Embed Size (px)
Citation preview
Research Collection
Doctoral Thesis
Ueber das Hederagenin
Author(s): Ehmann, Ludwig
Publication Date: 1936
Permanent Link: https://doi.org/10.3929/ethz-a-000095073
Rights / License: In Copyright - Non-Commercial Use Permitted
This page was generated automatically upon download from the ETH Zurich Research Collection. For moreinformation please consult the Terms of use.
ETH Library

über das Hederagenin
Von der
Eidgenössischen Technischen Hochschule
in Zürich
zur Erlangung der
Würde eines Doktors der technischen Wissenschaften
genehmigte
Promotionsarbeit
vorgelegt von
Ludwig Ehmann, dipl. Ingenieur-Chemiker
aus Ernetschwil (St. Gallen)
Referent: Herr Prof. Dr. L. Ruzicka
Korreferent: Herr Prof. Dr. H. E. Fierz
Weida i. Thür. 1936
Druck von Thomas & Hubert
Spezialdruckerei für Dissertationen

Leer - Vide - Empty

Meinen lieben Eltern
aus Dankbarkeit gewidmet.

Leer - Vide - Empty

Die vorliegende Arbeit wurde im Laboratorium für
organische Chemie an der Eidgenössischen Technischen
Hochschule in Zürich ausgeführt.Meinem hochverehrten Lehrer,
Herrn Prof. Dr. L Ruzicka,
möchte ich auch an dieser Stelle herzlich danken für die
Anregungen zu dieser Arbeit und für die wohlwollende
Unterstützung, die er ihr allzeit angedeihen ließ.

Leer - Vide - Empty

Theoretischer Teil.
Einleitung.
Über das Bauprinzip und Ausdehnung der Terpenchemie.
Die Natur produziert im tierischen und pflanzlichen Organismus
eine außerordentlich mannigfaltige Reihe von Substanzen, die man
vom Gesichtspunkt der chemischen Konstitution in der Gruppe
der Terpene zusammen zu fassen sucht.
Es sind dies Stoffe, die teils in flüssiger, teils in kristalliner Form
aus verschiedenen ätherischen ölen isoliert werden können. Sie
treten als wesentliche Bestandteile der Harze auf, sind als Farbstoffe
überall zu beobachten und haben in der Reihe der biologisch höchst
wichtigen Vitamine ebenfalls einige Vertreter.
Als erstes, sehr charakteristisches Merkmal des Molekülbaus der
Terpene erkannte man durch die Elementaranalyse das typische
Atomverhältnis von fünf Kohlenstoffatomen zu acht Wasserstoff¬
atomen, oder allgemeiner ausgedrückt die Bruttoformel
(C5Hx)yOz>
worin x = 6—9, y = 2—8 und z = 0—6 sein kann. Die Zahl der
Kohlenstoffatome ist somit immer durch fünf, diejenige der Wasser¬
stoffatome durch acht (oder eine davon nur wenig abweichende
Zahl) teilbar. Die Zahl der Sauerstoffatome ist sehr schwankend.
Der Index x gibt auch weiterhin Auskunft über den Sättigungs¬
zustand der Terpenkörper, bzw. über die Frage nach dem Kohlenstoff¬
gerüst, ob aliphatische, monozyklische, bi-, tri- usw. polyzyklische
Struktur vorhanden sei; Index y kennzeichnet die Molekulargröße:
Hemiterpene (y = i), Monoterpene C10, Sesqui-, Di-, Tri- und Poly-
terpene, während schließlich die Sauerstoffatome für die Terpene
weiter nicht charakteristisch sind und gänzlich fehlen können.
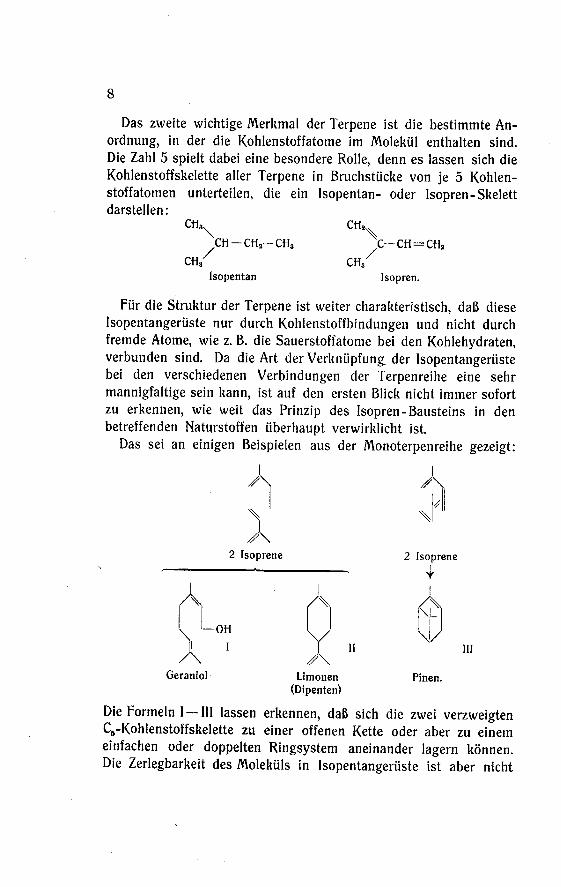
8
Das zweite wichtige Merkmal der Terpene ist die bestimmte An¬
ordnung, in der die Kohlenstoffatome im Molekül enthalten sind.
Die Zahl 5 spielt dabei eine besondere Rolle, denn es lassen sich die
KohlenstoffSkelette aller Terpene in Bruchstücke von je 5 Kohlen¬stoffatomen unterteilen, die ein Isopentan- oder Isopren-Skelettdarstellen:
Cfl,
CH,
CH —CHa—CHs
CHi
CH
C—CH=CH,
Isopentan Isopren.
Für die Struktur der Terpene ist weiter charakteristisch, daß diese
Isopentangerüste nur durch Kohlenstoffbindungen und nicht durch
fremde Atome, wie z. B. die Sauerstoffatome bei den Kohlehydraten,verbunden sind. Da die Art der Verknüpfung der Isopentangerüstebei den verschiedenen Verbindungen der Terpenreihe eine sehr
mannigfaltige sein kann, ist auf den ersten Blick nicht immer sofort
zu erkennen, wie weit das Prinzip des Isopren-Bausteins in den
betreffenden Naturstoffen überhaupt verwirklicht ist.
Das sei an einigen Beispielen aus der Monoterpenreihe gezeigt:
-/\
2 Isoprene
^
\
2 Isoprenei
-OH
I
Geraniol Limonen
(Dipenten)
\
/
Pinen.
III
Die Formeln I —III lassen erkennen, daß sich die zwei verzweigtenC6-Kohlenstoffskelette zu einer offenen Kette oder aber zu einem
einfachen oder doppelten Ringsystem aneinander lagern können.
Die Zerlegbarkeit des Moleküls in Isopentangerüste ist aber nicht

9
bloß eine schematische Betrachtungsweise. So wird bei der trockenen
Destillation von Kautschuk, sowie bei der thermischen Zersetzung
von Pinen Isopren erhalten, und umgekehrt läßt sich ein Aufbau
aus Isopren in manchen Fällen auch durchführen. Schon seit langem
war die Bildung des in der Natur vorkommenden Dipentens durch
Kondensation zweier Isoprene bekannt. Kürzlich gelang auch die Dar¬
stellung aliphatischer und zyklischer Terpenalkohole durch Polymeri¬
sation des Isoprens in Gegenwart eines wasseranlagernden Mittels.
Ferner stehen die drei oben angeführten Vertreter der Monoterpene
auch untereinander in genetischen Beziehungen. Sowohl aus dem
aliphatischen Alkohol I, wie aus der bizyklischen Verbindung III
kann durch Einwirkung starker Säuren ein monozyklischer Kohlen¬
wasserstoff II gewonnen werden. Diese Isomerisierungsreaktionen
sind für die ganze Körperklasse außerordentlich typisch, und es ist
naheliegend, daß auch in der Pflanze die gegenseitige Umwandlung
der Terpenderivate in Gegenwart der im Zellsaft gelösten Säuren
vor sich gehen kann.
Eine weitere Stütze für die Isoprenbausteintheorie bilden die
„Synthesen von Terpenen aus Isopren" vonTh. Wagner-Jauregg1,
wonach man beispielsweise die Formel des- Geraniols, durch
1,4-Addition von 2 Isoprenen und Absättigung der radikalartigen
Enden der auf diese Weise entstandenen Kohlenstoffskelette mit
den Resten des Wassers, rekonstruieren kann.
CH3 CH3 CH3
I I IC
/\CHa CH
I I> CHa CH2—OH
\CH
IIC
/\CH3 CH3
Das Prinzip der verzweigten C5-Kohlenstoffkette hat sich in der
Terpenchemie als sehr fruchtbar erwiesen. Die soeben angeführten
Formeln von Vertretern einfacher Terpene lassen schon die ungemein
C
CH2 CH11
C
/\CH2 CH
1 1
II
CH2 CH2
\CH
1
1 \
y CH2 CH2
\CH
111
C
/\CH2 CH3
II
c
/\CH8 CH2—
1 A. 496, 52 (1932).

10
große Mannigfaltigkeit der Terpenchemie erkennen, die durch die
unbegrenzte Variation des einfachen Bauprinzips bedingt werden.Die Natur macht aber von diesen Möglichkeiten nur geringen Ge¬
brauch. Die weitaus größte Zahl der Monoterpene und der Sesqui¬terpene enthält die Isoprenreste in Form einer regelmäßigen Kette,während nur in seltenen Fällen auch eine unregelmäßige Anein¬
anderreihung der Isoprenreste vorkommt.
Man versteht unter einer solchen regelmäßigen Kette die Ver¬
bindung der benachbarten Isoprenreste mit den entgegengesetztenEnden. Das hat zur Folge, daß sich in einer solchen, aus vielen
Isoprenresten aufgebauten, aliphatischen Verbindung, die als Seiten¬
ketten vorhandenen Methylgruppen in regelmäßigen Abständen folgen.Als einfache Beispiele seien die Alkohole Geraniol und Farnesol
sowie das Phytol angeführt, welches als Vertreter der höheren
Terpenreihe die natürliche Fortsetzung des Farnesols bildet.
Ctt2OHPhytol.
In der Reihe der höheren Terpene ist das Prinzip der regel¬mäßigen Isoprenkette nur noch teilweise verwirklicht. Dafür er¬
scheinen in manchen Fällen neue Ordnungsprinzipien, z. B. Symmetrie¬zentren. So ist der aliphatische Triterpenkohlenwasserstoff Squalenaus insgesamt 6 Isopentanresten aufgebaut, und zwar sind 3 Isopren¬reste in regelmäßiger Kette angeordnet und daran anschließend folgtin umgekehrter Reihenfolge wiederum eine Dreikette. Das Molekülist somit auf die Mitte symmetrisch gebaut:
/
//
/
A Squalen.
/

11
Die Konstitutionsaufklärung der Terpene
durch Abbau gelang nur in der Monoterpenreihe mit befriedigendem
Erfolg, während beispielsweise in der Reihe der Sesquiterpene die
Oxydation, mit Ausnahme weniger Fälle, zu nicht trennbaren
Gemischen führte oder überhaupt keine greifbaren höhermolekularen
Produkte als Essigsäure und Kohlendioxyd lieferte.
Nun ist aber, wie schon anfangs erwähnt, die Isomerisierungs-
reaktion für die ganze Körpergruppe der Terpene außerordentlich
typisch. Es gelingt, die Vertreter der einzelnen in der Natur vor¬
kommenden Terpengruppen ineinander umzuwandeln; es entstehen
im wesentlichen Ringschlüsse, zyklische Isomere, so daß es nahe
lag zu versuchen, daraus durch geeignete Dehydrierungsoperationen
die dazugehörigen aromatischen Kohlenwasserstoffe darzustellen.
„Diese sind fester gefügt als die hydrierten Abkömmlinge und
sollten dann eher charakteristische Abbauprodukte liefern und sich
auch leichter synthetisieren lassen. Die sekundäre Aufgabe, die
Ermittlung der Lage der Kohlenstoffdoppelbindungen, der quaternären,
bei der Dehydrierung abgespaltenen Methylgruppen und der sauer¬
stoffhaltigen Substituenten ließe sich dann anhand des bekannten
Ringsystems weit einfacher erledigen."1Dieses Arbeitsprogramm hat sich in der Folge für die Konstitutions¬
aufklärung der Terpene als außerordentlich fruchtbar erwiesen.
So ist für die Mehrzahl der Monoterpene das p-Cymol das
experimentell zugängliche Sinnbild des regelmäßigen Baues. Dieser
Benzolkohlenwasserstoff läßt sich auch tatsächlich aus allen be¬
kannten Monoterpenen dieses Typus herstellen.
i i
Monoterpen- Typus p-Cymol.
Bei der nächst höheren Terpengruppe, den Sesquiterpenen, wo
die Möglichkeit für Variationen natürlich größer ist, gelingt es alle
1 Aus L Ruzicka, Über Konstitution und Zusammenhänge in der Sesqui-
terpenreihe, S. 6—7.

12
Abkömmlinge auf zwei aromatische Grundkohlenwasserstoffe, dasCadalin und das Eudalin zurückzuführen. Man kann diese Kohlen¬wasserstoffe schematisch aus einer Drei-Isoprenkette, wie sie dieNatur im Farnesol bildet, durch Einfügung von Ringbindungen ab¬
leiten, und kommt im Falle des Cadalins zur folgenden Gleichung:
/\/\
/\
Cadalin.
Beachtet man ferner, da Eudalin nur 14 Kohlenstoffatome enthält,daß bei derDehydrierung eudalinliefernderSesquiterpen-Verbindungeneine quaternäre Methylgruppe abgespalten wird, so gelangt man
für die Bildung dieses Grundkohlenwasserstoffes zu folgender Um¬wandlungsreihe.
\_\ "T \/ /~\/V
Eudalin.
Die Diterpene zeichnen sich vor allen anderen Gruppen der
Terpene durch die größte Mannigfaltigkeit im Kohlenstoffgerüstaus. Dieses ist von kaum zehn in der Natur vorkommenden Di-
terpenen bekannt, und man hat dabei fünf verschiedene Bautypenangetroffen. Diese Typen haben alle wiederum das eine gemein¬same Merkmal der Aufteilbarkeit in Isoprenreste; die Art der Zu¬
sammensetzung zum Molekül der Diterpene ist jedoch in manchenEinzelheiten recht verschieden.
Der einfachste Fall einer regelmäßigen Kette von 4 Isoprenrestenist bisher im Phytol, der einen Alkoholkomponente des Chlorophylls,mit Sicherheit nachgewiesen worden.
Das Vitamin A enthält gegenüber dem Phytol ein neues, be¬merkenswertes Bauelement, einen Sechsring, wie er bisher bei denin der Natur vorkommenden Mono- und Sesquiterpenen noch nicht
angetroffen wurde, wohl aber in den künstlichen Veichenriechstoffen,den Jononen, enthalten ist.
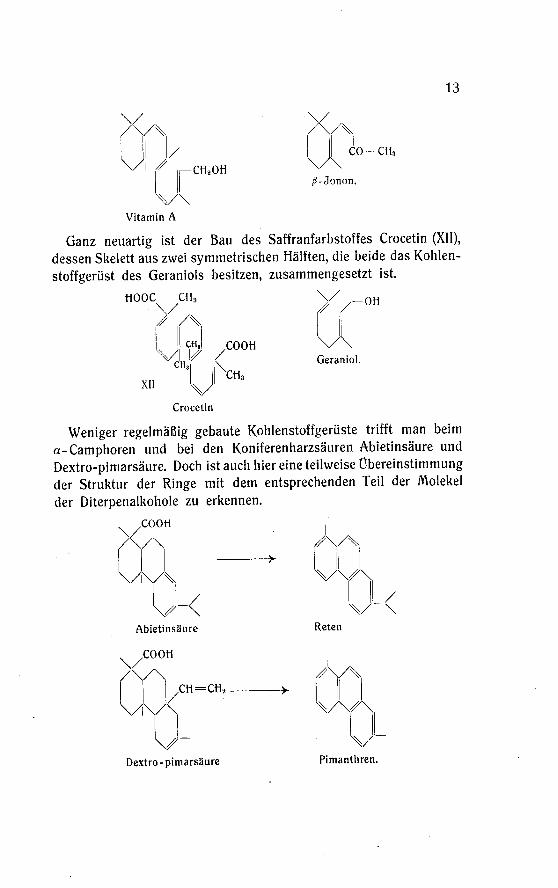
13
/ CH2OH
CO—CH3
i3-Jonon.
Vitamin A
Ganz neuartig ist der Bau des Saffranfarbstoffes Crocetin (XII),
dessen Skelett aus zwei symmetrischen Hälften, die beide das Kohlen¬
stoffgerüst des Geraniols besitzen, zusammengesetzt ist.
HOOC CH3\/
/—OH
\/CH,
XII
CH3
Crocetin
COOH
CH3
Geraniol.
Weniger regelmäßig gebaute Kohlenstoffgerüste trifft man beim
a-Camphoren und bei den Koniferenharzsäuren Abietinsäure und
Dextro-pimarsäure. Doch ist auch hier eine teilweise Übereinstimmung
der Struktur der Ringe mit dem entsprechenden Teil der Molekel
der Diterpenalkohole zu erkennen.
,coon
/\\/\
Abietinsäure
COOH
\/v\
I IL/V \
Reten
/\/\£H=CH2
\/\/\
\Y~
Dextro - pimarsäure Pimanthren.

14
Es gibt in der Diterpenreihe keine wichtigen aromatischen Grund¬
kohlenwasserstoffe, die wie das p-Cymol, das Cadalin und das
Eudalin einer größeren Reihe derselben zugrunde liegen würden.
Als gemeinsames Merkmal der Diterpene ist wohl der Phenanthren-
grundriß zu betrachten, wie er in den trizyklischen Vertretern dieser
Reihe verwirklicht ist, und in welcher Schreibweise auch die übrigenDiterpene bildlich dargestellt werden können.
Polyterpene mit 25 Kohlenstoffatomen sind bisher in der
Natur noch nicht aufgefunden worden. Es wird dies wohl kein
Zufall sein, sondern mit dem Bildungsmechanismus dieser Ver¬
bindungen in der Natur zusammenhängen.
Weit verbreitet sind dagegen in der Natur die Triterpene mit
30 Kohlenstoffatomen. Zu dieser Gruppe gehören die in den
meisten Milchsaft führenden Pflanzen enthaltenen Amyrine, das
Lupeol usw., sowie die zahlreichen Sapogenine. Die Kenntnis der
Struktur dieser Verbindungen ist mit Ausnahme des aliphatischenTriterpens Squalen sehr lückenhaft geblieben. Sie besitzen alle
ein ungefähr gleiches Verhältnis von Kohlenstoff zu Wasserstoff,die eine für ein tetra- oder pentazyklisches Triterpen mögliche Formel
verlangen würde. Es finden sich in der Literatur auch verschiedene
Meinungsäußerungen, daß diese Stoffe mit den Terpenen insbesondere
mit den Sesquiterpenen zusammenhängen sollen, oder vielleicht mit
den Sterinen oder Gallensäuren in verwandtschaftlichen Beziehungenstehen.
Nachdem sich nun die Dehydrierungsmethode bei der Konstitutions¬
aufklärung der Sesquiterpene als überaus wertvoll erwiesen hatte,versuchte man diese Methode auch zur Aufklärung des Kohlenstoff¬
gerüstes der Triterpene heran zu ziehen.
Als wichtigstes Dehydrierungsprodukte erhielt man bei allen diesen
Naturkörpern Trimethyl-naphthaline, und zwar 1,2,5- und 1,2,7-Tri-
methyl-naphthalin. Ferner gelang auch bei einigen pentazyklischenTypen die Isolierung eines Kohlenwasserstoffes von der Brutto¬
formel C25H20, dem die Grundstruktur eines Picens zuerteilt
wurde.
Die zwischen Di- und Triterpenen stehende, vorläufig noch nicht
aufgefundene Terpengruppe mit 25 Kohlenstoffatomen, sollte demnach
von einem Chrysengrundriß abgeleitet werden können.
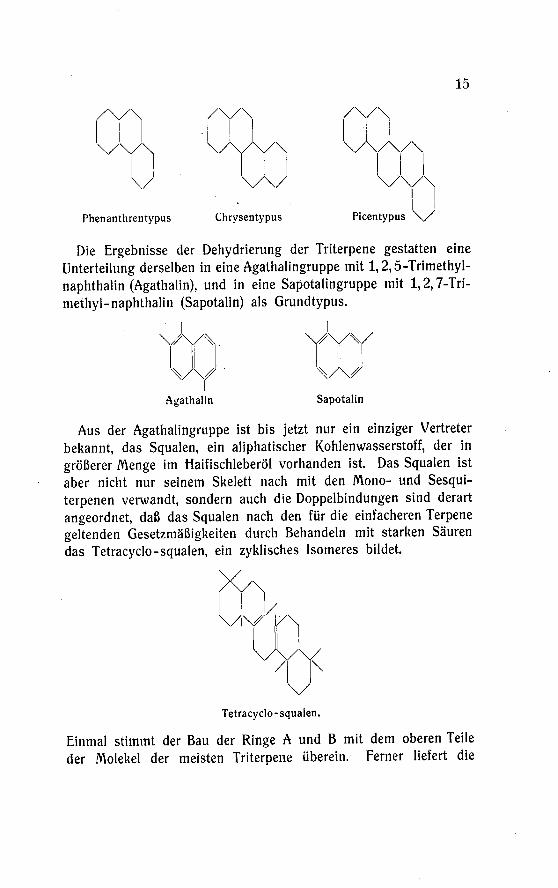
15
Phenanthrentypus Chrysentypus Picentypus \S
Die Ergebnisse der Dehydrierung der Triterpene gestatten eine
Unterteilung derselben in eine Agathalingruppe mit 1,2,5-Trimethyl-
naphthalin (Agathalin), und in eine Sapotalingruppe mit 1,2,7-Tri-
methyl-naphthalin (Sapotalin) als Grundtypus.
\J\/\
Agathalin
\^\/\y
Sapotalin
Aus der Agathalingruppe ist bis jetzt nur ein einziger Vertreter
bekannt, das Squalen, ein aliphatischer Kohlenwasserstoff, der in
größerer Menge im Haifischleberöl vorhanden ist. Das Squalen ist
aber nicht nur seinem Skelett nach mit den Mono- und Sesqui-
terpenen verwandt, sondern auch die Doppelbindungen sind derart
angeordnet, daß das Squalen nach den für die einfacheren Terpene
geltenden Gesetzmäßigkeiten durch Behandeln mit starken Säuren
das Tetracyclo-squalen, ein zyklisches Isomeres bildet.
Tetracyclo-squalen.
Einmal stimmt der Bau der Ringe A und B mit dem oberen Teile
der Molekel der meisten Triterpene überein. Ferner liefert die

16
Dehydrierung des Tetracyclo-squalens unter Sprengung des Mole¬
küls genau in der Mitte das 1,2,5-Trimethyl-naphthalin. Dieser
Kohlenwasserstoff wurde auch bei der Dehydrierung des Vitamins A
mit Selen erhalten1.
Höhermolekulare Dehydrierungsprodukte, wie methylierte Di-
naphthyläthane oder gar Picen, deren Bildung ja durchaus möglichwären und das ganze Grundskelett des Tetracyclo-squalens wider¬
spiegeln würden, konnten bisher nicht gefaßt werden.
Bei allen anderen nach der Selenmethode dehydrierten Triterpenenwurde dagegen das 1,2,7-Trimethyl-naphthalin (Sapotalin) erhalten,und neben verschiedenen anderen Produkten konnte in den meisten
Fällen, wie schon erwähnt, auch noch ein Kohlenwasserstoff isoliert
werden, der die Eigenschaften eines Trimethyl-picens aufweist.
Auffallend ist, daß fast alle mit Selen dehydrierten Triterpene in
der Hauptsache das gleiche Resultat ergeben haben, also wohl das
gleiche oder doch nur wenig voneinander abweichende Kohlenstoff¬
gerüste aufweisen werden. Diese Tatsache ist bemerkenswert, da eine
so große Mannigfaltigkeit in den Kohlenstoffgerüsten der Diterpen-reihe festgestellt worden ist.
In der Gruppe der Triterpene werden eine große Reihe von Sub¬
stanzen zusammengefaßt, die, wie schon erwähnt, auf Grund der Er¬
gebnisse der Selendehydrierung im wesentlichen denselben chemischen
Grundriß aufweisen müssen. Es sind dies Naturkörper, die mit
Ausnahme des im Fischleberöl vorkommenden aliphatischen Kohlen¬wasserstoffes Squalen ausschließlich im Pflanzenreich aufgefundenwerden. Sie lassen sich weiterhin nach der Art ihres Vorkommens
in der Natur unterscheiden in solche, die frei in der Pflanze auf¬
treten und andere, die an Zucker als Glukoside gebunden sind.
Zur ersten Gruppe gehören einmal die Triterpenalkohole Betulin,aus der Birkenrinde, die in den meisten Milchsaft führenden Pflanzen
enthaltenen Amyrine und Lupeol, ferner die Reihe der Harzsäuren,wie die Boswellinsäure aus Weihrauchharz, die Elemisäuren aus
Manila-Elemiharz, die Sia- und Suma-resinolsäuren aus Siam- bzw.
Sumatrabenzoe und schließlich die weit verbreitete Ursolsäure.
Die erwähnten Glukoside sind schon lange unter dem Namen
Saponine bekannt, eine Bezeichnung, die ihnen auf Grund ihrer
1In diesem Zusammenhang sei noch erwähnt, daß auch Agathendisäure, ein
Vertreter der Diterpenreihe, neben Pimanthren, 1, 2, 5-Trimethyl-naphthalin liefert.

17
charakteristischen Fähigkeit, mit Wasser ähnlich den Seifen einen
dauerhaften Schaum zu liefern, gegeben wurde. Sie vermögen noch
in großer Verdünnung Blutkörperchen aufzulösen. Diese besonders
hervortretende Hämolysewirkung wird durch Cholesterin und andere
Sterine durch Komplexbildung wieder aufgehoben. Parallel zur
Intensität der Hämolyse der verschiedenen Saponine läuft die Reiz¬
wirkung auf Auge und Geschmacksempfindung, sowie die toxische
Wirkung auf Fische.
Saponine sind in über 60 Pflanzenfamilien nachgewiesen worden.
Doch ist der Gehalt an diesen Stoffen in den Pflanzen sehr ver¬
schieden. Manche Vertreter enthalten in fast allen ihren Teilen
Saponine, während bei anderen das Vorkommen im wesentlichen auf
Blätter, Stengel, Früchte oder Wurzeln beschränkt ist. Bei der
hydrolytischen Spaltung der Saponine werden neben verschiedenen
Zuckerarten, wie Galactose, d-Glucose, 1-Arabinose, Rhamnose usw.,
die zuckerfreien Aglukone, die Sapogenine, erhalten, die auch in freiem
Zustand in verschiedenen Pflanzen aufgefunden werden. Die Ein¬
gangs erwähnte Unterteilung in frei vorkommende und glukosidisch
gebundene Triterpene ist somit nur rein schematisch haltbar, wie
auch eine große Zahl verschiedener Saponine auf ein und dasselbe
Aglukon zurückgeführt werden können.
Folgende Saponine bzw. Sapogenine, deren Aglukone bei der
Dehydrierung mit Selen Sapotalin liefern, sind bis jetzt bekannt
geworden :
Ascin bzw. Ascigenin aus der Roßkastanie Aesculuc hippocastanum.
Camelia-saponin bzw. -sapogenin aus Camelia japonica.
Cyclatnin bzw. Cyclamiretin aus dem Alpenveilchen Cyclamen
europaeum.
Glycyrrhizin bzw. Glycyrrhetin aus der Süßholzwurzel Glycyrrhiza
glabra.
Gypsophila-saponin (Albsaponin, Saponalbin) bzw. Gypsogenin, das
in der Seifenwurzel Gypsophila arrostii und panniculata vor¬
kommt.
Quillaja-saponin bzw. Quillajasäure aus Quillaja saponaria.
Oleanolsäure, die zu den verbreitesten Sapogeninen zählt und in
zahlreichen Pflanzen als Glukosid wie auch in freier Form
angetroffen wird; so in Olivenblättern, in der Zuckerrübe als
Ehmann. 2

18
Rübensaponin, in Gewürznelken, in Viskumarten als Viskutn-
säure, in der Guajakrinde, in Panax repens (Panax-sapogenin)und in Aralia-Arten vergesellschaftet mit Hederagenin.
Hederin (Aralin, Sapindus- oder Kalosaponin), dessen Vorkommen
für die Gruppe der Araliaceen wie Efeu fiedera helix, Aralia
japonica, Kalopanax ricinifolius und Sapindusarten wie Seifen¬
nuß Sapindus mukerossi charakteristisch ist, liefert bei der
sauren Hydrolyse Hederagenin (Sapindus- oder Kalosaponin).
Terpene mit 35 Kohlenstoffatomen sind in der Natur nicht
aufgefunden worden, dagegen einige wohl charakterisierte Vertreter
der nächst höheren und gleichzeitig höchsten bisher bekannt ge¬
wordenen Gruppe von Terpenen mit sicher ermittelter Molekular¬
größe. Die wichtigsten natürlichen Polyenfarbstoffe, das Carotin,das Xanthophyll und Lutein, die im Pflanzenreich so verbreiteten
Verbindungen mit 40 Kohlenstoffatomen im Molekül, ferner
der rote Farbstoff der Tomate, das dem Carotin isomere Lycopinund verschiedene andere Pflanzenpigmente gehören in diese Reihe.
/ /X
\//\
\/l /
Lycopin /S-Carotin.
Die Farbe dieser Verbindungen ist auf die lange Reihe kon¬
jugierter Doppelbindungen zurückzuführen. Typisch ist sowohl beim
Carotin wie beim Lycopin die Zusammensetzung aus zwei sym¬
metrischen Hälften, analog dem Squalen. Was den äußersten Teil
des Moleküls anbetrifft, so stehen Carotin und Lycopin zueinander
im gleichen Verhältnis wie Geraniol zu ß-Jonon, oder wie Phytolzu Vitamin A. Die aus Analogiegründen mit den einfacheren Terpen-verbindungen denkbare Umwandlung von Lycopin in Carotin oder
in ein noch stärker zyklisiertes Gebilde ist bisher, bei der großenEmpfindlichkeit dieser Körper gegenüber Säuren, nicht durchführbar.

19
Der Pflanze aber, die unter wesentlich milderen Bedingungen arbeitet,
wird dies durchaus möglich sein.
Den Abschluß der Reihe derTerpene bildet als höchstes bekanntes
Glied der Kautschuk, der ebenfalls aus einer regelmäßigen Isopren¬
kette besteht, dessen Molekülgröße bis jetzt aber nur annähernd
bekannt ist, sicher jedoch sehr groß sein muß.
....(-C=C-C=C-)X....
Untersuchungen über das Hederagenin.
Vorkommen und Eigenschaften des tiederagenins.
Hederagenin wurde erstmals von van der Haar1 aus dem, in
Efeublättern (Hedera helix) enthaltenen Saponin, Hederin rein dar¬
gestellt. Das Glukosid spaltet bei saurer Hydrolyse je ein Mol
Arabinose und Rhamnose ab und liefert das kristalline, bei 331°
schmelzende Hederagenin. Später wurde von W.A.Jacobs2 die
Identität des Hederagenins mit dem aus Seifennüssen (Sapindus
mukerossi) gewonnenen Sapindus-sapogenin festgestellt. Die Seifen¬
nüsse bilden bis heute wohl die leichtest zugängliche Quelle zur
Darstellung größerer Mengen von Hederagenin. Desgleichen stimmt
auch das Kalo-sapogenin aus Kalopanax ricinifolius3 in allen
Eigenschaften mit Hederagenin überein. Wahrscheinlich ist Hedera¬
genin in der Natur viel weiter verbreitet als bis anhin festgestellt
wurde.
Hederagenin ist eine Dioxy - monocarbonsäure. Die Carboxyl-
gruppe läßt sich in alkoholischer Lösung glatt titrieren. Die Alkali¬
salze des Hederagenins sind in Wasser nicht löslich, was die Un¬
löslichkeit des Sapogenins trotz der Carboxylgruppe erklärt. Die
Veresterung der Carboxylgruppe bietet keine Schwierigkeiten, man
erhält beispielsweise:
Hederagenin-methylester Fp. 238° und
Hederagenin-äthylester Fp. 190°.
1 Arch. Pharm. 250, 424 (1912); 251, 632 (1913).* 3. Biol. Chem. 63, 621 (1925).3 M. K o t a k e und K. T a g u c h i, Scient. Pap. Inst. Physical Chem. Res. 18,5 (1932);
Proc. Imp. Acad. Tokyo 8, 12 (1932).
2*

20
Die beiden Hydroxyle sind alkoholischer Natur und liefern mit
Acetanhydrid
fiederagenin-diacetat Fp. 176°.
Als weitere funktionelle Derivate seien erwähnt:
fiederagenin-methylester-diacetat . Fp. 190° und
Hederagenin-äthylester-diacetat . . Fp. 158°.
Herderagenin ist optisch aktiv und besitzt nach A. Winterstein
und Stein1 ein dg° = + 71,4° (in Chloroform-Methylalkohol).Auf Grund des Verlaufs der bekannten Sterin-Farbreaktionen (nach
Salkovski, Liebermann-Burchard usw.), sowie des Verhaltens
bei der Zinkstaubdestillation im Wasserstoffstrom, wurde das
Hederagenin schon von van der Haar2 den pflanzlichen Sterinen
zugeordnet, bzw. in naher Verwandtschaft mit jener Körperklassevermutet.
Die Bruttoformel des Hederagenins.
E. Winterstein und H. Blau8 stellten seinerzeit für das aus
Sapindus Mukorossi dargestellte Sapogenin die Formel C18H2803 auf.
Auf Grund von Brombestimmungen an einem Monobromderivat des
Sapogenins aus Sapindus Mukorossi gelangten A. Winterstein
und 3. Meyer* zu einem wesentlich höheren Molekulargewicht (480)als die erstere Formel verlangen würde. Sie postulierten die
Formel C81H5004 in Übereinstimmung mit den Untersuchungen von
W. A. Jacobs5 über das Sapogenin aus Sapindus saponaria L.
Dieselbe Bruttoformel wurde schon vor längerer Zeit von A.W. van
der Haar6, sowie von van der Haar und Tamburello' für das
aus Efeu (Hedera helix) dargestellte Hederagenin vorgeschlagen.Hederagenin wäre somit im Gegensatz zu anderen Vertretern der
Triterpenreihe, bei denen 30 C-Atome die Regel darstellen, die
einzige Verbindung mit 31 C-Atomen. Es ist natürlich außer¬
ordentlich schwierig durch Elementaranalyse eine zuverlässige Ent-
1 H. 211, 9 (1932).3 B. 55, 1054, 3041 (1922).3 Z. physiol. Chem. 75, 410 (1911).4 Z. physiol. Chem. 199, 37 (1931).6 3. biol. Chem. 63, 621 (1925).6 Arch. Pharm. 250, 430 (1912).7
B. 54, 3151 (1921).

21
Scheidung zu treffen, da die für die in Betracht kommenden Formeln
berechneten Werte für Kohlenstoff und Wasserstoff meist innerhalb
der Fehlergrenze der Analysenmethode liegen. So beträgt der
Unterschied der berechneten C-Werte von C80 auf C3l 0,27 und für
Wasserstoff 0,12%. Dagegen sind die Molekulargewichte um
14,0 Einheiten verschieden, so daß die Titration einen zuverlässigen
Entscheid zwischen den homologen Formeln erlauben sollte.
Die von M. Furter ausgeführten Analysen ergaben Werte, die
innerhalb folgender Grenzen schwankten:
C 76,43-76,44, H 10,30-10,46 °/0 und
Molekulargewicht: 479,8-485,5.
CaoHisOi. Ber.: C 76,21; H 10,24°/0; Molekulargewicht: 472,4.
C81H60O4. Ber.: C 76,48; H 10,36°/0; Molekulargewicht: 486,4.
Nach neueren Untersuchungen von A.Winterstein und G.Stein1
dagegen ist Hederagenin als eine Dioxy - triterpensäure C30H4804
anzusprechen. Die Verfasser stützen diese um ein Kohlenstoff
ärmere Formel einmal durch Elementaranalyse und Äquivalent¬
gewichtsbestimmungen, und ferner durch die auffällige Tatsache,
daß Oleanolsäure, welche eindeutig 30 Kohlenstoffatome besitzt, mit
Hederagenin in Aralia japonica vergesellschaftet aufgefunden wurde.
Dieses Ergebnis ist um so mehr zu begrüßen, als damit Hederagenin
seine Ausnahmestellung verlieren würde und sich zwangslos in die
Reihe der übrigen bekannten Triterpene einfügen läßt.
Die Anzahl der Doppelbindungen.
Nachdem nun die Bruttoformel des Hederagenins, ob 30 oder
31 Kohlenstoffatome, noch nicht eindeutig entschieden ist, erhebt
sich neben der Frage nach dem Kohlenstoffgerüst in erster Linie
die nach der Zahl der im Molekül anwesenden Kohlenstoffringe
bzw. Doppelbindungen. Die Prüfung des Sättigungszustandes kann
nach verschiedenen Methoden erfolgen, einmal durch katalytische
Hydrierung, durch die Ozonidbildung, durch Einwirkung von Benzo¬
persäure, durch Bestimmung der Molekular-Refraktion, durch die
Gelbfärbung mittels Tetranitromethan und durch Darstellung der
1 Z. physiol. Chem. 211, 8 (1932).

22
Bromlaktone nach Winterstein. Dabei ist aber zu beachten, daß
das Versagen der einen Reaktion kein sicherer Beweis für das
Fehlen von Doppelbindungen ist.
So ist im Fall des Amyrins auch unter ganz energischen Be¬
dingungen keine Hydrierung zu erzwingen, obgleich beim Behandeln
mit Ozon Ozonide erhalten werden, mit Benzopersäure Sauerstoff
verbraucht und mit Tetranitromethan eine deutliche Gelbfärbungbeobachtet wird1.
Nach Untersuchungen von A. W. van der Haar2, W.A. Jacobs3
und A. Winterstein und W. Wiegand4 scheint fiederagenin gegen¬über katalytisch erregtem Wasserstoff indifferent zu sein. Es verhält
sich in dieser Hinsicht analog wie das eben erwähnte Amyrin, die
Siaresinolsäure, die Sumaresinolsäure und die Oleanolsäure.
Aus Hederagenin, sowie aus den soeben angeführten Triterpenenerhält man durch Einwirkung von Ozon Ozonide, deren Analyseauf eine Aufnahme von 3—5 Mol Ozon hinweist, die aber durch¬
wegs halogenhaltig sind, so daß der diagnostische Wert dieser
Methode ein wenig zweifelhaft erscheint. Ferner gestattet der Sauer¬
stoffgehalt der Ozoneinwirkungsprodukte keine Rückschlüsse auf
die Zahl der Kohlenstoffdoppelbindungen. Man kann beispielsweisean das einfach ungesättigte Cholesterin nach Fürth und Felsen¬
reich8 bis 4 Mol Ozon anlagern und die zweifach ungesättigtenHarzsäuren Abietinsäure6und Dextro-primarsäure7 liefern Additions¬
produkte von 3 Mol Ozon.
Hederagenin selbst war infolge seiner Schwerlöslichkeit in ozon¬
beständigen Lösungsmitteln, wie Chloroform, Tetrachlorkohlenstoff,Äthylbromid, Essigester, Eisessig und dergleichen mehr, für diese
Versuche ungeeignet.Durch andauernde Einwirkung eines ozonhaltigen Sauerstoffstromes
auf Hederagenin-methylester in Chloroform oder Äthylbromid erhielt
man ein flockiges Ozonid, das auch nach monatelangem Trocknen
1Ruzicka und Mitarbeiter, A. 471, 21 (1929).
2Arch. Pharm. 252, 421 (1914); R. 44, 770 (1925).
3 3. of biol. Chem. 88, 153 (1930).4
Z. physiol. Chem. 199, 47 (1931).5 Bio. Ch. Z. 69, 416 (1915).6 Helv. 6, 685 (1923).' Helv. 5, 331 (1922).

23
im Hochvakuum nicht halogenfrei zu erhalten war. Die Analysen
zeigten einen Chlorgehalt von 17—18%-
Dieselben Erscheinungen waren auch bei der Ozonisation der
Siaresinolsäure1, des Betulins3 und des Amyrins3 zutage getreten,
wo der Chlorgehalt zwischen 12 und 14% schwankte. Ob dieser
Halogengehalt nur auf absorbiertes Lösungsmittel4 oder ob vielleicht
das Lösungsmittel selbst durch Einwirkung von Ozon Halogen ab¬
spaltet, und dieses dann ebenfalls in Reaktion tritt, ist zur Zeit
noch nicht aufgeklärt. Es scheint überhaupt, daß die Ozonide
zwecks Erfassung der Zahl der Doppelbindungen bei den Triterpenen
nicht in Frage kommen.
Als weiteres Kriterium, für das Vorhandensein einer aktiven
Doppelbindung bei den Triterpenen, ist die Titration mit Benzoper¬
säure herangezogen worden. Es ist aber wie bei den Ozoniden
zu beachten, daß die Zahl der aufgenommenen Sauerstoffatome
keineswegs der Zahl der Kohlenstoffdoppelbindungen äquivalent zu
sein braucht. So wurde von Ruzicka und Frank5 beim Dihydro-
dextro-primarsäure-rnethylester mit überschüssiger Benzopersäure
die Sauerstoffzahl 2 statt 1, wie zu erwarten war, festgestellt. Sie
konnten das zugehörige Dioxyd nicht isolieren, erhielten aber eine
isomere Dextro-pimarsäure. Offenbar ist dies der dehydrierenden
Wirkung der Benzopersäure zuzuschreiben, da Wind au s und
Lüttringhaus6 beim Ergostenol-acetat und dem „Dihydro-
ergosterin I" ebenfalls die entsprechenden Dehydroprodukte aus
dem Reaktionsgemisch isolieren konnten. In analoger Weise ließ
sich beim a-Amyrilen, das auch 2 Sauerstoffatome aufnahm,
nur ein Monoxyd isolieren'. Diese eigenartigen Resultate ver¬
mindern die Aussichten, in der Benzopersäure über ein allgemein
und gleichmäßig anwendbares Reagens in der Polyterpenreihe zu
verfügen. Es ist aus diesen Gründen ratsam, bei einem Verbrauch
1R. Egli, Ober die Siaresinolsäure, Diss. E. T. H. 1932, S. 20.
2 H. Brüngger, Über das Betulin, Diss. E. T. H, 1932, S. 18.
3Ruzicka, Huyser, Pfeiffer und Seidel, A. 471, 23 (1929).
4Beim Betulin-ozonid gelang es, den Halogengehalt von 13,11 °/0 Chlor durch
intensives Trocknen im Hochvakuum auf 1,69 °/0 herunter zu drücken.
5 Helv. 15, 1294 (1932).6 A. 481, 119 (1930).1 Ruzicka, Silbermann und Pieth, Helv. 15, 1285 (1932).

24
von über einem Sauerstoffatom, die Versuche nur noch qualitativzu deuten.
Es gelang A. Winterstein und G.Stein1 nach 200stündigerEinwirkungsdauer von Benzopersäure die Aufnahme von 1 Atom
Sauerstoff zu erzwingen. Wesentlich leichter dagegen verlief die
Benzopersäuretitration beim Diacetylhederabetulin2, welches schon
nach 15 Stunden die für eine Doppelbindung berechnete MengeSauerstoff aufnahm. Die Doppelbindung im Hederagenin wurde
somit durch die Kohlensäure-Abspaltung wesentlich reaktionsfähiger,was auch in der relativ leichten Hydrierbarkeit des Hederabetulins
deutlich zum Ausdruck kam.
Zur weiteren Aufklärung des Sättigungzustandes, beziehungsweiseder Ringzahl des Hederagenins, wurde auch die Ermittlung der
Molekular-Refraktion herangezogen. Man kennt in der Literatur
zahlreiche Verbindungen, deren Kohlenstoffdoppelbindungen gegen
katalytisch erregten Wasserstoff oder gegen Benz"opersäure reaktions¬
träge sind, die aber mittels der Molekular-Refraktion die Doppel¬bindung ebenso deutlich wie bei reaktionsfähigen Verbindungenerkennen lassen. Es erhebt sich die Frage, ob bei der Molekular-
Refraktion solcher Triterpene die als Doppelbindung gedeutetenExaltationen nicht etwa durch das besondere unbekannte poly¬zyklische System verursacht sein könnten. Es sind bisher noch
keine Fälle bekannt geworden, wo die Molekular-Refraktion poly¬zyklischer Kohlenstoffverbindungen, wenn nicht gerade aromatische
oder sonst besonders kumulierte Doppelbindungen anwesend sind,eine besonders starke Exaltation aufweisen würden. So wurde von
Tschugaeff3 beim Cholesterin und von Ruzicka und Rudolph4beim Cholatriensäureester, die höchstgliedrigen bisher untersuchten
Ringsysteme, eine auf vier Ringe hinweisende, also normale Mole¬
kular-Refraktion festgestellt.Für die Bestimmung der Dichte und des Brechungsindex kam
Hederagenin selbst, des hohen Schmelzpunktes wegen, nicht in Frage.
1Z. physiol. Chem. 199, 75 (1931).
s
Neuerdings haben A. Winterstein und G. Stein, Z. physiol. Chem. 211,10 (1932), für das dekarboxylierte Hederagenin den Namen Hederagenol vor¬
geschlagen.3 A. 385, 352 (1911).4A. 471, 39 (1929).

25
Es mußte deshalb nach Derivaten gesucht werden, deren Schmelz¬
punkt innerhalb des experimentell möglichen Temperaturbereichs fiel,
und deren Beständigkeit bei der üntersuchungstemperatur durchaus
sichergestellt war. Zur Untersuchung kamen erstens der schon von
van der Haar1 dargestellte Diacetyl-hederagenin-äthylester
(Fp.l50°) und zweitens das von A. Winterstein und G. Stein2 erst¬
mals beschriebene Dihydro-hederabetulin-diacetat (Fp. 133,5
bis 134,5°). Die gefundenen Werte der Molekular-Refraktion sind
in folgender Tabelle zum Vergleich gegen die berechneten Werte
zusammengestellt:
Diacetyl-hederagenin-äthylester. Temperatur 158,5°.
Molekular-Refraktion für C^kAi =ber.: 160,21. EMD +1,41.
„QeHaeOell „ 161,94. EMD - 0,32.
gef.: 161,62.
Molekular-Refraktion für C37rl5806 =ber.: 164,82. EMD +1,06.
„C3,ti580611 „
166,55. EMD -0,67.
gef.: 165,49.
Dihydro-hederabetulin-diacetat. Temperatur 144,7°.
Molekular-Refraktion für Ca3rl640i J>er.: 146,82. EMD +0,14.
„C33H54O4 |l „ 148,55. EMD - 1,59.
gef.: 146,96.
Molekular-Refraktion für C34H6604 =ber.: 151,53. EMD -0,58.
„C34H6604|l „
153,26. EMD -2,31.
gef.: 150,95.
Die obige Zusammenstellung der berechneten und gefundenen
Molekular-Refraktionen für Diacetyl-hederagenin-äthylester und für
Dihydro-hederabetulin-diacetat zeigt die gute Übereinstimmung mit
den von einem Cg0-Grundskelett abgeleiteten Werten.
Somit würde Hederagenin in Obereinstimmung mit neueren Be¬
funden A. Wintersteins die Bruttoformel C30rl48O4 besitzen, ferner
pentazyklische Struktur und eine Doppelbindung in der Molekel
aufweisen.
1 B. 54, 3418 (1921).s Z. physiol. Chem. 199, 75 (1931).

26
Für die ungesättigte Natur des Hederagenins, also für das Vor¬handensein von mindestens einer Doppelbindung, konnte außer der
Molekular-Refraktion noch ein weiterer Anhaltspunkt erhalten werden,nämlich die Gelbfärbung, welche das Hederagenin in Chloroform¬
lösung mit Tetranitromethan zeigt. Es gibt wohl Kohlenstoffdoppel¬bindungen, z. B. solche a-/?-ungesättigter Fettsäuren, Ketone oder
Aldehyde, welche mit Tetranitromethan keine Gelbfärbung geben.Umgekehrt hat man aber bisher noch keinen Fall beobachtet, beidem eine organische Verbindung ohne Doppelbindung Gelbfärbunggeben würde. Dagegen ist nach Untersuchungen von Ruzicka fest¬
gestellt worden, daß Verbindungen, bei denen die Anwesenheit
reaktionsträger Doppelbindungen sichergestellt ist, überraschender¬weise mit Tetranitromethan in Chloroform Gelbfärbungen zeigen,die ebenso deutlich zutage treten wie bei Verbindungen mit reaktions¬
fähigen Doppelbindungen. Der positive Ausfall der Reaktion mitTetranitromethan ist somit immer auf die Anwesenheit einer Kohlen¬
stoffdoppelbindung zurückzuführen, während der Ausfall der Gelb¬färbung keine Deutung zuläßt1.
Eindeutig und präparativ konnten schließlich A. Wintersteinund W. Wiegand2 den ungesättigten Charakter des Hederageninsdurch die Darstellung des Bromlactons nachweisen, in welchemdie Kohlenstoffdoppelbindung durch eine Bromatom- und eine Lacton-
bindung abgesättigt wird. Die genannten Autoren konnten zeigen,daß sich die Sapogeninsäuren, z. B. Hederagenin und Oleanolsäure,wie auch Sia- und Sumaresinolsäure, in Methylalkohol bequembromieren lassen, und zwar unter Bildung eines Lactonringes.Im Vergleich zu den Bougaultschen Jodlactonen ist dieserReaktionsverlauf durchaus verständlich,« und läßt darüber hinaus
zugleich einen Teil der Konstitution des Hederagenins erkennen,wonach in demselben eine ß,y- oder y, <5-ungesättigte Karbon¬säure vorliegt.
Diese Annahme wurde noch wesentlich gestützt dadurch, daß
Hederagenin selbst durch Einwirkung starker Säuren ebenfalls eineneutrale Verbindung, das Hederageninlacton, lieferte, welche alsodieselbe Bruttoformel wie das Ausgangsmaterial besitzt.
1
Vergleiche die Zusammenstellung von Ruzicka, Huyser, Pfeiffer undSeidel, A. 471, 25— 26 (1929).
3 Z. physiol. Chem. 199, 46 (1931).
27
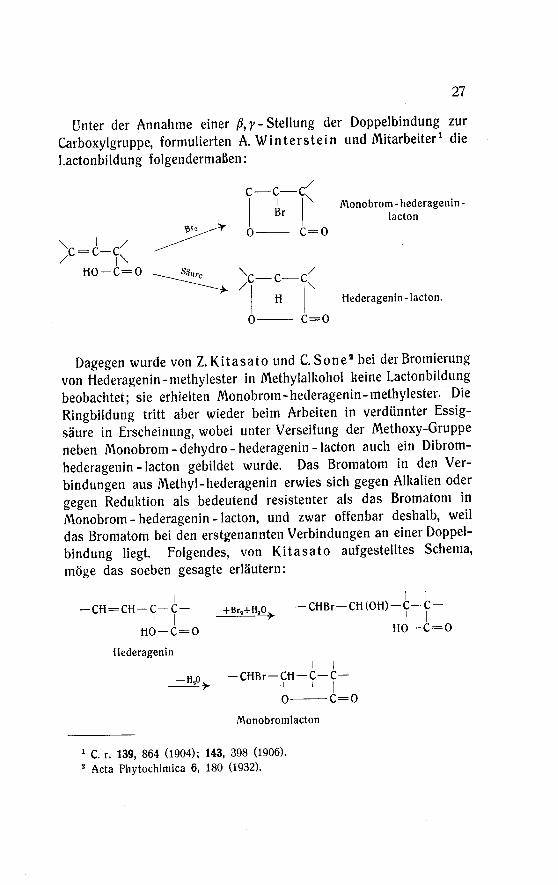
Unter der Annahme einer ß, y -Stellung der Doppelbindung zur
Carboxylgmppe, formulierten A. Winterstein und Mitarbeiter1 die
Lactonbildung folgendermaßen:
c—c—c;
Br
* /
\
-c=o
xc—c—cy\
-c=o
Monobrom - hederagenin -
lacton
Hederagenin-lacton.
Dagegen wurde von Z. Kitasato und C. Sone2 bei derBromierung
von Hederagenin-methylester in Methylalkohol keine Lactonbildung
beobachtet; sie erhielten Monobrom-hederagenin-methylester. Die
Ringbildung tritt aber wieder beim Arbeiten in verdünnter Essig¬
säure in Erscheinung, wobei unter Verseifung der Methoxy-Gruppe
neben Monobrom-dehydro-hederagenin-lacton auch ein Dibrotn-
hederagenin - lacton gebildet wurde. Das Bromatom in den Ver¬
bindungen aus Methyl-hederagenin erwies sich gegen Alkalien oder
gegen Reduktion als bedeutend resistenter als das Bromatom in
Monobrom-hederagenin-lacton, und zwar offenbar deshalb, weil
das Bromatom bei den erstgenannten Verbindungen an einer Doppel¬
bindung liegt. Folgendes, von Kitasato aufgestelltes Schema,
möge das soeben gesagte erläutern:
! I
-CH =CH-C— C—
1 IHO —C=0
Hederagenin
-CHBr—CH(OH) —C—C—' I
HO —C=0
I I
-H,o -CHBr—CH—C—C--J-> ! ! I
0- -c=o
Monobromlacton
1 C. r. 139, 864 (1904); 143, 398 (1906).2 Acta Phytochimica 6, 180 (1932).

28
1— CH= Crl — C-
!
1
-c—
|+ Bi•,+H,0
-
I 1
-CHBr—CrI(OH)-C—C-i i
CH30--c=o CHsO—O=0
Methyl - hederagenin
—CBr=CH-1 1
-c-c-1 1
+ Br2+H!C) —CBr(Br)--CH(OH)-I
-C-i
1-c
1
-H,0>
CH30—C=0 Crl30—C=0
Monobrom - hederagenin - methylester
-ch3oh —CBr(Br)—CH —C—C— _HBr —CBr=C—C—C-—> I 1 I > I 1 I
0 c=o 0 c=o
Dibrom - lacton Monobrom - dehydro - hederagenin -
lacton.
Durch Einwirkung von Zinkstaub-Eisessig wurde aus Monobrom-
hederagenin-lacton wieder Hederagenin regeneriert. Dagegen konnte
mittelst methylalkoholischer Kalilauge eine neue bromfreie Säure,sowie deren Methylester, erhalten werden, die offenbar, in Analogiezu den Bougaultschen Beobachtungen an den Jodlactonen, eine
y-Ketosäure bzw. deren Methylester darstellt.
1 1— CH2-CO—C—C—
1 1
1 1— CH,—CO—C—C—
1 1
CH30-C=0 HO—C=0
Keto - hederagenin - methylester y- Keto - hederagenin.
Schließlich konnte durch vorsichtige Oxydation von Diacetyl-hederagenin mit Chromsäure ein &-Keto-y-lacton, «5-Keto-diacetyl-hederagenin-y-lacton, erhalten werden, wodurch die Annahme der
7, (5-Stellung der Kohlenstoffdoppelbindung zur Carboxylgruppeweiterhin bestätigt werden konnte. Nach Kitasato verläuft die
Oxydation in der Weise, daß vorerst die —CH = CH—Gruppe in
—CH(OH)—CH(OH)— verwandelt wird, woraus unter Wasseraustrittan der y-Stelle und an der Carboxylgruppe ein <5-0xy- y -lacton
gebildet wird, dessen (5-Oxygruppe weiter zum Carbonyl oxydiertwird. Der Reaktionsverlauf sei durch das folgende Schema ver-

29
anschaulicht und kann ferner bequem mit der Bromlacton-Bildung
verglichen werden:
ll II
—CH=CH— C— C——Cti(OH)— CH(OH) —C—C—
, j V! |
HO—C=0 HO—C=0
—CH(OH)—CH—C-C— -CO—CH—C—C—
* I i I + j i |O C=0 0 C=0
—CH=CH —C—C— -CHBr-CH(OH)—C—C— —CHBr—CH-C—C—1 I
' ! > I ' I
HO —C=0 HO—C=0 0 C=0
Daß die Ketogruppe wirklich in ô-Stellung, bzw. die Doppel¬
bindung in /, d-Stellung zur Carboxylgruppe stehen muß, ergibt
sich weiter aus der für solche <3-Ketone charakteristischen Beständig¬
keit gegen Alkalien, und durch die Bildung eines Monobromderivates,
dessen Bromatom nach der Auffassung von Kitasato f-Stellung
zugewiesen wird.
Die Oxydation des Hederagenins.
Ein weiterer Beitrag zur Kenntnis der Feinstruktur des Hedera¬
genins wurde von W. A.Jacobs und Mitarbeitern1 in einer Reihe
von Untersuchungen über die Oxydationsprodukte des Hederagenin-
methylesters niedergelegt. W.A.Jacobs erhielt durch Einwirkung
eines Gemisches von Chrom- und Schwefelsäure auf Hederagenin-
methylester als Hauptprodukt den Hedragon- methylester (C31H4803)
und ferner unter Kohlensäure-Abspaltung einen Monoketo-dicarbon-
ester C8iH4805. Mittels Kaliumpermanganat in Aceton erhielt er
zwei weitere Abbauprodukte, Hederagon- methylester (CaaH50O4) und
Hederagin-methylester (C82HB00B). Die beiden mit Kaliumperman¬
ganat erhaltenen Oxydationsprodukte konnte er mittels Chromsäure
in Hedragon-methylester überführen.
1 3. biol. Chem. 63, 631 (1925); 69, 641 (1926).
30
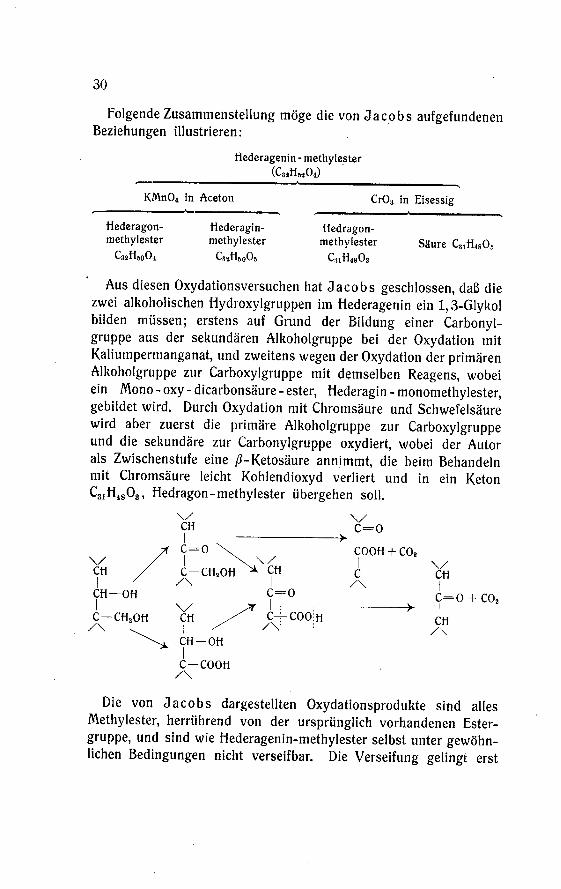
Folgende Zusammenstellung möge die von Jacobs aufgefundenenBeziehungen illustrieren:
Hederagenin - methylester(C32H52O1)
KMn04 in Aceton Cr03 in Eisessig
Hederagon-methylester
C32H50O4
Hederagin-methylester
C32H60O5
Hedragon-methylester
C11H48O3
Säure C3lH4805
Aus diesen Oxydationsversuchen hat Jacobs geschlossen, daß die
zwei alkoholischen Hydroxylgruppen im Hederagenin ein 1,3-Glykolbilden müssen; erstens auf Grund der Bildung einer Carbonyl-gruppe aus der sekundären Alkoholgruppe bei der Oxydation mit
Kaliumpermanganat, und zweitens wegen der Oxydation der primärenAlkoholgruppe zur Carboxylgruppe mit demselben Reagens, wobei
ein Mono-oxy-dicarbonsäure-ester, Hederagin-monomethylester,gebildet wird. Durch Oxydation mit Chromsäure und Schwefelsäurewird aber zuerst die primäre Alkoholgruppe zur Carboxylgruppeund die sekundäre zur Carbonylgruppe oxydiert, wobei der Autorals Zwischenstufe eine ß-Ketosäure annimmt, die beim Behandelnmit Chromsäure leicht Kohlendioxyd verliert und in ein KetonC31H4803, Hedragon-methylester übergehen soll.
\/CH
ICH—OH
IC-CH2OH
CH
Ic=o
C—CH2OH
CH
CH-OH
IC —COOH/\
c=o
C^COCVH/Y
c=o
COOH + C02IC/\
\/CH
IC=0 + C02
!CH/\
Die von Jacobs dargestellten Oxydationsprodukte sind alles
Methylester, herrührend von der ursprünglich vorhandenen Ester¬
gruppe, und sind wie Hederagenin-methylester selbst unter gewöhn¬lichen Bedingungen nicht verseifbar. Die Verseifung gelingt erst

31
mit einigermaßen befriedigender Ausbeute beim Erwärmen des
Reaktionsgemisches auf etwa 150°.
Kitasato erhielt beim Behandeln von Monobrom - hederagenin-
lacton mit Beckmannscher Mischung ein Oxydationsprodukt, das
nach dem von Jacobs angenommenen Reaktionsverlauf entstanden
sein muß, denn beim Kochen dieses neutralen, bromhaltigen
Oxydationsproduktes mit Zinkstaub-Eisessig wurde eine bromfreie
Säure erhalten, die nach erfolgter Methylierung mit Diazomethan
mit dem von Jacobs erhaltenen fiedragon-methylester identisch war.
Die schon erwähnte enorm schwere Verseifbarkeit desfiederagenin-
methylesters läßt erkennen, daß die Carboxylgruppe im Hederagenin-
molekül sicher nicht primärer Art ist, sondern daß diese Reaktions¬
trägheit1 bestimmt für sekundäre oder tertiäre Bindung spricht.
Im Hederagenin muß das Kohlenstoffatom in der a- oder ß-Stellung
zur Carboxylgruppe von besonderer Natur sein, so daß eine Art
sterischer Hinderung eintritt. Zur Klärung dieser Frage hat Jacobs
einen orientierenden Versuch angestellt. Er hat nämlich beobachtet,
daß die Estergruppe von dem neuen ß-Keto-ester (C31H4604), welcher
aus Methylhederagenin durch Oxydation mit Chromsäure und
Schwefelsäure, partielle Dehydrierung mit Schwefel und darauf¬
folgende Oxydation mit Kaliumpermanganat gebildet wird, durch
verdünnte Alkalilauge leicht verseifbar ist und die so frei gemachte
Säure mit lOVoigem Alkali leicht Kohlensäure abspaltet. Aus dieser
Tatsache hat er darauf aufmerksam gemacht, daß im Hederagenin-
methylester das Kohlenstoffatom an der ß-Stellung zur Carboxyl¬
gruppe von besonderer Natur ist.
Zur Frage des Kohlenstoffgerüsts des tiederagenins.
Es hat natürlich nicht an Versuchen gefehlt, durch energische
Abbaumethoden, wie sie die Oxydation mit Chromsäure oder Kalium¬
permanganat darstellen, kleinere Bruchstücke des Hederagenin-
Moleküls zu erhalten, deren Aufklärung wesentlich leichter durch¬
zuführen wäre.
1 Es sei in diesem Zusammenhang an die Verhältnisse bei den Diterpensäuren,
z. B. Abietinsäure, erinnert, wo sich die Carboxylgruppen sehr reaktionsträge
verhielten.

32
So wurde versucht durch trockene Destillation oder durch Zink¬
staubdestillation in das Dunkel der Konstitution einzudringen. Die
diesbezüglichen Versuche, die seinerzeit von van der Haar1, Kotake2und Winterstein ausgeführt worden sind, verliefen aber alle
praktisch ergebnislos.
Erst die Dielssche Methode der Selendehydrierung8, welche
mit Erfolg in der Chemie der Gallensäuren und des Cholesterins
angewandt wurde, brachte Klarheit über das Kohlenstoffgerüst des
Hederagenins, sowie der Triterpene überhaupt.
Die Dehydrierung mit Selen hat gegenüber der katalytischenDehydrierung oder der erschöpfenden Bromierung nach Bayer den
Vorteil, daß sie durch quaternäre Kohlenstoffatome nicht gehindertwird. Selen hat Schwefel gegenüber den weiteren Vorteil, daß die
Substanzen weniger verkohlen und daß daher die Ausbeuten an
Dehydrierungsprodukten erheblich gesteigert werden können. Auch
scheint die Gefahr der Bildung neuer Ringschlüsse viel geringerzu sein als beim Schwefel, der beispielsweise o,o'-Ditolyl bei 250°
glatt in Phenanthren überführt.
So wurde von Ruzicka und Huyser4 die Methode der Selen¬
dehydrierung auf das Gemisch der Amyrine angewandt, und mit
van Veen5 auf eine Reihe von zehn weiteren Triterpenen (Sapo-geninen, fiarzalkohole u. a. m.) übertragen, wobei fast ohne Aus¬
nahme vorerst überall derselbe Naphthalinkohlenwasserstoff C18H14erhalten wurde. Dieser Kohlenwasserstoff, der seiner besonderen
Bedeutung wegen Sapotalin getauft wurde, lieferte bei der Oxydationmit Kaliumferricyanid eine Naphthalin-tricarbonsäure, wodurch sich
für den Kohlenwasserstoff die Konstitution eines Trimethylnaph-thalins ergab.
Das Sapotalin, welches durch sein bei 128—129° schmelzendes
orangerotes Pikrat, sein bei 156—157° schmelzendes orangegelbesStyphnat und durch sein hellgelbes, bei 143° schmelzendes Trinitro-
benzolat näher charakterisiert wurde, erwies sich in der Folge als
1 B. 55, 1054 (1922).2
Proc. Imp. Acad. Japan 8, 14 (1932).3 A. 459, 1 (1927); 478, 129 (1930).4 A. 471, 35 (1929).5
Z. physiol. Chem. 184, 69 (1929).

33
identisch mit dem synthetisch dargestellten 1, 2, 7-Trimethyl-
naphthalin.Die sorgfältige Aufarbeitung größerer Mengen des mit Selen er¬
haltenen Dehydrierungsgemisches aus Hederagenin ließ erkennen,
daß das Sapotalin nicht das einzige charakteristische Spaltstück
dieses Naturstoffes darstellt. Es wurden noch weitere fünf Kohlen¬
wasserstoffe, sowie ein sauerstoffhaltiger Körper aufgefunden und
durch geeignete Derivate gekennzeichnet, worüber nachfolgende
Zusammenstellung orientiert:
Brutto¬
formel
Kristallisations¬
wertePikrat Styphnat
Trinitro-
benzolatChinon
Cioflu Kp. ca. 105-115°
(10 mm)
— — — 203-204° (Di-
bromverbindung)
CiaHis 96-97° 136-136,5° 158,5° 151-151,5° 114-115°
Cj3Hi4 Kp. ca. 135-140°
(11 mm)
131° 157° 143° 115-116°
^18*118 126—127° 165° 174° 174° 203-204°
Casiiao 306-307° — —— über 350° Zer¬
setzung
CnHn 245° — — ——
C,3rluO 157° 164° — — 89,5-90°(Methyläther).
Die folgende Aufarbeitungsmethode des Gemisches der Dehy¬
drierungsprodukte bestand zunächst darin, daß man durch wieder¬
holtes Ausziehen mit Äther die darin leichter löslichen Anteile
herausextrahierte. Dieser Extrakt, der ein dunkelbraunes, manchmal
grün fluoreszierendes und sehr unangenehm riechendes öl darstellte,
wurde nun der fraktionierten Destillation unterworfen. Eine scharfe
Trennung war aber keineswegs möglich, und es wurde deshalb nur
ganz roh in tiefer- und höhersiedende Anteile getrennt. Die tiefer¬
siedenden Fraktionen, bis 160° (10 mm), wurden nun zweckmäßiger¬
weise mit methylalkoholischer Pikrinsäurelösung behandelt, wodurch
eine Trennung der pikratbildenden aromatischen Kohlenwasserstoffe
von den nichtpikratfähigen und nicht aromatischen Anteilen erzielt
werden konnte. Das rohe, kristallisierte Pikratgemisch wurde wieder
zerlegt und die erhaltenen pikratbildenden Kohlenwasserstoffe einer
Ehmann. 3

34
sorgfältigen fraktionierten Destillation unterworfen. Eine einwand¬
freie Trennung der homologen Naphthalinkohlenwasserstoffe durch
fraktionierte Destillation war natürlich ausgeschlossen. Die Trennungwurde aber wesentlich erleichtert durch den Umstand, daß sich aus
manchen öligen Fraktionen durch Stehenlassen in der Kälte einzelne
Kohlenwasserstoffe in kristallisierter Form direkt ausschieden und
durch Abnutschen, eventuell nach vorherigem Digerieren mit Hexan,von den amorphen Beimengungen getrennt werden konnten. In
den übrigen Fällen wurden die öligen Fraktionen wieder in die
Pikrate zurückverwandelt und diese einer fraktionierten Kristallisationnach dem Dreieckschema unterworfen. Die Reindarstellung der
Kohlenwasserstoffe nach dieser Methode war natürlich nur mit sehr
schlechten Ausbeuten möglich. Die erhaltenen Spitzenfraktionenwurden nochmals zerlegt, die Kohlenwasserstoffe über Natrium
destilliert, und daraus wieder Pikrat, sowie Styphnat und Trinitro-
benzolat dargestellt, welche dann analysiert und mit synthetischenPräparaten, soweit dies möglich war, verglichen wurden.
Die höhermolekularen Kohlenwasserstoffe konnten aber nicht
nach dieser Pikrat-Methode gewonnen werden, da die Pikrinsäure-
Additionsprodukte zu unbeständig waren. In diesem Fall blieb einzigdie wiederholte fraktionierte Destillation, wobei die innerhalb einigerGrade siedenden Anteile entweder selbst teilweise kristallin er¬
starrten oder durch Anreiben mit einem dazu geeigneten Lösungs¬mittel zum Kristallisieren gebracht werden konnten.
Zur Isolierung der einzelnen Dehydrierungsprodukte ist noch
folgendes zu bemerken:
Den Anstoß zur Entdeckung dieses Kohlenwasserstoffes liefertendie Dehydrierungsergebnisse der Siaresinolsäure1 und des Betulins2,bei welchen das 1,2,3,4-Tetramethyl-benzol erstmalig isoliert werdenkonnte. Zu diesem Zweck wurden alle pikrat- und nicht pikrat-fähigen Vorläufe wieder vereinigt und wiederholt über Natriumdestilliert. Man erhielt eine wasserklare, stark lichtbrechende und
nicht mehr übelriechende Flüssigkeit, aus der schließlich eine zwischen
105—115° (10 mm) siedende Fraktion abgetrennt werden konnte.
1ttelv. 15, 1504 (1932).
2Helv. 15, 1501 (1932).

35
Diese Fraktion lieferte, mit Brom in Methylalkohol verrieben, ein
bei 203—204° schmelzendes Dibromid.
Ob es sich bei dem dazu gehörigen Kohlenwasserstoff um 1,2,3,4-
oder 1,2,3,5-Tetramethyl-benzol handelt, konnte noch nicht ent¬
schieden werden und wird erst durch Oxydation zur entsprechenden
Tetracarbonsäure seine endgültige Aufklärung finden.
Cläri12.
Dieser Kohlenwasserstoff wurde durch längeres Stehenlassen der
bei 130—138° (10 mm) siedenden Fraktion im Kühlschrank aus-
kristallisiert; er konnte aber auch bei sehr vorsichtiger fraktionierter
Destillation direkt kristallin erstarrend erhalten werden. Nach
Schmelzpunkt und Mischprobe war er mindern von Weißgerber
und Kruber beschriebenen 2, 7-Dimethyl-naphthalin1 identisch.
Auch die Pikrate, Styphnate, Trinitrobenzolate und Chinone erwiesen
sich nach Schmelzpunkt und Mischprobe als identisch.
Ci3nu.
Das Sapotalin war der erste Kohlenwasserstoff, der in Form seines
Pikrats aus dem Dehydrierungsgemisch isoliert werden konnte. Er
erwies sich, wie schon erwähnt, als identisch mit dem synthetisch
dargestellten 1,2,7-Trimethyl-naphthalin.Anläßlich der fraktionierten Kristallisation eines rohen Pikrat¬
gemisches [erhalten aus Fraktion 140—150 ° (10 mm)] konnte schlie߬
lich eine Spitzenfraktion erhalten werden, die bei 134 —134,5°
schmolz und deutlicher dunkel gefärbt war wie das normale Sapo-
talinpikrat oder gar wie das synthetische Präparat, dessen Analysen
trotzdem gut auf das Pikrat eines Kohlenwasserstoffes C13H14 stimmten,
welches auch mit Sapotalinpikrat gemischt keine Schmelzpunkts¬
depression zeigte. Es ist wahrscheinlich, daß dieses Pikrat aus
einem Gemisch von viel Sapotalinpikrat mit geringen Mengen des
höheren Homologen besteht. Dieses rote Pikrat deutet besonders
auf die Anwesenheit des Kohlenwasserstoffes CuHle, der aus den
verschiedensten Triterpenen erhalten worden war, dessen Isolierung
beim Hederagenin bisher noch nicht geglückt ist.
1 B. 52, 355 (1919).3*

36
Aus der Fraktion 140—190° (0,15 mm), welche eine rötliche, zäh¬
flüssige, gegen das Ende der Fraktion glasig erstarrende Substanz
darstellte, konnte durch Digerieren mit Hexan eine kristalline Ab¬
scheidung erhalten werden, die nach mehrmaligem Umkristallisierenaus Alkohol konstant bei 126—127° schmolz. Die Analyse deutete
vorerst auf die allgemeine Formel C„H„, die dann durch Mole-
kulargewichtsbestimmung als C18fl18 erkannt wurde, was schlie߬lich auch durch die Analyse des bei 203—204° schmelzendenChinons bekräftigt wurde. Weitere Derivate, wie das Pikrat, das bei165° schmolz, das Styphnat (174°) und das Trinitrobenzolat (174°),waren für die Analyse nicht geeignet, da sie zu große Neigungzeigten, wieder in ihre Komponenten zu zerfallen.
Die Bruttoformel des Kohlenwasserstoffes, sowie die des Chinonsdeuten auf das Vorliegen eines Phenanthren-Kohlenwasserstoffes,und zwar vermutlich eines Tetramethyl-phenanthrens.
Der beim Auskochen des rohen Dehydrierungsproduktes mit
Äther verbleibende Rückstand enthielt in dem durch tangelangesAuskochen mit Äther resultierenden Extrakt ein braunes, voluminöses
Pulver, das durch wiederholtes Umkristallisieren aus Cumol und
Pyridin gereinigt werden konnte. Man erhielt derbe, weiße Platten,deren Elementaranalyse auf die Bruttoformel C2BH20 hinwiesen. Zurweiteren Charakterisierung des Kohlenwasserstoffes konnte aus
diesem durch Oxydation mit Chromsäure ein Chinon erhalten werden,dessen Analyse ebenfalls auf die entsprechende Formel C25H1803hindeuteten.
C13HuO.
Dieses Naphthol wurde mit dem Kohlenwasserstoff C18H18 ver¬
gesellschaftet aufgefunden, von dem es sich durch seine bedeutendschwerere Löslichkeit in Hexan unterschied. Es löste sich glatt in
verdünnter Alkalilauge und konnte daraus durch Ansäuern wiederzurückerhalten werden, wodurch im Verein mit der Elementaranalyseder Phenolcharakter des Sauerstoffatoms eindeutig festgelegt war.
Das sehr zersetzliche dunkelrote Pikrat schmolz zirka bei 164°. Durch

37
Methylieren konnte der entsprechende, bei 89,5—90° schmelzende
Methyläther, und daraus das dazugehörige Trinitrobenzolat (Fp. 145
bis 146°) erhalten werden.
Es lag also in der Substanz wahrscheinlich einTrimethyl-naphthol
vor. Zur Klärung dieses Umstandes wurde eine Zinkstaubdestillation
ausgeführt, wobei ein Kohlenwasserstoff erhalten werden konnte,
der nach Schmelzpunkt und Mischprobe mit dem 1,2,7-Trimethyl-
naphthalin identisch war. Auch die entsprechenden Pikrate und
Trinitrobenzolate erwiesen sich als identisch. Weitere Versuche,
durch Oxydation die Konstitution des Oxy-sapotalins aufzuklären,
verliefen ergebnislos.
Versucht man nun zu erklären, wie eine 30 Kohlenstoffatome
enthaltende, pentazyklische Verbindung, wie sie das Hederagenin
zweifellos darstellt, die eben einzeln besprochenen Dehydrierungs¬
produkte liefern kann, so sei auf den einzigen bisher aufgeklärten
Fall eines Triterpens, das aliphatische Squalen, hingewiesen.
Dem Tetracyclo-squalen, das bei der Behandlung des Squalens
mit Ameisensäure gebildet wird, kommt offenbar die Formel I zu,
woraus mit Leichtigkeit das bei der Dehydrierung entstehende
1,2,5-Trimethyl-naphthalin seine Erklärung findet. Denkt man
sich in der Formel des Tetracyclo-squalens die zwischen beiden
Äthylenbindungen befindliche Lücke ebenfalls geschlossen, so gelangt
man zu einem pentazyklischen System, der ein hydrierter Picenring
zugrunde liegt.
Dieses hydrierte Picengerüst wäre als hypothetisches Kohlen¬
stoffskelett einer Triterpenverbindung durchaus denkbar, zumal es
die logische Fortsetzung des in der Diterpenreihe festgestellten
Phenanthrenringes darstellen würde.
S\/\
A B
\/V/
38
/\/\
\B V
/\/\/\l/NC I D
/\/\
\/
IV /
S\
LY
w\
</w
/\ /\
\/\/x
/v
\/
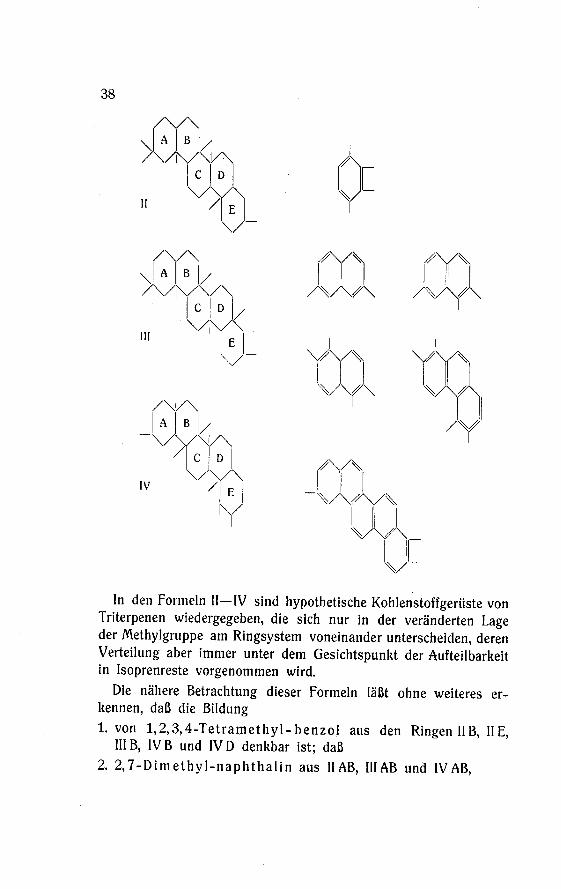
In den Formeln II—IV sind hypothetische Kohlenstoffgerüste von
Triterpenen wiedergegeben, die sich nur in der veränderten Lageder Methylgruppe am Ringsystem voneinander unterscheiden, deren
Verteilung aber immer unter dem Gesichtspunkt der Aufteilbarkeitin Isoprenreste vorgenommen wird.
Die nähere Betrachtung dieser Formeln läßt ohne weiteres er¬
kennen, daß die Bildung1. von 1,2,3,4-Tetramethyl-benzol aus den Ringen IIB, IIE,
HIB, IVB und IVD denkbar ist; daß
2. 2,7-Dimethyl-naphthalin aus HAB, III AB und IV AB,

39
3. 1,2,7-Trimethyl-naphthalin aus II AB, III AB, IV AB und IV DE,
4. 1,2,5, 6-Tetramethyl-naphthalin, dessen Bildung beim
Hederagenin noch nicht sicher ist, aus II DE, III BC und IV CD
gebildet werden können.
5. Rein schematisch könnte auch aus IVCDE die Bildung eines
Phenanthrens seine Erklärung finden und schließlich
6. durch einfaches Aromatisieren des Grundskeletts die Bildung eines
Trimethyl-picens gedeutet werden.

Experimenteller Teil.
Über die Bereitung des Hederagenins.
Die Gewinnung des Hederagenins erfolgte ausschließlich aus
Seifennüssen (Sapindus saponaria, Sapindus Mukorossi Gärtner,Sapindus Rarak De), welche durch Vermittlung der Firma R.Budden-
sieg (Paris) aus Algier bezogen wurden. Die gelieferten Nüsse waren
teilweise schon aufgeklopft und von den tiefschwarzen, außer-odentlich harten Steinen befreit. Da das Saponin ausschließlichin den Schalen enthalten ist, wurden die Steine von Hand möglichstvollständig heraus gelesen und die verbleibenden, klebrig an¬
zufühlenden Schalen in einem warmen Luftstrom getrocknet. DieSchalen ließen sich nun in einer Kugelmühle zu einem feinenPulver zermahlen. Aus diesem Pulver wurde mit wäßrigem Alkoholdas Saponin extrahiert und dieses ohne vorhergehende Isolierungauf Hederagenin verarbeitet. Die weiteren Einzelheiten der folgendenArbeitsvorschrift wurden den Angaben von W.A.Jacobs1 undA. Winterstein und J. Meyer2 entnommen.
Je 10 kg gemahlene Schalen wurden in eine Lösung von 24 LiterAlkohol (den., 96 % ig) un^ 14,4 Liter Wasser eingerührt, und dann12 Stunden unter Rückfluß gekocht. Man ließ über Nacht stehenund filtrierte anderntags durch große Steinzeugnutschen. DerRückstand auf der Nutsche wurde gut abgepreßt und nochmalsmit der gleichen Menge wäßrigen Alkohols (Mischung wie oben)
1 3. of biol. Chem. 63, 621 (1925).2
Z. physiol. Chem. 199, 40 (1931).

41
10 Stunden ausgekocht. Nach dem Erkalten wurde wieder filtriert
und abgepreßt; das nur noch hellbraun gefärbte Filtrat wurde zur
Extraktion einer neuen Portion Schalen verwendet. Das ausgekochte
Schalenpulver war nun praktisch saponinfrei; eine Probe mit Wasser
angefeuchtet war nicht mehr klebrig anzufühlen, und mit Wasser
aufgeschlämmt und dann mit Luft geschlagen bildete sich kein
Saponinschaum mehr.
Der erste Extakt wurde mit 200 g frisch entwässertem Carboraffin
8 Stunden rückflußgekocht, filtriert und nochmals mit der gleichen
Menge Kohle behandelt. Man erhielt schließlich einen nur noch
schwach hellbraun gefärbten Extrakt, in welchem das Saponin, von
Ballaststoffen weitgehend befreit, enthalten ist.
Zur Hydrolyse des Saponins wurden je 8 Liter des alkoholischen
Extraktes mit 250 ccm konzentrierter Schwefelsäure vermischt und
auf dem Dampfbad zum Sieden erhitzt. Es bildete sich ziemlich
bald eine Gallerte, die beim weiteren Kochen wieder zerkrümelte
und in das körnige Endsapogenin überging. Nach 8 stündigem
Kochen war die Hydrolyse beendet; eine Probe des ausgeschiedenen
Produktes destillierte im Glührohr ohne Verkohlung, war also zucker¬
frei. Das ausgeschiedene Roh-Sapogenin wurde auf der Nutsche mit
heißem Wasser säurefrei gewaschen und im Trockenschrank ge¬
trocknet. Man erhielt ca. 400 g Roh-Hederagenin aus 10 kg Schalen.
Das so gewonnene Hederagenin bildete ein hellbraunes, körniges
Pulver und wurde zur weiteren Reinigung in das Kaliumsalz über¬
geführt. Zu diesem Zweck wurden je 500 g Roh-Hederagenin in
5000 ccm Äthylalkohol (den., 96 °/0 >g) aufgeschwemmt, zum Sieden
erhitzt und bis zur klaren Lösung konzentrierte, wäßrige Kalilauge
zugetropft. Die braune Lösung des Kaliumsalzes wurde mit
Carboraffin entfärbt, und aus der nun klaren, nahezu farblosen
Lösung das Sapogenin mittels konzentrierter Salzsäure gefällt. Man
erhielt ein schneeweißes, feinkristallines Produkt, welches zwischen
300—305° (korr. Berl) schmolz. Aus 10 kg Schalen wurden durch¬
schnittlich 370 g Hederagenin gewonnen1, das in diesem Reinheits¬
grad für die weiteren Untersuchungen und Umsetzungen zur Ver¬
wendung gelangte.
1 A. Winterstein erhält ca. 4,5°/0 Hederagenin. Die Ausbeute schwankt
offenbar mit der Provenienz und Qualität der Seifennüsse.

42
Die Bruttoformel des Hederagenins.Für die Elementaranalyse wurde das Hederagenin insgesamt
fünfzehnmal aus reinem Dioxan1 umkristallisiert. Der Schmelz¬
punkt lag konstant zwischen 312—314° (nicht korrigiert). Die im
Vakuumexsikkator getrocknete Substanz verlor bei 10 stündigemErhitzen auf 115° (12 mm) nur 0,05% ihres Gewichtes, ohne dabei
eine äußere Veränderung zu zeigen. Zur Kontrolle der Elementar¬
analysen wie auch der Molekulargewichtsbestimmung durch Titration
wurden stets solche mit reinster Dextro-pimarsäure ausgeführteBestimmungen dazwischen geschaltet.
Analysen:
Dextro-pimarsäure.
3,078 mg Substanz gaben 8,95 mg C02 und 2,76 mg H20.
C20H30O2. Ber.: C 79,39; H 10,00 VGef.: C 79,30; H 10,03 °/0.
Hederagenin.
3,113 mg Substanz gaben 8,725 mg C02 und 2,91 mg H20.3,293 mg Substanz gaben 9,23 mg C02 und 3,03 mg H20.3,420 mg Substanz gaben 9,585 mg COs und 3,16 mg H20.
C31H50O4. Ber.: C 76,48
C3oH4804. Ber.: C 76,21
Gef.: C 76,44
C 76,44
C 76,43
H 10,36 °/o. Molekulargewicht: 486,4.H 10,24 °/o- Molekulargewicht: 472,4.
H 10,46 VH 10,30 VH 10,34 V
Titrationen.
Infolge der Schwerlöslichkeit des Hederagenins in Alkohol mußtewährend der Titration längere Zeit gekocht werden.
Dextro - pimarsäure 1) 21,075 mg Substanz verbrauchten 3,486 ccm 0,02-n. Kalilauge.Hederagenin .... 1) 39,76 mg Substanz verbrauchten 4,109 ccm 0,02-n. Kalilauge.Hederagenin . . . . 2) 42,91 mg Substanz verbrauchten 4,438 ccm 0,02-n. Kalilauge.Dextro-pimarsäure 2) 24,51 mg Substanz verbrauchten 4,027 ccm 0,02-n. Kalilauge.
Dextro-primarsäure. Gef. Molekulargewicht: 1) 302,2; 2) 304,3.Hederagenin. Gef. Molekulargewicht: 1) 485,5; 2) 483,5.
1
Dargestellt aus technischem Dioxan nach Angaben von E. Eigen berger,3. pr. Chem. 130, 75 (1931).

43
Es wurde noch eine neue Reihe von Titrationen ausgeführt, wobei
die Substanz durch genau 5 Minuten langes Kochen in 15 ccm
Alkohol gelöst wurde. Die Vergleichstitrationen mit Dextro-pimar-
säure wurden genau gleich ausgeführt. Während der Titration
wurde die Lösung dauernd in der Siedehitze gehalten.
Dextro-pimarsäure 1) 25,48 mg Substanz verbrauchten 4,215 ccm 0,02-n Kalilauge.
Hederagenin . . . . 1) 45,22 mg Substanz verbrauchten 4,708 ccm 0,02-n Kalilauge.
Dextro-pimarsäure 2) 26,58 mg Substanz verbrauchten 4,386ccm 0,02-n Kalilauge.
Hederagenin .... 2) 43,24 mg Substanz verbrauchten 4,506 ccm 0,02-n Kalilauge.
Dextro - pimarsäure 3) 26,89 mg Substanz verbrauchten 4,473 ccm 0,02-n Kalilauge.
Hederagenin .... 3) 42,83 mg Substanz verbrauchten 4,440 ccm 0,02-n Kalilauge.
Dextro-pimarsäure 4) 25,28 mg Substanz verbrauchten 4,159 ccm 0,02-n Kalilauge.
Hederagenin .... 4) 44,64 mg Substanz verbrauchten 4,620 ccm 0,02-n Kalilauge.
Hederagenin .... 5) 42,46 mg Substanz verbrauchten 4,392 ccm 0,02-n Kalilauge.
Dextro-pimarsäure 5) 27,43 mg Substanz verbrauchten 4,538 ccm 0,02-n Kalilauge.
Dextro-pimarsäure. Gef. Molekulargewicht: 302,3 303,0 300,6 303,9 302,3.
Hederagenin. Gef. Molekulargewicht: 480,2 479,8 482,2 483,1 483,4.
Molekular - Refraktion.
Zur Messung von Dichte und Berechnungsindex war Hederagenin
selbst nicht geeignet, da sein hoher Schmelzpunkt außerhalb dem
für diese Messungen experimentell möglichen Temperaturbereich
lag. Es wurde deshalb nach einem möglichst tiefschmelzenden
und möglichst einfachen Derivat des Hederagenins gesucht. Die
Wahl fiel auf den Athylester des Hederagenin-diacetats, der
schon von W. van der Haar und A. Tamburello1 dargestellt wurde
und bei 150° schmilzt.
Zur siedenden Lösung von 20 g Hederagenin und 3,5 g Atzkali
in 250 ccm Äthylalkohol wurden 14 g Diäthylsulfat zugefügt. Nach
einer Stunde Kochen unter Rückfluß wurde konzentrierte Kalilauge
bis zur bleibenden stark alkalischen Reaktion zugegeben, filtriert
und erkalten gelassen. Der Hederagenin-äthylester schied sich in
prächtigen, bei 201° schmelzenden Nadeln aus. Nach mehrmaligem
Umkristallisieren aus Äthylalkohol schmolz der Ester konstant
zwischen 207—209°. 15 g des auf diese Weise erhaltenen reinen
1 B. 54, 3148 (1921).

44
Esters wurden mit 250 ccm Acetanhydrid 3 Stunden rückflußgekocht.Das überschüssige Essigsäureanhydrid wurde darauf im Vakuum
abgesaugt, der zähe, klebrige Rückstand in Äthylalkohol aufgenommenund das Lösungsmittel wiederum im Vakuum verdampft. Diese
Operation wurde noch dreimal wiederholt, wonach der Rückstandfrei von Acetanhydrid war und kristallin erstarrte. Der acetylierteEster wurde aus Äthylalkohol umkristallisiert. Es bildeten sichderbe Nadeln, welche scharf bei 155° schmolzen und bei weiteremUmkristallisieren ihren Schmelzpunkt nicht mehr änderten. W. van
der Haar fand den Schmelzpunkt des Hederagenin-äthylester-diacetats bei 150°.
3,187 mg Substanz gaben 8,66 mg COs und 2,77 mg H20.
CsoHseOe. Ber : C 73,92 und H 9,66 VCs,HB8Oa. Ber.: C 74,19 und H9,77°/0.
Gef.: C 74,11 und H 9,73 °/0.
Molekular-Refraktion:
di58,5°= lj0185.
*
nJ58.5°= 1,47521 und n"1,7°= 1,47103.
Temperaturkoeffizient für n = 0,00032 pro 1°.
Für CetWW Ber.: MD = 161,94.
Gef.: MD58'5°= 161,92; EMD=-0,32.
Für C3,H58061. Ber.: MD= 166,55.
Gef.: MD58'5°= 165,49; EMD=+1,06.
Als weiteres tiefschmelzendes, wenn auch schon teilweise ab¬
gebautes Derivat des Hederagenins wurde das von A. Wintersteinund G. Stein1 erhaltene Dihydro-hedrabetulin-diacetat zur
Bestimmung der Molekular-Refraktion herangezogen.Die Darstellung erfolgte nach den von obigen Autoren gegebenen
Vorschriften. Der Schmelzpunkt lag nach mehrmaligem Um¬
kristallisieren aus Äthylalkohol konstant zwischen 133,5—134,5°.
1Z. physiol. Chem. 199, 75 (1931).

45
Molekular-Refraktion:
dj44'7^ 1,0032.
„139,5° ^ 148660 und n{)51'7°= 1,48276.
Temperaturkoeffizient für nD= 0,00032 pro 1°.
Daraus berechnet nj)44'7^ 1,48494.
Für C33H5404. Ber.: MD = 146,82.
Gef.: Md44'70^ 146,96. EMD = +0,14.
Für C31H6604. Ber.: MD = 151,53.
Gef.: Md44'7°= 150,95. EMD = -0,58.
Ozonide des Hederagenin-methytesters.
Da Hederagenin in einem gegen Ozon beständigen Lösungsmittel,wie Wasser, Äthylbromid, Chloroform, Tetrachlorkohlenstoff, Essig¬ester und dergleichen mehr, zu unlöslich war, wurde Hederagenin-
methylester, der in allen Lösungsmitteln wesentlich leichter löslich
ist, ozonisiert.
9 g Hederagenin-methylester wurden in 200 ccm alkoholfreiem
Chloroform gelöst und unter Eiskühlung während 3 Tagen mit einem
6—8°/0igen Ozonstrom behandelt. Nach zirka 24stündiger Versuchs¬
dauer bildete sich in der Umgebung des Einleitungsrohres eine
gelbliche, klebrige Masse, deren Menge langsam zunahm, während
die Chloroformlösung eine starke gelbgrüne Färbung annahm. Das
ausgeschiedene Produkt wurde auf einem Glasgoochtiegel gesammelt,mit wenig reinem Chloroform gewaschen und dann im Exsikkator
über Phosphorpentoxyd, Atzkali und Paraffinschnitzel bei Raum¬
temperatur und 0,1 mm getrocknet. Die Masse blähte im Vakuum
stark auf und verfärbte sich schwach nach bräunlich-gelb, obwohl
der Exsikkator die ganze Zeit über im Dunkeln belassen wurde.
Nach zirka 10 Wochen hatte das Präparat Gewichtskonstanz erreicht.
4,017 mg Substanz gaben 6,86 mg C02 und 1,85 mg H20.
Gef.: C 46,58 und H 5,15°/0.
Eine Probe der Substanz im Reagensglas mit Wasser erwärmt
ergab einen deutlichen Geruch nach Chloroform. Der Chlorgehalt

46
konnte auch deutlich mittels Kalk nachgewiesen werden. Das
Präparat wurde weitere drei Monate im Exsikkator getrocknet.
5,043 mg Substanz gaben 8,64 mg C02 und 2,26 mg ti20.
10,08 mg Substanz gaben 7,005 mg AgCl.
Gef.: C 46,72, H 5,02 und Cl 17,19 °/0.CaiHioO^CU. Ben: C 46,84, ti 5,08 und C! 17,84°/0.
Es wurde auch versucht, Hederagenin-methylester in Tetrachlor¬
kohlenstoff-Lösung der Einwirkung des Ozons zu unterwerfen. Der
Versuch scheiterte aber daran, daß bei 0° ca. 95% des bei 15°
gelösten Esters wieder auskristallisierte und auf diese Weise der
Reaktion entzogen wurde.
Dagegen erwies sich Äthylbromid als gutes, ozonbeständigesLösungsmittel für Hederagenin - methylester. 10 g dieses Esters
wurden in 500 ccm Äthylbromid gelöst und in oben beschriebener
Weise ozonisiert. Das ausgefallene Ozonid war aber nach inten¬
sivstem Trocknen immer noch bromhaltig.
Reduktion des Hederagenin-methylcsters nach
Bouveault- Blanc.
65 g Hederagenin-methylester wurden in 300 g Amylalkohol gelöstund auf einmal zu 50 g Natrium zulaufen gelassen. Unter Ab¬
scheidung einer weißen Masse trat eine nur sehr träge Reaktion ein;das Natrium wurde immer mehr von dieser Ausscheidung umhüllt,wodurch die Reduktion noch mehr verlangsamt wurde. Der Kolbenwurde darauf in ein schon auf 150° vorgewärmtes Ölbad eingetauchtund zur Anregung der Reduktion nochmals 500 ccm Amylalkoholzugegeben. Die ausgeschiedene weiße Masse löste sich dabei ziemlich
rasch wieder auf und unmittelbar darnach setzte eine sehr heftigeWasserstoff-Entwicklung ein. Die Badtemperatur stieg mittlerweile
auf 180° und wurde in dieser Höhe beibehalten. Nach dem Ab¬
flauen der heftigsten Reaktion wurde unter stetem Kochen nach und
nach soviel Amylalkohol zugegeben, bis nach 4 Stunden alles Natrium
gelöst war. Im ganzen wurde 1675 g absoluter Amylalkohol be¬
nötigt. Der Kolbeninhalt wurde noch eine Stunde unter Rückfluß
erhitzt. Darauf wurde das gebildete Natrium-amylat mit 60 g Wasser

47
(geringer Überschuß über die zur Zersetzung theoretisch nötige
Menge Wasser) zerlegt, das vorsichtig durch den Kühler zulaufen
gelassen wurde. Die anfänglich sehr stürmische Reaktion ging dabei
in ein gelindes Sieden über und wurde noch drei Stunden dabei
belassen. Aus dem Reaktionsgemisch wurde nun der Amylalkoholmittels Wasserdampf abgeblasen. Nachdem aller Alkohol entfernt
war, bildeten sich in der verbleibenden Lösung gelblich - weiße
Klumpen. Diese wurden über Glaswolle von der alkalischen Mutter¬
lauge getrennt und im Vakuum bei 100° getrocknet. Zwecks völliger
Verseifung etwa noch vorhandenen Hederagenin-methylesters wurde
der 55,8 g wiegende Rückstand mit einer Lösung von 30 g Ätz¬
natron in 400 ccm Methylalkohol im Autoklaven während 48 Stunden
auf 150—165° erhitzt. Die erhaltene völlig klare Lösung wurde
zur Entfernung des Methylalkohols mit Wasserdampf behandelt.
Es bildete sich wiederum eine weiße krümelige Masse, die auf der
Nutsche gesammelt, mit wenig heißem Wasser gewaschen und
getrocknet wurde.
Der trockene Rückstand wurde nun im Apparat mehrere Tagemit Äther ausgekocht.Aus der Ätherlösung schieden sich nach einiger Zeit geringe
Mengen eines weißen Pulvers aus, das auf einem Filter gesammeltwurde. Das Produkt schmolz zwischen 115—120° unter Bräunung
und wurde seiner geringen Menge wegen nicht weiter untersucht.
Der klare Ätherextrakt hinterließ beim Verdunsten des Lösungs¬mittels einen strohgelben, in der Kälte nahezu erstarrenden Rück¬
stand, der in Petroläther äußerst schwer, in Methyl- und Äthyl¬
alkohol, sowie Benzol wenig, in Chloroform, Aceton und Xylol etwas
besser löslich war. Aus keinem dieser Lösungsmittel gelang es das
Produkt in kristalliner Form zu erhalten, und es wurde deshalb eine
Reinigung durch Destillation versucht. Das Produkt siedete nach Ab¬
trennung eines geringfügigen Vorlaufs zwischen 285—290° (0,2 mm).
Destillationsrückstand war praktisch keiner vorhanden. Das er¬
haltene hellgelbe Destillat erstarrte beim Stehenlassen langsam ohne
zu kristallisieren. Das Produkt wurde zur weiteren Reinigung wieder¬
holt fraktioniert destilliert; die ersten Tropfen, welche jeweils bei
283—285° (0,2 mm) überdestillierten, wurden als Vorlauf abgetrennt
und die Mittelfraktion, Kp. 285—290° (0,2 mm), weiter fraktioniert.
Bei diesem sehr hohen Siedepunkt wurde als Heizbad eine Zinn-

48
Blei-Legierung verwendet. Die Siedepunktsgrenzen selbst konnten
bei dieser Temperatur natürlicherweiser nicht enger gezogen werden.
Die auf diese Weise gereinigte Mittelfraktion wurde nach dem Er¬
starren im Mörser zerrieben und schmolz zwischen 145—148°. Die
Analyse zeigte folgende Werte:
3,797 mg Substanz gaben 10,97 mg C02 und 3,71 mg H20.
C30H6o03. Ber.: C 78,54 und H 10,99 V
C31H5203. Ber.: C 78,74 und H 11,09°/0.
Gef.: C 78,80 und H 10,93 °/0.
Der ätherunlösliche Anteil wurde mit Äthylalkohol unter Rückfluß
gekocht, von Spuren Ungelöstem durch Filtrieren befreit und mit
konzentrierter Salzsäure angesäuert. Es fiel ein gallertartiger Nieder¬
schlag aus, der beim weiteren Kochen kristallin wurde. Nach dem
Erkalten nutschte man den Niederschlag ab und digerierte mehrmals
mit heißem Wasser. Das feinkristalline Pulver schmolz nach dem
Trocknen zwischen 300—310° (unkorr. Berl). Nach zweimaligemUmkristallisieren aus reinem Dioxan stieg der Schmelzpunkt auf
310 —315u (unkorr. Berl). Die Mischprobe mit Hederagenin ließ
keine Depression des Schmelzpunktes erkennen. Aus den Mutter¬
laugen fiel bei weiterem Zusatz verdünnter Salzsäure nochmals eine
Gallerte aus, die aber durch längeres Kochen nicht kristallin er¬
halten werden konnte.
Die Reduktion des Hederagenin-methylesters mit Natrium und
Äthylalkohol verlief ergebnislos: Es wurden 50 g Ester mit 65 g
Natrium in 3000 ccm absolutem Äthylalkohol in der oben be¬
schriebenen Weise zur Reaktion gebracht. Nach dem Zersetzen mit
200 ccm Wasser wurde der Kolbeninhalt im Autoklaven während
60 Stunden auf 160—165° erwärmt, darauf der Äthylalkohol mit
Wasserdampf abgeblasen, der ausgeschiedene, krümelig erstarrende
Klumpen auf der Nutsche gesammelt, gewaschen und getrocknet.Das Produkt wurde mit siedendem Äther mehrere Tage ausgekocht.Es konnten aber nur minimale Mengen ätherlösliche Anteile ge¬
wonnen werden. Der Extraktionsrückstand war in heißem Methyl¬alkohol glatt löslich und bildete nach Zusatz von konzentrierter
Salzsäure in der Hitze einen erst gallertigen, dann kristallin werdenden
Niederschlag. Dieser wurde aus Dioxan mehrmals umkristallisiert
und erwies sich nach Schmelzpunkt und Mischprobe unzweifelhaft

49
als fiederagenin. Insgesamt wurden ca. 95% des in die Reaktion
eingebrachten fiederagenin-methylesters in Form von Hederagenin
wieder zurückgewonnen.
Dehydrierung des Hederagenins mit Selen.
Die im Verlauf der Untersuchung der Dehydrierungsprodukte des
Hederagenins notwendig gewesenen Spaltungsversuche mit Selen
führten schließlich zu nachfolgend skizzierter Arbeitsweise:
Als Reaktionsgefäß diente ein Langhalsrundkolben aus Jenaerglas,
an welchen, nach dem Einfüllen des Ansatzes, ein ca. 15—20 mm
weites und ca. 100—150cm langes Rohr angeschmolzen war. Dieses
wurde an seinem oberen Ende zweimal umgebogen, derart, daß das
Mittelstück von ca. 20 cm Länge leicht nach schräg unten gerichtet
war. Das absteigende Rohr, ebenfalls ca. 20 cm lang, wurde durch
einen Gummistopfen mit einem geräumigen Reagensglas mit Ansatz
verbunden. Vom Ansatz führte eine Glasrohrleitung zum Ab¬
sorptionsturm, an den zur Sicherheit noch ein zweites Absorptions¬
gefäß angeschlossen war. Die Verbindung der Glasröhren wurde
mit dickem Vakuumschläuchen hergestellt, die Rohrenden mußten
sich berühren (Glas auf Glas). Alle Gummiverbindungen wurden
mit Cellon bestrichen, um Spuren Selenwassenstoff, welche durch
den Gummi diffundierten, zurückzuhalten. Die Absorptionstürme
wurden mit Chlorkalkbrocken beschickt, wobei darauf geachtet
wurde, daß die gebildeten Gase ohne allzu große Behinderung durch
die Gefäße hindurch entweichen konnten. Um zu verhindern, daß
die im Rohr kondensierte Flüssigkeit einfach an der Kolbenwand
zurückfließt und an der Berührungsstelle Bad —Glas wieder abspritzt,
wurde eine Tropfspitze eingeschmolzen, die das Destillat in die
Kolbenmitte tropfen ließ, wodurch eine vollständigere Dehydrierung
der leichtflüchtigen Anteile gewährleistet wurde. Als Wärmeüber¬
träger hatte sich ein elektrisch geheiztes Metallbad, englisches Löt¬
zinn enthaltend, am besten bewährt. Zwecks Konstanthaltung der
Temperatur wurde noch ein Kontakt -Thermoregulator in den Strom¬
kreis eingeschaltet.Maximal können auf diese Weise 200 g Substanz mit 400 g
Selen auf einmal zur Dehydrierung angesetzt werden. Größere
Ehmann. 4

50
Ansätze sind nicht ratsam, da ein gleichmäßiges Durchdehydriereninnerhalb nützlicher Frist nicht gewährleistet wird.
Das zur Verwendung gelangende rote, gefällte Selen, Provenienz
Kahlbaum, wurde zuerst im Trockenschrank bei 100° getrocknet,wobei das Selen zusammensinterte und in die schwarze Modifikation
überging.
Zur Einleitung der Reaktion wurde das Gemisch von einem Teil
Hederagenin und zwei Teilen Selen im Metallbad etwa 10 Minuten
lang auf 350° erhitzt. Es begann eine lebhafte Zersetzung, wobei
das gebildete Wasser mit einem leichtflüchtigen hellgelben öl ins
Reagensglas mit Ansatz überdestillierte. Im Chlorkalkturm wurde
der entwickelte Selenwasserstoff zu rotem Selen oxydiert. Man
mäßigte nun die Temperatur des Metallbades auf etwa 300°, wobei
das Dehydrierungsgemisch immer noch im kräftigen Sieden blieb und
noch sehr dünnflüssig aussah. Nach ungefähr 55—60 Stunden
Erhitzungsdauer begann die Masse dickflüssig zu werden, worauf
die Dehydrierung abgebrochen wurde. Man ließ erkalten und sogden in der Apparatur noch vorhandenen Selenwasserstoff durch
den Chlorkalkturm ab.
Der Reaktionskolben wurde nun unterhalb der Tropfspitze vom
Rohr abgeschnitten und die leicht löslichen Dehydrierungsproduktemittels Äther herausgelöst. Darauf wurde zwecks Gewinnung des
Dehydrierungsrückstandes der Kolben zerschlagen, der Rückstand
vom Selenregulus befreit und schließlich mit Äther ca. 14 Tageausgekocht. Dabei bildete sich im Ätherextrakt nach einiger Zeit
ein voluminöses, braunes Pulver, das auf einem Filter gesammelt,getrocknet und gesondert aufgearbeitet wurde.
Die in der beschriebenen Weise erhaltenen rotbraunen, dunkel¬
grünfluoreszierenden Ätherextrakte wurden vereinigt, mehrmals mit
Wasser gewaschen und schließlich einige Tage mit Wasser stehen
gelassen. Es bildete sich dabei an der Kolbenwandung ein roter,dünner Selenbeschlag. Man filtrierte das Äther-Wasser-Gemisch,trennte das Wasser möglichst vollständig ab und trocknete die so
erhaltene klare Ätherlösung über Natriumsulfat. Das nach dem
Verdunsten des Lösungsmittels verbleibende dunkelbraune, dick¬
flüssige öl wurde im Vakuum fraktioniert, wobei man folgendeAnteile erhielt:

51
1. Fraktion: —80° (14 mm), hellgelbes, sehr leicht beweglichesund höchst übelriechendes öl.
5-10 %-
2. Fraktion: 80-160° (14 mm), hellgelbes, dünnflüssiges Öl. 25 bis
30%.
3. Fraktion: 80-190° (0,2 mm), zähes rotbraunes öl. 15-20%.
4. Fraktion: 190—300° (0,2 mm), sehr zähes, gegen Ende der Fraktion
glasig erstarrendes öl. 25—30%-
5. Destillations-Rückstand, in der Kälte glasig erstarrende, dunkel¬
rotbraune Masse. 15—20%-
Die in obiger Zusammenstellung aufgeführten Prozentzahlen be¬
deuten die ungefähren Ausbeuten an einzelnen Fraktionen, bezogenauf die angewandte Extraktmenge.
Die Mengenverhältnisse der einzelnen fiauptfraktionen bewegten
sich bei den verschiedenen ausgeführten Dehydrierungen immer
innerhalb obiger Grenzen. Dagegen schwankte die Menge des er¬
haltenen Ätherextrakts erheblich, je nach den Bedingungen unter
denen die Dehydrierung durchgeführt wurde, und zwar zeigte es
sich, daß kurz andauernde, energische Spaltungen wohl reichlich
leichtflüchtige Anteile lieferten (Fraktionen 1—2), die aber nur wenig
pikratbildende öle enthielten und daß bei langandauernder Dehy¬
drierung (100 und mehr Stunden) starke Verkohlung eintrat. Im
letzteren Fall blieben bis zu 50% des eingesetzten flederagenins
als in Äther völlig unlösliches Produkt in der Extraktionshülse zurück.
Aus zahlreichen Dehydrierungsversuchen wurden, wie schon erwähnt,
bei ca. 50 stündiger Versuchsdauer die besten Resultate erhalten.
Ober die bei der Dehydrierung des flederagenins erhaltenen
Naphthalin - Kohlenwasserstoffe.
Zuerst wurde versucht durch fraktionierte Destillation die Fraktion 2
[Kp. 80—160° (14 mm)] in einzelne Teilfraktionen zu zerlegen. Der
Siedepunkt stieg aber dauernd; es konnten keine deutlichen Fraktionen
erkannt werden. Auch zeigten Proben der willkürlich von 10 zu
10 Grad genommenen Fraktionen mit alkoholischer Pikrinsäure starke
4*

52
Rotfärbung, was auf das Vorhandensein pikratfähiger Kohlenwasser¬
stoffe schließen ließ. Deshalb wurde die gesamte Fraktion 2 mit
dem gleichen Gewicht Pikrinsäure in Methylalkohol unter Erwärmen
gelöst. Es bildete sich eine tiefrote, klare Lösung, aus welcher beim
Stehen in der Kälte ein rotes Pikrat in langen, zu Büscheln ver¬
einigten Nadeln auskristallisierte. Das Pikrat wurde auf der Nutsche
gesammelt, gut abgepreßt und mit wenig kaltem Methylalkohol
nachgewaschen. Aus dem Filtrat wurde nach dem Einengen auf
ungefähr das halbe Volumen eine zweite Portion eines schon mehr
tiefroten Pikrats gewonnen.
Bei weiterem Konzentrieren des Filtrats bildeten sich nur noch sehr
wenige Pikrate nebst reichlichen Mengen Pikrinsäure, die mit einem
nun ebenfalls ausfallenden dunkelgefärbten öl verschmiert waren.
Die beiden ersten Pikratfraktionen wurden zusammen mit ver¬
dünntem wäßrigen Ammoniak unter gelindem Erwärmen zersetzt.
Das gebildete öl wurde in Petroläther aufgenommen, die ätherische
Lösung mit Wasser gründlich gewaschen, über Natriumsulfat ge¬
trocknet und das Lösungsmittel verdampft. Es wurden ca. 50 %des eingesetzten Materials als pikratfähiges öl zurück erhalten.
Dieses wurde bei einem Druck von 11 mm wiederholt über Natrium
destilliert, bis das Natrium im Kolben völlig blank zurück blieb.
Dann wurde wiederum bei 11 mm in folgende drei Fraktionen zerlegt:
Fraktion 2a 138° ca. 45°/0.Fraktion 2b
. .138—140° ca. 45 —50%.
Fraktion 2c 140° ca. 5%.
Fraktion 2a wurde im Kühlschrank mehrere Tage auf—15°
gekühlt. Dabei kristallisierten aus dem öl feine farblose Blättchen,die in der Kälte abgenuscht und mit wenig kaltem Methylalkoholnachgewaschen wurden. Zur weiteren Reinigung wurde das Präparataus 50 % igem Äthylalkohol mehrmals umkristallisiert. Man erhielt
kleine weiße Blättchen, die bei 96—97° schmolzen. Schließlich
wurde das Präparat noch sublimiert; der Schmelzpunkt änderte sich
aber nicht mehr.
3,160 mg Substanz gaben 10,675 mg C02 und 2,23 mg tt20.
2,960 mg Substanz gaben 10,01 mg C02 und 2,01 mg H20.
C12fl12. Ber.: C 92,25 und H7,75°/0.Gef.: C 92,13 und H 7,89 V
C 92,23 und H 7,60 °/0.

53
Aus dem Kohlenwasserstoff wurde mit der berechneten Menge
Pikrinsäure das Pikrat hergestellt, das aus Methylalkohol in gold¬
gelben, verfilzten Nädelchen kristallisierte. Fp. 136—136,5°.
3,830 mg Substanz gaben 7,91 mg C02 und 1,41 mg H20.
dsH^O,^. Ber.: C 56,10 und H 3,93 °/0,
Gef.: C 56,33 und H 4,12 °/0.
Das Styphnat kristallisierte aus Methylalkohol in blaßgelben,
feinen Nadeln, die bei 158—159,5° schmolzen.
3,957 mg Substanz gaben 7,84 mg C02 und 1,405 mg H20.
C19rl1608N3. Ber.: C 53,85 und H 3,77V
Gef.: C 54,04 und ti 3,97°/0.
Das in analoger Weise dargestellte Trinitrobenzolat kristallisierte
aus Äthylalkohol in kanariengelben, feinen Nädelchen, die bei
151—151,5° schmolzen.
Die Mischprobe des erhaltenen Kohlenwasserstoffs, des Pikrats,
Styphnats und Trinitrobenzolats, mit synthetisch dargestelltem
2,7-Dimethyl-naphthalin bzw. dessen Derivaten zeigte keine
Schmelzpunktsdepression.Die Mischprobe des Styphnats mit 1,2,7-Trimethyl-naphthalin-
(Sapotalin-) styphnat (Fp. 150°) gab eine Depression von ca. 10°, wo¬
gegen das Pikrat mit dem synthetischen 1,2,7-Trimethyl-naphthalin-
pikrat (Fp. 129°) keine Schmelzpunktserniedrigung erkennen ließ.
Fraktion 2b wurde mit der äquivalenten Menge Pikrinsäure in
Methylalkohol gelöst und das gebildete Pikrat wiederholt fraktioniert
kristallisiert. Man erhielt ausschließlich ein dunkel orangerotes,
in feinen Nädelchen kristallisierendes Pikrat, das bei 132—132,5°
schmolz.
4,395 mg Substanz gaben 9,21 mg C02 und 1,77 mg H20.
3,841 mg Substanz gaben 8,07 mg C02 und 1,51 mg H20.
C19H170,Na. Ber.: C 57,13 und H 4,28 °/0.
Gef.: C 57,15 und H 4,50 V
C 57,30 und H 4,40V
Aus der bei 132° schmelzenden Pikratfraktion wurde der Kohlen¬
wasserstoff regeniert und ins Styphnat übergeführt. Die orange-

54
gelben Nadeln schmolzen nach dem Umkristallisieren aus Methyl¬alkohol bei 156,5°.
3,850 mg Substanz gaben 7,76 mg C02 und 1,45 mg H20.
C19H1708N3. Ber.: C 54,92 und H 4,13 VGef.: C 54,97 und H 4,22 °/0.
Ferner wurde ein Teil des aus dem Pikrat regenerierten Kohlen¬
wasserstoffs mit Trinitrobenzol umgesetzt. Das erhaltene hellgelbeSapotalin-Trinitrobenzolat bildete in Methylalkohol feine
Nädelchen, die nach einmaligem Umkristallisieren scharf bei 143°
schmolzen.
Das Sapotalin erwies sich nach Schmelzpunkt und Mischprobedes Pikrats, des Styphnats und des Trinitrobenzolats unzweideutigals das 1,2,7-Trimethyl-naphthalin.
Fraktion 2c lieferte beim Behandeln mit Pikrinsäure ein Pikrat,aus dem durch wiederholtes fraktioniertes Kristallisieren aus Methyl¬alkohol nach dem Dreieck-Schema ein, gegenüber dem soeben be¬
schriebenen Sapotalin-Pikrat, bedeutend dunkler orange-rotes Pikrat
erhalten wurde. Der Schmelzpunkt lag scharf zwischen 134—134,5°.
3,830 mg Substanz gaben 7,91 mg C02 und 1,41 mg H20.
C18fl150,Ns. Ber.: C 56,10 und H 3,93 °/0.C19H1707N3. Ber.: C 57,12 und H 4,29 °/0.
Gef.: C 55,76 und H 4,01 °/0.
Die Mischprobe mit Sapotalin- sowie mit synthetischem 1,2,7-Tri-methyl-naphthalin-Pikrat gab keine Erniedrigung des Schmelzpunktes.
Ein Teil des erhaltenen Pikrats wurde zersetzt und der so ge¬
wonnene flüssige Kohlenwasserstoff mit Styphninsäure ins Styphnatübergeführt. Dieses kristallisierte aus Methylalkohol in feinen gelbenNädelchen, die bei 155° schmolzen und mit Sapotalin-Styphnatebenfalls keine Schmelzpunktsdepression erkennen ließen.
Die nicht pikratbildenden öle der Fraktion 2 wurden erneut mit
der gleichen Gewichtsmenge Selen dehydriert. Das Reaktionsgemischwurde in bekannter Weise mit Äther ausgezogen und destilliert
Neben einem geringfügigen Vor- und Nachlauf destillierte die Haupt¬menge bei 133° (11 mm) als farbloses öl. Dieses wurde wiederum

55
ins Pikrat verwandelt, das nach mehrmaligem Umkristallisieren aus
Methylalkohol orange-rote, bei 129,5—130° schmelzende Nadeln
lieferte, die nach der Mischprobe mit Sapotalin-Pikrat identisch
waren.
3,591 mg Substanz gaben 7,54 mg C02 und 1,41 mg H20.
CmHjANa. Ber.: C 57,12 und H 4,29 °/0.
Gef.: C 57,26 und H 4,39 V
Der aus dem Pikrat regenerierte Kohlenwasserstoff gab ein bei
156,5—157° schmelzendes, orange-gelbes Styphnat, das mit Sapotalin-
Styphnat keine Depression des Schmelzpunktes zeigte.
Untersuchung des Vorlaufs Fraktion 1.
[Kp. -80° (14 mm).]
Der Vorlauf Fraktion 1 wurde mit den aus Fraktion 2 stammenden
nicht-pikratfähigen ölen vereinigt und erneut destilliert, wobei
folgende Fraktionen erhalten wurden:
Fraktion la....
—95° (10 mm) ca. 45 — 50%.
Fraktion lb. .
95-115° (10 mm) ca. 30-35°/0.
Fraktion lc.115—145° (10 mm) ca. 20%.
Der Vorlauf war eine sehr leicht bewegliche, äußerst übelriechende
Flüssigkeit, die beim Erwärmen mit Natrium völlig zersetzt wurde.
Fraktion lb reagierte bedeutend weniger mit Natrium, und nach
zweimaliger Destillation über Natrium konnte ein farbloses, nahezu
geruchloses Öl gewonnen werden, dessen Siedepunkt zwischen
105— 115° (10 mm) lag. Eine Probe dieses Öls wurde mit methyl¬
alkoholischer Pikrinsäure auf dem Uhrglas Eindunsten gelassen. Es
bildete sich neben auskristallisierender Pikrinsäure ein blaßgelbes
Pikrat, das aber bei weiterem Umkristallisieren aus Methylalkohol
wieder gespalten wurde.
Eine weitere Probe wurde in Methylalkohol suspendiert und mit
wenig Brom verrieben. Dabei trat eine heftige Reaktion ein unter
Abscheidung eines weißen, festen Produktes. Es wurde noch weiter
soviel Brom hinzugefügt, daß dessen Farbe gerade bestehen blieb, und
das Ganze über Nacht sich selbst überlassen. Der ausgeschiedene

56
Bromkörper wurde abgenutscht und mehrmals aus Methylalkoholumkristallisiert, wonach das Produkt konstant zwischen 203—204°
schmolz.
3,388 mg Substanz gaben 5,080 mg C02 und 1,284 mg rl20.
Ci0ri12Br2. Ber.: C 41,11 und H4,13°/0.Gef.: C 40,90 und H 4,24 °/0.
Mit der bei 203 — 204° schmelzenden Dibromverbindung des
1,2,3,4-Tetramethyl-benzols konnte keine Schmelzpunktsdepressionbeobachtet werden.
Über die höhersiedenden Dehydrierungsprodukte des tiedera-
genins.
Fraktion 3, Kp. 80—190° (0,2 mm), wurde nochmals im Vakuum
fraktioniert destilliert, wobei folgende Anteile erhalten wurden:
Fraktion 3a: —120° (0,25 mm); dünnflüssig, nicht erstarrend,
hellbraun-gelb, ca. 20°/0.Fraktion 3b: 120—130° (0,25 mm); dünnflüssig, später teilweise
kristallisierend, ca. 10 %•Fraktion 3c: 130— 140° (0,20 mm); zur Hauptsache erstarrend,
hellbraun, ca. 10—15°/0.Fraktion 3d: 140—160° (0,15 mm); schon sehr dickflüssig, noch
reichliche Kristall - Bildung,dunkelbraungelb, ca. 15—20°/0.
Fraktion 3e: 160—190° (0,15 mm); sehr dickflüssig, rote Selen¬
schlieren, ca. 10 —15%-Fraktion 3f : 190—250° (0,13 mm); äußerst zähflüssig, glasig er¬
starrend, hellgelb, ca. 25—30%.
Die Fraktionen 3b—3e wurden mit Hexan versetzt und mit dem
Glasstab angerieben, wobei die öligen Anteile in Lösung gingenund die Kristalle als feinkörnige Masse sich auf dem Boden des
Gefäßes ansammelten. Man ließ die Fraktionen zur Vervollständigungder Ausscheidung im Kühlschrank bei —15° stehen und nutschte
darauf in der Kälte ab. Der Rückstand auf dem Filter wurde noch
mit ebenfalls gekühltem Hexan nachgewaschen und dann getrocknet.

57
Die einzelnen Fraktionen wurden auf ihren Schmelzpunkt unter¬
sucht, wobei sich zeigte, daß die aus den Fraktionen 3b und 3c
erhaltenen kristallinen Produkte übereinstimmend zwischen 138—141°
schmolzen, während der Schmelzpunkt aus Fraktion 3d auf 105—110°
und derjenige der Fraktion 3e bis auf 97—110° gesunken war.
In bezug auf die Löslichkeit in Hexan erwiesen sich die beiden
ersten Fraktionen als ziemlich schwer löslich, wogegen die beiden
letzten, zumal die letzte Fraktion bedeutend leichter löslich waren.
In Methyl- und Äthylalkohol lagen die Verhältnisse gerade um¬
gekehrt. Der Unterschied wurde noch besser im verschiedenen Ver¬
halten gegen verdünnte Kalilauge erkannt, worin die ersten beiden
Fraktionen (aus 3b und 3 c) glatt mit violetter Fluoreszenz löslich
waren und daraus durch verdünnte Säure wieder ausgefällt werden
konnten. Die Kristalle aus Fraktion 3d waren nur noch sehr wenig
löslich; die Fluoreszenz war kaum erkennbar, während Fraktion 3e
in verdünnter Kalilauge völlig unlöslich war.
Untersuchung der sauren Anteile aus Fraktion 3.
Die Fraktionen 3b und 3c wurden zusammen genommen und
aus Methylalkohol-Wasser umkristallisiert. Man erhielt kleine,
weiße Blättchen, die bei 157° schmolzen. Schließlich wurde das
Präparat noch sublimiert. Der Schmelzpunkt änderte sich aber
dabei nicht mehr.
3,509 mg Substanz gaben 10,78 mg C02 und 2,37 mg H20.
8,663 mg Substanz gaben 11,23 mg C02 und 2,51 mg H20.
CisHuO. Ber.: C 83,82 und H 7,58 °/0.
Gef.: C 83,79 und H 7,56 °/0.
C 83,61 und H 7,67 °/0.
In verdünnter wäßriger Kalilauge lösten sich die Kristalle glatt auf,
wobei die klare Lösung eine deutlich blaue Fluoreszenz zeigte.
Aus dem Trimethyl-naphthol wurde durch Umsetzung mit der
äquivatenten Menge Pikrinsäure in Methylalkohol das Pikrat her¬
gestellt, das in dunkelroten, derben Nädelchen kristallisierte. Das
Präparat schmolz bei 164° unter Zersetzung und zerfiel bei weiterem
Umkristallisieren aus Methylalkohol wieder in seine Komponenten.

58
Trimethyl - naphthol - methyläther.
Da die Umsetzung des Trimethyl-naphthols mit Diazomethan nur
äußerst langsam vor sich ging, wurde die Methylierung mitDimethyl-sulfat bewerkstelligt.Man löste das Naphthol in einem kleinen Überschuß Claisenscher
Lauge (350 g Kaliumhydroxyd und 250 ccm Wasser mit Methyl¬alkohol auf 1 Liter aufgefüllt), verdünnte es mit dem gleichen VolumenMethylalkohol und ließ in der Siedehitze 1 Mol Dimethylsulfat zu-
tropfen. Den Zusatz von Lauge und Dimethylsulfat wiederholte
man noch achtmal. Das Reaktionsgemisch wurde nun in Äther
aufgenommen und mit Claisenscher Lauge gut durchgeschüttelt. Der
erhaltene Methyläther schmolz nach mehrmaligem Umkristallisierenaus Methylalkohol bei 89,5— 90°. Die farblosen Blättchen fluores¬zierten im ultravioletten Licht hellblau.
2,988 mg Substanz gaben 9,23 mg C02 und 2,20 mg H80.
C14H160. Ber.: C 84,0 und H 8,0 °/0.Gef.: C 84,25 und H 8,24 °/0.
Das in methylalkoholischer Lösung bereitete Trinitrobenzolatkristallisierte aus dem gleichen Lösungsmittel in langen, roten
Nädelchen, die zwischen 145 —146° schmolzen.
3,373 mg Substanz gaben 7,26 mg COa und 1,27 mg H20.
C2oHi70,N3. Ber.: C 58,4 und H4,l°/0.Gef.: C 58,7 und H 4,21 °/0.
Zinkstaubdestillation des Trimethyl-naphthols.
Das Naphthol wurde mit der zwanzigfachen Menge reinem Zinkstaub
gemischt und in einem Rohre in ganz schwachem Stickstoffstrom15 Minuten auf 400° erhitzt. Den Rohrinhalt kochte man mit Ätherwiederholt aus und schüttelte die ätherische Lösung mit Natronlauge.Das im Äther gelöste neutrale öl destillierte man über Natrium und
versetzte es mit der berechneten Menge alkoholischer Pikrinsäure.Man erhielt ein orangefarbenes Pikrat, das bei 127—127,5° schmolzund nach Mischprobe mit Sapotalin-Pikrat identisch war.
Der aus dem Pikrat regenerierte Kohlenwasserstoff wurde mitTrinitrobenzol umgesetzt. Die erhaltene hellgelbe Additionsverbindung

59
schmolz nach dem Umkristallisieren aus Alkohol bei 143° und gab
mit dem bei der gleichen Temperatur schmelzenden, aus syn¬
thetischem 1, 2, 7-Trimethylnaphthalin bereiteten Trinitrobenzolat
gemischt keine Schmelzpunktsdepression.
Untersuchung der neutralen Anteile aus Fraktion 3.
Die kristallinen Anteile aus den Fraktionen 3d und 3e wurden
vereinigt und aus Äthylalkohol umkristallisiert. Man erhielt kleine,
derbe Prismen, die bei 126 — 127° schmolzen.
3,186 mg Substanz gaben 10,80 mg C02 und 2,17 mg rl20.
3,349 mg Substanz gaben 11,34 mg COa und 2,31 mg H20.
3,203 mg Substanz gaben 10,85 mg COa und 2,23 mg H20.
C17rl16. Ben: C 92,67 und H 7,33 °/0. Molekulargewicht: 220,13.
CiaH,9. Ber.: C 92,25 und H 7,75°/0. Molekulargewicht: 234,14.
C19H20. Ben: C 91,88 und H8,12°/0. Molekulargewicht: 248,16.
Gef.: C 92,35 und H 7,62 °/0.
C 92,35 und H 7,72 °/0.
C 92,37 und H 7,79 °/0.
0,962 mg Substanz und 4,692 mg Campher gaben ein J 31,8 und 31,4°.
Gef. Molekulargewicht: 258.
Aus dem Kohlenwasserstoff wurde mit der berechneten Menge
Pikrinsäure das Pikrat hergestellt, das aus Methylalkohol in feinen,
orangefarbenen Nädelchen kristallisierte. Schmelzpunkt 165—165,5°.
Nach einmaligem Umkristallisieren aus demselben Lösungsmittel sank
der Schmelzpunkt schon auf 160°. Das Präparat zeigte im Fluoreszenz¬
mikroskop eine Menge hellblau fluoreszierender Pünktchen; es war
somit schon Spaltung eingetreten. Nach mehrmaligem Umlösen
konnte schließlich der Kohlenwasserstoff wieder zurück erhalten
werden.
Das Styphnat kristallisierte beim Zusammengeben äquivalenter
Mengen Kohlenwasserstoff und Styphninsäure aus Methylalkohol
in feinen, orange-gelben Nädelchen, die bei 174—175° schmolzen.
Es zersetzte sich ebenfalls bei weiterem Umkristallisieren.
In analoger Weise wurde das Trinitrobenzolat, gelbe, lange
Nadeln, erhalten, welches zwischen 174—175° schmolz und eben¬
falls zur Zersetzung neigte.

60
Oxydation des Kohlenwasserstoffes mit Chromsäure.
0,2 g Kohlenwasserstoff wurden in 25 ccm reinstem Eisessiggelöst, mit 0,17 g Chromsäureanhydrid versetzt und auf dem Wasser¬
bad während zwei Stunden erwärmt. Nach einigen Minuten schon
färbt sich die Lösung violett-rot und scheidet nach dem Erkalten-
und Stehenlassen über Nacht kleine, braunrote Kriställchen ab, diezwischen 193—200° schmolzen. Das Oxydationsprodukt schmolznach zweimaligem Umkristallisieren konstant bei 203—204°.
3,274 mg Substanz gaben 9,805 mg C02 und 1,81 mg H20.
3,162 mg Substanz gaben 9,48 mg C02 und 1,74 mg H20.
C^HuO,. Ber.: C 81,56 und H5,64°/0.
Ci8H,eOä. Ber.: C 81,78 und H 6,11 °/0.
C19H1802. Ber.: C 81,97 und H 6,52 °/0.
Gef.: C 81,68 und H 6,19 °/0.
C 81,76 und H 6,15 °/0.
Untersuchung der Fraktion 4.
[Kp. 190—300° (0,2 mm)].
Fraktion 4 wurde in heißem Äthylalkohol gelöst und dann im
Kühlschrank mehrere Tage bei —15° stehen gelassen. Dabei schieden
sich geringe Mengen einer gelblich-weißen Masse aus, die aus
Benzol und darauf aus Dioxan umkristallisiert wurde. Man erhielt
kleine, weiße Blättchen, die bei 245° schmolzen.
3,563 mg Substanz gaben 12,05 mg C(>2 und 2,31 mg H20.
3,312 mg Substanz gaben 11,21 mg C02 und 2,08 mg H20.
Gef.: C 92,24 und H 7,26°/0.
\C 92,31 und H 7,03 °/0.
Das Präparat, das sehr wahrscheinlich einen Kohlenwasserstoff
darstellt, wurde zur weiteren Reinigung noch sublimiert. Der
Schmelzpunkt stieg dabei auf 250°.
1,481 mg Substanz gaben 5,00 mg C02 und 0,97 mg H20.
Gef.: C 92,07 und H 7,33 °/0.

61
Diese Analysenwerte entsprechen ungefähr der Formel Cntin (be¬
rechnet: C 92,3 und fi 7,7 °/0)- Es gelang nicht, ein Pikrat her¬
zustellen, da es zu große Neigung zeigte, wieder in die Komponenten
zu zerfallen.
Der beim Auskochen des rohen Dehydrierungsproduktes mit Äther
verbleibende Rückstand wurde seinerzeit mit Äther weiter er¬
schöpfend extrahiert. Dabei bildete sich nach einigen Tagen in dem
Atherextrakt ein voluminöser, brauner Niederschlag, der auf einer
Nutsche gesammelt und getrocknet wurde. Dieses wurde in der
nötigen Menge Cutnol heiß gelöst, die braune Lösung mit wenig
Carboraffin entfärbt, worauf beim Abkühlen derbe, weiße Platten
auskristallisierten. Der Schmelzpunkt lag bei 302—303°.
3,363 mg Substanz gaben 11,57 mg C02 und 5,92 mg H20.
C24H18. Ber.: C 94,07 und H 5,93 °/0.
CA. Ber.: C 93,70 und H 6,30°/0.
Gef.: C 93,83 und H 5,96 °/0.
Der Kohlenwasserstoff wurde weiter aus Pyridin umkristallisiert.
Der Schmelzpunkt blieb nach mehrmaligem Kristallisieren bei
306 — 307° konstant.
3,261 mg Substanz gaben 11,225 mg C02 und 1,785 mg H20.
3,511 mg Substanz gaben 12,07 mg C02 und 1,93 mg H20.
3,258 mg Substanz gaben 11,21 mg COa und 1,78 mg H20.
Gef.: C 93,88 und H 6,13 °/0.
C 93,76 und H 6,15 V
C 93,84 und H 6,11 V
Oxydation des Kohlenwasserstoffes mit Chromsäure.
10 g Trimethylpicen wurden in 600 ccm reinstem Eisessig auf¬
geschwemmt und in der Siedehitze langsam mit 14 g Chromsäure
versetzt. Nach dem Eintragen der letzten Chromsäureportion wurde
noch insgesamt 8 Stunden am Rückfluß gekocht, wobei der Kohlen¬
wasserstoff langsam teilweise in Lösung ging. Schließlich wurde
heiß filtriert und erkalten gelassen. Beim Stehenlassen über Nacht
kristallisierten kleine, tiefrote Drusen, die aber keinen Schmelzpunkt
aufwiesen, sondern lediglich über 350° Zersetzung erlitten. Zur

62
Analyse wurde das Präparat noch zweimal aus Eisessig umgefälltund im Hochvakuum scharf getrocknet.
3,430 mg Substanz gaben 10,76 mg C02 und 1,51 mg H20.
C24Hi602. Ber.: C 85,68 und H4,80°/o.C25H1802. Ben: C 85,68 und H5,180/0.C28H20O3. Ber.: C 85,68 und H 5,54%.
Gef.: C 85,61 und H 4,93 °/0.
* *
*
Die Analysen wurden im Mikrolaboratorium der EidgenössischenTechnischen Hochschule unter Leitung von Herrn Dr. M. Furter
ausgeführt.

Curriculum vitae.
Am 25. Juni 1905 wurde ich, Karl Ludwig Friedrich Ehmann,
Bürger von Ernetschwil (Kanton St. Gallen), Sohri des Karl Johann
Friedrich Ehmann und der Johanne Wilhelmine Auguste geb. Unterberg,
in St. Gallen geboren. Daselbst besuchte ich die städtischen Primar¬
und Sekundärschulen, und nach Absolvierung der technischen Ab¬
teilung der St. Gallischen Kantonsschule beschloß ich im Herbst 1924
die Mittelschulbildung mit der Reifeprüfung. Von der Zeit. ab
studierte ich an der chemischen Abteilung der EidgenössischenTechnischen Hochschule in Zürich und erhielt im Frühjahr 1928
das Diplom als Ingenieur-Chemiker. Ich arbeitete dann drei Semester
bei Herrn Prof. R. Kuhn. Hierauf wurde ich im Herbst 1929 bei
Herrn Prof. L. Ruzicka Saal-Assistent und habe nebenbei auch
vorliegende Untersuchungen über Hederagenin ausgeführt. Seit
Frühjahr 1933 arbeite ich in der pharmazeutisch-wissenschaftlichen
Abteilung der Gesellschaft für Chemische Industrie in Basel.
Basel, im Februar 1934.










![CONFLICT OF LAWS - docshare01.docshare.tipsdocshare01.docshare.tips/files/15748/157483295.pdf[substance v. procedural distinction] Cadalin v. POEA: if forum state has borrowing law](https://img.pdfslide.net/doc/110x75/5ad20d0a7f8b9a0f198bfcfa/conflict-of-laws-substance-v-procedural-distinction-cadalin-v-poea-if-forum.jpg)


















