Embed Size (px)
Citation preview

ORIGINAL PAPERJournal of PathologyJ Pathol 2010; 221: 264–274Published online 23 April 2010 in Wiley InterScience(www.interscience.wiley.com) DOI: 10.1002/path.2703
In vivo hepatic endoplasmic reticulum stress in patientswith chronic hepatitis CTarik Asselah,1 – 3* Ivan Bieche,4,5 Abdellah Mansouri,1,2 Ingrid Laurendeau,4 Dominique Cazals-Hatem,6Gerard Feldmann,1,2 Pierre Bedossa,1,2,6 Valerie Paradis,1,2,6 Michelle Martinot-Peignoux,1 – 3 Didier Lebrec,1 – 3
Cecile Guichard,1,2 Eric Ogier-Denis,1,2 Michel Vidaud,4,5 Zera Tellier,7 Vassili Soumelis,8 Patrick Marcellin1 – 3
and Richard Moreau1 – 3
1 INSERM U773, Centre de Recherche CRB3, Paris, 75018, France2 Universite Denis Diderot-Paris 7, Site Bichat, 75018, France3 Service d’Hepatologie, Hopital Beaujon, Clichy, 92118, France4 INSERM, U745, Universite Rene Descartes, Paris 75006, France5 Service de Genetique Moleculaire, Hopital Beaujon, Clichy, France6 Service d’Anatomie Pathologie, Hopital Beaujon, Clichy, France7 Laboratoire Francais du Fractionnement et des Biotechnologies, Courtaboeuf, France8 INSERM U653, Institut Curie, 75245 Paris, France
*Correspondence to: Tarik Asselah, Service d’Hepatologie, Hopital Beaujon, Clichy, 92118, France. e-mail: [email protected]
AbstractIn hepatocytes, the accumulation of unfolded proteins in the endoplasmic reticulum (ER) causes ER stress andthe unfolded protein response (UPR), mediated by the ER-resident stress sensors ATF-6, IRE1, and PERK. UPR-responsive genes are involved in the fate of ER-stressed cells. Cells carrying hepatitis C virus (HCV) subgenomicreplicons exhibit in vitro ER stress and suggest that HCV inhibits the UPR. Since in vivo ER homeostasis is unknownin livers with chronic HCV infection, we investigated ER stress and the UPR in liver samples from untreatedpatients with chronic hepatitis C (CHC), in comparison with normal livers. Electron microscopy, western blotting,and real-time RT-PCR were used in liver biopsy specimens. Electron microscopy identified features showing ERstress in hepatocyte samples from patients with CHC; however, ‘ER-stressed’ hepatocytes were found in clusters(3-5 cells) that were scattered in the liver parenchyma. Western blot analysis confirmed the existence of hepaticER stress by showing activation of the three ER stress sensors ATF-6, IRE1, and PERK in CHC. Real-time RT-PCRshowed no significant induction of UPR-responsive genes in CHC. In contrast, genes involved in the control ofdiffuse processes such as liver proliferation, inflammation, and apoptosis were significantly induced in CHC. Inconclusion, livers from patients with untreated CHC exhibit in vivo hepatocyte ER stress and activation of thethree UPR sensors without apparent induction of UPR-responsive genes. This lack of gene induction may beexplained by the inhibiting action of HCV per se (as suggested by in vitro studies) and/or by our finding of thelocalized nature of hepatocyte ER stress.Copyright 2010 Pathological Society of Great Britain and Ireland. Published by John Wiley & Sons, Ltd.
Keywords: electron microscopy; chronic hepatitis; CREB3L3; eIF-2-alpha; unfolded protein response; physiology; pathology; inflammation
Received 27 August 2009; Revised 12 February 2010; Accepted 16 February 2010
No conflicts of interest were declared.
Introduction
In cells, newly synthesized secretory and membrane-associated proteins are correctly folded and assem-bled in the endoplasmic reticulum (ER) [1–3]. OnceER homeostasis is perturbed by various pathologicalconditions, newly synthesized unfolded proteins accu-mulate in the ER, resulting in ER stress [1–3]. Tocope with accumulated unfolded ER proteins, mam-malian cells trigger a specific adaptive response calledthe unfolded protein response (UPR) [1–3] (Figure 1).There are three distinct signalling pathways that areinduced by ER stress mediated by three ER-residentstress sensors, ie the activating transcription factor 6
(ATF-6), the inositol requiring enzyme 1 (IRE1), andthe double-stranded RNA-activated protein kinase-likeER kinase (PERK) [4–6].
ATF-6 is synthesized as a precursor protein andanchored to the ER membrane, where it is retainedby the HSP70-class ER chaperone BiP [also known asglucose-related protein (GRP) 78] [1–3]. In response toER stress, ATF-6 is released from BiP and transportedto the Golgi complex, where ATF-6 undergoes regu-lated intramembrane proteolysis, ie sequential cleavageby two proteases, S1P and S2P [2]. The processed formof ATF-6, which may target genes since it is a tran-scription factor [belonging to the basic-region leucinezipper (bZIP) family [7]], translocates to the nucleus
Copyright 2010 Pathological Society of Great Britain and Ireland. J Pathol 2010; 221: 264–274Published by John Wiley & Sons, Ltd. www.pathsoc.org.uk www.thejournalofpathology.com

In vivo hepatic ER stress in patients with chronic hepatitis C 265
Figure 1. Adaptative signalling of the unfolded protein response (UPR). The three UPR transducers PERK, ATF-6, and IRE1 are associatedwith BiP in their monomeric inactive form. Upon ER stress-dependent accumulation of unfolded proteins in the ER lumen, Bip is releasedfrom these sensors, inducing their homodimerization and subsequent activation. Activated PERK phosphorylates the translation initiationfactor eIF-2α, which attenuates the general translation rate and induces the translation of selective mRNAs with inhibitory uORFs in their5′ UTR, such as the transcriptional factor ATF-4. GADD34, a target of ATF-4 activation, negatively regulates the eIF-2α-mediated inhibitionof translation. The release of BiP from ATF-6 induces its trafficking into the Golgi apparatus, where it is cleaved by proteases S1P/S2P andthereby enters the nucleus to activate the transcription of XBP1 and other UPR target genes. Activated IRE1 catalyses the splicing of XBP1mRNA to allow the translation of mature XBP-1 protein. XBP-1 mediates transcriptional up-regulation of numerous ER stress-dependentgenes. The downstream effectors of these three signalling pathways combinatorially induce the expression of genes involved in the ERprotein folding capacity, such as chaperones. In parallel, the ER-associated degradation (ERAD) process is activated to remove misfoldedproteins from the ER lumen in coordination with the proteasome machinery.
[1,5–7]. The second UPR branch involves IRE1 andX-box-binding protein 1 (XBP-1) [1–4]. IRE1 containsboth serine/threonine kinase and ribonuclease domains.Under normal conditions, XBP1 mRNA is translated,but its product is a weak transcriptional activator witha short protein half-life [2]. During ER stress, activatedIRE1 cuts 26 nucleotides from XBP1 mRNA to gener-ate spliced XBP1 mRNA, which encodes the more sta-ble and transcriptionally active XBP-1S protein (whichbelongs to the family of bZIP transcription factors [2]).The third UPR branch is mediated by PERK, whichis a serine/threonine protein kinase that phosphory-lates the alpha subunit of eukaryotic translation ini-tiation factor 2 (eIF2-alpha) [1–3]. Phosphorylation ofeIF2-alpha subsequently inhibits global protein synthe-sis [1–3]. Paradoxically, eIF2-alpha phosphorylationinduces translation of activating transcription factor 4(ATF4 ) mRNA into the bZIP transcription factor ATF-4 [2].
The UPR activates the transcription of certain genes(eg those encoding chaperones, lectins, and calciumpump) that serve to increase the ER’s protein folding
capacity as needed. UPR signalling can protect cellsfrom ER stress by expanding the amount of ER in thecell, enhancing the degradation of misfolded proteins(eg via XBP-1S induction of EDEM1 ), and reducingthe synthesis of new proteins (via eIF2-alpha phos-phorylation) [1–3]. In addition, the UPR may alsopromote an adaptive response, known as the ER over-load response (EOR), which involves IRE1-mediatedactivation of nuclear factor (NF)-kappaB, a transcrip-tion factor involved in anti-apoptotic responses [8].The EOR has also been shown to involve GSK3-beta, which inhibits p53-induced apoptosis [9]. How-ever, when the adaptive responses are not sufficient torelieve the ER stress, apoptotic pathway(s) are acti-vated via IRE1 or eIF2-alpha/ATF-4 pathways [1,2].IRE1 is associated with the apoptosis-regulating BCL-2 protein family members BAK and BAX [10]. IRE1signalling has been shown to activate the c-Jun NH2-terminal kinase (JNK) and/or caspase-12 [1,2]. ERstress-induced apoptosis may also result from theinduction of the ATF-4 target gene DDIT3 [which
Copyright 2010 Pathological Society of Great Britain and Ireland. J Pathol 2010; 221: 264–274Published by John Wiley & Sons, Ltd. www.pathsoc.org.uk www.thejournalofpathology.com

266 T Asselah et al
codes for C/EBP homologous protein-10 (CHOP), abZIP transcription factor [1,2,7]].
Chronic hepatitis C (CHC) is a leading cause ofcirrhosis and hepatocellular carcinoma worldwide [11].HCV is an enveloped flavivirus with a 9.6-kb single-strand RNA genome [11]. This genome serves asa template for replication and as a viral messengerRNA for production of the virus. HCV enzymes (ieNS2–3 and NS3–4A proteases, NS3 helicase, andNS5B RdRp) are essential for HCV replication [11].The development of a subgenomic HCV RNA repliconcapable of replication in the human hepatoma cell lineHuh7 has been a significant advance [12,13]. Recently,complete replication of HCV in cell culture has beenachieved [14].
HCV core synthesis, processing, and folding occurin the ER. Envelope proteins E1 and E2 reside inthe ER lumen and the viral replicase is assumed tolocalize on ER-derived membranes [15–17], leadingto the possibility of inducing the ER-resident stresssensors. In vitro studies have shown that ER stress maybe triggered by HCV subgenomic replicons [18,19],structural or non-structural proteins [20–23]. In vitrostudies also suggest that HCV may interfere withthe UPR to inhibit it [24]. Together, in vitro studiessuggest that HCV may trigger ER stress and inhibitthe resulting UPR. However, experimental models forHCV infection have limitations. For instance, thesemodels remained unable to release infectious HCVparticles [12,13]. Therefore, in vivo studies are ofmajor importance. To date, there is no information onin vivo ER homeostasis and the UPR in CHC. Thus,this study investigated in vivo ER stress and the UPRin liver samples from patients with CHC.
Materials and methods
Patients and tissue specimensPercutaneous liver biopsies from 28 untreated patientswith CHC were studied (Table 1); 13 had mild fibro-sis and 15 advanced fibrosis (Supporting informa-tion, Supplementary materials and methods). Percu-taneous liver-biopsy specimens were selected froma cohort of adult patients with untreated CHC fol-lowed at Beaujon Hospital (Clichy, France). All CHCpatients had antibodies against HCV (AxSYM Anti-HCV, Abbott) and detectable serum HCV RNA (TMA,Bayer’s Versant HCV RNA Qualitative Assay). HCVgenotyping was performed (sequencing) and serumHCV RNA was quantified [Bayer’s Versant HCVRNA 3.0 Assay (bDNA)] for all patients. No patienthad clinical evidence of hepatic decompensation. Thefollowing conditions were excluded: positive HBsAg,human immunodeficiency virus infection, autoimmunehepatitis, haemochromatosis, alpha1-antitrypsin defi-ciency, and Wilson’s disease. Characteristics of thesepatients are presented in Table 1. Pretreatment liverbiopsies from patients before decision to treat was
Table 1. Characteristics of 28 patients with chronic hepatitis CVariable Patients
No 28Gender (male, n) 19Age (years) 45.3 ± 8.2Source of infection (n)
Blood transfusion 9Intravenous drug use 9Unknown 10
Body mass index (kg/m2) (± SD) 21.3 ± 1.9Blood glucose (mmol/l) (± SD) 5.1 ± 1.2ALT (IU/l, median) 114 ± 43HCV genotype (n)
1 152 43 74 2
Viral load (mean, log10 IU/ml) 5.2 ± 0.4Stage
F1 13Stage (n)F3–F4 15
made, both immediately frozen liver tissue (stored at−80 ◦C) and fixed paraffin-embedded tissue (for his-tology), were available.
Ten patients had mild chronic hepatitis B. All ofthese patients had positive hepatitis B surface anti-gen (HBsAg), detectable serum HBV DNA [Bayer’sVersant HBV DNA 3.0 Assay (bDNA)], and wereuntreated. No patient had clinical evidence of hepaticdecompensation. Moreover, in the present study, thefollowing conditions were excluded: positive antibod-ies against HCV, human immunodeficiency virus infec-tion, autoimmune hepatitis, haemochromatosis, alpha1-antitrypsin deficiency, and Wilson’s disease.
Liver biopsies from seven adults with normal liverhistological aspects were studied [25].
Percutaneous liver-biopsy specimens were obtainedfrom adults with mild elevated alanine aminotrans-ferase activity, with no cause of liver disease (med-ication, alcohol, chronic viral hepatitis, autoimmuneprocesses, and metabolic disease). All of these adultsgave their informed consent for the study. All of theseliver tissue specimens were histologically normal (ieabsence of inflammation, fibrosis, and pathological pat-tern). The study was approved by the Local Committeeof Ethics and conformed to the ethical guidelines ofHelsinki. All patients gave their informed consent priorto liver biopsy.
Transmission electron microscopyBlinded electron microscopy was performed in 13patients with mild CHC and five histologically normalcontrols. In fact, since cirrhosis and advanced fibrosisare associated with several features (apoptosis, necro-sis, cholestasis, etc), we hypothesized that ER stressactivation might not be specific for HCV but relatedto the development of cirrhosis. Thus, we decided tostudy patients with chronic HCV infection and mildliver disease. Liver tissue was immediately fixed by
Copyright 2010 Pathological Society of Great Britain and Ireland. J Pathol 2010; 221: 264–274Published by John Wiley & Sons, Ltd. www.pathsoc.org.uk www.thejournalofpathology.com

In vivo hepatic ER stress in patients with chronic hepatitis C 267
Table 2. Unfolded protein response (UPR)-responsive genes in livers from patients with mild or advanced HCV-related fibrosis relative tonormal livers
Advanced p value p valueNormal Mild HCV-related fibrosis HCV-related Mild HCV-related Mild vs advanced
Gene (n = 7) (n = 13) fibrosis (n = 15) fibrosis vs normal HCV-related fibrosis
XBP1 1.17 (0.65–1.31) 0.87 (0.22–1.39) 0.58 (0.16–1.14) 0.19 0.19Spliced XBP1 1.19 (0.33–1.45) 1.29 (0.27–1.98) 0.79 (0.26–2.31) 0.75 0.31Ratio of spliced XBP1 to XBP1 1.02 (0.42–1.25) 1.25 (0.82–2.00) 1.36 (0.81–2.03) 0.02 0.56ATF6 1.00 (0.67–1.18) 1.09 (0.71–3.96) 1.23 (0.47–2.71) 0.15 0.91CREBL1 1.00 (0.86–1.26) 0.85 (1.06–2.90) 0.79 (0.41–1.96) 0.16 0.60ATF4 1.00 (0.80–1.24) 0.90 (0.67–2.61) 1.10 (0.56–2.19) 0.43 0.20HSPA5 0.96 (0.71–2.21) 0.83 (0.26–2.94) 0.84 (0.26–2.40) 0.51 0.91HSP90B1 1.05 (0.56–1.55) 0.87 (0.32–1.43) 0.83 (0.037–1.67) 0.55 0.71CANX 1.20 (0.70–1.54) 0.77 (0.14–2.26) 0.62 (0.27–1.40) 0.19 0.62CALR 1.20 (0.58–1.86) 1.02 (0.28–2.45) 1.01 (0.57–2.02) 0.69 0.98EDEM1 1.04 (0.56–1.51) 0.76 (0.28–1.66) 0.52 (0.33–1.28) 0.38 0.13ATP2A2 1.00 (0.44–2.45) 0.91 (0.36–1.48) 0.78 (0.51–3.48) 0.36 0.88PPP1R15A 1.03 (0.49–1.56) 0.81 (0.44–1.54) 1.21 (0.29–1.86) 0.04 0.19DDIT3 1.17 (0.00–1.95) 1.19 (0.06–2.47) 0.88 (0.31–2.72) 0.58 0.43NFE2L2 1.00 (0.44–2.45) 1.19 (0.66–1.78) 1.24 (0.42–2.54) 0.20 0.66
Values are medians with range. HCV = hepatitis C virus. For other abbreviations, see Supporting information, Supplementary Table 1. The Ntarget values of thesamples were subsequently normalized such that the mean of the normal histological liver Ntarget values was 1.
immersion in a 2% solution of glutaraldehyde bufferedwith 0.2 M cacodylate buffer and post-fixed in osmiumtetroxide before embedding in epoxy resin. Ultra-thinsections stained with uranyl acetate and lead citratewere examined with a Jeol 10 10 (Tokyo, Japan) elec-tron microscope.
Real-time RT-PCR
In previous studies using the same technologicalapproach, we have shown that several altered molecularpathways are involved in CHC [26,27]. The method ofreal-time quantitative reverse transcriptase-polymerasechain reaction (RT-PCR) has been described in detailelsewhere [26,27] and in the Supporting information,Supplementary materials and methods. By studyingthe literature, we selected 17 genes that have beenshown to be induced by ‘traditional’ ER stressors[under in vivo conditions (in particular, in the liver)or in vitro in hepatoma or any other human cells][28–31] or by HCV subgenomic replicons, structuralor non-structural proteins (under in vitro conditions inhepatoma cells) (Supporting information, Supplemen-tary Table 1). Oligonucleotide primer sequences usedare listed in Supporting information, SupplementaryTable 2.
Western blot analysis
The protein expression levels of EDEM, ATF-4,BiP/GRP78, eIF2-alpha, phosphorylated eIF2-alpha,and beta-actin were measured in liver samples. ATF-6 alpha, ATF-6 beta, XBP-1, PERK, eIF2-alpha,EDEM, ATF-4, BiP/GRP78, and beta-actin (Santa CruzBiotechnology, Inc, Santa Cruz, CA, USA), and anti-bodies against phospho-PERK (Thr980) and phospho-eIF2-alpha (Ser51) (Cell Signaling Technology, Bev-erly, MA, USA) were measured.
Statistical analysisValues (medians with range) were tested using theKruskall–Wallis test. Differences between three groupswere judged significant at confidence levels greaterthan 95% (p < 0.05). The relative mRNA levels shownin Table 2 (calculated as described in the Supportinginformation, Supplementary materials and methods)show the abundance of the target relative to the endoge-nous control (TBP ), in order to normalize the startingamount and quality of total RNA. Similar results wereobtained with a second endogenous control, RPLP0(also known as 36B4) (data not shown).
Results
Electron microscopy shows altered hepatocyte ERmorphology in livers from patients with mildHCV-related fibrosisElectron microscopy was used to compare livers frompatients with mild CHC with normal livers. The appear-ance of the ER in hepatocytes from normal livers wasnormal, with cisternae of rough ER regularly orga-nized into stacks around the nucleus, while smooth ERappeared as small vesicles or tubules on the periph-ery of the stacks. Cisternae lumens were narrow andon the external face, many groups of bound ribo-somes were visible (Figure 2A). In contrast, in liv-ers from most patients with mild CHC (9/13), bothparts of the ER (smooth and rough) were dilated anddisorganized (Figure 2B). However, many bound ribo-somes were still visible on the surface of the roughER (Figure 2B). These alterations differ from thoseknown to occur in necrotic hepatocytes, which alsohave dilated ER but are associated with a signifi-cant loss of ribosomes. Together, these morphologicalfindings are similar to those found elsewhere in ER-stressed pancreatic endocrine cells [32]. It should be
Copyright 2010 Pathological Society of Great Britain and Ireland. J Pathol 2010; 221: 264–274Published by John Wiley & Sons, Ltd. www.pathsoc.org.uk www.thejournalofpathology.com

268 T Asselah et al
Figure 2. Electron microscopy shows altered hepatocyte ER morphology in liver biopsy specimens from patients with mild HCV-relatedliver fibrosis. (A) The appearance of endoplasmic reticulum (ER) in hepatocytes from normal livers was normal, with cisternae of the roughER (RER) regularly organized in stacks around the nucleus. Lumens of the cisternae were narrow and on the external face, many groupsof bound ribosomes were visible (arrows). (B) In contrast, in hepatocytes from livers with mild hepatitis C, the two parts of the ER (roughand smooth, RER and SER) were dilated and disorganized. However, at the surface of the rough ER, bound ribosomes were clearly visible,with maintenance of many ribosomes (arrows). Fine granular deposits in the lumen of the RER could represent unfolded protein (stars). N= nucleus; M = mitochondria. (C) At this low magnification, electron microscopy shows a cluster of three hepatocytes (H) where the ER(arrows) is dilated.
noted that livers with ER-stressed hepatocytes exhib-ited changes that were not diffuse but clustered intosmall groups of 3–5 cells, with no preferential lobu-lar localization (Figure 2C). In addition, the proportionof cells with ER changes differed from one patientto another (range 10–30%), indicating inter-individualvariability. Interestingly, there was no significant cor-relation between viral load and the proportion of cellswith ER changes. Finally, the other structures of thehepatocytes such as the nucleus and the mitochondriawere normal in all patients. Moreover, there were nosignificant changes in ‘non-hepatocyte’ cells.
Livers from patients with HCV-related fibrosisexhibit activation of the three proximal ER-residentstress sensors
Next we investigated whether ER stress was associatedwith induction of UPR in livers from patients withmild or advanced HCV-related fibrosis compared withnormal livers.
ATF-6 pathway
There are two ATF-6 isoforms, alpha and beta (Sup-porting information, Supplementary Table 1). In ER-stressed cells, ATF-6 is released from the ER mem-brane and is converted (by cleavage due to regu-lated intra-membrane proteolysis) from a 90 kD pro-tein (p90ATF-6, alpha or beta) to a 50 kD protein(p50ATF-6, alpha or beta) which translocates to thenucleus [1–3]. Thus, we investigated whether ER stressinduces hepatic p50ATF-6 alpha and/or p50ATF-6 betaexpression in livers with mild HCV-related fibrosis.For this, we measured the expression level of the twoATF-6 isoforms using specific antibodies. In normallivers, there was no expression of the two isoforms,while in mild HCV-related fibrosis, the expression
levels of the two p50ATF-6 isoforms were signifi-cantly increased (Figure 3), a finding consistent withER stress-induced ATF-6 activation. Hepatic expres-sion levels of p50ATF-6 alpha and p50ATF-6 beta weresignificantly higher in advanced fibrosis compared withmild fibrosis (Figure 3).
IRE1 pathway
ER stress activates IRE1 [1–3]. Activated IRE1 cutsXBP1 mRNA into spliced XBP1 mRNA, encodingtranscriptionally active XBP-1S protein [1–3]. Thus,the relative hepatic mRNA expression of spliced XBP1was examined and the ratio of spliced XBP1 mRNA toXBP1 mRNA, which indirectly reflects IRE1 endori-bonuclease activity [34], was calculated. The ratio ofspliced XBP1 to XBP1 mRNAs in mild CHC wassignificantly increased compared with normal livers(Table 2), suggesting ER stress-elicited activation ofIRE1. However, there was no significant increase inthe protein expression levels of XBP-1S in mild fibro-sis (Figure 3). Although the ratio of spliced XBP1to XBP1 mRNAs was similarly increased in liverswith advanced fibrosis and in those with mild fibro-sis (Table 2), significant increases in XBP-1S proteinexpression levels were found in the former but not thelatter (Figure 3). There is no clear explanation for thesefindings in advanced fibrosis.
PERK pathway
ER stress activates the eIF2-alpha kinase PERK[1–3]. PERK activation is associated with its phos-phorylation on Thr980 [2] and results in eIF2-alphaphosphorylation on Ser21. Thus, we studied the hep-atic expression levels of total PERK, total eIF2-alpha,phosphorylated PERK (Thr980), and phosphorylated
Copyright 2010 Pathological Society of Great Britain and Ireland. J Pathol 2010; 221: 264–274Published by John Wiley & Sons, Ltd. www.pathsoc.org.uk www.thejournalofpathology.com
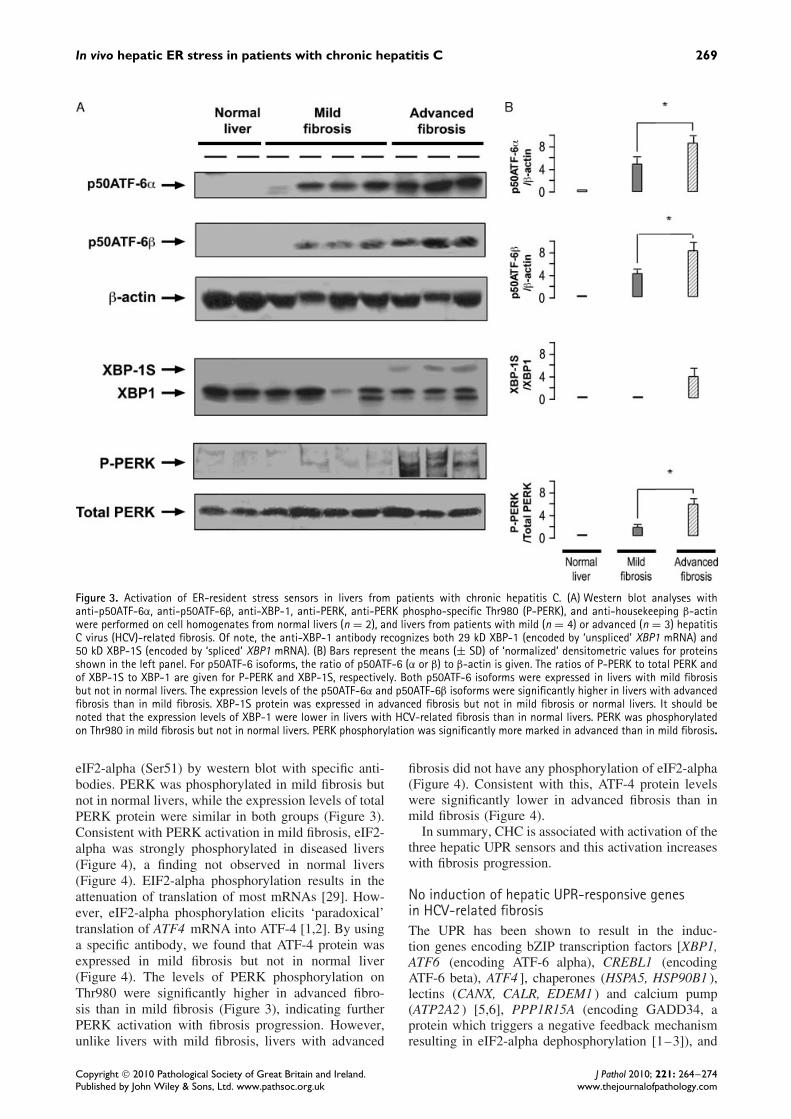
In vivo hepatic ER stress in patients with chronic hepatitis C 269
Figure 3. Activation of ER-resident stress sensors in livers from patients with chronic hepatitis C. (A) Western blot analyses withanti-p50ATF-6α, anti-p50ATF-6β, anti-XBP-1, anti-PERK, anti-PERK phospho-specific Thr980 (P-PERK), and anti-housekeeping β-actinwere performed on cell homogenates from normal livers (n = 2), and livers from patients with mild (n = 4) or advanced (n = 3) hepatitisC virus (HCV)-related fibrosis. Of note, the anti-XBP-1 antibody recognizes both 29 kD XBP-1 (encoded by ‘unspliced’ XBP1 mRNA) and50 kD XBP-1S (encoded by ‘spliced’ XBP1 mRNA). (B) Bars represent the means (± SD) of ‘normalized’ densitometric values for proteinsshown in the left panel. For p50ATF-6 isoforms, the ratio of p50ATF-6 (α or β) to β-actin is given. The ratios of P-PERK to total PERK andof XBP-1S to XBP-1 are given for P-PERK and XBP-1S, respectively. Both p50ATF-6 isoforms were expressed in livers with mild fibrosisbut not in normal livers. The expression levels of the p50ATF-6α and p50ATF-6β isoforms were significantly higher in livers with advancedfibrosis than in mild fibrosis. XBP-1S protein was expressed in advanced fibrosis but not in mild fibrosis or normal livers. It should benoted that the expression levels of XBP-1 were lower in livers with HCV-related fibrosis than in normal livers. PERK was phosphorylatedon Thr980 in mild fibrosis but not in normal livers. PERK phosphorylation was significantly more marked in advanced than in mild fibrosis.
eIF2-alpha (Ser51) by western blot with specific anti-bodies. PERK was phosphorylated in mild fibrosis butnot in normal livers, while the expression levels of totalPERK protein were similar in both groups (Figure 3).Consistent with PERK activation in mild fibrosis, eIF2-alpha was strongly phosphorylated in diseased livers(Figure 4), a finding not observed in normal livers(Figure 4). EIF2-alpha phosphorylation results in theattenuation of translation of most mRNAs [29]. How-ever, eIF2-alpha phosphorylation elicits ‘paradoxical’translation of ATF4 mRNA into ATF-4 [1,2]. By usinga specific antibody, we found that ATF-4 protein wasexpressed in mild fibrosis but not in normal liver(Figure 4). The levels of PERK phosphorylation onThr980 were significantly higher in advanced fibro-sis than in mild fibrosis (Figure 3), indicating furtherPERK activation with fibrosis progression. However,unlike livers with mild fibrosis, livers with advanced
fibrosis did not have any phosphorylation of eIF2-alpha(Figure 4). Consistent with this, ATF-4 protein levelswere significantly lower in advanced fibrosis than inmild fibrosis (Figure 4).
In summary, CHC is associated with activation of thethree hepatic UPR sensors and this activation increaseswith fibrosis progression.
No induction of hepatic UPR-responsive genesin HCV-related fibrosisThe UPR has been shown to result in the induc-tion genes encoding bZIP transcription factors [XBP1,ATF6 (encoding ATF-6 alpha), CREBL1 (encodingATF-6 beta), ATF4 ], chaperones (HSPA5, HSP90B1 ),lectins (CANX, CALR, EDEM1 ) and calcium pump(ATP2A2 ) [5,6], PPP1R15A (encoding GADD34, aprotein which triggers a negative feedback mechanismresulting in eIF2-alpha dephosphorylation [1–3]), and
Copyright 2010 Pathological Society of Great Britain and Ireland. J Pathol 2010; 221: 264–274Published by John Wiley & Sons, Ltd. www.pathsoc.org.uk www.thejournalofpathology.com

270 T Asselah et al
Figure 4. Altered expression of EDEM, ATF-4, phosphorylated eIF2-α in livers from patients with chronic hepatitis C according to theseverity of liver fibrosis. (A) Western blot analyses using anti-EDEM, anti-ATF-4, anti-BiP/GRP78, anti-eIF2-α, anti-eIF2-α phospho-specificSer51 (P-eIF2-α), and anti-housekeeping gene (β-actin) antibodies were performed on liver cell homogenates from patients with mild(n = 4) or advanced (n = 4) hepatitis C virus (HCV)-related fibrosis. Cell homogeneates from patients with normal livers (n = 3) werealso investigated. (B) Bars represent the means (± SD) of ‘normalized’ densitometric values for proteins shown in the left panel. Theexpression of EDEM, ATF-4, and BiP/GRP78 was normalized using corresponding β-actin. The expression of P-eIF2-α was normalized usingcorresponding values of total eIF2-α. The protein expression levels of EDEM and ATF-4 were significantly lower and the levels of eIF2-αphosphorylation significantly higher in advanced fibrosis than in mild fibrosis. There was also a trend in the decrease in the expressionlevel of BiP/GRP78.
DDIT3 (encoding the pro-apoptotic protein CHOP).The UPR may also be associated with the induc-tion of NFE2L2 [encoding nuclear factor erythroid2-related factor 2 (Nrf2), a Cap’n’Collar bZIP tran-scription factor that dimerizes with ATF-4 to acti-vate the antioxidant response element [30]]. Thus, therelative levels for all of these mRNAs were exam-ined in mild or advanced CHC compared with nor-mal livers. We found no significant induction of anyof these mRNAs in CHC compared with normal liv-ers (Table 2). PPP1R15A mRNA expression was evendown-regulated in diseased livers compared with nor-mal livers (Table 2). In addition, western blot analy-sis showed an accumulation of BiP/GRP78 (the pro-tein encoded by HSPA5 ) and EDEM1 in mild fibro-sis but not in normal liver (Figure 4). Together, thesefindings suggest that post-transcriptional mechanismsfavour the accumulation of BiP/GRP78 and EDEM1
proteins in mild CHC. Interestingly, the protein expres-sion levels of EDEM1 were significantly lower in liverswith advanced fibrosis compared with mild fibrosis(Figure 4).
Induction of genes involved in liver proliferation,inflammation, and apoptosis in HCV-related fibrosisWe then asked whether the expression of genesknown to be involved in diffuse processes suchas hepatocyte proliferation, liver inflammation, andapoptosis [11–26] was increased in diseased livers.Thus, we measured the expression mRNA levels ofMKi67 (proliferation), JUN (a result of JNK acti-vation involved in proliferation and inflammation),CREB3L3 mRNA (also known as CREBH, encodingCREB-H [31], a hepatocyte-specific bZIP transcriptionfactor of the ATF subfamily which serves as a linkbetween inflammation and the acute-phase response),NF-kappaB-induced anti-apoptotic mRNAs (cFLAR,
Copyright 2010 Pathological Society of Great Britain and Ireland. J Pathol 2010; 221: 264–274Published by John Wiley & Sons, Ltd. www.pathsoc.org.uk www.thejournalofpathology.com

In vivo hepatic ER stress in patients with chronic hepatitis C 271
Table 3. Expression of genes involved in the control of proliferation, inflammation, and apoptosis in livers from patients with mild oradvanced HCV-related fibrosis relative to normal livers
Mild HCV-related Advanced HCV-related p value p valueNormal fibrosis fibrosis Mild HCV-related Mild vs advanced
Gene (n = 7) (n = 13) (n = 15) fibrosis vs normal HCV-related fibrosis
MKI67 0.99 (0.69–2.20) 2.02 (0.13–3.81) 3.78 (1.63–24.39) 0.04 <0.01JUN 0.99 (0.69–1.40) 1.63 (0.78–3.46) 3.17 (1.61–19.16) 0.02 <0.01CREB3L3 1.00 (0.73–1.49) 1.64 (0.80–2.80) 1.80 (0.79–3.72) 0.02 0.79CFLAR 0.99 (0.29–1.33) 0.96 (0.27–1.98) 0.43 (0.03–0.98) 0.86 0.01GADD45B 1.15 (0.62–3.71) 0.37 (0.11–3.15) 0.50 (0.23–1.58) 0.08 0.71BCL2A1 1.00 (0.72–2.07) 2.35 (0.90–10.36) 4.71 (0.78–34.35) <0.01 0.20IER3 0.99 (0.18–2.42) 2.15 (0.48–33.32) 5.09 (1.83–22.22) 0.07 0.06IER3S 1.00 (0.82–1.46) 1.99 (0.86–5.12) 4.93 (1.84–14.14) <0.01 <0.01BBC3 0.99 (0.49–1.89) 1.56 (0.37–4.00) 2.49 (0.95–7.16) 0.11 0.23PMAIP1 1.00 (0.27–2.94) 0.68 (0.17–3.75) 0.99 (0.14–5.63) 0.63 0.79BAX 1.01 (0.71–1.54) 0.83 (0.29–2.16) 1.86 (0.83–2.81) 0.90 0.02FAS 1.00 (0.77–2.16) 1.82 (0.54–3.53) 3.19 (1.60–5.83) 0.07 <0.01
Values are medians with range. HCV = hepatitis C virus. For other abbreviations, see Supporting information, Supplementary Table 1. The Ntarget values of thesamples were subsequently normalized such that the mean of the normal histological liver Ntarget values was 1.
GADD45B, BCL2A1, and IRE3 ), the short variant ofIER3 called IER3S, and p53-inducible pro-apoptoticmRNAs (BBC3, PMAIP1, BAX, and FAS ).
In livers with mild CHC, there were significantincreases in the relative mRNA levels of MKi67,JUN, CREB3L3, BCL2A1, and IER3S (encoding thepro-apoptotic immediate early response 3 isoformshort variant) (Table 3). Compared with mild CHC,advanced CHC had significant increases in the mRNAlevels of MKi67, JUN, IER3S, BAX, and FAS (Table 3).In contrast, cFLAR expression was down-regulated(Table 3). Thus, our results confirm that real-time RT-PCR is able to detect changes in gene expressionrelated to diffuse intra-hepatic processes.
Hepatic mRNA expression associated with mild liverfibrosis may differ between HBV and HCV infectionsTo determine whether the genes that are deregulatedduring mild chronic HCV-induced fibrosis (the transi-tion from normal to mild fibrosis) are specific for HCVinfection, the mRNA expression levels of selectedgenes were measured in livers with mild HBV fibrosis.
UPR-responsive genes
Alterations observed in patients with HCV infec-tion [ie increased ratio of spliced XBP1 mRNA toXBP1 mRNA (indicating engagement of the IRE1pathway) and down-regulated PPP1R15A (see aboveand Table 4)] were not found in livers with mild HBVfibrosis (Table 4). In contrast, certain genes whoseexpression was unchanged in mild HCV fibrosis wereup-regulated in mild HBV fibrosis, including ATF6,ATF4, and NFE2L2 (Table 4).
Genes involved in liver proliferation, inflammation,and apoptosis
CREB3L3, which was up-regulated in mild HCVinfection, was not altered in patients with mild HBV-related fibrosis. FAS, whose expression was unchanged
in the early stage of chronic HCV infection, was up-regulated in patients with mild HBV-related fibrosis(Table 4). BCL2A1, IER3S, and JUN were up-regulatedin both groups of patients. Finally, GADD45B, BBC3,PMAIP1, BAX, and cFLAR were not induced in eithergroup of patients.
Discussion
In this study, hepatocytes in livers from untreatedpatients with mild CHC had dilated and disorganizedrough ER on electron microscopy. Experimental stud-ies have shown that dilation and disorganization ofrough ER occur when misfolded or unfolded proteinsaccumulate in the ER lumen, ie when there is ERstress [1–3]. Thus, our morphological findings showthat there is hepatocyte ER stress in mild CHC. More-over, we found evidence of the activation of the threeER-resident stress sensors, ATF-6, IRE1, and PERK,indicating proximal engagement of UPR pathways inthese livers. In addition, there was a more markedengagement of the three ER-resident stress sensors inadvanced CHC, suggesting an increase in ER stresswith fibrosis progression. Our results are supported byin vitro studies suggesting that HCV induces perturba-tion of ER homeostasis. HCV structural proteins E1and E2 may be disulphide-linked and form misfoldedaggregates in the ER lumen [21]. HepG2 cells express-ing the HCV core develop ER calcium depletion [20],a mechanism of ER stress [2]. There is indirect evi-dence of engagement of the UPR (ie ER stress) in cellsexpressing HCV subgenomic replicons [18,19], struc-tural proteins (ie core [20], E2 alone [21], E1 alone orcombined with E2 [22]), or the non-structural proteinNS4B [23].
It should be noted that in our study morphologicalevidence of ER stress was found only in some clus-ters of hepatocytes. Interestingly, studies have shownthat HCV infects only a limited number of hepatocytes[33,34]. Recently, by combining 2-photon microscopy
Copyright 2010 Pathological Society of Great Britain and Ireland. J Pathol 2010; 221: 264–274Published by John Wiley & Sons, Ltd. www.pathsoc.org.uk www.thejournalofpathology.com

272 T Asselah et al
Table 4. Gene expression in livers from patients with HBV mild fibrosis, relative to normal liversNormal Mild HBV-related p value
Genes (n = 7) fibrosis (n = 10) HBV vs normal
UPR-responsive genesXBP1 1.17 (0.65–1.31) 0.67 (0.21–1.38) 0.04Spliced XBP1 1.19 (0.33–1.45) 0.51 (0.20–2.00) 0.22Ratio of spliced XBP1 to XBP1 1.02 (0.42–1.25) 1.10 (0.49–1.62) 0.55ATF6 1.00 (0.67–1.18) 1.45 (1.09–2.82) <0.01CREBL1 1.00 (0.86–1.26) 1.01 (0.63–1.62) 0.96ATF4 1.00 (0.80–1.24) 1.37 (1.00–3.29) 0.02EDEM1 1.04 (0.56–1.51) 0.93 (0.40–1.83) 0.78HSPA5 0.96 (0.71–2.21) 0.99 (0.04–4.73) 0.45HSP90B1 1.05 (0.56–1.55) 0.88 (0.54–1.49) 0.75CANX 1.20 (0.70–1.54) 0.84 (0.33–1.76) 0.22CALR 1.20 (0.58–1.86) 0.73 (0.27–2.65) 0.25ATP2A2 1.00 (0.44–2.45) 0.82 (0.05–5.52) 0.45PPP1R15A 1.03 (0.49–1.56) 0.80 (0.15–1.26) 0.12DDIT3 1.17 (0.00–1.95) 1.09 (0.11–1.67) 0.84NFE2L2 1.00 (0.44–2.45) 1.32 (0.85–2.88) 0.02
Other genes∗
MKI67 0.99 (0.69–2.20) 2.33 (0.46–4.98) 0.05JUN 0.99 (0.69–1.40) 1.63 (0.71–3.20) 0.02CREB3L3 1.00 (0.73–1.49) 1.17 (0.81–5.84) 0.17CFLAR 0.99 (0.29–1.33) 0.50 (0.30–1.10) 0.07BCL2A1 1.00 (0.72–2.07) 4.51 (0.46–14.59) 0.02IER3 0.99 (0.18–2.42) 2.02 (0.24–11.22) 0.05IER3S 1.00 (0.82–1.46) 1.77 (0.80–12.02) 0.02GADD45B 1.15 (0.62–3.71) 0.62 (0.28–3.10) 0.20BBC3 0.99 (0.49–1.89) 1.07 (0.32–3.70) 0.62PMAIP1 1.00 (0.27–2.94) 2.45 (0.19–5.04) 0.28BAX 1.01 (0.71–1.54) 0.95 (0.02–2.17) 0.72FAS 1.00 (0.77–2.16) 1.96 (0.98–6.04) 0.03
Values are medians with range. HBV = hepatitis B virus. For other abbreviations, see Supporting information, Supplementary Table 1. The Ntarget values of thesamples were subsequently normalized such that the mean of the normal histological liver Ntarget values was 1. ∗Other genes are those which are involved in thecontrol of proliferation, inflammation, and apoptosis.
with virus-specific, fluorescent, semiconductor quan-tum dot probes, it has been shown that HCV involvesa limited number of hepatocytes [33]. Furthermore, thelow average levels of HCV-RNA in biopsy samplesmight be explained by focal distribution of infectedhepatocytes [34]. Thus, ER stress may exist only in asmall number of hepatocytes because of a ‘localized’pattern of HCV infection. Future studies are needed tocorrelate ER stress to the presence of HCV.
This study shows that although there was ER stressand activation of the three UPR sensors in livers withCHC, full induction of UPR-responsive genes wasnot found in these livers. This finding may have twoexplanations which are not mutually exclusive. First,in vitro studies suggest that HCV per se may inhibitthe UPR [24]. HCV subgenomic replicons activateATF-6, induce HSP5 mRNA coding for BiP butinhibit its translation [18]. HCV subgenomic repliconswere found to induce XBP1 mRNA splicing andalso to inhibit XBP-1S trans-activating activity, EDEMinduction, and degradation of unfolded proteins [19].E2 (but not E1) protein induces the HSP5 mRNAcoding for BiP but not the protein itself [21]. Inaddition, E2 proteins may inhibit the activity of PERK[35] or the interferon-induced, double-stranded RNA-activated protein kinase (PKR) [36]. Moreover, there
is evidence that initiation of protein synthesis by HCVis refractory to eIF2-alpha phosphorylation [37].
Our morphological findings may provide anotherexplanation for the lack of induction of UPR-responsivegenes in CHC liver-biopsy specimens. Indeed, the lackof gene induction may be related to the fact that onlyhepatocytes in small clusters were ‘ER-stressed’ andsurrounded by numerous cells that did not experienceER stress (see above). Instead, processes such as pro-liferation, inflammation, and apoptosis are known tobe diffuse in livers with CHC [11,26,38–40]. Inflam-mation is a diffuse feature in CHC driven by thepersistent expression of chemokines [26,38,40]. It isalso accepted that apoptosis of hepatocytes is a promi-nent feature of CHC [39]. Consistent with this, wefound that genes involved in the control of prolifer-ation, inflammation, and apoptosis were significantlyinduced in livers from patients with CHC.
Livers from mice with dietary-induced or genetic(ob/ob) obesity exhibit chronic ER stress which mayresult in hepatic insulin resistance [41]. Thus, inpatients with CHC, excess weight, but not HCV per se,may trigger hepatic ER stress. However, this hypoth-esis is unlikely since our patients had a normal bodymass index and normal blood glucose concentrations.
Finally, this study investigated gene expression pro-files in livers with mild HBV-related fibrosis. It is
Copyright 2010 Pathological Society of Great Britain and Ireland. J Pathol 2010; 221: 264–274Published by John Wiley & Sons, Ltd. www.pathsoc.org.uk www.thejournalofpathology.com

In vivo hepatic ER stress in patients with chronic hepatitis C 273
important to note that the expression gene profile dif-fered between HBV and HCV infection. Five genesincluding ATF6, ATF4, NFE2L2, IER3, and FAS wereup-regulated in livers with mild HBV-related fibrosisbut not in those with mild HCV-related fibrosis. Inter-estingly, although ATF6 and ATF4 are known to beinduced by XBP-1S, no evidence of IRE1 activation orinduction of other XBP-1S target mRNAs (eg CREBL1or EDEM1 ) was found. Moreover, it should be notedthat ATF6, ATF4, and NFE2L2 each encode a proteinthat belongs to the family of bZIP transcription fac-tors [7]. Further studies are needed to investigate the‘propensity’ of HBV infection to induce genes codingfor bZIP transcription factors.
In conclusion, livers from patients with CHC exhibitin vivo hepatocyte ER stress and activation of the threeER-resident UPR sensors without apparent inductionof UPR-responsive genes. This lack of gene inductionmay be explained by the inhibiting action of HCVper se and/or by our finding of the localized natureof hepatocyte ER stress.
Acknowledgment
We thank Alain Grodet for his technical assistancein electron microscopy and Jean-Pierre Lagneau formedical illustrations. This study was supported by theINSERM, LFB and the association pour la Recherchesur le cancer (ARC). CG received a Dame SheilaSherlock Fellowship from EASL. RM is in receipt ofan Interface INSERM-AP-HP Fellowship.
References
Note: References 42–44 are cited in the Supportinginformation to this article.
1. Ma Y, Hendershot LM. The role of the unfolded protein responsein tumour development: friend or foe? Nature Rev Cancer 2004;4: 966–977.
2. Schroder M, Kaufman RJ. The mammalian unfolded proteinresponse. Annu Rev Biochem 2005; 74: 739–789.
3. Ron D, Walter P. Signal integration in the endoplasmic reticulumunfolded protein response. Nature Rev Mol Cell Biol 2007; 8:519–529.
4. Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset ofendoplasmic reticulum resident chaperone genes in the unfoldedprotein response. Mol Cell Biol 2003; 23: 7448–7459.
5. Wu J, Rutkowski DT, Dubois M, Swathirajan J, Saunders T,Wang J, et al . ATF6alpha optimizes long-term endoplasmic retic-ulum function to protect cells from chronic stress. Dev Cell 2007;13: 351–364.
6. Yamamoto K, Sato T, Matsui T, Sato M, Okada T, Yoshida H,et al . Transcriptional induction of mammalian ER quality controlproteins is mediated by single or combined action of ATF6alphaand XBP1. Dev Cell 2007; 13: 365–376.
7. Newman JR, Keating AE. Comprehensive identification of humanbZIP interactions with coiled-coil arrays. Science 2003; 300:2097–2101.
8. Pahl HL, Baeuerle PA. The ER-overload response: activation ofNF-kappa B. Trends Biochem Sci 1997; 22: 63–67.
9. Qu L, Huang S, Baltzis D, Rivas-Estilla AM, Pluquet O, Hat-zoglou M, et al . Endoplasmic reticulum stress induces p53 cyto-plasmic localization and prevents p53-dependent apoptosis by apathway involving glycogen synthase kinase-3beta. Genes Dev
2004; 18: 261–277.10. Hetz C, Bernasconi P, Fisher J, Lee AH, Bassik MC, Antons-
son B, et al . Proapoptotic BAX and BAK modulate the unfoldedprotein response by a direct interaction with IRE1alpha. Science
2006; 312: 572–576.11. Rehermann B, Nascimbeni M. Immunology of hepatitis B virus
and hepatitis C virus infection. Nature Rev Immunol 2005; 5:215–229.
12. Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Barten-schlager R. Replication of subgenomic hepatitis C virus RNAs ina hepatoma cell line. Science 1999; 285: 110–113.
13. Blight KJ, Kolykhalov AA, Rice CM. Efficient initiation of HCVRNA replication in cell culture. Science 2000; 290: 1972–1974.
14. Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z,et al . Production of infectious hepatitis C virus in tissue culturefrom a cloned viral genome. Nature Med 2005; 11: 791–796.
15. Deleersnyder V, Pillez A, Wychowski C, Blight K, Xu J,Hahn YS, et al . Formation of native hepatitis C virus glycoproteincomplexes. J Virol 1997; 71: 697–704.
16. Dubuisson J., Penin F, Moradpour D. Interaction of hepatitis Cvirus proteins with host cell membranes and lipids. Trends Cell
Biol 2002; 12: 517–523.17. Miyanari Y, Atsuzawa K, Usuda N, Watashi K, Hishiki T,
Zayas M, et al . The lipid droplet is an important organelle forhepatitis C virus production. Nature Cell Biol 2007; 9: 1089–1097.
18. Tardif KD, Mori K, Siddiqui A. Hepatitis C virus subgenomicreplicons induce endoplasmic reticulum stress activating an intra-cellular signaling pathway. J Virol 2002; 76: 7453–7459.
19. Tardif KD, Mori K, Kaufman RJ, Siddiqui A. Hepatitis C virussuppresses the IRE1–XBP1 pathway of the unfolded proteinresponse. J Biol Chem 2004; 279: 17158–17164.
20. Benali-Furet NL, Chami M, Houel L, De Giorgi F, Vernejoul F,Lagorce D, et al . Hepatitis C virus core triggers apoptosis in livercells by inducing ER stress and ER calcium depletion. Oncogene
2005; 24: 4921–4933.21. Liberman E, Fong YL, Selby MJ, Choo QL, Cousens L,
Houghton M, et al . Activation of the grp78 and grp94 pro-moters by hepatitis C virus E2 envelope protein. J Virol 1999; 73:3718–3722.
22. Chan SW, Egan PA. Hepatitis C virus envelope proteins regulateCHOP via induction of the unfolded protein response. FASEB J
2005; 19: 1510–1512.23. Zheng Y, Gao B, Ye L, Kog L, Jing W, Yang X, et al . Hepatitis
C virus non-structural protein NS4B can modulate an unfoldedprotein response. J Microbiol 2005; 43: 529–536.
24. Tardif KD, Waris G, Siddiqui A. Hepatitis C virus, ER stress, andoxidative stress. Trends Microbiol 2005; 13: 159–163.
25. Asselah T, Bieche I, Laurendeau I, Martinot-Peignoux M, Par-adis V, Vidaud D, et al . Significant gene expression differencesin histologically ‘normal’ liver biopsies: implications for controltissue. Hepatology 2008; 48: 953–962.
26. Bieche I, Asselah T, Laurendeau I, Vidaud D, Degot C, Paradis V,et al . Molecular profiling of early stage liver fibrosis in patientswith chronic hepatitis C virus infection. Virology 2005; 332:130–144.
27. Asselah T, Bieche I, Laurendeau I, Paradis V, Vidaud D,Degott C, et al . Liver gene expression signature of mild fibrosisin patients with chronic hepatitis C. Gastroenterology 2005; 129:2064–2075.
28. Tazi KA, Bieche I, Paradis V, Guichard C, Laurendeau I,Dargere D, et al . In vivo altered unfolded protein response and
Copyright 2010 Pathological Society of Great Britain and Ireland. J Pathol 2010; 221: 264–274Published by John Wiley & Sons, Ltd. www.pathsoc.org.uk www.thejournalofpathology.com

274 T Asselah et al
apoptosis in livers from lipopolysaccharide-challenged cirrhoticrats. J Hepatol 2007; 46: 1075–1088.
29. Holcik M, Sonenberg N. Translational control in stress and apop-tosis. Nature Rev Mol Cell Biol 2005; 6: 318–327.
30. Hayes JD, McMahon M. The double-edged sword of Nrf2: sub-version of redox homeostasis during the evolution of cancer. Mol
Cell 2006; 21: 732–734.31. Zhang K, Shen X, Wu J, Sakaki K, Saunders T, Rutkowski DT,
et al . Endoplasmic reticulum stress activates cleavage of CREBHto induce a systemic inflammatory response. Cell 2006; 124:587–599.
32. Scheuner D, Vander Mierde D, Song B, Flamez D, Creemers JW,Tsukamoto K, et al . Control of mRNA translation preserves endo-plasmic reticulum function in beta cells and maintains glucosehomeostasis. Nature Med 2005; 11: 757–764.
33. Liang Y, Shilagard T, Xiao SY, Snyder N, Lau D, Cicalese L,et al . Visualizing hepatitis C virus infections in human liver bytwo-photon microscopy. Gastroenterology 2009; 137: 1448–1458.
34. Stiffler JD, Nguyen M, Sohn JA, Liu C, Kaplan D, Seeger C.Focal distribution of hepatitis C virus RNA in infected livers. PLoS
One 2009; 4: e6661.35. Pavio N, Romano PR, Graczyk TM, Feinstone SM, Taylor DR.
Protein synthesis and endoplasmic reticulum stress can be mod-ulated by the hepatitis C virus envelope protein E2 through theeukaryotic initiation factor 2alpha kinase PERK. J Virol 2003; 77:3578–3585.
36. Taylor DR, Shi ST, Romano PR, Barber GN, Lai MM. Inhibitionof the interferon-inducible protein kinase PKR by HCV E2 protein.Science 1999; 285: 107–110.
SUPPORTING INFORMATION ON THE INTERNET
The following supporting information may be found in the online version of this article.
Supplementary materials and methods. List of genes investigated and oligonucleotide primer sequences used.
Table S1. List of genes investigated in patients with HCV-related fibrosis.
Table S2. Oligonucleotide primer sequences used.
37. Robert F, Kapp LD, Khan SN, Acker MG, Kolitz S, Kazemi S,et al . Initiation of protein synthesis by hepatitis C virus isrefractory to reduced eIF2.GTP.Met-tRNA(i)(Met) ternary com-plex availability. Mol Biol Cell 2006; 17: 4632–4644.
38. Heydtmann M, Adams DH. Chemokines in the immunopathogen-esis of hepatitis C infection. Hepatology 2009; 49: 676–688.
39. Bantel H, Lugering A, Poremba C, Lugering N, Held J, Dom-schke W, et al . Caspase activation correlates with the degree ofinflammatory liver injury in chronic hepatitis C virus infection.Hepatology 2001; 34: 758–767.
40. Asselah T, Bieche I, Sabbagh A, Bedossa P, Moreau R, Valla D,et al . Gene expression and hepatitis C virus infection. Gut 2009;58: 846–858.
41. Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E,et al . Endoplasmic reticulum stress links obesity, insulin action,and type 2 diabetes. Science 2004; 306: 457–461.
42. Bedossa P, Poynard T. An algorithm for the grading of activity inchronic hepatitis C. Hepatology 1996; 24: 289–293.
43. Bieche I, Asselah T, Laurendeau I, Vidaud D, Degot C, Paradis V,et al . Molecular profiling of early stage liver fibrosis in patientswith chronic hepatitis C virus infection. Virology 2005; 332:130–144.
44. Asselah T, Bieche I, Laurendeau I, Paradis V, Vidaud D,Degott C, et al . Liver gene expression signature of mild fibrosisin patients with chronic hepatitis C. Gastroenterology 2005; 129:2064–2075.
Copyright 2010 Pathological Society of Great Britain and Ireland. J Pathol 2010; 221: 264–274Published by John Wiley & Sons, Ltd. www.pathsoc.org.uk www.thejournalofpathology.com


























![Endoplasmic reticulum[1]](https://img.pdfslide.net/doc/110x75/58ed5fc71a28aba1678b4611/endoplasmic-reticulum1.jpg)


