(motif)
+ Scerevisiae[[i]])
+ ))
29 38
Solution 2
30 38
Solution 3
31 38
SessionInfo
gt toLatex(sessionInfo())
I R version 292 RC (2009-08-17 r49312) i386-apple-darwin980
I Localeen_USUTF-8en_USUTF-8CCen_USUTF-8en_USUTF-8
I Base packages base datasets graphics grDevices grid methodsstats utils
I Other packages Biostrings 2128 BSgenome 1123BSgenomeScerevisiaeUCSCsacCer1 1313 classGraph 07-2graph 1222 IRanges 123 lattice 017-25 Rgraphviz 1234ShortRead 121
I Loaded via a namespace (and not attached) Biobase 241hwriter 11 tools 292
32 38
Some ShortRead classes
ShortRead
ShortReadQ
AlignedRead
33 38
Some Biostrings classes (single strings)
XString
BString DNAString RNAString AAString
34 38
Some Biostrings classes (sets of strings)
XStringSet
BStringSet DNAStringSet RNAStringSet AAStringSet PhredQualitySolexaQualityQualityScaledXStringSet
QualityScaledBStringSetQualityScaledDNAStringSetQualityScaledRNAStringSetQualityScaledAAStringSet
35 38

Alignment input FASTQ files
FASTQ files represent a common ldquoend-pointrdquo from the various sequencingplatforms (ie NCBI short read archivehttpwwwncbinlmnihgovTracessrasracgi) Qualities areencoded in ASCII and depending on the platform have slightly differentmeaningsranges and encoding More details can be found athttpenwikipediaorgwikiFASTQ_format
GA-EAS46_2_209DG61890752TTCTCTTAAGTCTTCTAGTTCTCTTCTTTCTCCACT+GA-EAS46_2_209DG61890752hhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhhGA-EAS46_2_209DG61905558TCTGGCTTAACTTCTTCTTTTTTTTCTTCTTCTTCT+GA-EAS46_2_209DG61905558hhhfhhhhhhhhhhhhhhhhhhhhhhhhehhGhhJh
4 38
Mapping reads to the transcriptome
Mapping reads to the transcriptome
Transcriptome
2^Genome
Reads
Genome
Illustration from Lior Patcher
Well established
5 38
Mapping reads to the transcriptome 2
Mapping transcripts
Genome
Transcript
Length in genome space
paired-end reads
6 38
Mapping reads to the transcriptome 3
Junction reads
chrX
Conservation
d_simulansd_sechelliad_yakubad_erectad_ananassaed_pseudoobscurad_persimilisd_willistonid_virilisd_mojavensisd_grimshawia_gambiaea_melliferat_castaneum
1067300010673500106740001067450010675000106755001067600010676500106770001067750010678000106785001067900010679500106800001068050010681000
CG8144_6lane_0MM
S2_DRSC_6_lanes
FlyBase Protein-Coding Genes
RefSeq Genes
12 Flies Mosquito Honeybee Beetle Multiz Alignments amp phastCons Scores
Repeating Elements by RepeatMasker
CG15211CG15211CG15211CG15211
Ant2Ant2
sesBsesBsesBsesB
CG15211CG15211CG15211CG15211
Ant2Ant2
sesBsesBsesBsesB
_ 1000
_ 0
_ 1000
_ 0
ps RNAi
S2 Untreated
Splice JunctionReads
GenomicReads
Splice JunctionReads
GenomicReads
Image from Brenton Gravely
Image courtesy of Brenton Graveley
7 38
Mapping reads to the transcriptome 4
Junction reads zoom
chrX
Conservation
d_simulansd_sechelliad_yakubad_erectad_ananassaed_pseudoobscurad_persimilisd_willistonid_virilisd_mojavensisd_grimshawia_gambiaea_melliferat_castaneum
1067300010673500106740001067450010675000106755001067600010676500106770001067750010678000106785001067900010679500106800001068050010681000
CG8144_6lane_0MM
S2_DRSC_6_lanes
FlyBase Protein-Coding Genes
RefSeq Genes
12 Flies Mosquito Honeybee Beetle Multiz Alignments amp phastCons Scores
Repeating Elements by RepeatMasker
CG15211CG15211CG15211CG15211
Ant2Ant2
sesBsesBsesBsesB
CG15211CG15211CG15211CG15211
Ant2Ant2
sesBsesBsesBsesB
_ 1000
_ 0
_ 1000
_ 0
ps RNAi
S2 Untreated
Splice JunctionReads
GenomicReads
Splice JunctionReads
GenomicReads
Image from Brenton Gravely
Image courtesy of Brenton Graveley
8 38
Sequencing errors Genome differences
Sample Genome
Ref Genome
True read
read with sequence error
Sample different from reference
Sequence errorMapping
Evidence suggests that Illumina sequencing does not introduce indels
9 38
Alignment Tools
The number of short read aligners have exploded but a couple tools haveemerged as the de facto standards
I Eland Illuminarsquos aligner quality aware fast paired end capable
I MAQ Good SNP caller quality aware paired end capable
I Bowtie Super fast offers different alignment strategies paired endcapable
I BWA Fast indel support paired ends qualities
I NovoAlign MAQ like speed many features
A superior overview of the different aligners is available athttpwwwsangeracukUserslh3NGSalignshtml Additionallya comparison of two of the best aligners can be found here httpwwwmassgenomicsorg200907maq-bwa-and-bowtie-comparedhtml
10 38
Common Alignment Strategies
I Use qualities (default for Bowtie and MAQ)
I Perfect match no repeats (Strict Lenient)
I Mismatches no repeats
I Paired end data (PET) (harder for RNA-Seq)
The standard Illumina protocol yields unstranded reads
Most aligners are evaluated in terms of ldquohow many reads are mappedrdquo Isthis the right objective
Watch out for the output of the program there are many differentconventions (0-based what happens to read hitting the reverse strandetc)
11 38
SAMBAM Formats
SAM (BAM) is a new general format for storing mapped reads It isdeveloped as part of the 1000 Genomes project and is quickly becoming akind of standard Some alignment tools output this format directlyotherwise there are scripts in samtools for doing it for most popularaligners Details at httpsamtoolssourceforgenetindexshtml
12 38
A comment
There are no great comprehensive tools for analyzing deep-sequence dataIt will involve a fair amount of coding and gluing together various tools
We will introduce some tools from Bioconductor that can be useful
I use a lot of shell scripting
13 38
Biostrings overview
I A package for working with large (biological) strings
I Two main types of objects A really long single string (thinkchromosome) or a set of shorter strings (think reads or genes)BString vs BStringSet
I These classes are implemented efficiently minimizing copy andmemory loadingunloading
I The BSgenome contains some infrastructure for whole genomes
I Methods for dealing with biological data including basicmanipulation (complementation translation etc) string searching(exactinexact matching Smith-Waterman PWMs)
I Fairly complicated class structure
Irsquom sorry to say that at least for me this has becomehopelessly confusing and I imagine that many other usersfeel the same ndash Simon Anders on bioc-sig-sequencing
Computing on genomes is not trivial The approach to a givencomputation is important
14 38
Strings in Biostrings
I BString (general) DNAString RNAString AAString (Amino Acid)all examples of XString
I complement reverse reverseComplement translate for the classeswhere ldquoit makes senserdquo
I Convert to and from a standard R character string
I Constructor DNAString(ACGGGGG)
I Support for IUPAC alphabet
I Subsetting subseq using the SEW format (two out of the threestartendwidth) Efficient
I StringSets are collections of Strings like DNAStringSet
15 38
BSgenomes
As examples we will use whole genomes Use availablegenomes to getavailable genomes Long package names but always a shorter objectname
gt library(BSgenomeScerevisiaeUCSCsacCer1)
gt Scerevisiae
gt Scerevisiae[[1]]
gt Scerevisiae[[chr1]]
A BSgenome may also have masks We will ignore this for now
16 38
Biostrings more
I alphabetFrequency oligonucleotideFrequency and others
gt alphabetFrequency(Scerevisiae[[chr1]])
gt oligonucleotideFrequency(Scerevisiae[[chr1]]
+ width = 3)
I chartr for character translation (ldquomake all As into Csrdquo)
I Various IO functions (also ShortRead)
17 38
Views
A view is a set of substrings of an XString stored and manipulated veryefficiently Example to store exon sequences one can just store thegenomic location of the exons
gt Views(Scerevisiae[[chr1]] start = c(300
+ 400 500) width = 50)
Views on a 230208-letter DNAString subjectsubject CCACACCACACCCAGTGGTGTGTGTGGGviews
start end width[1] 300 349 50 [CTGTTCTTAAATAAC][2] 400 449 50 [CCCTCACTAGTATAT][3] 500 549 50 [TCTCTCACCGGCACT]
A view is associated with a subject Internally they are essentiallyIRanges so they are very fast to compute with Views can in many casesbe treated exactly as other strings There are methods such as narrowtrim gaps restrict
18 38
Matching in Biostrings
There are various ways of matching or aligning strings to each other Weuse matching to denote searching for an exact match or possibly a matchwith a certain number of mismatches Alignment denotes a more generalstrategy eg Smith-Waterman
I matchPattern countPattern match 1 sequence to 1 sequence
I vmatchPattern vcountPattern match 1 sequence to manysequences
I pairwiseAlignment align many sequences to 1 sequence
I matchPDict countPDict match many sequences to 1 sequence(ldquodictrdquo indicates that the many sequences are preprocessed into adictionary)
I matchPWM trimLRpattern
The different functions are optimized for different situations They alsouse different algorithms which has a big impact EspeciallypairwiseAlignment is flexible and therefore has a complicated syntax
19 38
Example mapping probes to a genome
Get a list of Scerevisiae probes from the ldquoyeast2rdquo Affymetrix array
gt library(yeast2probe)
gt ids lt- scan(s_pombemsk skip = 2
+ what = list(probeset = character(0)
+ junk = character(0)))$probeset
gt probes lt- yeast2probe$sequence[yeast2probe$ProbeSetName in
+ ids]
gt probes lt- DNAStringSet(probes)
Mapping
gt require(BSgenomeScerevisiaeUCSCsacCer1)
gt dict0 lt- PDict(probes)
gt dict0r lt- PDict(reverseComplement(probes))
gt hits lt- matchPDict(dict0 Scerevisiae[[1]])
gt table(countIndex(hits))
gt table(countPDict(dict0 Scerevisiae[[1]]))
20 38
Example contrsquod
Over all chromosomes
gt allhits lt- lapply(116 function(i)
+ countPDict(dict0 Scerevisiae[[i]]) +
+ countPDict(dict0r Scerevisiae[[i]])
+ )
gt table(rowSums(docall(cbind allhits)))
21 38
ShortRead
ShortRead contains tools for
I Work with GERALDBUSTARD output
I Generate QA report on BUSTARD files
I Read a variety of short read data formats
22 38
Data
We will use data from (Lee Hansen Bullard Dudoit and Sherlock(2008) PLoS Genetics)
We are considering a wild-type and a mutant strain of yeast both grownin rich media The two strains were sequenced using an Illumina GenomeAnalyzer
We are using a subset of the data 1M reads from each of two lanes ofthe two strains
23 38
Reading the unaligned data
gt require(ShortRead)
gt fq lt- readFastq(seqdata mut_1_ffastq$)
gt fq
class ShortReadQlength 1000000 reads width 36 cycles
There are various simple accessor functions for this object especiallysread and quality
24 38
A brief look at the unaligned data
Using alphabetByCycle and as( rdquomatrixrdquo) (which creates a big read timescycle matrix) we can do
gt alp lt- alphabetByCycle(sread(fq))
gt matplot(t(proptable(alp[DNA_BASES
+ ] margin = 2)) type = l)
gt qaMatraw lt- as(quality(fq) matrix)
gt plot(colMeans(qaMatraw))
25 38
Aligning the data
We now align the data using Bowtie
binbash
BOWTIE_OPTS=-m 1 -v 2 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-m 1 -v 0 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 2 -k 2 -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 0 -k 1 -p 2 -q --quiet -3 10
for f in `ls seqdatafastq`dobowtie s_cerevisiae $BOWTIE_OPTS $f gt $fbowtiedone
How many hits do we get
26 38
Reading in the aligned data
We read the data into R as a 4-component list (one lane per component)
gt files lt- listfiles(seqdata
+ pattern = bowtie)
gt aligned lt- sapply(files function(f)
+ readAligned(seqdata pattern = f
+ type = Bowtie)
+ )
gt names(aligned) lt- gsub(_ffastqbowtie
+ names(aligned))
gt aligned[[1]]
gt save(aligned file = alignedrda)
27 38
Some exercises
1 Determine the number of times the motif ldquoTATAArdquo occurs in theyeast genome How often does it occur with one mismatch
2 The seqdata directory has an object called ste12 which is aposition weight matrix for the transcription factor ldquoSte12rdquo (obtainedfrom SCPD) Where does it occur in the genome
3 Compute the average quality for each cycle for the aligned reads andcompare to the qualities for the unaligned reads What is thedifference
28 38
Solution 1
gt sum(sapply(116 function(i)
+ motif lt- DNAString(TATAA)
+ countPattern(motif Scerevisiae[[i]]) +
+ countPattern(reverseComplement(motif)
+ Scerevisiae[[i]])
+ ))
29 38
Solution 2
30 38
Solution 3
31 38
SessionInfo
gt toLatex(sessionInfo())
I R version 292 RC (2009-08-17 r49312) i386-apple-darwin980
I Localeen_USUTF-8en_USUTF-8CCen_USUTF-8en_USUTF-8
I Base packages base datasets graphics grDevices grid methodsstats utils
I Other packages Biostrings 2128 BSgenome 1123BSgenomeScerevisiaeUCSCsacCer1 1313 classGraph 07-2graph 1222 IRanges 123 lattice 017-25 Rgraphviz 1234ShortRead 121
I Loaded via a namespace (and not attached) Biobase 241hwriter 11 tools 292
32 38
Some ShortRead classes
ShortRead
ShortReadQ
AlignedRead
33 38
Some Biostrings classes (single strings)
XString
BString DNAString RNAString AAString
34 38
Some Biostrings classes (sets of strings)
XStringSet
BStringSet DNAStringSet RNAStringSet AAStringSet PhredQualitySolexaQualityQualityScaledXStringSet
QualityScaledBStringSetQualityScaledDNAStringSetQualityScaledRNAStringSetQualityScaledAAStringSet
35 38
Mapping reads to the transcriptome
Mapping reads to the transcriptome
Transcriptome
2^Genome
Reads
Genome
Illustration from Lior Patcher
Well established
5 38
Mapping reads to the transcriptome 2
Mapping transcripts
Genome
Transcript
Length in genome space
paired-end reads
6 38
Mapping reads to the transcriptome 3
Junction reads
chrX
Conservation
d_simulansd_sechelliad_yakubad_erectad_ananassaed_pseudoobscurad_persimilisd_willistonid_virilisd_mojavensisd_grimshawia_gambiaea_melliferat_castaneum
1067300010673500106740001067450010675000106755001067600010676500106770001067750010678000106785001067900010679500106800001068050010681000
CG8144_6lane_0MM
S2_DRSC_6_lanes
FlyBase Protein-Coding Genes
RefSeq Genes
12 Flies Mosquito Honeybee Beetle Multiz Alignments amp phastCons Scores
Repeating Elements by RepeatMasker
CG15211CG15211CG15211CG15211
Ant2Ant2
sesBsesBsesBsesB
CG15211CG15211CG15211CG15211
Ant2Ant2
sesBsesBsesBsesB
_ 1000
_ 0
_ 1000
_ 0
ps RNAi
S2 Untreated
Splice JunctionReads
GenomicReads
Splice JunctionReads
GenomicReads
Image from Brenton Gravely
Image courtesy of Brenton Graveley
7 38
Mapping reads to the transcriptome 4
Junction reads zoom
chrX
Conservation
d_simulansd_sechelliad_yakubad_erectad_ananassaed_pseudoobscurad_persimilisd_willistonid_virilisd_mojavensisd_grimshawia_gambiaea_melliferat_castaneum
1067300010673500106740001067450010675000106755001067600010676500106770001067750010678000106785001067900010679500106800001068050010681000
CG8144_6lane_0MM
S2_DRSC_6_lanes
FlyBase Protein-Coding Genes
RefSeq Genes
12 Flies Mosquito Honeybee Beetle Multiz Alignments amp phastCons Scores
Repeating Elements by RepeatMasker
CG15211CG15211CG15211CG15211
Ant2Ant2
sesBsesBsesBsesB
CG15211CG15211CG15211CG15211
Ant2Ant2
sesBsesBsesBsesB
_ 1000
_ 0
_ 1000
_ 0
ps RNAi
S2 Untreated
Splice JunctionReads
GenomicReads
Splice JunctionReads
GenomicReads
Image from Brenton Gravely
Image courtesy of Brenton Graveley
8 38
Sequencing errors Genome differences
Sample Genome
Ref Genome
True read
read with sequence error
Sample different from reference
Sequence errorMapping
Evidence suggests that Illumina sequencing does not introduce indels
9 38
Alignment Tools
The number of short read aligners have exploded but a couple tools haveemerged as the de facto standards
I Eland Illuminarsquos aligner quality aware fast paired end capable
I MAQ Good SNP caller quality aware paired end capable
I Bowtie Super fast offers different alignment strategies paired endcapable
I BWA Fast indel support paired ends qualities
I NovoAlign MAQ like speed many features
A superior overview of the different aligners is available athttpwwwsangeracukUserslh3NGSalignshtml Additionallya comparison of two of the best aligners can be found here httpwwwmassgenomicsorg200907maq-bwa-and-bowtie-comparedhtml
10 38
Common Alignment Strategies
I Use qualities (default for Bowtie and MAQ)
I Perfect match no repeats (Strict Lenient)
I Mismatches no repeats
I Paired end data (PET) (harder for RNA-Seq)
The standard Illumina protocol yields unstranded reads
Most aligners are evaluated in terms of ldquohow many reads are mappedrdquo Isthis the right objective
Watch out for the output of the program there are many differentconventions (0-based what happens to read hitting the reverse strandetc)
11 38
SAMBAM Formats
SAM (BAM) is a new general format for storing mapped reads It isdeveloped as part of the 1000 Genomes project and is quickly becoming akind of standard Some alignment tools output this format directlyotherwise there are scripts in samtools for doing it for most popularaligners Details at httpsamtoolssourceforgenetindexshtml
12 38
A comment
There are no great comprehensive tools for analyzing deep-sequence dataIt will involve a fair amount of coding and gluing together various tools
We will introduce some tools from Bioconductor that can be useful
I use a lot of shell scripting
13 38
Biostrings overview
I A package for working with large (biological) strings
I Two main types of objects A really long single string (thinkchromosome) or a set of shorter strings (think reads or genes)BString vs BStringSet
I These classes are implemented efficiently minimizing copy andmemory loadingunloading
I The BSgenome contains some infrastructure for whole genomes
I Methods for dealing with biological data including basicmanipulation (complementation translation etc) string searching(exactinexact matching Smith-Waterman PWMs)
I Fairly complicated class structure
Irsquom sorry to say that at least for me this has becomehopelessly confusing and I imagine that many other usersfeel the same ndash Simon Anders on bioc-sig-sequencing
Computing on genomes is not trivial The approach to a givencomputation is important
14 38
Strings in Biostrings
I BString (general) DNAString RNAString AAString (Amino Acid)all examples of XString
I complement reverse reverseComplement translate for the classeswhere ldquoit makes senserdquo
I Convert to and from a standard R character string
I Constructor DNAString(ACGGGGG)
I Support for IUPAC alphabet
I Subsetting subseq using the SEW format (two out of the threestartendwidth) Efficient
I StringSets are collections of Strings like DNAStringSet
15 38
BSgenomes
As examples we will use whole genomes Use availablegenomes to getavailable genomes Long package names but always a shorter objectname
gt library(BSgenomeScerevisiaeUCSCsacCer1)
gt Scerevisiae
gt Scerevisiae[[1]]
gt Scerevisiae[[chr1]]
A BSgenome may also have masks We will ignore this for now
16 38
Biostrings more
I alphabetFrequency oligonucleotideFrequency and others
gt alphabetFrequency(Scerevisiae[[chr1]])
gt oligonucleotideFrequency(Scerevisiae[[chr1]]
+ width = 3)
I chartr for character translation (ldquomake all As into Csrdquo)
I Various IO functions (also ShortRead)
17 38
Views
A view is a set of substrings of an XString stored and manipulated veryefficiently Example to store exon sequences one can just store thegenomic location of the exons
gt Views(Scerevisiae[[chr1]] start = c(300
+ 400 500) width = 50)
Views on a 230208-letter DNAString subjectsubject CCACACCACACCCAGTGGTGTGTGTGGGviews
start end width[1] 300 349 50 [CTGTTCTTAAATAAC][2] 400 449 50 [CCCTCACTAGTATAT][3] 500 549 50 [TCTCTCACCGGCACT]
A view is associated with a subject Internally they are essentiallyIRanges so they are very fast to compute with Views can in many casesbe treated exactly as other strings There are methods such as narrowtrim gaps restrict
18 38
Matching in Biostrings
There are various ways of matching or aligning strings to each other Weuse matching to denote searching for an exact match or possibly a matchwith a certain number of mismatches Alignment denotes a more generalstrategy eg Smith-Waterman
I matchPattern countPattern match 1 sequence to 1 sequence
I vmatchPattern vcountPattern match 1 sequence to manysequences
I pairwiseAlignment align many sequences to 1 sequence
I matchPDict countPDict match many sequences to 1 sequence(ldquodictrdquo indicates that the many sequences are preprocessed into adictionary)
I matchPWM trimLRpattern
The different functions are optimized for different situations They alsouse different algorithms which has a big impact EspeciallypairwiseAlignment is flexible and therefore has a complicated syntax
19 38
Example mapping probes to a genome
Get a list of Scerevisiae probes from the ldquoyeast2rdquo Affymetrix array
gt library(yeast2probe)
gt ids lt- scan(s_pombemsk skip = 2
+ what = list(probeset = character(0)
+ junk = character(0)))$probeset
gt probes lt- yeast2probe$sequence[yeast2probe$ProbeSetName in
+ ids]
gt probes lt- DNAStringSet(probes)
Mapping
gt require(BSgenomeScerevisiaeUCSCsacCer1)
gt dict0 lt- PDict(probes)
gt dict0r lt- PDict(reverseComplement(probes))
gt hits lt- matchPDict(dict0 Scerevisiae[[1]])
gt table(countIndex(hits))
gt table(countPDict(dict0 Scerevisiae[[1]]))
20 38
Example contrsquod
Over all chromosomes
gt allhits lt- lapply(116 function(i)
+ countPDict(dict0 Scerevisiae[[i]]) +
+ countPDict(dict0r Scerevisiae[[i]])
+ )
gt table(rowSums(docall(cbind allhits)))
21 38
ShortRead
ShortRead contains tools for
I Work with GERALDBUSTARD output
I Generate QA report on BUSTARD files
I Read a variety of short read data formats
22 38
Data
We will use data from (Lee Hansen Bullard Dudoit and Sherlock(2008) PLoS Genetics)
We are considering a wild-type and a mutant strain of yeast both grownin rich media The two strains were sequenced using an Illumina GenomeAnalyzer
We are using a subset of the data 1M reads from each of two lanes ofthe two strains
23 38
Reading the unaligned data
gt require(ShortRead)
gt fq lt- readFastq(seqdata mut_1_ffastq$)
gt fq
class ShortReadQlength 1000000 reads width 36 cycles
There are various simple accessor functions for this object especiallysread and quality
24 38
A brief look at the unaligned data
Using alphabetByCycle and as( rdquomatrixrdquo) (which creates a big read timescycle matrix) we can do
gt alp lt- alphabetByCycle(sread(fq))
gt matplot(t(proptable(alp[DNA_BASES
+ ] margin = 2)) type = l)
gt qaMatraw lt- as(quality(fq) matrix)
gt plot(colMeans(qaMatraw))
25 38
Aligning the data
We now align the data using Bowtie
binbash
BOWTIE_OPTS=-m 1 -v 2 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-m 1 -v 0 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 2 -k 2 -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 0 -k 1 -p 2 -q --quiet -3 10
for f in `ls seqdatafastq`dobowtie s_cerevisiae $BOWTIE_OPTS $f gt $fbowtiedone
How many hits do we get
26 38
Reading in the aligned data
We read the data into R as a 4-component list (one lane per component)
gt files lt- listfiles(seqdata
+ pattern = bowtie)
gt aligned lt- sapply(files function(f)
+ readAligned(seqdata pattern = f
+ type = Bowtie)
+ )
gt names(aligned) lt- gsub(_ffastqbowtie
+ names(aligned))
gt aligned[[1]]
gt save(aligned file = alignedrda)
27 38
Some exercises
1 Determine the number of times the motif ldquoTATAArdquo occurs in theyeast genome How often does it occur with one mismatch
2 The seqdata directory has an object called ste12 which is aposition weight matrix for the transcription factor ldquoSte12rdquo (obtainedfrom SCPD) Where does it occur in the genome
3 Compute the average quality for each cycle for the aligned reads andcompare to the qualities for the unaligned reads What is thedifference
28 38
Solution 1
gt sum(sapply(116 function(i)
+ motif lt- DNAString(TATAA)
+ countPattern(motif Scerevisiae[[i]]) +
+ countPattern(reverseComplement(motif)
+ Scerevisiae[[i]])
+ ))
29 38
Solution 2
30 38
Solution 3
31 38
SessionInfo
gt toLatex(sessionInfo())
I R version 292 RC (2009-08-17 r49312) i386-apple-darwin980
I Localeen_USUTF-8en_USUTF-8CCen_USUTF-8en_USUTF-8
I Base packages base datasets graphics grDevices grid methodsstats utils
I Other packages Biostrings 2128 BSgenome 1123BSgenomeScerevisiaeUCSCsacCer1 1313 classGraph 07-2graph 1222 IRanges 123 lattice 017-25 Rgraphviz 1234ShortRead 121
I Loaded via a namespace (and not attached) Biobase 241hwriter 11 tools 292
32 38
Some ShortRead classes
ShortRead
ShortReadQ
AlignedRead
33 38
Some Biostrings classes (single strings)
XString
BString DNAString RNAString AAString
34 38
Some Biostrings classes (sets of strings)
XStringSet
BStringSet DNAStringSet RNAStringSet AAStringSet PhredQualitySolexaQualityQualityScaledXStringSet
QualityScaledBStringSetQualityScaledDNAStringSetQualityScaledRNAStringSetQualityScaledAAStringSet
35 38
Mapping reads to the transcriptome 2
Mapping transcripts
Genome
Transcript
Length in genome space
paired-end reads
6 38
Mapping reads to the transcriptome 3
Junction reads
chrX
Conservation
d_simulansd_sechelliad_yakubad_erectad_ananassaed_pseudoobscurad_persimilisd_willistonid_virilisd_mojavensisd_grimshawia_gambiaea_melliferat_castaneum
1067300010673500106740001067450010675000106755001067600010676500106770001067750010678000106785001067900010679500106800001068050010681000
CG8144_6lane_0MM
S2_DRSC_6_lanes
FlyBase Protein-Coding Genes
RefSeq Genes
12 Flies Mosquito Honeybee Beetle Multiz Alignments amp phastCons Scores
Repeating Elements by RepeatMasker
CG15211CG15211CG15211CG15211
Ant2Ant2
sesBsesBsesBsesB
CG15211CG15211CG15211CG15211
Ant2Ant2
sesBsesBsesBsesB
_ 1000
_ 0
_ 1000
_ 0
ps RNAi
S2 Untreated
Splice JunctionReads
GenomicReads
Splice JunctionReads
GenomicReads
Image from Brenton Gravely
Image courtesy of Brenton Graveley
7 38
Mapping reads to the transcriptome 4
Junction reads zoom
chrX
Conservation
d_simulansd_sechelliad_yakubad_erectad_ananassaed_pseudoobscurad_persimilisd_willistonid_virilisd_mojavensisd_grimshawia_gambiaea_melliferat_castaneum
1067300010673500106740001067450010675000106755001067600010676500106770001067750010678000106785001067900010679500106800001068050010681000
CG8144_6lane_0MM
S2_DRSC_6_lanes
FlyBase Protein-Coding Genes
RefSeq Genes
12 Flies Mosquito Honeybee Beetle Multiz Alignments amp phastCons Scores
Repeating Elements by RepeatMasker
CG15211CG15211CG15211CG15211
Ant2Ant2
sesBsesBsesBsesB
CG15211CG15211CG15211CG15211
Ant2Ant2
sesBsesBsesBsesB
_ 1000
_ 0
_ 1000
_ 0
ps RNAi
S2 Untreated
Splice JunctionReads
GenomicReads
Splice JunctionReads
GenomicReads
Image from Brenton Gravely
Image courtesy of Brenton Graveley
8 38
Sequencing errors Genome differences
Sample Genome
Ref Genome
True read
read with sequence error
Sample different from reference
Sequence errorMapping
Evidence suggests that Illumina sequencing does not introduce indels
9 38
Alignment Tools
The number of short read aligners have exploded but a couple tools haveemerged as the de facto standards
I Eland Illuminarsquos aligner quality aware fast paired end capable
I MAQ Good SNP caller quality aware paired end capable
I Bowtie Super fast offers different alignment strategies paired endcapable
I BWA Fast indel support paired ends qualities
I NovoAlign MAQ like speed many features
A superior overview of the different aligners is available athttpwwwsangeracukUserslh3NGSalignshtml Additionallya comparison of two of the best aligners can be found here httpwwwmassgenomicsorg200907maq-bwa-and-bowtie-comparedhtml
10 38
Common Alignment Strategies
I Use qualities (default for Bowtie and MAQ)
I Perfect match no repeats (Strict Lenient)
I Mismatches no repeats
I Paired end data (PET) (harder for RNA-Seq)
The standard Illumina protocol yields unstranded reads
Most aligners are evaluated in terms of ldquohow many reads are mappedrdquo Isthis the right objective
Watch out for the output of the program there are many differentconventions (0-based what happens to read hitting the reverse strandetc)
11 38
SAMBAM Formats
SAM (BAM) is a new general format for storing mapped reads It isdeveloped as part of the 1000 Genomes project and is quickly becoming akind of standard Some alignment tools output this format directlyotherwise there are scripts in samtools for doing it for most popularaligners Details at httpsamtoolssourceforgenetindexshtml
12 38
A comment
There are no great comprehensive tools for analyzing deep-sequence dataIt will involve a fair amount of coding and gluing together various tools
We will introduce some tools from Bioconductor that can be useful
I use a lot of shell scripting
13 38
Biostrings overview
I A package for working with large (biological) strings
I Two main types of objects A really long single string (thinkchromosome) or a set of shorter strings (think reads or genes)BString vs BStringSet
I These classes are implemented efficiently minimizing copy andmemory loadingunloading
I The BSgenome contains some infrastructure for whole genomes
I Methods for dealing with biological data including basicmanipulation (complementation translation etc) string searching(exactinexact matching Smith-Waterman PWMs)
I Fairly complicated class structure
Irsquom sorry to say that at least for me this has becomehopelessly confusing and I imagine that many other usersfeel the same ndash Simon Anders on bioc-sig-sequencing
Computing on genomes is not trivial The approach to a givencomputation is important
14 38
Strings in Biostrings
I BString (general) DNAString RNAString AAString (Amino Acid)all examples of XString
I complement reverse reverseComplement translate for the classeswhere ldquoit makes senserdquo
I Convert to and from a standard R character string
I Constructor DNAString(ACGGGGG)
I Support for IUPAC alphabet
I Subsetting subseq using the SEW format (two out of the threestartendwidth) Efficient
I StringSets are collections of Strings like DNAStringSet
15 38
BSgenomes
As examples we will use whole genomes Use availablegenomes to getavailable genomes Long package names but always a shorter objectname
gt library(BSgenomeScerevisiaeUCSCsacCer1)
gt Scerevisiae
gt Scerevisiae[[1]]
gt Scerevisiae[[chr1]]
A BSgenome may also have masks We will ignore this for now
16 38
Biostrings more
I alphabetFrequency oligonucleotideFrequency and others
gt alphabetFrequency(Scerevisiae[[chr1]])
gt oligonucleotideFrequency(Scerevisiae[[chr1]]
+ width = 3)
I chartr for character translation (ldquomake all As into Csrdquo)
I Various IO functions (also ShortRead)
17 38
Views
A view is a set of substrings of an XString stored and manipulated veryefficiently Example to store exon sequences one can just store thegenomic location of the exons
gt Views(Scerevisiae[[chr1]] start = c(300
+ 400 500) width = 50)
Views on a 230208-letter DNAString subjectsubject CCACACCACACCCAGTGGTGTGTGTGGGviews
start end width[1] 300 349 50 [CTGTTCTTAAATAAC][2] 400 449 50 [CCCTCACTAGTATAT][3] 500 549 50 [TCTCTCACCGGCACT]
A view is associated with a subject Internally they are essentiallyIRanges so they are very fast to compute with Views can in many casesbe treated exactly as other strings There are methods such as narrowtrim gaps restrict
18 38
Matching in Biostrings
There are various ways of matching or aligning strings to each other Weuse matching to denote searching for an exact match or possibly a matchwith a certain number of mismatches Alignment denotes a more generalstrategy eg Smith-Waterman
I matchPattern countPattern match 1 sequence to 1 sequence
I vmatchPattern vcountPattern match 1 sequence to manysequences
I pairwiseAlignment align many sequences to 1 sequence
I matchPDict countPDict match many sequences to 1 sequence(ldquodictrdquo indicates that the many sequences are preprocessed into adictionary)
I matchPWM trimLRpattern
The different functions are optimized for different situations They alsouse different algorithms which has a big impact EspeciallypairwiseAlignment is flexible and therefore has a complicated syntax
19 38
Example mapping probes to a genome
Get a list of Scerevisiae probes from the ldquoyeast2rdquo Affymetrix array
gt library(yeast2probe)
gt ids lt- scan(s_pombemsk skip = 2
+ what = list(probeset = character(0)
+ junk = character(0)))$probeset
gt probes lt- yeast2probe$sequence[yeast2probe$ProbeSetName in
+ ids]
gt probes lt- DNAStringSet(probes)
Mapping
gt require(BSgenomeScerevisiaeUCSCsacCer1)
gt dict0 lt- PDict(probes)
gt dict0r lt- PDict(reverseComplement(probes))
gt hits lt- matchPDict(dict0 Scerevisiae[[1]])
gt table(countIndex(hits))
gt table(countPDict(dict0 Scerevisiae[[1]]))
20 38
Example contrsquod
Over all chromosomes
gt allhits lt- lapply(116 function(i)
+ countPDict(dict0 Scerevisiae[[i]]) +
+ countPDict(dict0r Scerevisiae[[i]])
+ )
gt table(rowSums(docall(cbind allhits)))
21 38
ShortRead
ShortRead contains tools for
I Work with GERALDBUSTARD output
I Generate QA report on BUSTARD files
I Read a variety of short read data formats
22 38
Data
We will use data from (Lee Hansen Bullard Dudoit and Sherlock(2008) PLoS Genetics)
We are considering a wild-type and a mutant strain of yeast both grownin rich media The two strains were sequenced using an Illumina GenomeAnalyzer
We are using a subset of the data 1M reads from each of two lanes ofthe two strains
23 38
Reading the unaligned data
gt require(ShortRead)
gt fq lt- readFastq(seqdata mut_1_ffastq$)
gt fq
class ShortReadQlength 1000000 reads width 36 cycles
There are various simple accessor functions for this object especiallysread and quality
24 38
A brief look at the unaligned data
Using alphabetByCycle and as( rdquomatrixrdquo) (which creates a big read timescycle matrix) we can do
gt alp lt- alphabetByCycle(sread(fq))
gt matplot(t(proptable(alp[DNA_BASES
+ ] margin = 2)) type = l)
gt qaMatraw lt- as(quality(fq) matrix)
gt plot(colMeans(qaMatraw))
25 38
Aligning the data
We now align the data using Bowtie
binbash
BOWTIE_OPTS=-m 1 -v 2 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-m 1 -v 0 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 2 -k 2 -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 0 -k 1 -p 2 -q --quiet -3 10
for f in `ls seqdatafastq`dobowtie s_cerevisiae $BOWTIE_OPTS $f gt $fbowtiedone
How many hits do we get
26 38
Reading in the aligned data
We read the data into R as a 4-component list (one lane per component)
gt files lt- listfiles(seqdata
+ pattern = bowtie)
gt aligned lt- sapply(files function(f)
+ readAligned(seqdata pattern = f
+ type = Bowtie)
+ )
gt names(aligned) lt- gsub(_ffastqbowtie
+ names(aligned))
gt aligned[[1]]
gt save(aligned file = alignedrda)
27 38
Some exercises
1 Determine the number of times the motif ldquoTATAArdquo occurs in theyeast genome How often does it occur with one mismatch
2 The seqdata directory has an object called ste12 which is aposition weight matrix for the transcription factor ldquoSte12rdquo (obtainedfrom SCPD) Where does it occur in the genome
3 Compute the average quality for each cycle for the aligned reads andcompare to the qualities for the unaligned reads What is thedifference
28 38
Solution 1
gt sum(sapply(116 function(i)
+ motif lt- DNAString(TATAA)
+ countPattern(motif Scerevisiae[[i]]) +
+ countPattern(reverseComplement(motif)
+ Scerevisiae[[i]])
+ ))
29 38
Solution 2
30 38
Solution 3
31 38
SessionInfo
gt toLatex(sessionInfo())
I R version 292 RC (2009-08-17 r49312) i386-apple-darwin980
I Localeen_USUTF-8en_USUTF-8CCen_USUTF-8en_USUTF-8
I Base packages base datasets graphics grDevices grid methodsstats utils
I Other packages Biostrings 2128 BSgenome 1123BSgenomeScerevisiaeUCSCsacCer1 1313 classGraph 07-2graph 1222 IRanges 123 lattice 017-25 Rgraphviz 1234ShortRead 121
I Loaded via a namespace (and not attached) Biobase 241hwriter 11 tools 292
32 38
Some ShortRead classes
ShortRead
ShortReadQ
AlignedRead
33 38
Some Biostrings classes (single strings)
XString
BString DNAString RNAString AAString
34 38
Some Biostrings classes (sets of strings)
XStringSet
BStringSet DNAStringSet RNAStringSet AAStringSet PhredQualitySolexaQualityQualityScaledXStringSet
QualityScaledBStringSetQualityScaledDNAStringSetQualityScaledRNAStringSetQualityScaledAAStringSet
35 38
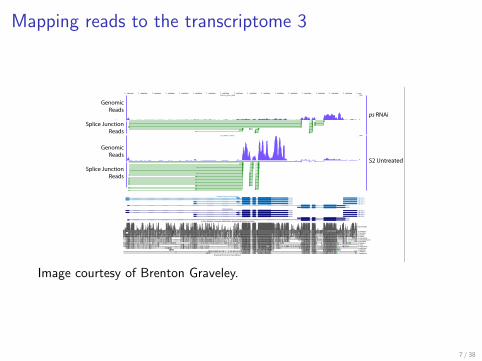
Mapping reads to the transcriptome 3
Junction reads
chrX
Conservation
d_simulansd_sechelliad_yakubad_erectad_ananassaed_pseudoobscurad_persimilisd_willistonid_virilisd_mojavensisd_grimshawia_gambiaea_melliferat_castaneum
1067300010673500106740001067450010675000106755001067600010676500106770001067750010678000106785001067900010679500106800001068050010681000
CG8144_6lane_0MM
S2_DRSC_6_lanes
FlyBase Protein-Coding Genes
RefSeq Genes
12 Flies Mosquito Honeybee Beetle Multiz Alignments amp phastCons Scores
Repeating Elements by RepeatMasker
CG15211CG15211CG15211CG15211
Ant2Ant2
sesBsesBsesBsesB
CG15211CG15211CG15211CG15211
Ant2Ant2
sesBsesBsesBsesB
_ 1000
_ 0
_ 1000
_ 0
ps RNAi
S2 Untreated
Splice JunctionReads
GenomicReads
Splice JunctionReads
GenomicReads
Image from Brenton Gravely
Image courtesy of Brenton Graveley
7 38
Mapping reads to the transcriptome 4
Junction reads zoom
chrX
Conservation
d_simulansd_sechelliad_yakubad_erectad_ananassaed_pseudoobscurad_persimilisd_willistonid_virilisd_mojavensisd_grimshawia_gambiaea_melliferat_castaneum
1067300010673500106740001067450010675000106755001067600010676500106770001067750010678000106785001067900010679500106800001068050010681000
CG8144_6lane_0MM
S2_DRSC_6_lanes
FlyBase Protein-Coding Genes
RefSeq Genes
12 Flies Mosquito Honeybee Beetle Multiz Alignments amp phastCons Scores
Repeating Elements by RepeatMasker
CG15211CG15211CG15211CG15211
Ant2Ant2
sesBsesBsesBsesB
CG15211CG15211CG15211CG15211
Ant2Ant2
sesBsesBsesBsesB
_ 1000
_ 0
_ 1000
_ 0
ps RNAi
S2 Untreated
Splice JunctionReads
GenomicReads
Splice JunctionReads
GenomicReads
Image from Brenton Gravely
Image courtesy of Brenton Graveley
8 38
Sequencing errors Genome differences
Sample Genome
Ref Genome
True read
read with sequence error
Sample different from reference
Sequence errorMapping
Evidence suggests that Illumina sequencing does not introduce indels
9 38
Alignment Tools
The number of short read aligners have exploded but a couple tools haveemerged as the de facto standards
I Eland Illuminarsquos aligner quality aware fast paired end capable
I MAQ Good SNP caller quality aware paired end capable
I Bowtie Super fast offers different alignment strategies paired endcapable
I BWA Fast indel support paired ends qualities
I NovoAlign MAQ like speed many features
A superior overview of the different aligners is available athttpwwwsangeracukUserslh3NGSalignshtml Additionallya comparison of two of the best aligners can be found here httpwwwmassgenomicsorg200907maq-bwa-and-bowtie-comparedhtml
10 38
Common Alignment Strategies
I Use qualities (default for Bowtie and MAQ)
I Perfect match no repeats (Strict Lenient)
I Mismatches no repeats
I Paired end data (PET) (harder for RNA-Seq)
The standard Illumina protocol yields unstranded reads
Most aligners are evaluated in terms of ldquohow many reads are mappedrdquo Isthis the right objective
Watch out for the output of the program there are many differentconventions (0-based what happens to read hitting the reverse strandetc)
11 38
SAMBAM Formats
SAM (BAM) is a new general format for storing mapped reads It isdeveloped as part of the 1000 Genomes project and is quickly becoming akind of standard Some alignment tools output this format directlyotherwise there are scripts in samtools for doing it for most popularaligners Details at httpsamtoolssourceforgenetindexshtml
12 38
A comment
There are no great comprehensive tools for analyzing deep-sequence dataIt will involve a fair amount of coding and gluing together various tools
We will introduce some tools from Bioconductor that can be useful
I use a lot of shell scripting
13 38
Biostrings overview
I A package for working with large (biological) strings
I Two main types of objects A really long single string (thinkchromosome) or a set of shorter strings (think reads or genes)BString vs BStringSet
I These classes are implemented efficiently minimizing copy andmemory loadingunloading
I The BSgenome contains some infrastructure for whole genomes
I Methods for dealing with biological data including basicmanipulation (complementation translation etc) string searching(exactinexact matching Smith-Waterman PWMs)
I Fairly complicated class structure
Irsquom sorry to say that at least for me this has becomehopelessly confusing and I imagine that many other usersfeel the same ndash Simon Anders on bioc-sig-sequencing
Computing on genomes is not trivial The approach to a givencomputation is important
14 38
Strings in Biostrings
I BString (general) DNAString RNAString AAString (Amino Acid)all examples of XString
I complement reverse reverseComplement translate for the classeswhere ldquoit makes senserdquo
I Convert to and from a standard R character string
I Constructor DNAString(ACGGGGG)
I Support for IUPAC alphabet
I Subsetting subseq using the SEW format (two out of the threestartendwidth) Efficient
I StringSets are collections of Strings like DNAStringSet
15 38
BSgenomes
As examples we will use whole genomes Use availablegenomes to getavailable genomes Long package names but always a shorter objectname
gt library(BSgenomeScerevisiaeUCSCsacCer1)
gt Scerevisiae
gt Scerevisiae[[1]]
gt Scerevisiae[[chr1]]
A BSgenome may also have masks We will ignore this for now
16 38
Biostrings more
I alphabetFrequency oligonucleotideFrequency and others
gt alphabetFrequency(Scerevisiae[[chr1]])
gt oligonucleotideFrequency(Scerevisiae[[chr1]]
+ width = 3)
I chartr for character translation (ldquomake all As into Csrdquo)
I Various IO functions (also ShortRead)
17 38
Views
A view is a set of substrings of an XString stored and manipulated veryefficiently Example to store exon sequences one can just store thegenomic location of the exons
gt Views(Scerevisiae[[chr1]] start = c(300
+ 400 500) width = 50)
Views on a 230208-letter DNAString subjectsubject CCACACCACACCCAGTGGTGTGTGTGGGviews
start end width[1] 300 349 50 [CTGTTCTTAAATAAC][2] 400 449 50 [CCCTCACTAGTATAT][3] 500 549 50 [TCTCTCACCGGCACT]
A view is associated with a subject Internally they are essentiallyIRanges so they are very fast to compute with Views can in many casesbe treated exactly as other strings There are methods such as narrowtrim gaps restrict
18 38
Matching in Biostrings
There are various ways of matching or aligning strings to each other Weuse matching to denote searching for an exact match or possibly a matchwith a certain number of mismatches Alignment denotes a more generalstrategy eg Smith-Waterman
I matchPattern countPattern match 1 sequence to 1 sequence
I vmatchPattern vcountPattern match 1 sequence to manysequences
I pairwiseAlignment align many sequences to 1 sequence
I matchPDict countPDict match many sequences to 1 sequence(ldquodictrdquo indicates that the many sequences are preprocessed into adictionary)
I matchPWM trimLRpattern
The different functions are optimized for different situations They alsouse different algorithms which has a big impact EspeciallypairwiseAlignment is flexible and therefore has a complicated syntax
19 38
Example mapping probes to a genome
Get a list of Scerevisiae probes from the ldquoyeast2rdquo Affymetrix array
gt library(yeast2probe)
gt ids lt- scan(s_pombemsk skip = 2
+ what = list(probeset = character(0)
+ junk = character(0)))$probeset
gt probes lt- yeast2probe$sequence[yeast2probe$ProbeSetName in
+ ids]
gt probes lt- DNAStringSet(probes)
Mapping
gt require(BSgenomeScerevisiaeUCSCsacCer1)
gt dict0 lt- PDict(probes)
gt dict0r lt- PDict(reverseComplement(probes))
gt hits lt- matchPDict(dict0 Scerevisiae[[1]])
gt table(countIndex(hits))
gt table(countPDict(dict0 Scerevisiae[[1]]))
20 38
Example contrsquod
Over all chromosomes
gt allhits lt- lapply(116 function(i)
+ countPDict(dict0 Scerevisiae[[i]]) +
+ countPDict(dict0r Scerevisiae[[i]])
+ )
gt table(rowSums(docall(cbind allhits)))
21 38
ShortRead
ShortRead contains tools for
I Work with GERALDBUSTARD output
I Generate QA report on BUSTARD files
I Read a variety of short read data formats
22 38
Data
We will use data from (Lee Hansen Bullard Dudoit and Sherlock(2008) PLoS Genetics)
We are considering a wild-type and a mutant strain of yeast both grownin rich media The two strains were sequenced using an Illumina GenomeAnalyzer
We are using a subset of the data 1M reads from each of two lanes ofthe two strains
23 38
Reading the unaligned data
gt require(ShortRead)
gt fq lt- readFastq(seqdata mut_1_ffastq$)
gt fq
class ShortReadQlength 1000000 reads width 36 cycles
There are various simple accessor functions for this object especiallysread and quality
24 38
A brief look at the unaligned data
Using alphabetByCycle and as( rdquomatrixrdquo) (which creates a big read timescycle matrix) we can do
gt alp lt- alphabetByCycle(sread(fq))
gt matplot(t(proptable(alp[DNA_BASES
+ ] margin = 2)) type = l)
gt qaMatraw lt- as(quality(fq) matrix)
gt plot(colMeans(qaMatraw))
25 38
Aligning the data
We now align the data using Bowtie
binbash
BOWTIE_OPTS=-m 1 -v 2 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-m 1 -v 0 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 2 -k 2 -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 0 -k 1 -p 2 -q --quiet -3 10
for f in `ls seqdatafastq`dobowtie s_cerevisiae $BOWTIE_OPTS $f gt $fbowtiedone
How many hits do we get
26 38
Reading in the aligned data
We read the data into R as a 4-component list (one lane per component)
gt files lt- listfiles(seqdata
+ pattern = bowtie)
gt aligned lt- sapply(files function(f)
+ readAligned(seqdata pattern = f
+ type = Bowtie)
+ )
gt names(aligned) lt- gsub(_ffastqbowtie
+ names(aligned))
gt aligned[[1]]
gt save(aligned file = alignedrda)
27 38
Some exercises
1 Determine the number of times the motif ldquoTATAArdquo occurs in theyeast genome How often does it occur with one mismatch
2 The seqdata directory has an object called ste12 which is aposition weight matrix for the transcription factor ldquoSte12rdquo (obtainedfrom SCPD) Where does it occur in the genome
3 Compute the average quality for each cycle for the aligned reads andcompare to the qualities for the unaligned reads What is thedifference
28 38
Solution 1
gt sum(sapply(116 function(i)
+ motif lt- DNAString(TATAA)
+ countPattern(motif Scerevisiae[[i]]) +
+ countPattern(reverseComplement(motif)
+ Scerevisiae[[i]])
+ ))
29 38
Solution 2
30 38
Solution 3
31 38
SessionInfo
gt toLatex(sessionInfo())
I R version 292 RC (2009-08-17 r49312) i386-apple-darwin980
I Localeen_USUTF-8en_USUTF-8CCen_USUTF-8en_USUTF-8
I Base packages base datasets graphics grDevices grid methodsstats utils
I Other packages Biostrings 2128 BSgenome 1123BSgenomeScerevisiaeUCSCsacCer1 1313 classGraph 07-2graph 1222 IRanges 123 lattice 017-25 Rgraphviz 1234ShortRead 121
I Loaded via a namespace (and not attached) Biobase 241hwriter 11 tools 292
32 38
Some ShortRead classes
ShortRead
ShortReadQ
AlignedRead
33 38
Some Biostrings classes (single strings)
XString
BString DNAString RNAString AAString
34 38
Some Biostrings classes (sets of strings)
XStringSet
BStringSet DNAStringSet RNAStringSet AAStringSet PhredQualitySolexaQualityQualityScaledXStringSet
QualityScaledBStringSetQualityScaledDNAStringSetQualityScaledRNAStringSetQualityScaledAAStringSet
35 38
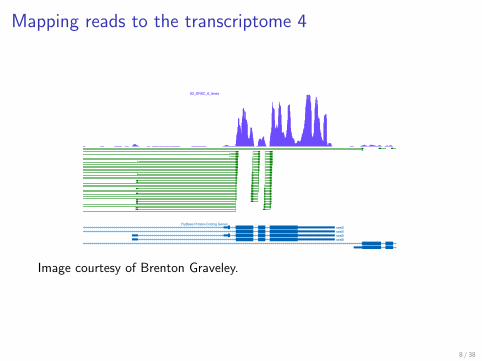
Mapping reads to the transcriptome 4
Junction reads zoom
chrX
Conservation
d_simulansd_sechelliad_yakubad_erectad_ananassaed_pseudoobscurad_persimilisd_willistonid_virilisd_mojavensisd_grimshawia_gambiaea_melliferat_castaneum
1067300010673500106740001067450010675000106755001067600010676500106770001067750010678000106785001067900010679500106800001068050010681000
CG8144_6lane_0MM
S2_DRSC_6_lanes
FlyBase Protein-Coding Genes
RefSeq Genes
12 Flies Mosquito Honeybee Beetle Multiz Alignments amp phastCons Scores
Repeating Elements by RepeatMasker
CG15211CG15211CG15211CG15211
Ant2Ant2
sesBsesBsesBsesB
CG15211CG15211CG15211CG15211
Ant2Ant2
sesBsesBsesBsesB
_ 1000
_ 0
_ 1000
_ 0
ps RNAi
S2 Untreated
Splice JunctionReads
GenomicReads
Splice JunctionReads
GenomicReads
Image from Brenton Gravely
Image courtesy of Brenton Graveley
8 38
Sequencing errors Genome differences
Sample Genome
Ref Genome
True read
read with sequence error
Sample different from reference
Sequence errorMapping
Evidence suggests that Illumina sequencing does not introduce indels
9 38
Alignment Tools
The number of short read aligners have exploded but a couple tools haveemerged as the de facto standards
I Eland Illuminarsquos aligner quality aware fast paired end capable
I MAQ Good SNP caller quality aware paired end capable
I Bowtie Super fast offers different alignment strategies paired endcapable
I BWA Fast indel support paired ends qualities
I NovoAlign MAQ like speed many features
A superior overview of the different aligners is available athttpwwwsangeracukUserslh3NGSalignshtml Additionallya comparison of two of the best aligners can be found here httpwwwmassgenomicsorg200907maq-bwa-and-bowtie-comparedhtml
10 38
Common Alignment Strategies
I Use qualities (default for Bowtie and MAQ)
I Perfect match no repeats (Strict Lenient)
I Mismatches no repeats
I Paired end data (PET) (harder for RNA-Seq)
The standard Illumina protocol yields unstranded reads
Most aligners are evaluated in terms of ldquohow many reads are mappedrdquo Isthis the right objective
Watch out for the output of the program there are many differentconventions (0-based what happens to read hitting the reverse strandetc)
11 38
SAMBAM Formats
SAM (BAM) is a new general format for storing mapped reads It isdeveloped as part of the 1000 Genomes project and is quickly becoming akind of standard Some alignment tools output this format directlyotherwise there are scripts in samtools for doing it for most popularaligners Details at httpsamtoolssourceforgenetindexshtml
12 38
A comment
There are no great comprehensive tools for analyzing deep-sequence dataIt will involve a fair amount of coding and gluing together various tools
We will introduce some tools from Bioconductor that can be useful
I use a lot of shell scripting
13 38
Biostrings overview
I A package for working with large (biological) strings
I Two main types of objects A really long single string (thinkchromosome) or a set of shorter strings (think reads or genes)BString vs BStringSet
I These classes are implemented efficiently minimizing copy andmemory loadingunloading
I The BSgenome contains some infrastructure for whole genomes
I Methods for dealing with biological data including basicmanipulation (complementation translation etc) string searching(exactinexact matching Smith-Waterman PWMs)
I Fairly complicated class structure
Irsquom sorry to say that at least for me this has becomehopelessly confusing and I imagine that many other usersfeel the same ndash Simon Anders on bioc-sig-sequencing
Computing on genomes is not trivial The approach to a givencomputation is important
14 38
Strings in Biostrings
I BString (general) DNAString RNAString AAString (Amino Acid)all examples of XString
I complement reverse reverseComplement translate for the classeswhere ldquoit makes senserdquo
I Convert to and from a standard R character string
I Constructor DNAString(ACGGGGG)
I Support for IUPAC alphabet
I Subsetting subseq using the SEW format (two out of the threestartendwidth) Efficient
I StringSets are collections of Strings like DNAStringSet
15 38
BSgenomes
As examples we will use whole genomes Use availablegenomes to getavailable genomes Long package names but always a shorter objectname
gt library(BSgenomeScerevisiaeUCSCsacCer1)
gt Scerevisiae
gt Scerevisiae[[1]]
gt Scerevisiae[[chr1]]
A BSgenome may also have masks We will ignore this for now
16 38
Biostrings more
I alphabetFrequency oligonucleotideFrequency and others
gt alphabetFrequency(Scerevisiae[[chr1]])
gt oligonucleotideFrequency(Scerevisiae[[chr1]]
+ width = 3)
I chartr for character translation (ldquomake all As into Csrdquo)
I Various IO functions (also ShortRead)
17 38
Views
A view is a set of substrings of an XString stored and manipulated veryefficiently Example to store exon sequences one can just store thegenomic location of the exons
gt Views(Scerevisiae[[chr1]] start = c(300
+ 400 500) width = 50)
Views on a 230208-letter DNAString subjectsubject CCACACCACACCCAGTGGTGTGTGTGGGviews
start end width[1] 300 349 50 [CTGTTCTTAAATAAC][2] 400 449 50 [CCCTCACTAGTATAT][3] 500 549 50 [TCTCTCACCGGCACT]
A view is associated with a subject Internally they are essentiallyIRanges so they are very fast to compute with Views can in many casesbe treated exactly as other strings There are methods such as narrowtrim gaps restrict
18 38
Matching in Biostrings
There are various ways of matching or aligning strings to each other Weuse matching to denote searching for an exact match or possibly a matchwith a certain number of mismatches Alignment denotes a more generalstrategy eg Smith-Waterman
I matchPattern countPattern match 1 sequence to 1 sequence
I vmatchPattern vcountPattern match 1 sequence to manysequences
I pairwiseAlignment align many sequences to 1 sequence
I matchPDict countPDict match many sequences to 1 sequence(ldquodictrdquo indicates that the many sequences are preprocessed into adictionary)
I matchPWM trimLRpattern
The different functions are optimized for different situations They alsouse different algorithms which has a big impact EspeciallypairwiseAlignment is flexible and therefore has a complicated syntax
19 38
Example mapping probes to a genome
Get a list of Scerevisiae probes from the ldquoyeast2rdquo Affymetrix array
gt library(yeast2probe)
gt ids lt- scan(s_pombemsk skip = 2
+ what = list(probeset = character(0)
+ junk = character(0)))$probeset
gt probes lt- yeast2probe$sequence[yeast2probe$ProbeSetName in
+ ids]
gt probes lt- DNAStringSet(probes)
Mapping
gt require(BSgenomeScerevisiaeUCSCsacCer1)
gt dict0 lt- PDict(probes)
gt dict0r lt- PDict(reverseComplement(probes))
gt hits lt- matchPDict(dict0 Scerevisiae[[1]])
gt table(countIndex(hits))
gt table(countPDict(dict0 Scerevisiae[[1]]))
20 38
Example contrsquod
Over all chromosomes
gt allhits lt- lapply(116 function(i)
+ countPDict(dict0 Scerevisiae[[i]]) +
+ countPDict(dict0r Scerevisiae[[i]])
+ )
gt table(rowSums(docall(cbind allhits)))
21 38
ShortRead
ShortRead contains tools for
I Work with GERALDBUSTARD output
I Generate QA report on BUSTARD files
I Read a variety of short read data formats
22 38
Data
We will use data from (Lee Hansen Bullard Dudoit and Sherlock(2008) PLoS Genetics)
We are considering a wild-type and a mutant strain of yeast both grownin rich media The two strains were sequenced using an Illumina GenomeAnalyzer
We are using a subset of the data 1M reads from each of two lanes ofthe two strains
23 38
Reading the unaligned data
gt require(ShortRead)
gt fq lt- readFastq(seqdata mut_1_ffastq$)
gt fq
class ShortReadQlength 1000000 reads width 36 cycles
There are various simple accessor functions for this object especiallysread and quality
24 38
A brief look at the unaligned data
Using alphabetByCycle and as( rdquomatrixrdquo) (which creates a big read timescycle matrix) we can do
gt alp lt- alphabetByCycle(sread(fq))
gt matplot(t(proptable(alp[DNA_BASES
+ ] margin = 2)) type = l)
gt qaMatraw lt- as(quality(fq) matrix)
gt plot(colMeans(qaMatraw))
25 38
Aligning the data
We now align the data using Bowtie
binbash
BOWTIE_OPTS=-m 1 -v 2 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-m 1 -v 0 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 2 -k 2 -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 0 -k 1 -p 2 -q --quiet -3 10
for f in `ls seqdatafastq`dobowtie s_cerevisiae $BOWTIE_OPTS $f gt $fbowtiedone
How many hits do we get
26 38
Reading in the aligned data
We read the data into R as a 4-component list (one lane per component)
gt files lt- listfiles(seqdata
+ pattern = bowtie)
gt aligned lt- sapply(files function(f)
+ readAligned(seqdata pattern = f
+ type = Bowtie)
+ )
gt names(aligned) lt- gsub(_ffastqbowtie
+ names(aligned))
gt aligned[[1]]
gt save(aligned file = alignedrda)
27 38
Some exercises
1 Determine the number of times the motif ldquoTATAArdquo occurs in theyeast genome How often does it occur with one mismatch
2 The seqdata directory has an object called ste12 which is aposition weight matrix for the transcription factor ldquoSte12rdquo (obtainedfrom SCPD) Where does it occur in the genome
3 Compute the average quality for each cycle for the aligned reads andcompare to the qualities for the unaligned reads What is thedifference
28 38
Solution 1
gt sum(sapply(116 function(i)
+ motif lt- DNAString(TATAA)
+ countPattern(motif Scerevisiae[[i]]) +
+ countPattern(reverseComplement(motif)
+ Scerevisiae[[i]])
+ ))
29 38
Solution 2
30 38
Solution 3
31 38
SessionInfo
gt toLatex(sessionInfo())
I R version 292 RC (2009-08-17 r49312) i386-apple-darwin980
I Localeen_USUTF-8en_USUTF-8CCen_USUTF-8en_USUTF-8
I Base packages base datasets graphics grDevices grid methodsstats utils
I Other packages Biostrings 2128 BSgenome 1123BSgenomeScerevisiaeUCSCsacCer1 1313 classGraph 07-2graph 1222 IRanges 123 lattice 017-25 Rgraphviz 1234ShortRead 121
I Loaded via a namespace (and not attached) Biobase 241hwriter 11 tools 292
32 38
Some ShortRead classes
ShortRead
ShortReadQ
AlignedRead
33 38
Some Biostrings classes (single strings)
XString
BString DNAString RNAString AAString
34 38
Some Biostrings classes (sets of strings)
XStringSet
BStringSet DNAStringSet RNAStringSet AAStringSet PhredQualitySolexaQualityQualityScaledXStringSet
QualityScaledBStringSetQualityScaledDNAStringSetQualityScaledRNAStringSetQualityScaledAAStringSet
35 38
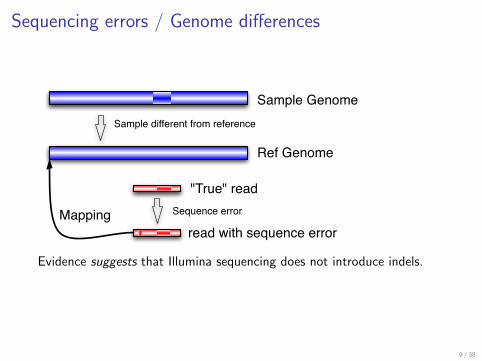
Sequencing errors Genome differences
Sample Genome
Ref Genome
True read
read with sequence error
Sample different from reference
Sequence errorMapping
Evidence suggests that Illumina sequencing does not introduce indels
9 38
Alignment Tools
The number of short read aligners have exploded but a couple tools haveemerged as the de facto standards
I Eland Illuminarsquos aligner quality aware fast paired end capable
I MAQ Good SNP caller quality aware paired end capable
I Bowtie Super fast offers different alignment strategies paired endcapable
I BWA Fast indel support paired ends qualities
I NovoAlign MAQ like speed many features
A superior overview of the different aligners is available athttpwwwsangeracukUserslh3NGSalignshtml Additionallya comparison of two of the best aligners can be found here httpwwwmassgenomicsorg200907maq-bwa-and-bowtie-comparedhtml
10 38
Common Alignment Strategies
I Use qualities (default for Bowtie and MAQ)
I Perfect match no repeats (Strict Lenient)
I Mismatches no repeats
I Paired end data (PET) (harder for RNA-Seq)
The standard Illumina protocol yields unstranded reads
Most aligners are evaluated in terms of ldquohow many reads are mappedrdquo Isthis the right objective
Watch out for the output of the program there are many differentconventions (0-based what happens to read hitting the reverse strandetc)
11 38
SAMBAM Formats
SAM (BAM) is a new general format for storing mapped reads It isdeveloped as part of the 1000 Genomes project and is quickly becoming akind of standard Some alignment tools output this format directlyotherwise there are scripts in samtools for doing it for most popularaligners Details at httpsamtoolssourceforgenetindexshtml
12 38
A comment
There are no great comprehensive tools for analyzing deep-sequence dataIt will involve a fair amount of coding and gluing together various tools
We will introduce some tools from Bioconductor that can be useful
I use a lot of shell scripting
13 38
Biostrings overview
I A package for working with large (biological) strings
I Two main types of objects A really long single string (thinkchromosome) or a set of shorter strings (think reads or genes)BString vs BStringSet
I These classes are implemented efficiently minimizing copy andmemory loadingunloading
I The BSgenome contains some infrastructure for whole genomes
I Methods for dealing with biological data including basicmanipulation (complementation translation etc) string searching(exactinexact matching Smith-Waterman PWMs)
I Fairly complicated class structure
Irsquom sorry to say that at least for me this has becomehopelessly confusing and I imagine that many other usersfeel the same ndash Simon Anders on bioc-sig-sequencing
Computing on genomes is not trivial The approach to a givencomputation is important
14 38
Strings in Biostrings
I BString (general) DNAString RNAString AAString (Amino Acid)all examples of XString
I complement reverse reverseComplement translate for the classeswhere ldquoit makes senserdquo
I Convert to and from a standard R character string
I Constructor DNAString(ACGGGGG)
I Support for IUPAC alphabet
I Subsetting subseq using the SEW format (two out of the threestartendwidth) Efficient
I StringSets are collections of Strings like DNAStringSet
15 38
BSgenomes
As examples we will use whole genomes Use availablegenomes to getavailable genomes Long package names but always a shorter objectname
gt library(BSgenomeScerevisiaeUCSCsacCer1)
gt Scerevisiae
gt Scerevisiae[[1]]
gt Scerevisiae[[chr1]]
A BSgenome may also have masks We will ignore this for now
16 38
Biostrings more
I alphabetFrequency oligonucleotideFrequency and others
gt alphabetFrequency(Scerevisiae[[chr1]])
gt oligonucleotideFrequency(Scerevisiae[[chr1]]
+ width = 3)
I chartr for character translation (ldquomake all As into Csrdquo)
I Various IO functions (also ShortRead)
17 38
Views
A view is a set of substrings of an XString stored and manipulated veryefficiently Example to store exon sequences one can just store thegenomic location of the exons
gt Views(Scerevisiae[[chr1]] start = c(300
+ 400 500) width = 50)
Views on a 230208-letter DNAString subjectsubject CCACACCACACCCAGTGGTGTGTGTGGGviews
start end width[1] 300 349 50 [CTGTTCTTAAATAAC][2] 400 449 50 [CCCTCACTAGTATAT][3] 500 549 50 [TCTCTCACCGGCACT]
A view is associated with a subject Internally they are essentiallyIRanges so they are very fast to compute with Views can in many casesbe treated exactly as other strings There are methods such as narrowtrim gaps restrict
18 38
Matching in Biostrings
There are various ways of matching or aligning strings to each other Weuse matching to denote searching for an exact match or possibly a matchwith a certain number of mismatches Alignment denotes a more generalstrategy eg Smith-Waterman
I matchPattern countPattern match 1 sequence to 1 sequence
I vmatchPattern vcountPattern match 1 sequence to manysequences
I pairwiseAlignment align many sequences to 1 sequence
I matchPDict countPDict match many sequences to 1 sequence(ldquodictrdquo indicates that the many sequences are preprocessed into adictionary)
I matchPWM trimLRpattern
The different functions are optimized for different situations They alsouse different algorithms which has a big impact EspeciallypairwiseAlignment is flexible and therefore has a complicated syntax
19 38
Example mapping probes to a genome
Get a list of Scerevisiae probes from the ldquoyeast2rdquo Affymetrix array
gt library(yeast2probe)
gt ids lt- scan(s_pombemsk skip = 2
+ what = list(probeset = character(0)
+ junk = character(0)))$probeset
gt probes lt- yeast2probe$sequence[yeast2probe$ProbeSetName in
+ ids]
gt probes lt- DNAStringSet(probes)
Mapping
gt require(BSgenomeScerevisiaeUCSCsacCer1)
gt dict0 lt- PDict(probes)
gt dict0r lt- PDict(reverseComplement(probes))
gt hits lt- matchPDict(dict0 Scerevisiae[[1]])
gt table(countIndex(hits))
gt table(countPDict(dict0 Scerevisiae[[1]]))
20 38
Example contrsquod
Over all chromosomes
gt allhits lt- lapply(116 function(i)
+ countPDict(dict0 Scerevisiae[[i]]) +
+ countPDict(dict0r Scerevisiae[[i]])
+ )
gt table(rowSums(docall(cbind allhits)))
21 38
ShortRead
ShortRead contains tools for
I Work with GERALDBUSTARD output
I Generate QA report on BUSTARD files
I Read a variety of short read data formats
22 38
Data
We will use data from (Lee Hansen Bullard Dudoit and Sherlock(2008) PLoS Genetics)
We are considering a wild-type and a mutant strain of yeast both grownin rich media The two strains were sequenced using an Illumina GenomeAnalyzer
We are using a subset of the data 1M reads from each of two lanes ofthe two strains
23 38
Reading the unaligned data
gt require(ShortRead)
gt fq lt- readFastq(seqdata mut_1_ffastq$)
gt fq
class ShortReadQlength 1000000 reads width 36 cycles
There are various simple accessor functions for this object especiallysread and quality
24 38
A brief look at the unaligned data
Using alphabetByCycle and as( rdquomatrixrdquo) (which creates a big read timescycle matrix) we can do
gt alp lt- alphabetByCycle(sread(fq))
gt matplot(t(proptable(alp[DNA_BASES
+ ] margin = 2)) type = l)
gt qaMatraw lt- as(quality(fq) matrix)
gt plot(colMeans(qaMatraw))
25 38
Aligning the data
We now align the data using Bowtie
binbash
BOWTIE_OPTS=-m 1 -v 2 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-m 1 -v 0 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 2 -k 2 -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 0 -k 1 -p 2 -q --quiet -3 10
for f in `ls seqdatafastq`dobowtie s_cerevisiae $BOWTIE_OPTS $f gt $fbowtiedone
How many hits do we get
26 38
Reading in the aligned data
We read the data into R as a 4-component list (one lane per component)
gt files lt- listfiles(seqdata
+ pattern = bowtie)
gt aligned lt- sapply(files function(f)
+ readAligned(seqdata pattern = f
+ type = Bowtie)
+ )
gt names(aligned) lt- gsub(_ffastqbowtie
+ names(aligned))
gt aligned[[1]]
gt save(aligned file = alignedrda)
27 38
Some exercises
1 Determine the number of times the motif ldquoTATAArdquo occurs in theyeast genome How often does it occur with one mismatch
2 The seqdata directory has an object called ste12 which is aposition weight matrix for the transcription factor ldquoSte12rdquo (obtainedfrom SCPD) Where does it occur in the genome
3 Compute the average quality for each cycle for the aligned reads andcompare to the qualities for the unaligned reads What is thedifference
28 38
Solution 1
gt sum(sapply(116 function(i)
+ motif lt- DNAString(TATAA)
+ countPattern(motif Scerevisiae[[i]]) +
+ countPattern(reverseComplement(motif)
+ Scerevisiae[[i]])
+ ))
29 38
Solution 2
30 38
Solution 3
31 38
SessionInfo
gt toLatex(sessionInfo())
I R version 292 RC (2009-08-17 r49312) i386-apple-darwin980
I Localeen_USUTF-8en_USUTF-8CCen_USUTF-8en_USUTF-8
I Base packages base datasets graphics grDevices grid methodsstats utils
I Other packages Biostrings 2128 BSgenome 1123BSgenomeScerevisiaeUCSCsacCer1 1313 classGraph 07-2graph 1222 IRanges 123 lattice 017-25 Rgraphviz 1234ShortRead 121
I Loaded via a namespace (and not attached) Biobase 241hwriter 11 tools 292
32 38
Some ShortRead classes
ShortRead
ShortReadQ
AlignedRead
33 38
Some Biostrings classes (single strings)
XString
BString DNAString RNAString AAString
34 38
Some Biostrings classes (sets of strings)
XStringSet
BStringSet DNAStringSet RNAStringSet AAStringSet PhredQualitySolexaQualityQualityScaledXStringSet
QualityScaledBStringSetQualityScaledDNAStringSetQualityScaledRNAStringSetQualityScaledAAStringSet
35 38

Alignment Tools
The number of short read aligners have exploded but a couple tools haveemerged as the de facto standards
I Eland Illuminarsquos aligner quality aware fast paired end capable
I MAQ Good SNP caller quality aware paired end capable
I Bowtie Super fast offers different alignment strategies paired endcapable
I BWA Fast indel support paired ends qualities
I NovoAlign MAQ like speed many features
A superior overview of the different aligners is available athttpwwwsangeracukUserslh3NGSalignshtml Additionallya comparison of two of the best aligners can be found here httpwwwmassgenomicsorg200907maq-bwa-and-bowtie-comparedhtml
10 38
Common Alignment Strategies
I Use qualities (default for Bowtie and MAQ)
I Perfect match no repeats (Strict Lenient)
I Mismatches no repeats
I Paired end data (PET) (harder for RNA-Seq)
The standard Illumina protocol yields unstranded reads
Most aligners are evaluated in terms of ldquohow many reads are mappedrdquo Isthis the right objective
Watch out for the output of the program there are many differentconventions (0-based what happens to read hitting the reverse strandetc)
11 38
SAMBAM Formats
SAM (BAM) is a new general format for storing mapped reads It isdeveloped as part of the 1000 Genomes project and is quickly becoming akind of standard Some alignment tools output this format directlyotherwise there are scripts in samtools for doing it for most popularaligners Details at httpsamtoolssourceforgenetindexshtml
12 38
A comment
There are no great comprehensive tools for analyzing deep-sequence dataIt will involve a fair amount of coding and gluing together various tools
We will introduce some tools from Bioconductor that can be useful
I use a lot of shell scripting
13 38
Biostrings overview
I A package for working with large (biological) strings
I Two main types of objects A really long single string (thinkchromosome) or a set of shorter strings (think reads or genes)BString vs BStringSet
I These classes are implemented efficiently minimizing copy andmemory loadingunloading
I The BSgenome contains some infrastructure for whole genomes
I Methods for dealing with biological data including basicmanipulation (complementation translation etc) string searching(exactinexact matching Smith-Waterman PWMs)
I Fairly complicated class structure
Irsquom sorry to say that at least for me this has becomehopelessly confusing and I imagine that many other usersfeel the same ndash Simon Anders on bioc-sig-sequencing
Computing on genomes is not trivial The approach to a givencomputation is important
14 38
Strings in Biostrings
I BString (general) DNAString RNAString AAString (Amino Acid)all examples of XString
I complement reverse reverseComplement translate for the classeswhere ldquoit makes senserdquo
I Convert to and from a standard R character string
I Constructor DNAString(ACGGGGG)
I Support for IUPAC alphabet
I Subsetting subseq using the SEW format (two out of the threestartendwidth) Efficient
I StringSets are collections of Strings like DNAStringSet
15 38
BSgenomes
As examples we will use whole genomes Use availablegenomes to getavailable genomes Long package names but always a shorter objectname
gt library(BSgenomeScerevisiaeUCSCsacCer1)
gt Scerevisiae
gt Scerevisiae[[1]]
gt Scerevisiae[[chr1]]
A BSgenome may also have masks We will ignore this for now
16 38
Biostrings more
I alphabetFrequency oligonucleotideFrequency and others
gt alphabetFrequency(Scerevisiae[[chr1]])
gt oligonucleotideFrequency(Scerevisiae[[chr1]]
+ width = 3)
I chartr for character translation (ldquomake all As into Csrdquo)
I Various IO functions (also ShortRead)
17 38
Views
A view is a set of substrings of an XString stored and manipulated veryefficiently Example to store exon sequences one can just store thegenomic location of the exons
gt Views(Scerevisiae[[chr1]] start = c(300
+ 400 500) width = 50)
Views on a 230208-letter DNAString subjectsubject CCACACCACACCCAGTGGTGTGTGTGGGviews
start end width[1] 300 349 50 [CTGTTCTTAAATAAC][2] 400 449 50 [CCCTCACTAGTATAT][3] 500 549 50 [TCTCTCACCGGCACT]
A view is associated with a subject Internally they are essentiallyIRanges so they are very fast to compute with Views can in many casesbe treated exactly as other strings There are methods such as narrowtrim gaps restrict
18 38
Matching in Biostrings
There are various ways of matching or aligning strings to each other Weuse matching to denote searching for an exact match or possibly a matchwith a certain number of mismatches Alignment denotes a more generalstrategy eg Smith-Waterman
I matchPattern countPattern match 1 sequence to 1 sequence
I vmatchPattern vcountPattern match 1 sequence to manysequences
I pairwiseAlignment align many sequences to 1 sequence
I matchPDict countPDict match many sequences to 1 sequence(ldquodictrdquo indicates that the many sequences are preprocessed into adictionary)
I matchPWM trimLRpattern
The different functions are optimized for different situations They alsouse different algorithms which has a big impact EspeciallypairwiseAlignment is flexible and therefore has a complicated syntax
19 38
Example mapping probes to a genome
Get a list of Scerevisiae probes from the ldquoyeast2rdquo Affymetrix array
gt library(yeast2probe)
gt ids lt- scan(s_pombemsk skip = 2
+ what = list(probeset = character(0)
+ junk = character(0)))$probeset
gt probes lt- yeast2probe$sequence[yeast2probe$ProbeSetName in
+ ids]
gt probes lt- DNAStringSet(probes)
Mapping
gt require(BSgenomeScerevisiaeUCSCsacCer1)
gt dict0 lt- PDict(probes)
gt dict0r lt- PDict(reverseComplement(probes))
gt hits lt- matchPDict(dict0 Scerevisiae[[1]])
gt table(countIndex(hits))
gt table(countPDict(dict0 Scerevisiae[[1]]))
20 38
Example contrsquod
Over all chromosomes
gt allhits lt- lapply(116 function(i)
+ countPDict(dict0 Scerevisiae[[i]]) +
+ countPDict(dict0r Scerevisiae[[i]])
+ )
gt table(rowSums(docall(cbind allhits)))
21 38
ShortRead
ShortRead contains tools for
I Work with GERALDBUSTARD output
I Generate QA report on BUSTARD files
I Read a variety of short read data formats
22 38
Data
We will use data from (Lee Hansen Bullard Dudoit and Sherlock(2008) PLoS Genetics)
We are considering a wild-type and a mutant strain of yeast both grownin rich media The two strains were sequenced using an Illumina GenomeAnalyzer
We are using a subset of the data 1M reads from each of two lanes ofthe two strains
23 38
Reading the unaligned data
gt require(ShortRead)
gt fq lt- readFastq(seqdata mut_1_ffastq$)
gt fq
class ShortReadQlength 1000000 reads width 36 cycles
There are various simple accessor functions for this object especiallysread and quality
24 38
A brief look at the unaligned data
Using alphabetByCycle and as( rdquomatrixrdquo) (which creates a big read timescycle matrix) we can do
gt alp lt- alphabetByCycle(sread(fq))
gt matplot(t(proptable(alp[DNA_BASES
+ ] margin = 2)) type = l)
gt qaMatraw lt- as(quality(fq) matrix)
gt plot(colMeans(qaMatraw))
25 38
Aligning the data
We now align the data using Bowtie
binbash
BOWTIE_OPTS=-m 1 -v 2 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-m 1 -v 0 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 2 -k 2 -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 0 -k 1 -p 2 -q --quiet -3 10
for f in `ls seqdatafastq`dobowtie s_cerevisiae $BOWTIE_OPTS $f gt $fbowtiedone
How many hits do we get
26 38
Reading in the aligned data
We read the data into R as a 4-component list (one lane per component)
gt files lt- listfiles(seqdata
+ pattern = bowtie)
gt aligned lt- sapply(files function(f)
+ readAligned(seqdata pattern = f
+ type = Bowtie)
+ )
gt names(aligned) lt- gsub(_ffastqbowtie
+ names(aligned))
gt aligned[[1]]
gt save(aligned file = alignedrda)
27 38
Some exercises
1 Determine the number of times the motif ldquoTATAArdquo occurs in theyeast genome How often does it occur with one mismatch
2 The seqdata directory has an object called ste12 which is aposition weight matrix for the transcription factor ldquoSte12rdquo (obtainedfrom SCPD) Where does it occur in the genome
3 Compute the average quality for each cycle for the aligned reads andcompare to the qualities for the unaligned reads What is thedifference
28 38
Solution 1
gt sum(sapply(116 function(i)
+ motif lt- DNAString(TATAA)
+ countPattern(motif Scerevisiae[[i]]) +
+ countPattern(reverseComplement(motif)
+ Scerevisiae[[i]])
+ ))
29 38
Solution 2
30 38
Solution 3
31 38
SessionInfo
gt toLatex(sessionInfo())
I R version 292 RC (2009-08-17 r49312) i386-apple-darwin980
I Localeen_USUTF-8en_USUTF-8CCen_USUTF-8en_USUTF-8
I Base packages base datasets graphics grDevices grid methodsstats utils
I Other packages Biostrings 2128 BSgenome 1123BSgenomeScerevisiaeUCSCsacCer1 1313 classGraph 07-2graph 1222 IRanges 123 lattice 017-25 Rgraphviz 1234ShortRead 121
I Loaded via a namespace (and not attached) Biobase 241hwriter 11 tools 292
32 38
Some ShortRead classes
ShortRead
ShortReadQ
AlignedRead
33 38
Some Biostrings classes (single strings)
XString
BString DNAString RNAString AAString
34 38
Some Biostrings classes (sets of strings)
XStringSet
BStringSet DNAStringSet RNAStringSet AAStringSet PhredQualitySolexaQualityQualityScaledXStringSet
QualityScaledBStringSetQualityScaledDNAStringSetQualityScaledRNAStringSetQualityScaledAAStringSet
35 38

Common Alignment Strategies
I Use qualities (default for Bowtie and MAQ)
I Perfect match no repeats (Strict Lenient)
I Mismatches no repeats
I Paired end data (PET) (harder for RNA-Seq)
The standard Illumina protocol yields unstranded reads
Most aligners are evaluated in terms of ldquohow many reads are mappedrdquo Isthis the right objective
Watch out for the output of the program there are many differentconventions (0-based what happens to read hitting the reverse strandetc)
11 38
SAMBAM Formats
SAM (BAM) is a new general format for storing mapped reads It isdeveloped as part of the 1000 Genomes project and is quickly becoming akind of standard Some alignment tools output this format directlyotherwise there are scripts in samtools for doing it for most popularaligners Details at httpsamtoolssourceforgenetindexshtml
12 38
A comment
There are no great comprehensive tools for analyzing deep-sequence dataIt will involve a fair amount of coding and gluing together various tools
We will introduce some tools from Bioconductor that can be useful
I use a lot of shell scripting
13 38
Biostrings overview
I A package for working with large (biological) strings
I Two main types of objects A really long single string (thinkchromosome) or a set of shorter strings (think reads or genes)BString vs BStringSet
I These classes are implemented efficiently minimizing copy andmemory loadingunloading
I The BSgenome contains some infrastructure for whole genomes
I Methods for dealing with biological data including basicmanipulation (complementation translation etc) string searching(exactinexact matching Smith-Waterman PWMs)
I Fairly complicated class structure
Irsquom sorry to say that at least for me this has becomehopelessly confusing and I imagine that many other usersfeel the same ndash Simon Anders on bioc-sig-sequencing
Computing on genomes is not trivial The approach to a givencomputation is important
14 38
Strings in Biostrings
I BString (general) DNAString RNAString AAString (Amino Acid)all examples of XString
I complement reverse reverseComplement translate for the classeswhere ldquoit makes senserdquo
I Convert to and from a standard R character string
I Constructor DNAString(ACGGGGG)
I Support for IUPAC alphabet
I Subsetting subseq using the SEW format (two out of the threestartendwidth) Efficient
I StringSets are collections of Strings like DNAStringSet
15 38
BSgenomes
As examples we will use whole genomes Use availablegenomes to getavailable genomes Long package names but always a shorter objectname
gt library(BSgenomeScerevisiaeUCSCsacCer1)
gt Scerevisiae
gt Scerevisiae[[1]]
gt Scerevisiae[[chr1]]
A BSgenome may also have masks We will ignore this for now
16 38
Biostrings more
I alphabetFrequency oligonucleotideFrequency and others
gt alphabetFrequency(Scerevisiae[[chr1]])
gt oligonucleotideFrequency(Scerevisiae[[chr1]]
+ width = 3)
I chartr for character translation (ldquomake all As into Csrdquo)
I Various IO functions (also ShortRead)
17 38
Views
A view is a set of substrings of an XString stored and manipulated veryefficiently Example to store exon sequences one can just store thegenomic location of the exons
gt Views(Scerevisiae[[chr1]] start = c(300
+ 400 500) width = 50)
Views on a 230208-letter DNAString subjectsubject CCACACCACACCCAGTGGTGTGTGTGGGviews
start end width[1] 300 349 50 [CTGTTCTTAAATAAC][2] 400 449 50 [CCCTCACTAGTATAT][3] 500 549 50 [TCTCTCACCGGCACT]
A view is associated with a subject Internally they are essentiallyIRanges so they are very fast to compute with Views can in many casesbe treated exactly as other strings There are methods such as narrowtrim gaps restrict
18 38
Matching in Biostrings
There are various ways of matching or aligning strings to each other Weuse matching to denote searching for an exact match or possibly a matchwith a certain number of mismatches Alignment denotes a more generalstrategy eg Smith-Waterman
I matchPattern countPattern match 1 sequence to 1 sequence
I vmatchPattern vcountPattern match 1 sequence to manysequences
I pairwiseAlignment align many sequences to 1 sequence
I matchPDict countPDict match many sequences to 1 sequence(ldquodictrdquo indicates that the many sequences are preprocessed into adictionary)
I matchPWM trimLRpattern
The different functions are optimized for different situations They alsouse different algorithms which has a big impact EspeciallypairwiseAlignment is flexible and therefore has a complicated syntax
19 38
Example mapping probes to a genome
Get a list of Scerevisiae probes from the ldquoyeast2rdquo Affymetrix array
gt library(yeast2probe)
gt ids lt- scan(s_pombemsk skip = 2
+ what = list(probeset = character(0)
+ junk = character(0)))$probeset
gt probes lt- yeast2probe$sequence[yeast2probe$ProbeSetName in
+ ids]
gt probes lt- DNAStringSet(probes)
Mapping
gt require(BSgenomeScerevisiaeUCSCsacCer1)
gt dict0 lt- PDict(probes)
gt dict0r lt- PDict(reverseComplement(probes))
gt hits lt- matchPDict(dict0 Scerevisiae[[1]])
gt table(countIndex(hits))
gt table(countPDict(dict0 Scerevisiae[[1]]))
20 38
Example contrsquod
Over all chromosomes
gt allhits lt- lapply(116 function(i)
+ countPDict(dict0 Scerevisiae[[i]]) +
+ countPDict(dict0r Scerevisiae[[i]])
+ )
gt table(rowSums(docall(cbind allhits)))
21 38
ShortRead
ShortRead contains tools for
I Work with GERALDBUSTARD output
I Generate QA report on BUSTARD files
I Read a variety of short read data formats
22 38
Data
We will use data from (Lee Hansen Bullard Dudoit and Sherlock(2008) PLoS Genetics)
We are considering a wild-type and a mutant strain of yeast both grownin rich media The two strains were sequenced using an Illumina GenomeAnalyzer
We are using a subset of the data 1M reads from each of two lanes ofthe two strains
23 38
Reading the unaligned data
gt require(ShortRead)
gt fq lt- readFastq(seqdata mut_1_ffastq$)
gt fq
class ShortReadQlength 1000000 reads width 36 cycles
There are various simple accessor functions for this object especiallysread and quality
24 38
A brief look at the unaligned data
Using alphabetByCycle and as( rdquomatrixrdquo) (which creates a big read timescycle matrix) we can do
gt alp lt- alphabetByCycle(sread(fq))
gt matplot(t(proptable(alp[DNA_BASES
+ ] margin = 2)) type = l)
gt qaMatraw lt- as(quality(fq) matrix)
gt plot(colMeans(qaMatraw))
25 38
Aligning the data
We now align the data using Bowtie
binbash
BOWTIE_OPTS=-m 1 -v 2 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-m 1 -v 0 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 2 -k 2 -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 0 -k 1 -p 2 -q --quiet -3 10
for f in `ls seqdatafastq`dobowtie s_cerevisiae $BOWTIE_OPTS $f gt $fbowtiedone
How many hits do we get
26 38
Reading in the aligned data
We read the data into R as a 4-component list (one lane per component)
gt files lt- listfiles(seqdata
+ pattern = bowtie)
gt aligned lt- sapply(files function(f)
+ readAligned(seqdata pattern = f
+ type = Bowtie)
+ )
gt names(aligned) lt- gsub(_ffastqbowtie
+ names(aligned))
gt aligned[[1]]
gt save(aligned file = alignedrda)
27 38
Some exercises
1 Determine the number of times the motif ldquoTATAArdquo occurs in theyeast genome How often does it occur with one mismatch
2 The seqdata directory has an object called ste12 which is aposition weight matrix for the transcription factor ldquoSte12rdquo (obtainedfrom SCPD) Where does it occur in the genome
3 Compute the average quality for each cycle for the aligned reads andcompare to the qualities for the unaligned reads What is thedifference
28 38
Solution 1
gt sum(sapply(116 function(i)
+ motif lt- DNAString(TATAA)
+ countPattern(motif Scerevisiae[[i]]) +
+ countPattern(reverseComplement(motif)
+ Scerevisiae[[i]])
+ ))
29 38
Solution 2
30 38
Solution 3
31 38
SessionInfo
gt toLatex(sessionInfo())
I R version 292 RC (2009-08-17 r49312) i386-apple-darwin980
I Localeen_USUTF-8en_USUTF-8CCen_USUTF-8en_USUTF-8
I Base packages base datasets graphics grDevices grid methodsstats utils
I Other packages Biostrings 2128 BSgenome 1123BSgenomeScerevisiaeUCSCsacCer1 1313 classGraph 07-2graph 1222 IRanges 123 lattice 017-25 Rgraphviz 1234ShortRead 121
I Loaded via a namespace (and not attached) Biobase 241hwriter 11 tools 292
32 38
Some ShortRead classes
ShortRead
ShortReadQ
AlignedRead
33 38
Some Biostrings classes (single strings)
XString
BString DNAString RNAString AAString
34 38
Some Biostrings classes (sets of strings)
XStringSet
BStringSet DNAStringSet RNAStringSet AAStringSet PhredQualitySolexaQualityQualityScaledXStringSet
QualityScaledBStringSetQualityScaledDNAStringSetQualityScaledRNAStringSetQualityScaledAAStringSet
35 38

SAMBAM Formats
SAM (BAM) is a new general format for storing mapped reads It isdeveloped as part of the 1000 Genomes project and is quickly becoming akind of standard Some alignment tools output this format directlyotherwise there are scripts in samtools for doing it for most popularaligners Details at httpsamtoolssourceforgenetindexshtml
12 38
A comment
There are no great comprehensive tools for analyzing deep-sequence dataIt will involve a fair amount of coding and gluing together various tools
We will introduce some tools from Bioconductor that can be useful
I use a lot of shell scripting
13 38
Biostrings overview
I A package for working with large (biological) strings
I Two main types of objects A really long single string (thinkchromosome) or a set of shorter strings (think reads or genes)BString vs BStringSet
I These classes are implemented efficiently minimizing copy andmemory loadingunloading
I The BSgenome contains some infrastructure for whole genomes
I Methods for dealing with biological data including basicmanipulation (complementation translation etc) string searching(exactinexact matching Smith-Waterman PWMs)
I Fairly complicated class structure
Irsquom sorry to say that at least for me this has becomehopelessly confusing and I imagine that many other usersfeel the same ndash Simon Anders on bioc-sig-sequencing
Computing on genomes is not trivial The approach to a givencomputation is important
14 38
Strings in Biostrings
I BString (general) DNAString RNAString AAString (Amino Acid)all examples of XString
I complement reverse reverseComplement translate for the classeswhere ldquoit makes senserdquo
I Convert to and from a standard R character string
I Constructor DNAString(ACGGGGG)
I Support for IUPAC alphabet
I Subsetting subseq using the SEW format (two out of the threestartendwidth) Efficient
I StringSets are collections of Strings like DNAStringSet
15 38
BSgenomes
As examples we will use whole genomes Use availablegenomes to getavailable genomes Long package names but always a shorter objectname
gt library(BSgenomeScerevisiaeUCSCsacCer1)
gt Scerevisiae
gt Scerevisiae[[1]]
gt Scerevisiae[[chr1]]
A BSgenome may also have masks We will ignore this for now
16 38
Biostrings more
I alphabetFrequency oligonucleotideFrequency and others
gt alphabetFrequency(Scerevisiae[[chr1]])
gt oligonucleotideFrequency(Scerevisiae[[chr1]]
+ width = 3)
I chartr for character translation (ldquomake all As into Csrdquo)
I Various IO functions (also ShortRead)
17 38
Views
A view is a set of substrings of an XString stored and manipulated veryefficiently Example to store exon sequences one can just store thegenomic location of the exons
gt Views(Scerevisiae[[chr1]] start = c(300
+ 400 500) width = 50)
Views on a 230208-letter DNAString subjectsubject CCACACCACACCCAGTGGTGTGTGTGGGviews
start end width[1] 300 349 50 [CTGTTCTTAAATAAC][2] 400 449 50 [CCCTCACTAGTATAT][3] 500 549 50 [TCTCTCACCGGCACT]
A view is associated with a subject Internally they are essentiallyIRanges so they are very fast to compute with Views can in many casesbe treated exactly as other strings There are methods such as narrowtrim gaps restrict
18 38
Matching in Biostrings
There are various ways of matching or aligning strings to each other Weuse matching to denote searching for an exact match or possibly a matchwith a certain number of mismatches Alignment denotes a more generalstrategy eg Smith-Waterman
I matchPattern countPattern match 1 sequence to 1 sequence
I vmatchPattern vcountPattern match 1 sequence to manysequences
I pairwiseAlignment align many sequences to 1 sequence
I matchPDict countPDict match many sequences to 1 sequence(ldquodictrdquo indicates that the many sequences are preprocessed into adictionary)
I matchPWM trimLRpattern
The different functions are optimized for different situations They alsouse different algorithms which has a big impact EspeciallypairwiseAlignment is flexible and therefore has a complicated syntax
19 38
Example mapping probes to a genome
Get a list of Scerevisiae probes from the ldquoyeast2rdquo Affymetrix array
gt library(yeast2probe)
gt ids lt- scan(s_pombemsk skip = 2
+ what = list(probeset = character(0)
+ junk = character(0)))$probeset
gt probes lt- yeast2probe$sequence[yeast2probe$ProbeSetName in
+ ids]
gt probes lt- DNAStringSet(probes)
Mapping
gt require(BSgenomeScerevisiaeUCSCsacCer1)
gt dict0 lt- PDict(probes)
gt dict0r lt- PDict(reverseComplement(probes))
gt hits lt- matchPDict(dict0 Scerevisiae[[1]])
gt table(countIndex(hits))
gt table(countPDict(dict0 Scerevisiae[[1]]))
20 38
Example contrsquod
Over all chromosomes
gt allhits lt- lapply(116 function(i)
+ countPDict(dict0 Scerevisiae[[i]]) +
+ countPDict(dict0r Scerevisiae[[i]])
+ )
gt table(rowSums(docall(cbind allhits)))
21 38
ShortRead
ShortRead contains tools for
I Work with GERALDBUSTARD output
I Generate QA report on BUSTARD files
I Read a variety of short read data formats
22 38
Data
We will use data from (Lee Hansen Bullard Dudoit and Sherlock(2008) PLoS Genetics)
We are considering a wild-type and a mutant strain of yeast both grownin rich media The two strains were sequenced using an Illumina GenomeAnalyzer
We are using a subset of the data 1M reads from each of two lanes ofthe two strains
23 38
Reading the unaligned data
gt require(ShortRead)
gt fq lt- readFastq(seqdata mut_1_ffastq$)
gt fq
class ShortReadQlength 1000000 reads width 36 cycles
There are various simple accessor functions for this object especiallysread and quality
24 38
A brief look at the unaligned data
Using alphabetByCycle and as( rdquomatrixrdquo) (which creates a big read timescycle matrix) we can do
gt alp lt- alphabetByCycle(sread(fq))
gt matplot(t(proptable(alp[DNA_BASES
+ ] margin = 2)) type = l)
gt qaMatraw lt- as(quality(fq) matrix)
gt plot(colMeans(qaMatraw))
25 38
Aligning the data
We now align the data using Bowtie
binbash
BOWTIE_OPTS=-m 1 -v 2 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-m 1 -v 0 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 2 -k 2 -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 0 -k 1 -p 2 -q --quiet -3 10
for f in `ls seqdatafastq`dobowtie s_cerevisiae $BOWTIE_OPTS $f gt $fbowtiedone
How many hits do we get
26 38
Reading in the aligned data
We read the data into R as a 4-component list (one lane per component)
gt files lt- listfiles(seqdata
+ pattern = bowtie)
gt aligned lt- sapply(files function(f)
+ readAligned(seqdata pattern = f
+ type = Bowtie)
+ )
gt names(aligned) lt- gsub(_ffastqbowtie
+ names(aligned))
gt aligned[[1]]
gt save(aligned file = alignedrda)
27 38
Some exercises
1 Determine the number of times the motif ldquoTATAArdquo occurs in theyeast genome How often does it occur with one mismatch
2 The seqdata directory has an object called ste12 which is aposition weight matrix for the transcription factor ldquoSte12rdquo (obtainedfrom SCPD) Where does it occur in the genome
3 Compute the average quality for each cycle for the aligned reads andcompare to the qualities for the unaligned reads What is thedifference
28 38
Solution 1
gt sum(sapply(116 function(i)
+ motif lt- DNAString(TATAA)
+ countPattern(motif Scerevisiae[[i]]) +
+ countPattern(reverseComplement(motif)
+ Scerevisiae[[i]])
+ ))
29 38
Solution 2
30 38
Solution 3
31 38
SessionInfo
gt toLatex(sessionInfo())
I R version 292 RC (2009-08-17 r49312) i386-apple-darwin980
I Localeen_USUTF-8en_USUTF-8CCen_USUTF-8en_USUTF-8
I Base packages base datasets graphics grDevices grid methodsstats utils
I Other packages Biostrings 2128 BSgenome 1123BSgenomeScerevisiaeUCSCsacCer1 1313 classGraph 07-2graph 1222 IRanges 123 lattice 017-25 Rgraphviz 1234ShortRead 121
I Loaded via a namespace (and not attached) Biobase 241hwriter 11 tools 292
32 38
Some ShortRead classes
ShortRead
ShortReadQ
AlignedRead
33 38
Some Biostrings classes (single strings)
XString
BString DNAString RNAString AAString
34 38
Some Biostrings classes (sets of strings)
XStringSet
BStringSet DNAStringSet RNAStringSet AAStringSet PhredQualitySolexaQualityQualityScaledXStringSet
QualityScaledBStringSetQualityScaledDNAStringSetQualityScaledRNAStringSetQualityScaledAAStringSet
35 38

A comment
There are no great comprehensive tools for analyzing deep-sequence dataIt will involve a fair amount of coding and gluing together various tools
We will introduce some tools from Bioconductor that can be useful
I use a lot of shell scripting
13 38
Biostrings overview
I A package for working with large (biological) strings
I Two main types of objects A really long single string (thinkchromosome) or a set of shorter strings (think reads or genes)BString vs BStringSet
I These classes are implemented efficiently minimizing copy andmemory loadingunloading
I The BSgenome contains some infrastructure for whole genomes
I Methods for dealing with biological data including basicmanipulation (complementation translation etc) string searching(exactinexact matching Smith-Waterman PWMs)
I Fairly complicated class structure
Irsquom sorry to say that at least for me this has becomehopelessly confusing and I imagine that many other usersfeel the same ndash Simon Anders on bioc-sig-sequencing
Computing on genomes is not trivial The approach to a givencomputation is important
14 38
Strings in Biostrings
I BString (general) DNAString RNAString AAString (Amino Acid)all examples of XString
I complement reverse reverseComplement translate for the classeswhere ldquoit makes senserdquo
I Convert to and from a standard R character string
I Constructor DNAString(ACGGGGG)
I Support for IUPAC alphabet
I Subsetting subseq using the SEW format (two out of the threestartendwidth) Efficient
I StringSets are collections of Strings like DNAStringSet
15 38
BSgenomes
As examples we will use whole genomes Use availablegenomes to getavailable genomes Long package names but always a shorter objectname
gt library(BSgenomeScerevisiaeUCSCsacCer1)
gt Scerevisiae
gt Scerevisiae[[1]]
gt Scerevisiae[[chr1]]
A BSgenome may also have masks We will ignore this for now
16 38
Biostrings more
I alphabetFrequency oligonucleotideFrequency and others
gt alphabetFrequency(Scerevisiae[[chr1]])
gt oligonucleotideFrequency(Scerevisiae[[chr1]]
+ width = 3)
I chartr for character translation (ldquomake all As into Csrdquo)
I Various IO functions (also ShortRead)
17 38
Views
A view is a set of substrings of an XString stored and manipulated veryefficiently Example to store exon sequences one can just store thegenomic location of the exons
gt Views(Scerevisiae[[chr1]] start = c(300
+ 400 500) width = 50)
Views on a 230208-letter DNAString subjectsubject CCACACCACACCCAGTGGTGTGTGTGGGviews
start end width[1] 300 349 50 [CTGTTCTTAAATAAC][2] 400 449 50 [CCCTCACTAGTATAT][3] 500 549 50 [TCTCTCACCGGCACT]
A view is associated with a subject Internally they are essentiallyIRanges so they are very fast to compute with Views can in many casesbe treated exactly as other strings There are methods such as narrowtrim gaps restrict
18 38
Matching in Biostrings
There are various ways of matching or aligning strings to each other Weuse matching to denote searching for an exact match or possibly a matchwith a certain number of mismatches Alignment denotes a more generalstrategy eg Smith-Waterman
I matchPattern countPattern match 1 sequence to 1 sequence
I vmatchPattern vcountPattern match 1 sequence to manysequences
I pairwiseAlignment align many sequences to 1 sequence
I matchPDict countPDict match many sequences to 1 sequence(ldquodictrdquo indicates that the many sequences are preprocessed into adictionary)
I matchPWM trimLRpattern
The different functions are optimized for different situations They alsouse different algorithms which has a big impact EspeciallypairwiseAlignment is flexible and therefore has a complicated syntax
19 38
Example mapping probes to a genome
Get a list of Scerevisiae probes from the ldquoyeast2rdquo Affymetrix array
gt library(yeast2probe)
gt ids lt- scan(s_pombemsk skip = 2
+ what = list(probeset = character(0)
+ junk = character(0)))$probeset
gt probes lt- yeast2probe$sequence[yeast2probe$ProbeSetName in
+ ids]
gt probes lt- DNAStringSet(probes)
Mapping
gt require(BSgenomeScerevisiaeUCSCsacCer1)
gt dict0 lt- PDict(probes)
gt dict0r lt- PDict(reverseComplement(probes))
gt hits lt- matchPDict(dict0 Scerevisiae[[1]])
gt table(countIndex(hits))
gt table(countPDict(dict0 Scerevisiae[[1]]))
20 38
Example contrsquod
Over all chromosomes
gt allhits lt- lapply(116 function(i)
+ countPDict(dict0 Scerevisiae[[i]]) +
+ countPDict(dict0r Scerevisiae[[i]])
+ )
gt table(rowSums(docall(cbind allhits)))
21 38
ShortRead
ShortRead contains tools for
I Work with GERALDBUSTARD output
I Generate QA report on BUSTARD files
I Read a variety of short read data formats
22 38
Data
We will use data from (Lee Hansen Bullard Dudoit and Sherlock(2008) PLoS Genetics)
We are considering a wild-type and a mutant strain of yeast both grownin rich media The two strains were sequenced using an Illumina GenomeAnalyzer
We are using a subset of the data 1M reads from each of two lanes ofthe two strains
23 38
Reading the unaligned data
gt require(ShortRead)
gt fq lt- readFastq(seqdata mut_1_ffastq$)
gt fq
class ShortReadQlength 1000000 reads width 36 cycles
There are various simple accessor functions for this object especiallysread and quality
24 38
A brief look at the unaligned data
Using alphabetByCycle and as( rdquomatrixrdquo) (which creates a big read timescycle matrix) we can do
gt alp lt- alphabetByCycle(sread(fq))
gt matplot(t(proptable(alp[DNA_BASES
+ ] margin = 2)) type = l)
gt qaMatraw lt- as(quality(fq) matrix)
gt plot(colMeans(qaMatraw))
25 38
Aligning the data
We now align the data using Bowtie
binbash
BOWTIE_OPTS=-m 1 -v 2 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-m 1 -v 0 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 2 -k 2 -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 0 -k 1 -p 2 -q --quiet -3 10
for f in `ls seqdatafastq`dobowtie s_cerevisiae $BOWTIE_OPTS $f gt $fbowtiedone
How many hits do we get
26 38
Reading in the aligned data
We read the data into R as a 4-component list (one lane per component)
gt files lt- listfiles(seqdata
+ pattern = bowtie)
gt aligned lt- sapply(files function(f)
+ readAligned(seqdata pattern = f
+ type = Bowtie)
+ )
gt names(aligned) lt- gsub(_ffastqbowtie
+ names(aligned))
gt aligned[[1]]
gt save(aligned file = alignedrda)
27 38
Some exercises
1 Determine the number of times the motif ldquoTATAArdquo occurs in theyeast genome How often does it occur with one mismatch
2 The seqdata directory has an object called ste12 which is aposition weight matrix for the transcription factor ldquoSte12rdquo (obtainedfrom SCPD) Where does it occur in the genome
3 Compute the average quality for each cycle for the aligned reads andcompare to the qualities for the unaligned reads What is thedifference
28 38
Solution 1
gt sum(sapply(116 function(i)
+ motif lt- DNAString(TATAA)
+ countPattern(motif Scerevisiae[[i]]) +
+ countPattern(reverseComplement(motif)
+ Scerevisiae[[i]])
+ ))
29 38
Solution 2
30 38
Solution 3
31 38
SessionInfo
gt toLatex(sessionInfo())
I R version 292 RC (2009-08-17 r49312) i386-apple-darwin980
I Localeen_USUTF-8en_USUTF-8CCen_USUTF-8en_USUTF-8
I Base packages base datasets graphics grDevices grid methodsstats utils
I Other packages Biostrings 2128 BSgenome 1123BSgenomeScerevisiaeUCSCsacCer1 1313 classGraph 07-2graph 1222 IRanges 123 lattice 017-25 Rgraphviz 1234ShortRead 121
I Loaded via a namespace (and not attached) Biobase 241hwriter 11 tools 292
32 38
Some ShortRead classes
ShortRead
ShortReadQ
AlignedRead
33 38
Some Biostrings classes (single strings)
XString
BString DNAString RNAString AAString
34 38
Some Biostrings classes (sets of strings)
XStringSet
BStringSet DNAStringSet RNAStringSet AAStringSet PhredQualitySolexaQualityQualityScaledXStringSet
QualityScaledBStringSetQualityScaledDNAStringSetQualityScaledRNAStringSetQualityScaledAAStringSet
35 38

Biostrings overview
I A package for working with large (biological) strings
I Two main types of objects A really long single string (thinkchromosome) or a set of shorter strings (think reads or genes)BString vs BStringSet
I These classes are implemented efficiently minimizing copy andmemory loadingunloading
I The BSgenome contains some infrastructure for whole genomes
I Methods for dealing with biological data including basicmanipulation (complementation translation etc) string searching(exactinexact matching Smith-Waterman PWMs)
I Fairly complicated class structure
Irsquom sorry to say that at least for me this has becomehopelessly confusing and I imagine that many other usersfeel the same ndash Simon Anders on bioc-sig-sequencing
Computing on genomes is not trivial The approach to a givencomputation is important
14 38
Strings in Biostrings
I BString (general) DNAString RNAString AAString (Amino Acid)all examples of XString
I complement reverse reverseComplement translate for the classeswhere ldquoit makes senserdquo
I Convert to and from a standard R character string
I Constructor DNAString(ACGGGGG)
I Support for IUPAC alphabet
I Subsetting subseq using the SEW format (two out of the threestartendwidth) Efficient
I StringSets are collections of Strings like DNAStringSet
15 38
BSgenomes
As examples we will use whole genomes Use availablegenomes to getavailable genomes Long package names but always a shorter objectname
gt library(BSgenomeScerevisiaeUCSCsacCer1)
gt Scerevisiae
gt Scerevisiae[[1]]
gt Scerevisiae[[chr1]]
A BSgenome may also have masks We will ignore this for now
16 38
Biostrings more
I alphabetFrequency oligonucleotideFrequency and others
gt alphabetFrequency(Scerevisiae[[chr1]])
gt oligonucleotideFrequency(Scerevisiae[[chr1]]
+ width = 3)
I chartr for character translation (ldquomake all As into Csrdquo)
I Various IO functions (also ShortRead)
17 38
Views
A view is a set of substrings of an XString stored and manipulated veryefficiently Example to store exon sequences one can just store thegenomic location of the exons
gt Views(Scerevisiae[[chr1]] start = c(300
+ 400 500) width = 50)
Views on a 230208-letter DNAString subjectsubject CCACACCACACCCAGTGGTGTGTGTGGGviews
start end width[1] 300 349 50 [CTGTTCTTAAATAAC][2] 400 449 50 [CCCTCACTAGTATAT][3] 500 549 50 [TCTCTCACCGGCACT]
A view is associated with a subject Internally they are essentiallyIRanges so they are very fast to compute with Views can in many casesbe treated exactly as other strings There are methods such as narrowtrim gaps restrict
18 38
Matching in Biostrings
There are various ways of matching or aligning strings to each other Weuse matching to denote searching for an exact match or possibly a matchwith a certain number of mismatches Alignment denotes a more generalstrategy eg Smith-Waterman
I matchPattern countPattern match 1 sequence to 1 sequence
I vmatchPattern vcountPattern match 1 sequence to manysequences
I pairwiseAlignment align many sequences to 1 sequence
I matchPDict countPDict match many sequences to 1 sequence(ldquodictrdquo indicates that the many sequences are preprocessed into adictionary)
I matchPWM trimLRpattern
The different functions are optimized for different situations They alsouse different algorithms which has a big impact EspeciallypairwiseAlignment is flexible and therefore has a complicated syntax
19 38
Example mapping probes to a genome
Get a list of Scerevisiae probes from the ldquoyeast2rdquo Affymetrix array
gt library(yeast2probe)
gt ids lt- scan(s_pombemsk skip = 2
+ what = list(probeset = character(0)
+ junk = character(0)))$probeset
gt probes lt- yeast2probe$sequence[yeast2probe$ProbeSetName in
+ ids]
gt probes lt- DNAStringSet(probes)
Mapping
gt require(BSgenomeScerevisiaeUCSCsacCer1)
gt dict0 lt- PDict(probes)
gt dict0r lt- PDict(reverseComplement(probes))
gt hits lt- matchPDict(dict0 Scerevisiae[[1]])
gt table(countIndex(hits))
gt table(countPDict(dict0 Scerevisiae[[1]]))
20 38
Example contrsquod
Over all chromosomes
gt allhits lt- lapply(116 function(i)
+ countPDict(dict0 Scerevisiae[[i]]) +
+ countPDict(dict0r Scerevisiae[[i]])
+ )
gt table(rowSums(docall(cbind allhits)))
21 38
ShortRead
ShortRead contains tools for
I Work with GERALDBUSTARD output
I Generate QA report on BUSTARD files
I Read a variety of short read data formats
22 38
Data
We will use data from (Lee Hansen Bullard Dudoit and Sherlock(2008) PLoS Genetics)
We are considering a wild-type and a mutant strain of yeast both grownin rich media The two strains were sequenced using an Illumina GenomeAnalyzer
We are using a subset of the data 1M reads from each of two lanes ofthe two strains
23 38
Reading the unaligned data
gt require(ShortRead)
gt fq lt- readFastq(seqdata mut_1_ffastq$)
gt fq
class ShortReadQlength 1000000 reads width 36 cycles
There are various simple accessor functions for this object especiallysread and quality
24 38
A brief look at the unaligned data
Using alphabetByCycle and as( rdquomatrixrdquo) (which creates a big read timescycle matrix) we can do
gt alp lt- alphabetByCycle(sread(fq))
gt matplot(t(proptable(alp[DNA_BASES
+ ] margin = 2)) type = l)
gt qaMatraw lt- as(quality(fq) matrix)
gt plot(colMeans(qaMatraw))
25 38
Aligning the data
We now align the data using Bowtie
binbash
BOWTIE_OPTS=-m 1 -v 2 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-m 1 -v 0 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 2 -k 2 -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 0 -k 1 -p 2 -q --quiet -3 10
for f in `ls seqdatafastq`dobowtie s_cerevisiae $BOWTIE_OPTS $f gt $fbowtiedone
How many hits do we get
26 38
Reading in the aligned data
We read the data into R as a 4-component list (one lane per component)
gt files lt- listfiles(seqdata
+ pattern = bowtie)
gt aligned lt- sapply(files function(f)
+ readAligned(seqdata pattern = f
+ type = Bowtie)
+ )
gt names(aligned) lt- gsub(_ffastqbowtie
+ names(aligned))
gt aligned[[1]]
gt save(aligned file = alignedrda)
27 38
Some exercises
1 Determine the number of times the motif ldquoTATAArdquo occurs in theyeast genome How often does it occur with one mismatch
2 The seqdata directory has an object called ste12 which is aposition weight matrix for the transcription factor ldquoSte12rdquo (obtainedfrom SCPD) Where does it occur in the genome
3 Compute the average quality for each cycle for the aligned reads andcompare to the qualities for the unaligned reads What is thedifference
28 38
Solution 1
gt sum(sapply(116 function(i)
+ motif lt- DNAString(TATAA)
+ countPattern(motif Scerevisiae[[i]]) +
+ countPattern(reverseComplement(motif)
+ Scerevisiae[[i]])
+ ))
29 38
Solution 2
30 38
Solution 3
31 38
SessionInfo
gt toLatex(sessionInfo())
I R version 292 RC (2009-08-17 r49312) i386-apple-darwin980
I Localeen_USUTF-8en_USUTF-8CCen_USUTF-8en_USUTF-8
I Base packages base datasets graphics grDevices grid methodsstats utils
I Other packages Biostrings 2128 BSgenome 1123BSgenomeScerevisiaeUCSCsacCer1 1313 classGraph 07-2graph 1222 IRanges 123 lattice 017-25 Rgraphviz 1234ShortRead 121
I Loaded via a namespace (and not attached) Biobase 241hwriter 11 tools 292
32 38
Some ShortRead classes
ShortRead
ShortReadQ
AlignedRead
33 38
Some Biostrings classes (single strings)
XString
BString DNAString RNAString AAString
34 38
Some Biostrings classes (sets of strings)
XStringSet
BStringSet DNAStringSet RNAStringSet AAStringSet PhredQualitySolexaQualityQualityScaledXStringSet
QualityScaledBStringSetQualityScaledDNAStringSetQualityScaledRNAStringSetQualityScaledAAStringSet
35 38

Strings in Biostrings
I BString (general) DNAString RNAString AAString (Amino Acid)all examples of XString
I complement reverse reverseComplement translate for the classeswhere ldquoit makes senserdquo
I Convert to and from a standard R character string
I Constructor DNAString(ACGGGGG)
I Support for IUPAC alphabet
I Subsetting subseq using the SEW format (two out of the threestartendwidth) Efficient
I StringSets are collections of Strings like DNAStringSet
15 38
BSgenomes
As examples we will use whole genomes Use availablegenomes to getavailable genomes Long package names but always a shorter objectname
gt library(BSgenomeScerevisiaeUCSCsacCer1)
gt Scerevisiae
gt Scerevisiae[[1]]
gt Scerevisiae[[chr1]]
A BSgenome may also have masks We will ignore this for now
16 38
Biostrings more
I alphabetFrequency oligonucleotideFrequency and others
gt alphabetFrequency(Scerevisiae[[chr1]])
gt oligonucleotideFrequency(Scerevisiae[[chr1]]
+ width = 3)
I chartr for character translation (ldquomake all As into Csrdquo)
I Various IO functions (also ShortRead)
17 38
Views
A view is a set of substrings of an XString stored and manipulated veryefficiently Example to store exon sequences one can just store thegenomic location of the exons
gt Views(Scerevisiae[[chr1]] start = c(300
+ 400 500) width = 50)
Views on a 230208-letter DNAString subjectsubject CCACACCACACCCAGTGGTGTGTGTGGGviews
start end width[1] 300 349 50 [CTGTTCTTAAATAAC][2] 400 449 50 [CCCTCACTAGTATAT][3] 500 549 50 [TCTCTCACCGGCACT]
A view is associated with a subject Internally they are essentiallyIRanges so they are very fast to compute with Views can in many casesbe treated exactly as other strings There are methods such as narrowtrim gaps restrict
18 38
Matching in Biostrings
There are various ways of matching or aligning strings to each other Weuse matching to denote searching for an exact match or possibly a matchwith a certain number of mismatches Alignment denotes a more generalstrategy eg Smith-Waterman
I matchPattern countPattern match 1 sequence to 1 sequence
I vmatchPattern vcountPattern match 1 sequence to manysequences
I pairwiseAlignment align many sequences to 1 sequence
I matchPDict countPDict match many sequences to 1 sequence(ldquodictrdquo indicates that the many sequences are preprocessed into adictionary)
I matchPWM trimLRpattern
The different functions are optimized for different situations They alsouse different algorithms which has a big impact EspeciallypairwiseAlignment is flexible and therefore has a complicated syntax
19 38
Example mapping probes to a genome
Get a list of Scerevisiae probes from the ldquoyeast2rdquo Affymetrix array
gt library(yeast2probe)
gt ids lt- scan(s_pombemsk skip = 2
+ what = list(probeset = character(0)
+ junk = character(0)))$probeset
gt probes lt- yeast2probe$sequence[yeast2probe$ProbeSetName in
+ ids]
gt probes lt- DNAStringSet(probes)
Mapping
gt require(BSgenomeScerevisiaeUCSCsacCer1)
gt dict0 lt- PDict(probes)
gt dict0r lt- PDict(reverseComplement(probes))
gt hits lt- matchPDict(dict0 Scerevisiae[[1]])
gt table(countIndex(hits))
gt table(countPDict(dict0 Scerevisiae[[1]]))
20 38
Example contrsquod
Over all chromosomes
gt allhits lt- lapply(116 function(i)
+ countPDict(dict0 Scerevisiae[[i]]) +
+ countPDict(dict0r Scerevisiae[[i]])
+ )
gt table(rowSums(docall(cbind allhits)))
21 38
ShortRead
ShortRead contains tools for
I Work with GERALDBUSTARD output
I Generate QA report on BUSTARD files
I Read a variety of short read data formats
22 38
Data
We will use data from (Lee Hansen Bullard Dudoit and Sherlock(2008) PLoS Genetics)
We are considering a wild-type and a mutant strain of yeast both grownin rich media The two strains were sequenced using an Illumina GenomeAnalyzer
We are using a subset of the data 1M reads from each of two lanes ofthe two strains
23 38
Reading the unaligned data
gt require(ShortRead)
gt fq lt- readFastq(seqdata mut_1_ffastq$)
gt fq
class ShortReadQlength 1000000 reads width 36 cycles
There are various simple accessor functions for this object especiallysread and quality
24 38
A brief look at the unaligned data
Using alphabetByCycle and as( rdquomatrixrdquo) (which creates a big read timescycle matrix) we can do
gt alp lt- alphabetByCycle(sread(fq))
gt matplot(t(proptable(alp[DNA_BASES
+ ] margin = 2)) type = l)
gt qaMatraw lt- as(quality(fq) matrix)
gt plot(colMeans(qaMatraw))
25 38
Aligning the data
We now align the data using Bowtie
binbash
BOWTIE_OPTS=-m 1 -v 2 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-m 1 -v 0 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 2 -k 2 -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 0 -k 1 -p 2 -q --quiet -3 10
for f in `ls seqdatafastq`dobowtie s_cerevisiae $BOWTIE_OPTS $f gt $fbowtiedone
How many hits do we get
26 38
Reading in the aligned data
We read the data into R as a 4-component list (one lane per component)
gt files lt- listfiles(seqdata
+ pattern = bowtie)
gt aligned lt- sapply(files function(f)
+ readAligned(seqdata pattern = f
+ type = Bowtie)
+ )
gt names(aligned) lt- gsub(_ffastqbowtie
+ names(aligned))
gt aligned[[1]]
gt save(aligned file = alignedrda)
27 38
Some exercises
1 Determine the number of times the motif ldquoTATAArdquo occurs in theyeast genome How often does it occur with one mismatch
2 The seqdata directory has an object called ste12 which is aposition weight matrix for the transcription factor ldquoSte12rdquo (obtainedfrom SCPD) Where does it occur in the genome
3 Compute the average quality for each cycle for the aligned reads andcompare to the qualities for the unaligned reads What is thedifference
28 38
Solution 1
gt sum(sapply(116 function(i)
+ motif lt- DNAString(TATAA)
+ countPattern(motif Scerevisiae[[i]]) +
+ countPattern(reverseComplement(motif)
+ Scerevisiae[[i]])
+ ))
29 38
Solution 2
30 38
Solution 3
31 38
SessionInfo
gt toLatex(sessionInfo())
I R version 292 RC (2009-08-17 r49312) i386-apple-darwin980
I Localeen_USUTF-8en_USUTF-8CCen_USUTF-8en_USUTF-8
I Base packages base datasets graphics grDevices grid methodsstats utils
I Other packages Biostrings 2128 BSgenome 1123BSgenomeScerevisiaeUCSCsacCer1 1313 classGraph 07-2graph 1222 IRanges 123 lattice 017-25 Rgraphviz 1234ShortRead 121
I Loaded via a namespace (and not attached) Biobase 241hwriter 11 tools 292
32 38
Some ShortRead classes
ShortRead
ShortReadQ
AlignedRead
33 38
Some Biostrings classes (single strings)
XString
BString DNAString RNAString AAString
34 38
Some Biostrings classes (sets of strings)
XStringSet
BStringSet DNAStringSet RNAStringSet AAStringSet PhredQualitySolexaQualityQualityScaledXStringSet
QualityScaledBStringSetQualityScaledDNAStringSetQualityScaledRNAStringSetQualityScaledAAStringSet
35 38

BSgenomes
As examples we will use whole genomes Use availablegenomes to getavailable genomes Long package names but always a shorter objectname
gt library(BSgenomeScerevisiaeUCSCsacCer1)
gt Scerevisiae
gt Scerevisiae[[1]]
gt Scerevisiae[[chr1]]
A BSgenome may also have masks We will ignore this for now
16 38
Biostrings more
I alphabetFrequency oligonucleotideFrequency and others
gt alphabetFrequency(Scerevisiae[[chr1]])
gt oligonucleotideFrequency(Scerevisiae[[chr1]]
+ width = 3)
I chartr for character translation (ldquomake all As into Csrdquo)
I Various IO functions (also ShortRead)
17 38
Views
A view is a set of substrings of an XString stored and manipulated veryefficiently Example to store exon sequences one can just store thegenomic location of the exons
gt Views(Scerevisiae[[chr1]] start = c(300
+ 400 500) width = 50)
Views on a 230208-letter DNAString subjectsubject CCACACCACACCCAGTGGTGTGTGTGGGviews
start end width[1] 300 349 50 [CTGTTCTTAAATAAC][2] 400 449 50 [CCCTCACTAGTATAT][3] 500 549 50 [TCTCTCACCGGCACT]
A view is associated with a subject Internally they are essentiallyIRanges so they are very fast to compute with Views can in many casesbe treated exactly as other strings There are methods such as narrowtrim gaps restrict
18 38
Matching in Biostrings
There are various ways of matching or aligning strings to each other Weuse matching to denote searching for an exact match or possibly a matchwith a certain number of mismatches Alignment denotes a more generalstrategy eg Smith-Waterman
I matchPattern countPattern match 1 sequence to 1 sequence
I vmatchPattern vcountPattern match 1 sequence to manysequences
I pairwiseAlignment align many sequences to 1 sequence
I matchPDict countPDict match many sequences to 1 sequence(ldquodictrdquo indicates that the many sequences are preprocessed into adictionary)
I matchPWM trimLRpattern
The different functions are optimized for different situations They alsouse different algorithms which has a big impact EspeciallypairwiseAlignment is flexible and therefore has a complicated syntax
19 38
Example mapping probes to a genome
Get a list of Scerevisiae probes from the ldquoyeast2rdquo Affymetrix array
gt library(yeast2probe)
gt ids lt- scan(s_pombemsk skip = 2
+ what = list(probeset = character(0)
+ junk = character(0)))$probeset
gt probes lt- yeast2probe$sequence[yeast2probe$ProbeSetName in
+ ids]
gt probes lt- DNAStringSet(probes)
Mapping
gt require(BSgenomeScerevisiaeUCSCsacCer1)
gt dict0 lt- PDict(probes)
gt dict0r lt- PDict(reverseComplement(probes))
gt hits lt- matchPDict(dict0 Scerevisiae[[1]])
gt table(countIndex(hits))
gt table(countPDict(dict0 Scerevisiae[[1]]))
20 38
Example contrsquod
Over all chromosomes
gt allhits lt- lapply(116 function(i)
+ countPDict(dict0 Scerevisiae[[i]]) +
+ countPDict(dict0r Scerevisiae[[i]])
+ )
gt table(rowSums(docall(cbind allhits)))
21 38
ShortRead
ShortRead contains tools for
I Work with GERALDBUSTARD output
I Generate QA report on BUSTARD files
I Read a variety of short read data formats
22 38
Data
We will use data from (Lee Hansen Bullard Dudoit and Sherlock(2008) PLoS Genetics)
We are considering a wild-type and a mutant strain of yeast both grownin rich media The two strains were sequenced using an Illumina GenomeAnalyzer
We are using a subset of the data 1M reads from each of two lanes ofthe two strains
23 38
Reading the unaligned data
gt require(ShortRead)
gt fq lt- readFastq(seqdata mut_1_ffastq$)
gt fq
class ShortReadQlength 1000000 reads width 36 cycles
There are various simple accessor functions for this object especiallysread and quality
24 38
A brief look at the unaligned data
Using alphabetByCycle and as( rdquomatrixrdquo) (which creates a big read timescycle matrix) we can do
gt alp lt- alphabetByCycle(sread(fq))
gt matplot(t(proptable(alp[DNA_BASES
+ ] margin = 2)) type = l)
gt qaMatraw lt- as(quality(fq) matrix)
gt plot(colMeans(qaMatraw))
25 38
Aligning the data
We now align the data using Bowtie
binbash
BOWTIE_OPTS=-m 1 -v 2 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-m 1 -v 0 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 2 -k 2 -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 0 -k 1 -p 2 -q --quiet -3 10
for f in `ls seqdatafastq`dobowtie s_cerevisiae $BOWTIE_OPTS $f gt $fbowtiedone
How many hits do we get
26 38
Reading in the aligned data
We read the data into R as a 4-component list (one lane per component)
gt files lt- listfiles(seqdata
+ pattern = bowtie)
gt aligned lt- sapply(files function(f)
+ readAligned(seqdata pattern = f
+ type = Bowtie)
+ )
gt names(aligned) lt- gsub(_ffastqbowtie
+ names(aligned))
gt aligned[[1]]
gt save(aligned file = alignedrda)
27 38
Some exercises
1 Determine the number of times the motif ldquoTATAArdquo occurs in theyeast genome How often does it occur with one mismatch
2 The seqdata directory has an object called ste12 which is aposition weight matrix for the transcription factor ldquoSte12rdquo (obtainedfrom SCPD) Where does it occur in the genome
3 Compute the average quality for each cycle for the aligned reads andcompare to the qualities for the unaligned reads What is thedifference
28 38
Solution 1
gt sum(sapply(116 function(i)
+ motif lt- DNAString(TATAA)
+ countPattern(motif Scerevisiae[[i]]) +
+ countPattern(reverseComplement(motif)
+ Scerevisiae[[i]])
+ ))
29 38
Solution 2
30 38
Solution 3
31 38
SessionInfo
gt toLatex(sessionInfo())
I R version 292 RC (2009-08-17 r49312) i386-apple-darwin980
I Localeen_USUTF-8en_USUTF-8CCen_USUTF-8en_USUTF-8
I Base packages base datasets graphics grDevices grid methodsstats utils
I Other packages Biostrings 2128 BSgenome 1123BSgenomeScerevisiaeUCSCsacCer1 1313 classGraph 07-2graph 1222 IRanges 123 lattice 017-25 Rgraphviz 1234ShortRead 121
I Loaded via a namespace (and not attached) Biobase 241hwriter 11 tools 292
32 38
Some ShortRead classes
ShortRead
ShortReadQ
AlignedRead
33 38
Some Biostrings classes (single strings)
XString
BString DNAString RNAString AAString
34 38
Some Biostrings classes (sets of strings)
XStringSet
BStringSet DNAStringSet RNAStringSet AAStringSet PhredQualitySolexaQualityQualityScaledXStringSet
QualityScaledBStringSetQualityScaledDNAStringSetQualityScaledRNAStringSetQualityScaledAAStringSet
35 38

Biostrings more
I alphabetFrequency oligonucleotideFrequency and others
gt alphabetFrequency(Scerevisiae[[chr1]])
gt oligonucleotideFrequency(Scerevisiae[[chr1]]
+ width = 3)
I chartr for character translation (ldquomake all As into Csrdquo)
I Various IO functions (also ShortRead)
17 38
Views
A view is a set of substrings of an XString stored and manipulated veryefficiently Example to store exon sequences one can just store thegenomic location of the exons
gt Views(Scerevisiae[[chr1]] start = c(300
+ 400 500) width = 50)
Views on a 230208-letter DNAString subjectsubject CCACACCACACCCAGTGGTGTGTGTGGGviews
start end width[1] 300 349 50 [CTGTTCTTAAATAAC][2] 400 449 50 [CCCTCACTAGTATAT][3] 500 549 50 [TCTCTCACCGGCACT]
A view is associated with a subject Internally they are essentiallyIRanges so they are very fast to compute with Views can in many casesbe treated exactly as other strings There are methods such as narrowtrim gaps restrict
18 38
Matching in Biostrings
There are various ways of matching or aligning strings to each other Weuse matching to denote searching for an exact match or possibly a matchwith a certain number of mismatches Alignment denotes a more generalstrategy eg Smith-Waterman
I matchPattern countPattern match 1 sequence to 1 sequence
I vmatchPattern vcountPattern match 1 sequence to manysequences
I pairwiseAlignment align many sequences to 1 sequence
I matchPDict countPDict match many sequences to 1 sequence(ldquodictrdquo indicates that the many sequences are preprocessed into adictionary)
I matchPWM trimLRpattern
The different functions are optimized for different situations They alsouse different algorithms which has a big impact EspeciallypairwiseAlignment is flexible and therefore has a complicated syntax
19 38
Example mapping probes to a genome
Get a list of Scerevisiae probes from the ldquoyeast2rdquo Affymetrix array
gt library(yeast2probe)
gt ids lt- scan(s_pombemsk skip = 2
+ what = list(probeset = character(0)
+ junk = character(0)))$probeset
gt probes lt- yeast2probe$sequence[yeast2probe$ProbeSetName in
+ ids]
gt probes lt- DNAStringSet(probes)
Mapping
gt require(BSgenomeScerevisiaeUCSCsacCer1)
gt dict0 lt- PDict(probes)
gt dict0r lt- PDict(reverseComplement(probes))
gt hits lt- matchPDict(dict0 Scerevisiae[[1]])
gt table(countIndex(hits))
gt table(countPDict(dict0 Scerevisiae[[1]]))
20 38
Example contrsquod
Over all chromosomes
gt allhits lt- lapply(116 function(i)
+ countPDict(dict0 Scerevisiae[[i]]) +
+ countPDict(dict0r Scerevisiae[[i]])
+ )
gt table(rowSums(docall(cbind allhits)))
21 38
ShortRead
ShortRead contains tools for
I Work with GERALDBUSTARD output
I Generate QA report on BUSTARD files
I Read a variety of short read data formats
22 38
Data
We will use data from (Lee Hansen Bullard Dudoit and Sherlock(2008) PLoS Genetics)
We are considering a wild-type and a mutant strain of yeast both grownin rich media The two strains were sequenced using an Illumina GenomeAnalyzer
We are using a subset of the data 1M reads from each of two lanes ofthe two strains
23 38
Reading the unaligned data
gt require(ShortRead)
gt fq lt- readFastq(seqdata mut_1_ffastq$)
gt fq
class ShortReadQlength 1000000 reads width 36 cycles
There are various simple accessor functions for this object especiallysread and quality
24 38
A brief look at the unaligned data
Using alphabetByCycle and as( rdquomatrixrdquo) (which creates a big read timescycle matrix) we can do
gt alp lt- alphabetByCycle(sread(fq))
gt matplot(t(proptable(alp[DNA_BASES
+ ] margin = 2)) type = l)
gt qaMatraw lt- as(quality(fq) matrix)
gt plot(colMeans(qaMatraw))
25 38
Aligning the data
We now align the data using Bowtie
binbash
BOWTIE_OPTS=-m 1 -v 2 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-m 1 -v 0 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 2 -k 2 -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 0 -k 1 -p 2 -q --quiet -3 10
for f in `ls seqdatafastq`dobowtie s_cerevisiae $BOWTIE_OPTS $f gt $fbowtiedone
How many hits do we get
26 38
Reading in the aligned data
We read the data into R as a 4-component list (one lane per component)
gt files lt- listfiles(seqdata
+ pattern = bowtie)
gt aligned lt- sapply(files function(f)
+ readAligned(seqdata pattern = f
+ type = Bowtie)
+ )
gt names(aligned) lt- gsub(_ffastqbowtie
+ names(aligned))
gt aligned[[1]]
gt save(aligned file = alignedrda)
27 38
Some exercises
1 Determine the number of times the motif ldquoTATAArdquo occurs in theyeast genome How often does it occur with one mismatch
2 The seqdata directory has an object called ste12 which is aposition weight matrix for the transcription factor ldquoSte12rdquo (obtainedfrom SCPD) Where does it occur in the genome
3 Compute the average quality for each cycle for the aligned reads andcompare to the qualities for the unaligned reads What is thedifference
28 38
Solution 1
gt sum(sapply(116 function(i)
+ motif lt- DNAString(TATAA)
+ countPattern(motif Scerevisiae[[i]]) +
+ countPattern(reverseComplement(motif)
+ Scerevisiae[[i]])
+ ))
29 38
Solution 2
30 38
Solution 3
31 38
SessionInfo
gt toLatex(sessionInfo())
I R version 292 RC (2009-08-17 r49312) i386-apple-darwin980
I Localeen_USUTF-8en_USUTF-8CCen_USUTF-8en_USUTF-8
I Base packages base datasets graphics grDevices grid methodsstats utils
I Other packages Biostrings 2128 BSgenome 1123BSgenomeScerevisiaeUCSCsacCer1 1313 classGraph 07-2graph 1222 IRanges 123 lattice 017-25 Rgraphviz 1234ShortRead 121
I Loaded via a namespace (and not attached) Biobase 241hwriter 11 tools 292
32 38
Some ShortRead classes
ShortRead
ShortReadQ
AlignedRead
33 38
Some Biostrings classes (single strings)
XString
BString DNAString RNAString AAString
34 38
Some Biostrings classes (sets of strings)
XStringSet
BStringSet DNAStringSet RNAStringSet AAStringSet PhredQualitySolexaQualityQualityScaledXStringSet
QualityScaledBStringSetQualityScaledDNAStringSetQualityScaledRNAStringSetQualityScaledAAStringSet
35 38

Views
A view is a set of substrings of an XString stored and manipulated veryefficiently Example to store exon sequences one can just store thegenomic location of the exons
gt Views(Scerevisiae[[chr1]] start = c(300
+ 400 500) width = 50)
Views on a 230208-letter DNAString subjectsubject CCACACCACACCCAGTGGTGTGTGTGGGviews
start end width[1] 300 349 50 [CTGTTCTTAAATAAC][2] 400 449 50 [CCCTCACTAGTATAT][3] 500 549 50 [TCTCTCACCGGCACT]
A view is associated with a subject Internally they are essentiallyIRanges so they are very fast to compute with Views can in many casesbe treated exactly as other strings There are methods such as narrowtrim gaps restrict
18 38
Matching in Biostrings
There are various ways of matching or aligning strings to each other Weuse matching to denote searching for an exact match or possibly a matchwith a certain number of mismatches Alignment denotes a more generalstrategy eg Smith-Waterman
I matchPattern countPattern match 1 sequence to 1 sequence
I vmatchPattern vcountPattern match 1 sequence to manysequences
I pairwiseAlignment align many sequences to 1 sequence
I matchPDict countPDict match many sequences to 1 sequence(ldquodictrdquo indicates that the many sequences are preprocessed into adictionary)
I matchPWM trimLRpattern
The different functions are optimized for different situations They alsouse different algorithms which has a big impact EspeciallypairwiseAlignment is flexible and therefore has a complicated syntax
19 38
Example mapping probes to a genome
Get a list of Scerevisiae probes from the ldquoyeast2rdquo Affymetrix array
gt library(yeast2probe)
gt ids lt- scan(s_pombemsk skip = 2
+ what = list(probeset = character(0)
+ junk = character(0)))$probeset
gt probes lt- yeast2probe$sequence[yeast2probe$ProbeSetName in
+ ids]
gt probes lt- DNAStringSet(probes)
Mapping
gt require(BSgenomeScerevisiaeUCSCsacCer1)
gt dict0 lt- PDict(probes)
gt dict0r lt- PDict(reverseComplement(probes))
gt hits lt- matchPDict(dict0 Scerevisiae[[1]])
gt table(countIndex(hits))
gt table(countPDict(dict0 Scerevisiae[[1]]))
20 38
Example contrsquod
Over all chromosomes
gt allhits lt- lapply(116 function(i)
+ countPDict(dict0 Scerevisiae[[i]]) +
+ countPDict(dict0r Scerevisiae[[i]])
+ )
gt table(rowSums(docall(cbind allhits)))
21 38
ShortRead
ShortRead contains tools for
I Work with GERALDBUSTARD output
I Generate QA report on BUSTARD files
I Read a variety of short read data formats
22 38
Data
We will use data from (Lee Hansen Bullard Dudoit and Sherlock(2008) PLoS Genetics)
We are considering a wild-type and a mutant strain of yeast both grownin rich media The two strains were sequenced using an Illumina GenomeAnalyzer
We are using a subset of the data 1M reads from each of two lanes ofthe two strains
23 38
Reading the unaligned data
gt require(ShortRead)
gt fq lt- readFastq(seqdata mut_1_ffastq$)
gt fq
class ShortReadQlength 1000000 reads width 36 cycles
There are various simple accessor functions for this object especiallysread and quality
24 38
A brief look at the unaligned data
Using alphabetByCycle and as( rdquomatrixrdquo) (which creates a big read timescycle matrix) we can do
gt alp lt- alphabetByCycle(sread(fq))
gt matplot(t(proptable(alp[DNA_BASES
+ ] margin = 2)) type = l)
gt qaMatraw lt- as(quality(fq) matrix)
gt plot(colMeans(qaMatraw))
25 38
Aligning the data
We now align the data using Bowtie
binbash
BOWTIE_OPTS=-m 1 -v 2 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-m 1 -v 0 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 2 -k 2 -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 0 -k 1 -p 2 -q --quiet -3 10
for f in `ls seqdatafastq`dobowtie s_cerevisiae $BOWTIE_OPTS $f gt $fbowtiedone
How many hits do we get
26 38
Reading in the aligned data
We read the data into R as a 4-component list (one lane per component)
gt files lt- listfiles(seqdata
+ pattern = bowtie)
gt aligned lt- sapply(files function(f)
+ readAligned(seqdata pattern = f
+ type = Bowtie)
+ )
gt names(aligned) lt- gsub(_ffastqbowtie
+ names(aligned))
gt aligned[[1]]
gt save(aligned file = alignedrda)
27 38
Some exercises
1 Determine the number of times the motif ldquoTATAArdquo occurs in theyeast genome How often does it occur with one mismatch
2 The seqdata directory has an object called ste12 which is aposition weight matrix for the transcription factor ldquoSte12rdquo (obtainedfrom SCPD) Where does it occur in the genome
3 Compute the average quality for each cycle for the aligned reads andcompare to the qualities for the unaligned reads What is thedifference
28 38
Solution 1
gt sum(sapply(116 function(i)
+ motif lt- DNAString(TATAA)
+ countPattern(motif Scerevisiae[[i]]) +
+ countPattern(reverseComplement(motif)
+ Scerevisiae[[i]])
+ ))
29 38
Solution 2
30 38
Solution 3
31 38
SessionInfo
gt toLatex(sessionInfo())
I R version 292 RC (2009-08-17 r49312) i386-apple-darwin980
I Localeen_USUTF-8en_USUTF-8CCen_USUTF-8en_USUTF-8
I Base packages base datasets graphics grDevices grid methodsstats utils
I Other packages Biostrings 2128 BSgenome 1123BSgenomeScerevisiaeUCSCsacCer1 1313 classGraph 07-2graph 1222 IRanges 123 lattice 017-25 Rgraphviz 1234ShortRead 121
I Loaded via a namespace (and not attached) Biobase 241hwriter 11 tools 292
32 38
Some ShortRead classes
ShortRead
ShortReadQ
AlignedRead
33 38
Some Biostrings classes (single strings)
XString
BString DNAString RNAString AAString
34 38
Some Biostrings classes (sets of strings)
XStringSet
BStringSet DNAStringSet RNAStringSet AAStringSet PhredQualitySolexaQualityQualityScaledXStringSet
QualityScaledBStringSetQualityScaledDNAStringSetQualityScaledRNAStringSetQualityScaledAAStringSet
35 38

Matching in Biostrings
There are various ways of matching or aligning strings to each other Weuse matching to denote searching for an exact match or possibly a matchwith a certain number of mismatches Alignment denotes a more generalstrategy eg Smith-Waterman
I matchPattern countPattern match 1 sequence to 1 sequence
I vmatchPattern vcountPattern match 1 sequence to manysequences
I pairwiseAlignment align many sequences to 1 sequence
I matchPDict countPDict match many sequences to 1 sequence(ldquodictrdquo indicates that the many sequences are preprocessed into adictionary)
I matchPWM trimLRpattern
The different functions are optimized for different situations They alsouse different algorithms which has a big impact EspeciallypairwiseAlignment is flexible and therefore has a complicated syntax
19 38
Example mapping probes to a genome
Get a list of Scerevisiae probes from the ldquoyeast2rdquo Affymetrix array
gt library(yeast2probe)
gt ids lt- scan(s_pombemsk skip = 2
+ what = list(probeset = character(0)
+ junk = character(0)))$probeset
gt probes lt- yeast2probe$sequence[yeast2probe$ProbeSetName in
+ ids]
gt probes lt- DNAStringSet(probes)
Mapping
gt require(BSgenomeScerevisiaeUCSCsacCer1)
gt dict0 lt- PDict(probes)
gt dict0r lt- PDict(reverseComplement(probes))
gt hits lt- matchPDict(dict0 Scerevisiae[[1]])
gt table(countIndex(hits))
gt table(countPDict(dict0 Scerevisiae[[1]]))
20 38
Example contrsquod
Over all chromosomes
gt allhits lt- lapply(116 function(i)
+ countPDict(dict0 Scerevisiae[[i]]) +
+ countPDict(dict0r Scerevisiae[[i]])
+ )
gt table(rowSums(docall(cbind allhits)))
21 38
ShortRead
ShortRead contains tools for
I Work with GERALDBUSTARD output
I Generate QA report on BUSTARD files
I Read a variety of short read data formats
22 38
Data
We will use data from (Lee Hansen Bullard Dudoit and Sherlock(2008) PLoS Genetics)
We are considering a wild-type and a mutant strain of yeast both grownin rich media The two strains were sequenced using an Illumina GenomeAnalyzer
We are using a subset of the data 1M reads from each of two lanes ofthe two strains
23 38
Reading the unaligned data
gt require(ShortRead)
gt fq lt- readFastq(seqdata mut_1_ffastq$)
gt fq
class ShortReadQlength 1000000 reads width 36 cycles
There are various simple accessor functions for this object especiallysread and quality
24 38
A brief look at the unaligned data
Using alphabetByCycle and as( rdquomatrixrdquo) (which creates a big read timescycle matrix) we can do
gt alp lt- alphabetByCycle(sread(fq))
gt matplot(t(proptable(alp[DNA_BASES
+ ] margin = 2)) type = l)
gt qaMatraw lt- as(quality(fq) matrix)
gt plot(colMeans(qaMatraw))
25 38
Aligning the data
We now align the data using Bowtie
binbash
BOWTIE_OPTS=-m 1 -v 2 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-m 1 -v 0 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 2 -k 2 -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 0 -k 1 -p 2 -q --quiet -3 10
for f in `ls seqdatafastq`dobowtie s_cerevisiae $BOWTIE_OPTS $f gt $fbowtiedone
How many hits do we get
26 38
Reading in the aligned data
We read the data into R as a 4-component list (one lane per component)
gt files lt- listfiles(seqdata
+ pattern = bowtie)
gt aligned lt- sapply(files function(f)
+ readAligned(seqdata pattern = f
+ type = Bowtie)
+ )
gt names(aligned) lt- gsub(_ffastqbowtie
+ names(aligned))
gt aligned[[1]]
gt save(aligned file = alignedrda)
27 38
Some exercises
1 Determine the number of times the motif ldquoTATAArdquo occurs in theyeast genome How often does it occur with one mismatch
2 The seqdata directory has an object called ste12 which is aposition weight matrix for the transcription factor ldquoSte12rdquo (obtainedfrom SCPD) Where does it occur in the genome
3 Compute the average quality for each cycle for the aligned reads andcompare to the qualities for the unaligned reads What is thedifference
28 38
Solution 1
gt sum(sapply(116 function(i)
+ motif lt- DNAString(TATAA)
+ countPattern(motif Scerevisiae[[i]]) +
+ countPattern(reverseComplement(motif)
+ Scerevisiae[[i]])
+ ))
29 38
Solution 2
30 38
Solution 3
31 38
SessionInfo
gt toLatex(sessionInfo())
I R version 292 RC (2009-08-17 r49312) i386-apple-darwin980
I Localeen_USUTF-8en_USUTF-8CCen_USUTF-8en_USUTF-8
I Base packages base datasets graphics grDevices grid methodsstats utils
I Other packages Biostrings 2128 BSgenome 1123BSgenomeScerevisiaeUCSCsacCer1 1313 classGraph 07-2graph 1222 IRanges 123 lattice 017-25 Rgraphviz 1234ShortRead 121
I Loaded via a namespace (and not attached) Biobase 241hwriter 11 tools 292
32 38
Some ShortRead classes
ShortRead
ShortReadQ
AlignedRead
33 38
Some Biostrings classes (single strings)
XString
BString DNAString RNAString AAString
34 38
Some Biostrings classes (sets of strings)
XStringSet
BStringSet DNAStringSet RNAStringSet AAStringSet PhredQualitySolexaQualityQualityScaledXStringSet
QualityScaledBStringSetQualityScaledDNAStringSetQualityScaledRNAStringSetQualityScaledAAStringSet
35 38

Example mapping probes to a genome
Get a list of Scerevisiae probes from the ldquoyeast2rdquo Affymetrix array
gt library(yeast2probe)
gt ids lt- scan(s_pombemsk skip = 2
+ what = list(probeset = character(0)
+ junk = character(0)))$probeset
gt probes lt- yeast2probe$sequence[yeast2probe$ProbeSetName in
+ ids]
gt probes lt- DNAStringSet(probes)
Mapping
gt require(BSgenomeScerevisiaeUCSCsacCer1)
gt dict0 lt- PDict(probes)
gt dict0r lt- PDict(reverseComplement(probes))
gt hits lt- matchPDict(dict0 Scerevisiae[[1]])
gt table(countIndex(hits))
gt table(countPDict(dict0 Scerevisiae[[1]]))
20 38
Example contrsquod
Over all chromosomes
gt allhits lt- lapply(116 function(i)
+ countPDict(dict0 Scerevisiae[[i]]) +
+ countPDict(dict0r Scerevisiae[[i]])
+ )
gt table(rowSums(docall(cbind allhits)))
21 38
ShortRead
ShortRead contains tools for
I Work with GERALDBUSTARD output
I Generate QA report on BUSTARD files
I Read a variety of short read data formats
22 38
Data
We will use data from (Lee Hansen Bullard Dudoit and Sherlock(2008) PLoS Genetics)
We are considering a wild-type and a mutant strain of yeast both grownin rich media The two strains were sequenced using an Illumina GenomeAnalyzer
We are using a subset of the data 1M reads from each of two lanes ofthe two strains
23 38
Reading the unaligned data
gt require(ShortRead)
gt fq lt- readFastq(seqdata mut_1_ffastq$)
gt fq
class ShortReadQlength 1000000 reads width 36 cycles
There are various simple accessor functions for this object especiallysread and quality
24 38
A brief look at the unaligned data
Using alphabetByCycle and as( rdquomatrixrdquo) (which creates a big read timescycle matrix) we can do
gt alp lt- alphabetByCycle(sread(fq))
gt matplot(t(proptable(alp[DNA_BASES
+ ] margin = 2)) type = l)
gt qaMatraw lt- as(quality(fq) matrix)
gt plot(colMeans(qaMatraw))
25 38
Aligning the data
We now align the data using Bowtie
binbash
BOWTIE_OPTS=-m 1 -v 2 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-m 1 -v 0 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 2 -k 2 -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 0 -k 1 -p 2 -q --quiet -3 10
for f in `ls seqdatafastq`dobowtie s_cerevisiae $BOWTIE_OPTS $f gt $fbowtiedone
How many hits do we get
26 38
Reading in the aligned data
We read the data into R as a 4-component list (one lane per component)
gt files lt- listfiles(seqdata
+ pattern = bowtie)
gt aligned lt- sapply(files function(f)
+ readAligned(seqdata pattern = f
+ type = Bowtie)
+ )
gt names(aligned) lt- gsub(_ffastqbowtie
+ names(aligned))
gt aligned[[1]]
gt save(aligned file = alignedrda)
27 38
Some exercises
1 Determine the number of times the motif ldquoTATAArdquo occurs in theyeast genome How often does it occur with one mismatch
2 The seqdata directory has an object called ste12 which is aposition weight matrix for the transcription factor ldquoSte12rdquo (obtainedfrom SCPD) Where does it occur in the genome
3 Compute the average quality for each cycle for the aligned reads andcompare to the qualities for the unaligned reads What is thedifference
28 38
Solution 1
gt sum(sapply(116 function(i)
+ motif lt- DNAString(TATAA)
+ countPattern(motif Scerevisiae[[i]]) +
+ countPattern(reverseComplement(motif)
+ Scerevisiae[[i]])
+ ))
29 38
Solution 2
30 38
Solution 3
31 38
SessionInfo
gt toLatex(sessionInfo())
I R version 292 RC (2009-08-17 r49312) i386-apple-darwin980
I Localeen_USUTF-8en_USUTF-8CCen_USUTF-8en_USUTF-8
I Base packages base datasets graphics grDevices grid methodsstats utils
I Other packages Biostrings 2128 BSgenome 1123BSgenomeScerevisiaeUCSCsacCer1 1313 classGraph 07-2graph 1222 IRanges 123 lattice 017-25 Rgraphviz 1234ShortRead 121
I Loaded via a namespace (and not attached) Biobase 241hwriter 11 tools 292
32 38
Some ShortRead classes
ShortRead
ShortReadQ
AlignedRead
33 38
Some Biostrings classes (single strings)
XString
BString DNAString RNAString AAString
34 38
Some Biostrings classes (sets of strings)
XStringSet
BStringSet DNAStringSet RNAStringSet AAStringSet PhredQualitySolexaQualityQualityScaledXStringSet
QualityScaledBStringSetQualityScaledDNAStringSetQualityScaledRNAStringSetQualityScaledAAStringSet
35 38

Example contrsquod
Over all chromosomes
gt allhits lt- lapply(116 function(i)
+ countPDict(dict0 Scerevisiae[[i]]) +
+ countPDict(dict0r Scerevisiae[[i]])
+ )
gt table(rowSums(docall(cbind allhits)))
21 38
ShortRead
ShortRead contains tools for
I Work with GERALDBUSTARD output
I Generate QA report on BUSTARD files
I Read a variety of short read data formats
22 38
Data
We will use data from (Lee Hansen Bullard Dudoit and Sherlock(2008) PLoS Genetics)
We are considering a wild-type and a mutant strain of yeast both grownin rich media The two strains were sequenced using an Illumina GenomeAnalyzer
We are using a subset of the data 1M reads from each of two lanes ofthe two strains
23 38
Reading the unaligned data
gt require(ShortRead)
gt fq lt- readFastq(seqdata mut_1_ffastq$)
gt fq
class ShortReadQlength 1000000 reads width 36 cycles
There are various simple accessor functions for this object especiallysread and quality
24 38
A brief look at the unaligned data
Using alphabetByCycle and as( rdquomatrixrdquo) (which creates a big read timescycle matrix) we can do
gt alp lt- alphabetByCycle(sread(fq))
gt matplot(t(proptable(alp[DNA_BASES
+ ] margin = 2)) type = l)
gt qaMatraw lt- as(quality(fq) matrix)
gt plot(colMeans(qaMatraw))
25 38
Aligning the data
We now align the data using Bowtie
binbash
BOWTIE_OPTS=-m 1 -v 2 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-m 1 -v 0 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 2 -k 2 -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 0 -k 1 -p 2 -q --quiet -3 10
for f in `ls seqdatafastq`dobowtie s_cerevisiae $BOWTIE_OPTS $f gt $fbowtiedone
How many hits do we get
26 38
Reading in the aligned data
We read the data into R as a 4-component list (one lane per component)
gt files lt- listfiles(seqdata
+ pattern = bowtie)
gt aligned lt- sapply(files function(f)
+ readAligned(seqdata pattern = f
+ type = Bowtie)
+ )
gt names(aligned) lt- gsub(_ffastqbowtie
+ names(aligned))
gt aligned[[1]]
gt save(aligned file = alignedrda)
27 38
Some exercises
1 Determine the number of times the motif ldquoTATAArdquo occurs in theyeast genome How often does it occur with one mismatch
2 The seqdata directory has an object called ste12 which is aposition weight matrix for the transcription factor ldquoSte12rdquo (obtainedfrom SCPD) Where does it occur in the genome
3 Compute the average quality for each cycle for the aligned reads andcompare to the qualities for the unaligned reads What is thedifference
28 38
Solution 1
gt sum(sapply(116 function(i)
+ motif lt- DNAString(TATAA)
+ countPattern(motif Scerevisiae[[i]]) +
+ countPattern(reverseComplement(motif)
+ Scerevisiae[[i]])
+ ))
29 38
Solution 2
30 38
Solution 3
31 38
SessionInfo
gt toLatex(sessionInfo())
I R version 292 RC (2009-08-17 r49312) i386-apple-darwin980
I Localeen_USUTF-8en_USUTF-8CCen_USUTF-8en_USUTF-8
I Base packages base datasets graphics grDevices grid methodsstats utils
I Other packages Biostrings 2128 BSgenome 1123BSgenomeScerevisiaeUCSCsacCer1 1313 classGraph 07-2graph 1222 IRanges 123 lattice 017-25 Rgraphviz 1234ShortRead 121
I Loaded via a namespace (and not attached) Biobase 241hwriter 11 tools 292
32 38
Some ShortRead classes
ShortRead
ShortReadQ
AlignedRead
33 38
Some Biostrings classes (single strings)
XString
BString DNAString RNAString AAString
34 38
Some Biostrings classes (sets of strings)
XStringSet
BStringSet DNAStringSet RNAStringSet AAStringSet PhredQualitySolexaQualityQualityScaledXStringSet
QualityScaledBStringSetQualityScaledDNAStringSetQualityScaledRNAStringSetQualityScaledAAStringSet
35 38

ShortRead
ShortRead contains tools for
I Work with GERALDBUSTARD output
I Generate QA report on BUSTARD files
I Read a variety of short read data formats
22 38
Data
We will use data from (Lee Hansen Bullard Dudoit and Sherlock(2008) PLoS Genetics)
We are considering a wild-type and a mutant strain of yeast both grownin rich media The two strains were sequenced using an Illumina GenomeAnalyzer
We are using a subset of the data 1M reads from each of two lanes ofthe two strains
23 38
Reading the unaligned data
gt require(ShortRead)
gt fq lt- readFastq(seqdata mut_1_ffastq$)
gt fq
class ShortReadQlength 1000000 reads width 36 cycles
There are various simple accessor functions for this object especiallysread and quality
24 38
A brief look at the unaligned data
Using alphabetByCycle and as( rdquomatrixrdquo) (which creates a big read timescycle matrix) we can do
gt alp lt- alphabetByCycle(sread(fq))
gt matplot(t(proptable(alp[DNA_BASES
+ ] margin = 2)) type = l)
gt qaMatraw lt- as(quality(fq) matrix)
gt plot(colMeans(qaMatraw))
25 38
Aligning the data
We now align the data using Bowtie
binbash
BOWTIE_OPTS=-m 1 -v 2 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-m 1 -v 0 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 2 -k 2 -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 0 -k 1 -p 2 -q --quiet -3 10
for f in `ls seqdatafastq`dobowtie s_cerevisiae $BOWTIE_OPTS $f gt $fbowtiedone
How many hits do we get
26 38
Reading in the aligned data
We read the data into R as a 4-component list (one lane per component)
gt files lt- listfiles(seqdata
+ pattern = bowtie)
gt aligned lt- sapply(files function(f)
+ readAligned(seqdata pattern = f
+ type = Bowtie)
+ )
gt names(aligned) lt- gsub(_ffastqbowtie
+ names(aligned))
gt aligned[[1]]
gt save(aligned file = alignedrda)
27 38
Some exercises
1 Determine the number of times the motif ldquoTATAArdquo occurs in theyeast genome How often does it occur with one mismatch
2 The seqdata directory has an object called ste12 which is aposition weight matrix for the transcription factor ldquoSte12rdquo (obtainedfrom SCPD) Where does it occur in the genome
3 Compute the average quality for each cycle for the aligned reads andcompare to the qualities for the unaligned reads What is thedifference
28 38
Solution 1
gt sum(sapply(116 function(i)
+ motif lt- DNAString(TATAA)
+ countPattern(motif Scerevisiae[[i]]) +
+ countPattern(reverseComplement(motif)
+ Scerevisiae[[i]])
+ ))
29 38
Solution 2
30 38
Solution 3
31 38
SessionInfo
gt toLatex(sessionInfo())
I R version 292 RC (2009-08-17 r49312) i386-apple-darwin980
I Localeen_USUTF-8en_USUTF-8CCen_USUTF-8en_USUTF-8
I Base packages base datasets graphics grDevices grid methodsstats utils
I Other packages Biostrings 2128 BSgenome 1123BSgenomeScerevisiaeUCSCsacCer1 1313 classGraph 07-2graph 1222 IRanges 123 lattice 017-25 Rgraphviz 1234ShortRead 121
I Loaded via a namespace (and not attached) Biobase 241hwriter 11 tools 292
32 38
Some ShortRead classes
ShortRead
ShortReadQ
AlignedRead
33 38
Some Biostrings classes (single strings)
XString
BString DNAString RNAString AAString
34 38
Some Biostrings classes (sets of strings)
XStringSet
BStringSet DNAStringSet RNAStringSet AAStringSet PhredQualitySolexaQualityQualityScaledXStringSet
QualityScaledBStringSetQualityScaledDNAStringSetQualityScaledRNAStringSetQualityScaledAAStringSet
35 38

Data
We will use data from (Lee Hansen Bullard Dudoit and Sherlock(2008) PLoS Genetics)
We are considering a wild-type and a mutant strain of yeast both grownin rich media The two strains were sequenced using an Illumina GenomeAnalyzer
We are using a subset of the data 1M reads from each of two lanes ofthe two strains
23 38
Reading the unaligned data
gt require(ShortRead)
gt fq lt- readFastq(seqdata mut_1_ffastq$)
gt fq
class ShortReadQlength 1000000 reads width 36 cycles
There are various simple accessor functions for this object especiallysread and quality
24 38
A brief look at the unaligned data
Using alphabetByCycle and as( rdquomatrixrdquo) (which creates a big read timescycle matrix) we can do
gt alp lt- alphabetByCycle(sread(fq))
gt matplot(t(proptable(alp[DNA_BASES
+ ] margin = 2)) type = l)
gt qaMatraw lt- as(quality(fq) matrix)
gt plot(colMeans(qaMatraw))
25 38
Aligning the data
We now align the data using Bowtie
binbash
BOWTIE_OPTS=-m 1 -v 2 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-m 1 -v 0 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 2 -k 2 -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 0 -k 1 -p 2 -q --quiet -3 10
for f in `ls seqdatafastq`dobowtie s_cerevisiae $BOWTIE_OPTS $f gt $fbowtiedone
How many hits do we get
26 38
Reading in the aligned data
We read the data into R as a 4-component list (one lane per component)
gt files lt- listfiles(seqdata
+ pattern = bowtie)
gt aligned lt- sapply(files function(f)
+ readAligned(seqdata pattern = f
+ type = Bowtie)
+ )
gt names(aligned) lt- gsub(_ffastqbowtie
+ names(aligned))
gt aligned[[1]]
gt save(aligned file = alignedrda)
27 38
Some exercises
1 Determine the number of times the motif ldquoTATAArdquo occurs in theyeast genome How often does it occur with one mismatch
2 The seqdata directory has an object called ste12 which is aposition weight matrix for the transcription factor ldquoSte12rdquo (obtainedfrom SCPD) Where does it occur in the genome
3 Compute the average quality for each cycle for the aligned reads andcompare to the qualities for the unaligned reads What is thedifference
28 38
Solution 1
gt sum(sapply(116 function(i)
+ motif lt- DNAString(TATAA)
+ countPattern(motif Scerevisiae[[i]]) +
+ countPattern(reverseComplement(motif)
+ Scerevisiae[[i]])
+ ))
29 38
Solution 2
30 38
Solution 3
31 38
SessionInfo
gt toLatex(sessionInfo())
I R version 292 RC (2009-08-17 r49312) i386-apple-darwin980
I Localeen_USUTF-8en_USUTF-8CCen_USUTF-8en_USUTF-8
I Base packages base datasets graphics grDevices grid methodsstats utils
I Other packages Biostrings 2128 BSgenome 1123BSgenomeScerevisiaeUCSCsacCer1 1313 classGraph 07-2graph 1222 IRanges 123 lattice 017-25 Rgraphviz 1234ShortRead 121
I Loaded via a namespace (and not attached) Biobase 241hwriter 11 tools 292
32 38
Some ShortRead classes
ShortRead
ShortReadQ
AlignedRead
33 38
Some Biostrings classes (single strings)
XString
BString DNAString RNAString AAString
34 38
Some Biostrings classes (sets of strings)
XStringSet
BStringSet DNAStringSet RNAStringSet AAStringSet PhredQualitySolexaQualityQualityScaledXStringSet
QualityScaledBStringSetQualityScaledDNAStringSetQualityScaledRNAStringSetQualityScaledAAStringSet
35 38

Reading the unaligned data
gt require(ShortRead)
gt fq lt- readFastq(seqdata mut_1_ffastq$)
gt fq
class ShortReadQlength 1000000 reads width 36 cycles
There are various simple accessor functions for this object especiallysread and quality
24 38
A brief look at the unaligned data
Using alphabetByCycle and as( rdquomatrixrdquo) (which creates a big read timescycle matrix) we can do
gt alp lt- alphabetByCycle(sread(fq))
gt matplot(t(proptable(alp[DNA_BASES
+ ] margin = 2)) type = l)
gt qaMatraw lt- as(quality(fq) matrix)
gt plot(colMeans(qaMatraw))
25 38
Aligning the data
We now align the data using Bowtie
binbash
BOWTIE_OPTS=-m 1 -v 2 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-m 1 -v 0 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 2 -k 2 -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 0 -k 1 -p 2 -q --quiet -3 10
for f in `ls seqdatafastq`dobowtie s_cerevisiae $BOWTIE_OPTS $f gt $fbowtiedone
How many hits do we get
26 38
Reading in the aligned data
We read the data into R as a 4-component list (one lane per component)
gt files lt- listfiles(seqdata
+ pattern = bowtie)
gt aligned lt- sapply(files function(f)
+ readAligned(seqdata pattern = f
+ type = Bowtie)
+ )
gt names(aligned) lt- gsub(_ffastqbowtie
+ names(aligned))
gt aligned[[1]]
gt save(aligned file = alignedrda)
27 38
Some exercises
1 Determine the number of times the motif ldquoTATAArdquo occurs in theyeast genome How often does it occur with one mismatch
2 The seqdata directory has an object called ste12 which is aposition weight matrix for the transcription factor ldquoSte12rdquo (obtainedfrom SCPD) Where does it occur in the genome
3 Compute the average quality for each cycle for the aligned reads andcompare to the qualities for the unaligned reads What is thedifference
28 38
Solution 1
gt sum(sapply(116 function(i)
+ motif lt- DNAString(TATAA)
+ countPattern(motif Scerevisiae[[i]]) +
+ countPattern(reverseComplement(motif)
+ Scerevisiae[[i]])
+ ))
29 38
Solution 2
30 38
Solution 3
31 38
SessionInfo
gt toLatex(sessionInfo())
I R version 292 RC (2009-08-17 r49312) i386-apple-darwin980
I Localeen_USUTF-8en_USUTF-8CCen_USUTF-8en_USUTF-8
I Base packages base datasets graphics grDevices grid methodsstats utils
I Other packages Biostrings 2128 BSgenome 1123BSgenomeScerevisiaeUCSCsacCer1 1313 classGraph 07-2graph 1222 IRanges 123 lattice 017-25 Rgraphviz 1234ShortRead 121
I Loaded via a namespace (and not attached) Biobase 241hwriter 11 tools 292
32 38
Some ShortRead classes
ShortRead
ShortReadQ
AlignedRead
33 38
Some Biostrings classes (single strings)
XString
BString DNAString RNAString AAString
34 38
Some Biostrings classes (sets of strings)
XStringSet
BStringSet DNAStringSet RNAStringSet AAStringSet PhredQualitySolexaQualityQualityScaledXStringSet
QualityScaledBStringSetQualityScaledDNAStringSetQualityScaledRNAStringSetQualityScaledAAStringSet
35 38

A brief look at the unaligned data
Using alphabetByCycle and as( rdquomatrixrdquo) (which creates a big read timescycle matrix) we can do
gt alp lt- alphabetByCycle(sread(fq))
gt matplot(t(proptable(alp[DNA_BASES
+ ] margin = 2)) type = l)
gt qaMatraw lt- as(quality(fq) matrix)
gt plot(colMeans(qaMatraw))
25 38
Aligning the data
We now align the data using Bowtie
binbash
BOWTIE_OPTS=-m 1 -v 2 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-m 1 -v 0 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 2 -k 2 -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 0 -k 1 -p 2 -q --quiet -3 10
for f in `ls seqdatafastq`dobowtie s_cerevisiae $BOWTIE_OPTS $f gt $fbowtiedone
How many hits do we get
26 38
Reading in the aligned data
We read the data into R as a 4-component list (one lane per component)
gt files lt- listfiles(seqdata
+ pattern = bowtie)
gt aligned lt- sapply(files function(f)
+ readAligned(seqdata pattern = f
+ type = Bowtie)
+ )
gt names(aligned) lt- gsub(_ffastqbowtie
+ names(aligned))
gt aligned[[1]]
gt save(aligned file = alignedrda)
27 38
Some exercises
1 Determine the number of times the motif ldquoTATAArdquo occurs in theyeast genome How often does it occur with one mismatch
2 The seqdata directory has an object called ste12 which is aposition weight matrix for the transcription factor ldquoSte12rdquo (obtainedfrom SCPD) Where does it occur in the genome
3 Compute the average quality for each cycle for the aligned reads andcompare to the qualities for the unaligned reads What is thedifference
28 38
Solution 1
gt sum(sapply(116 function(i)
+ motif lt- DNAString(TATAA)
+ countPattern(motif Scerevisiae[[i]]) +
+ countPattern(reverseComplement(motif)
+ Scerevisiae[[i]])
+ ))
29 38
Solution 2
30 38
Solution 3
31 38
SessionInfo
gt toLatex(sessionInfo())
I R version 292 RC (2009-08-17 r49312) i386-apple-darwin980
I Localeen_USUTF-8en_USUTF-8CCen_USUTF-8en_USUTF-8
I Base packages base datasets graphics grDevices grid methodsstats utils
I Other packages Biostrings 2128 BSgenome 1123BSgenomeScerevisiaeUCSCsacCer1 1313 classGraph 07-2graph 1222 IRanges 123 lattice 017-25 Rgraphviz 1234ShortRead 121
I Loaded via a namespace (and not attached) Biobase 241hwriter 11 tools 292
32 38
Some ShortRead classes
ShortRead
ShortReadQ
AlignedRead
33 38
Some Biostrings classes (single strings)
XString
BString DNAString RNAString AAString
34 38
Some Biostrings classes (sets of strings)
XStringSet
BStringSet DNAStringSet RNAStringSet AAStringSet PhredQualitySolexaQualityQualityScaledXStringSet
QualityScaledBStringSetQualityScaledDNAStringSetQualityScaledRNAStringSetQualityScaledAAStringSet
35 38

Aligning the data
We now align the data using Bowtie
binbash
BOWTIE_OPTS=-m 1 -v 2 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-m 1 -v 0 --all -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 2 -k 2 -p 2 -q --quiet -3 10BOWTIE_OPTS=-v 0 -k 1 -p 2 -q --quiet -3 10
for f in `ls seqdatafastq`dobowtie s_cerevisiae $BOWTIE_OPTS $f gt $fbowtiedone
How many hits do we get
26 38
Reading in the aligned data
We read the data into R as a 4-component list (one lane per component)
gt files lt- listfiles(seqdata
+ pattern = bowtie)
gt aligned lt- sapply(files function(f)
+ readAligned(seqdata pattern = f
+ type = Bowtie)
+ )
gt names(aligned) lt- gsub(_ffastqbowtie
+ names(aligned))
gt aligned[[1]]
gt save(aligned file = alignedrda)
27 38
Some exercises
1 Determine the number of times the motif ldquoTATAArdquo occurs in theyeast genome How often does it occur with one mismatch
2 The seqdata directory has an object called ste12 which is aposition weight matrix for the transcription factor ldquoSte12rdquo (obtainedfrom SCPD) Where does it occur in the genome
3 Compute the average quality for each cycle for the aligned reads andcompare to the qualities for the unaligned reads What is thedifference
28 38
Solution 1
gt sum(sapply(116 function(i)
+ motif lt- DNAString(TATAA)
+ countPattern(motif Scerevisiae[[i]]) +
+ countPattern(reverseComplement(motif)
+ Scerevisiae[[i]])
+ ))
29 38
Solution 2
30 38
Solution 3
31 38
SessionInfo
gt toLatex(sessionInfo())
I R version 292 RC (2009-08-17 r49312) i386-apple-darwin980
I Localeen_USUTF-8en_USUTF-8CCen_USUTF-8en_USUTF-8
I Base packages base datasets graphics grDevices grid methodsstats utils
I Other packages Biostrings 2128 BSgenome 1123BSgenomeScerevisiaeUCSCsacCer1 1313 classGraph 07-2graph 1222 IRanges 123 lattice 017-25 Rgraphviz 1234ShortRead 121
I Loaded via a namespace (and not attached) Biobase 241hwriter 11 tools 292
32 38
Some ShortRead classes
ShortRead
ShortReadQ
AlignedRead
33 38
Some Biostrings classes (single strings)
XString
BString DNAString RNAString AAString
34 38
Some Biostrings classes (sets of strings)
XStringSet
BStringSet DNAStringSet RNAStringSet AAStringSet PhredQualitySolexaQualityQualityScaledXStringSet
QualityScaledBStringSetQualityScaledDNAStringSetQualityScaledRNAStringSetQualityScaledAAStringSet
35 38

Reading in the aligned data
We read the data into R as a 4-component list (one lane per component)
gt files lt- listfiles(seqdata
+ pattern = bowtie)
gt aligned lt- sapply(files function(f)
+ readAligned(seqdata pattern = f
+ type = Bowtie)
+ )
gt names(aligned) lt- gsub(_ffastqbowtie
+ names(aligned))
gt aligned[[1]]
gt save(aligned file = alignedrda)
27 38
Some exercises
1 Determine the number of times the motif ldquoTATAArdquo occurs in theyeast genome How often does it occur with one mismatch
2 The seqdata directory has an object called ste12 which is aposition weight matrix for the transcription factor ldquoSte12rdquo (obtainedfrom SCPD) Where does it occur in the genome
3 Compute the average quality for each cycle for the aligned reads andcompare to the qualities for the unaligned reads What is thedifference
28 38
Solution 1
gt sum(sapply(116 function(i)
+ motif lt- DNAString(TATAA)
+ countPattern(motif Scerevisiae[[i]]) +
+ countPattern(reverseComplement(motif)
+ Scerevisiae[[i]])
+ ))
29 38
Solution 2
30 38
Solution 3
31 38
SessionInfo
gt toLatex(sessionInfo())
I R version 292 RC (2009-08-17 r49312) i386-apple-darwin980
I Localeen_USUTF-8en_USUTF-8CCen_USUTF-8en_USUTF-8
I Base packages base datasets graphics grDevices grid methodsstats utils
I Other packages Biostrings 2128 BSgenome 1123BSgenomeScerevisiaeUCSCsacCer1 1313 classGraph 07-2graph 1222 IRanges 123 lattice 017-25 Rgraphviz 1234ShortRead 121
I Loaded via a namespace (and not attached) Biobase 241hwriter 11 tools 292
32 38
Some ShortRead classes
ShortRead
ShortReadQ
AlignedRead
33 38
Some Biostrings classes (single strings)
XString
BString DNAString RNAString AAString
34 38
Some Biostrings classes (sets of strings)
XStringSet
BStringSet DNAStringSet RNAStringSet AAStringSet PhredQualitySolexaQualityQualityScaledXStringSet
QualityScaledBStringSetQualityScaledDNAStringSetQualityScaledRNAStringSetQualityScaledAAStringSet
35 38

Some exercises
1 Determine the number of times the motif ldquoTATAArdquo occurs in theyeast genome How often does it occur with one mismatch
2 The seqdata directory has an object called ste12 which is aposition weight matrix for the transcription factor ldquoSte12rdquo (obtainedfrom SCPD) Where does it occur in the genome
3 Compute the average quality for each cycle for the aligned reads andcompare to the qualities for the unaligned reads What is thedifference
28 38
Solution 1
gt sum(sapply(116 function(i)
+ motif lt- DNAString(TATAA)
+ countPattern(motif Scerevisiae[[i]]) +
+ countPattern(reverseComplement(motif)
+ Scerevisiae[[i]])
+ ))
29 38
Solution 2
30 38
Solution 3
31 38
SessionInfo
gt toLatex(sessionInfo())
I R version 292 RC (2009-08-17 r49312) i386-apple-darwin980
I Localeen_USUTF-8en_USUTF-8CCen_USUTF-8en_USUTF-8
I Base packages base datasets graphics grDevices grid methodsstats utils
I Other packages Biostrings 2128 BSgenome 1123BSgenomeScerevisiaeUCSCsacCer1 1313 classGraph 07-2graph 1222 IRanges 123 lattice 017-25 Rgraphviz 1234ShortRead 121
I Loaded via a namespace (and not attached) Biobase 241hwriter 11 tools 292
32 38
Some ShortRead classes
ShortRead
ShortReadQ
AlignedRead
33 38
Some Biostrings classes (single strings)
XString
BString DNAString RNAString AAString
34 38
Some Biostrings classes (sets of strings)
XStringSet
BStringSet DNAStringSet RNAStringSet AAStringSet PhredQualitySolexaQualityQualityScaledXStringSet
QualityScaledBStringSetQualityScaledDNAStringSetQualityScaledRNAStringSetQualityScaledAAStringSet
35 38

Solution 1
gt sum(sapply(116 function(i)
+ motif lt- DNAString(TATAA)
+ countPattern(motif Scerevisiae[[i]]) +
+ countPattern(reverseComplement(motif)
+ Scerevisiae[[i]])
+ ))
29 38
Solution 2
30 38
Solution 3
31 38
SessionInfo
gt toLatex(sessionInfo())
I R version 292 RC (2009-08-17 r49312) i386-apple-darwin980
I Localeen_USUTF-8en_USUTF-8CCen_USUTF-8en_USUTF-8
I Base packages base datasets graphics grDevices grid methodsstats utils
I Other packages Biostrings 2128 BSgenome 1123BSgenomeScerevisiaeUCSCsacCer1 1313 classGraph 07-2graph 1222 IRanges 123 lattice 017-25 Rgraphviz 1234ShortRead 121
I Loaded via a namespace (and not attached) Biobase 241hwriter 11 tools 292
32 38
Some ShortRead classes
ShortRead
ShortReadQ
AlignedRead
33 38
Some Biostrings classes (single strings)
XString
BString DNAString RNAString AAString
34 38
Some Biostrings classes (sets of strings)
XStringSet
BStringSet DNAStringSet RNAStringSet AAStringSet PhredQualitySolexaQualityQualityScaledXStringSet
QualityScaledBStringSetQualityScaledDNAStringSetQualityScaledRNAStringSetQualityScaledAAStringSet
35 38

Solution 2
30 38
Solution 3
31 38
SessionInfo
gt toLatex(sessionInfo())
I R version 292 RC (2009-08-17 r49312) i386-apple-darwin980
I Localeen_USUTF-8en_USUTF-8CCen_USUTF-8en_USUTF-8
I Base packages base datasets graphics grDevices grid methodsstats utils
I Other packages Biostrings 2128 BSgenome 1123BSgenomeScerevisiaeUCSCsacCer1 1313 classGraph 07-2graph 1222 IRanges 123 lattice 017-25 Rgraphviz 1234ShortRead 121
I Loaded via a namespace (and not attached) Biobase 241hwriter 11 tools 292
32 38
Some ShortRead classes
ShortRead
ShortReadQ
AlignedRead
33 38
Some Biostrings classes (single strings)
XString
BString DNAString RNAString AAString
34 38
Some Biostrings classes (sets of strings)
XStringSet
BStringSet DNAStringSet RNAStringSet AAStringSet PhredQualitySolexaQualityQualityScaledXStringSet
QualityScaledBStringSetQualityScaledDNAStringSetQualityScaledRNAStringSetQualityScaledAAStringSet
35 38

Solution 3
31 38
SessionInfo
gt toLatex(sessionInfo())
I R version 292 RC (2009-08-17 r49312) i386-apple-darwin980
I Localeen_USUTF-8en_USUTF-8CCen_USUTF-8en_USUTF-8
I Base packages base datasets graphics grDevices grid methodsstats utils
I Other packages Biostrings 2128 BSgenome 1123BSgenomeScerevisiaeUCSCsacCer1 1313 classGraph 07-2graph 1222 IRanges 123 lattice 017-25 Rgraphviz 1234ShortRead 121
I Loaded via a namespace (and not attached) Biobase 241hwriter 11 tools 292
32 38
Some ShortRead classes
ShortRead
ShortReadQ
AlignedRead
33 38
Some Biostrings classes (single strings)
XString
BString DNAString RNAString AAString
34 38
Some Biostrings classes (sets of strings)
XStringSet
BStringSet DNAStringSet RNAStringSet AAStringSet PhredQualitySolexaQualityQualityScaledXStringSet
QualityScaledBStringSetQualityScaledDNAStringSetQualityScaledRNAStringSetQualityScaledAAStringSet
35 38
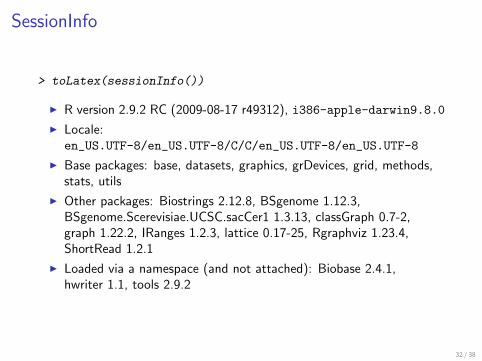
SessionInfo
gt toLatex(sessionInfo())
I R version 292 RC (2009-08-17 r49312) i386-apple-darwin980
I Localeen_USUTF-8en_USUTF-8CCen_USUTF-8en_USUTF-8
I Base packages base datasets graphics grDevices grid methodsstats utils
I Other packages Biostrings 2128 BSgenome 1123BSgenomeScerevisiaeUCSCsacCer1 1313 classGraph 07-2graph 1222 IRanges 123 lattice 017-25 Rgraphviz 1234ShortRead 121
I Loaded via a namespace (and not attached) Biobase 241hwriter 11 tools 292
32 38
Some ShortRead classes
ShortRead
ShortReadQ
AlignedRead
33 38
Some Biostrings classes (single strings)
XString
BString DNAString RNAString AAString
34 38
Some Biostrings classes (sets of strings)
XStringSet
BStringSet DNAStringSet RNAStringSet AAStringSet PhredQualitySolexaQualityQualityScaledXStringSet
QualityScaledBStringSetQualityScaledDNAStringSetQualityScaledRNAStringSetQualityScaledAAStringSet
35 38
Some ShortRead classes
ShortRead
ShortReadQ
AlignedRead
33 38
Some Biostrings classes (single strings)
XString
BString DNAString RNAString AAString
34 38
Some Biostrings classes (sets of strings)
XStringSet
BStringSet DNAStringSet RNAStringSet AAStringSet PhredQualitySolexaQualityQualityScaledXStringSet
QualityScaledBStringSetQualityScaledDNAStringSetQualityScaledRNAStringSetQualityScaledAAStringSet
35 38
Some Biostrings classes (single strings)
XString
BString DNAString RNAString AAString
34 38
Some Biostrings classes (sets of strings)
XStringSet
BStringSet DNAStringSet RNAStringSet AAStringSet PhredQualitySolexaQualityQualityScaledXStringSet
QualityScaledBStringSetQualityScaledDNAStringSetQualityScaledRNAStringSetQualityScaledAAStringSet
35 38
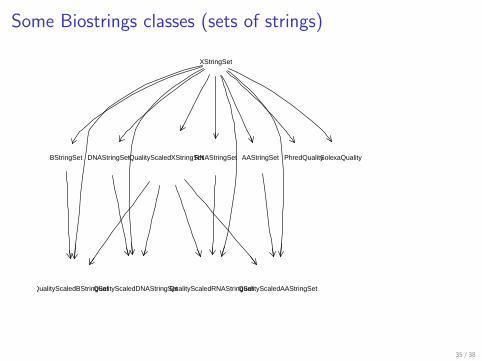
Some Biostrings classes (sets of strings)
XStringSet
BStringSet DNAStringSet RNAStringSet AAStringSet PhredQualitySolexaQualityQualityScaledXStringSet
QualityScaledBStringSetQualityScaledDNAStringSetQualityScaledRNAStringSetQualityScaledAAStringSet
35 38





























