Embed Size (px)
Citation preview

Identificación de nuevas
proteínas de membrana
externa implicadas en la
resistencia a antimicrobianos
en Klebsiella pneumoniae.
Laura García Sureda
Tesis Doctoral 2011

UNIVERSITAT DE LES ILLES BALEARS
Identificación de nuevas proteínas de membrana externa implicadas en la
resistencia a antimicrobianos en Klebsiella pneumoniae
Tesis Doctoral
Laura García Sureda
Diciembre 2011
Tesis Doctoral dirigida por el Dr. Sebastián Albertí Serrano. Área de Microbiología. Departamento de Biología.
Universitat de les Illes Balears


A la meva família, que passi el que passi sempre està a prop.


AGRAÏMENTS 5
AGRAÏMENTS. La vida és un llarg camí a recórrer, amb diferents etapes i en el que anam creixent i
evolucionant. Amb aquesta tesis s’acaba una d’aquestes etapes per donar-ne peu a una altra,
de moment plena d’incerteses, però plena d’esperança i ganes de seguir endavant, descobrint,
poc a poc, el que ens ofereix la vida.
Els meus inicis dins el món de la investigació foren fa uns set anys. Tot va començar amb una
beca de col·laboració amb el Dr. Sebastià Albertí Serrano, després vaig tenir una beca de la
Fundació Mateu Orfila, i posteriorment una beca FPI que m’ha permés realitzar aquest
doctorat. Aquests anys d’investigació, primer a Son Dureta i posteriorment a la UIB, han
suposat molts de canvis a la meva vida, a més d’anar aprenent com funciona el món de la
investigació i tot el necessari per fer-hi feina, he crescut com ha persona i m’he anat formant
en altres aspectes tant personals com professionals. Durant aquests anys són moltes les
persones amb les que m’he creuat a la vida, algunes encara són a prop i esper que hi restin
molta estona més, però d’altres han seguit el seu camí deixant, a vegades sense adonar-se’n,
una important petjada en mi. Per tot això aprofito aquesta oportunitat per agrair a tots ells el
que han fet per mi.
En primer terme voldria agrair al Dr. Sebastià Albertí Serrano l’oportunitat que em va donar
entrar a formar part del seu grup de recerca i així conèixer el món de la investigació. Agrair-li la
seva paciència per ensenyar-me tantes i tantes coses dins i fora del laboratori, per no tenir
peresa de posar-se la bata i donar un cop de mà si era necessari, per intentar sempre trobar la
millor manera d’enfocar i fer els experiments, i sobretot, per la seva ajuda i ànims durant
aquesta tesis. Gràcies pel teu suport i confiança.
Són moltes les persones que durant aquests anys han format part del nostre grup
d’investigació, de fet avui en dia ja no hi queda ningú d’aquelles amb les que ferem la mudança
des del laboratori de Son Dureta al de la UIB. Tot i això, vull agrair a na Maria Antònia Oliver, a
na Marta Franco i a na Mercedes Urdiain tot el que m’ensenyaren quan vaig arribar, les hores,
rialles i mal de caps compartits entre pipetes i bacteris. A n’Enma Padilla, en David Moranta i
n’Ivan López els bons moments que compartirem dins el laboratori. Però, sense cap dubte, les
persones amb qui més temps he estat fent feina i investigant han estat na Mariette Barbier i
n’Inmaculada Martínez Ramos. Mariette, arribats en aquest punt no tenc cap dubte que tot el
que hem compartit, dins i fora del la UIB, ha estat important per assolir aquest moment. A més

AGRAÏMENTS 6
d’aprendre tècniques d’investigació, a utilitzar programes informàtics, compartir viatges i
congressos, la teva ajuda i amistat han fet que creixés com a persona. Inma, la teva arribada fa
uns anys va suposar una recàrrega d’alegria i d’energia positiva. Aquests anys hem compartit
moltes coses, són moltes les vegades que ens hem rellevat en els experiments o que hem
compartit esperes. També són moltes les vegades que hem anat a ballar o hem sortit de festa
plegades. Es sorprenent com ets capaç de portar sempre un somriure dibuixat i transmetre
alegria i ànims als altres. No canviïs i gràcies per la teva amistat i alegria.
A més, vull donar les gràcies a tots i cada un dels membres de l’àrea de microbiologia de la
UIB. Són molts els moments que hem compartit tant en el departament de microbiologia com
en seminaris, berenars, etc. i més de dues i tres les vegades que he vengut a fer “d’okupa” al
vostre laboratori, a emprar aparells o que us he fet consultes de protocols. Gràcies a tots i a
totes.
També vull manifestar el meu agraïment al Dr. Antonio Oliver, al Dr. Carlos Juan, al Dr. Tomeu
Moyà i al Dr. Antonio Domenéch-Sanchez per la seva ajuda i col·laboració tant en la part
experimental com discutint resultats. Així com al Dr. Vivancos i a tots els membres del seu
grup, que m’acolliren a la Universitat Complutense de Madrid i m’ensenyaren les tècniques de
proteòmica bidimensional.
Per una altra banda no em puc oblidar de tota la gent que està per l’edifici cientificotècnic i
que han fet que el dia a dia fos més entretingut i més bo de dur. Gràcies a na Marga,
n’Antònia, en Guillem, na Marta, na Marina, na Joana, na Mar, na Maria, en Manu, na Trinitat,
na Maribel i en Juanmi per tot el que hem compartit tant a l’hora de dinar com pels
passadissos. En especial a na Trinitat per la seva ajuda amb les seqüències de ADN i a na Marta
i na Joana pels seus consells, sobretot aquests darrers mesos. No m’oblido tampoc de tots els
altres, tant la gent d’administració (na Magdalena, n’Antònia, na Jeroni i en Jesús), com d’en
Fernando i na Maria de consergeria o els tècnics de l’edifici (en Toni, en Raúl, en Jose, na Rosa,
i na Teresa) sense tots voltros el dia a dia no hagués estat el mateix.
Ja fora del món del la investigació no puc oblidar els meus amics Gerardo, Natalie, Jesús,
Alfonso, Xisca, Milton i Bàrbara. Són molts els anys que fa que ens coneixem, amb alguns més
de 15, i moltes les estones que m’heu sentit parlar de tesis i d’experiments, gràcies per tots els
moments que hem compartit i pels ànims els dies que les coses no sortien bé.

AGRAÏMENTS 7
A les amigues de la carrera de Biologia i als seus al·∙lots (n’Aina, n’Alicia, na Bàrbara, na Joana
C., na Joana F., na Juanamari, na Marga, na Yolanda, en Martí, en Biel, en Victor, en Juanma, en
Miquel i en Joan) també els vull agrair el haver estat a prop durant el transcurs d’aquesta
etapa. Hem compartit moltes coses plegats, i encara en queden moltes per compartir.
Sobretot vull donar les gràcies a na Joana C., en Juanma i a na Marga, trobaré a faltar els
berenars i dinars quan feim la xerradeta sobre la vida, les seves ventures i desventures. I de la
part de Bioquímica agraïr-li a na Maria Servera la seva amistat i els moments “Kinder Bueno”
que sempre han ajudat a agafar forces.
Gràcies també als amics de Sa Tropa, en especial a na Cris, n’Elisa, n’Olivia, n’Andrea i na Cata,
per totes ses estones de festa, sopars, les excursions i els moments de desconnexió que ajuden
a recarregar piles quan fa falta. Gràcies per ser com sou.
I en darrer terme, i no per això els menys importants vull donar les gràcies a la meva família, a
tots ells, des dels padrins i abuelos a tots als meus tios/ties i cosins/es, que molts de cops heu
escoltat ses meves aventures per la UIB o amb els meus “bitxos”, sense saber exactament en
que consistia tot això. Però sobretot vull donar-los les gràcies als meus pares Catalina i Toni, a
la meva germana Irene, al meu cunyat Jose, i al meu fillol Pau i la meva neboda Neus. Són els
que més han patit durant aquests anys els dies que les coses no sortien bé, els canvis d’ànims,
el haver de pujar a la UIB els caps de setmana i no poder passar més temps amb ells. Són els
que amb el seu suport, paciència i consells fan que qualsevol cosa sigui possible. Sabeu lo molt
que us estim, i sense voltros no seria el que soc. Gràcies per estar sempre al meu costat.
Laura García Sureda


ÍNDICE 9
ÍNDICE AGRADECIMIENTOS ...................................................................................................... 5 ÍNDICE .......................................................................................................................... 9 ABREVIATURAS ............................................................................................................ 11 1. INTRODUCCIÓN ................................................................................................................ 13 1.1 Klebsiella como patógeno ............................................................................... 15 1.1.1 Importancia clínica de K. pneumoniae ................................................... 16 1.1.2 Factores de virulencia de K. pneumoniae .............................................. 18 1.1.3 Mecanismos de resistencia a los antimicrobianos en K. pneumoniae ..... 25 1.1.3.1 Enzimas inactivadores de antibióticos: las β-lactamasas ................. 25 1.1.3.2 Expulsión activa ............................................................................ 35 1.1.3.3 Alteraciones de la permeabilidad .................................................. 37 1.2 La membrana externa de las bacterias Gram negativas ................................... 39 1.2.1 Proteínas de membrana externa ........................................................... 40 1.2.1.1 Porinas en K. pneumoniae .............................................................. 42 1.2.1.1.1 OmpK35 y OmpK36 ............................................................... 42 1.2.1.1.2 OmpA .................................................................................... 51 1.2.1.1.3 OmpK37 ............................................................................... 55 1.2.1.1.4 PhoE .................................................................................... 56 1.2.1.1.5 LamB .................................................................................... 59 1.2.1.1.6 La familia de las porinas KdgM .............................................. 62 2. HIPÓTESIS Y OBJETIVOS .......................................................................................... 67 3. MATERIAL Y MÉTODOS .......................................................................................... 71 3.1 Bacterias .................................................................................................. 73 3.2 Medios y condiciones de cultivo ..................................................................... 74 3.3 Plásmidos .................................................................................................. 74 3.4 Estudios de sensibilidad .................................................................................. 75 3.4.1 Agentes antimicrobianos ....................................................................... 75 3.4.2 Concentración mínima inhibitoria ......................................................... 75 3.5 Métodos de análisis y purificación de proteínas .............................................. 76
3.5.1 Aislamiento de proteínas de membrana externa .................................... 76 3.5.2 Purificación de la porina OmpK36 .......................................................... 77 3.5.3 Determinación de la concentración de proteínas ................................... 77 3.5.4 Electroforesis en gel de poliacrilamida con SDS (SDS-PAGE) ................... 77 3.5.5 Identificación de proteínas .................................................................... 78 3.5.6 Identificación de proteínas con anticuerpos ........................................... 78
Obtención de antisuero ........................................................................ 78 Western blot o transferencia Western ................................................. 79

ÍNDICE 10
3.6 Métodos de purificación, manipulación y análisis de ácidos nucleicos .............. 79
3.6.1 Purificación de ADN .............................................................................. 79 3.6.2 Purificación de ARN............................................................................... 80 3.6.3 Electroforesis en geles de agarosa ......................................................... 80 3.6.4 Southern blot o transferencia Southern ................................................. 80 3.6.5 Introducción de ADN plasmídico ............................................................ 81 3.6.6 Reacción en cadena de la polimerasa (PCR)............................................ 82 3.6.7 RT-PCR .................................................................................................. 83 3.6.8 Secuenciación ....................................................................................... 84 3.6.9 Construcción de mutantes delecionados mediante inserción-duplicación 84
3.7 Otros métodos ............................................................................................... 86 3.7.1 Curvas de crecimiento y cálculo del tiempo de duplicación ..................... 86 3.7.2 Experimentos de frecuencia de mutación ............................................... 86 3.7.3 Experimentos de competición in vitro .................................................... 87 3.7.4 Estudios de virulencia en un modelo de infección sistémica en ratón ...... 88
4. RESULTADOS Y DISCUSIÓN ...................................................................................... 91 4.1 Efectos de diferentes β-lactámicos sobre la frecuencia de aparición y selección de mutantes en K. pneumoniae. ................................................................................. 93
4.2 Papel de LamB en la resistencia a antimicrobianos ......................................... 100 4.3 Papel de OmpK26 en la resistencia a antimicrobianos. .................................... 106 Identificación de OmpK26 ............................................................................. 106 El papel de OmpK26 en la resistencia antimicrobiana .................................... 109 Coste biológico de la deficiencia en porinas de K. pneumoniae expresando OmpK26 ...................................................................................................... 111 5. COROLARIO ........................................................................................................ 115 6. CONCLUSIONES ..................................................................................................... 121 7. REFERENCIAS ........................................................................................................ 125 8. ANEXOS ................................................................................................................ 147 Recetas ............................................................................................................... 149 Secuencia de OmpK26 ......................................................................................... 155 Artículos .............................................................................................................. 157

ABREVIATURAS 11
ABREVIATURAS UTILIZADAS ADN ............ Ácido desoxiribonucléico AMK ............ Amikacina AMP ............ Ampicilina AMPc .......... Adenosín monofosfato cíclico APS ............. Persulfato amónico ARN ............. Ácido ribonucléico ARNm ......... ARN mensajero ARNr ........... ARN ribosómico ARNt ........... ARN de transferencia ATP ............. Adenosín trifosfato AZT .............. Aztreonam BLEA ............ β-lactamasa de amplio espectro BSA ............. Albúmina bovina sérica (bovine serum albumine) CA ............... Ácido clavulánico CAZ ............. Ceftazidima CEF .............. Cefalotina CIP............... Ciprofloxacino CIT ............... Cefotetan CLSI ............. Clinical and Laboratory Standards Institute CMI ............. Concentración mínima inhibitoria CPS ............. Cápsula csp .............. cantidad suficiente para CTX .............. Cefotaxima CHL ............. Cloranfenicol D.O. ............. Densidad óptica EDTA ........... Ácido etileno-diamino-tetra-acético ELISA ........... Ensayo por inmunoabsorción ligado a enzima ETP .............. Ertapenem FEP .............. Cefepime FOX ............. Cefoxitina GEN ............. Gentamicina H2Od ........... Agua destilada IC ................. Índice de competición IMP ............. Imipenem KAN ............. Kanamicina LB ................ Luria- Bertani LPS .............. Lipopolisacárido LVX ............. Levofloxacino MDR ............ Multi Drug Resistant ME .............. Membrana externa MEN ............ Meropenem

ABREVIATURAS 12
MH II ........... Mueller Hinton II NAL ............. Ácido nalidíxico NCCLS ......... National Committee for Clinical Laboratory Standards pb................ Pares de bases PBP ............. Proteína fijadora de penicilina (penicillin binding protein) PBS .............. Tampón fosfato salino (phosphate buffered saline) PDB ............. Base de datos de proteínas (Protein Data Base) PCR ............. Reacción en cadena de la polimerasa (polimerase chain reaction) PIP ............... Piperacilina PIR .............. Cefpirome PME ............ Proteínas de membrana externa RIF ............... Rifampicina RT-PCR ........ Reverse transcription–PCR (PCR de transcripción reversa) SDS .............. Dodecilsulfato sódico SDS-PAGE.... Electroforesis en geles de poliacrilamida con dodecilsulfato sódico TAE.............. Tris acético EDTA TEMED ........ N,N,N´,N´-tetrametil-etilendiamina TET .............. Tetraciclina TOB ............. Tobramicina TZB .............. Tazobactam UCI .............. Unidad de cuidados intensivos UFC ............. Unidades formadoras de colonias Las abreviaturas de los aminoácidos y los nucleótidos se han utilizado siguiendo el código IUPAC.

1. INTRODUCCIÓN.


INTRODUCCIÓN 15
1.1 Klebsiella como patógeno.
Klebsiella es un género de bacterias Gram negativas pertenecientes a la familia
Enterobacteriaceae, que se caracteriza por ser bacilos anaeróbicos facultativos no flagelados.
Este género está extendido de manera ubicua en la naturaleza y está asociado a gran variedad
de hábitats. Klebsiella se encuentra en aguas, suelo, superficies húmedas, vertidos industriales
y en la vegetación [1]. Además también es un microorganismo residente o transitorio de la
flora (principalmente de la flora del tracto gastrointestinal). Su importancia clínica se debe a
que Klebsiella es un patógeno oportunista para los humanos y otros animales.
Durante mucho tiempo, casi todos los aislamientos de Klebsiella se identificaron como una sola
especie, K. pneumoniae. Sin embargo, en la década de los 80 estudios fenotípicos y genotípicos
demostraron que estos aislamientos correspondían a cinco especies diferentes [2],
diferenciables mediante pruebas bioquímicas (Tabla 1.1): K. pneumoniae (con tres
subespecies: pneumoniae, ozaenae y rhinoscleromatis), K. oxytoca, K. terrigena, K.
ornithinolytica, y K. planticola. Recientemente, los estudios filogenéticos basados en la
comparación de los genes rrs, rpoB [3], gyrA y parC [4], han modificado esta clasificación. De
hecho K. planticola, K. terrígena y K. ornithinolytica se han transferido a un nuevo género,
Raoutella [3]. Por otra parte, Calymmatobacterium granulomatis, se ha visto que está
estrechamente relacionado con K. pneumoniae mediante comparación de las secuencias de
rrs (gen 16S rRNA), y es por ello que fue transferido al género Klebsiella como Klebsiella
granulomatis [5, 6]. Sin embargo no se sabe si K. granulomatis es una especie distinta a K.
pneumoniae.
Figura 1.1. A. Imagen de Klebsiella pneumoniae crecida sobre agar Mueller Hinton. B. Imagen coloreada de K. pneumoniae al microscopio electrónico de barrido.
INTRODUCCIÓN 16
La especie de mayor relevancia clínica es K. pneumoniae y, en mucho menor grado K. oxytoca.
A finales del siglo XX también se detectaron aislamientos clínicos pertenecientes a K. terrigena
y K. planticola, especies originalmente consideradas sin relevancia clínica y restringidas a los
ambientes acuáticos, a la vegetación y al suelo [2, 7-9]. De hecho, en determinados estudios K.
planticola representaba más del 20% de los aislados clínicos de Klebsiella, mientras que K.
terrigena se encontraba ocasionalmente y sólo representaba el 0,4% de los aislados de
Klebsiella [9, 10]. Cabe comentar que actualmente las especies K. terrígena, K. planticola y K.
ornithinolytica se han reclasificado en el género Raoultella.
Tabla 1.1. Reacciones bioquímicas de las especies de Klebsiellaa.
Característica
Klebsiella pneumoniae Klebsiella oxytoca
Klebsiella terrigenac
Klebsiella planticolac
Klebsiella ornithinolyticac subsp.
pneumoniae subsp.
ozaenae subsp.
rhinoscleromatis
Indol -b - - + - V +
Ornitina descarboxilasa - - - - - - +
Lisina descarboxilasa + V - + + + +
Degradación de pectato - - - + - - -
Gas de lactosa a 44,5ºC + - - - - - -
Crecimiento a 10ºC - - - + + + +
Ácido a partir de: D-melecitosa - - - V + - -
L-sorbosa V V V + + + -
Utilización de: m-Hidroxibenzoato - - - + + - -
Hidroxi-L-prolina V V V V V + -
Malonato + - + + + + +
Test de rojo de metileno - + + - + V +
Reacción Voges-Proskauer + - - + + + + a Datos recopilados de las referencias [11-18] b - reacción negativa, +, reacción positiva; V reacción variable. c Actualmente pertenecientes al género Raoultella.
1.1.1 Importancia clínica de K. pneumoniae.
Las infecciones causadas por K. pneumoniae van desde infecciones leves del tracto urinario a
bacteriemia, infecciones intra-abdominales y neumonías graves con una alta tasa de
mortalidad y morbilidad [19-21]. En la mayoría de los casos se caracterizan por un curso clínico
rápido complicado por abscesos pulmonares y de implicación multilobular, que deja escaso
INTRODUCCIÓN 17
margen de tiempo para poder establecer un tratamiento antibiótico eficaz. Además, K.
pneumoniae es uno de los microorganismos más comunes responsables de empiema [22].
Como se observa en la tabla 1.2, Klebsiella se encuentra entre los cinco primeros patógenos
nosocomiales que afectan al tracto urinario. Es responsable de entre un 6% y un 17% de dichas
infecciones y muestra incluso una incidencia mayor en grupos especiales de riesgo, como
pacientes con diabetes mellitus [23]. Como causa de bacteriemias nosocomiales producidas
por Gram negativos, Klebsiella es el segundo o tercer patógeno, únicamente por detrás de
Escherichia coli y, en ocasiones, miembros del género Enterobacter [24-27]. En el caso de las
neumonías Klebsiella está entre los cuatro patógenos nosocomiales más importantes,
representando entre un 7% y 14% de dichas infecciones.
Por otra parte, las tasas de porte de Klebsiella en el intestino grueso varían entre un 5% y un
38% y entre un 1% y un 6% en la nasofaringe, y se pueden incrementar con la hospitalización,
con la presencia de enfermedades severas subyacentes, como diabetes mellitus, y con el
consumo de antibióticos en adultos y niños [2, 28].
Las infecciones nosocomiales debidas a Klebsiella son especialmente problemáticas en las
unidades pediátricas, sobre todo en las unidades de cuidados intensivos y de neonatos. Las
especies del género Klebsiella son patógenos relacionados con la sepsis neonatal (Tabla 1.2),
tanto en infecciones neonatales tempranas como tardías [29, 30].
La aparición de aislados nosocomiales de K. pneumoniae multiresistentes a los antibióticos ha
acentuado el interés por el estudio de los factores de virulencia de Klebsiella, así como el de
los mecanismos que permiten a las bacterias adquirir resistencia a los antibióticos [31, 32].
Tabla 1.2. Infecciones nosocomiales más frecuentes provocadas por Klebsiella sp. [2]
Infección % Infecciones causadas por Klebsiella Posición Referencias
Infecciones del tracto urinario 6-17 5-7 [33-35] Neumonía 7-14 2-4 [33, 34] Infecciones de heridas 2-4 6-11 [34, 35] Infecciones nosocomiales en UCI 4-17 4-9 [33-35] Septicemia 4-15 3-8 [20, 25-27, 35, 36] Septicemia neonatal 3-20 2-8 [30, 37-40]

INTRODUCCIÓN 18
1.1.2 Factores de virulencia de K. pneumoniae
Los factores de virulencia participan principalmente en la superación de los mecanismos de
defensa innatos del huésped, aunque también pueden estar implicados en diferentes etapas
del proceso de infección. Su importancia hace que se sigan realizando esfuerzos para la
identificación de nuevos factores de virulencia, por ejemplo, a través del estudio de genotecas
realizadas por mutagénesis dirigida [41].
En la actualidad, y en base a los estudios realizados sobre la virulencia de K. pneumoniae, los
principales factores que contribuyen a su patogenicidad son: la cápsula, el lipopolisacárido, las
fimbrias y adhesinas, y los sideróforos.
Cápsula.
La cápsula (CPS), que también recibe el nombre de antígeno K, es la estructura más externa de
las envolturas celulares y, al estar en contacto directo con el medio, es el componente que
protege a la bacteria de las condiciones ambientales adversas.
K. pneumoniae normalmente desarrolla cápsulas prominentes compuestas de polisacáridos
acídicos complejos. Las unidades de repetición capsulares constan de cuatro a seis azúcares y,
a menudo, ácidos urónicos como componentes cargados negativamente. Actualmente se
conocen 77 serotipos diferentes [42]. La cápsula es esencial para la virulencia de K.
pneumoniae [43-49]. El material capsular forma estructuras fibrilares que cubren la superficie
de la bacteria formando capas [50], protegiendo
a la bacteria de la fagocitosis por granulocitos
polimorfonucleares y macrófagos alveolares [43,
45, 51-55]. Además de su función antifagocítica,
se ha descrito que la CPS de K. pneumoniae
inhibe la diferenciación y capacidad funcional de
los macrófagos in vitro [56, 57]. La CPS también
evita la opsonización y lisis de la bacteria por la
acción del complemento evitando la unión de
C3b a la bacteria [43, 54, 58]. [59]
Los estudios epidemiológicos demuestran que
los serotipos capsulares más frecuentemente
aislados a partir de pacientes con infecciones del tracto urinario, neumonía o bacteriemia son
K1 y K2 [60-62]. No se sabe a ciencia cierta a que se debe este hecho, pero una de las posibles
Figura 1.2. Fotografía de K. pneumoniae tomada con el microscopio electrónico de transmisión donde se puede apreciar la presencia de cápsula [59].

INTRODUCCIÓN 19
explicaciones es que el grado de virulencia conferido por un antígeno K determinado parece
estar relacionado con el contenido de manosas de la CPS, concretamente por las secuencias
manosa-α-2/3-manosa, ya que son las que determinan la eliminación de K. pneumoniae por
parte de las defensas del huésped [53, 63]. Así pues, los tipos capsulares de baja virulencia
como los antígenos K7 o K21a [52, 53, 64], contienen secuencias repetitivas de manosa-α-2/3-
manosa o L-ramnosa-α-2/3-L-ramnosa, reconocidas por una lectina superficial de los
macrófagos, que media la lectinofagocitosis, es decir, la fagocitosis independiente de
opsonización [65]. Los macrófagos con la lectina específica para la manosa-α-2/3-manosa o
receptor de manosa, reconocen, internalizan y matan a los serotipos de K. pneumoniae que
contienen CPS con secuencias repetitivas de manosa-α-2/3-manosa o L-ramnosa-α-2/3-L-
ramosa. Por el contrario, las cepas que carecen de estas secuencias repetidas no son
reconocidas por los macrófagos y, por tanto, no son fagocitadas, que es lo que sucede en los
tipos capsulares K1 y K2, ya que carecen completamente de estructuras manosa-α-2/3-manosa
[64, 66].
Lipopolisacárido
El lipopolisacárido (LPS) es una molécula glucolipídica anclada a la cara externa de la
membrana externa de la mayoría de bacterias Gram negativas que actúa como componente
estructural y está claramente implicado en la resistencia de muchos microorganismos a los
mecanismos de defensa del huésped. La cantidad de lipopolisacárido (LPS) producido varía
entre cepas y está influenciado por las condiciones del medio donde crece la bacteria [67].
El LPS está compuesto por el lípido A, un núcleo oligosacárido y una cadena polisacarídica que
recibe el nombre de antígeno O (figura 1.3).
- Lípido A o endotoxina, es una región compuesta por un disacárido de glucosamina unido a
varios ácidos grasos. Su principal actividad biológica es el mantenimiento de la integridad
de la membrana bacteriana. Su interés clínico reside en su función como endotoxina, ya
que provoca la activación de los macrófagos, induce la respuesta inmunológica y tiene un
efecto pirógeno.
- Núcleo, es una región oligosacárida central que contribuye de manera indirecta a servir de
unión del antígeno O, auténtico factor de virulencia. Se ha comprobado que las tres
regiones del LPS pueden actuar como inmunógenos, pero el núcleo presenta motivos
estructurales muy conservados y, por tanto, se considera una diana adecuada para la
inducción de anticuerpos de gran reactividad cruzada. En este sentido, se han obtenido
anticuerpos a través de la inmunización de ratones con K. pneumoniae que reconocen de

INTRODUCCIÓN 20
manera altamente específica el núcleo de la especie, con independencia del serotipo de
su antígeno O. Estos anticuerpos dirigidos contra el núcleo del LPS podrían utilizarse de
tratamiento en pacientes con sepsis causada por bacterias Gram negativas.
- Antígeno O, es una región polisacárida conocida como D-galactano I que está constituida
por la repetición de la unidad básica disacárida [→3-β-d-Galf-(1→3)-α-d-Galp-(1→]. El D-
galactano I puede modificarse por O-acetilación o por la adición de otros dominios
polisacáridos con una estructura básica diferente [68]. Estas diferencias en la composición
de azúcares no solo se encuentran entre las diferentes especies de una bacteria Gram
negativa, sino que también se encuentran entre las cepas de una misma especie
bacteriana. La variabilidad de esta estructura y sus propiedades inmunogénicas han
originado la descripción de hasta 9 serotipos diferentes de K. pneumoniae [69]. El serotipo
O1 es el antígeno O que más comúnmente se ha encontrado en los aislados clínicos de K.
pneumoniae (figura 1.3). De sa fig [70]
El reducido número de serotipos O en K. pneumoniae es una gran ventaja para su
aplicación como vacuna. Una vacuna multivalente compuesta por 8 tipos de LPS, o bien la
inclusión del antígeno O1 en una vacuna de polisacárido capsular sería una aproximación
prometedora [2, 71].
Figura 1.3. A. Representación esquemática de la estructura del LPS en K. pneumoniae. B. Estructuras de las diferentes cadenas de antígeno O aisladas de diferentes LPS desaminados procedentes de distintos serotipos de K. pneumoniae [70].
Lípido A
Núcleo
Antígeno O
n
A B -3) –β-Galp-α- Galp-(1- –β-Galf –(1- -3) –α-Galp-β- Galf-(1- -3) –α–Galp-PC
β-Galf- (1- -3) - α- Galp - (1-3)- β – Galf -(1- -3) α- Galp- PC
-5)–β-Galf-(1-2)-β-GlucNAc–(1- –β-Galf – (1- -3)–α-Galp-β- Galf-(1- -3)–α–Galp- PC
-2) –α – Man (1-2) - α- Man (1-3)-α-Man – (1- –3) – α – Man – (1-3) – α- Man - PC
α – KDO- (2-2)-β-Ribf- (1- -4) - α- Gal - (1-2)- β – Ribf -(1- -4) α- Gal – (1- PC
Me –3)–α–Man–(1- -3)-β-Man-(1-2)-α–Man-(1- -3)-α-Man–(1-3)–α–Man– (1- PC
β – KDO- (2-3)-α-Rha- (1- -3) - β- GlcNAc - (1-4)- α – Rha -(1- – (1- PC
3) - β – GlcNAc – (1-5) – α – KDO – (2-6) - anhMan
α-Hep - (1-4)
Serotipo
O1
O1, O2a, O2a,c O2a,c
O3
O4
O5
O12
n n
n
n n
n
n
n
n
PC: Parte común

INTRODUCCIÓN 21
En K. pneumoniae se ha podido demostrar la importancia del antígeno O para la acción
lítica del complemento. El mecanismo de resistencia al suero de dicho antígeno se basa
en:
- la presencia o ausencia de dicho antígeno. Se ha visto que los mutantes de
laboratorio desprovistos de este antígeno son sensibles a la acción lítica del
complemento, activando tanto la vía clásica como la vía alternativa [72-74]. En
cambio, las cepas en las que las cadenas laterales del antígeno O no están cubiertas
ni protegidas por la cápsula, el componente C3b del complemento al unirse
principalmente a las cadenas laterales O más largas, queda alejado de la membrana
bacteriana, evitando la formación del complejo de ataque a la membrana y la lisis
osmótica [75-78]. Así pues, las cadenas de antígeno O representan una barrera
física que impide la correcta actuación de la cascada de reacciones de las proteínas
del complemento y las cepas en las que el LPS presenta antígeno O únicamente
activan la vía alternativa.
- la cantidad de C3b depositada [72]. La activación de ambas vías por parte de las
cepas sensibles supone la deposición de elevados niveles de C3b y, por tanto, un
mayor daño y muerte celular, mientras que en las cepas resistentes los niveles de
C3b depositado son ineficaces para continuar la cascada del complemento [72].
Por lo tanto, la virulencia de las cepas de K. pneumoniae está fuertemente correlacionada
con la CPS y el antígeno O [79].
También se ha visto que, en relación al papel del antígeno O respecto a la patogenia de la
neumonía, dicho antígeno se presenta al huésped con una carga neutral [80]. Este
enmascaramiento de las cargas implica una disminución de la unión de las partículas
cargadas facilitando el establecimiento de la neumonía, por ejemplo, la unión entre los
péptidos antimicrobianos y la superficie de la bacteria queda alterada, así como la unión
con las defensinas del huésped [80]. Además, aunque se ha visto que la presencia del
antígeno O no afecta a la diseminación de la neumonía, si que afecta a la capacidad de
infección de Klebsiella, ya que cuando una cepa expresa antígeno O es más fácil de
eliminar por los mecanismos del sistema inmune presentes en el pulmón [45].
En resumen, el antígeno O facilita el proceso inicial de adhesión y confiere resistencia a la
bacteria contra la actividad bactericida del suero [2, 77].

INTRODUCCIÓN 22
Fimbrias y adhesinas.
Las fimbrias son proyecciones filamentosas no flagelares de la superficie bacteriana (figura
1.4). Estas estructuras tienen un diámetro entre 1 y 11 nm y pueden medir hasta 10 μm de
longitud. Las fimbrias están formadas por subunidades proteicas globulares poliméricas con un
peso molecular que oscila entre 15 y 26 kDa [81, 82]. Las propiedades de adhesión de las
enterobacterias dependen generalmente de los distintos tipos de fimbrias o pili. Así pues, las
fimbrias permiten al microorganismo mimetizar lo máximo posible las superficies mucosas del
huésped y mantenerse próximo mediante la adhesión a sus células, paso fundamental para
poder desarrollar un proceso infeccioso. Las fimbrias también determinan las propiedades de
hemaglutinación.
De los distintos tipos de fimbrias descritas en las enterobacterias, en K. pneumoniae
predominan los tipos 1 y 3. Ref fig [59]
Las fimbrias tipo 1 son las adhesinas mejor
estudiadas. Estas fimbrias se unen a receptores
glucoproteicos que contienen manósidos
presentes en las células del organismo huésped,
por lo que la manosa inhibe esta unión. Se ha
observado en diferentes estudios que la adhesión
mediada por fimbrias de tipo 1 es un importante
factor de virulencia en las infecciones del tracto
urinario causadas por K. pneumoniae, al permitir
la colonización de la vejiga urinaria [83]. Además,
la capacidad de unión de las fimbrias de tipo 1 a
glucoproteínas solubles presentes en la saliva
puede jugar un papel importante en la
colonización del tracto respiratorio superior [84].
Si bien este tipo de fimbrias favorecen el establecimiento inicial del microorganismo en las
superficies mucosas, su expresión en las etapas posteriores del proceso infeccioso resulta
perjudicial ya que es una estructura reconocida por los sistemas de defensa del organismo
huésped. Este tipo de fimbrias son reconocidas por receptores que contienen manosa
presentes en la superficie de los leucocitos y activan la lectinofagocitosis. Para evitar este
mecanismo de defensa la bacteria controla la expresión de fimbrias de tipo 1 pudiendo revertir
al fenotipo no fimbriado [85]. En este sentido, el paso de un estado a otro está regulado por
Figura 1.4. Fotografía de K. pneumoniae tomada con el microscopio electrónico de transmisión donde se puede apreciar la presencia de fimbrias [59].

INTRODUCCIÓN 23
los genes fim, concretamente por la inversión de un segmento de ADN que contiene el
promotor para el gen fimA. La orientación de dicho segmento de ADN invertible está
controlado por el producto de los genes fimB y fimE que, respectivamente, median el estado
de activación/inactivación o de solo inactivación de la expresión de los genes fim, dando como
resultado una bacteria con o sin fimbrias [59, 86].
Las fimbrias de tipo 3 se expresan en la mayoría de cepas de K. pneumoniae y facilitan su
adherencia a las células endoteliales, al epitelio del tracto respiratorio y a células uroepiteliales
[87-89]. A diferencia de las fimbrias de tipo 1, las fimbrias tipo 3 están relacionadas con la
formación de biofilms [90-92]. Estas fimbrias están codificadas por un grupo de 6 genes, los
genes mrk. El gen mrkA codifica para la subunidad de la fimbria que polimeriza para formar el
eje fimbrial, mientras que el gen mrkD codifica para la adhesina fimbrial y es importante para
la formación de biofilms. Por otra parte, los genes mrkB, mrkC y mrkE parecen estar implicados
en el ensamblaje del filamento fimbrial y en la regulación de su expresión. Por último, el
producto del gen mrkF es necesario para el mantenimiento de la estabilidad de la fimbria en la
superficie celular [90, 91, 93, 94].
Además de las fimbrias, en K. pneumoniae se han descrito otros tipos de adhesinas:
- la adhesina CF29K, codificada por el plásmido-R de K. pneumoniae, que promueve la
adherencia a las líneas celulares humanas Intestine-407 y CaCo-2. Este tipo de
adhesina parece idéntica a la CS31-A de las cepas de E. coli que provoca diarreas y
proviene de la familia de las adhesinas K88. Los datos sugieren que CF29K es producto
de la transferencia de los genes CS31-A desde E. coli a K. pneumoniae en el intestino
humano[95].
- la adhesina KPF-28, similar a las fimbrias, se encuentra en la mayoría de las cepas de K.
pneumoniae resistentes a β-lactámicos y, presuntamente, es un factor de colonización
del intestino humano [96].
- la CPS es otra macromolécula presente en la superficie bacteriana que puede
participar en el proceso de adhesión. A pesar de que diferentes estudios indican que la
CPS reduce la capacidad de adhesión de K. pneumoniae a células epiteliales
procedentes de diferentes tejidos [67, 97], la adhesión de bacterias capsuladas
únicamente se ve favorecida cuando interacciona con células epiteliales que presentan
compuestos mucoides en su superficie celular. Estudios recientes sugieren que la CPS
induce una respuesta inmunológica del huésped defectuosa al dificultar la unión de las
bacterias y su internalización [98].

INTRODUCCIÓN 24
- el LPS también se ha considerado un factor de adhesión en K. pneumoniae.
Concretamente, se ha demostrado que el antígeno del LPS se une, in vitro, al receptor
de manosa de los macrófagos, mediando la adhesión de este tipo de células [99].
Además, mutantes en el antígeno O del LPS presentan una reducción de adhesión a
células epiteliales [100].
Sideróforos
Los sideróforos son proteínas quelantes con gran afinidad por el hierro y de bajo peso
molecular, secretadas por las bacterias para asegurarse el aporte de hierro, elemento esencial
para el crecimiento bacteriano, y capaces de competir con las proteínas del huésped [101].
En las enterobacterias los tres sistemas de sideróforos más frecuentes son la enterobactina, la
aerobactina y la yersiniabactina. La enterobactina, también conocida como enteroquelina, fue
el primer sideróforo bacteriano descrito, es un sideróforo tipo catecolato y lo producen más
del 90% de los aislamientos enterobacterianos [102-104]. La aerobactina es un sideróforo tipo
hidroxamato, lo producen una pequeña fracción de enterobacterias y tiene menor afinidad al
hierro libre que la enterobactina o la yersiniabactina [105, 106]. El tercer tipo de sideróforo es
la yersinoabactina, descrito por primera vez en especies de Yersinia pero encontrado en otras
especies de enterobacterias, se cree que se transfiere por transferencia horizontal [107].
Hasta la fecha, se han realizado gran número de estudios para evaluar la regulación genética,
la función y la capacidad de captación del hierro de cada uno de estos sideróforos [106, 108-
110]. También se ha investigado como afecta la deficiencia de uno o más de estos sideróforos
en cepas de E. coli, tanto en su crecimiento en medios pobres en hierro como en su capacidad
infectiva. Se ha visto que en modelos de infección del tracto urinario en ratón, cepas
deficientes en aerobactina y enterobactina muestran una menor capacidad de diseminación en
el riñón, sin embargo, mutantes deficientes sólo en aerobactina o enterobactina no presentan
alteraciones ni en la capacidad de colonización ni en la diseminación [111].
En el caso de K. pneumoniae existe un importante número de estudios que han analizado la
distribución de los sideróforos en aislados clínicos y que han determinado que casi la totalidad
de dichos aislados producen enterobactina [112, 113], mientras que sólo un pequeño
porcentaje de los mismos producen aerobactina o yersiniabactina [112, 114-116]. Sin
embargo, no existen estudios concluyentes que hayan determinado el papel de cada uno de
estos sideróforos en la virulencia de K. pneumoniae.

INTRODUCCIÓN 25
1.1.3 Mecanismos de resistencia a los antimicrobianos en K. pneumoniae.
El interés por el estudio de K. pneumoniae ha aumentado ya que, además de ser uno de los
microorganismos más frecuentemente asociados a las infecciones nosocomiales, el número de
aislamientos resistentes a los antibióticos ha incrementado notablemente [117-120].
De hecho, el uso de antibióticos junto con la
prolongación de la estancia hospitalaria
incrementan notablemente la probabilidad de
infección por K. pneumoniae [118]. Además, la
alta morbilidad y la mortalidad de las
infecciones sistémicas graves producidas por
Klebsiella a pesar del uso de una terapia
antibiótica adecuada, hacen que se sigan
estudiando los mecanismos de resistencia que
presenta K. pneumoniae y también se
considere a este patógeno como una
importante fuente de transmisión de
resistencia antibiótica.
En general, los mecanismos de resistencia a los antimicrobianos que más habitualmente
presenta K. pneumoniae son tres (ver figura 1.5):
i) β-lactamasas que rompen los puentes amida del anillo β-lactámico.
ii) Alteración de la permeabilidad de la membrana externa afectando a la
penetración de los antibióticos.
iii) Bombas de expulsión o bombas de flujo que expulsan los antibióticos.
1.1.1.1 Enzimas inactivadores de antibióticos: las β-lactamasas.
El principal mecanismo de resistencia a los antibióticos β-lactámicos es la expresión de β-
lactamasas que inactivan estos antibióticos mediante la hidrólisis del anillo β-lactámico. La
resistencia se puede deber a la activación natural o a mutaciones en los genes cromosómicos,
o la adquisición de elementos genéticos extracromosómicos (plásmidos o transposones)
portadores de genes que codifican estos enzimas [121].
Figura 1.5. Figura esquemática de los mecanismos de resistencia presentes en K. pneumoniae.

INTRODUCCIÓN 26
K. pneumoniae es intrínsecamente resistente a penicilina, ampicilina, amoxicilina, oxacilina,
carbenicilina y ticarcilina debido a que expresa una β-lactamasa de forma constitutiva SHV-1,
cuyo gen se localiza en el cromosoma bacteriano [122]. Para convertirse en resistente a las
cefalosporinas y a otros β-lactámicos, K. pneumoniae necesita adquirir enzimas que hidrolizan
las cefalosporinas como son las β-lactamasas. Hay dos tipos de β-lactamasas: las β-lactamasas
de espectro reducido y las β-lactamasas de amplio espectro (BLEAs). Ambas se pueden
localizar en plásmidos transferibles que también codifican la resistencia a otros agentes
antimicrobianos, incluyendo cotrimoxazol (trimetoprima con sulfametoxazol), tetraciclinas,
aminoglicósidos y más recientemente fluoroquinolonas [123, 124]. Así pues, estas β-
lactamasas normalmente están codificadas por plásmidos de gran tamaño que contienen, al
mismo tiempo, otros genes de resistencia antibiótica. Su localización plasmídica facilita la
diseminación entre cepas y la acumulación de genes de resistencia. Por otra parte, hay que
tener en cuenta que la expansión del espectro de substrato de una β-lactamasas se consigue
mediante uno o dos puntos de mutación en el gen codificante de dicha β-lactamasa, en
algunos casos estas mutaciones provocan que una β-lactamasa de reducido espectro se
convierta en una BLEAs [125].
El número y tipo de β-lactamasas descubiertas ha ido creciendo a lo largo de los años.
Actualmente existen dos sistemas de clasificación de dichos enzimas, la clasificación molecular
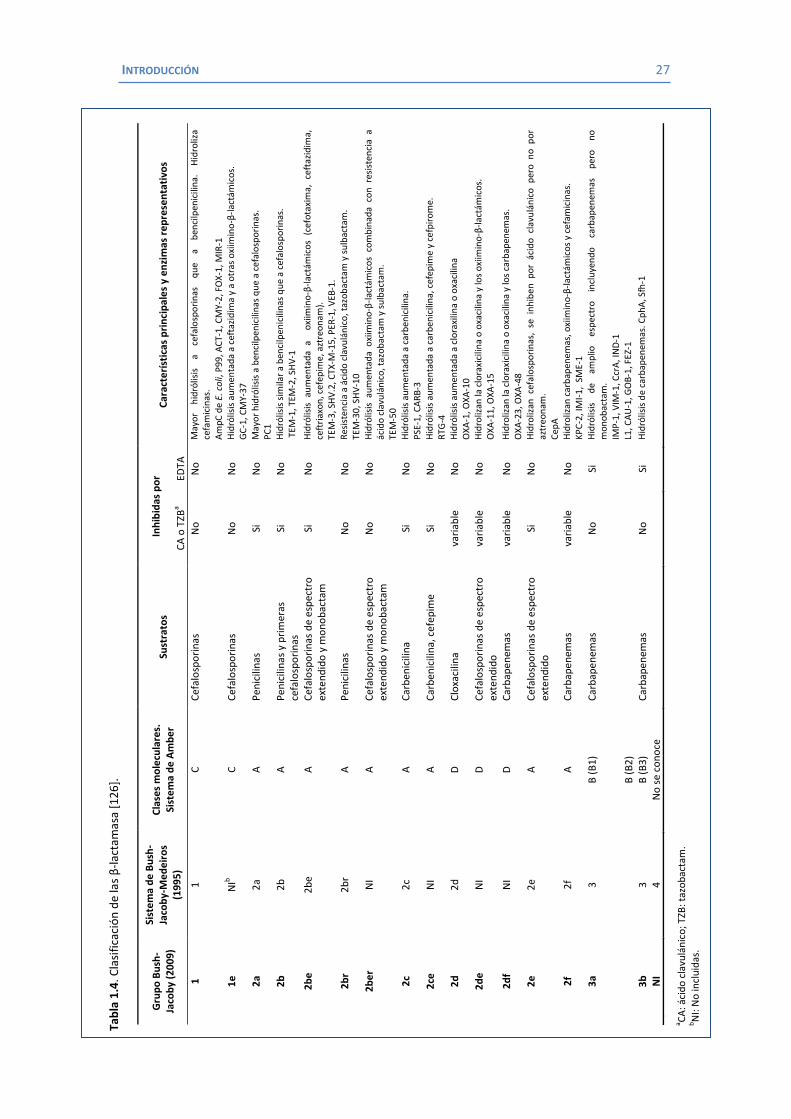
o clasificación de Amber y el sistema de clasificación de Bush y colaboradores (tabla 1.3). [126]
En el caso de K. pneumoniae las β-lactamasas más habituales pertenecen a los siguientes
grupos:
1.- β-lactamasas tipo TEM y SHV.
2.- β-lactamasas tipo AmpC.
3.- β-lactamasas tipo CTX-M.
4.- β-lactamasas de clase A: Carbapenemasas.
5.- β-lactamasas de clase D: oxacilinasas (OXA).
6.- β-lactamasas de clase B: Metalo- β-lactamasas (MBL).
Los tres primeros grupos están más implicados en la resistencia a cefalosporinas, mientras que
los tres últimos tienen gran importancia en la resistencia a los carbapenemas.
INTRODUCCIÓN 27
Gru
po B
ush-
Jaco
by (2
009)
Sist
ema
de B
ush-
Jaco
by-M
edei
ros
(199
5)
Clas
es m
olec
ular
es.
Sist
ema
de A
mbe
r Su
stra
tos
Inhi
bida
s por
Ca
ract
erís
ticas
prin
cipa
les y
enz
imas
repr
esen
tativ
os
CA o
TZB
a ED
TA
1 1
C Ce
falo
spor
inas
N
o N
o M
ayor
hi
dról
isis
a ce
falo
spor
inas
qu
e a
benc
ilpen
icili
na.
Hidr
oliz
a ce
fam
icin
as.
AmpC
de
E. co
li, P
99, A
CT-1
, CM
Y-2,
FO
X-1,
MIR
-1
1e
NIb
C Ce
falo
spor
inas
N
o N
o Hi
dról
isis a
umen
tada
a c
efta
zidim
a y
a ot
ras o
xiim
ino-β-
lact
ámic
os.
GC-1
, CM
Y-37
2a
2a
A
Peni
cilin
as
Si
No
May
or h
idró
lisis
a be
ncilp
enic
ilina
s que
a c
efal
ospo
rinas
. PC
1 2b
2b
A
Peni
cilin
as y
prim
eras
ce
falo
spor
inas
Si
N
o Hi
dról
isis s
imila
r a b
enci
lpen
icili
nas q
ue a
cef
alos
porin
as.
TEM
-1, T
EM-2
, SHV
-1
2be
2be
A Ce
falo
spor
inas
de
espe
ctro
ex
tend
ido
y m
onob
acta
m
Si
No
Hidr
ólisi
s au
men
tada
a
oxiim
ino-β-
lact
ámic
os (
cefo
taxi
ma,
cef
tazid
ima,
ce
ftria
xon,
cef
epim
e, a
ztre
onam
). TE
M-3
, SHV
.2, C
TX-M
-15,
PER
-1, V
EB-1
. 2b
r 2b
r A
Peni
cilin
as
No
No
Resis
tenc
ia a
áci
do c
lavu
láni
co, t
azob
acta
m y
sulb
acta
m.
TEM
-30,
SHV
-10
2ber
N
I A
Cefa
losp
orin
as d
e es
pect
ro
exte
ndid
o y
mon
obac
tam
N
o N
o Hi
dról
isis
aum
enta
da o
xiim
ino-β-
lact
ámic
os c
ombi
nada
con
res
isten
cia
a ác
ido
clav
ulán
ico,
tazo
bact
am y
sulb
acta
m.
TEM
-50
2c
2c
A Ca
rben
icili
na
Si
No
Hidr
ólisi
s aum
enta
da a
car
beni
cilin
a.
PSE-
1, C
ARB-
3 2c
e N
I A
Carb
enic
ilina
, cef
epim
e Si
N
o Hi
dról
isis a
umen
tada
a c
arbe
nici
lina,
cef
epim
e y
cefp
irom
e.
RTG-
4 2d
2d
D
Clox
acili
na
varia
ble
No
Hidr
ólisi
s aum
enta
da a
clo
raxi
lina
o ox
acili
na
OXA
-1, O
XA-1
0 2d
e N
I D
Cefa
losp
orin
as d
e es
pect
ro
exte
ndid
o va
riabl
e N
o Hi
drol
izan
la c
lora
xici
lina
o ox
acili
na y
los o
xiim
ino-β-
lact
ámic
os.
OXA
-11,
OXA
-15
2df
NI
D Ca
rbap
enem
as
varia
ble
No
Hidr
oliza
n la
clo
raxi
cilin
a o
oxac
ilina
y lo
s car
bape
nem
as.
OXA
-23,
OXA
-48
2e
2e
A Ce
falo
spor
inas
de
espe
ctro
ex
tend
ido
Si
No
Hidr
oliza
n ce
falo
spor
inas
, se
inh
iben
por
áci
do c
lavu
láni
co p
ero
no p
or
aztr
eona
m.
CepA
2f
2f
A
Carb
apen
emas
va
riabl
e N
o Hi
drol
izan
carb
apen
emas
, oxi
imin
o-β-
lact
ámic
os y
cef
amic
inas
. KP
C-2,
IMI-1
, SM
E-1
3a
3 B
(B1)
Ca
rbap
enem
as
No
Si
Hidr
ólisi
s de
am
plio
es
pect
ro
incl
uyen
do
carb
apen
emas
pe
ro
no
mon
obac
tam
. IM
P-1,
VIM
-1, C
crA,
IND-
1
B
(B2)
L1, C
AU-1
, GO
B-1,
FEZ
-1
3b
3 B
(B3)
Ca
rbap
enem
as
No
Si
Hidr
ólisi
s de
carb
apen
emas
. Cph
A, S
fh-1
N
I 4
No
se c
onoc
e
a CA
: áci
do c
lavi
láni
co; T
ZB: t
azob
acta
m.
b NI:
No
incl
uida
s.
Tabl
a 1.
4. C
lasif
icac
ión
de la
s β-la
ctam
asa
[126
].
a CA: á
cido
cla
vulá
nico
; TZB
: taz
obac
tam
. b N
I: N
o in
clui
das.

INTRODUCCIÓN 28
β-lactamasas tipo TEM y SHV.
La primera vez que se describió una cepa de K. pneumoniae con una β-lactamasa SHV fue en
1985. Kliebe y colaboradores [127] describieron una cepa de K. pneumoniae procedente de
Alemania con una β-lactamasa tipo SHV-2, mutante de SHV-1, que deriva de la β-lactamasa
cromosómica de clase A de K. pneumoniae, y que era capaz de hidrolizar oxiimino-
cefalosporinas. En el caso de las β-lactamasas tipo TEM aunque se cree que dicha familia tiene
un ancestro común, su origen es aún incierto. El número de β-lactamasa de ambos tipos
descubiertas ha ido aumentando a lo largo de los años y muchas veces debido a la aparición de
mutaciones sobre la β-lactamasa inicial. En 1987 que Sirot y colaboradores [128] describieron
varias cepas de diferentes especies de enterobacterias que presentaban una cefalosporinasa
derivada de TEM-1 mediante la mutación de tres aminoácidos y que llamaron TEM-3 [129].
Hay que tener en cuenta que la mayor causa de la resistencia a las cefalosporinas de tercera
generación en las Enterobacteriaceae son las β-lactamasas de clase A según la clasificación de
Amber (tabla 1.4). Son de especial relevancia, las variantes de las TEM-1, TEM-2 y SHV-1 que se
han generado por mutaciones de dichas β-lactamasas y les han permitido ampliar su
especificidad de sustrato a las cefalosporinas de tercera y cuarta generación, en concreto, a
cefotaxima, ceftriaxol y ceftazidima. Estas β-lactamasas muestran un amplio rango de
sustituciones aminoacídicas que conllevan a la ampliación de la especificidad del sustrato.
Estas mutaciones afectan al reconocimiento de los sustratos y al ratio de formación e hidrólisis
del complejo acil-enzima. Hay suficientes evidencias de que las β-lactamasas de clase A sufren
varios cambios conformacionales inducidos por la unión y la reacción con el sustrato.
Paralelamente, otros sustratos experimentan reorganizaciones químicas llevadas a cabo por
ataques enzimáticos que pueden dirigir cinéticas más complejas. La interacción de ambos
procesos tiene como resultado un espectro del sustrato ampliado y diferentes ratios de
hidrólisis para las diferentes cefalosporinas [130].
Actualmente, las variantes de TEM y SHV catalogadas ha crecido hasta 185 y más de 140
respectivamente, (ver http://www.lahey.org/Studies/) la mayoría de ellas con actividad contra
las oxiimino-cefalosporinas. Las primeras variantes de TEM y SHV, identificadas los años 1970 y
1980 se caracterizan por la hidrólisis de cefalosporinas de primera y de segunda generación y
se inhiben por ácido clavulánico y tazobactam; incluyen las enzimas TEM-1, TEM-2 y SHV-1 y
forman parte las serin β-lactamasas. Posteriormente se han encontrado β-lactamasas del tipo
TEM y SHV, como TEM-30, TEM-31, TEM-163, SHV-10 y SHV-72, que tienen el mismo espectro
de actividad frente a los β–lactámicos pero son resistentes al ácido clavulánico y al tazobactam
[126].

INTRODUCCIÓN 29
En K. pneumoniae los tipos TEM más extendidos son TEM-3 y TEM-4 aislados sobretodo en las
UCIs [131, 132]. También podemos encontrar cepas con TEM-1 e hiperproducción de ésta,
dando lugar a un fenotipo caracterizado por la resistencia al ácido clavulánico y a las
cefalosporinas de primera generación. En la familia de las SHV, uno de los enzimas más
prevalente en K. pneumoniae es el SHV-12 [133]. La presencia del correspondiente gen blaSHV-12
se ha asociado a elementos genéticos que confieren resistencia a fluoroquinolonas, (qnr), que
puede haber contribuido a su dispersión [134, 135]. También podemos encontrar cepas de
Klebsiella con expresión de altos niveles de SHV-1, presente de forma natural en cromosoma
de la mayoría de cepas clínicas de K. pneumoniae, que puede llegar a afecta a las
cefalosporinas de segunda generación e incluso a la ceftazidima.
β-lactamasas tipo AmpC.
El gen ampC, que está presente en el cromosoma de casi todas las enterobacterias excepto en
las especies de Klebsiella y Proteus, está regulado por un mecanismo complejo. Los
antibióticos β-lactámicos como la cefoxitina inducen la expresión de ampC mediante la unión a
transpeptidasas (proteínas de unión a penicilina) provocando un cambio en el balance de la
degradación de la mureina [136]. Los productos de degradación son transferidos al citoplasma
mediados por la porina AmpD [137]. La función de estos productos de degradación en el
citoplasma es la activación del regulador transcripcional AmpR causando un incremento de la
actividad del promotor. La amidasa AmpD contraresta esta activación mediante el incremento
de la degradación de muropéptidos. Diferentes mutaciones en el gen ampD suponen la
pérdida de la funcionalidad de AmpD y causan una expresión constitutiva de altos niveles de
ampC. Otra razón para la expresión permanente de ampC es una mutación específica de ampR
que reduce la habilidad de AmpR para unirse al represor muropeptido-UDP [138].
La expresión constitutiva de los genes ampC localizados en plásmidos media la resistencia a las
cefalosporinas de amplio espectro en especies sin genes propios ampC como K. pneumoniae.
El punto inicial es la movilización de los genes ampC del cromosoma de diferentes bacterias
entericas, como Citrobacter freundii o Enterobacter cloacae, y la transferencia horizontal a
otras especies [139]. Solo son probables unos pocos eventos de movilización, por ejemplo los
genes ampC de C. freundii parece que sólo se han movilizado una vez no hace mucho [140]. El
primer gen ampC de origen plasmídico, llamado CMY-1, fue identificado en 1989 [141],
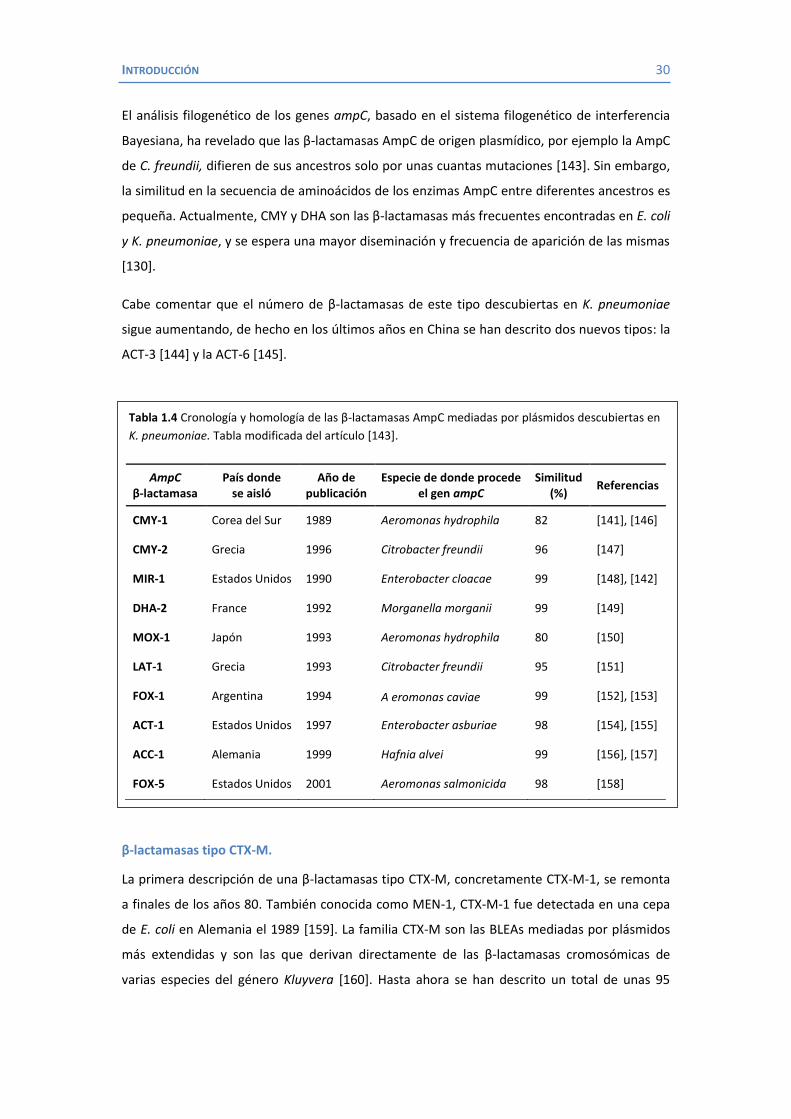
seguido por MIR-1 y CMY-2 [142]. Desde entonces se han descrito muchos otros tipos como
MOX-1, FOX-1, DHA-2, ACC-1, etc. (En la tabla 1.4 se pueden ver las BLEAs homólogas a AmpC
detectadas por primera vez en cepas de K. pneumoniae).
INTRODUCCIÓN 30
El análisis filogenético de los genes ampC, basado en el sistema filogenético de interferencia
Bayesiana, ha revelado que las β-lactamasas AmpC de origen plasmídico, por ejemplo la AmpC
de C. freundii, difieren de sus ancestros solo por unas cuantas mutaciones [143]. Sin embargo,
la similitud en la secuencia de aminoácidos de los enzimas AmpC entre diferentes ancestros es
pequeña. Actualmente, CMY y DHA son las β-lactamasas más frecuentes encontradas en E. coli
y K. pneumoniae, y se espera una mayor diseminación y frecuencia de aparición de las mismas
[130].
Cabe comentar que el número de β-lactamasas de este tipo descubiertas en K. pneumoniae
sigue aumentando, de hecho en los últimos años en China se han descrito dos nuevos tipos: la
ACT-3 [144] y la ACT-6 [145].
Tabla 1.4 Cronología y homología de las β-lactamasas AmpC mediadas por plásmidos descubiertas en K. pneumoniae. Tabla modificada del artículo [143].
AmpC β-lactamasa
País donde se aisló
Año de publicación
Especie de donde procede el gen ampC
Similitud (%) Referencias
CMY-1 Corea del Sur 1989 Aeromonas hydrophila 82 [141], [146]
CMY-2 Grecia 1996 Citrobacter freundii 96 [147]
MIR-1 Estados Unidos 1990 Enterobacter cloacae 99 [148], [142]
DHA-2 France 1992 Morganella morganii 99 [149]
MOX-1 Japón 1993 Aeromonas hydrophila 80 [150]
LAT-1 Grecia 1993 Citrobacter freundii 95 [151]
FOX-1 Argentina 1994 A eromonas caviae 99 [152], [153]
ACT-1 Estados Unidos 1997 Enterobacter asburiae 98 [154], [155]
ACC-1 Alemania 1999 Hafnia alvei 99 [156], [157]
FOX-5 Estados Unidos 2001 Aeromonas salmonicida 98 [158]
β-lactamasas tipo CTX-M.
La primera descripción de una β-lactamasas tipo CTX-M, concretamente CTX-M-1, se remonta
a finales de los años 80. También conocida como MEN-1, CTX-M-1 fue detectada en una cepa
de E. coli en Alemania el 1989 [159]. La familia CTX-M son las BLEAs mediadas por plásmidos
más extendidas y son las que derivan directamente de las β-lactamasas cromosómicas de
varias especies del género Kluyvera [160]. Hasta ahora se han descrito un total de unas 95

INTRODUCCIÓN 31
variantes diferentes según la base de datos de Lahey clinic. Estas variantes se clasifican en 5
grupos diferentes de CTX-Ms según la similitud de su secuencia aminoacídica: CTX-M-1, CTX-
M-2, CTX-M-8, CTX-M-9 y CTX-M-25. Pero los enzimas más ampliamente difundidos son CTX-
M-2, CTX-M-3, CTX-M-14 y CTX-M-15 [161].
La heterogeneidad entre los aislados clínicos de enterobacterias que presentan CTX-M,
demuestra que los genes blaCTX-M se han capturado de diferentes fuentes dentro del género de
Kluyvera y por múltiples eventos [162], en los que secuencias de inserción ISEcp1 e ISCR1
asociadas con integrones de clase 1, juegan un papel muy importante [161, 163, 164].
Durante los últimos 15 años este tipo de β-lactamasas han protagonizado una propagación
rápida y global, y determinados lugares las BLEAs tipo CTX-M son más numerosas que las β-
lactamasas clásicas de tipo TEM y SHV. En España, el índice de producción de este tipo de
enzimas es del 12,5% en aislados de K. pneumoniae, con el predominio del grupo 9 (CTX-M-9 y
CTX-M-14) y del grupo 1 (CTX-M-10) [133]. En el caso de CTX-M-1, la primera cepa de K.
pneumoniae multiresistente productora de dicha β-lactamasa se describió en 2006 en un brote
epidémico de una UCI española, concretamente en el Hospital Universitario de Son Dureta.
La incorporación de los genes blaCTX-M en las especies de Klebsiella provoca resistencia a
cefotaxima. La expansión del espectro hidrolítico a la ceftazidima se debe a mutaciones que
principalmente afectan al asa Ω de los enzimas [165-167]. Aunque los carbapenemas se
mantienen estables a los enzimas tipo CTX-M, ya se han publicado casos de la aparición de
resistencia a carbapenemas durante el tratamiento de estas cepas de K. pneumoniae
productoras de CTX-M-15 y CTX-M-1 debido a la pérdida de porinas [168, 169].
β-lactamasas de clase A: Carbapenemasas.
Recientemente se han conocido cuatro tipos diferentes de enzimas tipo A que presentan
actividad cabapenemasa:
- SME: prácticamente asociados de manera exclusiva con Serratia marcescens con solo tres
variantes descritas [170, 171];
- IMI (NUN-A): preferentemente presentes en E. cloacae (de Estados Unidos, Francia y
Argentina). Sin embargo, al estar flanqueado por tnpA de TN503, no se sabe si la blaIMI es
móvil [172]
- GES (cuyo nombre se les puso después de ser detectadas por primera vez en cepas de K.
pneumoniae procedentes de Guinea): hasta el momento se conocen 16 variantes y hay

INTRODUCCIÓN 32
indicios de que se localizan en integrones. Los enzimas GES se encuentran principalmente
en Pseudomonas aeruginosa, pero también en E. coli y K. pneumoniae tanto en Norte
América como en Asia [173].
- KPC (acrónimo en inglés de carbapenemasas de K. pneumoniae): hasta el momento se
conocen 10 variedades. KPC-2 y KPC-3 son las variedades más frecuentes a nivel mundial.
Los genes blaKPC están localizados en un nuevo transposón basado en Tn3, el transposón
Tn4401 [174]. KPC-2 fue por primera vez descrita en una cepa de K. pneumoniae
resistente a carbapenemas en Carolina del norte en 2001 [175]. Además KPC-2 fue
identificada en aislados de Salmonella enterica en Estados Unidos [176], en K. oxytoca
[177], en P. aeruginosa [178] en E. cloacae [179] y en C. freundii en España [180]. KPC-3
fue detectada en aislados nosocomiales de K. pneumoniae en el Noreste de Estados
Unidos y de Israel [179, 181]. También se han detectado cepas de K. pneumoniae
productoras de KPC en Europa [174] y en Finlandia [182], en América del Sur [178], en
China [183] y en España [184].
A modo de resumen, en la figura 1.6 se puede observar la distribución de cepas de K.
pneumoniae resistentes a carbapenemas en Europa en 2009.
Figura 1.6. Mapa de la distribución de cepas de K. pneumoniae resistentes a carbapenemas en Europa en 2009. Mapa obtenido de la European Centre for Disease Prevention and Control (www.ecdc.europa.eu)
Liechtenstein
Luxemburgo
Malta
< 1% Entre el 1% y < 5% Entre el 5% y < 10% Entre el 10% y < 25% Entre el 25% y < 50% ≥ 50% Sin datos o con datos de menos de 10 aislados Datos no incluidos
Porcentaje de resistencia

INTRODUCCIÓN 33
β-lactamasas de clase D: oxacilinasas (OXA).
Según la definición original, las β-lactamasas OXA se llaman así debido a su capacidad de
hidrolizar oxacilina. Este grupo, también incluye varias oxacilinasas con un espectro de
hidrólisis ampliado a las cefalosporinas debido a sustituciones aminoacídicas [185, 186]. La
mayoría de estas BLEAs tipo OXA derivan de OXA-10, por ejemplo: OXA-11, OXA-14, OXA-16,
OXA-17; o de OXA-15, como OXA-19 y OXA-28. Los derivados de OXA-2 son poco frecuentes
[187]. Las carbapenemasas OXA son diferentes enzimas de clase D de Amber, tales como OXA-
23-like, OXA-24-like, OXA-48, OXA-51-like y OXA-58-like [187]. Inicialmente las β-lactamasas
OXA se identificaron en P. aeruginosa pero en la actualidad también han sido descritas en
muchas otras bacterias Gram negativas incluidas en las Enterobacteriaceae. El reservorio
natural de los genes blaOXA son más probablemente las bacteria medioambientales Ralstonia
spp., Burkholderia spp. y Shewanell spp. [188]. La gran familia de carbapenemasas OXA es muy
diversa y los genes blaOXA están localizados tanto en el cromosoma como en plásmidos. Un
estudio basado en la filogenia bayesiana de los genes blaOXA revela que esta diversidad es
debida principalmente a eventos ancestrales, así como a la movilización del cromosoma a los
plásmidos ocurrida hace millones de años [189].
Los genes blaOXA codificados por el cromosoma juegan un papel importante en la resistencia en
Acinetobacter baumannii [130]. Aunque estos genes se están expandiendo a otras especies. En
los últimos años han sido varios los casos de aislamientos de K. pneumoniae que han
presentado carbapenemasas tipo OXA, por ejemplo en Turquía y Francia [190, 191], en los
Países Bajos [192], en Túnez [193] y en Marruecos [194]. Incluso ha habido aislamientos de K.
pneumoniae con dicho tipo de carbapenemasa en nuestro país [195, 196].
β-lactamasas de clase B: Metalo-β-lactamasas (MBL).
Las MBL pertenecen a una superfamilia de enzimas con una amplia diversidad catalítica
(oxidoreductasas, glioxilasas, fosoforil colinesterasas, etc. (ver revisión [197])). Estos enzimas
son capaces de hidrolizar todos los antibióticos β-lactámicos excepto los monobactámicos. En
base a los alineamientos de las secuencias de ADN de sus genes, las MBL están clasificadas en
3 subclases: B1, B2 y B3 [198]. Sin embargo, aunque hay un bajo grado de similitud entre las
diferentes subclases, estos agrupamientos están respaldados por los análisis cristalográficos de
los correspondientes enzimas. La habilidad de la producción de MBL no solo se ha detectado

INTRODUCCIÓN 34
en los patógenos bacterianos Gram negativos, sino también en un sorprendente número de
bacterias ambientales.
Los genes de las MBL de la subclase B1 (intrínsecos o adquiridos) se han encontrado en
muchas especies bacterianas. Recientemente se ha descubierto un nuevo enzima
perteneciente a las MBL, el NDM-1, que ha sido identificado en K. pneumoniae [199]. La
subclase B2, en cambio, se encuentra principalmente en diferentes especies de Aeromonas
[200]. La subclase B3 son enzimas intrínsecas a un número de diferentes especies ambientales
por lo que algunas se pueden convertir en patógenos nosocomiales de pacientes
inmunodeprimidos, por ejemplo Stenotrophomonas maltophila [201].
Las MBL más frecuentes adquiridas por patógenos bacterianos Gram negativos son las tipo
IMP y VIM. Estos enzimas fueron originalmente descritos en P. aeruginosa y A. baumannii,
pero hoy en día están presentes en la mayoría de Enterobacteriaceae [202, 203]. De los
enzimas tipo VIM se conocen más de 12 variantes alélicas [204]. La aparición de diferentes
subtipos VIM e IMP en diferentes áreas geográficas sugiere que sus correspondientes genes
son adquiridos independientemente de reservorios naturales desconocidos [130]. Esta idea
está apoyada por los diferentes estudios sobre las MBL más raramente adquiridas: SPM-1 en
América del Sur [205], GIM-1 en Alemania [206] y SIM-1 en Corea [207]. Los determinantes
génicos para la adquisición de estas MBL, en la mayoría de casos, son integrones de tipo 1 y 3
localizados en plásmidos de gran tamaño o en el cromosoma. Para los genes blaSPM-1 se ha
descrito una asociación con la secuencia de inserción ISCR44. Esta secuencia de inserción es
capaz de movilizar grandes secuencias de DNA adyacentes, probablemente mediante
mecanismos de replicación. Los elementos ISCR también se han descrito en P. aeruginosa y
están relacionados con los genes blaIMP y blaVIM [208].
En K. pneumoniae se empezaron a encontrar cepas que presentaban enzimas IMP a partir del
año 1999 en Singapur [209] y, posteriormente, se identificaron cepas que presentaban VIM en
Grecia [210]. Desde entonces se han ido extendiendo mundialmente y en nuestro país se
detectaron por primera vez aislados que presentaban enzimas VIM en 2005 [203].
[126]

INTRODUCCIÓN 35
1.1.1.2 Expulsión activa
Las bacterias pueden reducir la acumulación intracelular de los antibióticos no sólo reduciendo
la penetración sino también mediante mecanismos que producen la expulsión de las
sustancias hacia el medio extracelular. Estos mecanismos, al contrario que la penetración a
través de las porinas, requieren energía y son parte importante de la resistencia intrínseca de
E. coli y otras enterobacterias como K. pneumoniae a compuestos que están presentes en su
hábitat natural como son las sales biliares.
Las bombas de flujo o de expulsión activa son proteínas transportadoras responsables de la
expulsión de sustratos tóxicos del interior celular al ambiente. Se conoce al menos una bomba
por cada clase de antibiótico capaz de expulsarlo. Estas proteínas se encuentran tanto en
bacterias Gram positivas como en Gram negativas. El análisis de algunos de los genomas
bacterianos disponibles muestra que las bombas de flujo conocidas y putativas constituyen
entre un 6% y un 18% de todos los transportadores encontrados en cualquier célula bacteriana
[211]. Las bombas pueden ser específicas de un sustrato o bien transportar varios compuestos
no relacionados ni química ni estructuralmente del interior bacteriano al exterior y
consumiendo energía, pero sin la alteración o degradación de dichos compuestos. Cuando
estas bombas transportan antibióticos de múltiples clases están asociadas a fenómenos de
multiresistencia a antibióticos y reciben el nombre de bombas de flujo multidroga. Las bombas
de flujo multidroga son particularmente preocupantes desde el punto de vista clínico, ya que
pueden eliminar una gran variedad de compuestos estructuralmente muy diferentes.
Las bombas de flujo bacterianas se clasifican en 5 familias (figura 1.7):
1.- RND: “Resistance Nodulation Division” [212]
2.- MFS: “Major Facilitator Superfamily” [213, 214]
3.- SMR: “Staphylococcal Multiresistance” [215]
4.- MATE: “Multidrug and Toxic Compound Extursion” [216]
5.- ABC: “ATP Binding Cassette” [217]
Las bombas de flujo también se pueden clasificar como bombas de un componente o de
múltiples componentes. Las bombas de un solo componente transportan los sustratos a través
de la membrana citoplasmática, por ejemplo las MATE. En cambio, las bombas de múltiples
componentes, que están presentes tanto en los organismos Gram negativos y en los Gram

INTRODUCCIÓN 36
positivos, como las RND, necesitan estar asociadas a otras proteínas para ser funcionales, de
hecho se unen a proteínas de membrana externa, y expulsan los sustratos a través de toda la
envoltura celular [218]. Referencia fig.
En la mayoría de casos las bombas de flujo multidroga están codificadas cromosómicamente y,
por tanto, no son transferibles entre bacterias. Sin embargo, hay algunos ejemplos de
bacterias Gram negativas donde dichos genes se han encontrado en elementos móviles [219].
Aunque las bacterias Gram negativas tienen bombas de flujo de las 5 superfamilias, las bombas
RND son las que tienen un papel más importante en este tipo de bacterias. Estas bombas de
flujo además de jugar un papel principal tanto en la resistencia intrínseca como en la
resistencia adquirida de muchas Gram negativas a una variedad de antibióticos clínicamente
relevantes, también exportan biocidas, tintes, detergentes y solventes orgánicos. Este tipo de
bombas abarcan toda la envoltura celular de las bacterias Gram negativas y únicamente
contribuye a la resistencia antimicrobiana cuando se combinan con una reducción de la
permeabilidad de la membrana externa. Ésta debe ser la principal razón por la que sólo unos
pocos de los sistemas de flujo multidrogas que no pertenecen a la familia RND promueven la
resistencia a antibióticos clínicamente importantes [218] .
Dentro de las RND destacamos la bomba AcrAB-TolC. Dicho sistema muestra una amplia
especificidad de sustratos. Los sustratos para esta bomba incluye acriflavina, β-lactámicos,
Figura 1.7. Dibujo esquemático de los principales tipos de bombas de flujo. Las bombas representadas son NorA de Staphylococcus aureus, miembro de la superfamilia MFS; EmrE de E. coli, miembro de la superfamilia SMR; NorM de Vibrio parahaemolyticus, miembro de la superfamilia MATE; AcrAB–TolC de E. coli, miembro de la superfamilia RND; y LmrA de Lactococcus lactis, miembro de la superfamilia ABC [218].
MembranaExterna
MembranaInterna
Citoplasma
Antibiótico
Antibiótico
Antibiótico

INTRODUCCIÓN 37
sales biliares, cloramfenicol, cristal violeta, bromuro de etidio, ácidos grasos, macrólidos,
solventes orgánicos, fluoroquinolonas y dodecilsulfato sódico (SDS) [218].
La importancia de los mecanismos de expulsión activa en la resistencia también se ha
demostrado en K. pneumoniae, por ejemplo se ha observado que los sistemas AcrAB (principal
sistema de expulsión activa de E. coli) juega un papel importante en esta especie [220]. De
hecho en un estudio de aislados clínicos de K. pneumoniae se observó la sobreexpresión de
AcrAB en cepas resistentes [221]. Por otra parte se ha sugerido que, además de contribuir al
fenotipo de multiresistencia, la bomba de flujo AcrAB puede representar un nuevo factor de
virulencia necesario para que K. pneumoniae sea resistente a los mecanismos de defensa del
sistema inmune en el pulmón, facilitando el inicio de la neumonía [222].
1.1.1.3 Alteraciones de la permeabilidad.
La resistencia adquirida a los β-lactámicos de una serie de organismos Gram negativos,
incluyendo K. pneumoniae, también se ha atribuido a los cambios de la membrana externa
relacionados con un descenso de la permeabilidad.
Estudios teóricos y datos experimentales indican que los agentes antimicrobianos de pequeño
tamaño (β-lactámicos, tetraciclinas, cloramfenicol y fluoroquinolonas) utilizan principalmente
la vía de las porinas para penetrar en el interior celular, por ejemplo en las enterobacterias, ya
que éstas presentan porinas altamente permeables.
Las porinas son proteínas de la membrana externa que permiten la difusión inespecífica de
moléculas de pequeño tamaño al interior de la célula bacteriana. La pérdida de porinas se ha
relacionado con la resistencia antibiótica. Una disminución de la permeabilidad puede producir
cambios significativos de la resistencia que se pueden incrementar cuando se combinan con la
inactivación enzimática (ver más adelante).
Estudios recientes indican que a partir de cepas productoras de β-lactamasas es más fácil la
selección de cepas deficientes en las dos porinas principales, OmpK35 y OmpK36. Las
alteraciones de la expresión de las porinas en K. pneumoniae cooperan con la expresión de las
β-lactamasas para provocar resistencia aumentada a las cefalosporinas de amplio espectro. De
hecho, se ha demostrado la importancia de la asociación entre la pérdida de las porinas y la
producción de AmpC para la aparición de la resistencia a carbapenemas [223, 224].

INTRODUCCIÓN 38
La alteración de la permeabilidad por la modificación de las porinas está basada en mutaciones
en los genes que codifican estas proteínas [225]. Estas mutaciones pueden reducir la expresión
de las porinas por modificaciones puntuales de la región promotora del gen que regula la
transcripción del ARN mensajero (ARNm). Algunas modificaciones no alteran la expresión de la
proteína pero producen cambios en su estructura que afectan al tamaño del canal por donde
pasan los antibióticos [226-228]. Las mutaciones más drásticas suponen la pérdida completa
de la expresión de las porinas por mutaciones puntuales que alteran la pauta de lectura del
gen o por la inserción de secuencias de inserción que interrumpen el gen [168].
La expresión de las dos porinas principales en K. pneumoniae no parece que sea crucial para la
entrada del antibiótico, ya que mutantes producidos en el laboratorio que únicamente
expresan una de las dos porinas no muestran alteraciones de la sensibilidad antimicrobiana, a
diferencia de lo que sucede cuando se pierde la expresión de ambas porinas, produciéndose
un aumento importante de la resistencia [229, 230].
Entre los aislamientos resistentes a antibióticos destacan especialmente las cepas productoras
de BLEAs, que confieren resistencia a la mayoría de β-lactámicos disponibles hasta el
momento, o también a través de enzimas modificadores de aminoglicósidos. Un problema
añadido es la facilidad que tiene K. pneumoniae de acumular otros mecanismos de resistencia
que actúan conjuntamente con estos enzimas, disminuyendo las opciones terapéuticas. Por
tanto, K. pneumoniae aparte de tener un papel importante como bacilo Gram negativo
causante de infecciones nosocomiales, también es una fuente importante de transmisión de
resistencia antibiótica.

INTRODUCCIÓN 39
1.2 La membrana externa de las bacterias Gram negativas.
La envoltura celular de las bacterias Gram negativas se compone de dos membranas
diferentes, una interna y otra externa; separadas por el periplasma, que es un compartimento
hidrofílico que incluye una capa de peptidoglicano [231]. La composición lipídica de la
membrana interna es similar a la mayoría de membranas biológicas, incluyendo la membrana
citoplasmática de los organismo Gram positivos, pero la membrana externa (ME) es un poco
atípica [232] (ver figura 1.8).
La ME de las bacterias Gram negativas tiene un papel crucial dotando de una capa extra de
protección al organismo sin comprometer el intercambio de material necesario para el
mantenimiento de la vida. En esta doble capacidad, la ME aparece como un sofisticado
ensamblado macromolecular. La ME actúa como una barrera selectiva, combinando una
bicapa lipídica altamente hidrofóbica con proteínas formadoras de poros con propiedades
específicas de tamaño de exclusión. Las propiedades de permeabilidad de esta barrera, sin
embargo, tienen un mayor impacto en la susceptibilidad del microorganismo a los antibióticos,
que hasta la fecha, son esencialmente dianas de procesos intracelulares. Antibióticos
pequeños e hidrofílicos, tales como los β-lactámicos, usan las proteínas formadoras de canales
para tener acceso al interior celular, mientras que los macrólidos y otros antibióticos
hidrofóbicos difunden a través de la bicapa lipídica [233].
La ME es una bicapa lipídica asimétrica con fosfolípidos formando la cara interna y con LPS
formando la cara externa. El LPS, como se ha comentado anteriormente, está compuesto de
lípido A, un núcleo oligosacárido y un polisacárido que recibe el nombre de antígeno O. La
Figura 1.8. Esquema de la estructura de la envoltura celular de las bacterias Gram negativas.

INTRODUCCIÓN 40
presencia de LPS en la capa externa de la ME confiere unas propiedades de permeabilidad
específicas a esta membrana y contribuye a la efectividad de su función como barrera
permeable. Así pues de entre estas propiedades del LPS destacan:
i) la composición química del dominio alifático del LPS. El alto número de sustituyentes
grasos acilos totalmente saturados por molécula de lípido crea un interior lipídico
similar a gel con muy baja fluidez que contribuye a la baja permeabilidad de los solutos
hidrofóbicos a través de la ME;
ii) la fuerte carga (aniónica) natural del antígeno O y la interacción lateral entre las
moléculas de LPS mediante la acción de unión de los cationes divalentes también
contribuye a la baja permeabilidad del LPS [218, 234];
iii) la fuerte asociación del LPS a las proteínas de membrana externa. Por ejemplo la
asociación con FhuA, un receptor para la captación de hidroxamato férrico, cuya unión
al lípido A ofrece un modo adicional de interacción entre las moléculas de LPS vecinas
[235]. Como resultado de esto, las bacterias Gram negativas están protegidas de
muchos compuestos tóxicos como son las sales biliares, detergentes, antibióticos,
péptidos antimicrobianos y ambientes hostiles durante la colonización del huésped o
la infección [231].
1.2.1 Proteínas de membrana externa.
Alrededor del 50% de la masa de la membrana externa consiste en proteínas, en forma de
proteínas integrales de membrana o como lipoproteínas que se anclan a la membrana
mediante lípidos unidos en el extremo N-terminal, concretamente vía una N-acil-
diacilglicerilcisteina [236]. Las lipoproteínas están localizadas tanto en la membrana interna
como en la externa. Se han identificado hasta la actualidad más de 130 lipoproteínas de
diversa estructura, función y origen bacteriano [237].
Las proteínas de membrana externa (PME) son normalmente estructuras transmembrana en
forma de barriles β antiparalelos y que funcionan como porinas, canales, receptores u otras
maquinarias de transporte que median la comunicación con el entorno extracelular y, por
tanto, permiten la entrada de nutrientes y otras sustancias esenciales, y la eliminación de
productos de desecho [231, 238]. Además, las proteínas y lipoproteínas frecuentemente

INTRODUCCIÓN 41
juegan un papel importante en la interacción entre las bacterias simbiontes o patogénicas con
su huésped [231, 238].
Las porinas son una de las familias de proteínas más altamente representadas en la ME. La
primera proteína de este tipo fue descubierta en 1976 [239], proponiéndose el nombre de
porina para denominar específicamente a este tipo de canales de difusión no específicos. Tal
como era de esperar, las porinas fueron descubriéndose en todas las especies de bacterias
Gram negativas estudiadas e incluso en bacterias Gram positivas, como Corynebacterium,
Nocardia y Mycobacterium, que producen una pared celular rica en lípidos y con una
estructura similar a una bicapa.
Al mismo tiempo que se descubrieron las porinas bacterianas también se vio que existían
porinas en las mitocondrias [240]. Estas porinas, también llamadas canales aniónicos
dependientes de voltaje, presentan canales grandes ya que las mitocondrias no tienen la
necesidad de eliminar los tóxicos presentes en el medio ambiente. Además, presentan una
estructura general basada en un barril de láminas β, como las porinas bacterianas (ver más
adelante). Para profundizar en las porinas mitocondriales pueden revisarse las referencias
[241, 242]. Otros orgánulos eucariotas que presentan porinas son los peroxisomas [243-245].
El término “porina” ha sufrido modificaciones debido a su popularidad. En un principio se
definió para hacer referencia únicamente a proteínas formadoras de canales inespecíficos,
pero actualmente es general su uso en todas aquellas proteína formadores de canales, como
en el caso de LamB, llamada maltoporina a pesar de ser un canal específico, o PhoE, llamada
fosfoporina aunque no es específica para fosfatos sino simplemente un canal con preferencia
por los aniones.
Las porinas generalmente se dividen en dos clases según su función:
Porinas no específicas que forman canales de difusión no específicos permitiendo la
entrada de moléculas pequeñas y polares (<600 kDa) que difunden a través de la
membrana, tales como OmpC y OmpF en E. coli o OmpK35 y OmpK36 en K.
pneumoniae;
Y porinas específicas que facilitan la difusión de sustratos específicos, como por
ejemplo LamB (que permite la entrada de derivados de la maltosa y actúa como
receptor del fago lambda) y PhoE [246]. Las porinas también sirven como receptores
de bacteriofagos y bacteriocinas y, junto al peptidoglicano y el LPS, juegan un papel
importante en el mantenimiento de la integridad celular.

INTRODUCCIÓN 42
La mayoría de estudios de porinas se han hecho en la familia de las Enterobacteriaceae,
concretamente en E. coli, K. pneumoniae y Enterobacter spp. [247-249]. También hay
bastantes estudios hechos en P. aeruginosa y, más recientes y menos extendidos, en A.
baumannii [250, 251].
1.2.1.1 Porinas en K. pneumoniae.
Las tres porinas mayoritarias en la ME de K. pneumoniae son la porina OmpA (también llamada
OmpK34), la porina OmpK35 (análoga a OmpF de E. coli) y la porina OmpK36 (análoga OmpC
de E. coli), con pesos moleculares aparentes de 34, 35 and 36 kDa, respectivamente [223, 230,
249]. Además de estas tres porinas, K. pneumoniae también puede expresar la porina
quiescente OmpK37 y porinas específicas de sustrato como son PhoE, LamB, OmpG y la familia
de porinas KdgM.
1.2.1.1.1 OmpK35 y OmpK36.
Las dos principales porinas no específicas de K. pneumoniae son las proteínas de membrana
externa OmpK35 y OmpK36, homólogas a OmpF y OmpC de E. coli, respectivamente. OmpK35
y OmpK36 son proteínas que muestran capacidad de modificación térmica y resistencia a la
tripsina [249]. Los genes que codifican estas porinas tienen 945 pb y 1118 pb,
respectivamente.
Estructura.
La estructura de las porinas se conoce bien debido a los estudios de cristalización y a los
estudios de caracterización funcional [247, 248, 252, 253]. Los rasgos estructurales generales
de OmpK36 y OmpK35 son muy similares a los de OmpC y OmpF, respectivamente. La
estructura de OmpK36 de K.pneumoniae se ha determinado por cristalografía de rayos X
(código de acceso a la Protein Data Base (PDB) 1OSM) [252], así como la estructura de OmpC
[254] y OmpF [255] de E. coli. Sin embargo, no existen estudios cristalográficos de OmpK35,
aunque debido a su alta semejanza con OmpF se espera que mantenga sus mismas
características.
Estructuralmente OmpK35 y OmpK36 son homotrímeros de subunidades íntimamente
asociadas (figura 1.9). Ambos homotrímeros consisten en barriles β de 16 láminas antiparalelas
con 8 giros periplásmicos cortos y 8 asas extracelulares de diferente longitud. Los tres ejes del
barril son perpendiculares al plano de la membrana y, aproximadamente, paralelos unos a

INTRODUCCIÓN 43
otros. Cada subunidad contiene entre 250-450 residuos aminoacídicos y cada hebra está unida
a sus vecinas próximas por puentes de hidrógeno a lo largo de toda la cadena. Un rasgo
estructural importante de estas porinas es que el asa 3 (L3) está plegada dentro del barril y
causa la constricción del poro. Este poro define las propiedades de difusión [247, 248, 252,
253]. En el caso de OmpK36 el tamaño del poro es de 10 Å [253].
Existe una gran variabilidad en las secuencia de las asas extracelulares de las porinas, de hecho
en ellas se acumulan la mayoría de inserciones y deleciones [252] (figura 1.10). Comparando
OmpF y OmpK36 podemos decir que en OmpK36 las asas L1 y L6 son más cortas en 5 residuos
cada una y el asa L2 en 2 residuos. Las inserciones más largas, de 10 residuos, se han
encontrado en el asa L4. El asa L8 contiene 3 residuos adicionales [252]. A pesar de ello, el
empaquetamiento de estas asas extracelulares es similar [252]. Por otra parte, la zona de
constricción del poro, es decir, el asa 3, y la salida al espacio periplásmico están
estructuralmente conservadas. Los residuos clave y conservados en todas las porinas
enterobacterianas son: Lys16, Arg38, Glu58, Arg75, Asp106, Glu110 y Arg126. El asa 3 de
OmpK35, homóloga a OmpF, sólo contiene un residuo más que OmpF y OmpC de E. coli y
OmpK36 de K. pneumoniae [230].
A diferencia de la gran semejanza estructural, la distribución de los residuos ionizables entre
OmpF, homóloga a OmpK35, y OmpK36 muestra un número importante de diferencias. En
OmpK36, el inusual grupo de residuos de ácido aspártico en el asa 1 (L1), dos de ellos dan al
A B
Figura 1.9. A y B. Vista frontal y lateral, respectivamente, de la estructura trimérica de la proteína OmpK36 de K. pneumoniae basada en su cristalización (código PDB 1OSM). En la imagen se observa la disposición de cada monómero (de diferente color) para formar la porina trimérica.

INTRODUCCIÓN 44
poro (Asp26, Asp33), y la pérdida de una lisina (Lys 243) en el extremo del asa 6 (L6) respecto a
OmpF le da a la entrada de OmpK36 más negatividad que a la de OmpF. Cerca de la zona de
constricción del poro, la Lys314 de OmpK36 reemplaza un residuo no cargado de isoleucina en
OmpF e introduce una carga positiva, pero también se producen dos reemplazamientos más,
la Lys80 por Trp y la Val18 por Asp, estos cambios rebajan la carga en dos unidades. Los
residuos de ácido aspártico 127 y 256, que en OmpF están ocultos y probablemente sin carga,
son residuos de asparagina en OmpK36. OmpC de E. coli muestra la misma distribución de
cargas en el poro que OmpK36, de manera que los potenciales electroestáticos son similares
[252].
La segregación de residuos ácidos y básicos a lo largo del poro crea un fuerte campo eléctrico
transversal en la zona de constricción del poro y es la que determina el potencial electrostático
de la porina [248]. De acuerdo con los cálculos electrostáticos, el potencial de OmpF y OmpK36
son muy similares [252].
La diferencia entre el flujo de solutos a través de las porinas OmpF y OmpC de E. coli se ha
atribuido a la ligera diferencia de tamaño del poro entre ambas porinas. Así pues, OmpC
presenta una considerable menor conductancia que OmpF [256-258]. Además, OmpC parece
ser más selectivo para los cationes [257]. El incremento de la selectividad para cationes de
OmpK36 (y OmpC) puede ser importante para el retraso de ciertos agentes tóxicos tales como
antibióticos, pero la selectividad iónica es más alta dependiendo de la concentración de sales
[252].
A B
Figura 1.10. A y B. Vista frontal y lateral, respectivamente, del monómero que forma la porina trimérica OmpK36 de K. pneumoniae basada en su cristalización (código PDB 1OSM). En la imagen se observan la disposición de cada asa: L1 (azul oscuro), L2 (azul claro), L3 (cian) L4 (verde claro), L5 (verde oscuro), L6 (amarillo), L7 (naranja), y L8 (rojo).
90ο

INTRODUCCIÓN 45
Regulación de la expresión.
Estudios recientes realizados en Enterobacteriaceae, principalmente en E. coli, Salmonella
enterica y Enterobacter aerogenes muestran la complejidad de la regulación de las porinas
principales de dichas bacterias, homólogas a OmpK35 y OmpK36 de K. pneumoniae. Así pues a
continuación se exponen los principales reguladores conocidos y su importancia, partiendo de
la base de que los mecanismos descritos en otras enterobacterias sean válidos para K.
pneumoniae.
Inicialmente se creía que el regulador OmpR era el principal regulador de los genes de las
porinas pero se ha visto que hay muchos otros reguladores implicados. A continuación se
explican los más importantes.
OmpR es un regulador de respuesta citoplasmática de dos componentes que también incluye
EnvZ, un sensor histidina quinasa de la membrana interna. OmpR es un regulador pleiotrópico
que controla la expresión de los genes de las porinas y procesos celulares tales como la
quimiotaxis y la virulencia [259-265]. La fosforilación de esta proteína, que es crucial para la
regulación de los genes de las porinas, la lleva a cabo EnvZ, que actúa como quinasa y fosfatasa
de OmpR [266]. Las señales ambientales que hacen que EnvZ se active como quinasa son la
osmolaridad, el pH, la temperatura, los nutrientes y las toxinas [267-269]. Al estar fosforilado
OmpR está en su forma activa e incrementa su unión a los promotores de los genes OmpC y
OmpF [270-272]. Por ejemplo, en E. coli la alta osmolaridad provoca una alta concentración de
OmpR-P. Este regulador se une a la región reguladora de ompC, activando su transcripción, y a
la zona reguladora de ompF, reprimiendo su transcripción [267]. El funcionamiento de este
regulador en otras enterobacterias aun no se conoce bien, aunque se han identificado zonas
de unión a OmpR en la región reguladora de las porinas OmpX de E. aerogenes [273] y OmpS1
y OmpS2 de S. enterica [274, 275]. En K. pneumoniae está descrita la presencia de dicho
regulador pero sus mecanismos de funcionamiento están sin estudiar.
El locus mar (acrónimo inglés de multiple antibiotic resistance) en E. coli es un operón contiguo
de 1335 pb con dos unidades transcripcionales divergentes marC y marRAB separadas por el
operador marO. El gen marC codifica para una proteína putativa de la membrana interna cuya
función parece no estar implicada en el fenotipo de multiresistencia a antimicrobianos [276,
277]. marR codifica un represor de 144 aminoácidos que actúa previniendo la expresión de la
cascada de regulación bajo condiciones de no estrés. marA codifica un activador
transcripcional que actúa como el origen de la cascada genética desencadenando numerosos

INTRODUCCIÓN 46
mecanismos de mutiresistencia a antimicrobianos. marB codifica una pequeña proteína de la
membrana interna con función desconocida [278, 279]. El operón marRAB muestra una
organización genética preservada entre las Enterobactereaceae [278].
MarA es un importante regulador implicado en la adaptación al ambiente y la protección
contra agresiones externas, induciendo de manera directa o indirecta más de 60 genes [280,
281]. La expresión de marA induce una disminución en OmpF en E. coli. Esta regulación
negativa se lleva a cabo mediante la transcripción de un ARN antisentido no traducido llamado
micF localizado corriente arriba del gen codificante de la porina OmpC. El promotor de micF
lleva una secuencia consenso tipo caja mar. Además, MarA activa la expresión de OmpX
implicado en el control de la expresión de las porinas [273, 282]. La expresión de estos
reguladores actúa de manera sinérgica para reducir la expresión de las porinas y limitar la
entrada de los antibióticos.
Diferentes estudios muestran la importancia del operón mar en K. pneumoniae y su relación
con la aparición de cepas multiresistentes [278, 283-285].
El efector del regulón soxRS, de respuesta global al superóxido, es SoxS. SoxS muestra un 50%
de homología con MarA y también pertenece a la familia AraC-XylS de activadores
transcripcionales [286]. En presencia de agentes activadores, tales como peróxido de
hidrógeno, SoxR cambia del estado reducido al oxidado desencadenando la transcripción del
gen soxS [286]. SoxS puede inducir la transcripción de micF y acrAB. Las dos cajas mar también
son secuencias diana para la unión de SoxS y el fenotipo inducido por SoxS es similar al
inducido por MarA. Se han identificado mutaciones en el activador del gen soxR, tanto en
aislamientos clínicos de K. pneumoniae como en experimentos in vitro, que las relacionan con
el fenotipo de resistencia a diferentes antimicrobianos [283, 284, 287].
La proteína Rob también es un miembro de la familia AraC-XylS y regula los genes implicados
en la resistencia a antibióticos, a solventes orgánicos y a metales pesados [288, 289]. La
sobreexpresión de Rob en E. coli produce tanto un incremento en la tolerancia de solventes
orgánicos como un bajo nivel de resistencia ante múltiples agentes antimicrobianos, debido al
incremento de la expresión de la bomba de expulsión activa AcrAB [290]. A diferencia de MarA
y de SoxS, Rob se expresa de manera constitutiva, y solo su supresión confiere el fenotipo de
multiresistencia a antimicrobianos [290]. Hay un considerable solapamiento entre las diana de
Rob y las que están bajo el control de Mar y Sox, pero Rob tiene un efecto limitado en los
niveles de expresión [291].

INTRODUCCIÓN 47
Se sabe que en E. coli tanto MarA como SoxS y Rob disminuyen la expresión de OmpF
activando la transcripción de micF en respuesta a diferentes estímulos ambientales [292, 293].
RamA es una proteína reguladora de 113 aminoácidos perteneciente a la familia de
activadores transcripcionales AraC-XylS, descrita por primera vez en un mutante
multiresistente a antimicrobianos de K. pneumoniae [294]. RamA comparte un 45% de
identidad con MarA con un alto grado de conservación en los motivos esenciales de unión a
ADN para su función de regulación. Así que se espera que RamA y MarA reconozcan un
conjunto de secuencias operadoras solapadas. El locus ramA clonado en E. coli provoca un alto
nivel de resistencia a diferentes antibióticos (cloramfenicol, tetraciclina, tigeciclina,
fluoroquinolonas, trimetoprima), una disminución de la expresión de OmpF y una activación
de las bombas de flujo [295, 296]. También se ha demostrado que RamA se une al operón mar
en E.coli y aumenta su transcripción, sugiriendo una interacción entre estos dos sistemas [297,
298]. Hay otros trabajos que relacionan la importancia entre el fenotipo de multiresistencia y
RamA, tanto en la sobreexpresión de bombas de flujo como en la modificación de los niveles
de expresión de las porinas [287, 295, 299, 300].
OmpX es una pequeña PME (18 kDa) que actúa como represor de la síntesis de porinas. La
sobreexpresión de OmpX se ha asociado con una disminución de la expresión de Omp36,
principal porina de E. aerogenes, y disminuye la susceptibilidad a los β-lactámicos [301].
Estudios recientes también indican que OmpX está influenciada por un número importante de
factores ambientales y por el regulador global mar [301, 302]. Por otra parte se ha
hipotetizado que esta proteína juega un papel central en una cascada reguladora alternativa e
independiente a mar para modular la permeabilidad de la membrana [282]. En un estudio de
aislamientos clínicos de K. pneumoniae se determinó la expresión de OmpX aunque no se
vieron cambios significativos que se pudieran relacionar con cambios en la expresión de
OmpK36 o OmpK35 [303].
Una clase emergente de reguladores de la expresión de los genes bacterianos y que,
frecuentemente, se han encontrado implicados en el control de la expresión de las PME son
los ARNs no codificantes y de pequeño tamaño. En E. coli se conocen varios de estos ARNs que
regulan las PME: micA, micC, micF, omrA, omrB, rybB, rseX, invR, cyaR y ipeX [304, 305] y se ha
demostrado la presencia de los 6 primeros en K. pneumoniae [305].
De estos ARNs micF es el más estudiado. micF es un gen presente en todas las enterobacterias
y filogenéticamente muy conservado. El gen micF de K. pneumoniae muestra un 96%
homología cuando se compara con la secuencia de micF de E. coli. Corriente arriba de micF las
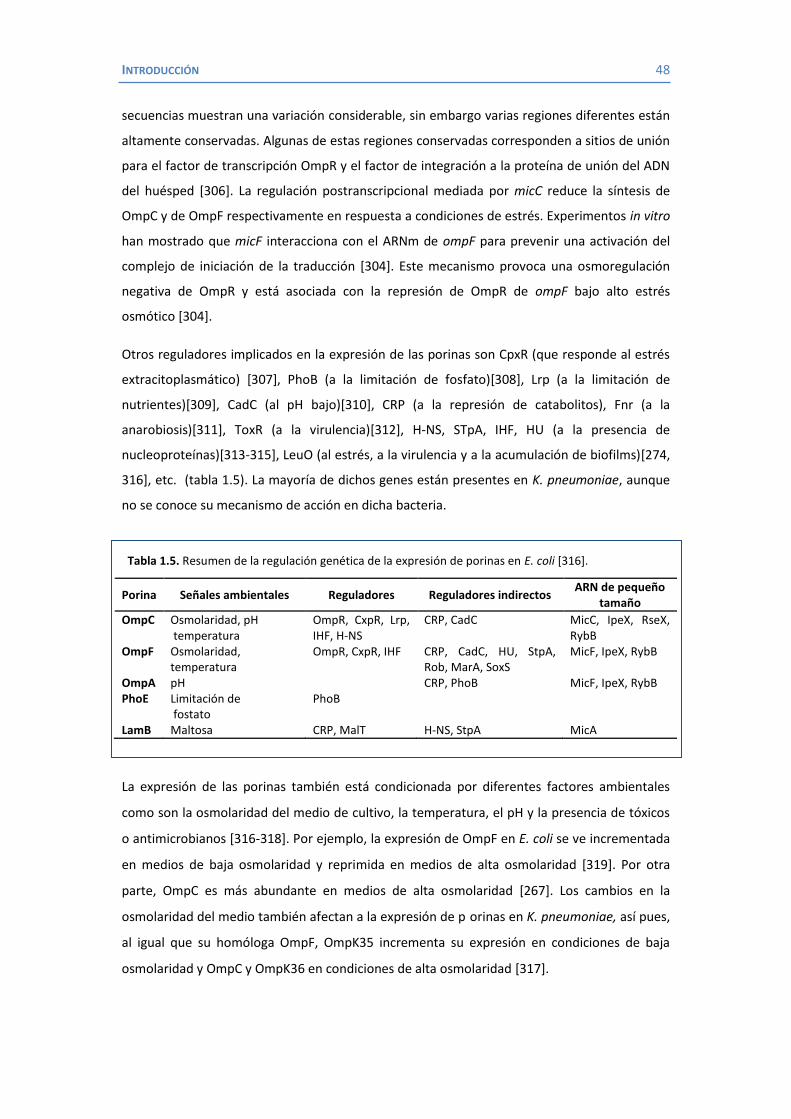
INTRODUCCIÓN 48
secuencias muestran una variación considerable, sin embargo varias regiones diferentes están
altamente conservadas. Algunas de estas regiones conservadas corresponden a sitios de unión
para el factor de transcripción OmpR y el factor de integración a la proteína de unión del ADN
del huésped [306]. La regulación postranscripcional mediada por micC reduce la síntesis de
OmpC y de OmpF respectivamente en respuesta a condiciones de estrés. Experimentos in vitro
han mostrado que micF interacciona con el ARNm de ompF para prevenir una activación del
complejo de iniciación de la traducción [304]. Este mecanismo provoca una osmoregulación
negativa de OmpR y está asociada con la represión de OmpR de ompF bajo alto estrés
osmótico [304].
Otros reguladores implicados en la expresión de las porinas son CpxR (que responde al estrés
extracitoplasmático) [307], PhoB (a la limitación de fosfato)[308], Lrp (a la limitación de
nutrientes)[309], CadC (al pH bajo)[310], CRP (a la represión de catabolitos), Fnr (a la
anarobiosis)[311], ToxR (a la virulencia)[312], H-NS, STpA, IHF, HU (a la presencia de
nucleoproteínas)[313-315], LeuO (al estrés, a la virulencia y a la acumulación de biofilms)[274,
316], etc. (tabla 1.5). La mayoría de dichos genes están presentes en K. pneumoniae, aunque
no se conoce su mecanismo de acción en dicha bacteria.
Tabla 1.5. Resumen de la regulación genética de la expresión de porinas en E. coli [316].
La expresión de las porinas también está condicionada por diferentes factores ambientales
como son la osmolaridad del medio de cultivo, la temperatura, el pH y la presencia de tóxicos
o antimicrobianos [316-318]. Por ejemplo, la expresión de OmpF en E. coli se ve incrementada
en medios de baja osmolaridad y reprimida en medios de alta osmolaridad [319]. Por otra
parte, OmpC es más abundante en medios de alta osmolaridad [267]. Los cambios en la
osmolaridad del medio también afectan a la expresión de p orinas en K. pneumoniae, así pues,
al igual que su homóloga OmpF, OmpK35 incrementa su expresión en condiciones de baja
osmolaridad y OmpC y OmpK36 en condiciones de alta osmolaridad [317].
Porina Señales ambientales Reguladores Reguladores indirectos ARN de pequeño tamaño
OmpC Osmolaridad, pH temperatura
OmpR, CxpR, Lrp, IHF, H-NS
CRP, CadC MicC, IpeX, RseX, RybB
OmpF Osmolaridad, temperatura
OmpR, CxpR, IHF CRP, CadC, HU, StpA, Rob, MarA, SoxS
MicF, IpeX, RybB
OmpA pH CRP, PhoB MicF, IpeX, RybB PhoE Limitación de
fostato PhoB
LamB Maltosa CRP, MalT H-NS, StpA MicA

INTRODUCCIÓN 49
Funciones y propiedades de OmpK35 y OmpK36.
Las porinas OmpK35 y OmpK36 son canales de difusión no específicos que permiten la entrada
moléculas pequeñas y polares (<600 kDa) que difunden a través de la membrana, tales como
nutrientes, tóxicos, antibióticos, etc.
Además, la porina OmpK36 de K. pneumoniae es el principal activador de la vía clásica del
complemento en la ME de dicho patógeno bacteriano. La activación ocurre después de la
unión de C1q por su región globular a la porina OmpK36 y es dependiente de la fuerza iónica
[252]. El mismo motivo de disposición linear de dos grupos básicos y uno ácido se encuentra
en IgM y que está implicado en las interacciones C1q-IgM [320] ha sido identificado
previamente en un modelo de homología de OmpK36 [249]. Este motivo se localiza en la
superficie externa del barril cerca del asa extracelular y está formado por la Lys279 y Lys281
(pertenecientes a la lámina β 13) y Asp282 (del asa L7). Al presentar una densidad electrónica
es débil, las cadenas laterales parecen ser flexibles. Una comparación estructural de OmpK36
con la estructura de Fc [321] muestra que los tres residuos se superponen bien, pero que la
estructura de las superficies vecinas muestra poca semejanza [249].
La pérdida de OmpK36 en K. pneumoniae provoca un incremento de la resistencia a
antimicrobianos, un incremento en la susceptibilidad a la fagocitosis por parte de los
neutrófilos, un incremento de la resistencia al suero y una reducción de la virulencia [322].
La pérdida de las dos porinas podría afectar significativamente a la capacidad metabólica de K.
pneumoniae, y su menor tasa de crecimiento podría causar que la eliminación de dicha
bacteria in vivo por parte del sistema inmune fuese más fácil [323].
OmpK35 y OmpK36 en la resistencia a antibióticos.
La mayoría de cepas de K. pneumoniae no productoras de β-lactamasas expresan las porinas
OmpK36 como OmpK35, mientras que la mayoría de las cepas clínicas de K. pneumoniae
productoras de BLEAs expresan sólo una de las dos porinas [317]. Este hecho determina que la
pérdida de expresión de porinas sea más probable en estas cepas y así adquieran un mayor
grado de resistencia a determinados antimicrobianos.
El primer estudio que demostró claramente que la pérdida de OmpK36 causaba un incremento
en la resistencia a determinados antibióticos fue llevado a cabo en 1996 por Martinez-
Martinez y colaboradores [324]. En dicho trabajo se estudiaron cuatro aislados de K.
pneumoniae (LB1, LB2, LB3 y LB4) obtenidos de un mismo paciente. Los cuatro aislados eran
indistinguibles en su biotipo, contenido de plásmidos, lipopolisacárido, y en el análisis de su
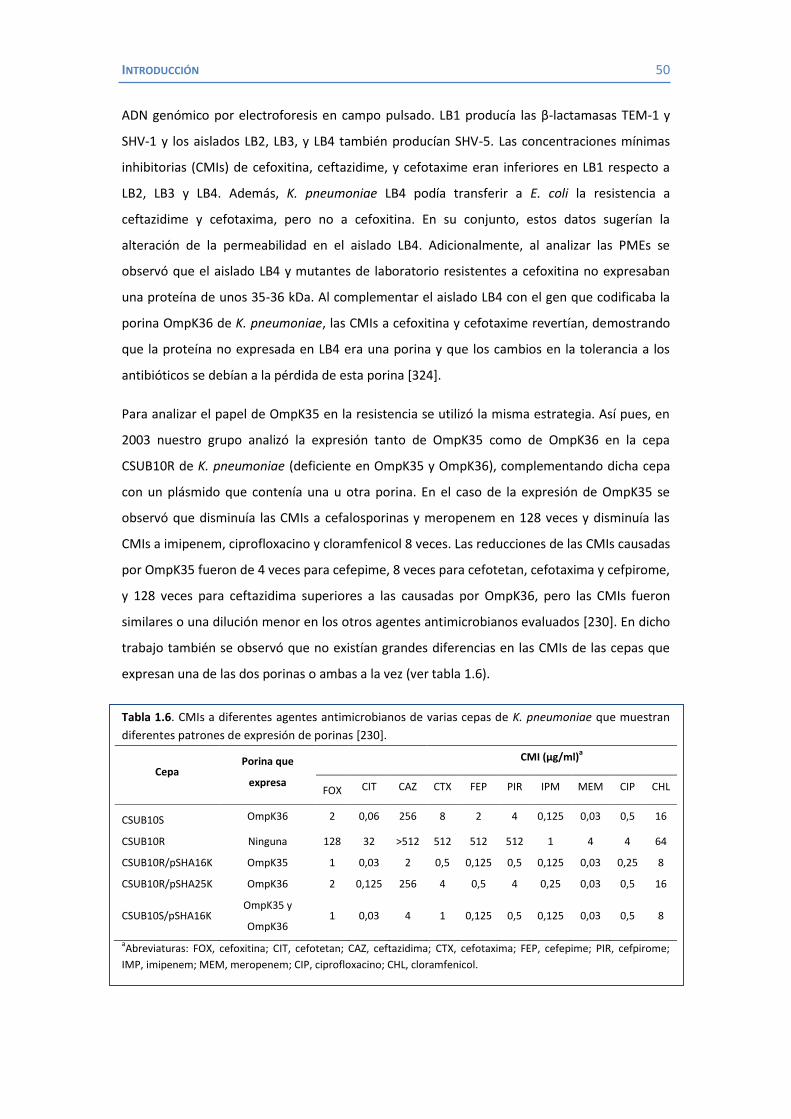
INTRODUCCIÓN 50
ADN genómico por electroforesis en campo pulsado. LB1 producía las β-lactamasas TEM-1 y
SHV-1 y los aislados LB2, LB3, y LB4 también producían SHV-5. Las concentraciones mínimas
inhibitorias (CMIs) de cefoxitina, ceftazidime, y cefotaxime eran inferiores en LB1 respecto a
LB2, LB3 y LB4. Además, K. pneumoniae LB4 podía transferir a E. coli la resistencia a
ceftazidime y cefotaxima, pero no a cefoxitina. En su conjunto, estos datos sugerían la
alteración de la permeabilidad en el aislado LB4. Adicionalmente, al analizar las PMEs se
observó que el aislado LB4 y mutantes de laboratorio resistentes a cefoxitina no expresaban
una proteína de unos 35-36 kDa. Al complementar el aislado LB4 con el gen que codificaba la
porina OmpK36 de K. pneumoniae, las CMIs a cefoxitina y cefotaxime revertían, demostrando
que la proteína no expresada en LB4 era una porina y que los cambios en la tolerancia a los
antibióticos se debían a la pérdida de esta porina [324].
Para analizar el papel de OmpK35 en la resistencia se utilizó la misma estrategia. Así pues, en
2003 nuestro grupo analizó la expresión tanto de OmpK35 como de OmpK36 en la cepa
CSUB10R de K. pneumoniae (deficiente en OmpK35 y OmpK36), complementando dicha cepa
con un plásmido que contenía una u otra porina. En el caso de la expresión de OmpK35 se
observó que disminuía las CMIs a cefalosporinas y meropenem en 128 veces y disminuía las
CMIs a imipenem, ciprofloxacino y cloramfenicol 8 veces. Las reducciones de las CMIs causadas
por OmpK35 fueron de 4 veces para cefepime, 8 veces para cefotetan, cefotaxima y cefpirome,
y 128 veces para ceftazidima superiores a las causadas por OmpK36, pero las CMIs fueron
similares o una dilución menor en los otros agentes antimicrobianos evaluados [230]. En dicho
trabajo también se observó que no existían grandes diferencias en las CMIs de las cepas que
expresan una de las dos porinas o ambas a la vez (ver tabla 1.6).
Tabla 1.6. CMIs a diferentes agentes antimicrobianos de varias cepas de K. pneumoniae que muestran diferentes patrones de expresión de porinas [230].
Cepa Porina que
expresa
CMI (µg/ml)a
FOX CIT CAZ CTX FEP PIR IPM MEM CIP CHL
CSUB10S OmpK36 2 0,06 256 8 2 4 0,125 0,03 0,5 16
CSUB10R Ninguna 128 32 >512 512 512 512 1 4 4 64
CSUB10R/pSHA16K OmpK35 1 0,03 2 0,5 0,125 0,5 0,125 0,03 0,25 8
CSUB10R/pSHA25K OmpK36 2 0,125 256 4 0,5 4 0,25 0,03 0,5 16
CSUB10S/pSHA16K OmpK35 y
OmpK36 1 0,03 4 1 0,125 0,5 0,125 0,03 0,5 8
aAbreviaturas: FOX, cefoxitina; CIT, cefotetan; CAZ, ceftazidima; CTX, cefotaxima; FEP, cefepime; PIR, cefpirome; IMP, imipenem; MEM, meropenem; CIP, ciprofloxacino; CHL, cloramfenicol.

INTRODUCCIÓN 51
Así pues, la pérdida de porinas en cepas de K. pneumoniae que producen BLEAs causan
resistencia a cefoxitina, aumento de la resistencia a las cefalosporinas y a los monobactámicos
de tercera generación, y disminución a fluoroquinolonas [324].
A partir de estos trabajos son numerosos los estudios que utilizando cepas clínicas isogénicas
han confirmado el papel de las porinas en la resistencia [324, 325]. Un ejemplo reciente es el
caso del brote epidémico provocado por una cepa de K. pneumoniae productora de CTX-M-1
que se detectó en 2006 en la UCI del Hospital Universitario de Son Dureta (Mallorca, España).
En dos de los pacientes afectados se desarrolló una resistencia a carbapenem (CMI de 8 a 12, y
< 32 µg/mL para imipenem, meropenem y ertapenem, respectivamente). El análisis de las PME
de los aislados susceptibles o resistentes a carbapenemas revelaron que los primeros
expresaban sólo una de las dos porinas mayoritarias, OmpK36, mientras que los últimos no
expresaban ninguna de ellas. En uno de los casos, se demostró que la pérdida de la expresión
de OmpK36 era mediada por la interrupción de la secuencia codificante por la secuencia de
inserción IS26 [168]. La relación entre la pérdida de la porina OmpK36 y la resistencia o la
reducción de la susceptibilidad a los carbapenemas en cepas de K. pneumoniae que producen
BLEAs o β-lactamasas tipo AmpC también se ha visto en otros trabajos [326, 327].
1.2.1.1.2 OmpA.
La proteína OmpA, a diferencia de otros componentes expuestos en la superficie de las
envueltas bacterianas, es una proteína altamente conservada en la familia de las
Enterobacteriaceae y a través de la evolución [328]. Además, la OmpA de E. coli es una de las
PME más extensamente estudiadas, tanto experimentalmente como a nivel computacional.
OmpA es también una de las pocas proteínas de membrana cuya estructura ha sido resuelta
mediante cristalografía de rayos X [329-331] y mediante resonancia magnética nuclear de la
proteína en micelas de detergentes en solución [332].
OmpA es ubicua en las células de E. coli con alrededor de 100.000 copias por célula. Casi todas
las bacterias Gram negativas expresan homólogos de OmpA a altos niveles. En K. pneumoniae
OmpA también recibe el nombre de Omp34 o de KpOmpA, y es una proteína de 34 kDa de 356
aminoácidos [333, 334].

INTRODUCCIÓN 52
Estructura
OmpA es una de las PME mejor caracterizadas, sobretodo en E. coli. Es una proteína
monomérica compuesta por dos dominios, uno N-terminal y otro C-terminal, conectados por
18 residuos que forman una región bisagra rica en prolinas [335]. El dominio N-terminal,
situado en la ME, es de aproximadamente 171 residuos y forma un barril de láminas β [329]. El
domino C-terminal de OmpA está compuesto por unos 150 residuos y es globular con una alta
proporción de hélices α y está localizado en el espacio periplásmico [335-337]. La estructura
cristalina del domino N-terminal ha sido determinada con una resolución de 1,65 Å y consiste
en un barril de 8 láminas β antiparalelas unidas por giros cortos en el lado periplásmico y por
asas más largas en la cara extracelular. Los estudios de cristalización muestran que las láminas
β están inclinadas unos 45º respecto al eje del barril. El barril tiene un diámetro seccional de
26 Å y una longitud cilíndrica de 57 Å [330]. La estructura cristalina de OmpA en K. pneumoniae
es prácticamente la misma que en E. coli , con pequeños cambios [338] (Figura 1.11).
Figura 1.11. Estructura terciaria de OmpA en K. pneumoniae. A Vista lateral de OmpA. Los caracteres estructurales principales OmpA consisten en dos tiras de residuos aromáticos (coloreados en dorado) y una tira lineal de residuos no polares (coloreados en verde). Los residuos sin asignar están coloreados en gris. B Vista centrada del interior del barril en la que se aprecia los puentes salinos entre la Glu69 y la Arg160, flanqueados por la Phe57 y la Tyr111 que constituyen la barrera más importante para el paso de iones a través del poro. También se representan los posibles puentes salinos entre la Glu162 y la Arg113/Lys24 [338].
A B

INTRODUCCIÓN 53
El paso de iones y moléculas de pequeño tamaño a través del poro está controlado por los
puentes salinos que se establecen entre varios residuos del interior del mismo. Mediante
estudios de resonancia magnética nuclear se ha demostrado que cuando la Arg138 forma una
fuerte interacción electrostática con el Glu52, el puente salino que se forma atravesando el
interior del poro bloquea el paso de iones. En cambio, cuando la Lys82 interfiere en esta
interacción y se produce un movimiento estocástico de este residuo hacia Glu52, la interacción
entre estos dos residuos hace que el Glu52 no bloquee el canal y permita el paso de iones a
través del poro [339, 340]. Cabe comentar que solo un 2-3% de las moléculas de OmpA
presentan un poro en conformación abierta [341] y que permite la entrada de solutos pero con
muy poca eficiencia, unas 100 veces menos que las porinas triméricas no específicas [231].
En K. pneumoniae OmpA presenta el mismo funcionamiento, aunque los puentes salinos se
establecen entre residuos homólogos, por ejemplo, entre el Glu69 y la Arg160 (homólogos al
Glu52 y la Arg138 de E. coli) [338].
Estudios recientes han demostrado que el mecanismo de funcionamiento de OmpA es más
complejo. Se ha propuesto que la proteína puede adoptar una conformación alternativa que
consiste en un barril β formado por 16 hebras que incluye un domino C-terminal de 8 láminas
β adicionales. Este modelo ha recibido el apoyo de estudios recientes de la conductancia de
OmpA a través de membranas bilipídicas [332, 342].
Regulación de la expresión.
OmpA es una proteína abundante y un antígeno predominante en las ME de las
enterobacterias [343]. Dado el coste que tiene expresar OmpA a tan alto nivel, no es de
extrañar que su expresión génica esté altamente regulada y responda a factores ambientales
[339].
El control de la renovación del ARNm de ompA ha sido intensamente estudiado y es un
paradigma de la regulación a nivel del ribosoma. La síntesis de OmpA y la vida media de su
ARNm son dependiente de la tasa de crecimiento [344, 345]. Durante las condiciones óptimas
de crecimiento la vida media del mensajero de ompA es de unos 15 min pero con una tasa de
crecimiento más lenta, por ejemplo por bajas temperaturas, su vida media cae a unos 4 min
[345]. La estabilidad y la longevidad del ARNm de ompA se debe a la región 5’-no traducida y
altamente plegada del transcrito (5’-UTR) [346-348]. La región monohebra de la zona 5’-UTR
(ss1 y ss2) son las diana para la ARNasaE; sin embargo, la ocupación por parte del ribosoma del

INTRODUCCIÓN 54
sitio de unión al ribosoma (RBS) reduce la acción de dicha ARNasa. Por otra parte, la unión de
la proteína Hfs a ss2 provoca la desestabilización del transcrito de ompA y facilita su
degradación [349, 350].
Recientemente, también se ha demostrado la implicación de ARN de pequeño tamaño en el
control de la expresión de OmpA. MicA (o también llamado SraD) interacciona con Hfq y al
unirse en las proximidades del sitio de unión al ribosoma del ARNm de ompA, provoca la
inhibición de la traducción y la subsiguiente disminución de los niveles de dicho ARNm [351,
352]. La dependencia de la estabilidad del ARNm a la fase de crecimiento está reforzada por el
hecho de que los niveles de MicA están inversamente correlacionados con los del transcrito de
ompA. La expresión de MicA está positivamente regulada por el factor citoplasmático σ SigmaE
en respuesta al desarrollo de estrés, por ejemplo la sobreexpresión de PME [353]. El estrés
sobre la membrana reduce los niveles de OmpA vía SigmaE y MicA.
El transcrito de ompA también es una diana para la ARNasaR, una 3’-5’exoribonucleasa que se
sobrexpresa en respuesta a condiciones de estrés y en la entrada de la fase estacionaria [354].
Así pues, en ausencia de la ARNasaR, en fase estacionaria, se producen altos niveles del
transcrito de ompA (y consecuentemente de la proteína OmpA) [355].
Funciones y propiedades de OmpA.
Dado el número de copias y la localización de OmpA presentes en la ME no es sorprendente
que esta proteína tenga multitud de funciones. Entre las funciones atribuidas a OmpA incluyen
el mantenimiento estructural de la célula, un papel en la conjugación bacteriana y en la unión
de bacteriófagos como el fago K3, Ox2 y M1 [356-359].
Un alto número de estudios han destacado el papel de OmpA en la patogénesis. OmpA en E.
coli actúa en la adhesión y en la invasión de las células epiteliales y de los macrófagos [360,
361]. Además actúa en la resistencia al suero y protege a la bacteria contra la acción de las
colectinas SP-D y SP-A del pulmón [362-365]. Sin embargo, OmpA también ejerce de diana
para el sistema inmune innato. La elastasa de los neutrófilos degrada OmpA provocando la
muerte celular [366]. La proteína amiloidea serológica de fase aguda A se une a OmpA y esto
se asocia con una disminución de detección de la bacteria por parte de los neutrófilos y de los
macrófagos [367, 368].
Por otra parte, OmpA de diferentes especies induce una respuesta citotóxica [369] y humoral
específica en ausencia de adyuvantes [370-372], a diferencia de la mayoría de proteínas

INTRODUCCIÓN 55
solubles que inducen una leve respuesta inmune o tolerancia cuando se administra en
presencia de adyuvante [373]. Así pues, se ha propuesto utilizar OmpA para el diseño de varias
vacunas antibacterianas [362, 369-371, 374, 375]. Entre ellas, una OmpA recombinante de
40kDa de K. pneumoniae [374, 375]. También cabe comentar que OmpA confiere resistencia a
péptidos antimicrobianos [376]. En cuanto a su papel en la resistencia a los antimicrobianos,
no hay estudios realizados en K. pneumoniae.
1.2.1.1.3 OmpK37.
OmpK37 es una porina trimérica quiescente formada por monómeros de 353 aminoácidos y de
un peso molecular de 39,491 kDa. La comparación entre su secuencia aminoacídica y las
secuencias de las porinas de las enterobacterias conocidas indica que, aunque se trata de una
porina, no pertenece a la familia de porinas OmpC, OmpF, PhoE o Lc/NmpC [377], sino que
está relacionada con OmpS2 de Salmonella typhi y OmpN de E. coli [378]. Los porcentajes de
identidad y similitud de OmpK37 con OmpS2 y OmpN son de 80 y 88% y 77 y 85%,
respectivamente. Por el contrario, otras porinas de K. pneumoniae relacionadas presenta
valores de identidad y similitud menores de 70 y 78%, y 58 y 68% para OmpK36 y OmpK35,
respectivamente [246].
La estructura secundaria de OmpK37 se ha predicho en base a su alineamiento con OmpF y a la
estructura tridimensional de dicha porina. Así pues, se cree que OmpK37 consiste en un
trímero de barriles formados por 16 láminas β altamente conservadas en todas las porinas, 8
giros periplasmáticos cortos y 8 asas extracelulares con secuencias altamente variables. El asa
3 (L3) es el más conservada en las diferentes porinas, en cambio, el asa 4 (L4) es la menos
conservada y, en OmpK37, presenta una inserción de 16 aminoácidos en respecto a OmpF.
Además en OmpK37 están presentes tanto el motivo Pro-Glu-Phe-Gly-Gly-Asp, altamente
conservado en la familia de las porinas, como los residuos cargados Arg37, Arg75, Arg124 y
Asp106 y Glu110, que se encuentran enfrentados en el poro de OmpF.
La comparación detallada entre la estructura tridimensional de OmpK36 y la estructura
secundaria predicha para OmpK37 demuestra la presencia de un residuo voluminoso (Tyr118)
localizado en el asa 3 de OmpK37. Teniendo en cuenta que el asa 3 está íntimamente
involucrada en la formación del poro, esta inserción podría contribuir a la formación de un
poro menor en OmpK37, hecho que se puede correlacionar con la menor penetración de los
azúcares por dicha porina [246].

INTRODUCCIÓN 56
Los mecanismos que regulan la expresión de esta porina son desconocidos, pero se sabe que
OmpK37, al igual que OmpS2 y OmpN, se encuentra sujeta a un estricto control de su
expresión que, en condiciones de laboratorio, impiden su síntesis. Lo más probable es que la
regulación ocurra a nivel transcripcional mediante mecanismos de tipo trans, pues la
sustitución del promotor nativo permite una sobreexpresión de ompK37 [246]. Al ser una
porina difícil de sintetizar, es difícil atribuir a esta porina quiescente una función específica. Sin
embargo, esta porina podría expresarse bajo otras condiciones, por ejemplo en presencia de
antibióticos en cepas deficientes en porinas. En este sentido, el hecho de que OmpK37 haya
sido detectada preferentemente en cepas clínicas deficientes en porinas resistentes a
antibióticos β-lactámicos, hace suponer que juega algún papel en la resistencia a los agentes
antimicrobianos [246].
EL menor tamaño del poro de OmpK37 está correlacionado con la susceptibilidad a ciertos
antibióticos β-lactámicos, ya que las cepas de K. pneumoniae que expresan OmpK37 pero no
expresan OmpK36 ni OmpK35, son menos susceptibles a los antibióticos β-lactámicos que las
mismas cepas expresando o bien OmpK36 o bien OmpK35, aunque siguen siendo sensibles a
los carbapenemas [246]. Así pues, OmpK37 es esencialmente impermeable a las cefalosporinas
y permeable a los carbapenemas [246]. La relación de OmpK37 con la resistencia a los
carbapenemas también se ha observado al analizar la expresión del ARNm de dicho gen en dos
cepas isogénicas, una sensible y otra resistente a los carbapenemas, donde la cepa sensible
mostraba una mayor expresión de ompK37 que la cepa resistente [283].
La presencia del gen que codifica para OmpK37 ha sido analizada en otros estudios con cepas
clínicas, aunque su expresión no ha podido ser detectada [175, 379].
1.2.1.1.4 PhoE.
PhoE, también llamada fosfoporina, es un canal con preferencia por los aniones aunque no es
específica para fosfatos, sin embargo su producción se induce bajo limitaciones de fosfatos
[231]. Esta proteína también recibe el nombre de proteína Ic [380], proteína e [319], proteína E
[381], o proteína NmpAB [382]. Además actúa de receptor para el fago TC45 [382-384].
PhoE comparte un 63% de identidad en su secuencia con OmpF [385]. También tiene un
tamaño que exclusión similar unos 600 kDa aunque difieren en otros aspectos, como que PhoE

INTRODUCCIÓN 57
Figura 1.12 Vista lateral de la estructura cristalográfica de un monómero de PhoE (código de acceso a la PDB: 1PHO). En amarillo se marcan las láminas β, en blanco las asas y los giros, en fucsia la hélice α presente en el asa 3 (L3) y en lila la hélice α perteneciente al asa L4.
es débilmente selectiva para los aniones, al contrario de OmpC y OmpF que son selectivas para
cationes [256].
La gran mayoría de datos sobre esta proteína se han obtenido con PhoE de E. coli, aunque
dada su homología con PhoE de K. pneumoniae, es probable que compartan características
comunes.
La estructura tridimensional de PhoE fue determinada al mismo tiempo que la de OmpF de E.
coli [255]. Así pues, esta porina, al igual que OmpK35 y OmpK36 de K. pneumoniae es
homotrímero de barriles formados por 16 láminas β antiparalelas. La inclinación de las hebras
respecto al eje del barril oscila entre 35º y 40º. El grupo N-terminal y C-terminal están unidos
por un puente salino en la lámina β decimosexta (L16), formando una estructura pseudocíclica.
Las hebras están conectadas por giros
cortos en la zona periplásmica y por asas de
diferente longitud en la superficie expuesta
del lado de la membrana. La inclinación del
poro respecto al eje del barril es de unos
16º y su tamaño está determinado por la
tercera asa (L3), que se encuentra plegada
dentro del barril, y constriñe dicho poro a
media altura (figura 1.12). Las diferencias
entre los esquemas de plegamiento de
OmpF y PhoE, consecuencia de las
inserciones y deleciones producidas durante
la evolución de las dos proteínas, se
concentran en las asas largas y en uno de los
giros cortos. Un cambio puntual en la
conformación del esqueleto de PhoE
causado por la inserción de la Ser121A en el
asa 3 (L3) permite la existencia de la Lys131
(Gly en OmpC)[255].
Los amplios estudios de mutaciones puntuales de PhoE, que muestran alteraciones en la unión
de fagos y anticuerpos a la proteína, han permitido que el modelo topológico concuerde con la
estructura tridimensional [255].

INTRODUCCIÓN 58
La presencia de grupos de cargas negativas y positivas, localizadas en el asa L3 y en la pared
opuesta del barril a L3, respectivamente, crean un fuerte campo electrostático transversal en
la zona de constricción del poro [255, 386, 387], el cual determina en gran medida la
permeabilidad y la selectividad iónica del poro [388-390]. Dicho poro tiene una sección elíptica
de 7 x 11 Å [255]. La reconstrucción de este canal proteico en bicapas lipídicas planares revela
que puede cerrarse por la aplicación de potencial entre ciertos umbrales de valores [391, 392].
Las diferencias en la selectividad, la débil especificidad para los cationes de OmpC y la
preferencia por la difusión de aniones en PhoE [256], se explican por la presencia en PhoE de
una Lys sustituyendo una Gly131 en OmpF que sobresale en el poro y extiende la fila de
residuos básicos en la zona de constricción [255]. Este residuo influye de manera crucial en la
selectividad de PhoE. La Lys69, residuo situado en la obertura de PhoE, también tienen algún
efecto sobre la selectividad, mientras que residuos localizados después de la constricción del
poro como la Lys18 tienen un efecto mínimo.
El crecimiento de bacterias bajo condiciones de limitación de fosfato provoca una inducción de
la expresión de PhoE [393, 394]. Así pues, las bacterias bajo concentraciones limitantes de
fosfato remplazan considerables cantidades de OmpF y OmpC por la proteína PhoE [395].
La síntesis de la proteína PhoE está cooregulada con la de la fosfatasa alcalina [396], el
producto del gen phoA. Ambos genes están bajo el control del regulón pho. Dicho regulón está
controlado por los genes reguladores phoB y phoR, cuyo mecanismo de funcionamiento es
similar en todas las enterobacterias con pequeñas diferencias [394, 397].
PhoR-PhoB es un sistema regulador de dos componentes que detecta la concentración de
fosfato del medio, y así, responde a la falta de fosfato. El sensor quinasa PhoR detecta una baja
concentración de fosfatos en el medio y activa mediante fosforilación a PhoB, el regulador de
la respuesta [398]. PhoB forforilado activa directamente la transcripción de gen phoE por la
unión a dos cajas Pho. Además de PhoE, PhoB regula negativamente a OmpT, OmpU y OmpA
principales porinas de Vibrio cholerae [308].
Hasta la fecha solo existe un artículo en el que los autores demuestren la relación entre PhoE y
la resistencia a antibióticos. En dicho trabajo, se compararon dos cepas isogénicas,
provenientes del mismo paciente, una sensible y otra resistente a carbapenemas. Aunque
ambas cepas eran deficientes en OmpK35 y OmpK36, la cepa resistente mostraba una
represión del gen phoE. Al complementar dicha cepa con un plásmido que contenía el gen de

INTRODUCCIÓN 59
phoE bajo el promotor heterólogo lacZ, dicha cepa revirtió la resistencia a carbapenemas. En
cambio, al complementarla con un plásmido que contenía el gen phoE con su promotor
natural, no hubo cambios en la resistencia. Así pues, la deficiencia en las porinas principales en
la cepa sensible estaba compensada por la sobreexpresión de PhoE y que su represión
incrementaba la resistencia a carbapenemas [283].
1.2.1.1.5 LamB.
LamB, es una de las proteínas que forman parte del sistema maltosa/maltodextrina. LamB fue
descubierta por primera vez en el año 1973 por el grupo de Schwartz, como receptor del fago
lambda en E. coli, de aquí su nombre original de LamB [399]. Sólo dos años después, el grupo
de Maurice Hofnung descubrió que LamB realmente era un transportador de ME para las
maltodextrinas [400], es por ello que también recibe el nombre de maltoporina, ya que es un
canal de difusión específico para maltosacáridos. Algunos estudios de mutación muestran que
ciertos mutantes resistentes al fago lambda no tienen afectada su adquisición de maltosa,
sugiriendo que los sitios de unión a maltosacáridos y al fago lambda son diferentes [401].
El gen lamB de K. pneumoniae codifica para el precursor de la proteína de LamB, un
polipéptido de 429 aminoácidos que revela un alto grado de homología con LamB de E. coli,
con 325 aminoácidos estrictamente idénticos. Los estudios sobre la estructura de LamB han
sido realizado en E. coli, donde también se ha analizado su interacción con diferentes azúcares
para poder entender el mecanismo de funcionamiento del sistema maltosa/maltodextrina.
Figura 1.13. Estructura cristalina de un
monómero de la proteína LamB. La parte
superior daría al exterior celular y la parte
inferior al periplasma. Las láminas β están
representadas por flechas. Las láminas están
conectadas con sus vecinas por asas o giros
regulares. Las asas L1 (en azul), L3 (en rojo) y L6
(en verde) se pliegan hacia dentro del barril.

INTRODUCCIÓN 60
LamB, al igual que otras PME, es un homotrímero de barriles de láminas β amfipáticas que
permiten la creación de una cara hidrofóbica hacia los lípidos y un interior hidrofílico en el
barril [249]. Cada monómero está formado por 18 láminas β transmembranales, a diferencia
de las 16 de las porinas clásicas. Tal como ocurre en el caso de las porinas, el asa 3 se pliega
hacia el interior del poro provocando una constricción del mismo (0,5-0,6 nm de diámetro). La
mitad exterior también es más estrecha en el caso de LamB, debido a pliegues de las asas 1 y
6 (ver figura 1.13).
Al comparar las secuencias de LamB de E. coli y de K. pneumoniae se ha visto que la tercera
región N-terminal de la proteína es la parte de la molécula más conservada, ya que esta parte
constituye el dominio del poro localizado en el interior de la membrana y que no es accesible
desde la superficie celular. Las diferencias entre las dos proteínas se encuentran,
principalmente, agrupadas en seis regiones que comprenden los residuos 145-167, 173-187,
197-226, 237-300, 311-329 y 367-387 (de la secuencia de LamB de K. pneumoniae). Los
cambios más importantes se han encontrado en las regiones plegadas predichas por el modelo
bidimensional de LamB que forman asas en la superficie [402].
Los estudios de cristalización con diferentes azúcares (maltotetraosa, maltosa, maltotriosa y
maltohexosa) han permitido determinar a lo largo de la pared del poro encontramos seis
residuos aromáticos alineados que son los que interaccionan los residuos del azúcar [403]. La
cristalización en presencia de sacarosa, trehalosa y melibiosa demostró que las dos últimas
podían unirse a los residuos aromáticos y ser transportadas, aunque cada interacción
únicamente implica a una unidad glucídica del disacárido [404]. En el caso de la sacarosa, sin
embargo, la fructosa impide la entrada del azúcar en la zona de constricción e impide su
translocación, lo que corrobora los resultados de experimentos de difusión con liposomas,
donde se ha visto que tanto la trehalosa como la melibiosa penetran por el poro, pero no así la
sacarosa [403]. La estructura del poro también predice que LamB es capaz de catalizar la
entrada de glucosa, hecho que ha sido confirmado in vitro [405].
A nivel genético LamB, al formar parte del sistema maltosa/maltodextrina, está codificado por
el regulón de maltosa, comprendido por dos regiones no consecutivas en el genoma de E. coli
y K. pneumoniae, MalA and MalB. El gen lamB es parte del operón malK-lamB de la región
MalB. En esta región también se encuentran el resto de genes del sistema de transporte
(malE, malF, and malG) [406] (figura 1.14).

INTRODUCCIÓN 61
Figura 1.14. Representación esquemática de la distribución de los genes pertenecientes al sistema maltosa/maltodextrina en K. pneumoniae.
En E. coli, la expresión de los genes de MalB, incluyendo lamB, está regulada de manera
positiva por el producto del gen malT, localizado en MalA, y por el complejo receptor-AMPc
[406]. Desde que se sabe que MalK está implicado en la regulación del sistema, y que el
receptor lambda es también necesario para la difusión de otros carbohidratos además de
maltodextrinas [407, 408], se debe considerar una regulación diferencial de estos dos genes
(en comparación con los genes del operon malEFG). Además, se puede inducir el control
traduccional de lamB cuando hay una necesidad de adquisición de carbohidratos bajo
condiciones de falta de nutrientes [409-412]. La función de LamB en la difusión de
maltodextrinas se ha analizado en un amplio número de estudios [413-418]. Todos estos
estudios demuestran la presencia de un sitio de unión a maltodextrinas que es esencial para
facilitar el proceso de difusión de las maltodextrinas. La principal contribución al
entendimiento de las bases moleculares del transporte a través de los canales de las porinas
llegó con la determinación de la estructura tridimensional de LamB, la cual se realizó en
presencia de diferentes maltooligosacáridos. En K. pneumoniae no se conoce el mecanismo de
regulación de este gen, pero algunas cepas expresan LamB de manera constitutiva.
En cuanto a su función, LamB es una de las principales proteínas que constituyen la ME de E.
coli y K. pneumoniae. La proteína LamB actúa, principalmente, de receptor para el fago lambda
pero también para otros fagos de E. coli [419]. Sin embargo, hay estudios que demuestran que
ninguno de los fagos específicos de LamB de E. coli son capaces de infectar a K. pneumoniae.
Esto puede deberse a la variabilidad de las secuencia de LamB expuestas en la superficie
bacteriana [402].
In vivo las propiedades del transporte de maltosa y maltodextrina de E. coli K12 y de las cepas
de K. pneumoniae son idénticas [402]. Así pues, LamB participa en transporte de maltosa,
maltotriosa y otros carbohidratos a través de la membrana como parte del sistema
maltosa/maltodextrina [413-418] . Además la expresión del gen lamB se puede inducir por la
maltosa y reprimir por la glucosa.

INTRODUCCIÓN 62
Aunque la maltosa puede entrar a través de OmpC y OmpF en E. coli, o a través de OmpK36 y
OmpK35 en K. pneumoniae, la tasa de difusión a través de las porinas es proporcional a la
diferencia de concentraciones a través de la membrana, de manera que cuando la
concentración extracelular desciende claramente por debajo del orden milimolar esta tasa es
bajísima. Así pues, la maltosa difunde a través de OmpF con una tasa de difusión 200 veces
menor que la difusión de la arabinosa. Las enterobacterias necesitan LamB obtener la fuente
de carbono más abundante en su hábitat natural, la maltosa, ya que la amilasa intestinal
degrada el almidón originando precisamente este compuesto. Además, estudios recientes han
demostrado que LamB es también importante para la captación de otros azúcares [403].
1.2.1.1.6 La familia de porinas KdgM.
Según los estudios en base a predicciones de láminas β, los experimentos de marcaje de
cisteínas, y otras caracterizaciones bioquímicas, la familia de porinas KdgM se engloba y
caracteriza dentro de la superfamilia de OmpG [420]. Esta superfamilia comprende un nuevo
grupo estructural de porinas con rasgos diferentes a la familia de las porinas clásicas.
Concretamente, estos canales son monoméricos y forman barriles de pequeño tamaño,
típicamente formados por 12 o 14 láminas β. A dicha superfamilia pertenecen las proteínas de
membrana externa de E. coli OmpG (un putativo canal específico para oligosacáridos)[421,
422] y Tsx (específica para nucleósidos)[423] y las proteínas implicadas en la captación de
compuestos hidrofóbicos, concretamente para la captación de ácidos grasos de cadena larga:
FadL en E. coli [424], TodX de P. putida y TbuX de Ralstonia pickettii [425].
Las porinas pertenecientes a la familia de KdgM son las porinas más pequeñas caracterizadas,
son monómeros que forman barriles de pequeño tamaño, típicamente formados por 12 o 14
láminas β transmembranas y con una media de tamaño de proteína madura de unos 220
aminoácidos. Los miembros de esta familia mejor caracterizados y cuyas estructuras han sido
cristalizadas son KdgM, KdgN de D. dadantii [420, 426] y NanC de E. coli [427].
KdgM, KdgN y NanC son estructuralmente más similares a OmpG que a OmpF, esto es
consecuente con el hecho que la familia de porinas KdgM pertenece a la superfamilia OmpG
[428, 429]. Los tamaños y los ratios de los poros de KdgM, KdgN y NanC son similares al de
OmpG, que tiene unas dimensiones de 26 Å para el eje mayor y 18 Å para el eje menor, dando
un ratio axial de 1,4 [426] (figura 1.15).

INTRODUCCIÓN 63
KdgM es la principal proteína monomérica de Dickeya dadantii (anteriormente, Erwinia
chrysanthemi), un fitopatógeno con importancia en la agricultura y el primer miembro que se
describió de esta familia de porinas [428]. Se han identificado otros homólogos a KdgM en
otras gammaproteobacterias. Todos ellos definen una gran familia de más de 100 miembros
distribuidos en, al menos, 50 especies, incluyendo los principales patógenos humanos Yersinia
pestis, S. enterica serovar thyphimurium, E. coli, Vibrio haliotiysis, en especies de Pseudomonas
y en K. pneumoniae [430]. El conocimiento actual de esta familia de canales es escaso y la
mayoría está relacionado con la información genética, como son el análisis del contexto
genético, que frecuentemente corresponde a islas de patogenicidad, y los posibles modelos de
regulación de la expresión génica [420]. En las Enterobacteriaceae, se encuentran
normalmente entre 2 y como máximo 4 secuencias homólogas, subrayando la ventaja
biológica para la bacteria de mantener varios parálogos. La razón de tal redundancia no está
clara, pero datos iniciales de KdgN, un homólogo próximo a KdgM de D. dadantii, revelan
diferencias en su regulación [431].
La proteína de esta familia mejor estudiada es NanC. NanC se pliega como un barril de láminas
β de 28 Å, con una sección elipsoidal de unos 26 Å x 20 Å. El barril está compuesto por unas 12
láminas β antiparalelas. La estructura de NanC muestra rasgos típicos de las PME. Las asas que
conectan las láminas β son más largas en la cara extracelular, con la excepción de la L1 y la L3,
mientras que los giros cortos se encuentran en el periplasma. Rodeando el barril se
encuentran dos líneas de residuos aromáticos que delimitan la superficie hidrofóbica de NanC
cuando dicha porina se encuentra embebida dentro de la membrana in vivo. La geometría del
poro, con un radio de unos 3,3 Å, contrasta con la descrita para las porinas clásicas, ya que no
hay ninguna asa que provoque la constricción de dicho poro en su interior. Por otra parte, si
que comparte con el resto de porinas bacterianas el hecho de que el poro está revestido
predominantemente con residuos cargados y polares [427].
Figura 1.15. Monómeros de KdgM, NanC, KdgN, OmpG y OmpF. La escala represente 25 Å. (A–C): Los monómeros de KdgM (A), NanC (B) y KdgN (C) se han calculado a partir de crio-imágenes de ME. KdgM, NanC y KdgN presentan una resolución de 7 Å. (D) Los cálculos del monómero OmpG provienen del archivo PDB 2IWW de una resolución de 4 Å. (E) El monómero OmpF procede del archivo PDB 2OMF de una resolución de 4 Å [426].

INTRODUCCIÓN 64
No existen muchos estudios realizados sobre la regulación de la expresión de los miembros de
la familia KdgM. Los mecanismos reguladores más estudiados son los de kdgM de D. dadantii y
su regulación en el metabolismo de las pectinas [428] y nanC de E.coli y su relación con la
expresión de fimB [429].
La expresión de kdgM de D. dadantii está controlada por el represor general de los genes
pectinolíticos, KdgR, por el represor de los genes del catabolismo de hexuronato, ExuR, por el
represor del gen de la peptinasa, PecS, y mediante represión catabólica a través del activador
transcripcional de la proteína receptora del AMP cíclico (CRP)[428]. KdgR, que pertenece a la
familia de reguladores transcripcionales IclR, es una proteína reguladora muy conservada en
las Enterobacteriaceaae, que se ha visto que está implicada en la regulación del catabolismo
de los galacturonatos en diferentes enterobacterias [430].
Por otra parte, NanC en E. coli, proteína homóloga a KdgM y coficada por el gen yjhA, no está
controlada por KdgR [429]. yjhA es el primer gen del presunto operón yjhATS. La función de
YjhT y YjhS no se conoce, y no tienen ninguna homología con proteínas de función conocida.
Este operón se trascribe divergentemente a fimB, gen cuyo producto controla la transcripción
de los genes fimbriales estructurales (desde fimA a fimH) y situado a continuación del operón
yjhATS [432]. La región intergénica yjhA-fimB es excepcionalmente larga para E. coli (1,4 kb).
Además, los elementos que regulan fimB y, que podrían regular potencialmente a yjhA, se han
identificado cerca de un sitio de unión a NanR. NanR es un regulador del metabolismo del
ácido N-acetylneuroamínico (Neu5Ac o ácido siálico) en E. coli. Los ácidos siálicos son una
familia de más de 40 monosacáridos de 9 carbonos presentes en la mayoría de los eucariotas,
donde juegan un papel en las interacciones célula-célula y célula-molécula [433]. Así pues se
ha visto que NanC está inducida por Neu5Ac y modulada por N-acetilglucosamina. Esta
regulación ocurre a través de los reguladores NanR y NagC, que también controlan la expresión
de fimB. La expresión de nanC también se activa por los reguladores del activador
transcripcional de la proteína receptora del AMP cíclico, OmpR y CpxR. [429].
Tanto KdgM como KdgN y NanC son proteínas implicadas en el metabolismo de los azúcares.
Los sustratos identificados de los miembro de la familia KdgM son los oligogalacturonatos,
oligohamnogalacturonatos, el alginato y el Neu5Ac [428, 430].
KdgM es el canal específico que permite la captación de los oligogalacturonatos largos
resultantes de la degradación de las pectinas en las infecciones causadas por D. dadantii.
KdgN, otro miembro de esta familia de porinas presente en D. dadantii, también está
implicada con el transporte de oligogalaturonatos [431].

INTRODUCCIÓN 65
Desde el punto de vista electrofisiológico KdgM, al igual que las porinas clásicas, se comporta
como una porina dependiente de voltaje que tiene una débil selectividad por los aniones y
muestra un rápido bloqueo por trigalacturonato [428]. Aunque NanC y KdgN también forman
canales de alta conductancia en la ME, y su conductancia y selectividad por los aniones son
similares a la de KdgM [429], cabe destacar que KdgM sólo se puede cerrar por un potencial
positivo, mientras que NanC muestra un cierre en ambas polaridades [428].
E. coli es capaz de utilizar el Neu5Ac como fuente de carbono y de nitrógeno para su
crecimiento. Se ha demostrado que NanC es el poro que permite la translocación del Neu5A, el
ácido siálico más abundante [433]. De hecho el Neu5A induce fuertemente la expresión de
NanC en ausencia de OmpF y OmpC [429]. Estudios recientes también han sugerido un papel
de NanC en la señalización del balance entre la relación huésped-patógeno [434, 435].


2. HIPÓTESIS Y OBJETIVOS.


HIPÓTESIS Y OBJETIVOS 69
En los últimos años la frecuencia con la que se aíslan cepas clínicas de K. pneumoniae
multiresistentes ha aumentado notablemente. En este patógeno oportunista, el fenotipo de
multiresistencia es el resultado de la combinación de varios mecanismos de resistencia a
antimicrobianos en la misma cepa, entre los que destacan la producción de una o varias BLEAs,
la expresión de bombas de expulsión y la alteración de la permeabilidad de la membrana
externa por pérdida de las principales porinas OmpK35 y OmpK36. La aparición de cepas
multiresistentes compromete la eficacia terapéutica de los antibióticos de los que disponemos
actualmente, incluyendo los carbapenémicos, que representan una de las últimas opciones
para combatir las infecciones causadas por cepas multiresistentes de K. pneumoniae.
El tratamiento prolongado con antibióticos conlleva, eventualmente, a la selección de cepas
deficientes en la expresión de las porinas principales. Sin embargo, es probable que no todos
los antibióticos utilizados habitualmente en el tratamiento de las infecciones por K.
pneumoniae tengan la misma capacidad para seleccionar cepas deficientes en porinas y que
utilicen mecanismos genéticos distintos para lograrlo.
Por otro lado, dado que las porinas principales, OmpK35 y OmpK36, representan la principal
puerta de entrada de solutos y nutrientes en la bacteria, es razonable plantear la hipótesis de
que su pérdida debe verse compensada por la expresión de porinas alternativas que aseguren
la viabilidad bacteriana en presencia de antibióticos.
Atendiendo a estas hipótesis, nuestros objetivos han sido:
- Investigar la capacidad de diferentes β-lactámicos, habitualmente utilizados para el
tratamiento de infecciones por K. pneumoniae, para seleccionar mutantes deficientes
en OmpK35 y OmpK36.
- Estudiar los mecanismos genéticos que causan la interrupción de la expresión de
OmpK36 en los mutantes deficientes en esta porina obtenidos por selección con
distintos antibióticos β-lactámicos.
- Averiguar si existen y cuáles son las porinas alternativas capaces de compensar la
pérdida de las porinas OmpK35 y OmpK36 en cepas de K. pneumoniae
multiresistentes.
- Investigar el coste biológico, tanto in vitro como in vivo, que supone la pérdida de las
porinas principales en K. pneumoniae.


3. MATERIALES Y MÉTODOS.

MATERIALES Y MÉTODOS 73
3.1. Bacterias.
Las cepas bacterianas de K. pneumoniae y E. coli utilizadas durante el desarrollo de esta tesis
se recogen en la tabla 3.1.
Tabla 3.1. Cepas bacterianas utilizadas en este estudio
Cepa Características más relevantes Origen/Referencia
K. pneumoniae CSUB10S Aislamiento clínico productor de la β-lactamasa SHV-2
que expresa OmpK36 y es deficiente en OmpK35. [325]
CSUB10R Aislamiento clínico derivado de CSUB10S productor de la β-lactamasa SHV-2 y deficiente en OmpK35 y OmpK36
[325]
CSUB10SΔLamB Mutante isogénico de CSUB10S deficiente en la porina LamB. Este trabajo
CSUB10RΔLamB Mutante isogénico de CSUB10R deficiente en la porina LamB. Este trabajo
KpCS-1 Aislamiento clínico productor de la β-lactamasa CTX-M-1 que expresa OmpK36 y es deficiente en OmpK35. [168]
KpCR-1 Aislamiento clínico derivado de KpCS-1 productor de la β-lactamasa CTX-M-1 y deficiente en OmpK35 y OmpK36.
[168]
Kpn-3 Aislamiento clínico homoresistente a carbapenemas productor de las β-lactamasas VIM-1 y SHV-12, que expresa OmpK36 y es deficiente en OmpK35
[436]
Kpn-17 Aislamiento clínico derivado de Kpn-3 heteroresistente a carbapenemas, productor de las β-lactamasas VIM-1 y SHV-12 y deficiente en OmpK35 y OmpK36
[436]
KpCR-1 pSHA2 Cepa KpCR-1 complementada con el plásmido pSHA2, portador del gen ompK36 Este trabajo
KpCR-1ΔOmpK26 Mutante isogénico de KpCR-1 deficiente en la porina OmpK26 Este trabajo
KT707 Cepa de laboratorio que expresa OmpK36 y es deficiente en OmpK35 y OmpA. [74]
E. coli
DH5α Cepa de laboratorio φ80dlacZΔM15, recA1, endA1, gyrA96, thi-1, hsdR17 (rK-,mK+), supE44, relA1, deoR, Δ(lacZYA-argF) U169
[437]
S17-1 λpir Cepa para mantenimiento de plásmidos dependientes de la proteína π [438]
MATERIALES Y MÉTODOS 74
3.2. Medios y condiciones de cultivo
Las cepas bacterianas se crecieron en caldo de Luria Bertani (LB) (Sharlau, Madrid, España) o
caldo Mueller Hinton II (MH II) (Becton, Dickinson and Company, Sparks, Maryland, EE.UU.), al
que se les añadió agar al 1,5% (Sigma-Aldrich, Madrid, España) para los crecimientos en placa.
Además, cuando fue necesario, se añadieron marcadores selectivos: cefoxitina (4, 8 o 16
µg/ml), meropenem (0,06, 0,12 o 0,24 µg/ml), imipenem (0,12, 0,24 o 0,48 µg/ml), ertapenem
(0,094, 0,188 o 0,367 µg/ml), kanamicina (50 µg/ml) o telurito (10 µg/ml). Todos los
crecimientos se realizaron a 37ºC.
En algunos de los experimentos las cepas bacterianas se crecieron en orina o suero humano. La
orina consistió en una mezcla de orinas procedente de varios voluntarios sanos, que se
sometió a centrifugación (10 min a 4500 x g) para descartar las posibles células presentes y
posteriormente se pasó por un filtro de 0,22 µm. El suero, a su vez, consistió en una mezcla de
sueros procedentes de varios voluntarios, que se recogió mediante tubos DB Vacutainer SST TM
y tras 30 min de coagulación se recuperó el sobrenadante y se congeló a -80ºC.
3.3. Plásmidos
Los plásmidos utilizados en este trabajo, así como sus características más relevantes y el origen
de los mismos, se describen en la tabla 3.2.
Tabla 3.2. Plásmidos utilizados en este estudio.
Nombre Características más relevantes Origen o Referencia
pGEM-T Easy Vector de clonación TA para clonar productos de PCR. Invitrogen
pFS100 Plásmido de integración dependiente de la proteína π, KanR y procedente de pGP704.
[439]
pDT1558 Plásmido que presenta el casete de resistencia a telurito kilA-Te. [440] pGEM-T LamB Plásmido pGEM-T Easy con un fragmento interno del gen lamB (PCR
del gen lamB, realizada con los cebadores Lamb F1 y Lamb R1). Este trabajo
pFSLamB Plásmido de integración procedente de pFS100 que contiene un fragmento interno del gen lamB.
Este trabajo
pGEM-T YjhA Plásmido pGEM-T Easy con un fragmento interno del gen ompK26 (PCR del gen ompK26, realizada con los cebadores YjhA F3 y YjhA R2).
Este trabajo
pFSOmpK26 Plásmido de integración procedente de pFS100 que contiene un fragmento interno del gen yjhA.
Este trabajo
pFSOmpK26-Te Plásmido de integración procedente de pFS100 que contiene un fragmento interno del gen yjhA y el casete de resistencia a telurito kilA-Te.
Este trabajo
pSHA2 Plásmido con el gen completo de ompK36 de K. pneumoniae y el casete de resistencia a telurito kilA-Te.
[324]
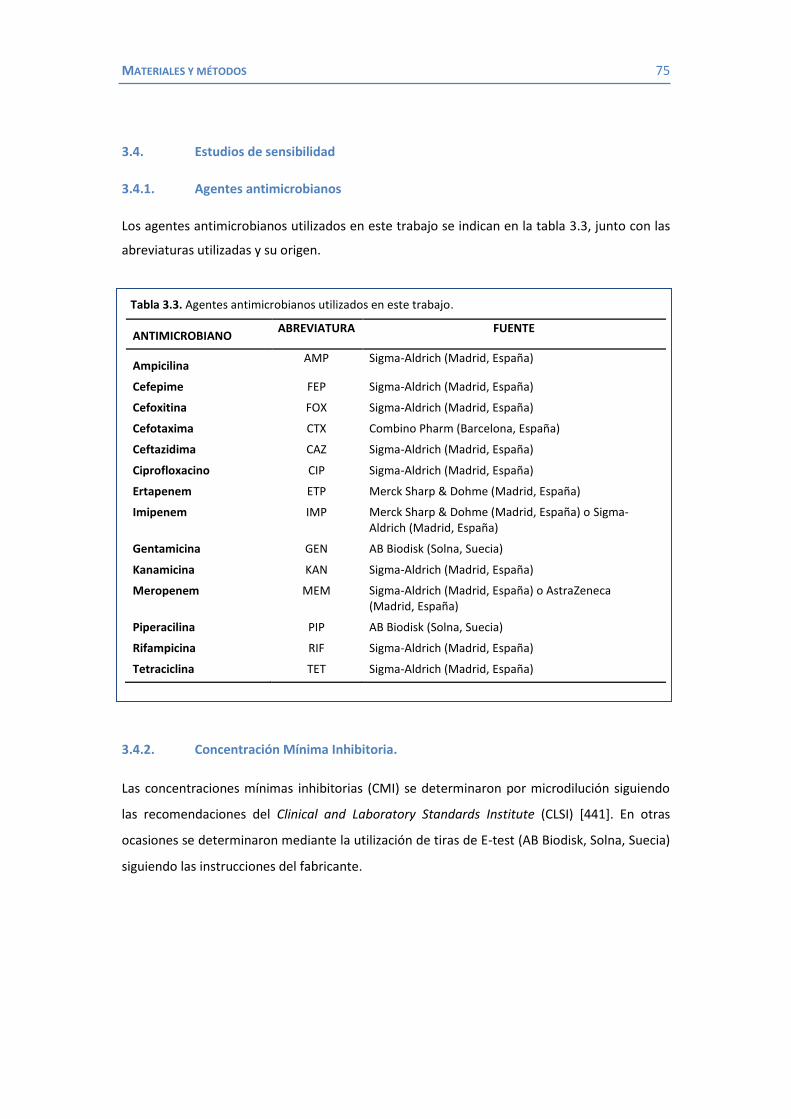
MATERIALES Y MÉTODOS 75
3.4. Estudios de sensibilidad
3.4.1. Agentes antimicrobianos
Los agentes antimicrobianos utilizados en este trabajo se indican en la tabla 3.3, junto con las
abreviaturas utilizadas y su origen.
Tabla 3.3. Agentes antimicrobianos utilizados en este trabajo.
ANTIMICROBIANO ABREVIATURA FUENTE
Ampicilina AMP Sigma-Aldrich (Madrid, España)
Cefepime FEP Sigma-Aldrich (Madrid, España)
Cefoxitina FOX Sigma-Aldrich (Madrid, España)
Cefotaxima CTX Combino Pharm (Barcelona, España)
Ceftazidima CAZ Sigma-Aldrich (Madrid, España)
Ciprofloxacino CIP Sigma-Aldrich (Madrid, España)
Ertapenem ETP Merck Sharp & Dohme (Madrid, España)
Imipenem IMP Merck Sharp & Dohme (Madrid, España) o Sigma-Aldrich (Madrid, España)
Gentamicina GEN AB Biodisk (Solna, Suecia)
Kanamicina KAN Sigma-Aldrich (Madrid, España)
Meropenem MEM Sigma-Aldrich (Madrid, España) o AstraZeneca (Madrid, España)
Piperacilina PIP AB Biodisk (Solna, Suecia)
Rifampicina RIF Sigma-Aldrich (Madrid, España)
Tetraciclina TET Sigma-Aldrich (Madrid, España)
3.4.2. Concentración Mínima Inhibitoria.
Las concentraciones mínimas inhibitorias (CMI) se determinaron por microdilución siguiendo
las recomendaciones del Clinical and Laboratory Standards Institute (CLSI) [441]. En otras
ocasiones se determinaron mediante la utilización de tiras de E-test (AB Biodisk, Solna, Suecia)
siguiendo las instrucciones del fabricante.

MATERIALES Y MÉTODOS 76
3.5. Métodos de análisis y purificación de proteínas.
3.5.1. Aislamiento de proteínas de membrana externa.
Para el aislamiento de las PME proteínas las bacterias se crecieron a 37°C y en agitación durante 16-20 h. Una vez recogidas por centrifugación, las bacterias se lavaron dos veces utilizando el tampón 10 mM Tris pH 7,2 - 5 mM MgCl2 (8.000 x g, 15 min). La lisis bacteriana se produjo mediante la utilización de una prensa de French (Fred S. Carver Inc., Menomonee Falls, WI, EE.UU.) a 10-15.000 libras·pulgada-2. Después de lisar las bacterias, se centrifugaron a 3.000 x g durante 10 min a 4°C, a fin de eliminar las bacterias intactas. Seguidamente se ultracentrifugó la muestra a 100.000 x g durante 1 h a 4°C, obteniéndose las envolturas bacterianas en el precipitado.
Para obtener la membrana externa, las envolturas bacterianas se solubilizaron durante 20 min
a temperatura ambiente en lauril sarcosinato sódico al 2% en 10 mM Tris pH 7,2 - 5 mM MgCl2
[442]. Posteriormente se ultracentrifugaron a 100.000 x g y, tras decantar el sobrenadante, se repitió el mismo proceso para solubilizar totalmente la membrana interna. Por último, se resuspendió el precipitado en un volumen adecuado de H2O destilada (H2Od) mediante sonicación (Soniprep 150 (MSE Ltd., Crawley, Sussex, EE.UU)).
El procedimiento descrito anteriormente resulta bastante laborioso ya que requiere del uso de ultracentrífuga e importantes volúmenes de cultivo bacteriano, así que se adaptó para volúmenes de muestra reducidos y que permitieran el uso microcentrífuga, aislando cantidad suficiente de PME para su posterior análisis.
Así pues, para análisis de PME de manera más rutinaria, las bacterias de 1,5 mL de cultivo de 16 -20 h se recogieron por centrifugación (13.000 x g durante 3 min), se resuspendieron con 1mL de tampón 10 mM Tris pH 7,2 - 5 mM MgCl2 y se centrifugaron 5 min a 13.000 x g. Seguidamente, estas bacterias lavadas se resuspendieron en 1 ml de este tampón y se sonicaron en un sonicador Soniprep 150 (MSE Ltd., Crawley, Sussex, EE.UU.) o Branson Sonicator 250 (Shanghái, China) a 4 °C, en 3 ciclos de 30 s, teniendo cada ciclo intervalos de 5 s de sonicación y 1 s de reposo (amplitud 18 μm). Las bacterias enteras se eliminaron por centrifugación en microcentrífuga (5.000 x g, 4 min), y el sobrenadante se centrifugó de nuevo a 13.000 x g durante 30 min en una microcentrífuga para recoger las envolturas bacterianas. Para obtener la membrana externa se trataron las envolturas con 1 mL de 10 mM Tris pH 7,2 - 5 mM MgCl2-lauril sarcosinato sódico 2% durante al menos 20 min. Una vez solubilizada la
membrana interna, se precipitó la membrana externa en una microcentrífuga durante 30 min a 13.000 x g, resuspendiéndose finalmente en 40 μL de tampón de Laemmli.

MATERIALES Y MÉTODOS 77
3.5.2. Purificación de la porina OmpK36.
Para la purificación de OmpK36, en primer lugar se realizó una purificación de porinas. Para
ello, se obtuvieron las envolturas bacterianas de manera similar al caso de las PME, siendo en
este caso el tampón utilizado para los lavados 50 mM Tris pH 7,7. Seguidamente a las
envolturas bacterianas se les añadió una solución de SDS al 2% y se dejaron incubar 1 h a 32ºC.
El material insoluble se recuperó por centrifugación a 100.000 x g durante 1 h, se trató de
nuevo con SDS al 2% y se volvió a centrifugar en las mismas condiciones. A continuación se
solubilizó el precipitado en una solución 50 mM Tris pH 7,7 conteniendo SDS al 1%, 400 mM de
NaCl, 5 mM de EDTA y β-mercaptoetanol al 0,05% y se sometió a una cromatografía de
filtración utilizando una columna de Sephacryl S-200 HR (Sigma-Aldrich, Madrid, España) de 1,6
x 90 cm. Para ello se añadió la muestra a la columna y se eluyó mediante una bomba
peristáltica P1 (Pharmacia Fine Chemicals, Upsala, Suecia). La cromatografía fue monitorizada
espectrofotométricamente a 280 nm (Ultrospect 6300 pro; Amersham Biosciences, Barcelona,
España), recogiéndose fracciones de 1 mL mediante un colector 2070 Ultrorac II Fraction
Collector (LKB, Bromma, Suecia). Las fracciones que contenían la porina OmpK36 se juntaron
en una sola fracción y dializaron frente a 3 mM de azida sódica mediante una membrana de
celulosa de 33 mm x 21 (Sigma-Aldrich, Madrid, España) a temperatura ambiente durante 3
días y luego a 4 °C durante 6 días. La porina OmpK36 totalmente purificada se guardó a 4°C.
3.5.3. Determinación de la concentración de proteínas.
La concentración de proteínas de las diferentes muestras se determinó mediante el método
Bradford utilizando el Bio-Rad protein Assay (Bio-Rad, Múnich, Alemania) y seroalbúmina
bovina como patrón [443].
3.5.4. Electroforesis en gel de poliacrilamida con SDS (SDS-PAGE).
El análisis de proteínas de membrana externa y porinas se realizó mediante electroforesis en
geles de poliacrilamida con dodecilsulfato sódico (SDS-PAGE). Los geles de 0,75 mm de grosor,
estaban compuestos en su zona de resolución por acrilamida al 11%, bisacrilamida al 0,5%, 375
mM Tris pH 8,8 y SDS al 0,1%. En la zona de carga de las muestras, el gel estaba compuesto por
acrilamida al 3,9%, bisacrilamida al 0,13%, 125 mM Tris pH 6,8 y SDS al 0,1%. Para catalizar la
polimerización de las acrilamidas se les añadió persulfato amónico (APS) al 0,25% (Sigma-
Aldrich, Madrid, España) y TEMED al 0,1% (N,N,N´,N´-tetrametil-etilendiamina, BioRad,
Madrid, España).

MATERIALES Y MÉTODOS 78
Las muestras, tanto de PME como de porinas, se hirvieron durante 5 min en tampón de Laemli
antes de la electroforesis. Los geles migraron a 70 V durante 20 min y posteriormente 1 h a
100V. Como tampón de electroforesis se utilizó 190 mM glicina, 25 mM Tris pH 8,3 y SDS al
0,1%. Finalmente, los geles se tiñeron durante al menos 1 h en solución de tinción de
Coomassie, se decoloraron en 45% metanol - 10% ácido acético y se secaron entre papel de
celulosa utilizando el sistema DryEase Mini-Gel Drying System (Novex, San Diego, CA., EE.UU.).
Como marcador de peso molecular se utilizó el Precision Plus Protein All Blue Standards (Bio-
Rad, Madrid, España).
Cuando fue necesario se tomaron imágenes de dichos geles y se cuantificó la cantidad de
proteína presente en los mismos mediante un densitómetro (GS-800™ Calibrated
Densitometer (Bio-Rad, Múnich, Alemania)).
3.5.5. Identificación de proteínas.
Para la identificación de proteínas se partió de geles SDS-PAGE de los que se escindió la
proteína a identificar. Posteriormente se realizó una digestión con tripsina y se analizó
mediante espectrometría de masas en tándem (MS/MS) por la Unidad de Proteómica de la
Universidad Complutense de Madrid, como se describe en otros trabajos [444]. Los resultados
para los péptidos filtrados se realizó utilizando el GPS Explorer v3.5 software con la versión 1.9
de MASCOT.
3.5.6. Identificación de proteínas con anticuerpos.
Obtención de antisuero.
Para la obtención del antisuero específico contra la porina OmpK36 se inmunizaron dos
conejos hembras raza New Zealand con OmpK36 purificada como se ha descrito en el apartado
3.5.2. Para ello se inyectaron por vía subcutánea 80 μg de la proteína purificada 3 veces con
intervalos de 2 semanas. Para incrementar la respuesta inmunológica la primera inmunización
se realizó en adyuvante de Freund completo (Sigma-Aldrich, Madrid, España) y las otras 2 en
adyuvante de Freund incompleto (Sigma-Aldrich, Madrid, España), en una proporción 1:1 de
OmpK36 y adyuvante. El animal se sangró transcurridas 2 semanas después de la última
inyección. El título de anticuerpos se determinó por ELISA [445].
Para evitar reacciones cruzadas con otros componentes de la ME, el antisuero se absorbió el
suero con la cepa de K. pneumoniae CSUB10R.

MATERIALES Y MÉTODOS 79
Western blot o transferencia Western.
La expresión de OmpK36 se analizó por la técnica de Western blot debido a su mayor
sensibilidad. Para ello se transfirieron geles SDS-PAGE a filtros de Immobilon-P (Millipore,
Bedford, MA, EE.UU.) mediante transferencia semiseca durante 20 min a 15 V (Transfer blot
SD Semidry transfer Cell (Bio-Rad, Múnich, Alemania) utilizando un tampón de pH 8,3 que
contenía 25 mM de Tris, 192 mM de glicina y metanol al 20% [446]. Los filtros se bloquearon
con albúmina sérica bovina (BSA) al 1% en tampón fosfato salino (PBS) o leche descremada al
5%. Tras 3 lavados con PBS, los filtros se incubaron secuencialmente con suero anti-OmpK36
diluído 1:1000 y con inmunoglobulina G contra Anti-IgG de conejo marcada con fosfatasa
alcalina (Sigma-Aldrich, Madrid, España). Finalmente los filtros se revelaron con Nitro Blue
Tetrazolium y 5-bromo-4-cloro-indolilfosfato (SIGMAFAST™ BCIP®/NBT, Sigma-Aldrich, Madrid).
Los anticuerpos se diluyeron y se incubaron en 1% BSA o 5% leche descremada – PBS a
temperatura ambiente durante 1 h.
3.6. Métodos de purificación, manipulación y análisis de ácidos nucleicos.
3.6.1. Purificación de ADN.
La extracción de ADN cromosómico se realizó según el método descrito por Dhaese y
colaboradores [447]. También se utilizaron los kits UltraCleanTM Microbial DNA Isolation Kit
(MoBio Laboratories Inc., CA, EE.UU.) y Wizard® Genomic DNA Purification Kit (Promega,
Madison, WI, EE.UU.) siguiendo las indicaciones de los fabricantes.
La obtención del ADN plasmídico se llevó a cabo según el método de la lisis alcalina [448],
utilizando el “UltraCleanTM Mini Plasmid Prep Kit” (MoBio Laboratories Inc., CA, EE.UU.) o el
“HiSpeedTM Plasmid Midi Kit” (QIAGEN GmbH, Hilden, Alemania) y siguiendo las indicaciones
del fabricante.
En aquellos casos en que fue necesario purificar un fragmento de ADN determinado se
preparó un gel con agarosa de bajo punto de fusión (Sigma-Aldrich, Madrid). Posteriormente a
la electroforesis, se recortó la zona de interés y se purificó el ADN con el kit comercial
QIAquick® Gel Extraction (QIAGEN GmbH, Hilden, Alemania).

MATERIALES Y MÉTODOS 80
3.6.2. Purificación de ARN.
El ARN se purificó de cultivos bacterianos en fase exponencial (crecidos 3 h a 37°C en
agitación), utilizando el Rneasy Mini Kit (QIAGEN Sciences, Mariland, EE.UU.) y siguiendo las
instrucciones del fabricante. Para eliminar las posibles contaminaciones de ADN se trató con 2
unidades de TURBO DNA-free™ (Ambion, Austin, TX, USA) durante 30 min a 37ºC. Finalmente,
se eluyó el ARN con H2O miliQ y se ajustó a una concentración final de 50 ng/μl con la ayuda de
un NanoDrop Spectrophotometer (Thermo Scientific, Wilmington, DE, EE.UU.).
3.6.3. Electroforesis en geles de agarosa.
Las moléculas de ácidos nucleicos se separaron en geles de agarosa de distintas
concentraciones en función de su tamaño. Estos geles se prepararon en tampón tris acético
EDTA (TAE). Los geles migraron a 30 V durante toda la noche en cubetas de Bio-Rad (Madrid,
España). En el caso de minigeles la electroforesis se realizó a 100 V durante 30-45 min
utilizando el sistema RunOneTM Electrophoresis Cell (Embitec, San Diego, CA, EE.UU.). Como
tampón de electroforesis se utilizó TAE. Finalmente, los geles se tiñeron durante al menos 15
min en una solución de bromuro de etidio 0,01 g/ml y se visualizaron con luz ultravioleta.
Cuando fue necesario se tomaron imágenes de dichos geles con el captador de imágenes
Molecular Imager ® Gel DocTM XR (Bio-Rad, Múnich, Alemania)).
Como marcador de peso molecular se utilizaron los marcadores de 1 Kb de Amersham
Biosciences (Uppsala, Suecia) y Bioron GmbH (Ludwigshafen, Alemania).
3.6.4. Southern blot o transferencia Southern.
Para la realización de la transferencia Southern, el ADN previamente digerido con el enzima de
restricción adecuado fue separado por electroforesis y visualizado por tinción con bromuro de
etidio. Seguidamente el gel se lavó dos veces durante 15 min con H2Od para eliminar los restos
de bromuro de etidio y se trató de forma secuencial, y durante 30 min en cada paso, con la
solución de depurinación (250 mM de HCl), la solución de desnaturalización (1,5 M de NaCl y
0,5 M de NaOH) y, finalmente, con la solución de neutralización (1,5 M de NaCl y 1 M
TrisBase). Posteriormente el ADN del gel se transfirió mediante capilaridad [448] o bien
mediante vacío con el aparato Vacum blotter Model 785 (Bio-Rad, Madrid, España) a una

MATERIALES Y MÉTODOS 81
membrana de nylon Hybond-N+ (Amersham Biosciences, Uppsala, Suecia). Una vez transferido
el ADN, éste se fijó mediante radiación ultravioleta (Spectrolinker XL-1000 UV Crosslinker,
Spectronics Corporation, NY, EE.UU).
Las hibridaciones con sondas específicas obtenidas mediante PCR se realizaron utilizando el kit
de marcaje y detección Amersham ECL DirectTM Nucleic Acid Labelling and Detection Systems
(GE Healthcare, Buckinghamshire, Inglaterra) siguiendo las instrucciones del fabricante.
3.6.5. Introducción de ADN plasmídico.
En el presente trabajo la introducción de moléculas de ADN en las bacterias se realizó
mediante conjugación, transformación o electroporación. En el caso de la conjugación, la cepa
donante y la receptora se crecieron durante toda la noche, se mezclaron en una proporción
1:1 y se depositaron sobre una placa de LB agar. Tras estar un mínimo de 12 h en contacto a 37
°C, se recogió la masa de conjugación y se sembró sobre placas de LB agar con los marcadores
de selección apropiados.
Para los experimentos de transformación, en primer lugar se prepararon células competentes
a partir de un cultivo exponencial de E. coli DH5α (DO550 nm = 0,3) mediante el método del Cl2Ca
y siguiendo los protocolos descritos por Sambrook y colaboradores [448] pero utilizando los
tampones de transformación 1 y 2 (ver anexo I). Una vez obtenidas las células competentes se
hicieron alícuotas de 300 µl conteniendo aproximadamente, 1.107 células competentes, y se
utilizaron inmediatamente o se congelaron a - 80°C. Para transformar dichas bacterias, se puso
en contacto una alícuota de células competentes con 20 µL del ADN (50 ng/µL), dicha mezcla
se incubó 1 h en hielo y se sometió a un choque térmico a 42 °C durante 90 s. Tras una corta
incubación de 10 min en hielo se añadió 1 mL de LB y se incubó durante 1 h a 37°C. Finalmente
se sembró la mezcla sobre placas de LB agar con los marcadores de selección apropiados.
En el caso de la electroporación, se prepararon células competentes a partir de un cultivo
exponencial de K. pneumoniae (DO600 nm = 0,5) que, para eliminar las sales, se lavó dos veces
con 50 mL de agua destilada estéril previamente enfriada en hielo. Tras poner en contacto las
bacterias con 20 µL del ADN a introducir (50 ng/µL) durante 1 min en hielo, se les sometió a un
pulso eléctrico de 1800 V durante 5 ms utilizando un electroporador Gene Pulser Xcell (Bio-
Rad, Múnich, Alemania). La mezcla se recogió en 1 mL de LB y se incubó a 37°C durante 1 h. En
último lugar, se sembraron sobre placas de LB agar con los marcadores de selección
apropiados.
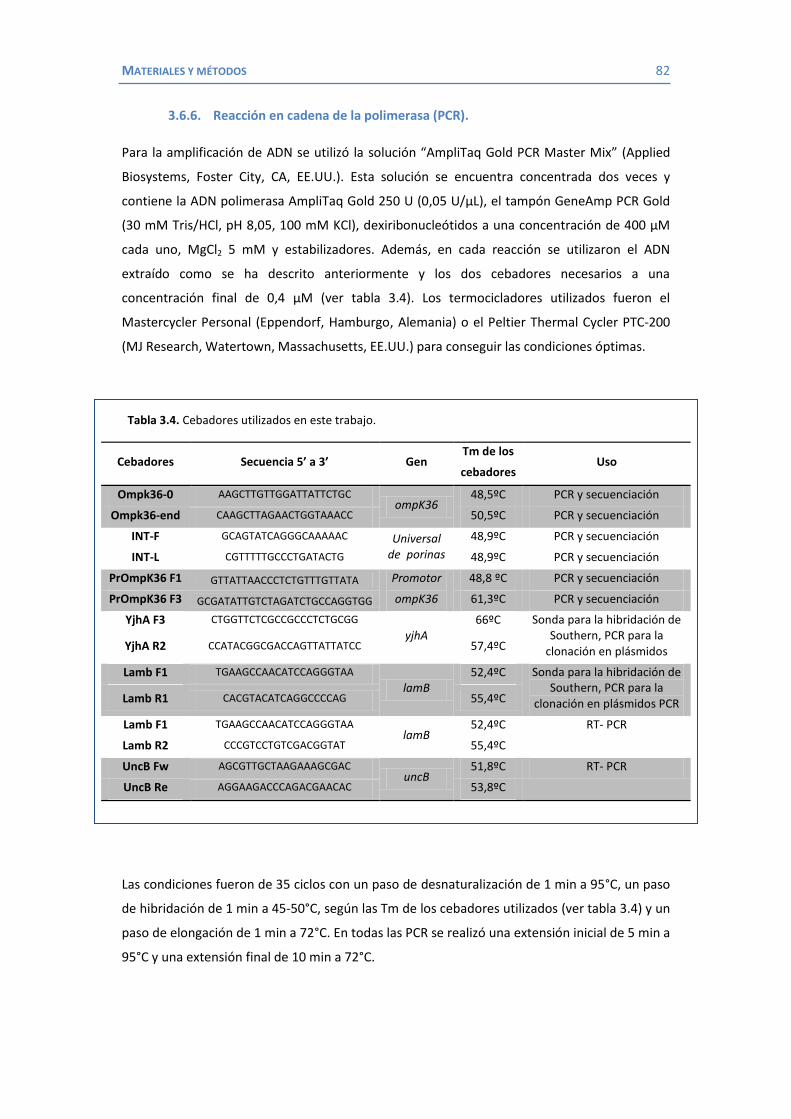
MATERIALES Y MÉTODOS 82
3.6.6. Reacción en cadena de la polimerasa (PCR).
Para la amplificación de ADN se utilizó la solución “AmpliTaq Gold PCR Master Mix” (Applied
Biosystems, Foster City, CA, EE.UU.). Esta solución se encuentra concentrada dos veces y
contiene la ADN polimerasa AmpliTaq Gold 250 U (0,05 U/μL), el tampón GeneAmp PCR Gold
(30 mM Tris/HCl, pH 8,05, 100 mM KCl), dexiribonucleótidos a una concentración de 400 μM
cada uno, MgCl2 5 mM y estabilizadores. Además, en cada reacción se utilizaron el ADN
extraído como se ha descrito anteriormente y los dos cebadores necesarios a una
concentración final de 0,4 μM (ver tabla 3.4). Los termocicladores utilizados fueron el
Mastercycler Personal (Eppendorf, Hamburgo, Alemania) o el Peltier Thermal Cycler PTC-200
(MJ Research, Watertown, Massachusetts, EE.UU.) para conseguir las condiciones óptimas.
Tabla 3.4. Cebadores utilizados en este trabajo.
Cebadores Secuencia 5’ a 3’ Gen Tm de los
cebadores Uso
Ompk36-0 AAGCTTGTTGGATTATTCTGC ompK36
48,5ºC PCR y secuenciación
Ompk36-end CAAGCTTAGAACTGGTAAACC 50,5ºC PCR y secuenciación
INT-F GCAGTATCAGGGCAAAAAC Universal de porinas
48,9ºC PCR y secuenciación
INT-L CGTTTTTGCCCTGATACTG 48,9ºC PCR y secuenciación
PrOmpK36 F1 GTTATTAACCCTCTGTTTGTTATA Promotor
ompK36
48,8 ºC PCR y secuenciación
PrOmpK36 F3 GCGATATTGTCTAGATCTGCCAGGTGG 61,3ºC PCR y secuenciación
YjhA F3 CTGGTTCTCGCCGCCCTCTGCGG yjhA
66ºC Sonda para la hibridación de Southern, PCR para la
clonación en plásmidos YjhA R2 CCATACGGCGACCAGTTATTATCC 57,4ºC
Lamb F1 TGAAGCCAACATCCAGGGTAA lamB
52,4ºC Sonda para la hibridación de Southern, PCR para la
clonación en plásmidos PCR Lamb R1 CACGTACATCAGGCCCCAG 55,4ºC
Lamb F1 TGAAGCCAACATCCAGGGTAA lamB
52,4ºC RT- PCR
Lamb R2 CCCGTCCTGTCGACGGTAT 55,4ºC
UncB Fw AGCGTTGCTAAGAAAGCGAC uncB
51,8ºC RT- PCR
UncB Re AGGAAGACCCAGACGAACAC 53,8ºC
Las condiciones fueron de 35 ciclos con un paso de desnaturalización de 1 min a 95°C, un paso
de hibridación de 1 min a 45-50°C, según las Tm de los cebadores utilizados (ver tabla 3.4) y un
paso de elongación de 1 min a 72°C. En todas las PCR se realizó una extensión inicial de 5 min a
95°C y una extensión final de 10 min a 72°C.

MATERIALES Y MÉTODOS 83
Para el diseño de los cebadores empleados en este trabajo se tomaron como base las
secuencias de genes depositadas en las bases de datos del European Bioinformatics Institute
(http://www.ebi.ac.uk/) y del “Genome Intitute” de la Universidad de Washington
(http://genome.wustl.edu/). Además se utilizaron las siguientes herramientas bioinformáticas:
BLAST, FASTA (http://www.ebi.ac.uk/), insilico.ehu.es [449], OligoClac [450] y OligoAnalyzer 3.1
(http://eu.idtdna.com/analyzer/Applications/OligoAnalyzer/Default.aspx).
3.6.7. RT-PCR.
En este trabajo la técnica de PCR de transcripción reversa (RT-PCR) a tiempo real se utilizó para
determinar los niveles de expresión de lamB en varias cepas de K. pneumoniae. Para ello se
siguió el protocolo descrito por Oh y colaboradores [451] con pequeñas modificaciones,
utilizando el kit QuantiTect® SYBR Green RT-PCR (QIAGEN, Hilden, Alemania) y las cantidades
indicadas en la tabla 3.5.
La reacción se llevó a cabo en un SmartCycler II™ (Cepheid, Sunnyvale, EE.UU.). Las
condiciones utilizadas fueron 40 ciclos con un paso de desnaturalización de 15 s a 95°C, un
paso de hibridación de 30 s a 50°C y un paso de elongación de 30 s a 72°C. Los cebadores
utilizados para el gen lamb fueron Lamb F1 y Lamb R2 (ver tabla 3.4). Como gen de referencia
se utilizó el gen uncB, que codifica para la subunidad A del ATPsintetasa F0 [452].
Tabla 3.5. Volúmenes utilizados para la RT-PCR en tiempo real
Reactivos Volumen por reacción (µL)
2x RT-PCR master mix 12,5
Quantitec RT mix 0,25
Cebador F (10 µM) 1
Cebador R (10 µM) 1
H2Od (libre de ARNasas) 9,5
ARN molde (50 ng/µL) 1
Volumen final 25,5
El cálculo de la expresión relativa se llevó a cabo como está detallado en la figura 3.1. En todos los casos se consideró el valor medio de ARNm obtenido en tres experimentos independientes en los que se realizaron las PCR por duplicado.

MATERIALES Y MÉTODOS 84
3.6.8. Secuenciación.
La reacción de secuenciación se realizó utilizando el kit “BigDie® Terminator v3.1” (Applied
Biosystems, Foster City, CA, EE.UU.) siguiendo las instrucciones del fabricante. Los productos
obtenidos se analizaron mediante un analizador genético ABI 3130 de Applied Biosystems
(Foster City, CA, EE.UU.). Finalmente las secuencias de nucleótidos fueron analizadas mediante
los programas informáticos Sequence Scanner 1.0 (Applied Biosystems, Foster City, CA, EE.UU.)
y el Bioedit Sequence Aligment Editor [410].
3.6.9. Construcción de mutantes delecionados mediante inserción-duplicación.
Los mutantes utilizados en este estudio fueron obtenidos por el método de inserción-
duplicación siguiendo el esquema de la figura 3.2. En primer lugar se obtuvo un fragmento
interno del gen a interrumpir mediante la amplificación con cebadores específicos. Dicho
fragmento fue clonado en el plásmido pGEM-T Easy (Promega, Madison, WI, EE.UU.) y después
transferido al pFS100 mediante digestión con EcoRI (Takara, Shinga, Japón) y ligación con la
ADN ligasa T4 (Promega, Madison, WI, EE.UU.) (ver figura 3.2 A). En algunos casos, el plásmido
resultante fue dotado con la resistencia a telurito procedente del plásmido pDT1558 [440] e
introducido en K. pneumoniae por conjugación. Mediante esta técnica se obtuvieron los
mutantes CSUB10SΔlamB, CSUB10RΔlamB y KpCR-1ΔompK26 (ver figura 3.2 B).
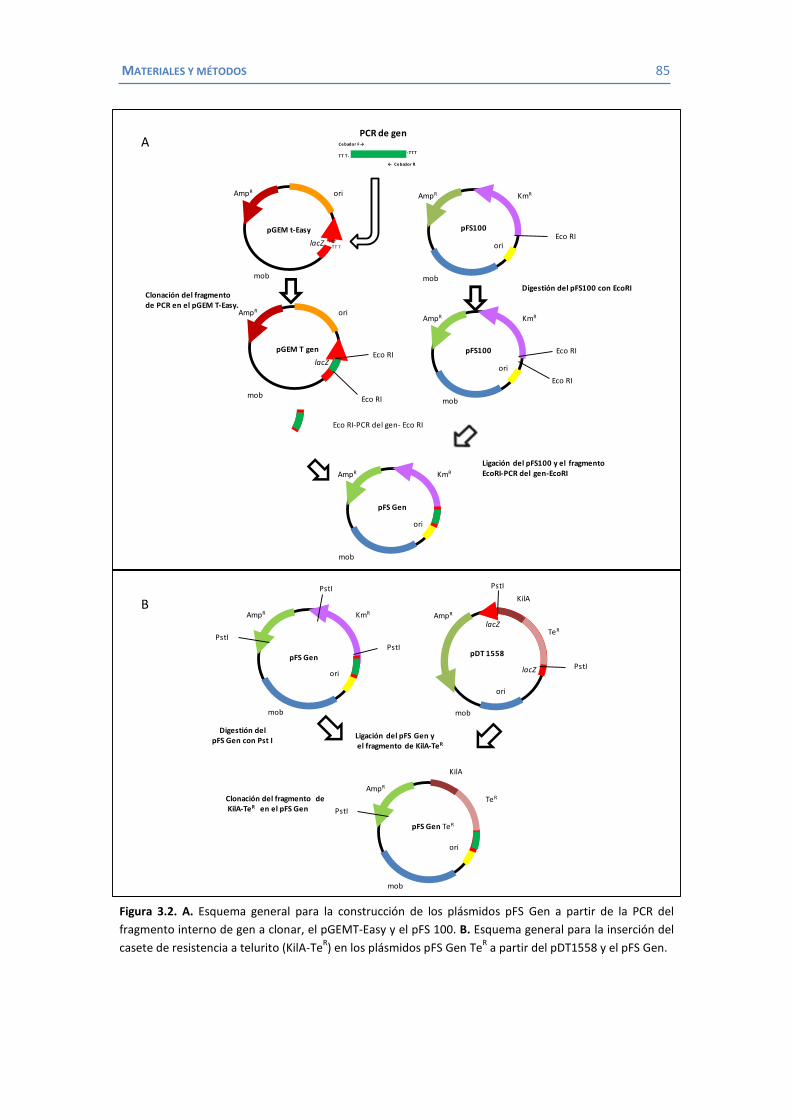
MATERIALES Y MÉTODOS 85
Clonación del fragmento de PCR en el pGEM T-Easy.
AmpR ori
mob
lacZpGEM t-Easy
AmpR ori
mob
lacZpGEM T gen
Eco RI
Eco RI
Eco RI-PCR del gen- Eco RI
Digestión del pFS100 con EcoRI
Ligación del pFS100 y el fragmento EcoRI-PCR del gen-EcoRI
AmpR KmR
mob
ori
pFS100Eco RI
AmpR KmR
mob
ori
pFS100 Eco RI
Eco RI
AmpR KmR
mob
ori
pFS Gen
PCR de gen
T T T--T TT
Cebador F →
← Cebador R
-T T T-T T T
Clonación del fragmento deKilA-TeR en el pFS Gen
Ligación del pFS Gen yel fragmento de KilA-TeR
Digestión del pFS Gen con Pst I
AmpR
TeR
mob
ori
pDT 1558
KilA
lacZ
lacZ PstI
PstI
AmpR KmR
mob
ori
pFS GenPstI
PstI
PstI
AmpR
mob
ori
pFS Gen TeR
PstI
KilA
TeR
Figura 3.2. A. Esquema general para la construcción de los plásmidos pFS Gen a partir de la PCR del fragmento interno de gen a clonar, el pGEMT-Easy y el pFS 100. B. Esquema general para la inserción del casete de resistencia a telurito (KilA-TeR) en los plásmidos pFS Gen TeR a partir del pDT1558 y el pFS Gen.
A
B

MATERIALES Y MÉTODOS 86
3.7. Otros métodos.
3.7.1. Curvas de crecimiento y cálculo del tiempo de duplicación.
Para calcular la tasa de crecimiento de cada cepa se realizó una curva de crecimiento y se
calculó el tiempo de duplicación en fase exponencial creciéndola en caldo LB a 37°C y en
agitación. Para ello se inocularon 500 µL de un cultivo en fase estacionaria de la cepa a
estudiar en un erlemeyer con 50 mL de medio LB. De dicho erlemeyer se determinó la
densidad óptica (D.O.) a 600 nm y se plaquearon diluciones seriada en LB agar y por duplicado
cada hora. Para el cálculo de los distintos parámetros de crecimiento se utilizaron las fórmulas
de la figura 3.3, con los resultados obtenidos en tres crecimientos independientes.
3.7.2. Experimentos de frecuencia de mutación.
A partir de un crecimiento fresco de la cepa de K. pneumoniae a estudiar se inoculó una
colonia en 4 mL de medio MH II y se dejó crecer durante 16 h. Posteriormente se diluyó dicho
cultivo inoculándose aproximadamente 102 UFC en 4 mL de medio MH II. A las 24 h, se
plaqueó dicho cultivo sobre MH II agar y MH II agar con la concentración de antibiótico
adecuada (figura 3.4).
La frecuencia de mutación se determinó calculando la proporción de unidades formadoras de
colonias (UFC) que crecieron en las placas con antibiótico respecto al número total de UFC que
crecieron en las placas sin selección (mediana del número de mutantes resistentes/mediana
del número total de células) como previamente se ha descrito en [453]. Los resultados
obtenidos proceden de al menos 8 experimentos independientes. Las comparaciones entre la
frecuencia de mutación de los diferentes antibióticos se realizó mediante un test t de Student
de dos colas.

MATERIALES Y MÉTODOS 87
2.1.1. Experimentos de competición in vitro.
Los experimentos de competición in vitro se realizaron según describió previamente por Moyà
y colaboradores [454]. En primer lugar se obtuvo un crecimiento en LB en fase exponencial de
cada una de las cepas que iban a competir. Seguidamente se mezclaron los cultivos en un ratio
de 1:1 y se diluyeron en Ringer. Aproximadamente 103 células de cada una de las mezclas se
inocularon en al menos ocho tubos de 10 mL de LB y se dejaron crecer a 37ºC en agitación
durante 16 h. Posteriormente se plaquearon diluciones seriadas, por duplicado, en LB y LB con
meropenem 0,75 μg/mL para detectar KpCR-1 o telurito 10 μg/mL para detectar KpCR-
1ΔOmpK26. Los recuentos se realizaron después de incubar las placas 20 h a 37ºC (figura 3.6
A).
Previamente a los experimentos de competición se comprobó que los marcadores selectivos
utilizados permitieran el crecimiento de todas las bacterias de la cepa resistente e inhibieran
por completo a la población sensible.
De forma alternativa, cuando el cociente entre el recuento de las placas LB agar y las de LB
agar con el marcador era menor de 2, se seleccionaron al azar 100 colonias de las placas sin
selección y se estriaron en LB agar y LB agar con el marcador selectivo adecuado (figura 3.6 A).
Después de incubar las placas a 37ºC durante 16-20 h, se diferenció entre fenotipo parental y
mutante por la ausencia/presencia de crecimiento en las placas de LB agar con el selector. El
índice de competición (IC) se definió como el ratio entre la cepa mutante y la parental. Los
cálculos de los valores de IC se realizaron de manera independiente para cada competición
Inocular 1 colonia de CSUB10S en 4 mL MH II ycrecer durante 16 h
Inocular 102 UFC en 10 mLMH II y crecer durante 24 h
Plaquear sobre MH II y MH II marcador selectivo
MH II MH IImarcador selectivo
Seleccionar 20 mutantes al azar
Análisis de la expresiónde OmpK36 mediante
Western Blot
Análisis de la resistencia a antibióticos mediante
CMI
Figura 3.4 Esquema general de los experimentos de frecuencia de mutación.

MATERIALES Y MÉTODOS 88
utilizando las fórmulas de la figura 3.5. El análisis estadístico de la distribución de los IC se
calculó utilizando un test t de Student, considerando significativos los valores de P<0.05.
2.1.1. Estudios de virulencia en un modelo de infección sistémica en ratón.
Los estudios de virulencia y adaptación de las cepas de K. pneumoniae se realizaron en ratones
machos ICR-CD1 de entre 20 y 30 g de peso (Harlan Ibérica, España). Para los estudios de
mortalidad, se inocularon intraperitonealmente (IP) aproximadamente 2. 108 UFC
resuspendidas en PBS, a grupos de 18 ratones. La mortalidad de los ratones fue monitorizada
cada día durante 7 días.
La adaptación in vivo fue evaluada por experimentos de competición en un modelo de
infección sistémica en ratón. Para este objetivo se inocularon intraperitonealmente 100 μL de
una mezcla 1:1 de cada par de cepas testadas (aproximadamente 5.107 UFC totales en fase
exponencial) a al menos 10 ratones por experimento. Los ratones fueron sacrificados 24 h
después de la inoculación y se les extrajo el bazo de manera aséptica. A continuación se
homogeneizó cada bazo en 5 mL de Ringer usando un homogeneizador (Polytron® PT 10-35,
Kinematica AG, Littau, Suiza) y se plaqueó el homogeneizado en LB agar y LB agar con el
marcador selectivo adecuado. El número de UFC de cada cepa y el valor del IC se determinó
utilizando las mismas fórmulas que en los experimentos in vitro (figura 3.5).
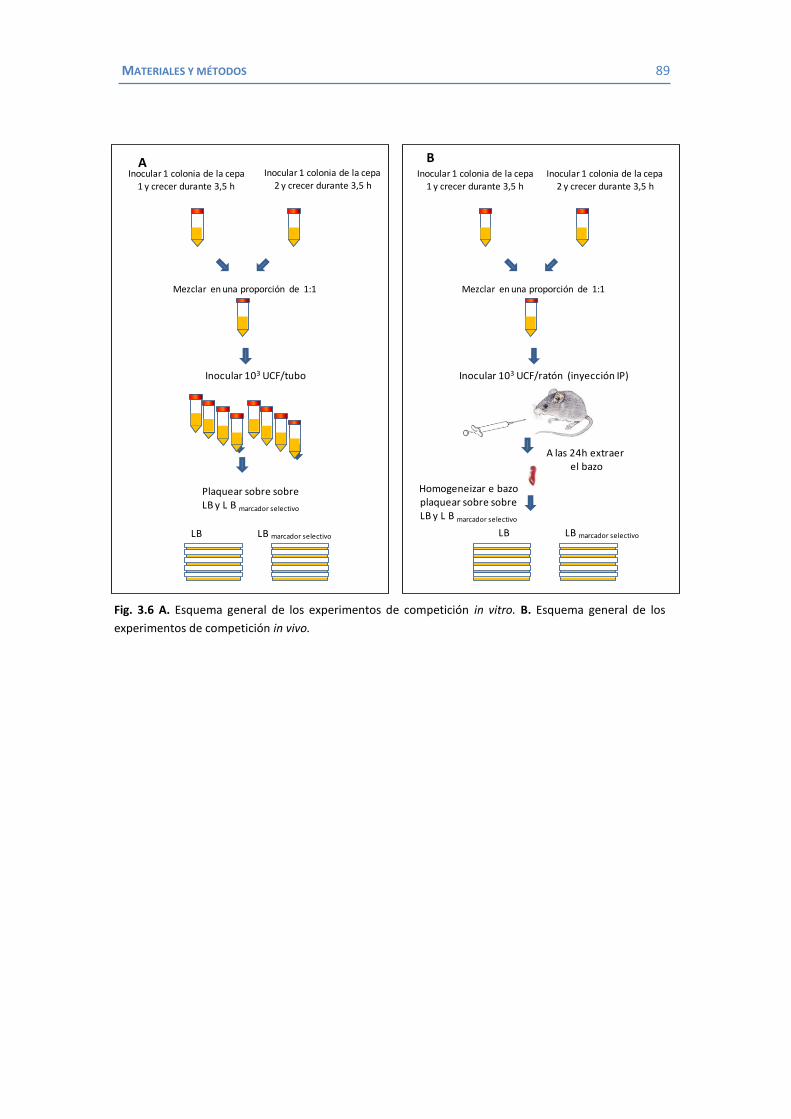
MATERIALES Y MÉTODOS 89
Fig. 3.6 A. Esquema general de los experimentos de competición in vitro. B. Esquema general de los experimentos de competición in vivo.
A B Inocular 1 colonia de la cepa
2 y crecer durante 3,5 h
Homogeneizar e bazo plaquear sobre sobre LB y L B marcador selectivo
LB LB marcador selectivo
Inocular 1 colonia de la cepa 1 y crecer durante 3,5 h
Inocular 1 colonia de la cepa 2 y crecer durante 3,5 h
Mezclar en una proporción de 1:1
Inocular 103 UCF/ratón (inyección IP)
A las 24h extraerel bazo
Inocular 1 colonia de la cepa 1 y crecer durante 3,5 h
Mezclar en una proporción de 1:1
Inocular 103 UCF/tubo
Plaquear sobre sobre LB y L B marcador selectivo
LB LB marcador selectivo


4. RESULTADOS Y DISCUSIÓN.


RESULTADOS Y DISCUSIÓN 93
4.1 Efectos de diferentes β-lactámicos sobre la frecuencia de aparición y selección de mutantes en K. pneumoniae.
K. pneumoniae es un patógeno oportunista que frecuentemente causa infecciones
nosocomiales, principalmente en pacientes inmunocomprometidos. El incremento de la
aparición de aislados nosocomiales de K. pneumoniae multiresistentes ha limitado las opciones
terapéuticas para el tratamiento de las infecciones intrahospitalarias causadas por este
patógeno oportunista [31, 32]. El fenotipo de multiresistencia es consecuencia de la progresiva
acumulación de diferentes mecanismos de resistencia en el mismo microorganismo,
incluyendo la producción de altos niveles de BLEAs y la deficiencia en la expresión de las dos
principales porinas, OmpK35 [230] y OmpK36 [249].
La revisión de los diferentes estudios realizados sobre la aparición de aislados clínicos
multiresistentes de K. pneumoniae nos muestra que la adquisición de este fenotipo, cuando
está basado en la pérdida de la expresión de las principales porinas, suele seguir unas pautas
muy conservadas. En este sentido, el tratamiento antibiótico prolongado administrado a un
paciente que presenta una cepa de K. pneumoniae que expresa una BLEA suele conllevar la
selección de mutantes deficientes en la expresión de la única de porina principal que expresa
incrementado significativamente el grado de resistencia a determinados antibióticos. Se ha
visto que este mecanismo opera en la resistencia a cefoxitina, a cefalosporinas de amplio
espectro, a monobactámicos y a las fluoroquinolonas [230, 283, 324, 455, 456]. Además,
estudios recientes muestran que también podrían tener un papel importante en la adquisición
de resistencia a los antibióticos carbapenémicos [168, 455, 457]. Sin embargo, existen pocos
estudios que hayan investigado el efecto de distintos antibióticos sobre la frecuencia de
aparición y selección de mutantes deficientes en porinas.
Para investigar el efecto de distintos antibióticos en la selección de mutantes deficientes en
porinas utilizamos como modelo la cepa K. pneumoniae CSUB10S. Esta cepa es un buen
ejemplo del proceso de adquisición de resistencia anteriormente descrito, ya que a partir de
ella, y después del tratamiento antibiótico, se obtuvo en el mismo paciente la cepa CSUB10R
[118, 325]. Aunque tanto CSUB10S como CSUB10R expresan la β-lactamasa SHV-2, su patrón
de sensibilidad a los antibióticos es diferente. En la tabla 4.1 se puede observar que, teniendo
en cuenta los puntos de corte establecidos por el CLSI (resistencia a cefoxitina 32 µg/mL,
resistencia a imipenem 16 µg/mL, resistencia a meropenem 16 µg/mL y resistencia a
ertapenem 8 µg/mL) [441], CSUB10S es sensible a cefoxitina, imipenem, meropenem y
ertapenem, sin embargo, CSUB10R presenta valores más altos de CMIs a estos cuatro
antibióticos, siendo resistente a cefoxitina y ertapenem (tabla 4.1). Este incremento en la
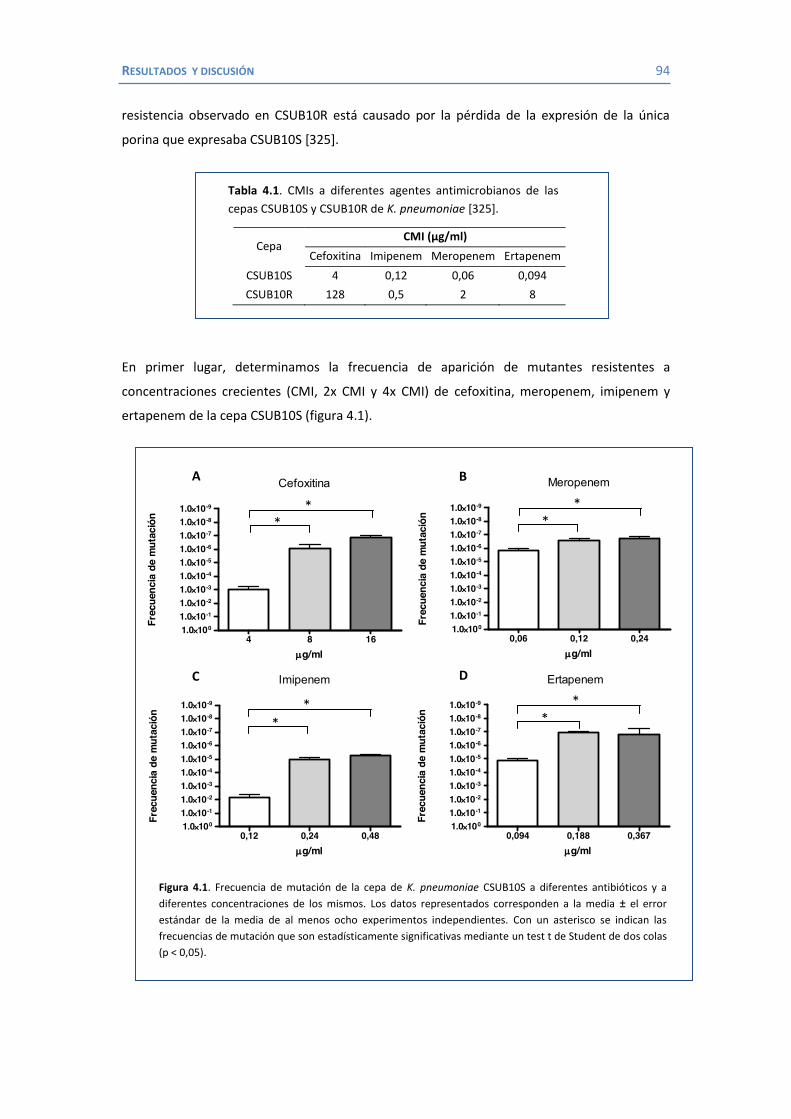
RESULTADOS Y DISCUSIÓN 94
resistencia observado en CSUB10R está causado por la pérdida de la expresión de la única
porina que expresaba CSUB10S [325].
En primer lugar, determinamos la frecuencia de aparición de mutantes resistentes a
concentraciones crecientes (CMI, 2x CMI y 4x CMI) de cefoxitina, meropenem, imipenem y
ertapenem de la cepa CSUB10S (figura 4.1).
Tabla 4.1. CMIs a diferentes agentes antimicrobianos de las cepas CSUB10S y CSUB10R de K. pneumoniae [325].
Cepa CMI (µg/ml)
Cefoxitina Imipenem Meropenem Ertapenem CSUB10S 4 0,12 0,06 0,094 CSUB10R 128 0,5 2 8
Figura 4.1. Frecuencia de mutación de la cepa de K. pneumoniae CSUB10S a diferentes antibióticos y a diferentes concentraciones de los mismos. Los datos representados corresponden a la media ± el error estándar de la media de al menos ocho experimentos independientes. Con un asterisco se indican las frecuencias de mutación que son estadísticamente significativas mediante un test t de Student de dos colas (p < 0,05).
A
* *
4 8 16
1.010-9
1.010-8
1.010-7
1.010-6
1.010-5
1.010-4
1.010-3
1.010-2
1.010-1
1.0100
Cefoxitina
g/ml
Frec
uenc
ia d
e m
utac
ión
B
* *
0,06 0,12 0,24
1.010-9
1.010-8
1.010-7
1.010-6
1.010-5
1.010-4
1.010-3
1.010-2
1.010-1
1.0100
g/ml
Meropenem
Frec
uenc
ia d
e m
utac
ión
C
0,12 0,24 0,48
1.010-9
1.010-8
1.010-7
1.010-6
1.010-5
1.010-4
1.010-3
1.010-2
1.010-1
1.0100
Imipenem
g/ml
Frec
uenc
ia d
e m
utac
ión *
*
D
* *
0,094 0,188 0,367
1.010-9
1.010-8
1.010-7
1.010-6
1.010-5
1.010-4
1.010-3
1.010-2
1.010-1
1.0100
Ertapenem
g/ml
Frec
uenc
ia d
e m
utac
ión
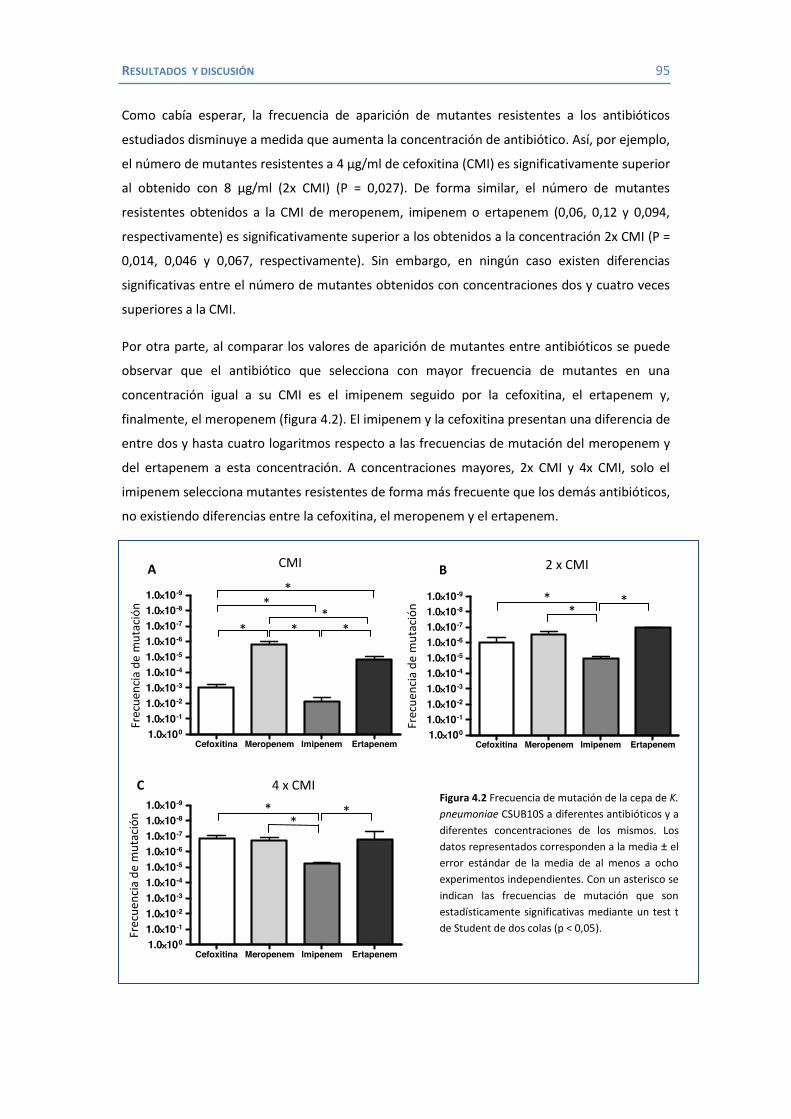
RESULTADOS Y DISCUSIÓN 95
Como cabía esperar, la frecuencia de aparición de mutantes resistentes a los antibióticos
estudiados disminuye a medida que aumenta la concentración de antibiótico. Así, por ejemplo,
el número de mutantes resistentes a 4 µg/ml de cefoxitina (CMI) es significativamente superior
al obtenido con 8 µg/ml (2x CMI) (P = 0,027). De forma similar, el número de mutantes
resistentes obtenidos a la CMI de meropenem, imipenem o ertapenem (0,06, 0,12 y 0,094,
respectivamente) es significativamente superior a los obtenidos a la concentración 2x CMI (P =
0,014, 0,046 y 0,067, respectivamente). Sin embargo, en ningún caso existen diferencias
significativas entre el número de mutantes obtenidos con concentraciones dos y cuatro veces
superiores a la CMI.
Por otra parte, al comparar los valores de aparición de mutantes entre antibióticos se puede
observar que el antibiótico que selecciona con mayor frecuencia de mutantes en una
concentración igual a su CMI es el imipenem seguido por la cefoxitina, el ertapenem y,
finalmente, el meropenem (figura 4.2). El imipenem y la cefoxitina presentan una diferencia de
entre dos y hasta cuatro logaritmos respecto a las frecuencias de mutación del meropenem y
del ertapenem a esta concentración. A concentraciones mayores, 2x CMI y 4x CMI, solo el
imipenem selecciona mutantes resistentes de forma más frecuente que los demás antibióticos,
no existiendo diferencias entre la cefoxitina, el meropenem y el ertapenem.
Figura 4.2 Frecuencia de mutación de la cepa de K. pneumoniae CSUB10S a diferentes antibióticos y a diferentes concentraciones de los mismos. Los datos representados corresponden a la media ± el error estándar de la media de al menos a ocho experimentos independientes. Con un asterisco se indican las frecuencias de mutación que son estadísticamente significativas mediante un test t de Student de dos colas (p < 0,05).
B
Frec
uenc
ia d
e m
utac
ión *
* *
Cefoxitina Meropenem Imipenem Ertapenem
1.010-9
1.010-8
1.010-7
1.010-6
1.010-5
1.010-4
1.010-3
1.010-2
1.010-1
1.0100
2 x CMI
C
* *
*
Frec
uenc
ia d
e m
utac
ión
Cefoxitina Meropenem Imipenem Ertapenem
1.010-9
1.010-8
1.010-7
1.010-6
1.010-5
1.010-4
1.010-3
1.010-2
1.010-1
1.0100
4 x CMI
A
Cefoxitina Meropenem Imipenem Ertapenem
1.010-9
1.010-8
1.010-7
1.010-6
1.010-5
1.010-4
1.010-3
1.010-2
1.010-1
1.0100
* * * *
* *
CMI
Frec
uenc
ia d
e m
utac
ión

RESULTADOS Y DISCUSIÓN 96
Para analizar la presencia de OmpK36 en los mutantes obtenidos con distintos antibióticos y
concentraciones utilizamos la técnica del Western blot y anticuerpos específicos anti-OmpK36.
Este ensayo, a pesar de ser más laborioso, es más específico y sensible que otras estrategias
que permiten analizar la expresión de OmpK36. Por un lado, el anticuerpo sólo detecta la
banda correspondiente a la porina que deseamos estudiar y evita los problemas en la
identificación de la proteínas que pueden aparecer al utilizar sólo la tinción con azul de
Coomassie de las proteínas de membrana externa de K. pneumoniae. Por otro lado, el uso de
anticuerpos incrementa notablemente la sensibilidad del método lo que permite asegurar que
la falta de reacción positiva es indicativa de la ausencia de OmpK36. El análisis se realizó
seleccionando al azar 480 mutantes, 40 por antibiótico y concentración ensayada. En la figura
4.3 se muestra un Western blot representativo de estos análisis, donde se observan las cepas
control CSUB10R y CSUB10S, dos mutantes de los analizados que expresan la misma cantidad
de OmpK36 que la cepa parental CSUB10S; dos mutantes analizados que expresan OmpK36
pero en menor cantidad que CSUB10S y dos mutantes que no expresan OmpK36.
Los resultados en conjunto del análisis de la presencia de OmpK36 en la membrana externa de
los 480 mutantes estudiados se recogen en la tabla 4.2.
Antibiótico CMI 2 x CMI 4 x CMI
Cefoxitina 45 % 67 % 100 %
Meropenem 65 % 60 % 100%
Imipenem 0 % 0 % 0 %
Ertapenem 48 % 81 % 100 %
Figura 4.3. Figura representativa del análisis de la expresión de OmpK36 mediante Western blot y anticuerpos anti-OmpK36. Carril 1: CSUB10R, Carril 2: CSUB10S; Carril 3 y 4: mutantes de CSUB10S que expresan la misma cantidad de OmpK36; Carril 5 y 6: mutantes de CSUB10S que expresan la menor cantidad de OmpK36; Carril 7 y 8: mutantes de CSUB10S que no expresan OmpK36. 1 2 3 4 5 6 7 8
Tabla 4.2. Porcentaje de mutantes deficientes en OmpK36 obtenidos con cada uno de los antibióticos y concentraciones utilizadas.

RESULTADOS Y DISCUSIÓN 97
El porcentaje de mutantes que no expresan OmpK36 fue incrementándose de forma
directamente proporcional a la concentración de cefoxitina o ertapenem sobre la que fueron
aislados. Todos los mutantes aislados a concentraciones iguales o superiores a cuatro veces el
valor de la CMI de estos antibióticos dejaron de expresar OmpK36, lo que sugiere que la
pérdida de la expresión de OmpK36 es un factor indispensable para la viabilidad bacteriana a
estas concentraciones. Sin embargo, a concentraciones inferiores deben existir otros
mecanismos que permiten a CSUB10S resistir la acción de la cefoxitina y el ertapenem sin
perder la capacidad de expresar OmpK36. Entre estos mecanismos podríamos incluir la
reducción de la expresión de la porina tal como describieron anteriormente Doménech-
Sánchez y colaboradores [458], o la alteración de la estructura de la propia porina como
describieron García-Fernández y colaboradores [226]. Ambos mecanismos podrían haber
pasado desapercibidos con la estrategia utilizada en nuestro estudio para determinar la
expresión de OmpK36. En cualquier caso la pérdida de OmpK36 en combinación con la
expresión de SHV-2 fue suficiente para que los valores de CMI de ambos antibióticos superaran
los establecidos por el CLSI para considerar a los aislados resistentes a cefoxitina y ertapenem.
Ninguno de los mutantes que aparecieron sobre las placas con distintas concentraciones de
imipenem fueron deficientes en la expresión de OmpK36. Por el contrario, un porcentaje
variable de los mutantes seleccionados sobre distintas concentraciones de meropenem, que
osciló entre el 60 y el 87 % de los mutantes analizados dependiendo de la concentración de
meropenem utilizada, no expresaban OmpK36. Estos datos sugieren que la ausencia de
porinas afecta más a la penetración del meropenem que a la del imipenem y es consistente
con los resultados por otros investigadores. En este sentido, Cornaglia y colaboradores
demostraron que la penetración del meropenem en los mutantes deficientes en porinas de
Enterobacter cloacae era 25 veces menos eficiente que en la cepa parental con porinas,
mientras que la penetración del imipenem solo lo era 7 veces menos [459]. Por otra parte,
Pragai y Nagy, utilizando cepas de K. pneumoniae con BLEA, fueron capaces de seleccionar in
vitro mutantes de deficientes en porinas utilizando meropenem como presión selectiva pero
no utilizado imipenem [460]. En su conjunto estos resultados indican que la penetración del
meropenem a través de OmpK36 es más eficiente que la del imipenem. De hecho, si bien
CSUB10R no es clínicamente resistente al meropenem, la pérdida de OmpK36 supuso un
notable incremento de la CMI de este antibiótico. No ocurrió lo mismo con el imipenem donde
la CMI apenas varió tras la pérdida de OmpK36 (ver tabla 4.2).
Mientras que al utilizar cefoxitina o ertapenem como presión selectiva se obtuvieron mutantes
clínicamente resistentes, el uso de meropenem, a pesar de seleccionar mutantes deficientes
en OmpK36, no permitió seleccionar ningún mutante resistente según el criterio establecido

RESULTADOS Y DISCUSIÓN 98
por el CLSI (CMI 16 µg/mL) (datos no mostrados). Este hecho contrasta con los resultados de
algunos estudios clínicos en los que la se ha asociado la ausencia de porinas con la resistencia
al meropenem. Un ejemplo es el estudio descrito por Mena y colaboradores en 2006 [168]. En
este estudio, la ausencia de OmpK36 en combinación con CTX-M-1 permitió a los aislados
KpCR-1 y KpCR-2 ser resistente a meropenem (CMI = 16 µg/ml) e incrementar su CMI a
imipenem alcanzando valores intermedios de resistencia (CMI = 8 µg/ml). Otro ejemplo más
reciente es el publicado por Findlay y colaboradores donde una cepa productora de CTX-M-15,
OXA-1, SHV-1 y TEM-1 susceptible a meropenem e imipenem pasó a ser resistente a
meropenem por la pérdida de las porinas [457]. Es probable que estas divergencias con
nuestro estudio sean debidas al tipo de BLEA presente en cada aislado.
Atendiendo a la frecuencia de mutación (figura 4.2) y al porcentaje de mutantes deficientes en
OmpK36 obtenidos (tabla 4.2), se podría afirmar que, en CSUB10S, la cefoxitina es el
antibiótico que con mayor eficiencia selecciona mutantes deficientes en OmpK36 seguido por
el ertapenem y finalmente el meropenem. El imipenem es el mejor antibiótico de los
ensayados para evitar la selección de mutantes deficientes en OmpK36.
Todos los mutantes deficientes en OmpK36, independientemente del antibiótico sobre el que
fueron seleccionados, fueron clínicamente resistentes a cefoxitina y ertapenem incluso al
utilizar concentraciones de ertapenem y meropenem lejanas de las del punto de corte de
resistencia del CLSI para su selección, lo que refleja el riesgo que supone el uso de estos
antibióticos para la selección de cepas multiresistentes.
Con el fin de investigar el mecanismo responsable de la deficiencia en la expresión de OmpK36
se analizó el gen ompk36 en 27 de los mutantes seleccionados al azar obtenidos tras la
selección con cefoxitina, ertapenem o meropenem (tabla 4.3). Independientemente del
antibiótico utilizado para la selección, los mecanismos más frecuentes de interrupción de la
expresión de ompk36 fueron las inserciones, ya sean de un solo nucleótido o de secuencias de
inserción, que interrumpieron la pauta de lectura del gen, y las deleciones. Las sustituciones
fueron las alteraciones responsables de la deficiencia de OmpK36 en el resto de los mutantes.
Es destacable que las mutaciones detectadas en los mutantes obtenidos in vitro en este
estudio no difieren de las que observaron en los mutantes obtenidos in vivo Doumith y
colaboradores [455]. La variedad en el tipo y localización de las mutaciones que causan la
interrupción de la expresión de ompk36 sugiere que estas interrupciones son el resultado de
acontecimientos distintos en cada mutante y que no parecen existir regiones dentro del gen en
las que sea más frecuente la mutación.
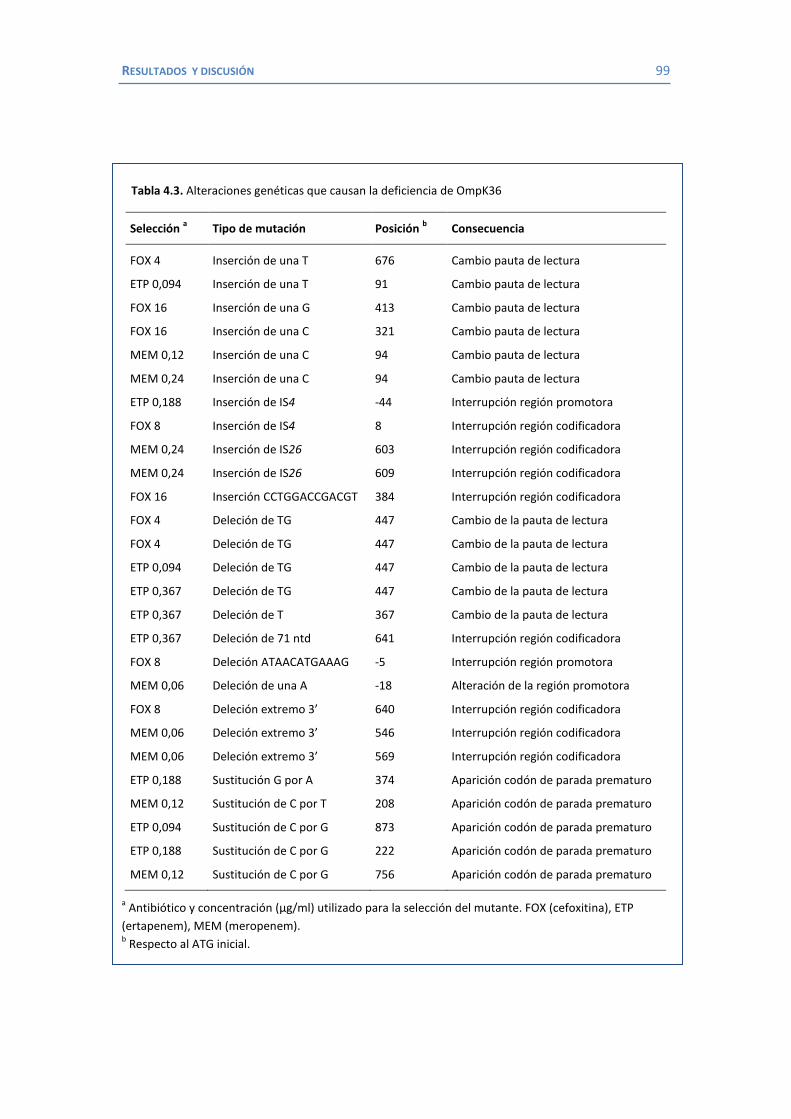
RESULTADOS Y DISCUSIÓN 99
Selección a Tipo de mutación Posición b Consecuencia
FOX 4 Inserción de una T 676 Cambio pauta de lectura
ETP 0,094 Inserción de una T 91 Cambio pauta de lectura
FOX 16 Inserción de una G 413 Cambio pauta de lectura
FOX 16 Inserción de una C 321 Cambio pauta de lectura
MEM 0,12 Inserción de una C 94 Cambio pauta de lectura
MEM 0,24 Inserción de una C 94 Cambio pauta de lectura
ETP 0,188 Inserción de IS4 -44 Interrupción región promotora
FOX 8 Inserción de IS4 8 Interrupción región codificadora
MEM 0,24 Inserción de IS26 603 Interrupción región codificadora
MEM 0,24 Inserción de IS26 609 Interrupción región codificadora
FOX 16 Inserción CCTGGACCGACGT 384 Interrupción región codificadora
FOX 4 Deleción de TG 447 Cambio de la pauta de lectura
FOX 4 Deleción de TG 447 Cambio de la pauta de lectura
ETP 0,094 Deleción de TG 447 Cambio de la pauta de lectura
ETP 0,367 Deleción de TG 447 Cambio de la pauta de lectura
ETP 0,367 Deleción de T 367 Cambio de la pauta de lectura
ETP 0,367 Deleción de 71 ntd 641 Interrupción región codificadora
FOX 8 Deleción ATAACATGAAAG -5 Interrupción región promotora
MEM 0,06 Deleción de una A -18 Alteración de la región promotora
FOX 8 Deleción extremo 3’ 640 Interrupción región codificadora
MEM 0,06 Deleción extremo 3’ 546 Interrupción región codificadora
MEM 0,06 Deleción extremo 3’ 569 Interrupción región codificadora
ETP 0,188 Sustitución G por A 374 Aparición codón de parada prematuro
MEM 0,12 Sustitución de C por T 208 Aparición codón de parada prematuro
ETP 0,094 Sustitución de C por G 873 Aparición codón de parada prematuro
ETP 0,188 Sustitución de C por G 222 Aparición codón de parada prematuro
MEM 0,12 Sustitución de C por G 756 Aparición codón de parada prematuro a Antibiótico y concentración (µg/ml) utilizado para la selección del mutante. FOX (cefoxitina), ETP (ertapenem), MEM (meropenem). b Respecto al ATG inicial.
Tabla 4.3. Alteraciones genéticas que causan la deficiencia de OmpK36

RESULTADOS Y DISCUSIÓN 100
4.2 Papel de LamB en la resistencia a antimicrobianos.
La pérdida o reducción de la expresión de las porinas principales OmpK35 y OmpK36, en los
aislados clínicos de K. pneumoniae resistentes a los antimicrobianos supone una ventaja para
dichas bacterias ya que así impiden o reducen la entrada de antibióticos a través de estos
canales. Pero esta deficiencia en la expresión de OmpK35 y/o OmpK36, implica, a su vez, una
reducción de la entrada de otras moléculas pequeñas y polares (<600 kDa) que difunden a
través de estas porinas y que actúan como nutrientes o elementos esenciales para el correcto
funcionamiento de la bacteria. Así pues, es lógico pensar que la bacteria necesita compensar la
pérdida de estas porinas expresando otras proteínas que le permitan adquirir las sustancias
necesarias para su desarrollo. En este sentido, trabajos previos de nuestro grupo han evaluado
la expresión génica diferencial entre los dos aislados clínicos isogénicos de K. pneumoniae,
CSUB10S y CSUB10R, mediante micromatrices de ADN de E. coli [461]. A partir de dicho trabajo
se observó que la cepa resistente (CSUB10R) sobreexpresaba, entre otros, un gran número de
genes ribosomales (rplB, rplC, rplD, rplE, rplF, rplW, rpsE, rpsJ, rpsK, rpsS) y genes implicados en
el metabolismo de ARNt (trmD, glyS). Pero el gen con mayor diferencia de expresión, con una
sobreexpresión de dos veces en la cepa resistente que en la cepa sensible, era el gen ycjV, gen
que forma parte de una bomba de flujo ABC putativa y está implicado en el metabolismo y
transporte de carbohidrato en E. coli [462]. A partir de esta información y mediante las
herramientas bioinformáticas BLAST y T-BLAST, se buscó la correspondencia de dicho gen en el
genoma y en el proteoma de K. pneumoniae MGH 78578.
Los resultados obtenidos nos indicaron que el gen ycjV de E. coli era homólogo al gen malK de
K. pneumoniae, con un 62 % de identidad génica, y un 49 % identidad y 67% similitud a nivel
proteico. Al analizar dicho gen con más profundidad, también se observó que malK formaba
parte del sistema de transporte maltosa/maltodextrina de K. pneumoniae, y dentro del mismo,
del operón malK-lamB. Todo ello añadido al hecho de que en estudios previos donde se
compararon los perfiles de PME de los aislado clínicos CSUB10S y CSUB10R se observara que
LamB podía ser una PME predominante en el aislado resistente, nos llevó a estudiar el papel
que podía tener el gen lamB en la resistencia a los antimicrobianos [325].
En primer lugar, se analizó la cantidad de LamB presente en la membrana externa de las cepas
CSUB10S y CUSB10R crecidas en medio MH II, en suero o en orina humana por SDS-PAGE como
se ha descrito previamente. Como se muestra en la figura 4.4 A, la membrana externa del
aislado resistente contiene mayor cantidad de LamB que el aislado susceptible, tanto en MH II
como en los fluidos fisiológicos. Este resultado se confirmó mediante el análisis densitométrico
de la intensidad de las bandas de LamB respecto a la banda de OmpA, de tres geles SDS-PAGE
independientes. En dicho análisis se observó que al crecer ambas cepas en MH II, la cepa
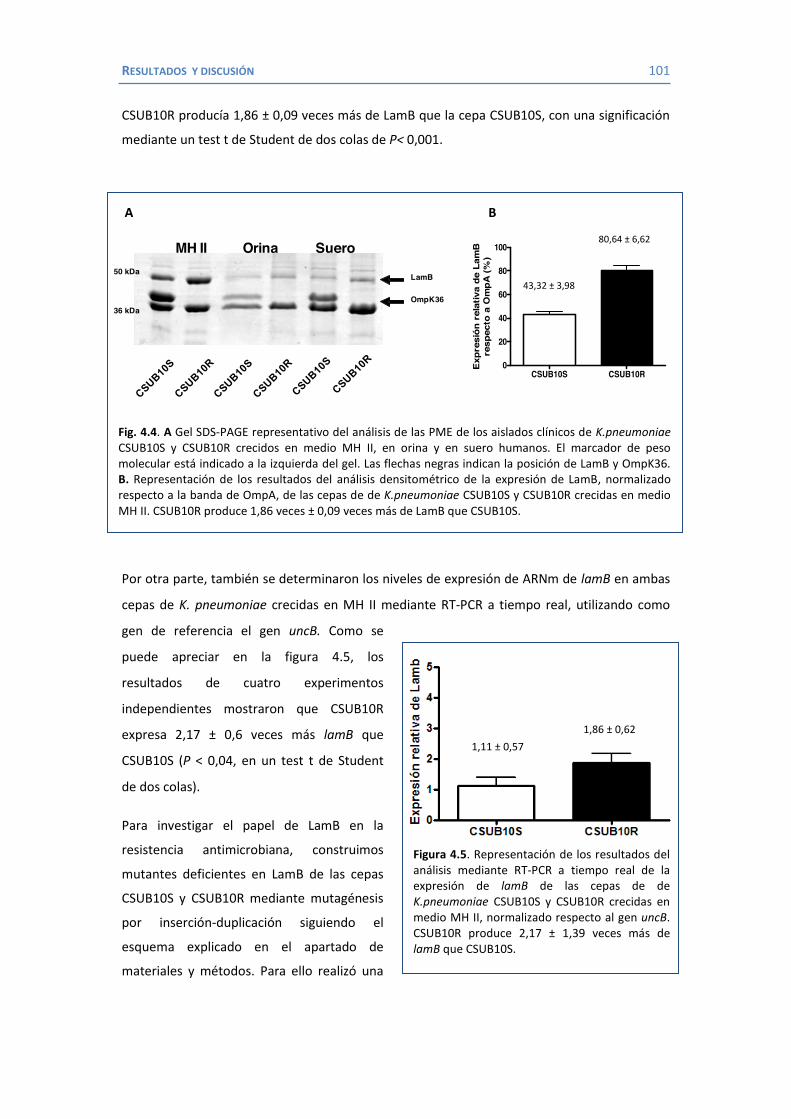
RESULTADOS Y DISCUSIÓN 101
CSUB10R producía 1,86 ± 0,09 veces más de LamB que la cepa CSUB10S, con una significación
mediante un test t de Student de dos colas de P< 0,001.
Por otra parte, también se determinaron los niveles de expresión de ARNm de lamB en ambas
cepas de K. pneumoniae crecidas en MH II mediante RT-PCR a tiempo real, utilizando como
gen de referencia el gen uncB. Como se
puede apreciar en la figura 4.5, los
resultados de cuatro experimentos
independientes mostraron que CSUB10R
expresa 2,17 ± 0,6 veces más lamB que
CSUB10S (P < 0,04, en un test t de Student
de dos colas).
Para investigar el papel de LamB en la
resistencia antimicrobiana, construimos
mutantes deficientes en LamB de las cepas
CSUB10S y CSUB10R mediante mutagénesis
por inserción-duplicación siguiendo el
esquema explicado en el apartado de
materiales y métodos. Para ello realizó una
Figura 4.5. Representación de los resultados del análisis mediante RT-PCR a tiempo real de la expresión de lamB de las cepas de de K.pneumoniae CSUB10S y CSUB10R crecidas en medio MH II, normalizado respecto al gen uncB. CSUB10R produce 2,17 ± 1,39 veces más de lamB que CSUB10S.
1,11 ± 0,57 1,86 ± 0,62
CSUB10S CSUB10R0
20
40
60
80
100
Exp
resi
ón r
elat
iva
de L
amB
resp
ecto
a O
mpA
(%
)
Fig. 4.4. A Gel SDS-PAGE representativo del análisis de las PME de los aislados clínicos de K.pneumoniae CSUB10S y CSUB10R crecidos en medio MH II, en orina y en suero humanos. El marcador de peso molecular está indicado a la izquierda del gel. Las flechas negras indican la posición de LamB y OmpK36. B. Representación de los resultados del análisis densitométrico de la expresión de LamB, normalizado respecto a la banda de OmpA, de las cepas de de K.pneumoniae CSUB10S y CSUB10R crecidas en medio MH II. CSUB10R produce 1,86 veces ± 0,09 veces más de LamB que CSUB10S.
B A
43,32 ± 3,98
80,64 ± 6,62 MH II Orina Suero
36 kDa
50 kDaLamB
OmpK36

RESULTADOS Y DISCUSIÓN 102
PCR de un fragmento interno del gen lamB utilizando los cebadores Lamb F1 y Lamb R1.
Seguidamente dicho fragmento se clonó en el plásmido pGEM-T Easy, obteniéndose el
plásmido pGEM-T LamB. Para la obtención del pFSLamB se realizó la transferencia del
fragmento lamB procedente del pGEM-T LamB al pFS100 mediante digestión con EcoRI y
posterior ligación con la ADN ligasa T4. El pFSLamB fue introducido en las cepas de K.
pneumoniae CSUB10S y CSUB10R mediante conjugación, dando como resultado los mutantes
isogénicos delecionados en lamB, CSUB10SΔlamB y CSUB10RΔlamB. Como se puede observar
en la figura 4.6, la comprobación de la correcta construcción de estos mutantes se realizó por
Southern blot con una sonda interna específica del gen lamB y por análisis del perfil de PME de
los mutantes mediante geles SDS-PAGE. En la imagen del Southern blot (figura 4.6 B) se puede
apreciar cómo, al utilizar una sonda específica de lamB, las cepas originales CSUB10S y
CSUB10R presentan una sola banda correspondiente al gen lamB y, en cambio, los mutantes
delecionados presentan dos bandas, correspondientes a los dos fragmentos de lamB
resultantes de su interrupción por el plásmido pFSLamB. En el panel C de la figura 4.6, se
observa la desaparición de la banda correspondiente a la proteína Lamb de los perfiles de PME
en CSUB10SΔlamB y CSUB10RΔlamB.
Tabla 4. CMIs de CSUB10S, CSUB10R y de sus mutantes isogénicos deficientes en lamB.
Figura 4.6. Construcción de los mutantes deficientes en LamB de las cepas CSUB10S y CSUB10R de K. pneumoniae. A Representación esquemática de la mutagénesis del gen lamB en K. pneumoniae CSUB10S y CSUB10R. En la parte superior se muestra el cromosoma de las cepas CSUB10S y CSUB10R, y en la parte inferior se muestra el cromosoma de sus mutantes isogénicos deficientes en lamB. El tamaño de los fragmentos de ADN en el esquema no está a escala. La línea de puntos en el genoma del mutante representa el plásmido integrado en el cromosoma. Las franjas negras indican la sonda utilizada en el análisis mediante Southern blot. Las flechas indican el promotor del operón. Debajo de la representación se indica el tamaño de los fragmentos en kilobases (kb) obtenidos por la digestión con Hind III que hibridan con la sonda anteriormente descrita. B Análisis mediante Southern blot de los cromosomas de K. pneumoniae CSUB10S y CSUB10R y sus mutantes isogénicos deficientes en lamB digeridos con Hind III. En el lateral izquierdo de la figura se muestra el marcador de peso molecular (en kilobases). C Análisis mediante gel de SDS-PAGE de las PME de las cepas de K. pneumoniae CSUB10S y CSUB10R y sus mutantes isogénicos deficientes en lamB. A la izquierda del gel se indica el marcador de peso molecular. Las flechas indican la posición de LamB.
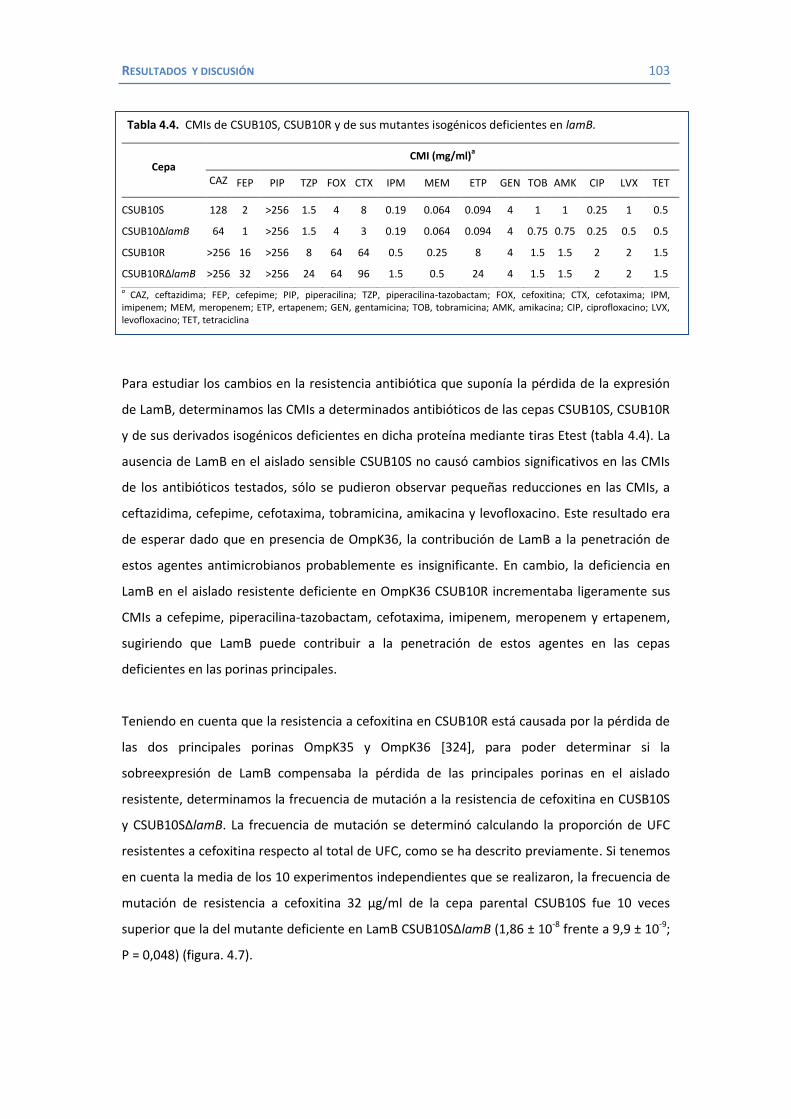
RESULTADOS Y DISCUSIÓN 103
a CAZ, ceftazidima; FEP, cefepime; PIP, piperacilina; TZP, piperacilina-tazobactam; FOX, cefoxitina; CTX, cefotaxima; IPM, imipenem; MEM, meropenem; ETP, ertapenem; GEN, gentamicina; TOB, tobramicina; AMK, amikacina; CIP, ciprofloxacino; LVX, levofloxacino; TET, tetraciclina
Para estudiar los cambios en la resistencia antibiótica que suponía la pérdida de la expresión
de LamB, determinamos las CMIs a determinados antibióticos de las cepas CSUB10S, CSUB10R
y de sus derivados isogénicos deficientes en dicha proteína mediante tiras Etest (tabla 4.4). La
ausencia de LamB en el aislado sensible CSUB10S no causó cambios significativos en las CMIs
de los antibióticos testados, sólo se pudieron observar pequeñas reducciones en las CMIs, a
ceftazidima, cefepime, cefotaxima, tobramicina, amikacina y levofloxacino. Este resultado era
de esperar dado que en presencia de OmpK36, la contribución de LamB a la penetración de
estos agentes antimicrobianos probablemente es insignificante. En cambio, la deficiencia en
LamB en el aislado resistente deficiente en OmpK36 CSUB10R incrementaba ligeramente sus
CMIs a cefepime, piperacilina-tazobactam, cefotaxima, imipenem, meropenem y ertapenem,
sugiriendo que LamB puede contribuir a la penetración de estos agentes en las cepas
deficientes en las porinas principales.
Teniendo en cuenta que la resistencia a cefoxitina en CSUB10R está causada por la pérdida de
las dos principales porinas OmpK35 y OmpK36 [324], para poder determinar si la
sobreexpresión de LamB compensaba la pérdida de las principales porinas en el aislado
resistente, determinamos la frecuencia de mutación a la resistencia de cefoxitina en CUSB10S
y CSUB10SΔlamB. La frecuencia de mutación se determinó calculando la proporción de UFC
resistentes a cefoxitina respecto al total de UFC, como se ha descrito previamente. Si tenemos
en cuenta la media de los 10 experimentos independientes que se realizaron, la frecuencia de
mutación de resistencia a cefoxitina 32 µg/ml de la cepa parental CSUB10S fue 10 veces
superior que la del mutante deficiente en LamB CSUB10SΔlamB (1,86 ± 10-8 frente a 9,9 ± 10-9;
P = 0,048) (figura. 4.7).
Cepa CMI (mg/ml)a
CAZ FEP PIP TZP FOX CTX IPM MEM ETP GEN TOB AMK CIP LVX TET
CSUB10S 128 2 >256 1.5 4 8 0.19 0.064 0.094 4 1 1 0.25 1 0.5
CSUB10ΔlamB 64 1 >256 1.5 4 3 0.19 0.064 0.094 4 0.75 0.75 0.25 0.5 0.5
CSUB10R >256 16 >256 8 64 64 0.5 0.25 8 4 1.5 1.5 2 2 1.5
CSUB10RΔlamB >256 32 >256 24 64 96 1.5 0.5 24 4 1.5 1.5 2 2 1.5
Tabla 4.4. CMIs de CSUB10S, CSUB10R y de sus mutantes isogénicos deficientes en lamB.
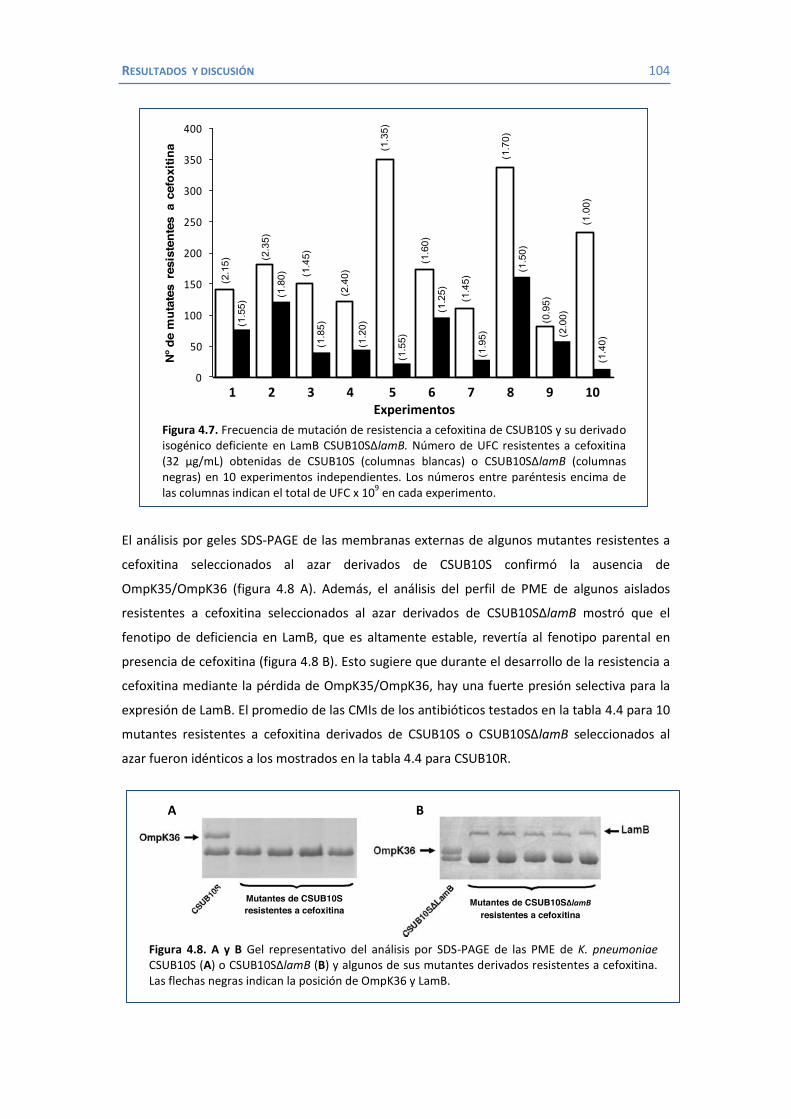
RESULTADOS Y DISCUSIÓN 104
El análisis por geles SDS-PAGE de las membranas externas de algunos mutantes resistentes a
cefoxitina seleccionados al azar derivados de CSUB10S confirmó la ausencia de
OmpK35/OmpK36 (figura 4.8 A). Además, el análisis del perfil de PME de algunos aislados
resistentes a cefoxitina seleccionados al azar derivados de CSUB10SΔlamB mostró que el
fenotipo de deficiencia en LamB, que es altamente estable, revertía al fenotipo parental en
presencia de cefoxitina (figura 4.8 B). Esto sugiere que durante el desarrollo de la resistencia a
cefoxitina mediante la pérdida de OmpK35/OmpK36, hay una fuerte presión selectiva para la
expresión de LamB. El promedio de las CMIs de los antibióticos testados en la tabla 4.4 para 10
mutantes resistentes a cefoxitina derivados de CSUB10S o CSUB10SΔlamB seleccionados al
azar fueron idénticos a los mostrados en la tabla 4.4 para CSUB10R.
Figura 4.8. A y B Gel representativo del análisis por SDS-PAGE de las PME de K. pneumoniae CSUB10S (A) o CSUB10SΔlamB (B) y algunos de sus mutantes derivados resistentes a cefoxitina. Las flechas negras indican la posición de OmpK36 y LamB.
Mutantes de CSUB10S resistentes a cefoxitina
Mutantes de CSUB10SΔlamB resistentes a cefoxitina
A B
0
50
100
150
200
250
300
350
400
Nº d
e m
utat
esre
sist
ente
s a
cefo
xitin
a
Experimentos
(2.1
5)
(1.5
5)
(1.4
5)
(1.9
5)
(2.3
5)(1
.80) (1
.45)
(1.8
5)
(2.4
0)
(1.2
0)
(1.3
5)
(1.5
5)
(1.6
0)
(1.2
5)
(1.7
0)
(1.5
0)
(0.9
5)
(2.0
0)
(1.0
0)(1
.40)
1 2 3 4 5 6 7 8 9 10 Experimentos
Figura 4.7. Frecuencia de mutación de resistencia a cefoxitina de CSUB10S y su derivado isogénico deficiente en LamB CSUB10SΔlamB. Número de UFC resistentes a cefoxitina (32 µg/mL) obtenidas de CSUB10S (columnas blancas) o CSUB10SΔlamB (columnas negras) en 10 experimentos independientes. Los números entre paréntesis encima de las columnas indican el total de UFC x 109 en cada experimento.

RESULTADOS Y DISCUSIÓN 105
Para investigar si LamB contribuía a la
penetración del meropenem en CSUB10R,
determinamos si la frecuencia de mutación a
meropenem 2 µg/mL se incrementaba en
ausencia de LamB. El mutante deficiente en
LamB CSUB10RΔlamB mostró una frecuencia
de mutación más alta (1,43 x 10-6) que la cepa
parental, CSUB10R, para la cual dicha
frecuencia fue indetectable (<1,0 x 10-11). El
análisis por SDS-PAGE de las PME para los
aislados resistentes a meropenem derivados
de CSUB10RΔlamB confirmó que el fenotipo
deficiente en LamB se conservaba, pero
también revelaba la presencia de una nueva
proteína de unos 26 kDa (Figura 4.9).
En este apartado hemos demostrado que la deficiencia de la porina OmpK36 está asociada a
una sobreexpresión de LamB. En otros estudios con mutantes de E.coli tolerantes a disolventes
orgánicos [463] y para algunos aislados clínicos de E. aerogenes [464] se han observado
resultados similares. A pesar del hecho de que esta asociación se puede explicar en parte por
la corregulación de LamB con la expresión de las porinas principales a nivel de transcripción
y/o a nivels de traducción [465, 466], la contribución de LamB a la resistencia antimicrobiana
no se ha estudiado detalladamente. Nuestros resultados utilizando mutantes isogénicos
deficientes en LamB demuestran que la sobreexpresión de esta porina es un mecanismo
esencial para compensar la pérdida de OmpK36 y permite a K. pneumoniae adquirir o poseer
altos niveles de resistencia a varias clases de antibióticos. Además, cuando hay una deficiencia
en las porinas principales, la pérdida de LamB promueve una disminución de la susceptibilidad
a los carbapenemas. Kaczmarek y colaboradores han obtenido resultados similares al estudiar
PhoE [283]. En su estudio, PhoE compensaba la pérdida de las porinas principales no
específicas, y la regulación a la baja del gen phoE estaba implicada en la resistencia a los
carbapenemas.
Figura 4.9. Análisis mediante geles de SDS-PAGE de las PME de K. pneumoniaeCSUB10RΔLamB y cuatro mutantes derivadosresistentes a meropenem D. El marcador depeso molecular se indica en kDa a la izquierdade cada gel. Las flechas indican la nuevaporina proteína de 26 kDa.
Mutantes de CSUB10RΔlamBresistentes a meropenem

RESULTADOS Y DISCUSIÓN 106
4.3 Papel de OmpK26 en la resistencia a antimicrobianos.
Identificación de OmpK26
La aparición de esta nueva proteína de 26 kDa en los mutantes resistentes a meropenem 2
µg/mL derivados de CSUB10RΔlamB descrita en el capítulo anterior nos hizo pensar que dicha
proteína podría estar implicada en la resistencia a carbapenemas. Así pues realizamos un
análisis y comparación los perfiles de PME de dos parejas de aislados clínicos isogénicos
susceptibles y resistentes a carbapenemas (figura 4.10). En las ME pertenecientes al aislado
susceptible KpCS-1 analizadas por geles SDS-PAGE se observó la presencia de una banda de
36kDa (indicada con una flecha en la figura 4.10), mientras que en las membranas
pertenecientes al aislado resistente KpCR-1 dicha banda no estaba presente. La confirmación
de que esta banda de 36 kDa presente en el aislado susceptible y ausente en el resistente
correspondía a la porina OmpK36 se realizó mediante análisis por Western blot utilizando un
anticuerpo específico contra OmpK36 y mediante análisis de secuenciación por espectrometría
de masas. La amplificación y secuenciación del gen ompK36 de la cepa KpCR-1 reveló la
presencia de la secuencia de inserción IS26 en la región codificante de ompK36 [168]. Además
de este cambio observado en el perfil de PME, las membranas de KpCR-1 contenían una
proteína de unos 26 kDa que no estaba presente en las membranas de KpCS-1 (indicada con
una flecha negra en la figura 4.10). Este resultado establece que el fenotipo de resistencia en
este aislado está correlacionado con la ausencia de la porina OmpK36 y con la presencia de
una nueva porina de unos 26 kDa en su ME. Un fenómeno similar se observó al comparar las
PME del otro par de aislados clínicos, uno heteroresistente (Kpn-3) y el otro homoresistente
(Kpn-17) a los carbapenemas. El aislado heteroresistente Kpn-3, que contiene solo una
subpoblación de bacterias capaces de crecer en presencia de altas concentraciones de
Figura 4.10. Análisis mediante gel SDS-PAGE de las PME de diferentes aislados clínicos de K. pneumoniae. En el carril 1 el aislado sensible a carbapenemas KpCS-1, en el carril 2 el aislado heteroresistente a carbapenemas Kpn-3, en el carril 3 el aislado clínico derivado de KpCS-1 resistente a carbapenemas KpCR-1 y en el carril 4 el derivado de Kpn-3 homoresistante a carbapenemas Kpn-17. El marcador de peso molecular está indicado en kDa en la parte izquierda del gel. Las flechas en blanco indican la posición de OmpK36 y las negras indican la posición de a nueva proteína de 26 kDa presente en los aislados resistentes.
36 kDa
25 kDa
KpCS-1
KpCR-1
Kpn-3
Kpn-17

RESULTADOS Y DISCUSIÓN 107
Figura 4.11. Resultado del análisis por MALDI TOF-TOF para la identificación de la proteína KdgM. En rojo se muestran los péptidos identificados. La identificación se realizó con un 67% de cobertura. Para la identificación mediante Mascot se utilizó el proteoma de la cepa de K. pneumoniae subsp. pneumoniae MHG 78758.
imipenem (32 µg/mL), expresaba OmpK36, mientras que el aislado homoresistente Kpn-17,
que es altamente resistente a imipenem (CMI>128 µg/mL), no expresaba OmpK36 y a su vez
expresaba una nueva proteína de 26 kDa (figura 4.10, carriles 3 y 4, respectivamente).
Para identificar la proteína de 26 kDa, la correspondiente banda fue escindida del gel y
sometida a análisis de espectrometría de masas. En cada aislado resistente, se encontró que la
banda correspondía a una putativa porina específica para oligogalacturonatos homóloga a
KdgM, designada como OmpK26. A su vez dicha proteína coincidía con la proteína
sobreexpresada en los mutantes de CSUB10RΔlamB resistentes a meropenem 2 µg/mL (figura
4.11). Por otra parte, una proteína con el mismo peso molecular había aparecido en dos
aislados de K. pneumoniae no susceptibles a carbapenemas aislados en un hospital chino
[327]. Los 15 aminoácidos N-terminales determinados de esta proteína mostraban un 100% de
similitud con la proteína identificada en nuestros aislados, sin embargo no se investigó su
contribución a la resistencia a los carbapenemas.
Al secuenciar el gen codificante de OmpK26, yjhA, de KpCR-1 y Kpn-17 este mostró un 100%
homología con el gen originario de K. pneumoniae subsp. pneumoniae MGH 78578 (GenBank
accession no. YP_001336895).
Para estudiar con más detalle esta nueva porina, la secuencia aminoacídica de OmpK26 de K.
pneumoniae se alineó con las secuencias de las porinas específicas de oligogalacturonatos
NanC de E. coli y KdgM de Dickeya dadantii, cuya estructura secundaria ya ha sido predicha
[426, 427], en base a la conservación de las láminas β y algunos de los residuos claves
(subrayados en negro en la figura 4.12) que están altamente conservados en esta nueva
familia de pequeños canales de membrana externa monoméricos. En base a este alineamiento
podemos decir que OmpK26 presenta una típica estructura de láminas β con cinco giros
periplásmicos cortos y seis asas extracelulares de longitud variable. Además también podemos
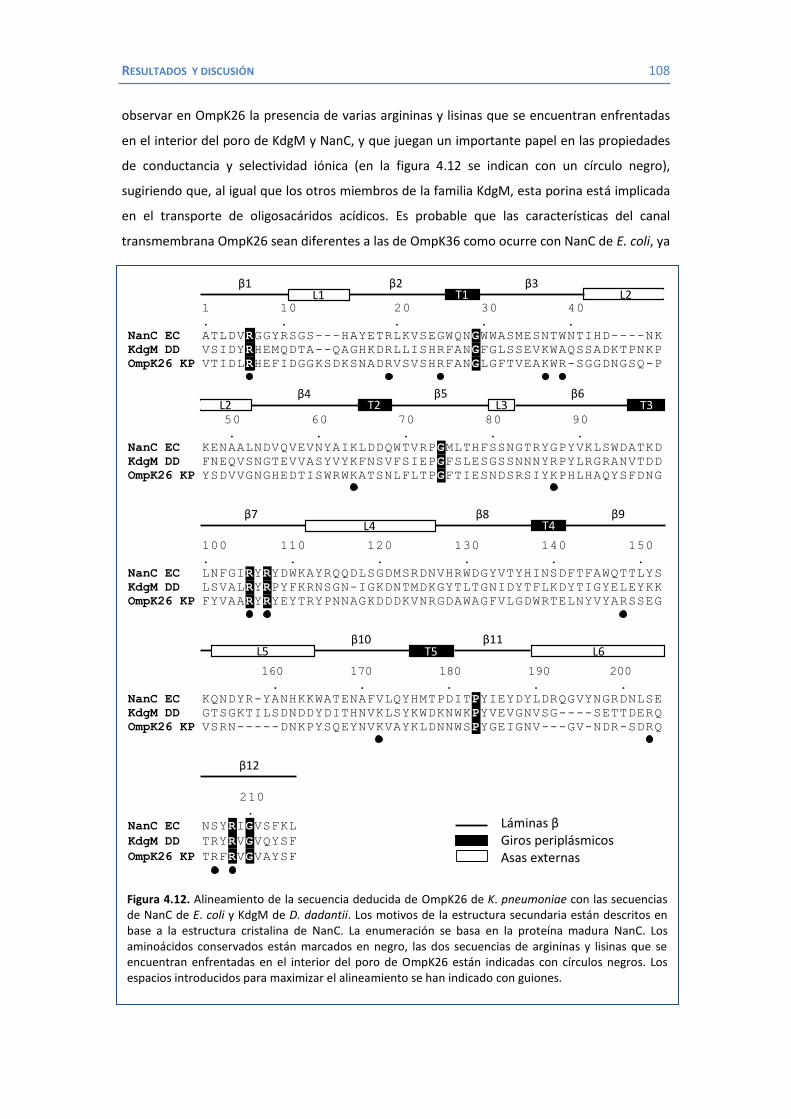
RESULTADOS Y DISCUSIÓN 108
observar en OmpK26 la presencia de varias argininas y lisinas que se encuentran enfrentadas
en el interior del poro de KdgM y NanC, y que juegan un importante papel en las propiedades
de conductancia y selectividad iónica (en la figura 4.12 se indican con un círculo negro),
sugiriendo que, al igual que los otros miembros de la familia KdgM, esta porina está implicada
en el transporte de oligosacáridos acídicos. Es probable que las características del canal
transmembrana OmpK26 sean diferentes a las de OmpK36 como ocurre con NanC de E. coli, ya
Figura 4.12. Alineamiento de la secuencia deducida de OmpK26 de K. pneumoniae con las secuencias de NanC de E. coli y KdgM de D. dadantii. Los motivos de la estructura secundaria están descritos en base a la estructura cristalina de NanC. La enumeración se basa en la proteína madura NanC. Los aminoácidos conservados están marcados en negro, las dos secuencias de argininas y lisinas que se encuentran enfrentadas en el interior del poro de OmpK26 están indicadas con círculos negros. Los espacios introducidos para maximizar el alineamiento se han indicado con guiones.
1 10 20 30 40 . . . . . NanC EC ATLDVRGGYRSGS---HAYETRLKVSEGWQNGWWASMESNTWNTIHD----NK KdgM DD VSIDYRHEMQDTA--QAGHKDRLLISHRFANGFGLSSEVKWAQSSADKTPNKP OmpK26 KP VTIDLRHEFIDGGKSDKSNADRVSVSHRFANGLGFTVEAKWR-SGGDNGSQ-P 50 60 70 80 90 . . . . . NanC EC KENAALNDVQVEVNYAIKLDDQWTVRPGMLTHFSSNGTRYGPYVKLSWDATKD KdgM DD FNEQVSNGTEVVASYVYKFNSVFSIEPGFSLESGSSNNNYRPYLRGRANVTDD OmpK26 KP YSDVVGNGHEDTISWRWKATSNLFLTPGFTIESNDSRSIYKPHLHAQYSFDNG 100 110 120 130 140 150 . . . . . . NanC EC LNFGIRYRYDWKAYRQQDLSGDMSRDNVHRWDGYVTYHINSDFTFAWQTTLYS KdgM DD LSVALRYRPYFKRNSGN-IGKDNTMDKGYTLTGNIDYTFLKDYTIGYELEYKK OmpK26 KP FYVAARYRYEYTRYPNNAGKDDDKVNRGDAWAGFVLGDWRTELNYVYARSSEG 160 170 180 190 200 . . . . . NanC EC KQNDYR-YANHKKWATENAFVLQYHMTPDITPYIEYDYLDRQGVYNGRDNLSE KdgM DD GTSGKTILSDNDDYDITHNVKLSYKWDKNWKPYVEVGNVSG----SETTDERQ OmpK26 KP VSRN-----DNKPYSQEYNVKVAYKLDNNWSPYGEIGNV---GV-NDR-SDRQ 210 . NanC EC NSYRIGVSFKL β s trands KdgM DD TRYRVGVQYSF Perip lasmic turns OmpK26 KP TRFRVGVAYSF External loops
β1 β2 β3 L2 L1 T1
L4 β7 β9 β8
T4
β6 β4 L3 L2 T2
β5 T3
L5 β10 β11
L6 T5
β12
Láminas β Giros periplásmicos Asas externas

RESULTADOS Y DISCUSIÓN 109
que esta última, en la zona de constricción del poro muestra un cuadrupolo electrostático en
lugar de la disposición dipolar descrita en OmpK36 [252, 427]. Esta diferencia estructural entre
OmpK36 y OmpK26 podría conducir a la menor penetración de los carbapenemas y causar la
reducción de su susceptibilidad en las cepas productoras de OmpK26 y deficientes en OmpK36.
El papel de OmpK26 en la resistencia antimicrobiana.
Para investigar el papel de esta porina en la resistencia a antimicrobianos, construimos un
mutante delecionado de la cepa KpCR-1 de K. pneumoniae mediante inserción-duplicación del
gen codificante de OmpK26, yjhA. Para ello se realizó una PCR de un fragmento interno del
gen yjhA utilizando los cebadores YjhA F3 y YjhA R2. Seguidamente, dicho fragmento se clonó
en el plásmido pGEM-T Easy, obteniéndose el plásmido pGEM-T YjhA. Para la obtención del
pFSOmpK26 se realizó la transferencia del fragmento yjhA procedente del pGEM-T YjhA al
pFS100 mediante digestión con EcoRI y posterior ligación con la ADN ligasa T4. Posteriormente
el plásmido resultante fue dotado con la resistencia a telurito procedente del plásmido
pDT1558 [440], obteniendo el plásmido pFSOmpK26-Te. La obtención del mutante isogénico
KpCR-1ΔOmpK26
KpCR-1
KpCS-1
yjhA
> 20 kb
> 20 kb ≈ 3.5 kb
21 kb21 kb
A B CH HH
HH HHH HH
ΔyjhA
36 kDa36 kDa
25 kDa25 kDa
OmpK36OmpK36
OmpK26OmpK26OmpK26OmpK26
KpCR-1ΔOmpK26
KpCR-1
4.2 kb4.2 kb3.5 kb3.5 kb
5.0 kb
Figura 4.13. Representación esquemática de la mutagénesis mediante inserción-duplicación del gen codificante de OmpK26, yjhA, en K. pneumoniae KpCR-1. A. El cromosoma de K. pneumoniae de la cepa KpCR-1 se muestra en la parte superior y el cromosoma de su mutante isogénico delecionado en yjhA está en la parte inferior. El tamaño de los fragmentos de ADN dibujados en el esquema no está a escala. La línea entre los fragmentos negros en el genoma del mutante representa el ADN del plásmido integrado en el cromosoma. Los fragmentos negros indican la sonda usada en el análisis de Southern blot. Los tamaños esperados de los fragmentos obtenidos con la digestión con Hind III que hibridan con la sonda descrita anteriormente esta en kilobases. B. Análisis por Southern blot de los cromosomas de la cepa de K. pneumoniae KpCR-1 y de su mutante isogénico deficiente de OmpK26, KpCR-1ΔOmpK26, digerido con Hind III El marcador de peso molecular (en kilobases) se muestra a la izquierda de la membrana. C. Análisis mediante gel de SDS-PAGE de las PME de la cepa de K. pneumoniae KpCS-1, KpCR-1 y el mutante isogénico deficiente en OmpK26 KpCR-1ΔOmpK26. El marcador de peso molecular está indicado a la izquierda en kDa. Las flechas indican la posición de OmpK36 y OmpK26.
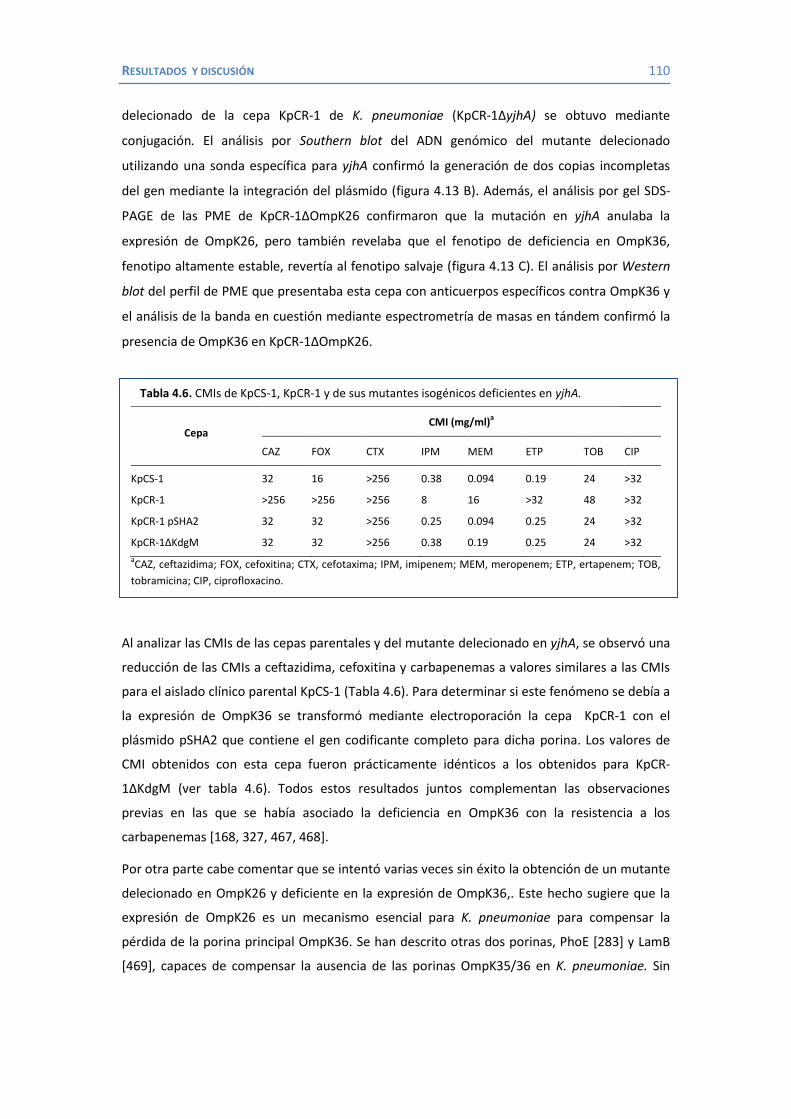
RESULTADOS Y DISCUSIÓN 110
delecionado de la cepa KpCR-1 de K. pneumoniae (KpCR-1ΔyjhA) se obtuvo mediante
conjugación. El análisis por Southern blot del ADN genómico del mutante delecionado
utilizando una sonda específica para yjhA confirmó la generación de dos copias incompletas
del gen mediante la integración del plásmido (figura 4.13 B). Además, el análisis por gel SDS-
PAGE de las PME de KpCR-1ΔOmpK26 confirmaron que la mutación en yjhA anulaba la
expresión de OmpK26, pero también revelaba que el fenotipo de deficiencia en OmpK36,
fenotipo altamente estable, revertía al fenotipo salvaje (figura 4.13 C). El análisis por Western
blot del perfil de PME que presentaba esta cepa con anticuerpos específicos contra OmpK36 y
el análisis de la banda en cuestión mediante espectrometría de masas en tándem confirmó la
presencia de OmpK36 en KpCR-1ΔOmpK26.
aCAZ, ceftazidima; FOX, cefoxitina; CTX, cefotaxima; IPM, imipenem; MEM, meropenem; ETP, ertapenem; TOB, tobramicina; CIP, ciprofloxacino.
Al analizar las CMIs de las cepas parentales y del mutante delecionado en yjhA, se observó una
reducción de las CMIs a ceftazidima, cefoxitina y carbapenemas a valores similares a las CMIs
para el aislado clínico parental KpCS-1 (Tabla 4.6). Para determinar si este fenómeno se debía a
la expresión de OmpK36 se transformó mediante electroporación la cepa KpCR-1 con el
plásmido pSHA2 que contiene el gen codificante completo para dicha porina. Los valores de
CMI obtenidos con esta cepa fueron prácticamente idénticos a los obtenidos para KpCR-
1ΔKdgM (ver tabla 4.6). Todos estos resultados juntos complementan las observaciones
previas en las que se había asociado la deficiencia en OmpK36 con la resistencia a los
carbapenemas [168, 327, 467, 468].
Por otra parte cabe comentar que se intentó varias veces sin éxito la obtención de un mutante
delecionado en OmpK26 y deficiente en la expresión de OmpK36,. Este hecho sugiere que la
expresión de OmpK26 es un mecanismo esencial para K. pneumoniae para compensar la
pérdida de la porina principal OmpK36. Se han descrito otras dos porinas, PhoE [283] y LamB
[469], capaces de compensar la ausencia de las porinas OmpK35/36 en K. pneumoniae. Sin
Cepa CMI (mg/ml)a
CAZ FOX CTX IPM MEM ETP TOB CIP
KpCS-1 32 16 >256 0.38 0.094 0.19 24 >32
KpCR-1 >256 >256 >256 8 16 >32 48 >32
KpCR-1 pSHA2 32 32 >256 0.25 0.094 0.25 24 >32
KpCR-1ΔKdgM 32 32 >256 0.38 0.19 0.25 24 >32
Tabla 4.6. CMIs de KpCS-1, KpCR-1 y de sus mutantes isogénicos deficientes en yjhA.
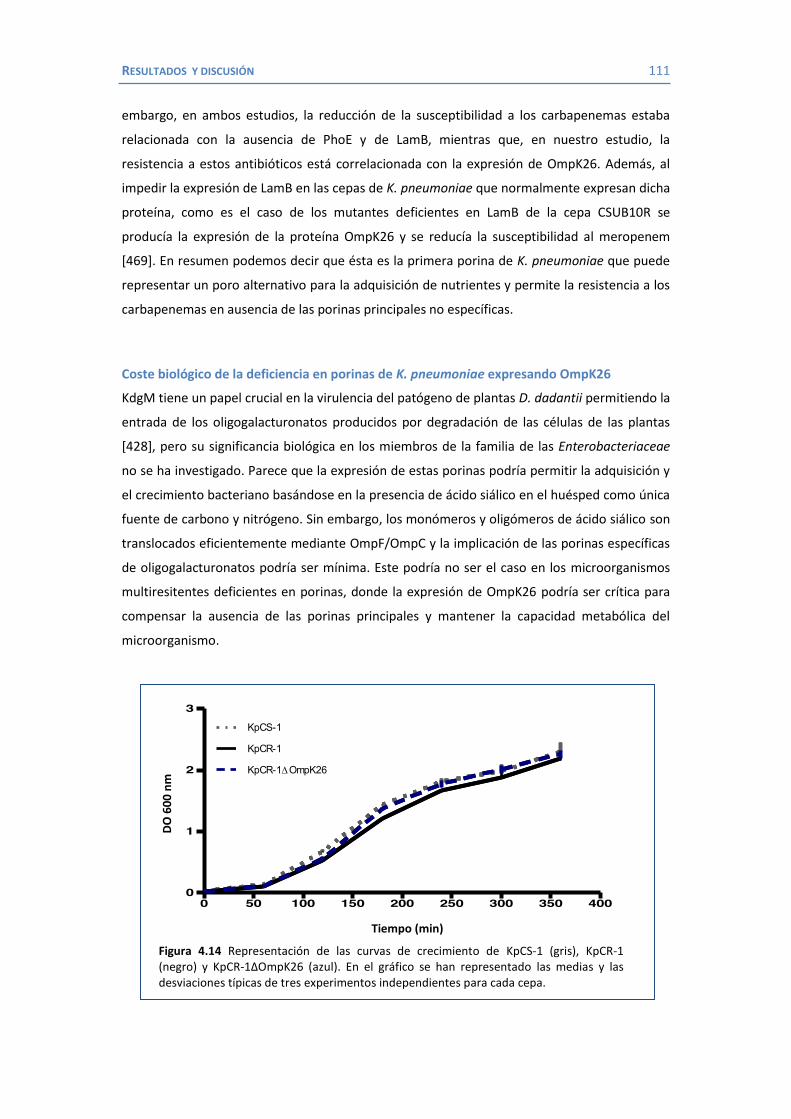
RESULTADOS Y DISCUSIÓN 111
0 50 100 150 200 250 300 350 4000
1
2
3KpCS-1
KpCR-1
KpCR-1OmpK26
Tiempo (min)
DO 60
0nm
embargo, en ambos estudios, la reducción de la susceptibilidad a los carbapenemas estaba
relacionada con la ausencia de PhoE y de LamB, mientras que, en nuestro estudio, la
resistencia a estos antibióticos está correlacionada con la expresión de OmpK26. Además, al
impedir la expresión de LamB en las cepas de K. pneumoniae que normalmente expresan dicha
proteína, como es el caso de los mutantes deficientes en LamB de la cepa CSUB10R se
producía la expresión de la proteína OmpK26 y se reducía la susceptibilidad al meropenem
[469]. En resumen podemos decir que ésta es la primera porina de K. pneumoniae que puede
representar un poro alternativo para la adquisición de nutrientes y permite la resistencia a los
carbapenemas en ausencia de las porinas principales no específicas.
Coste biológico de la deficiencia en porinas de K. pneumoniae expresando OmpK26
KdgM tiene un papel crucial en la virulencia del patógeno de plantas D. dadantii permitiendo la
entrada de los oligogalacturonatos producidos por degradación de las células de las plantas
[428], pero su significancia biológica en los miembros de la familia de las Enterobacteriaceae
no se ha investigado. Parece que la expresión de estas porinas podría permitir la adquisición y
el crecimiento bacteriano basándose en la presencia de ácido siálico en el huésped como única
fuente de carbono y nitrógeno. Sin embargo, los monómeros y oligómeros de ácido siálico son
translocados eficientemente mediante OmpF/OmpC y la implicación de las porinas específicas
de oligogalacturonatos podría ser mínima. Este podría no ser el caso en los microorganismos
multiresitentes deficientes en porinas, donde la expresión de OmpK26 podría ser crítica para
compensar la ausencia de las porinas principales y mantener la capacidad metabólica del
microorganismo.
0 50 100 150 200 250 300 350 4000
1
2
3KpCS-1
KpCR-1
KpCR-1OmpK26
Tiempo (min)
DO
600
nm
Figura 4.14 Representación de las curvas de crecimiento de KpCS-1 (gris), KpCR-1 (negro) y KpCR-1ΔOmpK26 (azul). En el gráfico se han representado las medias y las desviaciones típicas de tres experimentos independientes para cada cepa.
Tiempo (min)
DO 6
00 n
m

RESULTADOS Y DISCUSIÓN 112
Así pues, para investigar el coste biológico de la ausencia de las principales porinas en cepas de
K. pneumoniae que expresaban OmpK26, llevamos a cabo una serie de experimentos de
competición in vitro e in vivo.
En primer lugar y como se puede observar en la figura 4.14, donde se representan las curvas
de crecimiento de KpCS-1, KpCR-1 y KpCR-1ΔKdgM, todas las cepas usadas en los
experimentos de competición mostraron curvas de crecimiento similares con diferencias no
significativas y tasas de duplicación comprendidas entre 33,3 min y 36,1 min.
En la figura 4.15 A se representan los resultados para los experimentos de competición in vitro.
Dichos resultados mostraron que, en el caso de la competición entre KpCR-1/KpCS-1, la cepa
KpCR-1 presentaba marcada menor capacidad de adaptación que la cepa KpCS-1 (mediana del
IC 0,035; P = 0,0036). De manera similar, el mutante KpCR-1ΔOmpK26 que expresa OmpK36
presentaba menor capacidad de adaptación que KpCR-1 (mediana del IC para la competición
KpCR-1/KpCR-1ΔOmpK26 = 0,101; P = 0,0023). En el tercer caso, la competición entre KpCR-
1ΔOmpK26/KpCS-1, el IC se aproximaba más a uno, lo que indicaba una recuperación de la
capacidad de competición de KpCR-1ΔOmpK26 frente a KpCS-1 (mediana del IC para la
competición KpCR-1ΔOmpK26/KpCS-1 = 0,679; P < 0,001). Sobre todo, los resultados de los
Figura 4.15. A. Resultados de los experimentos de competición in vitro. Las competiciones in vitro se realizaron en tubos con medio LB en los cuales las bacterias se crecieron a 37°C y 180 rpm durante 9 h como se describe en el apartado de materiales y métodos. Los valores de IC obtenidos se determinaron para cada experimento de manera independiente. La mediana del IC se muestra entre paréntesis. B. Resultados de los experimentos de competición in vivo. Los experimentos de competición in vivo se realizaron un modelo de infección sistémica de ratón donde se cuantificaron las bacterias aisladas de bazo a las 24 h después de la infección. Se determinaron, al menos, los valores de IC de 8 experimentos independientes. La mediana de los valores de IC se muestra ente paréntesis.
(0.004) (0.008)
(0.700)
(0.004) (0.008)
(0.700)
Índi
ce d
e co
mpe
tició
n (IC
)
(0.035)(0.101)
(0.679)
(0.035)(0.101)
(0.679)
Índi
ce d
e co
mpe
tició
n (IC
) A B
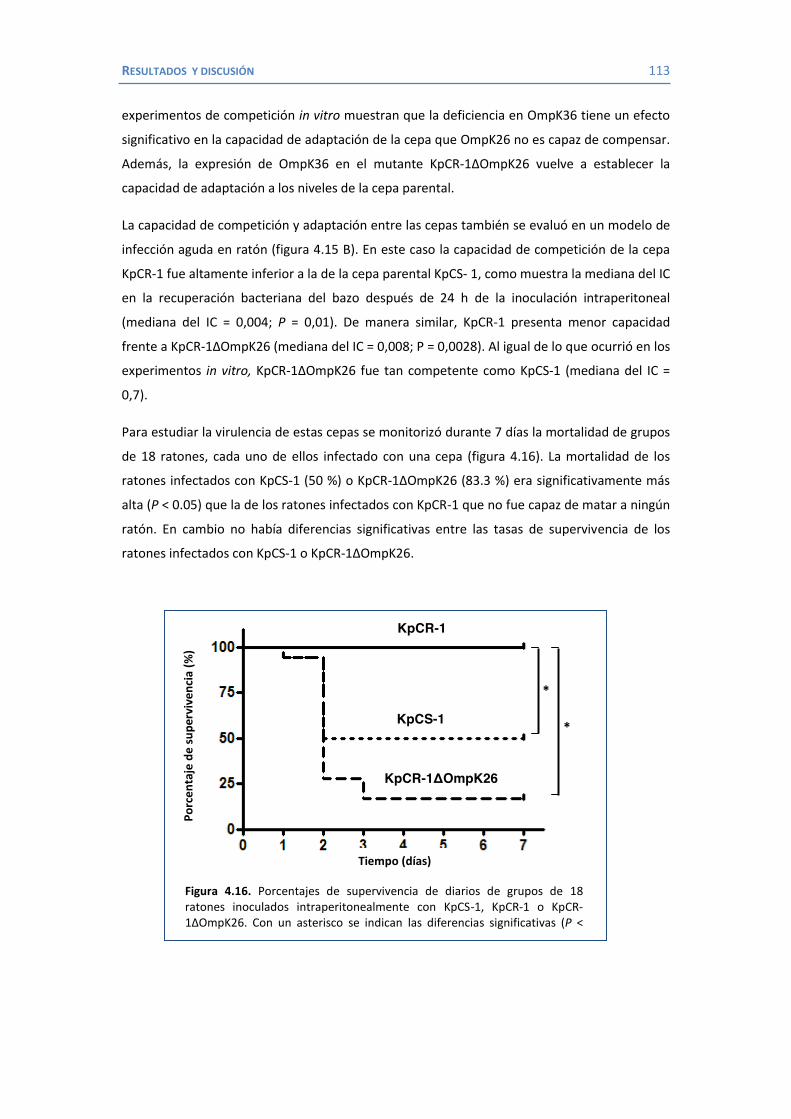
RESULTADOS Y DISCUSIÓN 113
experimentos de competición in vitro muestran que la deficiencia en OmpK36 tiene un efecto
significativo en la capacidad de adaptación de la cepa que OmpK26 no es capaz de compensar.
Además, la expresión de OmpK36 en el mutante KpCR-1ΔOmpK26 vuelve a establecer la
capacidad de adaptación a los niveles de la cepa parental.
La capacidad de competición y adaptación entre las cepas también se evaluó en un modelo de
infección aguda en ratón (figura 4.15 B). En este caso la capacidad de competición de la cepa
KpCR-1 fue altamente inferior a la de la cepa parental KpCS- 1, como muestra la mediana del IC
en la recuperación bacteriana del bazo después de 24 h de la inoculación intraperitoneal
(mediana del IC = 0,004; P = 0,01). De manera similar, KpCR-1 presenta menor capacidad
frente a KpCR-1ΔOmpK26 (mediana del IC = 0,008; P = 0,0028). Al igual de lo que ocurrió en los
experimentos in vitro, KpCR-1ΔOmpK26 fue tan competente como KpCS-1 (mediana del IC =
0,7).
Para estudiar la virulencia de estas cepas se monitorizó durante 7 días la mortalidad de grupos
de 18 ratones, cada uno de ellos infectado con una cepa (figura 4.16). La mortalidad de los
ratones infectados con KpCS-1 (50 %) o KpCR-1ΔOmpK26 (83.3 %) era significativamente más
alta (P < 0.05) que la de los ratones infectados con KpCR-1 que no fue capaz de matar a ningún
ratón. En cambio no había diferencias significativas entre las tasas de supervivencia de los
ratones infectados con KpCS-1 o KpCR-1ΔOmpK26.
Figura 4.16. Porcentajes de supervivencia de diarios de grupos de 18 ratones inoculados intraperitonealmente con KpCS-1, KpCR-1 o KpCR-1ΔOmpK26. Con un asterisco se indican las diferencias significativas (P < 0.05).
KpCS-1
KpCR-1ΔOmpK26
KpCR-1
Tiempo (días)
Porc
enta
je d
e su
perv
iven
cia
(%)
*
*

RESULTADOS Y DISCUSIÓN 114
Estos resultados indican que la deficiencia en OmpK35/36 reduce la virulencia de K.
pneumoniae en un modelo de infección sistémica en ratón, y son consistentes con los
resultados aportados recientemente por Bialek y colaboradores [470] y por Tsai y
colaboradores [323], en los que usando un modelo de Caenorhabditis elegans o un modelo de
ratón, respectivamente, muestran que la pérdida de la expresión de las porinas está asociado
con una disminución de la virulencia en K. pneumoniae. Sin embargo, los mecanismos
subyacentes a la atenuación de la virulencia causada por la ausencia de la expresión de las
porinas principales aún no se comprenden ni se conoce suficientemente. Los resultados de
nuestros experimentos in vitro sugieren que, además de la contribución potencial de las
porinas principales en la defensa bacteriana, contra el sistema inmune del huésped [323], su
expresión y función como transportadores de membrana es indispensable para mantener la
capacidad de supervivencia, adaptación y competición del microorganismo, esto puede ayudar
a entender lo que sucede con el fenotipo atenuado in vivo. Por tanto, aunque la bacteria con la
expresión de OmpK35/36 alterada representa una amenaza porque muestra niveles de
resistencia a β-lactámicos de amplio espectro más altos, en ausencia de la presión selectiva del
antibiótico, son menos competitivas que las cepas salvajes.
En resumen, hemos demostrado que la expresión de la porina específica de oligogalacturonato
OmpK26 compensa la ausencia de expresión de OmpK35/36 en la resistencia a carbapenemas
en K. pneumoniae pero no puede restablecer la capacidad de supervivencia ni de competición
del microorganismo ni in vitro ni in vivo.

5. COROLARIO


COROLARIO 117
El uso abusivo e inadecuado de los antibióticos es, probablemente, la principal causa del
incremento de la resistencia bacteriana a los agentes antimicrobianos, uno de los mayores
problemas actuales de salud pública. En este sentido, el incremento de la aparición de cepas
multiresistentes, como pueden ser algunos aislados nosocomiales de K. pneumoniae, limita
notablemente las opciones terapéuticas, aumentado las tasas de mortalidad y morbilidad de
los pacientes infectados.
Al utilizar un antibiótico para hacer frente a una infección bacteriana, lo importante es la
cantidad de principio activo que circula en sangre y llega al foco de la infección [471]. La
cantidad de antibiótico administrada se calcula en función de todos los procesos que
experimenta este antimicrobiano hasta llegar al foco de infección, así como de los procesos de
eliminación de dicho fármaco a través de los riñones o el hígado. Así pues, la concentración de
fármaco presente en sangre se mantiene entre el nivel de concentración mínima eficaz y el de
concentración tóxica. El mal uso de los antibióticos, como por ejemplo el no respetar la
frecuencia de administración o la dosis que se debe ingerir, hace que la concentración de
fármaco en sangre no sea la adecuada, y pueda estar por debajo de los niveles de eficacia. En
estas condiciones, las bacterias son capaces de sobrevivir y eventualmente se pueden
seleccionar mutantes o variantes de la cepa original que son capaces de resistir el efecto
bactericida de los antibióticos a concentraciones superiores de las que lo hacia la cepa original.
Nuestros resultados demuestran que la cefoxitina, el ertapenem y el meropenem son capaces
de seleccionar mutantes deficientes en la expresión de OmpK36. Dada la estrategia utilizada
en nuestro trabajo para obtener los mutantes deficientes en OmpK36, lo más probable es que
los resultados obtenidos se deben solo al efecto selectivo de estos antibióticos y no a un efecto
sobre la tasa de mutación por inducción de la vía de respuesta SOS y afectación de los sistemas
de recombinación intracromosómica de secuencias de ADN homólogo como se ha descrito que
tienen otros antimicrobianos como las fluoroquinolonas en E. coli [472].
Desde el punto de vista práctico, lo más preocupante es que concentraciones muy bajas de
ertapenem y meropenem son capaces de seleccionar mutantes deficientes en OmpK36
limitando las opciones terapéuticas que eviten la selección de cepas multiresistentes, siendo
en este caso, el imipenem, el antibiótico más recomendable para evitar esta selección. En
efecto, nuestros resultados sugieren que en ausencia de las porinas principales, el imipenem
penetra mejor que el resto de los antibióticos estudiados. Dado que las características
químicas de los tres carbapenémicos estudiados son muy similares, no se puede descartar que

COROLARIO 118
las diferencias observadas en nuestro trabajo respecto a la capacidad para seleccionar
mutantes deficientes en OmpK36 sean solo debidas a la capacidad de penetración de cada uno
de los carbapenémicos sino también a la eficiencia de hidrólisis de éstos por parte de la BLEA
presente en la cepa. En este sentido, a pesar de que las BLEAs de tipo CTX-M no tienen
actividad carbapenemasa, se han descrito aislados clínicos productores de CTX-M que al no
expresar las porinas principales OmpK36 y OmpK35 presentan un fenotipo clínico resistente al
meropenem e intermedio al imipenem [168].
La función natural de las porinas OmpK35 y OmpK36 de K. pneumoniae es permitir el paso de
solutos y nutrientes hacia el interior de la bacteria. Es razonable pensar que la deficiencia en
estas porinas como resultado de la presión selectiva de los antibióticos conlleve la expresión
de otras proteínas que actúen como canales para la entrada de nutrientes esenciales que
permitan mantener la actividad metabólica bacteriana.
En este trabajo hemos identificado a las proteínas LamB y OmpK26 como porinas que pueden
compensar la falta de las principales porinas en las cepas resistentes a antibióticos β-
lactámicos. Estos resultados no descartan la posibilidad de que existan otras proteínas de
membrana externa que puedan realizar la misma función. De hecho, el análisis de las proteínas
de membrana externa de otros aislados clínicos de K. pneumoniae deficientes en las porinas
OmpK35 y OmpK36 de las que disponemos en el laboratorio o descritos en la literatura solo ha
detectado la presencia de OmpK26 en algunos. Sería necesario disponer de un amplio número
de aislados deficientes en las porinas principales y un estudio detallado de las proteínas de
membrana externa para identificar nuevas porinas alternativas a las principales.
El análisis pormenorizado de la secuencia del genoma completo de K. pneumoniae permite
realizar una primera aproximación en la identificación de nuevas porinas alternativas a
OmpK35 y OmpK36. En este sentido, hemos encontrado un total de 54 genes que codifican
para proteínas que potencialmente podrían formar parte de la membrana externa de K.
pneumoniae. De ellas, 23 serían porinas o proteínas formadoras de canales, 7 formarían parte
de sistemas de expulsión y 9 proteínas son receptores o transportadores específicos con
función ya descrita. Las 15 proteínas restantes que hipotéticamente se localizarían en la
membrana externa, no tienen una función conocida. Si nos centramos en el grupo de los genes
que codifican proteínas formadoras de poro, 13 codifican porinas ya conocidas, entre las que
incluimos OmpK35, OmpK36, OmpA, LamB, OmpK26, PhoE y OmpK37, pero aún así existen 10
genes que codifican para porinas totalmente desconocidas y cuya función, estructura y

COROLARIO 119
regulación está sin estudiar. Esta observación denota el enorme potencial y versatilidad del
genoma K. pneumoniae capaz de dar respuesta a situaciones ambientales hostiles muy
diversas como la presencia de agentes antimicrobianos y el crecimiento en el cuerpo humano.
A pesar de elevado número de porinas que K. pneumoniae es capaz de producir, la mayor
parte de aislamientos clínicos y ambientales expresan OmpK35 u OmpK36, o ambas a la vez
[317] lo que denota la importancia de dichas porinas para la viabilidad bacteriana. Este estudio
junto con otros [323, 470] indica que su pérdida supone una disminución del fitness de K.
pneumoniae tanto in vitro como in vivo. Estos resultados aportan una prueba más de que, al
igual que pasa con otros patógenos oportunistas como P. aeruginosa [454], S. enterica [473] y
E. coli [474], la adquisición del fenotipo multiresistente comporta la reducción de la capacidad
de la bacteria por crecer en el huésped, es decir, reduce la virulencia. Esta observación permite
sugerir que la aparición de cepas de K. pneumoniae multiresistentes puede estar limitada a los
pacientes que presenta el sistema inmune más comprometido, por lo que la investigación de
los mecanismos que subyacen en la pérdida del fitness de las bacterias multiresistentes en el
huésped permitiría establecer nuevas estrategias preventivas dirigidas hacia la potenciación de
aquellos componentes del sistema inmune claves para evitar la aparición de cepas
multiresistentes.


6. CONCLUSIONES


CONCLUSIONES 123
1. En nuestro estudio, la cefoxitina es el antibiótico que con mayor frecuencia selecciona mutantes
deficientes en OmpK36 seguido por el ertapenem y finalmente el meropenem. El imipenem es el
mejor antibiótico de los ensayados para evitar la selección de mutantes deficientes en OmpK36.
2. Los principales mecanismos genéticos responsables de la pérdida de expresión de OmpK36,
independientemente del antibiótico utilizado para la selección, son las inserciones y las deleciones,
y en último lugar las mutaciones puntuales. Así mismo, la variedad en el tipo y localización de las
mutaciones que causan la interrupción del gen ompk36 muestran que estas interrupciones son el
resultado de acontecimientos distintos en cada mutante y que no existen regiones dentro del gen
en las que sea más frecuente la mutación.
3. La pérdida de porinas principales OmpK35 y OmpK36, en cepas de K. pneumoniae resistentes a los
antimicrobianos como CSUB10R, puede compensarse por la sobreexpresión de LamB.
4. La pérdida de LamB en cepas de K. pneumoniae desprovistas de las porinas principales OmpK35 y
OmpK36 disminuye la susceptibilidad a los carbapenémicos.
5. El fenotipo de resistencia en los aislados clínicos resistentes a carbapenémicos, KpCR-1 y Kpn-17, se
correlaciona con la ausencia de la porina OmpK36 y la presencia de una nueva proteína de
membrana externa de 26 kDa denominada OmpK26.
6. OmpK26 pertenece a la familia de porinas KdgM específicas para oligogalacturonatos y presenta
una estructura secundaria de barril de láminas β con 5 giros periplasmáticos cortos y 6 asas
extracelulares de longitud variable. Las diferencias en la disposición de los residuos básicos y ácidos
en el interior del poro entre OmpK26 y OmpK36 podrían explicar la menor penetración de los
carbapenémicos a través del poro de OmpK26.
7. La ausencia de las porinas principales, OmpK35 y OmpK36, reduce la capacidad de adaptación o
fitness de las cepas tanto in vitro como in vivo.
8. OmpK26 puede representar una porina alternativa para la entrada de nutrientes que compensa la
deficiencia de OmpK35/36 en los principales aislados de K. pneumoniae deficientes en porinas y
resistentes a carbapenémicos, pero no es capaz de restablecer el coste biológico de la pérdida de
las porinas principales ni in vitro ni in vivo.


7. Referencias


REFERENCIAS 127
1. Bagley, S.T., Habitat association of Klebsiella species. Infect Control, 1985. 6(2): p. 52-8. 2. Podschun, R. and U. Ullmann, Klebsiella spp. as nosocomial pathogens: epidemiology, taxonomy, typing
methods, and pathogenicity factors. Clin Microbiol Rev, 1998. 11(4): p. 589-603. 3. Drancourt, M., et al., Phylogenetic analyses of Klebsiella species delineate Klebsiella and Raoultella gen.
nov., with description of Raoultella ornithinolytica comb. nov., Raoultella terrigena comb. nov. and Raoultella planticola comb. nov. Int J Syst Evol Microbiol, 2001. 51(Pt 3): p. 925-32.
4. Brisse, S. and J. Verhoef, Phylogenetic diversity of Klebsiella pneumoniae and Klebsiella oxytoca clinical isolates revealed by randomly amplified polymorphic DNA, gyrA and parC genes sequencing and automated ribotyping. Int J Syst Evol Microbiol, 2001. 51(Pt 3): p. 915-24.
5. Carter, J.S., et al., Phylogenetic evidence for reclassification of Calymmatobacterium granulomatis as Klebsiella granulomatis comb. nov. Int J Syst Bacteriol, 1999. 49 Pt 4: p. 1695-700.
6. Kharsany, A.B., et al., Phylogenetic analysis of Calymmatobacterium granulomatis based on 16S rRNA gene sequences. J Med Microbiol, 1999. 48(9): p. 841-7.
7. Podschun, R. and U. Ullmann, Isolation of Klebsiella terrigena from clinical specimens. Eur J Clin Microbiol Infect Dis, 1992. 11(4): p. 349-52.
8. Podschun, R., et al., Isolation of Klebsiella planticola from newborns in a neonatal ward. J Clin Microbiol, 1998. 36(8): p. 2331-2.
9. Westbrook, G.L., et al., Incidence and identification of Klebsiella planticola in clinical isolates with emphasis on newborns. J Clin Microbiol, 2000. 38(4): p. 1495-7.
10. Sahly, H., R. Podschun, and U. Ullmann, Klebsiella infections in the immunocompromised host. Adv Exp Med Biol, 2000. 479: p. 237-49.
11. Bagley, S.T., R.J. Seidler, and D.J. Brenner, Klebsiella planticola sp. nov.: a new species of Enterobacteriaceae found primarily in nonclinical environments. Curr Microbiol, 1981. 6(105-109).
12. Farmer, J.J., 3rd, et al., Biochemical identification of new species and biogroups of Enterobacteriaceae isolated from clinical specimens. J Clin Microbiol, 1985. 21(1): p. 46-76.
13. Izard, D., et al., Klebsiella terrigena, a new species from soil and water. Int. J. Antimicrob. Agents, 1981. 31: p. 116-127.
14. Monnet, D. and J. Freney, Method for differentiating Klebsiella planticola and Klebsiella terrigena from other Klebsiella species. J Clin Microbiol, 1994. 32(4): p. 1121-2.
15. Monnet, D., et al., Difficulties in identifying Klebsiella strains of clinical origin. Zentralbl Bakteriol, 1991. 274(4): p. 456-64.
16. Orskov, I., Genus Klebsiella. Bergey's manual of systematic bacteriology. N. R. Krieg and J. G. Holt. Vol. 1. 1984, Baltimore, Md: The Williams & Wilkins Co. 461-465.
17. Sakazaki, R., et al., Klebsiella ornithinolytica sp. nov., formerly known as ornithine-positive Klebsiella oxytoca. . Curr Microbiol, 1989. 18: p. 201-206.
18. Stenzel, W., H. Burger, and W. Mannheim, [Classification and differential diagnosis of the Klebsiella group with special reference to the so-called oxytocum types]. Zentralbl Bakteriol Orig A, 1972. 219(2): p. 193-203.
19. Bartlett, J.G., et al., Bacteriology of hospital-acquired pneumonia. Arch Intern Med, 1986. 146(5): p. 868-71.
20. Garcia de la Torre, M., et al., Klebsiella bacteremia: an analysis of 100 episodes. Rev Infect Dis, 1985. 7(2): p. 143-50.
21. Jarvis, W.R., et al., The epidemiology of nosocomial infections caused by Klebsiella pneumoniae. Infect Control, 1985. 6(2): p. 68-74.
22. Bouza, E. and E. Cercenado, Klebsiella and enterobacter: antibiotic resistance and treatment implications. Semin Respir Infect, 2002. 17(3): p. 215-30.
23. Lye, W.C., et al., Urinary tract infections in patients with diabetes mellitus. J Infect, 1992. 24(2): p. 169-74. 24. Canton, R., et al., Quality control for beta-lactam susceptibility testing with a well-defined collection of
Enterobacteriaceae and Pseudomonas aeruginosa strains in Spain. J Clin Microbiol, 2003. 41(5): p. 1912-8. 25. Duggan, J.M., G.S. Oldfield, and H.K. Ghosh, Septicaemia as a hospital hazard. J Hosp Infect, 1985. 6(4): p.
406-12. 26. Pittet, D., N. Li, and R.P. Wenzel, Association of secondary and polymicrobial nosocomial bloodstream
infections with higher mortality. Eur J Clin Microbiol Infect Dis, 1993. 12(11): p. 813-9. 27. Yinnon, A.M., et al., Klebsiella bacteraemia: community versus nosocomial infection. Qjm, 1996. 89(12): p.
933-41.

REFERENCIAS 128
28. Montgomerie, J.Z., Epidemiology of Klebsiella and hospital-associated infections. Rev Infect Dis, 1979. 1(5): p. 736-53.
29. Gotoff, S.P., Sepsis in the newborn., ed. S.L.K. S. Krugman, A. A. Gershon, and C. M. Wilfert. Vol. Infection diseases of children 9th ed. Mosby-Year Book. 1992, St. Louis, Mo. 402-418.
30. Hervas, J.A., et al., Neonatal sepsis and meningitis in Mallorca, Spain, 1977-1991. Clin Infect Dis, 1993. 16(5): p. 719-24.
31. Meyer, K.S., et al., Nosocomial outbreak of Klebsiella infection resistant to late-generation cephalosporins. Ann Intern Med, 1993. 119(5): p. 353-8.
32. Rice, L.B., et al., Outbreak of ceftazidime resistance caused by extended-spectrum beta-lactamases at a Massachusetts chronic-care facility. Antimicrob Agents Chemother, 1990. 34(11): p. 2193-9.
33. Bergogne-Berezin, E., Nosocomial infections: new agents, incidence, prevention. Presse Med, 1995. 24(2): p. 89-97.
34. Horan, T., et al., Pathogens causing nosocomial infections. Antimicrobic Newsl, 1988. 5: p. 65-67. 35. Ullmann, U., [Bacterial infection agents in hospitalized patients]. Zentralbl Bakteriol Mikrobiol Hyg B,
1986. 183(2-3): p. 103-13. 36. Bryan, C.S., K.L. Reynolds, and E.R. Brenner, Analysis of 1,186 episodes of gram-negative bacteremia in
non-university hospitals: the effects of antimicrobial therapy. Rev Infect Dis, 1983. 5(4): p. 629-38. 37. Ohlsson, A., T. Bailey, and F. Takieddine, Changing etiology and outcome of neonatal septicemia in Riyadh,
Saudi Arabia. Acta Paediatr Scand, 1986. 75(4): p. 540-4. 38. Tessin, I., B. Trollfors, and K. Thiringer, Incidence and etiology of neonatal septicaemia and meningitis in
western Sweden 1975-1986. Acta Paediatr Scand, 1990. 79(11): p. 1023-30. 39. Tullus, K., et al., Epidemiology of fecal strains of the family Enterobacteriaceae in 22 neonatal wards and
influence of antibiotic policy. J Clin Microbiol, 1988. 26(6): p. 1166-70. 40. Vesikari, T., et al., Neonatal septicaemia in Finland 1981-85. Predominance of group B streptococcal
infections with very early onset. Acta Paediatr Scand, 1989. 78(1): p. 44-50. 41. Struve, C., C. Forestier, and K.A. Krogfelt, Application of a novel multi-screening signature-tagged
mutagenesis assay for identification of Klebsiella pneumoniae genes essential in colonization and infection. Microbiology, 2003. 149(Pt 1): p. 167-76.
42. Orskov, I. and F. Orskov, Serotyping of Klebsiella. Methods Microbiol. , 1984. 14 p. 143-164. 43. Alvarez, D., et al., Capsular polysaccharide is a major complement resistance factor in lipopolysaccharide O
side chain-deficient Klebsiella pneumoniae clinical isolates. Infect Immun, 2000. 68(2): p. 953-5. 44. Baer, H. and L. Ehrenworth, The pathogenicity of Klebsiella pneumoniae for mice: the relationship to the
quantity and rate of production of type-specific capsular polysaccharide. J Bacteriol, 1956. 72(5): p. 713-7. 45. Cortes, G., et al., Molecular analysis of the contribution of the capsular polysaccharide and the
lipopolysaccharide O side chain to the virulence of Klebsiella pneumoniae in a murine model of pneumonia. Infect Immun, 2002. 70(5): p. 2583-90.
46. Cryz, S.J., Jr., F. Furer, and R. Germanier, Experimental Klebsiella pneumoniae burn wound sepsis: role of capsular polysaccharide. Infect Immun, 1984. 43(1): p. 440-1.
47. Domenico, P., W.G. Johanson, Jr., and D.C. Straus, Lobar pneumonia in rats produced by clinical isolates of Klebsiella pneumoniae. Infect Immun, 1982. 37(1): p. 327-35.
48. Highsmith, A.K. and W.R. Jarvis, Klebsiella pneumoniae: selected virulence factors that contribute to pathogenicity. Infect Control, 1985. 6(2): p. 75-7.
49. Keynan, Y. and E. Rubinstein, The changing face of Klebsiella pneumoniae infections in the community. Int J Antimicrob Agents, 2007. 30(5): p. 385-9.
50. Amako, K., Y. Meno, and A. Takade, Fine structures of the capsules of Klebsiella pneumoniae and Escherichia coli K1. J Bacteriol, 1988. 170(10): p. 4960-2.
51. Cortes, G., et al., Role of lung epithelial cells in defense against Klebsiella pneumoniae pneumonia. Infect Immun, 2002. 70(3): p. 1075-80.
52. Podschun, R., I. Penner, and U. Ullmann, Interaction of Klebsiella capsule type 7 with human polymorphonuclear leucocytes. Microb Pathog, 1992. 13(5): p. 371-9.
53. Podschun, R. and U. Ullmann, Klebsiella capsular type K7 in relation to toxicity, susceptibility to phagocytosis and resistance to serum. J Med Microbiol, 1992. 36(4): p. 250-4.
54. Simoons-Smit, A.M., et al., Chemiluminescence of human leukocytes stimulated by clinical isolates of Klebsiella. J Med Microbiol, 1985. 19(3): p. 333-8.
55. Simoons-Smit, A.M., A.M. Verweij-van Vught, and D.M. MacLaren, The role of K antigens as virulence factors in Klebsiella. J Med Microbiol, 1986. 21(2): p. 133-7.

REFERENCIAS 129
56. Yokochi, T., I. Nakashima, and N. Kato, Effect of capsular polysaccharide of Klebsiella pneumoniae on the differentiation and functional capacity of macrophages cultured in vitro. Microbiol Immunol, 1977. 21(10): p. 601-10.
57. Yokochi, T., I. Nakashima, and N. Kato, Further studies on generation of macrophages in in vitro cultures of mouse spleen cells and its inhibition by the capsular polysaccharide of Klebsiella pneumoniae. Microbiol Immunol, 1979. 23(6): p. 487-99.
58. Williams, P., et al., The role of the O and K antigens in determining the resistance of Klebsiella aerogenes to serum killing and phagocytosis. J Gen Microbiol, 1983. 129(7): p. 2181-91.
59. Schembri, M.A., et al., Capsule and fimbria interaction in Klebsiella pneumoniae. Infect Immun, 2005. 73(8): p. 4626-33.
60. Yeh, K.M., et al., Capsular serotype K1 or K2, rather than magA and rmpA, is a major virulence determinant for Klebsiella pneumoniae liver abscess in Singapore and Taiwan. J Clin Microbiol, 2007. 45(2): p. 466-71.
61. Edmondson, A.S., et al., A comparison of the properties of Klebsiella strains isolated from different sources. J Med Microbiol, 1980. 13(4): p. 541-50.
62. Podschun, R., Phenotypic properties of Klebsiella pneumoniae and K. oxytoca isolated from different sources. Zentralbl Hyg Umweltmed, 1990. 189(6): p. 527-35.
63. Kabha, K., et al., SP-A enhances phagocytosis of Klebsiella by interaction with capsular polysaccharides and alveolar macrophages. Am J Physiol, 1997. 272(2 Pt 1): p. L344-52.
64. Ofek, I., et al., Genetic exchange of determinants for capsular polysaccharide biosynthesis between Klebsiella pneumoniae strains expressing serotypes K2 and K21a. Infect Immun, 1993. 61(10): p. 4208-16.
65. Athamna, A., et al., Lectinophagocytosis of encapsulated Klebsiella pneumoniae mediated by surface lectins of guinea pig alveolar macrophages and human monocyte-derived macrophages. Infect Immun, 1991. 59(5): p. 1673-82.
66. Kabha, K., et al., Relationships among capsular structure, phagocytosis, and mouse virulence in Klebsiella pneumoniae. Infect Immun, 1995. 63(3): p. 847-52.
67. Favre-Bonte, S., B. Joly, and C. Forestier, Consequences of reduction of Klebsiella pneumoniae capsule expression on interactions of this bacterium with epithelial cells. Infect Immun, 1999. 67(2): p. 554-61.
68. Kos, V. and C. Whitfield, A membrane-located glycosyltransferase complex required for biosynthesis of the D-galactan I lipopolysaccharide O antigen in Klebsiella pneumoniae. J Biol Chem. 285(25): p. 19668-87.
69. Hansen, D.S., et al., Klebsiella pneumoniae lipopolysaccharide O typing: revision of prototype strains and O-group distribution among clinical isolates from different sources and countries. J Clin Microbiol, 1999. 37(1): p. 56-62.
70. Vinogradov, E., et al., Structures of lipopolysaccharides from Klebsiella pneumoniae. Eluicidation of the structure of the linkage region between core and polysaccharide O chain and identification of the residues at the non-reducing termini of the O chains. J Biol Chem, 2002. 277(28): p. 25070-81.
71. Clements, A., et al., Targeting subcapsular antigens for prevention of Klebsiella pneumoniae infections. Vaccine, 2008. 26(44): p. 5649-53.
72. Alberti, S., et al., Analysis of complement C3 deposition and degradation on Klebsiella pneumoniae. Infect Immun, 1996. 64(11): p. 4726-32.
73. Alberti, S., et al., C1q binding and activation of the complement classical pathway by Klebsiella pneumoniae outer membrane proteins. Infect Immun, 1993. 61(3): p. 852-60.
74. Tomas, J.M., et al., Role of capsule and O antigen in resistance of Klebsiella pneumoniae to serum bactericidal activity. Infect Immun, 1986. 54(1): p. 85-9.
75. Taylor, P.W., Bactericidal and bacteriolytic activity of serum against gram-negative bacteria. Microbiol Rev, 1983. 47(1): p. 46-83.
76. Ramm, L.E., M.B. Whitlow, and M.M. Mayer, Size distribution and stability of the trans-membrane channels formed by complement complex C5b-9. Mol Immunol, 1983. 20(2): p. 155-60.
77. Merino, S., et al., Mechanisms of Klebsiella pneumoniae resistance to complement-mediated killing. Infect Immun, 1992. 60(6): p. 2529-35.
78. Taylor, P.W. and H.P. Kroll, Effect of lethal doses of complement on the functional integrity of target enterobacteria. Curr Top Microbiol Immunol, 1985. 121: p. 135-58.
79. Kostina, E., et al., Noncapsulated Klebsiella pneumoniae bearing mannose-containing O antigens is rapidly eradicated from mouse lung and triggers cytokine production by macrophages following opsonization with surfactant protein D. Infect Immun, 2005. 73(12): p. 8282-90.

REFERENCIAS 130
80. Clements, A., et al., The major surface-associated saccharides of Klebsiella pneumoniae contribute to host cell association. PLoS One, 2008. 3(11): p. e3817.
81. Jones, G.W. and R.E. Isaacson, Proteinaceous bacterial adhesins and their receptors. Crit Rev Microbiol, 1983. 10(3): p. 229-60.
82. Ofek, I. and D.R. J., Bacterial adhesion to cells and tissues., in L. Chapman & Hall, Editor 1994: London, U.K. 83. Podschun, R., et al., Serotypes, hemagglutinins, siderophore synthesis, and serum resistance of Klebsiella
isolates causing human urinary tract infections. J Infect Dis, 1993. 168(6): p. 1415-21. 84. Ayars, G.H., L.C. Altman, and M.D. Fretwell, Effect of decreased salivation and pH on the adherence of
Klebsiella species to human buccal epithelial cells. Infect Immun, 1982. 38(1): p. 179-82. 85. Maayan, M.C., et al., Population shift in mannose-specific fimbriated phase of Klebsiella pneumoniae
during experimental urinary tract infection in mice. Infect Immun, 1985. 49(3): p. 785-9. 86. Olsen, P.B. and P. Klemm, Localization of promoters in the fim gene cluster and the effect of H-NS on the
transcription of fimB and fimE. FEMS Microbiol Lett, 1994. 116(1): p. 95-100. 87. Hornick, D.B., et al., Adherence to respiratory epithelia by recombinant Escherichia coli expressing
Klebsiella pneumoniae type 3 fimbrial gene products. Infect Immun, 1992. 60(4): p. 1577-88. 88. Tarkkanen, A.M., et al., Binding of the type 3 fimbriae of Klebsiella pneumoniae to human endothelial and
urinary bladder cells. Infect Immun, 1997. 65(4): p. 1546-9. 89. Wurker, M., et al., Type of fimbriation determines adherence of Klebsiella bacteria to human epithelial
cells. Zentralbl Bakteriol, 1990. 274(2): p. 239-45. 90. Jagnow, J. and S. Clegg, Klebsiella pneumoniae MrkD-mediated biofilm formation on extracellular matrix-
and collagen-coated surfaces. Microbiology, 2003. 149(Pt 9): p. 2397-405. 91. Langstraat, J., M. Bohse, and S. Clegg, Type 3 fimbrial shaft (MrkA) of Klebsiella pneumoniae, but not the
fimbrial adhesin (MrkD), facilitates biofilm formation. Infect Immun, 2001. 69(9): p. 5805-12. 92. Schroll, C., et al., Role of type 1 and type 3 fimbriae in Klebsiella pneumoniae biofilm formation. BMC
Microbiol, 2010. 10: p. 179. 93. Allen, B.L., G.F. Gerlach, and S. Clegg, Nucleotide sequence and functions of mrk determinants necessary
for expression of type 3 fimbriae in Klebsiella pneumoniae. J Bacteriol, 1991. 173(2): p. 916-20. 94. Hornick, D.B., et al., Adherence properties of an mrkD-negative mutant of Klebsiella pneumoniae. Infect
Immun, 1995. 63(5): p. 2026-32. 95. Darfeuille-Michaud, A., et al., R-plasmid-encoded adhesive factor in Klebsiella pneumoniae strains
responsible for human nosocomial infections. Infect Immun, 1992. 60(1): p. 44-55. 96. Di Martino, P., et al., A new fimbrial antigen harbored by CAZ-5/SHV-4-producing Klebsiella pneumoniae
strains involved in nosocomial infections. Infect Immun, 1996. 64(6): p. 2266-73. 97. Sahly, H., et al., Capsule impedes adhesion to and invasion of epithelial cells by Klebsiella pneumoniae.
Infect Immun, 2000. 68(12): p. 6744-9. 98. Evrard, B., et al., Roles of capsule and lipopolysaccharide O antigen in interactions of human monocyte-
derived dendritic cells and Klebsiella pneumoniae. Infect Immun, 2010. 78(1): p. 210-9. 99. Zamze, S., et al., Recognition of bacterial capsular polysaccharides and lipopolysaccharides by the
macrophage mannose receptor. J Biol Chem, 2002. 277(44): p. 41613-23. 100. Merino, S., et al., Cloning and sequencing of the Klebsiella pneumoniae O5 wb gene cluster and its role in
pathogenesis. Infect Immun, 2000. 68(5): p. 2435-40. 101. Griffiths, E., The iron-uptake systems of pathogenic bacteria, ed. B.a.E. Griffiths1987, New York: John
Wiley & Sons. 69-137. 102. Mokracka, J., R. Koczura, and A. Kaznowski, Yersiniabactin and other siderophores produced by clinical
isolates of Enterobacter spp. and Citrobacter spp. FEMS Immunol Med Microbiol, 2004. 40(1): p. 51-5. 103. Raymond, K.N., E.A. Dertz, and S.S. Kim, Enterobactin: an archetype for microbial iron transport. Proc Natl
Acad Sci U S A, 2003. 100(7): p. 3584-8. 104. Visca, P., et al., Siderophore production by Salmonella species isolated from different sources. FEMS
Microbiol Lett, 1991. 63(2-3): p. 225-31. 105. Brock, J.H., et al., Relative availability of transferrin-bound iron and cell-derived iron to aerobactin-
producing and enterochelin-producing strains of Escherichia coli and to other microorganisms. Infect Immun, 1991. 59(9): p. 3185-90.
106. Perry, R.D., et al., Yersiniabactin from Yersinia pestis: biochemical characterization of the siderophore and its role in iron transport and regulation. Microbiology, 1999. 145 ( Pt 5): p. 1181-90.
107. Bach, S., A. de Almeida, and E. Carniel, The Yersinia high-pathogenicity island is present in different members of the family Enterobacteriaceae. FEMS Microbiol Lett, 2000. 183(2): p. 289-94.

REFERENCIAS 131
108. Crosa, J.H., Genetics and molecular biology of siderophore-mediated iron transport in bacteria. Microbiol Rev, 1989. 53(4): p. 517-30.
109. Fetherston, J.D., S.W. Bearden, and R.D. Perry, YbtA, an AraC-type regulator of the Yersinia pestis pesticin/yersiniabactin receptor. Mol Microbiol, 1996. 22(2): p. 315-25.
110. Fetherston, J.D., J.W. Lillard, Jr., and R.D. Perry, Analysis of the pesticin receptor from Yersinia pestis: role in iron-deficient growth and possible regulation by its siderophore. J Bacteriol, 1995. 177(7): p. 1824-33.
111. Torres, A.G., et al., TonB-dependent systems of uropathogenic Escherichia coli: aerobactin and heme transport and TonB are required for virulence in the mouse. Infect Immun, 2001. 69(10): p. 6179-85.
112. Podschun, R., A. Fischer, and U. Ullmann, Siderophore production of Klebsiella species isolated from different sources. Zentralbl Bakteriol, 1992. 276(4): p. 481-6.
113. Reissbrodt, R. and W. Rabsch, Further differentiation of Enterobacteriaceae by means of siderophore-pattern analysis. Zentralbl Bakteriol Mikrobiol Hyg A, 1988. 268(3): p. 306-17.
114. Koczura, R. and A. Kaznowski, Occurrence of the Yersinia high-pathogenicity island and iron uptake systems in clinical isolates of Klebsiella pneumoniae. Microb Pathog, 2003. 35(5): p. 197-202.
115. Schubert, S., et al., High-pathogenicity island of Yersinia pestis in enterobacteriaceae isolated from blood cultures and urine samples: prevalence and functional expression. J Infect Dis, 2000. 182(4): p. 1268-71.
116. Williams, P., et al., Novel aerobactin receptor in Klebsiella pneumoniae. J Gen Microbiol, 1989. 135(12): p. 3173-81.
117. Coudron, P.E., N.D. Hanson, and M.W. Climo, Occurrence of extended-spectrum and AmpC beta-lactamases in bloodstream isolates of Klebsiella pneumoniae: isolates harbor plasmid-mediated FOX-5 and ACT-1 AmpC beta-lactamases. J Clin Microbiol, 2003. 41(2): p. 772-7.
118. Pena, C., et al., Epidemiology and successful control of a large outbreak due to Klebsiella pneumoniae producing extended-spectrum beta-lactamases. Antimicrob Agents Chemother, 1998. 42(1): p. 53-8.
119. Babini, G.S. and D.M. Livermore, Antimicrobial resistance amongst Klebsiella spp. collected from intensive care units in Southern and Western Europe in 1997-1998. J Antimicrob Chemother, 2000. 45(2): p. 183-9.
120. Rebuck, J.A., et al., Characterization of an outbreak due to extended-spectrum beta-lactamase-producing Klebsiella pneumoniae in a pediatric intensive care unit transplant population. Clin Infect Dis, 2000. 31(6): p. 1368-72.
121. Livermore, D.M., beta-Lactamases: quantity and resistance. Clin Microbiol Infect, 1997. 3 Suppl 4: p. S10-S19.
122. Haeggman, S., et al., Diversity and evolution of the class A chromosomal beta-lactamase gene in Klebsiella pneumoniae. Antimicrob Agents Chemother, 2004. 48(7): p. 2400-8.
123. Jacoby, G.A., N. Chow, and K.B. Waites, Prevalence of plasmid-mediated quinolone resistance. Antimicrob Agents Chemother, 2003. 47(2): p. 559-62.
124. Rice, L.B., S.H. Marshall, and L.L. Carias, Tn5381, a conjugative transposon identifiable as a circular form in Enterococcus faecalis. J Bacteriol, 1992. 174(22): p. 7308-15.
125. Bradford, P.A., Extended-spectrum beta-lactamases in the 21st century: characterization, epidemiology, and detection of this important resistance threat. Clin Microbiol Rev, 2001. 14(4): p. 933-51, table of contents.
126. Bush, K. and G.A. Jacoby, Updated functional classification of beta-lactamases. Antimicrob Agents Chemother, 2010. 54(3): p. 969-76.
127. Kliebe, C., et al., Evolution of plasmid-coded resistance to broad-spectrum cephalosporins. Antimicrob Agents Chemother, 1985. 28(2): p. 302-7.
128. Sirot, D., et al., Transferable resistance to third-generation cephalosporins in clinical isolates of Klebsiella pneumoniae: identification of CTX-1, a novel beta-lactamase. J Antimicrob Chemother, 1987. 20(3): p. 323-34.
129. Sougakoff, W., et al., Plasmid-mediated resistance to third-generation cephalosporins caused by point mutations in TEM-type penicillinase genes. Rev Infect Dis, 1988. 10(4): p. 879-84.
130. Pfeifer, Y., A. Cullik, and W. Witte, Resistance to cephalosporins and carbapenems in Gram-negative bacterial pathogens. Int J Med Microbiol, 2010. 300(6): p. 371-9.
131. Asensio, A., et al., Outbreak of a multiresistant Klebsiella pneumoniae strain in an intensive care unit: antibiotic use as risk factor for colonization and infection. Clin Infect Dis, 2000. 30(1): p. 55-60.
132. De Champs, C., et al., A 1998 survey of extended-spectrum beta-lactamases in Enterobacteriaceae in France. The French Study Group. Antimicrob Agents Chemother, 2000. 44(11): p. 3177-9.
133. Canton, R., et al., Prevalence and spread of extended-spectrum beta-lactamase-producing Enterobacteriaceae in Europe. Clin Microbiol Infect, 2008. 14 Suppl 1: p. 144-53.

REFERENCIAS 132
134. Wu, J.J., et al., Prevalence of plasmid-mediated quinolone resistance determinants QnrA, QnrB, and QnrS among clinical isolates of Enterobacter cloacae in a Taiwanese hospital. Antimicrob Agents Chemother, 2007. 51(4): p. 1223-7.
135. Poirel, L., C. Leviandier, and P. Nordmann, Prevalence and genetic analysis of plasmid-mediated quinolone resistance determinants QnrA and QnrS in Enterobacteriaceae isolates from a French university hospital. Antimicrob Agents Chemother, 2006. 50(12): p. 3992-7.
136. Jacobs, C., J.M. Frere, and S. Normark, Cytosolic intermediates for cell wall biosynthesis and degradation control inducible beta-lactam resistance in gram-negative bacteria. Cell, 1997. 88(6): p. 823-32.
137. Normark, S., beta-Lactamase induction in gram-negative bacteria is intimately linked to peptidoglycan recycling. Microb Drug Resist, 1995. 1(2): p. 111-4.
138. Kuga, A., R. Okamoto, and M. Inoue, ampR gene mutations that greatly increase class C beta-lactamase activity in Enterobacter cloacae. Antimicrob Agents Chemother, 2000. 44(3): p. 561-7.
139. Philippon, A., G. Arlet, and G.A. Jacoby, Plasmid-determined AmpC-type beta-lactamases. Antimicrob Agents Chemother, 2002. 46(1): p. 1-11.
140. Barlow, M. and B.G. Hall, Origin and evolution of the AmpC beta-lactamases of Citrobacter freundii. Antimicrob Agents Chemother, 2002. 46(5): p. 1190-8.
141. Bauernfeind, A., Y. Chong, and S. Schweighart, Extended broad spectrum beta-lactamase in Klebsiella pneumoniae including resistance to cephamycins. Infection, 1989. 17(5): p. 316-21.
142. Papanicolaou, G.A., A.A. Medeiros, and G.A. Jacoby, Novel plasmid-mediated beta-lactamase (MIR-1) conferring resistance to oxyimino- and alpha-methoxy beta-lactams in clinical isolates of Klebsiella pneumoniae. Antimicrob Agents Chemother, 1990. 34(11): p. 2200-9.
143. Jacoby, G.A., AmpC beta-lactamases. Clin Microbiol Rev, 2009. 22(1): p. 161-82, Table of Contents. 144. Chen, Y., et al., ACT-3, a novel plasmid-encoded class C beta-lactamase in a Klebsiella pneumoniae isolate
from China. Int J Antimicrob Agents, 2009. 33(1): p. 95-6. 145. Zhu, Y.L., et al., ACT-6, a novel plasmid-encoded class C beta-lactamase in a Klebsiella pneumoniae isolate
from China. J Antibiot (Tokyo), 2011. 64(4): p. 317-20. 146. Bauernfeind, A., et al., Comparative characterization of the cephamycinase blaCMY-1 gene and its
relationship with other beta-lactamase genes. Antimicrob Agents Chemother, 1996. 40(8): p. 1926-30. 147. Bauernfeind, A., et al., Characterization of the plasmidic beta-lactamase CMY-2, which is responsible for
cephamycin resistance. Antimicrob Agents Chemother, 1996. 40(1): p. 221-4. 148. Jacoby, G.A. and J. Tran, Sequence of the MIR-1 beta-lactamase gene. Antimicrob Agents Chemother,
1999. 43(7): p. 1759-60. 149. Fortineau, N., L. Poirel, and P. Nordmann, Plasmid-mediated and inducible cephalosporinase DHA-2 from
Klebsiella pneumoniae. J Antimicrob Chemother, 2001. 47(2): p. 207-10. 150. Horii, T., et al., Plasmid-mediated AmpC-type beta-lactamase isolated from Klebsiella pneumoniae confers
resistance to broad-spectrum beta-lactams, including moxalactam. Antimicrob Agents Chemother, 1993. 37(5): p. 984-90.
151. Tzouvelekis, L.S., et al., Identification of a novel plasmid-mediated beta-lactamase with chromosomal cephalosporinase characteristics from Klebsiella pneumoniae. J Antimicrob Chemother, 1993. 31(5): p. 645-54.
152. Fosse, T., et al., Sequence analysis and biochemical characterisation of chromosomal CAV-1 (Aeromonas caviae), the parental cephalosporinase of plasmid-mediated AmpC 'FOX' cluster. FEMS Microbiol Lett, 2003. 222(1): p. 93-8.
153. Gonzalez Leiza, M., et al., Gene sequence and biochemical characterization of FOX-1 from Klebsiella pneumoniae, a new AmpC-type plasmid-mediated beta-lactamase with two molecular variants. Antimicrob Agents Chemother, 1994. 38(9): p. 2150-7.
154. Bradford, P.A., et al., Imipenem resistance in Klebsiella pneumoniae is associated with the combination of ACT-1, a plasmid-mediated AmpC beta-lactamase, and the foss of an outer membrane protein. Antimicrob Agents Chemother, 1997. 41(3): p. 563-9.
155. Rottman, M., et al., Chromosomal ampC genes in Enterobacter species other than Enterobacter cloacae, and ancestral association of the ACT-1 plasmid-encoded cephalosporinase to Enterobacter asburiae. FEMS Microbiol Lett, 2002. 210(1): p. 87-92.
156. Bauernfeind, A., et al., A novel type of AmpC beta-lactamase, ACC-1, produced by a Klebsiella pneumoniae strain causing nosocomial pneumonia. Antimicrob Agents Chemother, 1999. 43(8): p. 1924-31.

REFERENCIAS 133
157. Girlich, D., et al., Biochemical-genetic characterization and regulation of expression of an ACC-1-like chromosome-borne cephalosporinase from Hafnia alvei. Antimicrob Agents Chemother, 2000. 44(6): p. 1470-8.
158. Queenan, A.M., S. Jenkins, and K. Bush, Cloning and biochemical characterization of FOX-5, an AmpC-type plasmid-encoded beta-lactamase from a New York City Klebsiella pneumoniae clinical isolate. Antimicrob Agents Chemother, 2001. 45(11): p. 3189-94.
159. Bauernfeind, A., H. Grimm, and S. Schweighart, A new plasmidic cefotaximase in a clinical isolate of Escherichia coli. Infection, 1990. 18(5): p. 294-8.
160. Canton, R. and T.M. Coque, The CTX-M beta-lactamase pandemic. Curr Opin Microbiol, 2006. 9(5): p. 466-75.
161. Bonnet, R., Growing group of extended-spectrum beta-lactamases: the CTX-M enzymes. Antimicrob Agents Chemother, 2004. 48(1): p. 1-14.
162. Barlow, M., et al., High rate of mobilization for blaCTX-Ms. Emerg Infect Dis, 2008. 14(3): p. 423-8. 163. Eckert, C., et al., Dissemination of CTX-M-type beta-lactamases among clinical isolates of
Enterobacteriaceae in Paris, France. Antimicrob Agents Chemother, 2004. 48(4): p. 1249-55. 164. Poirel, L., et al., ISEcp1B-mediated transposition of blaCTX-M in Escherichia coli. Antimicrob Agents
Chemother, 2005. 49(1): p. 447-50. 165. Poirel, L., M. Gniadkowski, and P. Nordmann, Biochemical analysis of the ceftazidime-hydrolysing
extended-spectrum beta-lactamase CTX-M-15 and of its structurally related beta-lactamase CTX-M-3. J Antimicrob Chemother, 2002. 50(6): p. 1031-4.
166. Cartelle, M., et al., High-level resistance to ceftazidime conferred by a novel enzyme, CTX-M-32, derived from CTX-M-1 through a single Asp240-Gly substitution. Antimicrob Agents Chemother, 2004. 48(6): p. 2308-13.
167. Novais, A., et al., Mutational events in cefotaximase extended-spectrum beta-lactamases of the CTX-M-1 cluster involved in ceftazidime resistance. Antimicrob Agents Chemother, 2008. 52(7): p. 2377-82.
168. Mena, A., et al., Characterization of a large outbreak by CTX-M-1-producing Klebsiella pneumoniae and mechanisms leading to in vivo carbapenem resistance development. J Clin Microbiol, 2006. 44(8): p. 2831-7.
169. Elliott, E., et al., In vivo development of ertapenem resistance in a patient with pneumonia caused by Klebsiella pneumoniae with an extended-spectrum beta-lactamase. Clin Infect Dis, 2006. 42(11): p. e95-8.
170. Queenan, A.M., et al., SME-type carbapenem-hydrolyzing class A beta-lactamases from geographically diverse Serratia marcescens strains. Antimicrob Agents Chemother, 2000. 44(11): p. 3035-9.
171. Queenan, A.M., et al., SME-3, a novel member of the Serratia marcescens SME family of carbapenem-hydrolyzing beta-lactamases. Antimicrob Agents Chemother, 2006. 50(10): p. 3485-7.
172. Rasmussen, B.A., et al., Characterization of IMI-1 beta-lactamase, a class A carbapenem-hydrolyzing enzyme from Enterobacter cloacae. Antimicrob Agents Chemother, 1996. 40(9): p. 2080-6.
173. Poirel, L., et al., Biochemical sequence analyses of GES-1, a novel class A extended-spectrum beta-lactamase, and the class 1 integron In52 from Klebsiella pneumoniae. Antimicrob Agents Chemother, 2000. 44(3): p. 622-32.
174. Naas, T., et al., Genetic structures at the origin of acquisition of the beta-lactamase bla KPC gene. Antimicrob Agents Chemother, 2008. 52(4): p. 1257-63.
175. Yigit, H., et al., Novel carbapenem-hydrolyzing beta-lactamase, KPC-1, from a carbapenem-resistant strain of Klebsiella pneumoniae. Antimicrob Agents Chemother, 2001. 45(4): p. 1151-61.
176. Miriagou, V., et al., Imipenem resistance in a Salmonella clinical strain due to plasmid-mediated class A carbapenemase KPC-2. Antimicrob Agents Chemother, 2003. 47(4): p. 1297-300.
177. Yigit, H., et al., Carbapenem-resistant strain of Klebsiella oxytoca harboring carbapenem-hydrolyzing beta-lactamase KPC-2. Antimicrob Agents Chemother, 2003. 47(12): p. 3881-9.
178. Villegas, M.V., et al., First identification of Pseudomonas aeruginosa isolates producing a KPC-type carbapenem-hydrolyzing beta-lactamase. Antimicrob Agents Chemother, 2007. 51(4): p. 1553-5.
179. Bratu, S., et al., Detection of KPC carbapenem-hydrolyzing enzymes in Enterobacter spp. from Brooklyn, New York. Antimicrob Agents Chemother, 2005. 49(2): p. 776-8.
180. Gomez-Gil, M.R., et al., Detection of KPC-2-producing Citrobacter freundii isolates in Spain. J Antimicrob Chemother. 65(12): p. 2695-7.
181. Navon-Venezia, S., et al., First report on a hyperepidemic clone of KPC-3-producing Klebsiella pneumoniae in Israel genetically related to a strain causing outbreaks in the United States. Antimicrob Agents Chemother, 2009. 53(2): p. 818-20.

REFERENCIAS 134
182. Osterblad, M., et al., First isolations of KPC-2-carrying ST258 Klebsiella pneumoniae strains in Finland, June and August 2009. Euro Surveill, 2009. 14(40).
183. Cai, J.C., et al., Emergence of Serratia marcescens, Klebsiella pneumoniae, and Escherichia coli Isolates possessing the plasmid-mediated carbapenem-hydrolyzing beta-lactamase KPC-2 in intensive care units of a Chinese hospital. Antimicrob Agents Chemother, 2008. 52(6): p. 2014-8.
184. Curiao, T., et al., Emergence of bla KPC-3-Tn4401a associated with a pKPN3/4-like plasmid within ST384 and ST388 Klebsiella pneumoniae clones in Spain. J Antimicrob Chemother, 2010. 65(8): p. 1608-14.
185. Naas, T. and P. Nordmann, OXA-type beta-lactamases. Curr Pharm Des, 1999. 5(11): p. 865-79. 186. Bush, K., Recent developments in beta-lactamase research and their implications for the future. Rev Infect
Dis, 1988. 10(4): p. 681-90. 187. Walther-Rasmussen, J. and N. Hoiby, OXA-type carbapenemases. J Antimicrob Chemother, 2006. 57(3): p.
373-83. 188. Heritier, C., L. Poirel, and P. Nordmann, Genetic and biochemical characterization of a chromosome-
encoded carbapenem-hydrolyzing ambler class D beta-lactamase from Shewanella algae. Antimicrob Agents Chemother, 2004. 48(5): p. 1670-5.
189. Barlow, M. and B.G. Hall, Phylogenetic analysis shows that the OXA beta-lactamase genes have been on plasmids for millions of years. J Mol Evol, 2002. 55(3): p. 314-21.
190. Levast, M., et al., Transfer of OXA-48-positive carbapenem-resistant Klebsiella pneumoniae from Turkey to France. J Antimicrob Chemother, 2011. 66(4): p. 944-5.
191. Poirel, L., et al., OXA-163, an OXA-48-related class D {beta}-lactamase with an extended activity toward expanded-spectrum cephalosporins. Antimicrob Agents Chemother, 2011.
192. Kalpoe, J.S., et al., Detection of an Ambler class D OXA-48-type {beta}-lactamase in a Klebsiella pneumoniae strain in The Netherlands. J Med Microbiol, 2011. 60(Pt 5): p. 677-8.
193. Ktari, S., et al., Spread of Klebsiella pneumoniae isolates producing OXA-48 {beta}-lactamase in a Tunisian university hospital. J Antimicrob Chemother, 2011.
194. Barguigua, A., et al., Characterization of ESBL-producing Escherichia coli and Klebsiella pneumoniae isolates from community in Morocco. J Med Microbiol, 2011.
195. Perez-Moreno, M.O., et al., Molecular epidemiology and resistance mechanisms involved in reduced susceptibility to amoxicillin/clavulanic acid in Klebsiella pneumoniae isolates from a chronic care centre. Int J Antimicrob Agents, 2011. 37(5): p. 462-6.
196. Ruiz, E., et al., Outbreak caused by a multi-resistant Klebsiella pneumoniae strain of new sequence type ST341 carrying new genetic environments of aac(6')-Ib-cr and qnrS1 genes in a neonatal intensive care unit in Spain. Int J Med Microbiol, 2010. 300(7): p. 464-9.
197. Bebrone, C., Metallo-beta-lactamases (classification, activity, genetic organization, structure, zinc coordination) and their superfamily. Biochem Pharmacol, 2007. 74(12): p. 1686-701.
198. Garau, G., et al., Update of the standard numbering scheme for class B beta-lactamases. Antimicrob Agents Chemother, 2004. 48(7): p. 2347-9.
199. Yong, D., et al., Characterization of a new metallo-beta-lactamase gene, bla(NDM-1), and a novel erythromycin esterase gene carried on a unique genetic structure in Klebsiella pneumoniae sequence type 14 from India. Antimicrob Agents Chemother, 2009. 53(12): p. 5046-54.
200. Walsh, T.R., et al., Enzyme kinetics and biochemical analysis of ImiS, the metallo-beta-lactamase from Aeromonas sobria 163a. J Antimicrob Chemother, 1996. 37(3): p. 423-31.
201. Walsh, T.R., et al., Sequence analysis of the L1 metallo-beta-lactamase from Xanthomonas maltophilia. Biochim Biophys Acta, 1994. 1218(2): p. 199-201.
202. Poirel, L., et al., Characterization of VIM-2, a carbapenem-hydrolyzing metallo-beta-lactamase and its plasmid- and integron-borne gene from a Pseudomonas aeruginosa clinical isolate in France. Antimicrob Agents Chemother, 2000. 44(4): p. 891-7.
203. Tortola, M.T., et al., First detection of a carbapenem-hydrolyzing metalloenzyme in two enterobacteriaceae isolates in Spain. Antimicrob Agents Chemother, 2005. 49(8): p. 3492-4.
204. Walsh, T.R., et al., Metallo-beta-lactamases: the quiet before the storm? Clin Microbiol Rev, 2005. 18(2): p. 306-25.
205. Toleman, M.A., et al., Molecular characterization of SPM-1, a novel metallo-beta-lactamase isolated in Latin America: report from the SENTRY antimicrobial surveillance programme. J Antimicrob Chemother, 2002. 50(5): p. 673-9.
206. Castanheira, M., et al., Molecular characterization of a beta-lactamase gene, blaGIM-1, encoding a new subclass of metallo-beta-lactamase. Antimicrob Agents Chemother, 2004. 48(12): p. 4654-61.

REFERENCIAS 135
207. Lee, K., et al., Novel acquired metallo-beta-lactamase gene, bla(SIM-1), in a class 1 integron from Acinetobacter baumannii clinical isolates from Korea. Antimicrob Agents Chemother, 2005. 49(11): p. 4485-91.
208. Toleman, M.A., P.M. Bennett, and T.R. Walsh, ISCR elements: novel gene-capturing systems of the 21st century? Microbiol Mol Biol Rev, 2006. 70(2): p. 296-316.
209. Koh, T.H., et al., Carbapenem-hydrolysing IMP-1 beta-lactamase in Klebsiella pneumoniae from Singapore. Lancet, 1999. 353(9170): p. 2162.
210. Giakkoupi, P., et al., VIM-1 Metallo-beta-lactamase-producing Klebsiella pneumoniae strains in Greek hospitals. J Clin Microbiol, 2003. 41(8): p. 3893-6.
211. Paulsen, I.T., M.K. Sliwinski, and M.H. Saier, Jr., Microbial genome analyses: global comparisons of transport capabilities based on phylogenies, bioenergetics and substrate specificities. J Mol Biol, 1998. 277(3): p. 573-92.
212. Saier, M.H., Jr., et al., Two novel families of bacterial membrane proteins concerned with nodulation, cell division and transport. Mol Microbiol, 1994. 11(5): p. 841-7.
213. Pao, S.S., I.T. Paulsen, and M.H. Saier, Jr., Major facilitator superfamily. Microbiol Mol Biol Rev, 1998. 62(1): p. 1-34.
214. Marger, M.D. and M.H. Saier, Jr., A major superfamily of transmembrane facilitators that catalyse uniport, symport and antiport. Trends Biochem Sci, 1993. 18(1): p. 13-20.
215. Paulsen, I.T., et al., The SMR family: a novel family of multidrug efflux proteins involved with the efflux of lipophilic drugs. Mol Microbiol, 1996. 19(6): p. 1167-75.
216. Brown, M.H., I.T. Paulsen, and R.A. Skurray, The multidrug efflux protein NorM is a prototype of a new family of transporters. Mol Microbiol, 1999. 31(1): p. 394-5.
217. van Veen, H.W. and W.N. Konings, The ABC family of multidrug transporters in microorganisms. Biochim Biophys Acta, 1998. 1365(1-2): p. 31-6.
218. Kumar, A. and H.P. Schweizer, Bacterial resistance to antibiotics: active efflux and reduced uptake. Adv Drug Deliv Rev, 2005. 57(10): p. 1486-513.
219. Butaye, P., A. Cloeckaert, and S. Schwarz, Mobile genes coding for efflux-mediated antimicrobial resistance in Gram-positive and Gram-negative bacteria. Int J Antimicrob Agents, 2003. 22(3): p. 205-10.
220. Martinez-Martinez, L., et al., Energy-dependent accumulation of fluoroquinolones in quinolone-resistant Klebsiella pneumoniae strains. Antimicrob Agents Chemother, 1998. 42(7): p. 1850-2.
221. Mazzariol, A., et al., AcrAB Efflux System: Expression and Contribution to Fluoroquinolone Resistance in Klebsiella spp. Antimicrob Agents Chemother, 2002. 46(12): p. 3984-6.
222. Padilla, E., et al., Klebsiella pneumoniae AcrAB efflux pump contributes to antimicrobial resistance and virulence. Antimicrob Agents Chemother, 2010. 54(1): p. 177-83.
223. Hernandez-Alles, S., et al., Relationship between outer membrane alterations and susceptibility to antimicrobial agents in isogenic strains of Klebsiella pneumoniae. J Antimicrob Chemother, 2000. 46(2): p. 273-7.
224. Padilla, E., et al., Effect of porins and plasmid-mediated AmpC beta-lactamases on the efficacy of beta-lactams in rat pneumonia caused by Klebsiella pneumoniae. Antimicrob Agents Chemother, 2006. 50(6): p. 2258-60.
225. Hernandez-Alles, S., et al., Development of resistance during antimicrobial therapy caused by insertion sequence interruption of porin genes. Antimicrob Agents Chemother, 1999. 43(4): p. 937-9.
226. Garcia-Fernandez, A., et al., An ertapenem-resistant ESBL-producing Klebsiella pneumoniae clone carries a novel OmpK36 porin variant. Antimicrob Agents Chemother, 2010.
227. Bredin, J., et al., Alteration of pore properties of Escherichia coli OmpF induced by mutation of key residues in anti-loop 3 region. Biochem J, 2002. 363(Pt 3): p. 521-8.
228. Thiolas, A., et al., Resistance to imipenem, cefepime, and cefpirome associated with mutation in Omp36 osmoporin of Enterobacter aerogenes. Biochem Biophys Res Commun, 2004. 317(3): p. 851-6.
229. Martinez-Martinez, L., et al., Roles of beta-lactamases and porins in activities of carbapenems and cephalosporins against Klebsiella pneumoniae. Antimicrob Agents Chemother, 1999. 43(7): p. 1669-73.
230. Domenech-Sanchez, A., et al., Role of Klebsiella pneumoniae OmpK35 porin in antimicrobial resistance. Antimicrob Agents Chemother, 2003. 47(10): p. 3332-5.
231. Nikaido, H., Molecular basis of bacterial outer membrane permeability revisited. Microbiol Mol Biol Rev, 2003. 67(4): p. 593-656.
232. Martinez-Martinez, L., Extended-spectrum beta-lactamases and the permeability barrier. Clin Microbiol Infect, 2008. 14 Suppl 1: p. 82-9.

REFERENCIAS 136
233. Delcour, A.H., Outer membrane permeability and antibiotic resistance. Biochim Biophys Acta, 2009. 1794(5): p. 808-16.
234. Wiese, A., et al., The dual role of lipopolysaccharide as effector and target molecule. Biol Chem, 1999. 380(7-8): p. 767-84.
235. Ferguson, A.D., et al., A conserved structural motif for lipopolysaccharide recognition by procaryotic and eucaryotic proteins. Structure, 2000. 8(6): p. 585-92.
236. Bos, M.P., V. Robert, and J. Tommassen, Biogenesis of the gram-negative bacterial outer membrane. Annu Rev Microbiol, 2007. 61: p. 191-214.
237. Hantke, K. and V. Braun, Covalent binding of lipid to protein. Diglyceride and amide-linked fatty acid at the N-terminal end of the murein-lipoprotein of the Escherichia coli outer membrane. Eur J Biochem, 1973. 34(2): p. 284-96.
238. Nikaido, H., Proteins forming large channels from bacterial and mitochondrial outer membranes: porins and phage lambda receptor protein. Methods Enzymol, 1983. 97: p. 85-100.
239. Nakae, T., Outer membrane of Salmonella. Isolation of protein complex that produces transmembrane channels. J Biol Chem, 1976. 251(7): p. 2176-8.
240. Schein, S.J., M. Colombini, and A. Finkelstein, Reconstitution in planar lipid bilayers of a voltage-dependent anion-selective channel obtained from paramecium mitochondria. J Membr Biol, 1976. 30(2): p. 99-120.
241. Blachly-Dyson, E. and M. Forte, VDAC channels. IUBMB Life, 2001. 52(3-5): p. 113-8. 242. Mannella, C.A., M. Forte, and M. Colombini, Toward the molecular structure of the mitochondrial channel,
VDAC. J Bioenerg Biomembr, 1992. 24(1): p. 7-19. 243. Reumann, S., et al., The membrane of leaf peroxisomes contains a porin-like channel. J Biol Chem, 1995.
270(29): p. 17559-65. 244. Reumann, S., et al., A specific porin is involved in the malate shuttle of leaf peroxisomes. Biochem Soc
Trans, 1996. 24(3): p. 754-7. 245. Reumann, S., et al., Permeability properties of the porin of spinach leaf peroxisomes. Eur J Biochem, 1998.
251(1-2): p. 359-66. 246. Domenech-Sanchez, A., et al., Identification and characterization of a new porin gene of Klebsiella
pneumoniae: its role in beta-lactam antibiotic resistance. J Bacteriol, 1999. 181(9): p. 2726-32. 247. Jap, B.K. and P.J. Walian, Structure and functional mechanism of porins. Physiol Rev, 1996. 76(4): p. 1073-
88. 248. Schirmer, T., General and specific porins from bacterial outer membranes. J Struct Biol, 1998. 121(2): p.
101-9. 249. Alberti, S., et al., A porin from Klebsiella pneumoniae: sequence homology, three-dimensional model, and
complement binding. Infect Immun, 1995. 63(3): p. 903-10. 250. McGowan, J.E., Jr., Resistance in nonfermenting gram-negative bacteria: multidrug resistance to the
maximum. Am J Infect Control, 2006. 34(5 Suppl 1): p. S29-37; discussion S64-73. 251. Poirel, L. and P. Nordmann, Carbapenem resistance in Acinetobacter baumannii: mechanisms and
epidemiology. Clin Microbiol Infect, 2006. 12(9): p. 826-36. 252. Dutzler, R., et al., Crystal structure and functional characterization of OmpK36, the osmoporin of Klebsiella
pneumoniae. Structure, 1999. 7(4): p. 425-34. 253. Schulz, G.E., Porins: general to specific, native to engineered passive pores. Curr Opin Struct Biol, 1996.
6(4): p. 485-90. 254. Basle, A., et al., Crystal structure of osmoporin OmpC from E. coli at 2.0 A. J Mol Biol, 2006. 362(5): p. 933-
42. 255. Cowan, S.W., et al., Crystal structures explain functional properties of two E. coli porins. Nature, 1992.
358(6389): p. 727-33. 256. Benz, R., Uptake of solutes through bacterial outer membranes. In Bacterial Cell Walls, ed. G. J.M. and H.
R.1994, Amsterdam: Elsevier. 397-424. 257. Lakey, J.H., J.P. Watts, and E.J. Lea, Characterisation of channels induced in planar bilayer membranes by
detergent solubilised Escherichia coli porins. Biochim Biophys Acta, 1985. 817(2): p. 208-16. 258. Prilipov, A., et al., Identification and characterization of two quiescent porin genes, nmpC and ompN, in
Escherichia coli BE. J Bacteriol, 1998. 180(13): p. 3388-92. 259. Brzostek, K., et al., OmpR negatively regulates expression of invasin in Yersinia enterocolitica.
Microbiology, 2007. 153(Pt 8): p. 2416-25.

REFERENCIAS 137
260. Chatfield, S.N., et al., Role of ompR-dependent genes in Salmonella typhimurium virulence: mutants deficient in both ompC and ompF are attenuated in vivo. Infect Immun, 1991. 59(1): p. 449-52.
261. Garmendia, J., et al., The roles of SsrA-SsrB and OmpR-EnvZ in the regulation of genes encoding the Salmonella typhimurium SPI-2 type III secretion system. Microbiology, 2003. 149(Pt 9): p. 2385-96.
262. Lee, A.K., C.S. Detweiler, and S. Falkow, OmpR regulates the two-component system SsrA-ssrB in Salmonella pathogenicity island 2. J Bacteriol, 2000. 182(3): p. 771-81.
263. Park, D. and S. Forst, Co-regulation of motility, exoenzyme and antibiotic production by the EnvZ-OmpR-FlhDC-FliA pathway in Xenorhabdus nematophila. Mol Microbiol, 2006. 61(6): p. 1397-412.
264. Shin, S. and C. Park, Modulation of flagellar expression in Escherichia coli by acetyl phosphate and the osmoregulator OmpR. J Bacteriol, 1995. 177(16): p. 4696-702.
265. Slauch, J.M. and T.J. Silhavy, Genetic analysis of the switch that controls porin gene expression in Escherichia coli K-12. J Mol Biol, 1989. 210(2): p. 281-92.
266. Qin, L., T. Yoshida, and M. Inouye, The critical role of DNA in the equilibrium between OmpR and phosphorylated OmpR mediated by EnvZ in Escherichia coli. Proc Natl Acad Sci U S A, 2001. 98(3): p. 908-13.
267. Pratt, L.A., et al., From acids to osmZ: multiple factors influence synthesis of the OmpF and OmpC porins in Escherichia coli. Mol Microbiol, 1996. 20(5): p. 911-7.
268. Forst, S. and M. Inouye, Environmentally regulated gene expression for membrane proteins in Escherichia coli. Annu Rev Cell Biol, 1988. 4: p. 21-42.
269. Liu, X. and T. Ferenci, An analysis of multifactorial influences on the transcriptional control of ompF and ompC porin expression under nutrient limitation. Microbiology, 2001. 147(Pt 11): p. 2981-9.
270. Forst, S., J. Delgado, and M. Inouye, Phosphorylation of OmpR by the osmosensor EnvZ modulates expression of the ompF and ompC genes in Escherichia coli. Proc Natl Acad Sci U S A, 1989. 86(16): p. 6052-6.
271. Head, C.G., A. Tardy, and L.J. Kenney, Relative binding affinities of OmpR and OmpR-phosphate at the ompF and ompC regulatory sites. J Mol Biol, 1998. 281(5): p. 857-70.
272. Huang, K.J., C.Y. Lan, and M.M. Igo, Phosphorylation stimulates the cooperative DNA-binding properties of the transcription factor OmpR. Proc Natl Acad Sci U S A, 1997. 94(7): p. 2828-32.
273. Dupont, M., et al., Enterobacter aerogenes OmpX, a cation-selective channel mar- and osmo-regulated. FEBS Lett, 2004. 569(1-3): p. 27-30.
274. Fernandez-Mora, M., J.L. Puente, and E. Calva, OmpR and LeuO positively regulate the Salmonella enterica serovar Typhi ompS2 porin gene. J Bacteriol, 2004. 186(10): p. 2909-20.
275. Oropeza, R., et al., Negative and positive regulation of the non-osmoregulated ompS1 porin gene in Salmonella typhi: a novel regulatory mechanism that involves OmpR. Mol Microbiol, 1999. 32(2): p. 243-52.
276. McDermott, P.F., et al., The marC gene of Escherichia coli is not involved in multiple antibiotic resistance. Antimicrob Agents Chemother, 2008. 52(1): p. 382-3.
277. Goldman, J.D., D.G. White, and S.B. Levy, Multiple antibiotic resistance (mar) locus protects Escherichia coli from rapid cell killing by fluoroquinolones. Antimicrob Agents Chemother, 1996. 40(5): p. 1266-9.
278. Cohen, S.P., H. Hachler, and S.B. Levy, Genetic and functional analysis of the multiple antibiotic resistance (mar) locus in Escherichia coli. J Bacteriol, 1993. 175(5): p. 1484-92.
279. Hachler, H., S.P. Cohen, and S.B. Levy, marA, a regulated locus which controls expression of chromosomal multiple antibiotic resistance in Escherichia coli. J Bacteriol, 1991. 173(17): p. 5532-8.
280. Barbosa, T.M. and S.B. Levy, Differential expression of over 60 chromosomal genes in Escherichia coli by constitutive expression of MarA. J Bacteriol, 2000. 182(12): p. 3467-74.
281. Schneiders, T., et al., The Escherichia coli transcriptional regulator MarA directly represses transcription of purA and hdeA. J Biol Chem, 2004. 279(10): p. 9037-42.
282. Dupont, M., et al., An early response to environmental stress involves regulation of OmpX and OmpF, two enterobacterial outer membrane pore-forming proteins. Antimicrob Agents Chemother, 2007. 51(9): p. 3190-8.
283. Kaczmarek, F.M., et al., High-level carbapenem resistance in a Klebsiella pneumoniae clinical isolate is due to the combination of bla(ACT-1) beta-lactamase production, porin OmpK35/36 insertional inactivation, and down-regulation of the phosphate transport porin phoe. Antimicrob Agents Chemother, 2006. 50(10): p. 3396-406.

REFERENCIAS 138
284. Bratu, S., et al., Correlation of the expression of acrB and the regulatory genes marA, soxS and ramA with antimicrobial resistance in clinical isolates of Klebsiella pneumoniae endemic to New York City. J Antimicrob Chemother, 2009. 64(2): p. 278-83.
285. Tavio, M.M., et al., Enhanced active efflux, repression of porin synthesis and development of Mar phenotype by diazepam in two enterobacteria strains. J Med Microbiol, 2004. 53(Pt 11): p. 1119-22.
286. Gaudu, P., N. Moon, and B. Weiss, Regulation of the soxRS oxidative stress regulon. Reversible oxidation of the Fe-S centers of SoxR in vivo. J Biol Chem, 1997. 272(8): p. 5082-6.
287. Bialek-Davenet, S., et al., In vitro selection of ramR and soxR mutants overexpressing efflux systems by fluoroquinolones as well as cefoxitin in Klebsiella pneumoniae. Antimicrob Agents Chemother, 2011.
288. Ariza, R.R., et al., Activation of multiple antibiotic resistance and binding of stress-inducible promoters by Escherichia coli Rob protein. J Bacteriol, 1995. 177(7): p. 1655-61.
289. Nakajima, H., et al., Overexpression of the robA gene increases organic solvent tolerance and multiple antibiotic and heavy metal ion resistance in Escherichia coli. Appl Environ Microbiol, 1995. 61(6): p. 2302-7.
290. Jair, K.W., et al., Transcriptional activation of promoters of the superoxide and multiple antibiotic resistance regulons by Rob, a binding protein of the Escherichia coli origin of chromosomal replication. J Bacteriol, 1996. 178(9): p. 2507-13.
291. Bennik, M.H., et al., Defining a rob regulon in Escherichia coli by using transposon mutagenesis. J Bacteriol, 2000. 182(13): p. 3794-801.
292. Delihas, N. and S. Forst, MicF: an antisense RNA gene involved in response of Escherichia coli to global stress factors. J Mol Biol, 2001. 313(1): p. 1-12.
293. Miller, P.F. and M.C. Sulavik, Overlaps and parallels in the regulation of intrinsic multiple-antibiotic resistance in Escherichia coli. Mol Microbiol, 1996. 21(3): p. 441-8.
294. George, A.M., R.M. Hall, and H.W. Stokes, Multidrug resistance in Klebsiella pneumoniae: a novel gene, ramA, confers a multidrug resistance phenotype in Escherichia coli. Microbiology, 1995. 141 ( Pt 8): p. 1909-20.
295. Schneiders, T., S.G. Amyes, and S.B. Levy, Role of AcrR and ramA in fluoroquinolone resistance in clinical Klebsiella pneumoniae isolates from Singapore. Antimicrob Agents Chemother, 2003. 47(9): p. 2831-7.
296. Keeney, D., A. Ruzin, and P.A. Bradford, RamA, a transcriptional regulator, and AcrAB, an RND-type efflux pump, are associated with decreased susceptibility to tigecycline in Enterobacter cloacae. Microb Drug Resist, 2007. 13(1): p. 1-6.
297. Yassien, M.A., et al., Molecular cloning and characterization of the Salmonella enterica Serovar Paratyphi B rma Gene, which confers multiple drug resistance in Escherichia coli. Antimicrob Agents Chemother, 2002. 46(2): p. 360-6.
298. Chollet, R., et al., RamA is an alternate activator of the multidrug resistance cascade in Enterobacter aerogenes. Antimicrob Agents Chemother, 2004. 48(7): p. 2518-23.
299. Kallman, O., et al., Cefuroxime non-susceptibility in multidrug-resistant Klebsiella pneumoniae overexpressing ramA and acrA and expressing ompK35 at reduced levels. J Antimicrob Chemother, 2008. 62(5): p. 986-90.
300. Ruzin, A., et al., Influence of transcriptional activator RamA on expression of multidrug efflux pump AcrAB and tigecycline susceptibility in Klebsiella pneumoniae. Antimicrob Agents Chemother, 2005. 49(3): p. 1017-22.
301. Bertin, P., et al., H-NS and H-NS-like proteins in Gram-negative bacteria and their multiple role in the regulation of bacterial metabolism. Biochimie, 2001. 83(2): p. 235-41.
302. Masi, M., et al., The eefABC multidrug efflux pump operon is repressed by H-NS in Enterobacter aerogenes. J Bacteriol, 2005. 187(11): p. 3894-7.
303. Hasdemir, U.O., et al., Detection and prevalence of active drug efflux mechanism in various multidrug-resistant Klebsiella pneumoniae strains from Turkey. J Clin Microbiol, 2004. 42(6): p. 2701-6.
304. Guillier, M., S. Gottesman, and G. Storz, Modulating the outer membrane with small RNAs. Genes Dev, 2006. 20(17): p. 2338-48.
305. Vogel, J. and K. Papenfort, Small non-coding RNAs and the bacterial outer membrane. Curr Opin Microbiol, 2006. 9(6): p. 605-11.
306. Esterling, L. and N. Delihas, The regulatory RNA gene micF is present in several species of gram-negative bacteria and is phylogenetically conserved. Mol Microbiol, 1994. 12(4): p. 639-46.
307. Batchelor, E., et al., The Escherichia coli CpxA-CpxR envelope stress response system regulates expression of the porins ompF and ompC. J Bacteriol, 2005. 187(16): p. 5723-31.

REFERENCIAS 139
308. von Kruger, W.M., et al., The phosphate-starvation response in Vibrio cholerae O1 and phoB mutant under proteomic analysis: disclosing functions involved in adaptation, survival and virulence. Proteomics, 2006. 6(5): p. 1495-511.
309. Ferrario, M., et al., The leucine-responsive regulatory protein of Escherichia coli negatively regulates transcription of ompC and micF and positively regulates translation of ompF. J Bacteriol, 1995. 177(1): p. 103-13.
310. Lee, Y.H., et al., CadC has a global translational effect during acid adaptation in Salmonella enterica serovar Typhimurium. J Bacteriol, 2007. 189(6): p. 2417-25.
311. Santiviago, C.A., et al., The Salmonella enterica sv. Typhimurium smvA, yddG and ompD (porin) genes are required for the efficient efflux of methyl viologen. Mol Microbiol, 2002. 46(3): p. 687-98.
312. Miller, V.L. and J.J. Mekalanos, A novel suicide vector and its use in construction of insertion mutations: osmoregulation of outer membrane proteins and virulence determinants in Vibrio cholerae requires toxR. J Bacteriol, 1988. 170(6): p. 2575-83.
313. Deighan, P., A. Free, and C.J. Dorman, A role for the Escherichia coli H-NS-like protein StpA in OmpF porin expression through modulation of micF RNA stability. Mol Microbiol, 2000. 38(1): p. 126-39.
314. Painbeni, E., M. Caroff, and J. Rouviere-Yaniv, Alterations of the outer membrane composition in Escherichia coli lacking the histone-like protein HU. Proc Natl Acad Sci U S A, 1997. 94(13): p. 6712-7.
315. Ramani, N., L. Huang, and M. Freundlich, In vitro interactions of integration host factor with the ompF promoter-regulatory region of Escherichia coli. Mol Gen Genet, 1992. 231(2): p. 248-55.
316. De la Cruz, M.A. and E. Calva, The complexities of porin genetic regulation. J Mol Microbiol Biotechnol, 2010. 18(1): p. 24-36.
317. Hernandez-Alles, S., et al., Porin expression in clinical isolates of Klebsiella pneumoniae. Microbiology, 1999. 145 ( Pt 3): p. 673-9.
318. Biro, I., et al., Comparing the temperature-dependent conductance of the two structurally similar E. coli porins OmpC and OmpF. Biophys J, 2010. 98(9): p. 1830-9.
319. Lugtenberg, B., et al., Pore protein e of the outer membrane of Escherichia coli K12. FEBS Lett, 1978. 96(1): p. 99-105.
320. Duncan, A.R. and G. Winter, The binding site for C1q on IgG. Nature, 1988. 332(6166): p. 738-40. 321. Deisenhofer, J., Crystallographic refinement and atomic models of a human Fc fragment and its complex
with fragment B of protein A from Staphylococcus aureus at 2.9- and 2.8-A resolution. Biochemistry, 1981. 20(9): p. 2361-70.
322. Chen, J.H., et al., Contribution of outer membrane protein K36 to antimicrobial resistance and virulence in Klebsiella pneumoniae. J Antimicrob Chemother, 2010. 65(5): p. 986-90.
323. Tsai, Y.K., et al., Klebsiella pneumoniae Outer Membrane Porins OmpK35 and OmpK36 Play Roles in both Antimicrobial Resistance and Virulence. Antimicrob Agents Chemother, 2011. 55(4): p. 1485-93.
324. Martinez-Martinez, L., et al., In vivo selection of porin-deficient mutants of Klebsiella pneumoniae with increased resistance to cefoxitin and expanded-spectrum-cephalosporins. Antimicrob Agents Chemother, 1996. 40(2): p. 342-8.
325. Ardanuy, C., et al., Outer membrane profiles of clonally related Klebsiella pneumoniae isolates from clinical samples and activities of cephalosporins and carbapenems. Antimicrob Agents Chemother, 1998. 42(7): p. 1636-40.
326. Skurnik, D., et al., Development of ertapenem resistance in a patient with mediastinitis caused by Klebsiella pneumoniae producing an extended-spectrum beta-lactamase. J Med Microbiol, 2010. 59(Pt 1): p. 115-9.
327. Wang, X.D., et al., Reduced susceptibility to carbapenems in Klebsiella pneumoniae clinical isolates associated with plasmid-mediated beta-lactamase production and OmpK36 porin deficiency. J Med Microbiol, 2009. 58(Pt 9): p. 1196-202.
328. Tappero, J.W., et al., Immunogenicity of 2 serogroup B outer-membrane protein meningococcal vaccines: a randomized controlled trial in Chile. Jama, 1999. 281(16): p. 1520-7.
329. Pautsch, A. and G.E. Schulz, Structure of the outer membrane protein A transmembrane domain. Nat Struct Biol, 1998. 5(11): p. 1013-7.
330. Pautsch, A. and G.E. Schulz, High-resolution structure of the OmpA membrane domain. J Mol Biol, 2000. 298(2): p. 273-82.
331. Pautsch, A., et al., Strategy for membrane protein crystallization exemplified with OmpA and OmpX. Proteins, 1999. 34(2): p. 167-72.

REFERENCIAS 140
332. Arora, A., et al., Structure of outer membrane protein A transmembrane domain by NMR spectroscopy. Nat Struct Biol, 2001. 8(4): p. 334-8.
333. Wu, K.M., et al., Genome sequencing and comparative analysis of Klebsiella pneumoniae NTUH-K2044, a strain causing liver abscess and meningitis. J Bacteriol, 2009. 191(14): p. 4492-501.
334. Fouts, D.E., et al., Complete genome sequence of the N2-fixing broad host range endophyte Klebsiella pneumoniae 342 and virulence predictions verified in mice. PLoS Genet, 2008. 4(7): p. e1000141.
335. Khalid, S., et al., OmpA: gating and dynamics via molecular dynamics simulations. Biochim Biophys Acta, 2008. 1778(9): p. 1871-80.
336. Bremer, E., et al., Export of a protein into the outer membrane of Escherichia coli K12. Stable incorporation of the OmpA protein requires less than 193 amino-terminal amino-acid residues. Eur J Biochem, 1982. 122(1): p. 223-31.
337. Sugawara, E., et al., Secondary structure of the outer membrane proteins OmpA of Escherichia coli and OprF of Pseudomonas aeruginosa. J Bacteriol, 1996. 178(20): p. 6067-9.
338. Renault, M., et al., Solution state NMR structure and dynamics of KpOmpA, a 210 residue transmembrane domain possessing a high potential for immunological applications. J Mol Biol, 2009. 385(1): p. 117-30.
339. Smith, S.G., et al., A molecular Swiss army knife: OmpA structure, function and expression. FEMS Microbiol Lett, 2007. 273(1): p. 1-11.
340. Hong, H., G. Szabo, and L.K. Tamm, Electrostatic couplings in OmpA ion-channel gating suggest a mechanism for pore opening. Nat Chem Biol, 2006. 2(11): p. 627-35.
341. Sugawara, E. and H. Nikaido, OmpA protein of Escherichia coli outer membrane occurs in open and closed channel forms. J Biol Chem, 1994. 269(27): p. 17981-7.
342. Stathopoulos, C., An alternative topological model for Escherichia coli OmpA. Protein Sci, 1996. 5(1): p. 170-3.
343. Koebnik, R., K.P. Locher, and P. Van Gelder, Structure and function of bacterial outer membrane proteins: barrels in a nutshell. Mol Microbiol, 2000. 37(2): p. 239-53.
344. Lugtenberg, B., et al., Influence of cultural conditions and mutations on the composition of the outer membrane proteins of Escherichia coli. Mol Gen Genet, 1976. 147(3): p. 251-62.
345. Nilsson, G., et al., Growth-rate dependent regulation of mRNA stability in Escherichia coli. Nature, 1984. 312(5989): p. 75-7.
346. Emory, S.A. and J.G. Belasco, The ompA 5' untranslated RNA segment functions in Escherichia coli as a growth-rate-regulated mRNA stabilizer whose activity is unrelated to translational efficiency. J Bacteriol, 1990. 172(8): p. 4472-81.
347. Emory, S.A., P. Bouvet, and J.G. Belasco, A 5'-terminal stem-loop structure can stabilize mRNA in Escherichia coli. Genes Dev, 1992. 6(1): p. 135-48.
348. Hansen, M.J., et al., The ompA 5' untranslated region impedes a major pathway for mRNA degradation in Escherichia coli. Mol Microbiol, 1994. 12(5): p. 707-16.
349. Vytvytska, O., et al., Host factor I, Hfq, binds to Escherichia coli ompA mRNA in a growth rate-dependent fashion and regulates its stability. Proc Natl Acad Sci U S A, 1998. 95(24): p. 14118-23.
350. Vytvytska, O., et al., Hfq (HF1) stimulates ompA mRNA decay by interfering with ribosome binding. Genes Dev, 2000. 14(9): p. 1109-18.
351. Rasmussen, A.A., et al., Regulation of ompA mRNA stability: the role of a small regulatory RNA in growth phase-dependent control. Mol Microbiol, 2005. 58(5): p. 1421-9.
352. Udekwu, K.I., et al., Hfq-dependent regulation of OmpA synthesis is mediated by an antisense RNA. Genes Dev, 2005. 19(19): p. 2355-66.
353. Udekwu, K.I. and E.G. Wagner, Sigma E controls biogenesis of the antisense RNA MicA. Nucleic Acids Res, 2007. 35(4): p. 1279-88.
354. Zuo, Y. and M.P. Deutscher, Exoribonuclease superfamilies: structural analysis and phylogenetic distribution. Nucleic Acids Res, 2001. 29(5): p. 1017-26.
355. Andrade, J.M., F. Cairrao, and C.M. Arraiano, RNase R affects gene expression in stationary phase: regulation of ompA. Mol Microbiol, 2006. 60(1): p. 219-28.
356. Schwarz, H., et al., Degrees of relatedness of T-even type E. coli phages using different or the same receptors and topology of serologically cross-reacting sites. Embo J, 1983. 2(3): p. 375-80.
357. Morona, R., M. Klose, and U. Henning, Escherichia coli K-12 outer membrane protein (OmpA) as a bacteriophage receptor: analysis of mutant genes expressing altered proteins. J Bacteriol, 1984. 159(2): p. 570-8.

REFERENCIAS 141
358. Riede, I., M.L. Eschbach, and U. Henning, DNA sequence heterogeneity in the genes of T-even type Escherichia coli phages encoding the receptor recognizing protein of the long tail fibers. Mol Gen Genet, 1984. 195(1-2): p. 144-52.
359. Montag, D., et al., Receptor-recognizing proteins of T-even type bacteriophages. Constant and hypervariable regions and an unusual case of evolution. J Mol Biol, 1987. 196(1): p. 165-74.
360. Prasadarao, N.V., et al., Outer membrane protein A of Escherichia coli contributes to invasion of brain microvascular endothelial cells. Infect Immun, 1996. 64(1): p. 146-53.
361. Sukumaran, S.K., H. Shimada, and N.V. Prasadarao, Entry and intracellular replication of Escherichia coli K1 in macrophages require expression of outer membrane protein A. Infect Immun, 2003. 71(10): p. 5951-61.
362. Jeannin, P., et al., Outer membrane protein A (OmpA): a new pathogen-associated molecular pattern that interacts with antigen presenting cells-impact on vaccine strategies. Vaccine, 2002. 20 Suppl 4: p. A23-7.
363. Weiser, J.N. and E.C. Gotschlich, Outer membrane protein A (OmpA) contributes to serum resistance and pathogenicity of Escherichia coli K-1. Infect Immun, 1991. 59(7): p. 2252-8.
364. Prasadarao, N.V., et al., A novel interaction of outer membrane protein A with C4b binding protein mediates serum resistance of Escherichia coli K1. J Immunol, 2002. 169(11): p. 6352-60.
365. Wu, H., et al., Surfactant proteins A and D inhibit the growth of Gram-negative bacteria by increasing membrane permeability. J Clin Invest, 2003. 111(10): p. 1589-602.
366. Belaaouaj, A., K.S. Kim, and S.D. Shapiro, Degradation of outer membrane protein A in Escherichia coli killing by neutrophil elastase. Science, 2000. 289(5482): p. 1185-8.
367. Hari-Dass, R., et al., Serum amyloid A protein binds to outer membrane protein A of gram-negative bacteria. J Biol Chem, 2005. 280(19): p. 18562-7.
368. Shah, C., R. Hari-Dass, and J.G. Raynes, Serum amyloid A is an innate immune opsonin for Gram-negative bacteria. Blood, 2006. 108(5): p. 1751-7.
369. Kim, S.K., et al., Direct detection and magnetic isolation of Chlamydia trachomatis major outer membrane protein-specific CD8+ CTLs with HLA class I tetramers. J Immunol, 2000. 165(12): p. 7285-92.
370. Poolman, J.T., Bacterial outer membrane protein vaccines. The meningococcal example. Adv Exp Med Biol, 1996. 397: p. 73-7.
371. Zhang, Y.X., et al., Protective monoclonal antibodies recognize epitopes located on the major outer membrane protein of Chlamydia trachomatis. J Immunol, 1987. 138(2): p. 575-81.
372. Karch, H. and K. Nixdorff, Antibody-producing cell responses to an isolated outer membrane protein and to complexes of this antigen with lipopolysaccharide or with vesicles of phospholipids from Proteus mirabilis. Infect Immun, 1981. 31(3): p. 862-7.
373. Mondino, A., A. Khoruts, and M.K. Jenkins, The anatomy of T-cell activation and tolerance. Proc Natl Acad Sci U S A, 1996. 93(6): p. 2245-52.
374. Goetsch, L., et al., Targeting of nasal mucosa-associated antigen-presenting cells in vivo with an outer membrane protein A derived from Klebsiella pneumoniae. Infect Immun, 2001. 69(10): p. 6434-44.
375. Rauly, I., et al., Carrier properties of a protein derived from outer membrane protein A of Klebsiella pneumoniae. Infect Immun, 1999. 67(11): p. 5547-51.
376. Llobet, E., et al., Klebsiella pneumoniae OmpA confers resistance to antimicrobial peptides. Antimicrob Agents Chemother, 2009. 53(1): p. 298-302.
377. Jeanteur, D., J.H. Lakey, and F. Pattus, The bacterial porin superfamily: sequence alignment and structure prediction. Mol Microbiol, 1991. 5(9): p. 2153-64.
378. Benedi, V.J., J. Jofre, and J.M. Tomas, Klebsiella pneumoniae C3 lipopolysaccharide mutants obtained by resistance to bacteriophage FC3-9. Ann Inst Pasteur Microbiol, 1986. 137B(1): p. 29-36.
379. Rasheed, J.K., et al., Characterization of the extended-spectrum beta-lactamase reference strain, Klebsiella pneumoniae K6 (ATCC 700603), which produces the novel enzyme SHV-18. Antimicrob Agents Chemother, 2000. 44(9): p. 2382-8.
380. Henning, U., W. Schmidmayr, and I. Hindennach, Major proteins of the outer cell envelope membrane of Escherichia coli K-12: multiple species of protein I. Mol Gen Genet, 1977. 154(3): p. 293-8.
381. Foulds, J. and T.J. Chai, New major outer membrane proteins found in an Escherichia coli tolF mutant resistant to bacteriophage TuIb. J Bacteriol, 1978. 133(3): p. 1478-83.
382. Pugsley, A.P., D.R. Lee, and C.A. Schnaitman, Genes affecting the major outer membrane proteins of Escherichia coli K-12: mutations at nmpA and nmpB. Mol Gen Genet, 1980. 177(4): p. 681-9.
383. Chai, T.J. and J. Foulds, Two bacteriophages which utilize a new Escherichia coli major outer membrane protein as part of their receptor. J Bacteriol, 1978. 135(1): p. 164-70.

REFERENCIAS 142
384. Chai, T.J. and J. Foulds, Inactivation of bacteriophages by protein E, a new major membrane protein isolated from an Escherichia coli mutant. J Bacteriol, 1979. 137(1): p. 226-33.
385. Mizuno, T., M.Y. Chou, and M. Inouye, A comparative study on the genes for three porins of the Escherichia coli outer membrane. DNA sequence of the osmoregulated ompC gene. J Biol Chem, 1983. 258(11): p. 6932-40.
386. Weiss, M.S., et al., Molecular architecture and electrostatic properties of a bacterial porin. Science, 1991. 254(5038): p. 1627-30.
387. Karshikoff, A., et al., Electrostatic properties of two porin channels from Escherichia coli. J Mol Biol, 1994. 240(4): p. 372-84.
388. Saint, N., et al., Structural and functional characterization of OmpF porin mutants selected for larger pore size. II. Functional characterization. J Biol Chem, 1996. 271(34): p. 20676-80.
389. Van Gelder, P., et al., Voltage sensing in the PhoE and OmpF outer membrane porins of Escherichia coli: role of charged residues. J Mol Biol, 1997. 269(4): p. 468-72.
390. Bauer, K., et al., One single lysine residue is responsible for the special interaction between polyphosphate and the outer membrane porin PhoE of Escherichia coli. J Biol Chem, 1989. 264(28): p. 16393-8.
391. Dargent, B., et al., The selectivity filter of voltage-dependent channels formed by phosphoporin (PhoE protein) from E. coli. Embo J, 1986. 5(4): p. 773-8.
392. Lakey, J.H., Voltage gating in porin channels. FEBS Lett, 1987. 211(1): p. 1-4. 393. Overbeeke, N. and B. Lugtenberg, Expression of outer membrane protein e of Escherichia coli K12 by
phosphate limitation. FEBS Lett, 1980. 112(2): p. 229-32. 394. Tommassen, J. and B. Lugtenberg, Localization of phoE, the structural gene for outer membrane protein e
in Escherichia coli K-12. J Bacteriol, 1981. 147(1): p. 118-23. 395. Overbeeke, N. and B. Lugtenberg, Recognition site for phosphorus-containing compounds and other
negatively charged solutes on the PhoE protein pore of the outer membrane of Escherichia coli K12. Eur J Biochem, 1982. 126(1): p. 113-8.
396. Tommassen, J. and B. Lugtenberg, Outer membrane protein e of Escherichia coli K-12 is co-regulated with alkaline phosphatase. J Bacteriol, 1980. 143(1): p. 151-7.
397. Lee, T.Y., et al., Phosphate regulon in members of the family Enterobacteriaceae: comparison of the phoB-phoR operons of Escherichia coli, Shigella dysenteriae, and Klebsiella pneumoniae. J Bacteriol, 1989. 171(12): p. 6593-9.
398. Wanner, B., Phosphorus assimilation and control of the phosphate regulon, in Escherichia coli and Salmonella : Cellular and Molecular Biology., I.J. Neidhardt FC, Lin ECC, Low KB, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE, Editor 1996, Am Soc Microbiol: Washington. p. 1357–1381.
399. Randall-Hazelbauer, L. and M. Schwartz, Isolation of the bacteriophage lambda receptor from Escherichia coli. J Bacteriol, 1973. 116(3): p. 1436-46.
400. Szmelcman, S. and M. Hofnung, Maltose transport in Escherichia coli K-12: involvement of the bacteriophage lambda receptor. J Bacteriol, 1975. 124(1): p. 112-8.
401. Charbit, A., et al., Maltose transport and starch binding in phage-resistant point mutants of maltoporin. Functional and topological implications. J Mol Biol, 1988. 201(3): p. 487-96.
402. Werts, C., et al., DNA sequence analysis of the lamB gene from Klebsiella pneumoniae: implications for the topology and the pore functions in maltoporin. Mol Gen Genet, 1992. 233(3): p. 372-8.
403. Luckey, M. and H. Nikaido, Diffusion of solutes through channels produced by phage lambda receptor protein of Escherichia coli: inhibition by higher oligosaccharides of maltose series. Biochem Biophys Res Commun, 1980. 93(1): p. 166-71.
404. Wang, Y.F., et al., Channel specificity: structural basis for sugar discrimination and differential flux rates in maltoporin. J Mol Biol, 1997. 272(1): p. 56-63.
405. Luckey, M. and H. Nikaido, Specificity of diffusion channels produced by lambda phage receptor protein of Escherichia coli. Proc Natl Acad Sci U S A, 1980. 77(1): p. 167-71.
406. Boos, W. and H. Shuman, Maltose/maltodextrin system of Escherichia coli: transport, metabolism, and regulation. Microbiol Mol Biol Rev, 1998. 62(1): p. 204-29.
407. Death, A., L. Notley, and T. Ferenci, Derepression of LamB protein facilitates outer membrane permeation of carbohydrates into Escherichia coli under conditions of nutrient stress. J Bacteriol, 1993. 175(5): p. 1475-83.
408. Klein, W. and W. Boos, Induction of the lambda receptor is essential for effective uptake of trehalose in Escherichia coli. J Bacteriol, 1993. 175(6): p. 1682-6.

REFERENCIAS 143
409. de Smit, M.H. and J. van Duin, Control of translation by mRNA secondary structure in Escherichia coli. A quantitative analysis of literature data. J Mol Biol, 1994. 244(2): p. 144-50.
410. Hall, M.N., et al., A role for mRNA secondary structure in the control of translation initiation. Nature, 1982. 295(5850): p. 616-8.
411. Death, A. and T. Ferenci, Between feast and famine: endogenous inducer synthesis in the adaptation of Escherichia coli to growth with limiting carbohydrates. J Bacteriol, 1994. 176(16): p. 5101-7.
412. Notley, L. and T. Ferenci, Differential expression of mal genes under cAMP and endogenous inducer control in nutrient-stressed Escherichia coli. Mol Microbiol, 1995. 16(1): p. 121-9.
413. Benz, R., et al., Investigation of the selectivity of maltoporin channels using mutant LamB proteins: mutations changing the maltodextrin binding site. Biochim Biophys Acta, 1992. 1104(2): p. 299-307.
414. Ferenci, T., et al., Lambda receptor in the outer membrane of Escherichia coli as a binding protein for maltodextrins and starch polysaccharides. J Bacteriol, 1980. 142(2): p. 521-6.
415. Freundlieb, S., U. Ehmann, and W. Boos, Facilitated diffusion of p-nitrophenyl-alpha-D-maltohexaoside through the outer membrane of Escherichia coli. Characterization of LamB as a specific and saturable channel for maltooligosaccharides. J Biol Chem, 1988. 263(1): p. 314-20.
416. Jordy, M., et al., Rate constants of sugar transport through two LamB mutants of Escherichia coli: comparison with wild-type maltoporin and LamB of Salmonella typhimurium. J Mol Biol, 1996. 259(4): p. 666-78.
417. Luckey, M. and H. Nikaido, Bacteriophage lambda receptor protein in Escherichia coli K-12: lowered affinity of some mutant proteins for maltose-binding protein in vitro. J Bacteriol, 1983. 153(2): p. 1056-9.
418. Nekolla, S., C. Andersen, and R. Benz, Noise analysis of ion current through the open and the sugar-induced closed state of the LamB channel of Escherichia coli outer membrane: evaluation of the sugar binding kinetics to the channel interior. Biophys J, 1994. 66(5): p. 1388-97.
419. de Vries, G.E., C.K. Raymond, and R.A. Ludwig, Extension of bacteriophage lambda host range: selection, cloning, and characterization of a constitutive lambda receptor gene. Proc Natl Acad Sci U S A, 1984. 81(19): p. 6080-4.
420. Pellinen, T., et al., Topology of the Erwinia chrysanthemi oligogalacturonate porin KdgM. Biochem J, 2003. 372(Pt 2): p. 329-34.
421. Subbarao, G.V. and B. van den Berg, Crystal structure of the monomeric porin OmpG. J Mol Biol, 2006. 360(4): p. 750-9.
422. Yildiz, O., et al., Structure of the monomeric outer-membrane porin OmpG in the open and closed conformation. Embo J, 2006. 25(15): p. 3702-13.
423. Ye, J. and B. van den Berg, Crystal structure of the bacterial nucleoside transporter Tsx. Embo J, 2004. 23(16): p. 3187-95.
424. van den Berg, B., et al., Crystal structure of the long-chain fatty acid transporter FadL. Science, 2004. 304(5676): p. 1506-9.
425. Hearn, E.M., D.R. Patel, and B. van den Berg, Outer-membrane transport of aromatic hydrocarbons as a first step in biodegradation. Proc Natl Acad Sci U S A, 2008. 105(25): p. 8601-6.
426. Signorell, G.A., et al., Projection maps of three members of the KdgM outer membrane protein family. J Struct Biol, 2007. 160(3): p. 395-403.
427. Wirth, C., et al., NanC crystal structure, a model for outer-membrane channels of the acidic sugar-specific KdgM porin family. J Mol Biol, 2009. 394(4): p. 718-31.
428. Blot, N., et al., The oligogalacturonate-specific porin KdgM of Erwinia chrysanthemi belongs to a new porin family. J Biol Chem, 2002. 277(10): p. 7936-44.
429. Condemine, G., et al., Function and expression of an N-acetylneuraminic acid-inducible outer membrane channel in Escherichia coli. J Bacteriol, 2005. 187(6): p. 1959-65.
430. Rodionov, D.A., M.S. Gelfand, and N. Hugouvieux-Cotte-Pattat, Comparative genomics of the KdgR regulon in Erwinia chrysanthemi 3937 and other gamma-proteobacteria. Microbiology, 2004. 150(Pt 11): p. 3571-90.
431. Condemine, G. and A. Ghazi, Differential regulation of two oligogalacturonate outer membrane channels, KdgN and KdgM, of Dickeya dadantii (Erwinia chrysanthemi). J Bacteriol, 2007. 189(16): p. 5955-62.
432. El-Labany, S., et al., Distant cis-active sequences and sialic acid control the expression of fimB in Escherichia coli K-12. Mol Microbiol, 2003. 49(4): p. 1109-18.
433. Vimr, E.R., et al., Diversity of microbial sialic acid metabolism. Microbiol Mol Biol Rev, 2004. 68(1): p. 132-53.

REFERENCIAS 144
434. Sohanpal, B.K., et al., Integrated regulatory responses of fimB to N-acetylneuraminic (sialic) acid and GlcNAc in Escherichia coli K-12. Proc Natl Acad Sci U S A, 2004. 101(46): p. 16322-7.
435. Sohanpal, B.K., et al., Multiple co-regulatory elements and IHF are necessary for the control of fimB expression in response to sialic acid and N-acetylglucosamine in Escherichia coli K-12. Mol Microbiol, 2007. 63(4): p. 1223-36.
436. Tato, M., et al., Carbapenem Heteroresistance in VIM-1-producing Klebsiella pneumoniae isolates belonging to the same clone: consequences for routine susceptibility testing. J Clin Microbiol, 2010. 48(11): p. 4089-93.
437. Grant, S.G., et al., Differential plasmid rescue from transgenic mouse DNAs into Escherichia coli methylation-restriction mutants. Proc Natl Acad Sci U S A, 1990. 87(12): p. 4645-9.
438. Herrero, M., V. de Lorenzo, and K.N. Timmis, Transposon vectors containing non-antibiotic resistance selection markers for cloning and stable chromosomal insertion of foreign genes in gram-negative bacteria. J Bacteriol, 1990. 172(11): p. 6557-67.
439. Rubires, X., et al., A gene (wbbL) from Serratia marcescens N28b (O4) complements the rfb-50 mutation of Escherichia coli K-12 derivatives. J Bacteriol, 1997. 179(23): p. 7581-6.
440. Walter, E.G. and D.E. Taylor, Comparison of tellurite resistance determinants from the IncP alpha plasmid RP4Ter and the IncHII plasmid pHH1508a. J Bacteriol, 1989. 171(4): p. 2160-5.
441. Institute, C.a.L.S., Performance standards for antimicrobial susceptibility testing. 16th informational supplement M100-S162006: Clinical and Laboratory Standards Institute, Wayne, PA.
442. Filip, C., et al., Solubilization of the cytoplasmic membrane of Escherichia coli by the ionic detergent sodium-lauryl sarcosinate. J Bacteriol, 1973. 115(3): p. 717-22.
443. Bradford, M.M., A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem, 1976. 72: p. 248-54.
444. Cabezon, V., et al., Analysis of Candida albicans plasma membrane proteome. Proteomics, 2009. 9(20): p. 4770-86.
445. Alcantar-Curiel, M.D., E. Garcia-Latorre, and J.I. Santos, Klebsiella pneumoniae 35 and 36 kDa porins are common antigens in different serotypes and induce opsonizing antibodies. Arch Med Res, 2000. 31(1): p. 28-36.
446. Towbin, H., T. Staehelin, and J. Gordon, Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A, 1979. 76(9): p. 4350-4.
447. Dhaese, P., et al., Rapid mapping of transposon insertion and deletion mutations in the large Ti-plasmids of Agrobacterium tumefaciens. Nucleic Acids Res, 1979. 7(7): p. 1837-49.
448. Sambrook, J. and D.W. Russel, Molecular cloning. A laboratory manual.2001, New York: Cold Spring Harbor Laboratory Press.
449. Bikandi, J., et al., In silico analysis of complete bacterial genomes: PCR, AFLP-PCR and endonuclease restriction. Bioinformatics, 2004. 20(5): p. 798-9.
450. Kibbe, W.A., OligoCalc: an online oligonucleotide properties calculator. Nucleic Acids Res, 2007. 35(Web Server issue): p. W43-6.
451. Oh, H., et al., Role of efflux pumps and mutations in genes for topoisomerases II and IV in fluoroquinolone-resistant Pseudomonas aeruginosa strains. Microb Drug Resist, 2003. 9(4): p. 323-8.
452. Li, D.W., et al., Properties and expression of a multidrug efflux pump AcrAB-KocC from Klebsiella pneumoniae. Biol Pharm Bull, 2008. 31(4): p. 577-82.
453. Crane, G.J., S.M. Thomas, and M.E. Jones, A modified Luria-Delbruck fluctuation assay for estimating and comparing mutation rates. Mutat Res, 1996. 354(2): p. 171-82.
454. Moya, B., et al., Benefit of having multiple ampD genes for acquiring beta-lactam resistance without losing fitness and virulence in Pseudomonas aeruginosa. Antimicrob Agents Chemother, 2008. 52(10): p. 3694-700.
455. Doumith, M., et al., Molecular mechanisms disrupting porin expression in ertapenem-resistant Klebsiella and Enterobacter spp. clinical isolates from the UK. J Antimicrob Chemother, 2009. 63(4): p. 659-67.
456. Jacoby, G.A., D.M. Mills, and N. Chow, Role of beta-lactamases and porins in resistance to ertapenem and other beta-lactams in Klebsiella pneumoniae. Antimicrob Agents Chemother, 2004. 48(8): p. 3203-6.
457. Findlay, J., et al., Rapid acquisition of decreased carbapenem susceptibility in a strain of Klebsiella pneumoniae arising during meropenem therapy. Clin Microbiol Infect, 2011.
458. Crowley, B., V.J. Benedi, and A. Domenech-Sanchez, Expression of SHV-2 beta-lactamase and of reduced amounts of OmpK36 porin in Klebsiella pneumoniae results in increased resistance to cephalosporins and carbapenems. Antimicrob Agents Chemother, 2002. 46(11): p. 3679-82.

REFERENCIAS 145
459. Cornaglia, G., et al., Relative importances of outer membrane permeability and group 1 beta-lactamase as determinants of meropenem and imipenem activities against Enterobacter cloacae. Antimicrob Agents Chemother, 1995. 39(2): p. 350-5.
460. Pragai, Z. and E. Nagy, In-vitro selection of porin-deficient mutants of two strains of Klebsiella pneumoniae with reduced susceptibilities to meropenem, but not to imipenem. J Antimicrob Chemother, 1998. 42(6): p. 821-4.
461. Domenech-Sanchez, A., et al., Evaluation of differential gene expression in susceptible and resistant clinical isolates of Klebsiella pneumoniae by DNA microarray analysis. Clin Microbiol Infect, 2006. 12(9): p. 936-40.
462. Jeong, H., et al., Genome sequences of Escherichia coli B strains REL606 and BL21(DE3). J Mol Biol, 2009. 394(4): p. 644-52.
463. Aono, R., N. Tsukagoshi, and M. Yamamoto, Involvement of outer membrane protein TolC, a possible member of the mar-sox regulon, in maintenance and improvement of organic solvent tolerance of Escherichia coli K-12. J Bacteriol, 1998. 180(4): p. 938-44.
464. Gayet, S., et al., Modification of outer membrane protein profile and evidence suggesting an active drug pump in Enterobacter aerogenes clinical strains. Antimicrob Agents Chemother, 2003. 47(5): p. 1555-9.
465. Click, E.M. and C.A. Schnaitman, Export-defective lamB protein is a target for translational control caused by ompC porin overexpression. J Bacteriol, 1989. 171(1): p. 616-9.
466. Diedrich, D.L. and J.A. Fralick, Relationship between the OmpC and LamB proteins of Escherichia coli and its influence on the protein mass of the outer membrane. J Bacteriol, 1982. 149(1): p. 156-60.
467. Kitchel, B., et al., Genetic factors associated with elevated carbapenem resistance in KPC-producing Klebsiella pneumoniae. Antimicrob Agents Chemother. 54(10): p. 4201-7.
468. Landman, D., S. Bratu, and J. Quale, Contribution of OmpK36 to carbapenem susceptibility in KPC-producing Klebsiella pneumoniae. J Med Microbiol, 2009. 58(Pt 10): p. 1303-8.
469. Garcia-Sureda, L., et al., Role of Klebsiella pneumoniae LamB Porin in antimicrobial resistance. Antimicrob Agents Chemother, 2011. 55(4): p. 1803-5.
470. Bialek, S., et al., Membrane efflux and influx modulate both multidrug resistance and virulence of Klebsiella pneumoniae in a Caenorhabditis elegans model. Antimicrob Agents Chemother, 2010. 54(10): p. 4373-8.
471. Nicolau, D.P., Predicting antibacterial response from pharmacodynamic and pharmacokinetic profiles. Infection, 2001. 29 Suppl 2: p. 11-5.
472. Lopez, E. and J. Blazquez, Effect of subinhibitory concentrations of antibiotics on intrachromosomal homologous recombination in Escherichia coli. Antimicrob Agents Chemother, 2009. 53(8): p. 3411-5.
473. Folkesson, A., et al., Components of the peptidoglycan-recycling pathway modulate invasion and intracellular survival of Salmonella enterica serovar Typhimurium. Cell Microbiol, 2005. 7(1): p. 147-55.
474. Martin, N.J., In search of fungus artifactus. Radiol Technol, 1992. 63(3): p. 205.


8. ANEXOS

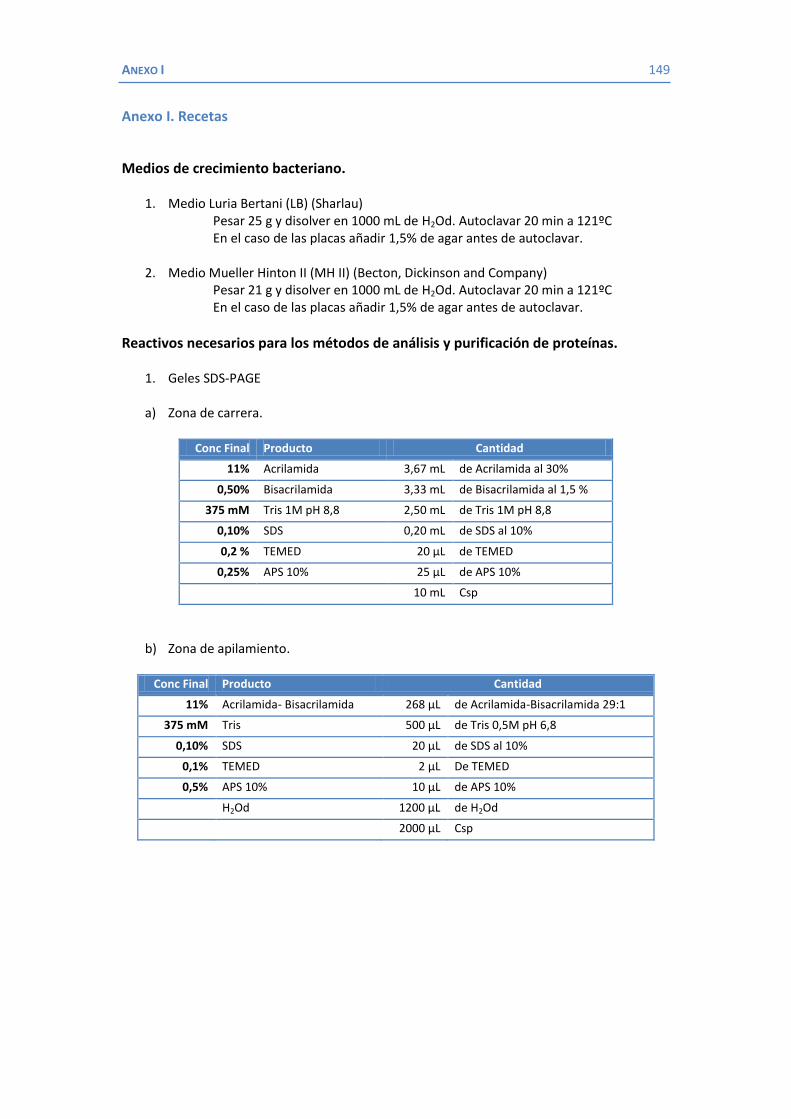
ANEXO I 149
Anexo I. Recetas
Medios de crecimiento bacteriano.
1. Medio Luria Bertani (LB) (Sharlau) Pesar 25 g y disolver en 1000 mL de H2Od. Autoclavar 20 min a 121ºC En el caso de las placas añadir 1,5% de agar antes de autoclavar.
2. Medio Mueller Hinton II (MH II) (Becton, Dickinson and Company)
Pesar 21 g y disolver en 1000 mL de H2Od. Autoclavar 20 min a 121ºC En el caso de las placas añadir 1,5% de agar antes de autoclavar.
Reactivos necesarios para los métodos de análisis y purificación de proteínas.
1. Geles SDS-PAGE
a) Zona de carrera.
Conc Final Producto Cantidad
11% Acrilamida 3,67 mL de Acrilamida al 30%
0,50% Bisacrilamida 3,33 mL de Bisacrilamida al 1,5 %
375 mM Tris 1M pH 8,8 2,50 mL de Tris 1M pH 8,8
0,10% SDS 0,20 mL de SDS al 10%
0,2 % TEMED 20 µL de TEMED
0,25% APS 10% 25 µL de APS 10%
10 mL Csp
b) Zona de apilamiento.
Conc Final Producto Cantidad
11% Acrilamida- Bisacrilamida 268 µL de Acrilamida-Bisacrilamida 29:1
375 mM Tris 500 µL de Tris 0,5M pH 6,8
0,10% SDS 20 µL de SDS al 10%
0,1% TEMED 2 µL De TEMED
0,5% APS 10% 10 µL de APS 10%
H2Od 1200 µL de H2Od
2000 µL Csp
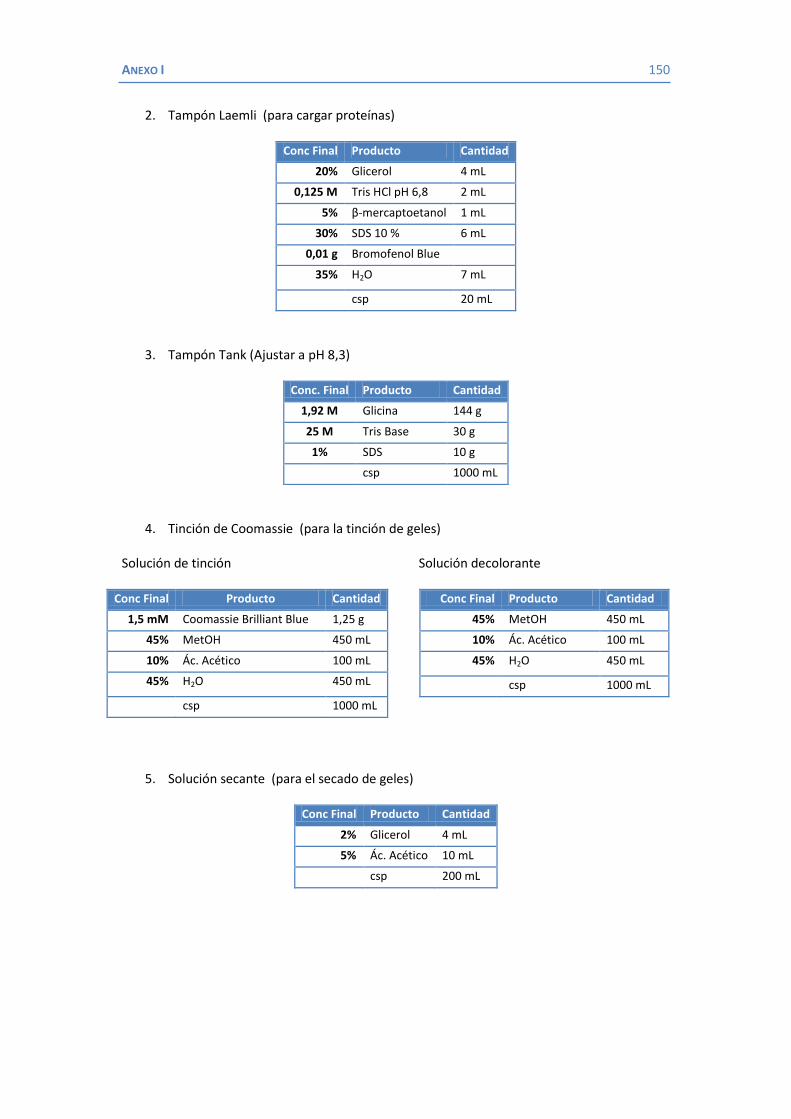
ANEXO I 150
2. Tampón Laemli (para cargar proteínas)
Conc Final Producto Cantidad
20% Glicerol 4 mL
0,125 M Tris HCl pH 6,8 2 mL
5% β-mercaptoetanol 1 mL
30% SDS 10 % 6 mL
0,01 g Bromofenol Blue
35% H2O 7 mL
csp 20 mL
3. Tampón Tank (Ajustar a pH 8,3)
Conc. Final Producto Cantidad
1,92 M Glicina 144 g
25 M Tris Base 30 g
1% SDS 10 g
csp 1000 mL
4. Tinción de Coomassie (para la tinción de geles) Solución de tinción
Conc Final Producto Cantidad
1,5 mM Coomassie Brilliant Blue 1,25 g
45% MetOH 450 mL
10% Ác. Acético 100 mL
45% H2O 450 mL
csp 1000 mL
Solución decolorante
Conc Final Producto Cantidad
45% MetOH 450 mL
10% Ác. Acético 100 mL
45% H2O 450 mL
csp 1000 mL
5. Solución secante (para el secado de geles)
Conc Final Producto Cantidad
2% Glicerol 4 mL
5% Ác. Acético 10 mL
csp 200 mL

ANEXO I 151
6. Tampón de Cromatografía (para la purificación de proteínas)
Conc Final Producto Cantidad
50 mM Tris 15,76 g
0,4 M NaCl 46,75 g
5 mM EDTA 3,72 g
3mM azida sódica 0,39 g
0,05% β-mercaptoetanol 1 mL
1% SDS 20 g
csp 2000 mL
7. Solución de revelado del ELISA
Para el revelado mezclar:
5 mL de H2O
5 mL Tampón Coating *
30 µL MgCl2 3M
2 pastillas de p-nitrofenilfosfato
8. Tampón Towbin (para la transferencia de geles por Western blot).
Conc. Final Producto Cantidad
25mM TrisBase 3,06 g
190 mM Glicina 14,3 g
20% MetOH 200 mL
csp 1000 mL
Reactivos necesarios para los métodos de análisis y purificación de ácidos nucleicos.
9. Tampón de carga de ADN
Conc. Final Producto Cantidad
0,10% Azul de bromofenol 0,1 g
0,1 M EDTA-Na2 4 mL de EDTA 0,25 M
0,10% SDS 100 µL de SDS 10%
50% Glicerol 5 mL
de H2O 900 mL
Tampón Coating (pH 9,5)
Na2CO3 1,59 g
NaHCO3 2,93 g
csp 1000 mL
Guardar a 4ºC un máximo de 15 días
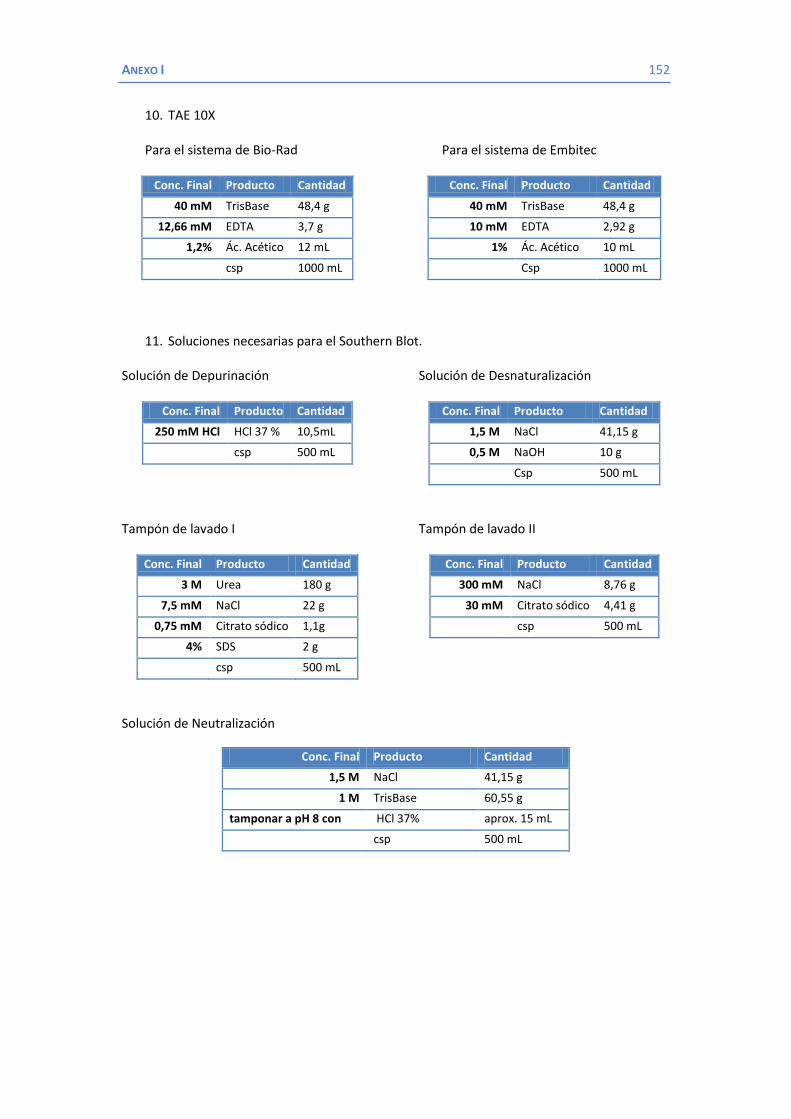
ANEXO I 152
10. TAE 10X
Para el sistema de Bio-Rad
Conc. Final Producto Cantidad
40 mM TrisBase 48,4 g
12,66 mM EDTA 3,7 g
1,2% Ác. Acético 12 mL
csp 1000 mL
Para el sistema de Embitec
Conc. Final Producto Cantidad
40 mM TrisBase 48,4 g
10 mM EDTA 2,92 g
1% Ác. Acético 10 mL
Csp 1000 mL
11. Soluciones necesarias para el Southern Blot. Solución de Depurinación
Conc. Final Producto Cantidad
250 mM HCl HCl 37 % 10,5mL
csp 500 mL
Solución de Desnaturalización
Conc. Final Producto Cantidad
1,5 M NaCl 41,15 g
0,5 M NaOH 10 g
Csp 500 mL
Tampón de lavado I
Conc. Final Producto Cantidad
3 M Urea 180 g
7,5 mM NaCl 22 g
0,75 mM Citrato sódico 1,1g
4% SDS 2 g
csp 500 mL
Tampón de lavado II
Conc. Final Producto Cantidad
300 mM NaCl 8,76 g
30 mM Citrato sódico 4,41 g
csp 500 mL
Solución de Neutralización
Conc. Final Producto Cantidad
1,5 M NaCl 41,15 g
1 M TrisBase 60,55 g
tamponar a pH 8 con HCl 37% aprox. 15 mL
csp 500 mL
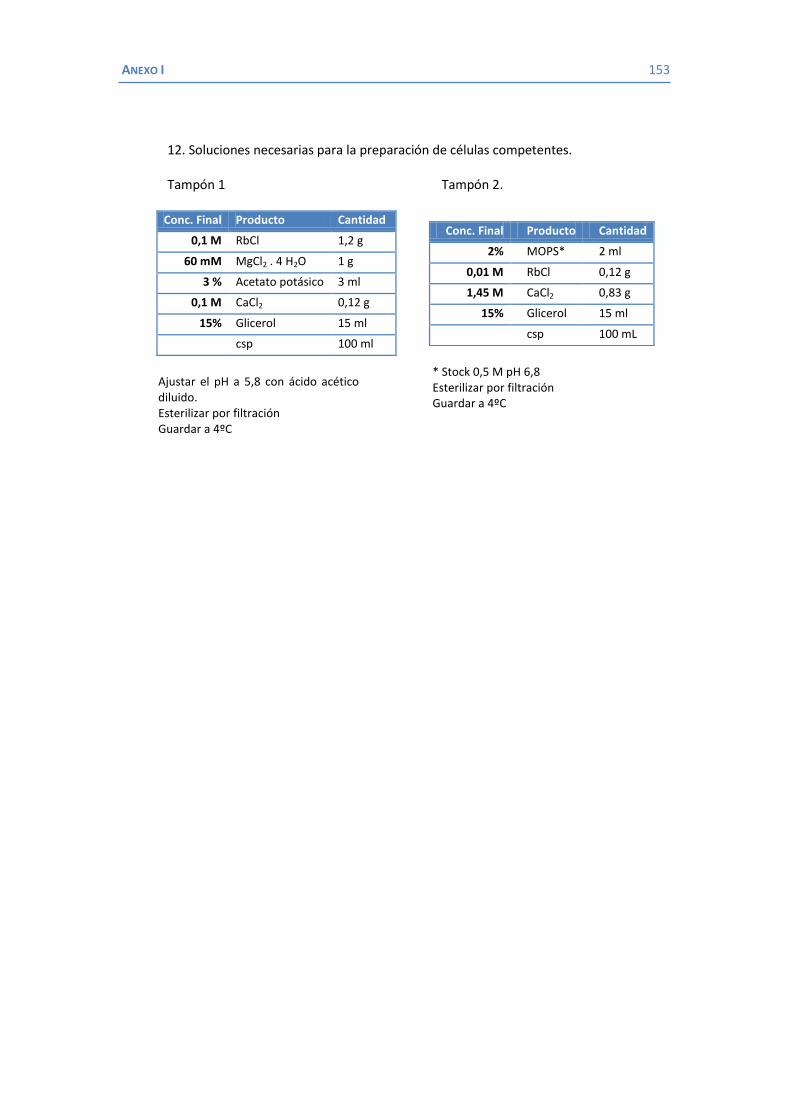
ANEXO I 153
12. Soluciones necesarias para la preparación de células competentes.
Tampón 1
Conc. Final Producto Cantidad
0,1 M RbCl 1,2 g
60 mM MgCl2 . 4 H2O 1 g
3 % Acetato potásico 3 ml
0,1 M CaCl2 0,12 g
15% Glicerol 15 ml
csp 100 ml
Ajustar el pH a 5,8 con ácido acético diluido. Esterilizar por filtración Guardar a 4ºC
Tampón 2.
* Stock 0,5 M pH 6,8 Esterilizar por filtración Guardar a 4ºC
Conc. Final Producto Cantidad
2% MOPS* 2 ml
0,01 M RbCl 0,12 g
1,45 M CaCl2 0,83 g
15% Glicerol 15 ml
csp 100 mL


ANEXO II 155
Anexo II.
Secuencia de nucleótidos del gen yjhA, el gen anterior (yqeF) y el gen posterior (tolB).
3’- ATGAAGGATGTCGTGATCGTCGGCGCGCTGCGAACGCCGATTGGCTGCTTTCAGGGCGCATTATCGCGCCATTCAGCCGTC GAGCTGGGGAGCGTGGTGGTGAAGGCGCTGGTGGAGAAGACGGGAATCGATCCGCACAGCGTGGATGAGGTGATCCTCGGCCAGGTGCTGACCGCGGGGACCGGACAAAATCCGGCGCGTCAGTCCGCTATTCGCGGTGGGCTGCCTAACACCGTATCGGCTATCACCATTAACGATGTCTGCGGTTCCGGCCTGAAGGCTCTGCACCTCGCTACCCAGGCGATCCAGTGCGGGGAAGCGGACGTGGTGATCGCTGGCGGCCAGGAGAATATGAGCCGCGCTCCGCATGTGCTTACCGACAGCCGCACCGGCGCCCAGCTGATTGACAGTCTGGTGCATGATGGGCTGTGGGATGCTTTCAACGATTATCATATGGGCGTCACGGCGGAAAACCTGGCCCGGGAATTTGGCATCAGTCGCGAGCTGCAGGACGCCTGGGCGCTCAGCTCACAGCATAAGGCGCGCAAGGCGATTGATTCCGGTCGCTTTCGCGATGAGATCGTCCCGGTTGTCACCGAGCATAACGGCGCGGCGCGCACCGTGGATACCGATGAACAACCGCGGGTGGATGCCAGCGCGGAAGGGCTGGCCAGCCTGCAGCCCACCTTCGATCGCCTTGGATCGGTAACCGCCGGCAACGCCTCAAGCATCAATGACGGCGCGGCGGCGGTAATGATGATGAGCGAGGCTAAAGCGCTGGAGCTGGGGCTGCCGATCCTTGCGCGGATCCGCGCCTTCGCCAGCGTCGGCGTCGATCCGGCGCTGATGGGCATTGCCCCGGTGCATGCCACCCGTCGCTGTCTGGAGCGGGCCGGCTGGCGGCTGGACGACGTCGATCTGATCGAAGCCAATGAGGCCTTTGCCGCTCAGGCGATCTCGGTTGGCCGGGTACTGGAGTGGGATGAGCGCCGGGTCAACGTCAACGGCGGCGCGATCGCCCTCGGCCACCCCATCGGCGCCTCCGGCTGCCGGATTCTGGTGTCGCTGGTGCATGAAATGATCAAGCGCGATGCCCGCAAAGGTCTGGCGACGCTGTGCATTGGCGGCGGTCAGGGCGTGGCGCTGGCGGTAGAACGCGCATAGCGCTATACCTCGATTTCCATGGCCTCCTGCGGGAGGCCTTTTTTATGCGCCGTGCTTTGCCGAACCCTTATTTTTCCCTCCCTCTTTCACTCTCCCCGGCGAAATTATTGAGCTTCGTCACGTCTTGTTGTGCCACCTTTTTTGAAACAAATAACTACGATCCTGGTCACTAAAACTATGGTGCCTAAAAAATAAAATGATGTTTCATAAAAACGAAATGTCATTTTATTTATTGTAAGGGTACCCTATGTTAAAACGCTCTCTGGTTCTCGCCGCCCTCTGCGGTATGTCTTTTGCAGCAACCGCGGTAACGATAGATTTACGTCATGAATTTATTGATGGCGGAAAGAGTGATAAAAGTAATGCTGACCGCGTCTCCGTTTCTCACCGCTTTGCCAACGGCTTAGGCTTTACCGTCGAGGCCAAATGGCGCTCAGGCGGCGACAACGGTTCGCAACCCTATTCGGACGTGGTGGGCAATGGTCATGAAGACACCATCAGCTGGCGCTGGAAAGCGACGTCGAACTTGTTCCTCACCCCCGGGTTTACCATTGAGAGCAACGACAGCCGCTCCATCTACAAGCCGCACCTGCACGCGCAGTACAGTTTCGACAACGGCTTCTATGTCGCCGCTCGCTATCGCTATGAATACACCCGCTATCCGAACAACGCCGGCAAAGATGATGACAAAGTTAACCGCGGCGACGCGTGGGCCGGATTTGTCCTCGGCGACTGGCGTACAGAGCTGAACTATGTCTATGCCCGCAGCTCGGAAGGGGTCAGCCGCAACGACAATAAACCCTACTCGCAGGAATATAACGTGAAGGTGGCCTATAAGCTGGATAATAACTGGTCGCCGTATGGCGAAATTGGCAACGTCGGGGTCAATGACCGCAGCGATCGCCAGACCCGCTTCCGCGTTGGGGTGGCCTACTCGTTCTGACCGACCATTCATCCCTCATCGTTTGTTTTCCCCGGCGCCGTCAGCCGGGGTTTTTTTATGGGGGAAAGAGAAGGGACAAAAAAAGCCCTCCATGAAGGAGGGCAAGGCCAGTTACAGAAAAAAGTGTGGCGTGGATCAGGCGCTCAGCATTGACGGGTGCGCGGGTAATTTCGCGATATAGATGGCCGGTTTACCGTCTTTATCGGAGCTGAACAGAATGGCGCTATCGTCCGGGGTAAATGAGGGGTGCGGGTGCGTTACCTGGCGGCTGTTGGCCACCGTCGCCCACGAGGTATCATGGCGCGCGACGCGGAAGTAGCGCTTTTCCGCCACGTTAAAGACATACAGCCAGGGATCGTTATCAATGGAGTAGCCGCCGGTATCCTTGACATCCACCGGTGTACCTGAGCCATCGCCTACCAGCAGCGTGCCATCGAAATTGCTCATCAGATGCGAGCAGGCCGGCATCGTCATCAGGGCCTCGTTGACGCCGCTTTCGGGATCAAAGCGATAGATCGTCCGCCCCTGCTGGCCTTTCAGATAGGAGACGTAGATCAGCGCCGAACCGTCCGGCACCCAAAATTCATGAGTACAGCTTTCGCCTGGAGCATGCGCTTTGACTTTGCGAACATGGCTGCCATCCTCGTTGACCAGCCACATGCGGGCGTCCACCAGATCGTGCGGCCCTTCATGGCAGAAGGCGACGGTATGGTCGTCGAAGGGGCGATAGATCGGGTGTCCCAGCCAGATTTTTTCCTCATGGATCACCTGGCTCGCACCGCTGCGCAGGTCGACGCGCAGCAGGCGGCAGTGCGGTCCTTTATGGAAGAAATCATGGAAAATCTGCCAGTCGTTGAGTGGCGTCCAGTCGCTTTTGGCAATCTCAATGCCCACCAGTTTGCTGCAATCGCTGTTTGCCACCCAGGTGCCATACCCTACCCAGTCATCGCTAACGCGATAAACTTCGCGCTCGGCGAGGGTGGTCAGGTTGACTTCCAGCAGGATACGGTCATTTTTCACGTAATAAAGCGACTTATCGTCCGGGGAGAGGAAGCCGCCGAAGGTGTTATCCCCGGCGCCTTCGGTTAGCTGGACGGCCTCGGCGCTGGCGATATTCAGCAGATAGTAGTTCCAGTGGCCGTCAAACTCGCCGGCGAACAGTAAATGGCTGCCATCGTTAAAAAAGCACTTCTGATAGAAGTAGTTGCGATGACAGGTGACCTCCGGGGGGGTTAAGCGGGTGACTTCCGCCCCGGTATCCGGATCGTGGCTGACGTGATAATTCAGTTTGACCCGCATGCCTTTAGCCAT – 5’
Leyenda Genes: yjhA: en azul yqeF gen anterior a yjhA: en rojo tolB (gen posterior a yjhA): en verde
Secuencias reguladoras: Sitio de unión de OmpR Pribnow box Posibles secuencias Shine Dalgarno
NOTA: La secuencia de nucleótidos corresponde a la hebra de ADN complementaria.


ANEXO III 157
Anexo III Artículos
Garcia-Sureda, L., Juan, C., Domenech-Sanchez, A., Alberti, S. Role of Klebsiella pneumoniae LamB Porin in antimicrobial resistance. Antimicrob Agents Chemother, 2011. 55(4): p. 1803-5. Garcia-Sureda, L., Domenech-Sanchez, A., Barbier, M., Juan, C., Gasco, J., Alberti, S. OmpK26, a novel porin associated with carbapenem resistance in Klebsiella pneumoniae. Antimicrob Agents Chemother, 2011. 55(10): p. 4742-7. Mena, A., Plasencia, V., Garcia, L., Hidalgo, O., Ayestaran, J. I., Alberti, S., Borrell, N., Perez, J. L., Oliver, A.et al., Characterization of a large outbreak by CTX-M-1-producing Klebsiella pneumoniae and mechanisms leading to in vivo carbapenem resistance development. J Clin Microbiol, 2006. 44(8): p. 2831-7. Tato, M., Morosini, M., Garcia, L., Alberti, S., Coque, M. T., Canton, R Carbapenem Heteroresistance in VIM-1-producing Klebsiella pneumoniae isolates belonging to the same clone: consequences for routine susceptibility testing. J Clin Microbiol, 2010. 48(11): p. 4089-93


ANTIMICROBIAL AGENTS AND CHEMOTHERAPY, Apr. 2011, p. 1803–1805 Vol. 55, No. 40066-4804/11/$12.00 doi:10.1128/AAC.01441-10Copyright © 2011, American Society for Microbiology. All Rights Reserved.
Role of Klebsiella pneumoniae LamB Porin in Antimicrobial Resistance!
Laura García-Sureda,1 Carlos Juan,1,2 Antonio Domenech-Sanchez,1 and Sebastian Albertí1*Instituto Universitario de Investigaciones en Ciencias de la Salud, Universidad de las Islas Baleares, Palma de Mallorca, Spain,1
and Servicio de Microbiología y Unidad de Investigacion, Hospital Universitario Son Dureta, Palma de Mallorca, Spain2
Received 19 October 2010/Returned for modification 23 November 2010/Accepted 21 January 2011
To investigate the contribution of LamB in Klebsiella pneumoniae antimicrobial resistance, we determined theMICs of various antibiotics and the frequency of mutation to increased cefoxitin or meropenem resistance ofthe strains CSUB10S (expressing only OmpK36), CSUB10R (lacking OmpK35 and OmpK36), and theirderived isogenic insertion-duplication mutants deficient in LamB. Expression of LamB was indispensable inorder for CSUB10S to lose OmpK36 and become resistant to cefoxitin, while in CSUB10R, LamB deficiencypromoted increased resistance to carbapenem.
An increasing emergence of multidrug resistance amongKlebsiella pneumoniae nosocomial isolates has limited the ther-apeutic options for the treatment of the intrahospital infec-tions caused by this opportunistic pathogen. A multidrug re-sistance phenotype results from the progressive accumulationof different mechanisms of resistance in the same microorgan-ism, including high-level production of extended-spectrum!-lactamases (ESBLs) and porin deficiency. K. pneumoniaeproduces two major porins, OmpK35 (8) and OmpK36 (1).However, most ESBL-expressing K. pneumoniae clinical iso-lates produce only OmpK36 (11). Clinical and experimentalevidences indicate that loss of these major porins in K. pneu-moniae strains producing ESBLs causes resistance to cefoxitin,increased resistance to expanded-spectrum cephalosporins andmonobactams, and decreased susceptibility to fluoroquinolo-nes (8, 9, 12–14, 15).
Besides the major porins, K. pneumoniae may express otherporins, such as PhoE, OmpK37, and LamB (3, 7, 13). Thesealternative porins may be crucial for the microorganism in theabsence of OmpK35/36, although their contribution to antimi-crobial resistance has been poorly investigated. In a previousstudy, the outer membrane protein (OMP) profile of the sus-ceptible K. pneumoniae clinical isolate CSUB10S (expressingonly OmpK36) and that of its resistant clinical derivativeCSUB10R (lacking both OmpK35 and OmpK36) were com-pared (3). It was observed that LamB may be a predominantOMP in the resistant isolate compared to the susceptible one,suggesting that LamB may contribute to resistance in K. pneu-moniae. To verify this result, the amounts of LamB present inthe outer membrane of CSUB10S and CSUB10R grown incation-supplemented Mueller-Hinton broth (MH) or in humanpools of urine or serum were analyzed by SDS-PAGE as pre-viously described (11). As shown in Fig. 1, the outer membraneof the resistant isolate contained larger amounts of LamB thanthat of the susceptible one, both in MH and in the physiolog-ical fluids. Densitometric analysis of the intensity of the bandof LamB normalized for the band of OmpA of three indepen-
dent gels demonstrated that in MH, CSUB10R produces 1.86-fold " 0.09-fold more LamB than CSUB10S (P # 0.001, two-tailed t test). Furthermore, reverse transcription-PCR (RT-PCR) quantification of the amount of lamB mRNA in bothstrains showed that CSUB10R expresses 2.17-fold " 0.6-foldmore lamB than CSUB10S (P # 0.04, two-tailed t test).
In order to investigate the role of LamB in K. pneumoniaeantimicrobial resistance, we used insertion-duplication mu-tagenesis to construct LamB-deficient knockouts from bothCSUB10S and CSUB10R, following the strategy previouslydescribed (16) (Fig. 2A). Southern blot analysis of the genomicDNA of both knockouts using a specific probe for lamB con-firmed that two incomplete copies of the gene were generatedby the integration of the plasmid (Fig. 2B). SDS-PAGE anal-ysis confirmed that the lamB mutation abolished the expres-sion of LamB in both knockouts (Fig. 2C).
The susceptibilities of the clinical isolates CSUB10S andCSUB10R and their derived isogenic LamB-deficient knock-outs, CSUB10S$lamB and CSUB10R$lamB, to a number ofantibiotics were determined using Etest strips (bioMerieux,Marcy l’Etoile, France), following the manufacturer’s instruc-tions (Table 1). The absence of LamB in the susceptible isolateCSUB10S did not caused significant changes in the MICs ofthe agents tested. Only modest reductions in the MICs ofceftazidime, cefepime, cefotaxime, tobramycin, amikacin, andlevofloxacin were observed. This was an expected result giventhat in the presence of OmpK36, the contribution of LamB tothe penetration of these antimicrobial agents is probably neg-
* Corresponding author. Mailing address: Edificio Científico-Tec-nico, CAMPUS-UIB, Ctra. Valldemosa, km 7.5, Palma de Mallorca07122, Spain. Phone: 34-971-173353. Fax: 34-971-259501. E-mail:[email protected].
! Published ahead of print on 31 January 2011.
FIG. 1. Representative SDS-PAGE analysis of the OMPs of K.pneumoniae CSUB10S and CSUB10R grown in cation-supplementedMH, urine, or serum. Molecular markers are indicated on the left.Black arrows indicate the positions of LamB and OmpK36.
1803
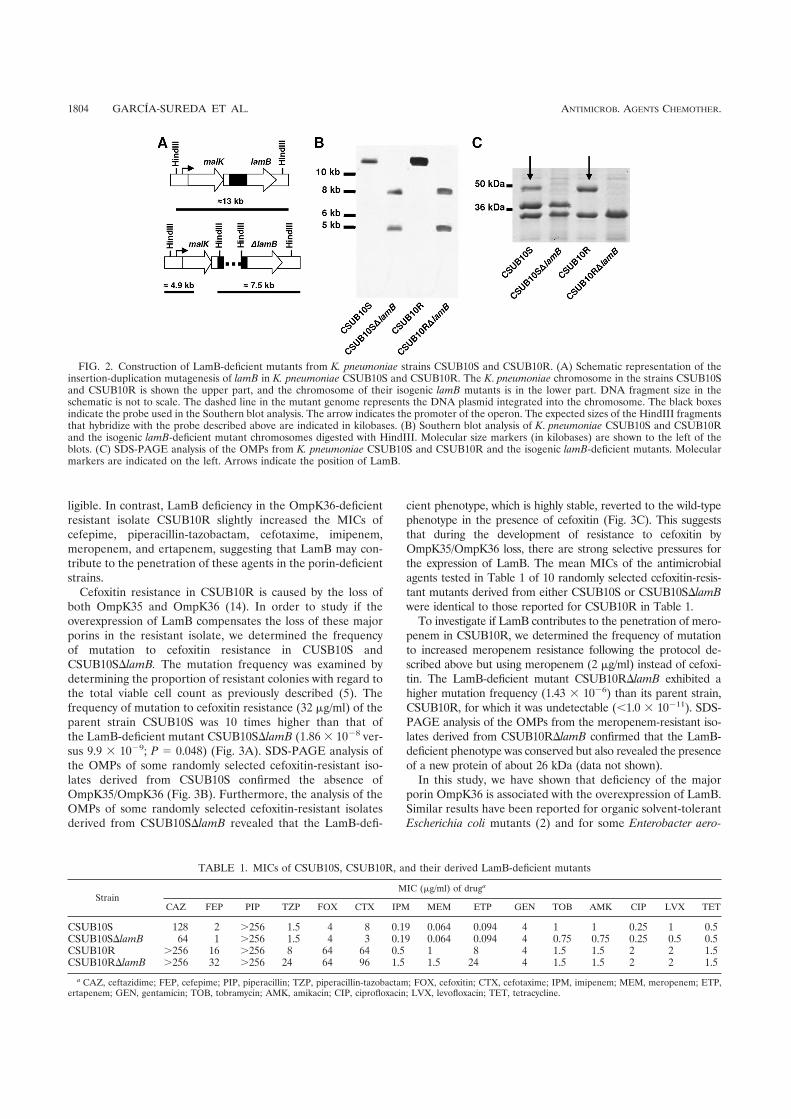
ligible. In contrast, LamB deficiency in the OmpK36-deficientresistant isolate CSUB10R slightly increased the MICs ofcefepime, piperacillin-tazobactam, cefotaxime, imipenem,meropenem, and ertapenem, suggesting that LamB may con-tribute to the penetration of these agents in the porin-deficientstrains.
Cefoxitin resistance in CSUB10R is caused by the loss ofboth OmpK35 and OmpK36 (14). In order to study if theoverexpression of LamB compensates the loss of these majorporins in the resistant isolate, we determined the frequencyof mutation to cefoxitin resistance in CUSB10S andCSUB10S$lamB. The mutation frequency was examined bydetermining the proportion of resistant colonies with regard tothe total viable cell count as previously described (5). Thefrequency of mutation to cefoxitin resistance (32 %g/ml) of theparent strain CSUB10S was 10 times higher than that ofthe LamB-deficient mutant CSUB10S$lamB (1.86 & 10'8 ver-sus 9.9 & 10'9; P # 0.048) (Fig. 3A). SDS-PAGE analysis ofthe OMPs of some randomly selected cefoxitin-resistant iso-lates derived from CSUB10S confirmed the absence ofOmpK35/OmpK36 (Fig. 3B). Furthermore, the analysis of theOMPs of some randomly selected cefoxitin-resistant isolatesderived from CSUB10S$lamB revealed that the LamB-defi-
cient phenotype, which is highly stable, reverted to the wild-typephenotype in the presence of cefoxitin (Fig. 3C). This suggeststhat during the development of resistance to cefoxitin byOmpK35/OmpK36 loss, there are strong selective pressures forthe expression of LamB. The mean MICs of the antimicrobialagents tested in Table 1 of 10 randomly selected cefoxitin-resis-tant mutants derived from either CSUB10S or CSUB10S$lamBwere identical to those reported for CSUB10R in Table 1.
To investigate if LamB contributes to the penetration of mero-penem in CSUB10R, we determined the frequency of mutationto increased meropenem resistance following the protocol de-scribed above but using meropenem (2 %g/ml) instead of cefoxi-tin. The LamB-deficient mutant CSUB10R$lamB exhibited ahigher mutation frequency (1.43 & 10'6) than its parent strain,CSUB10R, for which it was undetectable ((1.0 & 10'11). SDS-PAGE analysis of the OMPs from the meropenem-resistant iso-lates derived from CSUB10R$lamB confirmed that the LamB-deficient phenotype was conserved but also revealed the presenceof a new protein of about 26 kDa (data not shown).
In this study, we have shown that deficiency of the majorporin OmpK36 is associated with the overexpression of LamB.Similar results have been reported for organic solvent-tolerantEscherichia coli mutants (2) and for some Enterobacter aero-
FIG. 2. Construction of LamB-deficient mutants from K. pneumoniae strains CSUB10S and CSUB10R. (A) Schematic representation of theinsertion-duplication mutagenesis of lamB in K. pneumoniae CSUB10S and CSUB10R. The K. pneumoniae chromosome in the strains CSUB10Sand CSUB10R is shown the upper part, and the chromosome of their isogenic lamB mutants is in the lower part. DNA fragment size in theschematic is not to scale. The dashed line in the mutant genome represents the DNA plasmid integrated into the chromosome. The black boxesindicate the probe used in the Southern blot analysis. The arrow indicates the promoter of the operon. The expected sizes of the HindIII fragmentsthat hybridize with the probe described above are indicated in kilobases. (B) Southern blot analysis of K. pneumoniae CSUB10S and CSUB10Rand the isogenic lamB-deficient mutant chromosomes digested with HindIII. Molecular size markers (in kilobases) are shown to the left of theblots. (C) SDS-PAGE analysis of the OMPs from K. pneumoniae CSUB10S and CSUB10R and the isogenic lamB-deficient mutants. Molecularmarkers are indicated on the left. Arrows indicate the position of LamB.
TABLE 1. MICs of CSUB10S, CSUB10R, and their derived LamB-deficient mutants
StrainMIC (%g/ml) of druga
CAZ FEP PIP TZP FOX CTX IPM MEM ETP GEN TOB AMK CIP LVX TET
CSUB10S 128 2 )256 1.5 4 8 0.19 0.064 0.094 4 1 1 0.25 1 0.5CSUB10S$lamB 64 1 )256 1.5 4 3 0.19 0.064 0.094 4 0.75 0.75 0.25 0.5 0.5CSUB10R )256 16 )256 8 64 64 0.5 1 8 4 1.5 1.5 2 2 1.5CSUB10R$lamB )256 32 )256 24 64 96 1.5 1.5 24 4 1.5 1.5 2 2 1.5
a CAZ, ceftazidime; FEP, cefepime; PIP, piperacillin; TZP, piperacillin-tazobactam; FOX, cefoxitin; CTX, cefotaxime; IPM, imipenem; MEM, meropenem; ETP,ertapenem; GEN, gentamicin; TOB, tobramycin; AMK, amikacin; CIP, ciprofloxacin; LVX, levofloxacin; TET, tetracycline.
1804 GARCIA-SUREDA ET AL. ANTIMICROB. AGENTS CHEMOTHER.
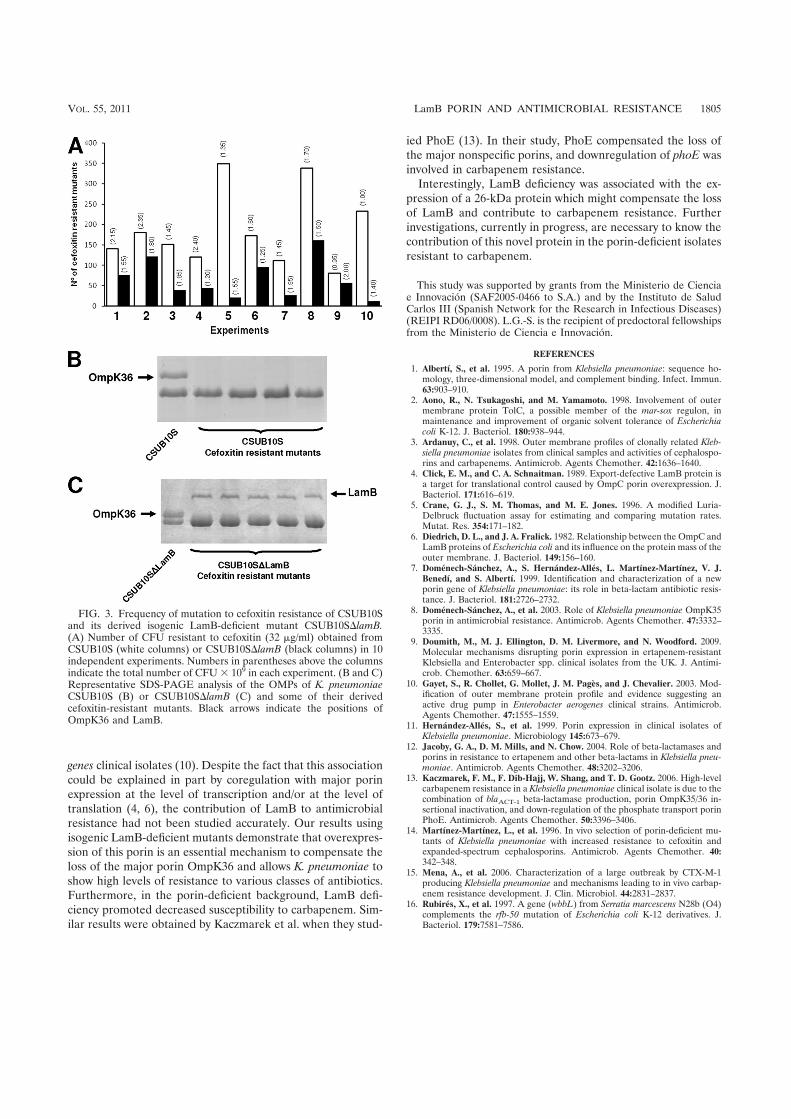
genes clinical isolates (10). Despite the fact that this associationcould be explained in part by coregulation with major porinexpression at the level of transcription and/or at the level oftranslation (4, 6), the contribution of LamB to antimicrobialresistance had not been studied accurately. Our results usingisogenic LamB-deficient mutants demonstrate that overexpres-sion of this porin is an essential mechanism to compensate theloss of the major porin OmpK36 and allows K. pneumoniae toshow high levels of resistance to various classes of antibiotics.Furthermore, in the porin-deficient background, LamB defi-ciency promoted decreased susceptibility to carbapenem. Sim-ilar results were obtained by Kaczmarek et al. when they stud-
ied PhoE (13). In their study, PhoE compensated the loss ofthe major nonspecific porins, and downregulation of phoE wasinvolved in carbapenem resistance.
Interestingly, LamB deficiency was associated with the ex-pression of a 26-kDa protein which might compensate the lossof LamB and contribute to carbapenem resistance. Furtherinvestigations, currently in progress, are necessary to know thecontribution of this novel protein in the porin-deficient isolatesresistant to carbapenem.
This study was supported by grants from the Ministerio de Cienciae Innovacion (SAF2005-0466 to S.A.) and by the Instituto de SaludCarlos III (Spanish Network for the Research in Infectious Diseases)(REIPI RD06/0008). L.G.-S. is the recipient of predoctoral fellowshipsfrom the Ministerio de Ciencia e Innovacion.
REFERENCES
1. Albertí, S., et al. 1995. A porin from Klebsiella pneumoniae: sequence ho-mology, three-dimensional model, and complement binding. Infect. Immun.63:903–910.
2. Aono, R., N. Tsukagoshi, and M. Yamamoto. 1998. Involvement of outermembrane protein TolC, a possible member of the mar-sox regulon, inmaintenance and improvement of organic solvent tolerance of Escherichiacoli K-12. J. Bacteriol. 180:938–944.
3. Ardanuy, C., et al. 1998. Outer membrane profiles of clonally related Kleb-siella pneumoniae isolates from clinical samples and activities of cephalospo-rins and carbapenems. Antimicrob. Agents Chemother. 42:1636–1640.
4. Click, E. M., and C. A. Schnaitman. 1989. Export-defective LamB protein isa target for translational control caused by OmpC porin overexpression. J.Bacteriol. 171:616–619.
5. Crane, G. J., S. M. Thomas, and M. E. Jones. 1996. A modified Luria-Delbruck fluctuation assay for estimating and comparing mutation rates.Mutat. Res. 354:171–182.
6. Diedrich, D. L., and J. A. Fralick. 1982. Relationship between the OmpC andLamB proteins of Escherichia coli and its influence on the protein mass of theouter membrane. J. Bacteriol. 149:156–160.
7. Domenech-Sanchez, A., S. Hernandez-Alles, L. Martínez-Martínez, V. J.Benedí, and S. Albertí. 1999. Identification and characterization of a newporin gene of Klebsiella pneumoniae: its role in beta-lactam antibiotic resis-tance. J. Bacteriol. 181:2726–2732.
8. Domenech-Sanchez, A., et al. 2003. Role of Klebsiella pneumoniae OmpK35porin in antimicrobial resistance. Antimicrob. Agents Chemother. 47:3332–3335.
9. Doumith, M., M. J. Ellington, D. M. Livermore, and N. Woodford. 2009.Molecular mechanisms disrupting porin expression in ertapenem-resistantKlebsiella and Enterobacter spp. clinical isolates from the UK. J. Antimi-crob. Chemother. 63:659–667.
10. Gayet, S., R. Chollet, G. Mollet, J. M. Pages, and J. Chevalier. 2003. Mod-ification of outer membrane protein profile and evidence suggesting anactive drug pump in Enterobacter aerogenes clinical strains. Antimicrob.Agents Chemother. 47:1555–1559.
11. Hernandez-Alles, S., et al. 1999. Porin expression in clinical isolates ofKlebsiella pneumoniae. Microbiology 145:673–679.
12. Jacoby, G. A., D. M. Mills, and N. Chow. 2004. Role of beta-lactamases andporins in resistance to ertapenem and other beta-lactams in Klebsiella pneu-moniae. Antimicrob. Agents Chemother. 48:3202–3206.
13. Kaczmarek, F. M., F. Dib-Hajj, W. Shang, and T. D. Gootz. 2006. High-levelcarbapenem resistance in a Klebsiella pneumoniae clinical isolate is due to thecombination of blaACT-1 beta-lactamase production, porin OmpK35/36 in-sertional inactivation, and down-regulation of the phosphate transport porinPhoE. Antimicrob. Agents Chemother. 50:3396–3406.
14. Martínez-Martínez, L., et al. 1996. In vivo selection of porin-deficient mu-tants of Klebsiella pneumoniae with increased resistance to cefoxitin andexpanded-spectrum cephalosporins. Antimicrob. Agents Chemother. 40:342–348.
15. Mena, A., et al. 2006. Characterization of a large outbreak by CTX-M-1producing Klebsiella pneumoniae and mechanisms leading to in vivo carbap-enem resistance development. J. Clin. Microbiol. 44:2831–2837.
16. Rubires, X., et al. 1997. A gene (wbbL) from Serratia marcescens N28b (O4)complements the rfb-50 mutation of Escherichia coli K-12 derivatives. J.Bacteriol. 179:7581–7586.
FIG. 3. Frequency of mutation to cefoxitin resistance of CSUB10Sand its derived isogenic LamB-deficient mutant CSUB10S$lamB.(A) Number of CFU resistant to cefoxitin (32 %g/ml) obtained fromCSUB10S (white columns) or CSUB10S$lamB (black columns) in 10independent experiments. Numbers in parentheses above the columnsindicate the total number of CFU & 109 in each experiment. (B and C)Representative SDS-PAGE analysis of the OMPs of K. pneumoniaeCSUB10S (B) or CSUB10S$lamB (C) and some of their derivedcefoxitin-resistant mutants. Black arrows indicate the positions ofOmpK36 and LamB.
VOL. 55, 2011 LamB PORIN AND ANTIMICROBIAL RESISTANCE 1805

!

ANTIMICROBIAL AGENTS AND CHEMOTHERAPY, Oct. 2011, p. 4742–4747 Vol. 55, No. 100066-4804/11/$12.00 doi:10.1128/AAC.00309-11Copyright © 2011, American Society for Microbiology. All Rights Reserved.
OmpK26, a Novel Porin Associated with CarbapenemResistance in Klebsiella pneumoniae!
Laura García-Sureda,1 Antonio Domenech-Sanchez,1 Mariette Barbier,1 Carlos Juan,1,2
Joan Gasco,3 and Sebastian Albertı1*Instituto Universitario de Investigaciones en Ciencias de la Salud, Universidad de las Islas Baleares, Palma de
Mallorca, Spain1; Servicio de Microbiología y Unidad de Investigacion, Hospital Universitario Son Espases,Palma de Mallorca, Spain2; and Servicio de Nefrología, Hospital Universitario Son Espases,
Palma de Mallorca, Spain3
Received 5 March 2011/Returned for modification 17 June 2011/Accepted 22 July 2011
Clinical isolates of Klebsiella pneumoniae resistant to carbapenems are being isolated with increasingfrequency. Loss of the expression of the major nonspecific porins OmpK35/36 is a frequent feature in theseisolates. In this study, we looked for porins that could compensate for the loss of the major porins incarbapenem-resistant organisms. Comparison of the outer membrane proteins from two K. pneumoniae clinicalisogenic isolates that are susceptible (KpCS-1) and resistant (KpCR-1) to carbapenems revealed the absenceof OmpK35/36 and the presence of a new 26-kDa protein in the resistant isolate. An identical result wasobtained when another pair of isogenic isolates that are homoresistant (Kpn-3) and heteroresistant (Kpn-17)to carbapenems were compared. Mass spectrometry and DNA sequencing analysis demonstrated that this newprotein, designated OmpK26, is a small monomeric oligogalacturonate-specific porin that belongs to the KdgMfamily of porins. Insertion-duplication mutagenesis of the OmpK26 coding gene, yjhA, in the carbapenem-resistant, porin-deficient isolate KpCR-1 caused the expression of OmpK36 and the reversion to the carbap-enem-susceptible phenotype, suggesting that OmpK26 is indispensable for KpCR-1 to lose OmpK36 andbecome resistant to these antibiotics. Moreover, loss of the major porin and expression of OmpK26 reducedin vitro fitness and attenuated virulence in a murine model of acute systemic infection. Altogether, these resultsindicate that expression of the oligogalacturonate-specific porin OmpK26 compensates for the absence ofOmpK35/36 and allows carbapenem resistance in K. pneumoniae but cannot restore the fitness of themicroorganism.
Klebsiella pneumoniae is one of the most prevalent nosoco-mial enterobacterial pathogens, causing infections that rangefrom mild urinary infections to severe bacteremia and pneu-monia with a high rate of mortality and morbidity (19). Anincreasing worldwide emergence of multidrug resistanceamong K. pneumoniae nosocomial isolates has limited the ther-apeutic options for treatment of these infections (4, 6, 13, 17,22). The multidrug resistance phenotype results from the pro-gressive accumulation of different mechanisms of resistance inthe same microorganism, including high-level production ofextended-spectrum !-lactamases (ESBLs), overproduction ofefflux pumps, and porin deficiency.
K. pneumoniae encodes two major nonspecific porins calledOmpK35 and OmpK36 through which nutrients and otherhydrophilic molecules, such as !-lactams, penetrate into thebacteria (11). ESBL-producing strains usually are deficient inone of these porins, which, in turn, makes them good candi-dates for developing a major increase in antimicrobial resis-tance by losing the expression of the remaining porin (11).Clinical and experimental evidence indicates that a loss ofporins in K. pneumoniae strains producing ESBLs causes re-
sistance to cefoxitin, increased resistance to oxyimino cepha-losporins and monobactams, and decreased susceptibility tofluoroquinolones (7, 8, 12, 13, 16, 17). Additionally, in strainsproducing some CTX-M type !-lactamases or AmpC !-lacta-mases, porin loss causes reduced susceptibility to carbapenems(6, 17).
Besides the major porins, K. pneumoniae may express otherporins, such as PhoE, LamB, and OmpK37, which might becrucial in the absence of OmpK36/35. Kaczmarek et al. andGarcía-Sureda et al. reported that PhoE and LamB, respec-tively, represented alternative porins that could compensatefor the function of the major porins in OmpK36/K35-deficientK. pneumoniae clinical isolates, avoiding the penetration ofcertain antibiotics (10, 13). In contrast, OmpK37, a quiescentporin, played a minimal role in antimicrobial resistance (7).
To identify new porins that could compensate for the loss ofthe major porins in carbapenem-resistant isolates, we con-ducted a comparative study of the outer membrane proteinsfrom a pair of isogenic clinical isolates, one susceptible and oneresistant. Interestingly, we detected a novel porin, which wasexclusively present in the porin-deficient resistant isolate. Thiswork describes the identification of this porin and its contri-bution to K. pneumoniae antimicrobial resistance.
MATERIALS AND METHODS
Bacterial strains, plasmids, and media. K. pneumoniae carbapenem-suscepti-ble clinical strain KpCS-1 and its isogenic carbapenem-resistant derivative
* Corresponding author. Mailing address: Edificio Científico-Tec-nico, CAMPUS-UIB, Crta. Valldemosa, km 7.5, Palma de Mallorca07122, Spain. Phone: 34-971-173353. Fax: 34-971-259501. E-mail:[email protected].
! Published ahead of print on 1 August 2011.
4742

KpCR-1 were isolated from the same patient before and after receiving carbap-enem treatment, respectively (17). The pulsed-field gel electrophoresis macro-restriction pattern of KpCR-1 was found to be identical to that of KpCS-1.
The carbapenem-heteroresistant clinical strain Kpn-3 and the carbapenem-homoresistant strain Kpn-17 are isogenic epidemic clones isolated at HospitalRamon y Cajal in Madrid (23). Escherichia coli strains used in the cloningexperiments were DH5! and strain S17-1 "pir, which encodes protein # from thepir gene essential for replication of plasmid pFS100 (20). Plasmid pGEM-T Easy(Invitrogen) is the TA cloning vector used for cloning PCR products. PlasmidspFS100 (19) and pDT1558 (25) were used to create insertion-duplication muta-tions by homologous recombination and as a source of the tellurite resistancecassette kilA-Ter, respectively. Plasmid pSHA2, coding the OmpK36 porin, waspreviously described (16). Bacterial cells were grown in cation-supplementedMueller-Hinton (MH) or Luria-Bertani (LB) broth at 37°C with shaking or weresolidified with 1.5% agar. When necessary, media were supplemented with am-picillin (50 $g/ml), kanamycin (50 $g/ml), or tellurite (10 $g/ml).
Susceptibility testing. MICs of several antibiotics were determined by usingEtest strips (bioMerieux, Marcy l’Etoile, France), following the manufacturer’srecommendations.
Isolation, analysis, and identification of outer membrane proteins. Isolation ofouter membrane proteins (OMP) were performed as previously described (11).Cell envelopes were isolated from K. pneumoniae strains by centrifugation at100,000 % g for 1 h at 4°C after French press cell lysis. OMP were isolated assodium lauryl sarcosinate-insoluble material. Electrophoretic analysis of OMP bysodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) wasperformed with 11% acrylamide-0.35% bisacrylamide-0.1% SDS by using Laem-mli’s buffer and Coomassie blue staining. Samples were boiled for 5 min insample buffer before electrophoresis. Western blot analysis of SDS-PAGE-sep-arated OMP was carried out with the buffers and conditions described byHernandez-Alles et al. (11), Immobilon P membranes (Millipore), rabbit anti-serum raised against purified OmpK36 porin, and alkaline phosphatase-labeledanti-rabbit immunoglobulin G (Sigma). Enzyme was detected with 5-bromo-4-chloro-3-indolylphosphate toluidinium–nitroblue tetrazolium. Selected proteinspots were excised from the gels, trypsin digested, and identified by microcapil-lary liquid chromatography-mass spectrometry ($LC-MS) and tandem massspectrometry (MS-MS) by the Unidad de Proteomica at the Universidad Com-plutense of Madrid, as described elsewhere (5). The search for filtered peptideswas performed using GPS Explorer v3.5 software with a licensed version 1.9 ofMASCOT.
DNA procedures. Plasmid DNA was isolated using the Wizard Miniprep kit(Promega) according to the manufacturer’s instructions. Isolation of genomicDNA, transformation, and conjugations were carried out by standard techniques(1). T4 DNA ligase and restriction endonucleases were used following the man-ufacturer’s recommendations (GE Healthcare). DNA fragments prepared byrestriction enzyme digestion were separated by agarose gel electrophoresis andvisualized by ethidium bromide staining. PCRs were performed according tostandard techniques (1). For Southern blot analysis and probe labeling anddetection, we used the ECL kit (GE Healthcare) according to the manufacturer’sprotocol.
The internal fragment of the OmpK26 coding gene, yjhA, used to generate aknockout mutant and as a probe in the Southern blot experiments, was obtainedby PCR amplification of the K. pneumoniae KpCR-1 genomic DNA with primersYjhaF3 (5&-CTGGTTCTCGCCGCCCTCTGCGG-3&) and YjhaR2 (5&-CCATACGGCGACCAGTTATTATCC-3&).
DNA sequencing was performed using the BigDye Terminator kit (PE-Ap-plied Biosystems), and sequences were analyzed with the ABI Prism 3100 DNAsequencer (PE-Applied Biosystems). Alignments were generated by ClustalWand subsequently rectified to position insertions and deletions in loop regions.
Construction of an OmpK26 knockout mutant from strain KpCR-1. To gen-erate an OmpK26 knockout mutant from KpCR-1, an internal fragment of thegene yjhA, obtained by PCR as described above, was first cloned into pGEM-TEasy and then transferred into the # protein-dependent shuttle vector pFS100digested with EcoRI to generate plasmid pFSOmpK26, which was endowed withthe tellurite resistance from pDT1558 and introduced in K. pneumoniae byconjugation. One integrant of plasmid pFSOmpK26-Te into the K. pneumoniaechromosome, which disrupted expression of yjhA, was further investigated.
In vitro competition experiments. In vitro competition experiments were per-formed as previously described (18). Briefly, exponentially growing cells of thecorresponding strains in LB broth were mixed in a 1:1 ratio and diluted in Ringersolution. Approximately, 103 cells from each of the mixtures were inoculated intoeight 10-ml LB broth tubes and grown at 37°C and 180 rpm for 9 h. Serial 10-folddilutions were plated in duplicate onto LB agar alone and LB agar with theselective markers (meropenem [0.75 $g/ml] for KpCR-1 or tellurite [10 $g/ml]
for KpCR-1'OmpK26), in order to determine the total CFU and the CFU of themutant, respectively, after overnight incubation at 37°C. Alternatively, when thedifference in the numbers of colonies between the LB plates with agar alone andthe LB plates with the selective marker was low (less than 2-fold), 100 randomlyselected colonies from the LB plates were replicated in LB plates with theappropriate selection; mutant and wild-type colonies were recognized bythe presence or absence of growth in these plates after overnight incubation. Thecompetition index (CI) was defined as the mutant/wild-type ratio. CI values werecalculated for each of the independent competitions, and the median values wererecorded. Statistical analysis of the distribution of the CI values was performedusing the Student’s t test. P values of (0.05 were considered to be statisticallysignificant. During the standardization of the procedure, the absence of sponta-neous loss of meropenem resistance was confirmed by plating KpCR-1 on LBagar plates with or without meropenem. Moreover, competition experimentsbetween strains KpCR-1 and KpCR-1/pSHA4 (harboring a vector with the tel-lurite resistance cassette) were performed; the CI values obtained (median, 0.94)ruled out any significant effect of the tellurite resistance cassette on bacterialfitness. To assess the growth rates under noncompetitive conditions, the doublingtimes of exponentially growing cells in LB broth at 37°C and 180 rpm weredetermined by plating serial 10-fold dilutions on LB at 1-h intervals. Threeindependent experiments were performed for each of the strains, and the resultswere compared using Student’s t test.
Virulence and fitness in a murine model of systemic infection. Virulence andfitness studies were performed with male ICR-CD1 mice, each weighing 20 to30 g. For the mouse lethality studies, approximately 2 % 108 CFU of K. pneu-moniae from an early-log-phase broth culture was administered by intraperito-neal injection to groups of 18 mice. Mortality was monitored daily for 7 days.
In vivo fitness was assessed by competition experiments in the mouse model ofsystemic infection. For this purpose, 1:1 mixtures of each of the pairs of testedstrains containing a total of approximately 5 % 107 exponentially growing cellswere inoculated intraperitoneally into at least 10 mice. Mice were sacrificed 24 h
FIG. 1. SDS-PAGE analysis (A) and Western blotting with anti-OmpK36 serum (B) of the outer membrane proteins of K. pneumoniaecarbapenem-susceptible clinical strain KpCS-1 and the carbapenem-heteroresistant strain Kpn-3 and their derivative carbapenem-resistantand -homoresistant isolates KpCR-1 and Kpn-17. Molecular sizemarkers are indicated in kDa on the left. White arrows indicate theposition of OmpK36, and the black arrow indicates the position of thenovel protein present in the resistant isolates.
VOL. 55, 2011 OmpK26 PORIN AND ANTIMICROBIAL RESISTANCE 4743

after inoculation, and their spleens were aseptically extracted and homogenizedin 5 ml of 0.9% saline solution. The number of CFU of each strain and the CIvalues were determined as described for in vitro competition experiments.
RESULTS AND DISCUSSION
Identification of OmpK26. In order to seek for porins thatcould compensate for the loss of the major porins in carbap-enem-resistant organisms, we compared the outer membraneproteins from a carbapenem-susceptible isolate and its derivedcarbapenem-resistant isogenic clinical isolate. As shown in Fig.1A, outer membranes prepared from the susceptible isolateKpCS-1 analyzed by SDS-PAGE contained visible amounts ofthe major porin OmpK36 (indicated with white arrows in Fig.1A), while membranes prepared from the resistant isolateKpCR-1 did not. The porin nature of the 36-kDa proteinpresent in the susceptible isolate and absent in the resistantone was confirmed by Western blot analysis using specific an-tibodies against OmpK36 and MS analysis (Fig. 1B). PCRamplification and sequencing of the ompK36 gene fromKpCR-1 revealed the presence of IS26 in the ompK36 coding
region (17). In contrast, membranes from KpCR-1 contained aprotein of about 26 kDa that was not present in the membranesof KpCS-1 (indicated with a black arrow in Fig. 1A). This resultestablishes that the resistance phenotype in this isolate corre-lates with the absence of OmpK36 porin and the presence of anovel protein of about 26 kDa in the outer membrane. Asimilar phenomenon was observed when we compared theouter membrane proteins of a pair of clinical isolates that areheteroresistant (Kpn-3) and homoresistant (Kpn-17) to car-bapenems. The heteroresistant Kpn-3 isolate, which containsonly a subpopulation of a few cells that grow in the presence ofa high concentration of imipenem (32 !g/ml), expressedOmpK36, while the homoresistant isolate Kpn-17, which ishighly resistant to imipenem (MIC, "128 !g/ml), failed toexpress OmpK36 and expressed a novel protein of about 26kDa (Fig. 1, lanes 3 and 4, respectively).
To identify the 26-kDa protein, the corresponding band wasexcised from the gel and the protein was subjected to MSanalysis. In each resistant isolate, the band was found to cor-respond to a putative oligogalacturonate-specific porin homol-
FIG. 2. Alignment of the deduced OmpK26 sequence from K. pneumoniae with the sequences of NanC from E. coli and KdgM from D. dadantii.Secondary structure motifs are described on the basis of the crystal structure of NanC. The numbering is based on the structure of the matureNanC. Conserved amino acids (highlighted in black), the two facing strings of arginines and lysines inside the OmpK26 pore (black circles) andgaps introduced to maximize alignment (hyphens) are indicated.
4744 GARCIA-SUREDA ET AL. ANTIMICROB. AGENTS CHEMOTHER.
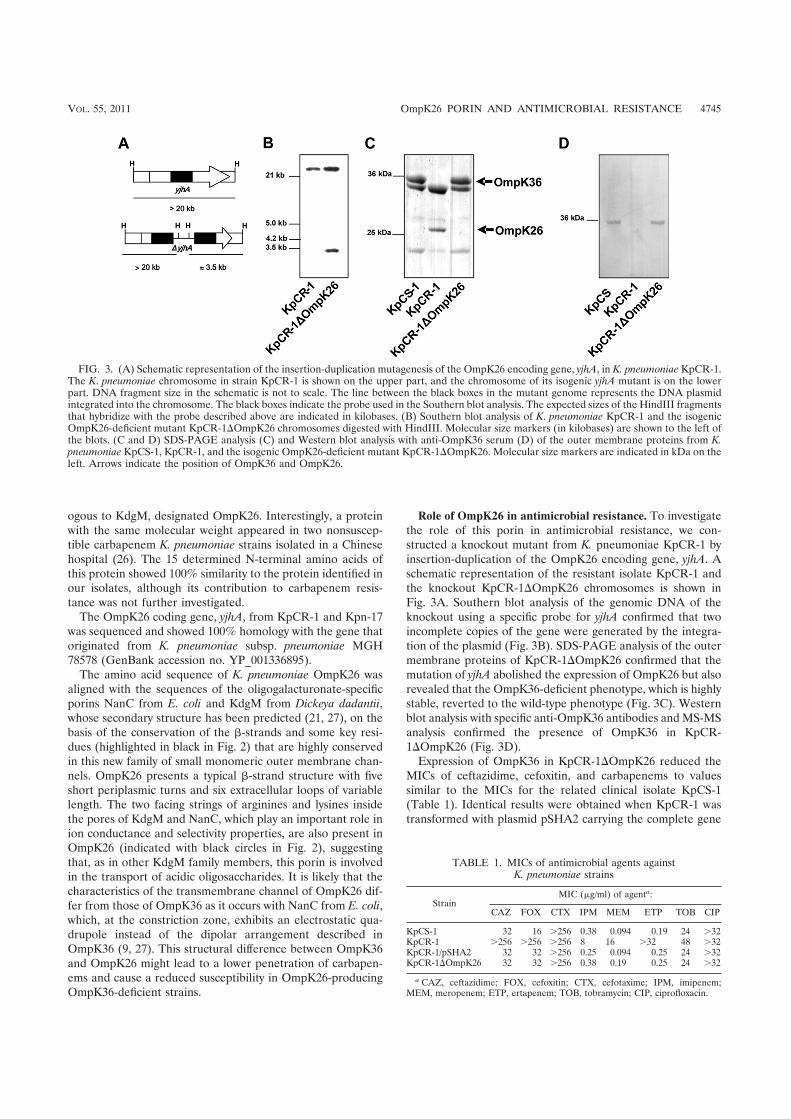
ogous to KdgM, designated OmpK26. Interestingly, a proteinwith the same molecular weight appeared in two nonsuscep-tible carbapenem K. pneumoniae strains isolated in a Chinesehospital (26). The 15 determined N-terminal amino acids ofthis protein showed 100% similarity to the protein identified inour isolates, although its contribution to carbapenem resis-tance was not further investigated.
The OmpK26 coding gene, yjhA, from KpCR-1 and Kpn-17was sequenced and showed 100% homology with the gene thatoriginated from K. pneumoniae subsp. pneumoniae MGH78578 (GenBank accession no. YP_001336895).
The amino acid sequence of K. pneumoniae OmpK26 wasaligned with the sequences of the oligogalacturonate-specificporins NanC from E. coli and KdgM from Dickeya dadantii,whose secondary structure has been predicted (21, 27), on thebasis of the conservation of the !-strands and some key resi-dues (highlighted in black in Fig. 2) that are highly conservedin this new family of small monomeric outer membrane chan-nels. OmpK26 presents a typical !-strand structure with fiveshort periplasmic turns and six extracellular loops of variablelength. The two facing strings of arginines and lysines insidethe pores of KdgM and NanC, which play an important role inion conductance and selectivity properties, are also present inOmpK26 (indicated with black circles in Fig. 2), suggestingthat, as in other KdgM family members, this porin is involvedin the transport of acidic oligosaccharides. It is likely that thecharacteristics of the transmembrane channel of OmpK26 dif-fer from those of OmpK36 as it occurs with NanC from E. coli,which, at the constriction zone, exhibits an electrostatic qua-drupole instead of the dipolar arrangement described inOmpK36 (9, 27). This structural difference between OmpK36and OmpK26 might lead to a lower penetration of carbapen-ems and cause a reduced susceptibility in OmpK26-producingOmpK36-deficient strains.
Role of OmpK26 in antimicrobial resistance. To investigatethe role of this porin in antimicrobial resistance, we con-structed a knockout mutant from K. pneumoniae KpCR-1 byinsertion-duplication of the OmpK26 encoding gene, yjhA. Aschematic representation of the resistant isolate KpCR-1 andthe knockout KpCR-1"OmpK26 chromosomes is shown inFig. 3A. Southern blot analysis of the genomic DNA of theknockout using a specific probe for yjhA confirmed that twoincomplete copies of the gene were generated by the integra-tion of the plasmid (Fig. 3B). SDS-PAGE analysis of the outermembrane proteins of KpCR-1"OmpK26 confirmed that themutation of yjhA abolished the expression of OmpK26 but alsorevealed that the OmpK36-deficient phenotype, which is highlystable, reverted to the wild-type phenotype (Fig. 3C). Westernblot analysis with specific anti-OmpK36 antibodies and MS-MSanalysis confirmed the presence of OmpK36 in KpCR-1"OmpK26 (Fig. 3D).
Expression of OmpK36 in KpCR-1"OmpK26 reduced theMICs of ceftazidime, cefoxitin, and carbapenems to valuessimilar to the MICs for the related clinical isolate KpCS-1(Table 1). Identical results were obtained when KpCR-1 wastransformed with plasmid pSHA2 carrying the complete gene
FIG. 3. (A) Schematic representation of the insertion-duplication mutagenesis of the OmpK26 encoding gene, yjhA, in K. pneumoniae KpCR-1.The K. pneumoniae chromosome in strain KpCR-1 is shown on the upper part, and the chromosome of its isogenic yjhA mutant is on the lowerpart. DNA fragment size in the schematic is not to scale. The line between the black boxes in the mutant genome represents the DNA plasmidintegrated into the chromosome. The black boxes indicate the probe used in the Southern blot analysis. The expected sizes of the HindIII fragmentsthat hybridize with the probe described above are indicated in kilobases. (B) Southern blot analysis of K. pneumoniae KpCR-1 and the isogenicOmpK26-deficient mutant KpCR-1"OmpK26 chromosomes digested with HindIII. Molecular size markers (in kilobases) are shown to the left ofthe blots. (C and D) SDS-PAGE analysis (C) and Western blot analysis with anti-OmpK36 serum (D) of the outer membrane proteins from K.pneumoniae KpCS-1, KpCR-1, and the isogenic OmpK26-deficient mutant KpCR-1"OmpK26. Molecular size markers are indicated in kDa on theleft. Arrows indicate the position of OmpK36 and OmpK26.
TABLE 1. MICs of antimicrobial agents againstK. pneumoniae strains
StrainMIC (#g/ml) of agenta:
CAZ FOX CTX IPM MEM ETP TOB CIP
KpCS-1 32 16 $256 0.38 0.094 0.19 24 $32KpCR-1 $256 $256 $256 8 16 $32 48 $32KpCR-1/pSHA2 32 32 $256 0.25 0.094 0.25 24 $32KpCR-1"OmpK26 32 32 $256 0.38 0.19 0.25 24 $32
a CAZ, ceftazidime; FOX, cefoxitin; CTX, cefotaxime; IPM, imipenem;MEM, meropenem; ETP, ertapenem; TOB, tobramycin; CIP, ciprofloxacin.
VOL. 55, 2011 OmpK26 PORIN AND ANTIMICROBIAL RESISTANCE 4745
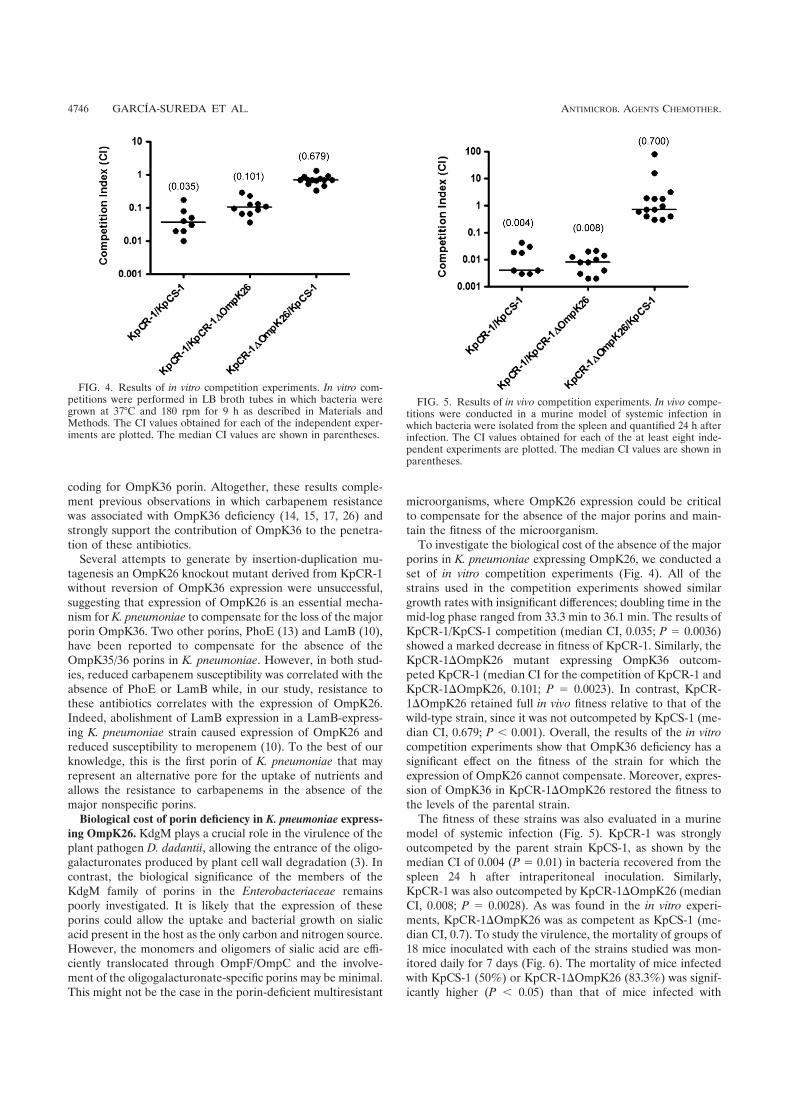
coding for OmpK36 porin. Altogether, these results comple-ment previous observations in which carbapenem resistancewas associated with OmpK36 deficiency (14, 15, 17, 26) andstrongly support the contribution of OmpK36 to the penetra-tion of these antibiotics.
Several attempts to generate by insertion-duplication mu-tagenesis an OmpK26 knockout mutant derived from KpCR-1without reversion of OmpK36 expression were unsuccessful,suggesting that expression of OmpK26 is an essential mecha-nism for K. pneumoniae to compensate for the loss of the majorporin OmpK36. Two other porins, PhoE (13) and LamB (10),have been reported to compensate for the absence of theOmpK35/36 porins in K. pneumoniae. However, in both stud-ies, reduced carbapenem susceptibility was correlated with theabsence of PhoE or LamB while, in our study, resistance tothese antibiotics correlates with the expression of OmpK26.Indeed, abolishment of LamB expression in a LamB-express-ing K. pneumoniae strain caused expression of OmpK26 andreduced susceptibility to meropenem (10). To the best of ourknowledge, this is the first porin of K. pneumoniae that mayrepresent an alternative pore for the uptake of nutrients andallows the resistance to carbapenems in the absence of themajor nonspecific porins.
Biological cost of porin deficiency in K. pneumoniae express-ing OmpK26. KdgM plays a crucial role in the virulence of theplant pathogen D. dadantii, allowing the entrance of the oligo-galacturonates produced by plant cell wall degradation (3). Incontrast, the biological significance of the members of theKdgM family of porins in the Enterobacteriaceae remainspoorly investigated. It is likely that the expression of theseporins could allow the uptake and bacterial growth on sialicacid present in the host as the only carbon and nitrogen source.However, the monomers and oligomers of sialic acid are effi-ciently translocated through OmpF/OmpC and the involve-ment of the oligogalacturonate-specific porins may be minimal.This might not be the case in the porin-deficient multiresistant
microorganisms, where OmpK26 expression could be criticalto compensate for the absence of the major porins and main-tain the fitness of the microorganism.
To investigate the biological cost of the absence of the majorporins in K. pneumoniae expressing OmpK26, we conducted aset of in vitro competition experiments (Fig. 4). All of thestrains used in the competition experiments showed similargrowth rates with insignificant differences; doubling time in themid-log phase ranged from 33.3 min to 36.1 min. The results ofKpCR-1/KpCS-1 competition (median CI, 0.035; P ! 0.0036)showed a marked decrease in fitness of KpCR-1. Similarly, theKpCR-1"OmpK26 mutant expressing OmpK36 outcom-peted KpCR-1 (median CI for the competition of KpCR-1 andKpCR-1"OmpK26, 0.101; P ! 0.0023). In contrast, KpCR-1"OmpK26 retained full in vivo fitness relative to that of thewild-type strain, since it was not outcompeted by KpCS-1 (me-dian CI, 0.679; P # 0.001). Overall, the results of the in vitrocompetition experiments show that OmpK36 deficiency has asignificant effect on the fitness of the strain for which theexpression of OmpK26 cannot compensate. Moreover, expres-sion of OmpK36 in KpCR-1"OmpK26 restored the fitness tothe levels of the parental strain.
The fitness of these strains was also evaluated in a murinemodel of systemic infection (Fig. 5). KpCR-1 was stronglyoutcompeted by the parent strain KpCS-1, as shown by themedian CI of 0.004 (P ! 0.01) in bacteria recovered from thespleen 24 h after intraperitoneal inoculation. Similarly,KpCR-1 was also outcompeted by KpCR-1"OmpK26 (medianCI, 0.008; P ! 0.0028). As was found in the in vitro experi-ments, KpCR-1"OmpK26 was as competent as KpCS-1 (me-dian CI, 0.7). To study the virulence, the mortality of groups of18 mice inoculated with each of the strains studied was mon-itored daily for 7 days (Fig. 6). The mortality of mice infectedwith KpCS-1 (50%) or KpCR-1"OmpK26 (83.3%) was signif-icantly higher (P # 0.05) than that of mice infected with
FIG. 4. Results of in vitro competition experiments. In vitro com-petitions were performed in LB broth tubes in which bacteria weregrown at 37°C and 180 rpm for 9 h as described in Materials andMethods. The CI values obtained for each of the independent exper-iments are plotted. The median CI values are shown in parentheses.
FIG. 5. Results of in vivo competition experiments. In vivo compe-titions were conducted in a murine model of systemic infection inwhich bacteria were isolated from the spleen and quantified 24 h afterinfection. The CI values obtained for each of the at least eight inde-pendent experiments are plotted. The median CI values are shown inparentheses.
4746 GARCIA-SUREDA ET AL. ANTIMICROB. AGENTS CHEMOTHER.

KpCR-1, which was unable to kill any mice. In contrast, therewere no significant differences in the survival rates between themice infected with KpCS-1 and those infected with KpCR-1!OmpK26.
These results indicate that OmpK35/36 deficiency reducesthe virulence of K. pneumoniae in a mouse model of systemicinfection, and they are consistent with those recently reportedby Bialek et al. (2) and Tsai et al. (24) which, using a Caeno-rhabditis elegans model and a mouse model, respectively,showed that lack of OmpK35/36 expression was associated witha decrease in the virulence of K. pneumoniae. However, themechanisms underlying the attenuated virulence caused by theabsence of the expression of the major porins are poorly un-derstood. The results from our in vitro experiments suggestthat, besides the potential contribution of the major porins tothe bacterial defense against the immune system of the host(24), their expression and function as membrane transportersare indispensable to maintain the fitness of the microorganism,which may help in explaining the attenuated phenotype in vivo.Therefore, although bacteria with altered OmpK35/36 expres-sion represent a threat because they exhibit increased levelsand extended spectra of "-lactam resistance, in the absence ofantibiotic selective pressure, they are less competitive than thewild-type strains.
In summary, we showed that the expression of the oligoga-lacturonate-specific porin OmpK26 compensated for the ab-sence of the expression of OmpK35/36 in carbapenem-resis-tant K. pneumoniae but could not restore the fitness of themicroorganism either in vitro or in vivo.
ACKNOWLEDGMENTS
This study was supported by grants from the Ministerio de Cienciae Innovacion (SAF2005-0466 to S.A.) and by Instituto de Salud CarlosIII (Spanish Network for the Research in Infectious Diseases (REIPIRD06/0008).) L.G.S. and C.J. are the recipients of, respectively, apredoctoral fellowship and a Juan de la Cierva contract from theMinisterio de Ciencia e Innovacion.
REFERENCES1. Ausubel, F. M., et al. 1997. Current protocols in molecular biology. Wiley
Interscience, New York, NY.2. Bialek, S., et al. 2010. Membrane efflux and influx modulated both multidrug
resistance and virulence of Klebsiella pneumoniae in a Caenorhabditis elegansmodel. Antimicrob. Agents Chemother. 54:4373–4378.
3. Blot, N., C. Berrier, N. Hugouvieux-Cottet-Pattat, A. Ghazi, and G. Conde-mine. 2002. The oligogalacturonate-specific porin KdgM of Erwinia chrysan-themi belongs to a new porin family. J. Biol. Chem. 277:7936–7944.
4. Bradford, P. A., et al. 1997. Imipenem resistance in Klebsiella pneumoniae isassociated with the combination of ACT-1, a plasmid-mediated AmpC "-lac-tamase, and the loss of an outer membrane protein. Antimicrob. AgentsChemother. 41:563–569.
5. Cabezon, V., A. Llama-Palacios, C. Nombela, L. Monteoliva, and C. Gil.2009. Analysis of Candida albicans plasma membrane proteome. Proteomics9:4770–4786.
6. Cao, V. T. B., et al. 2000. Emergence of imipenem resistance in Klebsiellapneumoniae owing to combination of plasmid-mediated CMY-4 and perme-ability alteration. J. Antimicrob. Chemother. 46:895–900.
7. Domenech-Sanchez, A., et al. 2003. Role of Klebsiella pneumoniae OmpK35porin in antimicrobial resistance. Antimicrob. Agents Chemother. 47:3332–3335.
8. Doumith, M., M. J. Ellington, D. M. Livermore, and N. Woodford. 2009.Molecular mechanisms disrupting porin expression in ertapenem-resistantKlebsiella and Enterobacter spp. clinical isolates from the UK. J. Antimi-crob. Chemother. 63:659–667.
9. Dutzler, R., et al. 1999. Crystal structure and functional characterization ofOmpK36, the osmoporin of Klebsiella pneumoniae. Structure 7:425–434.
10. García-Sureda, L., C. Juan, A. Domenech-Sanchez, and S. Albertí. 31 Jan-uary 2011. Role of Klebsiella pneumoniae LamB porin in antimicrobial re-sistance. Antimicrob. Agents Chemother. doi:10.1128/AAC.01441–10.
11. Hernandez-Alles, S., et al. 1999. Porin expression in clinical isolates ofKlebsiella pneumoniae. Microbiology 145:673–679.
12. Jacoby, G. A., D. M. Mills, and N. Chow. 2004. Role of "-lactamases andporins in resistance to ertapenem and other "-lactams in Klebsiella pneu-moniae. Antimicrob. Agents Chemother. 48:3202–3206.
13. Kaczmarek, F. M., F. Dib-Hajj, W. Shang, and T. D. Gootz. 2006. High-levelcarbapenem resistance in a Klebsiella pneumoniae clinical isolate is due to thecombination of blaACT-1 "-lactamase production, porin OmpK35/36 inser-tional inactivation, and down-regulation of the phosphate transport porinPhoE. Antimicrob. Agents Chemother. 50:3396–3406.
14. Kitchel, B., et al. 2010. Genetic factors associated with elevated carbapenemresistance in KPC-producing Klebsiella pneumoniae. Antimicrob. AgentsChemother. 54:4201–4207.
15. Landman, D., S. Bratu, and J. Quale. 2009. Contribution of OmpK36 tocarbapenem susceptibility in KPC-producing Klebsiella pneumoniae. J. Med.Microbiol. 58:1303–1308.
16. Martínez-Martínez, L., et al. 1996. In vivo selection of porin-deficient mu-tants of Klebsiella pneumoniae with increased resistance to cefoxitin andexpanded-spectrum cephalosporins. Antimicrob. Agents Chemother. 40:342–348.
17. Mena, A., et al. 2006. Characterization of a large outbreak by CTX-M-1producing Klebsiella pneumoniae and mechanisms leading to in vivo carbap-enem resistance development. J. Clin. Microbiol. 44:2831–2837.
18. Moya, B., C. Juan, S. Albertí, J. L. Perez, and A. Oliver. 2008. Benefit ofhaving multiple ampD genes for acquiring "-lactam resistance without losingfitness and virulence in Pseudomonas aeruginosa. Antimicrob. Agents Che-mother. 52:3694–3700.
19. Podschun, R., and U. Ullman. 1998. Klebsiella spp. as nosocomial pathogens:epidemiology, taxonomy, typing methods, and pathogenicity factors. Clin.Microbiol. Rev. 11:589–603.
20. Rubires, X., et al. 1997. A gene (wbbL) from Serratia marcescens N28b (O4)complements the rfb-50 mutation of Escherichia coli K-12 derivatives. J.Bacteriol. 179:7581–7586.
21. Signorell, G. A., et al. 2007. Projection maps of three members of the KdgMouter membrane protein family. J. Struct. Biol. 160:395–403.
22. Song, W., et al. 2009. In vivo selection of carbapenem-resistant Klebsiellapneumoniae by OmpK36 loss during meropenem treatment. Diagn. Micro-biol. Infect. Dis. 65:447–449.
23. Tato, M., et al. 2010. Carbapenem heteroresistance in VIM-1 producing-Klebsiella pneumoniae isolates belonging to the same clone: consequences forroutine susceptibility testing. J. Clin. Microbiol. 48:4089–4093.
24. Tsai, Y. K., et al. 31 January 2011. The Klebsiella pneumoniae outer mem-brane porins OmpK35 and OmpK36 play roles in both antimicrobial resis-tance and virulence. Antimicrob. Agents Chemother. doi:10.1128/AAC.01275–10.
25. Walter, E. G., and D. E. Taylor. 1989. Comparison of tellurite resistancedeterminants from the IncPa plasmid RP4Ter and the IncHII plasmidPHH1508a. J. Bacteriol. 171:2160–2165.
26. Wang, X. D., J. C. Cai, H. W. Zhou, R. Zhang, and G. X. Chen. 2009.Klebsiella pneumoniae clinical isolates associated with plasmid-mediated be-ta-lactamase production and OmpK36 porin deficiency. J. Med. Microbiol.58:1196–1202.
27. Wirth, C., et al. 2009. NanC crystal structure, a model for outer-membranechannels of the acidic sugar-specific KdgM porin family. J. Mol. Biol. 394:718–731.
FIG. 6. Percentages of survival over time for groups of 18 miceinoculated intraperitoneally with KpCS-1, KpCR-1, or KpCR-1!OmpK26. Significant differences are indicated with an asterisk (P #0.05).
VOL. 55, 2011 OmpK26 PORIN AND ANTIMICROBIAL RESISTANCE 4747

JOURNAL OF CLINICAL MICROBIOLOGY, Aug. 2006, p. 2831–2837 Vol. 44, No. 80095-1137/06/$08.00!0 doi:10.1128/JCM.00418-06Copyright © 2006, American Society for Microbiology. All Rights Reserved.
Characterization of a Large Outbreak by CTX-M-1-ProducingKlebsiella pneumoniae and Mechanisms Leading to
In Vivo Carbapenem Resistance DevelopmentAna Mena,1 Virginia Plasencia,1 Laura Garcıa,2 Olga Hidalgo,3 Jose Ignacio Ayestaran,4
Sebastian Alberti,2 Nuria Borrell,1 Jose L. Perez,1 and Antonio Oliver1*Servicio de Microbiologıa, Hospital Son Dureta and Instituto Universitario de Investigacion en Ciencias de la Salud,1 Area de
Microbiologıa and Instituto Universitario de Investigacion en Ciencias de la Salud, Universidad de las Islas Baleares,2Servicio de Medicina Preventiva, Hospital Son Dureta,3 and Servicio de Medicina Intensiva,
Hospital Son Dureta,4 Palma de Mallorca, Spain
Received 26 February 2006/Returned for modification 25 April 2006/Accepted 2 June 2006
All extended-spectrum !-lactamase (ESBL)-producing Enterobacteriaceae isolates from patients admit-ted to and adult intensive care unit were prospectively documented from 2002 to 2005, when a largeoutbreak (51 patients affected) of multiresistant ESBL-producing Klebsiella pneumoniae infection wasdetected. The involvement of a single K. pneumoniae clone was demonstrated by pulsed-field gel electro-phoresis. In addition to the ESBL-mediated resistance, the epidemic strain uniformly showed cross-resistance to ciprofloxacin, gentamicin, tobramycin, trimethoprim-sulfamethoxazole, and tetracycline,whereas resistance to the !-lactam–!-lactamase inhibitor combinations was variable. The ESBL involvedwas CTX-M-1, as demonstrated by isoelectric focusing, PCR amplification, and sequencing. CTX-M-1 aswell as the aminoglycoside resistance determinants were encoded in a 50-kb plasmid that could betransferred to Escherichia coli only by transformation. In two of the infected patients, carbapenemresistance development (MICs of 8 to 12, 16, and >32 "g/ml for imipenem, meropenem, and ertapenem,respectively) was documented, both in clinical samples and in intestinal colonization studies. The analysisof the outer membrane proteins of the carbapenem-susceptible and -resistant isolates revealed that theformer expressed only one of the two major porins, OmpK36, whereas the latter did not express either ofthem. In one of the cases, the lack of expression of OmpK36 was demonstrated to be mediated by theinterruption of the coding sequence by the insertion sequence IS26. This is the first report of a largeoutbreak of CTX-M-1-producing Enterobacteriaceae and, curiously, the first documented description in theliterature of CTX-M-1 in K. pneumoniae, despite the fact that this enzyme has been found in multiplespecies. Furthermore, we document and characterize for the first time carbapenem resistance developmentin CTX-M-1-producing Enterobacteriaceae.
Since plasmid-mediated extended-spectrum "-lactamases(ESBLs) were first detected in a Klebsiella pneumoniae isolatein 1983 in Germany (22), they have been increasingly reportedworldwide (5). The classical ESBLs are those derived from thebroad-spectrum enzymes TEM-1, TEM-2, and SHV-1 by theacquisition of specific point mutations which expand theirspectrum of hydrolysis to oxyimino-cephalosporins and aztreo-nam (7). Nevertheless, the most widespread plasmid-mediatedESBLs nowadays are the CTX-M enzymes, which are directlyderived from the chromosomal "-lactamases of several speciesof the genus Kluyvera (3). Five different groups of CTX-Mscontaining a total of over 30 different variants have been de-scribed so far, with CTX-M-1, CTX-M-2, CTX-M-8, CTX-M-9, and CTX-M-25 being the first reported representatives.CTX-M-1 (also known as MEN-1) was the first reported variant,detected from an Escherichia coli strain in 1989 in Germany (2),but CTX-M-2, CTX-M-3, CTX-M-14, and CTX-M-15 are themost widespread enzymes (3).
The production of ESBLs in Enterobacteriaceae confers re-sistance to all penicillins and cephalosporins (with the excep-tion of cephamycins), with the organism generally remainingsusceptible only to "-lactam–"-lactamase inhibitor combina-tions, such as amoxicillin-clavulanate, and the carbapenems,which are frequently the only therapeutic options available fortreatment of hospital-acquired severe infections caused bythese microorganisms (25). Coresistance to non-"-lactam an-tibiotics is also frequent, either by the cotransfer of the resis-tance determinants in the same genetic elements (such as ami-noglycoside resistance) or simply by the coselection of bothresistance mechanisms, as occurs with fluoroquinolones (32).
In this work we characterized a large outbreak caused by aCTX-M-1-producing multiresistant K. pneumoniae strain in aSpanish intensive care unit (ICU). Although CTX-M-1 was thefirst CTX-M described, over 15 years ago, and has been de-tected in various countries, including at least Germany andFrance (14), Italy (31), and recently, in a single E. coli isolatefrom cattle milk, Spain (6), and in several species, including E.coli, Proteus mirabilis, Morganella morganii, and Citrobacteramalonaticus (28), reports of CTX-M-1-producing K. pneu-moniae strains have not yet been published, and this is the firstreport of a large outbreak of CTX-M-1-producing Enterobac-
* Corresponding author. Mailing address: Servicio de Microbiolo-gıa, Hospital Son Dureta, C. Andrea Doria no. 55, 07014 Palma deMallorca, Spain. Phone: 34 971 175 185. Fax: 34 971 175 185. E-mail:[email protected].
2831

teriaceae infection. Furthermore, in this work we documentand characterize for the first time in vivo resistance develop-ment to carbapenems in CTX-M-1-producing Enterobacteria-ceae due to the selection of mutations leading to the lack ofexpression of porins.
MATERIALS AND METHODS
Clinical strains and antibiotic susceptibility testing. All ESBL-producingEnterobacteriaceae isolates from patients admitted to the adult ICU (30 bedsdivided in three contiguous units) from Hospital Son Dureta (900 beds;reference public hospital from the Island of Mallorca, Spain) have beenprospectively documented from 2002 to 2005, when a large outbreak causedby multiresistant ESBL-producing K. pneumoniae was detected. Bacterialidentification and initial susceptibility testing were performed with theWIDER semiautomatic system (Francisco Soria Melguizo, Madrid, Spain)(8). Double-disk synergy testing (DDST) for the detection of ESBL produc-tion was performed using amoxicillin-clavulanate, cefotaxime, ceftazidime,cefepime, and aztreonam disks that were applied 30 and/or 20 mm apart (20).Additionally, the MICs of several antibiotics were determined for selectedisolates by using Etest strips (AB Biodisk, Solna, Sweden), following themanufacturer’s recommendations. For the screening of intestinal coloniza-tion by the epidemic K. pneumoniae ESBL-producing strain, from May toDecember 2005, rectal swabs, collected weekly, were plated on MacConkeyagar supplemented with 2 !g/ml of cefotaxime and 10 !g/ml of gentamicin.The screening for intestinal colonization was carried out for all patients withpositive clinical samples as well as those patients present in the same unit atthe time of isolation. For those patients, weekly rectal swabs were obtaineduntil discharge or until three consecutive negative colonization results.
Molecular strain typing. The clonal relationship between the different isolateswas studied by pulsed-field gel electrophoresis (PFGE). Agarose plugs contain-ing total bacterial DNA were prepared as described elsewhere (21). Plugs werethen digested with XbaI and loaded into a 1% Megabase agarose (Bio-Rad, LaJolla, Calif.) gel. DNA separation was performed in a CHEF-DRIII apparatus(Bio-Rad, La Jolla, Calif.) under the following conditions: 6 V/cm2 for 20 h at14°C, with initial and final pulse times of 2 s and 35 s, respectively. The resultswere interpreted following the criteria of Tenover et al. (37).
Characterization of !-lactamases and their genes. The pIs of the "-lactamaseswere determined by isoelectric focusing (IEF), applying the supernatants ofcrude sonic cell extracts to Phast gels (Pharmacia AB, Uppsala, Sweden) with apH gradient of 3 to 9 in a Phast system (Pharmacia). "-Lactamases with knownpI values (TEM-1, TEM-2, TEM-4, TEM-3, SHV-1, CTX-M-10, and CTX-M-1)were included as controls. Gels were stained with 500 !g/ml of nitrocefin (Oxoid)to identify the bands corresponding to "-lactamases. PCRs for genes encodingTEM, SHV, CTX-M-9, and CTX-M-10 "-lactamases were performed usingprimers and conditions described previously (10). Primers CTX-MF (5#-GACTATTCATGTTGTTGTTATTTC-3#) and CTX-MR (5#-TTACAAACCGTTGGTGACG-3#) were used for the amplification of a 923-bp DNA fragment (nucle-otides $48 to the stop codon) from the genes encoding CTX-M-1 or closelyrelated ESBLs. Taq Gold polymerase (PE-Applied Biosystems) was used forPCR amplification under the following conditions: 12 min at 94°C, 35 cycles ofamplification (1 min at 94°C, 1 min at 58°C, and 1 min at 72°C), and a finalextension step of 10 min at 72°C. Two independent PCR products were se-quenced on both strands, using the BigDye terminator kit (PE-Applied Biosys-tems) for performing the sequencing reactions, which were analyzed with theABI Prism 3100 DNA sequencer (PE-Applied Biosystems).
Conjugation, transformation, and plasmid analysis. Conjugation experimentswere performed by filter mating using a rifampin-resistant mutant of E. coli strainHB101 as the recipient in 1:1 ratio. Alternatively, triparenteral crossings usingthe same recipient and E. coli HB101(pRK2013) as a mobilization helper (12) ina 1:1:1 ratio were attempted. Transconjugants were selected in Luria-Bertani(LB) agar plates containing 100 !g/ml of rifampin and 2 !g/ml of cefotaxime. Fortransformation experiments, plasmid DNAs from the ESBL-producing K. pneu-moniae isolates obtained using the QIAGEN plasmid Midi kit (QIAGEN,Hilden, Germany) were transformed into the E. coli XL1-Blue strain madecompetent by CaCl2 treatment. Transformants were selected in LB agar platescontaining 50 !g/ml of ampicillin. Transconjugants or transformants werechecked by DDST followed by IEF, PCR amplification of the appropriate ESBL-encoding gene, and Etest testing of susceptibility to all "-lactams and non-"-lactams (to determine the resistance determinants cotransferred with the ESBL)defined above. For the estimation of the plasmid size, plasmid DNA obtainedfrom one of the transformants was digested with EcoRI, BamHI, and EcoRI plus
BamHI, and the restriction fragments were separated by electrophoresis in a 1%agarose gel. The sizes of the different fragments were estimated by plottingagainst the standard curve obtained with the Superladder-Mid1 100-bp laddermolecular weight markers (Gensura) and the lambda HindIII digest (NewEngland Biolabs).
Isolation and analysis of OMP. Cell envelopes were isolated from K. pneu-moniae strains grown in LB medium by centrifugation at 100,000 % g for 1 h at4°C after French press cell lysis. Outer membrane proteins (OMP) were isolatedas sodium lauryl sarcosynate-insoluble material (15). Electrophoretic analysis ofOMP by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) was performed with 11% acrylamide–0.35% bisacrylamide–0.1% SDS byusing Laemmli’s buffers (24) and Coomassie blue staining. Samples were boiledfor 5 min in sample buffer before electrophoresis. Western blot analysis ofSDS-PAGE-separated OMP was carried out with the buffers and conditionsdescribed by Towbin et al. (38), Immobilon P membranes (Millipore), rabbitantiserum raised against purified OmpK36 porin (1), and alkaline phosphatase-labeled anti-rabbit immunoglobulin G (Sigma). Enzyme was detected with 5-bromo-4-chloro-3-indolylphosphate toluidinium–nitroblue tetrazolium. K. pneumoniaeLB1, which expresses only one porin, was used as a control (26).
PCR amplification of porin genes. The primers used to amplify the OmpK36porin gene were OmpK36-0 (5# AAGCTTGTTGGATTATTCTGC-3#) andOmpK36-end (5#-CAAGCTTAGAACTGGTAAACC-3#). They anneal specifi-cally to sequences of ompK36 located 116 bp upstream and 1,084 bp downstreamof the ompK36 start codon (26), respectively. PCR amplifications were per-formed in a Thermoline Amplitron 1 thermal cycler by using Taq polymerase(Pharmacia) with 30 cycles of amplification (1 min at 94°C, 1 min at 55°C, and 1min at 72°C). Amplicons were visualized by agarose gel electrophoresis andethidium bromide staining and sequenced as described above.
RESULTS
All ESBL-producing Enterobacteriaceae isolates from pa-tients admitted to an adult ICU were prospectively docu-mented from 2002 to 2005, when a large outbreak of multi-resistant ESBL-producing K. pneumoniae infection was detected.The incidence of ESBL-producing Enterobacteriaceae isolatedduring this period is shown in Fig. 1A. As can be observed, adramatic increase in the number of patients infected by ESBL-producing K. pneumoniae was documented during 2005, with atotal of 52 cases, in contrast to the 1 case documented in either2003 or 2004 and the 0 cases documented in 2002. Figure 1Bshows the distribution of new cases on a monthly basis during2005, clearly showing that the epidemic started in Februaryand reached its peak during May and June, with 13 and 12 newcases, respectively. From then on, the incidence of new caseson a monthly basis fluctuated from zero to six new cases permonth. Since five new cases were detected in December, theoutbreak still cannot be considered to have been controlled atthe end of the year. In May 2005, in addition to measures forthe contact isolation of infected patients, a protocol for routinedetection and isolation of patients with intestinal colonizationwas established; rectal swabs were collected weekly and platedin selective media, and ESBL-producing K. pneumoniae wasidentified as described in Materials and Methods. The out-break had a dramatic impact on the prevalence of ESBL-producing K. pneumoniae in the ICU, reaching 60.3% of pa-tients infected (not including patients with only intestinalcolonization) by K. pneumoniae during 2005, in contrast to 0%,4.5%, and 3.8% for the years 2002, 2003, and 2004, respec-tively.
All but one of the K. pneumoniae isolates recovered from the52 patients had the same pattern of multiresistance as initiallydocumented with the WIDER microdilution-based semiauto-matic system for bacterial identification and susceptibility test-ing (8), showing, in addition to the ESBL-mediated resistance
2832 MENA ET AL. J. CLIN. MICROBIOL.
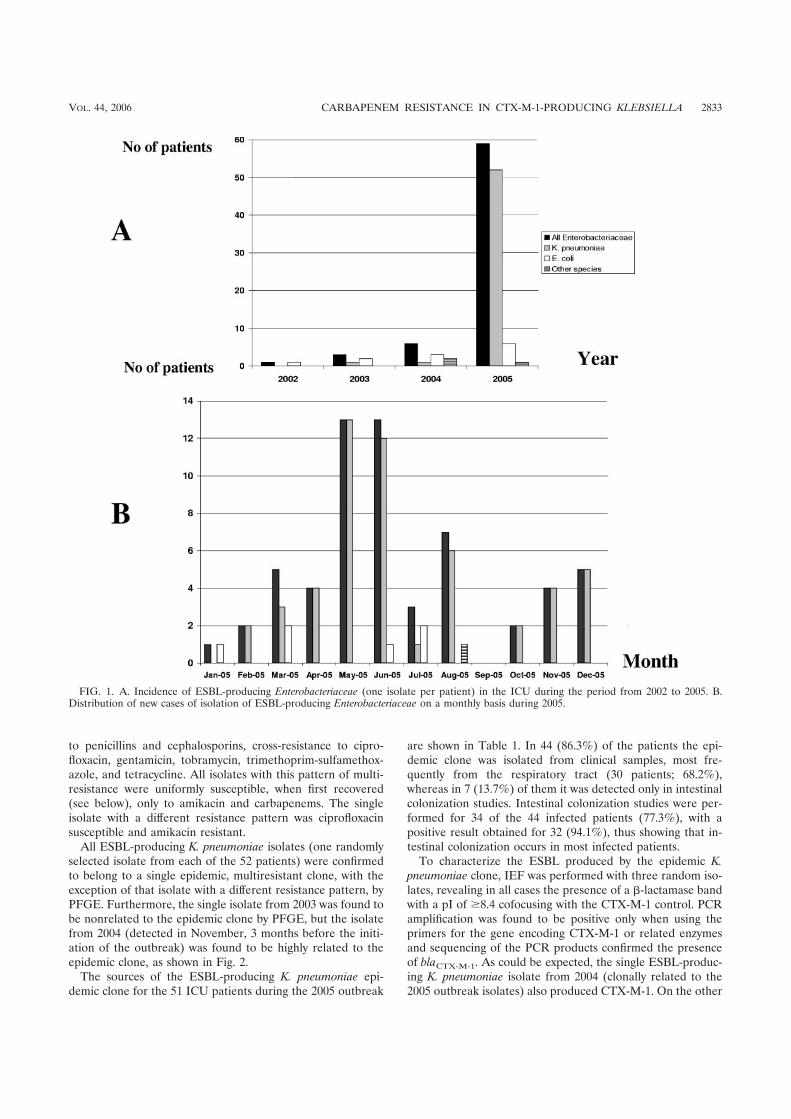
to penicillins and cephalosporins, cross-resistance to cipro-floxacin, gentamicin, tobramycin, trimethoprim-sulfamethox-azole, and tetracycline. All isolates with this pattern of multi-resistance were uniformly susceptible, when first recovered(see below), only to amikacin and carbapenems. The singleisolate with a different resistance pattern was ciprofloxacinsusceptible and amikacin resistant.
All ESBL-producing K. pneumoniae isolates (one randomlyselected isolate from each of the 52 patients) were confirmedto belong to a single epidemic, multiresistant clone, with theexception of that isolate with a different resistance pattern, byPFGE. Furthermore, the single isolate from 2003 was found tobe nonrelated to the epidemic clone by PFGE, but the isolatefrom 2004 (detected in November, 3 months before the initi-ation of the outbreak) was found to be highly related to theepidemic clone, as shown in Fig. 2.
The sources of the ESBL-producing K. pneumoniae epi-demic clone for the 51 ICU patients during the 2005 outbreak
are shown in Table 1. In 44 (86.3%) of the patients the epi-demic clone was isolated from clinical samples, most fre-quently from the respiratory tract (30 patients; 68.2%),whereas in 7 (13.7%) of them it was detected only in intestinalcolonization studies. Intestinal colonization studies were per-formed for 34 of the 44 infected patients (77.3%), with apositive result obtained for 32 (94.1%), thus showing that in-testinal colonization occurs in most infected patients.
To characterize the ESBL produced by the epidemic K.pneumoniae clone, IEF was performed with three random iso-lates, revealing in all cases the presence of a !-lactamase bandwith a pI of !8.4 cofocusing with the CTX-M-1 control. PCRamplification was found to be positive only when using theprimers for the gene encoding CTX-M-1 or related enzymesand sequencing of the PCR products confirmed the presenceof blaCTX-M-1. As could be expected, the single ESBL-produc-ing K. pneumoniae isolate from 2004 (clonally related to the2005 outbreak isolates) also produced CTX-M-1. On the other
FIG. 1. A. Incidence of ESBL-producing Enterobacteriaceae (one isolate per patient) in the ICU during the period from 2002 to 2005. B.Distribution of new cases of isolation of ESBL-producing Enterobacteriaceae on a monthly basis during 2005.
VOL. 44, 2006 CARBAPENEM RESISTANCE IN CTX-M-1-PRODUCING KLEBSIELLA 2833

hand, the 2003 isolate produced an ESBL from the CTX-M-9group.
Several attempts to transfer blaCTX-M-1 by conjugation to theE. coli recipient consistently failed. Nevertheless, the plasmidlocation of blaCTX-M-1was demonstrated by transformation ex-periments. Plasmid DNA was transformed into E. coli, andthree transformants growing on LB agar plates containing 50!g/ml of ampicillin were first checked by DDST followed byIEF, which revealed the presence of a single "-lactamase bandwith a pI of !8.4, and then by PCR amplification with thespecific primers. The size of the plasmid determined by theanalysis of the EcoRI and BamHI restriction fragments wasestimated to be approximately 50 kb.
In two of the patients infected by the epidemic multiresistantK. pneumoniae clone, development of carbapenem (imipenem,meropenem, and ertapenem) resistance was documented. Forone of them, the carbapenem-resistant isolate was recoveredfrom respiratory samples after treatment with imipenem, andfor the other the isolate was recovered from urine after treat-ment with meropenem. In both patients, the carbapenem-re-sistant isolates were additionally detected in the intestinal col-onization studies. PFGE macrorestriction patterns of the twocarbapenem-resistant isolates were found to be identical to
those of the carbapenem-susceptible isolates, and the acquisi-tion of additional "-lactamases was ruled out by IEF.
Table 2 shows the MICs (determined by Etest) of several"-lactam and non-"-lactam antibiotics for five representativeisolates of the epidemic clone recovered from different pa-tients and during different periods and for the two carbap-enem-resistant isolates. The MICs in both resistant isolateswere slightly higher for ertapenem (MIC of #32 !g/ml), fol-lowed by meropenem (MIC of 16 !g/ml) and imipenem (MICof 8 to 12 !g/ml). The MICs for the E. coli transformantharboring the blaCTX-M-1-carrying plasmid (pAMR-1) are alsoshown in Table 2. As can be observed, in addition to theESBL-mediated resistance pattern, aminoglycoside (gentami-cin and tobramycin) resistance was cotransferred in the sameplasmid. As for carbapenems, pAMR-1 significantly raised theMICs for E. coli XL1-Blue of ertapenem (from 0.012 to 0.125!g/ml) and, to a lesser extent, of meropenem (from 0.016 to0.047 !g/ml) but not of imipenem (0.38 !g/ml) (harboring or notpAMR-1). Nevertheless, the MICs of the three carbapenemsremained far below the resistance breakpoints.
In order to investigate whether carbapenem resistance wascaused by alterations of the outer membrane permeability, weanalyzed the outer membrane proteins of susceptible and re-sistant pairs of K. pneumoniae clinical isolates from two pa-tients. SDS-PAGE analysis of the OMP of K. pneumoniaegrown in LB showed only two major proteins, of about 33 and36 kDa, in the 31- to 45-kDa range for the carbapenem-sus-ceptible isolates, corresponding to OmpA and OmpK36, re-spectively. On the other hand, the 36-kDa protein was absentin both carbapenem-resistant isolates (Fig. 3A). The porinnature of the 36-kDa protein present in the carbapenem-sus-ceptible isolates and absent in the resistant ones was confirmedby Western blot analysis using specific antibodies (Fig. 3B).The specific antiserum reacted with the band present in thesusceptible isolates and the control K. pneumoniae LB1 butfailed to detect the protein in the resistant isolates.
To investigate the cause of the porin deficiency observed inthe carbapenem-resistant isolates, we amplified the entire
FIG. 2. XbaI PFGE patterns of ESBL-producing K. pneumoniae isolates from ICU patients. The single ESBL-producing K. pneumoniae isolatesdetected in 2003 and 2004 are represented in lanes 1 and 2, respectively, whereas lanes 3 to 22 contain representative isolates from the 2005epidemic. MWM, molecular weight markers.
TABLE 1. Sources of the ESBL-producing K. pneumoniae epidemicclone for the 51 ICU patients during the 2005 outbreak
Source No. (%) ofpatients
Intestinal colonization only ........................................................ 7 (13.7)
Isolation from clinical samples ..................................................44 (86.3)Respiratory tract......................................................................30 (68.2)Urinary tract............................................................................. 7 (15.9)Wound infection ......................................................................11 (25.0)Vascular catheter..................................................................... 8 (18.2)Blood......................................................................................... 4 (9.1)Others ....................................................................................... 4 (9.1)!2 sources................................................................................15 (34.1)
2834 MENA ET AL. J. CLIN. MICROBIOL.
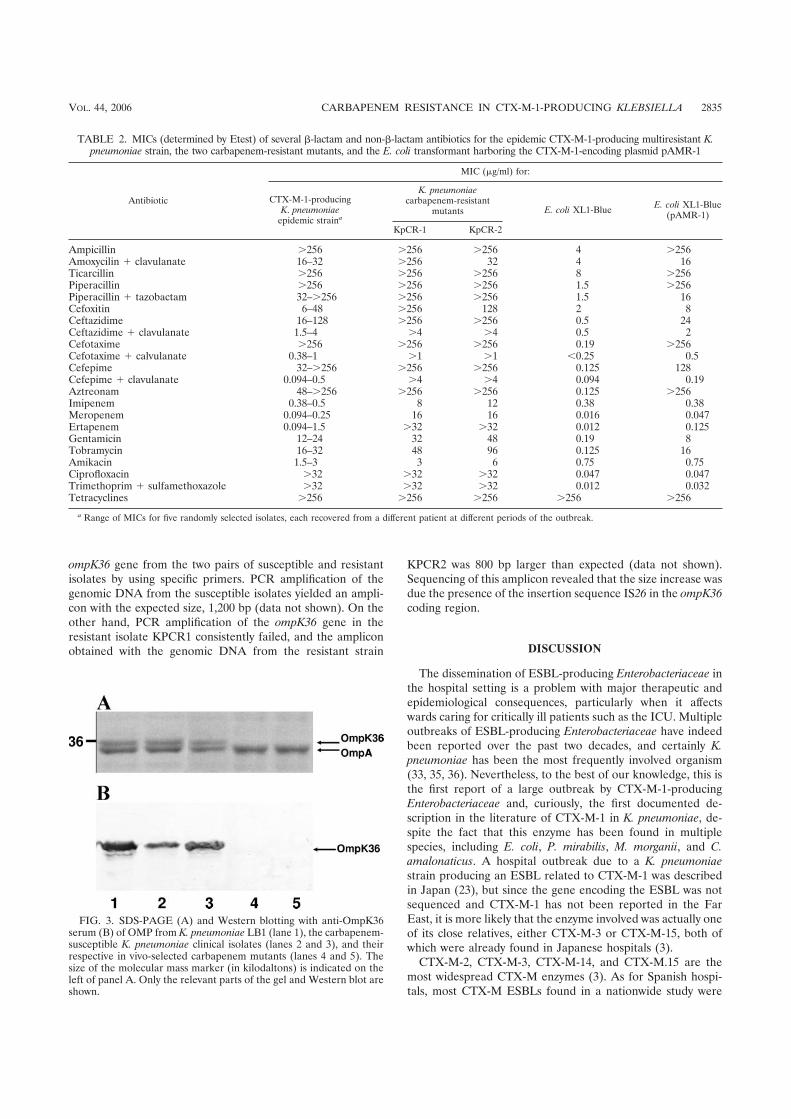
ompK36 gene from the two pairs of susceptible and resistantisolates by using specific primers. PCR amplification of thegenomic DNA from the susceptible isolates yielded an ampli-con with the expected size, 1,200 bp (data not shown). On theother hand, PCR amplification of the ompK36 gene in theresistant isolate KPCR1 consistently failed, and the ampliconobtained with the genomic DNA from the resistant strain
KPCR2 was 800 bp larger than expected (data not shown).Sequencing of this amplicon revealed that the size increase wasdue the presence of the insertion sequence IS26 in the ompK36coding region.
DISCUSSION
The dissemination of ESBL-producing Enterobacteriaceae inthe hospital setting is a problem with major therapeutic andepidemiological consequences, particularly when it affectswards caring for critically ill patients such as the ICU. Multipleoutbreaks of ESBL-producing Enterobacteriaceae have indeedbeen reported over the past two decades, and certainly K.pneumoniae has been the most frequently involved organism(33, 35, 36). Nevertheless, to the best of our knowledge, this isthe first report of a large outbreak by CTX-M-1-producingEnterobacteriaceae and, curiously, the first documented de-scription in the literature of CTX-M-1 in K. pneumoniae, de-spite the fact that this enzyme has been found in multiplespecies, including E. coli, P. mirabilis, M. morganii, and C.amalonaticus. A hospital outbreak due to a K. pneumoniaestrain producing an ESBL related to CTX-M-1 was describedin Japan (23), but since the gene encoding the ESBL was notsequenced and CTX-M-1 has not been reported in the FarEast, it is more likely that the enzyme involved was actually oneof its close relatives, either CTX-M-3 or CTX-M-15, both ofwhich were already found in Japanese hospitals (3).
CTX-M-2, CTX-M-3, CTX-M-14, and CTX-M.15 are themost widespread CTX-M enzymes (3). As for Spanish hospi-tals, most CTX-M ESBLs found in a nationwide study were
FIG. 3. SDS-PAGE (A) and Western blotting with anti-OmpK36serum (B) of OMP from K. pneumoniae LB1 (lane 1), the carbapenem-susceptible K. pneumoniae clinical isolates (lanes 2 and 3), and theirrespective in vivo-selected carbapenem mutants (lanes 4 and 5). Thesize of the molecular mass marker (in kilodaltons) is indicated on theleft of panel A. Only the relevant parts of the gel and Western blot areshown.
TABLE 2. MICs (determined by Etest) of several !-lactam and non-!-lactam antibiotics for the epidemic CTX-M-1-producing multiresistant K.pneumoniae strain, the two carbapenem-resistant mutants, and the E. coli transformant harboring the CTX-M-1-encoding plasmid pAMR-1
Antibiotic
MIC ("g/ml) for:
CTX-M-1-producingK. pneumoniae
epidemic straina
K. pneumoniaecarbapenem-resistant
mutants E. coli XL1-Blue E. coli XL1-Blue(pAMR-1)
KpCR-1 KpCR-2
Ampicillin #256 #256 #256 4 #256Amoxycilin $ clavulanate 16–32 #256 32 4 16Ticarcillin #256 #256 #256 8 #256Piperacillin #256 #256 #256 1.5 #256Piperacillin $ tazobactam 32–#256 #256 #256 1.5 16Cefoxitin 6–48 #256 128 2 8Ceftazidime 16–128 #256 #256 0.5 24Ceftazidime $ clavulanate 1.5–4 #4 #4 0.5 2Cefotaxime #256 #256 #256 0.19 #256Cefotaxime $ calvulanate 0.38–1 #1 #1 %0.25 0.5Cefepime 32–#256 #256 #256 0.125 128Cefepime $ clavulanate 0.094–0.5 #4 #4 0.094 0.19Aztreonam 48–#256 #256 #256 0.125 #256Imipenem 0.38–0.5 8 12 0.38 0.38Meropenem 0.094–0.25 16 16 0.016 0.047Ertapenem 0.094–1.5 #32 #32 0.012 0.125Gentamicin 12–24 32 48 0.19 8Tobramycin 16–32 48 96 0.125 16Amikacin 1.5–3 3 6 0.75 0.75Ciprofloxacin #32 #32 #32 0.047 0.047Trimethoprim $ sulfamethoxazole #32 #32 #32 0.012 0.032Tetracyclines #256 #256 #256 #256 #256
a Range of MICs for five randomly selected isolates, each recovered from a different patient at different periods of the outbreak.
VOL. 44, 2006 CARBAPENEM RESISTANCE IN CTX-M-1-PRODUCING KLEBSIELLA 2835

either CTX-M-14 or its close relative CTX-M-9 (16), whereasCTX-M-10, which is closely related to CTX-M-1, was highlydisseminated among multiple Enterobacteriaceae species in sin-gle hospital from Madrid (10, 30). At this stage, the onlypublished documentation of CTX-M-1-producing Enterobacte-riaceae in Spain is the report of a single E. coli isolate detectedin cattle milk (6).
Carbapenems are frequently the only therapeutic optionsavailable for treatment of hospital-acquired severe infectionscaused by multiresistant ESBL-producing Enterobacteriaceaesuch as the strain described in this work. Nevertheless, univer-sal susceptibility to these last-line antimicrobials in Enterobac-teriaceae is no longer guaranteed. Indeed, several carbapen-emases have been described as occurring in Enterobacteriaceae,including representatives from classes A, such as the highlydisseminated KPC enzymes; B, such as IMP or VIM metallo-!-lactamases; or D, such as OXA-48 (29, 34, 39). Additionally,carbapenem resistance development in strains producingESBLs or AmpC !-lactamases due to the selection of mutantswith reduced permeability to these antimicrobials is being in-creasingly reported (4, 9). Previous work has shown that theexpression of both major K. pneumoniae porins, OmpK35 andOmpK36, plays a role in the susceptibility to carbapenem an-tibiotics and that mutants lacking expression of both outermembrane proteins show reduced susceptibility to these anti-microbials, especially when combined with AmpC enzymes andsome class A ESBLs (11, 13, 19, 27). Nevertheless, in this workwe document and characterize for the first time the develop-ment of in vivo resistance to carbapenems in CTX-M-1-pro-ducing Enterobacteriaceae due to the selection of mutationsleading to the lack of porin expression. As frequently occurs inclinical ESBL-producing K. pneumoniae isolates (18), the epi-demic strain described in this work did not express OmpK35,favoring carbapenem resistance development through the in-activation of OmpK36. Inactivation of OmpK36 in one of thestrains was demonstrated to be mediated by the interruption ofthe coding sequence by an insertion sequence, as has beenpreviously reported for other clinical strains (17). As for thesecond carbapenem-resistant isolate, the failure of ompK36amplification could suggest that the mechanism for inactiva-tion was either a deletion of part or the complete codingsequence or its interruption by an insertion sequence too largeto be amplified by conventional PCR.
Overall, CTX-M-1-producing K. pneumoniae isolates from 2of the 44 infected patients (4.5%) developed carbapenem re-sistance in clinical samples. Nevertheless, both patients hadintestinal colonization by the carbapenem-resistant isolatewhen it was detected in the clinical samples, which could sug-gest that resistance development may have taken place in theintestines of the colonized patients. Since the development ofcarbapenem resistance was not systematically explored in theintestinal colonization studies, we may indeed be underesti-mating the magnitude of the potential problem of carbapenemresistance development in colonized patients as a source ofcarbapenem-resistant isolates.
In summary, we have described a large outbreak due to amultiresistant CTX-M-1-producing and OmpK35-deficient K.pneumoniae strain in an ICU and have characterized the mech-anisms leading to in vivo carbapenem resistance development
through the inactivation of OmpK36 in two of the infectedpatients.
ACKNOWLEDGMENTS
We are grateful to Enrique Ruiz de Gopegui for collecting thedata on the prevalence of ESBL-producing K. pneumoniae infec-tions in the ICU.
This work was partially supported by the “Red Espanola de Inves-tigacion en Patologıa Infecciosa” (C03-014) and grant PI040339 fromthe Fondo de Investigaciones Sanitarias from the Ministerio deSanidad and grant SAF2005-01466 from the Ministerio de Educaciony Ciencia of Spain.
REFERENCES
1. Albertı, S., F. Rodrıguez-Quinones, T. Schirmer, G. Rummel, J. M. Tomas,J. P. Rosenbusch, and V. J. Benedı. 1995. A porin from Klebsiella pneu-moniae: sequence homology, three-dimensional structure, and complementbinding. Infect. Immun. 63:903–910.
2. Bauernfeind, A., H. Grimm, and S. Schweighart. 1990. A new plasmidiccefotaximase in a clinical isolate of Escherichia coli. Infection 18:294–298.
3. Bonnet, R. 2004. Growing group of extended-spectrum !-lactamases: theCTX-M enzymes. Antimicrob. Agents Chemother. 48:1–14.
4. Bornet, C., A. Davin-Regli, C. Bosi, J. M. Pages, and C. Bollet. 2000. Imi-penem resistance of Enterobacter aerogenes mediated by outer membranepermeability. J. Clin. Microbiol. 38:1048–1052.
5. Bradford, P. A. 2001. Extended-spectrum !-lactamases in the 21st century:characterization, epidemiology, and detection of this important resistantthreat. Clin. Microbiol. Rev. 14:933–951.
6. Brinas, L., M. A. Moreno, T. Teshager, Y. Saenz, M. C. Porrero, L.Domınguez, and C. Torres. 2005. Monitoring and characterization of extended-spectrum !-lactamases in Escherichia coli strains from healthy and sickanimals in Spain in 2003. Antimicrob. Agents Chemother. 49:1262–1264.
7. Bush, K., G. A. Jacoby, and A. A. Medeiros. 1995. A functional classificationscheme for !-lactamases and its correlation with molecular structure. Anti-microb. Agents Chemother. 39:1211–1233.
8. Canton, R., M. Perez-Vazquez, A. Oliver, B. Sanchez Del Saz, M. O. Gutierrez,M. Martinez-Ferrer, and F. Baquero. 2000. Evaluation of the WIDER sys-tem, a new computer-assisted image-processing device for bacterial identi-fication and susceptibility testing. J. Clin. Microbiol. 38:1339–1346.
9. Cao, V. T. B., G. Arlet, B. M. Ericsson, A. Tammelin, P. Courvalin, and T.Lambert. 2000. Emergence of imipenem resistance in Klebsiella pneumoniaeowing to combination of plasmid-mediated CMY-4 and permeability alter-ation. J. Antimicrob. Chemother. 46:895–900.
10. Coque, T. M., A. Oliver, J. C. Perez-Diaz, F. Baquero, and R. Canton. 2002.Genes encoding TEM-4, SHV-2, and CTX-M-10 extended-spectrum beta-lactamases are carried by multiple Klebsiella pneumoniae clones in a singlehospital (Madrid 1989 to 2000). Antimicrob. Agents Chemother. 46:500–510.
11. Crowley, B., V. J. Benedi, and A. Domenech-Sanchez. 2002. Expression ofSHV-2 !-lactamase and of reduced amounts of OmpK36 porin in Klebsiellapneumoniae results in increased resistance to cephalosporins and carbapenems.Antimicrob. Agents Chemother. 46:3679–3682.
12. Davison, M. S., and A. O. Summers. 1983. Wide-host-range plasmids in thegenus Thiobacillus. Appl. Environ. Microbiol. 46:565–572.
13. Domenech-Sanchez, A., L. Martınez-Martınez, S. Hernandez-Alles, M. C.Conejo, A. Pascual, J. M. Tomas, S. Albertı, and V. J. Benedı. 2003. Role ofKlebsiella pneumoniae OmpK35 porin in antimicrobial resistance. Antimi-crob. Agents Chemother. 47:3332–3335.
14. Dutour, C., R. Bonnet, H. Marchandin, M. Boyer, C. Chanal, D. Sirot, andJ. Sirot. 2002. CTX-M-1, CTX-M-3, and CTX-M-14 beta-lactamases fromEnterobacteriaceae isolated in France. Antimicrob. Agents Chemother. 46:534–537.
15. Filip, C., G. Fletcher, J. L. Wulf, and C. F. Earhart. 1973. Solubilization ofthe cytoplasmic membrane of Escherichia coli by the ionic detergent sodiumlauryl sarcosinate. J. Bacteriol. 115:717–722.
16. Hernandez, J. R., L. Martınez-Martınez, R. Canton, T. M. Coque, A.Pascual, and the Spanish Group for Nosocomial Infections. 2005. Nation-wide study of Escherichia coli and Klebsiella pneumoniae producing extended-spectrum !-lactamases in Spain. Antimicrob. Agents Chemother. 49:2122–2125.
17. Hernandez-Alles, S., V. J. Benedı, L. Martınez-Martınez, A. Pascual, A.Aguilar, J. M. Tomas, and S. Alberti. 1999. Development of resistanceduring antimicrobial therapy caused by insertion sequence interruption ofporin genes. Antimicrob. Agents Chemother. 43:937–939.
18. Hernandez-Alles, S., M. Conejo, A. Pascual, J. M. Tomas, V. J. Benedı, andL. Martınez-Martınez. 2000. Relationship between outer membrane alter-ations and susceptibility to antimicrobial agents in isogenic strains of Kleb-siella pneumoniae. J. Antimicrob. Chemother. 46:273–277.
19. Jacoby, G. A., D. M. Mills, and N. Chow. 2004. Role of !-lactamases and
2836 MENA ET AL. J. CLIN. MICROBIOL.

porins in resistance to ertapenem and other !-lactams in Klebsiella pneu-moniae. Antimicrob. Agents Chemother. 48:3203–3206.
20. Jarlier, V., M. Nicolas, G. Fournier, and A. Philippon. 1988. Extendedbroad-spectrum !-lactamases conferring transferable resistance to newer!-lactam agents in Enterobacteriaceae: hospital prevalence and susceptibil-ity patterns. Rev. Infect. Dis. 10:867–878.
21. Kaufmann, M. E. 1998. Pulsed-field gel electrophoresis. Methods Mol. Med.15:17–31.
22. Kliebe, C., B. A. Nies, J. F. Meyer, R. M. Toixdorff-Neutzling, and B. Wiedemann.1985. Evolution of plasmid-coded resistance to broad-spectrum cephalosporins.Antimicrob. Agents Chemother. 28:302–307.
23. Komatsu, M., N. Ikeda, M. Aihara, Y. Nakamachi, S. Kinoshita, K.Yamasaki, and K. Shimakawa. 2000. Hospital outbreak of MEN-1-derivedextended spectrum beta-lactamase-producing Klebsiella pneumoniae. J. In-fect. Chemother. 7:94–101.
24. Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly ofthe head of bacteriophage T4. Nature 277:680–685.
25. Livermore, D. M. 1997. !-Lactamases in laboratory and clinical resistance.Clin. Microbiol. Rev. 8:557–584.
26. Martınez-Martınez, L., S. Hernandez-Alles, S. Albertı, J. M. Tomas, V. J.Benedı, and G. A. Jacoby. 1996. In vivo selection of porin-deficient mutantsof Klebsiella pneumoniae with increased resistance to cefoxitin and expanded-spectrum cephalosporins. Antimicrob. Agents Chemother. 40:342–348.
27. Martınez-Martınez, L., A. Pascual, S. Hernandez-Alles, D. Alvarez-Dıaz,A. I. Suarez, J. Tran, V. J. Benedı, and G. A. Jacoby. 1999. Roles of !-lac-tamases and porins in activities of carbapenems and cephalosporins againstKlebsiella pneumoniae. Antimicrob. Agents Chemother. 43:1669–1673.
28. Mugnaioli, C., F. Luzzaro, F. De Luca, G. Brigante, G. Amicosante, andG. M. Rossolini. 2005. Dissemination of CTX-M-type extended-spectrum!-lactamase genes to inusual hosts. J. Clin. Microbiol. 43:4183–4185.
29. Nordmann, P., and L. Poirel. 2002. Emerging carbapenemases in gram-negative aerobes. Clin. Microbiol. Infect. 8:321–331.
30. Oliver, A., T. M. Coque, D. Alonso, A. Valverde, F. Baquero, and R. Canton.2005. CTX-M-10 linked to a phage-related element is widely disseminatedamong Enterobacteriaceae in a Spanish hospital. Antimicrob. Agents Che-mother. 49:1567–1571.
31. Pagani, L., E. Dell’Amico, R. Migliavacca, M. M. D’Andrea, E. Giacobone,
G. Amicosante, E. Romero, and G. M. Rossolini. 2003. CTX-M-type extended-spectrum !-lactamases in nosocomial isolates of Enterobacteriaceae from ahospital in northern Italy. J. Clin. Microbiol. 41:4264–4269.
32. Paterson, D. L., L. Mulazimoglu, J. M. Casellas, W. C. Ko, H. Goossens, A.Von Gotteberg, S. Mohapatra, G. M. Trenholme, K. P. Kugman, J. G.McCormack, and V. L. Yu. 2000. Epidemiology of ciprofloxacin resistanceand its relationship to extended-spectrum beta-lactamase production inKlebsiella pneumoniae isolates causing bacteremia. Clin. Infect. Dis. 30:473–478.
33. Pena, C., M. Pujol, C. Ardanuy, A. Ricart, R. Pallares, J. Linares, J. Ariza,and F. Gudiol. 1998. Epidemiology and successful control of a large outbreakdue to Klebsiella pneumoniae producing extended-spectrum beta-lactamases.Antimicrob. Agents Chemother. 42:53–58.
34. Poirel, L., C. Heritier, V. Tolun, and P. Nordmann. 2004. Emergence ofoxacillinase-mediated resistance to imipenem in Klebsiella pneumoniae. An-timicrob. Agents Chemother. 48:15–22.
35. Quale, J. M., D. Landman, P. A. Bradford, M. Visalli, J. Ravishankar, C.Flores, D. Mayorga, K. Vangala, and A. Adedeji. 2002. Molecular epidemi-ology of a citywide outbreak of extended-spectrum beta-lactamase-produc-ing Klebsiella pneumoniae infection. Clin. Infect. Dis. 35:834–841.
36. Rebuck, J. A., K. M. Olsen, P. D. Fey, A. N. Langnas, and M. E. Rupp. 2000.Characterization of an outbreak due to extended-spectrum beta-lactamaseproducing Klebsiella pneumoniae in a pediatric intensive care unit transplantpopulation. Clin. Infect. Dis. 31:1368–1372.
37. Tenover, F. C., R. D. Arbeit, R. V. Goering, P. A. Mickelsen, B. E. Murray,D. H. Persing, and B. Swaminathan. 1995. Interpreting chromosomal DNArestriction patterns produced by pulsed-field gel electrophoresis: criteria forbacterial strain typing. J. Clin. Microbiol. 33:2233–2239.
38. Towbin, H., T. Staehelin, and J. Gordon. 1979. Electrophoretic transfer ofproteins from polyacrylamide gels to nitrocellulose sheets: procedure andsome applications. Proc. Natl. Acad. Sci. USA 76:4350–4354.
39. Yigit, H., A. M. Queenan, G. J. Anderson, A. Domenech-Sanchez, J. W.Biddle, C. D. Steward, S. Alberti, K. Bush, and F. C. Tenover. 2001. Novelcarbapenem-hydrolyzing beta-lactamase, KPC-1, from a carbapenem-resis-tant strain of Klebsiella pneumoniae. Antimicrob. Agents Chemother. 45:1151–1161.
VOL. 44, 2006 CARBAPENEM RESISTANCE IN CTX-M-1-PRODUCING KLEBSIELLA 2837

!

JOURNAL OF CLINICAL MICROBIOLOGY, Nov. 2010, p. 4089–4093 Vol. 48, No. 110095-1137/10/$12.00 doi:10.1128/JCM.01130-10Copyright © 2010, American Society for Microbiology. All Rights Reserved.
Carbapenem Heteroresistance in VIM-1-Producing Klebsiella pneumoniaeIsolates Belonging to the Same Clone: Consequences
for Routine Susceptibility Testing!
M. Tato,1 M. Morosini,1 L. García,3 S. Albertı,3 M. T. Coque,1,2 and R. Canton1,2*Servicio de Microbiología, CIBER en Epidemiología y Salud Publica (CIBERESP) Hospital Universitario Ramon y Cajal, and
Instituto Ramon y Cajal de Investigacion Sanitaria (IRYCIS),1 and Unidad de Resistencia Asociada al Consejo Superior deInvestigaciones Científicas (CSIC),2 Madrid, and Instituto Universitario de Investigacion en Ciencias de la Salud,
Universidad de las Islas Baleares, Palma de Mallorca,3 Spain
Received 3 June 2010/Returned for modification 23 July 2010/Accepted 8 September 2010
Susceptibility results with low reproducibility by the same or different methods have been observed formetallo-beta-lactamase (MBL)-producing Enterobacteriaceae. Eighteen VIM-1-producing Klebsiella pneumoniaeisolates (one per patient) belonging to a single epidemic clone in our hospital (2005 to 2008) but with differentsusceptibilities to carbapenems were studied. Imipenem MICs ranged from 8 to >128 mg/liter by standardCLSI microdilution, from <1 to >8 mg/liter by the semiautomatic Wider system, and from 0.75 to >32 mg/literby Etest. Meropenem MICs ranged from 0.5 to 128, <1 to >8, and 0.38 to >32 mg/liter, respectively.Ertapenem MICs by CLSI microdilution and Etest ranged from 1 to 64 and 0.75 to >32 mg/liter, respectively.The rates of essential agreement (!1 log2 dilution) for imipenem and meropenem MICs between the Widersystem and the reference microdilution method were 45% and 49%, respectively. Those between Etest and thereference microdilution method for imipenem, meropenem, and ertapenem MICs were 33%, 67%, and 84%. Therates of very major errors for the Wider system and Etest were 33% and 28% for imipenem and 25% and 75%for meropenem, respectively. Low MIC reproducibility was observed even when the same inoculum was used(differences up to 4-fold dilutions). Heteroresistance was suspected due to the presence of colonies in the Etestinhibition zone. It was confirmed by population analysis profiles of 4 isolates displaying different imipenemMICs, with the exception of an OmpK36-porin-deficient isolate that homogeneously expressed carbapenemresistance (MIC, >128 mg/liter). Low carbapenem MIC reproducibility could be due to the presence ofresistant subpopulations and variable expression of the resistance mechanisms. Since carbapenem MICs arenot good markers of MBL production, reliable and reproducible phenotypic methods are needed to detect thepresence of this mechanism.
Resistance to carbapenems due to metallo-beta-lactamases(MBLs) in Enterobacteriaceae is increasingly recognized, anddifferent levels of resistance to carbapenems have been ob-served in these isolates (18). The detection of MBL-carryingisolates in clinical laboratories presents practical difficultieswhen routine susceptibility testing is performed (10, 28). Au-tomated systems do not accurately detect enzyme-mediatedcarbapenem resistance when it is expressed at low levels, andMBL-carrying organisms can even appear to be fully suscepti-ble to these compounds (9, 28). Additionally, reproducibleresults within the same method or among different methods,such as disk diffusion or Etest, are not always obtained, anddiscrepancies in the susceptibility to imipenem and mero-penem of carbapenemase (KPC or VIM types)-producingKlebsiella pneumoniae isolates have already been reported(10, 28).
In addition, some studies describing heteroresistance to car-bapenems in Acinetobacter baumannii isolates demonstrate itspresence by the growth of colonies in the inhibition zone of theEtest strip (12, 19). This phenomenon has also been reported
for Klebsiella pneumoniae (28), Pseudomonas aeruginosa (20),and, more recently, Enterobacter aerogenes isolates (11).
The objective of this work was to analyze the carbapenemsusceptibility profiles of VIM-1-producing K. pneumoniae iso-lates by different susceptibility testing methods. These isolatesbelong to a single clone that was involved in an epidemic in ourhospital (2005 to 2008) affecting 18 patients in different wards(25).
MATERIALS AND METHODS
Bacterial strains. Eighteen VIM-1-producing K. pneumoniae isolates (oneisolate per patient) belonging to an epidemic clone were studied. All isolateswere collected from 2005 through 2008 at Ramon y Cajal University Hospital inMadrid, Spain. Isolates were identified using the semiautomatic Wider system(Fco. Soria Melguizo, Madrid, Spain) (1).
Clonal relatedness. Clonality among K. pneumoniae isolates was demonstratedby comparison of their XbaI-digested genomic DNA patterns obtained bypulsed-field gel electrophoresis (PFGE) as described previously (25). Electro-phoresis conditions were 24 h at 6 V/cm and 14°C, with initial and final pulsetimes of 10 and 40 s, using a CHEF-DRIII system (Bio-Rad, Hemel Hempstead,United Kingdom). Clonality was assessed based on the criteria of Tenover et al.(27).
Susceptibility testing. Imipenem, meropenem, and ertapenem MICs weredetermined by the broth microdilution method (range of concentrations tested,0.06 to 128 mg/liter) using BBL Mueller-Hinton broth (lot 6139218; BectonDickinson, Sparks, MD) (3). Imipenem and ertapenem powders were providedby Merck, Sharp & Dohme (Spain), and meropenem was provided by AstraZeneca (Spain). Microdilution (Wider system) (1) and Etest (bioMerieux,
* Corresponding author. Mailing address: Servicio de Microbiología,Hospital Universitario Ramon y Cajal, Carretera de Colmenar Km 9,1,28034 Madrid. Spain. Phone: 34-913368330. Fax: 34-913368809. E-mail:[email protected].
! Published ahead of print on 15 September 2010.
4089
Marcy-l’Etoile, France) were performed according to the manufacturers’ instruc-tions by using Mueller-Hinton agar plates (BBL Mueller Hinton broth and Bactoagar [lots 6139218 and 6188520, respectively]; Becton Dickinson, Sparks, MD)made in-house. In addition, 4 selected isolates were tested in triplicate forsusceptibility to carbapenems either by the reference microdilution method orwith Etest strips by using the same and different inocula. The reference strainEnterococcus faecalis ATCC 29212 (imipenem MIC, 0.5 to 2 mg/liter; mero-penem MIC, 2 to 8 mg/liter; ertapenem MIC, 4 to 16 mg/liter) was included forquality control in all tests. All MIC values were read by two persons and, ifnecessary, by a third person to resolve disagreements.
Analysis of susceptibility testing results. Carbapenem MICs obtained by stan-dard microdilution, the Wider semiautomatic system (panel reference C095-31/W/REV2; Fco. Soria Melguizo, Madrid, Spain), and Etest were compared. EtestMIC values were converted to the double-dilution scale currently used in con-ventional methods (rounded up to the next highest dilution when necessary).Qualitative interpretive categories were defined using the breakpoints newlyapproved by the CLSI for imipenem and meropenem (susceptible, !1 mg/liter;resistant, "4 mg/liter) and ertapenem (susceptible, !0.25 mg/liter; resistant, "1mg/liter) (4). Ertapenem MICs obtained by the Wider system were ignored in theanalysis, since the concentrations included in the susceptibility testing panel werenot adequate for the new CLSI breakpoints.
Essential agreement was defined as identical MIC results, or MIC resultsagreeing within !1 log2 dilution, by the test method and the reference method.Categorical agreement and errors in the clinical interpretive categories weredetermined by using standard broth microdilution as the reference method.Categorical agreement was defined as identical interpretive categories (suscep-tible, intermediate, or resistant) obtained by the two methods. A very major errorwas considered to be present when the isolate was categorized as susceptible bythe test method and as resistant by the reference method. A major error occurredwhen the test method results fell into the resistant category but the result by thereference microdilution method was in the susceptible category. A minor erroroccurred when the isolate was considered susceptible or resistant either by thetest method or by the reference method but intermediate by the other method.Denominators for calculating error rates, based on broth microdilution results,were as follows: the number of resistant isolates (very major error rate), thenumber of susceptible isolates (major error rate), and the total number ofisolates tested (minor error rate).
Study of mechanisms contributing to carbapenem resistance. The presence ofMBL was inferred from the double-disk synergy test using a blank disk coatedwith 10 "l of 0.5 M EDTA versus imipenem and ceftazidime disks (each 15 mmaway from the EDTA-containing disk) and was confirmed by PCR amplificationand further sequencing as described previously (25). The porin expression of fiveselected strains was also studied by isolation and analysis of outer membraneproteins (OMP) as described previously (17).
PAP. Heteroresistance was determined by population analysis profile (PAP)for 4 selected strains displaying different imipenem MIC values and for the
carbapenem-susceptible K. pneumoniae isolate ATCC 13883. For this purpose, 1colony from a culture grown overnight on Columbia agar plates was inoculatedinto 5 ml of LB broth. After 24 h of incubation, 10#1, 10#3, and 10#5 dilutionsof this culture were prepared in saline. Volumes (100 "l) were inoculated ontoLB agar plates containing 0, 0.25, 0.5, 1, 2, 4, 6, 8, 12, 16, and 32 mg/liter ofimipenem. After 48 h of incubation at 35°C, the colonies were counted, and thelog10 CFU/ml was plotted against the antibiotic concentration.
RESULTS
Susceptibility testing. Eighteen VIM-1-producing K. pneu-moniae isolates belonging to one clone according to PFGEresults (data not shown) were tested for resistance to imi-penem, meropenem, and ertapenem by broth microdilution,Etest, and Wider conventional susceptibility panels. The sus-ceptibility testing results obtained by each method and theinterpretive criteria are shown in Table 1. In general, the imi-penem, meropenem, and ertapenem results obtained by theWider system and Etest were lower than the respective valuesobtained by the reference broth microdilution method. Mero-penem MICs were lower than the MICs obtained for imi-penem and ertapenem using standard broth microdilution. Itshould be noted that, in contrast to meropenem, all isolateswere resistant to imipenem and ertapenem by standard brothmicrodilution with the new CLSI breakpoints for these antibi-otics (Table 1).
The levels of essential agreement between the results ob-tained by the Etest method or the Wider system and thoseobtained by the broth microdilution test for susceptibility toimipenem, meropenem, and ertapenem are shown in Table 2.The rate of agreement within 1 2-fold dilution between Etestand the broth microdilution reference method was lower forimipenem (33%) than for meropenem (67%) or ertapenem(84%). The rate of essential agreement between the Widersystem and the broth microdilution method was 45% for imi-penem and 49% for meropenem. The levels of interpretivecategorical agreement between the Wider system and the ref-erence method were 67% for imipenem and 33% for mero-penem. With Etest, these values were 39% for imipenem, 17%
TABLE 1. Susceptibility testing results obtained by each method and interpretative criteria for the 18VIM-1-producing K. pneumoniae isolates
Method andantibiotic
No. of isolates inhibited at the following concn (mg/liter)a: MIC (mg/liter)b No. of isolates within the followinginterpretive criterionc:
0.5 1 2 4 8 16 32 64 128 $128 Range 50% 90% Susceptible Intermediate Resistant
MicrodilutionImipenem % 11 5 1 & 1 8–$128 8 32 0 0 18Meropenem %1 3 10 3 1 1& 0.5–128 2 4 4 10 4Ertapenem % 2 13 1 1 & 1–64 2 8 0 0 18
EtestImipenem 5 3 6 2 & 2 0.75–$32 3 $32 5 6 7Meropenem 3 13 1 & 1 0.38–$32 1 1.5 16 1 1Ertapenem 1 9 6 1 & 1 0.75–$32 2 12 0 1 17
WiderImipenem %6 5 3& 4 !1–$8 4 $8 6 0 12Meropenem %8 2 3 3& 2 !1–$8 2 $8 8 2 8a Left and right brackets indicate the lowest and highest concentrations tested, respectively.b 50% and 90%, MICs at which 50 and 90% of the isolates, respectively, were inhibited.c The CLSI breakpoints for susceptibility and resistance were !1 mg/liter and "4 mg/liter for imipenem and meropenem and !0.25 mg/liter and "1 mg/liter for
ertapenem, respectively.
4090 TATO ET AL. J. CLIN. MICROBIOL.
for meropenem, and 72% for ertapenem. Interpretive categor-ical error rates with current CLSI breakpoints for each com-pound and method are shown in Table 3.
Low reproducibility of MICs was observed when the same ordifferent inocula were used. Differences of !4-fold in dilutionswere observed with the Etest for imipenem (1.5 to !32 mg/liter), meropenem (0.5 to !32 mg/liter), and ertapenem (8 to!32 mg/liter). With microdilution, these ranges were 16 to!128 mg/liter, 0.5 to 4 mg/liter, and 1 to 8 mg/liter, respec-tively.
Porin expression analysis. In order to investigate whethercarbapenem resistance could be due to mechanisms other thanVIM-1 production, 5 selected strains with different MICs wereanalyzed for OMP. Only one isolate with a high level of imi-penem resistance (MIC, !128 mg/liter) failed to express theOmpK36 porin.
Population analysis profile. Heteroresistance for imipenemwas initially suspected due to the recurrent growth of colonieswithin the inhibition zones of Etest strips. Heteroresistancewas confirmed by the PAP results (Fig. 1) obtained for allselected strains (Kpn 3, Kpn 8, and Kpn 12), with the exceptionof the porin-deficient K. pneumoniae isolate that expressedcarbapenem resistance homogeneously (Kpn 17). Heteroresis-tant isolates (Kpn 3, Kpn 8, and Kpn 12) contained resistantsubpopulations that grew in the presence of imipenem at con-centrations as high as 32 mg/liter. The proportions of resistantsubpopulations that were able to grow at the highest drugconcentration ranged from 3 " 10#7 to 5 " 10#6.
DISCUSSION
It is well known that heteroresistance, a phenomenon longobserved in Gram-positive pathogenic bacteria, affects suscep-tibility testing, particularly when routine automatic systems areused. From a clinical point of view, its importance is a matterof debate (22, 23). However, it has been less well documentedin Gram-negative isolates.
In this study we confirm that it is difficult to detect carbap-enem resistance in VIM-producing K. pneumoniae isolates,since different susceptibility testing results for either imi-penem, meropenem, or ertapenem were obtained by differentmethods. Problems in detecting carbapenem resistance withautomatic systems and Etest strips are a matter of concern, asdeduced from the high error rates in clinical category inter-pretation by use of microdilution as the reference method. Theautomatic system categorized six isolates and one isolate asfully susceptible to imipenem and meropenem (MICs, !1 mg/liter), respectively, whereas these isolates were categorized asresistant by the reference method. Likewise, the Etest failed todetect imipenem and meropenem resistance in five and threeisolates, respectively. This inconsistency is produced even withthe new breakpoints of the CLSI, which recommended valueslower than those previously used (3, 4). Although the objectiveof breakpoints is the clinical categorization of susceptibilitytesting results, our study demonstrates that current break-points still do not capture all carbapenemase-producing iso-lates. This is important because most of the laboratories, at
TABLE 2. Numbers and percentages of isolates tested by Etest and the WIDER system showing differences in log2 dilutionsfrom the results of the reference broth microdilution method
Antibioticand test
No. (%) of isolates showing a log2 dilution difference of:
!#3 #3 #2 #1 0 1 2 3 !3
ImipenemEtest 3 (17) 4 (22) 5 (28) 4 (22) 2 (11)WIDER 6 (33) 1 (6) 3 (17) 1 (6) 5 (28) 2 (11)
MeropenemEtest 5 (28) 9 (50) 3 (17) 1 (6)WIDER 1 (6) 3 (17) 3 (17) 4 (22) 2 (11) 4 (22) 1 (6)
Ertapenem, Etest 1 (6) 1 (6) 9 (50) 5 (28) 1 (6) 1 (6)
TABLE 3. Error rates for imipenem, meropenem, and ertapenem with the Wider system and Etest compared with the results of the brothmicrodilution reference methoda for the 18 VIM-1-producing K. pneumoniae isolates
Test method
No. (%) of errors for:
Imipenem Meropenem Ertapenem
Minor (n $ 18)b Major (n $ 0) Very major(n $ 18) Minor (n $ 18) Major (n $ 4) Very major
(n $ 4) Minor (n $ 18) Major (n $ 0) Very major(n $ 18)
Etest 6 (33) 0 5 (28) 11 (61) 0 3 (75) 1 (6) 0 0Wider 0 0 6 (33) 10 (56) 1 (25) 1 (25) NAc NA NA
a According to the CLSI (3, 4).b n, denominator used for calculating error rates, based on broth microdilution results. Denominators are the total number of isolates tested (minor error rate), the
number of susceptible isolates (major error rate), and the number of resistant isolates (very major error rate).c NA, not available.
VOL. 48, 2010 CARBAPENEM HETERORESISTANCE IN K. PNEUMONIAE 4091
least in our country, commonly use an automated system forroutine susceptibility testing and a second testing method, usu-ally Etest strips, to confirm dubious results. Discrepancies inresistance to carbapenems as determined by automated sus-ceptibility testing systems have been described previously (11,28). It has been shown that false resistance results were due tothe degradation of the drug, whereas false susceptibility resultscan be caused by underinoculation of the panels. On the otherhand, the growth of colonies within the inhibition zones of theEtest strips can make the MIC reading difficult. Nevertheless,in our case, the Etest MIC values were lower than those ob-tained by the reference method, even when one considers theintrahalo colonies, as recommended by the manufacturer, forthe Etest MIC reading. Another unexpected result is the lowreproducibility of the MIC results either by reference microdi-lution or with Etest strips, even by use of the same inoculum.
The diversity in carbapenem resistance among VIM-1-pro-ducing strains can be due to the variable expression of thisresistance mechanism affecting carbapenems or to the pres-ence of other resistance mechanisms, such as porin deficiency(15). The level of expression of a resistance gene can be influ-enced by the position of the cassette in the insert region and bydifferences in promoter strength (5, 8). In this study, all thestrains belonged to the same clone, and in all of them, theblaVIM cassette was located in the first position of the integronand was expressed from a P1/Pant promoter variant that hasbeen considered a hybrid of the weak and the strong versionsof Pant, showing an intermediate strength (14, 26; also M.Tato, M. T. Coque and R. Canton, unpublished data). Anothersource of MIC variability could be related to the potentialunstable plasmids containing the blaVIM gene. In this case, theplasmid copy number could differ in each passage of the iso-late. We even observed variability in the Etest susceptibilityresults when the same inoculum was used. Furthermore, theisolate with a homogeneously high level of carbapenem resis-tance (MIC, !16 mg/liter by the 3 methods) was the only onethat did not express the OmpK36 porin. The lack of this porinhas also been shown to affect the cephalosporin and carbap-enem MICs, particularly for extended-spectrum beta-lacta-mase or KPC producers (13, 16, 17). Another possible expla-
nation, at least in part, of the variable phenotypic expression ofcarbapenem resistance in VIM-producing K. pneumoniae iso-lates, despite the fact that they belong to the same clone, couldbe the presence of subpopulations expressing different resis-tance levels. In fact, population analyses revealed that a pro-portion of the initial bacterial inoculum can grow at high imi-penem concentrations, up to 32 mg/liter. This heterogeneousmode of growth against carbapenems has been described for A.baumannii, E. aerogenes, Bacillus cereus, and KPC-producingK. pneumoniae isolates (11, 19, 24, 28). Moreover, heteroresis-tance to carbapenems has also been confirmed by PAP analysisfor several Gram-negative rods, such as A. baumannii, P.aeruginosa, and K. pneumoniae (2, 12, 20, 21).
The unpredictable phenotypic expression of resistance inVIM– producing K. pneumoniae isolates and the consequentfailures in detecting resistance to carbapenems in these isolatesmay have implications for the clinical outcome of patients andfor the implementation of infection control procedures. Theadequate treatment of the patients infected with these micro-organisms is currently under debate (2). One issue is whetherthe infections caused by carbapenem-susceptible isolates thatproduce a carbapenemase can be treated with a carbapenem ornot, either alone or in combination with other active drugs (2,6). Some studies suggested that carbapenem resistance is anindependent predictor of adverse outcomes and that carbap-enems may provide some therapeutic benefits against MBL-positive carbapenem-susceptible isolates (7, 8). According tothis, it seems that carbapenem susceptibility in MBL-produc-ing isolates should be reported in accordance with the MICvalues, which is in agreement with current CLSI and EUCASTrecommendations (4; http://www.eucast.org). Despite slightdifferences between these committees on carbapenem break-points for Enterobacteriaceae, our results are particularly rele-vant for routine testing that may have important clinical con-sequences. According to the current recommendations, it is nolonger necessary to test for the presence of carbapenemases(i.e., modified Hodge test), and susceptibility testing resultsshould be reported as found. Nevertheless, the inaccuracy ofroutine methods may produce inadequate clinical categoriza-tion of carbapenems, with unacceptable rates of very major
FIG. 1. PAPs of 5 K. pneumoniae isolates: 3 VIM-1 producers (Kpn 3, Kpn 8, and Kpn 11), 1 VIM-1 producer deficient in OmpK36 porin (Kpn17), and 1 susceptible isolate (Kpn ATCC 13883).
4092 TATO ET AL. J. CLIN. MICROBIOL.

errors for carbapenems. Moreover, and according to the dataobtained in our study, it is remarkably difficult to obtain re-producible carbapenem MIC values with these isolates, partic-ularly by use of routine systems. From a practical point of view,all this supports the need to identify carbapenemase produc-tion as well as to determine the MIC.
In summary, this study shows that in the absence of moreclinical data, an alert should be released from clinical labora-tories when carbapenemase-producing isolates are found, in-dependently of their MIC values, since the correct identifica-tion of carbapenemase production also has implications for theimplementation of infection control measures to control theirdissemination (2). One limitation of our study is that the re-sults observed were obtained with isolates belonging to a singleVIM-1-producing K. pneumoniae clone, but low reproducibilityhas been noted by other authors with different isolates andsusceptibility testing methods (6, 9, 10, 18, 28). Despite thispotential limitation, we stressed discrepancies in carbapenemsusceptibility results even when the isolates belong to the sameclone.
ACKNOWLEDGMENTS
M.T. was supported by the Fondo de Investigaciones Sanitarias,which belongs to the Instituto de Salud Carlos III, Spanish Ministry ofScience and Innovation (reference no. CM06/00044, Rio Hortega pro-gram). This study was funded by research grants from the EuropeanCommission (LSHM-CT-2009-223031), the CIBERESP Network forBiomedical Research in Epidemiology and Public Health (referenceno. CB06/02/0053), and the Spanish Network for Research in Infec-tious Diseases (REIPI RD06/0008), supported by the Instituto deSalud Carlos III, Spanish Ministry of Health (co-financed by the Eu-ropean Development Regional Fund [“A way to achieve Europe”ERDF]).
We declare no conflict of interest.
REFERENCES
1. Canton, R., M. Perez-Vazquez, A. Oliver, B. Sanchez Del Saz, M. O. Guti-errez, M. Martínez-Ferrer, and F. Baquero. 2000. Evaluation of the Widersystem, a new computer-assisted image-processing device for bacterial iden-tification and susceptibility testing. J. Clin. Microbiol. 38:1339–1346.
2. Carmeli, Y., M. Akova, G. Cornaglia, G. L. Daikos, J. Garau, S. Harbarth,G. M. Rossolini, M. Souli, and H. Giamarellou. 2010. Controlling the spreadof carbapenemase-producing Gram-negatives: therapeutic approach and in-fection control. Clin. Microbiol. Infect. 16:102–111.
3. Clinical and Laboratory Standards Institute. 2010. Performance standardsfor antimicrobial susceptibility testing: 20th international Supplement.M100–S20. CLSI, Wayne, PA.
4. Clinical and Laboratory Standards Institute. 2010. Performance standardsfor antimicrobial susceptibility testing: 20th international supplement.M100–S20. June update. CLSI, Wayne, PA.
5. Collis, C. M., and R. M. Hall. 1995. Expression of antibiotic resistance genesin the integrated cassettes of integrons. Antimicrob. Agents Chemother.39:155–162.
6. Cornaglia, G., M. Akova, G. Amicosante, R. Canton, R. Cauda, J. D. Doc-quier, M. Edelstein, J. M. Frere, M. Fuzi, M. Galleni, H. Giamarellou, M.Gniadkowski, R. Koncan, B. Libisch, F. Luzzaro, V. Miriagou, F. Navarro, P.Nordmann, L. Pagani, L. Peixe, L. Poirel, M. Souli, E. Tacconelli, A. Vato-poulos, and G. M. Rossolini; ESCMID Study Group for Antimicrobial Re-sistance Surveillance (ESGARS). 2007. Metallo-beta-lactamases as emerg-ing resistant determinants in Gram-negative pathogens: open issues. Int. J.Antimicrob. Agents 29:380–388.
7. Daikos, G. L., P. Petrikkos, M. Psichogiou, C. Kosmidis, E. Vryonis, A.Skoutelis, K. Georgousi, L. S. Tzouvelekis, P. T. Tassios, C. Bamia, and G.Petrikkos. 2009. Prospective observational study of the impact of VIM-1metallo-!-lactamase on the outcome of patients with Klebsiella pneumoniaebloodstream infections. Antimicrob. Agents Chemother. 53:1868–1873.
8. Falcone, M., M. L. Mezzatesta, M. Perilli, C. Forcella, A. Giordano, V.
Cafiso, G. Amicosante, S. Stefani, and M. Venditti. 2009. Infections withVIM-1 metallo-!-lactamase-producing Enterobacter cloacae and their corre-lation with clinical outcome. J. Clin. Microbiol. 47:3514–3519.
9. Franklin, C., L. Liolios, and A. Y. Peleg. 2006. Phenotypic detection ofcarbapenem-susceptible metallo-beta-lactamase-producing gram-negativebacilli in the clinical laboratory. J. Clin. Microbiol. 44:3139–3144.
10. Giakkoupi, P., L. S. Tzouvelekis, G. L. Daikos, V. Miriagou, G. Petrikkos,N. J. Legakis, and A. C. Vatopoulos. 2005. Discrepancies and interpretationproblems in susceptibility testing of VIM-1-producing Klebsiella pneumoniaeisolates. J. Clin. Microbiol. 43:494–496.
11. Gordon, N. C., and D. W. Wareham. 2009. Failure of the MicroScan Walk-Away System to detect heteroresistance to carbapenems in a patient withEnterobacter aerogenes bacteremia. J. Clin. Microbiol. 47:3024–3025.
12. Ikonomidis, A., E. Neou, V. Gogou, G. Vrioni, A. Tsakris, and S. Pournaras.2009. Heteroresistance to meropenem in carbapenem-susceptible Acineto-bacter baumanii. J. Clin. Microbiol. 47:4055–4059.
13. Kitchel, B., J. K. Rasheed, A. Endimiani, A. M. Hujer, K. F. Anderson, R. A.Bonomo, and J. B. Patel. 26 July 2010. Genetic factors associated withelevated carbapenem resistance in KPC-producing Klebsiella pneumoniae.Antimicrob. Agents Chemother. [Epub ahead of print.] doi:10.1128/AAC.00008-10.
14. Levesque, C., S. Brassard, and P. H. Roy. 1994. Diversity and relativestrength of tandem promoters for the antibiotic-resistance genes of severalintegrons. Gene 142:49–54.
15. Loli, A., L. S. Tzouvelekis, E. Tzelepi, A. Carattoli, A. C. Vatopoulos, P. T.Tassios, and V. Miriagou. 2006. Sources of diversity of carbapenem resis-tance levels in Klebsiella pneumoniae carrying blaVIM-1. J. Antimicrob. Che-mother. 58:669–672.
16. Martínez-Martínez, L., A. Pascual, S. Hernandez-Alles, D. Alvarez-Díaz,A. I. Suarez, J. Tran, V. J. Benedí, and G. A. Jacoby. 1999. Roles of !-lac-tamases and porins in activities of carbapenems and cephalosporins againstKlebsiella pneumoniae. Antimicrob. Agents Chemother. 43:1669–1673.
17. Mena, A., V. Plasencia, L. García, O. Hidalgo, J. I. Ayestaran, S. Alberti, N.Borrell, J. L. Perez, and A. Oliver. 2006. Characterization of a large outbreakby CTX-M-1-producing Klebsiella pneumoniae and mechanisms leading to invivo carbapenem resistance development. J. Clin. Microbiol. 44:2831–2837.
18. Miriagou, V., G. Cornaglia, M. Edelstein, I. Galani, C. G. Giske, M. Gniad-kowski, E. Malamou-Lada, L. Martinez-Martinez, F. Navarro, P. Nord-mann, L. Peixe, S. Pournaras, G. M. Rossolini, A. Tsakris, A. Vatopoulos,and R. Canton. 2010. Acquired carbapenemases in gram-negative bacterialpathogens: detection and surveillance issues. Clin. Microbiol. Infect. 16:112–122.
19. Pournaras, S., A. Ikonomidis, A. Markogiannakis, A. N. Maniatis, and A.Tsakris. 2005. Heteroresistance to carbapenems in Acinetobacter baumannii.J. Antimicrob. Chemother. 55:1055–1056.
20. Pournaras, S., A. Ikonomidis, A. Markogiannakis, N. Spanakis, A. N. Ma-niatis, and A. Tsakris. 2007. Characterization of clinical isolates of Pseudo-monas aeruginosa heterogeneously resistant to carbapenems. J. Med. Micro-biol. 56:66–70.
21. Pournaras, S., I. Kristo, G. Vrioni, A. Ikonomidis, A. Poulou, D. Petropou-lou, and A. Tsakris. 2010. Characteristics of meropenem heteroresistance inKlebsiella pneumoniae carbapenemase (KPC)-producing clinical isolates ofK. pneumoniae. J. Clin. Microbiol. 48:2601–2604.
22. Rinder, H. 2001. Hetero-resistance: an underrecognised confounder in di-agnosis and therapy? J. Med. Microbiol. 50:1018–1020.
23. Sakoulas, G., and R. C. Moellering, Jr. 2008. Increasing antibiotic resistanceamong methicillin-resistant Staphylococcus aureus strains. Clin. Infect. Dis.46:S360–367.
24. Savini, V., M. Favaro, C. Fontana, C. Catavitello, A. Balbinot, M. Talia, F.Febbo, and D. D’Antonio. 2009. Bacillus cereus heteroresistance to carbap-enems in a cancer patient. J. Hosp. Infect. 71:288–290.
25. Tato, M., T. M. Coque, P. Ruíz-Garbajosa, V. Pintado, J. Cobo, H. S. Sader,R. N. Jones, F. Baquero, and R. Canton. 2007. Complex clonal and plasmidepidemiology in the first outbreak of Enterobacteriaceae infection involvingVIM-1 metallo-beta-lactamase in Spain: toward endemicity? Clin. Infect.Dis. 45:1171–1178.
26. Tato, M., T. M. Coque, F. Baquero, and R. Canton. 2010. Dispersal ofcarbapenemase blaVIM-1 gene associated with different Tn402 variants, mer-cury transposons, and conjugative plasmids in Enterobacteriaceae andPseudomonas aeruginosa. Antimicrob. Agents Chemother. 54:320–327.
27. Tenover, F. C., R. D. Arbeit, R. V. Goering, P. A. Mickelsen, B. E. Murray,D. H. Persing, and B. Swaminathan. 1995. Interpreting chromosomal DNArestriction patterns produced by pulsed-field gel electrophoresis: criteria forbacterial strain typing. J. Clin. Microbiol. 33:2233–2239.
28. Tenover, F. C., R. K. Kalsi, P. P. Williams, R. B. Carey, S. Stocker, D.Lonsway, J. K. Rasheed, J. W. Biddle, J. E. McGowan, Jr., and B. Hanna.2006. Carbapenem resistance in Klebsiella pneumoniae not detected by au-tomated susceptibility testing. Emerg. Infect. Dis. 12:1209–1213.
VOL. 48, 2010 CARBAPENEM HETERORESISTANCE IN K. PNEUMONIAE 4093






























