Embed Size (px)
Citation preview

Available online at www.sciencedirect.com
www.elsevier.com/locate/brainres
b r a i n r e s e a r c h 1 5 7 7 ( 2 0 1 4 ) 6 9 – 7 6
http://dx.doi.org/100006-8993/& 2014 Pu
nCorresponding annCorresponding
PR China.E-mail addresses1These authors
Research Report
HO-1 attenuates hippocampal neurons injury via theactivation of BDNF–TrkB–PI3K/Akt signaling pathwayin stroke
Dashi Qia,1, Changjie Ouyangb,1, Yulan Wangb,1, Shichun Zhangb,1,Xijuan Mad, YuanJian Songa, HongLi Yua, Jiali Tange, Wei Fuf, Lei Shengg,Lihua Yanga, Mei Wanga, Weihao Zhanga, Lei Miaoa, Tengteng Lia,Xiaojing Huangc,n, Hongyan Donga,c,nn
aResearch Center for Neurobiology and Department of Neurobiology, Xuzhou Medical College, 209 Tongshan Road,Xuzhou, Jiangsu 221004, PR ChinabDepartment of Human anatomy, Xuzhou Medical College, PR ChinacSchool of Public Health, Xuzhou Medical College, PR ChinadDepartment of Radiology, Xuzhou Central Hospital, PR ChinaeSchool of Life Science and Technology, China Pharmaceutical University, PR ChinafDepartment of General Surgery, Affiliated Hospital of Xuzhou Medical College, PR ChinagDepartment of Clinical Pharmacology, Zhong Shan Hospital, Fudan University, PR China
a r t i c l e i n f o
Article history:
Accepted 25 June 2014
Although recent studies have found that HO-1 plays an important role in neuronal
survival, little is known about the precise mechanisms occurring during cerebral ische-
Available online 2 July 2014
Keywords:
HO-1
Stroke
Brain-derived neurotrophic factor
(BNDF)
TrkB
PI3K/Akt
Apoptosis
.1016/j.brainres.2014.06.03blished by Elsevier B.V.
uthor.author at: Research Cent
: [email protected] (X. Hcontributed equally to th
a b s t r a c t
mia/reperfusion (I/R). Therefore, the aim of this study was to investigate the neuroprotec-
tive mechanisms of HO-1 against ischemic brain injury induced by cerebral I/R and to
explore whether the BDNF–TrkB–PI3K/Akt signaling pathway contributed to the protection
provided by HO-1. Over-expressed HO-1 plasmids were employed to induce the over-
expression of HO-1 through hippocampi CA1 injection 5 days before the cerebral I/R animal
model was induced by four-vessel occlusion for 15 min transient ischemia and followed by
reperfusion in Sprague-Dawley rats. Immunoblotting was carried out to examine the
expression of the related proteins, and HE-staining was used to detect the percentage of
living neurons in the hippocampal CA1 region. The results showed that over-expressed
HO-1 could significantly protect neurons against cerebral I/R. Furthermore, the protein
expression of BDNF, TrkB and p-Akt also increased in the rats treated with over-expressed
HO-1 plasmids. However, treatment with tropomyosin receptor kinase B (TrkB) receptor
antagonist (K252a) reversed the HO-1-induced increase in BDNF and p-Akt protein levels
and decreased the level of cleaved caspase-3 protein in I/R rats. In summary, our results
1
er for Neurobiology, Xuzhou Medical College, 209 Tongshan Road, Xuzhou, Jiangsu 221004,
uang), [email protected] (H. Dong).is work.

b r a i n r e s e a r c h 1 5 7 7 ( 2 0 1 4 ) 6 9 – 7 670
imply that HO-1 can decrease cell apoptosis in the I/R rat brain and that the mechanism
may be related to the activation of the BDNF–TrkB–PI3K/Akt signaling pathway.
& 2014 Published by Elsevier B.V.
1. Introduction
The risk of ischemic stroke, a major cause of disability andmortality worldwide, occurs when a cerebral blood vessel isruptured or occluded, contributing to a variety of acute andchronic diseases of the brain (Brima et al., 2013; Hankey,2006). The World Health Organization has estimated thatapproximately 15 million people worldwide suffer fromstroke annually (Hoffmann et al., 2012). Therefore, a safeand effective therapy that can target multiple pathways forneuroprotection is urgently needed.
Hemoxygenase (HO) can catalyze the first and rate-limitingenzymatic step of heme degradation and produces carbonmonoxide (CO), iron and biliverdin (BVD), which are convertedinto bilirubin (BR) via biliverdin reductase (Banerjee et al., 2011).HO can be classified as three genetically distinct HO isoforms:HO-1, HO-2, and HO-3 (Lin et al., 2010). Among the three knownoxygenases, HO-1 is the only inducible isoform, whereas HO-2and HO-3 are constitutively expressed (de Sousa et al., 2011).HO-1 is the major inducible lowmolecular weight stress proteinof mammalian cells and tissues, and its expression is increasedseveral folds in response to a variety of cellular stresses andstimuli, including ischemia, hypoxia, oxidative stress andinflammatory cytokines, suggesting an important role of thisenzyme in tissue protection (Morse et al., 2009). Accumulatingevidence indicates that HO-1 over-expression protects againstinjury in a variety of tissues, while reduced HO-1 levels increasesusceptibility to injury in a variety of stress conditions (Pachoriet al., 2007). For example, cerebral ischemic injury is amplifiedin HO-1 knockout mice (Choi and Kim, 2008). In addition, HO-1is an important protective protein in many clinically relevantdisease states, such as Alzheimer's disease, hypertension,atherosclerosis, and diabetes (Calabrese et al., 2005).
Brain-derived neurotrophic factor (BDNF), a member ofneurotrophin family protein, exerts a neuroprotective effectagainst ischemic brain injury in vivo via its effects on anti-excitatory amino acids, inhibiting inflammatory factor anddecreasing apoptosis (Fanaei et al., 2014; Harada et al., 2012).Interventions that increased BDNF levels in the perilesionalareas are most often associated with improved recovery offunction (Bejot et al., 2011). However, attenuating BDNF levels orits effects following cerebral ischemia reduces neuroplasticchanges or recovery of function, either spontaneously orinduced by rehabilitation (Ploughman et al., 2009). Variousstudies have shown that BDNF represents one of the mostimportant signaling molecules for adaptive brain plasticity afterstroke (Cowansage et al., 2010). Based on these data, pharma-cological strategies for post-ischemic cerebral BDNF productionprovide a new experimental foundation for treatment, whichappears to be a promising option in the treatment of stroke.
Cell apoptosis is generally thought to play a crucial role inthe process of neuronal death after cerebral ischemia(Broughton et al., 2009; Vakili et al., 2014). Increasing
experimental studies have shown that the oxidative load inmitochondria leads to the release of cytochrome c and thetranslocation of Bax from cytosol to mitochondria, which iscontrolled by the Bcl-2 family proteins (Kuwana et al., 2002).The release of cytochrome c into the cytosol causes theformation of apoptosome, which permits the autoactivationof procaspase-9 (Ferraro et al., 2008). After the autoactivationof procaspase-9, procaspase-3 is activated and results in DNAfragmentation (Li et al., 1997). In addition, it has been shownthat apoptosis contributes to the development of ischemicinfarction with DNA fragmentation (Yao et al., 2001). Thus,the ideal preventive or therapeutic approach would targetapoptosis after cerebral ischemia.
Some researchers have demonstrated that HO-1 overexpres-sion induces upregulation of BDNF mRNA levels in Rat C6Glioma Cells (Morita et al., 2009) and that HO-1 exerts neuro-protection through BDNF induction. Therefore, we investigatedthe relations between the neuroprotective effects of HO-1 andBDNF in the rat brain after stroke. In this study, we hypothe-sized that the overexpression of HO-1 may protect neuronsagainst cerebral I/R injury through the BDNF–TrkB–PI3K/Aktsignaling pathway and can suppress downstream pro-apoptosissignaling molecules in the rat brain after stroke.
2. Results
2.1. The neuroprotective effects of HO-1 againstI/R-induced neuronal loss
To determine whether HO-1 has a neuroprotective effectagainst neuronal injury induced by reperfusion following ische-mia, we investigated the effect of HO-1 overexpression on thesurvival of CA1 pyramidal neurons in the rat hippocampus,where neurons were particularly vulnerable to ischemic injury,at 5 days of reperfusion. HE-staining was used to examine thesurviving neurons after 5 days of reperfusion following 15minischemia. Shrunken cells with pyknotic nuclei after ischemiawere counted as dead cells. As shown in Fig. 1, transientcerebral ischemia followed by 5 days of reperfusion inducessevere neuronal death. However, pretreatment with HO-1 over-expression plasmids significantly decreased the neuronaldegeneration, whereas in the controls, nonsense oligonucleo-tides had no effect on cell death during cerebral I/R in thehippocampal CA1 region. The results suggest that HO-1 mayprotect neurons against ischemic brain injury.
2.2. HO-1 increased the protein levels of BDNF and TrkBand p-Akt
To confirm the effects of HO-1 overexpression plasmids andHO-1-NON on the protein levels of HO-1, we examined the
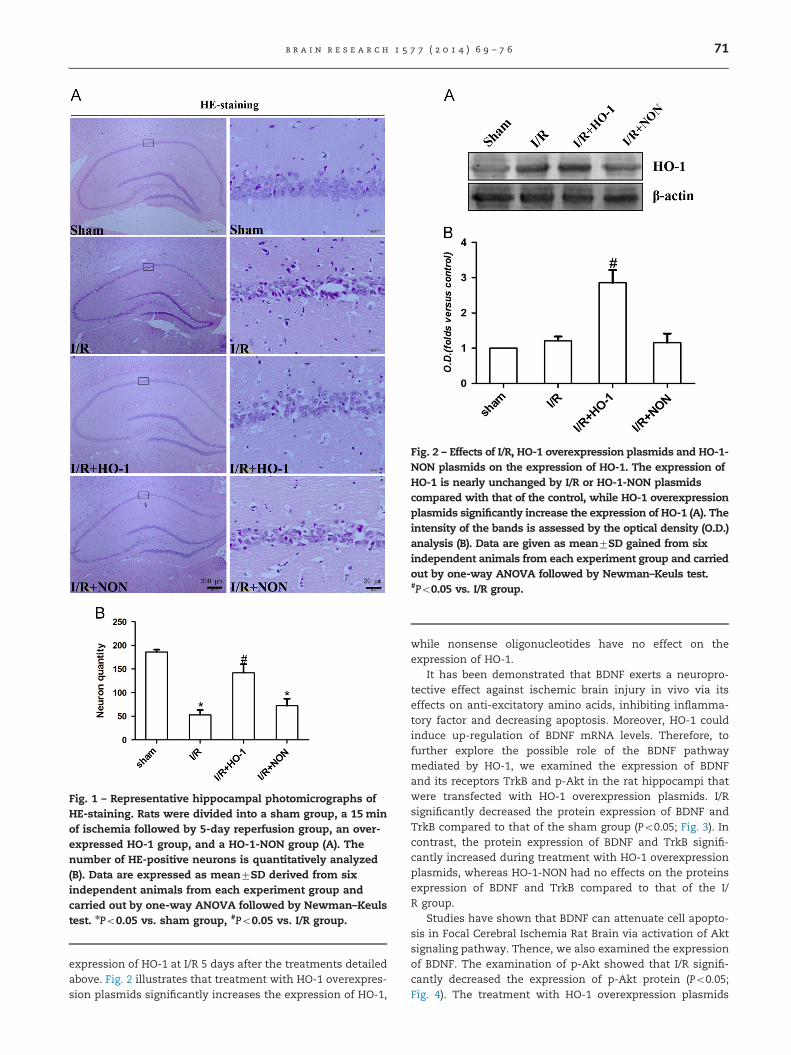
Fig. 1 – Representative hippocampal photomicrographs ofHE-staining. Rats were divided into a sham group, a 15 minof ischemia followed by 5-day reperfusion group, an over-expressed HO-1 group, and a HO-1-NON group (A). Thenumber of HE-positive neurons is quantitatively analyzed(B). Data are expressed as mean7SD derived from sixindependent animals from each experiment group andcarried out by one-way ANOVA followed by Newman–Keulstest. nPo0.05 vs. sham group, #Po0.05 vs. I/R group.
Fig. 2 – Effects of I/R, HO-1 overexpression plasmids and HO-1-NON plasmids on the expression of HO-1. The expression ofHO-1 is nearly unchanged by I/R or HO-1-NON plasmidscompared with that of the control, while HO-1 overexpressionplasmids significantly increase the expression of HO-1 (A). Theintensity of the bands is assessed by the optical density (O.D.)analysis (B). Data are given as mean7SD gained from sixindependent animals from each experiment group and carriedout by one-way ANOVA followed by Newman–Keuls test.#Po0.05 vs. I/R group.
b r a i n r e s e a r c h 1 5 7 7 ( 2 0 1 4 ) 6 9 – 7 6 71
expression of HO-1 at I/R 5 days after the treatments detailedabove. Fig. 2 illustrates that treatment with HO-1 overexpres-sion plasmids significantly increases the expression of HO-1,
while nonsense oligonucleotides have no effect on theexpression of HO-1.
It has been demonstrated that BDNF exerts a neuropro-tective effect against ischemic brain injury in vivo via itseffects on anti-excitatory amino acids, inhibiting inflamma-tory factor and decreasing apoptosis. Moreover, HO-1 couldinduce up-regulation of BDNF mRNA levels. Therefore, tofurther explore the possible role of the BDNF pathwaymediated by HO-1, we examined the expression of BDNFand its receptors TrkB and p-Akt in the rat hippocampi thatwere transfected with HO-1 overexpression plasmids. I/Rsignificantly decreased the protein expression of BDNF andTrkB compared to that of the sham group (Po0.05; Fig. 3). Incontrast, the protein expression of BDNF and TrkB signifi-cantly increased during treatment with HO-1 overexpressionplasmids, whereas HO-1-NON had no effects on the proteinsexpression of BDNF and TrkB compared to that of the I/R group.
Studies have shown that BDNF can attenuate cell apopto-sis in Focal Cerebral Ischemia Rat Brain via activation of Aktsignaling pathway. Thence, we also examined the expressionof BDNF. The examination of p-Akt showed that I/R signifi-cantly decreased the expression of p-Akt protein (Po0.05;Fig. 4). The treatment with HO-1 overexpression plasmids
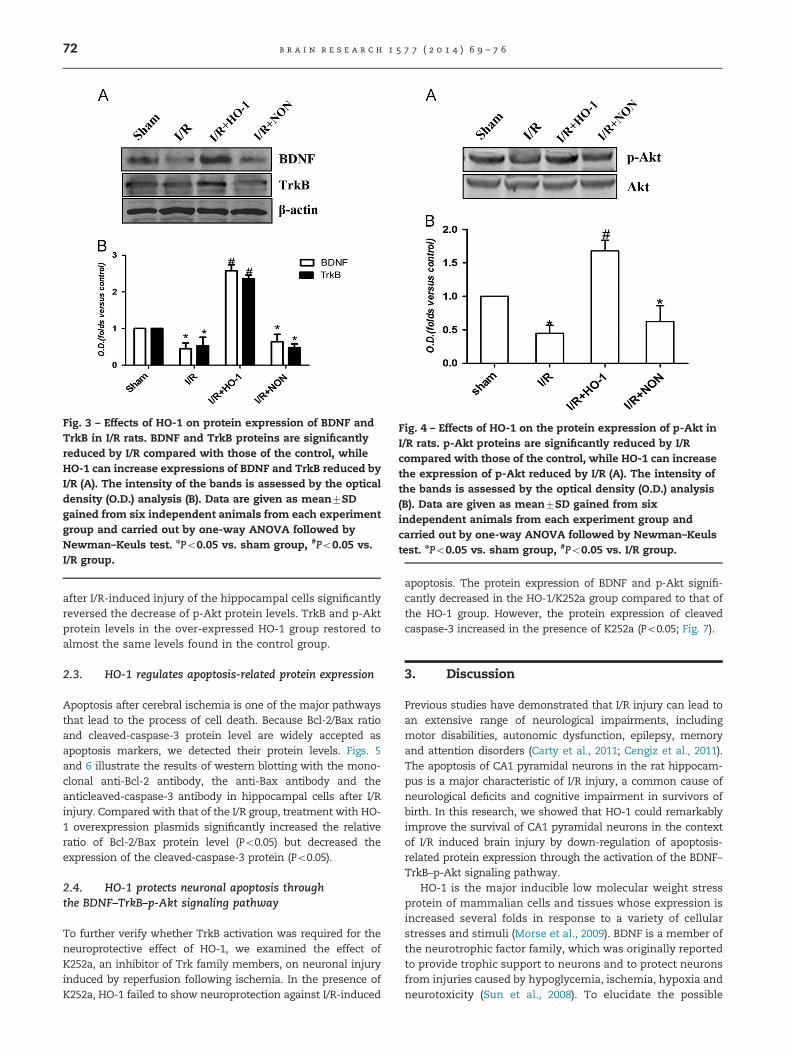
Fig. 3 – Effects of HO-1 on protein expression of BDNF andTrkB in I/R rats. BDNF and TrkB proteins are significantlyreduced by I/R compared with those of the control, whileHO-1 can increase expressions of BDNF and TrkB reduced byI/R (A). The intensity of the bands is assessed by the opticaldensity (O.D.) analysis (B). Data are given as mean7SDgained from six independent animals from each experimentgroup and carried out by one-way ANOVA followed byNewman–Keuls test. nPo0.05 vs. sham group, #Po0.05 vs.I/R group.
Fig. 4 – Effects of HO-1 on the protein expression of p-Akt inI/R rats. p-Akt proteins are significantly reduced by I/Rcompared with those of the control, while HO-1 can increasethe expression of p-Akt reduced by I/R (A). The intensity ofthe bands is assessed by the optical density (O.D.) analysis(B). Data are given as mean7SD gained from sixindependent animals from each experiment group andcarried out by one-way ANOVA followed by Newman–Keulstest. nPo0.05 vs. sham group, #Po0.05 vs. I/R group.
b r a i n r e s e a r c h 1 5 7 7 ( 2 0 1 4 ) 6 9 – 7 672
after I/R-induced injury of the hippocampal cells significantlyreversed the decrease of p-Akt protein levels. TrkB and p-Aktprotein levels in the over-expressed HO-1 group restored toalmost the same levels found in the control group.
2.3. HO-1 regulates apoptosis-related protein expression
Apoptosis after cerebral ischemia is one of the major pathwaysthat lead to the process of cell death. Because Bcl-2/Bax ratioand cleaved-caspase-3 protein level are widely accepted asapoptosis markers, we detected their protein levels. Figs. 5and 6 illustrate the results of western blotting with the mono-clonal anti-Bcl-2 antibody, the anti-Bax antibody and theanticleaved-caspase-3 antibody in hippocampal cells after I/Rinjury. Compared with that of the I/R group, treatment with HO-1 overexpression plasmids significantly increased the relativeratio of Bcl-2/Bax protein level (Po0.05) but decreased theexpression of the cleaved-caspase-3 protein (Po0.05).
2.4. HO-1 protects neuronal apoptosis throughthe BDNF–TrkB–p-Akt signaling pathway
To further verify whether TrkB activation was required for theneuroprotective effect of HO-1, we examined the effect ofK252a, an inhibitor of Trk family members, on neuronal injuryinduced by reperfusion following ischemia. In the presence ofK252a, HO-1 failed to show neuroprotection against I/R-induced
apoptosis. The protein expression of BDNF and p-Akt signifi-cantly decreased in the HO-1/K252a group compared to that ofthe HO-1 group. However, the protein expression of cleavedcaspase-3 increased in the presence of K252a (Po0.05; Fig. 7).
3. Discussion
Previous studies have demonstrated that I/R injury can lead toan extensive range of neurological impairments, includingmotor disabilities, autonomic dysfunction, epilepsy, memoryand attention disorders (Carty et al., 2011; Cengiz et al., 2011).The apoptosis of CA1 pyramidal neurons in the rat hippocam-pus is a major characteristic of I/R injury, a common cause ofneurological deficits and cognitive impairment in survivors ofbirth. In this research, we showed that HO-1 could remarkablyimprove the survival of CA1 pyramidal neurons in the contextof I/R induced brain injury by down-regulation of apoptosis-related protein expression through the activation of the BDNF–TrkB–p-Akt signaling pathway.
HO-1 is the major inducible low molecular weight stressprotein of mammalian cells and tissues whose expression isincreased several folds in response to a variety of cellularstresses and stimuli (Morse et al., 2009). BDNF is a member ofthe neurotrophic factor family, which was originally reportedto provide trophic support to neurons and to protect neuronsfrom injuries caused by hypoglycemia, ischemia, hypoxia andneurotoxicity (Sun et al., 2008). To elucidate the possible
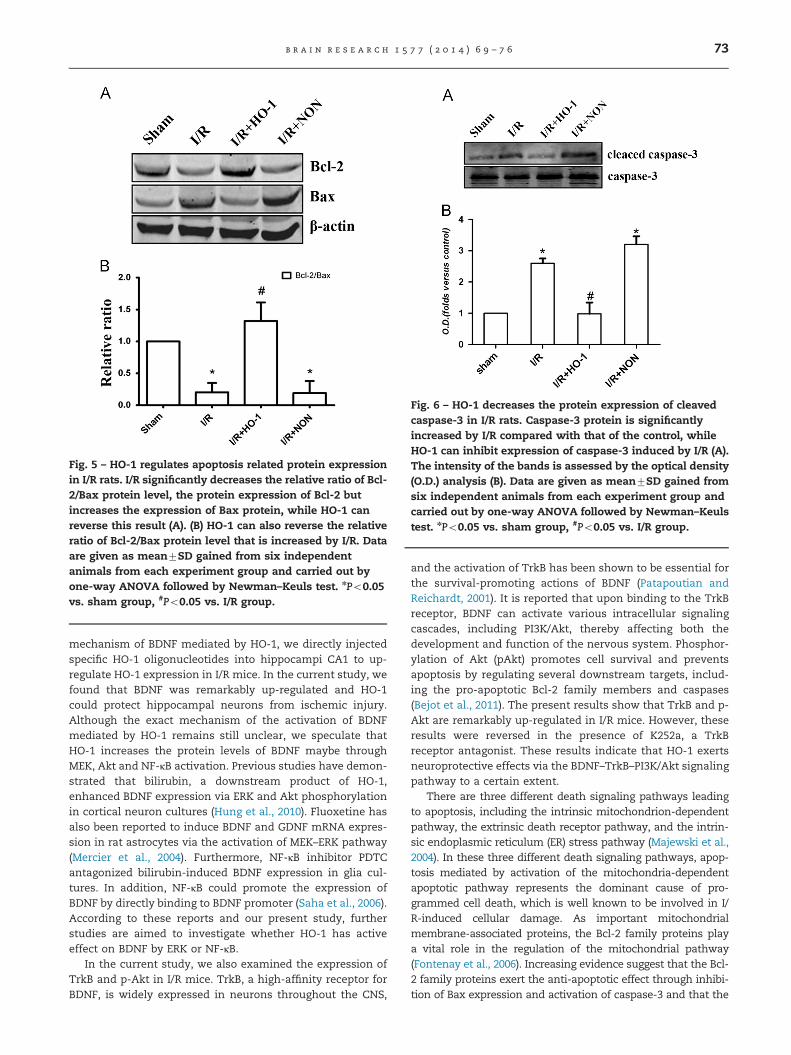
Fig. 5 – HO-1 regulates apoptosis related protein expressionin I/R rats. I/R significantly decreases the relative ratio of Bcl-2/Bax protein level, the protein expression of Bcl-2 butincreases the expression of Bax protein, while HO-1 canreverse this result (A). (B) HO-1 can also reverse the relativeratio of Bcl-2/Bax protein level that is increased by I/R. Dataare given as mean7SD gained from six independentanimals from each experiment group and carried out byone-way ANOVA followed by Newman–Keuls test. nPo0.05vs. sham group, #Po0.05 vs. I/R group.
Fig. 6 – HO-1 decreases the protein expression of cleavedcaspase-3 in I/R rats. Caspase-3 protein is significantlyincreased by I/R compared with that of the control, whileHO-1 can inhibit expression of caspase-3 induced by I/R (A).The intensity of the bands is assessed by the optical density(O.D.) analysis (B). Data are given as mean7SD gained fromsix independent animals from each experiment group andcarried out by one-way ANOVA followed by Newman–Keulstest. nPo0.05 vs. sham group, #Po0.05 vs. I/R group.
b r a i n r e s e a r c h 1 5 7 7 ( 2 0 1 4 ) 6 9 – 7 6 73
mechanism of BDNF mediated by HO-1, we directly injectedspecific HO-1 oligonucleotides into hippocampi CA1 to up-regulate HO-1 expression in I/R mice. In the current study, wefound that BDNF was remarkably up-regulated and HO-1could protect hippocampal neurons from ischemic injury.Although the exact mechanism of the activation of BDNFmediated by HO-1 remains still unclear, we speculate thatHO-1 increases the protein levels of BDNF maybe throughMEK, Akt and NF-κB activation. Previous studies have demon-strated that bilirubin, a downstream product of HO-1,enhanced BDNF expression via ERK and Akt phosphorylationin cortical neuron cultures (Hung et al., 2010). Fluoxetine hasalso been reported to induce BDNF and GDNF mRNA expres-sion in rat astrocytes via the activation of MEK–ERK pathway(Mercier et al., 2004). Furthermore, NF-κB inhibitor PDTCantagonized bilirubin-induced BDNF expression in glia cul-tures. In addition, NF-κB could promote the expression ofBDNF by directly binding to BDNF promoter (Saha et al., 2006).According to these reports and our present study, furtherstudies are aimed to investigate whether HO-1 has activeeffect on BDNF by ERK or NF-κB.
In the current study, we also examined the expression ofTrkB and p-Akt in I/R mice. TrkB, a high-affinity receptor forBDNF, is widely expressed in neurons throughout the CNS,
and the activation of TrkB has been shown to be essential forthe survival-promoting actions of BDNF (Patapoutian andReichardt, 2001). It is reported that upon binding to the TrkBreceptor, BDNF can activate various intracellular signalingcascades, including PI3K/Akt, thereby affecting both thedevelopment and function of the nervous system. Phosphor-ylation of Akt (pAkt) promotes cell survival and preventsapoptosis by regulating several downstream targets, includ-ing the pro-apoptotic Bcl-2 family members and caspases(Bejot et al., 2011). The present results show that TrkB and p-Akt are remarkably up-regulated in I/R mice. However, theseresults were reversed in the presence of K252a, a TrkBreceptor antagonist. These results indicate that HO-1 exertsneuroprotective effects via the BDNF–TrkB–PI3K/Akt signalingpathway to a certain extent.
There are three different death signaling pathways leadingto apoptosis, including the intrinsic mitochondrion-dependentpathway, the extrinsic death receptor pathway, and the intrin-sic endoplasmic reticulum (ER) stress pathway (Majewski et al.,2004). In these three different death signaling pathways, apop-tosis mediated by activation of the mitochondria-dependentapoptotic pathway represents the dominant cause of pro-grammed cell death, which is well known to be involved in I/R-induced cellular damage. As important mitochondrialmembrane-associated proteins, the Bcl-2 family proteins playa vital role in the regulation of the mitochondrial pathway(Fontenay et al., 2006). Increasing evidence suggest that the Bcl-2 family proteins exert the anti-apoptotic effect through inhibi-tion of Bax expression and activation of caspase-3 and that the
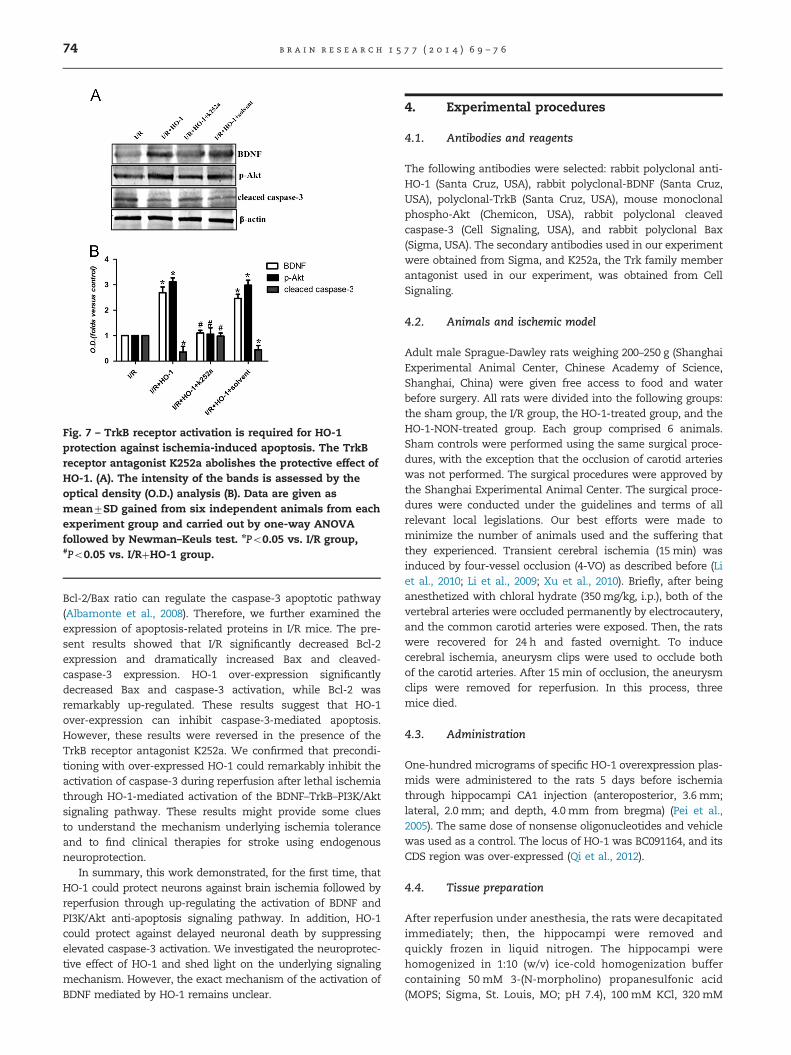
Fig. 7 – TrkB receptor activation is required for HO-1protection against ischemia-induced apoptosis. The TrkBreceptor antagonist K252a abolishes the protective effect ofHO-1. (A). The intensity of the bands is assessed by theoptical density (O.D.) analysis (B). Data are given asmean7SD gained from six independent animals from eachexperiment group and carried out by one-way ANOVAfollowed by Newman–Keuls test. nPo0.05 vs. I/R group,#Po0.05 vs. I/RþHO-1 group.
b r a i n r e s e a r c h 1 5 7 7 ( 2 0 1 4 ) 6 9 – 7 674
Bcl-2/Bax ratio can regulate the caspase-3 apoptotic pathway(Albamonte et al., 2008). Therefore, we further examined theexpression of apoptosis-related proteins in I/R mice. The pre-sent results showed that I/R significantly decreased Bcl-2expression and dramatically increased Bax and cleaved-caspase-3 expression. HO-1 over-expression significantlydecreased Bax and caspase-3 activation, while Bcl-2 wasremarkably up-regulated. These results suggest that HO-1over-expression can inhibit caspase-3-mediated apoptosis.However, these results were reversed in the presence of theTrkB receptor antagonist K252a. We confirmed that precondi-tioning with over-expressed HO-1 could remarkably inhibit theactivation of caspase-3 during reperfusion after lethal ischemiathrough HO-1-mediated activation of the BDNF–TrkB–PI3K/Aktsignaling pathway. These results might provide some cluesto understand the mechanism underlying ischemia toleranceand to find clinical therapies for stroke using endogenousneuroprotection.
In summary, this work demonstrated, for the first time, thatHO-1 could protect neurons against brain ischemia followed byreperfusion through up-regulating the activation of BDNF andPI3K/Akt anti-apoptosis signaling pathway. In addition, HO-1could protect against delayed neuronal death by suppressingelevated caspase-3 activation. We investigated the neuroprotec-tive effect of HO-1 and shed light on the underlying signalingmechanism. However, the exact mechanism of the activation ofBDNF mediated by HO-1 remains unclear.
4. Experimental procedures
4.1. Antibodies and reagents
The following antibodies were selected: rabbit polyclonal anti-HO-1 (Santa Cruz, USA), rabbit polyclonal-BDNF (Santa Cruz,USA), polyclonal-TrkB (Santa Cruz, USA), mouse monoclonalphospho-Akt (Chemicon, USA), rabbit polyclonal cleavedcaspase-3 (Cell Signaling, USA), and rabbit polyclonal Bax(Sigma, USA). The secondary antibodies used in our experimentwere obtained from Sigma, and K252a, the Trk family memberantagonist used in our experiment, was obtained from CellSignaling.
4.2. Animals and ischemic model
Adult male Sprague-Dawley rats weighing 200–250 g (ShanghaiExperimental Animal Center, Chinese Academy of Science,Shanghai, China) were given free access to food and waterbefore surgery. All rats were divided into the following groups:the sham group, the I/R group, the HO-1-treated group, and theHO-1-NON-treated group. Each group comprised 6 animals.Sham controls were performed using the same surgical proce-dures, with the exception that the occlusion of carotid arterieswas not performed. The surgical procedures were approved bythe Shanghai Experimental Animal Center. The surgical proce-dures were conducted under the guidelines and terms of allrelevant local legislations. Our best efforts were made tominimize the number of animals used and the suffering thatthey experienced. Transient cerebral ischemia (15 min) wasinduced by four-vessel occlusion (4-VO) as described before (Liet al., 2010; Li et al., 2009; Xu et al., 2010). Briefly, after beinganesthetized with chloral hydrate (350 mg/kg, i.p.), both of thevertebral arteries were occluded permanently by electrocautery,and the common carotid arteries were exposed. Then, the ratswere recovered for 24 h and fasted overnight. To inducecerebral ischemia, aneurysm clips were used to occlude bothof the carotid arteries. After 15min of occlusion, the aneurysmclips were removed for reperfusion. In this process, threemice died.
4.3. Administration
One-hundred micrograms of specific HO-1 overexpression plas-mids were administered to the rats 5 days before ischemiathrough hippocampi CA1 injection (anteroposterior, 3.6 mm;lateral, 2.0 mm; and depth, 4.0 mm from bregma) (Pei et al.,2005). The same dose of nonsense oligonucleotides and vehiclewas used as a control. The locus of HO-1 was BC091164, and itsCDS region was over-expressed (Qi et al., 2012).
4.4. Tissue preparation
After reperfusion under anesthesia, the rats were decapitatedimmediately; then, the hippocampi were removed andquickly frozen in liquid nitrogen. The hippocampi werehomogenized in 1:10 (w/v) ice-cold homogenization buffercontaining 50 mM 3-(N-morpholino) propanesulfonic acid(MOPS; Sigma, St. Louis, MO; pH 7.4), 100 mM KCl, 320 mM

b r a i n r e s e a r c h 1 5 7 7 ( 2 0 1 4 ) 6 9 – 7 6 75
sucrose, 50 mM NaF, 0.5 mM MgCl2, 0.2 mM dithiothreitol(DTT), 1 mM EDTA, 1 mM EGTA, 1 mM Na3VO4 (Sigma),20 mM sodium pyrophosphate, 20 mM β-phosphoglycerol,1 mM p-nitrophenyl phosphate (PNPP), 1 mM benzamidine,1 mM phenylmethylsulfonyl fluoride(PMSF), and 5 μg/ml eachof leupeptin, aprotinin, and pepstatin A. Then, they werecentrifuged at 12,000g for 15 min at 4 1C. The supernatants,including nuclear parts, were collected, and the proteinconcentrations were determined by the Lowry method. Thesamples were stored at �80 1C and were thawed only once.
4.5. Immunoblotting
Proteins were separated on polyacrylamide gels and thenelectrotransferred onto a nitrocellulose membrane (Amer-sham, Buckinghamshire, United Kingdom). The membraneswere blocked for 3 h in Tris-buffered saline with 0.1% Tween20 (TBST) and 3% bovine serum albumin (BSA) and thenincubated overnight at 4 1C with primary antibodies in TBSTcontaining 1% BSA. The membranes were then washed andincubated with alkaline phosphatase-conjugated secondaryantibodies in TBST for 2 h and developed with NBT/BCIP colorsubstrate (Promega, Madison, WI). The densities of the bandson the membrane were scanned and analyzed using animage analyzer (LabWorks Software, Upland, CA).
4.6. Hematoxylin–eosin staining (HE-staining)
The rats subjected to 5 days of reperfusion were perfusion-fixedwith 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4)under anesthesia. The paraffin-embedded brain sections (5 μm)were prepared and stained with hematoxylin and eosin. Histo-logical evaluations were performed with HE-staining for theassessment of neuronal damage in the hippocampus. An initialdissector frame was positioned randomly in the hippocampalsector and cells in every 10th section throughout the entirehippocampus. The cell numbers in the hippocampus wereassessed using previously published unbiased stereologicaltechniques. In brief, cell counts were performed at 400�magnification with the use of an Olympus BH-2 microscopeconnected to a Sony charge-coupled device video camera, amotorized stage system, and commercial stereology software.
4.7. Statistical evaluation
Six animals were independently selected as samples in allgroups for immunoblotting and histology assays. Image J(Version 1.30v) analysis software was used to conduct quan-titative analysis of the bands. Values were expressed asmean7SD. Statistical analysis of the results was carried outby one-way ANOVA followed by Newman–Keuls test. P-values of Po0.05 were considered significant.
Acknowledgments
This work was supported by the foundation of XuzhouMedical College (2011KJ11).
r e f e r e n c e s
Albamonte, M.S., Willis, M.A., Albamonte, M.I., Jensen, F.,Espinosa, M.B., Vitullo, A.D., 2008. The developing humanovary: immunohistochemical analysis of germ-cell-specificVASA protein, BCL-2/BAX expression balance and apoptosis.Hum. Reprod. 23, 1895–1901.
Banerjee, P., Basu, A., Datta, D., Gasser, M., Waaga-Gasser, A.M.,Pal, S., 2011. The heme oxygenase-1 protein is overexpressedin human renal cancer cells following activation of the Ras-Raf-ERK pathway and mediates anti-apoptotic signal. J. Biol.Chem. 286, 33580–33590.
Bejot, Y., Prigent-Tessier, A., Cachia, C., Giroud, M., Mossiat, C.,Bertrand, N., Garnier, P., Marie, C., 2011. Time-dependentcontribution of non neuronal cells to BDNF production afterischemic stroke in rats. Neurochem. Int. 58, 102–111.
Brima, T., Otahal, J., Mares, P., 2013. Increased susceptibility topentetrazol-induced seizures in developing rats after corticalphotothrombotic ischemic stroke at P7. Brain Res. 1507,146–153.
Broughton, B.R., Reutens, D.C., Sobey, C.G., 2009. Apoptoticmechanisms after cerebral ischemia. Stroke 40, e331–e339.
Calabrese, V., Lodi, R., Tonon, C., D’Agata, V., Sapienza, M.,Scapagnini, G., Mangiameli, A., Pennisi, G., Stella, A.M.,Butterfield, D.A., 2005. Oxidative stress, mitochondrialdysfunction and cellular stress response in Friedreich’s ataxia.J. Neurol. Sci. 233, 145–162.
Carty, M.L., Wixey, J.A., Reinebrant, H.E., Gobe, G., Colditz, P.B.,Buller, K.M., 2011. Ibuprofen inhibits neuroinflammation andattenuates white matter damage following hypoxia-ischemiain the immature rodent brain. Brain Res. 1402, 9–19.
Cengiz, P., Uluc, K., Kendigelen, P., Akture, E., Hutchinson, E.,Song, C., Zhang, L., Lee, J., Budoff, G.E., Meyerand, E., Sun, D.,Ferrazzano, P., 2011. Chronic neurological deficits in mice afterperinatal hypoxia and ischemia correlate with hemispherictissue loss and white matter injury detected by MRI. Dev.Neurosci. 33, 270–279.
Choi, B.M., Kim, B.R., 2008. Upregulation of heme oxygenase-1 bybrazilin via the phosphatidylinositol 3-kinase/Akt and ERKpathways and its protective effect against oxidative injury.Eur. J. Pharmacol. 580, 12–18.
Cowansage, K.K., LeDoux, J.E., Monfils, M.H., 2010. Brain-derivedneurotrophic factor: a dynamic gatekeeper of neural plasticity.Curr. Mol. Pharmacol. 3, 12–29.
de Sousa, Oliveira Vanderlei E., de Araujo, I.W., Quindere, A.L.,Fontes, B.P., Eloy, Y.R., Rodrigues, J.A., e Silva, A.A., Chaves, H.V., Jorge, R.J., de Menezes, D.B., Evangelista, J.S., Bezerra, M.M.,Benevides, N.M., 2011. The involvement of the HO-1 pathwayin the anti-inflammatory action of a sulfated polysaccharideisolated from the red seaweed Gracilaria birdiae. Inflamm.Res. 60, 1121–1130.
Fanaei, H., Karimian, S.M., Sadeghipour, H.R., Hassanzade, G.,Kasaeian, A., Attari, F., Khayat, S., Ramezani, V., Javadimehr,M., 2014. Testosterone enhances functional recovery afterstroke through promotion of antioxidant defenses, BDNFlevels and neurogenesis in male rats. Brain Res. 1558,74–83.
Ferraro, E., Pulicati, A., Cencioni, M.T., Cozzolino, M., Navoni, F., diMartino, S., Nardacci, R., Carri, M.T., Cecconi, F., 2008.Apoptosome-deficient cells lose cytochrome c throughproteasomal degradation but survive by autophagy-dependent glycolysis. Mol. Biol. Cell. 19, 3576–3588.
Fontenay, M., Cathelin, S., Amiot, M., Gyan, E., Solary, E., 2006.Mitochondria in hematopoiesis and hematological diseases.Oncogene 25, 4757–4767.
Hankey, G.J., 2006. Potential new risk factors for ischemic stroke:what is their potential? Stroke 37, 2181–2188.

b r a i n r e s e a r c h 1 5 7 7 ( 2 0 1 4 ) 6 9 – 7 676
Harada, S., Fujita-Hamabe, W., Tokuyama, S., 2012. Amelioratingeffect of hypothalamic brain-derived neurotrophic factoragainst impaired glucose metabolism after cerebral ischemicstress in mice. J. Pharmacol. Sci. 118, 109–116.
Hoffmann, A., Zhu, G., Wintermark, M., 2012. Advancedneuroimaging in stroke patients: prediction of tissue fate andhemorrhagic transformation. Expert Rev. Cardiovasc. Ther. 10,515–524.
Hung, S.Y., Liou, H.C., Fu, W.M., 2010. The mechanism of hemeoxygenase-1 action involved in the enhancement ofneurotrophic factor expression. Neuropharmacology 58,321–329.
Kuwana, T., Mackey, M.R., Perkins, G., Ellisman, M.H., Latterich,M., Schneiter, R., Green, D.R., Newmeyer, D.D., 2002. Bid, Bax,and lipids cooperate to form supramolecular openings in theouter mitochondrial membrane. Cell 111, 331–342.
Li, C., Xu, B., Wang, W.W., Yu, X.J., Zhu, J., Yu, H.M., Han, D., Pei, D.S., Zhang, G.Y., 2010. Coactivation of GABA receptors inhibitsthe JNK3 apoptotic pathway via disassembly of GluR6-PSD-95-MLK3 signaling module in KA-induced seizure. Epilepsia 51,391–403.
Li, P., Nijhawan, D., Budihardjo, I., Srinivasula, S.M., Ahmad, M.,Alnemri, E.S., Wang, X., 1997. Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiatesan apoptotic protease cascade. Cell 91, 479–489.
Li, T., Yu, H.M., Sun, Y.F., Song, Y.J., Zhang, G.Y., Pei, D.S., 2009.Inhibition of cerebral ischemia/reperfusion-induced injury byadenovirus expressed C-terminal amino acids of GluR6. BrainRes. 1300, 169–176.
Lin, T.H., Tang, C.H., Hung, S.Y., Liu, S.H., Lin, Y.M., Fu, W.M.,Yang, R.S., 2010. Upregulation of heme oxygenase-1 inhibitsthe maturation and mineralization of osteoblasts. J. CellPhysiol. 222, 757–768.
Majewski, N., Nogueira, V., Robey, R.B., Hay, N., 2004. Akt inhibitsapoptosis downstream of BID cleavage via a glucose-dependent mechanism involving mitochondrial hexokinases.Mol. Cell Biol. 24, 730–740.
Mercier, G., Lennon, A.M., Renouf, B., Dessouroux, A., Ramauge,M., Courtin, F., Pierre, M., 2004. MAP kinase activation byfluoxetine and its relation to gene expression in cultured ratastrocytes. J. Mol. Neurosci. 24, 207–216.
Morita, K., Lee, M.S., Her, S., 2009. Possible relation of hemin-induced HO-1 expression to the upregulation of VEGF andBDNF mRNA levels in rat C6 glioma cells. J. Mol. Neurosci. 38,31–40.
Morse, D., Lin, L., Choi, A.M., Ryter, S.W., 2009. Heme oxygenase-1, a critical arbitrator of cell death pathways in lung injury anddisease. Free Radic. Biol. Med. 47, 1–12.
Pachori, A.S., Smith, A., McDonald, P., Zhang, L., Dzau, V.J., Melo,L.G., 2007. Heme-oxygenase-1-induced protection againsthypoxia/reoxygenation is dependent on biliverdin reductaseand its interaction with PI3K/Akt pathway. J. Mol. Cell Cardiol.43, 580–592.
Patapoutian, A., Reichardt, L.F., 2001. Trk receptors: mediators ofneurotrophin action. Curr. Opin. Neurobiol. 11, 272–280.
Pei, D.S., Guan, Q.H., Sun, Y.F., Zhang, Q.X., Xu, T.L., Zhang, G.Y.,2005. Neuroprotective effects of GluR6 antisenseoligodeoxynucleotides on transient brain ischemia/reperfusion-induced neuronal death in rat hippocampal CA1region. J. Neurosci. Res. 82, 642–649.
Ploughman, M., Windle, V., Mac, L, ellan, C.L., White, N., Dore, J.J.,Corbett, D., 2009. Brain-derived neurotrophic factorcontributes to recovery of skilled reaching after focal ischemiain rats. Stroke 40, 1490–1495.
Qi, D., Liu, H., Niu, J., Fan, X., Wen, X., Du, Y., Mou, J., Pei, D., Liu,Z., Zong, Z., Wei, X., Song, Y., 2012. Heat shock protein 72inhibits c-Jun N-terminal kinase 3 signaling pathway via Akt1during cerebral ischemia. J. Neurol. Sci. 317, 123–129.
Saha, R.N., Liu, X., Pahan, K., 2006. Up-regulation of BDNF inastrocytes by TNF-alpha: a case for the neuroprotective role ofcytokine. J. Neuroimmune Pharmacol. 1, 212–222.
Sun, X., Zhou, H., Luo, X., Li, S., Yu, D., Hua, J., Mu, D., Mao, M.,2008. Neuroprotection of brain-derived neurotrophic factoragainst hypoxic injury in vitro requires activation ofextracellular signal-regulated kinase and phosphatidylinositol3-kinase. Int. J. Dev. Neurosci. 26, 363–370.
Vakili, A., Sharifat, S., Akhavan, M.M., Bandegi, A.R., 2014. Effectof lavender oil (Lavandula angustifolia) on cerebral edema andits possible mechanisms in an experimental model of stroke.Brain Res. 1548, 56–62.
Xu, J., Liu, Z.A., Pei, D.S., Xu, T.J., 2010. Calcium/calmodulin-dependent kinase II facilitated GluR6 subunit serinephosphorylation through GluR6-PSD95-CaMKII signalingmodule assembly in cerebral ischemia injury. Brain Res. 1366,197–203.
Yao, H., Takasawa, R., Fukuda, K., Shiokawa, D., Sadanaga-Akiyoshi, F., Ibayashi, S., Tanuma, S., Uchimura, H., 2001. DNAfragmentation in ischemic core and penumbra in focalcerebral ischemia in rats. Brain Res. Mol. Brain Res. 91,112–118.

本文献由“学霸图书馆-文献云下载”收集自网络,仅供学习交流使用。
学霸图书馆(www.xuebalib.com)是一个“整合众多图书馆数据库资源,
提供一站式文献检索和下载服务”的24 小时在线不限IP
图书馆。
图书馆致力于便利、促进学习与科研,提供最强文献下载服务。
图书馆导航:
图书馆首页 文献云下载 图书馆入口 外文数据库大全 疑难文献辅助工具





























