Embed Size (px)
Citation preview

AGENCIA VALENCIANA DE LA SALUD DG de Farmacia y Productos Sanitarios
1
IDENTIFICACIÓN DEL MEDICAMENTO Y AUTORES DEL INFORM E
− Medicamento: Rilpivirina. Antivirales para uso sistémico (antivirales de acción directa),
inhibidor de la transcriptasa inversa no análogo de nucleósidos (ITINN).
− Indicación clínica: tratamiento de la infección por el virus de la inmunodeficiencia
humana tipo 1 (VIH-1) en pacientes adultos con una carga viral (CV) ≤100.000
copias/mL que no han recibido tratamiento antirretroviral previamente (pacientes naïve).
− Autores / Revisores: Mª de los Ángeles Pena Pardo, Pedro Zapater Hernández, Ana
María Peiró Peiró y José Francisco Horga de la Parte. Unidad de Farmacología Clínica.
Hospital General Universitario de Alicante.
− Declaración de conflicto de intereses de los autores: los autores declaran la ausencia
de conflictos de interés.

AGENCIA VALENCIANA DE LA SALUD DG de Farmacia y Productos Sanitarios
2
DESCRIPCIÓN DEL MEDICAMENTO
En la siguiente tabla se resumen las características de la especialidad farmacéutica objeto
de evaluación en este informe.
Tabla 1: Características principales de la especial idad farmacéutica EDURANT.
Nombre genérico: Rilpivirina [1]
Nombre comercial: EDURANT
Laboratorio: Janssen-Cilag International NV
Grupo terapéutico: J05AG05 Antivirales para uso sistémico (antivirales de acción directa), ITINN (inhibidor de la transcriptasa inversa no análogo de nucleósidos)
Vía de administración: oral
Tipo de dispensación: Medicamento sujeto a prescripción médica restringida y de uso hospitalario
Vía de registro: procedimiento centralizado en la EMA (Agencia Europea de Medicamentos)
ACCIÓN FARMACOLÓGICA
Mecanismo de acción:
Es un fármaco diarilpirimidínico, no análogo de nucleósidos, inhibidor de la transcriptasa
inversa del VIH-1 (ITINN). Su actividad se basa en una inhibición no competitiva de la
transcriptasa inversa (TI) del VIH-1. Rilpivirina no inhibe las polimerasas α, β y γ del ADN
celular humano [1].
Indicación clínica autorizada:
En combinación con otros medicamentos antirretrovirales, en el tratamiento de la infección
por el virus de la inmunodeficiencia humana tipo 1 (VIH-1) en pacientes adultos con una
carga viral ≤100.000 copias/mL y que no han recibido tratamiento antirretroviral
previamente (naïve) [1].
Fecha de autorización: 13/01/2012.
Posología, forma de preparación y administración [1]:
Rilpivirina se debe administrar siempre en combinación con otros medicamentos
antirretrovirales. La dosis recomendada es de un comprimido de 25 mg una vez al día. Se
debe tomar con una comida. Se recomienda que el comprimido recubierto con película de
rilpivirina se trague entero con agua y no se mastique ni triture.

AGENCIA VALENCIANA DE LA SALUD DG de Farmacia y Productos Sanitarios
3
• Dosis omitidas
Si el paciente olvida tomar una dosis de rilpivirina dentro de las 12 horas desde la hora
habitual de la toma, debe tomarla con una comida lo antes posible y reanudar la pauta
posológica habitual. Si el paciente olvida tomar una dosis de rilpivirina y ya han trascurrido
más de 12 horas desde la hora habitual de la toma, no debe tomar la dosis omitida, sino
reanudar la pauta posológica habitual.
Si un paciente vomita dentro de las 4 horas desde la toma de rilpivirina, debe tomar otro
comprimido con una comida. Si un paciente vomita pasadas más de 4 horas después de
la toma de rilpivirina, no es necesario que el paciente tome otra dosis hasta la próxima
dosis establecida.
Farmacocinética:
A continuación se muestran resumidas las características farmacocinéticas del fármaco
evaluado [1,2]:
Tabla 2: Características farmacocinéticas de la esp ecialidad farmacéutica EDURANT.
Parámetro farmacocinético Rilpivirina [1,2]
Biodisponibilidad absoluta Se desconoce.
Las concentraciones plasmáticas no aumentan proporcionalmente con la dosis.
Profármaco No
Metabolito activo –
Tmax 4-5 horas
Efecto de la comida rica en grasa En los ensayos clínicos la biodisponibilidad de rilpivirina fue en general menor en los pacientes infectados por el VIH-1 que en los voluntarios sanos .
Su biodisponibilidad es aproximadamente un 40% menor en ayunas en comparación con una comida calórica normal (533 kcal) o con una comida hipercalórica y rica en grasas (928 kcal). Con una bebida nutricional rica en proteínas, la biodsiponibilidad es un 50% menor que con una comida.
Unión a proteínas plasmáticas (%) 99,7 (fundamentalmente se une a albúmina)
Vida media de eliminación (horas) 34-55 horas
Metabolismo oxidativo (CYP3A)
Tmax = tiempo hasta alcanzar la concentración máxima.
Las propiedades farmacocinéticas de rilpivirina se han evaluado en adultos sanos y en

AGENCIA VALENCIANA DE LA SALUD DG de Farmacia y Productos Sanitarios
4
pacientes adultos naïve infectados por el VIH-1. La farmacocinética de rilpivirina en los
pacientes pediátricos se encuentra en fase de investigación.
Tras la administración oral, la concentración plasmática máxima de rilpivirina se alcanza
por lo general en el plazo de 4-5 horas. Se desconoce su biodisponibilidad absoluta. Las
concentraciones plasmáticas no aumentan proporcionalmente con la dosis.
La cantidad de rilpivirina que alcanzó la circulación sistémica (biodisponibilidad) fue
aproximadamente un 40% menor cuando esta se administró en ayunas en comparación
con una comida calórica normal (533 kcal) o con una comida hipercalórica y rica en
grasas (928 kcal). Cuando se tomó con solo una bebida nutricional rica en proteínas, la
biodsiponibilidad fue un 50% menor que cuando se tomó con una comida. Por ello, para
lograr una absorción óptima rilpivirina se debe tomar con una comida y evitar tomarla tras
una bebida nutricional rica en proteínas.
Rilpivirina se une aproximadamente en un 99,7% a las proteínas plasmáticas in vitro,
principalmente a la albúmina. No se ha evaluado en seres humanos la distribución de
rilpivirina en otros compartimentos que no sean el plasma (por ejemplo, líquido
cefalorraquídeo y secreciones genitales).
Los estudios in vitro indican que rilpivirina experimenta fundamentalmente un metabolismo
oxidativo mediado por el sistema del citocromo P450 (CYP3A).
La eliminación de rilpivirina del plasma es lenta y su semivida de eliminación terminal es
de 34-55 horas. Tras la administración oral de dosis únicas de 14C-rilpivirina, se recuperó
en las heces y la orina un promedio del 85% y el 6,1% de la radiactividad,
respectivamente. En las heces, se detectó un promedio del 25% de la dosis administrada
de rilpivirina. En la orina se detectaron únicamente cantidades mínimas de rilpivirina
intacta (<1% de la dosis).
Actualmente se están desarrollando diferentes formulaciones de rilpivirina (tres
formulaciones pediátricas -granular, solución y suspensión- , una formulación inyectable
de acción prolongada).
Características comparadas de otros medicamentos co n la misma indicación:
En la siguiente tabla se compara rilpivirina con efavirenz ya que este último ha sido el
fármaco control (o referencia) en los ensayos clínicos.

AGENCIA VALENCIANA DE LA SALUD DG de Farmacia y Productos Sanitarios
5
Tabla 3: Características comparadas de rilpivirina frente a efavirenz.
Nombre del principio activo Rilpivirina [1] Efavirenz [3]
Presentación Comprimidos recubiertos con película de 25 mg
Comprimidos recubiertos con película de 600 mg
Cápsulas duras de 50 mg, de 100 mg y de 200 mg
Posología (en adultos) 25 mg/día (1 comprimido/día) 600 mg/día (1 comprimido/día)
Otras características comparadas
Grupo ATC: J05AG05
Es un inhibidor de la transcriptasa inversa del VIH-1 (ITINN), no análogo de nucleósidos.
Está indicado en combinación con otros medicamentos antirretrovirales, en el tratamiento de la infección por el virus de la inmunodeficiencia humana tipo 1 en pacientes adultos con una carga viral ≤100.000 copias/mL y que no han recibido tratamiento antirretroviral previamente.
Grupo ATC: J05AG03
Es un inhibidor no competitivo de la transcriptasa inversa del VIH-1, no análogo de nucleósidos.
Está indicado en el tratamiento antirretroviral combinado del virus de la inmunodeficiencia humana-1 en adultos infectados, adolescentes y niños de 3 años de edad y mayores.
Algunas consideraciones sobre la terapia antirretro viral en nuestro país para
adultos infectados por el VIH-1:
Desde que en 1995 la Secretaría del Plan Nacional sobre el SIDA (SPNS) y su Consejo
Asesor Clínico editaran las primeras “Recomendaciones de tratamiento antirretroviral en el
adulto”, este organismo junto al Grupo de Estudio de SIDA (GESIDA) de la Sociedad
Española de Enfermedades Infecciosas y Microbiología Clínica (SEIMC) las han ido
actualizando anualmente y las han publicado en la revista Enfermedades Infecciosas y
Microbiología Clínica y en sus respectivas páginas web.
En la actualidad las recomendaciones del documento de consenso del Grupo GESIDA de
la SEIMC/Plan Nacional sobre el SIDA (actualización de enero 2013) del tratamiento de la
infección por el VIH-1 en pacientes adultos son las siguientes [4]:
- Las estrategias terapéuticas deben ser individualizadas para conseguir en
cualquier circunstancia una carga viral plasmática (CVP) indetectable, para lo cual
el cumplimiento terapéutico es fundamental.
- El tratamiento antirretroviral de elección para la infección por el VIH-1 actualmente
lo constituye una combinación de al menos tres fármacos que incluyan dos
inhibidores de la transcriptasa inversa análogos de nucleósidos o nucleótidos
(ITIAN) asociado a tercer fármaco antirretroviral (a un inhibidor de la proteasa

AGENCIA VALENCIANA DE LA SALUD DG de Farmacia y Productos Sanitarios
6
potenciado con dosis reducidas de ritonavir o a un ITINN o a un inhibidor de la
integrasa).
- El inicio del TAR debe ser individualizado y debe basarse en las manifestaciones
clínicas, el número de CD4, la carga viral plasmática y la presencia de
comorbilidades o características del paciente.
- Se aconseja el uso de fármacos coformulados para facilitar el cumplimiento
terapéutico.
- Se deben estudiar las mutaciones de resistencia (mediante prueba de resistencias
genotípicas) ya que su conocimiento permite un mejor uso de los fármacos.
- El tratamiento antirretroviral debe ser indefinido.
EVALUACIÓN DE LA EFICACIA
Ensayos clínicos disponibles para la indicación clí nica evaluada (descripción de la
búsqueda bibliográfica: criterios y resultados de l a misma):
Se realizó una búsqueda bibliográfica sistemática de ensayos clínicos aleatorizados de
rilpivirina en MEDLINE (PubMed).
Las palabras clave empleadas fueron “rilpivirine”, “randomized controlled trial” y “Ovid full-
text available” y “rilpivirine” y “review articles”. Se encontraron 2.441 artículos y 173
artículos (muchos de ellos estaban repetidos), respectivamente. De todos ellos
seleccionamos los de acceso libre los cuales son los expuestos en este informe.
Igualmente se ha recuperado el dossier de la EMA en el que se identificaban los 2
ensayos clínicos en los que se basó su autorización por la EMA. Se ha analizado también
la información proporcionada por la industria y la ficha técnica aprobada por la EMA.
Además se localizaron los ensayos clínicos con rilpivirina en las bases de registro de
ensayos del Instituto Nacional de Salud de Estados Unidos y de la Unión Europea de
registro de ensayos clínicos (http://www.clinicaltrials.gov./ y
https://www.clinicaltrialsregister.eu/).
En la siguiente tabla se muestran los dos ensayos clínicos fase III llevados a cabo a
efectos de evaluar la eficacia y seguridad y apoyar la autorización de comercialización por
parte de las agencias reguladoras de este nuevo inhibidor de la transcriptasa inversa no
análogo de nucleósidos (ITINN) en la indicación referida.

AGENCIA VALENCIANA DE LA SALUD DG de Farmacia y Productos Sanitarios
7
Tabla 4: Ensayos clínicos fase III realizados con r ilpivirina.
FARMACO EN ESTUDIO
ACRÓNIMO DEL ENSAYO
CÓDIGO DE REGISTRO
ClinicalTrials.gov (FDA)
COMPARADOR ACTIVO
MEDICAMENTOS ANTIRRETROVIRALES DE BASE
ECHO NCT00540449 efavirenz 600 mg (una vez al día)
tenofovir (disoproxil fumarato) + emtricitabina
RILPIVIRINA (EDURANT) 25 mg (una vez al día) THRIVE NCT00543725 efavirenz
600 mg (una vez al día)
- tenofovir (disoproxil fumarato) + emtricitabina
- o zidovudina + lamivudina - o abacavir + lamivudina
ECHO (MC278-C209 against HIV in a once-daily regimen versus efavirenz). THRIVE (Efficacy Comparison in Treatment-naive, HIV-infected Subjects of TMC278 and Efavirenz
A continuación se resumen las características principales de los ensayos clínicos de
rilpivirina en el tratamiento antirretroviral combinado de la infección por el VIH-1 en
pacientes adultos naïve con una carga viral ≤100.000 copias/mL.
Tabla 5: Características principales de los ensayos clínicos fase III realizados con rilpivirina.
RILPIVIRINA (HIDROCLURURO)
Ensayo clínico (nº de pacientes aleatorizados)
ECHO (346) [5]
THRIVE (340) [6]
Fármaco en estudio Dosis
rilpivirina 25 mg oral (una vez al día)
Fármaco comparador Dosis
efavirenz 600 mg oral (una vez al día)
Fármacos antirretrovirales de base
tenofovir (disoproxil fumarato) + emtricitabina
tenofovir (disoproxil fumarato) + emtricitabina, o zidovudina + lamivudina, o abacavir + lamivudina
Diseño EC, R, DC, DS, de grupos paralelos, no inferioridad
Características de los pacientes
Mujeres rilpivirina: 78 (23%) efavirenz: 69 (20%)
rilpivirina: 90 (26%) efavirenz: 94 (28%)
Edad (media, rango) rilpivirina: 36 (18-78) años efavirenz: 36 (19-67) años
rilpivirina: 36 (19-62, 29-42) años efavirenz: 36 (19-69, 29-43) años
Carga viral (mediana, rango)
rilpivirina: 5 (2-7) log10 copias/mL efavirenz: 5 (3-7) log10 copias/mL
rilpivirina: 5 (3-7; 4,5-5,3) log10 copias/mL efavirenz: 5 (3-7; 4,5-5,4) log10 copias/mL
Recuento CD4 (mediana, rango)
rilpivirina: 240 (1-888) células/µL efavirenz: 257 (1-757) células/µL
rilpivirina: 263 (2-744, 177-342) células/µL efavirenz: 263 (1-1.137, 171-353) células/µL

AGENCIA VALENCIANA DE LA SALUD DG de Farmacia y Productos Sanitarios
8
Criterios de inclusión
Pacientes ≥18 años, con infección VIH-1 documentada, nunca tratados con vacuna frente al VIH o fármacos antirretrovirales antes del screening, con una carga viral >5.000 copias/mL (medida por PCR), sensibles al tratamiento con tenofovir y emtricitabina y que estaban de acuerdo en no iniciar el tratamiento antirretroviral antes de la visita basal del ensayo.
Pacientes ≥18 años, con infección VIH-1 documentada, nunca tratados con vacuna frente al VIH o fármacos antirretrovirales antes del screening, con una carga viral >5.000 copias/mL (medida por PCR), sensibles al tratamiento con dos ITIAN y que estaban de acuerdo en no iniciar el tratamiento antirretroviral antes de la visita basal del ensayo y con HLA-B*5701 negativo (si se iba a incluir abacavir en el tratamiento).
Criterios de exclusión
Uso previo de cualquier fármaco antirretroviral, cualquier evidencia documentada de mutaciones asociadas a resistencia a ITINN, categoría C definitoria de SIDA (excepto, sarcoma de Kaposi estable y síndrome de fatiga no progresivo, neumonia por Pneumocistis carinni no curada, tuberculosis activa, alergia o hipersensibilidad a cualquiera de los fármacos del ensayo, toxicidad específica grado 3 o 4, deterioro renal (aclaramiento de creatinina calculado <50 mL/min).
Análisis estadístico primario
Por ITT
Variable principal (de eficacia):
Respuesta virológica: porcentaje de pacientes que recibieron al menos una dosis de la medicación del ensayo con respuesta confirmada (carga viral <50 copias/mL, definida por un algoritmo de análisis, por intención de tratar, de tiempo hasta la pérdida de respuesta virológica (TLOVR) a las 48 semanas.
Variables secundarias:
duración de la actividad antiviral, cambios del recuento de células CD4 respecto a la situación basal, la seguridad, la tolerabilidad, características genotípicas y fenotípicas VIH (en los fracasos virológicos), cumplimiento terapéutico (medido con el cuestionario M-MASRI), farmacocinética y relaciones farmacocinética-farmacodinámicas.
EC = ensayo clínico. R = randomizado (aleatorizado). DC = doble ciego. DS = doble simulación, con placebo. ITT = por Intención de Tratar (Intention-To-Treat). M-MASRI (Modified Medication Adherence Self-Report Inventory o Inventario modificado de autoinforme de adherencia a la medicación). Carga viral = ARN del VIH-1 en plasma. TLOVR (en inglés) = Tiempo hasta la pérdida de respuesta virológica. Esta variable (TLOVR) básicamente se trata de un análisis por intención de tratar (se incluyen todos los pacientes aleatorizados) en el que se considera fracaso cualquier discontinuación del tratamiento (fallecimiento, pérdida de seguimiento o cambio de tratamiento) y la pérdida de control virológico confirmada en dos determinaciones consecutivas (ignorando las visitas perdidas e incluyendo todas las determinaciones realizadas, incluso fuera de visita).
La eficacia y la seguridad de rilpivirina en esta indicación se ha evaluado en estos dos
ensayos clínicos fase III y en un metaanálisis de los mismos:
- ECHO (MC278-C209 against HIV in a once-daily regimen versus efavirenz) [5], -THRIVE (Efficacy Comparison in Treatment-naive, HIV-infected Subjects of TMC278 and Efavirenz ) [6] - y “pooled analysis of ECHO and THRIVE trials” [7]
En ambos ensayos se prohibieron los medicamentos que pudiesen reducir la
biodisponibilidad de rilpivirina (como inductores potentes del CYP3A4 e inhibidores de la
bomba de protones), los medicamentos no permitidos con los antirretrovirales de base,

AGENCIA VALENCIANA DE LA SALUD DG de Farmacia y Productos Sanitarios
9
cualquier otro tratamiento anti-VIH y los medicamentos en investigación. Y se permitió el
uso concomitante de antiácidos (pero administrados al menos 2 horas antes o al menos 4
horas después de rilpivirina) y de antagonistas del receptor antihistamínico H2 (pero
administrados al menos 12 horas antes o al menos 4 horas después de rilpivirina).
También se permitió el cambio de ITIAN si se producía intolerancia.
En ambos estudios el análisis estadístico primario realizado fue por Intención de Tratar
(ITT o Intention-to-treat) y fue un análisis de no inferioridad. También se efectuó un
análisis por protocolo. La elección del margen de no inferioridad se efectuó de acuerdo a
las recomendaciones de la Food and Drug Administration (FDA) para el desarrollo de
medicamentos antirretrovirales [8]. Se consideraría que las condiciones de tratamiento
comparadas en el ensayo resultarían ser no inferiores si la diferencia con rilpivirina ± el
intervalo de confianza (IC) al 95% no resultaba ser inferior al 12% (para la variable
principal) y al 10% (para la variable secundaria) del límite inferior de ese intervalo de
confianza con respecto al tratamiento comparador (efavirenz).
En los dos ensayos se estratificó la asignación del antirretroviral en estudio en función de
la carga viral en el momento del screening (≤100.000 copias/mL, >100.000 copias/mL o
>500.000 copias/mL). Además en el ensayo THRIVE también se hizo una estratificación
en función del tratamiento antirretroviral ITIAN de base administrado (tenofovir-disoproxil-
fumarato y emtricitabina o zidovudina y lamivudina o abacavir y lamivudina).
Resultados de los ensayos clínicos:
Aunque aquí se narran los resultados de los ensayos, al final de este informe se resumen
en forma de tablas “Resultados de los ensayos clínicos disponibles”.
En los dos ensayos (ECHO y THRIVE) se demostró, en pacientes naïve infectados por
VIH-1, la no-inferioridad para los márgenes 12% y 10% del intervalo de confianza al 95%
en términos de eficacia (porcentaje de pacientes que habiendo recibido al menos una
dosis de rilpivirina o de efavirenz presentaron una respuesta virológica confirmada
definida por la variable “tiempo hasta la pérdida de respuesta virológica” -TLOVR- a las
48 semanas del tratamiento). En definitiva, se realizó un análisis estadístico en el que se
consideraron todos los pacientes aleatorizados a uno de los dos grupos de tratamiento de
los ensayos, fracaso por cualquier discontinuación del tratamiento (independientemente
del motivo de discontinuación) y pérdida de control virológico confirmada en dos

AGENCIA VALENCIANA DE LA SALUD DG de Farmacia y Productos Sanitarios
10
determinaciones consecutivas (independientemente de si se realizaban todas las visitas y
de si las determinaciones se realizaban fuera de visita).
Tabla 6: Resultados de eficacia a las 48 semanas en los pacientes tratados con rilpivirina y los trata dos con efavirenz.
Rilpivirina Efavirenz Dif. (IC 95%)
nº de pacientes
aleatorizados
nº de pacientes
con la variable
evaluada (%)
nº de pacientes
aleatorizados
nº de pacientes
con la variable
evaluada (%)
Variable principal
Porcentaje de pacientes con respuesta confirmada (con un margen de no inferioridad del 12%)
ECHO 346 287 (83) 344 285 (83) 0,1 (-5,5 a 5,7)
THRIVE 340 291 (86) 338 276 (82) 3,9 (-1,6 a 9,5)
ECHO + THRIVE 686 578 (84) 682 561 (82) 2,0 (-2,0 a 6,0)
Modelo de predicción de respuesta
ECHO* (83) (84) -0,4 (-5,9 a 5,2)
THRIVE** (87) (83) 3,5 (-1,7 a 8,8)
ECHO + THRIVE*** (86) (84) 1,6 (-2,2 a 5,3) Para la elaboración de este modelo de predicción de respuesta se realizó una regresión logística (log10) ajustada por: *carga viral basal, **carga viral basal y régimen antirretroviral de base, ***carga viral basal, régimen antirretroviral de base y ensayo. Dif. = diferencia observada. IC 95% = intervalo de confianza al 95%.
Tabla 7: Resultados de eficacia a las 48 semanas en el subgrupo de pacientes con CV basal ≤100.000 copias/mL tratados con
rilpivirina y los tratados con efavirenz.
Rilpivirina Efavirenz Dif. (IC 95%)
nº de pacientes
aleatorizados
nº de pacientes
con la variable evaluada
(%)
nº de pacientes
aleatorizados
nº de pacientes
con la variable evaluada
(%)
Porcentaje de pacientes con CV basal ≤100.000 copias/mL y con respuesta confirmada (con un margen de no inferioridad del 12%)
ECHO 181 162 (90) 163 136 (83) --------
THRIVE 187 170 (91) 167 140 (84) --------
ECHO + THRIVE 368 332 (90) 330 276 (84) 6,6 (1,6 a 11,5)

AGENCIA VALENCIANA DE LA SALUD DG de Farmacia y Productos Sanitarios
11
Dif. = diferencia observada. IC95% = intervalo de confianza al 95%.
El porcentaje de fracaso virológico debido a resistencias fue ligeramente mayor (pero
estadísticamente no significativo) en el grupo de tratamiento con rilpivirina que en el grupo
efavirenz. Esta diferencia de porcentaje de fracaso virológico a favor de efavirenz fue más
marcada en el ensayo ECHO. Los autores de los ensayos explican este resultado por una
menor respuesta en los pacientes con rilpivirina con una carga viral basal mayor y por el
impacto negativo que podría haber tenido el diseño de doble simulación (con toma de la
medicación dos veces al día) sobre el cumplimiento del tratamiento y también porque los
pacientes no hubiesen seguido la recomendación de tomar la medicación con comida
(con la consiguiente absorción subóptima de rilpivirina).
En el ensayo THRIVE el tipo de régimen de tratamiento ITIAN de base no tuvo efecto
significativo en la respuesta.
Tabla 8: Resultados de fracaso virológico y resiste ncias a las 48 semanas.
Rilpivirina Efavirenz
nº de pacientes aleatorizados
nº de pacientes con la variable evaluada
(%)
nº de pacientes aleatorizados
nº de pacientes con la variable evaluada (%)
Fracaso virológico (FV)
ECHO 346 45 (13) 344 19 (6)
THRIVE 340 24 (7) 338 18 (5)
ECHO + THRIVE 686 72 (10) 682 39 (6)
FV con resistencias a ITNN y a ITIAN(t)
ECHO + THRIVE 686 62 682 28
FV con resistencias a ITNN ECHO + THRIVE
686
39/62 (63)
682
15/28 (54)
FV con resistencias a ITIAN(t) ECHO + THRIVE
42/62 (68)
9/28 (32)
Fracaso virológico (FV) = Incluye pacientes que presentaron un repunte (carga viral confirmada ≥50 copias/mL después de ser respondedores), o pacientes en los que nunca llegó a alcanzarse la supresión viral (carga viral <50 copias/mL no confirmada, en pacientes con tratamiento en curso o que lo interrumpieron debido a falta o pérdida de eficacia). Resistencia = cualquier resistencia asociada a mutaciones emergentes ocurridas en el momento del fracaso. No se ha

AGENCIA VALENCIANA DE LA SALUD DG de Farmacia y Productos Sanitarios
12
realizado en esta tabla una diferenciación en función de carga viral. Algunos de los FV no pudieron ser genotipados y fenotipados debido a una baja carga viral. ITIAN(t) = inhibidores de la transcriptasa inversa análogos de nucleósidos.
El análisis conjunto de todos los datos de resistencia a las 48 semanas de tratamiento se
ha presentado separadamente en una publicación [9]. En la misma se objetivó que
desarrollaron resistencia asociada a los ITIAN zidovudina y lamivudina/emtricitabina más
pacientes en tratamiento con rilpivirina que los pacientes en tratamiento con efavirenz.
Con una carga viral inicial baja (≤100.000 copias/mL) la resistencia cruzada al ITINN
etravirina fue menor. En pacientes con una CV inicial >100.000 copias/mL, la proporción
de fracasos virológicos fue mayor con rilpivirina que con efavirenz (17% vs 7%). El
número absoluto de fracasos virológicos con mutación asociada a resistencia emergente
a ITINN fue mayor con rilpivirina que con efavirenz, aunque las proporciones relativas
fueron similares (63% vs 54%), y la mutación más frecuente con rilpivirina fue E138K
(cuya prevalencia en la práctica clínica rutinaria es baja <1%). En comparación con
efavirenz más fracasos virológicos con rilpivirina tuvieron mutación asociada a resistencia
emergente a ITIAN (rilpivirina 68% vs 32% efavirenz), fundamentalmente las mutaciones
M184I y M184V. Resumiendo, el fracaso virológico (de acuerdo a la definición del mismo
considerada en los dos ensayos ECHO y THRIVE) y el fracaso virológico con mutación
asociada a resistencia emergente fueron similares en cargas virales basales bajas
(≤100.000 copias/mL), pero más frecuentes con rilpivirina que con efavirenz al partir de
cargas virales basales altas (>100.000 copias/mL).
También se efectuó un análisis (previamente planeado en el plan de análisis estadístico)
del conjunto de los datos de los dos ensayos para evaluar subgrupos de pacientes (en
función de la carga viral inicial, del régimen de tratamiento antirretroviral de base, del
género, de la raza, del recuento de linfocitos CD4+ y del genotipo) y predictores de
respuesta y de fracaso virológico [7].
En este análisis se agrupó a los pacientes en función de la evaluación del cumplimiento
del tratamiento medido como M-MASRI (≤95% vs >95%) y se concluye que :
− Las respuestas en los análisis con la variable de eficacia principal a las 48
semanas en los ensayos por separado y el conjunto de los datos de los dos
ensayos son consistentes. En el análisis conjunto la respuesta a las 48 semanas
fue similar (84% con rilpivirina y 82% con efavirenz, IC95%: -2,0-6,0%).

AGENCIA VALENCIANA DE LA SALUD DG de Farmacia y Productos Sanitarios
13
− Dichas respuestas son semejantes al analizarlas en función del régimen de ITIAN
de base, del género, de la raza y de genotipos pareados (especies emparentadas).
En los dos grupos de tratamiento el porcentaje de respuesta fue menor en los
pacientes blancos y africano-americanos y mayor en los pacientes asiáticos con
genotipo CRF01_AE.
− El cumplimiento terapéutico subóptimo (M-MASRI≤95%), la carga viral inicial mayor
(>500.000 copias/mL) y el recuento <50 CD4+/mm3 se asocian a menos
respuestas, a más fracaso virológico y al desarrollo de resistencias (incluso
resistencias cruzadas) en ambos grupos de tratamiento (rilpivirina y efavirenz).
− Los efectos de fracasos virológicos son más aparentes con rilpivirina.
− El pobre cumplimiento del tratamiento HIV-1 es un predictor importante del fracaso
virológico, pero por sí solo no explica la diferencia encontrada en el fracaso
virológico para la variable de eficacia entre los dos grupos de tratamiento.
Evaluación de la validez y de la utilidad práctica de los resultados:
Al final de este informe se resumen en forma de tablas (“Análisis de validez interna de
los ensayos” y “Cuestionario para la valoración global de la valid ez externa o
aplicabilidad de los ensayos clínicos” ) los análisis de las evaluaciones de la validez
interna y externa (o de aplicabilidad) de los ensayos .
Revisiones sistemáticas publicadas y sus conclusion es:
Actualmente las publicaciones existentes al respecto de rilpivirina se basan en los datos
de eficacia y de seguridad de los dos ensayos clínicos fase III mencionados en este
informe.
Evaluaciones de fuentes secundarias (como guías de práctica clínica, evaluaciones
de organismos independientes y opiniones de experto s):
Son las recogidas en otros apartados de este informe (apartado “Características
comparadas de otros medicamentos con la misma indicación” y apartado “DESCRIPCIÓN
ECONÓMICA”).

AGENCIA VALENCIANA DE LA SALUD DG de Farmacia y Productos Sanitarios
14
EVALUACIÓN DE LA SEGURIDAD
La evaluación de la seguridad se basa en los datos conjuntos de 1.368 pacientes que
participaron en los ensayos fase III controlados ECHO y THRIVE, realizados en pacientes
adultos naïve infectados por el VIH-1.
Descripción de los efectos adversos más significati vos:
En las siguientes tablas se resumen los acontecimientos adversos (AA) notificados en los
ensayos.
Tabla 9: Descripción conjunta de los acontecimiento s adversos notificados en los ensayos ECHO y THRIVE .
AA en pacientes adultos naïve infectados por el VIH-1 y tratados con rilpivirina + medicación antirretroviral de base (ECHO + THRIVE, n = 686)
Órgano y sistema afectado Frecuencia Descripción del AA
Trastornos de la sangre y del sistema linfático
frecuentes (≥1/100 a <1/10)
disminución del recuento de leucocitos descenso de hemoblobina disminución del recuento de plaquetas
Trastorno del sistema inmunológico
poco frecuente (≥1/1.000 a <1/100)
síndrome de reconstitución inmune
muy frecuentes (≥1/10)
elevación del colesterol total (en ayunas) elevación del colesterol LDL (en ayunas)
Trastornos del metabolismo y de la nutrición
frecuentes (≥1/100 a <1/10)
disminución del apetito hipertrigliceridemia (en ayunas)
Trastornos psiquiátricos Frecuentes (≥1/100 a <1/10)
sueños anormales insomnio depresión trastornos del sueño estado anímico deprimido
muy frecuentes (≥1/10)
cefalea Trastornos del sistema nervioso
frecuentes (≥1/100 a <1/10)
mareos somnolencia
muy frecuentes (≥1/10)
náuseas aumento de amilasa pancreática
Trastornos gastrointestinales
frecuentes (≥1/100 a <1/10)
dolor abdominal vómitos aumento de la lipasa molestias abdominales sequedad de boca
muy frecuente (≥1/10)
elevación de las transaminasas Trastornos hepatobiliares
frecuente (≥1/100 a <1/10)
elevación de la bilirrubina
Trastornos de la piel y del tejido frecuente erupción cutánea

AGENCIA VALENCIANA DE LA SALUD DG de Farmacia y Productos Sanitarios
15
subcutáneo
(≥1/100 a <1/10)
Trastornos generales y alteraciones en el lugar de administración
frecuente (≥1/100 a <1/10)
cansancio
En los dos ensayos hubo un comité independiente e internacional para la monitorización
de los datos y la seguridad. Los acontecimientos adversos se codificaron mediante el
MedDRA (Diccionario Médico para Actividades Reguladoras, versión 11.0) y se estableció
la gravedad de los mismos a través de la versión 1 de la escala de graduación para
adultos y niños de la División del SIDA del Instituto Nacional de Alergia y Enfermedades
Infecciosas del Instituto de Salud de USA (Division of Acquired Immunodeficiency
Syndrome grading scale). En el análisis de seguridad se tuvo en cuenta la evaluación de
acontecimientos adversos neurológicos y psiquiátricos de interés de los ITINN y la posible
afectación de la función renal y del intervalo QT del electrocardiograma (ECG). En este
análisis se incluyeron los datos de los pacientes tratados más allá de las 48 semanas.
Según esta escala los AA se clasifican en grado 1 (leve, síntomas que no causan o
causan una mínima interferencia en las actividades funcionales y sociales usuales), grado
2 (moderado, síntomas que causan más que una mínima interferencia en las actividades
funcionales y sociales usuales), grado 3 (grave, síntomas que causan incapacidad para
llevar a cabo las actividades funcionales y sociales usuales) y grado 4 (potencialmente
mortal o que pone en peligro la vida si no se trata, síntomas que provocan incapacidad
para realizar funciones básicas de auto-cuidado o intervención médica u operativa
indicadas para prevenir daño permanente, incapacidad persistente o muerte).
Tabla 10: Análisis individual y conjunto de los aco ntecimientos adversos notificados en los ensayos EC HO y THRIVE.
Acontecimientos Adversos (AA)
ECHO Rilpivirina
(n= 346)
Efavirenz
(n= 344)
AA graves 23 (7%) 31 (9%)
AA relacionado de grado ≥ 2 55 (16%) 108 (31%)
AA que conllevaron abandono permanente del tratamiento 8 (2%) 27 (8%)
AA analíticos grado 3-4 34 (10%) 55 (16%)

AGENCIA VALENCIANA DE LA SALUD DG de Farmacia y Productos Sanitarios
16
erupción cutánea relacionada de cualquier grado 12 (4%) 50 (15%)
THRIVE Rilpivirina
(n= 340)
Efavirenz
(n= 338)
AA graves 22 (7%) 24 (7%)
AA relacionado de grado ≥ 2 54 (16%) 104 (31%)
AA que conllevaron abandono permanente del tratamiento 15 (4%) 25 (7%)
AA analíticos grado 3-4 41 (12%) 63 (19%)
erupción cutánea relacionada de cualquier grado 9 (3%) 43 (13%)
ECHO + THRIVE Rilpivirina
(n= 686)
Efavirenz
(n= 682)
AA graves 45 (7%) 55 (8%)
AA relacionado de grado ≥ 2 109 (16%) 212 (31%)
AA que conllevaron abandono permanente del tratamiento 23 (3%) 52 (8%)
AA analíticos grado 3-4 75 (11%) 118 (18%)
erupción cutánea relacionada de cualquier grado 21 (3%) 93 (14%)
Al final de este informe se resume, en forma de tablas (“Resultados de seguridad” ), el
análisis de los acontecimientos adversos más significativos en los ensayos clínicos (de
forma individual y en conjunto). Y a continuación se describen en detalle.
Abandono de la medicación por acontecimientos adversos
En el análisis de seguridad del conjunto de los datos de los dos ensayos clínicos fase III
se concluye que la incidencia de acontecimientos adversos que conllevan el abandono
permanente del tratamiento es menor en el grupo de rilpivirina (3% vs 8%), siendo estos
los más frecuentes erupción cutánea y depresión [6].
Acontecimientos adversos posiblemente relacionados con la medicación grado 2 o más
En el análisis conjunto de los dos ensayos fase III, la incidencia de acontecimientos
adversos posiblemente relacionados con la medicación al menos grado 2 o más fue
menor con rilpivirina (16% vs 31%) y los más frecuentes en el ensayo ECHO (2% o más

AGENCIA VALENCIANA DE LA SALUD DG de Farmacia y Productos Sanitarios
17
de los pacientes) fueron mareos, trastornos del sueño (sueños anómalos y pesadillas,
insomnio), náuseas y cualquier erupción cutánea [6].
Acontecimientos adversos graves
En los dos ensayos, la incidencia de acontecimientos adversos graves fue similar en
ambos grupos de tratamiento (en el ensayo ECHO: 7% con rilpivirina vs 9% con efavirenz,
en el ensayo THRIVES: 7% vs 7% y en el análisis conjunto de los dos ensayos: 7% vs
8%) y las muertes que hubo (en el ensayo ECHO: una en el grupo de efavirenz debido a
un linfoma de Burkitt y en el ensayo THRIVE, una en el grupo de ripilvirina debida a una
bronconeumonia y 3 en el grupo de efavirenz: una toxoplasmosis cerebral y disentería, un
accidente cerebrovascular y un fallo respiratorio) no se relacionaron con la medicación.
Erupción cutánea
En el análisis conjunto de los dos ensayos la incidencia de erupción cutánea de cualquier
grado relacionada con el tratamiento fue inferior (y estadísticamente significativa) en el
brazo de rilpivirina que en el de efavirenz (3% vs 14%). La mayoría fueron de grado 1 o 2
y desaparecieron al continuar con la medicación.
Anomalías de laboratorio
Las alteraciones analíticas (básicamente alteraciones en la bioquímica y el
hemograma; parámetros de valoración de la función hepática y renal, iones en sangre,
perfil lipídico) grado 3 o 4 fueron más frecuentes con efavirenz (en el ensayo ECHO: 16%
frente al 10%y en el ensayo THRIVE, 19% vs 12%). La única excepción fue la
hipofosfatemia (más frecuente con rilpivirina, 2% frente a 1%) en el ensayo ECHO.
En cuanto a las alteraciones del perfil lipídico, en los dos ensayos rilpivirina se asoció a
unos incrementos (a las 48 semanas de tratamiento respecto a la situación basal)
menores de colesterol total (en el ensayo ECHO: 1/346 vs 6/339, en el ensayo THRIVE:
0/340 vs 11/329 y en el análisis conjunto de estos dos ensayos: 34/686 y 122/682) y del
colesterol HDL).
Anomalías de la función renal y del intervalo QT

AGENCIA VALENCIANA DE LA SALUD DG de Farmacia y Productos Sanitarios
18
En general, en los dos ensayos no hubo diferencias estadísticamente significativas entre
los dos grupos de tratamiento (rilpivirina y efavirenz) en cuanto a cambios a las 48
semanas del tratamiento respecto a la situación basal de la función renal (creatinina
sérica y tasa de filtración glomerular estimada) y del intervalo QT de los pacientes
(evaluado de acuerdo a la fórmula corregida de Fridericia, donde QTcF = QT/ RR1/3).
Sí que cabe destacar que en el ensayo THRIVE, se retiró del mismo a un paciente del
grupo de rilpivirina, a pesar de estar asintomático, debido a una prolongación del intervalo
QT grado 3 (aumento del QTcF >0,06 segundos en la semana 48 respecto a la situación
basal). Este paciente no estaba tomando medicación concomitante que pudiese causar un
alargamiento de este intervalo.
Acontecimientos adversos de especial interés
En ambos ensayos los acontecimientos adversos al menos posiblemente relacionados (y
de cualquier grado) con el tratamiento de especial interés en el caso de los ITINN tanto
neurológicos (incluyendo entre otros: cefaleas, mareos, vértigo, visión borrosa, trastornos
de la atención, fotofobia, neuropatía, sensación de presión en el oído y somnolencia)
como psiquiátricos (como sueños anómalos, trastornos afectivos, agresión, agitación,
ansiedad, estado confusional, depresión, euforia, nerviosismo, ideas homicidas y
disminución de la libido) fueron menos frecuentes con rilpivirina (en el ensayo ECHO,
eventos neurológicos: 16% vs 37%; eventos psiquiátricos: 15% vs 25%; y en el ensayo
THRIVE, eventos neurológicos: 18% vs 39%; eventos psiquiátricos: 15% vs 20%). La
mayoría de ellos fueron de grado 1 o 2.
Ensayos clínicos comparativos de seguridad:
Hoy por hoy los datos de seguridad de rilpivirina se basan en los datos de los dos
ensayos clínicos fase III mencionados en este informe y en comparación con efavirenz.
Fuentes secundarias sobre seguridad (como evaluacio nes de organismos
independientes, alertas de la AEMPS, EMA y FDA, cen tros de Farmacovigilancia y
opiniones de expertos):
Las únicas fuentes por ahora son las recogidas en otros apartados de este informe y
provenientes de los análisis (individual y conjunto) de seguridad de los dos ensayos

AGENCIA VALENCIANA DE LA SALUD DG de Farmacia y Productos Sanitarios
19
clínicos fase III y como subapartado de las evaluaciones de organismos independientes.
Dada la escasa experiencia de uso en la práctica clínica no hay datos al respecto de la
seguridad postcomercialización de rilpivirina (alertas y Centros de Farmacovigilancia).
Contraindicaciones, prevención de errores y precauc iones en casos especiales [1]:
• La hipersensibilidad al principio activo o a alguno de los excipientes es una
contraindicación para el tratamiento con rilpivirina.
• Se debe informar a los pacientes de que el tratamiento antirretroviral actual no cura
la infección por VIH y mientras tomen rilpivirina, aún existe el riesgo de transmitir la
infección del VIH a otras personas por contacto sexual o por contacto con sangre
contaminada. Los pacientes deben continuar utilizando las precauciones
adecuadas para impedir la transmisión del VIH.
• Sobredosis: No hay un antídoto específico para la sobredosis de rilpivirina. La
experiencia en relación con dicha sobredosis en el ser humano es limitada. El
tratamiento de la misma consiste en medidas de soporte generales, que
comprenden vigilancia de las constantes vitales y el ECG (intervalo QT) y
observación del estado clínico del paciente. También puede administrarse carbón
activado para facilitar la eliminación del principio activo no absorbido. Dado que
rilpivirina presenta una gran afinidad por las proteínas plasmáticas, no es probable
que la diálisis logre la eliminación de cantidades significativas del principio activo.
• Embarazo, lactancia, población pediátrica y fertilidad: No existen datos en humanos
disponibles acerca de los efectos de rilpivirina sobre el embarazo y el feto, la
lactancia y la fertilidad. Rilpivirina aún está en fase de investigación en la población
pediátrica. Los estudios realizados en animales no han mostrado toxicidad para la
reproducción y han mostrado un paso limitado a placenta. No se ha observado
teratogenia con rilpivirina en ratas ni conejos. Por ello, que se ha clasificado por la
FDA como medicamento con categoría B en el embarazo (los estudios de
reproducción en animales no han mostrado efectos adversos sobre el feto, pero no

AGENCIA VALENCIANA DE LA SALUD DG de Farmacia y Productos Sanitarios
20
hay ensayos clínicos controlados y apropiados en mujeres embarazadas o
animales que hayan demostrado efectos adversos sobre el feto).
• Otras poblaciones especiales:
Pacientes de edad avanzada: La información sobre el uso de rilpivirina en pacientes
mayores de 65 años es limitada.
Insuficiencia hepática: La información sobre el uso de rilpivirina en pacientes con
insuficiencia hepática leve o moderada (clases A o B de Child-Pugh) también es limitada.
No se ha estudiado en pacientes con insuficiencia hepática grave (clase C de Child-
Pugh).
Insuficiencia renal: No hay datos del uso de rilpivirina en los pacientes con insuficiencia
renal grave o nefropatía terminal. La combinación con un inhibidor potente del CYP3A (por
ejemplo, un inhibidor de la proteasa del VIH potenciado con ritonavir) únicamente se debe
utilizar en pacientes con insuficiencia renal grave o nefropatía terminal si el beneficio
supera el riesgo.
Pacientes coinfectados por los virus de la hepatitis B (VHB) y/o C (VHC)
En los pacientes coinfectados por los virus de la hepatitis B o C tratados con rilpivirina, la
incidencia de elevaciones de las enzimas hepáticas en los ensayos clínicos fue mayor que
en los pacientes no coinfectados. Esto mismo se observó en los ensayos en el brazo de
tratamiento con efavirenz. La biodisponibilidad farmacocinética de rilpivirina fue similar en
los pacientes coinfectados y en los no coinfectados.
• Fracaso virológico y desarrollo de resistencia
Las siguientes mutaciones asociadas a resistencia a rilpivirina, si están presentes en el
momento basal, pueden afectar a la actividad de rilpivirina: K101E, K101P, E138A,
E138G, E138K, E138R, E138Q, V179L, Y181C, Y181I, Y181V, H221Y, F227C, M230I y
M230L. Sólo estas mutaciones asociadas a resistencia a rilpivirina deben guiar su uso en
el tratamiento de pacientes naïve. Estas mutaciones asociadas a resistencia fueron
obtenidas a partir de datos in vivo de pacientes naïve y, por lo tanto, no se pueden usar
como predicción de la actividad de rilpivirina en pacientes en fracaso virológico.
Como con otros antirretrovirales, la prueba de resistencias debe guiar el uso de rilpivirina.
En el análisis conjunto de los ensayos fase III, los pacientes tratados con rilpivirina que

AGENCIA VALENCIANA DE LA SALUD DG de Farmacia y Productos Sanitarios
21
presentaban al inicio una carga viral >100.000 copias/mL tuvieron un mayor riesgo de
fracaso virológico (15,1% con rilpivirina frente a 6,3% del brazo de efavirenz) en
comparación con los pacientes que partían de una carga viral basal ≤100.000 copias/mL
(3,8% con rilpivirina frente a 3,3% del brazo de efavirenz). Los pacientes con una carga
viral basal >100.000 copias/mL que experimentaron fracaso virológico mostraron una
frecuencia más elevada de aparición de resistencias a los ITINN. El desarrollo de
resistencia asociada a lamivudina/emtricitabina fue mayor en los pacientes que fracasaron
virológicamente a rilpivirina que en los que fracasaron virológicamente a efavirenz.
• Precauciones especiales
- Efectos cardiovasculares (prolongación del espacio QTc): debido a la constancia en
ensayos clínicos de la prolongación del intervalo QTc tanto con efavirenz como con
rilpivirina se debe tener en cuenta esta posibilidad y realizar los controles oportunos sobre
todo cuando se administra conjuntamente con medicamentos con riesgo conocido
de Torsade de Pointes.
- Efectos en el tejido adiposo (lipodistrofia): La terapia antirretroviral combinada (TARC) se
ha asociado con una redistribución de la grasa corporal (lipodistrofia) en pacientes con
infección por el VIH. En la actualidad se desconocen las consecuencias a largo plazo de
estos acontecimientos. El mecanismo no se conoce por completo. El riesgo de padecer
lipodistrofia aumenta con factores personales tales como la edad avanzada, y con
factores relacionados con el fármaco, tales como una duración prolongada del tratamiento
antirretroviral y trastornos metabólicos asociados. La exploración física de estos pacientes
debe incluir la evaluación de los signos físicos de redistribución del tejido adiposo.
- Efectos inmunológicos (síndrome de reconstitución inmune): En los pacientes infectados
por el VIH que presentan una inmunodeficiencia grave, cuando se instaura la TARC,
puede aparecer una reacción inflamatoria a patógenos oportunistas asintomáticas o
latentes y provocar cuadros clínicos graves o empeoramiento de los síntomas. Por lo
general, estas reacciones se observan en las primeras semanas o meses siguientes al
comienzo de la TARC. Por ello, se deben evaluar los posibles síntomas inflamatorios y
establecer un tratamiento cuando sea necesario.
- Efectos sobre la capacidad de conducir y utilización de máquinas: No se han llevado a

AGENCIA VALENCIANA DE LA SALUD DG de Farmacia y Productos Sanitarios
22
cabo estudios sobre los efectos de rilpivirina sobre la capacidad para conducir o utilizar
máquinas. Se han notificado casos de fatiga, mareos y somnolencia en algunos de los
pacientes que tomaban rilpivirina.
Interacciones farmacológicas:
A continuación se describen las principales interacciones farmacológicas con los efectos
tanto de la rilpivirina sobre otros medicamentos y preparados como con los efectos de
otros medicamentos y preparados sobre la propia rilpivirina. Y además se repasan las
interacciones con otros medicamentos que se utilizan habitualmente es pacientes con
infección VIH y SIDA [1, 2].
• Medicamentos y productos de herboristería que afectan a la biodisponibilidad de
rilpivirina
Rilpivirina se metabolizada principalmente a través de las enzimas CYP3A del citocromo
P450. Los medicamentos que inducen o inhiben el CYP3A pueden afectar a la eliminación
de rilpivirina.
La administración conjunta de rilpivirina y medicamentos que inducen (en otras
palabras, que aumentan la actividad de enzimas y por tanto, la metabolización de
fármacos es más rápida) el CYP3A disminuye las concentraciones plasmáticas de
rilpivirina, lo que podría reducir el efecto terapéutico de rilpivirina.
Por el contrario, la administración conjunta de rilpivirina y medicamentos que inhiben el
CYP3A aumenta las concentraciones plasmáticas de rilpivirina.
Así mismo la administración conjunta de rilpivirina con medicamentos que elevan el pH
gástrico puede disminuir las concentraciones plasmáticas de rilpivirina, lo que podría
reducir el efecto terapéutico de rilpivirina. Por ejemplo, no se debe administrar
conjuntamente con los siguientes medicamentos, dado que se pueden producir
descensos significativos de las concentraciones plasmáticas de rilpivirina como
consecuencia de la inducción enzimática del CYP3A o del aumento del pH gástrico, lo que
puede causar la pérdida del efecto terapéutico de rilpivirina:
- antiepilépticos como carbamacepina, oxcarbacepina, fenobarbital y fenitoína
- antimicobacterianos como rifabutina, rifampicina y rifapentina
- inhibidores de la bomba de protones (como omeprazol, esomeprazol, lansoprazol,
pantoprazol y rabeprazol) y otros antiácidos (como famotidina)

AGENCIA VALENCIANA DE LA SALUD DG de Farmacia y Productos Sanitarios
23
- el glucocorticoide sistémico, dexametasona, excepto como tratamiento de dosis única
- hierba de San Juan o Hipérico (Hypericum perforatum).
• Medicamentos afectados por el uso de rilpivirina
Por lo que se sabe, no es probable que rilpivirina a dosis de 25 mg una vez al día tenga
un efecto clínicamente relevante sobre la biodisponibilidad de los medicamentos
metabolizados por las enzimas del CYP.
Rilpivirina inhibe la glucoproteína-P in vitro. Así, podría inhibir la glucoproteína-P intestinal
y afectar a los medicamentos que son transportados por la glucoproteína-P en el intestino,
tales como dabigatrán. Este hecho puede resultar en una elevación de las
concentraciones plasmáticas de dichos medicamentos.
Rilpivirina inhibe la secreción tubular renal activa de creatinina. Mediante el mismo
mecanismo, la biodisponibilidad de metformina puede aumentar. Los pacientes deben ser
vigilados estrechamente cuando se inicie o suspenda la administración concomitante de
rilpivirina y metformina.
• Interacciones con fármacos clave en el manejo del VIH/SIDA [2].
Es importante tener en mente que estos pacientes con frecuencia son tratados con varias
medicaciones simultáneamente las cuales son necesarias para el manejo de su
enfermedad y que pueden originar interacciones farmacológicas (incluidas interacciones
clínicamente relevantes). A continuación se describen algunas de estas interacciones que
se han estudiado en ensayos en humanos (en general son ensayos clínicos con pocos
sujetos y además sanos VIH-negativos).
- Fármacos para el tratamiento de la tuberculosis (como rifampicina y rifabutina)
En un estudio farmacocinético de interacción se constató que al administrar a 16
voluntarios VIH-negativos durante 7 días rifampicina 600 mg una vez al día junto con
rilpivirina 150 mg una vez al día, disminuían los valores de rilpivirina de AUC24h (un 80%),
Cmax (un 69%) y Cmin (un 89%). En otro estudio farmacocinético de interacción en 18
sujetos VIH-negativos se comprobó que al administrar durante 11 días rifabutina 300 mg
una vez al día junto con rilpivirina 150 mg una vez al día, disminuían los valores de
rilpivirina de AUC24h (un 46%), Cmax (un 35%) y Cmin (un 49%).
- Antirretrovirales (como tenofovir y la combinación de darunavir/ritonavir)

AGENCIA VALENCIANA DE LA SALUD DG de Farmacia y Productos Sanitarios
24
En un estudio en 16 voluntarios sanos en el que se coadministraron rilpivirina y tenofovir,
aunque se objetivó un aumento del AUC24h de rilpivirina del 24% (estadísticamente
significativo) este no se consideró clínicamente relevante.
En otro estudio donde se administró a 16 voluntarios VIH-negativos rilpivirina 150 mg una
vez al día junto con darunavir/ritonavir 800/100 mg una vez al día, se halló un aumento
importante de la biodisponibilidad de rilpivirina (AUC24h 130%), Cmax 79%) y Cmin 178%).
Esto mismo se confirmó en un segundo estudio y se concluyó que era debido a una
inhibición del CYP3A4.
- Antifúngicos (como ketonazol). Ketoconazol es un inhibidor del CYP3A4 y al
coadministrarse a 16 voluntarios VIH-negativos a dosis de 400 mg junto con rilpivirina
150 mg una vez al día se produjo un aumento de los valores de rilpivirina de AUC24h (un
49%), Cmax (un 30%) y Cmin (un 76%) y una disminución de los valores de ketoconazol de
AUC24h (un 24%), Cmax (un 15%) y Cmin (un 66%).
- Metadona. En un estudio al coadministrar dosis individualizadas de metadona y rilpivirina
25 mg una vez al día a 13 voluntarios VIH-negativos la farmacocinética de rilpivirina no
cambio y la de maetadona sufrió cambios modestos. No obstante, a raíz de este estudio
se recomienda la monitorización clínica de los síntomas por retirada de metadona.
• Otras interacciones evaluadas en ensayos.
Además de los medicamentos anteriormente reseñados, también se ha estudiado la
posible interacción con otros grupos farmacológicos y con otros medicamentos concretos
(como sildenafilo, atorvastatina y paracetamol).
Cabe reseñar que se ha estudiado la posible interacción con algunos anticonceptivos
orales (etinilestradiol y noretindrona). En un estudio se administró, a 18 mujeres VIH-
negativas, el anticonceptivo oral compuesto (una vez al día) por etiniliestradiol 0,035 mg y
noretindrona 1 mg junto con rilpivirina (25 mg, una vez al día) durante tres ciclos
menstruales y no se encontraron cambios farmacocinéticos significativos en ninguno de
los tres principios activos. Aunque se evidenció un aumento del 17% en la Cmax de
etinilestradiol, no se consideró clínicamente relevante (los niveles séricos de
progesterona, de la hormona luteinizante y de la hormona folículo-estimulante en los días
1 y 14 del ciclo estuvieron dentro de los rangos normales) y por ello, no se requieren
ajustes de dosis.

AGENCIA VALENCIANA DE LA SALUD DG de Farmacia y Productos Sanitarios
25
DESCRIPCIÓN ECONÓMICA
Comparación con la terapia de referencia o alternat iva a dosis usuales:
En la siguiente tabla se recogen los costes (PVL) en nuestro país del nuevo antirretroviral
inhibidor de la transcriptasa inversa no análogo de nucleósidos y del fármaco control en
los dos ensayos clínicos fase III descritos en este informe.
Tabla 11: Costes de las especialidades farmacéutica s EDURANT (rilpivirina ) y SUSTIVA (efavirenz) en n uestro país.
Fármaco Dosis Coste unitario (PVL)
Coste diario Coste total del tratamiento/año
Rilpivirina (EDURANT)
25 mg/día (1 comprimido/día)
# 238,53 euros EDURANT 25 mg (rilpivirina) comprimidos recubiertos con película (30x1)
# 7,95 euros
# 2.902 euros
Efavirenz (SUSTIVA)
600 mg/día (1 comprimido/día)
265 euros comprimidos recubiertos (30x1)
8,83 euros 3.223 euros
En todo caso, debe tenerse en cuenta que se trata de un tratamiento crónico y combinado (al menos tres fármacos antirretrovirales) y que además de fármacos antirretrovirales no genéricos también hay antirretrovirales genéricos. #A fecha 23/01/2013 el Ministerio de Sanidad, Servicios Sociales e Igualdad ha fijado estos precios para EDURANT (rilpivirina): 238,53 euros/unidad (PVL), 289,44 (PVP) y 301,02 (PVP+IVA) . Existe también una combinación de emtricitabina, rilpivirina y tenofovir disoproxil fumarato (EVIPLERA, 200/25/245 mg, comprimidos recubiertos con película) para la que se ha fijado estos precios: 697,63 euros/unidad (PVL), 753,54 (PVP) y 783,68 (PVP+IVA).
Coste Eficacia/Efectividad Incremental (CEI):
En este momento no disponemos de estudios farmacoeconómicos publicados de
rilpivirina desde la perspectiva del Sistema Nacional de Salud (SNS) Español para la
indicación aprobada.
Hasta ahora se han difundido estudios farmacoeconómicos de rilpivirina en forma de
póster en Congresos y Conferencias desde la perspectiva de diferentes Sistemas
Nacionales de Salud (SNS): SNS Español ([10]) y Británico ([11]).
Según los datos de los ensayos clínicos ECHO y THRIVE, la rilpivirina (EDURANT®)
sería más coste-eficaz que la terapia de referencia, no suponiendo ningún coste

AGENCIA VALENCIANA DE LA SALUD DG de Farmacia y Productos Sanitarios
26
incremental respecto a efavirenz.
Coste por paciente con carga viral basal ≤ 100.000 copias/mL que alcanza carga viral indetectable (CVP<50 copias/mL) a las 48 semanas
ARV
Coste del tratmiento
antirretroviral
(48 semanas)*
Pacientes con carga viral
indetectable en la
semana 48
(según EC pivotales **)
(IC 95%)
Coste por paciente con carga viral
≤ 100,000 copias/mL con carga
viral indetectable a las 48
semanas*** (IC 95%)
Rilpivirina
(EDURANT®)2.791,83 € 90,2% (86,7%-93,1%) 3.095,16 € (2.998,75 € - 3.220,11 €)
Efavirenz 3.102,00 € 83,6% (79,2%-87,5%) 3.710,53 € (3.545,14 € - 3.916,67 €)
*PVL - 7,5% + 4% IVA
** ECHO/THRIVE Pooled data analisis
*** Coste del tratamiento anual/ % pacientes con CV indetectable en la semana 48 Coste Eficacia Incremental
Referencia Tipo de resultado Variable evaluableMedicamento con que se
comparaNNT (IC 95%)
Coste
Incremental
Eficacia
Incremental (IC
95%)
CEI (IC 95%)
Pooled data
(ECHO/THRIVE)
Subgrupo CV basal ≤ 100,000 copias/ml
CV<50 copias/ml EFV15 (13 a 17) -310,16 € 6,6% (5,6%-7,5%) Dominado
Cabe destacar que el mejor perfil de tolerabilidad de rilpivirina (EDURANT) frente a
efavirenz no se ha tenido en cuenta en este ejercicio y podría traducirse en: a) reducción
de los costes asociados del manejo de efectos adversos; b) reducción de los costes de
seguimiento programado de los pacientes; c) reducción de atenciones en urgencias e
ingresos hospitalarios; y d) menores tasas de abandono de tratamiento por efectos
adversos, mejorando la adherencia y retrasando en el tiempo la instauración de segundas
líneas de tratamiento generalmente más costosas.
La primera agencia europea evaluadora en realizar una informe económico acerca de
EDURANT ha sido Scottish Medicines Consortium [12]. Según su informe:
� Los ensayos han demostrado que rilpivirina (en combinación con otros
antirretrovirales) reduce los niveles del VIH circulando en el torrente sanguíneo a
igual que otro fármaco similar en pacientes no tratados previamente.
� En los ensayos, experimentan un número menor de efectos secundarios los
pacientes en tratamiento con rilpivirina en comparación con los pacientes que
recibieron el otro medicamento (efavirenz).
� Se acepta la rilpivirina en combinación con otros antirretrovirales para el
tratamiento de la infección por el VIH-1 porque se considera que ofrece valor por el
dinero invertido.

AGENCIA VALENCIANA DE LA SALUD DG de Farmacia y Productos Sanitarios
27
Aunque hay publicado un análisis de costes y de la relación coste/eficacia de las
“pautas preferentes” de GESIDA/Plan Nacional sobre el SIDA en 2012 para el
tratamiento antirretroviral inicial en adultos infectados por el VIH, este análisis no
incluye a ripilvirina [13].
Coste estimado anual en centro sanitario:
De momento no se dispone de datos más allá de los referidos en los estudios
farmacoeconómicos (principalmente modelizaciones y sin datos reales de lo que sucede
en nuestro entorno).
Estimación del impacto económico global (en hospita les y ambulatorios) a nivel
autonómico:
Sucede lo mismo que con el coste estimado anual en centro sanitario.
CONCLUSIONES
Resumen de los aspectos más significativos y propue sta:
En base a los dos ensayos clínicos fase III se puede decir que en el tratamiento
antirretroviral combinado de la infección por el VIH-1 en pacientes adultos naïve con una
carga viral ≤100.000 copias/mL, ripilvirina 25 mg oral no es inferior (porcentaje de
pacientes que habiendo recibido al menos una dosis de rilpivirina o de efavirenz
presentaron una respuesta virológica con carga viral <50 copias/mL confirmada en dos
determinaciones consecutivas y definida por la variable “tiempo hasta la pérdida de
respuesta virológica” a las 48 semanas del tratamiento) a efavirenz 600 mg.
A partir del análisis de seguridad conjunto de los dos ensayos clínicos fase III se puede
decir que la incidencia de acontecimientos adversos que conllevan el abandono
permanente del tratamiento es menor en el grupo de rilpivirina (3% vs 8%), lo que podría
redundar en una buena relación beneficio-riesgo (las reacciones adversas son la primera
causa de abandono permanente del tratamiento antirretroviral). También en este análisis
conjunto la incidencia de erupción cutánea de cualquier grado relacionada con el
tratamiento es inferior (y además estadísticamente significativa, incluso también en el

AGENCIA VALENCIANA DE LA SALUD DG de Farmacia y Productos Sanitarios
28
análisis de cada uno de los dos ensayos por separado) en el brazo de rilpivirina que en el
de efavirenz (3% vs 14%). Respecto a las reacciones adversas consideradas de especial
interés en los ITINN (efectos neurológicos y psiquiátricos), estas en los dos ensayos fase
III fueron menos frecuentes con rilpivirina (en el ensayo ECHO, eventos neurológicos:
16% vs 37%; eventos psiquiátricos: 15% vs 25%; y en el ensayo THRIVE, eventos
neurológicos: 18% vs 39%; eventos psiquiátricos: 15% vs 20%).
No se conocen los efectos del tratamiento con este medicamento a largo plazo. Y dada la
inexperiencia de uso actual en la práctica clínica en nuestro país tampoco hay estudios
económicos de coste-efectividad con rilpivirina desde la perspectiva del SNS español.
En este momento, tras haberse fijado su precio, la eficiencia de rilpivirina para la
indicación de ficha técnica (en combinación con otros medicamentos antirretrovirales en la
indicación del tratamiento de la infección por el VIH-1 en pacientes adultos naïve con una
carga viral ≤100.000 copias/mL) es mayor frente a efavirenz, a expensas de confirmarse
los datos de seguridad durante el periodo de Farmacovigilancia postcomercialización.
Lugar en la terapéutica:
Su lugar en la terapéutica se ha establecido siguiendo el algoritmo para la asignación de
la calificación final de un nuevo medicamento establecido en la resolución de 28 de enero
de 2011, de la Gerencia de la Agencia Valenciana de Salud [DOGV. 6459, 14/02/2011 ].
En dos ensayos clínicos fase III rilpivirina en combinación con otros antirretrovirales y en
comparación con efavirenz en el tratamiento de pacientes adultos naïve infectados por el
VHI-1 con una carga viral ≤100.000 copias/mL fue no inferior en cuanto a eficacia y
presentó menos acontecimientos adversos que conllevaron el abandono permanente del
tratamiento.
En definitiva, rilpivirina en combinación con otros medicamentos antirretrovirales y en
base a los ensayos clínicos realizados a medio plazo, representa una opción terapéutica
para aquellos pacientes adultos infectados por el VIH-1 que presentan una carga viral
≤100.000 copias/mL y que no han sido tratados previamente (naïve) cuando por motivos
de seguridad (básicamente neurológica y psiquiátrica ) pueda considerarse aconsejable
no iniciar el tratamiento con efavirenz. Es probable que el mejor perfil de seguridad
observado con rilpivirina en los ensayos obedezca al hecho de que no inhibe la

AGENCIA VALENCIANA DE LA SALUD DG de Farmacia y Productos Sanitarios
29
polimerasa γ (“gamma”) del ADN celular humano. Hay datos publicados que sugieren que
la inhibición de la polimerasa γ del ADN celular humano se asocia a la toxicidad de
clase, debida en parte a la inhibición de la síntesis del ADN mitocondrial, de los
inhibidores de la transcriptasa inversa (fundamentalmente anemia, granulocitopenia,
miopatía, neuropatía periférica y pancreatitis) [14].
También es importante considerar esta alternativa en el caso de mujeres fértiles que no
usen al menos dos métodos anticonceptivos eficaces, ya que está clasificado por la FDA
como medicamento de categoría B de la FDA de riesgo farmacológico en el embarazo
(efavirenz pertenece a la categoría D: existe evidencia de riesgo para el feto basada en
datos de investigación, datos postcomercialización, registros de reacciones adversas o
estudios en humanos).
No debe olvidarse que su administración oral, aunque cómoda, no garantiza el
cumplimiento terapéutico y que no se conocen los efectos del tratamiento con este
medicamento antirretroviral a largo plazo, en poblaciones más heterogéneas y en
subgrupos de pacientes con ciertas comorbilidades no estudiadas en los ensayos clínicos.
Indicaciones y servicios aprobados:
Rilpivirina está autorizada para uso hospitalario en combinación con otros medicamentos
antirretrovirales en la indicación del tratamiento de la infección por el VIH-1 en pacientes
adultos con una carga viral ≤100.000 copias/mL y que no han recibido tratamiento
antirretroviral previamente (naïve).
Especificar si la inclusión del fármaco va acompaña da con la propuesta de retirada
de algún otro fármaco:
No procede.
Especificar si se produce algún cambio en el PIT (P rograma de Intercambio
Terapéutico):
No procede.
Clasificación del avance terapéutico:
Rilpivirina comparado frente a efavirenz (ambos en combinación con tratamiento
antirretroviral de base):

AGENCIA VALENCIANA DE LA SALUD DG de Farmacia y Productos Sanitarios
30
Calificación 2. *Aporta en situaciones concretas: en combinación con otros medicamentos
antirretrovirales en la indicación del tratamiento de la infección por el VIH-1 en pacientes
adultos naïve con una carga viral ≤100.000 copias/mL. Puede presenta ventajas en
cuanto a la seguridad (fundamentalmente menor incidencia de acontecimientos adversos
que conllevan el abandono permanente del tratamiento).
BIBLIOGRAFÍA
[1] Ficha técnica de Edurant®. European Medicines Agency. http://www.ema.europa.eu [2] Ford N, Lee J, Andrieux-Meyer I, Calmy A. Safety, efficacy and pharmacokinetics of rilpivirine:
systematic review with an emphasis on resource-limited settings. HIV/AIDS-Research and Palliative care 2011; 3: 35-44.
[3] Ficha técnica de Sustiva®.Centro de Información online de Medicamentos de la AEMPS – CIMA. http://www.aemps.gob.es/cima
[4] Panel de de Expertos de GESIDA y Plan Nacional sobre el SIDA. Documento de consenso de GESIDA/ Plan Nacional sobre el SIDA respecto al tratamiento antirretroviral en adultos infectados por el virus de la inmunodeficiencia humana (actualización enero 2013). Disponible en: http://www.msc.es/
[5] Molina J-M, Cahn P, Grinsztejn B, Lazzarin A, Mills A, Saag M, Supparatpinyo K, Walmsley S, Crauwels H, Rimsky LT, Vanveggel S, Boven K, on behalf of the ECHO study group. Rilpivirine versus efavirenz with tenofovir and emtricitabine in treatment-naive adults infected with HIV-1 (ECHO): a phase 3 randomised duoble-blind active-controlled trial. Lancet 2011; 378: 238–46.
[6] Cohen JC, Andrade-Villanueva J, Clotet B, Fourie J, Johnson MA, Ruxrungtham K, Wu H, Zorrilla C, Crauwels H, Rimsky LT, Vanveggel S, Boven K, on behalf of the THRIVE study group. Rilpivirine versus efavirenz with two background nucleoside or nucleotide reverse transcriptase inhibitirs in treatment-naive adults infected with HIV-1 (THRIVE): a phase 3, randomised, non-inferiority trial. Lancet 2011; 378: 229–37.
[7] Cohen JC, Molina J-M, Cahn P, Clotet B, Fourie J, Grinsztejn B, Wu H, Johnson MA, Saag M, Supparatpinyo K, Crauwels H, Lefebvre E, Rimsky LT, Vanveggel S, Williams P, Boven K, on behalf of the ECHO AND THRIVE Study Groups. Efficacy and Safety of Rilpivirine (TMC278) Versus Efavirenz at 48 Weeks in Treatment-Naive HIV-1-Infected Patients: Pooled Results From the Phase 3 Duoble-Blind Randomized ECHO and THRIVE Trials. J Acquir Immune Defic Syndr 2012; 60: 33–42.
[8] Center for Drug Evaluation and Research. Guidance for industry: antiretroviral drugs using plasma HIV RNA measurements—clinical considerations for accelerated and traditional approval.Silver Spring, MD: US Food and Drug Administration, 2002.
[9] Rimsky L, Vingerhoets J, Van Eygen V, et al. Genotypic and phenotypic characterization of HIV-1 isolates obtained from patients on rilpivirine therapy experiencing virologic failure in the phase 3 ECHO and THRIVE studies: 48-week analysis. J Acquir Immune Defic Syndr 2012; 59: 39–46.
[10] Gostkorzewick J, Ledesma F. How should we target cost-efficacy in the treatment of hiv-1 naïve patients with recommended therapies based on nnrtis in Spain?. ISPOR Congress, Berlin, 3-7 November 2012.
[11] Girod I, Bell M, Adriaenssen I, Knight K. Cost-effectiveness of rilpivirine- or efavirenz-based regimens for treatment-naïve, HIV-infected patients: NHS England perspective. Poster presented at the 18th Annual Conference of the British HIV Association (BHIVA), Birmingham, UK, 18-20 April 2012.
[12] Scottish Medicines Consortium advice to NHSScotland. SMC briefing note. Number 51. February 2012.http://www.scottishmedicines.org/files/briefingnotes/2012/Briefing_note_SMC_February12.pdf.
[13] Antonio Javier Blasco, José Ramón Arribas, Vicente Boix, Bonaventura Clotet, Pere Domingo,

AGENCIA VALENCIANA DE LA SALUD DG de Farmacia y Productos Sanitarios
31
Juan González-García, Hernando Knobel, Juan Carlos López, Josep M. Llibre, Fernando Lozano, José M. Miró, Daniel Podzamczer, Juan Miguel Santamaría, Montserrat Tuset, Laura Zamora, Pablo Lázaro y Josep M. Gatell, en representación de GESIDA. Análisis de costes y de coste/eficacia de las pautas preferentes de GESIDA/Plan Nacional sobre el SIDA en 2012 para el tratamiento antirretroviral inicial en adultos infectados por el virus de la inmunodeficiencia humana (VIH). Enferm Infecc Microbiol Clin 2012; 30(6): 283-293.
[14] Brunton LL. Goodman & Gilman's pharmacological basis of therapeutics 12th ed. Flexner C. Capter 59: Antiretroviral Agents and Treatment of HIV. Page 1631.
SÍNTESIS DE LA EVIDENCIA [DOGV. 6459, 14/02/2011 ]
En las últimas páginas de este informe se sintetizan (en forma de tablas) las evidencias
disponibles de este medicamento provenientes de ensayos clínicos conforme a los
formatos de la resolución de 28 de enero de 2011, de la Gerencia de la Agencia
Valenciana de Salud, por la que se aprueba el protocolo normalizado de trabajo para la
evaluación de novedades terapéuticas y la estructura de los informes técnicos de la

AGENCIA VALENCIANA DE LA SALUD DG de Farmacia y Productos Sanitarios
32
evaluación [DOGV. 6459, 14/02/2011 ].
A continuación se realizan algunas consideraciones que son importantes a la hora de
interpretar los datos que figuran en las tablas resumen que aparecen al final de este
informe.
La validez interna de un estudio hace referencia a cómo de fiables son los datos
recogidos y por tanto, de los que partimos para hacer el análisis estadístico de los
resultados. Y la validez externa de un estudio se refiere a si estos resultados son o no
extrapolables a la población general.
La decisión de si un tratamiento (o una intervención médica) es o no más eficaz que otro
con el que se compara no debe basarse únicamente en el resultado de una prueba de
significación estadística (p). Con el valor de p, se demuestra si existe o no efecto, pero
no su relevancia clínica . Es importante la estimación de la magnitud del efecto (en
forma de medidas absolutas o relativas de variables) y la precisión (a través de los
intervalos de confianza). El intervalo de confianza (IC) es aquel intervalo entre cuyos
límites se tiene un “X”% (por ejemplo, 95%) de confianza de que se encuentre la
verdadera magnitud del efecto.
Por ello, los resultados estadísticos de los ensayos se deben presentar como medidas
(magnitud) junto con un intervalo de confianza (precisión). Las medidas absolutas se
basan en calcular la diferencia entre frecuencias del resultado observado en cada grupo
de sujetos, mientras que las medidas relativas se basan en cociente entre dichas
frecuencias (cuántas veces es más probable que ocurra un suceso en un grupo que en
otro). Las medidas relativas no tienen en cuenta el riesgo de base.
Algunos conceptos básicos a conocer y comprender al respecto son:
� RAR (Reducción Absoluta del Riesgo o Diferencias de Ries go o Riesgo
Atribuible ) = Riesgo (incidencia, proporción) en el grupo experimental – Riesgo
(incidencia, proporción) en el grupo control (o riesgo de base). Es la diferencia de
proporciones de un suceso en el grupo control y el grupo experimental. Y es la
forma más sencilla de expresar la diferencia de eficacia entre los grupos
estudiados.
� RR (Riesgo Relativo o Razón de Riesgos ) = Riesgo (incidencia, proporción) en

AGENCIA VALENCIANA DE LA SALUD DG de Farmacia y Productos Sanitarios
33
el grupo control / Riesgo (incidencia, proporción) en el grupo experimental. Es un
cociente (o relación de proporciones) entre el grupo control y el grupo experimental.
Toma valores entre 0 e infinito. El valor 1 es el valor neutro e indica que no hay
diferencias entre los grupos comparados.
� RRR (Reducción Relativa del Riesgo ) = 1-RR
� Odss = Es una razón en la que el numerador es la probabilidad de que ocurra un
suceso y el denominador es la probabilidad de que el mismo no suceda.
� OR (Odds Ratio ) = casos/no casos del grupo experimental “entre” casos/no casos
del grupo control. Es la división de una odds por otra odds (cociente entre dos
grupos).
� NNT (Número Necesario a Tratar ) = 1/RAR. También se puede expresar en
porcentajes (100/RAR). Es el número necesario de sujetos a tratar para producir
una unidad adicional de eficacia (o dicho de otra manera, el grado de “esfuerzo a
realizar para conseguir una unidad adicional de eficacia, un resultado). Suele hacer
referencia a una intervención terapéutica. A menor NNT mayor eficacia (hay que
tratar a menos sujetos para conseguir un resultado concreto).
� NNH (Number Needed to Harm o número necesario para dañar ). Hace
referencia a un efecto perjudicial (dañino) y es una medida que indica cuántos
sujetos necesitan estar expuestos a un factor de riesgo en un periodo de tiempo
concreto para causar daño a un sujeto que de otra manera no hubiera salido
perjudicado.
Cuando se emplea como medida del efecto una diferencia (por ejemplo, RAR o RRR), si
el IC incluye el 0, el resultado no es estadísticamente significativo, y viceversa. Por el
contrario, cuando se utiliza un cociente (como RR u OR) si el IC fijado incluye el valor 1 el
resultado no es estadísticamente significativo, y viceversa.

Tablas “Resultados de los ensayos clínicos disponib les”
Modelo general de tabla de resultados de eficacia:
Referencia: ECHO (NCT00540449)
Breve descripción del ensayo, haciendo constar los aspectos más relevantes sobre: - Nº de pacientes: 694 pacientes aleatorizados (346 en el grupo de rilpivirina y 348 en el grupo de efavirenz), 690 pacientes tratados (346 en el grupo de rilpivirina y 344 en el grupo de efavirenz) y 584 pacientes completados (296 en el grupo de rilpivirina y 288 en el grupo de efavirenz). - Diseño: Fase del ensayo, aleatorización, ciego o abierto, etc.: ensayo clínico (EC) fase III, multicéntrico, internacional, aleatorizado, doble-ciego, double-dummy (doble simulación, simulación con placebo), de grupos paralelos y controlado con comparador activo en pacientes con infección por VIH-1 en pacientes adultos que no han recibido tratamiento antirretroviral previamente (pacientes naïve). Estudio de no-inferioridad. Estudio de eficacia y seguridad. Participaron 91 centros. - Tratamientos (grupo activo y grupo control): rilpivirina 25 mg + tratamiento antirretroviral de base: tenofovir (disoproxil-fumarato) y emtricitabina (en el grupo activo) Y efavirenz 600 mg + tratamiento antirretroviral de base: tenofovir (disoproxil-fumarato) y emtricitabina (en el grupo control). Duración total del tratamiento en ambos casos: 96 semanas. Periodo de seguimiento de 48 semanas. - Criterios de inclusión: Pacientes ≥18 años, con infección VIH-1 documentada, nunca tratados con vacuna frente al VIH o fármacos antirretrovirales antes del screening, con una carga viral >5.000 copias/mL, sensibles al tratamiento con tenofovir y emtricitabina y que estaban de acuerdo en no iniciar el tratamiento antirretroviral antes de la visita basal del ensayo. - Criterios de exclusión (los criterios seleccionados): Uso previo de cualquier fármaco antirretroviral, cualquier evidencia documentada de mutaciones asociadas a resistencia a ITINN, categoría C definitoria de SIDA (excepto, sarcoma de Kaposi estable y síndrome de fatiga no progresivo, neumonia por Pneumocistis carinni no curada, tuberculosis activa, alergia o hipersensibilidad a cualquiera de los fármacos del ensayo, toxicidad específica grado 3 o 4, deterioro renal (aclaramiento de creatinina calculado <50 mL/min). - Pérdidas: 50 pacientes en el grupo de rilpivirina y 56 pacientes en el grupo de efavirenz interrumpieron el tratamiento. - Tipo de análisis: Análisis principal: No-inferioridad/ por intención de tratar.
Resultados
Variable evaluada en el estudio
Variable principal (de eficacia): Respuesta virológica = porcentaje de pacientes que recibieron al menos una dosis de la medicación del ensayo con respuesta confirmada (carga viral <50 copias/mL, definida por un algoritmo de análisis, por intención de tratar, de tiempo hasta la pérdida de respuesta virológica (TLOVR) a las 48 semanas. No-inferioridad con un margen del 12% para el límite inferior del IC95%. Variables secundarias (algunas de ellas): - Respuesta virológica: No-inferioridad con un margen del 10%. - Seguridad (AAGrave). - AA asociados a abandono permanente del tratamiento.
Trat estudiado N
(nº paciente)**
287/346 (83%)
285/346 (82%) 23/346 (7%) 8/346 (2%)
Trat control N
(nº paciente)**
285/344 (83%)
281/344 (82%) 31/344 (9%) 27/344 (8%)

Resultado principal - Breve descripción variable Variable principal (de eficacia): Respuesta virológica (No-inferioridad con un margen del 12%)
RAR (vs efavirenz):
0,1% (IC 95% −5,5 a 5,7) –es la diferencia de eficacia entre los grupos estudiados.
RR (vs efavirenz):
1 (IC 95% 1,07 a 0,94) –no hay diferencias.
Resultados secundarios de interés - Breve descripción variable Respuesta virológica: No-inferioridad con un margen del 10%. Resultados por subgrupos - Breve descripción variable Respuesta virológica a las 48 semanas en pacientes con CV basal ≤100.000 copias/mL en ECHO + THRIVE
332/368 (90%)
276/330 (84%)
RAR (vs efavirenz):
0,7% (IC 95% −5 a 6,4)
RAR (vs efavirenz):
6,6% (IC 95% 1,6 a 11,5)
NNT (vs efavirenz):
15
Calculadoras para variables binarias: RAR y NNT y sus IC 95%. CASPe. SIGN:
Calculadora para variables continuas: R. Saracho.

Modelo general de tabla de resultados de eficacia:
Referencia: THRIVE (NCT00543725)
Breve descripción del ensayo, haciendo constar los aspectos más relevantes sobre: - Nº de pacientes: 680 pacientes aleatorizados (340 en el grupo de rilpivirina y 340 en el grupo de efavirenz), 678 pacientes tratados (340 en el grupo de rilpivirina y 338 en el grupo de efavirenz) y 578 pacientes completados (296 en el grupo de rilpivirina y 282 en el grupo de efavirenz). - Diseño: Fase del ensayo, aleatorización, ciego o abierto, etc.: ensayo clínico (EC) fase III, multicéntrico, internacional, aleatorizado, doble-ciego, double-dummy, de grupos paralelos y controlado con comparador activo en pacientes con infección por VIH-1 en pacientes adultos que no han recibido tratamiento antirretroviral previamente (pacientes naïve). Estudio de no-inferioridad. Estudio de eficacia y seguridad. Participaron 83 centros. - Tratamientos (grupo activo y grupo control): rilpivirina 25 mg + tratamiento antirretroviral de base: tenofovir (disoproxil-fumarato) y emtricitabina (en el grupo activo) Y efavirenz 600 mg + tratamiento antirretroviral de base: tenofovir (disoproxil-fumarato) + emtricitabina o zidovudina + lamivudina o abacavir + lamivudina (en el grupo control). Duración total del tratamiento en ambos casos: 96 semanas. Periodo de seguimiento de 48 semanas. tenofovir (disoproxil fumarato) + emtricitabina, - Criterios de inclusión: Pacientes ≥18 años, con infección VIH-1 documentada, nunca tratados con vacuna frente al VIH o fármacos antirretrovirales antes del screening, con una carga viral > 5.000 copias de ARN/mL, sensibles al tratamiento con tenofovir y emtricitabina y que estaban de acuerdo en no iniciar el tratamiento antirretroviral antes de la visita basal del ensayo. - Criterios de exclusión (los criterios seleccionados): Uso previo de cualquier fármaco antirretroviral, cualquier evidencia documentada de mutaciones asociadas a resistencia a ITINN, categoría C definitoria de SIDA (excepto, sarcoma de Kaposi estable y síndrome de fatiga no progresivo, neumonia por Pneumocistis carinni no curada, tuberculosis activa, alergia o hipersensibilidad a cualquiera de los fármacos del ensayo, toxicidad específica grado 3 o 4, deterioro renal (aclaramiento de creatinina calculado <50 mL/min). - Pérdidas: 44 pacientes en el grupo de rilipivirina y 56 pacientes en el grupo de efavirenz interrumpieron el tratamiento. - Tipo de análisis: Análisis principal: No-inferioridad/ por intención de tratar.
Resultados
Variable evaluada en el estudio
Variable principal (de eficacia): Respuesta virológica = porcentaje de pacientes que recibieron al menos una dosis de la medicación del ensayo con respuesta confirmada (carga viral <50 copias/mL, definida por un algoritmo de análisis, por intención de tratar, de tiempo hasta la pérdida de respuesta virológica (TLOVR) a las 48 semanas. No-inferioridad con un margen del 12% para el límite inferior del IC95%. Variables secundarias: - Respuesta virológica: No-inferioridad con un margen del 10%. - Seguridad (AAGrave). - AA asociados a abandono permanente del tratamiento.
Trat estudiado N
(nº paciente)**
291/340 (86%)
281/340 (83%) 22/340 (6%) 15/340 (4%)
Trat control N
(nº paciente)**
276/338 (82%)
265/338 (78%) 24/338 (7%) 25/338 (7%)

Resultado principal - Breve descripción variable Variable principal (de eficacia): - Respuesta virológica: No-inferioridad con un margen del 12%.
RAR (vs efavirenz):
3,9% (IC 95% −1,6 a 9,5)
RR (vs efavirenz):
1,05 (IC 95% 1,12-0,98)
NNT (vs efavirenz):
25 (IC 95% 61 a -11) – habrá que tratar a 25 pacientes para tener un resultado (una unidad adicional de eficacia), es decir, un paciente más respondedor.
Resultados secundarios de interés - Breve descripción variable Respuesta virológica: No-inferioridad con un margen del 10%.
RAR (vs efavirenz):
0,4% (IC 95% −1,7 a 10,2)
Calculadoras para variables binarias: RAR y NNT y sus IC 95%. CASPe. SIGN:
Calculadora para variables continuas: R. Saracho.

Tablas “Resultados de seguridad”
Referencia: ECHO (NCT00540449)
Breve descripción del ensayo y diseño: en tabla previa
Resultados de seguridad
Variable de seguridad
evaluada en el estudio
Rilpivirina
total nº
pacientes =
346
Efavirenz
total nº
pacientes =
344
RAR (IC 95%)
Diferencia riesgo
absoluto
p NNH o
NND
(IC 95%)
- AA con abandono del tratamiento 8 (2%) 27 (8%) --- --- --- - AA relacionado grado ≥ 2 55 (16 %) 108 (31%) 16% (IC95: 9,3% a 21,7%) <0,0001 6 (11 a 5) - AA grave 23 (7 %) 31 (9%) --- --- --- - Erupción cutánea relacionada 12 (4%) 50 (15%) 11% (IC95: 6,9% a 15,3%) <0,0001 9 (15 a 7) - AA analíticos grado 3-4 34 (10%) 55 (16%) --- --- --- - AA de especial interés Neurológicos 55 (16%) 126 (37%) 21% (IC95: 14,3% a 27,1%) <0,0001 5 (7 a 4) Psiquiátricos 50 (15%) 86 (25%) 11 % (IC95: 4,7% a 16,4%) 0,0006 9 (21 a 6)
(*) RAR y NNT con IC 95% se exponen en la tabla sólo si p<0,05
AA grave: incluida la muerte. Erupción cutánea: de cualquier grado.
Referencia: THRIVE (NCT00543725)
Breve descripción del ensayo y diseño: en tabla previa
Resultados de seguridad
Variable de seguridad
evaluada en el estudio
Rilpivirina
total nº
pacientes =
340
Efavirenz
total nº
pacientes =
338
RAR (IC 95%)
Diferencia riesgo
absoluto
p NNH o
NND
(IC 95%)
- AA con abandono del tratamiento 15 (4%) 25 (7%) --- --- --- - AA relacionado grado ≥ 2 54 (16%) 104 (31%) 15% (IC95: 8,6% a 21,2%) <0,001 7 (12 a 5) - AA grave 22 (7%) 24 (7%) --- --- --- - Erupción cutánea relacionada 9 (3%) 43 (13%) 10% (IC95: 6,1% a 14%) <0,001 10 (16 a 7) - AA analíticos grado 3-4 41 (12%) 63 (19%) --- --- --- - AA de especial interés Neurológicos 62 (18%) 132 (39%) 21% (IC95: 14,2% a 27,4%) <0,001 5 (7 a 4) Psiquiátricos 52 (15%) 69 (20%) --- --- ---
(*) RAR y NNT con IC 95% se exponen en la tabla sólo si p<0,05
AA grave: incluida la muerte. Erupción cutánea: de cualquier grado.

Referencia: ECHO + THRIVE (análisis conjunto)
Breve descripción del ensayo y diseño: en las tablas previas
Resultados de seguridad
Variable de seguridad
evaluada en el estudio
Rilpivirina
total nº
pacientes =
686
Efavirenz
total nº
pacientes =
682
RAR (IC 95%)
Diferencia riesgo
absoluto
p NNH o
NND
(IC 95%)
- AA con abandono del tratamiento 23 (3%) 52 (8%) 4% (IC95: 1,9% a 6,7%) 0,0005 23 (54 a15) - AA relacionado grado ≥ 2 109 (16%) 212 (31 %) 15% (IC95: 10,8% a 19,6%) <0,0001 7 (9 a 5) - AA grave 45 (7 %) 55 (8%) --- --- --- - Erupción cutánea 21 (3%) 93 (14%) 11% (IC95: 7,7% a 13,5%) <0,0001 9 (13 a 7) - AA analíticos grado 3-4 /685 (10,9%) /670 (17,6%) 6% (IC95: 2,7% a 10%) <0,001 16 (37 a 10) - AA de especial interés más frecuentes Neurológicos (mareos) 55 (8%) 179 (26%) 18% (IC95: 14,4% a 22,1%) <0,0001 5 (7 a 5) Psiquiátricos 56 (8%) 87 (13%) 5% (IC95: 1,4% a 7,8%) 0,006 22 (74 a 13) (sueños anómalos/pesadillas)
(*) RAR y NNT con IC 95% se exponen en la tabla sólo si p<0,05
AA grave: incluida la muerte. Erupción cutánea: de cualquier tipo.

Tablas “Análisis de validez interna de los ensayos”
Cuestionario sobre la validez de un ensayo de bioequivalencia o no inferioridad
ECHO (NCT00540449) Sí/No Justificar -¿Está claramente definido el objetivo como un estudio de no inferioridad o de equivalencia?
SI Estudio de no inferioridad.
- ¿El comparador es adecuado? SI Efavirenz (comparador activo con la misma indicación).
- ¿Se ha establecido un margen de equivalencia? SI Margen de no-inferioridad pre-especificado del 12 % para la variable principal.
-¿El seguimiento ha sido completo? SI Se especifican los pacientes aleatorizados, las pérdidas de pacientes, los que completan el ensayo y los que se tienen en cuenta en el análisis estadístico.
- ¿Se analizan los resultados según análisis por ITT y también per protocolo?
SI Análisis estadístico principal: por ITT .
- ¿El intervalo de confianza permite asegurar la equivalencia?
SI Para la variable principal (de eficacia): Dif. o RAR: −0,4% (IC 95% −5,9 a 5,2; p<0,0001)
- Otros sesgos o limitaciones encontradas en el estudio SI - Únicamente se administró un régimen de ITIAN de base (tenofovir y emtricitabina). - No tiene el poder estadístico suficiente como para establecer comparaciones de eficacia en varios subgrupos de pacientes. No obstante, este no era el objetivo principal del ensayo. - Podría haberse sobrestimado el porcentaje de pacientes respondedores (respondedores en los ensayos frente a los respondedores reales en la práctica clínica) porque se excluyeron los pacientes con evidencia de resistencia asociada a mutación de al menos un ITINN. - La forma de medir la variable principal mediante el tiempo hasta la pérdida de respuesta virológica (o TLOVR) no está exenta de críticas por inducir a la sobrevaloración de los fracasos y porque además en la práctica clínica hay repuntes virológicos que son temporales. Un alternativa a este análisis es el análisis “snapshot” (o de foto fija) que consiste en la valoración de las cargas virales en una visita concreta determinada con un intervalo de de tiempo.
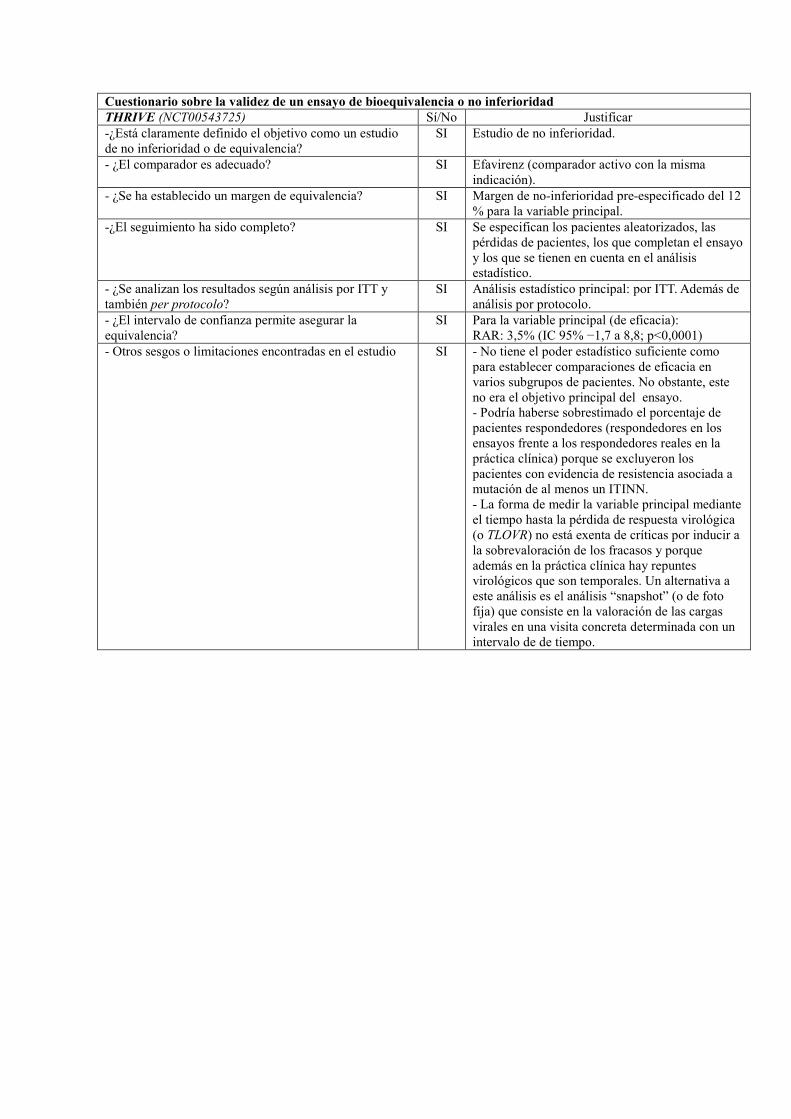
Cuestionario sobre la validez de un ensayo de bioequivalencia o no inferioridad
THRIVE (NCT00543725) Sí/No Justificar -¿Está claramente definido el objetivo como un estudio de no inferioridad o de equivalencia?
SI Estudio de no inferioridad.
- ¿El comparador es adecuado? SI Efavirenz (comparador activo con la misma indicación).
- ¿Se ha establecido un margen de equivalencia? SI Margen de no-inferioridad pre-especificado del 12 % para la variable principal.
-¿El seguimiento ha sido completo? SI Se especifican los pacientes aleatorizados, las pérdidas de pacientes, los que completan el ensayo y los que se tienen en cuenta en el análisis estadístico.
- ¿Se analizan los resultados según análisis por ITT y también per protocolo?
SI Análisis estadístico principal: por ITT. Además de análisis por protocolo.
- ¿El intervalo de confianza permite asegurar la equivalencia?
SI Para la variable principal (de eficacia): RAR: 3,5% (IC 95% −1,7 a 8,8; p<0,0001)
- Otros sesgos o limitaciones encontradas en el estudio SI - No tiene el poder estadístico suficiente como para establecer comparaciones de eficacia en varios subgrupos de pacientes. No obstante, este no era el objetivo principal del ensayo. - Podría haberse sobrestimado el porcentaje de pacientes respondedores (respondedores en los ensayos frente a los respondedores reales en la práctica clínica) porque se excluyeron los pacientes con evidencia de resistencia asociada a mutación de al menos un ITINN. - La forma de medir la variable principal mediante el tiempo hasta la pérdida de respuesta virológica (o TLOVR) no está exenta de críticas por inducir a la sobrevaloración de los fracasos y porque además en la práctica clínica hay repuntes virológicos que son temporales. Un alternativa a este análisis es el análisis “snapshot” (o de foto fija) que consiste en la valoración de las cargas virales en una visita concreta determinada con un intervalo de de tiempo.

AGENCIA VALENCIANA DE LA SALUD DG de Farmacia y Productos Sanitarios
42
Tablas “Cuestionario para la valoración global de l a validez externa o aplicabilidad de los
ensayos clínicos”
Estudio (referencia) ECHO (NCT00540449) Fecha de revisión: diciembre 2012 Nombre(s) del revisor(es): Mª Ángeles Pena Pardo Sí/No Justificar -¿Considera adecuado el comparador? SI Efavirenz (comparador activo con la misma indicación). - ¿Considera adecuada la variable de medida? SI La variable de medida fue la respuesta virológica (en
términos de porcentaje de pacientes que recibieron al menos una dosis de la medicación del ensayo y con carga viral plasmática <50 copias/mL , definida por un algoritmo de análisis, por intención de tratar, de tiempo hasta la pérdida de respuesta virológica a las 48 semanas). Y uno de los principales motivos para iniciar el tratamiento antirretroviral es precisamente la supresión completa y duradera de la replicación del VIH-1 medida como carga viral plasmática.
- ¿Considera adecuados los criterios de inclusión y/o exclusión de los pacientes?
SI Son pacientes con diagnóstico documentado de infección por VIH-1 nunca antes tratados con antirretrovirales (pacientes naïve) y sin SIDA, pero se han excluido a pacientes con cualquier evidencia documentada de mutaciones asociadas a resistencia a ITINN. Ello, podría haber llevado a sobrestimar el porcentaje de pacientes respondedores (respondedores en los ensayos frente a los respondedores reales en la práctica clínica).
-¿Cree que los resultados pueden ser aplicados directamente a la práctica clínica? Incluir NNT (cuando proceda)
SI
- ¿Se analizan los resultados según análisis por ITT y también per protocolo?
SI Análisis estadístico principal por ITT.
- Comentarios: otros sesgos o limitaciones encontradas en el estudio
SI - Únicamente se administró un régimen de ITIAN de base (tenofovir y emtricitabina). - No tiene el poder estadístico suficiente como para establecer comparaciones de eficacia en varios subgrupos de pacientes. Pero este no era el objetivo principal del ensayo y además se realizó una predicción de respuesta ajustada y estratificada por carga viral basal. - Podría haberse sobrestimado el porcentaje de pacientes respondedores (respondedores en los ensayos frente a los respondedores reales en la práctica clínica) porque se excluyeron los pacientes con evidencia de resistencia asociada a mutación de al menos un ITINN.

AGENCIA VALENCIANA DE LA SALUD DG de Farmacia y Productos Sanitarios
43
Estudio (referencia) THRIVE (NCT00543725) Fecha de revisión: diciembre 2012 Nombre(s) del revisor(es): Mª Ángeles Pena Pardo Sí/No Justificar -¿Considera adecuado el comparador? SI Efavirenz (comparador activo con la misma indicación). - ¿Considera adecuada la variable de medida? SI La variable de medida fue la respuesta virológica (en
términos de porcentaje de pacientes que recibieron al menos una dosis de la medicación del ensayo y con carga viral plasmática <50 copias/mL , definida por un algoritmo de análisis, por intención de tratar, de tiempo hasta la pérdida de respuesta virológica a las 48 semanas). Y uno de los principales motivos para iniciar el tratamiento antirretroviral es precisamente la supresión completa y duradera de la replicación del VIH-1 medida como carga viral plasmática.
- ¿Considera adecuados los criterios de inclusión y/o exclusión de los pacientes?
SI Son pacientes con diagnóstico documentado de infección por VIH-1 nunca antes tratados con antirretrovirales (pacientes naïve) y sin SIDA, pero se han excluido a pacientes con cualquier evidencia documentada de mutaciones asociadas a resistencia a ITINN. Ello, podría haber llevado a sobrestimar el porcentaje de pacientes respondedores (respondedores en los ensayos frente a los respondedores reales en la práctica clínica).
-¿Cree que los resultados pueden ser aplicados directamente a la práctica clínica? Incluir NNT (cuando proceda)
SÍ NNT: 25
- ¿Se analizan los resultados según análisis por ITT y también per protocolo?
SI Análisis principal por ITT.
- Comentarios: otros sesgos o limitaciones encontradas en el estudio
SI - No tiene el poder estadístico suficiente como para establecer comparaciones de eficacia en varios subgrupos de pacientes. Pero este no era el objetivo principal del ensayo y además se realizó una predicción de respuesta ajustada y estratificada por carga viral basal y régimen antirretroviral de base. - Podría haberse sobrestimado el porcentaje de pacientes respondedores (respondedores en los ensayos frente a los respondedores reales en la práctica clínica) porque se excluyeron los pacientes con evidencia de resistencia asociada a mutación de al menos un ITINN.





























