Embed Size (px)
Citation preview

INFECTION AND IMMUNITY, Dec. 2009, p. 5612–5622 Vol. 77, No. 120019-9567/09/$12.00 doi:10.1128/IAI.00618-09Copyright © 2009, American Society for Microbiology. All Rights Reserved.
Toll-Like Receptor Signaling in AirborneBurkholderia thailandensis Infection�
T. Eoin West,1* Thomas R. Hawn,2 and Shawn J. Skerrett1
Division of Pulmonary and Critical Care Medicine1 and Division of Allergy and Infectious Diseases,2
Department of Medicine, University of Washington School of Medicine, Seattle, Washington
Received 1 June 2009/Returned for modification 2 July 2009/Accepted 14 September 2009
Melioidosis is a tropical disease endemic in southeast Asia and northern Australia caused by the gram-negative soil saprophyte Burkholderia pseudomallei. Although infection is often systemic, the lung is frequentlyinvolved. B. thailandensis is a closely related organism that at high doses causes lethal pneumonia in mice. Weexamined the role of Toll-like receptors (TLRs), essential components of innate immunity, in vitro and in vivoduring murine B. thailandensis pneumonia. TLR2, TLR4, and TLR5 mediate NF-�B activation by B. thailan-densis in transfected HEK293 or CHO cells. In macrophages, TLR4 and the adaptor molecule MyD88, but notTLR2 or TLR5, are required for tumor necrosis factor alpha production induced by B. thailandensis. Inlow-dose airborne infection, TLR4 is needed for early, but not late, bacterial containment, and MyD88 isessential for control of infection and host survival. TLR2 and TLR5 are not necessary to contain low-doseinfection. In high-dose airborne infection, TLR2 deficiency confers a slight survival advantage. Lung andsystemic inflammatory responses are induced by low-dose inhaled B. thailandensis independently of individualTLRs or MyD88. These findings suggest that redundancy in TLR signaling or other MyD88-dependentpathways may be important in pneumonic B. thailandensis infection but that MyD88-independent mechanismsof inflammation are also activated. TLR signaling in B. thailandensis infection is substantially comparable tosignaling induced by virulent B. pseudomallei. These studies provide additional insights into the host-pathogeninteraction in pneumonic Burkholderia infection.
Burkholderia pseudomallei is a gram-negative, flagellated soilsaprophyte that causes the tropical disease melioidosis. En-demic in southeast Asia and northern Australia, melioidosisresults from the inhalation or cutaneous inoculation of B.pseudomallei. Although there is a myriad of clinical presenta-tions, the lung is the organ most commonly involved (30). B.pseudomallei is considered a CDC category B pathogen be-cause of concerns about its use as a bioweapon (5). Burkhold-eria thailandensis is a closely related organism that also isisolated from soil in the tropics (24) and is largely avirulent tohumans but is lethal at high doses to mice (28, 29). Study of B.thailandensis does not require the strict biocontainment con-ditions necessary for research with B. pseudomallei, and be-cause of its genomic similarity with B. pseudomallei (33), hasbeen used as a surrogate organism to study melioidosis.
Toll-like receptors (TLRs) are membrane-associated patho-gen recognition receptors that provide essential host defenseagainst infection by inducing nuclear transcription factor(NF)-�B translocation and activation of a proinflammatoryresponse (17). TLR2, in conjunction with TLR1 or TLR6, isstimulated by bacterial cell wall lipopeptides and peptidogly-can. TLR4, in association with coreceptors CD14 and MD-2,recognizes the lipid A component of lipopolysaccharide (LPS)of most gram-negative organisms. TLR5 is activated by bacte-rial flagellin. All these TLRs signal via adaptor moleculeMyD88, although TLR4 may alternatively signal through
TRIF. Various TLRs and their downstream adaptor moleculesare important in both experimental and human bacterial lunginfections (10, 14, 18, 19, 21). We and others have previouslyshown that TLR2 and TLR4 mediate host recognition of B.pseudomallei in vitro (15, 27, 31), and we have shown that B.pseudomallei LPS (and subcomponent lipid A) is a TLR4 ag-onist (27). However, despite the lethality of B. thailandensiswhen aerosolized at high doses to mice (28), little is knownabout innate immunity in B. thailandensis infection.
In this study of B. thailandensis infection, we examined theroles of three TLRs that recognize conserved motifs of gram-negative, flagellated bacteria—TLR2, TLR4, and TLR5—aswell as the TLR adaptor molecule MyD88. In vitro, we foundthat TLR2, TLR4, and TLR5 recognize B. thailandensis andmediate activation of NF-�B, but that only TLR4 or MyD88deficiency impairs tumor necrosis factor alpha (TNF-�) secre-tion by macrophages. In vivo, TLR4 is required only for earlybacterial control in low-dose infection. Absence of TLR2 pro-longs survival slightly after high-dose infection. MyD88 defi-ciency results in exponential bacterial replication, dissemina-tion, and death. Lack of TLR2, TLR4, TLR5, or MyD88 doesnot impair the host inflammatory response to inhaled B. thai-landensis.
MATERIALS AND METHODS
Bacterial strains and growth conditions. Burkholderia thailandensis E264 wasprovided by Donald Woods (University of Calgary). Bacteria were grown fromfrozen glycerol stock in LB broth at 37°C for 9 (log phase) or 18 (stationaryphase) hours, isolated by centrifugation, washed twice, and suspended in Dul-becco’s phosphate-buffered saline (PBS) to the desired concentration. An opticaldensity of 0.20 at 600 nm yielded approximately 1 � 108 CFU/ml. Bacteria wereheat-killed by heating to 65°C for 40 to 45 min.
* Corresponding author. Mailing address: Division of Pulmonaryand Critical Care Medicine, Harborview Medical Center, Box 359640,325 9th Ave., Seattle, WA 98104. Phone: (206) 897-5271. Fax: (206)897-5392. E-mail: [email protected].
� Published ahead of print on 21 September 2009.
5612
on May 22, 2020 by guest
http://iai.asm.org/
Dow
nloaded from

Cell transfections and stimulations. HEK293 cells were used as previouslydescribed (27). Cells were cultured in a 96-well flat-bottomed tissue culture plateat �5 � 104 cells/well in Dulbecco’s modified Eagle’s medium plus 10% fetalbovine serum (FBS). The following day, cells were transiently transfected with 5�l of transfection reagent comprised of a 1:1 mix of 0.25 M CaCl2 containing 2�BBS buffer [50 mM N,N-bis(2-hydroxyethyl)-2-aminoethanesulfonic acid (BES),280 mM NaCl, and 1.5 mM NaH2PO4] and the following DNA: NF-�B-depen-dent firefly ELAM luciferase and control �-actin-dependent Renilla luciferase,and murine CD14 and MD-2, with TLR2, TLR2 and TLR1, TLR2 and TLR6, orTLR4. CD14 enhances TLR2-dependent responses to B. pseudomallei (31) and,when cotransfected with CD14, MD-2 is necessary for TLR4-dependent signal-ing (27). For simplicity, both coreceptors were transfected together with eitherTLR2 or TLR4. DNA was added in the following amounts to each well: 0.0025�g MD-2, 0.0025 �g CD14, 0.01 �g ELAM luciferase, and 0.0003 �g Renillaluciferase. When transfected alone, the following amounts of DNA were added:0.0025 �g TLR2 or 0.0003 �g TLR4. When cotransfected with TLR1 or TLR6,0.0025 �g TLR2 was used in addition to 0.025 �g TLR1 or 0.0025 �g TLR6. Alltransfections were normalized to 0.05 �g total DNA with the addition of anempty vector. Transfected cells were washed once after 4 hours and were stim-ulated the following day with experimental ligands, nonspecific stimulus recom-binant human interleukin-1� (IL-1�; Pierce Endogen, Rockford, IL), controlTLR4 ligand ultrapure Escherichia coli O111:B4 LPS (Invivogen, San Diego,CA), or control TLR2 ligand Pam3CSK4 (Invivogen and EMC Microcollections,Tubingen, Germany). After 4 hours, cells were lysed with passive lysis buffer(Promega, Madison, WI), and NF-�B activation was determined in 10 �l oflysate by the ratio of firefly to Renilla luciferase light emission by the use of theDual-Luciferase reporter system (Promega, Madison, WI). CHO cells stablytransfected with human TLR5 were used as previously described (14). Cellpopulations expressing TLR5, NF-�B-dependent firefly ELAM luciferase, andcontrol thymidine kinase-driven Renilla luciferase maintained in Ham’s F-12medium with L-glutamine, 10% FBS, and 100 �g/ml G418 were washed andadded onto a 96-well plate at �6 � 104 cells/well before stimulation the followingday with experimental ligands, recombinant human IL-1�, E. coli O111:B4 LPS,or control TLR5 ligand Salmonella enterica serovar Typhimurium FliC flagellinpurified as previously described (12). After 5 hours, cells were lysed, and NF-�Bactivation was determined as described above.
Animals. TLR2�/�, TLR4�/�, TLR5�/�, and MyD88�/� mice were derivedby Shizuo Akira (University of Osaka) and backcrossed for 8 to 10 generationsto C57BL/6 (16, 25, 26). Specific-pathogen-free C57BL/6 mice were obtainedfrom Jackson Laboratories (Bar Harbor, ME). All animals were housed inlaminar flow cages and were permitted ad lib access to sterile food and water.Euthanasia was performed by injection of intraperitoneal pentobarbital followedby exsanguination from a cardiac puncture. The Institutional Animal Care andUse Committee of the University of Washington approved all experimentalprocedures.
Macrophage stimulations. Femurs and tibias were harvested under sterileconditions from euthanized mice. Marrow was flushed out using a 26-gaugeneedle through a 0.2-�m strainer and cultured in petri dishes in RPMI mediumsupplemented with 1% L-glutamine, 100 IU/ml penicillin, 100 �g/ml streptomy-cin, 10% FBS, and 20% L929 cell-conditioned medium at 37°C under 5% CO2
for 5 to 10 days to allow macrophages to predominate. The monolayer waswashed twice with Hank’s balanced salt solution or medium, and macrophageswere resuspended in RPMI medium supplemented with 1% L-glutamine, 1%HEPES, and 10% FBS. Cells were added to a 96-well flat-bottomed tissueculture plate at 3 � 104 to 2 � 105 cells/well, depending on the experiment. Thefollowing day, cells were stimulated with experimental ligands in fresh mediumadded to each well. Alveolar macrophages were harvested by cannulating thetracheas of euthanized mice with a 20-gauge catheter and lavaging both lungswith 4 ml 0.9% NaCl-0.6 mM EDTA in divided aliquots. Cells were pelleted bycentrifugation, resuspended in RPMI medium supplemented with 1% L-glu-tamine, 1% HEPES, 100 IU/ml penicillin, 100 �g/ml streptomycin, and 10% FBSand were added to a 96-well plate at 7 � 104 cells/well. The following day, cellswere washed three times and stimulated with experimental ligands or controlSalmonella serovar Typhimurium FliC flagellin in fresh antibiotic-free medium.After stimulation for 16 to 24 h, macrophage supernatants were removed andstored at �80°C until assayed.
Murine model of pneumonia. Mice were exposed to aerosolized bacteria bythe use of a snout-only inhalation system (In-Tox Products, Moriarty, NM) aspreviously described (28). Aerosols were generated from a MiniHeart Hi-Flonebulizer (Westmed, Tucson, AZ) driven at 40 lb/in2. Airflow through the systemwas maintained for 10 min at 24 liters/min followed by 5 minutes purge with air.Bacterial deposition in each experiment was determined from a quantitativeculture of lung tissue from sentinel mice sacrificed immediately after infection.
Animals were examined one to three times daily for illness or death, and ab-dominal surface temperatures were measured using a Ranger MX4P digitalinfrared thermometer (Raytek, Santa Cruz, CA). Ill animals with temperaturesof �22.5°C or ruffled fur, eye crusting, hunched posture, and lack of resistanceto handling were deemed terminal and were euthanized (spontaneous death wasnot required as an endpoint). At specific time points after infection, mice weresacrificed, and the left pulmonary hilum was tied off; the left lung, medianhepatic lobe, and spleen were each homogenized in 1 ml sterile Dulbecco’s PBS,and serial dilutions were plated onto LB agar. Colonies were counted after 2 to4 days of incubation at 37°C in humid air under 5% CO2. Bronchoalveolar lavage(BAL) was performed by cannulating the trachea with a 20-gauge catheter andlavaging the right lung with four 0.5-ml aliquots of 0.9% NaCl-0.6 mM EDTA.Cell counts in BAL fluid specimens were measured in a hemocytometer. Differ-entials were determined from examination of cytocentrifuge slides (ThermoShandon, Pittsburgh, PA) that were stained with a modified Wright-Giemsatechnique (Diff-Quik; Dade Behring, Dudingen, Switzerland). Left lung homog-enates in Dulbecco’s PBS were diluted 1:1 in lysis buffer containing 2� proteaseinhibitor cocktail (Roche Diagnostics, Mannheim, Germany), incubated on icefor 30 min, and then centrifuged at 1,500 � g. Supernatants were harvested andstored at �80°C until assayed for cytokines. Whole blood was centrifuged, andserum was removed and stored at �80°C until assayed.
Measurements of cytokines. TNF-� was quantified in the supernatants ofstimulated macrophages by using the DuoSet enzyme-linked immunosorbentassay (R&D Systems, Minneapolis, MN). TNF-�, IL-1�, IL-6, KC, monocytechemoattractant protein 1 (MCP-1), and MIP-2 levels in lung homogenates andsera were measured using a multiplex bead assay (Luminex, Austin, TX) andreagents purchased from R&D Systems.
Statistical analyses. Combined data that follow a normal distribution arereported as mean standard deviation. Comparisons between two groups ofnormally distributed data were made using the t test and between three or moregroups by using analysis of variance (ANOVA) and either the Dunnett orBonferroni post-tests. Data displayed on a logarithmic scale in Fig. 4 to 7 werefirst transformed to log10 before testing. Survival analyses were performed withthe log rank test. All statistics were performed with Stata 9.0 (College Station,TX) or GraphPad Prism 4.0 (San Diego, CA). A two-sided P value of �0.05 wasconsidered significant.
RESULTS
TLR2 and TLR4 mediate activation of NF-�B by B. thailan-densis. To determine whether B. thailandensis stimulates TLR2and TLR4, we transfected HEK293 cells with TLR2, TLR2/1,TLR2/6, or TLR4, and CD14/MD-2. We stimulated the cellswith stationary-phase, heat-killed B. thailandensis and quanti-fied NF-�B activation by using a luciferase reporter assay (Fig.1A). We observed 2.4- to 7.4-fold TLR2- and 6.2- to 8.8-foldTLR4-mediated induction of NF-�B activation upon stimula-tion with 105 to 107 CFU/ml B. thailandensis. The responseswere dose dependent, and TLR2-mediated signaling was en-hanced by cotransfection of either TLR1 or TLR6.
TLR5 mediates activation of NF-�B by B. thailandensis. Todetermine whether B. thailandensis activates NF-�B in aTLR5-dependent manner, we stimulated TLR5-transfectedCHO cells with heat-killed B. thailandensis and quantifiedNF-�B activation by light emission (Fig. 1B). Because of con-cern that B. pseudomallei may lose or reduce expression offlagella at stationary phase (Sharon Peacock, personal commu-nication), we used both log- and stationary-phase B. thailan-densis cells. We detected NF-�B activation in transfected cellsin response to both preparations of B. thailandensis, indicatingthat TLR5 mediates recognition of B. thailandensis. The signalwas more robust on average (16.6- to 21.4-fold) but morevariable (2.8- to 17.5-fold) in response to 105 to 107 CFU/mllog-phase bacteria than to 107 to 109 CFU/ml stationary-phasebacteria, suggesting diminished presence of flagellin in station-ary-phase organisms. Similar TLR5-dependent activation of
VOL. 77, 2009 TOLL-LIKE RECEPTORS IN B. THAILANDENSIS PNEUMONIA 5613
on May 22, 2020 by guest
http://iai.asm.org/
Dow
nloaded from
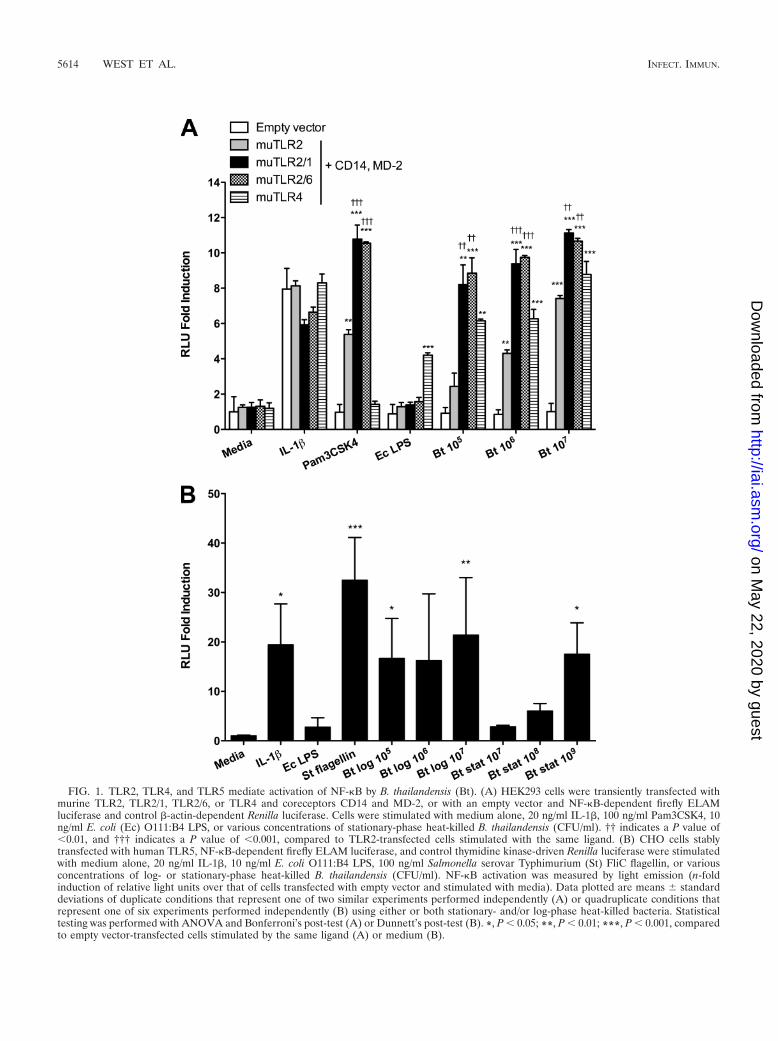
FIG. 1. TLR2, TLR4, and TLR5 mediate activation of NF-�B by B. thailandensis (Bt). (A) HEK293 cells were transiently transfected withmurine TLR2, TLR2/1, TLR2/6, or TLR4 and coreceptors CD14 and MD-2, or with an empty vector and NF-�B-dependent firefly ELAMluciferase and control �-actin-dependent Renilla luciferase. Cells were stimulated with medium alone, 20 ng/ml IL-1�, 100 ng/ml Pam3CSK4, 10ng/ml E. coli (Ec) O111:B4 LPS, or various concentrations of stationary-phase heat-killed B. thailandensis (CFU/ml). †† indicates a P value of�0.01, and ††† indicates a P value of �0.001, compared to TLR2-transfected cells stimulated with the same ligand. (B) CHO cells stablytransfected with human TLR5, NF-�B-dependent firefly ELAM luciferase, and control thymidine kinase-driven Renilla luciferase were stimulatedwith medium alone, 20 ng/ml IL-1�, 10 ng/ml E. coli O111:B4 LPS, 100 ng/ml Salmonella serovar Typhimurium (St) FliC flagellin, or variousconcentrations of log- or stationary-phase heat-killed B. thailandensis (CFU/ml). NF-�B activation was measured by light emission (n-foldinduction of relative light units over that of cells transfected with empty vector and stimulated with media). Data plotted are means standarddeviations of duplicate conditions that represent one of two similar experiments performed independently (A) or quadruplicate conditions thatrepresent one of six experiments performed independently (B) using either or both stationary- and/or log-phase heat-killed bacteria. Statisticaltesting was performed with ANOVA and Bonferroni’s post-test (A) or Dunnett’s post-test (B). *, P � 0.05; **, P � 0.01; ***, P � 0.001, comparedto empty vector-transfected cells stimulated by the same ligand (A) or medium (B).
5614 WEST ET AL. INFECT. IMMUN.
on May 22, 2020 by guest
http://iai.asm.org/
Dow
nloaded from
NF-�B was observed upon stimulation with live B. thailanden-sis (data not shown).
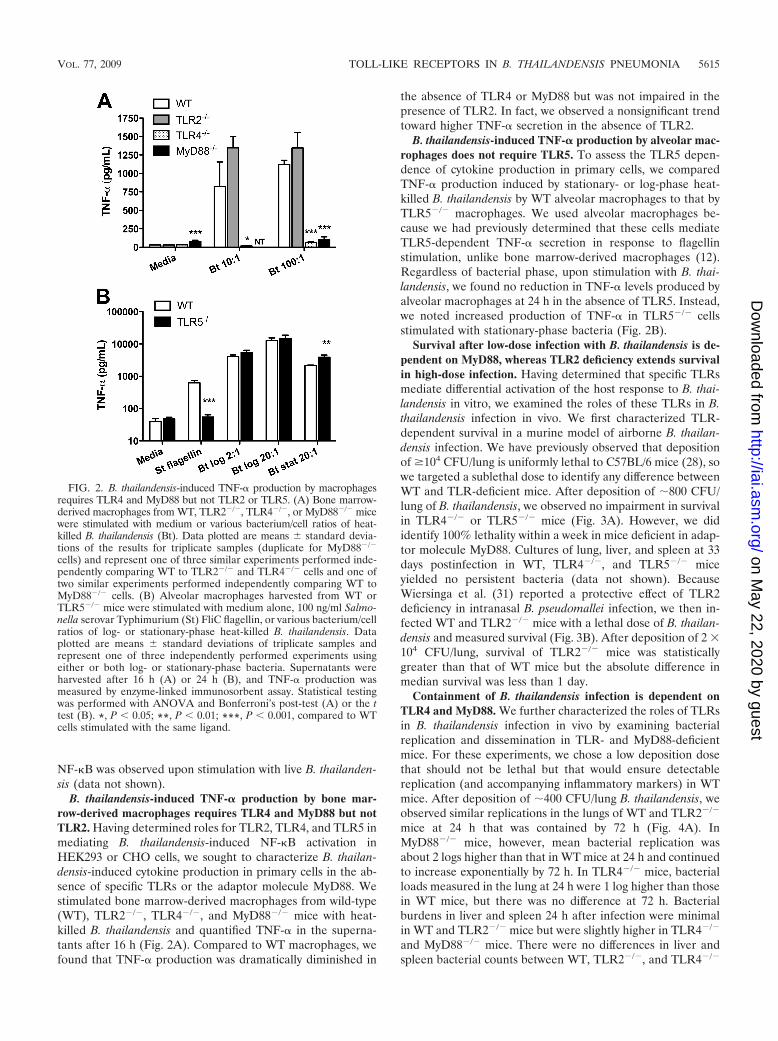
B. thailandensis-induced TNF-� production by bone mar-row-derived macrophages requires TLR4 and MyD88 but notTLR2. Having determined roles for TLR2, TLR4, and TLR5 inmediating B. thailandensis-induced NF-�B activation inHEK293 or CHO cells, we sought to characterize B. thailan-densis-induced cytokine production in primary cells in the ab-sence of specific TLRs or the adaptor molecule MyD88. Westimulated bone marrow-derived macrophages from wild-type(WT), TLR2�/�, TLR4�/�, and MyD88�/� mice with heat-killed B. thailandensis and quantified TNF-� in the superna-tants after 16 h (Fig. 2A). Compared to WT macrophages, wefound that TNF-� production was dramatically diminished in
the absence of TLR4 or MyD88 but was not impaired in thepresence of TLR2. In fact, we observed a nonsignificant trendtoward higher TNF-� secretion in the absence of TLR2.
B. thailandensis-induced TNF-� production by alveolar mac-rophages does not require TLR5. To assess the TLR5 depen-dence of cytokine production in primary cells, we comparedTNF-� production induced by stationary- or log-phase heat-killed B. thailandensis by WT alveolar macrophages to that byTLR5�/� macrophages. We used alveolar macrophages be-cause we had previously determined that these cells mediateTLR5-dependent TNF-� secretion in response to flagellinstimulation, unlike bone marrow-derived macrophages (12).Regardless of bacterial phase, upon stimulation with B. thai-landensis, we found no reduction in TNF-� levels produced byalveolar macrophages at 24 h in the absence of TLR5. Instead,we noted increased production of TNF-� in TLR5�/� cellsstimulated with stationary-phase bacteria (Fig. 2B).
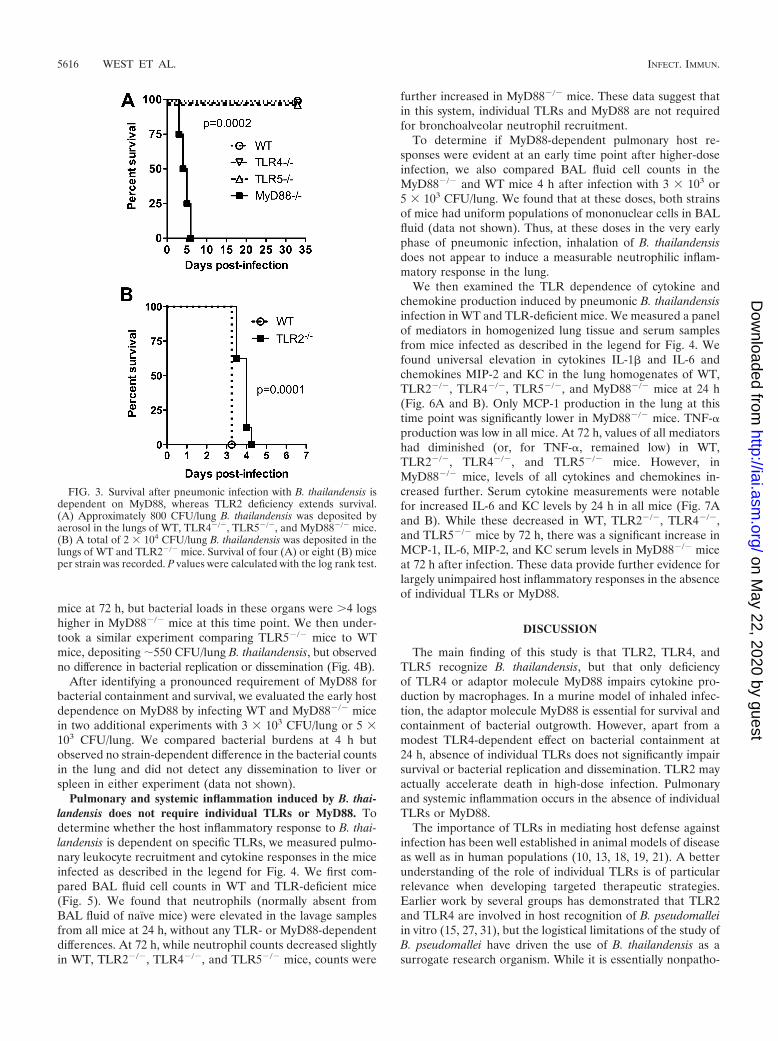
Survival after low-dose infection with B. thailandensis is de-pendent on MyD88, whereas TLR2 deficiency extends survivalin high-dose infection. Having determined that specific TLRsmediate differential activation of the host response to B. thai-landensis in vitro, we examined the roles of these TLRs in B.thailandensis infection in vivo. We first characterized TLR-dependent survival in a murine model of airborne B. thailan-densis infection. We have previously observed that depositionof �104 CFU/lung is uniformly lethal to C57BL/6 mice (28), sowe targeted a sublethal dose to identify any difference betweenWT and TLR-deficient mice. After deposition of �800 CFU/lung of B. thailandensis, we observed no impairment in survivalin TLR4�/� or TLR5�/� mice (Fig. 3A). However, we dididentify 100% lethality within a week in mice deficient in adap-tor molecule MyD88. Cultures of lung, liver, and spleen at 33days postinfection in WT, TLR4�/�, and TLR5�/� miceyielded no persistent bacteria (data not shown). BecauseWiersinga et al. (31) reported a protective effect of TLR2deficiency in intranasal B. pseudomallei infection, we then in-fected WT and TLR2�/� mice with a lethal dose of B. thailan-densis and measured survival (Fig. 3B). After deposition of 2 �104 CFU/lung, survival of TLR2�/� mice was statisticallygreater than that of WT mice but the absolute difference inmedian survival was less than 1 day.
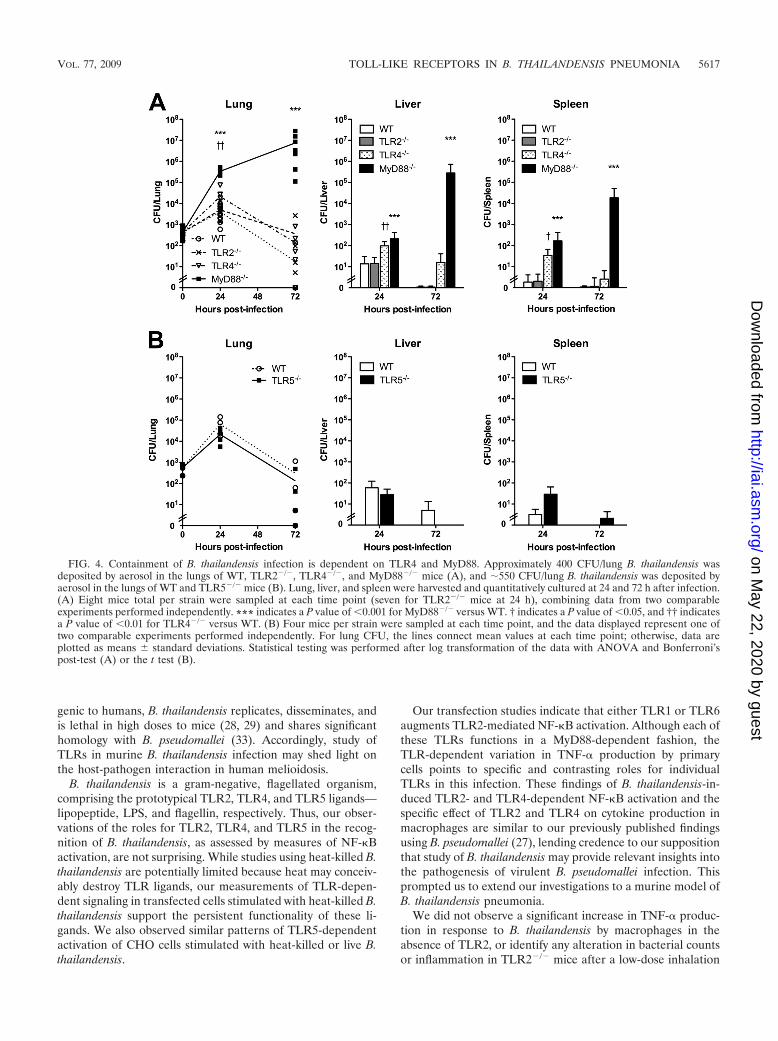
Containment of B. thailandensis infection is dependent onTLR4 and MyD88. We further characterized the roles of TLRsin B. thailandensis infection in vivo by examining bacterialreplication and dissemination in TLR- and MyD88-deficientmice. For these experiments, we chose a low deposition dosethat should not be lethal but that would ensure detectablereplication (and accompanying inflammatory markers) in WTmice. After deposition of �400 CFU/lung B. thailandensis, weobserved similar replications in the lungs of WT and TLR2�/�
mice at 24 h that was contained by 72 h (Fig. 4A). InMyD88�/� mice, however, mean bacterial replication wasabout 2 logs higher than that in WT mice at 24 h and continuedto increase exponentially by 72 h. In TLR4�/� mice, bacterialloads measured in the lung at 24 h were 1 log higher than thosein WT mice, but there was no difference at 72 h. Bacterialburdens in liver and spleen 24 h after infection were minimalin WT and TLR2�/� mice but were slightly higher in TLR4�/�
and MyD88�/� mice. There were no differences in liver andspleen bacterial counts between WT, TLR2�/�, and TLR4�/�
FIG. 2. B. thailandensis-induced TNF-� production by macrophagesrequires TLR4 and MyD88 but not TLR2 or TLR5. (A) Bone marrow-derived macrophages from WT, TLR2�/�, TLR4�/�, or MyD88�/� micewere stimulated with medium or various bacterium/cell ratios of heat-killed B. thailandensis (Bt). Data plotted are means standard devia-tions of the results for triplicate samples (duplicate for MyD88�/�
cells) and represent one of three similar experiments performed inde-pendently comparing WT to TLR2�/� and TLR4�/� cells and one oftwo similar experiments performed independently comparing WT toMyD88�/� cells. (B) Alveolar macrophages harvested from WT orTLR5�/� mice were stimulated with medium alone, 100 ng/ml Salmo-nella serovar Typhimurium (St) FliC flagellin, or various bacterium/cellratios of log- or stationary-phase heat-killed B. thailandensis. Dataplotted are means standard deviations of triplicate samples andrepresent one of three independently performed experiments usingeither or both log- or stationary-phase bacteria. Supernatants wereharvested after 16 h (A) or 24 h (B), and TNF-� production wasmeasured by enzyme-linked immunosorbent assay. Statistical testingwas performed with ANOVA and Bonferroni’s post-test (A) or the ttest (B). *, P � 0.05; **, P � 0.01; ***, P � 0.001, compared to WTcells stimulated with the same ligand.
VOL. 77, 2009 TOLL-LIKE RECEPTORS IN B. THAILANDENSIS PNEUMONIA 5615
on May 22, 2020 by guest
http://iai.asm.org/
Dow
nloaded from
mice at 72 h, but bacterial loads in these organs were 4 logshigher in MyD88�/� mice at this time point. We then under-took a similar experiment comparing TLR5�/� mice to WTmice, depositing �550 CFU/lung B. thailandensis, but observedno difference in bacterial replication or dissemination (Fig. 4B).
After identifying a pronounced requirement of MyD88 forbacterial containment and survival, we evaluated the early hostdependence on MyD88 by infecting WT and MyD88�/� micein two additional experiments with 3 � 103 CFU/lung or 5 �103 CFU/lung. We compared bacterial burdens at 4 h butobserved no strain-dependent difference in the bacterial countsin the lung and did not detect any dissemination to liver orspleen in either experiment (data not shown).
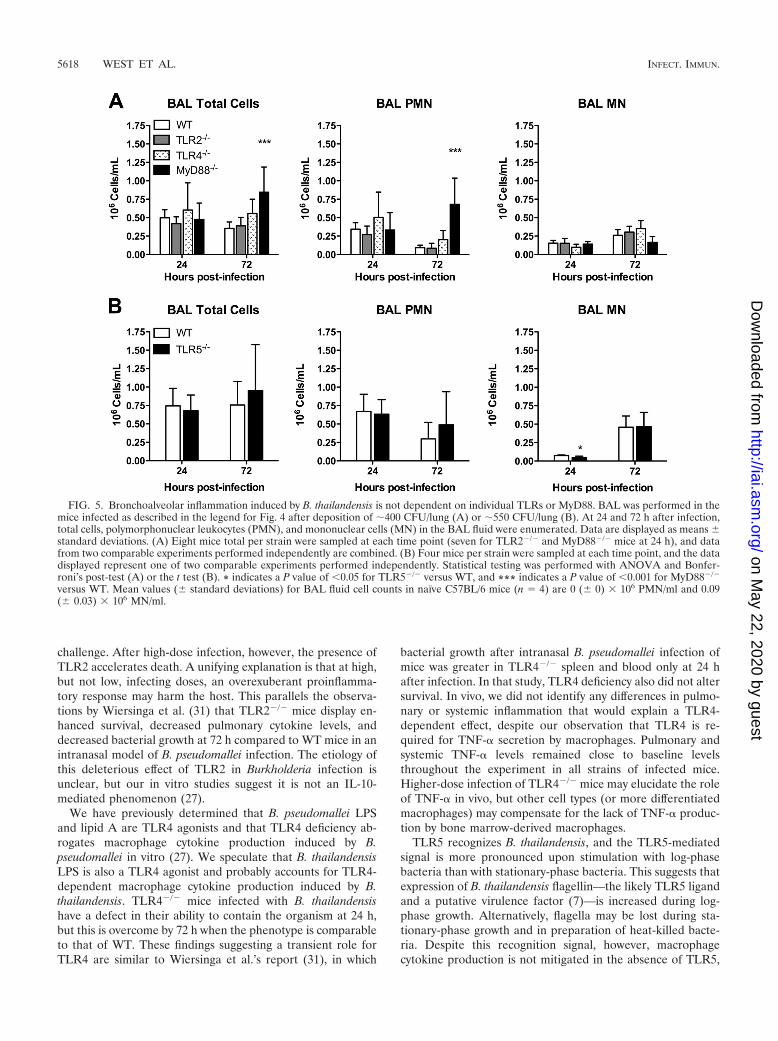
Pulmonary and systemic inflammation induced by B. thai-landensis does not require individual TLRs or MyD88. Todetermine whether the host inflammatory response to B. thai-landensis is dependent on specific TLRs, we measured pulmo-nary leukocyte recruitment and cytokine responses in the miceinfected as described in the legend for Fig. 4. We first com-pared BAL fluid cell counts in WT and TLR-deficient mice(Fig. 5). We found that neutrophils (normally absent fromBAL fluid of naïve mice) were elevated in the lavage samplesfrom all mice at 24 h, without any TLR- or MyD88-dependentdifferences. At 72 h, while neutrophil counts decreased slightlyin WT, TLR2�/�, TLR4�/�, and TLR5�/� mice, counts were
further increased in MyD88�/� mice. These data suggest thatin this system, individual TLRs and MyD88 are not requiredfor bronchoalveolar neutrophil recruitment.
To determine if MyD88-dependent pulmonary host re-sponses were evident at an early time point after higher-doseinfection, we also compared BAL fluid cell counts in theMyD88�/� and WT mice 4 h after infection with 3 � 103 or5 � 103 CFU/lung. We found that at these doses, both strainsof mice had uniform populations of mononuclear cells in BALfluid (data not shown). Thus, at these doses in the very earlyphase of pneumonic infection, inhalation of B. thailandensisdoes not appear to induce a measurable neutrophilic inflam-matory response in the lung.
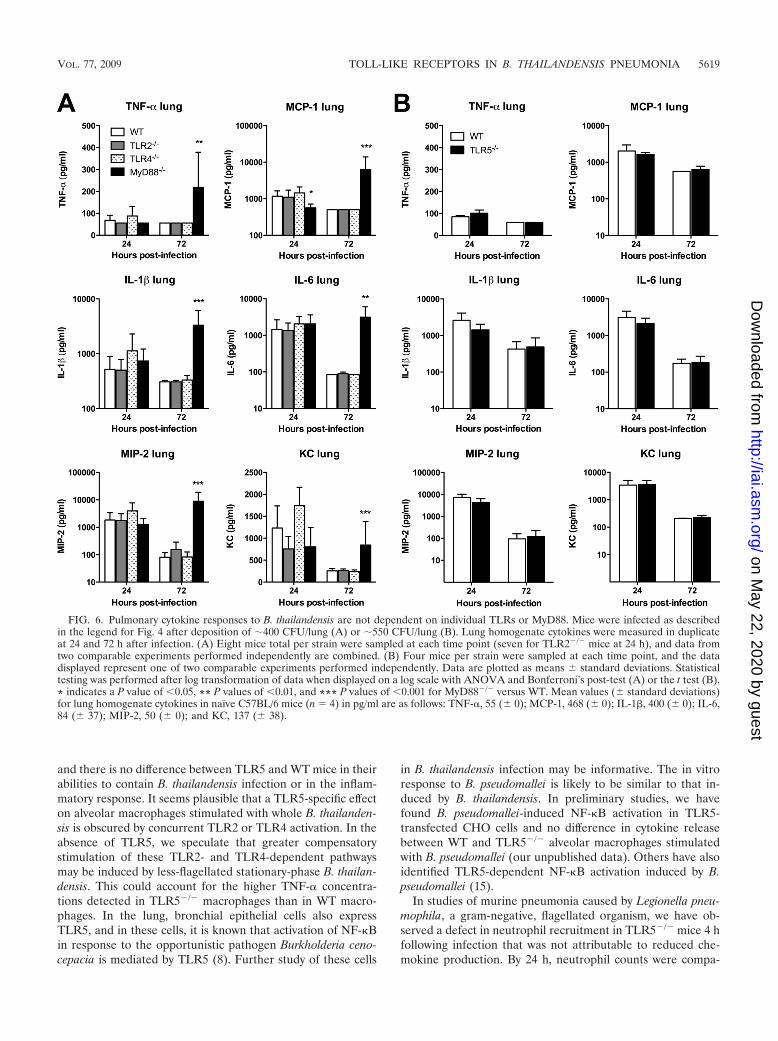
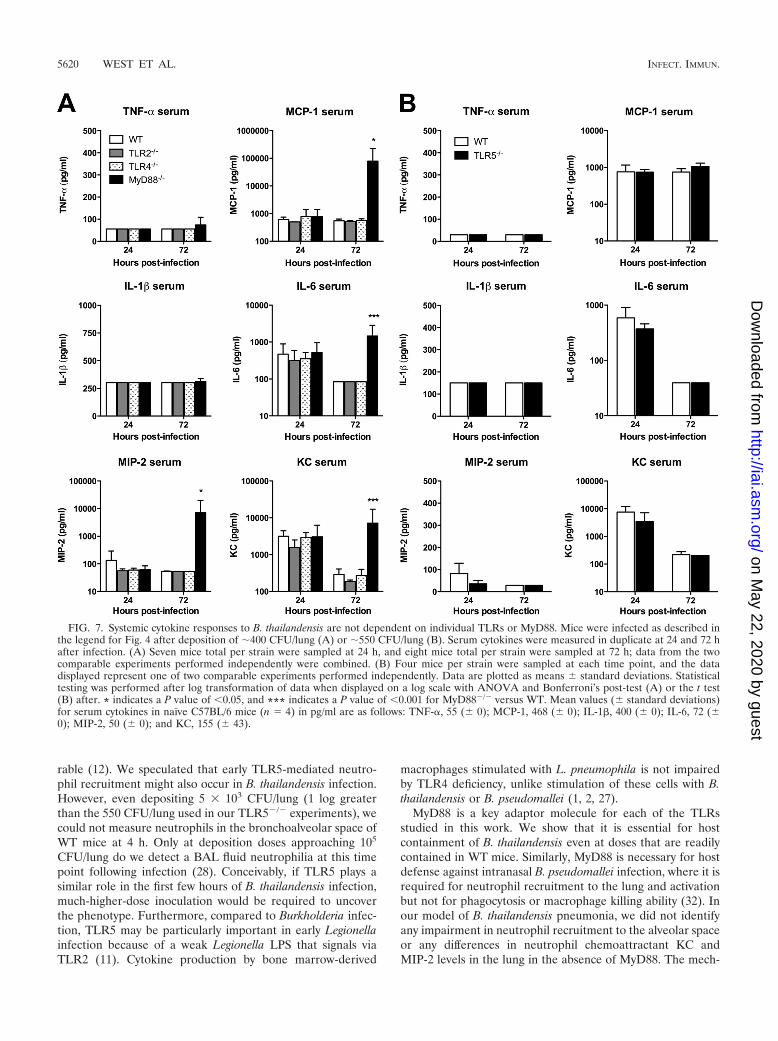
We then examined the TLR dependence of cytokine andchemokine production induced by pneumonic B. thailandensisinfection in WT and TLR-deficient mice. We measured a panelof mediators in homogenized lung tissue and serum samplesfrom mice infected as described in the legend for Fig. 4. Wefound universal elevation in cytokines IL-1� and IL-6 andchemokines MIP-2 and KC in the lung homogenates of WT,TLR2�/�, TLR4�/�, TLR5�/�, and MyD88�/� mice at 24 h(Fig. 6A and B). Only MCP-1 production in the lung at thistime point was significantly lower in MyD88�/� mice. TNF-�production was low in all mice. At 72 h, values of all mediatorshad diminished (or, for TNF-�, remained low) in WT,TLR2�/�, TLR4�/�, and TLR5�/� mice. However, inMyD88�/� mice, levels of all cytokines and chemokines in-creased further. Serum cytokine measurements were notablefor increased IL-6 and KC levels by 24 h in all mice (Fig. 7Aand B). While these decreased in WT, TLR2�/�, TLR4�/�,and TLR5�/� mice by 72 h, there was a significant increase inMCP-1, IL-6, MIP-2, and KC serum levels in MyD88�/� miceat 72 h after infection. These data provide further evidence forlargely unimpaired host inflammatory responses in the absenceof individual TLRs or MyD88.
DISCUSSION
The main finding of this study is that TLR2, TLR4, andTLR5 recognize B. thailandensis, but that only deficiencyof TLR4 or adaptor molecule MyD88 impairs cytokine pro-duction by macrophages. In a murine model of inhaled infec-tion, the adaptor molecule MyD88 is essential for survival andcontainment of bacterial outgrowth. However, apart from amodest TLR4-dependent effect on bacterial containment at24 h, absence of individual TLRs does not significantly impairsurvival or bacterial replication and dissemination. TLR2 mayactually accelerate death in high-dose infection. Pulmonaryand systemic inflammation occurs in the absence of individualTLRs or MyD88.
The importance of TLRs in mediating host defense againstinfection has been well established in animal models of diseaseas well as in human populations (10, 13, 18, 19, 21). A betterunderstanding of the role of individual TLRs is of particularrelevance when developing targeted therapeutic strategies.Earlier work by several groups has demonstrated that TLR2and TLR4 are involved in host recognition of B. pseudomalleiin vitro (15, 27, 31), but the logistical limitations of the study ofB. pseudomallei have driven the use of B. thailandensis as asurrogate research organism. While it is essentially nonpatho-
FIG. 3. Survival after pneumonic infection with B. thailandensis isdependent on MyD88, whereas TLR2 deficiency extends survival.(A) Approximately 800 CFU/lung B. thailandensis was deposited byaerosol in the lungs of WT, TLR4�/�, TLR5�/�, and MyD88�/� mice.(B) A total of 2 � 104 CFU/lung B. thailandensis was deposited in thelungs of WT and TLR2�/� mice. Survival of four (A) or eight (B) miceper strain was recorded. P values were calculated with the log rank test.
5616 WEST ET AL. INFECT. IMMUN.
on May 22, 2020 by guest
http://iai.asm.org/
Dow
nloaded from
genic to humans, B. thailandensis replicates, disseminates, andis lethal in high doses to mice (28, 29) and shares significanthomology with B. pseudomallei (33). Accordingly, study ofTLRs in murine B. thailandensis infection may shed light onthe host-pathogen interaction in human melioidosis.
B. thailandensis is a gram-negative, flagellated organism,comprising the prototypical TLR2, TLR4, and TLR5 ligands—lipopeptide, LPS, and flagellin, respectively. Thus, our obser-vations of the roles for TLR2, TLR4, and TLR5 in the recog-nition of B. thailandensis, as assessed by measures of NF-�Bactivation, are not surprising. While studies using heat-killed B.thailandensis are potentially limited because heat may conceiv-ably destroy TLR ligands, our measurements of TLR-depen-dent signaling in transfected cells stimulated with heat-killed B.thailandensis support the persistent functionality of these li-gands. We also observed similar patterns of TLR5-dependentactivation of CHO cells stimulated with heat-killed or live B.thailandensis.
Our transfection studies indicate that either TLR1 or TLR6augments TLR2-mediated NF-�B activation. Although each ofthese TLRs functions in a MyD88-dependent fashion, theTLR-dependent variation in TNF-� production by primarycells points to specific and contrasting roles for individualTLRs in this infection. These findings of B. thailandensis-in-duced TLR2- and TLR4-dependent NF-�B activation and thespecific effect of TLR2 and TLR4 on cytokine production inmacrophages are similar to our previously published findingsusing B. pseudomallei (27), lending credence to our suppositionthat study of B. thailandensis may provide relevant insights intothe pathogenesis of virulent B. pseudomallei infection. Thisprompted us to extend our investigations to a murine model ofB. thailandensis pneumonia.
We did not observe a significant increase in TNF-� produc-tion in response to B. thailandensis by macrophages in theabsence of TLR2, or identify any alteration in bacterial countsor inflammation in TLR2�/� mice after a low-dose inhalation
FIG. 4. Containment of B. thailandensis infection is dependent on TLR4 and MyD88. Approximately 400 CFU/lung B. thailandensis wasdeposited by aerosol in the lungs of WT, TLR2�/�, TLR4�/�, and MyD88�/� mice (A), and �550 CFU/lung B. thailandensis was deposited byaerosol in the lungs of WT and TLR5�/� mice (B). Lung, liver, and spleen were harvested and quantitatively cultured at 24 and 72 h after infection.(A) Eight mice total per strain were sampled at each time point (seven for TLR2�/� mice at 24 h), combining data from two comparableexperiments performed independently. *** indicates a P value of �0.001 for MyD88�/� versus WT. † indicates a P value of �0.05, and †† indicatesa P value of �0.01 for TLR4�/� versus WT. (B) Four mice per strain were sampled at each time point, and the data displayed represent one oftwo comparable experiments performed independently. For lung CFU, the lines connect mean values at each time point; otherwise, data areplotted as means standard deviations. Statistical testing was performed after log transformation of the data with ANOVA and Bonferroni’spost-test (A) or the t test (B).
VOL. 77, 2009 TOLL-LIKE RECEPTORS IN B. THAILANDENSIS PNEUMONIA 5617
on May 22, 2020 by guest
http://iai.asm.org/
Dow
nloaded from
challenge. After high-dose infection, however, the presence ofTLR2 accelerates death. A unifying explanation is that at high,but not low, infecting doses, an overexuberant proinflamma-tory response may harm the host. This parallels the observa-tions by Wiersinga et al. (31) that TLR2�/� mice display en-hanced survival, decreased pulmonary cytokine levels, anddecreased bacterial growth at 72 h compared to WT mice in anintranasal model of B. pseudomallei infection. The etiology ofthis deleterious effect of TLR2 in Burkholderia infection isunclear, but our in vitro studies suggest it is not an IL-10-mediated phenomenon (27).
We have previously determined that B. pseudomallei LPSand lipid A are TLR4 agonists and that TLR4 deficiency ab-rogates macrophage cytokine production induced by B.pseudomallei in vitro (27). We speculate that B. thailandensisLPS is also a TLR4 agonist and probably accounts for TLR4-dependent macrophage cytokine production induced by B.thailandensis. TLR4�/� mice infected with B. thailandensishave a defect in their ability to contain the organism at 24 h,but this is overcome by 72 h when the phenotype is comparableto that of WT. These findings suggesting a transient role forTLR4 are similar to Wiersinga et al.’s report (31), in which
bacterial growth after intranasal B. pseudomallei infection ofmice was greater in TLR4�/� spleen and blood only at 24 hafter infection. In that study, TLR4 deficiency also did not altersurvival. In vivo, we did not identify any differences in pulmo-nary or systemic inflammation that would explain a TLR4-dependent effect, despite our observation that TLR4 is re-quired for TNF-� secretion by macrophages. Pulmonary andsystemic TNF-� levels remained close to baseline levelsthroughout the experiment in all strains of infected mice.Higher-dose infection of TLR4�/� mice may elucidate the roleof TNF-� in vivo, but other cell types (or more differentiatedmacrophages) may compensate for the lack of TNF-� produc-tion by bone marrow-derived macrophages.
TLR5 recognizes B. thailandensis, and the TLR5-mediatedsignal is more pronounced upon stimulation with log-phasebacteria than with stationary-phase bacteria. This suggests thatexpression of B. thailandensis flagellin—the likely TLR5 ligandand a putative virulence factor (7)—is increased during log-phase growth. Alternatively, flagella may be lost during sta-tionary-phase growth and in preparation of heat-killed bacte-ria. Despite this recognition signal, however, macrophagecytokine production is not mitigated in the absence of TLR5,
FIG. 5. Bronchoalveolar inflammation induced by B. thailandensis is not dependent on individual TLRs or MyD88. BAL was performed in themice infected as described in the legend for Fig. 4 after deposition of �400 CFU/lung (A) or �550 CFU/lung (B). At 24 and 72 h after infection,total cells, polymorphonuclear leukocytes (PMN), and mononuclear cells (MN) in the BAL fluid were enumerated. Data are displayed as means standard deviations. (A) Eight mice total per strain were sampled at each time point (seven for TLR2�/� and MyD88�/� mice at 24 h), and datafrom two comparable experiments performed independently are combined. (B) Four mice per strain were sampled at each time point, and the datadisplayed represent one of two comparable experiments performed independently. Statistical testing was performed with ANOVA and Bonfer-roni’s post-test (A) or the t test (B). * indicates a P value of �0.05 for TLR5�/� versus WT, and *** indicates a P value of �0.001 for MyD88�/�
versus WT. Mean values ( standard deviations) for BAL fluid cell counts in naïve C57BL/6 mice (n � 4) are 0 ( 0) � 106 PMN/ml and 0.09( 0.03) � 106 MN/ml.
5618 WEST ET AL. INFECT. IMMUN.
on May 22, 2020 by guest
http://iai.asm.org/
Dow
nloaded from
and there is no difference between TLR5 and WT mice in theirabilities to contain B. thailandensis infection or in the inflam-matory response. It seems plausible that a TLR5-specific effecton alveolar macrophages stimulated with whole B. thailanden-sis is obscured by concurrent TLR2 or TLR4 activation. In theabsence of TLR5, we speculate that greater compensatorystimulation of these TLR2- and TLR4-dependent pathwaysmay be induced by less-flagellated stationary-phase B. thailan-densis. This could account for the higher TNF-� concentra-tions detected in TLR5�/� macrophages than in WT macro-phages. In the lung, bronchial epithelial cells also expressTLR5, and in these cells, it is known that activation of NF-�Bin response to the opportunistic pathogen Burkholderia ceno-cepacia is mediated by TLR5 (8). Further study of these cells
in B. thailandensis infection may be informative. The in vitroresponse to B. pseudomallei is likely to be similar to that in-duced by B. thailandensis. In preliminary studies, we havefound B. pseudomallei-induced NF-�B activation in TLR5-transfected CHO cells and no difference in cytokine releasebetween WT and TLR5�/� alveolar macrophages stimulatedwith B. pseudomallei (our unpublished data). Others have alsoidentified TLR5-dependent NF-�B activation induced by B.pseudomallei (15).
In studies of murine pneumonia caused by Legionella pneu-mophila, a gram-negative, flagellated organism, we have ob-served a defect in neutrophil recruitment in TLR5�/� mice 4 hfollowing infection that was not attributable to reduced che-mokine production. By 24 h, neutrophil counts were compa-
FIG. 6. Pulmonary cytokine responses to B. thailandensis are not dependent on individual TLRs or MyD88. Mice were infected as describedin the legend for Fig. 4 after deposition of �400 CFU/lung (A) or �550 CFU/lung (B). Lung homogenate cytokines were measured in duplicateat 24 and 72 h after infection. (A) Eight mice total per strain were sampled at each time point (seven for TLR2�/� mice at 24 h), and data fromtwo comparable experiments performed independently are combined. (B) Four mice per strain were sampled at each time point, and the datadisplayed represent one of two comparable experiments performed independently. Data are plotted as means standard deviations. Statisticaltesting was performed after log transformation of data when displayed on a log scale with ANOVA and Bonferroni’s post-test (A) or the t test (B).* indicates a P value of �0.05, ** P values of �0.01, and *** P values of �0.001 for MyD88�/� versus WT. Mean values ( standard deviations)for lung homogenate cytokines in naïve C57BL/6 mice (n � 4) in pg/ml are as follows: TNF-�, 55 ( 0); MCP-1, 468 ( 0); IL-1�, 400 ( 0); IL-6,84 ( 37); MIP-2, 50 ( 0); and KC, 137 ( 38).
VOL. 77, 2009 TOLL-LIKE RECEPTORS IN B. THAILANDENSIS PNEUMONIA 5619
on May 22, 2020 by guest
http://iai.asm.org/
Dow
nloaded from
rable (12). We speculated that early TLR5-mediated neutro-phil recruitment might also occur in B. thailandensis infection.However, even depositing 5 � 103 CFU/lung (1 log greaterthan the 550 CFU/lung used in our TLR5�/� experiments), wecould not measure neutrophils in the bronchoalveolar space ofWT mice at 4 h. Only at deposition doses approaching 105
CFU/lung do we detect a BAL fluid neutrophilia at this timepoint following infection (28). Conceivably, if TLR5 plays asimilar role in the first few hours of B. thailandensis infection,much-higher-dose inoculation would be required to uncoverthe phenotype. Furthermore, compared to Burkholderia infec-tion, TLR5 may be particularly important in early Legionellainfection because of a weak Legionella LPS that signals viaTLR2 (11). Cytokine production by bone marrow-derived
macrophages stimulated with L. pneumophila is not impairedby TLR4 deficiency, unlike stimulation of these cells with B.thailandensis or B. pseudomallei (1, 2, 27).
MyD88 is a key adaptor molecule for each of the TLRsstudied in this work. We show that it is essential for hostcontainment of B. thailandensis even at doses that are readilycontained in WT mice. Similarly, MyD88 is necessary for hostdefense against intranasal B. pseudomallei infection, where it isrequired for neutrophil recruitment to the lung and activationbut not for phagocytosis or macrophage killing ability (32). Inour model of B. thailandensis pneumonia, we did not identifyany impairment in neutrophil recruitment to the alveolar spaceor any differences in neutrophil chemoattractant KC andMIP-2 levels in the lung in the absence of MyD88. The mech-
FIG. 7. Systemic cytokine responses to B. thailandensis are not dependent on individual TLRs or MyD88. Mice were infected as described inthe legend for Fig. 4 after deposition of �400 CFU/lung (A) or �550 CFU/lung (B). Serum cytokines were measured in duplicate at 24 and 72 hafter infection. (A) Seven mice total per strain were sampled at 24 h, and eight mice total per strain were sampled at 72 h; data from the twocomparable experiments performed independently were combined. (B) Four mice per strain were sampled at each time point, and the datadisplayed represent one of two comparable experiments performed independently. Data are plotted as means standard deviations. Statisticaltesting was performed after log transformation of data when displayed on a log scale with ANOVA and Bonferroni’s post-test (A) or the t test(B) after. * indicates a P value of �0.05, and *** indicates a P value of �0.001 for MyD88�/� versus WT. Mean values ( standard deviations)for serum cytokines in naïve C57BL/6 mice (n � 4) in pg/ml are as follows: TNF-�, 55 ( 0); MCP-1, 468 ( 0); IL-1�, 400 ( 0); IL-6, 72 (0); MIP-2, 50 ( 0); and KC, 155 ( 43).
5620 WEST ET AL. INFECT. IMMUN.
on May 22, 2020 by guest
http://iai.asm.org/
Dow
nloaded from

anism underlying the MyD88-dependent mortality in B. thai-landensis infection therefore remains unclear. We have ob-served a comparable essential dependence on MyD88 in ourstudies of inhaled Pseudomonas aeruginosa, another gram-neg-ative, flagellated pathogen (23). This contrasts with inhaledStaphylococcus aureus infection, which is contained andcleared by the murine host independently of MyD88. However,the absence of individual MyD88-dependent TLRs (apart fromthe modest phenotype observed in TLR4�/� mice) does notexplain the MyD88�/� phenotype in pneumonic B. thailanden-sis infection. Furthermore, the slight survival advantage in theabsence of TLR2 is opposite of the effect of MyD88 deficiency.This suggests a possible role for TLR9 in host defense againstB. thailandensis infection. TLR9 recognizes CpG DNA andalso signals via MyD88 (17). Preliminary studies of TLR9 in B.pseudomallei infection by others do not support this specula-tion (32), but pulmonary host defense in infection with gram-negative pathogens L. pneumophila or Klebsiella pneumoniae ispartially TLR9 dependent (3, 4). Our observations also pointto likely critical redundancy in multiple coordinated TLR sig-naling pathways, and a better understanding of these mecha-nisms may require systematic study of combined TLR genedeletions. MyD88 serves as an adaptor molecule for IL-1- andIL-18-dependent signaling pathways, so the pronounced phe-notype may alternatively be explained by a dependence onthese receptors. In support of this hypothesis, caspase-1, acomponent of the host inflammasome that processes IL-1� andIL-18 production, promotes resistance to B. pseudomallei in-fection (6).
Although MyD88 is essential for bacterial containment andhost survival in vivo, and for TNF-� production by macro-phages, the host inflammatory response in MyD88�/� mice at24 h is largely the same as those in WT and TLR-deficientmice. This is in part explained by the greater bacterial burdensin the MyD88�/� mice, but is notable because of the presenceof substantial MyD88-independent inflammation. The sole me-diator that was significantly lower in the lungs of MyD88�/�
mice at 24 h was MCP-1, a C-C chemokine produced by avariety of cell types that governs migration and infiltration ofmonocytes, memory T lymphocytes, and NK cells (9). MCP-1induction may be MyD88 dependent or independent in differ-ent experimental systems (20, 22). We did not observe a re-duction in bronchoalveolar macrophage recruitment at thistime point, however. More marked pulmonary and systemicinflammation is apparent in MyD88�/� mice at 72 h whenbacterial loads have increased further. Substantial pulmonaryinflammation has also been reported following intranasal B.pseudomallei infection in the absence of MyD88 (32), yet wehave observed abrogated inflammation in the lungs of highlysusceptible MyD88�/� mice infected with P. aeruginosa (23).Thus, non-MyD88-mediated pathways of host defense seem tobe particularly relevant in airborne Burkholderia infection andmay involve cell types other than macrophages.
In conclusion, our studies add to the growing literatureabout B. thailandensis, specifically demonstrating the distinctand complex roles of TLR2, TLR4, TLR5, and adaptor mol-ecule MyD88 in host defense against this organism. Many ofour findings with B. thailandensis mirror published reports ofvirulent B. pseudomallei, confirming the utility of B. thailan-
densis as a surrogate research organism for B. pseudomallei inelucidating the host-pathogen interaction in melioidosis.
ACKNOWLEDGMENTS
This work was supported by NIH grants U54 AI057141 (T.E.W. andS.J.S.), K08 HL094759 (T.E.W.), and R01 AI061464 (T.R.H.) and bya Parker B. Francis Fellowship for Pulmonary Research (T.E.W.).
Malinka Jansson-Hutson and Ravi Iyer provided expert technicalassistance. Christopher Wilson and Alan Aderem provided the TLR-deficient mice.
We declare that we have no conflicts of interest.
REFERENCES
1. Akamine, M., F. Higa, N. Arakaki, K. Kawakami, K. Takeda, S. Akira, andA. Saito. 2005. Differential roles of Toll-like receptors 2 and 4 in in vitroresponses of macrophages to Legionella pneumophila. Infect. Immun. 73:352–361.
2. Archer, K. A., and C. R. Roy. 2006. MyD88-dependent responses involvingToll-like receptor 2 are important for protection and clearance of Legionellapneumophila in a mouse model of Legionnaires’ disease. Infect. Immun.74:3325–3333.
3. Bhan, U., N. W. Lukacs, J. J. Osterholzer, M. W. Newstead, X. Zeng, T. A.Moore, T. R. McMillan, A. M. Krieg, S. Akira, and T. J. Standiford. 2007.TLR9 is required for protective innate immunity in gram-negative bacterialpneumonia: role of dendritic cells. J. Immunol. 179:3937–3946.
4. Bhan, U., G. Trujillo, K. Lyn-Kew, M. W. Newstead, X. Zeng, C. M. Hoga-boam, A. M. Krieg, and T. J. Standiford. 2008. Toll-like receptor 9 regulatesthe lung macrophage phenotype and host immunity in murine pneumoniacaused by Legionella pneumophila. Infect. Immun. 76:2895–2904.
5. Bossi, P., A. Tegnell, A. Baka, F. Van Loock, J. Hendriks, A. Werner, H.Maidhof, and G. Gouvras. 2004. Bichat guidelines for the clinical manage-ment of glanders and melioidosis and bioterrorism-related glanders andmelioidosis. Euro Surveill. 9:E17–E18.
6. Breitbach, K., G. W. Sun, J. Kohler, K. Eske, P. Wongprompitak, G. Tan, Y.Liu, Y. H. Gan, and I. Steinmetz. 2009. Caspase-1 mediates resistance inmurine melioidosis. Infect. Immun. 77:1589–1595.
7. Chua, K. L., Y. Y. Chan, and Y. H. Gan. 2003. Flagella are virulencedeterminants of Burkholderia pseudomallei. Infect. Immun. 71:1622–1629.
8. de C. Ventura, G. M., R. Le Goffic, V. Balloy, M. C. Plotkowski, M. Chignard,and M. Si-Tahar. 2008. TLR 5, but neither TLR2 nor TLR4, is involved inlung epithelial cell response to Burkholderia cenocepacia. FEMS Immunol.Med. Microbiol. 54:37–44.
9. Deshmane, S. L., S. Kremlev, S. Amini, and B. E. Sawaya. 2009. Monocytechemoattractant protein-1 (MCP-1): an overview. J. Interferon CytokineRes. 29:313–326.
10. Gerold, G., A. Zychlinsky, and J. L. de Diego. 2007. What is the role ofToll-like receptors in bacterial infections? Semin. Immunol. 19:41–47.
11. Girard, R., T. Pedron, S. Uematsu, V. Balloy, M. Chignard, S. Akira, and R.Chaby. 2003. Lipopolysaccharides from Legionella and Rhizobium stimulatemouse bone marrow granulocytes via Toll-like receptor 2. J. Cell Sci. 116:293–302.
12. Hawn, T. R., W. R. Berrington, I. A. Smith, S. Uematsu, S. Akira, A. Aderem,K. D. Smith, and S. J. Skerrett. 2007. Altered inflammatory responses inTLR5-deficient mice infected with Legionella pneumophila. J. Immunol.179:6981–6987.
13. Hawn, T. R., E. A. Misch, S. J. Dunstan, G. E. Thwaites, N. T. Lan, H. T.Quy, T. T. Chau, S. Rodrigues, A. Nachman, M. Janer, T. T. Hien, J. J.Farrar, and A. Aderem. 2007. A common human TLR1 polymorphism reg-ulates the innate immune response to lipopeptides. Eur. J. Immunol. 37:2280–2289.
14. Hawn, T. R., A. Verbon, K. D. Lettinga, L. P. Zhao, S. S. Li, R. J. Laws, S. J.Skerrett, B. Beutler, L. Schroeder, A. Nachman, A. Ozinsky, K. D. Smith,and A. Aderem. 2003. A common dominant TLR5 stop codon polymorphismabolishes flagellin signaling and is associated with susceptibility to legion-naires’ disease. J. Exp. Med. 198:1563–1572.
15. Hii, C. S., G. W. Sun, J. W. Goh, J. Lu, M. P. Stevens, and Y. H. Gan. 2008.Interleukin-8 induction by Burkholderia pseudomallei can occur withoutToll-like receptor signaling but requires a functional type III secretion sys-tem. J. Infect. Dis. 197:1537–1547.
16. Kawai, T., O. Adachi, T. Ogawa, K. Takeda, and S. Akira. 1999. Unrespon-siveness of MyD88-deficient mice to endotoxin. Immunity 11:115–122.
17. Kawai, T., and S. Akira. 2006. TLR signaling. Cell Death Differ. 13:816–825.18. Khor, C. C., S. J. Chapman, F. O. Vannberg, A. Dunne, C. Murphy, E. Y.
Ling, A. J. Frodsham, A. J. Walley, O. Kyrieleis, A. Khan, C. Aucan, S. Segal,C. E. Moore, K. Knox, S. J. Campbell, C. Lienhardt, A. Scott, P. Aaby, O. Y.Sow, R. T. Grignani, J. Sillah, G. Sirugo, N. Peshu, T. N. Williams, K.Maitland, R. J. Davies, D. P. Kwiatkowski, N. P. Day, D. Yala, D. W. Crook,K. Marsh, J. A. Berkley, L. A. O’Neill, and A. V. Hill. 2007. A Mal functionalvariant is associated with protection against invasive pneumococcal disease,bacteremia, malaria and tuberculosis. Nat. Genet. 39:523–528.
VOL. 77, 2009 TOLL-LIKE RECEPTORS IN B. THAILANDENSIS PNEUMONIA 5621
on May 22, 2020 by guest
http://iai.asm.org/
Dow
nloaded from

19. Korbel, D. S., B. E. Schneider, and U. E. Schaible. 2008. Innate immunity intuberculosis: myths and truth. Microbes Infect. 10:995–1004.
20. Moon, S. K., J. I. Woo, H. Y. Lee, R. Park, J. Shimada, H. Pan, R. Gellibo-lian, and D. J. Lim. 2007. Toll-like receptor 2-dependent NF-�B activationis involved in nontypeable Haemophilus influenzae-induced monocyte che-motactic protein 1 up-regulation in the spiral ligament fibrocytes of the innerear. Infect. Immun. 75:3361–3372.
21. Salomao, R., P. S. Martins, M. K. Brunialti, L. Fernandes Mda, L. S.Martos, M. E. Mendes, N. E. Gomes, and O. Rigato. 2008. TLR signalingpathway in patients with sepsis. Shock 30(Suppl 1):73–77.
22. Serbina, N. V., W. Kuziel, R. Flavell, S. Akira, B. Rollins, and E. G. Pamer.2003. Sequential MyD88-independent and -dependent activation of innateimmune responses to intracellular bacterial infection. Immunity 19:891–901.
23. Skerrett, S. J., H. D. Liggitt, A. M. Hajjar, and C. B. Wilson. 2004. Cuttingedge: myeloid differentiation factor 88 is essential for pulmonary host de-fense against Pseudomonas aeruginosa but not Staphylococcus aureus. J. Im-munol. 172:3377–3381.
24. Smith, M. D., B. J. Angus, V. Wuthiekanun, and N. J. White. 1997. Arabi-nose assimilation defines a nonvirulent biotype of Burkholderia pseudomallei.Infect. Immun. 65:4319–4321.
25. Takeuchi, O., K. Hoshino, T. Kawai, H. Sanjo, H. Takada, T. Ogawa, K.Takeda, and S. Akira. 1999. Differential roles of TLR2 and TLR4 in recog-nition of gram-negative and gram-positive bacterial cell wall components.Immunity 11:443–451.
26. Uematsu, S., M. H. Jang, N. Chevrier, Z. Guo, Y. Kumagai, M. Yamamoto,H. Kato, N. Sougawa, H. Matsui, H. Kuwata, H. Hemmi, C. Coban, T.Kawai, K. J. Ishii, O. Takeuchi, M. Miyasaka, K. Takeda, and S. Akira. 2006.Detection of pathogenic intestinal bacteria by Toll-like receptor 5 on intes-tinal CD11c� lamina propria cells. Nat. Immunol. 7:868–874.
27. West, T. E., R. K. Ernst, M. J. Jansson-Hutson, and S. J. Skerrett. 2008.Activation of Toll-like receptors by Burkholderia pseudomallei. BMC Im-munol. 9:46.
28. West, T. E., C. W. Frevert, H. D. Liggitt, and S. J. Skerrett. 2008. Inhalationof Burkholderia thailandensis results in lethal necrotizing pneumonia inmice: a surrogate model for pneumonic melioidosis. Trans. R. Soc. Trop.Med. Hyg. 102(Suppl. 1):S119–S126.
29. Wiersinga, W. J., A. F. de Vos, R. de Beer, C. W. Wieland, J. J. Roelofs, D. E.Woods, and T. van der Poll. 2008. Inflammation patterns induced by differ-ent Burkholderia species in mice. Cell. Microbiol. 10:81–87.
30. Wiersinga, W. J., T. van der Poll, N. J. White, N. P. Day, and S. J. Peacock.2006. Melioidosis: insights into the pathogenicity of Burkholderia pseudoma-llei. Nat. Rev. Microbiol. 4:272–282.
31. Wiersinga, W. J., C. W. Wieland, M. C. Dessing, N. Chantratita, A. C.Cheng, D. Limmathurotsakul, W. Chierakul, M. Leendertse, S. Florquin,A. F. de Vos, N. White, A. M. Dondorp, N. P. Day, S. J. Peacock, and T. vander Poll. 2007. Toll-like receptor 2 impairs host defense in gram-negativesepsis caused by Burkholderia pseudomallei (melioidosis). PLoS Med.4:e248.
32. Wiersinga, W. J., C. W. Wieland, J. J. Roelofs, and T. van der Poll. 2008.MyD88 dependent signaling contributes to protective host defense againstBurkholderia pseudomallei. PLoS ONE 3:e3494.
33. Yu, Y., H. S. Kim, H. H. Chua, C. H. Lin, S. H. Sim, D. Lin, A. Derr, R.Engels, D. DeShazer, B. Birren, W. C. Nierman, and P. Tan. 2006. Genomicpatterns of pathogen evolution revealed by comparison of Burkholderiapseudomallei, the causative agent of melioidosis, to avirulent Burkholderiathailandensis. BMC Microbiol. 6:46.
Editor: J. L. Flynn
5622 WEST ET AL. INFECT. IMMUN.
on May 22, 2020 by guest
http://iai.asm.org/
Dow
nloaded from





























