Embed Size (px)
Citation preview

Rocco, Diomedes A. Logothetis, Jeanne M. Nerbonne and Joseph A. HillBarry London, Weinong Guo, Xiang-hua Pan, Joon S. Lee, Vladimir Shusterman, Christopher J.
and Resistance to Drug-Induced QT ProlongationK,slowISensitive Component of −Targeted Replacement of Kv1.5 in the Mouse Leads to Loss of the 4-Aminopyridine
Print ISSN: 0009-7330. Online ISSN: 1524-4571 Copyright © 2001 American Heart Association, Inc. All rights reserved.is published by the American Heart Association, 7272 Greenville Avenue, Dallas, TX 75231Circulation Research
doi: 10.1161/hh0901.0909292001;88:940-946; originally published online April 27, 2001;Circ Res.
http://circres.ahajournals.org/content/88/9/940World Wide Web at:
The online version of this article, along with updated information and services, is located on the
http://circres.ahajournals.org//subscriptions/
is online at: Circulation Research Information about subscribing to Subscriptions:
http://www.lww.com/reprints Information about reprints can be found online at: Reprints:
document. Permissions and Rights Question and Answer about this process is available in the
located, click Request Permissions in the middle column of the Web page under Services. Further informationEditorial Office. Once the online version of the published article for which permission is being requested is
can be obtained via RightsLink, a service of the Copyright Clearance Center, not theCirculation Researchin Requests for permissions to reproduce figures, tables, or portions of articles originally publishedPermissions:
by guest on February 23, 2013http://circres.ahajournals.org/Downloaded from

Targeted Replacement of Kv1.5 in the Mouse Leads to Lossof the 4-Aminopyridine–Sensitive Component ofI K,slow and
Resistance to Drug-Induced QT ProlongationBarry London, Weinong Guo, Xiang-hua Pan, Joon S. Lee, Vladimir Shusterman,
Christopher J. Rocco, Diomedes A. Logothetis, Jeanne M. Nerbonne, Joseph A. Hill
Abstract—The K1 channel mKv1.5 is thought to encode a 4-aminopyridine (4-AP)–sensitive component of the currentIK,slow in the mouse heart. We used gene targeting to replace mKv1.5 with the 4-AP–insensitive channel rKv1.1 (SWAPmice) and directly test the role of Kv1.5 in the mouse ventricle. Kv1.5 RNA and protein were undetectable, rKv1.1 wasexpressed, and Kv2.1 protein was upregulated in homozygous SWAP hearts. The density of the K1 current IK,slow
(depolarizations to140 mV, pA/pF) was similar in left ventricular myocytes isolated from SWAP homozygotes (1761,n527) and littermate controls (1662, n519). The densities and properties ofIpeak, I to,f, I to,s, andIss were also unchanged.In homozygous SWAP myocytes, the 50-mmol/L 4-AP–sensitive component ofIK,slow was absent (n56), the density ofthe 20-mmol/L tetraethylammonium-sensitive component ofIK,slow was increased (961 versus 561,P,0.05), and no100- to 200-nmol/La-dendrotoxin–sensitive current was found (n58). APD90 in SWAP myocytes was similar tocontrols at baseline but did not prolong in response to 30mmol/L 4-AP. Similarly, QTc (ms) was not prolonged inanesthetized SWAP mice (6462, homozygotes, n59; 6262, controls, n59), and injection with 4-AP prolonged QTconly in controls (6361, homozygotes; 7262, controls;P,0.05). SWAP mice had no increase in arrhythmias duringambulatory telemetry monitoring. Thus, Kv1.5 encodes the 4-AP–sensitive component ofIK,slow in the mouse ventricleand confers sensitivity to 4-AP–induced prolongation of APD and QTc. Compensatory upregulation of Kv2.1 mayexplain the phenotypic differences between SWAP mice and the previously described transgenic mice expressing atruncated dominant-negative Kv1.1 construct.(Circ Res. 2001;88:940-946.)
Key Words: potassium channelsn heart n genetically engineered micen drug-induced long-QT syndromen arrhythmias
Transgenic and gene-targeting technologies have led tomarked advances in the understanding of the molecular
basis of cardiac repolarization in the mouse.1,2 Dominant-negative transgenic mice overexpressing mutated Kv1.x,Kv2.x, Kv4.x, and HERGa subunits have less repolarizingK1 current, varying degrees of cardiac action potentialduration (APD) and QT prolongation, and arrhythmias.3–7 Inthese experiments, the transgene may interact with severalrelated K1 channels in the heart, the phenotype may dependon the details of the transgene design,5,6 and the relationshipof individual gene products to the phenotype may be unclear.Gene targeting of K1 channels using embryonic stem (ES)cells circumvents several of these difficulties by directlyknocking out a single gene product.8–13 However, genetargeting usually leads to loss of the gene in multiple organsand throughout development and is still subject to compen-satory upregulation of other genes. Cross-mating lines of
mice with different mutations provides one mechanism to sortout these interactions.14
We previously reported dominant-negative transgenic micethat overexpress in the heart an N-terminal fragment of the ratbrain K1 channel rKv1.1, have QT prolongation, and lack arapidly activating, slowly inactivating, 4-aminopyridine (4-AP)–sensitive K1 current, IK,slow, in their ventricular myo-cytes.3,15 These mice have both spontaneous and inducibleventricular arrhythmias, attributable at least in part to in-creased dispersion of repolarization and refractoriness be-tween the apex and the base of the heart.16,17 Although thesemice have decreased protein levels of Kv1.5, the transgenemay disrupt other cardiac K1 channels, including Kv1.4,which has been shown to encodeI to,s.9,14Thus, the precise roleof the loss of Kv1.5 in the pathogenesis of the phenotype isuncertain. In addition, nothing is known about the relation-ship of Kv1.5 to Kv2.x, the subunits responsible for the4-AP–resistant component ofIK,slow.4
Original received August 11, 2000; revision received April 3, 2001; accepted April 3, 2001.From the Cardiovascular Institute (B.L., X.-h.P., J.S.L., V.S., C.J.R.), University of Pittsburgh, Pittsburgh, Pa; Washington University School of
Medicine (W.G., J.M.N.), St. Louis, Mo; Mount Sinai School of Medicine (D.A.L.), New York, NY; and University of Iowa College of Medicine andDepartment of Veterans Affairs (J.A.H.), Iowa City, Iowa.
Correspondence to Barry London, MD, PhD, Cardiovascular Institute, University of Pittsburgh, BST 1744, 200 Lothrop St, Pittsburgh, PA 15213.E-mail [email protected]
© 2001 American Heart Association, Inc.
Circulation Researchis available at http://www.circresaha.org
940 by guest on February 23, 2013http://circres.ahajournals.org/Downloaded from

Here we report gene-targeted mice in which mKv1.5 isreplaced by the 4-AP–insensitive channel subunit rKv1.1(SWAP mice). The 4-AP–sensitive component ofIK,slow isabsent in ventricular myocytes isolated from these animals,proving definitively that Kv1.5 underlies this current. Ofnote, totalIK,slow is unchanged at least in part because ofthe upregulation of the tetraethylammonium (TEA)-sensitive component encoded by Kv2.1. As a result, SWAPmice have normal cellular APDs and QT intervals onbaseline electrocardiograms (EKGs) and resistance todrug-induced prolongation of APD and QT intervals afterexposure to 4-AP.
Materials and MethodsAll animal experiments were approved by the Institutional AnimalCare and Use Committees at the University of Pittsburgh orWashington University School of Medicine.
Gene TargetingThe mouse Kv1.5 gene (mKv1.5) was cloned (genomic SV129library, Stratagene), restriction-mapped, and sequenced. A targetingconstruct was engineered using a 2-kb 59 arm consisting of thepromoter and 59-untranslated region (UTR) ofmKv1.5, the rat Kv1.1K1 channel (rKv1.1) tagged with the 9-amino-acid hemagglutinintag (HA) and cloned into the SmaI site located at position26 ofmKv1.5 (relative to the ATG start codon), a neomycin resistancecassette (NeoR), a 3-kb 39arm starting at the XbaI site in the 39-UTRof mKv1.5, and the thymidine kinase gene (TK) for negativeselection (Figure 1A). Homologous recombination with this con-struct should yield rKv1.1 driven by the mKv1.5 promoter, althoughthe effect of the NeoR cassette is unknown, and any 39regulatoryelements may be lost.
Electroporation of ES cells, identification of ES cell lines het-erozygous for the targeted allele, blastocyst injection to obtainchimeras, and mating with C57BL/6 mice to obtain germ-linetransmission and mice heterozygous for the targeted allele (SWAPheterozygotes) were done as previously described.8 Mice werebackcrossed into the C57BL/6 line two additional generations. Maleand female SWAP heterozygotes were then mated to yield the 2- to7-month-old SWAP homozygotes, SWAP heterozygotes, and wild-type littermate controls used in the subsequent experiments. All micewere genotyped using genomic Southern blots.
ElectrophysiologyThe coding region of mKv1.5 was cloned into the high-expressionoocyte vector pGH19K after addition of a Kozak consensus se-quence.18 The HA epitope was added to the 39end of rKv1.1 bypolymerase chain reaction (PCR) (rKv1.1-HA). Channel propertieswere tested inXenopusoocytes injected with in vitro transcribedcRNA (50 ng/oocyte) using the 2-microelectrode voltage-clamptechnique as previously described.19
Single ventricular myocytes were isolated either from the entireleft ventricle or separately from the left ventricular apex and septum,and whole-cell voltage- and current-clamp studies were performed atroom temperature or 35°C using a Dagan 3900A (Dagan Corpora-tion) or an Axopatch-1D (Axon Instruments) patch-clamp amplifierinterfaced to a microcomputer equipped with a Digidata 1200 Seriesanalog-to-digital interface and pClamp 7 software (Axon Instru-ments).9,20 Membrane capacitance and series resistance were com-pensated electronically (.85%). Voltage errors were,6 mV and notcorrected. Only data from cells with input resistances.0.7 GVwereanalyzed.
EKGs and Ambulatory TelemetryThree lead EKGs (leads I, II, and AP) were performed on miceanesthetized with avertin (0.5 mg/kg) using subcutaneous electrodes,a differential amplifier (Warner DP301), and an analog-to-digital
converter (MacLab, ADInstruments). Signals were digitized at 1 kHzand stored on computer. QT interval (ms) was determined as the timeto 95% return to baseline by an individual blinded to genotype andcorrected for heart rate using the formula QTc5QT/(= RR/100).3,21
At least 6 days after implantation of the telemetry device(TA10ETA-F20, Data Sciences), 24-hour ambulatory EKG record-ings were performed digitized at 400 Hz, stored on disk, andanalyzed by hand for arrhythmias and using custom softwaredesigned to determine heart rate and heart rate variability.
Data AnalysisVoltage-clamp data were analyzed using Clampfit 6.0.5 (AxonInstruments), and current densities, normalized for cell capacitance(pA/pF), are reported. The amplitudes of theIK,slow, I to,f, I to,s, and ISS
were determined by fitting the decay phase of the outward K1
currents to the sum of two or three exponentials, as describedpreviously.9,14,20 Correlation coefficients were determined, and thex2 test was used to assess the quality of the fits. The amplitudes ofthe drug-sensitive currents were obtained by offline digital subtrac-tion of the records obtained before and after drug application. Alldata are presented as mean6SEM. Statistical significance wasdetermined using one-way ANOVA followed by Student-Neuman-Keuls test for comparison between groups or Student’st test withcorrection for multiple comparisons, withP,0.05 defined assignificant.
Results
Expression of mKv1.5 and rKv1.1-HA In VitroIn vitro transcribed cRNA from mKv1.5 yielded delayedrectifier currents with minimal inactivation (Figure 1B).Steady-state inactivation curves (generated in 50 mmol/Lrubidium chloride from isochronal tail current measurements)gave a total gating valence (z54.660.1 e2) and a voltage of10% activation (V10%523262 mV) (n56). This gatingvalence is 23% lower and V10% is shifted 20 mV to the rightcompared with rKv1.1 (P,0.01).
Addition of the 9-amino-acid HA epitope to rKv1.1 did notaffect the currents when injected intoXenopusoocytes. Wesubcloned rKv1.1-HA into the prokaryotic/eukaryotic vectorpBK-CMV (Stratagene, California) and transfected the con-struct into COS cells. The HA epitope was easily detectedusing monoclonal antibodies both on Western blots and byimmunofluorescence (data not shown).
Properties of SWAP MiceTwo lines of targeted ES cells were identified (SWAPL77and SWAPL46; Figure 1C). Additional insertion sites of therKv1.1 transgene were excluded by genomic Southern blotusing a probe within the transgene. The targeted band in theSWAPL46 ES line was reproducibly weaker than the nativeband, suggesting that the targeted allele was less abundant.This could result from either a mixed population of ES cellsor a degree of aneuploidy. Both ES lines underwent germ-linetransmission, however, and heterozygotes had one copy ofeach allele in equal abundance. Experimental results wereconfirmed on both lines of mice.
SWAP homozygotes from both lines appeared phenotypi-cally normal, and there was no evidence of increased mortal-ity. Hearts were not hypertrophied, and histology of the heartswas normal (data not shown). No overt neurological pheno-type was evident in the mice.
London et al IK,slow and QT Interval in Kv1.5-Targeted Mice 941
by guest on February 23, 2013http://circres.ahajournals.org/Downloaded from

K 1 Channel Expression in SWAP MiceKv1.5 RNA and protein were absent in hearts from SWAPhomozygotes (Figures 1D and 1E). KV1.1 RNA was detected inSWAP homozygotes by Northern blot (Figure 1D), and reversetranscriptase (RT)-PCR was used to confirm the expression of thetransgene (Figure 1F). Using antibodies against the HA epitope, we
detected a specific band at'62 kDa in the hearts of SWAP micethat was identical in size to the band produced by transfecting ratneonatal cardiac myocytes with rKv1.1-HA (Figure 1E). We didnot detect rKv1.1-HA protein in the brain, although native mKv1.1protein was quite abundant and runs at'72 kDa, as previouslydescribed (data not shown).22
Figure 1. Targeted replacement of mKv1.5 by rKv1.1. A, Schematic representation of the mouse Kv1.5 genomic clone (top) and thetargeting construct (bottom). The coding regions of Kv1.5 and Kv1.1 are labeled, and the heavier black line indicates the untranslatedregions of Kv1.5 (59-UTR and 39-UTR). NEO and TK represent the neomycin resistance cassette and the thymidine kinase cassette,respectively. B, Expression of rKv1.1 and mKv1.5 in Xenopus oocytes; 100-ms depolarizing steps to the voltage indicated were used.C, Genomic Southern blots of BglII-digested DNA from 3 NEO- and TK-resistant cell lines (top left) and from mouse tails (top right). Aschematic diagram showing the expected band sizes and location of the probes is shown below. Probe A (A*) was used for the blotshown. Note that only lines L77 and L46 underwent targeting events and that the targeted allele for the L46 cell line appears less abun-dant than the wild-type allele (see text). Mouse tail DNA for the L77 line comes from an F2 cross of 2 heterozygotes, whereas the tailDNA for the L46 line comes from the F1 and F2 offspring. D, Northern blot analysis with 25 mg/lane of total RNA from the heart andbrain of a wild-type mouse (WT) and a SWAP homozygote (SW) probed with cDNA fragments from the N-terminal part of the Kv1.5coding region (left), the Kv1.1 coding region (middle), and the 39 end of Kv2.1 (right). Arrows point to native Kv1.5 expression and torKv1.1 transgene expression, whereas the arrowhead points to expression of the native mKv1.1 gene. Note that faint expression ofmKv1.1 is present in the whole-heart RNA; this may represent Kv1.1 in cells other than myocytes. Results were repeated using at least3 mice from each line. To correct for loading during quantitative measurements, Kv2.1 signals were measured on a PhosphorImagerand normalized to the signals obtained when the blots were reprobed with a-actin. E, Representative Western blot using a rabbit poly-clonal anti-Kv1.5 antibody (Upstate Biotechnology, New York, NY), an antibody to the HA epitope (anti-HA, Sigma Immunochemicals,St Louis, Mo), and an anti-Kv2.1 antibody (Alomone Laboratories, Jerusalem, Israel) on crude membrane preparations (100 to 200 mgof protein/lane) from hearts and brains of a wild-type mice, SWAP homozygotes, and cultured rat neonatal cardiac myocytes trans-fected with either an empty plasmid (2) or a construct containing rKv1.1-HA driven by the a-myosin heavy chain promoter (1).3,8 Iden-tical results were obtained on hearts from both lines of mice. Arrows indicate the bands specific for Kv1.5, Kv1.1-HA, and Kv2.1,whereas asterisks indicate nonspecific bands. Equal protein loading from tissue samples is confirmed by Coomassie blue–stained gels.F, RT-PCR on cDNA from hearts of wild-type mice, SWAP heterozygotes (SW-Het), and SWAP homozygotes (SW-Hom). PCR was per-formed using a sense primer in the 59-UTR of Kv1.5 and an antisense primer in the coding region of Kv1.1, followed by Southern trans-fer of the products and probing with a radiolabeled oligonucleotide in the 59-UTR of Kv1.5. Parallel reactions were performed in theabsence of RT to exclude genomic contamination. Note that only Kv1.1 expression driven by the transgene would be detected by thistechnique.
942 Circulation Research May 11, 2001
by guest on February 23, 2013http://circres.ahajournals.org/Downloaded from
Kv2.1 RNA expression was not significantly changed inhomozygous SWAP compared with control ventricles(0.9560.14, n53 each; Figure 1D), but Kv2.1 protein wasincreased by Western blot analysis (n53 each; Figure 1E).
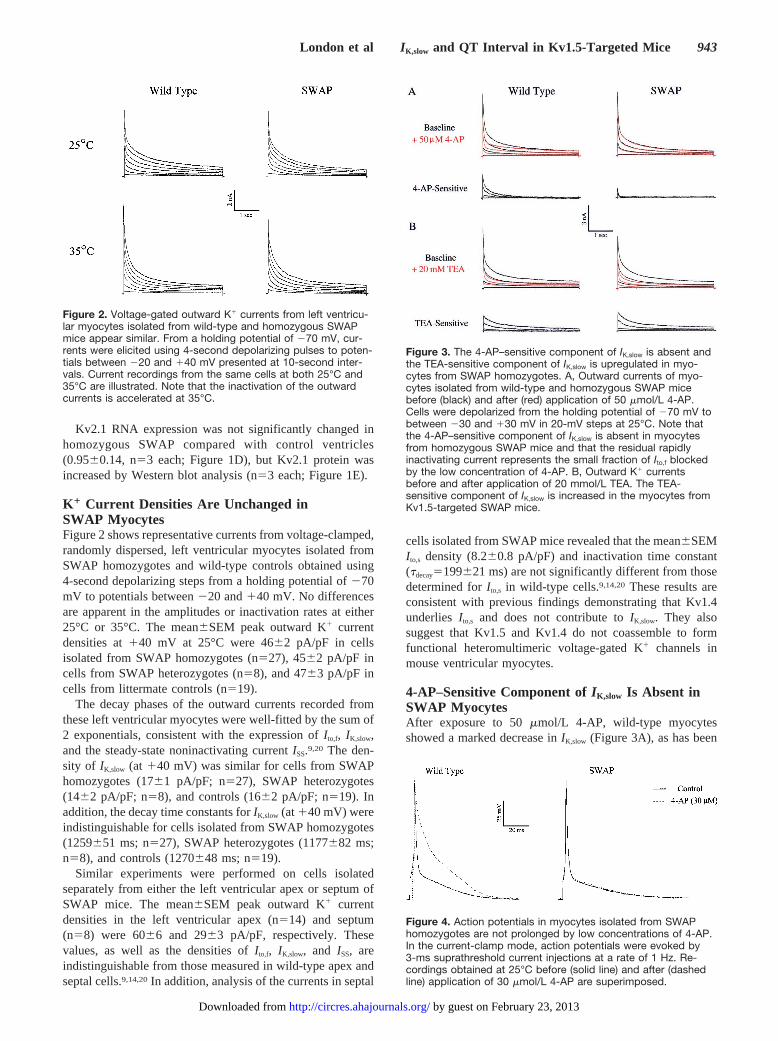
K 1 Current Densities Are Unchanged inSWAP MyocytesFigure 2 shows representative currents from voltage-clamped,randomly dispersed, left ventricular myocytes isolated fromSWAP homozygotes and wild-type controls obtained using4-second depolarizing steps from a holding potential of270mV to potentials between220 and140 mV. No differencesare apparent in the amplitudes or inactivation rates at either25°C or 35°C. The mean6SEM peak outward K1 currentdensities at140 mV at 25°C were 4662 pA/pF in cellsisolated from SWAP homozygotes (n527), 4562 pA/pF incells from SWAP heterozygotes (n58), and 4763 pA/pF incells from littermate controls (n519).
The decay phases of the outward currents recorded fromthese left ventricular myocytes were well-fitted by the sum of2 exponentials, consistent with the expression ofI to,f, IK,slow,and the steady-state noninactivating currentISS.9,20 The den-sity of IK,slow (at 140 mV) was similar for cells from SWAPhomozygotes (1761 pA/pF; n527), SWAP heterozygotes(1462 pA/pF; n58), and controls (1662 pA/pF; n519). Inaddition, the decay time constants forIK,slow (at140 mV) wereindistinguishable for cells isolated from SWAP homozygotes(1259651 ms; n527), SWAP heterozygotes (1177682 ms;n58), and controls (1270648 ms; n519).
Similar experiments were performed on cells isolatedseparately from either the left ventricular apex or septum ofSWAP mice. The mean6SEM peak outward K1 currentdensities in the left ventricular apex (n514) and septum(n58) were 6066 and 2963 pA/pF, respectively. Thesevalues, as well as the densities ofI to,f, IK,slow, and ISS, areindistinguishable from those measured in wild-type apex andseptal cells.9,14,20In addition, analysis of the currents in septal
cells isolated from SWAP mice revealed that the mean6SEMI to,s density (8.260.8 pA/pF) and inactivation time constant(tdecay5199621 ms) are not significantly different from thosedetermined forI to,s in wild-type cells.9,14,20 These results areconsistent with previous findings demonstrating that Kv1.4underlies I to,s and does not contribute toIK,slow. They alsosuggest that Kv1.5 and Kv1.4 do not coassemble to formfunctional heteromultimeric voltage-gated K1 channels inmouse ventricular myocytes.
4-AP–Sensitive Component ofIK,slow Is Absent inSWAP MyocytesAfter exposure to 50mmol/L 4-AP, wild-type myocytesshowed a marked decrease inIK,slow (Figure 3A), as has been
Figure 2. Voltage-gated outward K1 currents from left ventricu-lar myocytes isolated from wild-type and homozygous SWAPmice appear similar. From a holding potential of 270 mV, cur-rents were elicited using 4-second depolarizing pulses to poten-tials between 220 and 140 mV presented at 10-second inter-vals. Current recordings from the same cells at both 25°C and35°C are illustrated. Note that the inactivation of the outwardcurrents is accelerated at 35°C.
Figure 3. The 4-AP–sensitive component of IK,slow is absent andthe TEA-sensitive component of IK,slow is upregulated in myo-cytes from SWAP homozygotes. A, Outward currents of myo-cytes isolated from wild-type and homozygous SWAP micebefore (black) and after (red) application of 50 mmol/L 4-AP.Cells were depolarized from the holding potential of 270 mV tobetween 230 and 130 mV in 20-mV steps at 25°C. Note thatthe 4-AP–sensitive component of IK,slow is absent in myocytesfrom homozygous SWAP mice and that the residual rapidlyinactivating current represents the small fraction of Ito,f blockedby the low concentration of 4-AP. B, Outward K1 currentsbefore and after application of 20 mmol/L TEA. The TEA-sensitive component of IK,slow is increased in the myocytes fromKv1.5-targeted SWAP mice.
Figure 4. Action potentials in myocytes isolated from SWAPhomozygotes are not prolonged by low concentrations of 4-AP.In the current-clamp mode, action potentials were evoked by3-ms suprathreshold current injections at a rate of 1 Hz. Re-cordings obtained at 25°C before (solid line) and after (dashedline) application of 30 mmol/L 4-AP are superimposed.
London et al IK,slow and QT Interval in Kv1.5-Targeted Mice 943
by guest on February 23, 2013http://circres.ahajournals.org/Downloaded from
shown in previous studies.3,4,9,15,20This low concentration of4-AP also blockedI to,f by '20%, an observation also consis-tent with previous studies.20 The 50-mmol/L 4-AP–sensitivecomponent ofIK,slow, however, was completely absent inmyocytes isolated from SWAP homozygotes (n56). Thisprovides strong evidence that Kv1.5 encodes ana subunitnecessary for this current. In contrast, the component ofIK,slow
sensitive to 20 mmol/L TEA (at140 mV; Figure 3B) wasincreased in myocytes from SWAP homozygotes (961 pA/pF, n511) compared with controls (561 pA/pF, n56;P,0.05). At this concentration, TEA partially blocks thecomponent ofIK,slow encoded by Kv2.1 channels.4,20 Togetherwith the Western blot data (Figure 1E), these findings suggestthat functional Kv2.1 channels are upregulated in response tothe loss of Kv1.5.
We were unable to identify any currents in myocytes fromeither wild-type or SWAP mice that were sensitive to 100 to200 nmol/L a-dentrotoxin (DTX, n58). Because Kv1.1 ishighly sensitive to DTX, this suggests that functional Kv1.1channels are not expressed at significant levels in SWAPmyocytes.
Action Potentials in SWAP Myocytes AreInsensitive to 4-APAPD90 values determined at 1-Hz stimulation were similar incurrent-clamped myocytes isolated from SWAP homozy-gotes (3562 ms, n58, at 25°C; 2763 ms, n57, at 35°C) andwild-type controls (3663 ms, n516, at 25°C; 2561 ms,n513; P5NS). As previously reported, wild-type myocytesshow marked APD prolongation in response to low concen-trations of 4-AP (Figure 4).15,20 As predicted from thevoltage-clamp studies, myocytes from SWAP mice wereinsensitive to 30mmol/L 4-AP (n53).
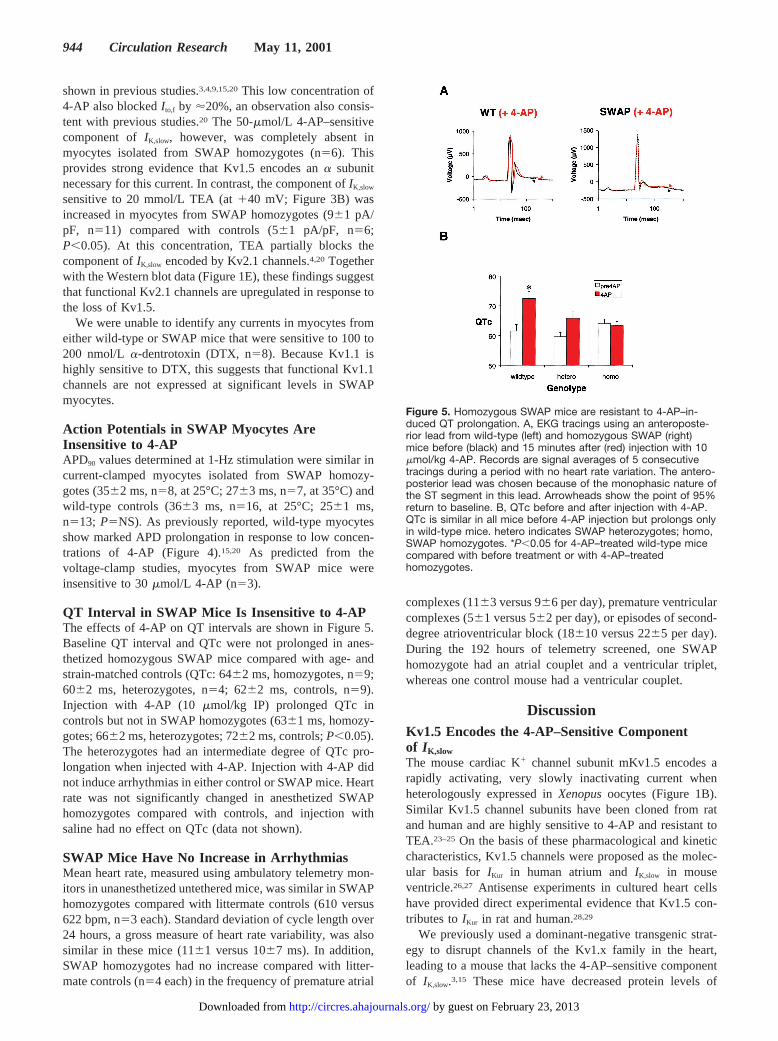
QT Interval in SWAP Mice Is Insensitive to 4-APThe effects of 4-AP on QT intervals are shown in Figure 5.Baseline QT interval and QTc were not prolonged in anes-thetized homozygous SWAP mice compared with age- andstrain-matched controls (QTc: 6462 ms, homozygotes, n59;6062 ms, heterozygotes, n54; 6262 ms, controls, n59).Injection with 4-AP (10 mmol/kg IP) prolonged QTc incontrols but not in SWAP homozygotes (6361 ms, homozy-gotes; 6662 ms, heterozygotes; 7262 ms, controls;P,0.05).The heterozygotes had an intermediate degree of QTc pro-longation when injected with 4-AP. Injection with 4-AP didnot induce arrhythmias in either control or SWAP mice. Heartrate was not significantly changed in anesthetized SWAPhomozygotes compared with controls, and injection withsaline had no effect on QTc (data not shown).
SWAP Mice Have No Increase in ArrhythmiasMean heart rate, measured using ambulatory telemetry mon-itors in unanesthetized untethered mice, was similar in SWAPhomozygotes compared with littermate controls (610 versus622 bpm, n53 each). Standard deviation of cycle length over24 hours, a gross measure of heart rate variability, was alsosimilar in these mice (1161 versus 1067 ms). In addition,SWAP homozygotes had no increase compared with litter-mate controls (n54 each) in the frequency of premature atrial
complexes (1163 versus 966 per day), premature ventricularcomplexes (561 versus 562 per day), or episodes of second-degree atrioventricular block (18610 versus 2265 per day).During the 192 hours of telemetry screened, one SWAPhomozygote had an atrial couplet and a ventricular triplet,whereas one control mouse had a ventricular couplet.
DiscussionKv1.5 Encodes the 4-AP–Sensitive Componentof IK,slow
The mouse cardiac K1 channel subunit mKv1.5 encodes arapidly activating, very slowly inactivating current whenheterologously expressed inXenopusoocytes (Figure 1B).Similar Kv1.5 channel subunits have been cloned from ratand human and are highly sensitive to 4-AP and resistant toTEA.23–25 On the basis of these pharmacological and kineticcharacteristics, Kv1.5 channels were proposed as the molec-ular basis for IKur in human atrium andIK,slow in mouseventricle.26,27 Antisense experiments in cultured heart cellshave provided direct experimental evidence that Kv1.5 con-tributes toIKur in rat and human.28,29
We previously used a dominant-negative transgenic strat-egy to disrupt channels of the Kv1.x family in the heart,leading to a mouse that lacks the 4-AP–sensitive componentof IK,slow.3,15 These mice have decreased protein levels of
Figure 5. Homozygous SWAP mice are resistant to 4-AP–in-duced QT prolongation. A, EKG tracings using an anteroposte-rior lead from wild-type (left) and homozygous SWAP (right)mice before (black) and 15 minutes after (red) injection with 10mmol/kg 4-AP. Records are signal averages of 5 consecutivetracings during a period with no heart rate variation. The antero-posterior lead was chosen because of the monophasic nature ofthe ST segment in this lead. Arrowheads show the point of 95%return to baseline. B, QTc before and after injection with 4-AP.QTc is similar in all mice before 4-AP injection but prolongs onlyin wild-type mice. hetero indicates SWAP heterozygotes; homo,SWAP homozygotes. *P,0.05 for 4-AP–treated wild-type micecompared with before treatment or with 4-AP–treatedhomozygotes.
944 Circulation Research May 11, 2001
by guest on February 23, 2013http://circres.ahajournals.org/Downloaded from

Kv1.5, probably because of increased degradation. The trans-gene does, however, affect other channels, and these experi-ments do not completely prove the relationship betweenmKv1.5 andIK,slow. In the present study, we have used genetargeting to selectively eliminate mKv1.5 and showed theselective loss of the 4-AP–sensitive portion ofIK,slow. Wechose the strategy to replace Kv1.5 with the pharmacologi-cally different channel Kv1.1 with the intention of minimiz-ing other potential changes. The findings presented heredefinitively link Kv1.5 to a component ofIK,slow in the mouseventricle.
Ectopic Expression of Kv1.1 in the Hearts ofSWAP MiceKv1.1 mRNA and protein are expressed in the hearts oftransgenic SWAP mice under the control of the Kv1.5promoter. (Figures 1D through 1F). We were not able todetect any DTX-sensitive currents, however. This suggeststhat very few functional Kv1.1 channels are present on theextracellular membranes of mouse ventricular myocytes andthat the mouse is functionally acting as a Kv1.5 knockout,although we cannot fully exclude the possibility that hetero-multimeric channels containing Kv1.1 are not DTX-sensitive.The smaller size of the rKv1.1 protein expressed in the mouseheart (62 kDa) compared with native mKv1.1 in the brain (72kDa) points to tissue-specific differences in posttranslationalprocessing.22 Recent studies have highlighted the importanceof helper proteins andb subunits in transporting K1 channelsto the surface membrane.30,31Kv1.1 is not normally expressedat high levels in the heart (Figure 1D), and the mechanism toprocess and successfully insert physiological levels of ex-pressed channels into the surface membrane may be absent.
Kv2.1 Upregulation Compensates for the Loss ofKv1.5 in the Hearts of SWAP MiceDespite the loss of the 50mmol/L 4-AP component ofIK,slow
in myocytes isolated from SWAP mice, there was no decreasein the overall density ofIK,slow or of the total outward currentcompared with controls. SWAP myocytes had an increaseddensity of the 20 mmol/L TEA-sensitive component ofIK,slow,and Western blots showed increased Kv2.1 protein in SWAPhearts. Kv2.1a subunits produce slowly activating, slowlyinactivating, or noninactivating K1 currents when expressedheterologously in tissue culture, and antibodies to Kv2.1block currents of this type in hippocampal neurons.32 Previ-ous studies have shown that the TEA-sensitive component ofthe rapidly activating, slowly inactivating cardiac currentIK,slow is selectively eliminated in transgenic mice overexpress-ing the dominant-negative Kv2.1N216Flag construct in theheart.4 Taken together, these data suggest that upregulation ofKv2.1 is one compensatory mechanism for the loss of Kv1.5in the ventricles of the SWAP mice. We cannot be certain thatthe entire compensation inIK,slow is attributable to upregulationof Kv2.1. In addition, the differences in the time- andvoltage-dependent properties of Kv2.1 between tissue culturecells, hippocampal neurons, and cardiac myocytes likelyreflect differences in accessory subunits or posttranslationalprocessing.
Kv2.1 RNA levels are not changed in the SWAP mice,whereas protein levels are increased. The mechanism bywhich the loss of Kv1.5 leads to posttranscriptional upregu-lation of Kv2.1 is unknown. The feedback could be based onthe action potential shape and ionic currents. Alternatively,proteins that bind to the ion channel subunits could directlymitigate subunit processing, transport to the membrane, orstability.
Difference Between Dominant-Negative Transgenicand Gene-Targeted MiceBoth Kv1.x dominant-negative transgenic and Kv1.5 ho-mozygous SWAP mice lack the 4-AP–sensitive componentof IK,slow.3,15 The Kv1.x dominant-negative transgenic micehave QT prolongation and arrhythmias.3,15–17 In this study,we show that targeted mice lackingmKv1.5 have no QTinterval prolongation and no arrhythmias. In fact, these miceare resistant to QT prolongation on exposure to the Kv1.5-blocking agent 4-AP. Likely explanations for the differencesinclude both the effect of the Kv1.x transgene on othercardiac K1 channels known to be important for repolariza-tion, such as Kv1.4,8,9,14 and compensatory regulation ofother K1 channels, such as Kv2.1, in the SWAP mouse.Clearly, loss of Kv1.5 alone is insufficient to lead to a highlyarrhythmogenic phenotype. These findings highlight the factthat transgenic and gene-targeting techniques give differentand complementary information on the role of ion channels incardiac function.
Additional study of both models, along with mating ofdifferent strains to form double mutants, should lead to botha better understanding of the role of individual genes inrepolarization and susceptibility to arrhythmias and to in-sights into the mechanisms by which K1 channel geneexpression is regulated in vivo in the heart.
AcknowledgmentsThese studies were supported in part by National Heart, Lung, andBlood Institute grants R01 HL58030 (to B.L.), R01 34161 (toJ.M.N.), and K08 HL03908 (to J.A.H.); a Grant-in-Aid (to B.L.),Scientist Development Grant (to V.S.), and Postdoctoral ResearchFellowship (to W.G.) from the American Heart Association; andgrants from the Veterans Administration, Procter & Gamble Corpo-ration, and the Roy J. Carver Charitable Trust (to J.A.H.). Thesequence of themKv1.5 genomic clone is available in GenBank(accession No. AF302768). We would like to thank Dr AlexandreF.R. Stewart for his assistance and suggestions.
References1. London B. Use of transgenic and gene-targeted mice to study K1 channel
function in the cardiovascular system. In: Archer SA, Rusch NJ, ed.Potassium Channels in Cardiovascular Biology. New York, NY: PlenumPublishing. In press.
2. Nerbonne JM. Molecular basis of the functional voltage-gated K1 channeldiversity in mammalian myocardium.J Physiol (Lond). 2000;525:285–298.
3. London B, Jeron A, Zhou J, Buckett P, Han X, Mitchell GF, Koren G.Long QT and ventricular arrhythmias in transgenic mice expressing the Nterminus and first transmembrane segment of a voltage-gated potassiumchannel.Proc Natl Acad Sci U S A. 1998;95:2926–2931.
4. Xu H, Barry DM, Li H, Brunet S, Guo W, Nerbonne JM. Attenuation ofthe slow component of the delayed rectification, action potential prolon-gation, and triggered activity in mice expressing a dominant-negative Kv2a subunit.Circ Res. 1999;85:623–633.
London et al IK,slow and QT Interval in Kv1.5-Targeted Mice 945
by guest on February 23, 2013http://circres.ahajournals.org/Downloaded from

5. Barry DM, Xu H, Schussler RB, Nerbonne JM. Functional knockout ofthe transient outward current, long QT syndrome, and cardiac remodelingin mice expressing a dominant-negative Kv4a subunit.Circ Res. 1998;83:560–567.
6. Wickenden AD, Lee P, Sah R, Huang Q, Fishman GE, Backx PH.Targeted expression of a dominant-negative Kv4.2 K1 channel subunit inthe mouse heart.Circ Res. 1999;85:1067–1076.
7. Babij P, Askew GR, Nieuwenhuijsen B, Su CM, Bridal TR, Jow B,Argentieri TM, Kulik J, DeGennaro LJ, Spinelli W, Colatsky TJ. Inhi-bition of cardiac delayed rectifier K1 current by overexpression of thelong-QT syndrome HERG G628S mutation in transgenic mice.Circ Res.1998;83:668–678.
8. London B, Wang DW, Hill JA, Bennett PB. The transient outward currentin mice lacking the potassium channel geneKv1.4. J Physiol (Lond).1998;509:171–182.
9. Guo W, Xu H, London B, Nerbonne JM. Molecular basis of transientoutward current diversity in mouse ventricular myocytes.J Physiol(Lond). 1999;521:587–599.
10. Wickman K, Nemec J, Gendler SJ, Clapham DE. Abnormal heart rateregulation in GIRK4 knockout mice.Neuron. 1998;20:103–114.
11. Drici MD. Arrighi I, Chouabe C, Mann JR. Lazdunski M, Romey G,Barhanin J. Involvement of IsK-associated K1 channel in heart ratecontrol of repolarization in a murine engineered model of Jervell andLange-Nielsen syndrome.Circ Res. 1998;83:95–102.
12. Kupershmidt S, Yang T, Anderson ME, Wessels A, Niswender KD,Magnuson MA, Roden DM. Replacement by homologous recombinationof the minK gene with lacZ reveals restriction of minK expression to themouse cardiac conduction system.Circ Res. 1999;84:146–152.
13. Zaritsky JJ, Schwarz TL. Targeted disruption of the Kir2.1 and Kir2.2genes in mice and the physiological consequences in the heart.Circu-lation. 1999;100(suppl I):I-351. Abstract.
14. Guo W, Li H, London B, Nerbonne JM. Functional elimination ofI to, f andI to, s: early afterdepolarizations, atrioventricular block and ventriculararrhythmias in mice lacking Kv1.4 and expressing a dominant-negativeKv4 a subunit.Circ Res. 2000;87:73–79.
15. Zhou J, Jeron A, London B, Han X, Koren G. Characterization of a slowlyinactivating outward current in adult mouse ventricular myocytes.CircRes. 1998;83:806–814.
16. Jeron A, Mitchell GF, Zhou J, Murata M, London B, Buckett P, WiviottSD, Koren G. Inducible polymorphic ventricular tachycardia in atransgenic mouse model with a long Q-T phenotype.Am J Physiol.2000;278:H1891–H1898.
17. Baker LC, London B, Choi B-R, Koren G, Salama G. Enhanced dis-persion of repolarization and refractoriness in transgenic mouse heartspromotes reentrant ventricular tachycardia.Circ Res. 2000;86:396–407.
18. London B, Trudeau MC, Newton KP, Beyer AK, Copeland NG, GilbertDJ, Jenkins NA, Satler CA, Robertson GA. Two isoforms of the mouse
ether-a-go-go-related gene coassemble to form channels with propertiessimilar to the rapidly activating component of the cardiac delayed rectifierK1 current.Circ Res. 1998;81:870–878.
19. Logothetis DE, Kammen BF, Lindpaintner K, Bisbas D, Nadal-Ginard B.Gating charge differences between two voltage-gated K1 channels aredue to the specific charge content of their respective S4 regions.Neuron.1993;10:1121–1129.
20. Xu H, Guo W, Nerbonne JM. Four kinetically distinct depolarization-ac-tivated K1 currents in adult mouse ventricular myocytes.J Gen Physiol.1999;113:661–678.
21. Mitchell GF, Jeron A, Koren G. Measurement of heart rate and Q-Tinterval in the conscious mouse.Am J Physiol. 1998;274:H747–H751.
22. Wang H, Kunkel DD, Martin TM, Schwartzkroin PA, Temple BL. Het-eromultimeric K1 channels in terminal and juxtranodal regions ofneurons.Nature. 1993;365:75–79.
23. Fedida D, Wible B, Wang Z, Fermini B, Faust F, Nattel S, Brown AM.Identity of a novel delayed rectifier current from human heart with acloned K1 channel current.Circ Res. 1993;73:210–216.
24. Snyders DJ, Tamkun MM, Bennett PB. A rapidly activating and slowlyinactivating potassium channel cloned from human heart: functional anal-ysis after stable mammalian cell culture expression.J Gen Physiol.1993;101:513–543.
25. Matsubara H, Liman ER, Hess P, Koren G. Pretranslational mechanismsdetermine the type of potassium channels expressed in the rat skeletal andcardiac muscles.J Biol Chem. 1991;266:13324–13328.
26. Wang Z, Fermini B, Nattel S. Sustained depolarization-induced outwardcurrent in human atrial myocytes: evidence for a novel delayed rectifierK1 current similar to Kv1.5 cloned channel currents.Circ Res. 1993;73:1061–1076.
27. Fiset C, Clark RB, Larsen TS, Giles WR. A rapidly activating sustainedK1 current modulates repolarization and excitation-contraction couplingin adult mouse ventricle.J Physiol (Lond). 1997;50:557–563.
28. Feng J, Wible B, Li GR, Wang Z, Nattel S. Antisense oligodeoxynucle-otides directed against Kv1.5 mRNA specifically inhibited ultrarapiddelayed rectifier K1 current in cultured human atrial myocytes.Circ Res.1997;80:572–579.
29. Bou-Abboud E, Nerbonne JM. Molecular correlates of the calcium-independent, depolarization-activated K1 currents in rat atrial myocytes.J Physiol (Lond). 1999;517:407–420.
30. Wible BA, Yang Q, Kuryshev YA, Accili EA, Brown AM. Cloning andexpression of a novel K1 channel regulatory protein, KChAP.J BiolChem. 1998;273:11745–11751.
31. Li D, Takimoto K, Levitan ES. Surface expression of Kv1 channels isgoverned by a C-terminal motif.J Biol Chem. 2000;275:11597–11602.
32. Murakoshi H, Trimmer JS. Identification of the Kv2.1 K1 channel as amajor component of the delayed rectifier K1 current in rat hippocampalneurons.J Neurosci. 2000;19:1728–1735.
946 Circulation Research May 11, 2001
by guest on February 23, 2013http://circres.ahajournals.org/Downloaded from





























