Embed Size (px)
Citation preview

Il ruolo del genetista nei programmi di diagnosi prenatale
Dr.ssa Eva Pompilii
GynePro Medical Group
Bologna
U.O. Ostetricia e Ginecologia Policlinico S. Orsola Malpighi Bologna
www.gynepro.it

Consulenza genetica.......
“ il ruolo del genetista clinico è quello di stabilire (o verificare) una diagnosi accurata su cui basare la consulenza e definire prognosi e follow-up, identificare il rischio di trasmettere la malattia ed le modalità con le quali questa può essere prevenuta o controllata. Questo processo è di tipo comunicativo e deve essere in grado di fornire adeguato supporto nella comprensione del significato della malattia genetica e sulle conseguenti decisioni da affrontare” (mod. da Kingston, 1994)
.........è un vero e proprio atto medico

CONSULENZA GENETICA PRENATALE
Positività/alterazione test combinato
Presenza di anomalie all’esame cromosomico eseguito su villi coriali/liquido amniotico
Presenza di malformazioni all’ecografia
Rischio teratogeno in seguito ad assunzione di
farmaci/esposizione a radiazioni ionizzanti

- Il riscontro di una condizione genetica (o non genetica) nel feto non permette generalmente alcuna terapia in utero
- Può, in alcuni casi, essere utile per una migliore gestione del caso in epoca
neonatale
- Nella maggioranza dei casi, quindi, pone la questione dell’interruzione (volontaria / “terapeutica”) della gravidanza (L. 194/78)
Diagnosi prenatale
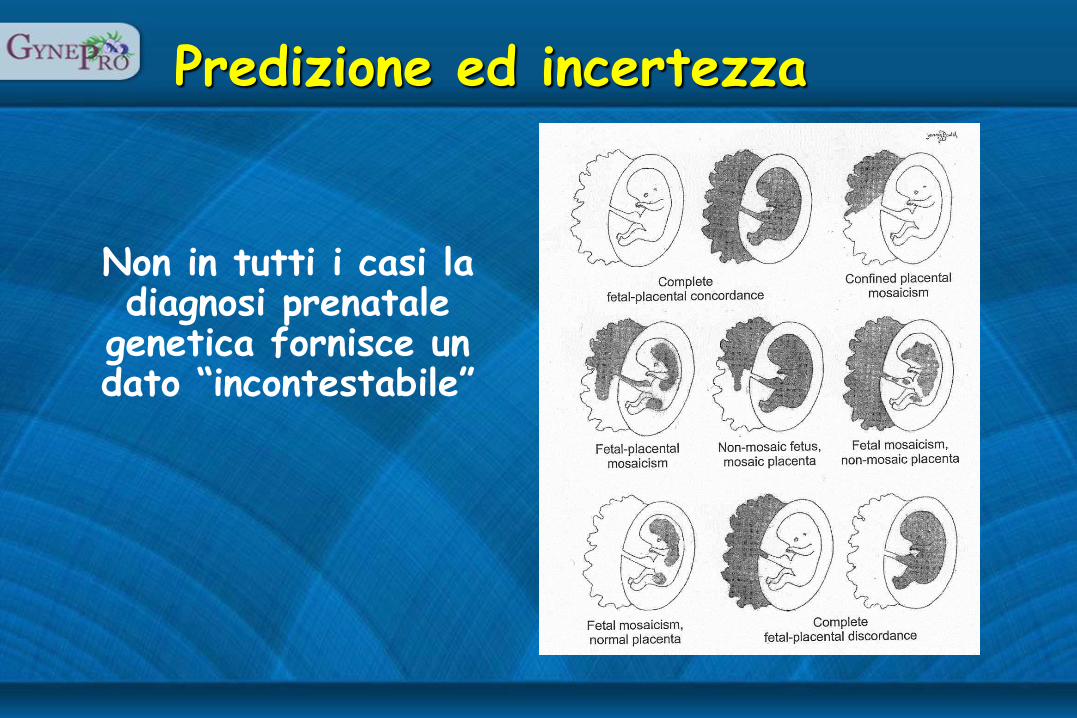
Predizione ed incertezza
Non in tutti i casi la diagnosi prenatale
genetica fornisce un dato “incontestabile”

Predizione ed incertezza
Non in tutti i casi la diagnosi prenatale genetica
“incontestabile” comporta una prognosi “prevedibile” (malattia/non malattia)

Mosaico cromosomico
Donna di 39 anni, si sottopone a villocentesi per rischio età materna
Esito del cariotipo fetale: cariotipo maschile a mosaico costituito da linea cellulare normale e da
linea cellulare primaria (16%)
Esegue Amniocentesi: 47,XY,+21[4]/46,XY[29] (12%)
Esegue Funicolocentesi: 47,XY,+21[6]/46,XY[94]
(6%)
Counselling genetico: rischio Down 6% circa …?

Predizione ed incertezza
Non in tutti i casi la certezza di malattia è percepibile come una
disabilità “insopportabile”

Espressività variabile
Donna di 25 anni, si sottopone a CVS per rischio di ricorrenza di neurofibromatosi di tipo 1 (50%). Il coniuge è affetto: presenta chiazze
caffè-latte multiple, un numero moderato di neurofibromi (8), amartomi iridei.
La diagnosi prenatale conferma la presenza della mutazione paterna.
Prognosi: ?
(50% disturbi di apprendimento; 10% tumori maligni dei nervi periferici; 2% displasia tibiale;
2% glioma ottico sintomatico)
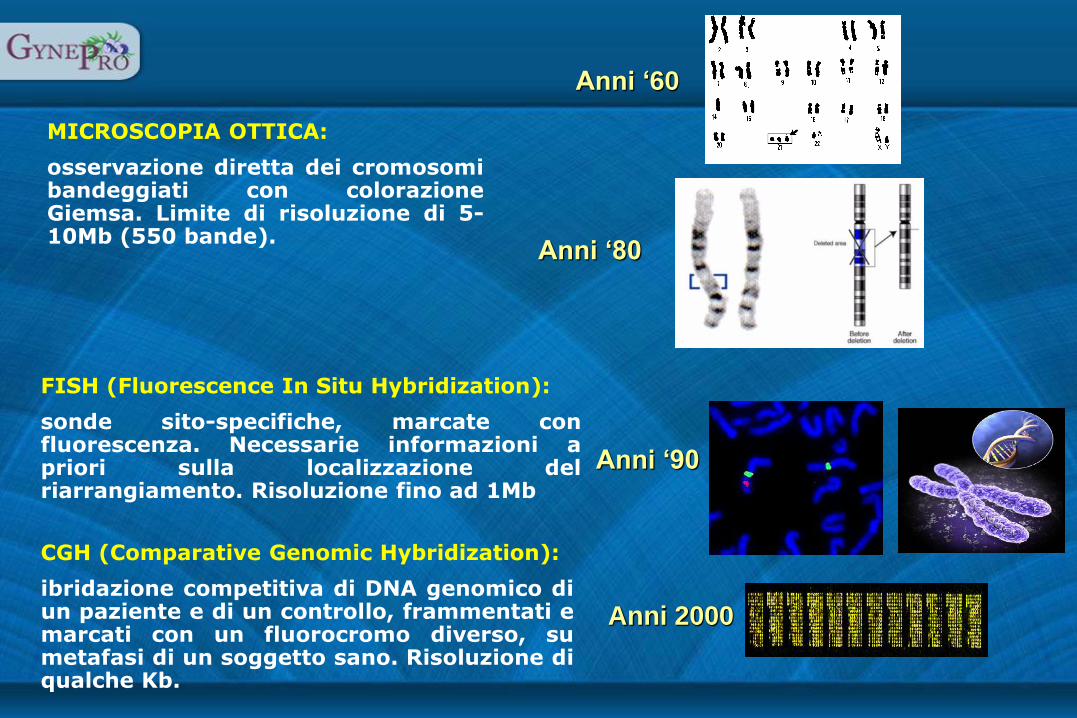
MICROSCOPIA OTTICA:
osservazione diretta dei cromosomi bandeggiati con colorazione Giemsa. Limite di risoluzione di 5-10Mb (550 bande).
FISH (Fluorescence In Situ Hybridization):
sonde sito-specifiche, marcate con fluorescenza. Necessarie informazioni a priori sulla localizzazione del riarrangiamento. Risoluzione fino ad 1Mb
CGH (Comparative Genomic Hybridization):
ibridazione competitiva di DNA genomico di un paziente e di un controllo, frammentati e marcati con un fluorocromo diverso, su metafasi di un soggetto sano. Risoluzione di qualche Kb.
Anni80‘
Anni 2000
Anni90‘
Anni60‘
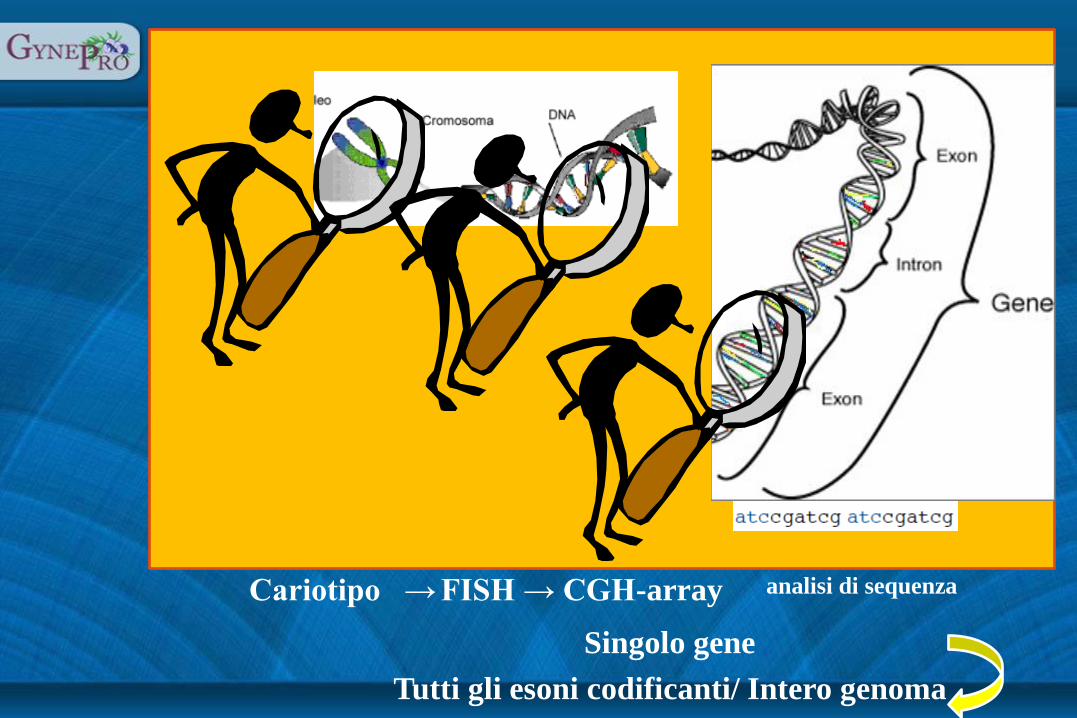
Cariotipo → FISH → CGH-array
Singolo gene
Tutti gli esoni codificanti/ Intero genoma
analisi di sequenza

CARIOTIPO STANDARD
→ Identifica anomalie strutturali bilanciate e sbilanciate
(traslocazioni, inversioni, cromosomi ad anello, isocromosomi, delezioni, duplicazioni)
e anomalie numeriche
ma
→ risoluzione di circa 5-8Mb
E’ POSSIBILE UNA
RISOLUZIONE CITOGENETICA
MAGGIORE?
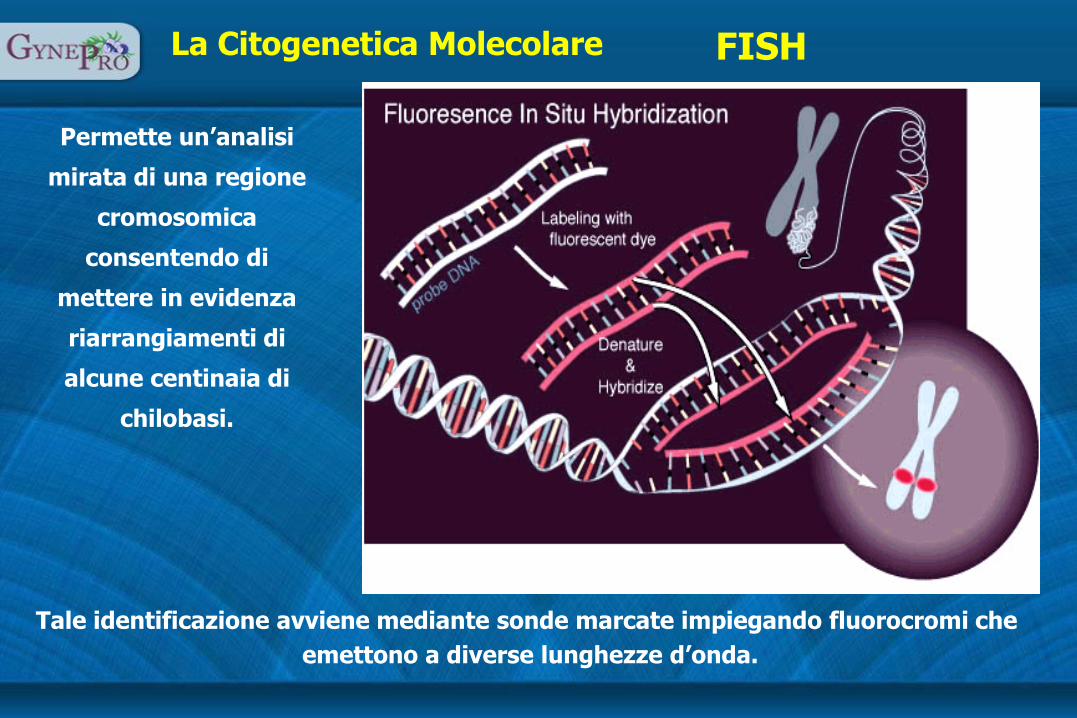
La Citogenetica Molecolare FISH
Permette un’analisi
mirata di una regione
cromosomica
consentendo di
mettere in evidenza
riarrangiamenti di
alcune centinaia di
chilobasi.
Tale identificazione avviene mediante sonde marcate impiegando fluorocromi che
emettono a diverse lunghezze d’onda.

Principali applicazioni diagnostiche della FISH
1. Diagnosi delle sindromi da microdelezione o da microduplicazione
2. Studio delle delezioni subtelomeriche
3. Caratterizzazione di marker cromosomici
4. Caratterizzazione di riarrangiamenti cromosomici, sia bilanciati che sbilanciati
5. Determinazione della percentuale di mosaicismi cromosomici
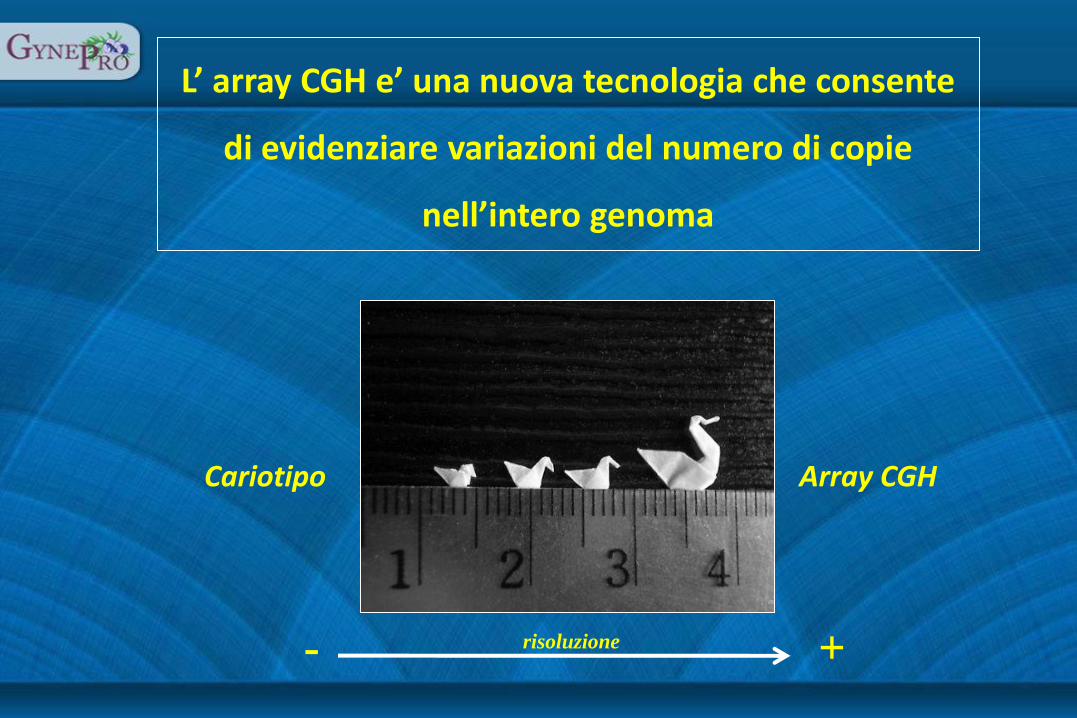
L’ array CGH e’ una nuova tecnologia che consente
di evidenziare variazioni del numero di copie
nell’intero genoma
Cariotipo Array CGH
risoluzione + -

CNV = Copy Number Variants = Copy Number Variation
Segmenti di DNA >1kb presenti nel genoma con un
numero variabile di copie (delezioni/duplicazioni)
Il 5-10% del genoma umano e’variabile nel numero di copie
in individui NORMALI
La risoluzione elevata dell’ array CGH ha determinato una
maggiore identificazione di varianti complicando talvolta
l’interpretazione dei risultati
http://projects.tcag.ca/variation/

La complessità del genoma rende difficile stabilire la natura delle CNVs:
differenze fenotipiche tra gli individui
ruolo evolutivo
patogenesi di malattie, sia mendeliane che
multigeniche
predisposizione ad alcune malattie
BENIGNE
PATOLOGICHE
PREDISPONENTI
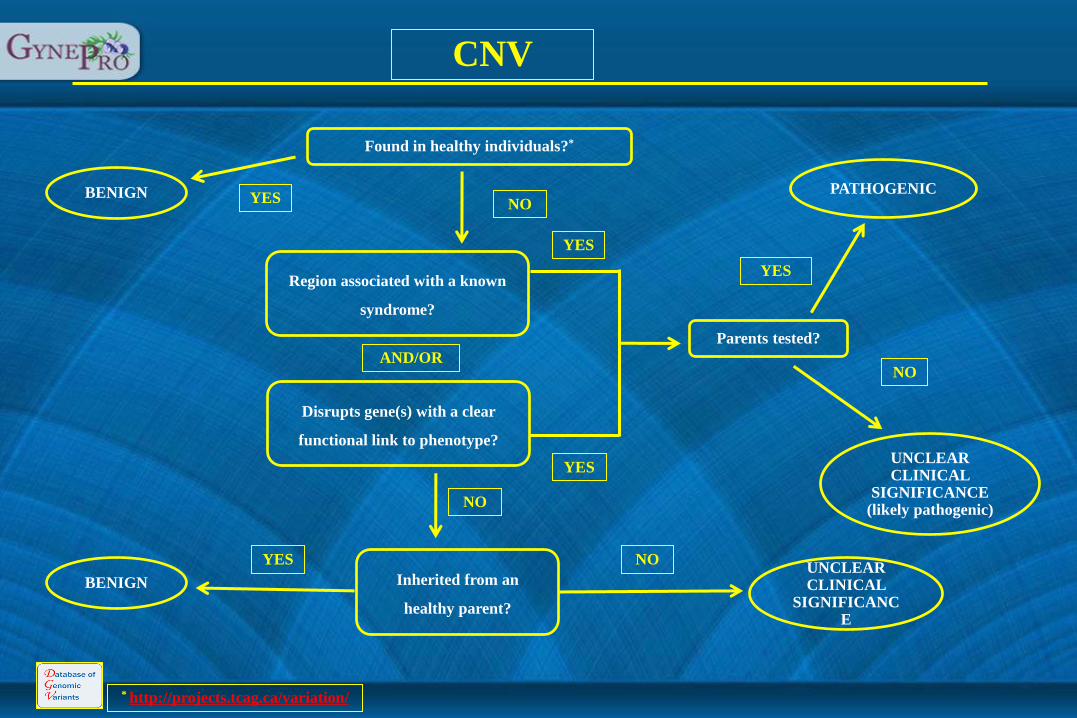
* http://projects.tcag.ca/variation/
Found in healthy individuals?*
Region associated with a known
syndrome?
Disrupts gene(s) with a clear
functional link to phenotype?
Inherited from an
healthy parent?
BENIGN
BENIGN
PATHOGENIC
UNCLEAR CLINICAL
SIGNIFICANCE (likely pathogenic)
UNCLEAR CLINICAL
SIGNIFICANCE
Parents tested?
YES
YES
YES
YES
YES NO
NO
NO
NO
CNV
AND/OR

Novel microdeletion/duplication syndromes:
Rearrangement Size Phenotype
1 del / dup 1q21.1 1.35 Mb
ASSOCIATED with variable pediatric
phenotype (MR, congenital anomalies,
autism)
2 del 1q21.1 500 kb
ASSOCIATED with TAR
(Thrombocytopenia Absent Radius)
syndrome
3 del 15q13.1-13.3 1.5-3.9 Mb MR, dysmorphisms, epilepsy
4 del 15q24.1-q24.3 1.5-3.9 Mb MR, multiple congenital anomalies
5 del / dup 16p13.3 1.65Mb
MR, autism, dysmorphisms, behavioral
problems
(Rubinstein-Taybi region)
6 del/dup 16p11.2 593 kb
ASSOCIATED with ASD, severe early-onset
obesity (incomplete penetrance and
variable phenotype
7 del / dup 17q12 1.45 Mb Diabetes, renal disease, epilepsy
8 del 17q21.31 500 kb MR, congenital anomalies, epilepsy

-I punti di rottura del riarrangiamento interrompono un gene che determina una malattia a
trasmissione autosomica dominante.
-Effetto di posizione.
-Il riarrangiamento è solo apparentemente bilanciato ma nasconde una perdita o una acquisizione di
materiale genomico non visibile al cariotipo standard.
-Problemi legati alla presenza di UPD.
Quali possono essere le cause della presenza di un quadro clinico in un individuo portatore di
un riarrangiamento de novo bilanciato?

Limiti della consulenza in caso di riarrangiamenti bilanciati de novo
Si può dare solo un rischio empirico
Non è possibile definire un fenotipo e conseguentemente una prognosi
Tempi stretti… sono possibili caratterizzazioni ulteriori a livello molecolare dei punti di rottura?
Se necessario offrire la ricerca della Disomia Uniparentale
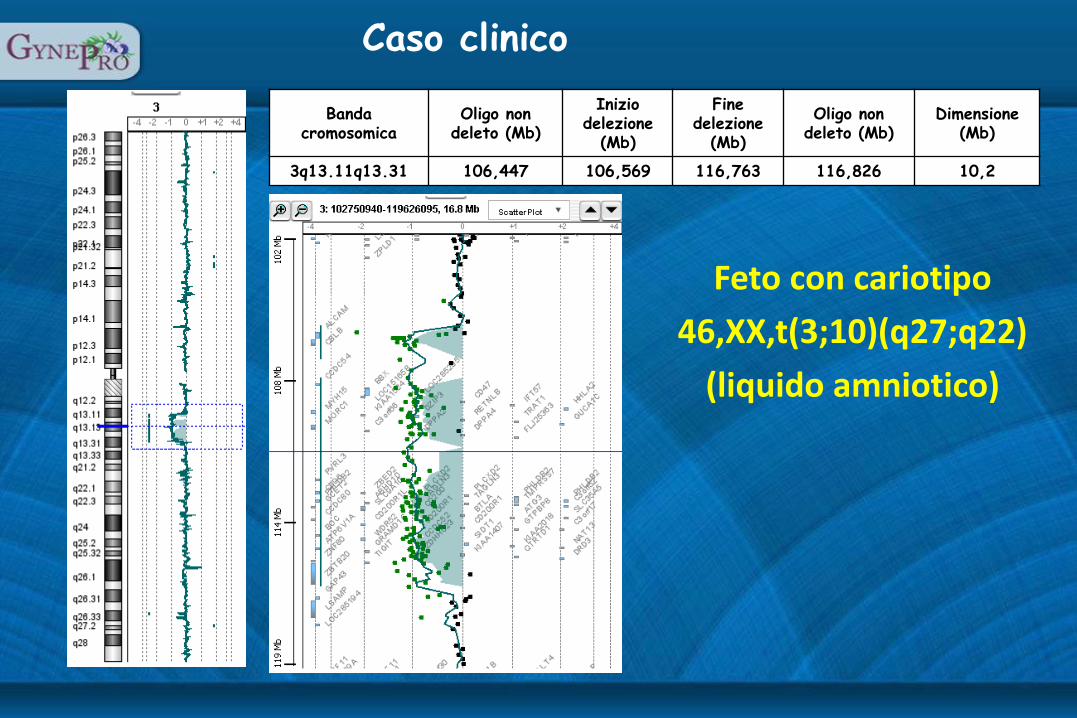
Banda cromosomica
Oligo non deleto (Mb)
Inizio delezione
(Mb)
Fine delezione
(Mb)
Oligo non deleto (Mb)
Dimensione (Mb)
3q13.11q13.31 106,447 106,569 116,763 116,826 10,2
Feto con cariotipo
46,XX,t(3;10)(q27;q22)
(liquido amniotico)
Caso clinico

Donna di 37 anni , PARA 2012
1° grav. Esegue tri test: pos
AF: dup 8 confermata con FISH
46,XY,?dup(8)(p23.2)ish(8)( wcp8+,D8S504+)
Eseguito cariotipo nei coniugi : signora portatrice medesima duplicazione
Bimbo riferito in buona salute
2° grav. AF: dup 8 materna
Bimba riferita in buona salute
3° grav. Aborto spontaneo

4° grav. CVS in altro lab pertanto eseguita conferma cariotipo sulla mamma No duplicazione cr. 8
Eseguito Array-CGH: delezione interstiziale del braccio lungo del cromosoma 7 a livello della banda 7q11.22
Eseguito Array su CVS: confermata delezione
gene AUTS2, variazioni nucleotidiche in tale gene, alcune delle quali riguardanti solo regioni non codificanti, sono associate a disordini dello spettro autistico (ASDs) e disordini neurologici, quali deficit di attenzione, iperattività, epilessia, dislessia, disturbi motori, ritardo del linguaggio, microcefalia e il consumo di alcol.

Donna di 33 anni, in buona salute, primigravida
Non consanguineità con il partner; storia familiare negativa per ritardo mentale, malformazioni congenite, poliabortività.
Villocentesi presso un centro privato senza specifica indicazione:
Riscontro di traslocazione apparentemente bilanciata tra cromosoma 1 e 8, con formula
46,XY,t(1;8)(p36.3;q24.1)
Cariotipo dei genitori nella norma (traslocazione de novo)
Caso clinico

Effettuato array-CGH con risoluzione media di 250 Kb su DNA fetale estratto
dai villi:
Riscontro di una delezione di circa 390Kb nella regione 8q23.3q24.11 (a livello di uno dei breakpoint), contenente un singolo gene
(EIF3H)
Quali implicazioni???

• EIF3H codifica per la subunità H del fattore di inizio traduzione EIF3.
• Ruolo cruciale nel processo di traduzione dell'mRNA in proteina ed è infatti altamente conservato nel corso dell'evoluzione.
• Database: la delezione in questione non è descritta né come polimorfismo né come associata a quadri patologici.
• Pubmed: attribuito un ruolo patogenetico all'over-espressione del gene, ma solamente in associazione ad alcune forme tumorali (cancro colon-
rettale).
• Nessuna segnalazione riguardo la delezione/inattivazione del gene nell'uomo...
• Un solo articolo associa l'inattivazione del gene a problemi nello sviluppo embrionale su un modello animale (zebrafish), ma si tratta di
una loss of function completa, mentre la delezione nel caso in esame è in eterozigosi...

Donna di 43 anni, in buona salute, quartigravida
Non consanguineità con il partner; storia familiare negativa per ritardo mentale, malformazioni congenite, poliabortività.
I grav: nasce una bimba che muore a 6 mesi di vita con diagnosi di RENE POLICISTICO RECESSIVO (25% rischio per
ogni gravidanza)
Test genetico nella bimba: conferma la diagnosi
Confermato stato di portatori nei genitori
II grav: CVS per cariotipo e diagnosi molecolare per Rene Policstico
Bimbo affetto da Rene Policistico Infantile e riscontro di Trisomia 21
III grav: aborto spontaneo
IV grav: in occasione del villo coriale riscontro ecografico di Oloprosencefalia lobare; cvs mostra Trisomia 18 e l’analisi molecolare
mostra un feto affetto da Rene Policistico
Quale potrà essere la percezione del rischio per i signori???

Conclusioni
La diagnosi genetica prenatale:
a. è un percorso individuale (= di coppia)
b. non ammette generalizzazioni
c. necessita di un’appropriata informazione (counselling) pre- e post-test
d. ha implicazioni differenti secondo la categoria di malattie per cui viene richiesta
e. avrà certamente importanti cambiamenti negli anni futuri (più dati da gestire!)

Mente genetica
in un’era genomica
GRAZIE!!!





























