Embed Size (px)
Citation preview

Aalborg Universitet
Improving Solvation Energy Predictions Using The SMD Solvation Method andSemiempirical Electronic Structure Methods
Kromann Jimmy Charnley Svendsen Casper Steinmann Jensen Jan Halborg
Published inJournal of Chemical Physics
DOI (link to publication from Publisher)1026434chemrxiv6708554v110106315047273
Publication date2018
Document VersionAccepted author manuscript peer reviewed version
Link to publication from Aalborg University
Citation for published version (APA)Kromann J C Svendsen C S amp Jensen J H (2018) Improving Solvation Energy Predictions Using TheSMD Solvation Method and Semiempirical Electronic Structure Methods Journal of Chemical Physics 149(10)[104102 ] httpsdoiorg1026434chemrxiv6708554v1 httpsdoiorg10106315047273
General rightsCopyright and moral rights for the publications made accessible in the public portal are retained by the authors andor other copyright ownersand it is a condition of accessing publications that users recognise and abide by the legal requirements associated with these rights
Users may download and print one copy of any publication from the public portal for the purpose of private study or research You may not further distribute the material or use it for any profit-making activity or commercial gain You may freely distribute the URL identifying the publication in the public portal
Take down policyIf you believe that this document breaches copyright please contact us at vbnaubaaudk providing details and we will remove access tothe work immediately and investigate your claim
Improving Solvation Energy Predictions Using The SMD SolvationMethod and Semiempirical Electronic Structure Methods
Jimmy C Kromann1 Casper Steinmann2 and Jan H Jensen1
1Department of Chemistry University of Copenhagen Copenhagen Denmark2Department of Chemistry and Bioscience Aalborg University Aalborg Denmark
The PM6 implementation in the GAMESS program is extended to elements requiring d-integralsand interfaced with the conducter-like polarized continuum model (C-PCM) of solvation in-cluding gradients The accuracy of aqueous solvation energies computed using AM1 PM3PM6 and DFTB and the SMD continuum solvation model is tested using the MNSOL dataset The errors in SMD solvation energies predicted using NDDO-based methods is considerablylarger than when using DFT and HF with RMSE values of 34-59 (neutrals) and 6-15 kcalmol(ions) compared to 24 and ca 5 kcalmol for HF6-31G(d) For the NDDO-based methods theerrors are especially large for cations and considerably higher than the corresponding COSMOresults which suggests that the NDDOSMD results can be improved by re-parameterizing theSMD parameters focusing on ions We found the best results are obtained by changing onlythe radii for hydrogen carbon oxygen nitrogen and sulfur and this leads to RMSE valuesfor PM3 (neutrals 28ions ca 5 kcalmol) PM6 (47ca 5 kcalmol) and DFTB (39ca 5kcalmol) that are more comparable to HF6-31G(d) (24ca 5 kcalmol) Though the radiiare optimized to reproduce aqueous solvation energies they also lead more accurate predictionsfor other polar solvents such as DMSO acetonitrile and methanol while the improvements fornon-polar solvents are negligible
IntroductionAccurate yet computationally efficient models of aqueous solvation represents an impor-tant challenge to molecular modeling Continuum solvation models such as the polarizedcontinuum model (PCM)[1] the conductor-like screening model (COSMO)[2] and theuniversal SMx models[3] offer a computational efficient model of solvation for moleculestreated with electronic structure methods While the accuracy of these and related meth-ods have been studied extensively using DFT and wavefunction-based methods[4 5 67 8 9] there has been relatively little work on corresponding studies with semiempir-ical methods[2 10 11 12 13] In this paper we study the accuracy of using AM1[14]PM3[15] PM6[16] and DFTB[17] together with the SMD[9] continuum solvation modelusing the MNSOL data set[18 19] and present new parameters that increase the accuracyconsiderably for polar solvents We chose the SMD method over methods with system-dependent radii since SMD generally applicable to cases such as transition states wheresuch a parameterization can become ill defined
This manuscript is organized as follows After a presentation of our computationalmethodology we discuss the accuracy of the semiempirical SMD calculations using theradii optimized for ab initio methods We then compare several sets of reoptimized radiiand select the best set We show that the new radii lead to more accurate pKa predictions
1
as well as more accurate solvation free energies for DMSO acetonitrile and methanolFinally we present a summary and an outlook based on the results presented in themanuscript
Computational DetailsFor reference we use the MNSOL[18 19] data set which contains experimental solvationfree energies of neutral and singly-charged molecules with atom-types of H C N O FSi P S Cl Br and I Here we focus mainly on the aqueous solvation subset of thatdata set Because iodine is not supported by 6-31G(d) we removed compounds test2018test4001-4 and test4007-9 from the reference data set Compounds c091 and i091 are rad-icals and are not included because PM6 is only implemented for RHF in GAMESS[20]Our final reference set of molecules then consists of 522 aqueous solvation free energies ofwhich 81 are for anions and 60 are for cations and we refer to this as the MNSOL data set
When parameterizing SMD Coulomb radii we use the a subset of the MNSOL data set(the SMD data set) which was also used by Marenich et al [9] This subset consists of 384solvation free energies of which 59 are for anions and 52 are for cations Following theoriginal SMD implementation we used mono-aquo microsolvated species for small ionswith atoms carrying large (partial) charges The SMD method is re-parameterized usingthe gas-phase structures optimized at the respective levels of theory
The integration of d-integrals for semiempirical methods and interface to the conducter-like polarizable continuum model (C-PCM)[21 22] is implemented in a locally modifiedversion of GAMESS using integral code donated by James Stewart The PM6PCMinterface follows the implementation of Steinmann et al[23] except that we use the semi-numerical gradient approach for the PCM gradient that is also used for the gas phasegradient The DFTB calculations are done using the DFTBPCM interface developed byNishimoto [24] in GAMESS and using version 3ob-3-1 of the 3OB DFTB parameter set[17 25 26 27] All SMD[9] calculations were carried out with the GAMESS programand COSMO[2] calculations were done with MOPAC2016[28]
The original SMD parameterization was done with the Gauss-Bonet[29] tessellation schemewhile the default tessellation scheme in GAMESS is FIXPVA[30] While this differencein tessellation scheme has a negligible effect on the solvation free energies of neutralmolecules it can have a rather larger effect for ions Thus for calculations involvingthe original SMD radii we use the Gauss-Bonet tessellation scheme (mthall=1 in the$tescav input group) while we use the more numerically stable FIXPVA scheme whenoptimizing the radii The original SMD parameterization was also done with integral-equation-formalism PCM (IEF-PCM)[31] while we chose the C-PCM method as it iscomputationally significantly more efficient for semiempirical calculations COSMO isused with AM1 PM3 and PM6 as implemented in the MOPAC program[28] The nullmodel is constructed by setting all the predicted solvation free energies to the average ofall the reference energies and the error bars reflect 95 confidence limits [32 33 34]
The pKa predictions are performed as described by Jensen et al [35] using the same dataset ie a modified version of the one used by Eckert and Klamt [36] Following Jensen
2
et al [35] cefadroxil has been removed due to proton transfer and piroxicam has alsobeen removed since we discovered that the wrong tautomeric form was used
Results and DiscussionTable 1 and Figure 1 shows the computed solvation free energy results obtained usingHF6-31G(d) AM1 PM3 PM6 and DFTB using the implicit solvation models SMDand COSMO using the MNSOL data set described in the Computational Details sectionWe compute the root mean square error (RMSE) mean signed error (MSE) and meanunsigned error (MUE) in order to properly quantify the accuracy of our results
HFSMDAM1SMD
PM3SMDPM6SMD
DFTBSMD
AM1COSMO
PM3COSMO
PM6COSMO null0
2
4
6
8
10
12
14
16
RMSE
[kca
lmol
]
Figure 1 RMSE (in kcalmol) of SMD aqueous solvation energies computed using HF6-31G(d) AM1 PM3 PM6 and DFTB and the original SMD Coulomb radii for the MNSOLdata set Corresponding RMSE values for the COSMO method are included for comparisonThe MNSOL data set is further split into the full set of molecules (black) neutral molecules(green) cations (red) and anions (blue)
We present the combined results (labeled rdquoallrdquo) for all molecules in the data set but alsopresent individual observations on neutral and singly charged (both anions and cations)species
SMD gives RMSE values of 34 56 55 86 kcalmol for HF AM1 PM3 and PM6respectively For ions the RMSE values are 50 and 61 kcalmol for cations and 56 and69 kcalmol for anions for HF and DFTB respectively For AM1 PM3 and PM6 theerror is significantly higher with RMSE values of 114 101 115 kcalmol for cations and74 78 147 kcalmol for anions respectively The NDDO-based method systematicallyunderestimate the solvation free energy of anions with MSE (mean-signed-error) valuesof -51 -54 -126 kcalmol and overestimate the solvation free energy of cations withMSE values of 108 92 109 kcalmol respectively
3
Table 1 RMSE MUE and MSE (in kcalmol) of SMD aqueous solvation energies computedusing HF6-31G(d) AM1 PM3 PM6 and DFTB and the original SMD radii set for theMNSOL data set COSMO solvation energies are included for comparison
SMD COSMO
HF AM1 PM3 PM6 DFTB AM1 PM3 PM6 nullAll
RMSE 34 56 55 86 50 52 48 50 309MUE 21 38 38 62 37 36 34 39 268MSE 03 03 06 -32 -16 00 05 -18 -00Max 178 179 245 302 183 201 213 211 875
NeutralRMSE 24 34 35 59 43 41 36 47 44MUE 13 21 24 40 31 26 25 35 34MSE -09 -01 06 -35 -21 -18 -10 -34 00Max 178 178 245 302 183 201 213 211 158
CationsRMSE 50 113 101 115 61 74 74 73 101MUE 36 108 93 109 48 60 59 60 73MSE 31 108 92 109 45 59 56 59 -00Max 140 179 166 204 161 176 186 183 409
AnionsRMSE 56 74 78 148 69 75 68 45 104MUE 46 65 66 132 58 64 58 38 79MSE 39 -51 -54 -126 -37 45 37 03 -00Max 143 144 168 263 155 148 152 105 300
4
COSMO-predicted solvation free energies have RMSE values for the full set of 52 48and 50 kcalmol for AM1 PM3 and PM6 respectively which are quite similar to thecorresponding SMD values for AM1 and PM3 However the accuracy of COSMO forthe ions are significantly better especially for cations with RMSE values of 73 74 and73 kcalmol for cations and 75 68 45 kcalmol for anions The COSMO results showthat is is possible to significantly improve the accuracy of the NDDOSMD calculationsand the comparatively high errors for ions indicate that the focus should be on the polarpart of the solvation free energy
Optimizing SMD Coulomb RadiiBased on the results in the previous subsection we optimize the Coulomb radii for PM3PM6 and DFTB independently while leaving the non-polar solvation free energy con-tribution unchanged We use the Nelder-Mead simplex method[37] to optimize the radiiusing the SMD data set with the SciPy[38] package using a custom optimization script[39]Optimization of the radii is done by minimizing the RMSE with respect to the radii
Initial tests showed that the solvation free energy does not vary greatly with a changein radii for neutral molecules and the flatness of the energy surface caused problems forthe Nelder-Mead method Furthermore the error for the ions is considerably larger thanfor the neutral molecules Thus only radii of the ions are optimized but when testingthe performance of the new radii neutral molecules are included This means that theoptimum parameters obtained for the ions will not necessarily lead to the lowest possibleerror for neutral molecules We therefore test the effect of optimizing a subset of theradii but include all molecules when testing the overall performance These results arepresented in Tables 2 and 3 and in Figure 2
For PM3 the results are relatively insensitive to the type of atoms chosen with RMSElowast
values of 44 to 47 kcalmol (Table 2 and Figure 2) (Note that the RMSE value arecomputed using different numbers of charged species so the RMSElowast does not necessarilydecrease when more parameters are optimized) Similarly the RMSE values computedusing all the molecules in the MNSOL data set range from 29 to 32 kcalmol as pre-sented in Table 3 The cations are most sensitive to the choice of atom types with anRMSE range of 50 to 62 kcalmol and the HCON- and HCONS-optimized radii set givingthe best and nearly identical accuracy For anions the RMSE for the HCON radii (42kcalmol) is lower than that for HCONS (45 kcalmol) and nearly identical to the lowestRMSE observed (41 kcalmol) The HCON-optimized radii are therefore the best com-promise
For PM6 the RMSElowast for HCO (32 kcalmol) is about 1 kcalmol lower than for the otherradii set However the RMSE values computed using all molecules are very similar witha range of 36 to 38 kcalmol with HCO and HCON tied for first place HCO does lead tothe lowest error for neutral molecules and anions but also an RMSE that is significantlylarger for cations (62 kcalmol) where PM6-HCONS performs best
When testing the PM6-HCON and PM6-HCONS radii on pKa prediction (see below) wefound that several solution-phase geometry optimizations failed because some X-H bonds
5
Table 2 Optimized Coulomb radii (in A) for PM3 PM6 and DFTB for different atom typesand original SMD radii parameters for comparison RMSESMD denotes the RMSE using theoriginal parameters and RMSElowast denotes RMSE after optimization The RMSE is given inkcalmol and is only calculated for the subset of ions in the SMD subset containing thosespecific atom types
H C N O F S Cl Br RMSESMD RMSElowast
SMD120 185 189 152 173 249 238 306
PM3029 172 165 96 44042 175 176 164 109 43062 176 172 162 258 107 46058 175 172 164 179 255 253 335 106 47
PM6066 187 173 123 32017 185 178 174 130 41039 178 182 175 262 127 43070 182 178 174 140 255 237 388 123 44
DFTB070 183 191 164 95 35
broke and the proton started moving into the solvent This indicated that the H radiusmight be too small leading to overpolarization of the H atom due to the solvent andwe tested several larger H radii for PM6-HCONS since the H radius was larger than forPM6-HCON We found that the overpolarization problem disappeared with a H radius of06 A and that using this value has a negligible 01 kcalmol effect of the accuracy ofthe solvation energies (HCONSh in Table 3) Thus we use PM6-HCONSh radii set goingforward
6
PM3PM3HCO
PM3HCON
PM3HCONSPM3all PM6
PM6HCO
PM6HCON
PM6HCONS
PM6HCONShPM6all
DFTB
DFTBHCON0
2
4
6
8
10
12
14
RMSE
[kca
lmol
]
Figure 2 RMSE values (in kcalmol) computed using SMD subset of the MNSOL data setand the Coulomb radii set shown in Table 2 HCO indicates that hydrogen carbon and oxygenradii parameters are changed (ie second and sixth entry in Table 2 The reference set is splitinto full set (black) neutral (green) cations (red) and anions (blue)
7
Table 3 RMSE MUE and MSE values (in kcalmol) computed using the SMD subset of the MNSOL data set and the Coulomb radii set shownin Table 2 HCO indicates that hydrogen carbon and oxygen radii parameters are changed (ie second and sixth row in Table 2)
PM3 PM6 DFTB
SMD HCO HCON HCONS all SMD HCO HCON HCONS HCONSh all SMD HCONAll
RMSE 50 32 29 30 32 75 36 36 38 38 37 41 33MUE 35 23 21 22 23 54 25 27 30 30 28 31 26MSE 10 13 10 10 13 -24 -04 -13 -17 -16 -14 -09 -08Max 168 116 113 126 116 226 184 186 169 169 176 155 109
NeutralRMSE 24 20 19 19 20 40 28 34 36 35 34 31 30MUE 18 15 15 15 15 30 19 24 29 28 25 24 24MSE 11 11 10 11 11 -26 -15 -21 -26 -26 -22 -11 -14Max 87 80 72 74 80 164 184 186 169 169 176 108 109
CationsRMSE 92 62 51 50 62 103 63 43 40 41 41 49 29MUE 85 55 40 39 55 100 58 36 34 35 35 40 24MSE 84 53 32 25 53 100 58 29 30 31 28 36 02Max 150 116 113 119 116 168 138 115 113 113 112 138 91
AnionsRMSE 79 41 42 45 41 142 36 43 46 46 46 67 44MUE 66 32 33 35 32 128 28 32 35 34 32 56 34MSE -61 -11 -13 -07 -11 -126 -06 -12 -14 -12 -13 -39 12Max 168 92 99 126 92 226 88 121 149 147 173 155 104
8
From the results discussed above and presented in Table 2 optimizing for atom typesbeyond HCON showed no significant increase in accuracy for either PM3 or PM6 There-fore the same procedure (optimizing HCON radii) was applied to DFTB For DFTB theRMSE values of the full set of molecules the subset of cation and the subset of anionsgoes from 41 49 and 67 kcalmol to 33 29 and 44 kcalmol respectively
In summary we find that optimizing the SMD radii in combination with either PM3 orPM6 gives appreciable decreases in RMSE accuracy asymp 7 kcalmol in both cases comparedto the reference results We also find that no significant improvement is obtained whenattempting to optimize the radii of other atoms than what was done with HCON subsetof radii For this subset DFTB also saw an increase in accuracy for ions of about asymp 6kcalmol Because of this we found the best choice was change the radii for hydrogencarbon oxygen and nitrogen (HCON) for PM3 and DFTB methods For PM6 we optimizedincluding sulfur and changed the radius of hydrogen to 06 A to remove overpolarizationproblems These methods are denoted as SMDdagger going forward
Comparison with Other Solvation MethodsUsing the new parameters for PM3SMDdagger PM6SMDdagger and DFTBSMDdagger we com-pare the obtained RMSE with other approaches to obtain solvent free energies such asHF6-31G(d)SMD (one of the levels of theory used in the original SMD parameteriza-tion) PM3COSMO and PM6COSMO for the MNSOL data set (Table 4 and Figure 3)
HFSMD
PM3SMDPM6SMD
DFTBSMD
PM3COSMO
PM6COSMO0
2
4
6
8
10
RMSE
[kca
lmol
]
Figure 3 RMSE (in kcalmol) of aqueous solvation energies computed with SMD using HF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicates that the Radiihas been reoptimized Corresponding RMSE values for the COSMO method are included forcomparison The dataset is split into full set (black) neutral (green) cations (red) and anions(blue)
The overall accuracy of PM3SMDdagger is now similar to HFSMD although the RMSEvalue is 04 and 06 kcalmol higher for neutral molecules and cations while the RMSE
9
is 07 kcalmol lower for anions For PM6 the overall RMSE is 14 kcalmol higher thanHF and the RMSE for neutral molecules is 23 kcalmol higher On the other hand theRMSE for cations is only 01 kcalmol higher while the RMSE for anions is 08 kcal-mol lower compared to HF Finally for DFTB the overall RMSE value (41 kcalmol)is in between those for PM3 and PM6 as is the RMSE values for neutral molecules (39kcalmol) But for cations the RMSE (37 kcalmol) is significantly lower than the othermethods while the RMSE for anions (51 kcalmol) is very similar
While the solvation free energies of neutral molecules can generally be measured with anaccuracy of ca 02 kcalmol the corresponding solvation free energies of ions are inferredfrom other experimental values with a typical accuracy about 30 kcalmol[40] Thus theaccuracy of the semiempirical SMDdagger is for all intents and purposes identical to HFSMDfor ions but significantly worse for neutral ions in the case of PM6 and DFTB Thesource of this error is most likely due to a poorer description of the electrostatic proper-ties compared to HF For example the error in dipole moments for HCNO compounds islarger for PM6 than for PM3[16]
Table 4 also lists values for PM3COSMO and PM6COSMO as implemented in MOPACfor comparison This COSMO implementation only evaluates the polar part of the solva-tion free energy and will not be as accurate as properly parameterized COSMO-RS[5] val-ues For PM3 the SMDdagger results are significantly lower than the corresponding COSMOresults while for PM6 there is only a significant improvement for cations
10
Table 4 RMSE MUE and MSE (in kcalmol) of aqueous solvation energies computed withSMD using HF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicatesthat the Radii has been reoptimized Corresponding RMSE values for the COSMO method areincluded for comparison The reference set is split into full set neutral cations and anions
SMD SMDdagger COSMO
HF PM3 PM6 DFTB PM3 PM6All
RMSE 34 36 48 41 48 50MUE 21 26 35 31 34 39MSE 03 10 -17 -11 05 -18Max 178 155 231 160 213 211
NeutralRMSE 24 28 47 39 36 47MUE 13 20 34 29 25 35MSE -09 09 -29 -21 -10 -34Max 178 155 231 160 213 211
CationsRMSE 50 56 51 37 74 73MUE 36 44 42 29 59 60MSE 31 36 38 10 56 59Max 140 127 124 91 186 183
AnionsRMSE 56 49 48 51 68 45MUE 46 37 36 40 58 38MSE 39 -03 -05 18 37 03Max 143 146 147 147 152 105
11
pKa PredictionPart of the impetus for this work were the relatively large error in pKa values predictedusing PM3COSMO compared to PM3SMD with the original SMD radii observed byJensen et al [35] so we use the new SMD radii to compute the pKa value using the samedata set The data is presented in Figure 4 and Table 5 and shows that the RMSE dropsfrom 15plusmn03 to 09plusmn03 pH units for PM3SMDdagger and 20plusmn04 to 16plusmn0304 pH unitsfor DFTBSMDdagger In both cases the improvement is primarily a result of the decreasein mean error (ME) ie the original radii led to a fairly systematic underestimation ofthe pKa values that has now been removed by the reparameterization For DFTB theincreased accuracy is also due to a larger number of low error predictions as is evident inFigure 4 In the case of PM3SMDdagger the accuracy is now very similar to PM3COSMOThe RMSE for PM6SMDdagger is similar to PM6COSMO and larger than for PM3SMDin analogy with the corresponding results for COSMO
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6
err
or
Figure 4 Plot of the errors in the predicted pKa values (pKaminus pKexpa )
12
Table 5 Root-Mean-Square-Error (RMSE) man error (ME) and Pearson correlation (r) for predicted pKa values relative to experiment togetherwith their statistical uncertainty (95 confidence limits)
PM3COSMO PM3SMD PM3SMDdagger PM6COSMO PM6SMDdagger DFTBSMD DFTBSMDdaggerRMSE 10 15 09 18 16 20 1695 conf plusmn 0202 plusmn 0303 plusmn 0202 plusmn 0304 plusmn 0304 plusmn 0404 plusmn 0304ME -05 plusmn 02 -12 plusmn 03 -02 plusmn 02 -13 plusmn 04 -08 plusmn 04 -07 plusmn 05 -01 plusmn 04r 095 093 093 090 086 087 08695 conf plusmn 002004 plusmn 003005 plusmn 003005 plusmn 004007 plusmn 006009 plusmn 005008 plusmn 006009
13
Non-aqueous SolventsThe new radii were optimized explicitly for water so it is not a priori clear whether theyoffer more accurate results for other solvents Table 6 shows the RMSE values for allneutral cations and anions for the other polar solvents in the MNSOL data set acetoni-trile DMSO and methanol for the original SMD radii and the corresponding re-optimizedradii (dagger) In addition corresponding combined RMSE values are given for the remaining88 mostly non-polar solvents in the MNSOL data set for which only data is availablefor neutral molecules It is clear from this data that the new radii sets lowers the errorof the predicted solvation free energies for the other polar solvents and has a negligibleeffect on the accuracy for the non-polar solvents
As for acetonitrile and DMSO the improvement is greatest for the ions where the RMSEdecreases by 3-12 kcalmol The improvement is more modest (05-3 kcalmol) for theneutral molecules For methanol which contains only ions the RMSE is similarly de-creased with 2-13 kcalmol
We thus recommend that the new radii optimized for water also be used for other polarsolvents when doing semiempirical SMD calculations For non-polar solvents either setof radii can be used
14
Table 6 RMSE (in kcalmol) of non-aqueous solvation energies computed with SMD usingHF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicates that theradii has been reoptimized Corresponding RMSE values for the COSMO method are includedfor comparison The reference set is split into full set neutral cations and anions
All Neu Cat Aniacetonitrile
HFSMD 112 29 105 131PM3SMD 194 41 162 246PM3SMDdagger 140 37 113 182PM6SMD 219 33 177 284PM6SMDdagger 130 12 114 160DFTBSMD 150 23 119 197DFTBSMDdagger 109 22 90 140PM3COSMO 130 31 128 147PM6COSMO 137 23 135 155
dimethylsulfoxideHFSMD 119 30 106 126PM3SMD 202 42 153 214PM3SMDdagger 170 35 90 182PM6SMD 255 45 172 272PM6SMDdagger 177 17 99 189DFTBSMD 169 23 108 181DFTBSMDdagger 125 21 76 134PM3COSMO 125 34 107 132PM6COSMO 150 31 119 159
methanolHFSMD 40 00 41 39PM3SMD 126 00 108 135PM3SMDdagger 66 00 60 69PM6SMD 163 00 120 183PM6SMDdagger 52 00 54 52DFTBSMD 84 00 63 94DFTBSMDdagger 47 00 44 49PM3COSMO 59 00 68 54PM6COSMO 54 00 67 46
otherHFSMD 42 42 00 00PM3SMD 51 51 00 00PM3SMDdagger 51 51 00 00PM6SMD 35 35 00 00PM6SMDdagger 34 34 00 00DFTBSMD 30 30 00 00DFTBSMDdagger 29 29 00 00PM3COSMO 36 36 00 00PM6COSMO 27 27 00 00
15
ConclusionThe PM6 method in GAMESS was extended to elements requiring d-integrals and inter-faced with the polarized continuum model (PCM) of solvation including semi-numericalgradients However the accuracy of aqueous solvation energies computed using AM1PM3 PM6 and DFTB and the SMD continuum solvation model was tested using a sub-set of molecules from the MNSOL data set which showed that the errors in SMD solvationenergies predicted using NDDO-based methods was considerably larger than when usingDFT and HF with RMSE values of 50 to 86 kcalmol compared to 34 kcalmol forHF6-31G(d) For the NDDO-based methods the errors were especially large for cationsand in the case of PM6 also for anions with RMSE values of 101 to 148 kcalmol incomparison with to 50 to 57 kcalmol for HF6-31G(d)SMD Corresponding COSMOresults (where the maximum RMSE is 75 kcalmol) suggested that the NDDOSMDresults could be improved by re-parameterizing the SMD Coulomb radii for the NDDOmethods
The fact that the NDDOSMD errors are largest for ions suggest that the problem iswith the polar solvation energy so we focus on optimizing the values of the Coulombradii while leaving all parameters associated with the non-polar solvation free energy un-changed We optimize the radii only for the ionic species on a subset of the MNSOL dataset previously used to parametrize RHF6-31G(d)SMD but include neutral moleculeswhen testing how well SMD with the new Coulomb radii perform We found the bestresults are obtained by changing only the radii for hydrogen carbon oxygen nitrogenand sulfur and this leads to RMSE values for PM3 (neutrals 28ions ca 5 kcalmol)PM6 (47ca 5 kcalmol) and DFTB (39ca 5 kcalmol) that are more comparable toHF6-31G(d) (24ca 5 kcalmol) We note that the SMD parameterization is done us-ing mono-aquo microsolvated species for small ions with atoms carrying large (partial)charges while the MNSOL data set also contains data for the non-microsolvated equiv-alents Comparison to the HF6-31G(d) MSEs reported by Marenich et al [8] reportedfor the selectively microsolvated ions indicates that the inclusion of non-microsolvatedspecies leads an MSE increase of about 1 kcalmol
Though the radii are optimized to reproduce aqueous solvation energies they also leadmore accurate predictions for other polar solvents such as DMSO acetonitrile andmethanol while the improvement for non-polar solvents are negligible
Supplementary MaterialSee supplementary material for Tables S1-S3 and Figures S1-S3 which are referred toin the text In addition we also provide a table of dielectric constants used for thecalculations and a table of aqueous solvation free energies obtained using the optimizedparameters
AcknowledgmentsJCK and JHJ would like to acknowledge Jimmy (Mr MOPAC) Stewart for providingsource code and guidance for the PM6 GAMESS implementation Also thanks to Donald
16
Truhlar and Christopher Cramer for sharing the MNSOL data set and for insightfulcomments on the manuscript CS thanks the Danish Council for Independent Research(the Sapere Aude program) for financial support (Grant no DFF 4181-00370)
References[1] J Tomasi B Mennucci and R Cammi ldquoQuantum mechanical continuum solvation
modelsrdquo Chem Rev vol 105 pp 2999ndash3093 2005
[2] A Klamt and G Schuurmann ldquoCOSMO a new approach to dielectric screeningin solvents with explicit expressions for the screening energy and its gradientrdquo JChem Soc Perkin Trans vol 2 pp 799ndash805 1993
[3] C J Cramer and D G Truhlar ldquoA universal approach to solvation modelingrdquoAccounts of Chemical Research vol 41 no 6 pp 760ndash768 jun 2008 doi101021ar800019z
[4] V Barone M Cossi and J Tomasi ldquoA new definition of cavities for thecomputation of solvation free energies by the polarizable continuum modelrdquo JChem Phys vol 107 no 8 pp 3210ndash3221 aug 1997 doi 1010631474671
[5] F Eckert and A Klamt ldquoFast solvent screening via quantum chemistryCOSMO-RS approachrdquo AIChE J vol 48 no 2 pp 369ndash385 feb 2002 doi101002aic690480220
[6] C Curutchet A Bidon-Chanal I Soteras M Orozco and F J Luque ldquoMSTcontinuum study of the hydration free energies of monovalent ionic speciesrdquo JPhys Chem B vol 109 no 8 pp 3565ndash3574 mar 2005 doi 101021jp047197s
[7] Y Takano and K N Houk ldquoBenchmarking the conductor-like polarizablecontinuum model (CPCM) for aqueous solvation free energies of neutral and ionicorganic moleculesrdquo J Chem Theory Comput vol 1 no 1 pp 70ndash77 jan 2005doi 101021ct049977a
[8] A V Marenich R M Olson C P Kelly C J Cramer and D G TruhlarldquoSelf-consistent reaction field model for aqueous and nonaqueous solutions based onaccurate polarized partial chargesrdquo Journal of Chemical Theory and Computationvol 3 no 6 pp 2011ndash2033 nov 2007 doi 101021ct7001418
[9] A V Marenich C J Cramer and D G Truhlar ldquoUniversal solvation model basedon solute electron density and on a continuum model of the solvent defined by thebulk dielectric constant and atomic surface tensionsrdquo J Phys Chem B vol 113no 18 pp 6378ndash6396 may 2009 doi 101021jp810292n
[10] G E Chudinov D V Napolov and M V Basilevsky ldquoQuantum-chemicalcalculations of the hydration energies of organic cations and anions in the frameworkof a continuum solvent approximationrdquo Chem Phys vol 160 no 1 pp 41 ndash 541992 doi 1010160301-0104(92)87090-V
[11] M J Negre M Orozco and F J Luque ldquoA new strategy for the representation ofenvironment effects in semi-empirical calculations based on dewarrsquos hamiltoniansrdquoChem Phys Lett vol 196 no 1 pp 27ndash36 1992
17
[12] F J Luque M J Negre and M Orozco ldquoAn am1-scrf approach to the study ofchanges in molecular properties induced by solventrdquo J Phys Chem vol 97 no 17pp 4386ndash4391 1993
[13] M Caricato B Mennucci and J Tomasi ldquoSolvent effects on the electronic spectraAn extension of the polarizable continuum model to the zindo methodrdquo J PhysChem A vol 108 no 29 pp 6248ndash6256 2004
[14] M J S Dewar E G Zoebisch E F Healy and J J P Stewart ldquoDevelopmentand use of quantum mechanical molecular models 76 AM1 a new general purposequantum mechanical molecular modelrdquo J A Chem Soc vol 10 pp 3902ndash39091985 doi 101021ja00299a024
[15] J J P Stewart ldquoOptimization of Parameters for Semi-Empirical Methods I-Methodrdquo J Comp Chem vol 10 pp 209ndash220 1989 doi 101002jcc540100208
[16] J J P Stewart ldquoOptimization of parameters for semiempirical methods V Modi-fication of NDDO approximations and application to 70 elementsrdquo J Mol Modelvol 13 pp 1172ndash1213 2007
[17] M Gaus Q Cui and M Elstner ldquoDFTB3 Extension of the Self-Consistent-Charge Density-Functional Tight-Binding Method (SCC-DFTB)rdquo J Chem TheoryComput vol 7 no 4 pp 931ndash948 apr 2011 doi 101021ct100684s
[18] A V Marenich C P Kelly J D Thompson G D Hawkins C C ChambersD J Giesen P P Winget C J Cramer and D G Truhlar ldquoMinnesota solvationdatabaserdquo 2012
[19] ldquoMinnesota solvation databaserdquo accessed November 24th 2017 [Online] Availablehttpscompchemumnedumnsol
[20] M W Schmidt K K Baldridge J A Boatz S T Elbert M S Gordon J HJensen S Koseki N Matsunaga K A Nguyen S Su T L Windus M Dupuisand JAMontgomery ldquoGeneral atomic and molecular electronic structure systemrdquoJ Comput Chem vol 14 pp 1347ndash1363 1993
[21] V Barone and M Cossi ldquoQuantum calculation of molecular energies and energygradients in solution by a conductor solvent modelrdquo The Journal of PhysicalChemistry A vol 102 no 11 pp 1995ndash2001 mar 1998 doi 101021jp9716997
[22] M Cossi N Rega G Scalmani and V Barone ldquoEnergies structures andelectronic properties of molecules in solution with the c-PCM solvation modelrdquoJournal of Computational Chemistry vol 24 no 6 pp 669ndash681 apr 2003 doi101002jcc10189
[23] C Steinmann K L Blaeligdel A S Christensen and J H Jensen ldquoInterface ofthe polarizable continuum model of solvation with semi-empirical methods in thegamess programrdquo PLoS ONE vol 8 no 7 p e67725 07 2013 doi 101371jour-nalpone0067725
[24] Y Nishimoto ldquoDFTBPCM applied to ground and excited state potential energysurfacesrdquo J Phys Chem A vol 120 no 5 pp 771ndash784 feb 2016 doi101021acsjpca5b10732
18
[25] M Gaus X Lu M Elstner and Q Cui ldquoParameterization of DFTB33OB forSulfur and Phosphorus for Chemical and Biological Applicationsrdquo J Chem TheoryComput vol 10 no 4 pp 1518ndash1537 apr 2014 doi 101021ct401002w
[26] X Lu M Gaus M Elstner and Q Cui ldquoParametrization of DFTB33OBfor Magnesium and Zinc for Chemical and Biological Applicationsrdquo The Journalof Physical Chemistry B vol 119 no 3 pp 1062ndash1082 jan 2015 doi101021jp506557r
[27] M Kubillus T Kubar M Gaus J Rezac and M Elstner ldquoParameterization ofthe DFTB3 Method for Br Ca Cl F I K and Na in Organic and BiologicalSystemsrdquo J Chem Theory Comput vol 11 no 1 pp 332ndash342 jan 2015 doi101021ct5009137
[28] J J P Stewart ldquoMOPAC2012rdquo 2016 Stewart Computational Chemistry ColoradoSprings CO USA [Online] Available httpopenmopacnet
[29] J L Pascual-Ahuir E Silla J Tomasi and R Bonaccorsi ldquoElectrostaticinteraction of a solute with a continuum improved description of the cavity and ofthe surface cavity bound charge distributionrdquo Journal of Computational Chemistryvol 8 no 6 pp 778ndash787 sep 1987 doi 101002jcc540080605
[30] P Su and H Li ldquoContinuous and smooth potential energy surface for conductorlikescreening solvation model using fixed points with variable areasrdquo The Journal ofChemical Physics vol 130 no 7 p 074109 feb 2009 doi 10106313077917
[31] J Tomasi B Mennucci and E Cances ldquoThe IEF version of the PCM solvationmethod an overview of a new method addressed to study molecular solutes at theQM ab initio levelrdquo Journal of Molecular Structure THEOCHEM vol 464 no1-3 pp 211ndash226 may 1999 doi 101016s0166-1280(98)00553-3
[32] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 1 the calculation of confidence intervalsrdquo J Comput Aided MolDes vol 28 no 9 pp 887ndash918 Sep 2014 doi 101007s10822-014-9753-z
[33] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 2 comparing methodsrdquo J Comput Aided Mol Des vol 30 no 2pp 103ndash126 Feb 2016 doi 101007s10822-016-9904-5
[34] J H Jensen ldquoWhich method is more accurate or errors have error barsrdquo PeerJPreprints vol 5 p e2693v1 2017 doi 107287peerjpreprints2693v1
[35] J H Jensen C J Swain and L Olsen ldquoPrediction of pKa values for druglikemolecules using semiempirical quantum chemical methodsrdquo The Journal of PhysicalChemistry A vol 121 no 3 pp 699ndash707 jan 2017 doi 101021acsjpca6b10990
[36] F Eckert and A Klamt ldquoAccurate prediction of basicity in aqueous solution withCOSMO-RSrdquo Journal of Computational Chemistry vol 27 no 1 pp 11ndash19 2005doi 101002jcc20309
[37] J A Nelder and R Mead ldquoA simplex method for function minimizationrdquo TheComputer Journal vol 7 no 4 pp 308ndash313 1965 doi 101093comjnl74308
19
[38] E Jones T Oliphant P Peterson et al ldquoSciPy Open source scientifictools for Pythonrdquo 2001 accessed November 24th 2017 [Online] Availablehttpwwwscipyorgrdquo
[39] J Charnley 2017 accessed November 24th 2017 [Online] Available httpsgithubcomcharnleyoptimize gamess parameters
[40] C P Kelly C J Cramer and D G Truhlar ldquoAqueous solvation free energies ofions and ion-water clusters based on an accurate value for the absolute aqueoussolvation free energy of the protonrdquo The Journal of Physical Chemistry B vol 110no 32 pp 16 066ndash16 081 aug 2006 doi 101021jp063552y
20
HFSMDAM1SMD
PM3SMDPM6SMD
DFTBSMD
AM1COSMO
PM3COSMO
PM6COSMO null0
2
4
6
8
10
12
14
16RM
SE [k
calm
ol]
PM3PM3HCO
PM3HCON
PM3HCONSPM3all PM6
PM6HCO
PM6HCON
PM6HCONS
PM6HCONShPM6all
DFTB
DFTBHCON0
2
4
6
8
10
12
14RM
SE [k
calm
ol]
HFSMD
PM3SMDPM6SMD
DFTBSMD
PM3COSMO
PM6COSMO0
2
4
6
8
10RM
SE [k
calm
ol]
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6err
or

Improving Solvation Energy Predictions Using The SMD SolvationMethod and Semiempirical Electronic Structure Methods
Jimmy C Kromann1 Casper Steinmann2 and Jan H Jensen1
1Department of Chemistry University of Copenhagen Copenhagen Denmark2Department of Chemistry and Bioscience Aalborg University Aalborg Denmark
The PM6 implementation in the GAMESS program is extended to elements requiring d-integralsand interfaced with the conducter-like polarized continuum model (C-PCM) of solvation in-cluding gradients The accuracy of aqueous solvation energies computed using AM1 PM3PM6 and DFTB and the SMD continuum solvation model is tested using the MNSOL dataset The errors in SMD solvation energies predicted using NDDO-based methods is considerablylarger than when using DFT and HF with RMSE values of 34-59 (neutrals) and 6-15 kcalmol(ions) compared to 24 and ca 5 kcalmol for HF6-31G(d) For the NDDO-based methods theerrors are especially large for cations and considerably higher than the corresponding COSMOresults which suggests that the NDDOSMD results can be improved by re-parameterizing theSMD parameters focusing on ions We found the best results are obtained by changing onlythe radii for hydrogen carbon oxygen nitrogen and sulfur and this leads to RMSE valuesfor PM3 (neutrals 28ions ca 5 kcalmol) PM6 (47ca 5 kcalmol) and DFTB (39ca 5kcalmol) that are more comparable to HF6-31G(d) (24ca 5 kcalmol) Though the radiiare optimized to reproduce aqueous solvation energies they also lead more accurate predictionsfor other polar solvents such as DMSO acetonitrile and methanol while the improvements fornon-polar solvents are negligible
IntroductionAccurate yet computationally efficient models of aqueous solvation represents an impor-tant challenge to molecular modeling Continuum solvation models such as the polarizedcontinuum model (PCM)[1] the conductor-like screening model (COSMO)[2] and theuniversal SMx models[3] offer a computational efficient model of solvation for moleculestreated with electronic structure methods While the accuracy of these and related meth-ods have been studied extensively using DFT and wavefunction-based methods[4 5 67 8 9] there has been relatively little work on corresponding studies with semiempir-ical methods[2 10 11 12 13] In this paper we study the accuracy of using AM1[14]PM3[15] PM6[16] and DFTB[17] together with the SMD[9] continuum solvation modelusing the MNSOL data set[18 19] and present new parameters that increase the accuracyconsiderably for polar solvents We chose the SMD method over methods with system-dependent radii since SMD generally applicable to cases such as transition states wheresuch a parameterization can become ill defined
This manuscript is organized as follows After a presentation of our computationalmethodology we discuss the accuracy of the semiempirical SMD calculations using theradii optimized for ab initio methods We then compare several sets of reoptimized radiiand select the best set We show that the new radii lead to more accurate pKa predictions
1
as well as more accurate solvation free energies for DMSO acetonitrile and methanolFinally we present a summary and an outlook based on the results presented in themanuscript
Computational DetailsFor reference we use the MNSOL[18 19] data set which contains experimental solvationfree energies of neutral and singly-charged molecules with atom-types of H C N O FSi P S Cl Br and I Here we focus mainly on the aqueous solvation subset of thatdata set Because iodine is not supported by 6-31G(d) we removed compounds test2018test4001-4 and test4007-9 from the reference data set Compounds c091 and i091 are rad-icals and are not included because PM6 is only implemented for RHF in GAMESS[20]Our final reference set of molecules then consists of 522 aqueous solvation free energies ofwhich 81 are for anions and 60 are for cations and we refer to this as the MNSOL data set
When parameterizing SMD Coulomb radii we use the a subset of the MNSOL data set(the SMD data set) which was also used by Marenich et al [9] This subset consists of 384solvation free energies of which 59 are for anions and 52 are for cations Following theoriginal SMD implementation we used mono-aquo microsolvated species for small ionswith atoms carrying large (partial) charges The SMD method is re-parameterized usingthe gas-phase structures optimized at the respective levels of theory
The integration of d-integrals for semiempirical methods and interface to the conducter-like polarizable continuum model (C-PCM)[21 22] is implemented in a locally modifiedversion of GAMESS using integral code donated by James Stewart The PM6PCMinterface follows the implementation of Steinmann et al[23] except that we use the semi-numerical gradient approach for the PCM gradient that is also used for the gas phasegradient The DFTB calculations are done using the DFTBPCM interface developed byNishimoto [24] in GAMESS and using version 3ob-3-1 of the 3OB DFTB parameter set[17 25 26 27] All SMD[9] calculations were carried out with the GAMESS programand COSMO[2] calculations were done with MOPAC2016[28]
The original SMD parameterization was done with the Gauss-Bonet[29] tessellation schemewhile the default tessellation scheme in GAMESS is FIXPVA[30] While this differencein tessellation scheme has a negligible effect on the solvation free energies of neutralmolecules it can have a rather larger effect for ions Thus for calculations involvingthe original SMD radii we use the Gauss-Bonet tessellation scheme (mthall=1 in the$tescav input group) while we use the more numerically stable FIXPVA scheme whenoptimizing the radii The original SMD parameterization was also done with integral-equation-formalism PCM (IEF-PCM)[31] while we chose the C-PCM method as it iscomputationally significantly more efficient for semiempirical calculations COSMO isused with AM1 PM3 and PM6 as implemented in the MOPAC program[28] The nullmodel is constructed by setting all the predicted solvation free energies to the average ofall the reference energies and the error bars reflect 95 confidence limits [32 33 34]
The pKa predictions are performed as described by Jensen et al [35] using the same dataset ie a modified version of the one used by Eckert and Klamt [36] Following Jensen
2
et al [35] cefadroxil has been removed due to proton transfer and piroxicam has alsobeen removed since we discovered that the wrong tautomeric form was used
Results and DiscussionTable 1 and Figure 1 shows the computed solvation free energy results obtained usingHF6-31G(d) AM1 PM3 PM6 and DFTB using the implicit solvation models SMDand COSMO using the MNSOL data set described in the Computational Details sectionWe compute the root mean square error (RMSE) mean signed error (MSE) and meanunsigned error (MUE) in order to properly quantify the accuracy of our results
HFSMDAM1SMD
PM3SMDPM6SMD
DFTBSMD
AM1COSMO
PM3COSMO
PM6COSMO null0
2
4
6
8
10
12
14
16
RMSE
[kca
lmol
]
Figure 1 RMSE (in kcalmol) of SMD aqueous solvation energies computed using HF6-31G(d) AM1 PM3 PM6 and DFTB and the original SMD Coulomb radii for the MNSOLdata set Corresponding RMSE values for the COSMO method are included for comparisonThe MNSOL data set is further split into the full set of molecules (black) neutral molecules(green) cations (red) and anions (blue)
We present the combined results (labeled rdquoallrdquo) for all molecules in the data set but alsopresent individual observations on neutral and singly charged (both anions and cations)species
SMD gives RMSE values of 34 56 55 86 kcalmol for HF AM1 PM3 and PM6respectively For ions the RMSE values are 50 and 61 kcalmol for cations and 56 and69 kcalmol for anions for HF and DFTB respectively For AM1 PM3 and PM6 theerror is significantly higher with RMSE values of 114 101 115 kcalmol for cations and74 78 147 kcalmol for anions respectively The NDDO-based method systematicallyunderestimate the solvation free energy of anions with MSE (mean-signed-error) valuesof -51 -54 -126 kcalmol and overestimate the solvation free energy of cations withMSE values of 108 92 109 kcalmol respectively
3
Table 1 RMSE MUE and MSE (in kcalmol) of SMD aqueous solvation energies computedusing HF6-31G(d) AM1 PM3 PM6 and DFTB and the original SMD radii set for theMNSOL data set COSMO solvation energies are included for comparison
SMD COSMO
HF AM1 PM3 PM6 DFTB AM1 PM3 PM6 nullAll
RMSE 34 56 55 86 50 52 48 50 309MUE 21 38 38 62 37 36 34 39 268MSE 03 03 06 -32 -16 00 05 -18 -00Max 178 179 245 302 183 201 213 211 875
NeutralRMSE 24 34 35 59 43 41 36 47 44MUE 13 21 24 40 31 26 25 35 34MSE -09 -01 06 -35 -21 -18 -10 -34 00Max 178 178 245 302 183 201 213 211 158
CationsRMSE 50 113 101 115 61 74 74 73 101MUE 36 108 93 109 48 60 59 60 73MSE 31 108 92 109 45 59 56 59 -00Max 140 179 166 204 161 176 186 183 409
AnionsRMSE 56 74 78 148 69 75 68 45 104MUE 46 65 66 132 58 64 58 38 79MSE 39 -51 -54 -126 -37 45 37 03 -00Max 143 144 168 263 155 148 152 105 300
4
COSMO-predicted solvation free energies have RMSE values for the full set of 52 48and 50 kcalmol for AM1 PM3 and PM6 respectively which are quite similar to thecorresponding SMD values for AM1 and PM3 However the accuracy of COSMO forthe ions are significantly better especially for cations with RMSE values of 73 74 and73 kcalmol for cations and 75 68 45 kcalmol for anions The COSMO results showthat is is possible to significantly improve the accuracy of the NDDOSMD calculationsand the comparatively high errors for ions indicate that the focus should be on the polarpart of the solvation free energy
Optimizing SMD Coulomb RadiiBased on the results in the previous subsection we optimize the Coulomb radii for PM3PM6 and DFTB independently while leaving the non-polar solvation free energy con-tribution unchanged We use the Nelder-Mead simplex method[37] to optimize the radiiusing the SMD data set with the SciPy[38] package using a custom optimization script[39]Optimization of the radii is done by minimizing the RMSE with respect to the radii
Initial tests showed that the solvation free energy does not vary greatly with a changein radii for neutral molecules and the flatness of the energy surface caused problems forthe Nelder-Mead method Furthermore the error for the ions is considerably larger thanfor the neutral molecules Thus only radii of the ions are optimized but when testingthe performance of the new radii neutral molecules are included This means that theoptimum parameters obtained for the ions will not necessarily lead to the lowest possibleerror for neutral molecules We therefore test the effect of optimizing a subset of theradii but include all molecules when testing the overall performance These results arepresented in Tables 2 and 3 and in Figure 2
For PM3 the results are relatively insensitive to the type of atoms chosen with RMSElowast
values of 44 to 47 kcalmol (Table 2 and Figure 2) (Note that the RMSE value arecomputed using different numbers of charged species so the RMSElowast does not necessarilydecrease when more parameters are optimized) Similarly the RMSE values computedusing all the molecules in the MNSOL data set range from 29 to 32 kcalmol as pre-sented in Table 3 The cations are most sensitive to the choice of atom types with anRMSE range of 50 to 62 kcalmol and the HCON- and HCONS-optimized radii set givingthe best and nearly identical accuracy For anions the RMSE for the HCON radii (42kcalmol) is lower than that for HCONS (45 kcalmol) and nearly identical to the lowestRMSE observed (41 kcalmol) The HCON-optimized radii are therefore the best com-promise
For PM6 the RMSElowast for HCO (32 kcalmol) is about 1 kcalmol lower than for the otherradii set However the RMSE values computed using all molecules are very similar witha range of 36 to 38 kcalmol with HCO and HCON tied for first place HCO does lead tothe lowest error for neutral molecules and anions but also an RMSE that is significantlylarger for cations (62 kcalmol) where PM6-HCONS performs best
When testing the PM6-HCON and PM6-HCONS radii on pKa prediction (see below) wefound that several solution-phase geometry optimizations failed because some X-H bonds
5
Table 2 Optimized Coulomb radii (in A) for PM3 PM6 and DFTB for different atom typesand original SMD radii parameters for comparison RMSESMD denotes the RMSE using theoriginal parameters and RMSElowast denotes RMSE after optimization The RMSE is given inkcalmol and is only calculated for the subset of ions in the SMD subset containing thosespecific atom types
H C N O F S Cl Br RMSESMD RMSElowast
SMD120 185 189 152 173 249 238 306
PM3029 172 165 96 44042 175 176 164 109 43062 176 172 162 258 107 46058 175 172 164 179 255 253 335 106 47
PM6066 187 173 123 32017 185 178 174 130 41039 178 182 175 262 127 43070 182 178 174 140 255 237 388 123 44
DFTB070 183 191 164 95 35
broke and the proton started moving into the solvent This indicated that the H radiusmight be too small leading to overpolarization of the H atom due to the solvent andwe tested several larger H radii for PM6-HCONS since the H radius was larger than forPM6-HCON We found that the overpolarization problem disappeared with a H radius of06 A and that using this value has a negligible 01 kcalmol effect of the accuracy ofthe solvation energies (HCONSh in Table 3) Thus we use PM6-HCONSh radii set goingforward
6
PM3PM3HCO
PM3HCON
PM3HCONSPM3all PM6
PM6HCO
PM6HCON
PM6HCONS
PM6HCONShPM6all
DFTB
DFTBHCON0
2
4
6
8
10
12
14
RMSE
[kca
lmol
]
Figure 2 RMSE values (in kcalmol) computed using SMD subset of the MNSOL data setand the Coulomb radii set shown in Table 2 HCO indicates that hydrogen carbon and oxygenradii parameters are changed (ie second and sixth entry in Table 2 The reference set is splitinto full set (black) neutral (green) cations (red) and anions (blue)
7
Table 3 RMSE MUE and MSE values (in kcalmol) computed using the SMD subset of the MNSOL data set and the Coulomb radii set shownin Table 2 HCO indicates that hydrogen carbon and oxygen radii parameters are changed (ie second and sixth row in Table 2)
PM3 PM6 DFTB
SMD HCO HCON HCONS all SMD HCO HCON HCONS HCONSh all SMD HCONAll
RMSE 50 32 29 30 32 75 36 36 38 38 37 41 33MUE 35 23 21 22 23 54 25 27 30 30 28 31 26MSE 10 13 10 10 13 -24 -04 -13 -17 -16 -14 -09 -08Max 168 116 113 126 116 226 184 186 169 169 176 155 109
NeutralRMSE 24 20 19 19 20 40 28 34 36 35 34 31 30MUE 18 15 15 15 15 30 19 24 29 28 25 24 24MSE 11 11 10 11 11 -26 -15 -21 -26 -26 -22 -11 -14Max 87 80 72 74 80 164 184 186 169 169 176 108 109
CationsRMSE 92 62 51 50 62 103 63 43 40 41 41 49 29MUE 85 55 40 39 55 100 58 36 34 35 35 40 24MSE 84 53 32 25 53 100 58 29 30 31 28 36 02Max 150 116 113 119 116 168 138 115 113 113 112 138 91
AnionsRMSE 79 41 42 45 41 142 36 43 46 46 46 67 44MUE 66 32 33 35 32 128 28 32 35 34 32 56 34MSE -61 -11 -13 -07 -11 -126 -06 -12 -14 -12 -13 -39 12Max 168 92 99 126 92 226 88 121 149 147 173 155 104
8
From the results discussed above and presented in Table 2 optimizing for atom typesbeyond HCON showed no significant increase in accuracy for either PM3 or PM6 There-fore the same procedure (optimizing HCON radii) was applied to DFTB For DFTB theRMSE values of the full set of molecules the subset of cation and the subset of anionsgoes from 41 49 and 67 kcalmol to 33 29 and 44 kcalmol respectively
In summary we find that optimizing the SMD radii in combination with either PM3 orPM6 gives appreciable decreases in RMSE accuracy asymp 7 kcalmol in both cases comparedto the reference results We also find that no significant improvement is obtained whenattempting to optimize the radii of other atoms than what was done with HCON subsetof radii For this subset DFTB also saw an increase in accuracy for ions of about asymp 6kcalmol Because of this we found the best choice was change the radii for hydrogencarbon oxygen and nitrogen (HCON) for PM3 and DFTB methods For PM6 we optimizedincluding sulfur and changed the radius of hydrogen to 06 A to remove overpolarizationproblems These methods are denoted as SMDdagger going forward
Comparison with Other Solvation MethodsUsing the new parameters for PM3SMDdagger PM6SMDdagger and DFTBSMDdagger we com-pare the obtained RMSE with other approaches to obtain solvent free energies such asHF6-31G(d)SMD (one of the levels of theory used in the original SMD parameteriza-tion) PM3COSMO and PM6COSMO for the MNSOL data set (Table 4 and Figure 3)
HFSMD
PM3SMDPM6SMD
DFTBSMD
PM3COSMO
PM6COSMO0
2
4
6
8
10
RMSE
[kca
lmol
]
Figure 3 RMSE (in kcalmol) of aqueous solvation energies computed with SMD using HF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicates that the Radiihas been reoptimized Corresponding RMSE values for the COSMO method are included forcomparison The dataset is split into full set (black) neutral (green) cations (red) and anions(blue)
The overall accuracy of PM3SMDdagger is now similar to HFSMD although the RMSEvalue is 04 and 06 kcalmol higher for neutral molecules and cations while the RMSE
9
is 07 kcalmol lower for anions For PM6 the overall RMSE is 14 kcalmol higher thanHF and the RMSE for neutral molecules is 23 kcalmol higher On the other hand theRMSE for cations is only 01 kcalmol higher while the RMSE for anions is 08 kcal-mol lower compared to HF Finally for DFTB the overall RMSE value (41 kcalmol)is in between those for PM3 and PM6 as is the RMSE values for neutral molecules (39kcalmol) But for cations the RMSE (37 kcalmol) is significantly lower than the othermethods while the RMSE for anions (51 kcalmol) is very similar
While the solvation free energies of neutral molecules can generally be measured with anaccuracy of ca 02 kcalmol the corresponding solvation free energies of ions are inferredfrom other experimental values with a typical accuracy about 30 kcalmol[40] Thus theaccuracy of the semiempirical SMDdagger is for all intents and purposes identical to HFSMDfor ions but significantly worse for neutral ions in the case of PM6 and DFTB Thesource of this error is most likely due to a poorer description of the electrostatic proper-ties compared to HF For example the error in dipole moments for HCNO compounds islarger for PM6 than for PM3[16]
Table 4 also lists values for PM3COSMO and PM6COSMO as implemented in MOPACfor comparison This COSMO implementation only evaluates the polar part of the solva-tion free energy and will not be as accurate as properly parameterized COSMO-RS[5] val-ues For PM3 the SMDdagger results are significantly lower than the corresponding COSMOresults while for PM6 there is only a significant improvement for cations
10
Table 4 RMSE MUE and MSE (in kcalmol) of aqueous solvation energies computed withSMD using HF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicatesthat the Radii has been reoptimized Corresponding RMSE values for the COSMO method areincluded for comparison The reference set is split into full set neutral cations and anions
SMD SMDdagger COSMO
HF PM3 PM6 DFTB PM3 PM6All
RMSE 34 36 48 41 48 50MUE 21 26 35 31 34 39MSE 03 10 -17 -11 05 -18Max 178 155 231 160 213 211
NeutralRMSE 24 28 47 39 36 47MUE 13 20 34 29 25 35MSE -09 09 -29 -21 -10 -34Max 178 155 231 160 213 211
CationsRMSE 50 56 51 37 74 73MUE 36 44 42 29 59 60MSE 31 36 38 10 56 59Max 140 127 124 91 186 183
AnionsRMSE 56 49 48 51 68 45MUE 46 37 36 40 58 38MSE 39 -03 -05 18 37 03Max 143 146 147 147 152 105
11
pKa PredictionPart of the impetus for this work were the relatively large error in pKa values predictedusing PM3COSMO compared to PM3SMD with the original SMD radii observed byJensen et al [35] so we use the new SMD radii to compute the pKa value using the samedata set The data is presented in Figure 4 and Table 5 and shows that the RMSE dropsfrom 15plusmn03 to 09plusmn03 pH units for PM3SMDdagger and 20plusmn04 to 16plusmn0304 pH unitsfor DFTBSMDdagger In both cases the improvement is primarily a result of the decreasein mean error (ME) ie the original radii led to a fairly systematic underestimation ofthe pKa values that has now been removed by the reparameterization For DFTB theincreased accuracy is also due to a larger number of low error predictions as is evident inFigure 4 In the case of PM3SMDdagger the accuracy is now very similar to PM3COSMOThe RMSE for PM6SMDdagger is similar to PM6COSMO and larger than for PM3SMDin analogy with the corresponding results for COSMO
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6
err
or
Figure 4 Plot of the errors in the predicted pKa values (pKaminus pKexpa )
12
Table 5 Root-Mean-Square-Error (RMSE) man error (ME) and Pearson correlation (r) for predicted pKa values relative to experiment togetherwith their statistical uncertainty (95 confidence limits)
PM3COSMO PM3SMD PM3SMDdagger PM6COSMO PM6SMDdagger DFTBSMD DFTBSMDdaggerRMSE 10 15 09 18 16 20 1695 conf plusmn 0202 plusmn 0303 plusmn 0202 plusmn 0304 plusmn 0304 plusmn 0404 plusmn 0304ME -05 plusmn 02 -12 plusmn 03 -02 plusmn 02 -13 plusmn 04 -08 plusmn 04 -07 plusmn 05 -01 plusmn 04r 095 093 093 090 086 087 08695 conf plusmn 002004 plusmn 003005 plusmn 003005 plusmn 004007 plusmn 006009 plusmn 005008 plusmn 006009
13
Non-aqueous SolventsThe new radii were optimized explicitly for water so it is not a priori clear whether theyoffer more accurate results for other solvents Table 6 shows the RMSE values for allneutral cations and anions for the other polar solvents in the MNSOL data set acetoni-trile DMSO and methanol for the original SMD radii and the corresponding re-optimizedradii (dagger) In addition corresponding combined RMSE values are given for the remaining88 mostly non-polar solvents in the MNSOL data set for which only data is availablefor neutral molecules It is clear from this data that the new radii sets lowers the errorof the predicted solvation free energies for the other polar solvents and has a negligibleeffect on the accuracy for the non-polar solvents
As for acetonitrile and DMSO the improvement is greatest for the ions where the RMSEdecreases by 3-12 kcalmol The improvement is more modest (05-3 kcalmol) for theneutral molecules For methanol which contains only ions the RMSE is similarly de-creased with 2-13 kcalmol
We thus recommend that the new radii optimized for water also be used for other polarsolvents when doing semiempirical SMD calculations For non-polar solvents either setof radii can be used
14
Table 6 RMSE (in kcalmol) of non-aqueous solvation energies computed with SMD usingHF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicates that theradii has been reoptimized Corresponding RMSE values for the COSMO method are includedfor comparison The reference set is split into full set neutral cations and anions
All Neu Cat Aniacetonitrile
HFSMD 112 29 105 131PM3SMD 194 41 162 246PM3SMDdagger 140 37 113 182PM6SMD 219 33 177 284PM6SMDdagger 130 12 114 160DFTBSMD 150 23 119 197DFTBSMDdagger 109 22 90 140PM3COSMO 130 31 128 147PM6COSMO 137 23 135 155
dimethylsulfoxideHFSMD 119 30 106 126PM3SMD 202 42 153 214PM3SMDdagger 170 35 90 182PM6SMD 255 45 172 272PM6SMDdagger 177 17 99 189DFTBSMD 169 23 108 181DFTBSMDdagger 125 21 76 134PM3COSMO 125 34 107 132PM6COSMO 150 31 119 159
methanolHFSMD 40 00 41 39PM3SMD 126 00 108 135PM3SMDdagger 66 00 60 69PM6SMD 163 00 120 183PM6SMDdagger 52 00 54 52DFTBSMD 84 00 63 94DFTBSMDdagger 47 00 44 49PM3COSMO 59 00 68 54PM6COSMO 54 00 67 46
otherHFSMD 42 42 00 00PM3SMD 51 51 00 00PM3SMDdagger 51 51 00 00PM6SMD 35 35 00 00PM6SMDdagger 34 34 00 00DFTBSMD 30 30 00 00DFTBSMDdagger 29 29 00 00PM3COSMO 36 36 00 00PM6COSMO 27 27 00 00
15
ConclusionThe PM6 method in GAMESS was extended to elements requiring d-integrals and inter-faced with the polarized continuum model (PCM) of solvation including semi-numericalgradients However the accuracy of aqueous solvation energies computed using AM1PM3 PM6 and DFTB and the SMD continuum solvation model was tested using a sub-set of molecules from the MNSOL data set which showed that the errors in SMD solvationenergies predicted using NDDO-based methods was considerably larger than when usingDFT and HF with RMSE values of 50 to 86 kcalmol compared to 34 kcalmol forHF6-31G(d) For the NDDO-based methods the errors were especially large for cationsand in the case of PM6 also for anions with RMSE values of 101 to 148 kcalmol incomparison with to 50 to 57 kcalmol for HF6-31G(d)SMD Corresponding COSMOresults (where the maximum RMSE is 75 kcalmol) suggested that the NDDOSMDresults could be improved by re-parameterizing the SMD Coulomb radii for the NDDOmethods
The fact that the NDDOSMD errors are largest for ions suggest that the problem iswith the polar solvation energy so we focus on optimizing the values of the Coulombradii while leaving all parameters associated with the non-polar solvation free energy un-changed We optimize the radii only for the ionic species on a subset of the MNSOL dataset previously used to parametrize RHF6-31G(d)SMD but include neutral moleculeswhen testing how well SMD with the new Coulomb radii perform We found the bestresults are obtained by changing only the radii for hydrogen carbon oxygen nitrogenand sulfur and this leads to RMSE values for PM3 (neutrals 28ions ca 5 kcalmol)PM6 (47ca 5 kcalmol) and DFTB (39ca 5 kcalmol) that are more comparable toHF6-31G(d) (24ca 5 kcalmol) We note that the SMD parameterization is done us-ing mono-aquo microsolvated species for small ions with atoms carrying large (partial)charges while the MNSOL data set also contains data for the non-microsolvated equiv-alents Comparison to the HF6-31G(d) MSEs reported by Marenich et al [8] reportedfor the selectively microsolvated ions indicates that the inclusion of non-microsolvatedspecies leads an MSE increase of about 1 kcalmol
Though the radii are optimized to reproduce aqueous solvation energies they also leadmore accurate predictions for other polar solvents such as DMSO acetonitrile andmethanol while the improvement for non-polar solvents are negligible
Supplementary MaterialSee supplementary material for Tables S1-S3 and Figures S1-S3 which are referred toin the text In addition we also provide a table of dielectric constants used for thecalculations and a table of aqueous solvation free energies obtained using the optimizedparameters
AcknowledgmentsJCK and JHJ would like to acknowledge Jimmy (Mr MOPAC) Stewart for providingsource code and guidance for the PM6 GAMESS implementation Also thanks to Donald
16
Truhlar and Christopher Cramer for sharing the MNSOL data set and for insightfulcomments on the manuscript CS thanks the Danish Council for Independent Research(the Sapere Aude program) for financial support (Grant no DFF 4181-00370)
References[1] J Tomasi B Mennucci and R Cammi ldquoQuantum mechanical continuum solvation
modelsrdquo Chem Rev vol 105 pp 2999ndash3093 2005
[2] A Klamt and G Schuurmann ldquoCOSMO a new approach to dielectric screeningin solvents with explicit expressions for the screening energy and its gradientrdquo JChem Soc Perkin Trans vol 2 pp 799ndash805 1993
[3] C J Cramer and D G Truhlar ldquoA universal approach to solvation modelingrdquoAccounts of Chemical Research vol 41 no 6 pp 760ndash768 jun 2008 doi101021ar800019z
[4] V Barone M Cossi and J Tomasi ldquoA new definition of cavities for thecomputation of solvation free energies by the polarizable continuum modelrdquo JChem Phys vol 107 no 8 pp 3210ndash3221 aug 1997 doi 1010631474671
[5] F Eckert and A Klamt ldquoFast solvent screening via quantum chemistryCOSMO-RS approachrdquo AIChE J vol 48 no 2 pp 369ndash385 feb 2002 doi101002aic690480220
[6] C Curutchet A Bidon-Chanal I Soteras M Orozco and F J Luque ldquoMSTcontinuum study of the hydration free energies of monovalent ionic speciesrdquo JPhys Chem B vol 109 no 8 pp 3565ndash3574 mar 2005 doi 101021jp047197s
[7] Y Takano and K N Houk ldquoBenchmarking the conductor-like polarizablecontinuum model (CPCM) for aqueous solvation free energies of neutral and ionicorganic moleculesrdquo J Chem Theory Comput vol 1 no 1 pp 70ndash77 jan 2005doi 101021ct049977a
[8] A V Marenich R M Olson C P Kelly C J Cramer and D G TruhlarldquoSelf-consistent reaction field model for aqueous and nonaqueous solutions based onaccurate polarized partial chargesrdquo Journal of Chemical Theory and Computationvol 3 no 6 pp 2011ndash2033 nov 2007 doi 101021ct7001418
[9] A V Marenich C J Cramer and D G Truhlar ldquoUniversal solvation model basedon solute electron density and on a continuum model of the solvent defined by thebulk dielectric constant and atomic surface tensionsrdquo J Phys Chem B vol 113no 18 pp 6378ndash6396 may 2009 doi 101021jp810292n
[10] G E Chudinov D V Napolov and M V Basilevsky ldquoQuantum-chemicalcalculations of the hydration energies of organic cations and anions in the frameworkof a continuum solvent approximationrdquo Chem Phys vol 160 no 1 pp 41 ndash 541992 doi 1010160301-0104(92)87090-V
[11] M J Negre M Orozco and F J Luque ldquoA new strategy for the representation ofenvironment effects in semi-empirical calculations based on dewarrsquos hamiltoniansrdquoChem Phys Lett vol 196 no 1 pp 27ndash36 1992
17
[12] F J Luque M J Negre and M Orozco ldquoAn am1-scrf approach to the study ofchanges in molecular properties induced by solventrdquo J Phys Chem vol 97 no 17pp 4386ndash4391 1993
[13] M Caricato B Mennucci and J Tomasi ldquoSolvent effects on the electronic spectraAn extension of the polarizable continuum model to the zindo methodrdquo J PhysChem A vol 108 no 29 pp 6248ndash6256 2004
[14] M J S Dewar E G Zoebisch E F Healy and J J P Stewart ldquoDevelopmentand use of quantum mechanical molecular models 76 AM1 a new general purposequantum mechanical molecular modelrdquo J A Chem Soc vol 10 pp 3902ndash39091985 doi 101021ja00299a024
[15] J J P Stewart ldquoOptimization of Parameters for Semi-Empirical Methods I-Methodrdquo J Comp Chem vol 10 pp 209ndash220 1989 doi 101002jcc540100208
[16] J J P Stewart ldquoOptimization of parameters for semiempirical methods V Modi-fication of NDDO approximations and application to 70 elementsrdquo J Mol Modelvol 13 pp 1172ndash1213 2007
[17] M Gaus Q Cui and M Elstner ldquoDFTB3 Extension of the Self-Consistent-Charge Density-Functional Tight-Binding Method (SCC-DFTB)rdquo J Chem TheoryComput vol 7 no 4 pp 931ndash948 apr 2011 doi 101021ct100684s
[18] A V Marenich C P Kelly J D Thompson G D Hawkins C C ChambersD J Giesen P P Winget C J Cramer and D G Truhlar ldquoMinnesota solvationdatabaserdquo 2012
[19] ldquoMinnesota solvation databaserdquo accessed November 24th 2017 [Online] Availablehttpscompchemumnedumnsol
[20] M W Schmidt K K Baldridge J A Boatz S T Elbert M S Gordon J HJensen S Koseki N Matsunaga K A Nguyen S Su T L Windus M Dupuisand JAMontgomery ldquoGeneral atomic and molecular electronic structure systemrdquoJ Comput Chem vol 14 pp 1347ndash1363 1993
[21] V Barone and M Cossi ldquoQuantum calculation of molecular energies and energygradients in solution by a conductor solvent modelrdquo The Journal of PhysicalChemistry A vol 102 no 11 pp 1995ndash2001 mar 1998 doi 101021jp9716997
[22] M Cossi N Rega G Scalmani and V Barone ldquoEnergies structures andelectronic properties of molecules in solution with the c-PCM solvation modelrdquoJournal of Computational Chemistry vol 24 no 6 pp 669ndash681 apr 2003 doi101002jcc10189
[23] C Steinmann K L Blaeligdel A S Christensen and J H Jensen ldquoInterface ofthe polarizable continuum model of solvation with semi-empirical methods in thegamess programrdquo PLoS ONE vol 8 no 7 p e67725 07 2013 doi 101371jour-nalpone0067725
[24] Y Nishimoto ldquoDFTBPCM applied to ground and excited state potential energysurfacesrdquo J Phys Chem A vol 120 no 5 pp 771ndash784 feb 2016 doi101021acsjpca5b10732
18
[25] M Gaus X Lu M Elstner and Q Cui ldquoParameterization of DFTB33OB forSulfur and Phosphorus for Chemical and Biological Applicationsrdquo J Chem TheoryComput vol 10 no 4 pp 1518ndash1537 apr 2014 doi 101021ct401002w
[26] X Lu M Gaus M Elstner and Q Cui ldquoParametrization of DFTB33OBfor Magnesium and Zinc for Chemical and Biological Applicationsrdquo The Journalof Physical Chemistry B vol 119 no 3 pp 1062ndash1082 jan 2015 doi101021jp506557r
[27] M Kubillus T Kubar M Gaus J Rezac and M Elstner ldquoParameterization ofthe DFTB3 Method for Br Ca Cl F I K and Na in Organic and BiologicalSystemsrdquo J Chem Theory Comput vol 11 no 1 pp 332ndash342 jan 2015 doi101021ct5009137
[28] J J P Stewart ldquoMOPAC2012rdquo 2016 Stewart Computational Chemistry ColoradoSprings CO USA [Online] Available httpopenmopacnet
[29] J L Pascual-Ahuir E Silla J Tomasi and R Bonaccorsi ldquoElectrostaticinteraction of a solute with a continuum improved description of the cavity and ofthe surface cavity bound charge distributionrdquo Journal of Computational Chemistryvol 8 no 6 pp 778ndash787 sep 1987 doi 101002jcc540080605
[30] P Su and H Li ldquoContinuous and smooth potential energy surface for conductorlikescreening solvation model using fixed points with variable areasrdquo The Journal ofChemical Physics vol 130 no 7 p 074109 feb 2009 doi 10106313077917
[31] J Tomasi B Mennucci and E Cances ldquoThe IEF version of the PCM solvationmethod an overview of a new method addressed to study molecular solutes at theQM ab initio levelrdquo Journal of Molecular Structure THEOCHEM vol 464 no1-3 pp 211ndash226 may 1999 doi 101016s0166-1280(98)00553-3
[32] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 1 the calculation of confidence intervalsrdquo J Comput Aided MolDes vol 28 no 9 pp 887ndash918 Sep 2014 doi 101007s10822-014-9753-z
[33] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 2 comparing methodsrdquo J Comput Aided Mol Des vol 30 no 2pp 103ndash126 Feb 2016 doi 101007s10822-016-9904-5
[34] J H Jensen ldquoWhich method is more accurate or errors have error barsrdquo PeerJPreprints vol 5 p e2693v1 2017 doi 107287peerjpreprints2693v1
[35] J H Jensen C J Swain and L Olsen ldquoPrediction of pKa values for druglikemolecules using semiempirical quantum chemical methodsrdquo The Journal of PhysicalChemistry A vol 121 no 3 pp 699ndash707 jan 2017 doi 101021acsjpca6b10990
[36] F Eckert and A Klamt ldquoAccurate prediction of basicity in aqueous solution withCOSMO-RSrdquo Journal of Computational Chemistry vol 27 no 1 pp 11ndash19 2005doi 101002jcc20309
[37] J A Nelder and R Mead ldquoA simplex method for function minimizationrdquo TheComputer Journal vol 7 no 4 pp 308ndash313 1965 doi 101093comjnl74308
19
[38] E Jones T Oliphant P Peterson et al ldquoSciPy Open source scientifictools for Pythonrdquo 2001 accessed November 24th 2017 [Online] Availablehttpwwwscipyorgrdquo
[39] J Charnley 2017 accessed November 24th 2017 [Online] Available httpsgithubcomcharnleyoptimize gamess parameters
[40] C P Kelly C J Cramer and D G Truhlar ldquoAqueous solvation free energies ofions and ion-water clusters based on an accurate value for the absolute aqueoussolvation free energy of the protonrdquo The Journal of Physical Chemistry B vol 110no 32 pp 16 066ndash16 081 aug 2006 doi 101021jp063552y
20
HFSMDAM1SMD
PM3SMDPM6SMD
DFTBSMD
AM1COSMO
PM3COSMO
PM6COSMO null0
2
4
6
8
10
12
14
16RM
SE [k
calm
ol]
PM3PM3HCO
PM3HCON
PM3HCONSPM3all PM6
PM6HCO
PM6HCON
PM6HCONS
PM6HCONShPM6all
DFTB
DFTBHCON0
2
4
6
8
10
12
14RM
SE [k
calm
ol]
HFSMD
PM3SMDPM6SMD
DFTBSMD
PM3COSMO
PM6COSMO0
2
4
6
8
10RM
SE [k
calm
ol]
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6err
or

as well as more accurate solvation free energies for DMSO acetonitrile and methanolFinally we present a summary and an outlook based on the results presented in themanuscript
Computational DetailsFor reference we use the MNSOL[18 19] data set which contains experimental solvationfree energies of neutral and singly-charged molecules with atom-types of H C N O FSi P S Cl Br and I Here we focus mainly on the aqueous solvation subset of thatdata set Because iodine is not supported by 6-31G(d) we removed compounds test2018test4001-4 and test4007-9 from the reference data set Compounds c091 and i091 are rad-icals and are not included because PM6 is only implemented for RHF in GAMESS[20]Our final reference set of molecules then consists of 522 aqueous solvation free energies ofwhich 81 are for anions and 60 are for cations and we refer to this as the MNSOL data set
When parameterizing SMD Coulomb radii we use the a subset of the MNSOL data set(the SMD data set) which was also used by Marenich et al [9] This subset consists of 384solvation free energies of which 59 are for anions and 52 are for cations Following theoriginal SMD implementation we used mono-aquo microsolvated species for small ionswith atoms carrying large (partial) charges The SMD method is re-parameterized usingthe gas-phase structures optimized at the respective levels of theory
The integration of d-integrals for semiempirical methods and interface to the conducter-like polarizable continuum model (C-PCM)[21 22] is implemented in a locally modifiedversion of GAMESS using integral code donated by James Stewart The PM6PCMinterface follows the implementation of Steinmann et al[23] except that we use the semi-numerical gradient approach for the PCM gradient that is also used for the gas phasegradient The DFTB calculations are done using the DFTBPCM interface developed byNishimoto [24] in GAMESS and using version 3ob-3-1 of the 3OB DFTB parameter set[17 25 26 27] All SMD[9] calculations were carried out with the GAMESS programand COSMO[2] calculations were done with MOPAC2016[28]
The original SMD parameterization was done with the Gauss-Bonet[29] tessellation schemewhile the default tessellation scheme in GAMESS is FIXPVA[30] While this differencein tessellation scheme has a negligible effect on the solvation free energies of neutralmolecules it can have a rather larger effect for ions Thus for calculations involvingthe original SMD radii we use the Gauss-Bonet tessellation scheme (mthall=1 in the$tescav input group) while we use the more numerically stable FIXPVA scheme whenoptimizing the radii The original SMD parameterization was also done with integral-equation-formalism PCM (IEF-PCM)[31] while we chose the C-PCM method as it iscomputationally significantly more efficient for semiempirical calculations COSMO isused with AM1 PM3 and PM6 as implemented in the MOPAC program[28] The nullmodel is constructed by setting all the predicted solvation free energies to the average ofall the reference energies and the error bars reflect 95 confidence limits [32 33 34]
The pKa predictions are performed as described by Jensen et al [35] using the same dataset ie a modified version of the one used by Eckert and Klamt [36] Following Jensen
2
et al [35] cefadroxil has been removed due to proton transfer and piroxicam has alsobeen removed since we discovered that the wrong tautomeric form was used
Results and DiscussionTable 1 and Figure 1 shows the computed solvation free energy results obtained usingHF6-31G(d) AM1 PM3 PM6 and DFTB using the implicit solvation models SMDand COSMO using the MNSOL data set described in the Computational Details sectionWe compute the root mean square error (RMSE) mean signed error (MSE) and meanunsigned error (MUE) in order to properly quantify the accuracy of our results
HFSMDAM1SMD
PM3SMDPM6SMD
DFTBSMD
AM1COSMO
PM3COSMO
PM6COSMO null0
2
4
6
8
10
12
14
16
RMSE
[kca
lmol
]
Figure 1 RMSE (in kcalmol) of SMD aqueous solvation energies computed using HF6-31G(d) AM1 PM3 PM6 and DFTB and the original SMD Coulomb radii for the MNSOLdata set Corresponding RMSE values for the COSMO method are included for comparisonThe MNSOL data set is further split into the full set of molecules (black) neutral molecules(green) cations (red) and anions (blue)
We present the combined results (labeled rdquoallrdquo) for all molecules in the data set but alsopresent individual observations on neutral and singly charged (both anions and cations)species
SMD gives RMSE values of 34 56 55 86 kcalmol for HF AM1 PM3 and PM6respectively For ions the RMSE values are 50 and 61 kcalmol for cations and 56 and69 kcalmol for anions for HF and DFTB respectively For AM1 PM3 and PM6 theerror is significantly higher with RMSE values of 114 101 115 kcalmol for cations and74 78 147 kcalmol for anions respectively The NDDO-based method systematicallyunderestimate the solvation free energy of anions with MSE (mean-signed-error) valuesof -51 -54 -126 kcalmol and overestimate the solvation free energy of cations withMSE values of 108 92 109 kcalmol respectively
3
Table 1 RMSE MUE and MSE (in kcalmol) of SMD aqueous solvation energies computedusing HF6-31G(d) AM1 PM3 PM6 and DFTB and the original SMD radii set for theMNSOL data set COSMO solvation energies are included for comparison
SMD COSMO
HF AM1 PM3 PM6 DFTB AM1 PM3 PM6 nullAll
RMSE 34 56 55 86 50 52 48 50 309MUE 21 38 38 62 37 36 34 39 268MSE 03 03 06 -32 -16 00 05 -18 -00Max 178 179 245 302 183 201 213 211 875
NeutralRMSE 24 34 35 59 43 41 36 47 44MUE 13 21 24 40 31 26 25 35 34MSE -09 -01 06 -35 -21 -18 -10 -34 00Max 178 178 245 302 183 201 213 211 158
CationsRMSE 50 113 101 115 61 74 74 73 101MUE 36 108 93 109 48 60 59 60 73MSE 31 108 92 109 45 59 56 59 -00Max 140 179 166 204 161 176 186 183 409
AnionsRMSE 56 74 78 148 69 75 68 45 104MUE 46 65 66 132 58 64 58 38 79MSE 39 -51 -54 -126 -37 45 37 03 -00Max 143 144 168 263 155 148 152 105 300
4
COSMO-predicted solvation free energies have RMSE values for the full set of 52 48and 50 kcalmol for AM1 PM3 and PM6 respectively which are quite similar to thecorresponding SMD values for AM1 and PM3 However the accuracy of COSMO forthe ions are significantly better especially for cations with RMSE values of 73 74 and73 kcalmol for cations and 75 68 45 kcalmol for anions The COSMO results showthat is is possible to significantly improve the accuracy of the NDDOSMD calculationsand the comparatively high errors for ions indicate that the focus should be on the polarpart of the solvation free energy
Optimizing SMD Coulomb RadiiBased on the results in the previous subsection we optimize the Coulomb radii for PM3PM6 and DFTB independently while leaving the non-polar solvation free energy con-tribution unchanged We use the Nelder-Mead simplex method[37] to optimize the radiiusing the SMD data set with the SciPy[38] package using a custom optimization script[39]Optimization of the radii is done by minimizing the RMSE with respect to the radii
Initial tests showed that the solvation free energy does not vary greatly with a changein radii for neutral molecules and the flatness of the energy surface caused problems forthe Nelder-Mead method Furthermore the error for the ions is considerably larger thanfor the neutral molecules Thus only radii of the ions are optimized but when testingthe performance of the new radii neutral molecules are included This means that theoptimum parameters obtained for the ions will not necessarily lead to the lowest possibleerror for neutral molecules We therefore test the effect of optimizing a subset of theradii but include all molecules when testing the overall performance These results arepresented in Tables 2 and 3 and in Figure 2
For PM3 the results are relatively insensitive to the type of atoms chosen with RMSElowast
values of 44 to 47 kcalmol (Table 2 and Figure 2) (Note that the RMSE value arecomputed using different numbers of charged species so the RMSElowast does not necessarilydecrease when more parameters are optimized) Similarly the RMSE values computedusing all the molecules in the MNSOL data set range from 29 to 32 kcalmol as pre-sented in Table 3 The cations are most sensitive to the choice of atom types with anRMSE range of 50 to 62 kcalmol and the HCON- and HCONS-optimized radii set givingthe best and nearly identical accuracy For anions the RMSE for the HCON radii (42kcalmol) is lower than that for HCONS (45 kcalmol) and nearly identical to the lowestRMSE observed (41 kcalmol) The HCON-optimized radii are therefore the best com-promise
For PM6 the RMSElowast for HCO (32 kcalmol) is about 1 kcalmol lower than for the otherradii set However the RMSE values computed using all molecules are very similar witha range of 36 to 38 kcalmol with HCO and HCON tied for first place HCO does lead tothe lowest error for neutral molecules and anions but also an RMSE that is significantlylarger for cations (62 kcalmol) where PM6-HCONS performs best
When testing the PM6-HCON and PM6-HCONS radii on pKa prediction (see below) wefound that several solution-phase geometry optimizations failed because some X-H bonds
5
Table 2 Optimized Coulomb radii (in A) for PM3 PM6 and DFTB for different atom typesand original SMD radii parameters for comparison RMSESMD denotes the RMSE using theoriginal parameters and RMSElowast denotes RMSE after optimization The RMSE is given inkcalmol and is only calculated for the subset of ions in the SMD subset containing thosespecific atom types
H C N O F S Cl Br RMSESMD RMSElowast
SMD120 185 189 152 173 249 238 306
PM3029 172 165 96 44042 175 176 164 109 43062 176 172 162 258 107 46058 175 172 164 179 255 253 335 106 47
PM6066 187 173 123 32017 185 178 174 130 41039 178 182 175 262 127 43070 182 178 174 140 255 237 388 123 44
DFTB070 183 191 164 95 35
broke and the proton started moving into the solvent This indicated that the H radiusmight be too small leading to overpolarization of the H atom due to the solvent andwe tested several larger H radii for PM6-HCONS since the H radius was larger than forPM6-HCON We found that the overpolarization problem disappeared with a H radius of06 A and that using this value has a negligible 01 kcalmol effect of the accuracy ofthe solvation energies (HCONSh in Table 3) Thus we use PM6-HCONSh radii set goingforward
6
PM3PM3HCO
PM3HCON
PM3HCONSPM3all PM6
PM6HCO
PM6HCON
PM6HCONS
PM6HCONShPM6all
DFTB
DFTBHCON0
2
4
6
8
10
12
14
RMSE
[kca
lmol
]
Figure 2 RMSE values (in kcalmol) computed using SMD subset of the MNSOL data setand the Coulomb radii set shown in Table 2 HCO indicates that hydrogen carbon and oxygenradii parameters are changed (ie second and sixth entry in Table 2 The reference set is splitinto full set (black) neutral (green) cations (red) and anions (blue)
7
Table 3 RMSE MUE and MSE values (in kcalmol) computed using the SMD subset of the MNSOL data set and the Coulomb radii set shownin Table 2 HCO indicates that hydrogen carbon and oxygen radii parameters are changed (ie second and sixth row in Table 2)
PM3 PM6 DFTB
SMD HCO HCON HCONS all SMD HCO HCON HCONS HCONSh all SMD HCONAll
RMSE 50 32 29 30 32 75 36 36 38 38 37 41 33MUE 35 23 21 22 23 54 25 27 30 30 28 31 26MSE 10 13 10 10 13 -24 -04 -13 -17 -16 -14 -09 -08Max 168 116 113 126 116 226 184 186 169 169 176 155 109
NeutralRMSE 24 20 19 19 20 40 28 34 36 35 34 31 30MUE 18 15 15 15 15 30 19 24 29 28 25 24 24MSE 11 11 10 11 11 -26 -15 -21 -26 -26 -22 -11 -14Max 87 80 72 74 80 164 184 186 169 169 176 108 109
CationsRMSE 92 62 51 50 62 103 63 43 40 41 41 49 29MUE 85 55 40 39 55 100 58 36 34 35 35 40 24MSE 84 53 32 25 53 100 58 29 30 31 28 36 02Max 150 116 113 119 116 168 138 115 113 113 112 138 91
AnionsRMSE 79 41 42 45 41 142 36 43 46 46 46 67 44MUE 66 32 33 35 32 128 28 32 35 34 32 56 34MSE -61 -11 -13 -07 -11 -126 -06 -12 -14 -12 -13 -39 12Max 168 92 99 126 92 226 88 121 149 147 173 155 104
8
From the results discussed above and presented in Table 2 optimizing for atom typesbeyond HCON showed no significant increase in accuracy for either PM3 or PM6 There-fore the same procedure (optimizing HCON radii) was applied to DFTB For DFTB theRMSE values of the full set of molecules the subset of cation and the subset of anionsgoes from 41 49 and 67 kcalmol to 33 29 and 44 kcalmol respectively
In summary we find that optimizing the SMD radii in combination with either PM3 orPM6 gives appreciable decreases in RMSE accuracy asymp 7 kcalmol in both cases comparedto the reference results We also find that no significant improvement is obtained whenattempting to optimize the radii of other atoms than what was done with HCON subsetof radii For this subset DFTB also saw an increase in accuracy for ions of about asymp 6kcalmol Because of this we found the best choice was change the radii for hydrogencarbon oxygen and nitrogen (HCON) for PM3 and DFTB methods For PM6 we optimizedincluding sulfur and changed the radius of hydrogen to 06 A to remove overpolarizationproblems These methods are denoted as SMDdagger going forward
Comparison with Other Solvation MethodsUsing the new parameters for PM3SMDdagger PM6SMDdagger and DFTBSMDdagger we com-pare the obtained RMSE with other approaches to obtain solvent free energies such asHF6-31G(d)SMD (one of the levels of theory used in the original SMD parameteriza-tion) PM3COSMO and PM6COSMO for the MNSOL data set (Table 4 and Figure 3)
HFSMD
PM3SMDPM6SMD
DFTBSMD
PM3COSMO
PM6COSMO0
2
4
6
8
10
RMSE
[kca
lmol
]
Figure 3 RMSE (in kcalmol) of aqueous solvation energies computed with SMD using HF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicates that the Radiihas been reoptimized Corresponding RMSE values for the COSMO method are included forcomparison The dataset is split into full set (black) neutral (green) cations (red) and anions(blue)
The overall accuracy of PM3SMDdagger is now similar to HFSMD although the RMSEvalue is 04 and 06 kcalmol higher for neutral molecules and cations while the RMSE
9
is 07 kcalmol lower for anions For PM6 the overall RMSE is 14 kcalmol higher thanHF and the RMSE for neutral molecules is 23 kcalmol higher On the other hand theRMSE for cations is only 01 kcalmol higher while the RMSE for anions is 08 kcal-mol lower compared to HF Finally for DFTB the overall RMSE value (41 kcalmol)is in between those for PM3 and PM6 as is the RMSE values for neutral molecules (39kcalmol) But for cations the RMSE (37 kcalmol) is significantly lower than the othermethods while the RMSE for anions (51 kcalmol) is very similar
While the solvation free energies of neutral molecules can generally be measured with anaccuracy of ca 02 kcalmol the corresponding solvation free energies of ions are inferredfrom other experimental values with a typical accuracy about 30 kcalmol[40] Thus theaccuracy of the semiempirical SMDdagger is for all intents and purposes identical to HFSMDfor ions but significantly worse for neutral ions in the case of PM6 and DFTB Thesource of this error is most likely due to a poorer description of the electrostatic proper-ties compared to HF For example the error in dipole moments for HCNO compounds islarger for PM6 than for PM3[16]
Table 4 also lists values for PM3COSMO and PM6COSMO as implemented in MOPACfor comparison This COSMO implementation only evaluates the polar part of the solva-tion free energy and will not be as accurate as properly parameterized COSMO-RS[5] val-ues For PM3 the SMDdagger results are significantly lower than the corresponding COSMOresults while for PM6 there is only a significant improvement for cations
10
Table 4 RMSE MUE and MSE (in kcalmol) of aqueous solvation energies computed withSMD using HF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicatesthat the Radii has been reoptimized Corresponding RMSE values for the COSMO method areincluded for comparison The reference set is split into full set neutral cations and anions
SMD SMDdagger COSMO
HF PM3 PM6 DFTB PM3 PM6All
RMSE 34 36 48 41 48 50MUE 21 26 35 31 34 39MSE 03 10 -17 -11 05 -18Max 178 155 231 160 213 211
NeutralRMSE 24 28 47 39 36 47MUE 13 20 34 29 25 35MSE -09 09 -29 -21 -10 -34Max 178 155 231 160 213 211
CationsRMSE 50 56 51 37 74 73MUE 36 44 42 29 59 60MSE 31 36 38 10 56 59Max 140 127 124 91 186 183
AnionsRMSE 56 49 48 51 68 45MUE 46 37 36 40 58 38MSE 39 -03 -05 18 37 03Max 143 146 147 147 152 105
11
pKa PredictionPart of the impetus for this work were the relatively large error in pKa values predictedusing PM3COSMO compared to PM3SMD with the original SMD radii observed byJensen et al [35] so we use the new SMD radii to compute the pKa value using the samedata set The data is presented in Figure 4 and Table 5 and shows that the RMSE dropsfrom 15plusmn03 to 09plusmn03 pH units for PM3SMDdagger and 20plusmn04 to 16plusmn0304 pH unitsfor DFTBSMDdagger In both cases the improvement is primarily a result of the decreasein mean error (ME) ie the original radii led to a fairly systematic underestimation ofthe pKa values that has now been removed by the reparameterization For DFTB theincreased accuracy is also due to a larger number of low error predictions as is evident inFigure 4 In the case of PM3SMDdagger the accuracy is now very similar to PM3COSMOThe RMSE for PM6SMDdagger is similar to PM6COSMO and larger than for PM3SMDin analogy with the corresponding results for COSMO
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6
err
or
Figure 4 Plot of the errors in the predicted pKa values (pKaminus pKexpa )
12
Table 5 Root-Mean-Square-Error (RMSE) man error (ME) and Pearson correlation (r) for predicted pKa values relative to experiment togetherwith their statistical uncertainty (95 confidence limits)
PM3COSMO PM3SMD PM3SMDdagger PM6COSMO PM6SMDdagger DFTBSMD DFTBSMDdaggerRMSE 10 15 09 18 16 20 1695 conf plusmn 0202 plusmn 0303 plusmn 0202 plusmn 0304 plusmn 0304 plusmn 0404 plusmn 0304ME -05 plusmn 02 -12 plusmn 03 -02 plusmn 02 -13 plusmn 04 -08 plusmn 04 -07 plusmn 05 -01 plusmn 04r 095 093 093 090 086 087 08695 conf plusmn 002004 plusmn 003005 plusmn 003005 plusmn 004007 plusmn 006009 plusmn 005008 plusmn 006009
13
Non-aqueous SolventsThe new radii were optimized explicitly for water so it is not a priori clear whether theyoffer more accurate results for other solvents Table 6 shows the RMSE values for allneutral cations and anions for the other polar solvents in the MNSOL data set acetoni-trile DMSO and methanol for the original SMD radii and the corresponding re-optimizedradii (dagger) In addition corresponding combined RMSE values are given for the remaining88 mostly non-polar solvents in the MNSOL data set for which only data is availablefor neutral molecules It is clear from this data that the new radii sets lowers the errorof the predicted solvation free energies for the other polar solvents and has a negligibleeffect on the accuracy for the non-polar solvents
As for acetonitrile and DMSO the improvement is greatest for the ions where the RMSEdecreases by 3-12 kcalmol The improvement is more modest (05-3 kcalmol) for theneutral molecules For methanol which contains only ions the RMSE is similarly de-creased with 2-13 kcalmol
We thus recommend that the new radii optimized for water also be used for other polarsolvents when doing semiempirical SMD calculations For non-polar solvents either setof radii can be used
14
Table 6 RMSE (in kcalmol) of non-aqueous solvation energies computed with SMD usingHF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicates that theradii has been reoptimized Corresponding RMSE values for the COSMO method are includedfor comparison The reference set is split into full set neutral cations and anions
All Neu Cat Aniacetonitrile
HFSMD 112 29 105 131PM3SMD 194 41 162 246PM3SMDdagger 140 37 113 182PM6SMD 219 33 177 284PM6SMDdagger 130 12 114 160DFTBSMD 150 23 119 197DFTBSMDdagger 109 22 90 140PM3COSMO 130 31 128 147PM6COSMO 137 23 135 155
dimethylsulfoxideHFSMD 119 30 106 126PM3SMD 202 42 153 214PM3SMDdagger 170 35 90 182PM6SMD 255 45 172 272PM6SMDdagger 177 17 99 189DFTBSMD 169 23 108 181DFTBSMDdagger 125 21 76 134PM3COSMO 125 34 107 132PM6COSMO 150 31 119 159
methanolHFSMD 40 00 41 39PM3SMD 126 00 108 135PM3SMDdagger 66 00 60 69PM6SMD 163 00 120 183PM6SMDdagger 52 00 54 52DFTBSMD 84 00 63 94DFTBSMDdagger 47 00 44 49PM3COSMO 59 00 68 54PM6COSMO 54 00 67 46
otherHFSMD 42 42 00 00PM3SMD 51 51 00 00PM3SMDdagger 51 51 00 00PM6SMD 35 35 00 00PM6SMDdagger 34 34 00 00DFTBSMD 30 30 00 00DFTBSMDdagger 29 29 00 00PM3COSMO 36 36 00 00PM6COSMO 27 27 00 00
15
ConclusionThe PM6 method in GAMESS was extended to elements requiring d-integrals and inter-faced with the polarized continuum model (PCM) of solvation including semi-numericalgradients However the accuracy of aqueous solvation energies computed using AM1PM3 PM6 and DFTB and the SMD continuum solvation model was tested using a sub-set of molecules from the MNSOL data set which showed that the errors in SMD solvationenergies predicted using NDDO-based methods was considerably larger than when usingDFT and HF with RMSE values of 50 to 86 kcalmol compared to 34 kcalmol forHF6-31G(d) For the NDDO-based methods the errors were especially large for cationsand in the case of PM6 also for anions with RMSE values of 101 to 148 kcalmol incomparison with to 50 to 57 kcalmol for HF6-31G(d)SMD Corresponding COSMOresults (where the maximum RMSE is 75 kcalmol) suggested that the NDDOSMDresults could be improved by re-parameterizing the SMD Coulomb radii for the NDDOmethods
The fact that the NDDOSMD errors are largest for ions suggest that the problem iswith the polar solvation energy so we focus on optimizing the values of the Coulombradii while leaving all parameters associated with the non-polar solvation free energy un-changed We optimize the radii only for the ionic species on a subset of the MNSOL dataset previously used to parametrize RHF6-31G(d)SMD but include neutral moleculeswhen testing how well SMD with the new Coulomb radii perform We found the bestresults are obtained by changing only the radii for hydrogen carbon oxygen nitrogenand sulfur and this leads to RMSE values for PM3 (neutrals 28ions ca 5 kcalmol)PM6 (47ca 5 kcalmol) and DFTB (39ca 5 kcalmol) that are more comparable toHF6-31G(d) (24ca 5 kcalmol) We note that the SMD parameterization is done us-ing mono-aquo microsolvated species for small ions with atoms carrying large (partial)charges while the MNSOL data set also contains data for the non-microsolvated equiv-alents Comparison to the HF6-31G(d) MSEs reported by Marenich et al [8] reportedfor the selectively microsolvated ions indicates that the inclusion of non-microsolvatedspecies leads an MSE increase of about 1 kcalmol
Though the radii are optimized to reproduce aqueous solvation energies they also leadmore accurate predictions for other polar solvents such as DMSO acetonitrile andmethanol while the improvement for non-polar solvents are negligible
Supplementary MaterialSee supplementary material for Tables S1-S3 and Figures S1-S3 which are referred toin the text In addition we also provide a table of dielectric constants used for thecalculations and a table of aqueous solvation free energies obtained using the optimizedparameters
AcknowledgmentsJCK and JHJ would like to acknowledge Jimmy (Mr MOPAC) Stewart for providingsource code and guidance for the PM6 GAMESS implementation Also thanks to Donald
16
Truhlar and Christopher Cramer for sharing the MNSOL data set and for insightfulcomments on the manuscript CS thanks the Danish Council for Independent Research(the Sapere Aude program) for financial support (Grant no DFF 4181-00370)
References[1] J Tomasi B Mennucci and R Cammi ldquoQuantum mechanical continuum solvation
modelsrdquo Chem Rev vol 105 pp 2999ndash3093 2005
[2] A Klamt and G Schuurmann ldquoCOSMO a new approach to dielectric screeningin solvents with explicit expressions for the screening energy and its gradientrdquo JChem Soc Perkin Trans vol 2 pp 799ndash805 1993
[3] C J Cramer and D G Truhlar ldquoA universal approach to solvation modelingrdquoAccounts of Chemical Research vol 41 no 6 pp 760ndash768 jun 2008 doi101021ar800019z
[4] V Barone M Cossi and J Tomasi ldquoA new definition of cavities for thecomputation of solvation free energies by the polarizable continuum modelrdquo JChem Phys vol 107 no 8 pp 3210ndash3221 aug 1997 doi 1010631474671
[5] F Eckert and A Klamt ldquoFast solvent screening via quantum chemistryCOSMO-RS approachrdquo AIChE J vol 48 no 2 pp 369ndash385 feb 2002 doi101002aic690480220
[6] C Curutchet A Bidon-Chanal I Soteras M Orozco and F J Luque ldquoMSTcontinuum study of the hydration free energies of monovalent ionic speciesrdquo JPhys Chem B vol 109 no 8 pp 3565ndash3574 mar 2005 doi 101021jp047197s
[7] Y Takano and K N Houk ldquoBenchmarking the conductor-like polarizablecontinuum model (CPCM) for aqueous solvation free energies of neutral and ionicorganic moleculesrdquo J Chem Theory Comput vol 1 no 1 pp 70ndash77 jan 2005doi 101021ct049977a
[8] A V Marenich R M Olson C P Kelly C J Cramer and D G TruhlarldquoSelf-consistent reaction field model for aqueous and nonaqueous solutions based onaccurate polarized partial chargesrdquo Journal of Chemical Theory and Computationvol 3 no 6 pp 2011ndash2033 nov 2007 doi 101021ct7001418
[9] A V Marenich C J Cramer and D G Truhlar ldquoUniversal solvation model basedon solute electron density and on a continuum model of the solvent defined by thebulk dielectric constant and atomic surface tensionsrdquo J Phys Chem B vol 113no 18 pp 6378ndash6396 may 2009 doi 101021jp810292n
[10] G E Chudinov D V Napolov and M V Basilevsky ldquoQuantum-chemicalcalculations of the hydration energies of organic cations and anions in the frameworkof a continuum solvent approximationrdquo Chem Phys vol 160 no 1 pp 41 ndash 541992 doi 1010160301-0104(92)87090-V
[11] M J Negre M Orozco and F J Luque ldquoA new strategy for the representation ofenvironment effects in semi-empirical calculations based on dewarrsquos hamiltoniansrdquoChem Phys Lett vol 196 no 1 pp 27ndash36 1992
17
[12] F J Luque M J Negre and M Orozco ldquoAn am1-scrf approach to the study ofchanges in molecular properties induced by solventrdquo J Phys Chem vol 97 no 17pp 4386ndash4391 1993
[13] M Caricato B Mennucci and J Tomasi ldquoSolvent effects on the electronic spectraAn extension of the polarizable continuum model to the zindo methodrdquo J PhysChem A vol 108 no 29 pp 6248ndash6256 2004
[14] M J S Dewar E G Zoebisch E F Healy and J J P Stewart ldquoDevelopmentand use of quantum mechanical molecular models 76 AM1 a new general purposequantum mechanical molecular modelrdquo J A Chem Soc vol 10 pp 3902ndash39091985 doi 101021ja00299a024
[15] J J P Stewart ldquoOptimization of Parameters for Semi-Empirical Methods I-Methodrdquo J Comp Chem vol 10 pp 209ndash220 1989 doi 101002jcc540100208
[16] J J P Stewart ldquoOptimization of parameters for semiempirical methods V Modi-fication of NDDO approximations and application to 70 elementsrdquo J Mol Modelvol 13 pp 1172ndash1213 2007
[17] M Gaus Q Cui and M Elstner ldquoDFTB3 Extension of the Self-Consistent-Charge Density-Functional Tight-Binding Method (SCC-DFTB)rdquo J Chem TheoryComput vol 7 no 4 pp 931ndash948 apr 2011 doi 101021ct100684s
[18] A V Marenich C P Kelly J D Thompson G D Hawkins C C ChambersD J Giesen P P Winget C J Cramer and D G Truhlar ldquoMinnesota solvationdatabaserdquo 2012
[19] ldquoMinnesota solvation databaserdquo accessed November 24th 2017 [Online] Availablehttpscompchemumnedumnsol
[20] M W Schmidt K K Baldridge J A Boatz S T Elbert M S Gordon J HJensen S Koseki N Matsunaga K A Nguyen S Su T L Windus M Dupuisand JAMontgomery ldquoGeneral atomic and molecular electronic structure systemrdquoJ Comput Chem vol 14 pp 1347ndash1363 1993
[21] V Barone and M Cossi ldquoQuantum calculation of molecular energies and energygradients in solution by a conductor solvent modelrdquo The Journal of PhysicalChemistry A vol 102 no 11 pp 1995ndash2001 mar 1998 doi 101021jp9716997
[22] M Cossi N Rega G Scalmani and V Barone ldquoEnergies structures andelectronic properties of molecules in solution with the c-PCM solvation modelrdquoJournal of Computational Chemistry vol 24 no 6 pp 669ndash681 apr 2003 doi101002jcc10189
[23] C Steinmann K L Blaeligdel A S Christensen and J H Jensen ldquoInterface ofthe polarizable continuum model of solvation with semi-empirical methods in thegamess programrdquo PLoS ONE vol 8 no 7 p e67725 07 2013 doi 101371jour-nalpone0067725
[24] Y Nishimoto ldquoDFTBPCM applied to ground and excited state potential energysurfacesrdquo J Phys Chem A vol 120 no 5 pp 771ndash784 feb 2016 doi101021acsjpca5b10732
18
[25] M Gaus X Lu M Elstner and Q Cui ldquoParameterization of DFTB33OB forSulfur and Phosphorus for Chemical and Biological Applicationsrdquo J Chem TheoryComput vol 10 no 4 pp 1518ndash1537 apr 2014 doi 101021ct401002w
[26] X Lu M Gaus M Elstner and Q Cui ldquoParametrization of DFTB33OBfor Magnesium and Zinc for Chemical and Biological Applicationsrdquo The Journalof Physical Chemistry B vol 119 no 3 pp 1062ndash1082 jan 2015 doi101021jp506557r
[27] M Kubillus T Kubar M Gaus J Rezac and M Elstner ldquoParameterization ofthe DFTB3 Method for Br Ca Cl F I K and Na in Organic and BiologicalSystemsrdquo J Chem Theory Comput vol 11 no 1 pp 332ndash342 jan 2015 doi101021ct5009137
[28] J J P Stewart ldquoMOPAC2012rdquo 2016 Stewart Computational Chemistry ColoradoSprings CO USA [Online] Available httpopenmopacnet
[29] J L Pascual-Ahuir E Silla J Tomasi and R Bonaccorsi ldquoElectrostaticinteraction of a solute with a continuum improved description of the cavity and ofthe surface cavity bound charge distributionrdquo Journal of Computational Chemistryvol 8 no 6 pp 778ndash787 sep 1987 doi 101002jcc540080605
[30] P Su and H Li ldquoContinuous and smooth potential energy surface for conductorlikescreening solvation model using fixed points with variable areasrdquo The Journal ofChemical Physics vol 130 no 7 p 074109 feb 2009 doi 10106313077917
[31] J Tomasi B Mennucci and E Cances ldquoThe IEF version of the PCM solvationmethod an overview of a new method addressed to study molecular solutes at theQM ab initio levelrdquo Journal of Molecular Structure THEOCHEM vol 464 no1-3 pp 211ndash226 may 1999 doi 101016s0166-1280(98)00553-3
[32] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 1 the calculation of confidence intervalsrdquo J Comput Aided MolDes vol 28 no 9 pp 887ndash918 Sep 2014 doi 101007s10822-014-9753-z
[33] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 2 comparing methodsrdquo J Comput Aided Mol Des vol 30 no 2pp 103ndash126 Feb 2016 doi 101007s10822-016-9904-5
[34] J H Jensen ldquoWhich method is more accurate or errors have error barsrdquo PeerJPreprints vol 5 p e2693v1 2017 doi 107287peerjpreprints2693v1
[35] J H Jensen C J Swain and L Olsen ldquoPrediction of pKa values for druglikemolecules using semiempirical quantum chemical methodsrdquo The Journal of PhysicalChemistry A vol 121 no 3 pp 699ndash707 jan 2017 doi 101021acsjpca6b10990
[36] F Eckert and A Klamt ldquoAccurate prediction of basicity in aqueous solution withCOSMO-RSrdquo Journal of Computational Chemistry vol 27 no 1 pp 11ndash19 2005doi 101002jcc20309
[37] J A Nelder and R Mead ldquoA simplex method for function minimizationrdquo TheComputer Journal vol 7 no 4 pp 308ndash313 1965 doi 101093comjnl74308
19
[38] E Jones T Oliphant P Peterson et al ldquoSciPy Open source scientifictools for Pythonrdquo 2001 accessed November 24th 2017 [Online] Availablehttpwwwscipyorgrdquo
[39] J Charnley 2017 accessed November 24th 2017 [Online] Available httpsgithubcomcharnleyoptimize gamess parameters
[40] C P Kelly C J Cramer and D G Truhlar ldquoAqueous solvation free energies ofions and ion-water clusters based on an accurate value for the absolute aqueoussolvation free energy of the protonrdquo The Journal of Physical Chemistry B vol 110no 32 pp 16 066ndash16 081 aug 2006 doi 101021jp063552y
20
HFSMDAM1SMD
PM3SMDPM6SMD
DFTBSMD
AM1COSMO
PM3COSMO
PM6COSMO null0
2
4
6
8
10
12
14
16RM
SE [k
calm
ol]
PM3PM3HCO
PM3HCON
PM3HCONSPM3all PM6
PM6HCO
PM6HCON
PM6HCONS
PM6HCONShPM6all
DFTB
DFTBHCON0
2
4
6
8
10
12
14RM
SE [k
calm
ol]
HFSMD
PM3SMDPM6SMD
DFTBSMD
PM3COSMO
PM6COSMO0
2
4
6
8
10RM
SE [k
calm
ol]
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6err
or

et al [35] cefadroxil has been removed due to proton transfer and piroxicam has alsobeen removed since we discovered that the wrong tautomeric form was used
Results and DiscussionTable 1 and Figure 1 shows the computed solvation free energy results obtained usingHF6-31G(d) AM1 PM3 PM6 and DFTB using the implicit solvation models SMDand COSMO using the MNSOL data set described in the Computational Details sectionWe compute the root mean square error (RMSE) mean signed error (MSE) and meanunsigned error (MUE) in order to properly quantify the accuracy of our results
HFSMDAM1SMD
PM3SMDPM6SMD
DFTBSMD
AM1COSMO
PM3COSMO
PM6COSMO null0
2
4
6
8
10
12
14
16
RMSE
[kca
lmol
]
Figure 1 RMSE (in kcalmol) of SMD aqueous solvation energies computed using HF6-31G(d) AM1 PM3 PM6 and DFTB and the original SMD Coulomb radii for the MNSOLdata set Corresponding RMSE values for the COSMO method are included for comparisonThe MNSOL data set is further split into the full set of molecules (black) neutral molecules(green) cations (red) and anions (blue)
We present the combined results (labeled rdquoallrdquo) for all molecules in the data set but alsopresent individual observations on neutral and singly charged (both anions and cations)species
SMD gives RMSE values of 34 56 55 86 kcalmol for HF AM1 PM3 and PM6respectively For ions the RMSE values are 50 and 61 kcalmol for cations and 56 and69 kcalmol for anions for HF and DFTB respectively For AM1 PM3 and PM6 theerror is significantly higher with RMSE values of 114 101 115 kcalmol for cations and74 78 147 kcalmol for anions respectively The NDDO-based method systematicallyunderestimate the solvation free energy of anions with MSE (mean-signed-error) valuesof -51 -54 -126 kcalmol and overestimate the solvation free energy of cations withMSE values of 108 92 109 kcalmol respectively
3
Table 1 RMSE MUE and MSE (in kcalmol) of SMD aqueous solvation energies computedusing HF6-31G(d) AM1 PM3 PM6 and DFTB and the original SMD radii set for theMNSOL data set COSMO solvation energies are included for comparison
SMD COSMO
HF AM1 PM3 PM6 DFTB AM1 PM3 PM6 nullAll
RMSE 34 56 55 86 50 52 48 50 309MUE 21 38 38 62 37 36 34 39 268MSE 03 03 06 -32 -16 00 05 -18 -00Max 178 179 245 302 183 201 213 211 875
NeutralRMSE 24 34 35 59 43 41 36 47 44MUE 13 21 24 40 31 26 25 35 34MSE -09 -01 06 -35 -21 -18 -10 -34 00Max 178 178 245 302 183 201 213 211 158
CationsRMSE 50 113 101 115 61 74 74 73 101MUE 36 108 93 109 48 60 59 60 73MSE 31 108 92 109 45 59 56 59 -00Max 140 179 166 204 161 176 186 183 409
AnionsRMSE 56 74 78 148 69 75 68 45 104MUE 46 65 66 132 58 64 58 38 79MSE 39 -51 -54 -126 -37 45 37 03 -00Max 143 144 168 263 155 148 152 105 300
4
COSMO-predicted solvation free energies have RMSE values for the full set of 52 48and 50 kcalmol for AM1 PM3 and PM6 respectively which are quite similar to thecorresponding SMD values for AM1 and PM3 However the accuracy of COSMO forthe ions are significantly better especially for cations with RMSE values of 73 74 and73 kcalmol for cations and 75 68 45 kcalmol for anions The COSMO results showthat is is possible to significantly improve the accuracy of the NDDOSMD calculationsand the comparatively high errors for ions indicate that the focus should be on the polarpart of the solvation free energy
Optimizing SMD Coulomb RadiiBased on the results in the previous subsection we optimize the Coulomb radii for PM3PM6 and DFTB independently while leaving the non-polar solvation free energy con-tribution unchanged We use the Nelder-Mead simplex method[37] to optimize the radiiusing the SMD data set with the SciPy[38] package using a custom optimization script[39]Optimization of the radii is done by minimizing the RMSE with respect to the radii
Initial tests showed that the solvation free energy does not vary greatly with a changein radii for neutral molecules and the flatness of the energy surface caused problems forthe Nelder-Mead method Furthermore the error for the ions is considerably larger thanfor the neutral molecules Thus only radii of the ions are optimized but when testingthe performance of the new radii neutral molecules are included This means that theoptimum parameters obtained for the ions will not necessarily lead to the lowest possibleerror for neutral molecules We therefore test the effect of optimizing a subset of theradii but include all molecules when testing the overall performance These results arepresented in Tables 2 and 3 and in Figure 2
For PM3 the results are relatively insensitive to the type of atoms chosen with RMSElowast
values of 44 to 47 kcalmol (Table 2 and Figure 2) (Note that the RMSE value arecomputed using different numbers of charged species so the RMSElowast does not necessarilydecrease when more parameters are optimized) Similarly the RMSE values computedusing all the molecules in the MNSOL data set range from 29 to 32 kcalmol as pre-sented in Table 3 The cations are most sensitive to the choice of atom types with anRMSE range of 50 to 62 kcalmol and the HCON- and HCONS-optimized radii set givingthe best and nearly identical accuracy For anions the RMSE for the HCON radii (42kcalmol) is lower than that for HCONS (45 kcalmol) and nearly identical to the lowestRMSE observed (41 kcalmol) The HCON-optimized radii are therefore the best com-promise
For PM6 the RMSElowast for HCO (32 kcalmol) is about 1 kcalmol lower than for the otherradii set However the RMSE values computed using all molecules are very similar witha range of 36 to 38 kcalmol with HCO and HCON tied for first place HCO does lead tothe lowest error for neutral molecules and anions but also an RMSE that is significantlylarger for cations (62 kcalmol) where PM6-HCONS performs best
When testing the PM6-HCON and PM6-HCONS radii on pKa prediction (see below) wefound that several solution-phase geometry optimizations failed because some X-H bonds
5
Table 2 Optimized Coulomb radii (in A) for PM3 PM6 and DFTB for different atom typesand original SMD radii parameters for comparison RMSESMD denotes the RMSE using theoriginal parameters and RMSElowast denotes RMSE after optimization The RMSE is given inkcalmol and is only calculated for the subset of ions in the SMD subset containing thosespecific atom types
H C N O F S Cl Br RMSESMD RMSElowast
SMD120 185 189 152 173 249 238 306
PM3029 172 165 96 44042 175 176 164 109 43062 176 172 162 258 107 46058 175 172 164 179 255 253 335 106 47
PM6066 187 173 123 32017 185 178 174 130 41039 178 182 175 262 127 43070 182 178 174 140 255 237 388 123 44
DFTB070 183 191 164 95 35
broke and the proton started moving into the solvent This indicated that the H radiusmight be too small leading to overpolarization of the H atom due to the solvent andwe tested several larger H radii for PM6-HCONS since the H radius was larger than forPM6-HCON We found that the overpolarization problem disappeared with a H radius of06 A and that using this value has a negligible 01 kcalmol effect of the accuracy ofthe solvation energies (HCONSh in Table 3) Thus we use PM6-HCONSh radii set goingforward
6
PM3PM3HCO
PM3HCON
PM3HCONSPM3all PM6
PM6HCO
PM6HCON
PM6HCONS
PM6HCONShPM6all
DFTB
DFTBHCON0
2
4
6
8
10
12
14
RMSE
[kca
lmol
]
Figure 2 RMSE values (in kcalmol) computed using SMD subset of the MNSOL data setand the Coulomb radii set shown in Table 2 HCO indicates that hydrogen carbon and oxygenradii parameters are changed (ie second and sixth entry in Table 2 The reference set is splitinto full set (black) neutral (green) cations (red) and anions (blue)
7
Table 3 RMSE MUE and MSE values (in kcalmol) computed using the SMD subset of the MNSOL data set and the Coulomb radii set shownin Table 2 HCO indicates that hydrogen carbon and oxygen radii parameters are changed (ie second and sixth row in Table 2)
PM3 PM6 DFTB
SMD HCO HCON HCONS all SMD HCO HCON HCONS HCONSh all SMD HCONAll
RMSE 50 32 29 30 32 75 36 36 38 38 37 41 33MUE 35 23 21 22 23 54 25 27 30 30 28 31 26MSE 10 13 10 10 13 -24 -04 -13 -17 -16 -14 -09 -08Max 168 116 113 126 116 226 184 186 169 169 176 155 109
NeutralRMSE 24 20 19 19 20 40 28 34 36 35 34 31 30MUE 18 15 15 15 15 30 19 24 29 28 25 24 24MSE 11 11 10 11 11 -26 -15 -21 -26 -26 -22 -11 -14Max 87 80 72 74 80 164 184 186 169 169 176 108 109
CationsRMSE 92 62 51 50 62 103 63 43 40 41 41 49 29MUE 85 55 40 39 55 100 58 36 34 35 35 40 24MSE 84 53 32 25 53 100 58 29 30 31 28 36 02Max 150 116 113 119 116 168 138 115 113 113 112 138 91
AnionsRMSE 79 41 42 45 41 142 36 43 46 46 46 67 44MUE 66 32 33 35 32 128 28 32 35 34 32 56 34MSE -61 -11 -13 -07 -11 -126 -06 -12 -14 -12 -13 -39 12Max 168 92 99 126 92 226 88 121 149 147 173 155 104
8
From the results discussed above and presented in Table 2 optimizing for atom typesbeyond HCON showed no significant increase in accuracy for either PM3 or PM6 There-fore the same procedure (optimizing HCON radii) was applied to DFTB For DFTB theRMSE values of the full set of molecules the subset of cation and the subset of anionsgoes from 41 49 and 67 kcalmol to 33 29 and 44 kcalmol respectively
In summary we find that optimizing the SMD radii in combination with either PM3 orPM6 gives appreciable decreases in RMSE accuracy asymp 7 kcalmol in both cases comparedto the reference results We also find that no significant improvement is obtained whenattempting to optimize the radii of other atoms than what was done with HCON subsetof radii For this subset DFTB also saw an increase in accuracy for ions of about asymp 6kcalmol Because of this we found the best choice was change the radii for hydrogencarbon oxygen and nitrogen (HCON) for PM3 and DFTB methods For PM6 we optimizedincluding sulfur and changed the radius of hydrogen to 06 A to remove overpolarizationproblems These methods are denoted as SMDdagger going forward
Comparison with Other Solvation MethodsUsing the new parameters for PM3SMDdagger PM6SMDdagger and DFTBSMDdagger we com-pare the obtained RMSE with other approaches to obtain solvent free energies such asHF6-31G(d)SMD (one of the levels of theory used in the original SMD parameteriza-tion) PM3COSMO and PM6COSMO for the MNSOL data set (Table 4 and Figure 3)
HFSMD
PM3SMDPM6SMD
DFTBSMD
PM3COSMO
PM6COSMO0
2
4
6
8
10
RMSE
[kca
lmol
]
Figure 3 RMSE (in kcalmol) of aqueous solvation energies computed with SMD using HF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicates that the Radiihas been reoptimized Corresponding RMSE values for the COSMO method are included forcomparison The dataset is split into full set (black) neutral (green) cations (red) and anions(blue)
The overall accuracy of PM3SMDdagger is now similar to HFSMD although the RMSEvalue is 04 and 06 kcalmol higher for neutral molecules and cations while the RMSE
9
is 07 kcalmol lower for anions For PM6 the overall RMSE is 14 kcalmol higher thanHF and the RMSE for neutral molecules is 23 kcalmol higher On the other hand theRMSE for cations is only 01 kcalmol higher while the RMSE for anions is 08 kcal-mol lower compared to HF Finally for DFTB the overall RMSE value (41 kcalmol)is in between those for PM3 and PM6 as is the RMSE values for neutral molecules (39kcalmol) But for cations the RMSE (37 kcalmol) is significantly lower than the othermethods while the RMSE for anions (51 kcalmol) is very similar
While the solvation free energies of neutral molecules can generally be measured with anaccuracy of ca 02 kcalmol the corresponding solvation free energies of ions are inferredfrom other experimental values with a typical accuracy about 30 kcalmol[40] Thus theaccuracy of the semiempirical SMDdagger is for all intents and purposes identical to HFSMDfor ions but significantly worse for neutral ions in the case of PM6 and DFTB Thesource of this error is most likely due to a poorer description of the electrostatic proper-ties compared to HF For example the error in dipole moments for HCNO compounds islarger for PM6 than for PM3[16]
Table 4 also lists values for PM3COSMO and PM6COSMO as implemented in MOPACfor comparison This COSMO implementation only evaluates the polar part of the solva-tion free energy and will not be as accurate as properly parameterized COSMO-RS[5] val-ues For PM3 the SMDdagger results are significantly lower than the corresponding COSMOresults while for PM6 there is only a significant improvement for cations
10
Table 4 RMSE MUE and MSE (in kcalmol) of aqueous solvation energies computed withSMD using HF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicatesthat the Radii has been reoptimized Corresponding RMSE values for the COSMO method areincluded for comparison The reference set is split into full set neutral cations and anions
SMD SMDdagger COSMO
HF PM3 PM6 DFTB PM3 PM6All
RMSE 34 36 48 41 48 50MUE 21 26 35 31 34 39MSE 03 10 -17 -11 05 -18Max 178 155 231 160 213 211
NeutralRMSE 24 28 47 39 36 47MUE 13 20 34 29 25 35MSE -09 09 -29 -21 -10 -34Max 178 155 231 160 213 211
CationsRMSE 50 56 51 37 74 73MUE 36 44 42 29 59 60MSE 31 36 38 10 56 59Max 140 127 124 91 186 183
AnionsRMSE 56 49 48 51 68 45MUE 46 37 36 40 58 38MSE 39 -03 -05 18 37 03Max 143 146 147 147 152 105
11
pKa PredictionPart of the impetus for this work were the relatively large error in pKa values predictedusing PM3COSMO compared to PM3SMD with the original SMD radii observed byJensen et al [35] so we use the new SMD radii to compute the pKa value using the samedata set The data is presented in Figure 4 and Table 5 and shows that the RMSE dropsfrom 15plusmn03 to 09plusmn03 pH units for PM3SMDdagger and 20plusmn04 to 16plusmn0304 pH unitsfor DFTBSMDdagger In both cases the improvement is primarily a result of the decreasein mean error (ME) ie the original radii led to a fairly systematic underestimation ofthe pKa values that has now been removed by the reparameterization For DFTB theincreased accuracy is also due to a larger number of low error predictions as is evident inFigure 4 In the case of PM3SMDdagger the accuracy is now very similar to PM3COSMOThe RMSE for PM6SMDdagger is similar to PM6COSMO and larger than for PM3SMDin analogy with the corresponding results for COSMO
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6
err
or
Figure 4 Plot of the errors in the predicted pKa values (pKaminus pKexpa )
12
Table 5 Root-Mean-Square-Error (RMSE) man error (ME) and Pearson correlation (r) for predicted pKa values relative to experiment togetherwith their statistical uncertainty (95 confidence limits)
PM3COSMO PM3SMD PM3SMDdagger PM6COSMO PM6SMDdagger DFTBSMD DFTBSMDdaggerRMSE 10 15 09 18 16 20 1695 conf plusmn 0202 plusmn 0303 plusmn 0202 plusmn 0304 plusmn 0304 plusmn 0404 plusmn 0304ME -05 plusmn 02 -12 plusmn 03 -02 plusmn 02 -13 plusmn 04 -08 plusmn 04 -07 plusmn 05 -01 plusmn 04r 095 093 093 090 086 087 08695 conf plusmn 002004 plusmn 003005 plusmn 003005 plusmn 004007 plusmn 006009 plusmn 005008 plusmn 006009
13
Non-aqueous SolventsThe new radii were optimized explicitly for water so it is not a priori clear whether theyoffer more accurate results for other solvents Table 6 shows the RMSE values for allneutral cations and anions for the other polar solvents in the MNSOL data set acetoni-trile DMSO and methanol for the original SMD radii and the corresponding re-optimizedradii (dagger) In addition corresponding combined RMSE values are given for the remaining88 mostly non-polar solvents in the MNSOL data set for which only data is availablefor neutral molecules It is clear from this data that the new radii sets lowers the errorof the predicted solvation free energies for the other polar solvents and has a negligibleeffect on the accuracy for the non-polar solvents
As for acetonitrile and DMSO the improvement is greatest for the ions where the RMSEdecreases by 3-12 kcalmol The improvement is more modest (05-3 kcalmol) for theneutral molecules For methanol which contains only ions the RMSE is similarly de-creased with 2-13 kcalmol
We thus recommend that the new radii optimized for water also be used for other polarsolvents when doing semiempirical SMD calculations For non-polar solvents either setof radii can be used
14
Table 6 RMSE (in kcalmol) of non-aqueous solvation energies computed with SMD usingHF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicates that theradii has been reoptimized Corresponding RMSE values for the COSMO method are includedfor comparison The reference set is split into full set neutral cations and anions
All Neu Cat Aniacetonitrile
HFSMD 112 29 105 131PM3SMD 194 41 162 246PM3SMDdagger 140 37 113 182PM6SMD 219 33 177 284PM6SMDdagger 130 12 114 160DFTBSMD 150 23 119 197DFTBSMDdagger 109 22 90 140PM3COSMO 130 31 128 147PM6COSMO 137 23 135 155
dimethylsulfoxideHFSMD 119 30 106 126PM3SMD 202 42 153 214PM3SMDdagger 170 35 90 182PM6SMD 255 45 172 272PM6SMDdagger 177 17 99 189DFTBSMD 169 23 108 181DFTBSMDdagger 125 21 76 134PM3COSMO 125 34 107 132PM6COSMO 150 31 119 159
methanolHFSMD 40 00 41 39PM3SMD 126 00 108 135PM3SMDdagger 66 00 60 69PM6SMD 163 00 120 183PM6SMDdagger 52 00 54 52DFTBSMD 84 00 63 94DFTBSMDdagger 47 00 44 49PM3COSMO 59 00 68 54PM6COSMO 54 00 67 46
otherHFSMD 42 42 00 00PM3SMD 51 51 00 00PM3SMDdagger 51 51 00 00PM6SMD 35 35 00 00PM6SMDdagger 34 34 00 00DFTBSMD 30 30 00 00DFTBSMDdagger 29 29 00 00PM3COSMO 36 36 00 00PM6COSMO 27 27 00 00
15
ConclusionThe PM6 method in GAMESS was extended to elements requiring d-integrals and inter-faced with the polarized continuum model (PCM) of solvation including semi-numericalgradients However the accuracy of aqueous solvation energies computed using AM1PM3 PM6 and DFTB and the SMD continuum solvation model was tested using a sub-set of molecules from the MNSOL data set which showed that the errors in SMD solvationenergies predicted using NDDO-based methods was considerably larger than when usingDFT and HF with RMSE values of 50 to 86 kcalmol compared to 34 kcalmol forHF6-31G(d) For the NDDO-based methods the errors were especially large for cationsand in the case of PM6 also for anions with RMSE values of 101 to 148 kcalmol incomparison with to 50 to 57 kcalmol for HF6-31G(d)SMD Corresponding COSMOresults (where the maximum RMSE is 75 kcalmol) suggested that the NDDOSMDresults could be improved by re-parameterizing the SMD Coulomb radii for the NDDOmethods
The fact that the NDDOSMD errors are largest for ions suggest that the problem iswith the polar solvation energy so we focus on optimizing the values of the Coulombradii while leaving all parameters associated with the non-polar solvation free energy un-changed We optimize the radii only for the ionic species on a subset of the MNSOL dataset previously used to parametrize RHF6-31G(d)SMD but include neutral moleculeswhen testing how well SMD with the new Coulomb radii perform We found the bestresults are obtained by changing only the radii for hydrogen carbon oxygen nitrogenand sulfur and this leads to RMSE values for PM3 (neutrals 28ions ca 5 kcalmol)PM6 (47ca 5 kcalmol) and DFTB (39ca 5 kcalmol) that are more comparable toHF6-31G(d) (24ca 5 kcalmol) We note that the SMD parameterization is done us-ing mono-aquo microsolvated species for small ions with atoms carrying large (partial)charges while the MNSOL data set also contains data for the non-microsolvated equiv-alents Comparison to the HF6-31G(d) MSEs reported by Marenich et al [8] reportedfor the selectively microsolvated ions indicates that the inclusion of non-microsolvatedspecies leads an MSE increase of about 1 kcalmol
Though the radii are optimized to reproduce aqueous solvation energies they also leadmore accurate predictions for other polar solvents such as DMSO acetonitrile andmethanol while the improvement for non-polar solvents are negligible
Supplementary MaterialSee supplementary material for Tables S1-S3 and Figures S1-S3 which are referred toin the text In addition we also provide a table of dielectric constants used for thecalculations and a table of aqueous solvation free energies obtained using the optimizedparameters
AcknowledgmentsJCK and JHJ would like to acknowledge Jimmy (Mr MOPAC) Stewart for providingsource code and guidance for the PM6 GAMESS implementation Also thanks to Donald
16
Truhlar and Christopher Cramer for sharing the MNSOL data set and for insightfulcomments on the manuscript CS thanks the Danish Council for Independent Research(the Sapere Aude program) for financial support (Grant no DFF 4181-00370)
References[1] J Tomasi B Mennucci and R Cammi ldquoQuantum mechanical continuum solvation
modelsrdquo Chem Rev vol 105 pp 2999ndash3093 2005
[2] A Klamt and G Schuurmann ldquoCOSMO a new approach to dielectric screeningin solvents with explicit expressions for the screening energy and its gradientrdquo JChem Soc Perkin Trans vol 2 pp 799ndash805 1993
[3] C J Cramer and D G Truhlar ldquoA universal approach to solvation modelingrdquoAccounts of Chemical Research vol 41 no 6 pp 760ndash768 jun 2008 doi101021ar800019z
[4] V Barone M Cossi and J Tomasi ldquoA new definition of cavities for thecomputation of solvation free energies by the polarizable continuum modelrdquo JChem Phys vol 107 no 8 pp 3210ndash3221 aug 1997 doi 1010631474671
[5] F Eckert and A Klamt ldquoFast solvent screening via quantum chemistryCOSMO-RS approachrdquo AIChE J vol 48 no 2 pp 369ndash385 feb 2002 doi101002aic690480220
[6] C Curutchet A Bidon-Chanal I Soteras M Orozco and F J Luque ldquoMSTcontinuum study of the hydration free energies of monovalent ionic speciesrdquo JPhys Chem B vol 109 no 8 pp 3565ndash3574 mar 2005 doi 101021jp047197s
[7] Y Takano and K N Houk ldquoBenchmarking the conductor-like polarizablecontinuum model (CPCM) for aqueous solvation free energies of neutral and ionicorganic moleculesrdquo J Chem Theory Comput vol 1 no 1 pp 70ndash77 jan 2005doi 101021ct049977a
[8] A V Marenich R M Olson C P Kelly C J Cramer and D G TruhlarldquoSelf-consistent reaction field model for aqueous and nonaqueous solutions based onaccurate polarized partial chargesrdquo Journal of Chemical Theory and Computationvol 3 no 6 pp 2011ndash2033 nov 2007 doi 101021ct7001418
[9] A V Marenich C J Cramer and D G Truhlar ldquoUniversal solvation model basedon solute electron density and on a continuum model of the solvent defined by thebulk dielectric constant and atomic surface tensionsrdquo J Phys Chem B vol 113no 18 pp 6378ndash6396 may 2009 doi 101021jp810292n
[10] G E Chudinov D V Napolov and M V Basilevsky ldquoQuantum-chemicalcalculations of the hydration energies of organic cations and anions in the frameworkof a continuum solvent approximationrdquo Chem Phys vol 160 no 1 pp 41 ndash 541992 doi 1010160301-0104(92)87090-V
[11] M J Negre M Orozco and F J Luque ldquoA new strategy for the representation ofenvironment effects in semi-empirical calculations based on dewarrsquos hamiltoniansrdquoChem Phys Lett vol 196 no 1 pp 27ndash36 1992
17
[12] F J Luque M J Negre and M Orozco ldquoAn am1-scrf approach to the study ofchanges in molecular properties induced by solventrdquo J Phys Chem vol 97 no 17pp 4386ndash4391 1993
[13] M Caricato B Mennucci and J Tomasi ldquoSolvent effects on the electronic spectraAn extension of the polarizable continuum model to the zindo methodrdquo J PhysChem A vol 108 no 29 pp 6248ndash6256 2004
[14] M J S Dewar E G Zoebisch E F Healy and J J P Stewart ldquoDevelopmentand use of quantum mechanical molecular models 76 AM1 a new general purposequantum mechanical molecular modelrdquo J A Chem Soc vol 10 pp 3902ndash39091985 doi 101021ja00299a024
[15] J J P Stewart ldquoOptimization of Parameters for Semi-Empirical Methods I-Methodrdquo J Comp Chem vol 10 pp 209ndash220 1989 doi 101002jcc540100208
[16] J J P Stewart ldquoOptimization of parameters for semiempirical methods V Modi-fication of NDDO approximations and application to 70 elementsrdquo J Mol Modelvol 13 pp 1172ndash1213 2007
[17] M Gaus Q Cui and M Elstner ldquoDFTB3 Extension of the Self-Consistent-Charge Density-Functional Tight-Binding Method (SCC-DFTB)rdquo J Chem TheoryComput vol 7 no 4 pp 931ndash948 apr 2011 doi 101021ct100684s
[18] A V Marenich C P Kelly J D Thompson G D Hawkins C C ChambersD J Giesen P P Winget C J Cramer and D G Truhlar ldquoMinnesota solvationdatabaserdquo 2012
[19] ldquoMinnesota solvation databaserdquo accessed November 24th 2017 [Online] Availablehttpscompchemumnedumnsol
[20] M W Schmidt K K Baldridge J A Boatz S T Elbert M S Gordon J HJensen S Koseki N Matsunaga K A Nguyen S Su T L Windus M Dupuisand JAMontgomery ldquoGeneral atomic and molecular electronic structure systemrdquoJ Comput Chem vol 14 pp 1347ndash1363 1993
[21] V Barone and M Cossi ldquoQuantum calculation of molecular energies and energygradients in solution by a conductor solvent modelrdquo The Journal of PhysicalChemistry A vol 102 no 11 pp 1995ndash2001 mar 1998 doi 101021jp9716997
[22] M Cossi N Rega G Scalmani and V Barone ldquoEnergies structures andelectronic properties of molecules in solution with the c-PCM solvation modelrdquoJournal of Computational Chemistry vol 24 no 6 pp 669ndash681 apr 2003 doi101002jcc10189
[23] C Steinmann K L Blaeligdel A S Christensen and J H Jensen ldquoInterface ofthe polarizable continuum model of solvation with semi-empirical methods in thegamess programrdquo PLoS ONE vol 8 no 7 p e67725 07 2013 doi 101371jour-nalpone0067725
[24] Y Nishimoto ldquoDFTBPCM applied to ground and excited state potential energysurfacesrdquo J Phys Chem A vol 120 no 5 pp 771ndash784 feb 2016 doi101021acsjpca5b10732
18
[25] M Gaus X Lu M Elstner and Q Cui ldquoParameterization of DFTB33OB forSulfur and Phosphorus for Chemical and Biological Applicationsrdquo J Chem TheoryComput vol 10 no 4 pp 1518ndash1537 apr 2014 doi 101021ct401002w
[26] X Lu M Gaus M Elstner and Q Cui ldquoParametrization of DFTB33OBfor Magnesium and Zinc for Chemical and Biological Applicationsrdquo The Journalof Physical Chemistry B vol 119 no 3 pp 1062ndash1082 jan 2015 doi101021jp506557r
[27] M Kubillus T Kubar M Gaus J Rezac and M Elstner ldquoParameterization ofthe DFTB3 Method for Br Ca Cl F I K and Na in Organic and BiologicalSystemsrdquo J Chem Theory Comput vol 11 no 1 pp 332ndash342 jan 2015 doi101021ct5009137
[28] J J P Stewart ldquoMOPAC2012rdquo 2016 Stewart Computational Chemistry ColoradoSprings CO USA [Online] Available httpopenmopacnet
[29] J L Pascual-Ahuir E Silla J Tomasi and R Bonaccorsi ldquoElectrostaticinteraction of a solute with a continuum improved description of the cavity and ofthe surface cavity bound charge distributionrdquo Journal of Computational Chemistryvol 8 no 6 pp 778ndash787 sep 1987 doi 101002jcc540080605
[30] P Su and H Li ldquoContinuous and smooth potential energy surface for conductorlikescreening solvation model using fixed points with variable areasrdquo The Journal ofChemical Physics vol 130 no 7 p 074109 feb 2009 doi 10106313077917
[31] J Tomasi B Mennucci and E Cances ldquoThe IEF version of the PCM solvationmethod an overview of a new method addressed to study molecular solutes at theQM ab initio levelrdquo Journal of Molecular Structure THEOCHEM vol 464 no1-3 pp 211ndash226 may 1999 doi 101016s0166-1280(98)00553-3
[32] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 1 the calculation of confidence intervalsrdquo J Comput Aided MolDes vol 28 no 9 pp 887ndash918 Sep 2014 doi 101007s10822-014-9753-z
[33] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 2 comparing methodsrdquo J Comput Aided Mol Des vol 30 no 2pp 103ndash126 Feb 2016 doi 101007s10822-016-9904-5
[34] J H Jensen ldquoWhich method is more accurate or errors have error barsrdquo PeerJPreprints vol 5 p e2693v1 2017 doi 107287peerjpreprints2693v1
[35] J H Jensen C J Swain and L Olsen ldquoPrediction of pKa values for druglikemolecules using semiempirical quantum chemical methodsrdquo The Journal of PhysicalChemistry A vol 121 no 3 pp 699ndash707 jan 2017 doi 101021acsjpca6b10990
[36] F Eckert and A Klamt ldquoAccurate prediction of basicity in aqueous solution withCOSMO-RSrdquo Journal of Computational Chemistry vol 27 no 1 pp 11ndash19 2005doi 101002jcc20309
[37] J A Nelder and R Mead ldquoA simplex method for function minimizationrdquo TheComputer Journal vol 7 no 4 pp 308ndash313 1965 doi 101093comjnl74308
19
[38] E Jones T Oliphant P Peterson et al ldquoSciPy Open source scientifictools for Pythonrdquo 2001 accessed November 24th 2017 [Online] Availablehttpwwwscipyorgrdquo
[39] J Charnley 2017 accessed November 24th 2017 [Online] Available httpsgithubcomcharnleyoptimize gamess parameters
[40] C P Kelly C J Cramer and D G Truhlar ldquoAqueous solvation free energies ofions and ion-water clusters based on an accurate value for the absolute aqueoussolvation free energy of the protonrdquo The Journal of Physical Chemistry B vol 110no 32 pp 16 066ndash16 081 aug 2006 doi 101021jp063552y
20
HFSMDAM1SMD
PM3SMDPM6SMD
DFTBSMD
AM1COSMO
PM3COSMO
PM6COSMO null0
2
4
6
8
10
12
14
16RM
SE [k
calm
ol]
PM3PM3HCO
PM3HCON
PM3HCONSPM3all PM6
PM6HCO
PM6HCON
PM6HCONS
PM6HCONShPM6all
DFTB
DFTBHCON0
2
4
6
8
10
12
14RM
SE [k
calm
ol]
HFSMD
PM3SMDPM6SMD
DFTBSMD
PM3COSMO
PM6COSMO0
2
4
6
8
10RM
SE [k
calm
ol]
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6err
or
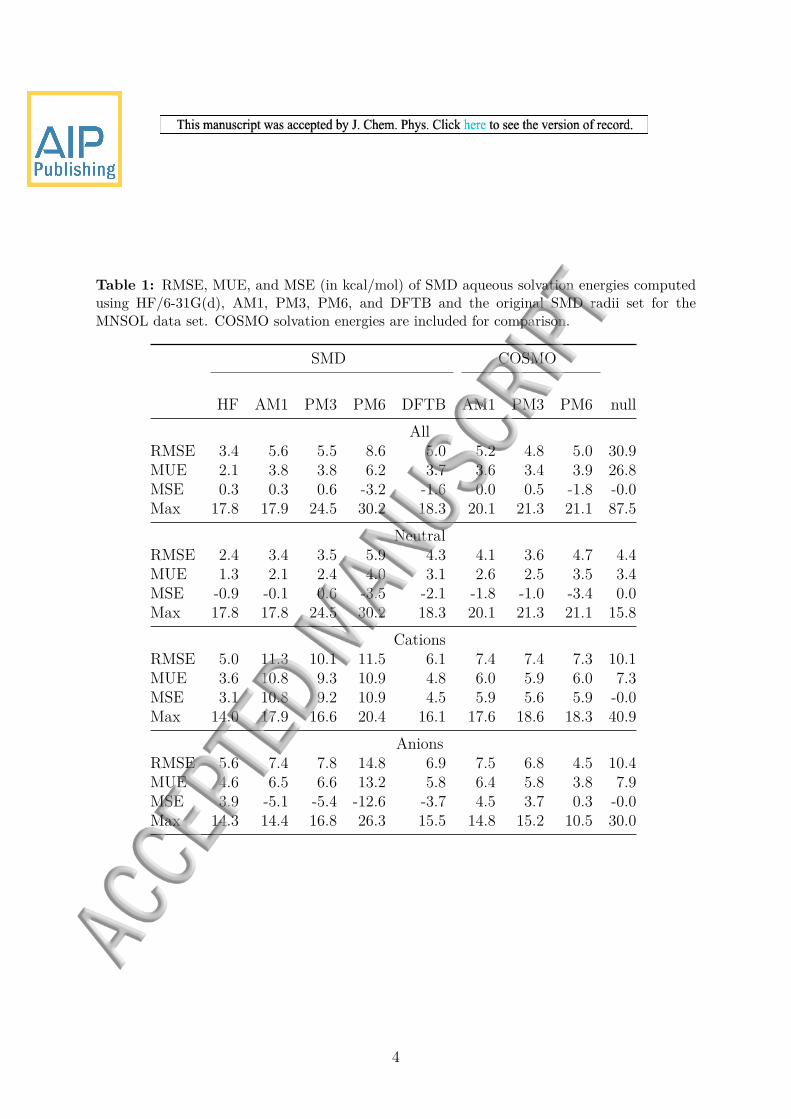
Table 1 RMSE MUE and MSE (in kcalmol) of SMD aqueous solvation energies computedusing HF6-31G(d) AM1 PM3 PM6 and DFTB and the original SMD radii set for theMNSOL data set COSMO solvation energies are included for comparison
SMD COSMO
HF AM1 PM3 PM6 DFTB AM1 PM3 PM6 nullAll
RMSE 34 56 55 86 50 52 48 50 309MUE 21 38 38 62 37 36 34 39 268MSE 03 03 06 -32 -16 00 05 -18 -00Max 178 179 245 302 183 201 213 211 875
NeutralRMSE 24 34 35 59 43 41 36 47 44MUE 13 21 24 40 31 26 25 35 34MSE -09 -01 06 -35 -21 -18 -10 -34 00Max 178 178 245 302 183 201 213 211 158
CationsRMSE 50 113 101 115 61 74 74 73 101MUE 36 108 93 109 48 60 59 60 73MSE 31 108 92 109 45 59 56 59 -00Max 140 179 166 204 161 176 186 183 409
AnionsRMSE 56 74 78 148 69 75 68 45 104MUE 46 65 66 132 58 64 58 38 79MSE 39 -51 -54 -126 -37 45 37 03 -00Max 143 144 168 263 155 148 152 105 300
4
COSMO-predicted solvation free energies have RMSE values for the full set of 52 48and 50 kcalmol for AM1 PM3 and PM6 respectively which are quite similar to thecorresponding SMD values for AM1 and PM3 However the accuracy of COSMO forthe ions are significantly better especially for cations with RMSE values of 73 74 and73 kcalmol for cations and 75 68 45 kcalmol for anions The COSMO results showthat is is possible to significantly improve the accuracy of the NDDOSMD calculationsand the comparatively high errors for ions indicate that the focus should be on the polarpart of the solvation free energy
Optimizing SMD Coulomb RadiiBased on the results in the previous subsection we optimize the Coulomb radii for PM3PM6 and DFTB independently while leaving the non-polar solvation free energy con-tribution unchanged We use the Nelder-Mead simplex method[37] to optimize the radiiusing the SMD data set with the SciPy[38] package using a custom optimization script[39]Optimization of the radii is done by minimizing the RMSE with respect to the radii
Initial tests showed that the solvation free energy does not vary greatly with a changein radii for neutral molecules and the flatness of the energy surface caused problems forthe Nelder-Mead method Furthermore the error for the ions is considerably larger thanfor the neutral molecules Thus only radii of the ions are optimized but when testingthe performance of the new radii neutral molecules are included This means that theoptimum parameters obtained for the ions will not necessarily lead to the lowest possibleerror for neutral molecules We therefore test the effect of optimizing a subset of theradii but include all molecules when testing the overall performance These results arepresented in Tables 2 and 3 and in Figure 2
For PM3 the results are relatively insensitive to the type of atoms chosen with RMSElowast
values of 44 to 47 kcalmol (Table 2 and Figure 2) (Note that the RMSE value arecomputed using different numbers of charged species so the RMSElowast does not necessarilydecrease when more parameters are optimized) Similarly the RMSE values computedusing all the molecules in the MNSOL data set range from 29 to 32 kcalmol as pre-sented in Table 3 The cations are most sensitive to the choice of atom types with anRMSE range of 50 to 62 kcalmol and the HCON- and HCONS-optimized radii set givingthe best and nearly identical accuracy For anions the RMSE for the HCON radii (42kcalmol) is lower than that for HCONS (45 kcalmol) and nearly identical to the lowestRMSE observed (41 kcalmol) The HCON-optimized radii are therefore the best com-promise
For PM6 the RMSElowast for HCO (32 kcalmol) is about 1 kcalmol lower than for the otherradii set However the RMSE values computed using all molecules are very similar witha range of 36 to 38 kcalmol with HCO and HCON tied for first place HCO does lead tothe lowest error for neutral molecules and anions but also an RMSE that is significantlylarger for cations (62 kcalmol) where PM6-HCONS performs best
When testing the PM6-HCON and PM6-HCONS radii on pKa prediction (see below) wefound that several solution-phase geometry optimizations failed because some X-H bonds
5
Table 2 Optimized Coulomb radii (in A) for PM3 PM6 and DFTB for different atom typesand original SMD radii parameters for comparison RMSESMD denotes the RMSE using theoriginal parameters and RMSElowast denotes RMSE after optimization The RMSE is given inkcalmol and is only calculated for the subset of ions in the SMD subset containing thosespecific atom types
H C N O F S Cl Br RMSESMD RMSElowast
SMD120 185 189 152 173 249 238 306
PM3029 172 165 96 44042 175 176 164 109 43062 176 172 162 258 107 46058 175 172 164 179 255 253 335 106 47
PM6066 187 173 123 32017 185 178 174 130 41039 178 182 175 262 127 43070 182 178 174 140 255 237 388 123 44
DFTB070 183 191 164 95 35
broke and the proton started moving into the solvent This indicated that the H radiusmight be too small leading to overpolarization of the H atom due to the solvent andwe tested several larger H radii for PM6-HCONS since the H radius was larger than forPM6-HCON We found that the overpolarization problem disappeared with a H radius of06 A and that using this value has a negligible 01 kcalmol effect of the accuracy ofthe solvation energies (HCONSh in Table 3) Thus we use PM6-HCONSh radii set goingforward
6
PM3PM3HCO
PM3HCON
PM3HCONSPM3all PM6
PM6HCO
PM6HCON
PM6HCONS
PM6HCONShPM6all
DFTB
DFTBHCON0
2
4
6
8
10
12
14
RMSE
[kca
lmol
]
Figure 2 RMSE values (in kcalmol) computed using SMD subset of the MNSOL data setand the Coulomb radii set shown in Table 2 HCO indicates that hydrogen carbon and oxygenradii parameters are changed (ie second and sixth entry in Table 2 The reference set is splitinto full set (black) neutral (green) cations (red) and anions (blue)
7
Table 3 RMSE MUE and MSE values (in kcalmol) computed using the SMD subset of the MNSOL data set and the Coulomb radii set shownin Table 2 HCO indicates that hydrogen carbon and oxygen radii parameters are changed (ie second and sixth row in Table 2)
PM3 PM6 DFTB
SMD HCO HCON HCONS all SMD HCO HCON HCONS HCONSh all SMD HCONAll
RMSE 50 32 29 30 32 75 36 36 38 38 37 41 33MUE 35 23 21 22 23 54 25 27 30 30 28 31 26MSE 10 13 10 10 13 -24 -04 -13 -17 -16 -14 -09 -08Max 168 116 113 126 116 226 184 186 169 169 176 155 109
NeutralRMSE 24 20 19 19 20 40 28 34 36 35 34 31 30MUE 18 15 15 15 15 30 19 24 29 28 25 24 24MSE 11 11 10 11 11 -26 -15 -21 -26 -26 -22 -11 -14Max 87 80 72 74 80 164 184 186 169 169 176 108 109
CationsRMSE 92 62 51 50 62 103 63 43 40 41 41 49 29MUE 85 55 40 39 55 100 58 36 34 35 35 40 24MSE 84 53 32 25 53 100 58 29 30 31 28 36 02Max 150 116 113 119 116 168 138 115 113 113 112 138 91
AnionsRMSE 79 41 42 45 41 142 36 43 46 46 46 67 44MUE 66 32 33 35 32 128 28 32 35 34 32 56 34MSE -61 -11 -13 -07 -11 -126 -06 -12 -14 -12 -13 -39 12Max 168 92 99 126 92 226 88 121 149 147 173 155 104
8
From the results discussed above and presented in Table 2 optimizing for atom typesbeyond HCON showed no significant increase in accuracy for either PM3 or PM6 There-fore the same procedure (optimizing HCON radii) was applied to DFTB For DFTB theRMSE values of the full set of molecules the subset of cation and the subset of anionsgoes from 41 49 and 67 kcalmol to 33 29 and 44 kcalmol respectively
In summary we find that optimizing the SMD radii in combination with either PM3 orPM6 gives appreciable decreases in RMSE accuracy asymp 7 kcalmol in both cases comparedto the reference results We also find that no significant improvement is obtained whenattempting to optimize the radii of other atoms than what was done with HCON subsetof radii For this subset DFTB also saw an increase in accuracy for ions of about asymp 6kcalmol Because of this we found the best choice was change the radii for hydrogencarbon oxygen and nitrogen (HCON) for PM3 and DFTB methods For PM6 we optimizedincluding sulfur and changed the radius of hydrogen to 06 A to remove overpolarizationproblems These methods are denoted as SMDdagger going forward
Comparison with Other Solvation MethodsUsing the new parameters for PM3SMDdagger PM6SMDdagger and DFTBSMDdagger we com-pare the obtained RMSE with other approaches to obtain solvent free energies such asHF6-31G(d)SMD (one of the levels of theory used in the original SMD parameteriza-tion) PM3COSMO and PM6COSMO for the MNSOL data set (Table 4 and Figure 3)
HFSMD
PM3SMDPM6SMD
DFTBSMD
PM3COSMO
PM6COSMO0
2
4
6
8
10
RMSE
[kca
lmol
]
Figure 3 RMSE (in kcalmol) of aqueous solvation energies computed with SMD using HF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicates that the Radiihas been reoptimized Corresponding RMSE values for the COSMO method are included forcomparison The dataset is split into full set (black) neutral (green) cations (red) and anions(blue)
The overall accuracy of PM3SMDdagger is now similar to HFSMD although the RMSEvalue is 04 and 06 kcalmol higher for neutral molecules and cations while the RMSE
9
is 07 kcalmol lower for anions For PM6 the overall RMSE is 14 kcalmol higher thanHF and the RMSE for neutral molecules is 23 kcalmol higher On the other hand theRMSE for cations is only 01 kcalmol higher while the RMSE for anions is 08 kcal-mol lower compared to HF Finally for DFTB the overall RMSE value (41 kcalmol)is in between those for PM3 and PM6 as is the RMSE values for neutral molecules (39kcalmol) But for cations the RMSE (37 kcalmol) is significantly lower than the othermethods while the RMSE for anions (51 kcalmol) is very similar
While the solvation free energies of neutral molecules can generally be measured with anaccuracy of ca 02 kcalmol the corresponding solvation free energies of ions are inferredfrom other experimental values with a typical accuracy about 30 kcalmol[40] Thus theaccuracy of the semiempirical SMDdagger is for all intents and purposes identical to HFSMDfor ions but significantly worse for neutral ions in the case of PM6 and DFTB Thesource of this error is most likely due to a poorer description of the electrostatic proper-ties compared to HF For example the error in dipole moments for HCNO compounds islarger for PM6 than for PM3[16]
Table 4 also lists values for PM3COSMO and PM6COSMO as implemented in MOPACfor comparison This COSMO implementation only evaluates the polar part of the solva-tion free energy and will not be as accurate as properly parameterized COSMO-RS[5] val-ues For PM3 the SMDdagger results are significantly lower than the corresponding COSMOresults while for PM6 there is only a significant improvement for cations
10
Table 4 RMSE MUE and MSE (in kcalmol) of aqueous solvation energies computed withSMD using HF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicatesthat the Radii has been reoptimized Corresponding RMSE values for the COSMO method areincluded for comparison The reference set is split into full set neutral cations and anions
SMD SMDdagger COSMO
HF PM3 PM6 DFTB PM3 PM6All
RMSE 34 36 48 41 48 50MUE 21 26 35 31 34 39MSE 03 10 -17 -11 05 -18Max 178 155 231 160 213 211
NeutralRMSE 24 28 47 39 36 47MUE 13 20 34 29 25 35MSE -09 09 -29 -21 -10 -34Max 178 155 231 160 213 211
CationsRMSE 50 56 51 37 74 73MUE 36 44 42 29 59 60MSE 31 36 38 10 56 59Max 140 127 124 91 186 183
AnionsRMSE 56 49 48 51 68 45MUE 46 37 36 40 58 38MSE 39 -03 -05 18 37 03Max 143 146 147 147 152 105
11
pKa PredictionPart of the impetus for this work were the relatively large error in pKa values predictedusing PM3COSMO compared to PM3SMD with the original SMD radii observed byJensen et al [35] so we use the new SMD radii to compute the pKa value using the samedata set The data is presented in Figure 4 and Table 5 and shows that the RMSE dropsfrom 15plusmn03 to 09plusmn03 pH units for PM3SMDdagger and 20plusmn04 to 16plusmn0304 pH unitsfor DFTBSMDdagger In both cases the improvement is primarily a result of the decreasein mean error (ME) ie the original radii led to a fairly systematic underestimation ofthe pKa values that has now been removed by the reparameterization For DFTB theincreased accuracy is also due to a larger number of low error predictions as is evident inFigure 4 In the case of PM3SMDdagger the accuracy is now very similar to PM3COSMOThe RMSE for PM6SMDdagger is similar to PM6COSMO and larger than for PM3SMDin analogy with the corresponding results for COSMO
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6
err
or
Figure 4 Plot of the errors in the predicted pKa values (pKaminus pKexpa )
12
Table 5 Root-Mean-Square-Error (RMSE) man error (ME) and Pearson correlation (r) for predicted pKa values relative to experiment togetherwith their statistical uncertainty (95 confidence limits)
PM3COSMO PM3SMD PM3SMDdagger PM6COSMO PM6SMDdagger DFTBSMD DFTBSMDdaggerRMSE 10 15 09 18 16 20 1695 conf plusmn 0202 plusmn 0303 plusmn 0202 plusmn 0304 plusmn 0304 plusmn 0404 plusmn 0304ME -05 plusmn 02 -12 plusmn 03 -02 plusmn 02 -13 plusmn 04 -08 plusmn 04 -07 plusmn 05 -01 plusmn 04r 095 093 093 090 086 087 08695 conf plusmn 002004 plusmn 003005 plusmn 003005 plusmn 004007 plusmn 006009 plusmn 005008 plusmn 006009
13
Non-aqueous SolventsThe new radii were optimized explicitly for water so it is not a priori clear whether theyoffer more accurate results for other solvents Table 6 shows the RMSE values for allneutral cations and anions for the other polar solvents in the MNSOL data set acetoni-trile DMSO and methanol for the original SMD radii and the corresponding re-optimizedradii (dagger) In addition corresponding combined RMSE values are given for the remaining88 mostly non-polar solvents in the MNSOL data set for which only data is availablefor neutral molecules It is clear from this data that the new radii sets lowers the errorof the predicted solvation free energies for the other polar solvents and has a negligibleeffect on the accuracy for the non-polar solvents
As for acetonitrile and DMSO the improvement is greatest for the ions where the RMSEdecreases by 3-12 kcalmol The improvement is more modest (05-3 kcalmol) for theneutral molecules For methanol which contains only ions the RMSE is similarly de-creased with 2-13 kcalmol
We thus recommend that the new radii optimized for water also be used for other polarsolvents when doing semiempirical SMD calculations For non-polar solvents either setof radii can be used
14
Table 6 RMSE (in kcalmol) of non-aqueous solvation energies computed with SMD usingHF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicates that theradii has been reoptimized Corresponding RMSE values for the COSMO method are includedfor comparison The reference set is split into full set neutral cations and anions
All Neu Cat Aniacetonitrile
HFSMD 112 29 105 131PM3SMD 194 41 162 246PM3SMDdagger 140 37 113 182PM6SMD 219 33 177 284PM6SMDdagger 130 12 114 160DFTBSMD 150 23 119 197DFTBSMDdagger 109 22 90 140PM3COSMO 130 31 128 147PM6COSMO 137 23 135 155
dimethylsulfoxideHFSMD 119 30 106 126PM3SMD 202 42 153 214PM3SMDdagger 170 35 90 182PM6SMD 255 45 172 272PM6SMDdagger 177 17 99 189DFTBSMD 169 23 108 181DFTBSMDdagger 125 21 76 134PM3COSMO 125 34 107 132PM6COSMO 150 31 119 159
methanolHFSMD 40 00 41 39PM3SMD 126 00 108 135PM3SMDdagger 66 00 60 69PM6SMD 163 00 120 183PM6SMDdagger 52 00 54 52DFTBSMD 84 00 63 94DFTBSMDdagger 47 00 44 49PM3COSMO 59 00 68 54PM6COSMO 54 00 67 46
otherHFSMD 42 42 00 00PM3SMD 51 51 00 00PM3SMDdagger 51 51 00 00PM6SMD 35 35 00 00PM6SMDdagger 34 34 00 00DFTBSMD 30 30 00 00DFTBSMDdagger 29 29 00 00PM3COSMO 36 36 00 00PM6COSMO 27 27 00 00
15
ConclusionThe PM6 method in GAMESS was extended to elements requiring d-integrals and inter-faced with the polarized continuum model (PCM) of solvation including semi-numericalgradients However the accuracy of aqueous solvation energies computed using AM1PM3 PM6 and DFTB and the SMD continuum solvation model was tested using a sub-set of molecules from the MNSOL data set which showed that the errors in SMD solvationenergies predicted using NDDO-based methods was considerably larger than when usingDFT and HF with RMSE values of 50 to 86 kcalmol compared to 34 kcalmol forHF6-31G(d) For the NDDO-based methods the errors were especially large for cationsand in the case of PM6 also for anions with RMSE values of 101 to 148 kcalmol incomparison with to 50 to 57 kcalmol for HF6-31G(d)SMD Corresponding COSMOresults (where the maximum RMSE is 75 kcalmol) suggested that the NDDOSMDresults could be improved by re-parameterizing the SMD Coulomb radii for the NDDOmethods
The fact that the NDDOSMD errors are largest for ions suggest that the problem iswith the polar solvation energy so we focus on optimizing the values of the Coulombradii while leaving all parameters associated with the non-polar solvation free energy un-changed We optimize the radii only for the ionic species on a subset of the MNSOL dataset previously used to parametrize RHF6-31G(d)SMD but include neutral moleculeswhen testing how well SMD with the new Coulomb radii perform We found the bestresults are obtained by changing only the radii for hydrogen carbon oxygen nitrogenand sulfur and this leads to RMSE values for PM3 (neutrals 28ions ca 5 kcalmol)PM6 (47ca 5 kcalmol) and DFTB (39ca 5 kcalmol) that are more comparable toHF6-31G(d) (24ca 5 kcalmol) We note that the SMD parameterization is done us-ing mono-aquo microsolvated species for small ions with atoms carrying large (partial)charges while the MNSOL data set also contains data for the non-microsolvated equiv-alents Comparison to the HF6-31G(d) MSEs reported by Marenich et al [8] reportedfor the selectively microsolvated ions indicates that the inclusion of non-microsolvatedspecies leads an MSE increase of about 1 kcalmol
Though the radii are optimized to reproduce aqueous solvation energies they also leadmore accurate predictions for other polar solvents such as DMSO acetonitrile andmethanol while the improvement for non-polar solvents are negligible
Supplementary MaterialSee supplementary material for Tables S1-S3 and Figures S1-S3 which are referred toin the text In addition we also provide a table of dielectric constants used for thecalculations and a table of aqueous solvation free energies obtained using the optimizedparameters
AcknowledgmentsJCK and JHJ would like to acknowledge Jimmy (Mr MOPAC) Stewart for providingsource code and guidance for the PM6 GAMESS implementation Also thanks to Donald
16
Truhlar and Christopher Cramer for sharing the MNSOL data set and for insightfulcomments on the manuscript CS thanks the Danish Council for Independent Research(the Sapere Aude program) for financial support (Grant no DFF 4181-00370)
References[1] J Tomasi B Mennucci and R Cammi ldquoQuantum mechanical continuum solvation
modelsrdquo Chem Rev vol 105 pp 2999ndash3093 2005
[2] A Klamt and G Schuurmann ldquoCOSMO a new approach to dielectric screeningin solvents with explicit expressions for the screening energy and its gradientrdquo JChem Soc Perkin Trans vol 2 pp 799ndash805 1993
[3] C J Cramer and D G Truhlar ldquoA universal approach to solvation modelingrdquoAccounts of Chemical Research vol 41 no 6 pp 760ndash768 jun 2008 doi101021ar800019z
[4] V Barone M Cossi and J Tomasi ldquoA new definition of cavities for thecomputation of solvation free energies by the polarizable continuum modelrdquo JChem Phys vol 107 no 8 pp 3210ndash3221 aug 1997 doi 1010631474671
[5] F Eckert and A Klamt ldquoFast solvent screening via quantum chemistryCOSMO-RS approachrdquo AIChE J vol 48 no 2 pp 369ndash385 feb 2002 doi101002aic690480220
[6] C Curutchet A Bidon-Chanal I Soteras M Orozco and F J Luque ldquoMSTcontinuum study of the hydration free energies of monovalent ionic speciesrdquo JPhys Chem B vol 109 no 8 pp 3565ndash3574 mar 2005 doi 101021jp047197s
[7] Y Takano and K N Houk ldquoBenchmarking the conductor-like polarizablecontinuum model (CPCM) for aqueous solvation free energies of neutral and ionicorganic moleculesrdquo J Chem Theory Comput vol 1 no 1 pp 70ndash77 jan 2005doi 101021ct049977a
[8] A V Marenich R M Olson C P Kelly C J Cramer and D G TruhlarldquoSelf-consistent reaction field model for aqueous and nonaqueous solutions based onaccurate polarized partial chargesrdquo Journal of Chemical Theory and Computationvol 3 no 6 pp 2011ndash2033 nov 2007 doi 101021ct7001418
[9] A V Marenich C J Cramer and D G Truhlar ldquoUniversal solvation model basedon solute electron density and on a continuum model of the solvent defined by thebulk dielectric constant and atomic surface tensionsrdquo J Phys Chem B vol 113no 18 pp 6378ndash6396 may 2009 doi 101021jp810292n
[10] G E Chudinov D V Napolov and M V Basilevsky ldquoQuantum-chemicalcalculations of the hydration energies of organic cations and anions in the frameworkof a continuum solvent approximationrdquo Chem Phys vol 160 no 1 pp 41 ndash 541992 doi 1010160301-0104(92)87090-V
[11] M J Negre M Orozco and F J Luque ldquoA new strategy for the representation ofenvironment effects in semi-empirical calculations based on dewarrsquos hamiltoniansrdquoChem Phys Lett vol 196 no 1 pp 27ndash36 1992
17
[12] F J Luque M J Negre and M Orozco ldquoAn am1-scrf approach to the study ofchanges in molecular properties induced by solventrdquo J Phys Chem vol 97 no 17pp 4386ndash4391 1993
[13] M Caricato B Mennucci and J Tomasi ldquoSolvent effects on the electronic spectraAn extension of the polarizable continuum model to the zindo methodrdquo J PhysChem A vol 108 no 29 pp 6248ndash6256 2004
[14] M J S Dewar E G Zoebisch E F Healy and J J P Stewart ldquoDevelopmentand use of quantum mechanical molecular models 76 AM1 a new general purposequantum mechanical molecular modelrdquo J A Chem Soc vol 10 pp 3902ndash39091985 doi 101021ja00299a024
[15] J J P Stewart ldquoOptimization of Parameters for Semi-Empirical Methods I-Methodrdquo J Comp Chem vol 10 pp 209ndash220 1989 doi 101002jcc540100208
[16] J J P Stewart ldquoOptimization of parameters for semiempirical methods V Modi-fication of NDDO approximations and application to 70 elementsrdquo J Mol Modelvol 13 pp 1172ndash1213 2007
[17] M Gaus Q Cui and M Elstner ldquoDFTB3 Extension of the Self-Consistent-Charge Density-Functional Tight-Binding Method (SCC-DFTB)rdquo J Chem TheoryComput vol 7 no 4 pp 931ndash948 apr 2011 doi 101021ct100684s
[18] A V Marenich C P Kelly J D Thompson G D Hawkins C C ChambersD J Giesen P P Winget C J Cramer and D G Truhlar ldquoMinnesota solvationdatabaserdquo 2012
[19] ldquoMinnesota solvation databaserdquo accessed November 24th 2017 [Online] Availablehttpscompchemumnedumnsol
[20] M W Schmidt K K Baldridge J A Boatz S T Elbert M S Gordon J HJensen S Koseki N Matsunaga K A Nguyen S Su T L Windus M Dupuisand JAMontgomery ldquoGeneral atomic and molecular electronic structure systemrdquoJ Comput Chem vol 14 pp 1347ndash1363 1993
[21] V Barone and M Cossi ldquoQuantum calculation of molecular energies and energygradients in solution by a conductor solvent modelrdquo The Journal of PhysicalChemistry A vol 102 no 11 pp 1995ndash2001 mar 1998 doi 101021jp9716997
[22] M Cossi N Rega G Scalmani and V Barone ldquoEnergies structures andelectronic properties of molecules in solution with the c-PCM solvation modelrdquoJournal of Computational Chemistry vol 24 no 6 pp 669ndash681 apr 2003 doi101002jcc10189
[23] C Steinmann K L Blaeligdel A S Christensen and J H Jensen ldquoInterface ofthe polarizable continuum model of solvation with semi-empirical methods in thegamess programrdquo PLoS ONE vol 8 no 7 p e67725 07 2013 doi 101371jour-nalpone0067725
[24] Y Nishimoto ldquoDFTBPCM applied to ground and excited state potential energysurfacesrdquo J Phys Chem A vol 120 no 5 pp 771ndash784 feb 2016 doi101021acsjpca5b10732
18
[25] M Gaus X Lu M Elstner and Q Cui ldquoParameterization of DFTB33OB forSulfur and Phosphorus for Chemical and Biological Applicationsrdquo J Chem TheoryComput vol 10 no 4 pp 1518ndash1537 apr 2014 doi 101021ct401002w
[26] X Lu M Gaus M Elstner and Q Cui ldquoParametrization of DFTB33OBfor Magnesium and Zinc for Chemical and Biological Applicationsrdquo The Journalof Physical Chemistry B vol 119 no 3 pp 1062ndash1082 jan 2015 doi101021jp506557r
[27] M Kubillus T Kubar M Gaus J Rezac and M Elstner ldquoParameterization ofthe DFTB3 Method for Br Ca Cl F I K and Na in Organic and BiologicalSystemsrdquo J Chem Theory Comput vol 11 no 1 pp 332ndash342 jan 2015 doi101021ct5009137
[28] J J P Stewart ldquoMOPAC2012rdquo 2016 Stewart Computational Chemistry ColoradoSprings CO USA [Online] Available httpopenmopacnet
[29] J L Pascual-Ahuir E Silla J Tomasi and R Bonaccorsi ldquoElectrostaticinteraction of a solute with a continuum improved description of the cavity and ofthe surface cavity bound charge distributionrdquo Journal of Computational Chemistryvol 8 no 6 pp 778ndash787 sep 1987 doi 101002jcc540080605
[30] P Su and H Li ldquoContinuous and smooth potential energy surface for conductorlikescreening solvation model using fixed points with variable areasrdquo The Journal ofChemical Physics vol 130 no 7 p 074109 feb 2009 doi 10106313077917
[31] J Tomasi B Mennucci and E Cances ldquoThe IEF version of the PCM solvationmethod an overview of a new method addressed to study molecular solutes at theQM ab initio levelrdquo Journal of Molecular Structure THEOCHEM vol 464 no1-3 pp 211ndash226 may 1999 doi 101016s0166-1280(98)00553-3
[32] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 1 the calculation of confidence intervalsrdquo J Comput Aided MolDes vol 28 no 9 pp 887ndash918 Sep 2014 doi 101007s10822-014-9753-z
[33] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 2 comparing methodsrdquo J Comput Aided Mol Des vol 30 no 2pp 103ndash126 Feb 2016 doi 101007s10822-016-9904-5
[34] J H Jensen ldquoWhich method is more accurate or errors have error barsrdquo PeerJPreprints vol 5 p e2693v1 2017 doi 107287peerjpreprints2693v1
[35] J H Jensen C J Swain and L Olsen ldquoPrediction of pKa values for druglikemolecules using semiempirical quantum chemical methodsrdquo The Journal of PhysicalChemistry A vol 121 no 3 pp 699ndash707 jan 2017 doi 101021acsjpca6b10990
[36] F Eckert and A Klamt ldquoAccurate prediction of basicity in aqueous solution withCOSMO-RSrdquo Journal of Computational Chemistry vol 27 no 1 pp 11ndash19 2005doi 101002jcc20309
[37] J A Nelder and R Mead ldquoA simplex method for function minimizationrdquo TheComputer Journal vol 7 no 4 pp 308ndash313 1965 doi 101093comjnl74308
19
[38] E Jones T Oliphant P Peterson et al ldquoSciPy Open source scientifictools for Pythonrdquo 2001 accessed November 24th 2017 [Online] Availablehttpwwwscipyorgrdquo
[39] J Charnley 2017 accessed November 24th 2017 [Online] Available httpsgithubcomcharnleyoptimize gamess parameters
[40] C P Kelly C J Cramer and D G Truhlar ldquoAqueous solvation free energies ofions and ion-water clusters based on an accurate value for the absolute aqueoussolvation free energy of the protonrdquo The Journal of Physical Chemistry B vol 110no 32 pp 16 066ndash16 081 aug 2006 doi 101021jp063552y
20
HFSMDAM1SMD
PM3SMDPM6SMD
DFTBSMD
AM1COSMO
PM3COSMO
PM6COSMO null0
2
4
6
8
10
12
14
16RM
SE [k
calm
ol]
PM3PM3HCO
PM3HCON
PM3HCONSPM3all PM6
PM6HCO
PM6HCON
PM6HCONS
PM6HCONShPM6all
DFTB
DFTBHCON0
2
4
6
8
10
12
14RM
SE [k
calm
ol]
HFSMD
PM3SMDPM6SMD
DFTBSMD
PM3COSMO
PM6COSMO0
2
4
6
8
10RM
SE [k
calm
ol]
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6err
or

COSMO-predicted solvation free energies have RMSE values for the full set of 52 48and 50 kcalmol for AM1 PM3 and PM6 respectively which are quite similar to thecorresponding SMD values for AM1 and PM3 However the accuracy of COSMO forthe ions are significantly better especially for cations with RMSE values of 73 74 and73 kcalmol for cations and 75 68 45 kcalmol for anions The COSMO results showthat is is possible to significantly improve the accuracy of the NDDOSMD calculationsand the comparatively high errors for ions indicate that the focus should be on the polarpart of the solvation free energy
Optimizing SMD Coulomb RadiiBased on the results in the previous subsection we optimize the Coulomb radii for PM3PM6 and DFTB independently while leaving the non-polar solvation free energy con-tribution unchanged We use the Nelder-Mead simplex method[37] to optimize the radiiusing the SMD data set with the SciPy[38] package using a custom optimization script[39]Optimization of the radii is done by minimizing the RMSE with respect to the radii
Initial tests showed that the solvation free energy does not vary greatly with a changein radii for neutral molecules and the flatness of the energy surface caused problems forthe Nelder-Mead method Furthermore the error for the ions is considerably larger thanfor the neutral molecules Thus only radii of the ions are optimized but when testingthe performance of the new radii neutral molecules are included This means that theoptimum parameters obtained for the ions will not necessarily lead to the lowest possibleerror for neutral molecules We therefore test the effect of optimizing a subset of theradii but include all molecules when testing the overall performance These results arepresented in Tables 2 and 3 and in Figure 2
For PM3 the results are relatively insensitive to the type of atoms chosen with RMSElowast
values of 44 to 47 kcalmol (Table 2 and Figure 2) (Note that the RMSE value arecomputed using different numbers of charged species so the RMSElowast does not necessarilydecrease when more parameters are optimized) Similarly the RMSE values computedusing all the molecules in the MNSOL data set range from 29 to 32 kcalmol as pre-sented in Table 3 The cations are most sensitive to the choice of atom types with anRMSE range of 50 to 62 kcalmol and the HCON- and HCONS-optimized radii set givingthe best and nearly identical accuracy For anions the RMSE for the HCON radii (42kcalmol) is lower than that for HCONS (45 kcalmol) and nearly identical to the lowestRMSE observed (41 kcalmol) The HCON-optimized radii are therefore the best com-promise
For PM6 the RMSElowast for HCO (32 kcalmol) is about 1 kcalmol lower than for the otherradii set However the RMSE values computed using all molecules are very similar witha range of 36 to 38 kcalmol with HCO and HCON tied for first place HCO does lead tothe lowest error for neutral molecules and anions but also an RMSE that is significantlylarger for cations (62 kcalmol) where PM6-HCONS performs best
When testing the PM6-HCON and PM6-HCONS radii on pKa prediction (see below) wefound that several solution-phase geometry optimizations failed because some X-H bonds
5
Table 2 Optimized Coulomb radii (in A) for PM3 PM6 and DFTB for different atom typesand original SMD radii parameters for comparison RMSESMD denotes the RMSE using theoriginal parameters and RMSElowast denotes RMSE after optimization The RMSE is given inkcalmol and is only calculated for the subset of ions in the SMD subset containing thosespecific atom types
H C N O F S Cl Br RMSESMD RMSElowast
SMD120 185 189 152 173 249 238 306
PM3029 172 165 96 44042 175 176 164 109 43062 176 172 162 258 107 46058 175 172 164 179 255 253 335 106 47
PM6066 187 173 123 32017 185 178 174 130 41039 178 182 175 262 127 43070 182 178 174 140 255 237 388 123 44
DFTB070 183 191 164 95 35
broke and the proton started moving into the solvent This indicated that the H radiusmight be too small leading to overpolarization of the H atom due to the solvent andwe tested several larger H radii for PM6-HCONS since the H radius was larger than forPM6-HCON We found that the overpolarization problem disappeared with a H radius of06 A and that using this value has a negligible 01 kcalmol effect of the accuracy ofthe solvation energies (HCONSh in Table 3) Thus we use PM6-HCONSh radii set goingforward
6
PM3PM3HCO
PM3HCON
PM3HCONSPM3all PM6
PM6HCO
PM6HCON
PM6HCONS
PM6HCONShPM6all
DFTB
DFTBHCON0
2
4
6
8
10
12
14
RMSE
[kca
lmol
]
Figure 2 RMSE values (in kcalmol) computed using SMD subset of the MNSOL data setand the Coulomb radii set shown in Table 2 HCO indicates that hydrogen carbon and oxygenradii parameters are changed (ie second and sixth entry in Table 2 The reference set is splitinto full set (black) neutral (green) cations (red) and anions (blue)
7
Table 3 RMSE MUE and MSE values (in kcalmol) computed using the SMD subset of the MNSOL data set and the Coulomb radii set shownin Table 2 HCO indicates that hydrogen carbon and oxygen radii parameters are changed (ie second and sixth row in Table 2)
PM3 PM6 DFTB
SMD HCO HCON HCONS all SMD HCO HCON HCONS HCONSh all SMD HCONAll
RMSE 50 32 29 30 32 75 36 36 38 38 37 41 33MUE 35 23 21 22 23 54 25 27 30 30 28 31 26MSE 10 13 10 10 13 -24 -04 -13 -17 -16 -14 -09 -08Max 168 116 113 126 116 226 184 186 169 169 176 155 109
NeutralRMSE 24 20 19 19 20 40 28 34 36 35 34 31 30MUE 18 15 15 15 15 30 19 24 29 28 25 24 24MSE 11 11 10 11 11 -26 -15 -21 -26 -26 -22 -11 -14Max 87 80 72 74 80 164 184 186 169 169 176 108 109
CationsRMSE 92 62 51 50 62 103 63 43 40 41 41 49 29MUE 85 55 40 39 55 100 58 36 34 35 35 40 24MSE 84 53 32 25 53 100 58 29 30 31 28 36 02Max 150 116 113 119 116 168 138 115 113 113 112 138 91
AnionsRMSE 79 41 42 45 41 142 36 43 46 46 46 67 44MUE 66 32 33 35 32 128 28 32 35 34 32 56 34MSE -61 -11 -13 -07 -11 -126 -06 -12 -14 -12 -13 -39 12Max 168 92 99 126 92 226 88 121 149 147 173 155 104
8
From the results discussed above and presented in Table 2 optimizing for atom typesbeyond HCON showed no significant increase in accuracy for either PM3 or PM6 There-fore the same procedure (optimizing HCON radii) was applied to DFTB For DFTB theRMSE values of the full set of molecules the subset of cation and the subset of anionsgoes from 41 49 and 67 kcalmol to 33 29 and 44 kcalmol respectively
In summary we find that optimizing the SMD radii in combination with either PM3 orPM6 gives appreciable decreases in RMSE accuracy asymp 7 kcalmol in both cases comparedto the reference results We also find that no significant improvement is obtained whenattempting to optimize the radii of other atoms than what was done with HCON subsetof radii For this subset DFTB also saw an increase in accuracy for ions of about asymp 6kcalmol Because of this we found the best choice was change the radii for hydrogencarbon oxygen and nitrogen (HCON) for PM3 and DFTB methods For PM6 we optimizedincluding sulfur and changed the radius of hydrogen to 06 A to remove overpolarizationproblems These methods are denoted as SMDdagger going forward
Comparison with Other Solvation MethodsUsing the new parameters for PM3SMDdagger PM6SMDdagger and DFTBSMDdagger we com-pare the obtained RMSE with other approaches to obtain solvent free energies such asHF6-31G(d)SMD (one of the levels of theory used in the original SMD parameteriza-tion) PM3COSMO and PM6COSMO for the MNSOL data set (Table 4 and Figure 3)
HFSMD
PM3SMDPM6SMD
DFTBSMD
PM3COSMO
PM6COSMO0
2
4
6
8
10
RMSE
[kca
lmol
]
Figure 3 RMSE (in kcalmol) of aqueous solvation energies computed with SMD using HF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicates that the Radiihas been reoptimized Corresponding RMSE values for the COSMO method are included forcomparison The dataset is split into full set (black) neutral (green) cations (red) and anions(blue)
The overall accuracy of PM3SMDdagger is now similar to HFSMD although the RMSEvalue is 04 and 06 kcalmol higher for neutral molecules and cations while the RMSE
9
is 07 kcalmol lower for anions For PM6 the overall RMSE is 14 kcalmol higher thanHF and the RMSE for neutral molecules is 23 kcalmol higher On the other hand theRMSE for cations is only 01 kcalmol higher while the RMSE for anions is 08 kcal-mol lower compared to HF Finally for DFTB the overall RMSE value (41 kcalmol)is in between those for PM3 and PM6 as is the RMSE values for neutral molecules (39kcalmol) But for cations the RMSE (37 kcalmol) is significantly lower than the othermethods while the RMSE for anions (51 kcalmol) is very similar
While the solvation free energies of neutral molecules can generally be measured with anaccuracy of ca 02 kcalmol the corresponding solvation free energies of ions are inferredfrom other experimental values with a typical accuracy about 30 kcalmol[40] Thus theaccuracy of the semiempirical SMDdagger is for all intents and purposes identical to HFSMDfor ions but significantly worse for neutral ions in the case of PM6 and DFTB Thesource of this error is most likely due to a poorer description of the electrostatic proper-ties compared to HF For example the error in dipole moments for HCNO compounds islarger for PM6 than for PM3[16]
Table 4 also lists values for PM3COSMO and PM6COSMO as implemented in MOPACfor comparison This COSMO implementation only evaluates the polar part of the solva-tion free energy and will not be as accurate as properly parameterized COSMO-RS[5] val-ues For PM3 the SMDdagger results are significantly lower than the corresponding COSMOresults while for PM6 there is only a significant improvement for cations
10
Table 4 RMSE MUE and MSE (in kcalmol) of aqueous solvation energies computed withSMD using HF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicatesthat the Radii has been reoptimized Corresponding RMSE values for the COSMO method areincluded for comparison The reference set is split into full set neutral cations and anions
SMD SMDdagger COSMO
HF PM3 PM6 DFTB PM3 PM6All
RMSE 34 36 48 41 48 50MUE 21 26 35 31 34 39MSE 03 10 -17 -11 05 -18Max 178 155 231 160 213 211
NeutralRMSE 24 28 47 39 36 47MUE 13 20 34 29 25 35MSE -09 09 -29 -21 -10 -34Max 178 155 231 160 213 211
CationsRMSE 50 56 51 37 74 73MUE 36 44 42 29 59 60MSE 31 36 38 10 56 59Max 140 127 124 91 186 183
AnionsRMSE 56 49 48 51 68 45MUE 46 37 36 40 58 38MSE 39 -03 -05 18 37 03Max 143 146 147 147 152 105
11
pKa PredictionPart of the impetus for this work were the relatively large error in pKa values predictedusing PM3COSMO compared to PM3SMD with the original SMD radii observed byJensen et al [35] so we use the new SMD radii to compute the pKa value using the samedata set The data is presented in Figure 4 and Table 5 and shows that the RMSE dropsfrom 15plusmn03 to 09plusmn03 pH units for PM3SMDdagger and 20plusmn04 to 16plusmn0304 pH unitsfor DFTBSMDdagger In both cases the improvement is primarily a result of the decreasein mean error (ME) ie the original radii led to a fairly systematic underestimation ofthe pKa values that has now been removed by the reparameterization For DFTB theincreased accuracy is also due to a larger number of low error predictions as is evident inFigure 4 In the case of PM3SMDdagger the accuracy is now very similar to PM3COSMOThe RMSE for PM6SMDdagger is similar to PM6COSMO and larger than for PM3SMDin analogy with the corresponding results for COSMO
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6
err
or
Figure 4 Plot of the errors in the predicted pKa values (pKaminus pKexpa )
12
Table 5 Root-Mean-Square-Error (RMSE) man error (ME) and Pearson correlation (r) for predicted pKa values relative to experiment togetherwith their statistical uncertainty (95 confidence limits)
PM3COSMO PM3SMD PM3SMDdagger PM6COSMO PM6SMDdagger DFTBSMD DFTBSMDdaggerRMSE 10 15 09 18 16 20 1695 conf plusmn 0202 plusmn 0303 plusmn 0202 plusmn 0304 plusmn 0304 plusmn 0404 plusmn 0304ME -05 plusmn 02 -12 plusmn 03 -02 plusmn 02 -13 plusmn 04 -08 plusmn 04 -07 plusmn 05 -01 plusmn 04r 095 093 093 090 086 087 08695 conf plusmn 002004 plusmn 003005 plusmn 003005 plusmn 004007 plusmn 006009 plusmn 005008 plusmn 006009
13
Non-aqueous SolventsThe new radii were optimized explicitly for water so it is not a priori clear whether theyoffer more accurate results for other solvents Table 6 shows the RMSE values for allneutral cations and anions for the other polar solvents in the MNSOL data set acetoni-trile DMSO and methanol for the original SMD radii and the corresponding re-optimizedradii (dagger) In addition corresponding combined RMSE values are given for the remaining88 mostly non-polar solvents in the MNSOL data set for which only data is availablefor neutral molecules It is clear from this data that the new radii sets lowers the errorof the predicted solvation free energies for the other polar solvents and has a negligibleeffect on the accuracy for the non-polar solvents
As for acetonitrile and DMSO the improvement is greatest for the ions where the RMSEdecreases by 3-12 kcalmol The improvement is more modest (05-3 kcalmol) for theneutral molecules For methanol which contains only ions the RMSE is similarly de-creased with 2-13 kcalmol
We thus recommend that the new radii optimized for water also be used for other polarsolvents when doing semiempirical SMD calculations For non-polar solvents either setof radii can be used
14
Table 6 RMSE (in kcalmol) of non-aqueous solvation energies computed with SMD usingHF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicates that theradii has been reoptimized Corresponding RMSE values for the COSMO method are includedfor comparison The reference set is split into full set neutral cations and anions
All Neu Cat Aniacetonitrile
HFSMD 112 29 105 131PM3SMD 194 41 162 246PM3SMDdagger 140 37 113 182PM6SMD 219 33 177 284PM6SMDdagger 130 12 114 160DFTBSMD 150 23 119 197DFTBSMDdagger 109 22 90 140PM3COSMO 130 31 128 147PM6COSMO 137 23 135 155
dimethylsulfoxideHFSMD 119 30 106 126PM3SMD 202 42 153 214PM3SMDdagger 170 35 90 182PM6SMD 255 45 172 272PM6SMDdagger 177 17 99 189DFTBSMD 169 23 108 181DFTBSMDdagger 125 21 76 134PM3COSMO 125 34 107 132PM6COSMO 150 31 119 159
methanolHFSMD 40 00 41 39PM3SMD 126 00 108 135PM3SMDdagger 66 00 60 69PM6SMD 163 00 120 183PM6SMDdagger 52 00 54 52DFTBSMD 84 00 63 94DFTBSMDdagger 47 00 44 49PM3COSMO 59 00 68 54PM6COSMO 54 00 67 46
otherHFSMD 42 42 00 00PM3SMD 51 51 00 00PM3SMDdagger 51 51 00 00PM6SMD 35 35 00 00PM6SMDdagger 34 34 00 00DFTBSMD 30 30 00 00DFTBSMDdagger 29 29 00 00PM3COSMO 36 36 00 00PM6COSMO 27 27 00 00
15
ConclusionThe PM6 method in GAMESS was extended to elements requiring d-integrals and inter-faced with the polarized continuum model (PCM) of solvation including semi-numericalgradients However the accuracy of aqueous solvation energies computed using AM1PM3 PM6 and DFTB and the SMD continuum solvation model was tested using a sub-set of molecules from the MNSOL data set which showed that the errors in SMD solvationenergies predicted using NDDO-based methods was considerably larger than when usingDFT and HF with RMSE values of 50 to 86 kcalmol compared to 34 kcalmol forHF6-31G(d) For the NDDO-based methods the errors were especially large for cationsand in the case of PM6 also for anions with RMSE values of 101 to 148 kcalmol incomparison with to 50 to 57 kcalmol for HF6-31G(d)SMD Corresponding COSMOresults (where the maximum RMSE is 75 kcalmol) suggested that the NDDOSMDresults could be improved by re-parameterizing the SMD Coulomb radii for the NDDOmethods
The fact that the NDDOSMD errors are largest for ions suggest that the problem iswith the polar solvation energy so we focus on optimizing the values of the Coulombradii while leaving all parameters associated with the non-polar solvation free energy un-changed We optimize the radii only for the ionic species on a subset of the MNSOL dataset previously used to parametrize RHF6-31G(d)SMD but include neutral moleculeswhen testing how well SMD with the new Coulomb radii perform We found the bestresults are obtained by changing only the radii for hydrogen carbon oxygen nitrogenand sulfur and this leads to RMSE values for PM3 (neutrals 28ions ca 5 kcalmol)PM6 (47ca 5 kcalmol) and DFTB (39ca 5 kcalmol) that are more comparable toHF6-31G(d) (24ca 5 kcalmol) We note that the SMD parameterization is done us-ing mono-aquo microsolvated species for small ions with atoms carrying large (partial)charges while the MNSOL data set also contains data for the non-microsolvated equiv-alents Comparison to the HF6-31G(d) MSEs reported by Marenich et al [8] reportedfor the selectively microsolvated ions indicates that the inclusion of non-microsolvatedspecies leads an MSE increase of about 1 kcalmol
Though the radii are optimized to reproduce aqueous solvation energies they also leadmore accurate predictions for other polar solvents such as DMSO acetonitrile andmethanol while the improvement for non-polar solvents are negligible
Supplementary MaterialSee supplementary material for Tables S1-S3 and Figures S1-S3 which are referred toin the text In addition we also provide a table of dielectric constants used for thecalculations and a table of aqueous solvation free energies obtained using the optimizedparameters
AcknowledgmentsJCK and JHJ would like to acknowledge Jimmy (Mr MOPAC) Stewart for providingsource code and guidance for the PM6 GAMESS implementation Also thanks to Donald
16
Truhlar and Christopher Cramer for sharing the MNSOL data set and for insightfulcomments on the manuscript CS thanks the Danish Council for Independent Research(the Sapere Aude program) for financial support (Grant no DFF 4181-00370)
References[1] J Tomasi B Mennucci and R Cammi ldquoQuantum mechanical continuum solvation
modelsrdquo Chem Rev vol 105 pp 2999ndash3093 2005
[2] A Klamt and G Schuurmann ldquoCOSMO a new approach to dielectric screeningin solvents with explicit expressions for the screening energy and its gradientrdquo JChem Soc Perkin Trans vol 2 pp 799ndash805 1993
[3] C J Cramer and D G Truhlar ldquoA universal approach to solvation modelingrdquoAccounts of Chemical Research vol 41 no 6 pp 760ndash768 jun 2008 doi101021ar800019z
[4] V Barone M Cossi and J Tomasi ldquoA new definition of cavities for thecomputation of solvation free energies by the polarizable continuum modelrdquo JChem Phys vol 107 no 8 pp 3210ndash3221 aug 1997 doi 1010631474671
[5] F Eckert and A Klamt ldquoFast solvent screening via quantum chemistryCOSMO-RS approachrdquo AIChE J vol 48 no 2 pp 369ndash385 feb 2002 doi101002aic690480220
[6] C Curutchet A Bidon-Chanal I Soteras M Orozco and F J Luque ldquoMSTcontinuum study of the hydration free energies of monovalent ionic speciesrdquo JPhys Chem B vol 109 no 8 pp 3565ndash3574 mar 2005 doi 101021jp047197s
[7] Y Takano and K N Houk ldquoBenchmarking the conductor-like polarizablecontinuum model (CPCM) for aqueous solvation free energies of neutral and ionicorganic moleculesrdquo J Chem Theory Comput vol 1 no 1 pp 70ndash77 jan 2005doi 101021ct049977a
[8] A V Marenich R M Olson C P Kelly C J Cramer and D G TruhlarldquoSelf-consistent reaction field model for aqueous and nonaqueous solutions based onaccurate polarized partial chargesrdquo Journal of Chemical Theory and Computationvol 3 no 6 pp 2011ndash2033 nov 2007 doi 101021ct7001418
[9] A V Marenich C J Cramer and D G Truhlar ldquoUniversal solvation model basedon solute electron density and on a continuum model of the solvent defined by thebulk dielectric constant and atomic surface tensionsrdquo J Phys Chem B vol 113no 18 pp 6378ndash6396 may 2009 doi 101021jp810292n
[10] G E Chudinov D V Napolov and M V Basilevsky ldquoQuantum-chemicalcalculations of the hydration energies of organic cations and anions in the frameworkof a continuum solvent approximationrdquo Chem Phys vol 160 no 1 pp 41 ndash 541992 doi 1010160301-0104(92)87090-V
[11] M J Negre M Orozco and F J Luque ldquoA new strategy for the representation ofenvironment effects in semi-empirical calculations based on dewarrsquos hamiltoniansrdquoChem Phys Lett vol 196 no 1 pp 27ndash36 1992
17
[12] F J Luque M J Negre and M Orozco ldquoAn am1-scrf approach to the study ofchanges in molecular properties induced by solventrdquo J Phys Chem vol 97 no 17pp 4386ndash4391 1993
[13] M Caricato B Mennucci and J Tomasi ldquoSolvent effects on the electronic spectraAn extension of the polarizable continuum model to the zindo methodrdquo J PhysChem A vol 108 no 29 pp 6248ndash6256 2004
[14] M J S Dewar E G Zoebisch E F Healy and J J P Stewart ldquoDevelopmentand use of quantum mechanical molecular models 76 AM1 a new general purposequantum mechanical molecular modelrdquo J A Chem Soc vol 10 pp 3902ndash39091985 doi 101021ja00299a024
[15] J J P Stewart ldquoOptimization of Parameters for Semi-Empirical Methods I-Methodrdquo J Comp Chem vol 10 pp 209ndash220 1989 doi 101002jcc540100208
[16] J J P Stewart ldquoOptimization of parameters for semiempirical methods V Modi-fication of NDDO approximations and application to 70 elementsrdquo J Mol Modelvol 13 pp 1172ndash1213 2007
[17] M Gaus Q Cui and M Elstner ldquoDFTB3 Extension of the Self-Consistent-Charge Density-Functional Tight-Binding Method (SCC-DFTB)rdquo J Chem TheoryComput vol 7 no 4 pp 931ndash948 apr 2011 doi 101021ct100684s
[18] A V Marenich C P Kelly J D Thompson G D Hawkins C C ChambersD J Giesen P P Winget C J Cramer and D G Truhlar ldquoMinnesota solvationdatabaserdquo 2012
[19] ldquoMinnesota solvation databaserdquo accessed November 24th 2017 [Online] Availablehttpscompchemumnedumnsol
[20] M W Schmidt K K Baldridge J A Boatz S T Elbert M S Gordon J HJensen S Koseki N Matsunaga K A Nguyen S Su T L Windus M Dupuisand JAMontgomery ldquoGeneral atomic and molecular electronic structure systemrdquoJ Comput Chem vol 14 pp 1347ndash1363 1993
[21] V Barone and M Cossi ldquoQuantum calculation of molecular energies and energygradients in solution by a conductor solvent modelrdquo The Journal of PhysicalChemistry A vol 102 no 11 pp 1995ndash2001 mar 1998 doi 101021jp9716997
[22] M Cossi N Rega G Scalmani and V Barone ldquoEnergies structures andelectronic properties of molecules in solution with the c-PCM solvation modelrdquoJournal of Computational Chemistry vol 24 no 6 pp 669ndash681 apr 2003 doi101002jcc10189
[23] C Steinmann K L Blaeligdel A S Christensen and J H Jensen ldquoInterface ofthe polarizable continuum model of solvation with semi-empirical methods in thegamess programrdquo PLoS ONE vol 8 no 7 p e67725 07 2013 doi 101371jour-nalpone0067725
[24] Y Nishimoto ldquoDFTBPCM applied to ground and excited state potential energysurfacesrdquo J Phys Chem A vol 120 no 5 pp 771ndash784 feb 2016 doi101021acsjpca5b10732
18
[25] M Gaus X Lu M Elstner and Q Cui ldquoParameterization of DFTB33OB forSulfur and Phosphorus for Chemical and Biological Applicationsrdquo J Chem TheoryComput vol 10 no 4 pp 1518ndash1537 apr 2014 doi 101021ct401002w
[26] X Lu M Gaus M Elstner and Q Cui ldquoParametrization of DFTB33OBfor Magnesium and Zinc for Chemical and Biological Applicationsrdquo The Journalof Physical Chemistry B vol 119 no 3 pp 1062ndash1082 jan 2015 doi101021jp506557r
[27] M Kubillus T Kubar M Gaus J Rezac and M Elstner ldquoParameterization ofthe DFTB3 Method for Br Ca Cl F I K and Na in Organic and BiologicalSystemsrdquo J Chem Theory Comput vol 11 no 1 pp 332ndash342 jan 2015 doi101021ct5009137
[28] J J P Stewart ldquoMOPAC2012rdquo 2016 Stewart Computational Chemistry ColoradoSprings CO USA [Online] Available httpopenmopacnet
[29] J L Pascual-Ahuir E Silla J Tomasi and R Bonaccorsi ldquoElectrostaticinteraction of a solute with a continuum improved description of the cavity and ofthe surface cavity bound charge distributionrdquo Journal of Computational Chemistryvol 8 no 6 pp 778ndash787 sep 1987 doi 101002jcc540080605
[30] P Su and H Li ldquoContinuous and smooth potential energy surface for conductorlikescreening solvation model using fixed points with variable areasrdquo The Journal ofChemical Physics vol 130 no 7 p 074109 feb 2009 doi 10106313077917
[31] J Tomasi B Mennucci and E Cances ldquoThe IEF version of the PCM solvationmethod an overview of a new method addressed to study molecular solutes at theQM ab initio levelrdquo Journal of Molecular Structure THEOCHEM vol 464 no1-3 pp 211ndash226 may 1999 doi 101016s0166-1280(98)00553-3
[32] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 1 the calculation of confidence intervalsrdquo J Comput Aided MolDes vol 28 no 9 pp 887ndash918 Sep 2014 doi 101007s10822-014-9753-z
[33] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 2 comparing methodsrdquo J Comput Aided Mol Des vol 30 no 2pp 103ndash126 Feb 2016 doi 101007s10822-016-9904-5
[34] J H Jensen ldquoWhich method is more accurate or errors have error barsrdquo PeerJPreprints vol 5 p e2693v1 2017 doi 107287peerjpreprints2693v1
[35] J H Jensen C J Swain and L Olsen ldquoPrediction of pKa values for druglikemolecules using semiempirical quantum chemical methodsrdquo The Journal of PhysicalChemistry A vol 121 no 3 pp 699ndash707 jan 2017 doi 101021acsjpca6b10990
[36] F Eckert and A Klamt ldquoAccurate prediction of basicity in aqueous solution withCOSMO-RSrdquo Journal of Computational Chemistry vol 27 no 1 pp 11ndash19 2005doi 101002jcc20309
[37] J A Nelder and R Mead ldquoA simplex method for function minimizationrdquo TheComputer Journal vol 7 no 4 pp 308ndash313 1965 doi 101093comjnl74308
19
[38] E Jones T Oliphant P Peterson et al ldquoSciPy Open source scientifictools for Pythonrdquo 2001 accessed November 24th 2017 [Online] Availablehttpwwwscipyorgrdquo
[39] J Charnley 2017 accessed November 24th 2017 [Online] Available httpsgithubcomcharnleyoptimize gamess parameters
[40] C P Kelly C J Cramer and D G Truhlar ldquoAqueous solvation free energies ofions and ion-water clusters based on an accurate value for the absolute aqueoussolvation free energy of the protonrdquo The Journal of Physical Chemistry B vol 110no 32 pp 16 066ndash16 081 aug 2006 doi 101021jp063552y
20
HFSMDAM1SMD
PM3SMDPM6SMD
DFTBSMD
AM1COSMO
PM3COSMO
PM6COSMO null0
2
4
6
8
10
12
14
16RM
SE [k
calm
ol]
PM3PM3HCO
PM3HCON
PM3HCONSPM3all PM6
PM6HCO
PM6HCON
PM6HCONS
PM6HCONShPM6all
DFTB
DFTBHCON0
2
4
6
8
10
12
14RM
SE [k
calm
ol]
HFSMD
PM3SMDPM6SMD
DFTBSMD
PM3COSMO
PM6COSMO0
2
4
6
8
10RM
SE [k
calm
ol]
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6err
or

Table 2 Optimized Coulomb radii (in A) for PM3 PM6 and DFTB for different atom typesand original SMD radii parameters for comparison RMSESMD denotes the RMSE using theoriginal parameters and RMSElowast denotes RMSE after optimization The RMSE is given inkcalmol and is only calculated for the subset of ions in the SMD subset containing thosespecific atom types
H C N O F S Cl Br RMSESMD RMSElowast
SMD120 185 189 152 173 249 238 306
PM3029 172 165 96 44042 175 176 164 109 43062 176 172 162 258 107 46058 175 172 164 179 255 253 335 106 47
PM6066 187 173 123 32017 185 178 174 130 41039 178 182 175 262 127 43070 182 178 174 140 255 237 388 123 44
DFTB070 183 191 164 95 35
broke and the proton started moving into the solvent This indicated that the H radiusmight be too small leading to overpolarization of the H atom due to the solvent andwe tested several larger H radii for PM6-HCONS since the H radius was larger than forPM6-HCON We found that the overpolarization problem disappeared with a H radius of06 A and that using this value has a negligible 01 kcalmol effect of the accuracy ofthe solvation energies (HCONSh in Table 3) Thus we use PM6-HCONSh radii set goingforward
6
PM3PM3HCO
PM3HCON
PM3HCONSPM3all PM6
PM6HCO
PM6HCON
PM6HCONS
PM6HCONShPM6all
DFTB
DFTBHCON0
2
4
6
8
10
12
14
RMSE
[kca
lmol
]
Figure 2 RMSE values (in kcalmol) computed using SMD subset of the MNSOL data setand the Coulomb radii set shown in Table 2 HCO indicates that hydrogen carbon and oxygenradii parameters are changed (ie second and sixth entry in Table 2 The reference set is splitinto full set (black) neutral (green) cations (red) and anions (blue)
7
Table 3 RMSE MUE and MSE values (in kcalmol) computed using the SMD subset of the MNSOL data set and the Coulomb radii set shownin Table 2 HCO indicates that hydrogen carbon and oxygen radii parameters are changed (ie second and sixth row in Table 2)
PM3 PM6 DFTB
SMD HCO HCON HCONS all SMD HCO HCON HCONS HCONSh all SMD HCONAll
RMSE 50 32 29 30 32 75 36 36 38 38 37 41 33MUE 35 23 21 22 23 54 25 27 30 30 28 31 26MSE 10 13 10 10 13 -24 -04 -13 -17 -16 -14 -09 -08Max 168 116 113 126 116 226 184 186 169 169 176 155 109
NeutralRMSE 24 20 19 19 20 40 28 34 36 35 34 31 30MUE 18 15 15 15 15 30 19 24 29 28 25 24 24MSE 11 11 10 11 11 -26 -15 -21 -26 -26 -22 -11 -14Max 87 80 72 74 80 164 184 186 169 169 176 108 109
CationsRMSE 92 62 51 50 62 103 63 43 40 41 41 49 29MUE 85 55 40 39 55 100 58 36 34 35 35 40 24MSE 84 53 32 25 53 100 58 29 30 31 28 36 02Max 150 116 113 119 116 168 138 115 113 113 112 138 91
AnionsRMSE 79 41 42 45 41 142 36 43 46 46 46 67 44MUE 66 32 33 35 32 128 28 32 35 34 32 56 34MSE -61 -11 -13 -07 -11 -126 -06 -12 -14 -12 -13 -39 12Max 168 92 99 126 92 226 88 121 149 147 173 155 104
8
From the results discussed above and presented in Table 2 optimizing for atom typesbeyond HCON showed no significant increase in accuracy for either PM3 or PM6 There-fore the same procedure (optimizing HCON radii) was applied to DFTB For DFTB theRMSE values of the full set of molecules the subset of cation and the subset of anionsgoes from 41 49 and 67 kcalmol to 33 29 and 44 kcalmol respectively
In summary we find that optimizing the SMD radii in combination with either PM3 orPM6 gives appreciable decreases in RMSE accuracy asymp 7 kcalmol in both cases comparedto the reference results We also find that no significant improvement is obtained whenattempting to optimize the radii of other atoms than what was done with HCON subsetof radii For this subset DFTB also saw an increase in accuracy for ions of about asymp 6kcalmol Because of this we found the best choice was change the radii for hydrogencarbon oxygen and nitrogen (HCON) for PM3 and DFTB methods For PM6 we optimizedincluding sulfur and changed the radius of hydrogen to 06 A to remove overpolarizationproblems These methods are denoted as SMDdagger going forward
Comparison with Other Solvation MethodsUsing the new parameters for PM3SMDdagger PM6SMDdagger and DFTBSMDdagger we com-pare the obtained RMSE with other approaches to obtain solvent free energies such asHF6-31G(d)SMD (one of the levels of theory used in the original SMD parameteriza-tion) PM3COSMO and PM6COSMO for the MNSOL data set (Table 4 and Figure 3)
HFSMD
PM3SMDPM6SMD
DFTBSMD
PM3COSMO
PM6COSMO0
2
4
6
8
10
RMSE
[kca
lmol
]
Figure 3 RMSE (in kcalmol) of aqueous solvation energies computed with SMD using HF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicates that the Radiihas been reoptimized Corresponding RMSE values for the COSMO method are included forcomparison The dataset is split into full set (black) neutral (green) cations (red) and anions(blue)
The overall accuracy of PM3SMDdagger is now similar to HFSMD although the RMSEvalue is 04 and 06 kcalmol higher for neutral molecules and cations while the RMSE
9
is 07 kcalmol lower for anions For PM6 the overall RMSE is 14 kcalmol higher thanHF and the RMSE for neutral molecules is 23 kcalmol higher On the other hand theRMSE for cations is only 01 kcalmol higher while the RMSE for anions is 08 kcal-mol lower compared to HF Finally for DFTB the overall RMSE value (41 kcalmol)is in between those for PM3 and PM6 as is the RMSE values for neutral molecules (39kcalmol) But for cations the RMSE (37 kcalmol) is significantly lower than the othermethods while the RMSE for anions (51 kcalmol) is very similar
While the solvation free energies of neutral molecules can generally be measured with anaccuracy of ca 02 kcalmol the corresponding solvation free energies of ions are inferredfrom other experimental values with a typical accuracy about 30 kcalmol[40] Thus theaccuracy of the semiempirical SMDdagger is for all intents and purposes identical to HFSMDfor ions but significantly worse for neutral ions in the case of PM6 and DFTB Thesource of this error is most likely due to a poorer description of the electrostatic proper-ties compared to HF For example the error in dipole moments for HCNO compounds islarger for PM6 than for PM3[16]
Table 4 also lists values for PM3COSMO and PM6COSMO as implemented in MOPACfor comparison This COSMO implementation only evaluates the polar part of the solva-tion free energy and will not be as accurate as properly parameterized COSMO-RS[5] val-ues For PM3 the SMDdagger results are significantly lower than the corresponding COSMOresults while for PM6 there is only a significant improvement for cations
10
Table 4 RMSE MUE and MSE (in kcalmol) of aqueous solvation energies computed withSMD using HF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicatesthat the Radii has been reoptimized Corresponding RMSE values for the COSMO method areincluded for comparison The reference set is split into full set neutral cations and anions
SMD SMDdagger COSMO
HF PM3 PM6 DFTB PM3 PM6All
RMSE 34 36 48 41 48 50MUE 21 26 35 31 34 39MSE 03 10 -17 -11 05 -18Max 178 155 231 160 213 211
NeutralRMSE 24 28 47 39 36 47MUE 13 20 34 29 25 35MSE -09 09 -29 -21 -10 -34Max 178 155 231 160 213 211
CationsRMSE 50 56 51 37 74 73MUE 36 44 42 29 59 60MSE 31 36 38 10 56 59Max 140 127 124 91 186 183
AnionsRMSE 56 49 48 51 68 45MUE 46 37 36 40 58 38MSE 39 -03 -05 18 37 03Max 143 146 147 147 152 105
11
pKa PredictionPart of the impetus for this work were the relatively large error in pKa values predictedusing PM3COSMO compared to PM3SMD with the original SMD radii observed byJensen et al [35] so we use the new SMD radii to compute the pKa value using the samedata set The data is presented in Figure 4 and Table 5 and shows that the RMSE dropsfrom 15plusmn03 to 09plusmn03 pH units for PM3SMDdagger and 20plusmn04 to 16plusmn0304 pH unitsfor DFTBSMDdagger In both cases the improvement is primarily a result of the decreasein mean error (ME) ie the original radii led to a fairly systematic underestimation ofthe pKa values that has now been removed by the reparameterization For DFTB theincreased accuracy is also due to a larger number of low error predictions as is evident inFigure 4 In the case of PM3SMDdagger the accuracy is now very similar to PM3COSMOThe RMSE for PM6SMDdagger is similar to PM6COSMO and larger than for PM3SMDin analogy with the corresponding results for COSMO
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6
err
or
Figure 4 Plot of the errors in the predicted pKa values (pKaminus pKexpa )
12
Table 5 Root-Mean-Square-Error (RMSE) man error (ME) and Pearson correlation (r) for predicted pKa values relative to experiment togetherwith their statistical uncertainty (95 confidence limits)
PM3COSMO PM3SMD PM3SMDdagger PM6COSMO PM6SMDdagger DFTBSMD DFTBSMDdaggerRMSE 10 15 09 18 16 20 1695 conf plusmn 0202 plusmn 0303 plusmn 0202 plusmn 0304 plusmn 0304 plusmn 0404 plusmn 0304ME -05 plusmn 02 -12 plusmn 03 -02 plusmn 02 -13 plusmn 04 -08 plusmn 04 -07 plusmn 05 -01 plusmn 04r 095 093 093 090 086 087 08695 conf plusmn 002004 plusmn 003005 plusmn 003005 plusmn 004007 plusmn 006009 plusmn 005008 plusmn 006009
13
Non-aqueous SolventsThe new radii were optimized explicitly for water so it is not a priori clear whether theyoffer more accurate results for other solvents Table 6 shows the RMSE values for allneutral cations and anions for the other polar solvents in the MNSOL data set acetoni-trile DMSO and methanol for the original SMD radii and the corresponding re-optimizedradii (dagger) In addition corresponding combined RMSE values are given for the remaining88 mostly non-polar solvents in the MNSOL data set for which only data is availablefor neutral molecules It is clear from this data that the new radii sets lowers the errorof the predicted solvation free energies for the other polar solvents and has a negligibleeffect on the accuracy for the non-polar solvents
As for acetonitrile and DMSO the improvement is greatest for the ions where the RMSEdecreases by 3-12 kcalmol The improvement is more modest (05-3 kcalmol) for theneutral molecules For methanol which contains only ions the RMSE is similarly de-creased with 2-13 kcalmol
We thus recommend that the new radii optimized for water also be used for other polarsolvents when doing semiempirical SMD calculations For non-polar solvents either setof radii can be used
14
Table 6 RMSE (in kcalmol) of non-aqueous solvation energies computed with SMD usingHF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicates that theradii has been reoptimized Corresponding RMSE values for the COSMO method are includedfor comparison The reference set is split into full set neutral cations and anions
All Neu Cat Aniacetonitrile
HFSMD 112 29 105 131PM3SMD 194 41 162 246PM3SMDdagger 140 37 113 182PM6SMD 219 33 177 284PM6SMDdagger 130 12 114 160DFTBSMD 150 23 119 197DFTBSMDdagger 109 22 90 140PM3COSMO 130 31 128 147PM6COSMO 137 23 135 155
dimethylsulfoxideHFSMD 119 30 106 126PM3SMD 202 42 153 214PM3SMDdagger 170 35 90 182PM6SMD 255 45 172 272PM6SMDdagger 177 17 99 189DFTBSMD 169 23 108 181DFTBSMDdagger 125 21 76 134PM3COSMO 125 34 107 132PM6COSMO 150 31 119 159
methanolHFSMD 40 00 41 39PM3SMD 126 00 108 135PM3SMDdagger 66 00 60 69PM6SMD 163 00 120 183PM6SMDdagger 52 00 54 52DFTBSMD 84 00 63 94DFTBSMDdagger 47 00 44 49PM3COSMO 59 00 68 54PM6COSMO 54 00 67 46
otherHFSMD 42 42 00 00PM3SMD 51 51 00 00PM3SMDdagger 51 51 00 00PM6SMD 35 35 00 00PM6SMDdagger 34 34 00 00DFTBSMD 30 30 00 00DFTBSMDdagger 29 29 00 00PM3COSMO 36 36 00 00PM6COSMO 27 27 00 00
15
ConclusionThe PM6 method in GAMESS was extended to elements requiring d-integrals and inter-faced with the polarized continuum model (PCM) of solvation including semi-numericalgradients However the accuracy of aqueous solvation energies computed using AM1PM3 PM6 and DFTB and the SMD continuum solvation model was tested using a sub-set of molecules from the MNSOL data set which showed that the errors in SMD solvationenergies predicted using NDDO-based methods was considerably larger than when usingDFT and HF with RMSE values of 50 to 86 kcalmol compared to 34 kcalmol forHF6-31G(d) For the NDDO-based methods the errors were especially large for cationsand in the case of PM6 also for anions with RMSE values of 101 to 148 kcalmol incomparison with to 50 to 57 kcalmol for HF6-31G(d)SMD Corresponding COSMOresults (where the maximum RMSE is 75 kcalmol) suggested that the NDDOSMDresults could be improved by re-parameterizing the SMD Coulomb radii for the NDDOmethods
The fact that the NDDOSMD errors are largest for ions suggest that the problem iswith the polar solvation energy so we focus on optimizing the values of the Coulombradii while leaving all parameters associated with the non-polar solvation free energy un-changed We optimize the radii only for the ionic species on a subset of the MNSOL dataset previously used to parametrize RHF6-31G(d)SMD but include neutral moleculeswhen testing how well SMD with the new Coulomb radii perform We found the bestresults are obtained by changing only the radii for hydrogen carbon oxygen nitrogenand sulfur and this leads to RMSE values for PM3 (neutrals 28ions ca 5 kcalmol)PM6 (47ca 5 kcalmol) and DFTB (39ca 5 kcalmol) that are more comparable toHF6-31G(d) (24ca 5 kcalmol) We note that the SMD parameterization is done us-ing mono-aquo microsolvated species for small ions with atoms carrying large (partial)charges while the MNSOL data set also contains data for the non-microsolvated equiv-alents Comparison to the HF6-31G(d) MSEs reported by Marenich et al [8] reportedfor the selectively microsolvated ions indicates that the inclusion of non-microsolvatedspecies leads an MSE increase of about 1 kcalmol
Though the radii are optimized to reproduce aqueous solvation energies they also leadmore accurate predictions for other polar solvents such as DMSO acetonitrile andmethanol while the improvement for non-polar solvents are negligible
Supplementary MaterialSee supplementary material for Tables S1-S3 and Figures S1-S3 which are referred toin the text In addition we also provide a table of dielectric constants used for thecalculations and a table of aqueous solvation free energies obtained using the optimizedparameters
AcknowledgmentsJCK and JHJ would like to acknowledge Jimmy (Mr MOPAC) Stewart for providingsource code and guidance for the PM6 GAMESS implementation Also thanks to Donald
16
Truhlar and Christopher Cramer for sharing the MNSOL data set and for insightfulcomments on the manuscript CS thanks the Danish Council for Independent Research(the Sapere Aude program) for financial support (Grant no DFF 4181-00370)
References[1] J Tomasi B Mennucci and R Cammi ldquoQuantum mechanical continuum solvation
modelsrdquo Chem Rev vol 105 pp 2999ndash3093 2005
[2] A Klamt and G Schuurmann ldquoCOSMO a new approach to dielectric screeningin solvents with explicit expressions for the screening energy and its gradientrdquo JChem Soc Perkin Trans vol 2 pp 799ndash805 1993
[3] C J Cramer and D G Truhlar ldquoA universal approach to solvation modelingrdquoAccounts of Chemical Research vol 41 no 6 pp 760ndash768 jun 2008 doi101021ar800019z
[4] V Barone M Cossi and J Tomasi ldquoA new definition of cavities for thecomputation of solvation free energies by the polarizable continuum modelrdquo JChem Phys vol 107 no 8 pp 3210ndash3221 aug 1997 doi 1010631474671
[5] F Eckert and A Klamt ldquoFast solvent screening via quantum chemistryCOSMO-RS approachrdquo AIChE J vol 48 no 2 pp 369ndash385 feb 2002 doi101002aic690480220
[6] C Curutchet A Bidon-Chanal I Soteras M Orozco and F J Luque ldquoMSTcontinuum study of the hydration free energies of monovalent ionic speciesrdquo JPhys Chem B vol 109 no 8 pp 3565ndash3574 mar 2005 doi 101021jp047197s
[7] Y Takano and K N Houk ldquoBenchmarking the conductor-like polarizablecontinuum model (CPCM) for aqueous solvation free energies of neutral and ionicorganic moleculesrdquo J Chem Theory Comput vol 1 no 1 pp 70ndash77 jan 2005doi 101021ct049977a
[8] A V Marenich R M Olson C P Kelly C J Cramer and D G TruhlarldquoSelf-consistent reaction field model for aqueous and nonaqueous solutions based onaccurate polarized partial chargesrdquo Journal of Chemical Theory and Computationvol 3 no 6 pp 2011ndash2033 nov 2007 doi 101021ct7001418
[9] A V Marenich C J Cramer and D G Truhlar ldquoUniversal solvation model basedon solute electron density and on a continuum model of the solvent defined by thebulk dielectric constant and atomic surface tensionsrdquo J Phys Chem B vol 113no 18 pp 6378ndash6396 may 2009 doi 101021jp810292n
[10] G E Chudinov D V Napolov and M V Basilevsky ldquoQuantum-chemicalcalculations of the hydration energies of organic cations and anions in the frameworkof a continuum solvent approximationrdquo Chem Phys vol 160 no 1 pp 41 ndash 541992 doi 1010160301-0104(92)87090-V
[11] M J Negre M Orozco and F J Luque ldquoA new strategy for the representation ofenvironment effects in semi-empirical calculations based on dewarrsquos hamiltoniansrdquoChem Phys Lett vol 196 no 1 pp 27ndash36 1992
17
[12] F J Luque M J Negre and M Orozco ldquoAn am1-scrf approach to the study ofchanges in molecular properties induced by solventrdquo J Phys Chem vol 97 no 17pp 4386ndash4391 1993
[13] M Caricato B Mennucci and J Tomasi ldquoSolvent effects on the electronic spectraAn extension of the polarizable continuum model to the zindo methodrdquo J PhysChem A vol 108 no 29 pp 6248ndash6256 2004
[14] M J S Dewar E G Zoebisch E F Healy and J J P Stewart ldquoDevelopmentand use of quantum mechanical molecular models 76 AM1 a new general purposequantum mechanical molecular modelrdquo J A Chem Soc vol 10 pp 3902ndash39091985 doi 101021ja00299a024
[15] J J P Stewart ldquoOptimization of Parameters for Semi-Empirical Methods I-Methodrdquo J Comp Chem vol 10 pp 209ndash220 1989 doi 101002jcc540100208
[16] J J P Stewart ldquoOptimization of parameters for semiempirical methods V Modi-fication of NDDO approximations and application to 70 elementsrdquo J Mol Modelvol 13 pp 1172ndash1213 2007
[17] M Gaus Q Cui and M Elstner ldquoDFTB3 Extension of the Self-Consistent-Charge Density-Functional Tight-Binding Method (SCC-DFTB)rdquo J Chem TheoryComput vol 7 no 4 pp 931ndash948 apr 2011 doi 101021ct100684s
[18] A V Marenich C P Kelly J D Thompson G D Hawkins C C ChambersD J Giesen P P Winget C J Cramer and D G Truhlar ldquoMinnesota solvationdatabaserdquo 2012
[19] ldquoMinnesota solvation databaserdquo accessed November 24th 2017 [Online] Availablehttpscompchemumnedumnsol
[20] M W Schmidt K K Baldridge J A Boatz S T Elbert M S Gordon J HJensen S Koseki N Matsunaga K A Nguyen S Su T L Windus M Dupuisand JAMontgomery ldquoGeneral atomic and molecular electronic structure systemrdquoJ Comput Chem vol 14 pp 1347ndash1363 1993
[21] V Barone and M Cossi ldquoQuantum calculation of molecular energies and energygradients in solution by a conductor solvent modelrdquo The Journal of PhysicalChemistry A vol 102 no 11 pp 1995ndash2001 mar 1998 doi 101021jp9716997
[22] M Cossi N Rega G Scalmani and V Barone ldquoEnergies structures andelectronic properties of molecules in solution with the c-PCM solvation modelrdquoJournal of Computational Chemistry vol 24 no 6 pp 669ndash681 apr 2003 doi101002jcc10189
[23] C Steinmann K L Blaeligdel A S Christensen and J H Jensen ldquoInterface ofthe polarizable continuum model of solvation with semi-empirical methods in thegamess programrdquo PLoS ONE vol 8 no 7 p e67725 07 2013 doi 101371jour-nalpone0067725
[24] Y Nishimoto ldquoDFTBPCM applied to ground and excited state potential energysurfacesrdquo J Phys Chem A vol 120 no 5 pp 771ndash784 feb 2016 doi101021acsjpca5b10732
18
[25] M Gaus X Lu M Elstner and Q Cui ldquoParameterization of DFTB33OB forSulfur and Phosphorus for Chemical and Biological Applicationsrdquo J Chem TheoryComput vol 10 no 4 pp 1518ndash1537 apr 2014 doi 101021ct401002w
[26] X Lu M Gaus M Elstner and Q Cui ldquoParametrization of DFTB33OBfor Magnesium and Zinc for Chemical and Biological Applicationsrdquo The Journalof Physical Chemistry B vol 119 no 3 pp 1062ndash1082 jan 2015 doi101021jp506557r
[27] M Kubillus T Kubar M Gaus J Rezac and M Elstner ldquoParameterization ofthe DFTB3 Method for Br Ca Cl F I K and Na in Organic and BiologicalSystemsrdquo J Chem Theory Comput vol 11 no 1 pp 332ndash342 jan 2015 doi101021ct5009137
[28] J J P Stewart ldquoMOPAC2012rdquo 2016 Stewart Computational Chemistry ColoradoSprings CO USA [Online] Available httpopenmopacnet
[29] J L Pascual-Ahuir E Silla J Tomasi and R Bonaccorsi ldquoElectrostaticinteraction of a solute with a continuum improved description of the cavity and ofthe surface cavity bound charge distributionrdquo Journal of Computational Chemistryvol 8 no 6 pp 778ndash787 sep 1987 doi 101002jcc540080605
[30] P Su and H Li ldquoContinuous and smooth potential energy surface for conductorlikescreening solvation model using fixed points with variable areasrdquo The Journal ofChemical Physics vol 130 no 7 p 074109 feb 2009 doi 10106313077917
[31] J Tomasi B Mennucci and E Cances ldquoThe IEF version of the PCM solvationmethod an overview of a new method addressed to study molecular solutes at theQM ab initio levelrdquo Journal of Molecular Structure THEOCHEM vol 464 no1-3 pp 211ndash226 may 1999 doi 101016s0166-1280(98)00553-3
[32] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 1 the calculation of confidence intervalsrdquo J Comput Aided MolDes vol 28 no 9 pp 887ndash918 Sep 2014 doi 101007s10822-014-9753-z
[33] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 2 comparing methodsrdquo J Comput Aided Mol Des vol 30 no 2pp 103ndash126 Feb 2016 doi 101007s10822-016-9904-5
[34] J H Jensen ldquoWhich method is more accurate or errors have error barsrdquo PeerJPreprints vol 5 p e2693v1 2017 doi 107287peerjpreprints2693v1
[35] J H Jensen C J Swain and L Olsen ldquoPrediction of pKa values for druglikemolecules using semiempirical quantum chemical methodsrdquo The Journal of PhysicalChemistry A vol 121 no 3 pp 699ndash707 jan 2017 doi 101021acsjpca6b10990
[36] F Eckert and A Klamt ldquoAccurate prediction of basicity in aqueous solution withCOSMO-RSrdquo Journal of Computational Chemistry vol 27 no 1 pp 11ndash19 2005doi 101002jcc20309
[37] J A Nelder and R Mead ldquoA simplex method for function minimizationrdquo TheComputer Journal vol 7 no 4 pp 308ndash313 1965 doi 101093comjnl74308
19
[38] E Jones T Oliphant P Peterson et al ldquoSciPy Open source scientifictools for Pythonrdquo 2001 accessed November 24th 2017 [Online] Availablehttpwwwscipyorgrdquo
[39] J Charnley 2017 accessed November 24th 2017 [Online] Available httpsgithubcomcharnleyoptimize gamess parameters
[40] C P Kelly C J Cramer and D G Truhlar ldquoAqueous solvation free energies ofions and ion-water clusters based on an accurate value for the absolute aqueoussolvation free energy of the protonrdquo The Journal of Physical Chemistry B vol 110no 32 pp 16 066ndash16 081 aug 2006 doi 101021jp063552y
20
HFSMDAM1SMD
PM3SMDPM6SMD
DFTBSMD
AM1COSMO
PM3COSMO
PM6COSMO null0
2
4
6
8
10
12
14
16RM
SE [k
calm
ol]
PM3PM3HCO
PM3HCON
PM3HCONSPM3all PM6
PM6HCO
PM6HCON
PM6HCONS
PM6HCONShPM6all
DFTB
DFTBHCON0
2
4
6
8
10
12
14RM
SE [k
calm
ol]
HFSMD
PM3SMDPM6SMD
DFTBSMD
PM3COSMO
PM6COSMO0
2
4
6
8
10RM
SE [k
calm
ol]
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6err
or
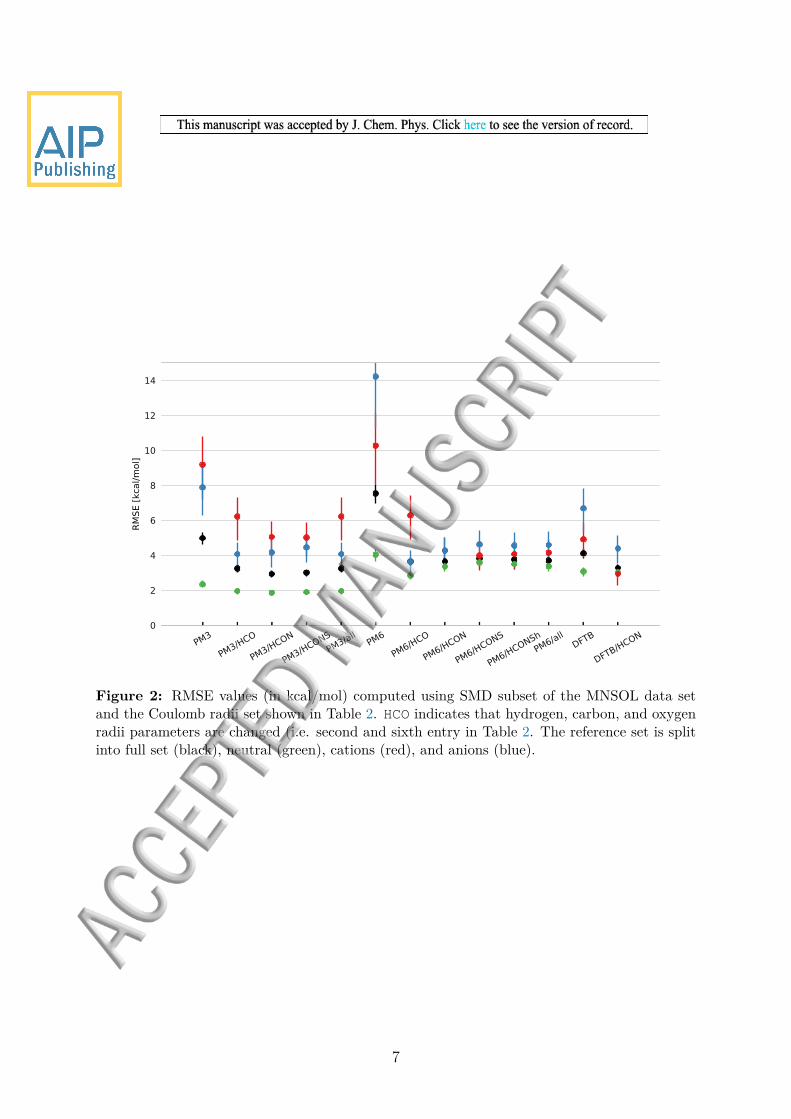
PM3PM3HCO
PM3HCON
PM3HCONSPM3all PM6
PM6HCO
PM6HCON
PM6HCONS
PM6HCONShPM6all
DFTB
DFTBHCON0
2
4
6
8
10
12
14
RMSE
[kca
lmol
]
Figure 2 RMSE values (in kcalmol) computed using SMD subset of the MNSOL data setand the Coulomb radii set shown in Table 2 HCO indicates that hydrogen carbon and oxygenradii parameters are changed (ie second and sixth entry in Table 2 The reference set is splitinto full set (black) neutral (green) cations (red) and anions (blue)
7
Table 3 RMSE MUE and MSE values (in kcalmol) computed using the SMD subset of the MNSOL data set and the Coulomb radii set shownin Table 2 HCO indicates that hydrogen carbon and oxygen radii parameters are changed (ie second and sixth row in Table 2)
PM3 PM6 DFTB
SMD HCO HCON HCONS all SMD HCO HCON HCONS HCONSh all SMD HCONAll
RMSE 50 32 29 30 32 75 36 36 38 38 37 41 33MUE 35 23 21 22 23 54 25 27 30 30 28 31 26MSE 10 13 10 10 13 -24 -04 -13 -17 -16 -14 -09 -08Max 168 116 113 126 116 226 184 186 169 169 176 155 109
NeutralRMSE 24 20 19 19 20 40 28 34 36 35 34 31 30MUE 18 15 15 15 15 30 19 24 29 28 25 24 24MSE 11 11 10 11 11 -26 -15 -21 -26 -26 -22 -11 -14Max 87 80 72 74 80 164 184 186 169 169 176 108 109
CationsRMSE 92 62 51 50 62 103 63 43 40 41 41 49 29MUE 85 55 40 39 55 100 58 36 34 35 35 40 24MSE 84 53 32 25 53 100 58 29 30 31 28 36 02Max 150 116 113 119 116 168 138 115 113 113 112 138 91
AnionsRMSE 79 41 42 45 41 142 36 43 46 46 46 67 44MUE 66 32 33 35 32 128 28 32 35 34 32 56 34MSE -61 -11 -13 -07 -11 -126 -06 -12 -14 -12 -13 -39 12Max 168 92 99 126 92 226 88 121 149 147 173 155 104
8
From the results discussed above and presented in Table 2 optimizing for atom typesbeyond HCON showed no significant increase in accuracy for either PM3 or PM6 There-fore the same procedure (optimizing HCON radii) was applied to DFTB For DFTB theRMSE values of the full set of molecules the subset of cation and the subset of anionsgoes from 41 49 and 67 kcalmol to 33 29 and 44 kcalmol respectively
In summary we find that optimizing the SMD radii in combination with either PM3 orPM6 gives appreciable decreases in RMSE accuracy asymp 7 kcalmol in both cases comparedto the reference results We also find that no significant improvement is obtained whenattempting to optimize the radii of other atoms than what was done with HCON subsetof radii For this subset DFTB also saw an increase in accuracy for ions of about asymp 6kcalmol Because of this we found the best choice was change the radii for hydrogencarbon oxygen and nitrogen (HCON) for PM3 and DFTB methods For PM6 we optimizedincluding sulfur and changed the radius of hydrogen to 06 A to remove overpolarizationproblems These methods are denoted as SMDdagger going forward
Comparison with Other Solvation MethodsUsing the new parameters for PM3SMDdagger PM6SMDdagger and DFTBSMDdagger we com-pare the obtained RMSE with other approaches to obtain solvent free energies such asHF6-31G(d)SMD (one of the levels of theory used in the original SMD parameteriza-tion) PM3COSMO and PM6COSMO for the MNSOL data set (Table 4 and Figure 3)
HFSMD
PM3SMDPM6SMD
DFTBSMD
PM3COSMO
PM6COSMO0
2
4
6
8
10
RMSE
[kca
lmol
]
Figure 3 RMSE (in kcalmol) of aqueous solvation energies computed with SMD using HF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicates that the Radiihas been reoptimized Corresponding RMSE values for the COSMO method are included forcomparison The dataset is split into full set (black) neutral (green) cations (red) and anions(blue)
The overall accuracy of PM3SMDdagger is now similar to HFSMD although the RMSEvalue is 04 and 06 kcalmol higher for neutral molecules and cations while the RMSE
9
is 07 kcalmol lower for anions For PM6 the overall RMSE is 14 kcalmol higher thanHF and the RMSE for neutral molecules is 23 kcalmol higher On the other hand theRMSE for cations is only 01 kcalmol higher while the RMSE for anions is 08 kcal-mol lower compared to HF Finally for DFTB the overall RMSE value (41 kcalmol)is in between those for PM3 and PM6 as is the RMSE values for neutral molecules (39kcalmol) But for cations the RMSE (37 kcalmol) is significantly lower than the othermethods while the RMSE for anions (51 kcalmol) is very similar
While the solvation free energies of neutral molecules can generally be measured with anaccuracy of ca 02 kcalmol the corresponding solvation free energies of ions are inferredfrom other experimental values with a typical accuracy about 30 kcalmol[40] Thus theaccuracy of the semiempirical SMDdagger is for all intents and purposes identical to HFSMDfor ions but significantly worse for neutral ions in the case of PM6 and DFTB Thesource of this error is most likely due to a poorer description of the electrostatic proper-ties compared to HF For example the error in dipole moments for HCNO compounds islarger for PM6 than for PM3[16]
Table 4 also lists values for PM3COSMO and PM6COSMO as implemented in MOPACfor comparison This COSMO implementation only evaluates the polar part of the solva-tion free energy and will not be as accurate as properly parameterized COSMO-RS[5] val-ues For PM3 the SMDdagger results are significantly lower than the corresponding COSMOresults while for PM6 there is only a significant improvement for cations
10
Table 4 RMSE MUE and MSE (in kcalmol) of aqueous solvation energies computed withSMD using HF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicatesthat the Radii has been reoptimized Corresponding RMSE values for the COSMO method areincluded for comparison The reference set is split into full set neutral cations and anions
SMD SMDdagger COSMO
HF PM3 PM6 DFTB PM3 PM6All
RMSE 34 36 48 41 48 50MUE 21 26 35 31 34 39MSE 03 10 -17 -11 05 -18Max 178 155 231 160 213 211
NeutralRMSE 24 28 47 39 36 47MUE 13 20 34 29 25 35MSE -09 09 -29 -21 -10 -34Max 178 155 231 160 213 211
CationsRMSE 50 56 51 37 74 73MUE 36 44 42 29 59 60MSE 31 36 38 10 56 59Max 140 127 124 91 186 183
AnionsRMSE 56 49 48 51 68 45MUE 46 37 36 40 58 38MSE 39 -03 -05 18 37 03Max 143 146 147 147 152 105
11
pKa PredictionPart of the impetus for this work were the relatively large error in pKa values predictedusing PM3COSMO compared to PM3SMD with the original SMD radii observed byJensen et al [35] so we use the new SMD radii to compute the pKa value using the samedata set The data is presented in Figure 4 and Table 5 and shows that the RMSE dropsfrom 15plusmn03 to 09plusmn03 pH units for PM3SMDdagger and 20plusmn04 to 16plusmn0304 pH unitsfor DFTBSMDdagger In both cases the improvement is primarily a result of the decreasein mean error (ME) ie the original radii led to a fairly systematic underestimation ofthe pKa values that has now been removed by the reparameterization For DFTB theincreased accuracy is also due to a larger number of low error predictions as is evident inFigure 4 In the case of PM3SMDdagger the accuracy is now very similar to PM3COSMOThe RMSE for PM6SMDdagger is similar to PM6COSMO and larger than for PM3SMDin analogy with the corresponding results for COSMO
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6
err
or
Figure 4 Plot of the errors in the predicted pKa values (pKaminus pKexpa )
12
Table 5 Root-Mean-Square-Error (RMSE) man error (ME) and Pearson correlation (r) for predicted pKa values relative to experiment togetherwith their statistical uncertainty (95 confidence limits)
PM3COSMO PM3SMD PM3SMDdagger PM6COSMO PM6SMDdagger DFTBSMD DFTBSMDdaggerRMSE 10 15 09 18 16 20 1695 conf plusmn 0202 plusmn 0303 plusmn 0202 plusmn 0304 plusmn 0304 plusmn 0404 plusmn 0304ME -05 plusmn 02 -12 plusmn 03 -02 plusmn 02 -13 plusmn 04 -08 plusmn 04 -07 plusmn 05 -01 plusmn 04r 095 093 093 090 086 087 08695 conf plusmn 002004 plusmn 003005 plusmn 003005 plusmn 004007 plusmn 006009 plusmn 005008 plusmn 006009
13
Non-aqueous SolventsThe new radii were optimized explicitly for water so it is not a priori clear whether theyoffer more accurate results for other solvents Table 6 shows the RMSE values for allneutral cations and anions for the other polar solvents in the MNSOL data set acetoni-trile DMSO and methanol for the original SMD radii and the corresponding re-optimizedradii (dagger) In addition corresponding combined RMSE values are given for the remaining88 mostly non-polar solvents in the MNSOL data set for which only data is availablefor neutral molecules It is clear from this data that the new radii sets lowers the errorof the predicted solvation free energies for the other polar solvents and has a negligibleeffect on the accuracy for the non-polar solvents
As for acetonitrile and DMSO the improvement is greatest for the ions where the RMSEdecreases by 3-12 kcalmol The improvement is more modest (05-3 kcalmol) for theneutral molecules For methanol which contains only ions the RMSE is similarly de-creased with 2-13 kcalmol
We thus recommend that the new radii optimized for water also be used for other polarsolvents when doing semiempirical SMD calculations For non-polar solvents either setof radii can be used
14
Table 6 RMSE (in kcalmol) of non-aqueous solvation energies computed with SMD usingHF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicates that theradii has been reoptimized Corresponding RMSE values for the COSMO method are includedfor comparison The reference set is split into full set neutral cations and anions
All Neu Cat Aniacetonitrile
HFSMD 112 29 105 131PM3SMD 194 41 162 246PM3SMDdagger 140 37 113 182PM6SMD 219 33 177 284PM6SMDdagger 130 12 114 160DFTBSMD 150 23 119 197DFTBSMDdagger 109 22 90 140PM3COSMO 130 31 128 147PM6COSMO 137 23 135 155
dimethylsulfoxideHFSMD 119 30 106 126PM3SMD 202 42 153 214PM3SMDdagger 170 35 90 182PM6SMD 255 45 172 272PM6SMDdagger 177 17 99 189DFTBSMD 169 23 108 181DFTBSMDdagger 125 21 76 134PM3COSMO 125 34 107 132PM6COSMO 150 31 119 159
methanolHFSMD 40 00 41 39PM3SMD 126 00 108 135PM3SMDdagger 66 00 60 69PM6SMD 163 00 120 183PM6SMDdagger 52 00 54 52DFTBSMD 84 00 63 94DFTBSMDdagger 47 00 44 49PM3COSMO 59 00 68 54PM6COSMO 54 00 67 46
otherHFSMD 42 42 00 00PM3SMD 51 51 00 00PM3SMDdagger 51 51 00 00PM6SMD 35 35 00 00PM6SMDdagger 34 34 00 00DFTBSMD 30 30 00 00DFTBSMDdagger 29 29 00 00PM3COSMO 36 36 00 00PM6COSMO 27 27 00 00
15
ConclusionThe PM6 method in GAMESS was extended to elements requiring d-integrals and inter-faced with the polarized continuum model (PCM) of solvation including semi-numericalgradients However the accuracy of aqueous solvation energies computed using AM1PM3 PM6 and DFTB and the SMD continuum solvation model was tested using a sub-set of molecules from the MNSOL data set which showed that the errors in SMD solvationenergies predicted using NDDO-based methods was considerably larger than when usingDFT and HF with RMSE values of 50 to 86 kcalmol compared to 34 kcalmol forHF6-31G(d) For the NDDO-based methods the errors were especially large for cationsand in the case of PM6 also for anions with RMSE values of 101 to 148 kcalmol incomparison with to 50 to 57 kcalmol for HF6-31G(d)SMD Corresponding COSMOresults (where the maximum RMSE is 75 kcalmol) suggested that the NDDOSMDresults could be improved by re-parameterizing the SMD Coulomb radii for the NDDOmethods
The fact that the NDDOSMD errors are largest for ions suggest that the problem iswith the polar solvation energy so we focus on optimizing the values of the Coulombradii while leaving all parameters associated with the non-polar solvation free energy un-changed We optimize the radii only for the ionic species on a subset of the MNSOL dataset previously used to parametrize RHF6-31G(d)SMD but include neutral moleculeswhen testing how well SMD with the new Coulomb radii perform We found the bestresults are obtained by changing only the radii for hydrogen carbon oxygen nitrogenand sulfur and this leads to RMSE values for PM3 (neutrals 28ions ca 5 kcalmol)PM6 (47ca 5 kcalmol) and DFTB (39ca 5 kcalmol) that are more comparable toHF6-31G(d) (24ca 5 kcalmol) We note that the SMD parameterization is done us-ing mono-aquo microsolvated species for small ions with atoms carrying large (partial)charges while the MNSOL data set also contains data for the non-microsolvated equiv-alents Comparison to the HF6-31G(d) MSEs reported by Marenich et al [8] reportedfor the selectively microsolvated ions indicates that the inclusion of non-microsolvatedspecies leads an MSE increase of about 1 kcalmol
Though the radii are optimized to reproduce aqueous solvation energies they also leadmore accurate predictions for other polar solvents such as DMSO acetonitrile andmethanol while the improvement for non-polar solvents are negligible
Supplementary MaterialSee supplementary material for Tables S1-S3 and Figures S1-S3 which are referred toin the text In addition we also provide a table of dielectric constants used for thecalculations and a table of aqueous solvation free energies obtained using the optimizedparameters
AcknowledgmentsJCK and JHJ would like to acknowledge Jimmy (Mr MOPAC) Stewart for providingsource code and guidance for the PM6 GAMESS implementation Also thanks to Donald
16
Truhlar and Christopher Cramer for sharing the MNSOL data set and for insightfulcomments on the manuscript CS thanks the Danish Council for Independent Research(the Sapere Aude program) for financial support (Grant no DFF 4181-00370)
References[1] J Tomasi B Mennucci and R Cammi ldquoQuantum mechanical continuum solvation
modelsrdquo Chem Rev vol 105 pp 2999ndash3093 2005
[2] A Klamt and G Schuurmann ldquoCOSMO a new approach to dielectric screeningin solvents with explicit expressions for the screening energy and its gradientrdquo JChem Soc Perkin Trans vol 2 pp 799ndash805 1993
[3] C J Cramer and D G Truhlar ldquoA universal approach to solvation modelingrdquoAccounts of Chemical Research vol 41 no 6 pp 760ndash768 jun 2008 doi101021ar800019z
[4] V Barone M Cossi and J Tomasi ldquoA new definition of cavities for thecomputation of solvation free energies by the polarizable continuum modelrdquo JChem Phys vol 107 no 8 pp 3210ndash3221 aug 1997 doi 1010631474671
[5] F Eckert and A Klamt ldquoFast solvent screening via quantum chemistryCOSMO-RS approachrdquo AIChE J vol 48 no 2 pp 369ndash385 feb 2002 doi101002aic690480220
[6] C Curutchet A Bidon-Chanal I Soteras M Orozco and F J Luque ldquoMSTcontinuum study of the hydration free energies of monovalent ionic speciesrdquo JPhys Chem B vol 109 no 8 pp 3565ndash3574 mar 2005 doi 101021jp047197s
[7] Y Takano and K N Houk ldquoBenchmarking the conductor-like polarizablecontinuum model (CPCM) for aqueous solvation free energies of neutral and ionicorganic moleculesrdquo J Chem Theory Comput vol 1 no 1 pp 70ndash77 jan 2005doi 101021ct049977a
[8] A V Marenich R M Olson C P Kelly C J Cramer and D G TruhlarldquoSelf-consistent reaction field model for aqueous and nonaqueous solutions based onaccurate polarized partial chargesrdquo Journal of Chemical Theory and Computationvol 3 no 6 pp 2011ndash2033 nov 2007 doi 101021ct7001418
[9] A V Marenich C J Cramer and D G Truhlar ldquoUniversal solvation model basedon solute electron density and on a continuum model of the solvent defined by thebulk dielectric constant and atomic surface tensionsrdquo J Phys Chem B vol 113no 18 pp 6378ndash6396 may 2009 doi 101021jp810292n
[10] G E Chudinov D V Napolov and M V Basilevsky ldquoQuantum-chemicalcalculations of the hydration energies of organic cations and anions in the frameworkof a continuum solvent approximationrdquo Chem Phys vol 160 no 1 pp 41 ndash 541992 doi 1010160301-0104(92)87090-V
[11] M J Negre M Orozco and F J Luque ldquoA new strategy for the representation ofenvironment effects in semi-empirical calculations based on dewarrsquos hamiltoniansrdquoChem Phys Lett vol 196 no 1 pp 27ndash36 1992
17
[12] F J Luque M J Negre and M Orozco ldquoAn am1-scrf approach to the study ofchanges in molecular properties induced by solventrdquo J Phys Chem vol 97 no 17pp 4386ndash4391 1993
[13] M Caricato B Mennucci and J Tomasi ldquoSolvent effects on the electronic spectraAn extension of the polarizable continuum model to the zindo methodrdquo J PhysChem A vol 108 no 29 pp 6248ndash6256 2004
[14] M J S Dewar E G Zoebisch E F Healy and J J P Stewart ldquoDevelopmentand use of quantum mechanical molecular models 76 AM1 a new general purposequantum mechanical molecular modelrdquo J A Chem Soc vol 10 pp 3902ndash39091985 doi 101021ja00299a024
[15] J J P Stewart ldquoOptimization of Parameters for Semi-Empirical Methods I-Methodrdquo J Comp Chem vol 10 pp 209ndash220 1989 doi 101002jcc540100208
[16] J J P Stewart ldquoOptimization of parameters for semiempirical methods V Modi-fication of NDDO approximations and application to 70 elementsrdquo J Mol Modelvol 13 pp 1172ndash1213 2007
[17] M Gaus Q Cui and M Elstner ldquoDFTB3 Extension of the Self-Consistent-Charge Density-Functional Tight-Binding Method (SCC-DFTB)rdquo J Chem TheoryComput vol 7 no 4 pp 931ndash948 apr 2011 doi 101021ct100684s
[18] A V Marenich C P Kelly J D Thompson G D Hawkins C C ChambersD J Giesen P P Winget C J Cramer and D G Truhlar ldquoMinnesota solvationdatabaserdquo 2012
[19] ldquoMinnesota solvation databaserdquo accessed November 24th 2017 [Online] Availablehttpscompchemumnedumnsol
[20] M W Schmidt K K Baldridge J A Boatz S T Elbert M S Gordon J HJensen S Koseki N Matsunaga K A Nguyen S Su T L Windus M Dupuisand JAMontgomery ldquoGeneral atomic and molecular electronic structure systemrdquoJ Comput Chem vol 14 pp 1347ndash1363 1993
[21] V Barone and M Cossi ldquoQuantum calculation of molecular energies and energygradients in solution by a conductor solvent modelrdquo The Journal of PhysicalChemistry A vol 102 no 11 pp 1995ndash2001 mar 1998 doi 101021jp9716997
[22] M Cossi N Rega G Scalmani and V Barone ldquoEnergies structures andelectronic properties of molecules in solution with the c-PCM solvation modelrdquoJournal of Computational Chemistry vol 24 no 6 pp 669ndash681 apr 2003 doi101002jcc10189
[23] C Steinmann K L Blaeligdel A S Christensen and J H Jensen ldquoInterface ofthe polarizable continuum model of solvation with semi-empirical methods in thegamess programrdquo PLoS ONE vol 8 no 7 p e67725 07 2013 doi 101371jour-nalpone0067725
[24] Y Nishimoto ldquoDFTBPCM applied to ground and excited state potential energysurfacesrdquo J Phys Chem A vol 120 no 5 pp 771ndash784 feb 2016 doi101021acsjpca5b10732
18
[25] M Gaus X Lu M Elstner and Q Cui ldquoParameterization of DFTB33OB forSulfur and Phosphorus for Chemical and Biological Applicationsrdquo J Chem TheoryComput vol 10 no 4 pp 1518ndash1537 apr 2014 doi 101021ct401002w
[26] X Lu M Gaus M Elstner and Q Cui ldquoParametrization of DFTB33OBfor Magnesium and Zinc for Chemical and Biological Applicationsrdquo The Journalof Physical Chemistry B vol 119 no 3 pp 1062ndash1082 jan 2015 doi101021jp506557r
[27] M Kubillus T Kubar M Gaus J Rezac and M Elstner ldquoParameterization ofthe DFTB3 Method for Br Ca Cl F I K and Na in Organic and BiologicalSystemsrdquo J Chem Theory Comput vol 11 no 1 pp 332ndash342 jan 2015 doi101021ct5009137
[28] J J P Stewart ldquoMOPAC2012rdquo 2016 Stewart Computational Chemistry ColoradoSprings CO USA [Online] Available httpopenmopacnet
[29] J L Pascual-Ahuir E Silla J Tomasi and R Bonaccorsi ldquoElectrostaticinteraction of a solute with a continuum improved description of the cavity and ofthe surface cavity bound charge distributionrdquo Journal of Computational Chemistryvol 8 no 6 pp 778ndash787 sep 1987 doi 101002jcc540080605
[30] P Su and H Li ldquoContinuous and smooth potential energy surface for conductorlikescreening solvation model using fixed points with variable areasrdquo The Journal ofChemical Physics vol 130 no 7 p 074109 feb 2009 doi 10106313077917
[31] J Tomasi B Mennucci and E Cances ldquoThe IEF version of the PCM solvationmethod an overview of a new method addressed to study molecular solutes at theQM ab initio levelrdquo Journal of Molecular Structure THEOCHEM vol 464 no1-3 pp 211ndash226 may 1999 doi 101016s0166-1280(98)00553-3
[32] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 1 the calculation of confidence intervalsrdquo J Comput Aided MolDes vol 28 no 9 pp 887ndash918 Sep 2014 doi 101007s10822-014-9753-z
[33] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 2 comparing methodsrdquo J Comput Aided Mol Des vol 30 no 2pp 103ndash126 Feb 2016 doi 101007s10822-016-9904-5
[34] J H Jensen ldquoWhich method is more accurate or errors have error barsrdquo PeerJPreprints vol 5 p e2693v1 2017 doi 107287peerjpreprints2693v1
[35] J H Jensen C J Swain and L Olsen ldquoPrediction of pKa values for druglikemolecules using semiempirical quantum chemical methodsrdquo The Journal of PhysicalChemistry A vol 121 no 3 pp 699ndash707 jan 2017 doi 101021acsjpca6b10990
[36] F Eckert and A Klamt ldquoAccurate prediction of basicity in aqueous solution withCOSMO-RSrdquo Journal of Computational Chemistry vol 27 no 1 pp 11ndash19 2005doi 101002jcc20309
[37] J A Nelder and R Mead ldquoA simplex method for function minimizationrdquo TheComputer Journal vol 7 no 4 pp 308ndash313 1965 doi 101093comjnl74308
19
[38] E Jones T Oliphant P Peterson et al ldquoSciPy Open source scientifictools for Pythonrdquo 2001 accessed November 24th 2017 [Online] Availablehttpwwwscipyorgrdquo
[39] J Charnley 2017 accessed November 24th 2017 [Online] Available httpsgithubcomcharnleyoptimize gamess parameters
[40] C P Kelly C J Cramer and D G Truhlar ldquoAqueous solvation free energies ofions and ion-water clusters based on an accurate value for the absolute aqueoussolvation free energy of the protonrdquo The Journal of Physical Chemistry B vol 110no 32 pp 16 066ndash16 081 aug 2006 doi 101021jp063552y
20
HFSMDAM1SMD
PM3SMDPM6SMD
DFTBSMD
AM1COSMO
PM3COSMO
PM6COSMO null0
2
4
6
8
10
12
14
16RM
SE [k
calm
ol]
PM3PM3HCO
PM3HCON
PM3HCONSPM3all PM6
PM6HCO
PM6HCON
PM6HCONS
PM6HCONShPM6all
DFTB
DFTBHCON0
2
4
6
8
10
12
14RM
SE [k
calm
ol]
HFSMD
PM3SMDPM6SMD
DFTBSMD
PM3COSMO
PM6COSMO0
2
4
6
8
10RM
SE [k
calm
ol]
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6err
or

Table 3 RMSE MUE and MSE values (in kcalmol) computed using the SMD subset of the MNSOL data set and the Coulomb radii set shownin Table 2 HCO indicates that hydrogen carbon and oxygen radii parameters are changed (ie second and sixth row in Table 2)
PM3 PM6 DFTB
SMD HCO HCON HCONS all SMD HCO HCON HCONS HCONSh all SMD HCONAll
RMSE 50 32 29 30 32 75 36 36 38 38 37 41 33MUE 35 23 21 22 23 54 25 27 30 30 28 31 26MSE 10 13 10 10 13 -24 -04 -13 -17 -16 -14 -09 -08Max 168 116 113 126 116 226 184 186 169 169 176 155 109
NeutralRMSE 24 20 19 19 20 40 28 34 36 35 34 31 30MUE 18 15 15 15 15 30 19 24 29 28 25 24 24MSE 11 11 10 11 11 -26 -15 -21 -26 -26 -22 -11 -14Max 87 80 72 74 80 164 184 186 169 169 176 108 109
CationsRMSE 92 62 51 50 62 103 63 43 40 41 41 49 29MUE 85 55 40 39 55 100 58 36 34 35 35 40 24MSE 84 53 32 25 53 100 58 29 30 31 28 36 02Max 150 116 113 119 116 168 138 115 113 113 112 138 91
AnionsRMSE 79 41 42 45 41 142 36 43 46 46 46 67 44MUE 66 32 33 35 32 128 28 32 35 34 32 56 34MSE -61 -11 -13 -07 -11 -126 -06 -12 -14 -12 -13 -39 12Max 168 92 99 126 92 226 88 121 149 147 173 155 104
8
From the results discussed above and presented in Table 2 optimizing for atom typesbeyond HCON showed no significant increase in accuracy for either PM3 or PM6 There-fore the same procedure (optimizing HCON radii) was applied to DFTB For DFTB theRMSE values of the full set of molecules the subset of cation and the subset of anionsgoes from 41 49 and 67 kcalmol to 33 29 and 44 kcalmol respectively
In summary we find that optimizing the SMD radii in combination with either PM3 orPM6 gives appreciable decreases in RMSE accuracy asymp 7 kcalmol in both cases comparedto the reference results We also find that no significant improvement is obtained whenattempting to optimize the radii of other atoms than what was done with HCON subsetof radii For this subset DFTB also saw an increase in accuracy for ions of about asymp 6kcalmol Because of this we found the best choice was change the radii for hydrogencarbon oxygen and nitrogen (HCON) for PM3 and DFTB methods For PM6 we optimizedincluding sulfur and changed the radius of hydrogen to 06 A to remove overpolarizationproblems These methods are denoted as SMDdagger going forward
Comparison with Other Solvation MethodsUsing the new parameters for PM3SMDdagger PM6SMDdagger and DFTBSMDdagger we com-pare the obtained RMSE with other approaches to obtain solvent free energies such asHF6-31G(d)SMD (one of the levels of theory used in the original SMD parameteriza-tion) PM3COSMO and PM6COSMO for the MNSOL data set (Table 4 and Figure 3)
HFSMD
PM3SMDPM6SMD
DFTBSMD
PM3COSMO
PM6COSMO0
2
4
6
8
10
RMSE
[kca
lmol
]
Figure 3 RMSE (in kcalmol) of aqueous solvation energies computed with SMD using HF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicates that the Radiihas been reoptimized Corresponding RMSE values for the COSMO method are included forcomparison The dataset is split into full set (black) neutral (green) cations (red) and anions(blue)
The overall accuracy of PM3SMDdagger is now similar to HFSMD although the RMSEvalue is 04 and 06 kcalmol higher for neutral molecules and cations while the RMSE
9
is 07 kcalmol lower for anions For PM6 the overall RMSE is 14 kcalmol higher thanHF and the RMSE for neutral molecules is 23 kcalmol higher On the other hand theRMSE for cations is only 01 kcalmol higher while the RMSE for anions is 08 kcal-mol lower compared to HF Finally for DFTB the overall RMSE value (41 kcalmol)is in between those for PM3 and PM6 as is the RMSE values for neutral molecules (39kcalmol) But for cations the RMSE (37 kcalmol) is significantly lower than the othermethods while the RMSE for anions (51 kcalmol) is very similar
While the solvation free energies of neutral molecules can generally be measured with anaccuracy of ca 02 kcalmol the corresponding solvation free energies of ions are inferredfrom other experimental values with a typical accuracy about 30 kcalmol[40] Thus theaccuracy of the semiempirical SMDdagger is for all intents and purposes identical to HFSMDfor ions but significantly worse for neutral ions in the case of PM6 and DFTB Thesource of this error is most likely due to a poorer description of the electrostatic proper-ties compared to HF For example the error in dipole moments for HCNO compounds islarger for PM6 than for PM3[16]
Table 4 also lists values for PM3COSMO and PM6COSMO as implemented in MOPACfor comparison This COSMO implementation only evaluates the polar part of the solva-tion free energy and will not be as accurate as properly parameterized COSMO-RS[5] val-ues For PM3 the SMDdagger results are significantly lower than the corresponding COSMOresults while for PM6 there is only a significant improvement for cations
10
Table 4 RMSE MUE and MSE (in kcalmol) of aqueous solvation energies computed withSMD using HF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicatesthat the Radii has been reoptimized Corresponding RMSE values for the COSMO method areincluded for comparison The reference set is split into full set neutral cations and anions
SMD SMDdagger COSMO
HF PM3 PM6 DFTB PM3 PM6All
RMSE 34 36 48 41 48 50MUE 21 26 35 31 34 39MSE 03 10 -17 -11 05 -18Max 178 155 231 160 213 211
NeutralRMSE 24 28 47 39 36 47MUE 13 20 34 29 25 35MSE -09 09 -29 -21 -10 -34Max 178 155 231 160 213 211
CationsRMSE 50 56 51 37 74 73MUE 36 44 42 29 59 60MSE 31 36 38 10 56 59Max 140 127 124 91 186 183
AnionsRMSE 56 49 48 51 68 45MUE 46 37 36 40 58 38MSE 39 -03 -05 18 37 03Max 143 146 147 147 152 105
11
pKa PredictionPart of the impetus for this work were the relatively large error in pKa values predictedusing PM3COSMO compared to PM3SMD with the original SMD radii observed byJensen et al [35] so we use the new SMD radii to compute the pKa value using the samedata set The data is presented in Figure 4 and Table 5 and shows that the RMSE dropsfrom 15plusmn03 to 09plusmn03 pH units for PM3SMDdagger and 20plusmn04 to 16plusmn0304 pH unitsfor DFTBSMDdagger In both cases the improvement is primarily a result of the decreasein mean error (ME) ie the original radii led to a fairly systematic underestimation ofthe pKa values that has now been removed by the reparameterization For DFTB theincreased accuracy is also due to a larger number of low error predictions as is evident inFigure 4 In the case of PM3SMDdagger the accuracy is now very similar to PM3COSMOThe RMSE for PM6SMDdagger is similar to PM6COSMO and larger than for PM3SMDin analogy with the corresponding results for COSMO
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6
err
or
Figure 4 Plot of the errors in the predicted pKa values (pKaminus pKexpa )
12
Table 5 Root-Mean-Square-Error (RMSE) man error (ME) and Pearson correlation (r) for predicted pKa values relative to experiment togetherwith their statistical uncertainty (95 confidence limits)
PM3COSMO PM3SMD PM3SMDdagger PM6COSMO PM6SMDdagger DFTBSMD DFTBSMDdaggerRMSE 10 15 09 18 16 20 1695 conf plusmn 0202 plusmn 0303 plusmn 0202 plusmn 0304 plusmn 0304 plusmn 0404 plusmn 0304ME -05 plusmn 02 -12 plusmn 03 -02 plusmn 02 -13 plusmn 04 -08 plusmn 04 -07 plusmn 05 -01 plusmn 04r 095 093 093 090 086 087 08695 conf plusmn 002004 plusmn 003005 plusmn 003005 plusmn 004007 plusmn 006009 plusmn 005008 plusmn 006009
13
Non-aqueous SolventsThe new radii were optimized explicitly for water so it is not a priori clear whether theyoffer more accurate results for other solvents Table 6 shows the RMSE values for allneutral cations and anions for the other polar solvents in the MNSOL data set acetoni-trile DMSO and methanol for the original SMD radii and the corresponding re-optimizedradii (dagger) In addition corresponding combined RMSE values are given for the remaining88 mostly non-polar solvents in the MNSOL data set for which only data is availablefor neutral molecules It is clear from this data that the new radii sets lowers the errorof the predicted solvation free energies for the other polar solvents and has a negligibleeffect on the accuracy for the non-polar solvents
As for acetonitrile and DMSO the improvement is greatest for the ions where the RMSEdecreases by 3-12 kcalmol The improvement is more modest (05-3 kcalmol) for theneutral molecules For methanol which contains only ions the RMSE is similarly de-creased with 2-13 kcalmol
We thus recommend that the new radii optimized for water also be used for other polarsolvents when doing semiempirical SMD calculations For non-polar solvents either setof radii can be used
14
Table 6 RMSE (in kcalmol) of non-aqueous solvation energies computed with SMD usingHF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicates that theradii has been reoptimized Corresponding RMSE values for the COSMO method are includedfor comparison The reference set is split into full set neutral cations and anions
All Neu Cat Aniacetonitrile
HFSMD 112 29 105 131PM3SMD 194 41 162 246PM3SMDdagger 140 37 113 182PM6SMD 219 33 177 284PM6SMDdagger 130 12 114 160DFTBSMD 150 23 119 197DFTBSMDdagger 109 22 90 140PM3COSMO 130 31 128 147PM6COSMO 137 23 135 155
dimethylsulfoxideHFSMD 119 30 106 126PM3SMD 202 42 153 214PM3SMDdagger 170 35 90 182PM6SMD 255 45 172 272PM6SMDdagger 177 17 99 189DFTBSMD 169 23 108 181DFTBSMDdagger 125 21 76 134PM3COSMO 125 34 107 132PM6COSMO 150 31 119 159
methanolHFSMD 40 00 41 39PM3SMD 126 00 108 135PM3SMDdagger 66 00 60 69PM6SMD 163 00 120 183PM6SMDdagger 52 00 54 52DFTBSMD 84 00 63 94DFTBSMDdagger 47 00 44 49PM3COSMO 59 00 68 54PM6COSMO 54 00 67 46
otherHFSMD 42 42 00 00PM3SMD 51 51 00 00PM3SMDdagger 51 51 00 00PM6SMD 35 35 00 00PM6SMDdagger 34 34 00 00DFTBSMD 30 30 00 00DFTBSMDdagger 29 29 00 00PM3COSMO 36 36 00 00PM6COSMO 27 27 00 00
15
ConclusionThe PM6 method in GAMESS was extended to elements requiring d-integrals and inter-faced with the polarized continuum model (PCM) of solvation including semi-numericalgradients However the accuracy of aqueous solvation energies computed using AM1PM3 PM6 and DFTB and the SMD continuum solvation model was tested using a sub-set of molecules from the MNSOL data set which showed that the errors in SMD solvationenergies predicted using NDDO-based methods was considerably larger than when usingDFT and HF with RMSE values of 50 to 86 kcalmol compared to 34 kcalmol forHF6-31G(d) For the NDDO-based methods the errors were especially large for cationsand in the case of PM6 also for anions with RMSE values of 101 to 148 kcalmol incomparison with to 50 to 57 kcalmol for HF6-31G(d)SMD Corresponding COSMOresults (where the maximum RMSE is 75 kcalmol) suggested that the NDDOSMDresults could be improved by re-parameterizing the SMD Coulomb radii for the NDDOmethods
The fact that the NDDOSMD errors are largest for ions suggest that the problem iswith the polar solvation energy so we focus on optimizing the values of the Coulombradii while leaving all parameters associated with the non-polar solvation free energy un-changed We optimize the radii only for the ionic species on a subset of the MNSOL dataset previously used to parametrize RHF6-31G(d)SMD but include neutral moleculeswhen testing how well SMD with the new Coulomb radii perform We found the bestresults are obtained by changing only the radii for hydrogen carbon oxygen nitrogenand sulfur and this leads to RMSE values for PM3 (neutrals 28ions ca 5 kcalmol)PM6 (47ca 5 kcalmol) and DFTB (39ca 5 kcalmol) that are more comparable toHF6-31G(d) (24ca 5 kcalmol) We note that the SMD parameterization is done us-ing mono-aquo microsolvated species for small ions with atoms carrying large (partial)charges while the MNSOL data set also contains data for the non-microsolvated equiv-alents Comparison to the HF6-31G(d) MSEs reported by Marenich et al [8] reportedfor the selectively microsolvated ions indicates that the inclusion of non-microsolvatedspecies leads an MSE increase of about 1 kcalmol
Though the radii are optimized to reproduce aqueous solvation energies they also leadmore accurate predictions for other polar solvents such as DMSO acetonitrile andmethanol while the improvement for non-polar solvents are negligible
Supplementary MaterialSee supplementary material for Tables S1-S3 and Figures S1-S3 which are referred toin the text In addition we also provide a table of dielectric constants used for thecalculations and a table of aqueous solvation free energies obtained using the optimizedparameters
AcknowledgmentsJCK and JHJ would like to acknowledge Jimmy (Mr MOPAC) Stewart for providingsource code and guidance for the PM6 GAMESS implementation Also thanks to Donald
16
Truhlar and Christopher Cramer for sharing the MNSOL data set and for insightfulcomments on the manuscript CS thanks the Danish Council for Independent Research(the Sapere Aude program) for financial support (Grant no DFF 4181-00370)
References[1] J Tomasi B Mennucci and R Cammi ldquoQuantum mechanical continuum solvation
modelsrdquo Chem Rev vol 105 pp 2999ndash3093 2005
[2] A Klamt and G Schuurmann ldquoCOSMO a new approach to dielectric screeningin solvents with explicit expressions for the screening energy and its gradientrdquo JChem Soc Perkin Trans vol 2 pp 799ndash805 1993
[3] C J Cramer and D G Truhlar ldquoA universal approach to solvation modelingrdquoAccounts of Chemical Research vol 41 no 6 pp 760ndash768 jun 2008 doi101021ar800019z
[4] V Barone M Cossi and J Tomasi ldquoA new definition of cavities for thecomputation of solvation free energies by the polarizable continuum modelrdquo JChem Phys vol 107 no 8 pp 3210ndash3221 aug 1997 doi 1010631474671
[5] F Eckert and A Klamt ldquoFast solvent screening via quantum chemistryCOSMO-RS approachrdquo AIChE J vol 48 no 2 pp 369ndash385 feb 2002 doi101002aic690480220
[6] C Curutchet A Bidon-Chanal I Soteras M Orozco and F J Luque ldquoMSTcontinuum study of the hydration free energies of monovalent ionic speciesrdquo JPhys Chem B vol 109 no 8 pp 3565ndash3574 mar 2005 doi 101021jp047197s
[7] Y Takano and K N Houk ldquoBenchmarking the conductor-like polarizablecontinuum model (CPCM) for aqueous solvation free energies of neutral and ionicorganic moleculesrdquo J Chem Theory Comput vol 1 no 1 pp 70ndash77 jan 2005doi 101021ct049977a
[8] A V Marenich R M Olson C P Kelly C J Cramer and D G TruhlarldquoSelf-consistent reaction field model for aqueous and nonaqueous solutions based onaccurate polarized partial chargesrdquo Journal of Chemical Theory and Computationvol 3 no 6 pp 2011ndash2033 nov 2007 doi 101021ct7001418
[9] A V Marenich C J Cramer and D G Truhlar ldquoUniversal solvation model basedon solute electron density and on a continuum model of the solvent defined by thebulk dielectric constant and atomic surface tensionsrdquo J Phys Chem B vol 113no 18 pp 6378ndash6396 may 2009 doi 101021jp810292n
[10] G E Chudinov D V Napolov and M V Basilevsky ldquoQuantum-chemicalcalculations of the hydration energies of organic cations and anions in the frameworkof a continuum solvent approximationrdquo Chem Phys vol 160 no 1 pp 41 ndash 541992 doi 1010160301-0104(92)87090-V
[11] M J Negre M Orozco and F J Luque ldquoA new strategy for the representation ofenvironment effects in semi-empirical calculations based on dewarrsquos hamiltoniansrdquoChem Phys Lett vol 196 no 1 pp 27ndash36 1992
17
[12] F J Luque M J Negre and M Orozco ldquoAn am1-scrf approach to the study ofchanges in molecular properties induced by solventrdquo J Phys Chem vol 97 no 17pp 4386ndash4391 1993
[13] M Caricato B Mennucci and J Tomasi ldquoSolvent effects on the electronic spectraAn extension of the polarizable continuum model to the zindo methodrdquo J PhysChem A vol 108 no 29 pp 6248ndash6256 2004
[14] M J S Dewar E G Zoebisch E F Healy and J J P Stewart ldquoDevelopmentand use of quantum mechanical molecular models 76 AM1 a new general purposequantum mechanical molecular modelrdquo J A Chem Soc vol 10 pp 3902ndash39091985 doi 101021ja00299a024
[15] J J P Stewart ldquoOptimization of Parameters for Semi-Empirical Methods I-Methodrdquo J Comp Chem vol 10 pp 209ndash220 1989 doi 101002jcc540100208
[16] J J P Stewart ldquoOptimization of parameters for semiempirical methods V Modi-fication of NDDO approximations and application to 70 elementsrdquo J Mol Modelvol 13 pp 1172ndash1213 2007
[17] M Gaus Q Cui and M Elstner ldquoDFTB3 Extension of the Self-Consistent-Charge Density-Functional Tight-Binding Method (SCC-DFTB)rdquo J Chem TheoryComput vol 7 no 4 pp 931ndash948 apr 2011 doi 101021ct100684s
[18] A V Marenich C P Kelly J D Thompson G D Hawkins C C ChambersD J Giesen P P Winget C J Cramer and D G Truhlar ldquoMinnesota solvationdatabaserdquo 2012
[19] ldquoMinnesota solvation databaserdquo accessed November 24th 2017 [Online] Availablehttpscompchemumnedumnsol
[20] M W Schmidt K K Baldridge J A Boatz S T Elbert M S Gordon J HJensen S Koseki N Matsunaga K A Nguyen S Su T L Windus M Dupuisand JAMontgomery ldquoGeneral atomic and molecular electronic structure systemrdquoJ Comput Chem vol 14 pp 1347ndash1363 1993
[21] V Barone and M Cossi ldquoQuantum calculation of molecular energies and energygradients in solution by a conductor solvent modelrdquo The Journal of PhysicalChemistry A vol 102 no 11 pp 1995ndash2001 mar 1998 doi 101021jp9716997
[22] M Cossi N Rega G Scalmani and V Barone ldquoEnergies structures andelectronic properties of molecules in solution with the c-PCM solvation modelrdquoJournal of Computational Chemistry vol 24 no 6 pp 669ndash681 apr 2003 doi101002jcc10189
[23] C Steinmann K L Blaeligdel A S Christensen and J H Jensen ldquoInterface ofthe polarizable continuum model of solvation with semi-empirical methods in thegamess programrdquo PLoS ONE vol 8 no 7 p e67725 07 2013 doi 101371jour-nalpone0067725
[24] Y Nishimoto ldquoDFTBPCM applied to ground and excited state potential energysurfacesrdquo J Phys Chem A vol 120 no 5 pp 771ndash784 feb 2016 doi101021acsjpca5b10732
18
[25] M Gaus X Lu M Elstner and Q Cui ldquoParameterization of DFTB33OB forSulfur and Phosphorus for Chemical and Biological Applicationsrdquo J Chem TheoryComput vol 10 no 4 pp 1518ndash1537 apr 2014 doi 101021ct401002w
[26] X Lu M Gaus M Elstner and Q Cui ldquoParametrization of DFTB33OBfor Magnesium and Zinc for Chemical and Biological Applicationsrdquo The Journalof Physical Chemistry B vol 119 no 3 pp 1062ndash1082 jan 2015 doi101021jp506557r
[27] M Kubillus T Kubar M Gaus J Rezac and M Elstner ldquoParameterization ofthe DFTB3 Method for Br Ca Cl F I K and Na in Organic and BiologicalSystemsrdquo J Chem Theory Comput vol 11 no 1 pp 332ndash342 jan 2015 doi101021ct5009137
[28] J J P Stewart ldquoMOPAC2012rdquo 2016 Stewart Computational Chemistry ColoradoSprings CO USA [Online] Available httpopenmopacnet
[29] J L Pascual-Ahuir E Silla J Tomasi and R Bonaccorsi ldquoElectrostaticinteraction of a solute with a continuum improved description of the cavity and ofthe surface cavity bound charge distributionrdquo Journal of Computational Chemistryvol 8 no 6 pp 778ndash787 sep 1987 doi 101002jcc540080605
[30] P Su and H Li ldquoContinuous and smooth potential energy surface for conductorlikescreening solvation model using fixed points with variable areasrdquo The Journal ofChemical Physics vol 130 no 7 p 074109 feb 2009 doi 10106313077917
[31] J Tomasi B Mennucci and E Cances ldquoThe IEF version of the PCM solvationmethod an overview of a new method addressed to study molecular solutes at theQM ab initio levelrdquo Journal of Molecular Structure THEOCHEM vol 464 no1-3 pp 211ndash226 may 1999 doi 101016s0166-1280(98)00553-3
[32] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 1 the calculation of confidence intervalsrdquo J Comput Aided MolDes vol 28 no 9 pp 887ndash918 Sep 2014 doi 101007s10822-014-9753-z
[33] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 2 comparing methodsrdquo J Comput Aided Mol Des vol 30 no 2pp 103ndash126 Feb 2016 doi 101007s10822-016-9904-5
[34] J H Jensen ldquoWhich method is more accurate or errors have error barsrdquo PeerJPreprints vol 5 p e2693v1 2017 doi 107287peerjpreprints2693v1
[35] J H Jensen C J Swain and L Olsen ldquoPrediction of pKa values for druglikemolecules using semiempirical quantum chemical methodsrdquo The Journal of PhysicalChemistry A vol 121 no 3 pp 699ndash707 jan 2017 doi 101021acsjpca6b10990
[36] F Eckert and A Klamt ldquoAccurate prediction of basicity in aqueous solution withCOSMO-RSrdquo Journal of Computational Chemistry vol 27 no 1 pp 11ndash19 2005doi 101002jcc20309
[37] J A Nelder and R Mead ldquoA simplex method for function minimizationrdquo TheComputer Journal vol 7 no 4 pp 308ndash313 1965 doi 101093comjnl74308
19
[38] E Jones T Oliphant P Peterson et al ldquoSciPy Open source scientifictools for Pythonrdquo 2001 accessed November 24th 2017 [Online] Availablehttpwwwscipyorgrdquo
[39] J Charnley 2017 accessed November 24th 2017 [Online] Available httpsgithubcomcharnleyoptimize gamess parameters
[40] C P Kelly C J Cramer and D G Truhlar ldquoAqueous solvation free energies ofions and ion-water clusters based on an accurate value for the absolute aqueoussolvation free energy of the protonrdquo The Journal of Physical Chemistry B vol 110no 32 pp 16 066ndash16 081 aug 2006 doi 101021jp063552y
20
HFSMDAM1SMD
PM3SMDPM6SMD
DFTBSMD
AM1COSMO
PM3COSMO
PM6COSMO null0
2
4
6
8
10
12
14
16RM
SE [k
calm
ol]
PM3PM3HCO
PM3HCON
PM3HCONSPM3all PM6
PM6HCO
PM6HCON
PM6HCONS
PM6HCONShPM6all
DFTB
DFTBHCON0
2
4
6
8
10
12
14RM
SE [k
calm
ol]
HFSMD
PM3SMDPM6SMD
DFTBSMD
PM3COSMO
PM6COSMO0
2
4
6
8
10RM
SE [k
calm
ol]
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6err
or

From the results discussed above and presented in Table 2 optimizing for atom typesbeyond HCON showed no significant increase in accuracy for either PM3 or PM6 There-fore the same procedure (optimizing HCON radii) was applied to DFTB For DFTB theRMSE values of the full set of molecules the subset of cation and the subset of anionsgoes from 41 49 and 67 kcalmol to 33 29 and 44 kcalmol respectively
In summary we find that optimizing the SMD radii in combination with either PM3 orPM6 gives appreciable decreases in RMSE accuracy asymp 7 kcalmol in both cases comparedto the reference results We also find that no significant improvement is obtained whenattempting to optimize the radii of other atoms than what was done with HCON subsetof radii For this subset DFTB also saw an increase in accuracy for ions of about asymp 6kcalmol Because of this we found the best choice was change the radii for hydrogencarbon oxygen and nitrogen (HCON) for PM3 and DFTB methods For PM6 we optimizedincluding sulfur and changed the radius of hydrogen to 06 A to remove overpolarizationproblems These methods are denoted as SMDdagger going forward
Comparison with Other Solvation MethodsUsing the new parameters for PM3SMDdagger PM6SMDdagger and DFTBSMDdagger we com-pare the obtained RMSE with other approaches to obtain solvent free energies such asHF6-31G(d)SMD (one of the levels of theory used in the original SMD parameteriza-tion) PM3COSMO and PM6COSMO for the MNSOL data set (Table 4 and Figure 3)
HFSMD
PM3SMDPM6SMD
DFTBSMD
PM3COSMO
PM6COSMO0
2
4
6
8
10
RMSE
[kca
lmol
]
Figure 3 RMSE (in kcalmol) of aqueous solvation energies computed with SMD using HF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicates that the Radiihas been reoptimized Corresponding RMSE values for the COSMO method are included forcomparison The dataset is split into full set (black) neutral (green) cations (red) and anions(blue)
The overall accuracy of PM3SMDdagger is now similar to HFSMD although the RMSEvalue is 04 and 06 kcalmol higher for neutral molecules and cations while the RMSE
9
is 07 kcalmol lower for anions For PM6 the overall RMSE is 14 kcalmol higher thanHF and the RMSE for neutral molecules is 23 kcalmol higher On the other hand theRMSE for cations is only 01 kcalmol higher while the RMSE for anions is 08 kcal-mol lower compared to HF Finally for DFTB the overall RMSE value (41 kcalmol)is in between those for PM3 and PM6 as is the RMSE values for neutral molecules (39kcalmol) But for cations the RMSE (37 kcalmol) is significantly lower than the othermethods while the RMSE for anions (51 kcalmol) is very similar
While the solvation free energies of neutral molecules can generally be measured with anaccuracy of ca 02 kcalmol the corresponding solvation free energies of ions are inferredfrom other experimental values with a typical accuracy about 30 kcalmol[40] Thus theaccuracy of the semiempirical SMDdagger is for all intents and purposes identical to HFSMDfor ions but significantly worse for neutral ions in the case of PM6 and DFTB Thesource of this error is most likely due to a poorer description of the electrostatic proper-ties compared to HF For example the error in dipole moments for HCNO compounds islarger for PM6 than for PM3[16]
Table 4 also lists values for PM3COSMO and PM6COSMO as implemented in MOPACfor comparison This COSMO implementation only evaluates the polar part of the solva-tion free energy and will not be as accurate as properly parameterized COSMO-RS[5] val-ues For PM3 the SMDdagger results are significantly lower than the corresponding COSMOresults while for PM6 there is only a significant improvement for cations
10
Table 4 RMSE MUE and MSE (in kcalmol) of aqueous solvation energies computed withSMD using HF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicatesthat the Radii has been reoptimized Corresponding RMSE values for the COSMO method areincluded for comparison The reference set is split into full set neutral cations and anions
SMD SMDdagger COSMO
HF PM3 PM6 DFTB PM3 PM6All
RMSE 34 36 48 41 48 50MUE 21 26 35 31 34 39MSE 03 10 -17 -11 05 -18Max 178 155 231 160 213 211
NeutralRMSE 24 28 47 39 36 47MUE 13 20 34 29 25 35MSE -09 09 -29 -21 -10 -34Max 178 155 231 160 213 211
CationsRMSE 50 56 51 37 74 73MUE 36 44 42 29 59 60MSE 31 36 38 10 56 59Max 140 127 124 91 186 183
AnionsRMSE 56 49 48 51 68 45MUE 46 37 36 40 58 38MSE 39 -03 -05 18 37 03Max 143 146 147 147 152 105
11
pKa PredictionPart of the impetus for this work were the relatively large error in pKa values predictedusing PM3COSMO compared to PM3SMD with the original SMD radii observed byJensen et al [35] so we use the new SMD radii to compute the pKa value using the samedata set The data is presented in Figure 4 and Table 5 and shows that the RMSE dropsfrom 15plusmn03 to 09plusmn03 pH units for PM3SMDdagger and 20plusmn04 to 16plusmn0304 pH unitsfor DFTBSMDdagger In both cases the improvement is primarily a result of the decreasein mean error (ME) ie the original radii led to a fairly systematic underestimation ofthe pKa values that has now been removed by the reparameterization For DFTB theincreased accuracy is also due to a larger number of low error predictions as is evident inFigure 4 In the case of PM3SMDdagger the accuracy is now very similar to PM3COSMOThe RMSE for PM6SMDdagger is similar to PM6COSMO and larger than for PM3SMDin analogy with the corresponding results for COSMO
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6
err
or
Figure 4 Plot of the errors in the predicted pKa values (pKaminus pKexpa )
12
Table 5 Root-Mean-Square-Error (RMSE) man error (ME) and Pearson correlation (r) for predicted pKa values relative to experiment togetherwith their statistical uncertainty (95 confidence limits)
PM3COSMO PM3SMD PM3SMDdagger PM6COSMO PM6SMDdagger DFTBSMD DFTBSMDdaggerRMSE 10 15 09 18 16 20 1695 conf plusmn 0202 plusmn 0303 plusmn 0202 plusmn 0304 plusmn 0304 plusmn 0404 plusmn 0304ME -05 plusmn 02 -12 plusmn 03 -02 plusmn 02 -13 plusmn 04 -08 plusmn 04 -07 plusmn 05 -01 plusmn 04r 095 093 093 090 086 087 08695 conf plusmn 002004 plusmn 003005 plusmn 003005 plusmn 004007 plusmn 006009 plusmn 005008 plusmn 006009
13
Non-aqueous SolventsThe new radii were optimized explicitly for water so it is not a priori clear whether theyoffer more accurate results for other solvents Table 6 shows the RMSE values for allneutral cations and anions for the other polar solvents in the MNSOL data set acetoni-trile DMSO and methanol for the original SMD radii and the corresponding re-optimizedradii (dagger) In addition corresponding combined RMSE values are given for the remaining88 mostly non-polar solvents in the MNSOL data set for which only data is availablefor neutral molecules It is clear from this data that the new radii sets lowers the errorof the predicted solvation free energies for the other polar solvents and has a negligibleeffect on the accuracy for the non-polar solvents
As for acetonitrile and DMSO the improvement is greatest for the ions where the RMSEdecreases by 3-12 kcalmol The improvement is more modest (05-3 kcalmol) for theneutral molecules For methanol which contains only ions the RMSE is similarly de-creased with 2-13 kcalmol
We thus recommend that the new radii optimized for water also be used for other polarsolvents when doing semiempirical SMD calculations For non-polar solvents either setof radii can be used
14
Table 6 RMSE (in kcalmol) of non-aqueous solvation energies computed with SMD usingHF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicates that theradii has been reoptimized Corresponding RMSE values for the COSMO method are includedfor comparison The reference set is split into full set neutral cations and anions
All Neu Cat Aniacetonitrile
HFSMD 112 29 105 131PM3SMD 194 41 162 246PM3SMDdagger 140 37 113 182PM6SMD 219 33 177 284PM6SMDdagger 130 12 114 160DFTBSMD 150 23 119 197DFTBSMDdagger 109 22 90 140PM3COSMO 130 31 128 147PM6COSMO 137 23 135 155
dimethylsulfoxideHFSMD 119 30 106 126PM3SMD 202 42 153 214PM3SMDdagger 170 35 90 182PM6SMD 255 45 172 272PM6SMDdagger 177 17 99 189DFTBSMD 169 23 108 181DFTBSMDdagger 125 21 76 134PM3COSMO 125 34 107 132PM6COSMO 150 31 119 159
methanolHFSMD 40 00 41 39PM3SMD 126 00 108 135PM3SMDdagger 66 00 60 69PM6SMD 163 00 120 183PM6SMDdagger 52 00 54 52DFTBSMD 84 00 63 94DFTBSMDdagger 47 00 44 49PM3COSMO 59 00 68 54PM6COSMO 54 00 67 46
otherHFSMD 42 42 00 00PM3SMD 51 51 00 00PM3SMDdagger 51 51 00 00PM6SMD 35 35 00 00PM6SMDdagger 34 34 00 00DFTBSMD 30 30 00 00DFTBSMDdagger 29 29 00 00PM3COSMO 36 36 00 00PM6COSMO 27 27 00 00
15
ConclusionThe PM6 method in GAMESS was extended to elements requiring d-integrals and inter-faced with the polarized continuum model (PCM) of solvation including semi-numericalgradients However the accuracy of aqueous solvation energies computed using AM1PM3 PM6 and DFTB and the SMD continuum solvation model was tested using a sub-set of molecules from the MNSOL data set which showed that the errors in SMD solvationenergies predicted using NDDO-based methods was considerably larger than when usingDFT and HF with RMSE values of 50 to 86 kcalmol compared to 34 kcalmol forHF6-31G(d) For the NDDO-based methods the errors were especially large for cationsand in the case of PM6 also for anions with RMSE values of 101 to 148 kcalmol incomparison with to 50 to 57 kcalmol for HF6-31G(d)SMD Corresponding COSMOresults (where the maximum RMSE is 75 kcalmol) suggested that the NDDOSMDresults could be improved by re-parameterizing the SMD Coulomb radii for the NDDOmethods
The fact that the NDDOSMD errors are largest for ions suggest that the problem iswith the polar solvation energy so we focus on optimizing the values of the Coulombradii while leaving all parameters associated with the non-polar solvation free energy un-changed We optimize the radii only for the ionic species on a subset of the MNSOL dataset previously used to parametrize RHF6-31G(d)SMD but include neutral moleculeswhen testing how well SMD with the new Coulomb radii perform We found the bestresults are obtained by changing only the radii for hydrogen carbon oxygen nitrogenand sulfur and this leads to RMSE values for PM3 (neutrals 28ions ca 5 kcalmol)PM6 (47ca 5 kcalmol) and DFTB (39ca 5 kcalmol) that are more comparable toHF6-31G(d) (24ca 5 kcalmol) We note that the SMD parameterization is done us-ing mono-aquo microsolvated species for small ions with atoms carrying large (partial)charges while the MNSOL data set also contains data for the non-microsolvated equiv-alents Comparison to the HF6-31G(d) MSEs reported by Marenich et al [8] reportedfor the selectively microsolvated ions indicates that the inclusion of non-microsolvatedspecies leads an MSE increase of about 1 kcalmol
Though the radii are optimized to reproduce aqueous solvation energies they also leadmore accurate predictions for other polar solvents such as DMSO acetonitrile andmethanol while the improvement for non-polar solvents are negligible
Supplementary MaterialSee supplementary material for Tables S1-S3 and Figures S1-S3 which are referred toin the text In addition we also provide a table of dielectric constants used for thecalculations and a table of aqueous solvation free energies obtained using the optimizedparameters
AcknowledgmentsJCK and JHJ would like to acknowledge Jimmy (Mr MOPAC) Stewart for providingsource code and guidance for the PM6 GAMESS implementation Also thanks to Donald
16
Truhlar and Christopher Cramer for sharing the MNSOL data set and for insightfulcomments on the manuscript CS thanks the Danish Council for Independent Research(the Sapere Aude program) for financial support (Grant no DFF 4181-00370)
References[1] J Tomasi B Mennucci and R Cammi ldquoQuantum mechanical continuum solvation
modelsrdquo Chem Rev vol 105 pp 2999ndash3093 2005
[2] A Klamt and G Schuurmann ldquoCOSMO a new approach to dielectric screeningin solvents with explicit expressions for the screening energy and its gradientrdquo JChem Soc Perkin Trans vol 2 pp 799ndash805 1993
[3] C J Cramer and D G Truhlar ldquoA universal approach to solvation modelingrdquoAccounts of Chemical Research vol 41 no 6 pp 760ndash768 jun 2008 doi101021ar800019z
[4] V Barone M Cossi and J Tomasi ldquoA new definition of cavities for thecomputation of solvation free energies by the polarizable continuum modelrdquo JChem Phys vol 107 no 8 pp 3210ndash3221 aug 1997 doi 1010631474671
[5] F Eckert and A Klamt ldquoFast solvent screening via quantum chemistryCOSMO-RS approachrdquo AIChE J vol 48 no 2 pp 369ndash385 feb 2002 doi101002aic690480220
[6] C Curutchet A Bidon-Chanal I Soteras M Orozco and F J Luque ldquoMSTcontinuum study of the hydration free energies of monovalent ionic speciesrdquo JPhys Chem B vol 109 no 8 pp 3565ndash3574 mar 2005 doi 101021jp047197s
[7] Y Takano and K N Houk ldquoBenchmarking the conductor-like polarizablecontinuum model (CPCM) for aqueous solvation free energies of neutral and ionicorganic moleculesrdquo J Chem Theory Comput vol 1 no 1 pp 70ndash77 jan 2005doi 101021ct049977a
[8] A V Marenich R M Olson C P Kelly C J Cramer and D G TruhlarldquoSelf-consistent reaction field model for aqueous and nonaqueous solutions based onaccurate polarized partial chargesrdquo Journal of Chemical Theory and Computationvol 3 no 6 pp 2011ndash2033 nov 2007 doi 101021ct7001418
[9] A V Marenich C J Cramer and D G Truhlar ldquoUniversal solvation model basedon solute electron density and on a continuum model of the solvent defined by thebulk dielectric constant and atomic surface tensionsrdquo J Phys Chem B vol 113no 18 pp 6378ndash6396 may 2009 doi 101021jp810292n
[10] G E Chudinov D V Napolov and M V Basilevsky ldquoQuantum-chemicalcalculations of the hydration energies of organic cations and anions in the frameworkof a continuum solvent approximationrdquo Chem Phys vol 160 no 1 pp 41 ndash 541992 doi 1010160301-0104(92)87090-V
[11] M J Negre M Orozco and F J Luque ldquoA new strategy for the representation ofenvironment effects in semi-empirical calculations based on dewarrsquos hamiltoniansrdquoChem Phys Lett vol 196 no 1 pp 27ndash36 1992
17
[12] F J Luque M J Negre and M Orozco ldquoAn am1-scrf approach to the study ofchanges in molecular properties induced by solventrdquo J Phys Chem vol 97 no 17pp 4386ndash4391 1993
[13] M Caricato B Mennucci and J Tomasi ldquoSolvent effects on the electronic spectraAn extension of the polarizable continuum model to the zindo methodrdquo J PhysChem A vol 108 no 29 pp 6248ndash6256 2004
[14] M J S Dewar E G Zoebisch E F Healy and J J P Stewart ldquoDevelopmentand use of quantum mechanical molecular models 76 AM1 a new general purposequantum mechanical molecular modelrdquo J A Chem Soc vol 10 pp 3902ndash39091985 doi 101021ja00299a024
[15] J J P Stewart ldquoOptimization of Parameters for Semi-Empirical Methods I-Methodrdquo J Comp Chem vol 10 pp 209ndash220 1989 doi 101002jcc540100208
[16] J J P Stewart ldquoOptimization of parameters for semiempirical methods V Modi-fication of NDDO approximations and application to 70 elementsrdquo J Mol Modelvol 13 pp 1172ndash1213 2007
[17] M Gaus Q Cui and M Elstner ldquoDFTB3 Extension of the Self-Consistent-Charge Density-Functional Tight-Binding Method (SCC-DFTB)rdquo J Chem TheoryComput vol 7 no 4 pp 931ndash948 apr 2011 doi 101021ct100684s
[18] A V Marenich C P Kelly J D Thompson G D Hawkins C C ChambersD J Giesen P P Winget C J Cramer and D G Truhlar ldquoMinnesota solvationdatabaserdquo 2012
[19] ldquoMinnesota solvation databaserdquo accessed November 24th 2017 [Online] Availablehttpscompchemumnedumnsol
[20] M W Schmidt K K Baldridge J A Boatz S T Elbert M S Gordon J HJensen S Koseki N Matsunaga K A Nguyen S Su T L Windus M Dupuisand JAMontgomery ldquoGeneral atomic and molecular electronic structure systemrdquoJ Comput Chem vol 14 pp 1347ndash1363 1993
[21] V Barone and M Cossi ldquoQuantum calculation of molecular energies and energygradients in solution by a conductor solvent modelrdquo The Journal of PhysicalChemistry A vol 102 no 11 pp 1995ndash2001 mar 1998 doi 101021jp9716997
[22] M Cossi N Rega G Scalmani and V Barone ldquoEnergies structures andelectronic properties of molecules in solution with the c-PCM solvation modelrdquoJournal of Computational Chemistry vol 24 no 6 pp 669ndash681 apr 2003 doi101002jcc10189
[23] C Steinmann K L Blaeligdel A S Christensen and J H Jensen ldquoInterface ofthe polarizable continuum model of solvation with semi-empirical methods in thegamess programrdquo PLoS ONE vol 8 no 7 p e67725 07 2013 doi 101371jour-nalpone0067725
[24] Y Nishimoto ldquoDFTBPCM applied to ground and excited state potential energysurfacesrdquo J Phys Chem A vol 120 no 5 pp 771ndash784 feb 2016 doi101021acsjpca5b10732
18
[25] M Gaus X Lu M Elstner and Q Cui ldquoParameterization of DFTB33OB forSulfur and Phosphorus for Chemical and Biological Applicationsrdquo J Chem TheoryComput vol 10 no 4 pp 1518ndash1537 apr 2014 doi 101021ct401002w
[26] X Lu M Gaus M Elstner and Q Cui ldquoParametrization of DFTB33OBfor Magnesium and Zinc for Chemical and Biological Applicationsrdquo The Journalof Physical Chemistry B vol 119 no 3 pp 1062ndash1082 jan 2015 doi101021jp506557r
[27] M Kubillus T Kubar M Gaus J Rezac and M Elstner ldquoParameterization ofthe DFTB3 Method for Br Ca Cl F I K and Na in Organic and BiologicalSystemsrdquo J Chem Theory Comput vol 11 no 1 pp 332ndash342 jan 2015 doi101021ct5009137
[28] J J P Stewart ldquoMOPAC2012rdquo 2016 Stewart Computational Chemistry ColoradoSprings CO USA [Online] Available httpopenmopacnet
[29] J L Pascual-Ahuir E Silla J Tomasi and R Bonaccorsi ldquoElectrostaticinteraction of a solute with a continuum improved description of the cavity and ofthe surface cavity bound charge distributionrdquo Journal of Computational Chemistryvol 8 no 6 pp 778ndash787 sep 1987 doi 101002jcc540080605
[30] P Su and H Li ldquoContinuous and smooth potential energy surface for conductorlikescreening solvation model using fixed points with variable areasrdquo The Journal ofChemical Physics vol 130 no 7 p 074109 feb 2009 doi 10106313077917
[31] J Tomasi B Mennucci and E Cances ldquoThe IEF version of the PCM solvationmethod an overview of a new method addressed to study molecular solutes at theQM ab initio levelrdquo Journal of Molecular Structure THEOCHEM vol 464 no1-3 pp 211ndash226 may 1999 doi 101016s0166-1280(98)00553-3
[32] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 1 the calculation of confidence intervalsrdquo J Comput Aided MolDes vol 28 no 9 pp 887ndash918 Sep 2014 doi 101007s10822-014-9753-z
[33] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 2 comparing methodsrdquo J Comput Aided Mol Des vol 30 no 2pp 103ndash126 Feb 2016 doi 101007s10822-016-9904-5
[34] J H Jensen ldquoWhich method is more accurate or errors have error barsrdquo PeerJPreprints vol 5 p e2693v1 2017 doi 107287peerjpreprints2693v1
[35] J H Jensen C J Swain and L Olsen ldquoPrediction of pKa values for druglikemolecules using semiempirical quantum chemical methodsrdquo The Journal of PhysicalChemistry A vol 121 no 3 pp 699ndash707 jan 2017 doi 101021acsjpca6b10990
[36] F Eckert and A Klamt ldquoAccurate prediction of basicity in aqueous solution withCOSMO-RSrdquo Journal of Computational Chemistry vol 27 no 1 pp 11ndash19 2005doi 101002jcc20309
[37] J A Nelder and R Mead ldquoA simplex method for function minimizationrdquo TheComputer Journal vol 7 no 4 pp 308ndash313 1965 doi 101093comjnl74308
19
[38] E Jones T Oliphant P Peterson et al ldquoSciPy Open source scientifictools for Pythonrdquo 2001 accessed November 24th 2017 [Online] Availablehttpwwwscipyorgrdquo
[39] J Charnley 2017 accessed November 24th 2017 [Online] Available httpsgithubcomcharnleyoptimize gamess parameters
[40] C P Kelly C J Cramer and D G Truhlar ldquoAqueous solvation free energies ofions and ion-water clusters based on an accurate value for the absolute aqueoussolvation free energy of the protonrdquo The Journal of Physical Chemistry B vol 110no 32 pp 16 066ndash16 081 aug 2006 doi 101021jp063552y
20
HFSMDAM1SMD
PM3SMDPM6SMD
DFTBSMD
AM1COSMO
PM3COSMO
PM6COSMO null0
2
4
6
8
10
12
14
16RM
SE [k
calm
ol]
PM3PM3HCO
PM3HCON
PM3HCONSPM3all PM6
PM6HCO
PM6HCON
PM6HCONS
PM6HCONShPM6all
DFTB
DFTBHCON0
2
4
6
8
10
12
14RM
SE [k
calm
ol]
HFSMD
PM3SMDPM6SMD
DFTBSMD
PM3COSMO
PM6COSMO0
2
4
6
8
10RM
SE [k
calm
ol]
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6err
or

is 07 kcalmol lower for anions For PM6 the overall RMSE is 14 kcalmol higher thanHF and the RMSE for neutral molecules is 23 kcalmol higher On the other hand theRMSE for cations is only 01 kcalmol higher while the RMSE for anions is 08 kcal-mol lower compared to HF Finally for DFTB the overall RMSE value (41 kcalmol)is in between those for PM3 and PM6 as is the RMSE values for neutral molecules (39kcalmol) But for cations the RMSE (37 kcalmol) is significantly lower than the othermethods while the RMSE for anions (51 kcalmol) is very similar
While the solvation free energies of neutral molecules can generally be measured with anaccuracy of ca 02 kcalmol the corresponding solvation free energies of ions are inferredfrom other experimental values with a typical accuracy about 30 kcalmol[40] Thus theaccuracy of the semiempirical SMDdagger is for all intents and purposes identical to HFSMDfor ions but significantly worse for neutral ions in the case of PM6 and DFTB Thesource of this error is most likely due to a poorer description of the electrostatic proper-ties compared to HF For example the error in dipole moments for HCNO compounds islarger for PM6 than for PM3[16]
Table 4 also lists values for PM3COSMO and PM6COSMO as implemented in MOPACfor comparison This COSMO implementation only evaluates the polar part of the solva-tion free energy and will not be as accurate as properly parameterized COSMO-RS[5] val-ues For PM3 the SMDdagger results are significantly lower than the corresponding COSMOresults while for PM6 there is only a significant improvement for cations
10
Table 4 RMSE MUE and MSE (in kcalmol) of aqueous solvation energies computed withSMD using HF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicatesthat the Radii has been reoptimized Corresponding RMSE values for the COSMO method areincluded for comparison The reference set is split into full set neutral cations and anions
SMD SMDdagger COSMO
HF PM3 PM6 DFTB PM3 PM6All
RMSE 34 36 48 41 48 50MUE 21 26 35 31 34 39MSE 03 10 -17 -11 05 -18Max 178 155 231 160 213 211
NeutralRMSE 24 28 47 39 36 47MUE 13 20 34 29 25 35MSE -09 09 -29 -21 -10 -34Max 178 155 231 160 213 211
CationsRMSE 50 56 51 37 74 73MUE 36 44 42 29 59 60MSE 31 36 38 10 56 59Max 140 127 124 91 186 183
AnionsRMSE 56 49 48 51 68 45MUE 46 37 36 40 58 38MSE 39 -03 -05 18 37 03Max 143 146 147 147 152 105
11
pKa PredictionPart of the impetus for this work were the relatively large error in pKa values predictedusing PM3COSMO compared to PM3SMD with the original SMD radii observed byJensen et al [35] so we use the new SMD radii to compute the pKa value using the samedata set The data is presented in Figure 4 and Table 5 and shows that the RMSE dropsfrom 15plusmn03 to 09plusmn03 pH units for PM3SMDdagger and 20plusmn04 to 16plusmn0304 pH unitsfor DFTBSMDdagger In both cases the improvement is primarily a result of the decreasein mean error (ME) ie the original radii led to a fairly systematic underestimation ofthe pKa values that has now been removed by the reparameterization For DFTB theincreased accuracy is also due to a larger number of low error predictions as is evident inFigure 4 In the case of PM3SMDdagger the accuracy is now very similar to PM3COSMOThe RMSE for PM6SMDdagger is similar to PM6COSMO and larger than for PM3SMDin analogy with the corresponding results for COSMO
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6
err
or
Figure 4 Plot of the errors in the predicted pKa values (pKaminus pKexpa )
12
Table 5 Root-Mean-Square-Error (RMSE) man error (ME) and Pearson correlation (r) for predicted pKa values relative to experiment togetherwith their statistical uncertainty (95 confidence limits)
PM3COSMO PM3SMD PM3SMDdagger PM6COSMO PM6SMDdagger DFTBSMD DFTBSMDdaggerRMSE 10 15 09 18 16 20 1695 conf plusmn 0202 plusmn 0303 plusmn 0202 plusmn 0304 plusmn 0304 plusmn 0404 plusmn 0304ME -05 plusmn 02 -12 plusmn 03 -02 plusmn 02 -13 plusmn 04 -08 plusmn 04 -07 plusmn 05 -01 plusmn 04r 095 093 093 090 086 087 08695 conf plusmn 002004 plusmn 003005 plusmn 003005 plusmn 004007 plusmn 006009 plusmn 005008 plusmn 006009
13
Non-aqueous SolventsThe new radii were optimized explicitly for water so it is not a priori clear whether theyoffer more accurate results for other solvents Table 6 shows the RMSE values for allneutral cations and anions for the other polar solvents in the MNSOL data set acetoni-trile DMSO and methanol for the original SMD radii and the corresponding re-optimizedradii (dagger) In addition corresponding combined RMSE values are given for the remaining88 mostly non-polar solvents in the MNSOL data set for which only data is availablefor neutral molecules It is clear from this data that the new radii sets lowers the errorof the predicted solvation free energies for the other polar solvents and has a negligibleeffect on the accuracy for the non-polar solvents
As for acetonitrile and DMSO the improvement is greatest for the ions where the RMSEdecreases by 3-12 kcalmol The improvement is more modest (05-3 kcalmol) for theneutral molecules For methanol which contains only ions the RMSE is similarly de-creased with 2-13 kcalmol
We thus recommend that the new radii optimized for water also be used for other polarsolvents when doing semiempirical SMD calculations For non-polar solvents either setof radii can be used
14
Table 6 RMSE (in kcalmol) of non-aqueous solvation energies computed with SMD usingHF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicates that theradii has been reoptimized Corresponding RMSE values for the COSMO method are includedfor comparison The reference set is split into full set neutral cations and anions
All Neu Cat Aniacetonitrile
HFSMD 112 29 105 131PM3SMD 194 41 162 246PM3SMDdagger 140 37 113 182PM6SMD 219 33 177 284PM6SMDdagger 130 12 114 160DFTBSMD 150 23 119 197DFTBSMDdagger 109 22 90 140PM3COSMO 130 31 128 147PM6COSMO 137 23 135 155
dimethylsulfoxideHFSMD 119 30 106 126PM3SMD 202 42 153 214PM3SMDdagger 170 35 90 182PM6SMD 255 45 172 272PM6SMDdagger 177 17 99 189DFTBSMD 169 23 108 181DFTBSMDdagger 125 21 76 134PM3COSMO 125 34 107 132PM6COSMO 150 31 119 159
methanolHFSMD 40 00 41 39PM3SMD 126 00 108 135PM3SMDdagger 66 00 60 69PM6SMD 163 00 120 183PM6SMDdagger 52 00 54 52DFTBSMD 84 00 63 94DFTBSMDdagger 47 00 44 49PM3COSMO 59 00 68 54PM6COSMO 54 00 67 46
otherHFSMD 42 42 00 00PM3SMD 51 51 00 00PM3SMDdagger 51 51 00 00PM6SMD 35 35 00 00PM6SMDdagger 34 34 00 00DFTBSMD 30 30 00 00DFTBSMDdagger 29 29 00 00PM3COSMO 36 36 00 00PM6COSMO 27 27 00 00
15
ConclusionThe PM6 method in GAMESS was extended to elements requiring d-integrals and inter-faced with the polarized continuum model (PCM) of solvation including semi-numericalgradients However the accuracy of aqueous solvation energies computed using AM1PM3 PM6 and DFTB and the SMD continuum solvation model was tested using a sub-set of molecules from the MNSOL data set which showed that the errors in SMD solvationenergies predicted using NDDO-based methods was considerably larger than when usingDFT and HF with RMSE values of 50 to 86 kcalmol compared to 34 kcalmol forHF6-31G(d) For the NDDO-based methods the errors were especially large for cationsand in the case of PM6 also for anions with RMSE values of 101 to 148 kcalmol incomparison with to 50 to 57 kcalmol for HF6-31G(d)SMD Corresponding COSMOresults (where the maximum RMSE is 75 kcalmol) suggested that the NDDOSMDresults could be improved by re-parameterizing the SMD Coulomb radii for the NDDOmethods
The fact that the NDDOSMD errors are largest for ions suggest that the problem iswith the polar solvation energy so we focus on optimizing the values of the Coulombradii while leaving all parameters associated with the non-polar solvation free energy un-changed We optimize the radii only for the ionic species on a subset of the MNSOL dataset previously used to parametrize RHF6-31G(d)SMD but include neutral moleculeswhen testing how well SMD with the new Coulomb radii perform We found the bestresults are obtained by changing only the radii for hydrogen carbon oxygen nitrogenand sulfur and this leads to RMSE values for PM3 (neutrals 28ions ca 5 kcalmol)PM6 (47ca 5 kcalmol) and DFTB (39ca 5 kcalmol) that are more comparable toHF6-31G(d) (24ca 5 kcalmol) We note that the SMD parameterization is done us-ing mono-aquo microsolvated species for small ions with atoms carrying large (partial)charges while the MNSOL data set also contains data for the non-microsolvated equiv-alents Comparison to the HF6-31G(d) MSEs reported by Marenich et al [8] reportedfor the selectively microsolvated ions indicates that the inclusion of non-microsolvatedspecies leads an MSE increase of about 1 kcalmol
Though the radii are optimized to reproduce aqueous solvation energies they also leadmore accurate predictions for other polar solvents such as DMSO acetonitrile andmethanol while the improvement for non-polar solvents are negligible
Supplementary MaterialSee supplementary material for Tables S1-S3 and Figures S1-S3 which are referred toin the text In addition we also provide a table of dielectric constants used for thecalculations and a table of aqueous solvation free energies obtained using the optimizedparameters
AcknowledgmentsJCK and JHJ would like to acknowledge Jimmy (Mr MOPAC) Stewart for providingsource code and guidance for the PM6 GAMESS implementation Also thanks to Donald
16
Truhlar and Christopher Cramer for sharing the MNSOL data set and for insightfulcomments on the manuscript CS thanks the Danish Council for Independent Research(the Sapere Aude program) for financial support (Grant no DFF 4181-00370)
References[1] J Tomasi B Mennucci and R Cammi ldquoQuantum mechanical continuum solvation
modelsrdquo Chem Rev vol 105 pp 2999ndash3093 2005
[2] A Klamt and G Schuurmann ldquoCOSMO a new approach to dielectric screeningin solvents with explicit expressions for the screening energy and its gradientrdquo JChem Soc Perkin Trans vol 2 pp 799ndash805 1993
[3] C J Cramer and D G Truhlar ldquoA universal approach to solvation modelingrdquoAccounts of Chemical Research vol 41 no 6 pp 760ndash768 jun 2008 doi101021ar800019z
[4] V Barone M Cossi and J Tomasi ldquoA new definition of cavities for thecomputation of solvation free energies by the polarizable continuum modelrdquo JChem Phys vol 107 no 8 pp 3210ndash3221 aug 1997 doi 1010631474671
[5] F Eckert and A Klamt ldquoFast solvent screening via quantum chemistryCOSMO-RS approachrdquo AIChE J vol 48 no 2 pp 369ndash385 feb 2002 doi101002aic690480220
[6] C Curutchet A Bidon-Chanal I Soteras M Orozco and F J Luque ldquoMSTcontinuum study of the hydration free energies of monovalent ionic speciesrdquo JPhys Chem B vol 109 no 8 pp 3565ndash3574 mar 2005 doi 101021jp047197s
[7] Y Takano and K N Houk ldquoBenchmarking the conductor-like polarizablecontinuum model (CPCM) for aqueous solvation free energies of neutral and ionicorganic moleculesrdquo J Chem Theory Comput vol 1 no 1 pp 70ndash77 jan 2005doi 101021ct049977a
[8] A V Marenich R M Olson C P Kelly C J Cramer and D G TruhlarldquoSelf-consistent reaction field model for aqueous and nonaqueous solutions based onaccurate polarized partial chargesrdquo Journal of Chemical Theory and Computationvol 3 no 6 pp 2011ndash2033 nov 2007 doi 101021ct7001418
[9] A V Marenich C J Cramer and D G Truhlar ldquoUniversal solvation model basedon solute electron density and on a continuum model of the solvent defined by thebulk dielectric constant and atomic surface tensionsrdquo J Phys Chem B vol 113no 18 pp 6378ndash6396 may 2009 doi 101021jp810292n
[10] G E Chudinov D V Napolov and M V Basilevsky ldquoQuantum-chemicalcalculations of the hydration energies of organic cations and anions in the frameworkof a continuum solvent approximationrdquo Chem Phys vol 160 no 1 pp 41 ndash 541992 doi 1010160301-0104(92)87090-V
[11] M J Negre M Orozco and F J Luque ldquoA new strategy for the representation ofenvironment effects in semi-empirical calculations based on dewarrsquos hamiltoniansrdquoChem Phys Lett vol 196 no 1 pp 27ndash36 1992
17
[12] F J Luque M J Negre and M Orozco ldquoAn am1-scrf approach to the study ofchanges in molecular properties induced by solventrdquo J Phys Chem vol 97 no 17pp 4386ndash4391 1993
[13] M Caricato B Mennucci and J Tomasi ldquoSolvent effects on the electronic spectraAn extension of the polarizable continuum model to the zindo methodrdquo J PhysChem A vol 108 no 29 pp 6248ndash6256 2004
[14] M J S Dewar E G Zoebisch E F Healy and J J P Stewart ldquoDevelopmentand use of quantum mechanical molecular models 76 AM1 a new general purposequantum mechanical molecular modelrdquo J A Chem Soc vol 10 pp 3902ndash39091985 doi 101021ja00299a024
[15] J J P Stewart ldquoOptimization of Parameters for Semi-Empirical Methods I-Methodrdquo J Comp Chem vol 10 pp 209ndash220 1989 doi 101002jcc540100208
[16] J J P Stewart ldquoOptimization of parameters for semiempirical methods V Modi-fication of NDDO approximations and application to 70 elementsrdquo J Mol Modelvol 13 pp 1172ndash1213 2007
[17] M Gaus Q Cui and M Elstner ldquoDFTB3 Extension of the Self-Consistent-Charge Density-Functional Tight-Binding Method (SCC-DFTB)rdquo J Chem TheoryComput vol 7 no 4 pp 931ndash948 apr 2011 doi 101021ct100684s
[18] A V Marenich C P Kelly J D Thompson G D Hawkins C C ChambersD J Giesen P P Winget C J Cramer and D G Truhlar ldquoMinnesota solvationdatabaserdquo 2012
[19] ldquoMinnesota solvation databaserdquo accessed November 24th 2017 [Online] Availablehttpscompchemumnedumnsol
[20] M W Schmidt K K Baldridge J A Boatz S T Elbert M S Gordon J HJensen S Koseki N Matsunaga K A Nguyen S Su T L Windus M Dupuisand JAMontgomery ldquoGeneral atomic and molecular electronic structure systemrdquoJ Comput Chem vol 14 pp 1347ndash1363 1993
[21] V Barone and M Cossi ldquoQuantum calculation of molecular energies and energygradients in solution by a conductor solvent modelrdquo The Journal of PhysicalChemistry A vol 102 no 11 pp 1995ndash2001 mar 1998 doi 101021jp9716997
[22] M Cossi N Rega G Scalmani and V Barone ldquoEnergies structures andelectronic properties of molecules in solution with the c-PCM solvation modelrdquoJournal of Computational Chemistry vol 24 no 6 pp 669ndash681 apr 2003 doi101002jcc10189
[23] C Steinmann K L Blaeligdel A S Christensen and J H Jensen ldquoInterface ofthe polarizable continuum model of solvation with semi-empirical methods in thegamess programrdquo PLoS ONE vol 8 no 7 p e67725 07 2013 doi 101371jour-nalpone0067725
[24] Y Nishimoto ldquoDFTBPCM applied to ground and excited state potential energysurfacesrdquo J Phys Chem A vol 120 no 5 pp 771ndash784 feb 2016 doi101021acsjpca5b10732
18
[25] M Gaus X Lu M Elstner and Q Cui ldquoParameterization of DFTB33OB forSulfur and Phosphorus for Chemical and Biological Applicationsrdquo J Chem TheoryComput vol 10 no 4 pp 1518ndash1537 apr 2014 doi 101021ct401002w
[26] X Lu M Gaus M Elstner and Q Cui ldquoParametrization of DFTB33OBfor Magnesium and Zinc for Chemical and Biological Applicationsrdquo The Journalof Physical Chemistry B vol 119 no 3 pp 1062ndash1082 jan 2015 doi101021jp506557r
[27] M Kubillus T Kubar M Gaus J Rezac and M Elstner ldquoParameterization ofthe DFTB3 Method for Br Ca Cl F I K and Na in Organic and BiologicalSystemsrdquo J Chem Theory Comput vol 11 no 1 pp 332ndash342 jan 2015 doi101021ct5009137
[28] J J P Stewart ldquoMOPAC2012rdquo 2016 Stewart Computational Chemistry ColoradoSprings CO USA [Online] Available httpopenmopacnet
[29] J L Pascual-Ahuir E Silla J Tomasi and R Bonaccorsi ldquoElectrostaticinteraction of a solute with a continuum improved description of the cavity and ofthe surface cavity bound charge distributionrdquo Journal of Computational Chemistryvol 8 no 6 pp 778ndash787 sep 1987 doi 101002jcc540080605
[30] P Su and H Li ldquoContinuous and smooth potential energy surface for conductorlikescreening solvation model using fixed points with variable areasrdquo The Journal ofChemical Physics vol 130 no 7 p 074109 feb 2009 doi 10106313077917
[31] J Tomasi B Mennucci and E Cances ldquoThe IEF version of the PCM solvationmethod an overview of a new method addressed to study molecular solutes at theQM ab initio levelrdquo Journal of Molecular Structure THEOCHEM vol 464 no1-3 pp 211ndash226 may 1999 doi 101016s0166-1280(98)00553-3
[32] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 1 the calculation of confidence intervalsrdquo J Comput Aided MolDes vol 28 no 9 pp 887ndash918 Sep 2014 doi 101007s10822-014-9753-z
[33] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 2 comparing methodsrdquo J Comput Aided Mol Des vol 30 no 2pp 103ndash126 Feb 2016 doi 101007s10822-016-9904-5
[34] J H Jensen ldquoWhich method is more accurate or errors have error barsrdquo PeerJPreprints vol 5 p e2693v1 2017 doi 107287peerjpreprints2693v1
[35] J H Jensen C J Swain and L Olsen ldquoPrediction of pKa values for druglikemolecules using semiempirical quantum chemical methodsrdquo The Journal of PhysicalChemistry A vol 121 no 3 pp 699ndash707 jan 2017 doi 101021acsjpca6b10990
[36] F Eckert and A Klamt ldquoAccurate prediction of basicity in aqueous solution withCOSMO-RSrdquo Journal of Computational Chemistry vol 27 no 1 pp 11ndash19 2005doi 101002jcc20309
[37] J A Nelder and R Mead ldquoA simplex method for function minimizationrdquo TheComputer Journal vol 7 no 4 pp 308ndash313 1965 doi 101093comjnl74308
19
[38] E Jones T Oliphant P Peterson et al ldquoSciPy Open source scientifictools for Pythonrdquo 2001 accessed November 24th 2017 [Online] Availablehttpwwwscipyorgrdquo
[39] J Charnley 2017 accessed November 24th 2017 [Online] Available httpsgithubcomcharnleyoptimize gamess parameters
[40] C P Kelly C J Cramer and D G Truhlar ldquoAqueous solvation free energies ofions and ion-water clusters based on an accurate value for the absolute aqueoussolvation free energy of the protonrdquo The Journal of Physical Chemistry B vol 110no 32 pp 16 066ndash16 081 aug 2006 doi 101021jp063552y
20
HFSMDAM1SMD
PM3SMDPM6SMD
DFTBSMD
AM1COSMO
PM3COSMO
PM6COSMO null0
2
4
6
8
10
12
14
16RM
SE [k
calm
ol]
PM3PM3HCO
PM3HCON
PM3HCONSPM3all PM6
PM6HCO
PM6HCON
PM6HCONS
PM6HCONShPM6all
DFTB
DFTBHCON0
2
4
6
8
10
12
14RM
SE [k
calm
ol]
HFSMD
PM3SMDPM6SMD
DFTBSMD
PM3COSMO
PM6COSMO0
2
4
6
8
10RM
SE [k
calm
ol]
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6err
or

Table 4 RMSE MUE and MSE (in kcalmol) of aqueous solvation energies computed withSMD using HF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicatesthat the Radii has been reoptimized Corresponding RMSE values for the COSMO method areincluded for comparison The reference set is split into full set neutral cations and anions
SMD SMDdagger COSMO
HF PM3 PM6 DFTB PM3 PM6All
RMSE 34 36 48 41 48 50MUE 21 26 35 31 34 39MSE 03 10 -17 -11 05 -18Max 178 155 231 160 213 211
NeutralRMSE 24 28 47 39 36 47MUE 13 20 34 29 25 35MSE -09 09 -29 -21 -10 -34Max 178 155 231 160 213 211
CationsRMSE 50 56 51 37 74 73MUE 36 44 42 29 59 60MSE 31 36 38 10 56 59Max 140 127 124 91 186 183
AnionsRMSE 56 49 48 51 68 45MUE 46 37 36 40 58 38MSE 39 -03 -05 18 37 03Max 143 146 147 147 152 105
11
pKa PredictionPart of the impetus for this work were the relatively large error in pKa values predictedusing PM3COSMO compared to PM3SMD with the original SMD radii observed byJensen et al [35] so we use the new SMD radii to compute the pKa value using the samedata set The data is presented in Figure 4 and Table 5 and shows that the RMSE dropsfrom 15plusmn03 to 09plusmn03 pH units for PM3SMDdagger and 20plusmn04 to 16plusmn0304 pH unitsfor DFTBSMDdagger In both cases the improvement is primarily a result of the decreasein mean error (ME) ie the original radii led to a fairly systematic underestimation ofthe pKa values that has now been removed by the reparameterization For DFTB theincreased accuracy is also due to a larger number of low error predictions as is evident inFigure 4 In the case of PM3SMDdagger the accuracy is now very similar to PM3COSMOThe RMSE for PM6SMDdagger is similar to PM6COSMO and larger than for PM3SMDin analogy with the corresponding results for COSMO
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6
err
or
Figure 4 Plot of the errors in the predicted pKa values (pKaminus pKexpa )
12
Table 5 Root-Mean-Square-Error (RMSE) man error (ME) and Pearson correlation (r) for predicted pKa values relative to experiment togetherwith their statistical uncertainty (95 confidence limits)
PM3COSMO PM3SMD PM3SMDdagger PM6COSMO PM6SMDdagger DFTBSMD DFTBSMDdaggerRMSE 10 15 09 18 16 20 1695 conf plusmn 0202 plusmn 0303 plusmn 0202 plusmn 0304 plusmn 0304 plusmn 0404 plusmn 0304ME -05 plusmn 02 -12 plusmn 03 -02 plusmn 02 -13 plusmn 04 -08 plusmn 04 -07 plusmn 05 -01 plusmn 04r 095 093 093 090 086 087 08695 conf plusmn 002004 plusmn 003005 plusmn 003005 plusmn 004007 plusmn 006009 plusmn 005008 plusmn 006009
13
Non-aqueous SolventsThe new radii were optimized explicitly for water so it is not a priori clear whether theyoffer more accurate results for other solvents Table 6 shows the RMSE values for allneutral cations and anions for the other polar solvents in the MNSOL data set acetoni-trile DMSO and methanol for the original SMD radii and the corresponding re-optimizedradii (dagger) In addition corresponding combined RMSE values are given for the remaining88 mostly non-polar solvents in the MNSOL data set for which only data is availablefor neutral molecules It is clear from this data that the new radii sets lowers the errorof the predicted solvation free energies for the other polar solvents and has a negligibleeffect on the accuracy for the non-polar solvents
As for acetonitrile and DMSO the improvement is greatest for the ions where the RMSEdecreases by 3-12 kcalmol The improvement is more modest (05-3 kcalmol) for theneutral molecules For methanol which contains only ions the RMSE is similarly de-creased with 2-13 kcalmol
We thus recommend that the new radii optimized for water also be used for other polarsolvents when doing semiempirical SMD calculations For non-polar solvents either setof radii can be used
14
Table 6 RMSE (in kcalmol) of non-aqueous solvation energies computed with SMD usingHF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicates that theradii has been reoptimized Corresponding RMSE values for the COSMO method are includedfor comparison The reference set is split into full set neutral cations and anions
All Neu Cat Aniacetonitrile
HFSMD 112 29 105 131PM3SMD 194 41 162 246PM3SMDdagger 140 37 113 182PM6SMD 219 33 177 284PM6SMDdagger 130 12 114 160DFTBSMD 150 23 119 197DFTBSMDdagger 109 22 90 140PM3COSMO 130 31 128 147PM6COSMO 137 23 135 155
dimethylsulfoxideHFSMD 119 30 106 126PM3SMD 202 42 153 214PM3SMDdagger 170 35 90 182PM6SMD 255 45 172 272PM6SMDdagger 177 17 99 189DFTBSMD 169 23 108 181DFTBSMDdagger 125 21 76 134PM3COSMO 125 34 107 132PM6COSMO 150 31 119 159
methanolHFSMD 40 00 41 39PM3SMD 126 00 108 135PM3SMDdagger 66 00 60 69PM6SMD 163 00 120 183PM6SMDdagger 52 00 54 52DFTBSMD 84 00 63 94DFTBSMDdagger 47 00 44 49PM3COSMO 59 00 68 54PM6COSMO 54 00 67 46
otherHFSMD 42 42 00 00PM3SMD 51 51 00 00PM3SMDdagger 51 51 00 00PM6SMD 35 35 00 00PM6SMDdagger 34 34 00 00DFTBSMD 30 30 00 00DFTBSMDdagger 29 29 00 00PM3COSMO 36 36 00 00PM6COSMO 27 27 00 00
15
ConclusionThe PM6 method in GAMESS was extended to elements requiring d-integrals and inter-faced with the polarized continuum model (PCM) of solvation including semi-numericalgradients However the accuracy of aqueous solvation energies computed using AM1PM3 PM6 and DFTB and the SMD continuum solvation model was tested using a sub-set of molecules from the MNSOL data set which showed that the errors in SMD solvationenergies predicted using NDDO-based methods was considerably larger than when usingDFT and HF with RMSE values of 50 to 86 kcalmol compared to 34 kcalmol forHF6-31G(d) For the NDDO-based methods the errors were especially large for cationsand in the case of PM6 also for anions with RMSE values of 101 to 148 kcalmol incomparison with to 50 to 57 kcalmol for HF6-31G(d)SMD Corresponding COSMOresults (where the maximum RMSE is 75 kcalmol) suggested that the NDDOSMDresults could be improved by re-parameterizing the SMD Coulomb radii for the NDDOmethods
The fact that the NDDOSMD errors are largest for ions suggest that the problem iswith the polar solvation energy so we focus on optimizing the values of the Coulombradii while leaving all parameters associated with the non-polar solvation free energy un-changed We optimize the radii only for the ionic species on a subset of the MNSOL dataset previously used to parametrize RHF6-31G(d)SMD but include neutral moleculeswhen testing how well SMD with the new Coulomb radii perform We found the bestresults are obtained by changing only the radii for hydrogen carbon oxygen nitrogenand sulfur and this leads to RMSE values for PM3 (neutrals 28ions ca 5 kcalmol)PM6 (47ca 5 kcalmol) and DFTB (39ca 5 kcalmol) that are more comparable toHF6-31G(d) (24ca 5 kcalmol) We note that the SMD parameterization is done us-ing mono-aquo microsolvated species for small ions with atoms carrying large (partial)charges while the MNSOL data set also contains data for the non-microsolvated equiv-alents Comparison to the HF6-31G(d) MSEs reported by Marenich et al [8] reportedfor the selectively microsolvated ions indicates that the inclusion of non-microsolvatedspecies leads an MSE increase of about 1 kcalmol
Though the radii are optimized to reproduce aqueous solvation energies they also leadmore accurate predictions for other polar solvents such as DMSO acetonitrile andmethanol while the improvement for non-polar solvents are negligible
Supplementary MaterialSee supplementary material for Tables S1-S3 and Figures S1-S3 which are referred toin the text In addition we also provide a table of dielectric constants used for thecalculations and a table of aqueous solvation free energies obtained using the optimizedparameters
AcknowledgmentsJCK and JHJ would like to acknowledge Jimmy (Mr MOPAC) Stewart for providingsource code and guidance for the PM6 GAMESS implementation Also thanks to Donald
16
Truhlar and Christopher Cramer for sharing the MNSOL data set and for insightfulcomments on the manuscript CS thanks the Danish Council for Independent Research(the Sapere Aude program) for financial support (Grant no DFF 4181-00370)
References[1] J Tomasi B Mennucci and R Cammi ldquoQuantum mechanical continuum solvation
modelsrdquo Chem Rev vol 105 pp 2999ndash3093 2005
[2] A Klamt and G Schuurmann ldquoCOSMO a new approach to dielectric screeningin solvents with explicit expressions for the screening energy and its gradientrdquo JChem Soc Perkin Trans vol 2 pp 799ndash805 1993
[3] C J Cramer and D G Truhlar ldquoA universal approach to solvation modelingrdquoAccounts of Chemical Research vol 41 no 6 pp 760ndash768 jun 2008 doi101021ar800019z
[4] V Barone M Cossi and J Tomasi ldquoA new definition of cavities for thecomputation of solvation free energies by the polarizable continuum modelrdquo JChem Phys vol 107 no 8 pp 3210ndash3221 aug 1997 doi 1010631474671
[5] F Eckert and A Klamt ldquoFast solvent screening via quantum chemistryCOSMO-RS approachrdquo AIChE J vol 48 no 2 pp 369ndash385 feb 2002 doi101002aic690480220
[6] C Curutchet A Bidon-Chanal I Soteras M Orozco and F J Luque ldquoMSTcontinuum study of the hydration free energies of monovalent ionic speciesrdquo JPhys Chem B vol 109 no 8 pp 3565ndash3574 mar 2005 doi 101021jp047197s
[7] Y Takano and K N Houk ldquoBenchmarking the conductor-like polarizablecontinuum model (CPCM) for aqueous solvation free energies of neutral and ionicorganic moleculesrdquo J Chem Theory Comput vol 1 no 1 pp 70ndash77 jan 2005doi 101021ct049977a
[8] A V Marenich R M Olson C P Kelly C J Cramer and D G TruhlarldquoSelf-consistent reaction field model for aqueous and nonaqueous solutions based onaccurate polarized partial chargesrdquo Journal of Chemical Theory and Computationvol 3 no 6 pp 2011ndash2033 nov 2007 doi 101021ct7001418
[9] A V Marenich C J Cramer and D G Truhlar ldquoUniversal solvation model basedon solute electron density and on a continuum model of the solvent defined by thebulk dielectric constant and atomic surface tensionsrdquo J Phys Chem B vol 113no 18 pp 6378ndash6396 may 2009 doi 101021jp810292n
[10] G E Chudinov D V Napolov and M V Basilevsky ldquoQuantum-chemicalcalculations of the hydration energies of organic cations and anions in the frameworkof a continuum solvent approximationrdquo Chem Phys vol 160 no 1 pp 41 ndash 541992 doi 1010160301-0104(92)87090-V
[11] M J Negre M Orozco and F J Luque ldquoA new strategy for the representation ofenvironment effects in semi-empirical calculations based on dewarrsquos hamiltoniansrdquoChem Phys Lett vol 196 no 1 pp 27ndash36 1992
17
[12] F J Luque M J Negre and M Orozco ldquoAn am1-scrf approach to the study ofchanges in molecular properties induced by solventrdquo J Phys Chem vol 97 no 17pp 4386ndash4391 1993
[13] M Caricato B Mennucci and J Tomasi ldquoSolvent effects on the electronic spectraAn extension of the polarizable continuum model to the zindo methodrdquo J PhysChem A vol 108 no 29 pp 6248ndash6256 2004
[14] M J S Dewar E G Zoebisch E F Healy and J J P Stewart ldquoDevelopmentand use of quantum mechanical molecular models 76 AM1 a new general purposequantum mechanical molecular modelrdquo J A Chem Soc vol 10 pp 3902ndash39091985 doi 101021ja00299a024
[15] J J P Stewart ldquoOptimization of Parameters for Semi-Empirical Methods I-Methodrdquo J Comp Chem vol 10 pp 209ndash220 1989 doi 101002jcc540100208
[16] J J P Stewart ldquoOptimization of parameters for semiempirical methods V Modi-fication of NDDO approximations and application to 70 elementsrdquo J Mol Modelvol 13 pp 1172ndash1213 2007
[17] M Gaus Q Cui and M Elstner ldquoDFTB3 Extension of the Self-Consistent-Charge Density-Functional Tight-Binding Method (SCC-DFTB)rdquo J Chem TheoryComput vol 7 no 4 pp 931ndash948 apr 2011 doi 101021ct100684s
[18] A V Marenich C P Kelly J D Thompson G D Hawkins C C ChambersD J Giesen P P Winget C J Cramer and D G Truhlar ldquoMinnesota solvationdatabaserdquo 2012
[19] ldquoMinnesota solvation databaserdquo accessed November 24th 2017 [Online] Availablehttpscompchemumnedumnsol
[20] M W Schmidt K K Baldridge J A Boatz S T Elbert M S Gordon J HJensen S Koseki N Matsunaga K A Nguyen S Su T L Windus M Dupuisand JAMontgomery ldquoGeneral atomic and molecular electronic structure systemrdquoJ Comput Chem vol 14 pp 1347ndash1363 1993
[21] V Barone and M Cossi ldquoQuantum calculation of molecular energies and energygradients in solution by a conductor solvent modelrdquo The Journal of PhysicalChemistry A vol 102 no 11 pp 1995ndash2001 mar 1998 doi 101021jp9716997
[22] M Cossi N Rega G Scalmani and V Barone ldquoEnergies structures andelectronic properties of molecules in solution with the c-PCM solvation modelrdquoJournal of Computational Chemistry vol 24 no 6 pp 669ndash681 apr 2003 doi101002jcc10189
[23] C Steinmann K L Blaeligdel A S Christensen and J H Jensen ldquoInterface ofthe polarizable continuum model of solvation with semi-empirical methods in thegamess programrdquo PLoS ONE vol 8 no 7 p e67725 07 2013 doi 101371jour-nalpone0067725
[24] Y Nishimoto ldquoDFTBPCM applied to ground and excited state potential energysurfacesrdquo J Phys Chem A vol 120 no 5 pp 771ndash784 feb 2016 doi101021acsjpca5b10732
18
[25] M Gaus X Lu M Elstner and Q Cui ldquoParameterization of DFTB33OB forSulfur and Phosphorus for Chemical and Biological Applicationsrdquo J Chem TheoryComput vol 10 no 4 pp 1518ndash1537 apr 2014 doi 101021ct401002w
[26] X Lu M Gaus M Elstner and Q Cui ldquoParametrization of DFTB33OBfor Magnesium and Zinc for Chemical and Biological Applicationsrdquo The Journalof Physical Chemistry B vol 119 no 3 pp 1062ndash1082 jan 2015 doi101021jp506557r
[27] M Kubillus T Kubar M Gaus J Rezac and M Elstner ldquoParameterization ofthe DFTB3 Method for Br Ca Cl F I K and Na in Organic and BiologicalSystemsrdquo J Chem Theory Comput vol 11 no 1 pp 332ndash342 jan 2015 doi101021ct5009137
[28] J J P Stewart ldquoMOPAC2012rdquo 2016 Stewart Computational Chemistry ColoradoSprings CO USA [Online] Available httpopenmopacnet
[29] J L Pascual-Ahuir E Silla J Tomasi and R Bonaccorsi ldquoElectrostaticinteraction of a solute with a continuum improved description of the cavity and ofthe surface cavity bound charge distributionrdquo Journal of Computational Chemistryvol 8 no 6 pp 778ndash787 sep 1987 doi 101002jcc540080605
[30] P Su and H Li ldquoContinuous and smooth potential energy surface for conductorlikescreening solvation model using fixed points with variable areasrdquo The Journal ofChemical Physics vol 130 no 7 p 074109 feb 2009 doi 10106313077917
[31] J Tomasi B Mennucci and E Cances ldquoThe IEF version of the PCM solvationmethod an overview of a new method addressed to study molecular solutes at theQM ab initio levelrdquo Journal of Molecular Structure THEOCHEM vol 464 no1-3 pp 211ndash226 may 1999 doi 101016s0166-1280(98)00553-3
[32] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 1 the calculation of confidence intervalsrdquo J Comput Aided MolDes vol 28 no 9 pp 887ndash918 Sep 2014 doi 101007s10822-014-9753-z
[33] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 2 comparing methodsrdquo J Comput Aided Mol Des vol 30 no 2pp 103ndash126 Feb 2016 doi 101007s10822-016-9904-5
[34] J H Jensen ldquoWhich method is more accurate or errors have error barsrdquo PeerJPreprints vol 5 p e2693v1 2017 doi 107287peerjpreprints2693v1
[35] J H Jensen C J Swain and L Olsen ldquoPrediction of pKa values for druglikemolecules using semiempirical quantum chemical methodsrdquo The Journal of PhysicalChemistry A vol 121 no 3 pp 699ndash707 jan 2017 doi 101021acsjpca6b10990
[36] F Eckert and A Klamt ldquoAccurate prediction of basicity in aqueous solution withCOSMO-RSrdquo Journal of Computational Chemistry vol 27 no 1 pp 11ndash19 2005doi 101002jcc20309
[37] J A Nelder and R Mead ldquoA simplex method for function minimizationrdquo TheComputer Journal vol 7 no 4 pp 308ndash313 1965 doi 101093comjnl74308
19
[38] E Jones T Oliphant P Peterson et al ldquoSciPy Open source scientifictools for Pythonrdquo 2001 accessed November 24th 2017 [Online] Availablehttpwwwscipyorgrdquo
[39] J Charnley 2017 accessed November 24th 2017 [Online] Available httpsgithubcomcharnleyoptimize gamess parameters
[40] C P Kelly C J Cramer and D G Truhlar ldquoAqueous solvation free energies ofions and ion-water clusters based on an accurate value for the absolute aqueoussolvation free energy of the protonrdquo The Journal of Physical Chemistry B vol 110no 32 pp 16 066ndash16 081 aug 2006 doi 101021jp063552y
20
HFSMDAM1SMD
PM3SMDPM6SMD
DFTBSMD
AM1COSMO
PM3COSMO
PM6COSMO null0
2
4
6
8
10
12
14
16RM
SE [k
calm
ol]
PM3PM3HCO
PM3HCON
PM3HCONSPM3all PM6
PM6HCO
PM6HCON
PM6HCONS
PM6HCONShPM6all
DFTB
DFTBHCON0
2
4
6
8
10
12
14RM
SE [k
calm
ol]
HFSMD
PM3SMDPM6SMD
DFTBSMD
PM3COSMO
PM6COSMO0
2
4
6
8
10RM
SE [k
calm
ol]
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6err
or

pKa PredictionPart of the impetus for this work were the relatively large error in pKa values predictedusing PM3COSMO compared to PM3SMD with the original SMD radii observed byJensen et al [35] so we use the new SMD radii to compute the pKa value using the samedata set The data is presented in Figure 4 and Table 5 and shows that the RMSE dropsfrom 15plusmn03 to 09plusmn03 pH units for PM3SMDdagger and 20plusmn04 to 16plusmn0304 pH unitsfor DFTBSMDdagger In both cases the improvement is primarily a result of the decreasein mean error (ME) ie the original radii led to a fairly systematic underestimation ofthe pKa values that has now been removed by the reparameterization For DFTB theincreased accuracy is also due to a larger number of low error predictions as is evident inFigure 4 In the case of PM3SMDdagger the accuracy is now very similar to PM3COSMOThe RMSE for PM6SMDdagger is similar to PM6COSMO and larger than for PM3SMDin analogy with the corresponding results for COSMO
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6
err
or
Figure 4 Plot of the errors in the predicted pKa values (pKaminus pKexpa )
12
Table 5 Root-Mean-Square-Error (RMSE) man error (ME) and Pearson correlation (r) for predicted pKa values relative to experiment togetherwith their statistical uncertainty (95 confidence limits)
PM3COSMO PM3SMD PM3SMDdagger PM6COSMO PM6SMDdagger DFTBSMD DFTBSMDdaggerRMSE 10 15 09 18 16 20 1695 conf plusmn 0202 plusmn 0303 plusmn 0202 plusmn 0304 plusmn 0304 plusmn 0404 plusmn 0304ME -05 plusmn 02 -12 plusmn 03 -02 plusmn 02 -13 plusmn 04 -08 plusmn 04 -07 plusmn 05 -01 plusmn 04r 095 093 093 090 086 087 08695 conf plusmn 002004 plusmn 003005 plusmn 003005 plusmn 004007 plusmn 006009 plusmn 005008 plusmn 006009
13
Non-aqueous SolventsThe new radii were optimized explicitly for water so it is not a priori clear whether theyoffer more accurate results for other solvents Table 6 shows the RMSE values for allneutral cations and anions for the other polar solvents in the MNSOL data set acetoni-trile DMSO and methanol for the original SMD radii and the corresponding re-optimizedradii (dagger) In addition corresponding combined RMSE values are given for the remaining88 mostly non-polar solvents in the MNSOL data set for which only data is availablefor neutral molecules It is clear from this data that the new radii sets lowers the errorof the predicted solvation free energies for the other polar solvents and has a negligibleeffect on the accuracy for the non-polar solvents
As for acetonitrile and DMSO the improvement is greatest for the ions where the RMSEdecreases by 3-12 kcalmol The improvement is more modest (05-3 kcalmol) for theneutral molecules For methanol which contains only ions the RMSE is similarly de-creased with 2-13 kcalmol
We thus recommend that the new radii optimized for water also be used for other polarsolvents when doing semiempirical SMD calculations For non-polar solvents either setof radii can be used
14
Table 6 RMSE (in kcalmol) of non-aqueous solvation energies computed with SMD usingHF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicates that theradii has been reoptimized Corresponding RMSE values for the COSMO method are includedfor comparison The reference set is split into full set neutral cations and anions
All Neu Cat Aniacetonitrile
HFSMD 112 29 105 131PM3SMD 194 41 162 246PM3SMDdagger 140 37 113 182PM6SMD 219 33 177 284PM6SMDdagger 130 12 114 160DFTBSMD 150 23 119 197DFTBSMDdagger 109 22 90 140PM3COSMO 130 31 128 147PM6COSMO 137 23 135 155
dimethylsulfoxideHFSMD 119 30 106 126PM3SMD 202 42 153 214PM3SMDdagger 170 35 90 182PM6SMD 255 45 172 272PM6SMDdagger 177 17 99 189DFTBSMD 169 23 108 181DFTBSMDdagger 125 21 76 134PM3COSMO 125 34 107 132PM6COSMO 150 31 119 159
methanolHFSMD 40 00 41 39PM3SMD 126 00 108 135PM3SMDdagger 66 00 60 69PM6SMD 163 00 120 183PM6SMDdagger 52 00 54 52DFTBSMD 84 00 63 94DFTBSMDdagger 47 00 44 49PM3COSMO 59 00 68 54PM6COSMO 54 00 67 46
otherHFSMD 42 42 00 00PM3SMD 51 51 00 00PM3SMDdagger 51 51 00 00PM6SMD 35 35 00 00PM6SMDdagger 34 34 00 00DFTBSMD 30 30 00 00DFTBSMDdagger 29 29 00 00PM3COSMO 36 36 00 00PM6COSMO 27 27 00 00
15
ConclusionThe PM6 method in GAMESS was extended to elements requiring d-integrals and inter-faced with the polarized continuum model (PCM) of solvation including semi-numericalgradients However the accuracy of aqueous solvation energies computed using AM1PM3 PM6 and DFTB and the SMD continuum solvation model was tested using a sub-set of molecules from the MNSOL data set which showed that the errors in SMD solvationenergies predicted using NDDO-based methods was considerably larger than when usingDFT and HF with RMSE values of 50 to 86 kcalmol compared to 34 kcalmol forHF6-31G(d) For the NDDO-based methods the errors were especially large for cationsand in the case of PM6 also for anions with RMSE values of 101 to 148 kcalmol incomparison with to 50 to 57 kcalmol for HF6-31G(d)SMD Corresponding COSMOresults (where the maximum RMSE is 75 kcalmol) suggested that the NDDOSMDresults could be improved by re-parameterizing the SMD Coulomb radii for the NDDOmethods
The fact that the NDDOSMD errors are largest for ions suggest that the problem iswith the polar solvation energy so we focus on optimizing the values of the Coulombradii while leaving all parameters associated with the non-polar solvation free energy un-changed We optimize the radii only for the ionic species on a subset of the MNSOL dataset previously used to parametrize RHF6-31G(d)SMD but include neutral moleculeswhen testing how well SMD with the new Coulomb radii perform We found the bestresults are obtained by changing only the radii for hydrogen carbon oxygen nitrogenand sulfur and this leads to RMSE values for PM3 (neutrals 28ions ca 5 kcalmol)PM6 (47ca 5 kcalmol) and DFTB (39ca 5 kcalmol) that are more comparable toHF6-31G(d) (24ca 5 kcalmol) We note that the SMD parameterization is done us-ing mono-aquo microsolvated species for small ions with atoms carrying large (partial)charges while the MNSOL data set also contains data for the non-microsolvated equiv-alents Comparison to the HF6-31G(d) MSEs reported by Marenich et al [8] reportedfor the selectively microsolvated ions indicates that the inclusion of non-microsolvatedspecies leads an MSE increase of about 1 kcalmol
Though the radii are optimized to reproduce aqueous solvation energies they also leadmore accurate predictions for other polar solvents such as DMSO acetonitrile andmethanol while the improvement for non-polar solvents are negligible
Supplementary MaterialSee supplementary material for Tables S1-S3 and Figures S1-S3 which are referred toin the text In addition we also provide a table of dielectric constants used for thecalculations and a table of aqueous solvation free energies obtained using the optimizedparameters
AcknowledgmentsJCK and JHJ would like to acknowledge Jimmy (Mr MOPAC) Stewart for providingsource code and guidance for the PM6 GAMESS implementation Also thanks to Donald
16
Truhlar and Christopher Cramer for sharing the MNSOL data set and for insightfulcomments on the manuscript CS thanks the Danish Council for Independent Research(the Sapere Aude program) for financial support (Grant no DFF 4181-00370)
References[1] J Tomasi B Mennucci and R Cammi ldquoQuantum mechanical continuum solvation
modelsrdquo Chem Rev vol 105 pp 2999ndash3093 2005
[2] A Klamt and G Schuurmann ldquoCOSMO a new approach to dielectric screeningin solvents with explicit expressions for the screening energy and its gradientrdquo JChem Soc Perkin Trans vol 2 pp 799ndash805 1993
[3] C J Cramer and D G Truhlar ldquoA universal approach to solvation modelingrdquoAccounts of Chemical Research vol 41 no 6 pp 760ndash768 jun 2008 doi101021ar800019z
[4] V Barone M Cossi and J Tomasi ldquoA new definition of cavities for thecomputation of solvation free energies by the polarizable continuum modelrdquo JChem Phys vol 107 no 8 pp 3210ndash3221 aug 1997 doi 1010631474671
[5] F Eckert and A Klamt ldquoFast solvent screening via quantum chemistryCOSMO-RS approachrdquo AIChE J vol 48 no 2 pp 369ndash385 feb 2002 doi101002aic690480220
[6] C Curutchet A Bidon-Chanal I Soteras M Orozco and F J Luque ldquoMSTcontinuum study of the hydration free energies of monovalent ionic speciesrdquo JPhys Chem B vol 109 no 8 pp 3565ndash3574 mar 2005 doi 101021jp047197s
[7] Y Takano and K N Houk ldquoBenchmarking the conductor-like polarizablecontinuum model (CPCM) for aqueous solvation free energies of neutral and ionicorganic moleculesrdquo J Chem Theory Comput vol 1 no 1 pp 70ndash77 jan 2005doi 101021ct049977a
[8] A V Marenich R M Olson C P Kelly C J Cramer and D G TruhlarldquoSelf-consistent reaction field model for aqueous and nonaqueous solutions based onaccurate polarized partial chargesrdquo Journal of Chemical Theory and Computationvol 3 no 6 pp 2011ndash2033 nov 2007 doi 101021ct7001418
[9] A V Marenich C J Cramer and D G Truhlar ldquoUniversal solvation model basedon solute electron density and on a continuum model of the solvent defined by thebulk dielectric constant and atomic surface tensionsrdquo J Phys Chem B vol 113no 18 pp 6378ndash6396 may 2009 doi 101021jp810292n
[10] G E Chudinov D V Napolov and M V Basilevsky ldquoQuantum-chemicalcalculations of the hydration energies of organic cations and anions in the frameworkof a continuum solvent approximationrdquo Chem Phys vol 160 no 1 pp 41 ndash 541992 doi 1010160301-0104(92)87090-V
[11] M J Negre M Orozco and F J Luque ldquoA new strategy for the representation ofenvironment effects in semi-empirical calculations based on dewarrsquos hamiltoniansrdquoChem Phys Lett vol 196 no 1 pp 27ndash36 1992
17
[12] F J Luque M J Negre and M Orozco ldquoAn am1-scrf approach to the study ofchanges in molecular properties induced by solventrdquo J Phys Chem vol 97 no 17pp 4386ndash4391 1993
[13] M Caricato B Mennucci and J Tomasi ldquoSolvent effects on the electronic spectraAn extension of the polarizable continuum model to the zindo methodrdquo J PhysChem A vol 108 no 29 pp 6248ndash6256 2004
[14] M J S Dewar E G Zoebisch E F Healy and J J P Stewart ldquoDevelopmentand use of quantum mechanical molecular models 76 AM1 a new general purposequantum mechanical molecular modelrdquo J A Chem Soc vol 10 pp 3902ndash39091985 doi 101021ja00299a024
[15] J J P Stewart ldquoOptimization of Parameters for Semi-Empirical Methods I-Methodrdquo J Comp Chem vol 10 pp 209ndash220 1989 doi 101002jcc540100208
[16] J J P Stewart ldquoOptimization of parameters for semiempirical methods V Modi-fication of NDDO approximations and application to 70 elementsrdquo J Mol Modelvol 13 pp 1172ndash1213 2007
[17] M Gaus Q Cui and M Elstner ldquoDFTB3 Extension of the Self-Consistent-Charge Density-Functional Tight-Binding Method (SCC-DFTB)rdquo J Chem TheoryComput vol 7 no 4 pp 931ndash948 apr 2011 doi 101021ct100684s
[18] A V Marenich C P Kelly J D Thompson G D Hawkins C C ChambersD J Giesen P P Winget C J Cramer and D G Truhlar ldquoMinnesota solvationdatabaserdquo 2012
[19] ldquoMinnesota solvation databaserdquo accessed November 24th 2017 [Online] Availablehttpscompchemumnedumnsol
[20] M W Schmidt K K Baldridge J A Boatz S T Elbert M S Gordon J HJensen S Koseki N Matsunaga K A Nguyen S Su T L Windus M Dupuisand JAMontgomery ldquoGeneral atomic and molecular electronic structure systemrdquoJ Comput Chem vol 14 pp 1347ndash1363 1993
[21] V Barone and M Cossi ldquoQuantum calculation of molecular energies and energygradients in solution by a conductor solvent modelrdquo The Journal of PhysicalChemistry A vol 102 no 11 pp 1995ndash2001 mar 1998 doi 101021jp9716997
[22] M Cossi N Rega G Scalmani and V Barone ldquoEnergies structures andelectronic properties of molecules in solution with the c-PCM solvation modelrdquoJournal of Computational Chemistry vol 24 no 6 pp 669ndash681 apr 2003 doi101002jcc10189
[23] C Steinmann K L Blaeligdel A S Christensen and J H Jensen ldquoInterface ofthe polarizable continuum model of solvation with semi-empirical methods in thegamess programrdquo PLoS ONE vol 8 no 7 p e67725 07 2013 doi 101371jour-nalpone0067725
[24] Y Nishimoto ldquoDFTBPCM applied to ground and excited state potential energysurfacesrdquo J Phys Chem A vol 120 no 5 pp 771ndash784 feb 2016 doi101021acsjpca5b10732
18
[25] M Gaus X Lu M Elstner and Q Cui ldquoParameterization of DFTB33OB forSulfur and Phosphorus for Chemical and Biological Applicationsrdquo J Chem TheoryComput vol 10 no 4 pp 1518ndash1537 apr 2014 doi 101021ct401002w
[26] X Lu M Gaus M Elstner and Q Cui ldquoParametrization of DFTB33OBfor Magnesium and Zinc for Chemical and Biological Applicationsrdquo The Journalof Physical Chemistry B vol 119 no 3 pp 1062ndash1082 jan 2015 doi101021jp506557r
[27] M Kubillus T Kubar M Gaus J Rezac and M Elstner ldquoParameterization ofthe DFTB3 Method for Br Ca Cl F I K and Na in Organic and BiologicalSystemsrdquo J Chem Theory Comput vol 11 no 1 pp 332ndash342 jan 2015 doi101021ct5009137
[28] J J P Stewart ldquoMOPAC2012rdquo 2016 Stewart Computational Chemistry ColoradoSprings CO USA [Online] Available httpopenmopacnet
[29] J L Pascual-Ahuir E Silla J Tomasi and R Bonaccorsi ldquoElectrostaticinteraction of a solute with a continuum improved description of the cavity and ofthe surface cavity bound charge distributionrdquo Journal of Computational Chemistryvol 8 no 6 pp 778ndash787 sep 1987 doi 101002jcc540080605
[30] P Su and H Li ldquoContinuous and smooth potential energy surface for conductorlikescreening solvation model using fixed points with variable areasrdquo The Journal ofChemical Physics vol 130 no 7 p 074109 feb 2009 doi 10106313077917
[31] J Tomasi B Mennucci and E Cances ldquoThe IEF version of the PCM solvationmethod an overview of a new method addressed to study molecular solutes at theQM ab initio levelrdquo Journal of Molecular Structure THEOCHEM vol 464 no1-3 pp 211ndash226 may 1999 doi 101016s0166-1280(98)00553-3
[32] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 1 the calculation of confidence intervalsrdquo J Comput Aided MolDes vol 28 no 9 pp 887ndash918 Sep 2014 doi 101007s10822-014-9753-z
[33] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 2 comparing methodsrdquo J Comput Aided Mol Des vol 30 no 2pp 103ndash126 Feb 2016 doi 101007s10822-016-9904-5
[34] J H Jensen ldquoWhich method is more accurate or errors have error barsrdquo PeerJPreprints vol 5 p e2693v1 2017 doi 107287peerjpreprints2693v1
[35] J H Jensen C J Swain and L Olsen ldquoPrediction of pKa values for druglikemolecules using semiempirical quantum chemical methodsrdquo The Journal of PhysicalChemistry A vol 121 no 3 pp 699ndash707 jan 2017 doi 101021acsjpca6b10990
[36] F Eckert and A Klamt ldquoAccurate prediction of basicity in aqueous solution withCOSMO-RSrdquo Journal of Computational Chemistry vol 27 no 1 pp 11ndash19 2005doi 101002jcc20309
[37] J A Nelder and R Mead ldquoA simplex method for function minimizationrdquo TheComputer Journal vol 7 no 4 pp 308ndash313 1965 doi 101093comjnl74308
19
[38] E Jones T Oliphant P Peterson et al ldquoSciPy Open source scientifictools for Pythonrdquo 2001 accessed November 24th 2017 [Online] Availablehttpwwwscipyorgrdquo
[39] J Charnley 2017 accessed November 24th 2017 [Online] Available httpsgithubcomcharnleyoptimize gamess parameters
[40] C P Kelly C J Cramer and D G Truhlar ldquoAqueous solvation free energies ofions and ion-water clusters based on an accurate value for the absolute aqueoussolvation free energy of the protonrdquo The Journal of Physical Chemistry B vol 110no 32 pp 16 066ndash16 081 aug 2006 doi 101021jp063552y
20
HFSMDAM1SMD
PM3SMDPM6SMD
DFTBSMD
AM1COSMO
PM3COSMO
PM6COSMO null0
2
4
6
8
10
12
14
16RM
SE [k
calm
ol]
PM3PM3HCO
PM3HCON
PM3HCONSPM3all PM6
PM6HCO
PM6HCON
PM6HCONS
PM6HCONShPM6all
DFTB
DFTBHCON0
2
4
6
8
10
12
14RM
SE [k
calm
ol]
HFSMD
PM3SMDPM6SMD
DFTBSMD
PM3COSMO
PM6COSMO0
2
4
6
8
10RM
SE [k
calm
ol]
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6err
or

Table 5 Root-Mean-Square-Error (RMSE) man error (ME) and Pearson correlation (r) for predicted pKa values relative to experiment togetherwith their statistical uncertainty (95 confidence limits)
PM3COSMO PM3SMD PM3SMDdagger PM6COSMO PM6SMDdagger DFTBSMD DFTBSMDdaggerRMSE 10 15 09 18 16 20 1695 conf plusmn 0202 plusmn 0303 plusmn 0202 plusmn 0304 plusmn 0304 plusmn 0404 plusmn 0304ME -05 plusmn 02 -12 plusmn 03 -02 plusmn 02 -13 plusmn 04 -08 plusmn 04 -07 plusmn 05 -01 plusmn 04r 095 093 093 090 086 087 08695 conf plusmn 002004 plusmn 003005 plusmn 003005 plusmn 004007 plusmn 006009 plusmn 005008 plusmn 006009
13
Non-aqueous SolventsThe new radii were optimized explicitly for water so it is not a priori clear whether theyoffer more accurate results for other solvents Table 6 shows the RMSE values for allneutral cations and anions for the other polar solvents in the MNSOL data set acetoni-trile DMSO and methanol for the original SMD radii and the corresponding re-optimizedradii (dagger) In addition corresponding combined RMSE values are given for the remaining88 mostly non-polar solvents in the MNSOL data set for which only data is availablefor neutral molecules It is clear from this data that the new radii sets lowers the errorof the predicted solvation free energies for the other polar solvents and has a negligibleeffect on the accuracy for the non-polar solvents
As for acetonitrile and DMSO the improvement is greatest for the ions where the RMSEdecreases by 3-12 kcalmol The improvement is more modest (05-3 kcalmol) for theneutral molecules For methanol which contains only ions the RMSE is similarly de-creased with 2-13 kcalmol
We thus recommend that the new radii optimized for water also be used for other polarsolvents when doing semiempirical SMD calculations For non-polar solvents either setof radii can be used
14
Table 6 RMSE (in kcalmol) of non-aqueous solvation energies computed with SMD usingHF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicates that theradii has been reoptimized Corresponding RMSE values for the COSMO method are includedfor comparison The reference set is split into full set neutral cations and anions
All Neu Cat Aniacetonitrile
HFSMD 112 29 105 131PM3SMD 194 41 162 246PM3SMDdagger 140 37 113 182PM6SMD 219 33 177 284PM6SMDdagger 130 12 114 160DFTBSMD 150 23 119 197DFTBSMDdagger 109 22 90 140PM3COSMO 130 31 128 147PM6COSMO 137 23 135 155
dimethylsulfoxideHFSMD 119 30 106 126PM3SMD 202 42 153 214PM3SMDdagger 170 35 90 182PM6SMD 255 45 172 272PM6SMDdagger 177 17 99 189DFTBSMD 169 23 108 181DFTBSMDdagger 125 21 76 134PM3COSMO 125 34 107 132PM6COSMO 150 31 119 159
methanolHFSMD 40 00 41 39PM3SMD 126 00 108 135PM3SMDdagger 66 00 60 69PM6SMD 163 00 120 183PM6SMDdagger 52 00 54 52DFTBSMD 84 00 63 94DFTBSMDdagger 47 00 44 49PM3COSMO 59 00 68 54PM6COSMO 54 00 67 46
otherHFSMD 42 42 00 00PM3SMD 51 51 00 00PM3SMDdagger 51 51 00 00PM6SMD 35 35 00 00PM6SMDdagger 34 34 00 00DFTBSMD 30 30 00 00DFTBSMDdagger 29 29 00 00PM3COSMO 36 36 00 00PM6COSMO 27 27 00 00
15
ConclusionThe PM6 method in GAMESS was extended to elements requiring d-integrals and inter-faced with the polarized continuum model (PCM) of solvation including semi-numericalgradients However the accuracy of aqueous solvation energies computed using AM1PM3 PM6 and DFTB and the SMD continuum solvation model was tested using a sub-set of molecules from the MNSOL data set which showed that the errors in SMD solvationenergies predicted using NDDO-based methods was considerably larger than when usingDFT and HF with RMSE values of 50 to 86 kcalmol compared to 34 kcalmol forHF6-31G(d) For the NDDO-based methods the errors were especially large for cationsand in the case of PM6 also for anions with RMSE values of 101 to 148 kcalmol incomparison with to 50 to 57 kcalmol for HF6-31G(d)SMD Corresponding COSMOresults (where the maximum RMSE is 75 kcalmol) suggested that the NDDOSMDresults could be improved by re-parameterizing the SMD Coulomb radii for the NDDOmethods
The fact that the NDDOSMD errors are largest for ions suggest that the problem iswith the polar solvation energy so we focus on optimizing the values of the Coulombradii while leaving all parameters associated with the non-polar solvation free energy un-changed We optimize the radii only for the ionic species on a subset of the MNSOL dataset previously used to parametrize RHF6-31G(d)SMD but include neutral moleculeswhen testing how well SMD with the new Coulomb radii perform We found the bestresults are obtained by changing only the radii for hydrogen carbon oxygen nitrogenand sulfur and this leads to RMSE values for PM3 (neutrals 28ions ca 5 kcalmol)PM6 (47ca 5 kcalmol) and DFTB (39ca 5 kcalmol) that are more comparable toHF6-31G(d) (24ca 5 kcalmol) We note that the SMD parameterization is done us-ing mono-aquo microsolvated species for small ions with atoms carrying large (partial)charges while the MNSOL data set also contains data for the non-microsolvated equiv-alents Comparison to the HF6-31G(d) MSEs reported by Marenich et al [8] reportedfor the selectively microsolvated ions indicates that the inclusion of non-microsolvatedspecies leads an MSE increase of about 1 kcalmol
Though the radii are optimized to reproduce aqueous solvation energies they also leadmore accurate predictions for other polar solvents such as DMSO acetonitrile andmethanol while the improvement for non-polar solvents are negligible
Supplementary MaterialSee supplementary material for Tables S1-S3 and Figures S1-S3 which are referred toin the text In addition we also provide a table of dielectric constants used for thecalculations and a table of aqueous solvation free energies obtained using the optimizedparameters
AcknowledgmentsJCK and JHJ would like to acknowledge Jimmy (Mr MOPAC) Stewart for providingsource code and guidance for the PM6 GAMESS implementation Also thanks to Donald
16
Truhlar and Christopher Cramer for sharing the MNSOL data set and for insightfulcomments on the manuscript CS thanks the Danish Council for Independent Research(the Sapere Aude program) for financial support (Grant no DFF 4181-00370)
References[1] J Tomasi B Mennucci and R Cammi ldquoQuantum mechanical continuum solvation
modelsrdquo Chem Rev vol 105 pp 2999ndash3093 2005
[2] A Klamt and G Schuurmann ldquoCOSMO a new approach to dielectric screeningin solvents with explicit expressions for the screening energy and its gradientrdquo JChem Soc Perkin Trans vol 2 pp 799ndash805 1993
[3] C J Cramer and D G Truhlar ldquoA universal approach to solvation modelingrdquoAccounts of Chemical Research vol 41 no 6 pp 760ndash768 jun 2008 doi101021ar800019z
[4] V Barone M Cossi and J Tomasi ldquoA new definition of cavities for thecomputation of solvation free energies by the polarizable continuum modelrdquo JChem Phys vol 107 no 8 pp 3210ndash3221 aug 1997 doi 1010631474671
[5] F Eckert and A Klamt ldquoFast solvent screening via quantum chemistryCOSMO-RS approachrdquo AIChE J vol 48 no 2 pp 369ndash385 feb 2002 doi101002aic690480220
[6] C Curutchet A Bidon-Chanal I Soteras M Orozco and F J Luque ldquoMSTcontinuum study of the hydration free energies of monovalent ionic speciesrdquo JPhys Chem B vol 109 no 8 pp 3565ndash3574 mar 2005 doi 101021jp047197s
[7] Y Takano and K N Houk ldquoBenchmarking the conductor-like polarizablecontinuum model (CPCM) for aqueous solvation free energies of neutral and ionicorganic moleculesrdquo J Chem Theory Comput vol 1 no 1 pp 70ndash77 jan 2005doi 101021ct049977a
[8] A V Marenich R M Olson C P Kelly C J Cramer and D G TruhlarldquoSelf-consistent reaction field model for aqueous and nonaqueous solutions based onaccurate polarized partial chargesrdquo Journal of Chemical Theory and Computationvol 3 no 6 pp 2011ndash2033 nov 2007 doi 101021ct7001418
[9] A V Marenich C J Cramer and D G Truhlar ldquoUniversal solvation model basedon solute electron density and on a continuum model of the solvent defined by thebulk dielectric constant and atomic surface tensionsrdquo J Phys Chem B vol 113no 18 pp 6378ndash6396 may 2009 doi 101021jp810292n
[10] G E Chudinov D V Napolov and M V Basilevsky ldquoQuantum-chemicalcalculations of the hydration energies of organic cations and anions in the frameworkof a continuum solvent approximationrdquo Chem Phys vol 160 no 1 pp 41 ndash 541992 doi 1010160301-0104(92)87090-V
[11] M J Negre M Orozco and F J Luque ldquoA new strategy for the representation ofenvironment effects in semi-empirical calculations based on dewarrsquos hamiltoniansrdquoChem Phys Lett vol 196 no 1 pp 27ndash36 1992
17
[12] F J Luque M J Negre and M Orozco ldquoAn am1-scrf approach to the study ofchanges in molecular properties induced by solventrdquo J Phys Chem vol 97 no 17pp 4386ndash4391 1993
[13] M Caricato B Mennucci and J Tomasi ldquoSolvent effects on the electronic spectraAn extension of the polarizable continuum model to the zindo methodrdquo J PhysChem A vol 108 no 29 pp 6248ndash6256 2004
[14] M J S Dewar E G Zoebisch E F Healy and J J P Stewart ldquoDevelopmentand use of quantum mechanical molecular models 76 AM1 a new general purposequantum mechanical molecular modelrdquo J A Chem Soc vol 10 pp 3902ndash39091985 doi 101021ja00299a024
[15] J J P Stewart ldquoOptimization of Parameters for Semi-Empirical Methods I-Methodrdquo J Comp Chem vol 10 pp 209ndash220 1989 doi 101002jcc540100208
[16] J J P Stewart ldquoOptimization of parameters for semiempirical methods V Modi-fication of NDDO approximations and application to 70 elementsrdquo J Mol Modelvol 13 pp 1172ndash1213 2007
[17] M Gaus Q Cui and M Elstner ldquoDFTB3 Extension of the Self-Consistent-Charge Density-Functional Tight-Binding Method (SCC-DFTB)rdquo J Chem TheoryComput vol 7 no 4 pp 931ndash948 apr 2011 doi 101021ct100684s
[18] A V Marenich C P Kelly J D Thompson G D Hawkins C C ChambersD J Giesen P P Winget C J Cramer and D G Truhlar ldquoMinnesota solvationdatabaserdquo 2012
[19] ldquoMinnesota solvation databaserdquo accessed November 24th 2017 [Online] Availablehttpscompchemumnedumnsol
[20] M W Schmidt K K Baldridge J A Boatz S T Elbert M S Gordon J HJensen S Koseki N Matsunaga K A Nguyen S Su T L Windus M Dupuisand JAMontgomery ldquoGeneral atomic and molecular electronic structure systemrdquoJ Comput Chem vol 14 pp 1347ndash1363 1993
[21] V Barone and M Cossi ldquoQuantum calculation of molecular energies and energygradients in solution by a conductor solvent modelrdquo The Journal of PhysicalChemistry A vol 102 no 11 pp 1995ndash2001 mar 1998 doi 101021jp9716997
[22] M Cossi N Rega G Scalmani and V Barone ldquoEnergies structures andelectronic properties of molecules in solution with the c-PCM solvation modelrdquoJournal of Computational Chemistry vol 24 no 6 pp 669ndash681 apr 2003 doi101002jcc10189
[23] C Steinmann K L Blaeligdel A S Christensen and J H Jensen ldquoInterface ofthe polarizable continuum model of solvation with semi-empirical methods in thegamess programrdquo PLoS ONE vol 8 no 7 p e67725 07 2013 doi 101371jour-nalpone0067725
[24] Y Nishimoto ldquoDFTBPCM applied to ground and excited state potential energysurfacesrdquo J Phys Chem A vol 120 no 5 pp 771ndash784 feb 2016 doi101021acsjpca5b10732
18
[25] M Gaus X Lu M Elstner and Q Cui ldquoParameterization of DFTB33OB forSulfur and Phosphorus for Chemical and Biological Applicationsrdquo J Chem TheoryComput vol 10 no 4 pp 1518ndash1537 apr 2014 doi 101021ct401002w
[26] X Lu M Gaus M Elstner and Q Cui ldquoParametrization of DFTB33OBfor Magnesium and Zinc for Chemical and Biological Applicationsrdquo The Journalof Physical Chemistry B vol 119 no 3 pp 1062ndash1082 jan 2015 doi101021jp506557r
[27] M Kubillus T Kubar M Gaus J Rezac and M Elstner ldquoParameterization ofthe DFTB3 Method for Br Ca Cl F I K and Na in Organic and BiologicalSystemsrdquo J Chem Theory Comput vol 11 no 1 pp 332ndash342 jan 2015 doi101021ct5009137
[28] J J P Stewart ldquoMOPAC2012rdquo 2016 Stewart Computational Chemistry ColoradoSprings CO USA [Online] Available httpopenmopacnet
[29] J L Pascual-Ahuir E Silla J Tomasi and R Bonaccorsi ldquoElectrostaticinteraction of a solute with a continuum improved description of the cavity and ofthe surface cavity bound charge distributionrdquo Journal of Computational Chemistryvol 8 no 6 pp 778ndash787 sep 1987 doi 101002jcc540080605
[30] P Su and H Li ldquoContinuous and smooth potential energy surface for conductorlikescreening solvation model using fixed points with variable areasrdquo The Journal ofChemical Physics vol 130 no 7 p 074109 feb 2009 doi 10106313077917
[31] J Tomasi B Mennucci and E Cances ldquoThe IEF version of the PCM solvationmethod an overview of a new method addressed to study molecular solutes at theQM ab initio levelrdquo Journal of Molecular Structure THEOCHEM vol 464 no1-3 pp 211ndash226 may 1999 doi 101016s0166-1280(98)00553-3
[32] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 1 the calculation of confidence intervalsrdquo J Comput Aided MolDes vol 28 no 9 pp 887ndash918 Sep 2014 doi 101007s10822-014-9753-z
[33] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 2 comparing methodsrdquo J Comput Aided Mol Des vol 30 no 2pp 103ndash126 Feb 2016 doi 101007s10822-016-9904-5
[34] J H Jensen ldquoWhich method is more accurate or errors have error barsrdquo PeerJPreprints vol 5 p e2693v1 2017 doi 107287peerjpreprints2693v1
[35] J H Jensen C J Swain and L Olsen ldquoPrediction of pKa values for druglikemolecules using semiempirical quantum chemical methodsrdquo The Journal of PhysicalChemistry A vol 121 no 3 pp 699ndash707 jan 2017 doi 101021acsjpca6b10990
[36] F Eckert and A Klamt ldquoAccurate prediction of basicity in aqueous solution withCOSMO-RSrdquo Journal of Computational Chemistry vol 27 no 1 pp 11ndash19 2005doi 101002jcc20309
[37] J A Nelder and R Mead ldquoA simplex method for function minimizationrdquo TheComputer Journal vol 7 no 4 pp 308ndash313 1965 doi 101093comjnl74308
19
[38] E Jones T Oliphant P Peterson et al ldquoSciPy Open source scientifictools for Pythonrdquo 2001 accessed November 24th 2017 [Online] Availablehttpwwwscipyorgrdquo
[39] J Charnley 2017 accessed November 24th 2017 [Online] Available httpsgithubcomcharnleyoptimize gamess parameters
[40] C P Kelly C J Cramer and D G Truhlar ldquoAqueous solvation free energies ofions and ion-water clusters based on an accurate value for the absolute aqueoussolvation free energy of the protonrdquo The Journal of Physical Chemistry B vol 110no 32 pp 16 066ndash16 081 aug 2006 doi 101021jp063552y
20
HFSMDAM1SMD
PM3SMDPM6SMD
DFTBSMD
AM1COSMO
PM3COSMO
PM6COSMO null0
2
4
6
8
10
12
14
16RM
SE [k
calm
ol]
PM3PM3HCO
PM3HCON
PM3HCONSPM3all PM6
PM6HCO
PM6HCON
PM6HCONS
PM6HCONShPM6all
DFTB
DFTBHCON0
2
4
6
8
10
12
14RM
SE [k
calm
ol]
HFSMD
PM3SMDPM6SMD
DFTBSMD
PM3COSMO
PM6COSMO0
2
4
6
8
10RM
SE [k
calm
ol]
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6err
or

Non-aqueous SolventsThe new radii were optimized explicitly for water so it is not a priori clear whether theyoffer more accurate results for other solvents Table 6 shows the RMSE values for allneutral cations and anions for the other polar solvents in the MNSOL data set acetoni-trile DMSO and methanol for the original SMD radii and the corresponding re-optimizedradii (dagger) In addition corresponding combined RMSE values are given for the remaining88 mostly non-polar solvents in the MNSOL data set for which only data is availablefor neutral molecules It is clear from this data that the new radii sets lowers the errorof the predicted solvation free energies for the other polar solvents and has a negligibleeffect on the accuracy for the non-polar solvents
As for acetonitrile and DMSO the improvement is greatest for the ions where the RMSEdecreases by 3-12 kcalmol The improvement is more modest (05-3 kcalmol) for theneutral molecules For methanol which contains only ions the RMSE is similarly de-creased with 2-13 kcalmol
We thus recommend that the new radii optimized for water also be used for other polarsolvents when doing semiempirical SMD calculations For non-polar solvents either setof radii can be used
14
Table 6 RMSE (in kcalmol) of non-aqueous solvation energies computed with SMD usingHF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicates that theradii has been reoptimized Corresponding RMSE values for the COSMO method are includedfor comparison The reference set is split into full set neutral cations and anions
All Neu Cat Aniacetonitrile
HFSMD 112 29 105 131PM3SMD 194 41 162 246PM3SMDdagger 140 37 113 182PM6SMD 219 33 177 284PM6SMDdagger 130 12 114 160DFTBSMD 150 23 119 197DFTBSMDdagger 109 22 90 140PM3COSMO 130 31 128 147PM6COSMO 137 23 135 155
dimethylsulfoxideHFSMD 119 30 106 126PM3SMD 202 42 153 214PM3SMDdagger 170 35 90 182PM6SMD 255 45 172 272PM6SMDdagger 177 17 99 189DFTBSMD 169 23 108 181DFTBSMDdagger 125 21 76 134PM3COSMO 125 34 107 132PM6COSMO 150 31 119 159
methanolHFSMD 40 00 41 39PM3SMD 126 00 108 135PM3SMDdagger 66 00 60 69PM6SMD 163 00 120 183PM6SMDdagger 52 00 54 52DFTBSMD 84 00 63 94DFTBSMDdagger 47 00 44 49PM3COSMO 59 00 68 54PM6COSMO 54 00 67 46
otherHFSMD 42 42 00 00PM3SMD 51 51 00 00PM3SMDdagger 51 51 00 00PM6SMD 35 35 00 00PM6SMDdagger 34 34 00 00DFTBSMD 30 30 00 00DFTBSMDdagger 29 29 00 00PM3COSMO 36 36 00 00PM6COSMO 27 27 00 00
15
ConclusionThe PM6 method in GAMESS was extended to elements requiring d-integrals and inter-faced with the polarized continuum model (PCM) of solvation including semi-numericalgradients However the accuracy of aqueous solvation energies computed using AM1PM3 PM6 and DFTB and the SMD continuum solvation model was tested using a sub-set of molecules from the MNSOL data set which showed that the errors in SMD solvationenergies predicted using NDDO-based methods was considerably larger than when usingDFT and HF with RMSE values of 50 to 86 kcalmol compared to 34 kcalmol forHF6-31G(d) For the NDDO-based methods the errors were especially large for cationsand in the case of PM6 also for anions with RMSE values of 101 to 148 kcalmol incomparison with to 50 to 57 kcalmol for HF6-31G(d)SMD Corresponding COSMOresults (where the maximum RMSE is 75 kcalmol) suggested that the NDDOSMDresults could be improved by re-parameterizing the SMD Coulomb radii for the NDDOmethods
The fact that the NDDOSMD errors are largest for ions suggest that the problem iswith the polar solvation energy so we focus on optimizing the values of the Coulombradii while leaving all parameters associated with the non-polar solvation free energy un-changed We optimize the radii only for the ionic species on a subset of the MNSOL dataset previously used to parametrize RHF6-31G(d)SMD but include neutral moleculeswhen testing how well SMD with the new Coulomb radii perform We found the bestresults are obtained by changing only the radii for hydrogen carbon oxygen nitrogenand sulfur and this leads to RMSE values for PM3 (neutrals 28ions ca 5 kcalmol)PM6 (47ca 5 kcalmol) and DFTB (39ca 5 kcalmol) that are more comparable toHF6-31G(d) (24ca 5 kcalmol) We note that the SMD parameterization is done us-ing mono-aquo microsolvated species for small ions with atoms carrying large (partial)charges while the MNSOL data set also contains data for the non-microsolvated equiv-alents Comparison to the HF6-31G(d) MSEs reported by Marenich et al [8] reportedfor the selectively microsolvated ions indicates that the inclusion of non-microsolvatedspecies leads an MSE increase of about 1 kcalmol
Though the radii are optimized to reproduce aqueous solvation energies they also leadmore accurate predictions for other polar solvents such as DMSO acetonitrile andmethanol while the improvement for non-polar solvents are negligible
Supplementary MaterialSee supplementary material for Tables S1-S3 and Figures S1-S3 which are referred toin the text In addition we also provide a table of dielectric constants used for thecalculations and a table of aqueous solvation free energies obtained using the optimizedparameters
AcknowledgmentsJCK and JHJ would like to acknowledge Jimmy (Mr MOPAC) Stewart for providingsource code and guidance for the PM6 GAMESS implementation Also thanks to Donald
16
Truhlar and Christopher Cramer for sharing the MNSOL data set and for insightfulcomments on the manuscript CS thanks the Danish Council for Independent Research(the Sapere Aude program) for financial support (Grant no DFF 4181-00370)
References[1] J Tomasi B Mennucci and R Cammi ldquoQuantum mechanical continuum solvation
modelsrdquo Chem Rev vol 105 pp 2999ndash3093 2005
[2] A Klamt and G Schuurmann ldquoCOSMO a new approach to dielectric screeningin solvents with explicit expressions for the screening energy and its gradientrdquo JChem Soc Perkin Trans vol 2 pp 799ndash805 1993
[3] C J Cramer and D G Truhlar ldquoA universal approach to solvation modelingrdquoAccounts of Chemical Research vol 41 no 6 pp 760ndash768 jun 2008 doi101021ar800019z
[4] V Barone M Cossi and J Tomasi ldquoA new definition of cavities for thecomputation of solvation free energies by the polarizable continuum modelrdquo JChem Phys vol 107 no 8 pp 3210ndash3221 aug 1997 doi 1010631474671
[5] F Eckert and A Klamt ldquoFast solvent screening via quantum chemistryCOSMO-RS approachrdquo AIChE J vol 48 no 2 pp 369ndash385 feb 2002 doi101002aic690480220
[6] C Curutchet A Bidon-Chanal I Soteras M Orozco and F J Luque ldquoMSTcontinuum study of the hydration free energies of monovalent ionic speciesrdquo JPhys Chem B vol 109 no 8 pp 3565ndash3574 mar 2005 doi 101021jp047197s
[7] Y Takano and K N Houk ldquoBenchmarking the conductor-like polarizablecontinuum model (CPCM) for aqueous solvation free energies of neutral and ionicorganic moleculesrdquo J Chem Theory Comput vol 1 no 1 pp 70ndash77 jan 2005doi 101021ct049977a
[8] A V Marenich R M Olson C P Kelly C J Cramer and D G TruhlarldquoSelf-consistent reaction field model for aqueous and nonaqueous solutions based onaccurate polarized partial chargesrdquo Journal of Chemical Theory and Computationvol 3 no 6 pp 2011ndash2033 nov 2007 doi 101021ct7001418
[9] A V Marenich C J Cramer and D G Truhlar ldquoUniversal solvation model basedon solute electron density and on a continuum model of the solvent defined by thebulk dielectric constant and atomic surface tensionsrdquo J Phys Chem B vol 113no 18 pp 6378ndash6396 may 2009 doi 101021jp810292n
[10] G E Chudinov D V Napolov and M V Basilevsky ldquoQuantum-chemicalcalculations of the hydration energies of organic cations and anions in the frameworkof a continuum solvent approximationrdquo Chem Phys vol 160 no 1 pp 41 ndash 541992 doi 1010160301-0104(92)87090-V
[11] M J Negre M Orozco and F J Luque ldquoA new strategy for the representation ofenvironment effects in semi-empirical calculations based on dewarrsquos hamiltoniansrdquoChem Phys Lett vol 196 no 1 pp 27ndash36 1992
17
[12] F J Luque M J Negre and M Orozco ldquoAn am1-scrf approach to the study ofchanges in molecular properties induced by solventrdquo J Phys Chem vol 97 no 17pp 4386ndash4391 1993
[13] M Caricato B Mennucci and J Tomasi ldquoSolvent effects on the electronic spectraAn extension of the polarizable continuum model to the zindo methodrdquo J PhysChem A vol 108 no 29 pp 6248ndash6256 2004
[14] M J S Dewar E G Zoebisch E F Healy and J J P Stewart ldquoDevelopmentand use of quantum mechanical molecular models 76 AM1 a new general purposequantum mechanical molecular modelrdquo J A Chem Soc vol 10 pp 3902ndash39091985 doi 101021ja00299a024
[15] J J P Stewart ldquoOptimization of Parameters for Semi-Empirical Methods I-Methodrdquo J Comp Chem vol 10 pp 209ndash220 1989 doi 101002jcc540100208
[16] J J P Stewart ldquoOptimization of parameters for semiempirical methods V Modi-fication of NDDO approximations and application to 70 elementsrdquo J Mol Modelvol 13 pp 1172ndash1213 2007
[17] M Gaus Q Cui and M Elstner ldquoDFTB3 Extension of the Self-Consistent-Charge Density-Functional Tight-Binding Method (SCC-DFTB)rdquo J Chem TheoryComput vol 7 no 4 pp 931ndash948 apr 2011 doi 101021ct100684s
[18] A V Marenich C P Kelly J D Thompson G D Hawkins C C ChambersD J Giesen P P Winget C J Cramer and D G Truhlar ldquoMinnesota solvationdatabaserdquo 2012
[19] ldquoMinnesota solvation databaserdquo accessed November 24th 2017 [Online] Availablehttpscompchemumnedumnsol
[20] M W Schmidt K K Baldridge J A Boatz S T Elbert M S Gordon J HJensen S Koseki N Matsunaga K A Nguyen S Su T L Windus M Dupuisand JAMontgomery ldquoGeneral atomic and molecular electronic structure systemrdquoJ Comput Chem vol 14 pp 1347ndash1363 1993
[21] V Barone and M Cossi ldquoQuantum calculation of molecular energies and energygradients in solution by a conductor solvent modelrdquo The Journal of PhysicalChemistry A vol 102 no 11 pp 1995ndash2001 mar 1998 doi 101021jp9716997
[22] M Cossi N Rega G Scalmani and V Barone ldquoEnergies structures andelectronic properties of molecules in solution with the c-PCM solvation modelrdquoJournal of Computational Chemistry vol 24 no 6 pp 669ndash681 apr 2003 doi101002jcc10189
[23] C Steinmann K L Blaeligdel A S Christensen and J H Jensen ldquoInterface ofthe polarizable continuum model of solvation with semi-empirical methods in thegamess programrdquo PLoS ONE vol 8 no 7 p e67725 07 2013 doi 101371jour-nalpone0067725
[24] Y Nishimoto ldquoDFTBPCM applied to ground and excited state potential energysurfacesrdquo J Phys Chem A vol 120 no 5 pp 771ndash784 feb 2016 doi101021acsjpca5b10732
18
[25] M Gaus X Lu M Elstner and Q Cui ldquoParameterization of DFTB33OB forSulfur and Phosphorus for Chemical and Biological Applicationsrdquo J Chem TheoryComput vol 10 no 4 pp 1518ndash1537 apr 2014 doi 101021ct401002w
[26] X Lu M Gaus M Elstner and Q Cui ldquoParametrization of DFTB33OBfor Magnesium and Zinc for Chemical and Biological Applicationsrdquo The Journalof Physical Chemistry B vol 119 no 3 pp 1062ndash1082 jan 2015 doi101021jp506557r
[27] M Kubillus T Kubar M Gaus J Rezac and M Elstner ldquoParameterization ofthe DFTB3 Method for Br Ca Cl F I K and Na in Organic and BiologicalSystemsrdquo J Chem Theory Comput vol 11 no 1 pp 332ndash342 jan 2015 doi101021ct5009137
[28] J J P Stewart ldquoMOPAC2012rdquo 2016 Stewart Computational Chemistry ColoradoSprings CO USA [Online] Available httpopenmopacnet
[29] J L Pascual-Ahuir E Silla J Tomasi and R Bonaccorsi ldquoElectrostaticinteraction of a solute with a continuum improved description of the cavity and ofthe surface cavity bound charge distributionrdquo Journal of Computational Chemistryvol 8 no 6 pp 778ndash787 sep 1987 doi 101002jcc540080605
[30] P Su and H Li ldquoContinuous and smooth potential energy surface for conductorlikescreening solvation model using fixed points with variable areasrdquo The Journal ofChemical Physics vol 130 no 7 p 074109 feb 2009 doi 10106313077917
[31] J Tomasi B Mennucci and E Cances ldquoThe IEF version of the PCM solvationmethod an overview of a new method addressed to study molecular solutes at theQM ab initio levelrdquo Journal of Molecular Structure THEOCHEM vol 464 no1-3 pp 211ndash226 may 1999 doi 101016s0166-1280(98)00553-3
[32] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 1 the calculation of confidence intervalsrdquo J Comput Aided MolDes vol 28 no 9 pp 887ndash918 Sep 2014 doi 101007s10822-014-9753-z
[33] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 2 comparing methodsrdquo J Comput Aided Mol Des vol 30 no 2pp 103ndash126 Feb 2016 doi 101007s10822-016-9904-5
[34] J H Jensen ldquoWhich method is more accurate or errors have error barsrdquo PeerJPreprints vol 5 p e2693v1 2017 doi 107287peerjpreprints2693v1
[35] J H Jensen C J Swain and L Olsen ldquoPrediction of pKa values for druglikemolecules using semiempirical quantum chemical methodsrdquo The Journal of PhysicalChemistry A vol 121 no 3 pp 699ndash707 jan 2017 doi 101021acsjpca6b10990
[36] F Eckert and A Klamt ldquoAccurate prediction of basicity in aqueous solution withCOSMO-RSrdquo Journal of Computational Chemistry vol 27 no 1 pp 11ndash19 2005doi 101002jcc20309
[37] J A Nelder and R Mead ldquoA simplex method for function minimizationrdquo TheComputer Journal vol 7 no 4 pp 308ndash313 1965 doi 101093comjnl74308
19
[38] E Jones T Oliphant P Peterson et al ldquoSciPy Open source scientifictools for Pythonrdquo 2001 accessed November 24th 2017 [Online] Availablehttpwwwscipyorgrdquo
[39] J Charnley 2017 accessed November 24th 2017 [Online] Available httpsgithubcomcharnleyoptimize gamess parameters
[40] C P Kelly C J Cramer and D G Truhlar ldquoAqueous solvation free energies ofions and ion-water clusters based on an accurate value for the absolute aqueoussolvation free energy of the protonrdquo The Journal of Physical Chemistry B vol 110no 32 pp 16 066ndash16 081 aug 2006 doi 101021jp063552y
20
HFSMDAM1SMD
PM3SMDPM6SMD
DFTBSMD
AM1COSMO
PM3COSMO
PM6COSMO null0
2
4
6
8
10
12
14
16RM
SE [k
calm
ol]
PM3PM3HCO
PM3HCON
PM3HCONSPM3all PM6
PM6HCO
PM6HCON
PM6HCONS
PM6HCONShPM6all
DFTB
DFTBHCON0
2
4
6
8
10
12
14RM
SE [k
calm
ol]
HFSMD
PM3SMDPM6SMD
DFTBSMD
PM3COSMO
PM6COSMO0
2
4
6
8
10RM
SE [k
calm
ol]
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6err
or

Table 6 RMSE (in kcalmol) of non-aqueous solvation energies computed with SMD usingHF6-31G(d) PM3 PM6 and DFTB for the MNSOL dataset Note that dagger indicates that theradii has been reoptimized Corresponding RMSE values for the COSMO method are includedfor comparison The reference set is split into full set neutral cations and anions
All Neu Cat Aniacetonitrile
HFSMD 112 29 105 131PM3SMD 194 41 162 246PM3SMDdagger 140 37 113 182PM6SMD 219 33 177 284PM6SMDdagger 130 12 114 160DFTBSMD 150 23 119 197DFTBSMDdagger 109 22 90 140PM3COSMO 130 31 128 147PM6COSMO 137 23 135 155
dimethylsulfoxideHFSMD 119 30 106 126PM3SMD 202 42 153 214PM3SMDdagger 170 35 90 182PM6SMD 255 45 172 272PM6SMDdagger 177 17 99 189DFTBSMD 169 23 108 181DFTBSMDdagger 125 21 76 134PM3COSMO 125 34 107 132PM6COSMO 150 31 119 159
methanolHFSMD 40 00 41 39PM3SMD 126 00 108 135PM3SMDdagger 66 00 60 69PM6SMD 163 00 120 183PM6SMDdagger 52 00 54 52DFTBSMD 84 00 63 94DFTBSMDdagger 47 00 44 49PM3COSMO 59 00 68 54PM6COSMO 54 00 67 46
otherHFSMD 42 42 00 00PM3SMD 51 51 00 00PM3SMDdagger 51 51 00 00PM6SMD 35 35 00 00PM6SMDdagger 34 34 00 00DFTBSMD 30 30 00 00DFTBSMDdagger 29 29 00 00PM3COSMO 36 36 00 00PM6COSMO 27 27 00 00
15
ConclusionThe PM6 method in GAMESS was extended to elements requiring d-integrals and inter-faced with the polarized continuum model (PCM) of solvation including semi-numericalgradients However the accuracy of aqueous solvation energies computed using AM1PM3 PM6 and DFTB and the SMD continuum solvation model was tested using a sub-set of molecules from the MNSOL data set which showed that the errors in SMD solvationenergies predicted using NDDO-based methods was considerably larger than when usingDFT and HF with RMSE values of 50 to 86 kcalmol compared to 34 kcalmol forHF6-31G(d) For the NDDO-based methods the errors were especially large for cationsand in the case of PM6 also for anions with RMSE values of 101 to 148 kcalmol incomparison with to 50 to 57 kcalmol for HF6-31G(d)SMD Corresponding COSMOresults (where the maximum RMSE is 75 kcalmol) suggested that the NDDOSMDresults could be improved by re-parameterizing the SMD Coulomb radii for the NDDOmethods
The fact that the NDDOSMD errors are largest for ions suggest that the problem iswith the polar solvation energy so we focus on optimizing the values of the Coulombradii while leaving all parameters associated with the non-polar solvation free energy un-changed We optimize the radii only for the ionic species on a subset of the MNSOL dataset previously used to parametrize RHF6-31G(d)SMD but include neutral moleculeswhen testing how well SMD with the new Coulomb radii perform We found the bestresults are obtained by changing only the radii for hydrogen carbon oxygen nitrogenand sulfur and this leads to RMSE values for PM3 (neutrals 28ions ca 5 kcalmol)PM6 (47ca 5 kcalmol) and DFTB (39ca 5 kcalmol) that are more comparable toHF6-31G(d) (24ca 5 kcalmol) We note that the SMD parameterization is done us-ing mono-aquo microsolvated species for small ions with atoms carrying large (partial)charges while the MNSOL data set also contains data for the non-microsolvated equiv-alents Comparison to the HF6-31G(d) MSEs reported by Marenich et al [8] reportedfor the selectively microsolvated ions indicates that the inclusion of non-microsolvatedspecies leads an MSE increase of about 1 kcalmol
Though the radii are optimized to reproduce aqueous solvation energies they also leadmore accurate predictions for other polar solvents such as DMSO acetonitrile andmethanol while the improvement for non-polar solvents are negligible
Supplementary MaterialSee supplementary material for Tables S1-S3 and Figures S1-S3 which are referred toin the text In addition we also provide a table of dielectric constants used for thecalculations and a table of aqueous solvation free energies obtained using the optimizedparameters
AcknowledgmentsJCK and JHJ would like to acknowledge Jimmy (Mr MOPAC) Stewart for providingsource code and guidance for the PM6 GAMESS implementation Also thanks to Donald
16
Truhlar and Christopher Cramer for sharing the MNSOL data set and for insightfulcomments on the manuscript CS thanks the Danish Council for Independent Research(the Sapere Aude program) for financial support (Grant no DFF 4181-00370)
References[1] J Tomasi B Mennucci and R Cammi ldquoQuantum mechanical continuum solvation
modelsrdquo Chem Rev vol 105 pp 2999ndash3093 2005
[2] A Klamt and G Schuurmann ldquoCOSMO a new approach to dielectric screeningin solvents with explicit expressions for the screening energy and its gradientrdquo JChem Soc Perkin Trans vol 2 pp 799ndash805 1993
[3] C J Cramer and D G Truhlar ldquoA universal approach to solvation modelingrdquoAccounts of Chemical Research vol 41 no 6 pp 760ndash768 jun 2008 doi101021ar800019z
[4] V Barone M Cossi and J Tomasi ldquoA new definition of cavities for thecomputation of solvation free energies by the polarizable continuum modelrdquo JChem Phys vol 107 no 8 pp 3210ndash3221 aug 1997 doi 1010631474671
[5] F Eckert and A Klamt ldquoFast solvent screening via quantum chemistryCOSMO-RS approachrdquo AIChE J vol 48 no 2 pp 369ndash385 feb 2002 doi101002aic690480220
[6] C Curutchet A Bidon-Chanal I Soteras M Orozco and F J Luque ldquoMSTcontinuum study of the hydration free energies of monovalent ionic speciesrdquo JPhys Chem B vol 109 no 8 pp 3565ndash3574 mar 2005 doi 101021jp047197s
[7] Y Takano and K N Houk ldquoBenchmarking the conductor-like polarizablecontinuum model (CPCM) for aqueous solvation free energies of neutral and ionicorganic moleculesrdquo J Chem Theory Comput vol 1 no 1 pp 70ndash77 jan 2005doi 101021ct049977a
[8] A V Marenich R M Olson C P Kelly C J Cramer and D G TruhlarldquoSelf-consistent reaction field model for aqueous and nonaqueous solutions based onaccurate polarized partial chargesrdquo Journal of Chemical Theory and Computationvol 3 no 6 pp 2011ndash2033 nov 2007 doi 101021ct7001418
[9] A V Marenich C J Cramer and D G Truhlar ldquoUniversal solvation model basedon solute electron density and on a continuum model of the solvent defined by thebulk dielectric constant and atomic surface tensionsrdquo J Phys Chem B vol 113no 18 pp 6378ndash6396 may 2009 doi 101021jp810292n
[10] G E Chudinov D V Napolov and M V Basilevsky ldquoQuantum-chemicalcalculations of the hydration energies of organic cations and anions in the frameworkof a continuum solvent approximationrdquo Chem Phys vol 160 no 1 pp 41 ndash 541992 doi 1010160301-0104(92)87090-V
[11] M J Negre M Orozco and F J Luque ldquoA new strategy for the representation ofenvironment effects in semi-empirical calculations based on dewarrsquos hamiltoniansrdquoChem Phys Lett vol 196 no 1 pp 27ndash36 1992
17
[12] F J Luque M J Negre and M Orozco ldquoAn am1-scrf approach to the study ofchanges in molecular properties induced by solventrdquo J Phys Chem vol 97 no 17pp 4386ndash4391 1993
[13] M Caricato B Mennucci and J Tomasi ldquoSolvent effects on the electronic spectraAn extension of the polarizable continuum model to the zindo methodrdquo J PhysChem A vol 108 no 29 pp 6248ndash6256 2004
[14] M J S Dewar E G Zoebisch E F Healy and J J P Stewart ldquoDevelopmentand use of quantum mechanical molecular models 76 AM1 a new general purposequantum mechanical molecular modelrdquo J A Chem Soc vol 10 pp 3902ndash39091985 doi 101021ja00299a024
[15] J J P Stewart ldquoOptimization of Parameters for Semi-Empirical Methods I-Methodrdquo J Comp Chem vol 10 pp 209ndash220 1989 doi 101002jcc540100208
[16] J J P Stewart ldquoOptimization of parameters for semiempirical methods V Modi-fication of NDDO approximations and application to 70 elementsrdquo J Mol Modelvol 13 pp 1172ndash1213 2007
[17] M Gaus Q Cui and M Elstner ldquoDFTB3 Extension of the Self-Consistent-Charge Density-Functional Tight-Binding Method (SCC-DFTB)rdquo J Chem TheoryComput vol 7 no 4 pp 931ndash948 apr 2011 doi 101021ct100684s
[18] A V Marenich C P Kelly J D Thompson G D Hawkins C C ChambersD J Giesen P P Winget C J Cramer and D G Truhlar ldquoMinnesota solvationdatabaserdquo 2012
[19] ldquoMinnesota solvation databaserdquo accessed November 24th 2017 [Online] Availablehttpscompchemumnedumnsol
[20] M W Schmidt K K Baldridge J A Boatz S T Elbert M S Gordon J HJensen S Koseki N Matsunaga K A Nguyen S Su T L Windus M Dupuisand JAMontgomery ldquoGeneral atomic and molecular electronic structure systemrdquoJ Comput Chem vol 14 pp 1347ndash1363 1993
[21] V Barone and M Cossi ldquoQuantum calculation of molecular energies and energygradients in solution by a conductor solvent modelrdquo The Journal of PhysicalChemistry A vol 102 no 11 pp 1995ndash2001 mar 1998 doi 101021jp9716997
[22] M Cossi N Rega G Scalmani and V Barone ldquoEnergies structures andelectronic properties of molecules in solution with the c-PCM solvation modelrdquoJournal of Computational Chemistry vol 24 no 6 pp 669ndash681 apr 2003 doi101002jcc10189
[23] C Steinmann K L Blaeligdel A S Christensen and J H Jensen ldquoInterface ofthe polarizable continuum model of solvation with semi-empirical methods in thegamess programrdquo PLoS ONE vol 8 no 7 p e67725 07 2013 doi 101371jour-nalpone0067725
[24] Y Nishimoto ldquoDFTBPCM applied to ground and excited state potential energysurfacesrdquo J Phys Chem A vol 120 no 5 pp 771ndash784 feb 2016 doi101021acsjpca5b10732
18
[25] M Gaus X Lu M Elstner and Q Cui ldquoParameterization of DFTB33OB forSulfur and Phosphorus for Chemical and Biological Applicationsrdquo J Chem TheoryComput vol 10 no 4 pp 1518ndash1537 apr 2014 doi 101021ct401002w
[26] X Lu M Gaus M Elstner and Q Cui ldquoParametrization of DFTB33OBfor Magnesium and Zinc for Chemical and Biological Applicationsrdquo The Journalof Physical Chemistry B vol 119 no 3 pp 1062ndash1082 jan 2015 doi101021jp506557r
[27] M Kubillus T Kubar M Gaus J Rezac and M Elstner ldquoParameterization ofthe DFTB3 Method for Br Ca Cl F I K and Na in Organic and BiologicalSystemsrdquo J Chem Theory Comput vol 11 no 1 pp 332ndash342 jan 2015 doi101021ct5009137
[28] J J P Stewart ldquoMOPAC2012rdquo 2016 Stewart Computational Chemistry ColoradoSprings CO USA [Online] Available httpopenmopacnet
[29] J L Pascual-Ahuir E Silla J Tomasi and R Bonaccorsi ldquoElectrostaticinteraction of a solute with a continuum improved description of the cavity and ofthe surface cavity bound charge distributionrdquo Journal of Computational Chemistryvol 8 no 6 pp 778ndash787 sep 1987 doi 101002jcc540080605
[30] P Su and H Li ldquoContinuous and smooth potential energy surface for conductorlikescreening solvation model using fixed points with variable areasrdquo The Journal ofChemical Physics vol 130 no 7 p 074109 feb 2009 doi 10106313077917
[31] J Tomasi B Mennucci and E Cances ldquoThe IEF version of the PCM solvationmethod an overview of a new method addressed to study molecular solutes at theQM ab initio levelrdquo Journal of Molecular Structure THEOCHEM vol 464 no1-3 pp 211ndash226 may 1999 doi 101016s0166-1280(98)00553-3
[32] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 1 the calculation of confidence intervalsrdquo J Comput Aided MolDes vol 28 no 9 pp 887ndash918 Sep 2014 doi 101007s10822-014-9753-z
[33] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 2 comparing methodsrdquo J Comput Aided Mol Des vol 30 no 2pp 103ndash126 Feb 2016 doi 101007s10822-016-9904-5
[34] J H Jensen ldquoWhich method is more accurate or errors have error barsrdquo PeerJPreprints vol 5 p e2693v1 2017 doi 107287peerjpreprints2693v1
[35] J H Jensen C J Swain and L Olsen ldquoPrediction of pKa values for druglikemolecules using semiempirical quantum chemical methodsrdquo The Journal of PhysicalChemistry A vol 121 no 3 pp 699ndash707 jan 2017 doi 101021acsjpca6b10990
[36] F Eckert and A Klamt ldquoAccurate prediction of basicity in aqueous solution withCOSMO-RSrdquo Journal of Computational Chemistry vol 27 no 1 pp 11ndash19 2005doi 101002jcc20309
[37] J A Nelder and R Mead ldquoA simplex method for function minimizationrdquo TheComputer Journal vol 7 no 4 pp 308ndash313 1965 doi 101093comjnl74308
19
[38] E Jones T Oliphant P Peterson et al ldquoSciPy Open source scientifictools for Pythonrdquo 2001 accessed November 24th 2017 [Online] Availablehttpwwwscipyorgrdquo
[39] J Charnley 2017 accessed November 24th 2017 [Online] Available httpsgithubcomcharnleyoptimize gamess parameters
[40] C P Kelly C J Cramer and D G Truhlar ldquoAqueous solvation free energies ofions and ion-water clusters based on an accurate value for the absolute aqueoussolvation free energy of the protonrdquo The Journal of Physical Chemistry B vol 110no 32 pp 16 066ndash16 081 aug 2006 doi 101021jp063552y
20
HFSMDAM1SMD
PM3SMDPM6SMD
DFTBSMD
AM1COSMO
PM3COSMO
PM6COSMO null0
2
4
6
8
10
12
14
16RM
SE [k
calm
ol]
PM3PM3HCO
PM3HCON
PM3HCONSPM3all PM6
PM6HCO
PM6HCON
PM6HCONS
PM6HCONShPM6all
DFTB
DFTBHCON0
2
4
6
8
10
12
14RM
SE [k
calm
ol]
HFSMD
PM3SMDPM6SMD
DFTBSMD
PM3COSMO
PM6COSMO0
2
4
6
8
10RM
SE [k
calm
ol]
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6err
or

ConclusionThe PM6 method in GAMESS was extended to elements requiring d-integrals and inter-faced with the polarized continuum model (PCM) of solvation including semi-numericalgradients However the accuracy of aqueous solvation energies computed using AM1PM3 PM6 and DFTB and the SMD continuum solvation model was tested using a sub-set of molecules from the MNSOL data set which showed that the errors in SMD solvationenergies predicted using NDDO-based methods was considerably larger than when usingDFT and HF with RMSE values of 50 to 86 kcalmol compared to 34 kcalmol forHF6-31G(d) For the NDDO-based methods the errors were especially large for cationsand in the case of PM6 also for anions with RMSE values of 101 to 148 kcalmol incomparison with to 50 to 57 kcalmol for HF6-31G(d)SMD Corresponding COSMOresults (where the maximum RMSE is 75 kcalmol) suggested that the NDDOSMDresults could be improved by re-parameterizing the SMD Coulomb radii for the NDDOmethods
The fact that the NDDOSMD errors are largest for ions suggest that the problem iswith the polar solvation energy so we focus on optimizing the values of the Coulombradii while leaving all parameters associated with the non-polar solvation free energy un-changed We optimize the radii only for the ionic species on a subset of the MNSOL dataset previously used to parametrize RHF6-31G(d)SMD but include neutral moleculeswhen testing how well SMD with the new Coulomb radii perform We found the bestresults are obtained by changing only the radii for hydrogen carbon oxygen nitrogenand sulfur and this leads to RMSE values for PM3 (neutrals 28ions ca 5 kcalmol)PM6 (47ca 5 kcalmol) and DFTB (39ca 5 kcalmol) that are more comparable toHF6-31G(d) (24ca 5 kcalmol) We note that the SMD parameterization is done us-ing mono-aquo microsolvated species for small ions with atoms carrying large (partial)charges while the MNSOL data set also contains data for the non-microsolvated equiv-alents Comparison to the HF6-31G(d) MSEs reported by Marenich et al [8] reportedfor the selectively microsolvated ions indicates that the inclusion of non-microsolvatedspecies leads an MSE increase of about 1 kcalmol
Though the radii are optimized to reproduce aqueous solvation energies they also leadmore accurate predictions for other polar solvents such as DMSO acetonitrile andmethanol while the improvement for non-polar solvents are negligible
Supplementary MaterialSee supplementary material for Tables S1-S3 and Figures S1-S3 which are referred toin the text In addition we also provide a table of dielectric constants used for thecalculations and a table of aqueous solvation free energies obtained using the optimizedparameters
AcknowledgmentsJCK and JHJ would like to acknowledge Jimmy (Mr MOPAC) Stewart for providingsource code and guidance for the PM6 GAMESS implementation Also thanks to Donald
16
Truhlar and Christopher Cramer for sharing the MNSOL data set and for insightfulcomments on the manuscript CS thanks the Danish Council for Independent Research(the Sapere Aude program) for financial support (Grant no DFF 4181-00370)
References[1] J Tomasi B Mennucci and R Cammi ldquoQuantum mechanical continuum solvation
modelsrdquo Chem Rev vol 105 pp 2999ndash3093 2005
[2] A Klamt and G Schuurmann ldquoCOSMO a new approach to dielectric screeningin solvents with explicit expressions for the screening energy and its gradientrdquo JChem Soc Perkin Trans vol 2 pp 799ndash805 1993
[3] C J Cramer and D G Truhlar ldquoA universal approach to solvation modelingrdquoAccounts of Chemical Research vol 41 no 6 pp 760ndash768 jun 2008 doi101021ar800019z
[4] V Barone M Cossi and J Tomasi ldquoA new definition of cavities for thecomputation of solvation free energies by the polarizable continuum modelrdquo JChem Phys vol 107 no 8 pp 3210ndash3221 aug 1997 doi 1010631474671
[5] F Eckert and A Klamt ldquoFast solvent screening via quantum chemistryCOSMO-RS approachrdquo AIChE J vol 48 no 2 pp 369ndash385 feb 2002 doi101002aic690480220
[6] C Curutchet A Bidon-Chanal I Soteras M Orozco and F J Luque ldquoMSTcontinuum study of the hydration free energies of monovalent ionic speciesrdquo JPhys Chem B vol 109 no 8 pp 3565ndash3574 mar 2005 doi 101021jp047197s
[7] Y Takano and K N Houk ldquoBenchmarking the conductor-like polarizablecontinuum model (CPCM) for aqueous solvation free energies of neutral and ionicorganic moleculesrdquo J Chem Theory Comput vol 1 no 1 pp 70ndash77 jan 2005doi 101021ct049977a
[8] A V Marenich R M Olson C P Kelly C J Cramer and D G TruhlarldquoSelf-consistent reaction field model for aqueous and nonaqueous solutions based onaccurate polarized partial chargesrdquo Journal of Chemical Theory and Computationvol 3 no 6 pp 2011ndash2033 nov 2007 doi 101021ct7001418
[9] A V Marenich C J Cramer and D G Truhlar ldquoUniversal solvation model basedon solute electron density and on a continuum model of the solvent defined by thebulk dielectric constant and atomic surface tensionsrdquo J Phys Chem B vol 113no 18 pp 6378ndash6396 may 2009 doi 101021jp810292n
[10] G E Chudinov D V Napolov and M V Basilevsky ldquoQuantum-chemicalcalculations of the hydration energies of organic cations and anions in the frameworkof a continuum solvent approximationrdquo Chem Phys vol 160 no 1 pp 41 ndash 541992 doi 1010160301-0104(92)87090-V
[11] M J Negre M Orozco and F J Luque ldquoA new strategy for the representation ofenvironment effects in semi-empirical calculations based on dewarrsquos hamiltoniansrdquoChem Phys Lett vol 196 no 1 pp 27ndash36 1992
17
[12] F J Luque M J Negre and M Orozco ldquoAn am1-scrf approach to the study ofchanges in molecular properties induced by solventrdquo J Phys Chem vol 97 no 17pp 4386ndash4391 1993
[13] M Caricato B Mennucci and J Tomasi ldquoSolvent effects on the electronic spectraAn extension of the polarizable continuum model to the zindo methodrdquo J PhysChem A vol 108 no 29 pp 6248ndash6256 2004
[14] M J S Dewar E G Zoebisch E F Healy and J J P Stewart ldquoDevelopmentand use of quantum mechanical molecular models 76 AM1 a new general purposequantum mechanical molecular modelrdquo J A Chem Soc vol 10 pp 3902ndash39091985 doi 101021ja00299a024
[15] J J P Stewart ldquoOptimization of Parameters for Semi-Empirical Methods I-Methodrdquo J Comp Chem vol 10 pp 209ndash220 1989 doi 101002jcc540100208
[16] J J P Stewart ldquoOptimization of parameters for semiempirical methods V Modi-fication of NDDO approximations and application to 70 elementsrdquo J Mol Modelvol 13 pp 1172ndash1213 2007
[17] M Gaus Q Cui and M Elstner ldquoDFTB3 Extension of the Self-Consistent-Charge Density-Functional Tight-Binding Method (SCC-DFTB)rdquo J Chem TheoryComput vol 7 no 4 pp 931ndash948 apr 2011 doi 101021ct100684s
[18] A V Marenich C P Kelly J D Thompson G D Hawkins C C ChambersD J Giesen P P Winget C J Cramer and D G Truhlar ldquoMinnesota solvationdatabaserdquo 2012
[19] ldquoMinnesota solvation databaserdquo accessed November 24th 2017 [Online] Availablehttpscompchemumnedumnsol
[20] M W Schmidt K K Baldridge J A Boatz S T Elbert M S Gordon J HJensen S Koseki N Matsunaga K A Nguyen S Su T L Windus M Dupuisand JAMontgomery ldquoGeneral atomic and molecular electronic structure systemrdquoJ Comput Chem vol 14 pp 1347ndash1363 1993
[21] V Barone and M Cossi ldquoQuantum calculation of molecular energies and energygradients in solution by a conductor solvent modelrdquo The Journal of PhysicalChemistry A vol 102 no 11 pp 1995ndash2001 mar 1998 doi 101021jp9716997
[22] M Cossi N Rega G Scalmani and V Barone ldquoEnergies structures andelectronic properties of molecules in solution with the c-PCM solvation modelrdquoJournal of Computational Chemistry vol 24 no 6 pp 669ndash681 apr 2003 doi101002jcc10189
[23] C Steinmann K L Blaeligdel A S Christensen and J H Jensen ldquoInterface ofthe polarizable continuum model of solvation with semi-empirical methods in thegamess programrdquo PLoS ONE vol 8 no 7 p e67725 07 2013 doi 101371jour-nalpone0067725
[24] Y Nishimoto ldquoDFTBPCM applied to ground and excited state potential energysurfacesrdquo J Phys Chem A vol 120 no 5 pp 771ndash784 feb 2016 doi101021acsjpca5b10732
18
[25] M Gaus X Lu M Elstner and Q Cui ldquoParameterization of DFTB33OB forSulfur and Phosphorus for Chemical and Biological Applicationsrdquo J Chem TheoryComput vol 10 no 4 pp 1518ndash1537 apr 2014 doi 101021ct401002w
[26] X Lu M Gaus M Elstner and Q Cui ldquoParametrization of DFTB33OBfor Magnesium and Zinc for Chemical and Biological Applicationsrdquo The Journalof Physical Chemistry B vol 119 no 3 pp 1062ndash1082 jan 2015 doi101021jp506557r
[27] M Kubillus T Kubar M Gaus J Rezac and M Elstner ldquoParameterization ofthe DFTB3 Method for Br Ca Cl F I K and Na in Organic and BiologicalSystemsrdquo J Chem Theory Comput vol 11 no 1 pp 332ndash342 jan 2015 doi101021ct5009137
[28] J J P Stewart ldquoMOPAC2012rdquo 2016 Stewart Computational Chemistry ColoradoSprings CO USA [Online] Available httpopenmopacnet
[29] J L Pascual-Ahuir E Silla J Tomasi and R Bonaccorsi ldquoElectrostaticinteraction of a solute with a continuum improved description of the cavity and ofthe surface cavity bound charge distributionrdquo Journal of Computational Chemistryvol 8 no 6 pp 778ndash787 sep 1987 doi 101002jcc540080605
[30] P Su and H Li ldquoContinuous and smooth potential energy surface for conductorlikescreening solvation model using fixed points with variable areasrdquo The Journal ofChemical Physics vol 130 no 7 p 074109 feb 2009 doi 10106313077917
[31] J Tomasi B Mennucci and E Cances ldquoThe IEF version of the PCM solvationmethod an overview of a new method addressed to study molecular solutes at theQM ab initio levelrdquo Journal of Molecular Structure THEOCHEM vol 464 no1-3 pp 211ndash226 may 1999 doi 101016s0166-1280(98)00553-3
[32] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 1 the calculation of confidence intervalsrdquo J Comput Aided MolDes vol 28 no 9 pp 887ndash918 Sep 2014 doi 101007s10822-014-9753-z
[33] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 2 comparing methodsrdquo J Comput Aided Mol Des vol 30 no 2pp 103ndash126 Feb 2016 doi 101007s10822-016-9904-5
[34] J H Jensen ldquoWhich method is more accurate or errors have error barsrdquo PeerJPreprints vol 5 p e2693v1 2017 doi 107287peerjpreprints2693v1
[35] J H Jensen C J Swain and L Olsen ldquoPrediction of pKa values for druglikemolecules using semiempirical quantum chemical methodsrdquo The Journal of PhysicalChemistry A vol 121 no 3 pp 699ndash707 jan 2017 doi 101021acsjpca6b10990
[36] F Eckert and A Klamt ldquoAccurate prediction of basicity in aqueous solution withCOSMO-RSrdquo Journal of Computational Chemistry vol 27 no 1 pp 11ndash19 2005doi 101002jcc20309
[37] J A Nelder and R Mead ldquoA simplex method for function minimizationrdquo TheComputer Journal vol 7 no 4 pp 308ndash313 1965 doi 101093comjnl74308
19
[38] E Jones T Oliphant P Peterson et al ldquoSciPy Open source scientifictools for Pythonrdquo 2001 accessed November 24th 2017 [Online] Availablehttpwwwscipyorgrdquo
[39] J Charnley 2017 accessed November 24th 2017 [Online] Available httpsgithubcomcharnleyoptimize gamess parameters
[40] C P Kelly C J Cramer and D G Truhlar ldquoAqueous solvation free energies ofions and ion-water clusters based on an accurate value for the absolute aqueoussolvation free energy of the protonrdquo The Journal of Physical Chemistry B vol 110no 32 pp 16 066ndash16 081 aug 2006 doi 101021jp063552y
20
HFSMDAM1SMD
PM3SMDPM6SMD
DFTBSMD
AM1COSMO
PM3COSMO
PM6COSMO null0
2
4
6
8
10
12
14
16RM
SE [k
calm
ol]
PM3PM3HCO
PM3HCON
PM3HCONSPM3all PM6
PM6HCO
PM6HCON
PM6HCONS
PM6HCONShPM6all
DFTB
DFTBHCON0
2
4
6
8
10
12
14RM
SE [k
calm
ol]
HFSMD
PM3SMDPM6SMD
DFTBSMD
PM3COSMO
PM6COSMO0
2
4
6
8
10RM
SE [k
calm
ol]
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6err
or

Truhlar and Christopher Cramer for sharing the MNSOL data set and for insightfulcomments on the manuscript CS thanks the Danish Council for Independent Research(the Sapere Aude program) for financial support (Grant no DFF 4181-00370)
References[1] J Tomasi B Mennucci and R Cammi ldquoQuantum mechanical continuum solvation
modelsrdquo Chem Rev vol 105 pp 2999ndash3093 2005
[2] A Klamt and G Schuurmann ldquoCOSMO a new approach to dielectric screeningin solvents with explicit expressions for the screening energy and its gradientrdquo JChem Soc Perkin Trans vol 2 pp 799ndash805 1993
[3] C J Cramer and D G Truhlar ldquoA universal approach to solvation modelingrdquoAccounts of Chemical Research vol 41 no 6 pp 760ndash768 jun 2008 doi101021ar800019z
[4] V Barone M Cossi and J Tomasi ldquoA new definition of cavities for thecomputation of solvation free energies by the polarizable continuum modelrdquo JChem Phys vol 107 no 8 pp 3210ndash3221 aug 1997 doi 1010631474671
[5] F Eckert and A Klamt ldquoFast solvent screening via quantum chemistryCOSMO-RS approachrdquo AIChE J vol 48 no 2 pp 369ndash385 feb 2002 doi101002aic690480220
[6] C Curutchet A Bidon-Chanal I Soteras M Orozco and F J Luque ldquoMSTcontinuum study of the hydration free energies of monovalent ionic speciesrdquo JPhys Chem B vol 109 no 8 pp 3565ndash3574 mar 2005 doi 101021jp047197s
[7] Y Takano and K N Houk ldquoBenchmarking the conductor-like polarizablecontinuum model (CPCM) for aqueous solvation free energies of neutral and ionicorganic moleculesrdquo J Chem Theory Comput vol 1 no 1 pp 70ndash77 jan 2005doi 101021ct049977a
[8] A V Marenich R M Olson C P Kelly C J Cramer and D G TruhlarldquoSelf-consistent reaction field model for aqueous and nonaqueous solutions based onaccurate polarized partial chargesrdquo Journal of Chemical Theory and Computationvol 3 no 6 pp 2011ndash2033 nov 2007 doi 101021ct7001418
[9] A V Marenich C J Cramer and D G Truhlar ldquoUniversal solvation model basedon solute electron density and on a continuum model of the solvent defined by thebulk dielectric constant and atomic surface tensionsrdquo J Phys Chem B vol 113no 18 pp 6378ndash6396 may 2009 doi 101021jp810292n
[10] G E Chudinov D V Napolov and M V Basilevsky ldquoQuantum-chemicalcalculations of the hydration energies of organic cations and anions in the frameworkof a continuum solvent approximationrdquo Chem Phys vol 160 no 1 pp 41 ndash 541992 doi 1010160301-0104(92)87090-V
[11] M J Negre M Orozco and F J Luque ldquoA new strategy for the representation ofenvironment effects in semi-empirical calculations based on dewarrsquos hamiltoniansrdquoChem Phys Lett vol 196 no 1 pp 27ndash36 1992
17
[12] F J Luque M J Negre and M Orozco ldquoAn am1-scrf approach to the study ofchanges in molecular properties induced by solventrdquo J Phys Chem vol 97 no 17pp 4386ndash4391 1993
[13] M Caricato B Mennucci and J Tomasi ldquoSolvent effects on the electronic spectraAn extension of the polarizable continuum model to the zindo methodrdquo J PhysChem A vol 108 no 29 pp 6248ndash6256 2004
[14] M J S Dewar E G Zoebisch E F Healy and J J P Stewart ldquoDevelopmentand use of quantum mechanical molecular models 76 AM1 a new general purposequantum mechanical molecular modelrdquo J A Chem Soc vol 10 pp 3902ndash39091985 doi 101021ja00299a024
[15] J J P Stewart ldquoOptimization of Parameters for Semi-Empirical Methods I-Methodrdquo J Comp Chem vol 10 pp 209ndash220 1989 doi 101002jcc540100208
[16] J J P Stewart ldquoOptimization of parameters for semiempirical methods V Modi-fication of NDDO approximations and application to 70 elementsrdquo J Mol Modelvol 13 pp 1172ndash1213 2007
[17] M Gaus Q Cui and M Elstner ldquoDFTB3 Extension of the Self-Consistent-Charge Density-Functional Tight-Binding Method (SCC-DFTB)rdquo J Chem TheoryComput vol 7 no 4 pp 931ndash948 apr 2011 doi 101021ct100684s
[18] A V Marenich C P Kelly J D Thompson G D Hawkins C C ChambersD J Giesen P P Winget C J Cramer and D G Truhlar ldquoMinnesota solvationdatabaserdquo 2012
[19] ldquoMinnesota solvation databaserdquo accessed November 24th 2017 [Online] Availablehttpscompchemumnedumnsol
[20] M W Schmidt K K Baldridge J A Boatz S T Elbert M S Gordon J HJensen S Koseki N Matsunaga K A Nguyen S Su T L Windus M Dupuisand JAMontgomery ldquoGeneral atomic and molecular electronic structure systemrdquoJ Comput Chem vol 14 pp 1347ndash1363 1993
[21] V Barone and M Cossi ldquoQuantum calculation of molecular energies and energygradients in solution by a conductor solvent modelrdquo The Journal of PhysicalChemistry A vol 102 no 11 pp 1995ndash2001 mar 1998 doi 101021jp9716997
[22] M Cossi N Rega G Scalmani and V Barone ldquoEnergies structures andelectronic properties of molecules in solution with the c-PCM solvation modelrdquoJournal of Computational Chemistry vol 24 no 6 pp 669ndash681 apr 2003 doi101002jcc10189
[23] C Steinmann K L Blaeligdel A S Christensen and J H Jensen ldquoInterface ofthe polarizable continuum model of solvation with semi-empirical methods in thegamess programrdquo PLoS ONE vol 8 no 7 p e67725 07 2013 doi 101371jour-nalpone0067725
[24] Y Nishimoto ldquoDFTBPCM applied to ground and excited state potential energysurfacesrdquo J Phys Chem A vol 120 no 5 pp 771ndash784 feb 2016 doi101021acsjpca5b10732
18
[25] M Gaus X Lu M Elstner and Q Cui ldquoParameterization of DFTB33OB forSulfur and Phosphorus for Chemical and Biological Applicationsrdquo J Chem TheoryComput vol 10 no 4 pp 1518ndash1537 apr 2014 doi 101021ct401002w
[26] X Lu M Gaus M Elstner and Q Cui ldquoParametrization of DFTB33OBfor Magnesium and Zinc for Chemical and Biological Applicationsrdquo The Journalof Physical Chemistry B vol 119 no 3 pp 1062ndash1082 jan 2015 doi101021jp506557r
[27] M Kubillus T Kubar M Gaus J Rezac and M Elstner ldquoParameterization ofthe DFTB3 Method for Br Ca Cl F I K and Na in Organic and BiologicalSystemsrdquo J Chem Theory Comput vol 11 no 1 pp 332ndash342 jan 2015 doi101021ct5009137
[28] J J P Stewart ldquoMOPAC2012rdquo 2016 Stewart Computational Chemistry ColoradoSprings CO USA [Online] Available httpopenmopacnet
[29] J L Pascual-Ahuir E Silla J Tomasi and R Bonaccorsi ldquoElectrostaticinteraction of a solute with a continuum improved description of the cavity and ofthe surface cavity bound charge distributionrdquo Journal of Computational Chemistryvol 8 no 6 pp 778ndash787 sep 1987 doi 101002jcc540080605
[30] P Su and H Li ldquoContinuous and smooth potential energy surface for conductorlikescreening solvation model using fixed points with variable areasrdquo The Journal ofChemical Physics vol 130 no 7 p 074109 feb 2009 doi 10106313077917
[31] J Tomasi B Mennucci and E Cances ldquoThe IEF version of the PCM solvationmethod an overview of a new method addressed to study molecular solutes at theQM ab initio levelrdquo Journal of Molecular Structure THEOCHEM vol 464 no1-3 pp 211ndash226 may 1999 doi 101016s0166-1280(98)00553-3
[32] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 1 the calculation of confidence intervalsrdquo J Comput Aided MolDes vol 28 no 9 pp 887ndash918 Sep 2014 doi 101007s10822-014-9753-z
[33] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 2 comparing methodsrdquo J Comput Aided Mol Des vol 30 no 2pp 103ndash126 Feb 2016 doi 101007s10822-016-9904-5
[34] J H Jensen ldquoWhich method is more accurate or errors have error barsrdquo PeerJPreprints vol 5 p e2693v1 2017 doi 107287peerjpreprints2693v1
[35] J H Jensen C J Swain and L Olsen ldquoPrediction of pKa values for druglikemolecules using semiempirical quantum chemical methodsrdquo The Journal of PhysicalChemistry A vol 121 no 3 pp 699ndash707 jan 2017 doi 101021acsjpca6b10990
[36] F Eckert and A Klamt ldquoAccurate prediction of basicity in aqueous solution withCOSMO-RSrdquo Journal of Computational Chemistry vol 27 no 1 pp 11ndash19 2005doi 101002jcc20309
[37] J A Nelder and R Mead ldquoA simplex method for function minimizationrdquo TheComputer Journal vol 7 no 4 pp 308ndash313 1965 doi 101093comjnl74308
19
[38] E Jones T Oliphant P Peterson et al ldquoSciPy Open source scientifictools for Pythonrdquo 2001 accessed November 24th 2017 [Online] Availablehttpwwwscipyorgrdquo
[39] J Charnley 2017 accessed November 24th 2017 [Online] Available httpsgithubcomcharnleyoptimize gamess parameters
[40] C P Kelly C J Cramer and D G Truhlar ldquoAqueous solvation free energies ofions and ion-water clusters based on an accurate value for the absolute aqueoussolvation free energy of the protonrdquo The Journal of Physical Chemistry B vol 110no 32 pp 16 066ndash16 081 aug 2006 doi 101021jp063552y
20
HFSMDAM1SMD
PM3SMDPM6SMD
DFTBSMD
AM1COSMO
PM3COSMO
PM6COSMO null0
2
4
6
8
10
12
14
16RM
SE [k
calm
ol]
PM3PM3HCO
PM3HCON
PM3HCONSPM3all PM6
PM6HCO
PM6HCON
PM6HCONS
PM6HCONShPM6all
DFTB
DFTBHCON0
2
4
6
8
10
12
14RM
SE [k
calm
ol]
HFSMD
PM3SMDPM6SMD
DFTBSMD
PM3COSMO
PM6COSMO0
2
4
6
8
10RM
SE [k
calm
ol]
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6err
or

[12] F J Luque M J Negre and M Orozco ldquoAn am1-scrf approach to the study ofchanges in molecular properties induced by solventrdquo J Phys Chem vol 97 no 17pp 4386ndash4391 1993
[13] M Caricato B Mennucci and J Tomasi ldquoSolvent effects on the electronic spectraAn extension of the polarizable continuum model to the zindo methodrdquo J PhysChem A vol 108 no 29 pp 6248ndash6256 2004
[14] M J S Dewar E G Zoebisch E F Healy and J J P Stewart ldquoDevelopmentand use of quantum mechanical molecular models 76 AM1 a new general purposequantum mechanical molecular modelrdquo J A Chem Soc vol 10 pp 3902ndash39091985 doi 101021ja00299a024
[15] J J P Stewart ldquoOptimization of Parameters for Semi-Empirical Methods I-Methodrdquo J Comp Chem vol 10 pp 209ndash220 1989 doi 101002jcc540100208
[16] J J P Stewart ldquoOptimization of parameters for semiempirical methods V Modi-fication of NDDO approximations and application to 70 elementsrdquo J Mol Modelvol 13 pp 1172ndash1213 2007
[17] M Gaus Q Cui and M Elstner ldquoDFTB3 Extension of the Self-Consistent-Charge Density-Functional Tight-Binding Method (SCC-DFTB)rdquo J Chem TheoryComput vol 7 no 4 pp 931ndash948 apr 2011 doi 101021ct100684s
[18] A V Marenich C P Kelly J D Thompson G D Hawkins C C ChambersD J Giesen P P Winget C J Cramer and D G Truhlar ldquoMinnesota solvationdatabaserdquo 2012
[19] ldquoMinnesota solvation databaserdquo accessed November 24th 2017 [Online] Availablehttpscompchemumnedumnsol
[20] M W Schmidt K K Baldridge J A Boatz S T Elbert M S Gordon J HJensen S Koseki N Matsunaga K A Nguyen S Su T L Windus M Dupuisand JAMontgomery ldquoGeneral atomic and molecular electronic structure systemrdquoJ Comput Chem vol 14 pp 1347ndash1363 1993
[21] V Barone and M Cossi ldquoQuantum calculation of molecular energies and energygradients in solution by a conductor solvent modelrdquo The Journal of PhysicalChemistry A vol 102 no 11 pp 1995ndash2001 mar 1998 doi 101021jp9716997
[22] M Cossi N Rega G Scalmani and V Barone ldquoEnergies structures andelectronic properties of molecules in solution with the c-PCM solvation modelrdquoJournal of Computational Chemistry vol 24 no 6 pp 669ndash681 apr 2003 doi101002jcc10189
[23] C Steinmann K L Blaeligdel A S Christensen and J H Jensen ldquoInterface ofthe polarizable continuum model of solvation with semi-empirical methods in thegamess programrdquo PLoS ONE vol 8 no 7 p e67725 07 2013 doi 101371jour-nalpone0067725
[24] Y Nishimoto ldquoDFTBPCM applied to ground and excited state potential energysurfacesrdquo J Phys Chem A vol 120 no 5 pp 771ndash784 feb 2016 doi101021acsjpca5b10732
18
[25] M Gaus X Lu M Elstner and Q Cui ldquoParameterization of DFTB33OB forSulfur and Phosphorus for Chemical and Biological Applicationsrdquo J Chem TheoryComput vol 10 no 4 pp 1518ndash1537 apr 2014 doi 101021ct401002w
[26] X Lu M Gaus M Elstner and Q Cui ldquoParametrization of DFTB33OBfor Magnesium and Zinc for Chemical and Biological Applicationsrdquo The Journalof Physical Chemistry B vol 119 no 3 pp 1062ndash1082 jan 2015 doi101021jp506557r
[27] M Kubillus T Kubar M Gaus J Rezac and M Elstner ldquoParameterization ofthe DFTB3 Method for Br Ca Cl F I K and Na in Organic and BiologicalSystemsrdquo J Chem Theory Comput vol 11 no 1 pp 332ndash342 jan 2015 doi101021ct5009137
[28] J J P Stewart ldquoMOPAC2012rdquo 2016 Stewart Computational Chemistry ColoradoSprings CO USA [Online] Available httpopenmopacnet
[29] J L Pascual-Ahuir E Silla J Tomasi and R Bonaccorsi ldquoElectrostaticinteraction of a solute with a continuum improved description of the cavity and ofthe surface cavity bound charge distributionrdquo Journal of Computational Chemistryvol 8 no 6 pp 778ndash787 sep 1987 doi 101002jcc540080605
[30] P Su and H Li ldquoContinuous and smooth potential energy surface for conductorlikescreening solvation model using fixed points with variable areasrdquo The Journal ofChemical Physics vol 130 no 7 p 074109 feb 2009 doi 10106313077917
[31] J Tomasi B Mennucci and E Cances ldquoThe IEF version of the PCM solvationmethod an overview of a new method addressed to study molecular solutes at theQM ab initio levelrdquo Journal of Molecular Structure THEOCHEM vol 464 no1-3 pp 211ndash226 may 1999 doi 101016s0166-1280(98)00553-3
[32] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 1 the calculation of confidence intervalsrdquo J Comput Aided MolDes vol 28 no 9 pp 887ndash918 Sep 2014 doi 101007s10822-014-9753-z
[33] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 2 comparing methodsrdquo J Comput Aided Mol Des vol 30 no 2pp 103ndash126 Feb 2016 doi 101007s10822-016-9904-5
[34] J H Jensen ldquoWhich method is more accurate or errors have error barsrdquo PeerJPreprints vol 5 p e2693v1 2017 doi 107287peerjpreprints2693v1
[35] J H Jensen C J Swain and L Olsen ldquoPrediction of pKa values for druglikemolecules using semiempirical quantum chemical methodsrdquo The Journal of PhysicalChemistry A vol 121 no 3 pp 699ndash707 jan 2017 doi 101021acsjpca6b10990
[36] F Eckert and A Klamt ldquoAccurate prediction of basicity in aqueous solution withCOSMO-RSrdquo Journal of Computational Chemistry vol 27 no 1 pp 11ndash19 2005doi 101002jcc20309
[37] J A Nelder and R Mead ldquoA simplex method for function minimizationrdquo TheComputer Journal vol 7 no 4 pp 308ndash313 1965 doi 101093comjnl74308
19
[38] E Jones T Oliphant P Peterson et al ldquoSciPy Open source scientifictools for Pythonrdquo 2001 accessed November 24th 2017 [Online] Availablehttpwwwscipyorgrdquo
[39] J Charnley 2017 accessed November 24th 2017 [Online] Available httpsgithubcomcharnleyoptimize gamess parameters
[40] C P Kelly C J Cramer and D G Truhlar ldquoAqueous solvation free energies ofions and ion-water clusters based on an accurate value for the absolute aqueoussolvation free energy of the protonrdquo The Journal of Physical Chemistry B vol 110no 32 pp 16 066ndash16 081 aug 2006 doi 101021jp063552y
20
HFSMDAM1SMD
PM3SMDPM6SMD
DFTBSMD
AM1COSMO
PM3COSMO
PM6COSMO null0
2
4
6
8
10
12
14
16RM
SE [k
calm
ol]
PM3PM3HCO
PM3HCON
PM3HCONSPM3all PM6
PM6HCO
PM6HCON
PM6HCONS
PM6HCONShPM6all
DFTB
DFTBHCON0
2
4
6
8
10
12
14RM
SE [k
calm
ol]
HFSMD
PM3SMDPM6SMD
DFTBSMD
PM3COSMO
PM6COSMO0
2
4
6
8
10RM
SE [k
calm
ol]
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6err
or

[25] M Gaus X Lu M Elstner and Q Cui ldquoParameterization of DFTB33OB forSulfur and Phosphorus for Chemical and Biological Applicationsrdquo J Chem TheoryComput vol 10 no 4 pp 1518ndash1537 apr 2014 doi 101021ct401002w
[26] X Lu M Gaus M Elstner and Q Cui ldquoParametrization of DFTB33OBfor Magnesium and Zinc for Chemical and Biological Applicationsrdquo The Journalof Physical Chemistry B vol 119 no 3 pp 1062ndash1082 jan 2015 doi101021jp506557r
[27] M Kubillus T Kubar M Gaus J Rezac and M Elstner ldquoParameterization ofthe DFTB3 Method for Br Ca Cl F I K and Na in Organic and BiologicalSystemsrdquo J Chem Theory Comput vol 11 no 1 pp 332ndash342 jan 2015 doi101021ct5009137
[28] J J P Stewart ldquoMOPAC2012rdquo 2016 Stewart Computational Chemistry ColoradoSprings CO USA [Online] Available httpopenmopacnet
[29] J L Pascual-Ahuir E Silla J Tomasi and R Bonaccorsi ldquoElectrostaticinteraction of a solute with a continuum improved description of the cavity and ofthe surface cavity bound charge distributionrdquo Journal of Computational Chemistryvol 8 no 6 pp 778ndash787 sep 1987 doi 101002jcc540080605
[30] P Su and H Li ldquoContinuous and smooth potential energy surface for conductorlikescreening solvation model using fixed points with variable areasrdquo The Journal ofChemical Physics vol 130 no 7 p 074109 feb 2009 doi 10106313077917
[31] J Tomasi B Mennucci and E Cances ldquoThe IEF version of the PCM solvationmethod an overview of a new method addressed to study molecular solutes at theQM ab initio levelrdquo Journal of Molecular Structure THEOCHEM vol 464 no1-3 pp 211ndash226 may 1999 doi 101016s0166-1280(98)00553-3
[32] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 1 the calculation of confidence intervalsrdquo J Comput Aided MolDes vol 28 no 9 pp 887ndash918 Sep 2014 doi 101007s10822-014-9753-z
[33] A Nicholls ldquoConfidence limits error bars and method comparison in molecularmodeling Part 2 comparing methodsrdquo J Comput Aided Mol Des vol 30 no 2pp 103ndash126 Feb 2016 doi 101007s10822-016-9904-5
[34] J H Jensen ldquoWhich method is more accurate or errors have error barsrdquo PeerJPreprints vol 5 p e2693v1 2017 doi 107287peerjpreprints2693v1
[35] J H Jensen C J Swain and L Olsen ldquoPrediction of pKa values for druglikemolecules using semiempirical quantum chemical methodsrdquo The Journal of PhysicalChemistry A vol 121 no 3 pp 699ndash707 jan 2017 doi 101021acsjpca6b10990
[36] F Eckert and A Klamt ldquoAccurate prediction of basicity in aqueous solution withCOSMO-RSrdquo Journal of Computational Chemistry vol 27 no 1 pp 11ndash19 2005doi 101002jcc20309
[37] J A Nelder and R Mead ldquoA simplex method for function minimizationrdquo TheComputer Journal vol 7 no 4 pp 308ndash313 1965 doi 101093comjnl74308
19
[38] E Jones T Oliphant P Peterson et al ldquoSciPy Open source scientifictools for Pythonrdquo 2001 accessed November 24th 2017 [Online] Availablehttpwwwscipyorgrdquo
[39] J Charnley 2017 accessed November 24th 2017 [Online] Available httpsgithubcomcharnleyoptimize gamess parameters
[40] C P Kelly C J Cramer and D G Truhlar ldquoAqueous solvation free energies ofions and ion-water clusters based on an accurate value for the absolute aqueoussolvation free energy of the protonrdquo The Journal of Physical Chemistry B vol 110no 32 pp 16 066ndash16 081 aug 2006 doi 101021jp063552y
20
HFSMDAM1SMD
PM3SMDPM6SMD
DFTBSMD
AM1COSMO
PM3COSMO
PM6COSMO null0
2
4
6
8
10
12
14
16RM
SE [k
calm
ol]
PM3PM3HCO
PM3HCON
PM3HCONSPM3all PM6
PM6HCO
PM6HCON
PM6HCONS
PM6HCONShPM6all
DFTB
DFTBHCON0
2
4
6
8
10
12
14RM
SE [k
calm
ol]
HFSMD
PM3SMDPM6SMD
DFTBSMD
PM3COSMO
PM6COSMO0
2
4
6
8
10RM
SE [k
calm
ol]
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6err
or

[38] E Jones T Oliphant P Peterson et al ldquoSciPy Open source scientifictools for Pythonrdquo 2001 accessed November 24th 2017 [Online] Availablehttpwwwscipyorgrdquo
[39] J Charnley 2017 accessed November 24th 2017 [Online] Available httpsgithubcomcharnleyoptimize gamess parameters
[40] C P Kelly C J Cramer and D G Truhlar ldquoAqueous solvation free energies ofions and ion-water clusters based on an accurate value for the absolute aqueoussolvation free energy of the protonrdquo The Journal of Physical Chemistry B vol 110no 32 pp 16 066ndash16 081 aug 2006 doi 101021jp063552y
20
HFSMDAM1SMD
PM3SMDPM6SMD
DFTBSMD
AM1COSMO
PM3COSMO
PM6COSMO null0
2
4
6
8
10
12
14
16RM
SE [k
calm
ol]
PM3PM3HCO
PM3HCON
PM3HCONSPM3all PM6
PM6HCO
PM6HCON
PM6HCONS
PM6HCONShPM6all
DFTB
DFTBHCON0
2
4
6
8
10
12
14RM
SE [k
calm
ol]
HFSMD
PM3SMDPM6SMD
DFTBSMD
PM3COSMO
PM6COSMO0
2
4
6
8
10RM
SE [k
calm
ol]
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6err
or
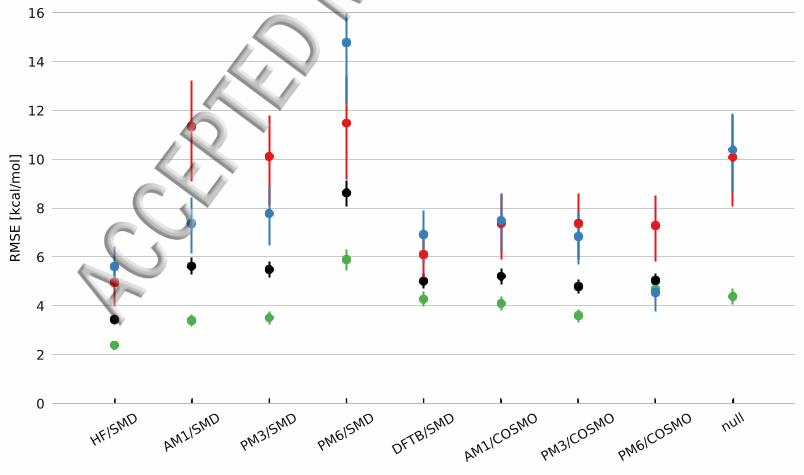
HFSMDAM1SMD
PM3SMDPM6SMD
DFTBSMD
AM1COSMO
PM3COSMO
PM6COSMO null0
2
4
6
8
10
12
14
16RM
SE [k
calm
ol]
PM3PM3HCO
PM3HCON
PM3HCONSPM3all PM6
PM6HCO
PM6HCON
PM6HCONS
PM6HCONShPM6all
DFTB
DFTBHCON0
2
4
6
8
10
12
14RM
SE [k
calm
ol]
HFSMD
PM3SMDPM6SMD
DFTBSMD
PM3COSMO
PM6COSMO0
2
4
6
8
10RM
SE [k
calm
ol]
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6err
or
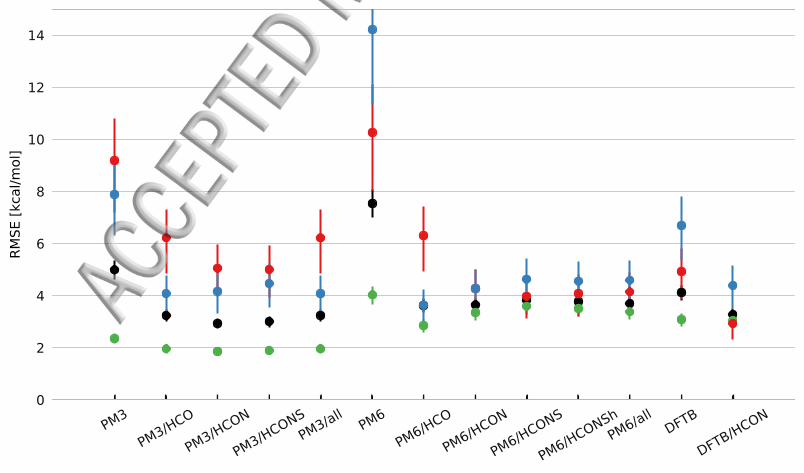
PM3PM3HCO
PM3HCON
PM3HCONSPM3all PM6
PM6HCO
PM6HCON
PM6HCONS
PM6HCONShPM6all
DFTB
DFTBHCON0
2
4
6
8
10
12
14RM
SE [k
calm
ol]
HFSMD
PM3SMDPM6SMD
DFTBSMD
PM3COSMO
PM6COSMO0
2
4
6
8
10RM
SE [k
calm
ol]
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6err
or
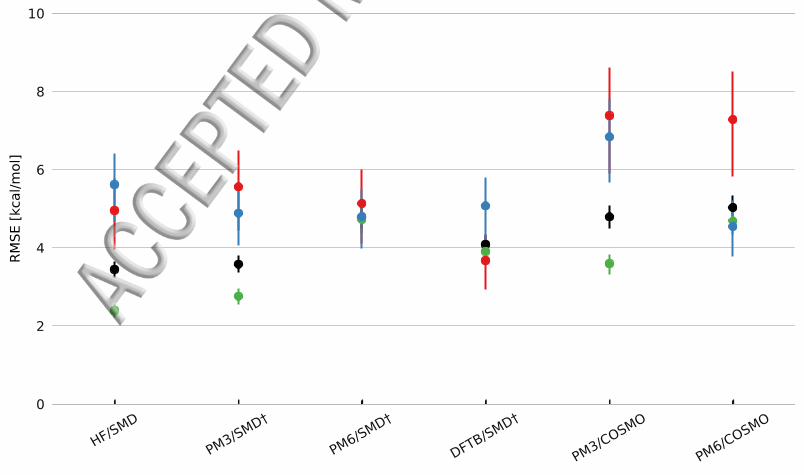
HFSMD
PM3SMDPM6SMD
DFTBSMD
PM3COSMO
PM6COSMO0
2
4
6
8
10RM
SE [k
calm
ol]
PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6err
or

PM3COSMO PM3SMD PM3SMD PM6COSMO PM6SMD DFTBSMD DFTBSMD
method
8
6
4
2
0
2
4
6err
or





























