Embed Size (px)
Citation preview

How Well Can We Predict Native Contacts in Proteins Basedon Decoy Structures and Their Energies?Jiang Zhu, Qianqian Zhu, Yunyu Shi,* and Haiyan Liu*Key Laboratory of Structural Biology, University of Science and Technology of China, Chinese Academy of Sciences, School ofLife Sciences, Hefei, Anhui, 230026, China
ABSTRACT One strategy for ab initio proteinstructure prediction is to generate a large numberof possible structures (decoys) and select the mostfitting ones based on a scoring or free energy func-tion. The conformational space of a protein is huge,and chances are rare that any heuristically gener-ated structure will directly fall in the neighborhoodof the native structure. It is desirable that, insteadof being thrown away, the unfitting decoy struc-tures can provide insights into native structures soprediction can be made progressively. First, wedemonstrate that a recently parameterized physics-based effective free energy function based on theGROMOS96 force field and a generalized Born/surface area solvent model is, as several other phys-ics-based and knowledge-based models, capable ofdistinguishing native structures from decoy struc-tures for a number of widely used decoy databases.Second, we observe a substantial increase in correla-tions of the effective free energies with the degree ofsimilarity between the decoys and the native struc-ture, if the similarity is measured by the content ofnative inter-residue contacts in a decoy structurerather than its root-mean-square deviation from thenative structure. Finally, we investigate the possibil-ity of predicting native contacts based on the fre-quency of occurrence of contacts in decoy struc-tures. For most proteins contained in the decoydatabases, a meaningful amount of native contactscan be predicted based on plain frequencies ofoccurrence at a relatively high level of accuracy.Relative to using plain frequencies, overwhelmingimprovements in sensitivity of the predictions areobserved for the 4_state_reduced decoy sets by apply-ing energy-dependent weighting of decoy struc-tures in determining the frequency. There, approxi-mately 80% native contacts can be predicted at anaccuracy of approximately 80% using energy-weighted frequencies. The sensitivity of the plainfrequency approach is much lower (20% to 40%).Such improvements are, however, not observed forthe other decoy databases. The rationalization andimplications of the results are discussed. Proteins2003;52:598–608. © 2003 Wiley-Liss, Inc.
Key words: decoy discrimination; generalized Bornmodel; solvent-accessible surface area;protein structure prediction
INTRODUCTION
Understanding the relationship between the sequence ofa protein and its unique three-dimensional structure is asubject that has intrigued scientists for decades.1 Intheory, genome projects will result in known sequences foralmost all proteins. The need to discover their structuresand functions highlights the importance of protein struc-ture prediction. In one possible scenario, a large number ofcandidate structures are generated for a peptide sequenceand evaluated with an energy function that can distin-guish the native structure from the mis-folded ones (de-coys). Toward the aim of developing such energy functions,two different types of approaches are currently underinvestigation. The first class, the so-called “knowledge-based potentials,” derived from databases of known pro-tein structures, usually represents interactions withinproteins at a low level of resolution. Many efficient poten-tials of this category have been widely used in comparativemodeling and fold recognition, and have been extensivelyreviewed.2–5 The second class is the so-called “physics-based potentials.”6–10 Until recently, the physics-basedall-atom molecular mechanics energy functions have notbeen as commonly used to distinguish protein folds as thestatistical models because their application needs struc-ture optimization at the atomic level, which is relativelyexpensive in terms of computational cost. Compared withthe statistical potentials, however, the physics-based mod-els have some significant advantages. Specifically, theymay possess more general applicability than the statisticalmodels, which can be biased by the databases. This isespecially relevant in discriminating native fold and de-coys, because many statistical models have been developedusing only knowledge of the native structures or folds, andone should not expect such models to produce reasonableenergies/scores for non-native-like decoy structures.
Grant sponsor: Chinese National Natural Science Foundation;Grant numbers: 30025013, 39990600; Grant sponsor: National BasicResearch Projects; Grant number: G1999075605.
J. Zhu’s present address is Department of Biochemistry and Molecu-lar Biophysics, Columbia University and the Howard Hughes MedicalInstitute, Black Building, 650 West, 168 Street, Room 221, New York,NY 10032. E-mail: [email protected]
*Correspondence to: Haiyan Liu or Yunyu Shi, School of LifeSciences, University of Science and Technology of China, Hefei, Anhui,230026, China. E-mail: [email protected] or [email protected]
Received 17 October 2002; Accepted 28 January 2003
PROTEINS: Structure, Function, and Genetics 52:598–608 (2003)
© 2003 WILEY-LISS, INC.

The ability of physical potentials as score functions todiscriminate protein structure has been under debate for along period.8,11–16 Recently, some research efforts wererefocused on this issue.7 A successful physics-based func-tion should contain all free energy components signifi-cantly contributing to the stabilization of the native struc-ture, including intramolecular bonded and non-bondedenergy terms as well as the free energies of solvation in anaqueous solution. Because the solvent environment hasprofound influences on the structures and functions ofproteins, the solvent effects are particularly important.Therefore, the treatment of solvent effects has been exten-sively studied and discussed.17,18 The explicit treatment ofwater molecules, although very useful in a range ofproblems, is not feasible for structure discrimination be-cause of the enormous requirement on computer time.19
The solvent effects can be considered by using varioustypes of implicit solvent models, including MMPB/SA(molecular mechanics, Poisson-Boltzmann and solvent ac-cessibility) or ES/IS (explicit solvent/implicit solvent),9,20,21
MMGB/SA (molecular mechanics, generalized-Born andsolvent accessibility)22–28 and other simpler empiricalsolvent models. In the MMPB/SA or ES/IS scheme, rela-tive free energies are estimated as the sum of the internalprotein energy, the polar components of the free energy ofsolvation obtained by solving the Poisson-Boltzmann equa-tion, and the hydrophobic components of the free energy ofsolvation calculated from the solvent-accessible surfaceareas. The polar components can also be computed byalternative approaches, e.g., the generalized Born model,to achieve higher computational efficiency without muchloss in accuracy. A number of other implicit solvent modelsuse either separate or combination models such as atomicsolvent-accessible surface areas,29–31 the Gaussian solvent-exclusion element approach,32 and empirically screened ormodified Coulombic interactions.33–37
If there were a structure generation approach guaran-teed to produce the native structure of a protein among alarge number of alternative structures, this, in combina-tion with a native/decoy discrimination energy function,would solve the ab initio structure prediction problem.However, because the conformational space of a protein isso huge, it is difficult for a heuristic structure generationprocedure to have any significant chance to produce thenative structure. It would be desirable to obtain insightsinto the native structure based on the decoy structures, sothat predictions can be made progressively. In fact, such aconsensus approach toward ab initio structure predictionhas been proposed by Samudrala et al.38–40 There dis-tance restrains were picked from a large set of modelstructures to refine a prediction of the native structure byconsensus criteria. It is of interest to investigate thefollowing questions: 1. What would be the most relevant“native characteristic” to extract from ensembles of decoystructures? 2. To what degree do consensus criteria applyto decoy sets generated by different approaches? 3. Howcan we take advantage of an energy function in suchprogressive predictions?
It is crucial for such progressive approaches that the“nativeness” of the candidate structures be somehowranked. Recently, Vorobjev and Hermans41 concluded thatthe free energy is not an accurate measure for ranking themost native-like structures among a set of models, if thesimilarity between decoys and their corresponding nativestructure is measured by positional root-mean-squaredeviations (RMSD). Although most of the previous decoydiscrimination studies used RMSD to quantify the differ-ences between decoys and native structures, this approachdoes not consider the flexibility of the native fold (i.e., thenative fold is treated as a single, frozen conformation incomputing the RMSD). Other criteria for measuring simi-larity to the native structure besides RMSD are possible. Ithas been shown that in X-ray structures, the inter-residuecontact preferences make dominant contributions to stabi-lizing native structures in globular proteins,3 which hasled to extensive studies on the construction of simplifiedcontact potentials.5,42–49
In a previous work,28 we have re-parameterized theGB/SA model proposed by Still et al.22,25 for the GRO-MOS96 force field with the specific goal of applying it inprotein simulations. The model parameters are deter-mined by a combined procedure of: 1. fitting to the freeenergies of solvation of amino-acid-like small molecules; 2.rationally considering the effects of the solvent model onhydrogen-bond interactions; and 3. most critically, perform-ing trial molecular dynamics simulations on two structur-ally distinct proteins and running the final test on a thirdprotein. The simulations with the GB/SA model showedexcellent agreement with crystal structures and explicitsolvent simulations. Particularly, although we have notoptimized the GB model specifically to reproduce PBresults, there exist strong correlations between the resultsof a numerical PB solver (FAMBE)10,21 and our GB modelresults for the free energies of solvation.
In the current work, we demonstrate that the GRO-MOS96-GB/SA energy function is capable of discriminat-ing between native and non-native folds among variousdecoy sets containing a large number of compact protein-like structures. These decoy databases have been gener-ated by others, with a variety of prediction algorithmsdesigned to cover the conformational space as much aspossible. Then, we examine the possibility of using theoccurrence of native inter-residue contacts to quantify the“nativeness” of a decoy structure. We further investigatethe possibility of identifying native contacts among allcontacts in a decoy set based on the frequencies of occur-rence of the contacts. Two approaches are compared, onewith each decoy structure energy-weighted when its con-tacts are counted, the other without weighting. A numberof related issues, including comparisons between physics-based and statistics-based potentials, and correlations andanti-correlations between energy components and thesimilarity with native structures, have been extensivelydiscussed in recent works by others.50 Because mostlysimilar results have been observed in our investigation,these aspects will not be discussed in detail here.
PREDICTING NATIVE CONTACTS IN PROTEINS 599

METHODSThe GROMOS96-GB/SA Energy Function
Here we are interested in the free energy differencesbetween different folded states of a protein. Such a freeenergy difference can be approximately expressed as thesum of two terms,
�Gtot � �Gint � ��Gsol (1)
where �Gint represents the contributions from intramolecu-lar degrees of freedom; ��Gsol is the difference in the freeenergies of solvation. Let us assume that the conforma-tional entropies of different folded states relative to a rigidmolecule are similar and their contributions to �Gint canbe neglected. Then the difference in free energies can befurther approximated as:
�Geff � �Uint � ��Gsol (2)
For each folded state, we compute Uint as the GRO-MOS96 intramolecular potential energies, �Gsol estimatedfrom our previously re-parameterized Still’s GB/SAmodel22,25 compatible with GROMOS96 solution forcefield (43 a1).28
Contact Analyses
Following the definition of Boczko and Brooks51 andSheinerman and Brooks,52 a contact was identified asbeing present if the centers of geometry of side-chains oftwo residues are within 6.5 Å from each other. In thenative structure, the contacts satisfying this definitionwere gathered as native contacts, which determine areference set for measuring the “distance” between a decoyand the native structure. Such a set of contacts can becalculated also for a decoy structure containing both nativeand non-native contacts.
Given a structure m, the state of each contact i collectedin the above way can be defined by a continuous switchingfunction,
xi�m� �1
1 � exp �20 � �di�m� � 6.75��(3)
The state of contact i, xi(m), is almost 1 (which indicatesthat the contact exists in structure m) if the distance di(m)between the centers of geometry of the side-chains in-volved is �6.5 Å, and almost 0 (which indicates that thecontact does not exist in structure m) if the distance is 7 Å.
Then, the fractional occurrence of native contacts occur-ring in a decoy structure is defined by:
�m� �
�i
pixi�m�
�i
pi(4)
where the weight pi is 1 if i is a native contact, 0 otherwise(a more reasonable definition may be estimating pi as thefrequency of contact i in a sufficiently long moleculardynamics simulation of the native state). The summationis over all contacts in the decoys and the native structures.
Given the target protein sequence, the decoy structurescan be seen as an ensemble of conformation samples fromthe conformational space. We can calculate the frequencyof occurrence of each contact in the decoy set by:
fi �
�m � 1
N
wmxi �m�
�m � 1
N
wm
(5)
where N is the total number of decoy structures in the set,wm a weighting factor for the decoy structure m. In thecurrent work, we examined two choices of the weightingfactors: one is unity for every m, the other is the followingGaussian form,
wm � exp��(�Geff,m � �Geff, native)2
s2 � �2 � (6)
which depends on the effective free energy of the decoy.Here � is the standard variation of the effective freeenergies of decoys within the set, and s is an empiricalscaling factor (set to 0.1). Because of the nature of theGaussian weighting, the decoy structures with low effec-tive free energies contribute most strongly.
MATERIALS
Five decoy sets are used in this work, including the4_state_reduced sets of Park and Levitt,8 the local-minimadecoy sets of Kesar and Levitt,53 the fisa and fisa_casp3sets of Simons et al.,54 and the lattice_ssfit sets of Sam-udrala et al.55
The 4_state_reduced decoy sets8 contain structural de-coys for seven small proteins containing from 54 to 75residues and having various topological forms. The proteinstructures have been generated by exhaustively enumerat-ing the backbone rotamer states of 10 selected residues ineach protein using an off-lattice model with four discretedihedral angles states per rotatable bond. Among hun-dreds of thousands of generated structures, only thosecompact structures scoring well using a variety of scoringfunctions and having reasonable RMSD from the nativestructure have been selected to form the decoy sets.
The local-minima decoy sets (lmds)53 also contain struc-tural decoys for seven small proteins. These decoys areconformations that occupy minima in a modified classicalENCAD force field using united and soft atoms. The initialdata sets are generated by randomly modifying only thedihedral angles in the loop regions. After minimization intorsional space, no more than 500 of the lowest energyconformations for each protein were kept to make up thefinal decoy sets.
The fisa and fisa_casp3 decoy sets54 together containseven proteins. The decoy structures are built by assem-bling together segments of unrelated protein structureswith similar local sequences (determined through a mul-tiple sequence alignment process). Low scoring structuresare identified using a simulated annealing procedure, in
600 J. ZHU ET AL.

which the torsion angles of a segment are replaced bythose of a different segment with a related amino acidsequence. The scoring function included terms for hydro-phobic burial, electrostatics, disulfide bonds, the packingof -helices and �-strands, and the formation of �-sheets.
The lattice_ssfit decoy sets55 contain six proteins. Thedecoy structures have been generated by a hierarchicalprediction strategy. First, simplified protein structureswere exhaustively enumerated by self-avoiding walk on atetrahedral lattice. A residue–residue contact energy func-tion was used to select the low energy conformations.Second, the predicted secondary structures of the targetsequence were fitted to the produced lattice models using agreedy growth algorithm and a four-state torsion anglemodel to generate all-atom structures. After energy mini-mization using ENCAD (energy calculations and dynam-ics),56–59 the all-atom structures were further subjected toscreening with three different scoring functions: RAPDF(residue-specific all-atom probability discriminatory func-tion),60 HCF (hydrophobic compactness function),55 andthe Shell energy function.16 As the last step in theprediction, a set of low energy conformations is kept toproduce a single cartesian structure using the consensus-based distance geometry.
RESULTS AND DISCUSSIONZ-Scores of the Respective Native Structures of theDecoy Sets Computed Using the GROMOS96-GB/SAEffective Free Energies
All decoy structures in the above sets and the correspond-ing native structures (from Protein Data Bank) wereenergy minimized with the GROMOS96-GB/SA energyfunction. The minimized structures differ slightly from theinitial structures by an average RMSD of 0.59 � 0.13 Å.The Z-score of a native structure is calculated to describehow far the effective energy of the native structure isseparated from the energy spectrum of the decoy struc-tures,
Z ��G � �G
�(7)
where �G is the effective free energy of the native struc-ture, and �G and � are the average and standard deviationof the energies, respectively, of the decoys in a set. TheZ-scores calculated for all decoy databases are presented inTable I. The computed Z-scores show that for the majorityof proteins considered, the native structure is widelyseparated from the bulk decoy structures by the effectivefree energy function. One exception is the protein 1fc2,which is contained in both the lmds and the fisa sets. Thesequence of 1fc2, however, corresponds to only a fragmentof an intact protein and is in complex with the immunoglo-bin Fc in the Protein Data Bank structure. In recent workreported by Felts et al.,50 the OPLS-AA/SGB energyfunction also failed in discriminating the native structureof 1fc2 from its decoy structures. The “native” structure of1fc2, stabilized by the hydrophobic contact between thefragment and immunoglobin Fc in the protein complex,may not be stable in the unbound state.
Correlations Between the Effective Energies andthe RMSD From the Native Structures
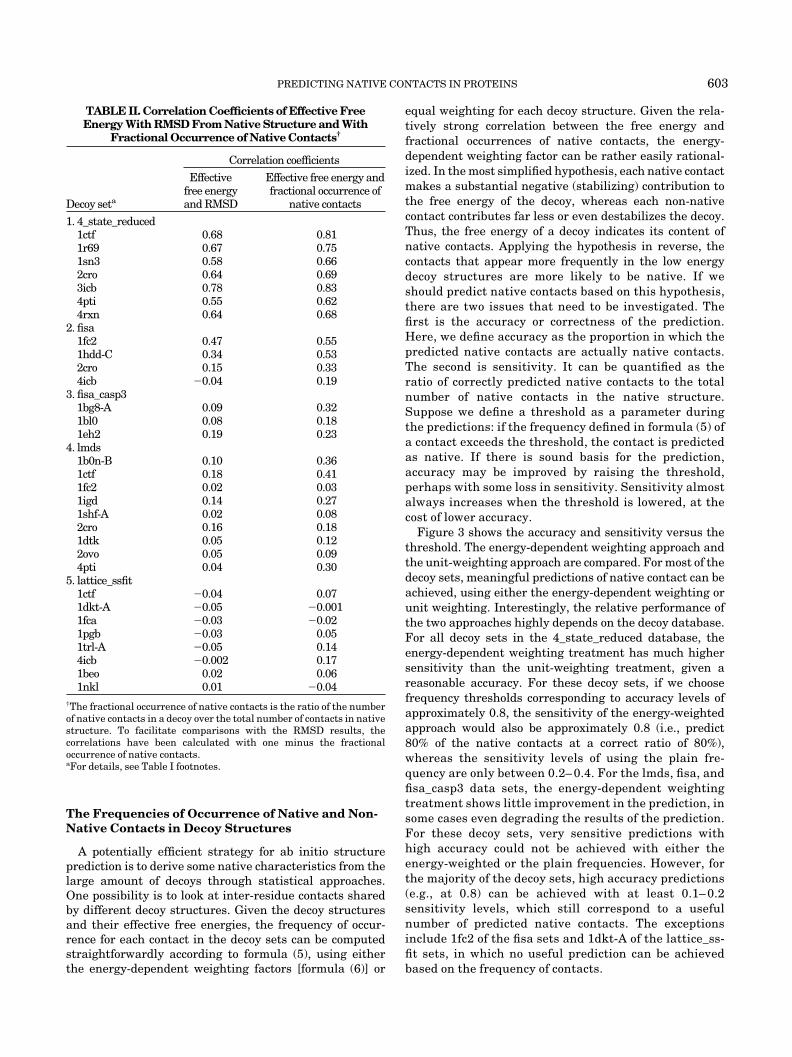
Following a number of previous studies, we investigatedthe correlation between the GROMOS96-GB/SA effectivefree energies and the RMSD of all atoms of the decoystructures from the native structures. Among the decoysets examined, only the 4_state_reduced data sets showstrong correlations between the energies and RMSD (seeFig. 1). This result is in line with studies reported byothers. To save space, the results for only one protein fromeach decoy database have been shown in Figure 1, andresults for proteins in the same decoy database are qualita-tively the same. In Table II, the effective free energy-RMSD correlation coefficients for the 4_state_reduced setsrange from 0.56 to 0.78. For the other decoy sets, the
TABLE I. Z-Scores† of Each Protein in the Decoy Sets
Decoy set Z-score
1. 4_state_reduceda
lctf �3.331r69 �3.631sn3 �5.702cro �3.553icb �1.974pti �5.094rxn �4.43
2. fisab
1fc2 �1.941hdd-C �2.862cro �4.544icb �3.00
3. fisa_casp3b
1bg8-A �2.531bl0 �2.851eh2 �3.07
4. lmdsc
1b0n-B �2.551ctf �4.381fc2 �0.221igd �5.801shf-A �7.532cro �5.971dtk �3.512ovo �4.514pti �7.04
5. lattice_ssfitd
1ctf �4.861dkt-A �3.221fca �5.891pgb �5.891trl-A �3.974icb �2.661beo �4.271nkl �1.85
†Z � (�G � �G)/�, where �G is the effective free energy of the nativestructure, �G and � are the average and standard deviation of theenergies of the decoys in a set.aData from Park and Levitt.8bData from Simons et al.54
cData from Samudrala and Levitt.53
dData from Xia et al.40 and Samudrala et al.55
PREDICTING NATIVE CONTACTS IN PROTEINS 601

correlation coefficients only range from �0.03 to 0.44. Thesignificant differences between the decoy sets are obvi-ously related to the methods by which the decoy structuresare generated.
Stronger Correlation Between GROMOS96-GB/SAEnergy and the Fractional Occurrence of NativeContacts
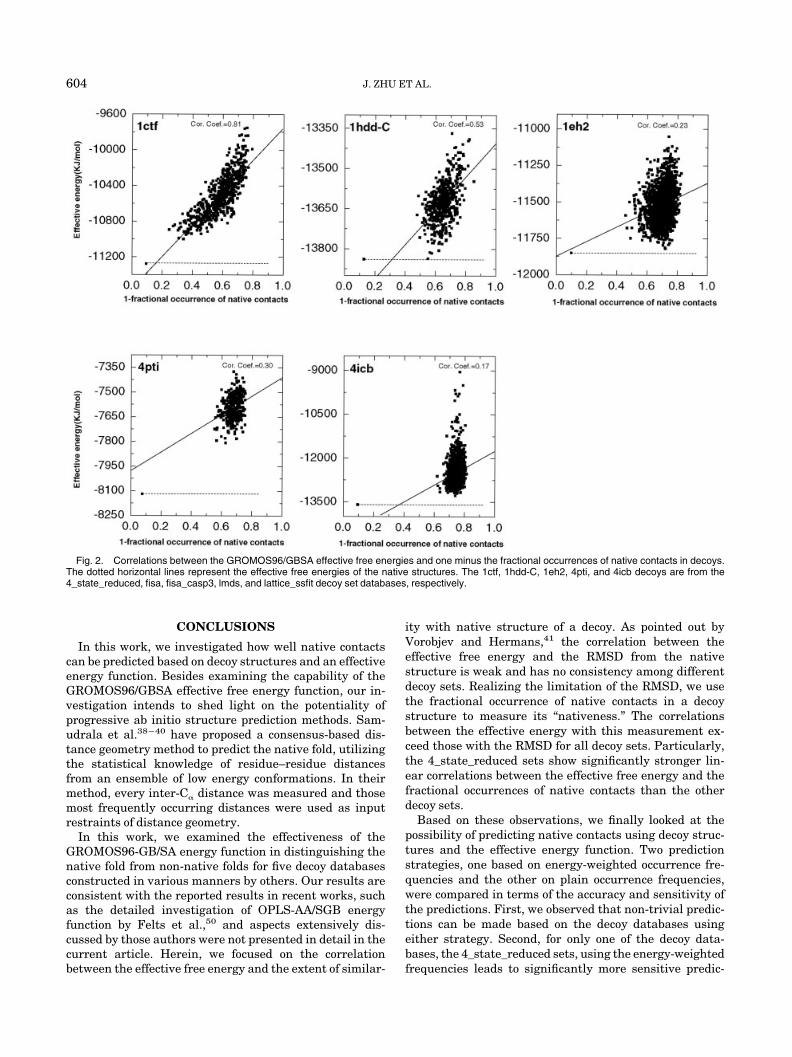
In Figure 2, the effective free energies of the decoystructures are plotted against one minus the fractionaloccurrences of native contacts in the decoys [the use of1-(m) instead of (m) is to facilitate comparisons with theenergy-RMSD correlations]. When this is compared withFigure 1, we see general improvement in correlations. Forthe 4_state_reduced sets, the averaged correlation coeffi-cient of the effective free energies with one minus thefractional occurrence of native contacts is 0.73, comparedwith 0.66 with the RMSD (Table II). For other decoy sets,although the correlations obtained using either RMSD orfractional occurrences of native contacts are weak, theimprovement by replacing the RMSD using the fractionaloccurrences of native contacts seems to be real: for none ofthe decoy sets investigated do we see a decrease in thecorrelation coefficients. Using the fractional occurrence
data obviously improves over the weak correlations be-tween the RMSD and the energies for nine decoy sets (1fc2,1hdd-C, and 2cro of the fisa sets, all three sets of fisa_casp3,and 1b0n-B, 1ctf, ligd of the lmds sets). Weak correlationsare observed for four decoy sets that do not show anycorrelations between the RMSD and energies (4icb of thefisa sets, 1dtk, 4pti of the lmds sets, and 1trl-A and 4icb ofthe lattice_ssfit sets). For the remaining nine decoy sets,the correlations generated using the fractional occurrencedo not show significant improvement.
In Figure 1, decoys of a given protein with energiesapproaching the energy of the native fold often span awide range of RMSD. In Figure 2, such decoys, if presentin the decoy set, are more likely to have similar frac-tional occurrence of native contacts when the energiesare similar, in agreement with the observed strongercorrelations in the latter plots. These results indicatethat the fractional occurrence of native contacts is abetter measurement of the “nativeness” of a decoystructure than the RMSD. Figure 2 also indicates thatthe decoy sets in several databases, especially the lmdsand the lattice_ssfit sets, tend to contain structures withsimilar degrees of “nativeness,” i.e., similar contents ofnative contacts.
Fig. 1. Correlations between the GROMOS96/GBSA effective free energies and the all-atom RMSD of decoys from their corresponding nativestructures. The dotted horizontal lines represent the effective free energies of the native structures. The 1ctf, 1hdd-C, 1eh2, 4pti, and 4icb decoys arefrom the 4_state_reduced, fisa, fisa_casp3, lmds, and lattice_ssfit decoy set databases, respectively.
602 J. ZHU ET AL.
The Frequencies of Occurrence of Native and Non-Native Contacts in Decoy Structures
A potentially efficient strategy for ab initio structureprediction is to derive some native characteristics from thelarge amount of decoys through statistical approaches.One possibility is to look at inter-residue contacts sharedby different decoy structures. Given the decoy structuresand their effective free energies, the frequency of occur-rence for each contact in the decoy sets can be computedstraightforwardly according to formula (5), using eitherthe energy-dependent weighting factors [formula (6)] or
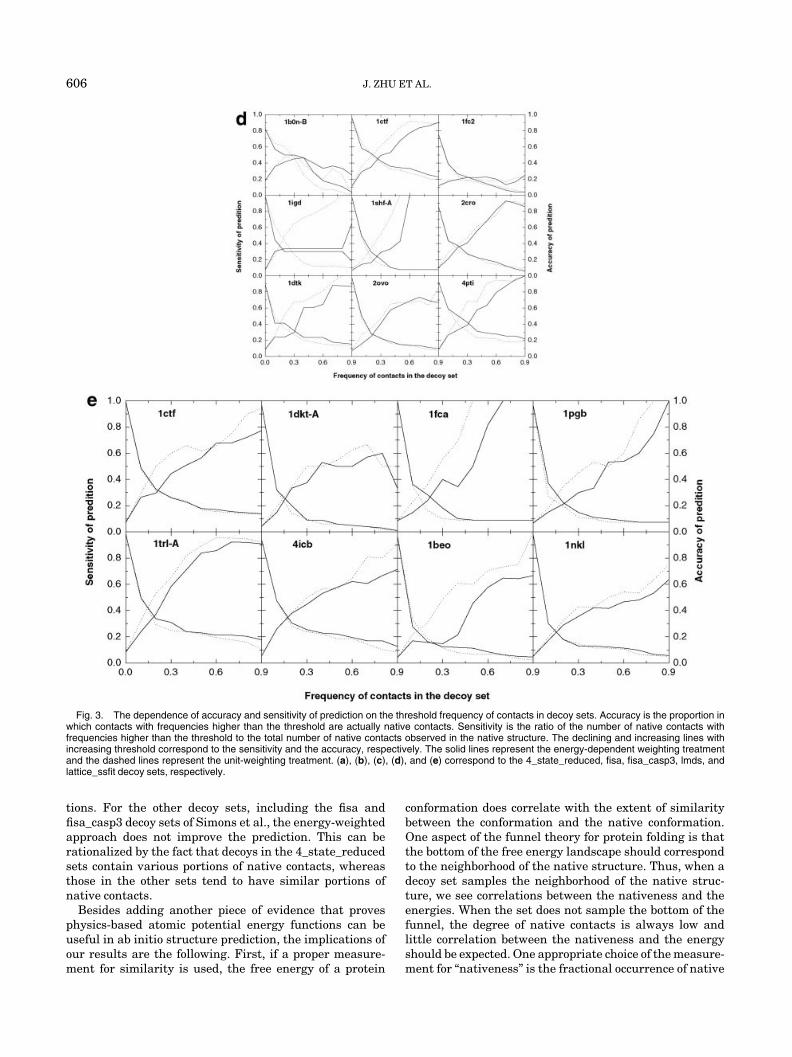
equal weighting for each decoy structure. Given the rela-tively strong correlation between the free energy andfractional occurrences of native contacts, the energy-dependent weighting factor can be rather easily rational-ized. In the most simplified hypothesis, each native contactmakes a substantial negative (stabilizing) contribution tothe free energy of the decoy, whereas each non-nativecontact contributes far less or even destabilizes the decoy.Thus, the free energy of a decoy indicates its content ofnative contacts. Applying the hypothesis in reverse, thecontacts that appear more frequently in the low energydecoy structures are more likely to be native. If weshould predict native contacts based on this hypothesis,there are two issues that need to be investigated. Thefirst is the accuracy or correctness of the prediction.Here, we define accuracy as the proportion in which thepredicted native contacts are actually native contacts.The second is sensitivity. It can be quantified as theratio of correctly predicted native contacts to the totalnumber of native contacts in the native structure.Suppose we define a threshold as a parameter duringthe predictions: if the frequency defined in formula (5) ofa contact exceeds the threshold, the contact is predictedas native. If there is sound basis for the prediction,accuracy may be improved by raising the threshold,perhaps with some loss in sensitivity. Sensitivity almostalways increases when the threshold is lowered, at thecost of lower accuracy.
Figure 3 shows the accuracy and sensitivity versus thethreshold. The energy-dependent weighting approach andthe unit-weighting approach are compared. For most of thedecoy sets, meaningful predictions of native contact can beachieved, using either the energy-dependent weighting orunit weighting. Interestingly, the relative performance ofthe two approaches highly depends on the decoy database.For all decoy sets in the 4_state_reduced database, theenergy-dependent weighting treatment has much highersensitivity than the unit-weighting treatment, given areasonable accuracy. For these decoy sets, if we choosefrequency thresholds corresponding to accuracy levels ofapproximately 0.8, the sensitivity of the energy-weightedapproach would also be approximately 0.8 (i.e., predict80% of the native contacts at a correct ratio of 80%),whereas the sensitivity levels of using the plain fre-quency are only between 0.2– 0.4. For the lmds, fisa, andfisa_casp3 data sets, the energy-dependent weightingtreatment shows little improvement in the prediction, insome cases even degrading the results of the prediction.For these decoy sets, very sensitive predictions withhigh accuracy could not be achieved with either theenergy-weighted or the plain frequencies. However, forthe majority of the decoy sets, high accuracy predictions(e.g., at 0.8) can be achieved with at least 0.1– 0.2sensitivity levels, which still correspond to a usefulnumber of predicted native contacts. The exceptionsinclude 1fc2 of the fisa sets and 1dkt-A of the lattice_ss-fit sets, in which no useful prediction can be achievedbased on the frequency of contacts.
TABLE II. Correlation Coefficients of Effective FreeEnergy With RMSD From Native Structure and With
Fractional Occurrence of Native Contacts†
Decoy seta
Correlation coefficients
Effectivefree energyand RMSD
Effective free energy andfractional occurrence of
native contacts
1. 4_state_reduced1ctf 0.68 0.811r69 0.67 0.751sn3 0.58 0.662cro 0.64 0.693icb 0.78 0.834pti 0.55 0.624rxn 0.64 0.68
2. fisa1fc2 0.47 0.551hdd-C 0.34 0.532cro 0.15 0.334icb �0.04 0.19
3. fisa_casp31bg8-A 0.09 0.321bl0 0.08 0.181eh2 0.19 0.23
4. lmds1b0n-B 0.10 0.361ctf 0.18 0.411fc2 0.02 0.031igd 0.14 0.271shf-A 0.02 0.082cro 0.16 0.181dtk 0.05 0.122ovo 0.05 0.094pti 0.04 0.30
5. lattice_ssfit1ctf �0.04 0.071dkt-A �0.05 �0.0011fca �0.03 �0.021pgb �0.03 0.051trl-A �0.05 0.144icb �0.002 0.171beo 0.02 0.061nkl 0.01 �0.04
†The fractional occurrence of native contacts is the ratio of the numberof native contacts in a decoy over the total number of contacts in nativestructure. To facilitate comparisons with the RMSD results, thecorrelations have been calculated with one minus the fractionaloccurrence of native contacts.aFor details, see Table I footnotes.
PREDICTING NATIVE CONTACTS IN PROTEINS 603
CONCLUSIONS
In this work, we investigated how well native contactscan be predicted based on decoy structures and an effectiveenergy function. Besides examining the capability of theGROMOS96/GBSA effective free energy function, our in-vestigation intends to shed light on the potentiality ofprogressive ab initio structure prediction methods. Sam-udrala et al.38–40 have proposed a consensus-based dis-tance geometry method to predict the native fold, utilizingthe statistical knowledge of residue–residue distancesfrom an ensemble of low energy conformations. In theirmethod, every inter-C distance was measured and thosemost frequently occurring distances were used as inputrestraints of distance geometry.
In this work, we examined the effectiveness of theGROMOS96-GB/SA energy function in distinguishing thenative fold from non-native folds for five decoy databasesconstructed in various manners by others. Our results areconsistent with the reported results in recent works, suchas the detailed investigation of OPLS-AA/SGB energyfunction by Felts et al.,50 and aspects extensively dis-cussed by those authors were not presented in detail in thecurrent article. Herein, we focused on the correlationbetween the effective free energy and the extent of similar-
ity with native structure of a decoy. As pointed out byVorobjev and Hermans,41 the correlation between theeffective free energy and the RMSD from the nativestructure is weak and has no consistency among differentdecoy sets. Realizing the limitation of the RMSD, we usethe fractional occurrence of native contacts in a decoystructure to measure its “nativeness.” The correlationsbetween the effective energy with this measurement ex-ceed those with the RMSD for all decoy sets. Particularly,the 4_state_reduced sets show significantly stronger lin-ear correlations between the effective free energy and thefractional occurrences of native contacts than the otherdecoy sets.
Based on these observations, we finally looked at thepossibility of predicting native contacts using decoy struc-tures and the effective energy function. Two predictionstrategies, one based on energy-weighted occurrence fre-quencies and the other on plain occurrence frequencies,were compared in terms of the accuracy and sensitivity ofthe predictions. First, we observed that non-trivial predic-tions can be made based on the decoy databases usingeither strategy. Second, for only one of the decoy data-bases, the 4_state_reduced sets, using the energy-weightedfrequencies leads to significantly more sensitive predic-
Fig. 2. Correlations between the GROMOS96/GBSA effective free energies and one minus the fractional occurrences of native contacts in decoys.The dotted horizontal lines represent the effective free energies of the native structures. The 1ctf, 1hdd-C, 1eh2, 4pti, and 4icb decoys are from the4_state_reduced, fisa, fisa_casp3, lmds, and lattice_ssfit decoy set databases, respectively.
604 J. ZHU ET AL.
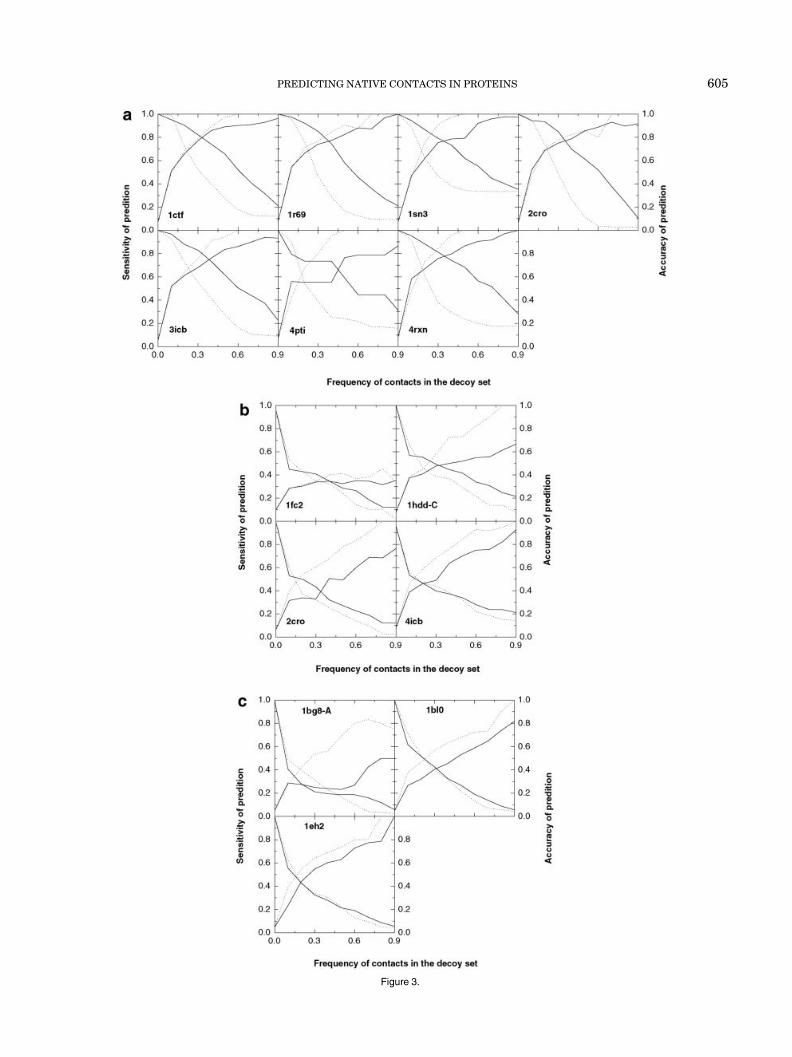
Figure 3.
PREDICTING NATIVE CONTACTS IN PROTEINS 605
tions. For the other decoy sets, including the fisa andfisa_casp3 decoy sets of Simons et al., the energy-weightedapproach does not improve the prediction. This can berationalized by the fact that decoys in the 4_state_reducedsets contain various portions of native contacts, whereasthose in the other sets tend to have similar portions ofnative contacts.
Besides adding another piece of evidence that provesphysics-based atomic potential energy functions can beuseful in ab initio structure prediction, the implications ofour results are the following. First, if a proper measure-ment for similarity is used, the free energy of a protein
conformation does correlate with the extent of similaritybetween the conformation and the native conformation.One aspect of the funnel theory for protein folding is thatthe bottom of the free energy landscape should correspondto the neighborhood of the native structure. Thus, when adecoy set samples the neighborhood of the native struc-ture, we see correlations between the nativeness and theenergies. When the set does not sample the bottom of thefunnel, the degree of native contacts is always low andlittle correlation between the nativeness and the energyshould be expected. One appropriate choice of the measure-ment for “nativeness” is the fractional occurrence of native
Fig. 3. The dependence of accuracy and sensitivity of prediction on the threshold frequency of contacts in decoy sets. Accuracy is the proportion inwhich contacts with frequencies higher than the threshold are actually native contacts. Sensitivity is the ratio of the number of native contacts withfrequencies higher than the threshold to the total number of native contacts observed in the native structure. The declining and increasing lines withincreasing threshold correspond to the sensitivity and the accuracy, respectively. The solid lines represent the energy-dependent weighting treatmentand the dashed lines represent the unit-weighting treatment. (a), (b), (c), (d), and (e) correspond to the 4_state_reduced, fisa, fisa_casp3, lmds, andlattice_ssfit decoy sets, respectively.
606 J. ZHU ET AL.

contacts. Secondly, iterative approaches to structure pre-diction can be designed based on this correlation, in whichnative characteristics may be statistically and progres-sively extracted from an ensemble of trial conformations.The optimum strategy of making use of the effective freeenergy function in such an approach, however, woulddepend on details of how the ensemble of trial conforma-tions would be sampled.
ACKNOWLEDGMENTS
H.Y. Liu acknowledges financial support from the Chi-nese National Natural Science Foundation (Grant30025013). Y.Y. Shi acknowledges support from the Na-tional Basic Research Projects (Grant G1999075605) andthe Chinese National Natural Science Foundation (Grant39990600). We are grateful to Professor Yaoqi Zhou forsuggesting that we investigate protein decoy structuresusing the GROMOS/GBSA model, sending us one of hismanuscripts before publication, and pointing out an errorin our manuscript. We are grateful to Professor W.F. vanGunsteren of the Department of Chemistry, ETH-Zurich,for providing the GROMOS96 package.
REFERENCES
1. Anfinsen CB. Principles that govern the folding of protein chains.Science 1973;181:223–230.
2. Sippl MJ. Knowledge-based potentials for proteins. Curr OpinStruct Biol 1995;5:229–235.
3. Jernigan RL, Bahar I. Structure-derived potentials and proteinsimulations. Curr Opin Struct Biol 1996;6:195–209.
4. Koppensteiner WA, Sippl MJ. Knowledge-based potentials: backto the roots. Biochemistry (Moscow) 1998;63:247–252.
5. Luthy R, Bowie JU, Eisenberg D. Assessment of protein modelswith three-dimensional profiles. Nature 1992;356:83–85.
6. Lazaridis T, Karplus M. Effective energy functions for proteinstructure prediction. Curr Opin Struct Biol 2000;10:139–145.
7. Feig M, Brooks CL III. Evaluating CASP4 predictions withphysical energy functions. Proteins 2002;49:232–245.
8. Park B, Levitt M. Energy functions that discriminate X-ray andnear-native folds from well-constructed decoys. J Mol Biol 1996;258:367–392.
9. Lee MR, Duan Y, Kollman PA. Use of MM-PB/SA in estimatingthe free energies of proteins: application to native, intermediates,and unfolded villin headpiece. Proteins 2000;39:309–316.
10. Vorobjev YN, Almagro JC, Hermans J. Discrimination betweennative and intentionally misfolded conformations of proteins:ES/IS, a new method for calculating conformational free energythat uses both dynamics simulations with an explicit solvent andan implicit solvent continuum model. Proteins 1998;32:399–413.
11. Novotny J, Bruccoleri RE, Karplus M. An analysis of incorrectlyfolded protein models: implications for structure prediction. J MolBiol 1984;177:787–818.
12. Novotny J, Rashin AA, Bruccoleri RE. Criteria that discriminatebetween native proteins and incorrectly folded models. Proteins1988;4:19–30.
13. Wang Y, Zhang H, Li W, Scott RA. Discriminating compactnonnative structures from the native structure of globular pro-teins. Proc Natl Acad Sci USA 1995;92:709–713.
14. Marti-Renom MA, Stote RH, Querol E, Aviles FX, Karplus M.Refolding of potato carboxypeptidase inhibitor by molecular dy-namics simulations with disulfide bond constraints. J Mol Biol1998;284:145–172.
15. Moult J. Comparison of database potentials and molecular mechan-ics force fields. Curr Opin Struct Biol 1997;7:194–199.
16. Park BH, Huang ES, Levitt M. Factors affecting the ability ofenergy functions to discriminate correct from incorrect folds. J MolBiol 1997;266:831–846.
17. Smith PE, Pettitt BM. Modeling solvent in biomolecular systems.J Phys Chem 1994;98:9700–9711.
18. Cramer CJ, Truhlar DG. Implicit solvation models: equilibria,structure, spectra, and dynamics. Chem Rev 1999;99:2161–2200.
19. DeBolt SE, Kollman PA. Investigation of structure, dynamics, andsolvation in 1-octanol and its water-saturated solution: moleculardynamics and free-energy perturbation studies. J Am Chem Soc1995;117:5316–5340.
20. Srinivasan J, Miller J, Kollman PA, Case DA. Continuum solventstudies of the stability of RNA hairpin loops and helices. J BiomolStruct Dyn 1998;16:671–682.
21. Vorobjev YN, Hermans J. ES/IS: estimation of conformational freeenergy by combining dynamics simulations with explicit solventwith an implicit solvent continuum model. Biophys Chem 1999;78:195–205.
22. Still WC, Tempczyk A, Hawley RC, Hendrickson T. Semianalyti-cal treatment of solvation for molecular mechanics and dynamics.J Am Chem Soc 1990;112:6127–6129.
23. Hawkins GD, Cramer CJ, Truhlar DG. Parametrized models ofaqueous free energies of solvation based on pairwise descreeningof solute atomic charges from a dielectric medium. J Phys Chem1996;100:19824–19839.
24. Ghosh A, Rapp CS, Friesner RA. Generalized Born model based ona surface integral formulation. J Phys Chem B 1998;102:10983–10990.
25. Qiu D, Shenkin PS, Hollinger FP, Still WC. The GB/SA continuummodel for solvation. A fast analytical method for the calculation ofapproximate Born radii. J Phys Chem A 1997;101:3005–3014.
26. Dominy BN, Brooks CL III. Development of a generalized Bornmodel parameterization for proteins and nucleic acids. J PhysChem B 1999;103:3765–3773.
27. Lee MS, Salsbury FR Jr, Brooks CL III. Novel generalized Bornmethods. J Chem Phys 2002;116:10606–10614.
28. Zhu J, Shi Y, Liu H. Parameterization of a generalized Born/solvent-accessible surface area model and applications to thesimulation of protein dynamics. J Phys Chem B 2002;106:4844–4853.
29. Wesson L, Eisenberg D. Atomic solvation parameters applied tomolecular dynamics of proteins in solution. Protein Sci 1992;1:227–235.
30. Klepeis JL, Floudas CA. A comparative study of global minimumenergy conformations of hydrated peptides. J Comp Chem 1999;20:636–654.
31. Ferrara P, Apostolakis J, Caflish A. Evaluation of a fast implicitsolvent model for molecular dynamics simulations. Proteins 2002;46:24–33.
32. Lazaridis T, Karplus M. Discrimination of the native from mis-folded protein models with an energy function including implicitsolvation. J Mol Biol 1999;288:477–487.
33. Daggett V, Kollman PA, Kuntz ID. Molecular dynamics simula-tions of small peptides: dependence on dielectric model and pH.Biopolymers 1991;31:285–304.
34. Guenot J, Kollman PA. Conformational and energetic effects oftruncating nonbonded interactions in an aqueous protein dynam-ics simulation. J Comp Chem 1993;14:295–311.
35. Hingerty BE, Ritchie RH, Ferrel TL, Turner JE. Dielectric effectsin biopolymers: the theory of ionic saturation revisited. Biopoly-mers 1985;24:427–439.
36. Ramstein J, Lavery R. Energetic coupling between DNA bendingand base pair opening. Proc Natl Acad Sci USA 1988;85:7231–7235.
37. Hill TL. Influence of electrolyte on effective dielectric constants inenzymes, proteins and other molecules. J Phys Chem 1956;60:253–255.
38. Huang ES, Samudrala R, Ponder JW. Distance geometry gener-ates native-like folds for small helical proteins using the consen-sus distances of predicted protein structures. Protein Sci 1998;7:1998–2003.
39. Samudrala R, Xia Y, Huang E, Levitt M. Ab initio proteinstructure prediction using a combined hierarchical approach.Proteins 1999;Suppl 3:194–198.
40. Xia Y, Huang ES, Levitt M, Samudrala R. Ab initio construction ofprotein tertiary structures using a hierarchical approach. J MolBiol 2000;300:171–185.
41. Vorobjev YN, Hermans J. Free energies of protein decoys provideinsight into determinants of protein stability. Protein Sci 2001;10:2498–2506.
42. Miyazawa S, Jernigan RL. Estimation of effective interresidue
PREDICTING NATIVE CONTACTS IN PROTEINS 607

contact energies from protein crystal structures: quasi-chemicalapproximation. Macromolecules 1985;18:534–552.
43. Miyazawa S, Jernigan RL. Residue–residue potentials with afavorable contact pair term and an unfavorable high packingdensity term, for simulation and threading. J Mol Biol 1996;256:623–644.
44. Sippl MJ. Calculation of conformational ensembles from poten-tials of mean force. An approach to the knowledge-based predic-tion of local structures in globular proteins. J Mol Biol 1990;213:859–883.
45. Hendlich M, Lackner P, Weitckus S, et al. Identification of nativeprotein folds amongst a large number of incorrect models. Thecalculation of low energy conformations from potentials of meanforce. J Mol Biol 1990;216:167–180.
46. Casari G, Sippl MJ. Structure-derived hydrophobic potential.Hydrophobic potential derived from X-ray structures of globularproteins is able to identify native folds. J Mol Biol 1992;224:725–732.
47. Bowie JU, Luthy R, Eisenberg D. A method to identify proteinsequences that fold into a known three-dimensional structure.Science 1991;253:164–170.
48. Wilmanns M, Eisenberg D. Three-dimensional profiles from resi-due-pair preferences: identification of sequences with beta/alpha-barrel fold. Proc Natl Acad Sci USA 1993;90:1379–1383.
49. Zhou H, Zhou Y. Distance-scaled, finite ideal-gas reference stateimproves structure-derived potentials of mean force for structureselection and stability prediction. Protein Sci 2002;11:2714–2726.
50. Felts AK, Gallicchio E, Wallqvist A, Levy RM. Distinguishingnative conformations of proteins from decoys with an effective freeenergy estimator based on the OPLS all-atom force field and the
surface generalized Born solvent model. Proteins 2002;48:404–422.
51. Boczko EM, Brooks CL III. First-principles calculation of thefolding free energy of a three-helix bundle protein. Science 1995;269:393–396.
52. Sheinerman FB, Brooks CL III. Calculations on folding of segmentB1 of streptococcal protein G. J Mol Biol 1998;278:439–456.
53. Samudrala R, Levitt M. Decoys ‘R’ Us: a database of incorrectconformations to improve protein structure prediction. Protein Sci2000;9:1399–1401.
54. Simons KT, Kooperberg C, Huang E, Baker D. Assembly of proteintertiary structures from fragments with similar local sequencesusing simulated annealing and Bayesian scoring functions. J MolBiol 1997;268:209–225.
55. Samudrala R, Xia Y, Levitt M, Huang ES. A combined approachfor ab initio construction of low resolution protein tertiary struc-tures from sequence. Pac Symp Biocomput 1999;505–516.
56. Levitt M, Lifson S. Refinement of protein conformations using amacromolecular energy minimization procedure. J Mol Biol 1969;46:269–279.
57. Levitt M. Energy refinement of hen egg-white lysozyme. J MolBiol 1974;82:393–420.
58. Levitt M. Molecular dynamics of native protein. II. Analysis andnature of motion. J Mol Biol 1983;168:595–620.
59. Levitt M, Hirshberg M, Sharon R, Daggett V. Potential energyfunction and parameters for simulations of the molecular dynam-ics of proteins and nucleic acids in solution. Comput Phys Com-mun 1995;91:215–231.
60. Samudrala R, Moult J. An all-atom distance-dependent condi-tional probability discriminatory function for protein structureprediction. J Mol Biol 1998;275:895–916.
608 J. ZHU ET AL.





























