Embed Size (px)
Citation preview

Identifying Structural Motifs in Proteins
Rohit Singh
Joint work with Mitul Saha
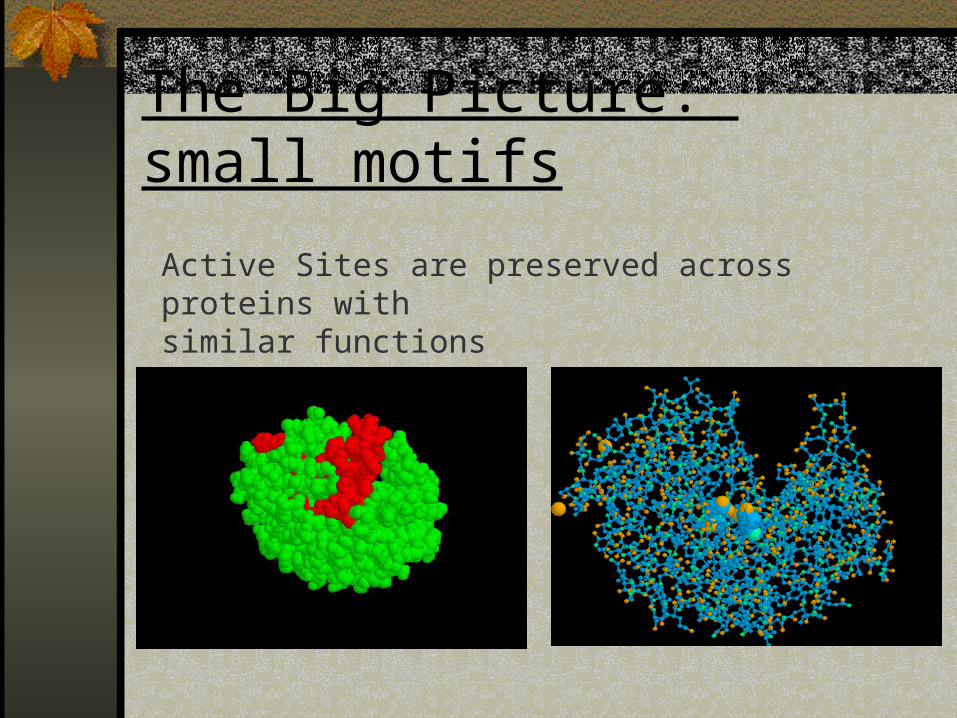
The Big Picture: small motifs
Active Sites are preserved across proteins withsimilar functions

The Big Picture: large motifs
Even bigger motifs are often conserved.

Oh, BTW…
There are two different issues here:
1. Find the best match for the motif in the protein
Extensively studied in vision/graphics
2. Is the match “significant” ? For small motifs a good match is more likely What is probability of a match against a
random protein being this good ? (cf. BLAST)
What’s in it for a CS guy ?
The problem of matching two point-sets has many applications
Most current algorithms geared towards points that are indistinguishable (e.g. points on a mesh)
There are few rigorous results on the significance of matches

So what have we done ?
Towards a more rigorous approach for scoring the quality of a match (between motif and protein)
Provide a method that is capable of finding the optimum match based on these criteria

Problem Description
Given a motif and a protein, for each point in the motif, find a corresponding point in the protein.
Given these correspondences, find the best transformation (rotation and translation only) of the motif that aligns it to the protein.
Optimize over all possible correspondences

Oh, BTW…
Given two sets of k points, easy to find the optimal rotation and translation that minimizes the least sum-of-squared error (also RMSD).
Boils down to finding the largest eigenvalue of a 4x4 matrix.
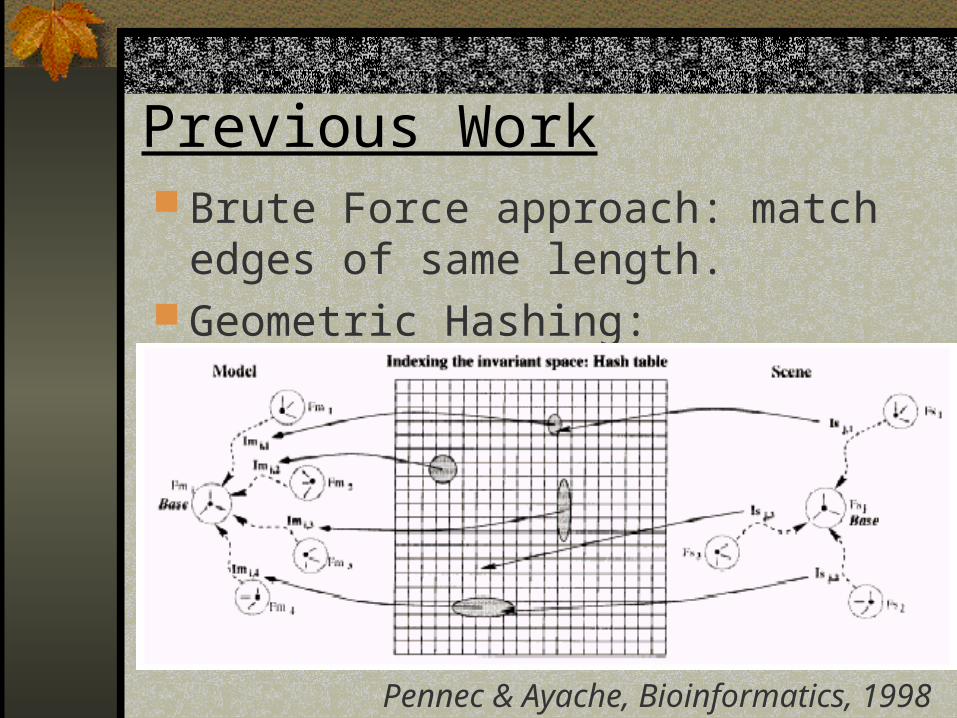
Previous Work Brute Force approach: match edges of
same length. Geometric Hashing:
Pennec & Ayache, Bioinformatics, 1998
What is missing ?
Ad hoc: Try to minimize a quantity that is only indirectly related to the least square error or RMSD.
Hard to evaluate the quality of partial matches Brute Force methods infeasible for larger
motifs Geometric Hashing requires significant
preprocessing
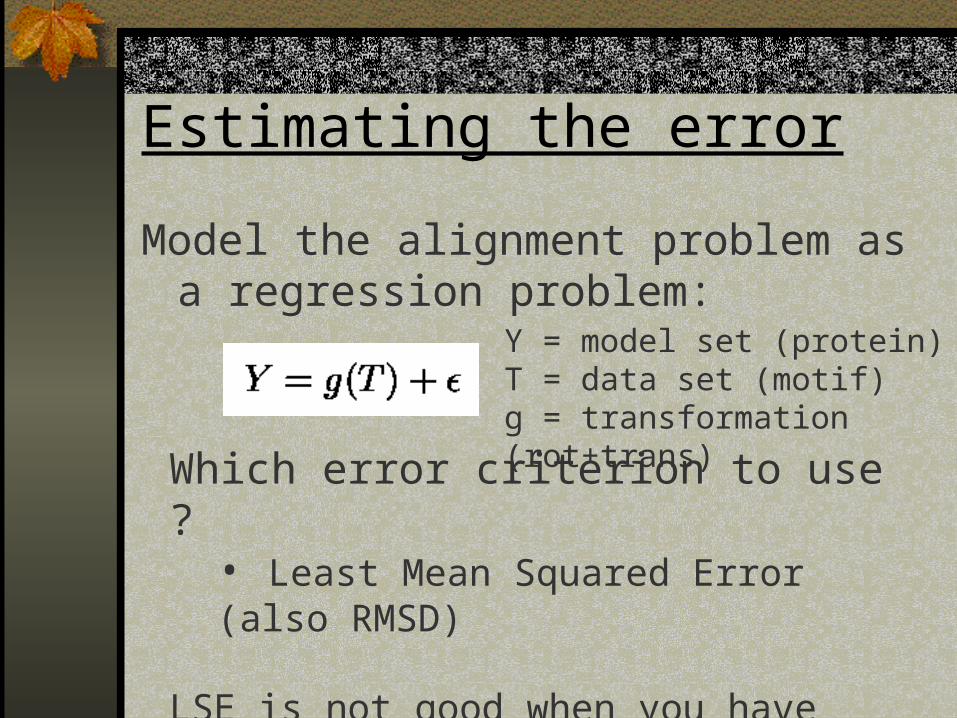
Estimating the error
Model the alignment problem as a regression problem:
Y = model set (protein)T = data set (motif)g = transformation (rot+trans)
Which error criterion to use ?• Least Mean Squared Error (also RMSD)
LSE is not good when you have outliers. what to do ?
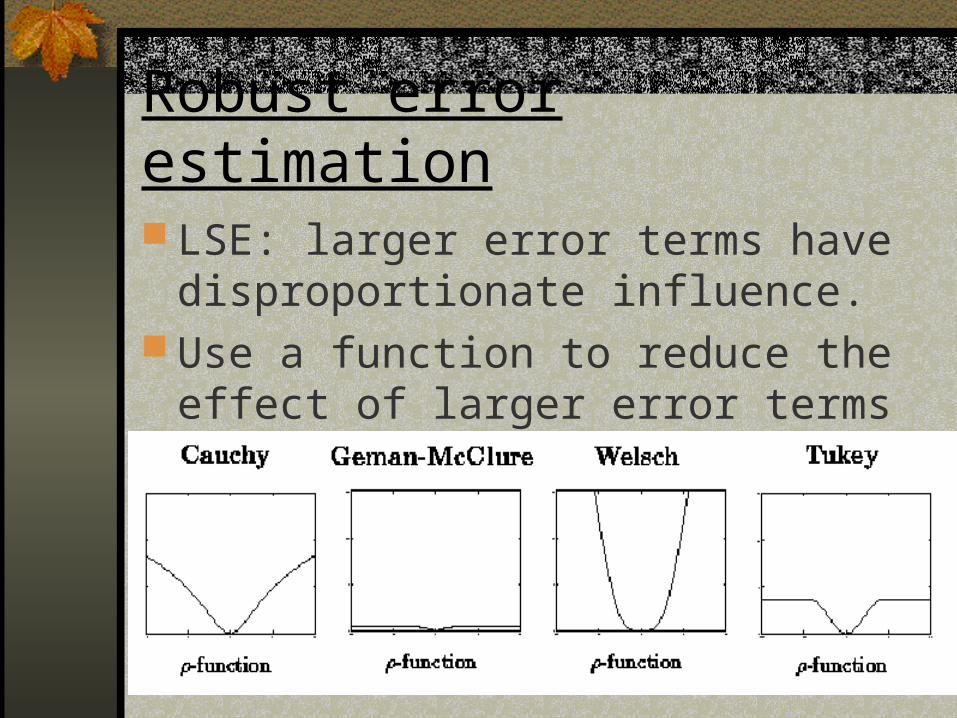
Robust error estimation
LSE: larger error terms have disproportionate influence.
Use a function to reduce the effect of larger error terms (M-estimators)
Its an optimization problem!
Consider the case of full matching:
Domain: set of all possible correspondences between points on the motif and points on the protein
Range: given a particular set of corresponding points, the minimum error in aligning those point sets.
Goal: find the global minimum of this function!
Looking for global minimum
Our approach: Prune the search space to a small and
plausible sub-space Find (most) of the local minima in this
sub-space quickly Choose the minimum over these local
minima
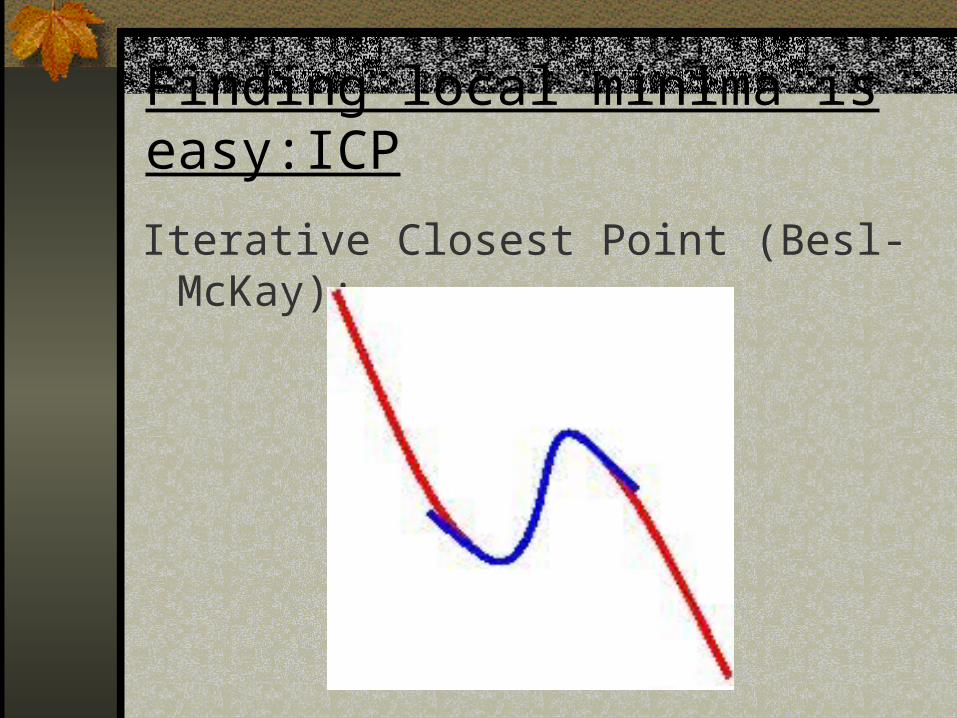
Finding local minima is easy:ICP
Iterative Closest Point (Besl-McKay):

ICP contd…
ICP is guaranteed to converge to a local minimum
But depends a lot on initial seeding Convergence is quick: ~4-5 iterations
ICP movie

Pruning the search space
Every point in motif/protein has some features: Amino acid type, element type, sec. structure,
hydrophobic/polar, ‘substitutable’ Assume: a point with feature X can only
match another point with feature X (or {Y,Z,W})
Assume: some features are more frequent than others

Our Approach
Find the feature that is least frequent in protein.
For each occurrence of the feature: Seed ICP appropriately. Find local
minimum. Look around a few more times
Return the best answer you have
Observations
Will always find a perfect match, if it exists.
Moreover, will find such a match quickly.
The error is directly interpretable in RMSD terms
Does it work ?
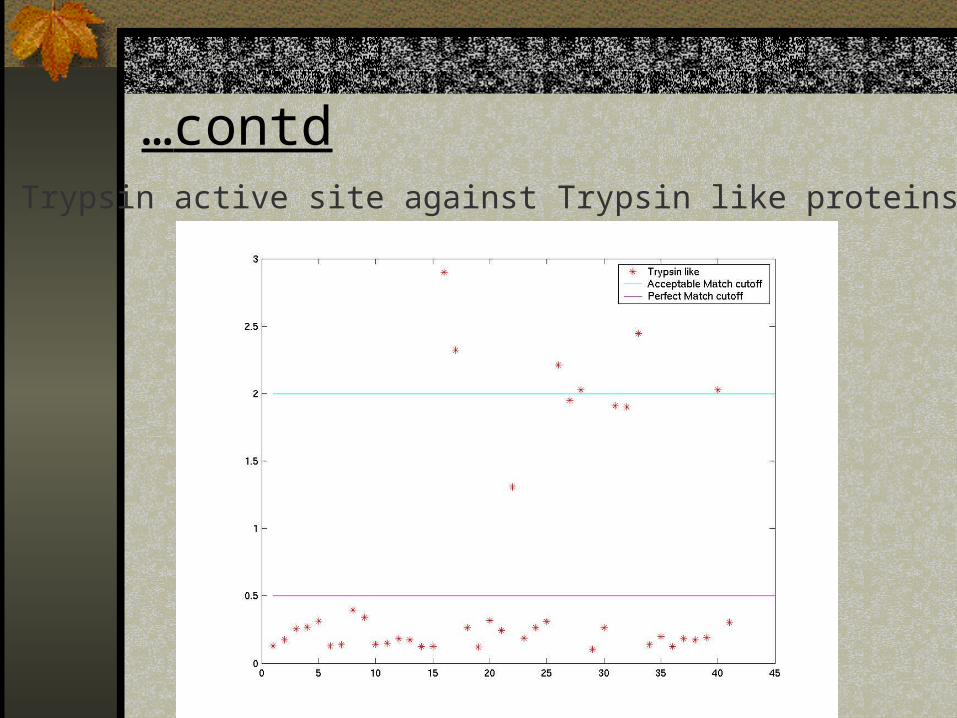
…contdTrypsin active site against Trypsin like proteins
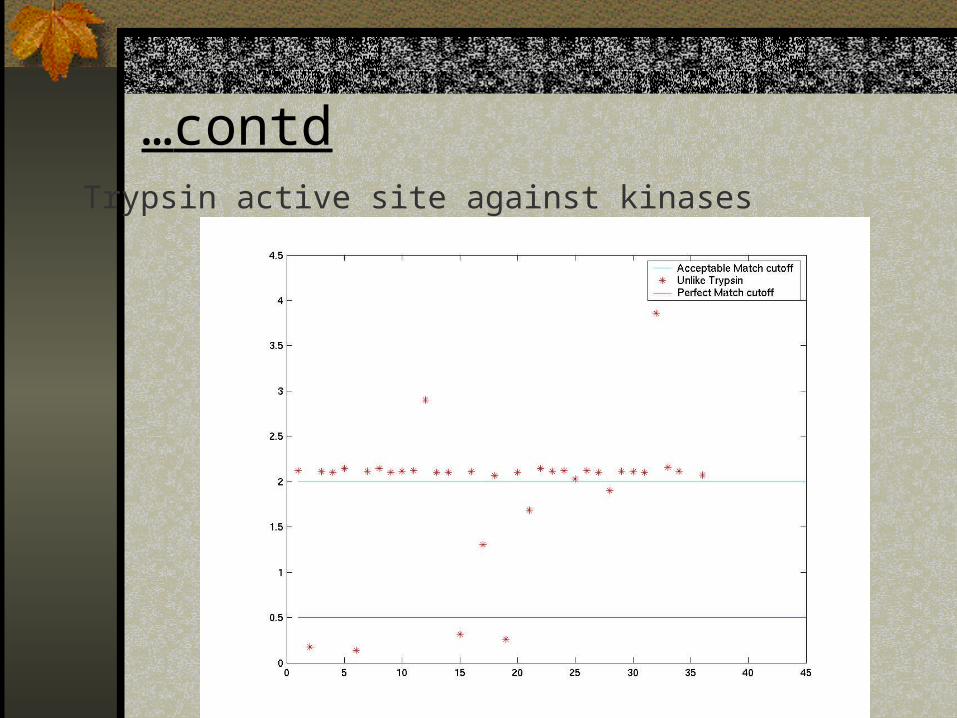
…contdTrypsin active site against kinases
What about partial matching ?
Basic idea is the same: pruning+ICP Replace least squared error estimates by M-
estimator based errors.
Problem: How to find the optimal rotation/translation that minimizes this new variety of error criterion?
Answer: weighted LSE ?
Is there a better way ?
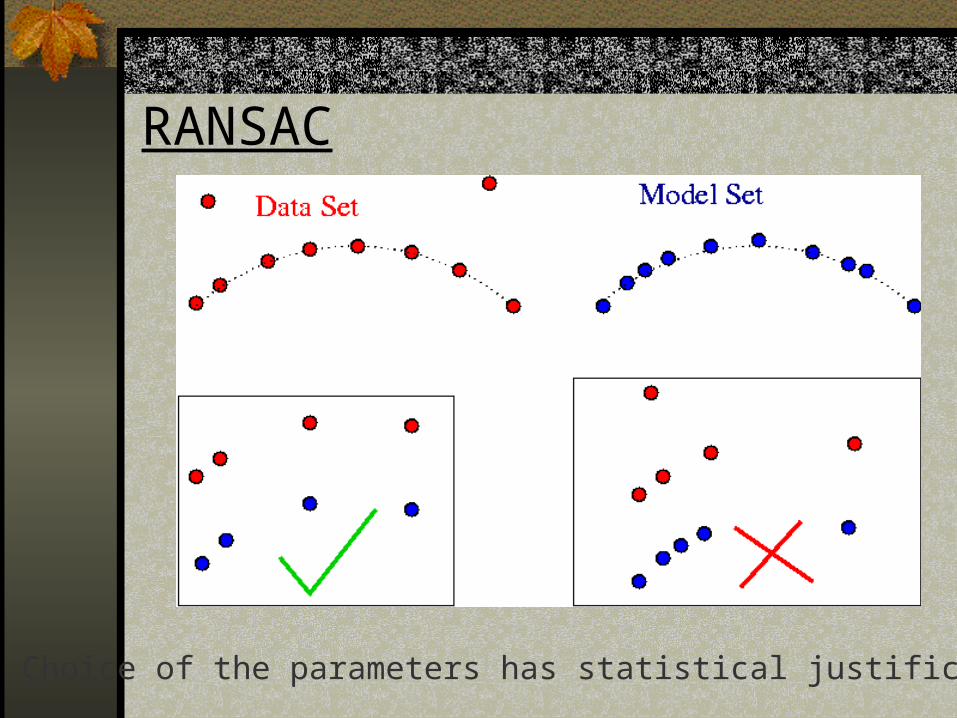
RANSAC
Choice of the parameters has statistical justification

Plain Vanilla (Least Squares):

M-estimator+ weighted LSE

M-estimator + RANSAC

…contdData for distorted trypsin active site against ten
different trypsins:

Future Work
Test on larger motifs: secondary structure elements
Choice of better features A theoretical guarantee about the
quality of results Explore different criteria for partial
matching

Thanks!





























