Embed Size (px)
Citation preview

Received: 30 May 2001Accepted: 5 February 2002Published online: 4 April 2002© Springer-Verlag 2002
Abstract Objectives: To study theeffect of hyperbaric oxygen therapyin alleviating acute lung injury in-duced by lipopolysaccharide (LPS)in rats. Design and interventions:The rats received an intraperitonealinjection of LPS (15 mg/kg). Ani-mals were either breathing air at1 ATA or subjected to hyperbaricoxygen (HBO2) therapy. The HBO2therapy was carried out in a hyper-baric chamber at a pressure of 3 ATAfor 90 min. In another two groups,LPS-treated rats also received intra-peritoneal injection of Nω-nitro-L-arginine (LNAME, 25 mg/kg) or L-N6-(iminoethyl)lysine (LNIL,10 ml/kg). Another two groups ofLPS-treated rats were subjected toHBO2 exposure after the injection ofL-NAME or L-NIL. Measurementsand main results: The bronchoalveo-lar lavage (BAL) was done into theleft lung at 7.5 h after intraperitonealinjection of LPS. Parts of the rightlung were excised for myeloperoxi-dase measurement, whereas the rest
was collected for wet/dry ratio deter-mination. LPS significantly in-creased the nitrite/nitrate (NOx
-) con-centration (34.4±15.7 vs4.5±3.1 µM), LDH activity (66±17vs 46±15 mAbs/min), and proteinconcentration (373±119 vs180±90 mg/l) in the BAL fluid.Treatment with HBO2 immediatelyafter the injection of LPS enhancedthe increase of NOx
- production, butreduced the LDH and protein inBAL fluid to the control levels. Pre-treatment with either L-NAME or L-NIL abolished the increase ofNOx
- in the BAL fluid and further el-evated the LDH level and proteinconcentration. Conclusion: Our re-sults suggested that HBO2 alleviatesthe LPS-induced acute lung injury,which may be related to the enhance-ment of nitric oxide production.
Keywords Lipopolysaccharide ·Acute lung injury · Hyperbaricoxygen · Nitric oxide
Intensive Care Med (2002) 28:636–641DOI 10.1007/s00134-002-1262-1 E X P E R I M E N TA L
M.-Y. LuB.-H. KangF.-J. WanC.-S. ChenK.-L. Huang
Hyperbaric oxygen attenuates lipopolysaccharide-induced acute lung injury
Introduction
Oxygen therapy remains the main treatment strategy foracute respiratory distress syndrome (ARDS). Refractoryhypoxemia is the key criteria for diagnosis of ARDS[1]. Improvement of arterial oxygenation (PaO2) indi-cates the effectiveness of treatment. Increased PaO2 de-livers adequate tissue oxygenation and may prevent pa-tients with ARDS from progressing to multiple organdysfunction. Although it is debatable whether an in-
crease in arterial oxygenation always results in a favor-able outcome for ARDS, much effort has been devotedto this mode of treatment [2, 3, 4, 5]. Ventilatory sup-port strategies have been extensively investigated aim-ing to improve PaO2 by a submaximal fraction of inspi-ratory oxygen [6,7]. Adjunctive pharmacological thera-pies targeting the systemic inflammation of early ARDShave also been widely studied [8, 9,10]. Despite a vari-ety of strategies, however, the mortality from ARDS re-mains stable.
M.-Y. Lu · B.-H. Kang · F.-J. WanK.-L. Huang (✉ )Institute of Undersea and Hyperbaric Medicine, National Defense Medical Center, 161 Ming-Chuan E. Road, Sec. 6, Taipei 114, Taiwan R.O.C.e-mail: [email protected].: +886-2-87924873Fax: +886-2-87924873
C.-S. Chen · K.-L. HuangDivision of Chest Medicine, Department of Internal Medicine, Tri-Service General Hospital, Taipei, Taiwan R.O.C.

Gram-negative sepsis is the most common risk factorfor the development of ARDS [11,12], although it maybe associated with a number of different conditions in-cluding major trauma, multiple transfusion, and aspira-tion of gastric contents [13]. Lipopolysaccharide (LPS),being unique to the Gram-negative bacteria, is a potentmicrobial product inducing a severe host inflammatoryresponse and the resultant tissue injur, including thelungs. LPS may produce its toxic effects on the lungs bydirect injury to endothelial cells and by indirect activa-tion of inflammatory cells and the production of cyto-kines as well as cytotoxic agents. LPS-induced lung inju-ry is an appropriate model for ARDS experiments [14].
Hyperbaric oxygen therapy (HBO2) is an intermittentadministration of 100% oxygen at a pressure greater thanat sea level. HBO2 therapy has been also recognized asan adjunctive treatment for several hypoxia-related dis-eases; however, it has not been tested in the treatment ofARDS. HBO2 provides a high partial pressure of oxygenin the inspiratory air which can facilitate oxygen diffu-sion through the edematous air-blood barrier and, theo-retically, may reverse hypoxemia as well as tissue hyp-oxia in ARDS. Intermittent administration is anothercharacteristic of HBO2 therapy which may be a solutionto the complications of oxygen ventilation. Furthermore,in an endothelial culture of human umbilical vein, Buraset al. [15] demonstrated that HBO2 enhanced eNOS ex-pression in the endothelial cells, inhibited expression ofintercellular adhesion molecules, and reduced neutrophiladhesion to the endothelial cells. This report suggestedthat HBO2 may reduce the neutrophil-dependent cellulardamage. However, a favorable outcome of ARDS hasnever been demonstrated after HBO2 therapy. In thepresent study, we investigated the effects of HBO2 onLPS-induced acute lung injury.
Material and methods
Animals
Male Sprague-Dawley rats weighing 250–350 g were used in thisstudy. All the experimental procedures were in accordance withthe Guiding Principles in the Care and Use of Animals approvedby the Institutional Animal Care and Use Committee.
Experimental protocols
The rats received an intraperitoneal injection of LPS (15 mg/kg).Animals were either breathing air at 1 ATA (LPS group, n=8) orsubjected to HBO2 exposure immediately after LPS injection(LPS/HBO group, n=6). The HBO group (n=7) received HBO2treatment after saline injection, whereas the control group (n=6)received only saline injection without HBO2. In the other twogroups, LPS-treated rats also received an intraperitoneal injectionof a nonselective inhibitor of nitric oxide synthase (NOS), Nω-nitro-L-arginine (25 mg/kg, LPS/LNAME group, n=6) or an inducible NOS inhibitor, L-N6-(iminoethyl)lysine (10 mg/kg,LPS/LNIL group, n=6). Another two groups of LPS-treated rats
were subjected to the HBO2 exposure after the injection of L-NAME (LPS/LNAME/HBO group, n=7) or L-NIL (LPS/LNIL/HBO group, n=6).
Rat was anesthetized by intraperitoneal injection of sodiumpentobarbital (50 mg/kg, i.p.) 7.5 h after the injection of LPS, fol-lowed by tracheal intubation and midline thoracotomy. The rightlung was clamped and excised for myeloperoxidase (MPO) mea-surement and the wet/dry weight ratio (W/D) determination. Bron-choalveolar lavage (BAL) was done to the left lung.
HBO2 exposure
The rats were placed in a hyperbaric chamber designed for animalstudies. The pressure inside the chamber was increased to threeabsolute atmospheres (ATA) at a rate of 2 ATA/min and was main-tained at that pressure for 90 min. The chamber was ventilatedwith 100% oxygen at a flow rate of 20 l/min to minimize CO2 ac-cumulation. The rats were then decompressed at a rate of1 ATA/min.
Bronchoalveolar lavage
BAL was performed to the left lung with 5 ml phosphate-balancedsaline in 2.5 ml aliquots after cannulation of the left trachea. Therecovered BAL fluid was centrifuged at 250 g for 10 min. Activityof LDH was measured by the method described by Vassault [16].In brief, part of the supernatant was incubated with 0.24 mMNADH in a Tris/NaCl pH 7.2 buffer at room temperature for5 min. The reaction was then started by the addition of 9.8 mMpyruvate and followed by using a spectrophotometer at 340 nm for2 min. The protein concentration of the supernatant was deter-mined using BCA protein assay reagents (Pierce, Rockford, Ill.,USA). Nitrate/nitrite (total NOx
-) concentration was measured byusing a chemiluminescence technique. Briefly, BAL fluid was de-proteinized with an equal volume of ice-cold absolute ethanol.The NOx
- in the supernatant was reduced with vanadium to NO,which in turn reacts with O3 to produce *NO2. The energy emittedfrom *NO can be detected by a chemiluminescence technique[17]. A spectrophotometric method [18] was used to determinemyeloperoxidase activity in the lung tissue. In brief, the specimenwas freeze-thawed and sonicated three times. Homogenates wascentrifuged at 15,000 g for 10 min at 4 °C. A 100-µl supernatantwas mixed with 900 µl of 50 mM phosphate buffer (pH 6.0) con-taining 0.167 mg/ml of o-dianisidine dihydrochloride and0.0005% hydrogen peroxide. One unit of peroxidase activityequals the amount of enzyme decomposing 1 µmol of hydrogenperoxide per min at 25 °C. Decomposition of hydrogen peroxide iscalculated from the oxidation of o-dianisidine using an absorptioncoefficient of 11.3·mM·cm at 460 nm.
Statistical analysis
The data were expressed as mean±SD. The differences betweentreatment groups, wet/dry ratio of lungs, and BLF analysis amonggroups were evaluated by using one-way ANOVA. When the vari-ables were found to be different, a multiple comparison test (Fisher’s PLSD) was performed utilizing StatView 5.0 softwarepackage (SAS Institute, Cary, N.C., USA). A value of P<0.05 wasaccepted as significant.
637
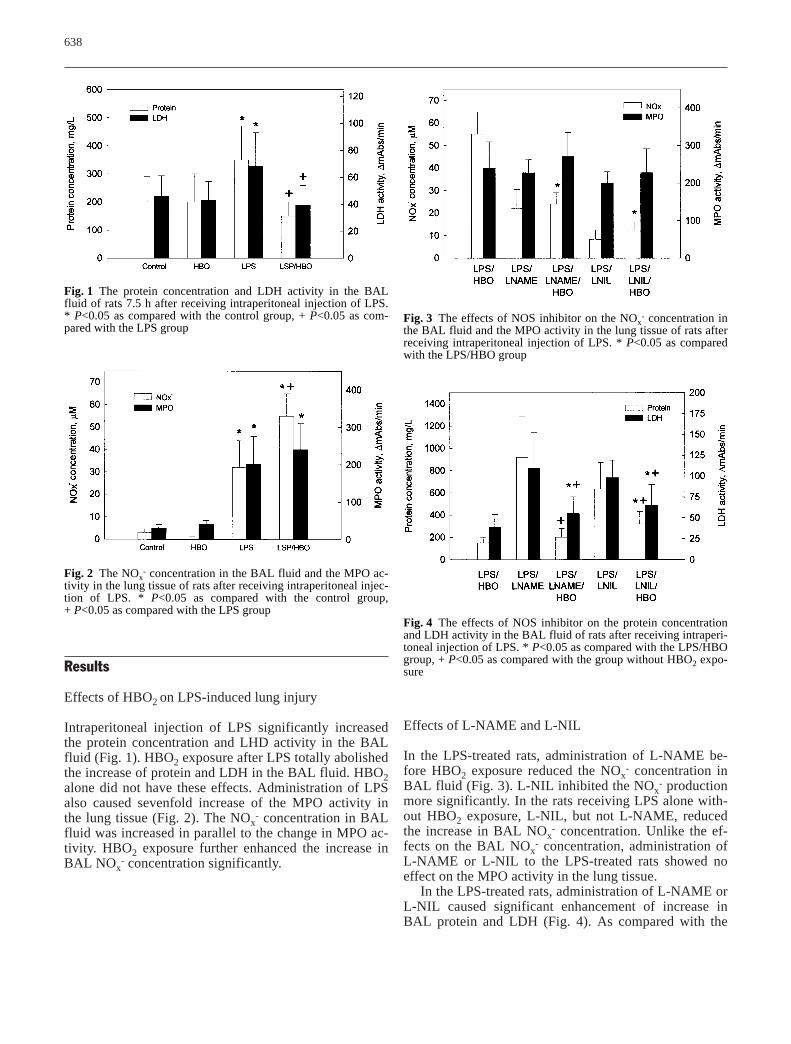
638
Results
Effects of HBO2 on LPS-induced lung injury
Intraperitoneal injection of LPS significantly increasedthe protein concentration and LHD activity in the BALfluid (Fig. 1). HBO2 exposure after LPS totally abolishedthe increase of protein and LDH in the BAL fluid. HBO2alone did not have these effects. Administration of LPSalso caused sevenfold increase of the MPO activity inthe lung tissue (Fig. 2). The NOx
- concentration in BALfluid was increased in parallel to the change in MPO ac-tivity. HBO2 exposure further enhanced the increase inBAL NOx
- concentration significantly.
Effects of L-NAME and L-NIL
In the LPS-treated rats, administration of L-NAME be-fore HBO2 exposure reduced the NOx
- concentration inBAL fluid (Fig. 3). L-NIL inhibited the NOx
- productionmore significantly. In the rats receiving LPS alone with-out HBO2 exposure, L-NIL, but not L-NAME, reducedthe increase in BAL NOx
- concentration. Unlike the ef-fects on the BAL NOx
- concentration, administration ofL-NAME or L-NIL to the LPS-treated rats showed noeffect on the MPO activity in the lung tissue.
In the LPS-treated rats, administration of L-NAME orL-NIL caused significant enhancement of increase inBAL protein and LDH (Fig. 4). As compared with the
Fig. 1 The protein concentration and LDH activity in the BALfluid of rats 7.5 h after receiving intraperitoneal injection of LPS.* P<0.05 as compared with the control group, + P<0.05 as com-pared with the LPS group
Fig. 2 The NOx- concentration in the BAL fluid and the MPO ac-
tivity in the lung tissue of rats after receiving intraperitoneal injec-tion of LPS. * P<0.05 as compared with the control group, + P<0.05 as compared with the LPS group
Fig. 3 The effects of NOS inhibitor on the NOx- concentration in
the BAL fluid and the MPO activity in the lung tissue of rats afterreceiving intraperitoneal injection of LPS. * P<0.05 as comparedwith the LPS/HBO group
Fig. 4 The effects of NOS inhibitor on the protein concentrationand LDH activity in the BAL fluid of rats after receiving intraperi-toneal injection of LPS. * P<0.05 as compared with the LPS/HBOgroup, + P<0.05 as compared with the group without HBO2 expo-sure

has been used as an indicator of cellular damage in sev-eral cultured cell models [19,20]. In other models of lunginjury, protein concentration and LDH level in the BALfluid have been widely used as an in vivo index of lunginjury [21, 22,23]. In the present study, LPS injectioncaused significant increases in concentrations of BALprotein and LDH activity, suggesting lung injury after in-traperitoneal injection of LPS. However, in the presentstudy, LPS did not cause a significant increase in theW/D weight ratio of the lung (Fig. 5). The lung injury in-duced by intraperitoneal injection of LPS in this studymay be in a stage earlier than than acute pulmonary ede-ma or ARDS. Normally, animals rarely survive an acutepulmonary edema without ventilation support. There-fore, we demonstrated an animal model of acute lung in-jury, but in an early stage.
Understanding the mechanism of LPS-induced lunginjury may pave an way for the effective treatment ofARDS. It has been well documented that the adhesionand activation of neutrophils [24,25] and the consequentrelease of cytotoxic agents play an important role in thepathogenesis of lung injury after severe sepsis [26,27].We demonstrated that intraperitoneal injection of LPSsignificantly increases the MPO activity in the lung tis-sue, indicating the increased sequestration of neutrophilsin the lung. However, HBO2 exposure did not reduce theMPO activity in the lung tissue although it did reducethe BAL LDH activity and protein concentration. Acti-vated neutrophils release not only protease and oxygenradicals causing tissue injury directly, but also many cy-tokine and chemokines amplifying the detrimental re-sponse [27]. On the other hand, administration of LPSmay induce expression and production of bioprotectionfactors such as heat shock proteins [28]. It remains to beinvestigated further whether HBO2 exposure alter theproduction of cytokines or heat shock proteins.
Nitric oxide plays a controversial role in the patho-genesis of acute lung injury. Evidence has shown thatNO in high concentrations may cause lung injury [29,30]either by itself or by the formation of peroxynitritewhich is a highly active radical [31]. Others have shownthat inhibition of NO synthesis by L-NAME enhancedLPS-induced acute lung injury [32]. In iNOS-deficientmice, Kobayashi et al. [33] demonstrated that iNOS in-duction serves as a protective mechanism to minimizelung injury after hyperoxic exposure. Our results showedthat injection of LPS increased NOx
- production in thebronchoalveolar space. Inhibition of NO production by anon-selective NOS inhibitor, L-NAME,or a selectiveiNOS inhibitor, L-NIL, significantly enhanced the LPS-induced lung injury. These results are compatible withKobayashi’s finding and suggest that production of NOx
-
may play a role counteracting the detrimental effect ofLPS to the lung.
HBO2 exposure enhanced the production of NOx- and
at the same time attenuated the LPS-induced lung injury.
639
LPS/LNAME group or the LPS/LNIL group, HBO2 ex-posure significantly attenuated these enhancements. Onthe other hand, as compared with the LPS/HBO group,administration of L-NAME or L-NIL caused significantincreases in the BAL protein and LDH activity.
Changes in W/D ratio of lung weight
Figure 5 summarizes the changes in the W/D ratio oflung weight. Intraperitoneal administration of LPScaused a slight, but not statistically significant, increaseof the W/D ratio. Neither L-NAME nor L-NIL enhancedthe lung weight response to LPS. Rats which only re-ceived HBO2 had a lower W/D ratio compared to thecontrol group. In the rats which received LPS or LPSand NOS inhibitors, however, HBO2 did not cause sig-nificant reduction in the W/D ratio.
Discussion
The present study demonstrated that intraperitoneal in-jection of LPS caused an acute lung injury as evidencedby increased MPO activity in lung tissue, and elevatedprotein concentration and LDH activity in BAL fluid.HBO2 therapy applied immediately after administrationof LPS reduced the increases in BAL protein and LDHactivity. This suggested that HBO2 therapy may alleviatethe acute lung injury caused by LPS.
Clinically, sepsis remains the most common cause ofacute lung injury or ARDS. It manifests a diffused pul-monary infiltration and refractory hypoxemia. In animalexperiments, the indexes of an acute lung injury includevascular permeability, wet/dry weight ratio of lung, andLDH activity and protein in BAL. The release of LDH
Fig. 5 The W/D ratio of lung weight of rats. * P<0.05 as com-pared with the control group

640
Ito et al. [34] demonstrated in rats that HBO2 at 3 ATAfor 2 h significantly increased the arginine metabolismsuggesting increase in NO generation during hyperbaricoxygen exposure. Using an in vivo microdialysis model,Elayan et al. [35] demonstrated in rat brain that HBO2treatment caused a sixfold increase in nitrite/nitrate. Al-though the authors suggested that the increase in NO lev-els may be responsible for the detrimental effects ofHBO2, they did not find a protective effect of L-NAMEpretreatment. Furthermore, Buras et al. [15] demonstrat-ed in an endothelial culture that HBO2 downregulatedthe expression ICAM-1 induced by hypoxia/hypoglyce-mia and the effects of HBO2 were attenuated by pretreat-
ment with L-NAME. Our results showed that in the ratstreated with L-NAME or L-NIL, in which intraperitonealinjection of LPS caused a significantly lung injury,HBO2 exposure reduced the increases in BAL proteinconcentration and LDH activity. These results suggestthat HBO2 may protect the lung from LPS-induced inju-ry by enhancement of NO production.
In summary, intraperitoneal injection of LPS caused asignificant lung injury in the rat. Exposure to HBO2 re-duced the acute lung injury caused by LPS injection. Thebeneficial effects of HBO2 were nullified by administra-tion of NOS inhibitor. These results suggest that HBO2may alleviate the LPS-induced acute lung injury in rats.
References
1. Bernard GR, Artigas A, Brigham KL,Carlet J, Falke K, Hudson L, Lamy M,Legall JR, Morris A, Spragg R (1994)The American-European ConsensusConference on ARDS. Definitions,mechanisms, relevant outcomes, andclinical trial coordination. Am J RespirCrit Care Med 149:818–824
2. Langer M, Mascheroni D, Marcolin R,Gattinoni L (1988) The prone positionin ARDS patients. A clinical study.Chest 94:103–107
3. Rossaint R, Falke KJ, Lopez F, SlamaK, Pison U, Zapol WM (1993) Inhalednitric oxide for the adult respiratorydistress syndrome. N Engl J Med328:399–405
4. Stocker R, Neff T, Stein S, Ecknauer E,Trentz O, Russi E (1997) Prone posi-tioning and low-volume pressure-limit-ed ventilation improve survival in pa-tients with severe ARDS. Chest111:1008–1017
5. Papazian L, Bregeon F, Gaillat F, Thirion X, Gainnier M, Gregoire R,Saux P, Gouin F, Jammes Y, Auffray JP(1998) Respective and combined ef-fects of prone position and inhaled ni-tric oxide in patients with acute respi-ratory distress syndrome. Am J RespirCrit Care Med 157:580–585
6. Brandstetter RD, Sharma KC, DellaBadia M, Cabreros LJ, KabinoffGS (1997) Adult respiratory distresssyndrome: a disorder in need of im-proved outcome. Heart Lung 26:3–14
7. McIntyre RC Jr, Pulido EJ, BensardDD, Shames BD, Abraham E (2000)Thirty years of clinical trials in acuterespiratory distress syndrome. CritCare Med 28:3314–3331
8. Payen DM, Gatecel C, Plaisance P(1993) Almitrine effect on nitric oxideinhalation in adult respiratory distresssyndrome. Lancet 341:1664
9. Jolliet P, Bulpa P, Ritz M, Ricou B,Lopez J, Chevrolet JC (1997) Additivebeneficial effects of the prone position,nitric oxide, and almitrine bismesylateon gas exchange and oxygen transportin acute respiratory distress syndrome.Crit Care Med 25:786–794
10. Weigelt JA, Gewertz BL, AurbakkenCM, Snyder WH 3rd (1982) Pharmaco-logic alterations in pulmonary arterypressure in the adult respiratory dis-tress syndrome. J Surg Res 32:243–248
11. Kaplan RL, Sahn SA, Petty TL (1979)Incidence and outcome of the respira-tory distress syndrome in Gram-nega-tive sepsis. Arch Intern Med139:867–869
12. Martin MA, Silverman HJ (1992)Gram-negative sepsis and the adult res-piratory distress syndrome. Clin InfectDis 14:1213–1228
13. Yamada H, Miyazaki H, Kikuchi T, Fujimoto J, Kudoh I (2000) Acid instil-lation enhanced the inflammatory re-sponses to subsequent lipopolysaccha-ride challenge in rats. Am J Respir CritCare Med 162:1366–1371
14. Schuster DP (1994) ARDS: Clinical lesions from the oleic acid model ofacute lung injury. Am J Respir CritCare Med 149:245–260
15. Buras JA, Stahl GL, Svoboda KK, Reenstra WR (2000) Hyperbaric oxy-gen downregulates ICAM-1 expressioninduced by hypoxia and hypoglycemia:the role of NOS. Am J Physiol CellPhysiol. 278:C292–C302
16. Vassault A (1983) Lactate debydrogen-ase. UV-method with pyruvate andNADH. In: Bergmeyer Hu (ed) Meth-ods of enzymatic analysis, 3rd edn. VolIII: enzymes I: oxidoreductase, trans-ferase. Verlag Chemie, Weinheim
17. Beckman JS, Conger K (1995) Directmeasurement of nitric oxide in solutionwith an ozone based chemilumines-cence detector. Methods 7:35–39
18. Day BJ, Shawen S, Liochev SI, CrapoJD (1995) A metalloporphyrin super-oxide dismutase mimetic protectsagainst paraquat-induced endothelialcell injury in vivo. J Pharmacol ExpTherap 275:1227–1232
19. Reindel JF, Hoorn CM, Wagner JG,Roth RA (1991) Comparison of re-sponse of bovine and porcine pulmona-ry arterial endothelial cells to monocro-taline pyrrole. Am J Physiol261:L406–L414
20. Huang KL, Wu JN, Lin HC, Mao SP,Kang B, Wan FJ (2000) Prolonged ex-posure to hyperbaric oxygen inducesneuronal damage in primary rat corticalcultures. Neurosci Lett 293:159–162
21. Smith LJ (1984) Lung damage inducedby butylated hydroxytoluene in mice.Biochemical, cellular, and morphologiccharacterization. Am Rev Respir Dis130:895–904
22. Denis M, Guojian L, Widmer M, Cantin A (1994) A mouse model oflung injury induced by microbial prod-ucts: implication of tumor necrosis fac-tor. Am J Respir Cell Mol Biol10:658–664
23. Tsuji C, Shioya S, Hirota Y, FukuyamaN, Kurita D, Tanigaki T, Ohta Y, Nakazawa H (2000) Increased produc-tion of nitrotyrosine in lung tissue ofrats with radiation-induced acute lunginjury. Am J Physiol Lung Cell MolPhysiol 278:L719-L725
24. Parsons PE, Gillespie MM, Moore EE,Moore FA, Worthen GS (1995) Neutro-phil response to endotoxin in the adultrespiratory distress syndrome: role ofCD14. Am J Respir Cell Mol Biol13:152–160

641
25. Makita H, Nishimura M, Miyamoto K,Nakano T, Tanino Y, Hirokawa J, Nishihira J, Kawakami Y (1998) Effectof anti-macrophage migration inhibito-ry factor antibody on lipopolysaccha-ride-induced pulmonary neutrophil ac-cumulation. Am J Respir Crit CareMed 158:573–579
26. Wollert PS, Menconi MJ, O’SullivanBP, Wang H, Larkin V, Fink MP (1993)LY255283, a novel leukotriene B4 re-ceptor antagonist, limits activation ofneutrophils and prevents acute lung in-jury induced by endotoxin in pigs. Sur-gery 114:191–198
27. Yamasawa H, Ishii Y, Kitamura S(1999) Cytokine-induced neutrophilchemoattractant in a rat model of lipo-polysaccharide-induced acute lung in-jury. Inflammation 23:263–274
28. Ofenstein JP, Heidemann S, Juett-Wilstermann A, Sarnaik A (2000)Expression of stress proteins HSP 72 &HSP 32 in response to endotoxemia.Ann Clin Lab Sci 30:92–98
29. Kristof AS, Goldberg P, Laubach V,Hussain SN (1998) Role of induciblenitric oxide synthase in endotoxin-induced acute lung injury. Am J RespirCrit Care Med 158:1883–1889
30. Matsuo N (1999) The role of intrapul-monary nitric oxide generation in thedevelopment of adult respiratory dis-tress syndrome. Surg Today29:1068–1074
31. Huie RE, Padmaja S (1993) The reac-tion of NO with superoxide. Free RadRes Comm 18:195–199
32. Walley KR, McDonald TE, Higashimoto Y, Hayashi S (1999)Modulation of proinflammatory cyto-kines by nitric oxide in murine acutelung injury. Am J Respir Crit Care Med160:698–704
33. Kobayashi H, Hataishi R, Mitsufuji H,Tanaka M, Jacobson M, Tomita T,Zapol WM, Jones RC (2001) Antiin-flammatory properties of inducible ni-tric oxide synthase in acute hyperoxiclung injury. Am J Respir Cell Mol Biol24:390–397
34. Ito T, Yufu K, Mori A, Packer L (1996)Oxidative stress alters arginine metabo-lism in rat brain: effect of sub-convul-sive hyperbaric oxygen exposure. Neurochem Int 29:187–195
35. Elayan IM, Axley MJ, Prasad PV, Ahlers ST, Auker CR (2000) Effect ofhyperbaric oxygen treatment on nitricoxide and oxygen free radicals in ratbrain. J Neurophysiol 83:2022–2029








![Original Article Salvianolic acid B attenuates lung ischemia ...ijcem.com/files/ijcem0062445.pdfcan relieve lipopolysaccharide-induced acute lung injury in mice [9]. However, the presence](https://img.pdfslide.net/doc/110x75/60ec9a292671437dd15da93b/original-article-salvianolic-acid-b-attenuates-lung-ischemia-ijcemcomfiles.jpg)




















