Embed Size (px)
Citation preview

Indian Journal of Chemistry Vo1 .39A, lan-March 2000, pp. 1 00- 1 05
Theoretical studies on nitro substitution in keto-enol and imine-enamine tautomeric processes
Prasad V Bharatam
Department of Chemistry, Guru Nanak Dev University, Amritsar 1 43 005, India
Received 2 October 1999; accepted 11 November 1999
Ab initio studies on the keto-enol tautomerism in nitroacetaldehyde and imine-enamine tautomerism in nitroacetaldimine are carried out by performing optimizations at HF/6-3 1 G* and MP2I6-3 1 G* levels. Final energies are obtained by performing MP4(SDTQ) single point energies on the MP2/6-3 1 G* geometries. The stabilities of the resultant nitrovinylalcohol and nitrovinyalamine have been estimated by studying the 1 ,5 hydrogen shift in these molecules. The 1 ,3-hydrogen shifts in nitroacetaldehyde and nitroacetaldimine to give acinitro tautomers are also analysed. The enol and enamine tautomers are stabilized due to intramolecular hydrogen bond upon nitro substitution. The tautomer energy differences are reduced in keto-enol tautomerism by about 8.7 kcallmol because of nitro substitution. The generally unstable enamine became more stable than imine by 2.5 kcallmol on nitro substitution.
Tautomerism occupies a prominent position in organic chemistry and underlies many important biological processes which occur by alpha substitution and condensation reactions ' ·5 . The subject of tautomerism is receiving renewed interest because of its recognized importance in biological processes and improved experimental and theoretical methods of analysis of the tautomerss. The comer stone in these processes is the ability for rapid proton transfer between e.g. , a (thermodynamically) more stable keto derivative and its (kinetically) more reactive enol isomer
s,x . The imine-enamine
tautomerism9• 10 is also well established but less dominant than the keto-enol system, while the thio group has received little focus. Other tautomeric equilibria involve the nitroso-oxime groups and the much less favourable proton shift between aliphatic nitro groups and their ad forms"· J 3 . These H-transfer processes and in particular the keto-enol equilibria extend to conjugated systems, but the quantitative information is trailing as compared to the parent systems.
Several theoretical studies have been reported on the important keto-enol, imine-enamine and nitro-acinitro tautomeric processes (Scheme I , Fig. I ), the results reported at the high accuracy G l and G2 levels are summarized in Table I (refs 1 0, 1 1 ) . The keto-enol tautomeric energy differences in acetaldehyde are 1 0.8 kcallmol, favouring the keto form. The acetaldimine tautomer is
more stable than the corresponding enamine by about 3 .9 kcal/mol . Nitro tautomer in nitromethane is about 14 . 1 kcal/mol more stable than the co
'rresponding aci
form. Recent experimental studies showed that the order of stabilities of the tautomers can be reversed with appropriate substituents. For example, Rappoport and co-workers have prepared several enols which are more stable than their keto tautomers7. Wu and Lienx have reported the substituent effects on the keto-enol tautomeric equilibrium in MeXC=O, where X=H, BH CH NH 2 ' 3 ' 2 ' OH, F, Cl, CN and NC, using HF/6-3 I G* and MP2/6-3 1 G* methods. They concluded that the 1t donating substituents show larger quantity of charge transfer, which facilitate the 1 ,3 hydrogen shift . During the studies on aromatic n itro compounds, we have observed that nitro group excerts a strong influence on the ortho substituents through intramolecular hydrogen bond formation '4. Also conjugation of the lone pair of electrons on N in vinylamine and isoelectronic compounds was shown to be very strongl 5 • This data indicate that nitro substitution in keto and imine compounds can stabilise the enol and enamine tautomers due to hydrogen bond- . ing and conjugation. In this article we report ab initio studies on nitroacetaldehyde, 1 and nitroacetaldimine, 2 and their tautomers to quantitatively estimate the effect of nitro substitution on keto-enol and imine-enamine tautomeric processes.

BHARATAM : THEORETICAL STUDIES ON NITRO SUBSTITUTION IN TAUTOMERIC PROCESSES 1 0 1
H :}-<: Keto-enol >={O
.. .. H H H H-H Imlne�narnine >={N-H --
Iii H H H H �N9 Mtro · acfnltro
>=�:o ... -
t'\i O H 0
Scheme I
Methods of Calculations Ab initio molecular orbital calculations 16 at HF/6-
3 I G*, MP2(full)/6-3 1 G* and MP4(SDTQ)/6-3 1 G*11 MP2(full)/6-3 1 G* levels have been carried out with the GAUSSIAN94WI7 program on a pair of PC pentium 1 00MHz computers. The compounds under investigation are nitroacetaldehyde la, its enol tautomer 1b, acinitro tautomer 1c, nitroacetaldimine 2a, enamine tautomer, 2b and the ad tautomer 2c. The transition states 1d and 2d for the 1 ,5 hydrogen shift in 1b and 2b are also studied. The important geometrical parameters are given in Fig. 2. The zero point vibrational energies (ZPE) are obtained by performing frequency calculations at HF/6-3 1 G* level and are scaled by a factor ofO.9 1 35 Ix . Absolute energies and ZPE values are given in Table 2 and the relative energies are given in Tables 3 and 4. Because the emphasis is on tautomeric processes, the trans and anti isomers and conformers of 1, 2 and their tautomers have not been studied. MP2(full)/6-3 1 G* geometries and MP4(SDTQ)/6-3 1 G*IIMP2(full)/6-3 1 G* (ZPE) energies are used in the paper unless otherwise specifically mentioned.
Results and Discussion
Geometries
Nitroacetaldehyde 1a and nitroacetaldimine 2a have C, symmetry (Fig. 2, Table 2) . Lammertsma and Prasad have shown that the geometrical parameters of the nitro group (i.e. C-N, N-O distances and O-N-O angles) can be properly estimated by theoretical methods only at the electron correlated levels I l a . The geometrical parameters of 1a and 2a confirm this observation; in Fig. 2 only the
o H,109.9 1 24.3,/ 1 090 ,'"=' C 1 .222 . C - C " ,"/ 1.502 \1 .1 09
H H
1 .095 H
1.026 N -H H �09.4 120.8/�09.7 1.091 . 'C'--'c 1 .283
" " J 1 .494 \1.099 H H
1.095 H
H ,107.1 0
;0 -
r1 "-::\ ,I 1 .090 . C - N . . .. ) 1 25.7 � ," J 1.485 � 0 H ' 1 .240 H 1 .087
H,\.974 108.1 (0 H 122.4 126.8/
1.086"'?-fc 1.367 1 .081/ 1 .336 \1.085
H H
H, 1 .0 15 �'" . , " "
H 122.0 126.1/N) 1 1 3.7 1 .086"'? -fc 1.400 1.082/ 1 .340 \1 .088
H H
0 0.981 H �16.3 13 1 .2 /�H 1.076 "? � 1 .425 1.077/ 1 .313 \1 .236
H 0 Fig. I - Important geometric parameters of acetaldehyde,
acetaldimine, nitromethane and their tautomers obtained at MP2(full)/6-3 1 G* level. Distances are in angstrom units and angles in degrees.
Table I - Tautomerization energies (kcallmol) for the prototypic tautomeric processes at various levels
of theoretical calculations,·h
Tautomeric pairs
keto-enol
imine-enamine
nitro-acinitro
MP4/6-3 1 G*//
MP2/6-3 1 G*( +ZPE)
1 4.4
6.0
22. 1
G I
1 0.9
3.8
1 4. 1
• Refs. 1 0, 1 1 a and 1 4 h Tautomeric energy differences.
G2
1 0.8
3 .9
1 4. 1
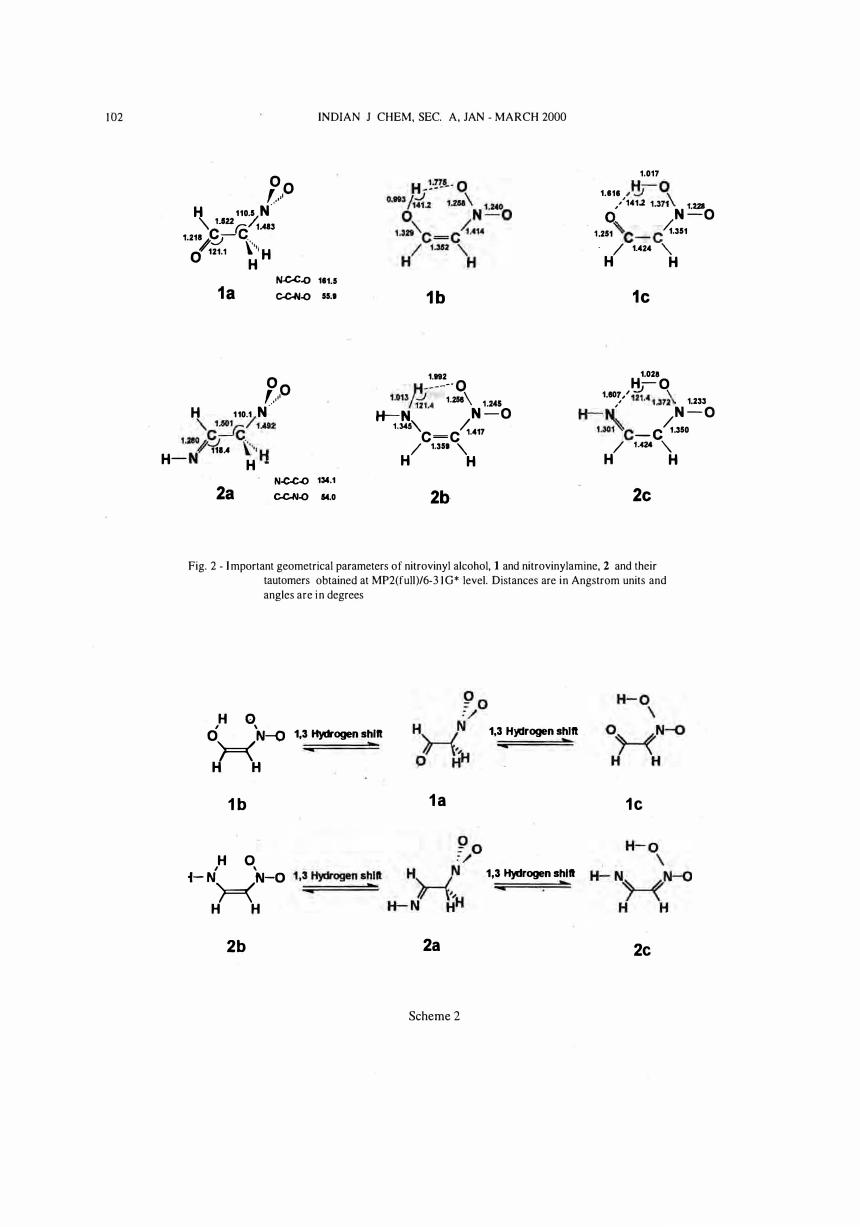
MP2(full)/6-3 I G* optimized parameters have been included for clarity. The C-N02 bond distances in 1a and 2a are 1 .483 and 1 .492 A respectively. The N-O bond distances are in the vicinity of 1 .23 A, in good agreement with the data obtained for nitromethanel la . Both the structures have anticlinical arrangement with N-CC-O torsional angle of 1 6 1 .5° in 1a and N-C-C-N torsional angle of 1 34 . 1 ° in 2a, corresponding synperiplanar structures are higher in energy. Nitrovinyl alcohol, 1b and nitrovinylamine 2b, have C, symmetry, the geometric parameters obtained for 2b are comparable to the X-ray c ry stal data reported for the trans-N,N-dimethyl-2-nitroethenamineIY . The most important observation is the short O . . . H nonbonded distances 1 .775 and 1 .992 A in 1b and 2b respectively, such small distances between 0 and H can be attributed to hydrogen bond. The O-H-O angle in 1b is 14 1 .2° and
1 02 INDIAN J CHEM, SEC. A, JAN - MARCH 2000
o " .. ,0
H 110.5 N' '\ "�/'Al3 ,.2'�S C ... "
o 121.1 \ H H 1 a
N.c.c-O 181.5 c..c.N-O 55.1
o ".,,0
H 110.1 N' '\ ':5r /U92 1.210//S q ..
H-tf IlIA \" " Ii H
2a N.c.c-O 134.1 c..c.N-O 54.0
1 b
1.1192 Ii - '
0 1.013/il,:--
1,251\ 1,2.45
H-N N -O 1,3045'\ /'0417 C=C / 1.351 '\
H H
2b
1.017 1.818 ��-q
,/141.2 1.371 \ 1.2.ZI
Q N -O - � /. 1.251 �C- C 1.351 - / 1.424 ,\ H H
1c
1.02' H- O 1 107 � � \ • ,,' 121.41.372\ 1.233 H- N. N -O
1.301'c _ C<350 / 1.424 '\ H H
2c
Fig, 2 - Important geometrical parameters of nitrovinyl alcohol, 1 and nitrovinylamine, 2 and their tautomers obtained at MP2(full)/6-3 1 G* level. Distances are in Angstrom units and angles are in degrees
H 0 I \ O,!=<N-o H H
1 b
H 0 I \ i- N N-O '!=< H H
2b
.. 1,3 H�rogen shIft ... 1,3 Hydrogen shIll ... -
1 a 1 c
1,3 Hydrogen shift ...
2a 2c
Scheme 2

BHARATAM : THEORETICAL STUDIES ON NITRO SUBSTITUTION IN TAUTOMERIC PROCESSES 103
H , --- H-o H 0 , 0 , \ , X)==(N-o
, \ X , ')N-O �
X N-O � H H H H H H
X=O 1 b 1 d 1 c X=NH 2b 2d 2c
Scheme 3
Table 2 - Absolute energies (in a.u.) of nitrovinyl alcohol, I and nitrovinylamine, 2, their tautomers
Structure NIF' HF/6-3 I G*
la C, 0 -356.37765
lb C 0 -356.37 1 29 , Ie C 0 -356.35703 , Id C -356.34804 ,
2a C, 0 336.54003
2b C 0 336.54755 , 2e C 0 336. 5 1 574 , 2d C 336.50763 , "Number of imaginary frequency hUsing MP2/6-3 1 G* geometries " Obtained at HF/6-3 1 G* level and scaled by 0.9 1 35
the N-H-O angle in 2b is 1 2 1 .4°. These values indicate that the hydrogen bond in lb is more stronger than the hydrogen bond in 2b. The C=C bond lengths 1 .352 and 1 .359 A in lb and 2b respectively at MP2(ful l)/ 6-3 1 G* level are longer than C=C bond in ethylene ( 1 .335 A) and in nitroethylene ( 1 .329 A) at the same level of theory. These values indicate that nitro substitution strengthens the C=C bond and further substitution by OH or NH, weakens the C=C bond in ethylene. The C-N bond distance in lb, 2b ( 1 .4 1 4 and 1 .4 1 7 A respectively) are shorter than the C-N d istance in nitromethane ( 1 .489 A) and nitroethylene ( 1 .46 1 A). All these factors indicate that there is a considerable amount
MP2/6-3 I G* MP4/6-3 1 G*h ZPE"
-357.366 1 7 -357.40566 37.3
-357.36069 -357.39829 38.5
-357.35329 -357.38967 37.9
-357. 349 1 1 -357.38392 35.3
337.5 1 459 337.55875 45.5
337.52 1 8 1 337.56267 45.5
337.49950 337.53963 45.5
337.49748 337.53609 42.8
of conjugation in the substituted ethylene system. Nitronic acid tautomers Ie and 2e are found to be true minima on the respective potential energy surfaces. Hydrogen bonds in Ie and 2e are characterized by small O . . . H distance ( 1 .6 1 6 A) in Ie and small N . . . H distance ( 1 .606 A) in 2e, smaller than those in lb and 2b. The tautomers Ie and 2e are highly conjugated as evidenced by distances. In Ie and 2e the N02H group adopts an anti O-N-O-H arrangement. In H2C=N02H, such an arrangement is found to be a transition state for rotation around N-O(H) bond l I u. This indicates that the intramolecular hydrogen bond in nitronic acids stabilises a formally unstable anti O-N-O-H arrangement.
1 04 INDIAN J CHEM, SEC. A, JAN - MARCH 2000
Table 3 - Relative energies ( in kcallmol) of nitrovinyl alcohol, I and nitrovinylamine, 2 and their tautomers.
ZPE corrected values are given in parentheses
Structure HF/6-3 I G* MP2/6-3 IG* MP4/6-3 IG*
I a 0.0 (0.0) 0.0 (0.0) 0.0 (0.0)
lb 4.0 (5 . 1 ) 3.4 (4.5) 4.6 (5.7)
Ie 1 2.9 ( 1 3.4) 8. 1 (8.6) 1 0.0 ( 1 0.5)
2a 0.0 (0.0) 0.0 (0.0) 0.0 (0.0)
2b -4.7 (-4.7) -4.5 (-4.5) -2.5 (-2.5)
2e 1 5.2 ( 1 5.2) 9.5 (9.5) 1 2.0 ( 1 2.0)
Energies 2-Nitroethanal, 1a is the global minimum on the PE
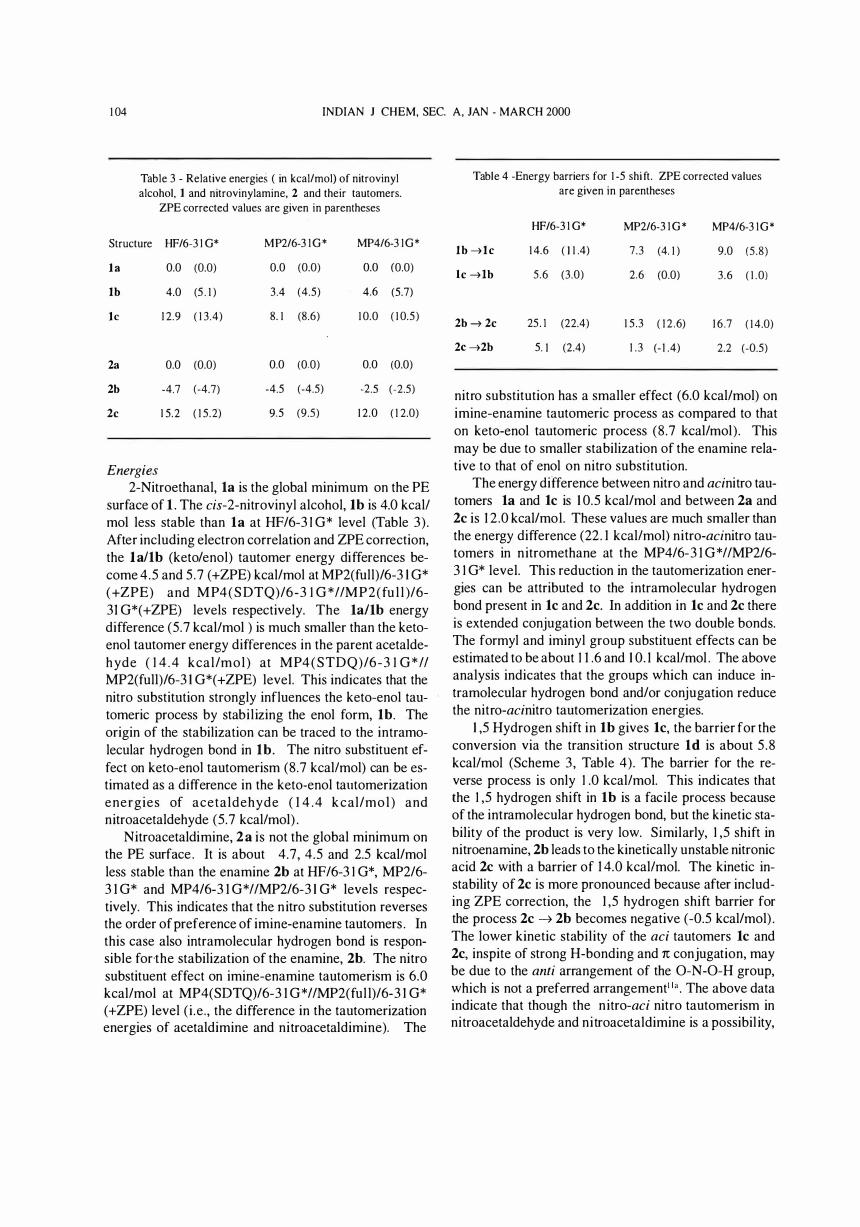
surface of 1 . The cis-2-nitrovinyl alcohol, 1b is 4.0 kcall mol less stable than 1a at HF/6-3 I G* level (Table 3) . After including electron correlation and ZPE correction, the 1a/1b (keto/enol) tautomer energy differences become 4.5 and 5 .7 (+ZPE) kcal/mol at MP2(full)/6-3 I G* (+ZPE) and MP4(SDTQ)/6-3 1 G*IIMP2(fu l l )/6-31 G*( +ZPE) levels respectively. The 1a11b energy difference (5.7 kcallmol ) is much smaller than the ketoenol tautomer energy differences in the parent acetaldehyde ( 1 4 .4 kcal/mol) at MP4(STDQ)/6-3 1 G*II MP2(full)/6-3 1 G*( +ZPE) level. This indicates that the nitro substitution strongly influences the keto-enol tautomeric process by stabilizing the enol form, lb. The origin of the stabilization can be traced to the intramolecular hydrogen bond in lb. The nitro substituent effect on keto-enol tautomerism (8.7 kcallmol) can be estimated as a difference in the keto-enol talltomerization energies of acetaldehyde ( 1 4 .4 kcal/mol ) and nitroacetaldehyde (5.7 kcallmol) .
Nitroacetaldimine, 2a is not the global minimum on the PE surface . It is about 4.7, 4.5 and 2.5 kcal/mol less stable than the enamine 2b at HF/6-3 1 G*, MP2/6-3 1 G* and MP4/6-3 1 G*IIMP2/6-3 I G* levels respectively. This indicates that the nitro substitution reverses the order of preference of imine-enamine tautomers . In this case also intramolecular hydrogen bond is responsible forthe stabilization of the enamine, 2b. The nitro substituent effect on imine-enamine tautomerism is 6.0 kcallmol at MP4(SDTQ)/6-3 1 G*IIMP2(ful l)/6-3 1 G* (+ZPE) level (i .e . , the difference in the tautomerization energies of acetaldimine and nitroacetaldimine). The
Table 4 -Energy barriers for 1 -5 shi ft. ZPE corrected values are given in parentheses
HF/6-3 I G* MP2/6-3 IG* MP4/6-3 IG*
Ib -7le 1 4.6 ( 1 1 .4) 7.3 (4. 1 ) 9.0 (5.8)
Ie -7Ib 5.6 (3.0) 2.6 (0.0) 3.6 ( 1 .0)
2b -7 2e 25. 1 (22.4) 1 5 .3 ( 1 2 .6) 1 6 .7 ( 1 4.0)
2c -72b 5. 1 (2.4) 1 .3 (- 1 .4) 2.2 (-0.5)
nitro substitution has a smaller effect (6.0 kcallmol) on imine-enamine tautomeric process as compared to that on keto-enol tautomeric process (8.7 kcallmol). This may be due to smaller stabilization of the enamine relative to that of enol on nitro substitution.
The energy difference between nitro and acinitro tautomers 1a and Ie is 1 0.5 kcal/mol and between 2a and 2e is 1 2 .0 kcal/mol. These values are much smaller than the energy difference (22. 1 kcallmol) nitro-acinitro tautomers in nitromethane at the MP4/6-3 1 G*IIMP2/6-3 1 G* level. This reduction in the tautomerization energies can be attributed to the intramolecular hydrogen bond present in Ie and 2e. In addition in Ie and 2e there is extended conjugation between the two double bonds. The formyl and iminyl group substituent effects can be estimated to be about 1 1 .6 and 1 0. 1 kcallmol . The above analysis indicates that the groups which can induce intramolecular hydrogen bond and/or conjugation reduce the nitro-acinitro tautomerization energies.
1 ,5 Hydrogen shift in Ib gives Ie, the barrier for the conversion via the transition structure 1d is about 5 .8 kcallmol (Scheme 3, Table 4). The barrier for the reverse process is only 1 .0 kcallmol. This indicates that the 1 ,5 hydrogen shift in 1b is a facile process because of the intramolecular hydrogen bond, but the kinetic stability of the product is very low. Similarly, 1 ,5 shift in nitroenamine, 2b leads to the kinetically unstable nitronic acid 2e with a barrier of 1 4.0 kcallmol. The kinetic instability of 2e is more pronounced because after including ZPE correction, the 1 ,5 hydrogen shift barrier for the process 2e � 2b becomes negative (-0.5 kcal/mol) . The lower kinetic stability of the ad tautomers Ie and 2e, inspite of strong H-bonding and 1t conjugation, may be due to the anti arrangement of the O-N-O-H group, which is not a preferred arrangement" " . The above data indicate that though the nitro-ad nitro tautomerism in nitroacetaldehyde and nitroacetaldimine is a possibil ity,

BHARATAM : THEORETICAL STUDIES ON NITRO SUBSTITUTION IN TAUTOMERIC PROCESSES 1 05
the kinetic stability of the corresponding acinitro compounds is small and it may not be possible to isolate these products. On the other hand, 1 ,3-hydrogen shift in la and 2a to give enol and vinylamine derivatives respectively becomes more feasible because of the substitution with nitro group, owing to the increased stability of these tautomers .
Conclusions Cis-nitro substitution at the � position stabilizes vi
nyl alcohol and vinylamine mainly due to the intramolecular hydrogen bond formation. This reduces the ketoenol and imine-enamine tautomer energy differences. The n itro substituent effect in these tautomeric processes can be estimated to be 8.7 and 6.0 kcaVmol. Nitroacin i tro tau tomeri sm i n n itroacetaldehyde and nitroacetaldimine gives kinetically unstable aci tautomers, though the formyl and iminyl substituent effects are about 1 1 .6 and 1 0 . 1 kcaVmol. 1 ,5 hydrogen shift in Ib and 2b requires 5 .8 and 14.0 kcallmol respectively.
Acknowledgement
The author thanks DST, New Delhi for financial support.
References 1 Ingold C K, Structure and mechanism in organic chemis
try ( Cornell University Press New York), 1 953. 2 Toullec J, Adv phys org Chern, 1 8 ( 1 982) I . 3 The chemistry of enols, edited by Z Rappoport (Wiley,
Chichester), 1 990. 4 Jemmis E D, Giju K T & Leszczynski J, J phys Chern A ,
10 1 ( 1997) 7389.
5 Apeloig Y, Arad D & Rappoport Z, J Am chern Soc, l I 2 ( 1990) 9 1 3 1 .
6 Smith B J, Nguyen M T, Bouma W J & Radom L J Am chern Soc , 1 1 3 ( 1 99 1 ) 6452.
7 Rappoport Z & Biali S E, Acc chern Res , 2 1 ( 1 988) 442. 8 Wu C C & Lien M H, J phys Chern, 1 02 ( 1 996) 594. 9 Smith B J & Radom L, J Am chern Soc, 1 14 ( 1 992) 36. 10 Lammertsma K & Prasad B V, J Am chern Soc, 1 1 6
( 1 994) 642. 1 1 (a) Lammertsma K & Prasad B V, J Am chern Soc, 1 1 5
( 1 993) 2348; (b) Harris N J & Lammertsma K, J Am chern Soc, 1 1 8 ( 1 996) 8048.
1 2 Bock H, Dienelt R, Schodel H, Halvas Z, Herdtweck E & Herrmann W A, Angew Chern Int Ed Engl, 32 ( 1 993) 1 758.
13 Beksic D, Bertran J, Lluch J M & Hynes J T, J phys Chern A 102 ( 1 998) 3977.
14 Bharatam P V & Lammertsma K (unpublished results). 1 5 (a) Prasad B V, Punam U & Bassi P S, Chern Phys Lett,
276 ( 1997) 3 1 ; (b) Prasad B V, Punam U, Grover G & Kaur D, THEOCHEM, 458 ( 1999) 227.
16 For an introduction of the methods employed, see:Hehre W J, Radom L, Schleyer P V R & Pople lA, Ab initio molecular orbital theory (Wiley, New York), 1 986.
17 Gaussian94, Revision B .2, Frisch M J, Trucks G W, Schlegel H B , Gill P M W, Johnson B G, Robb M A, Cheeseman J R, Keith T, Petersson G A, Montgomery J A, Raghavachari K, AI-Laham M A, Zakrzewski V G, . Ortiz J V, Foresman J B , Peng C Y, Ayala P Y, Chen W, Wong M W, Andres J L, Replogle E S , Gomperts R, Martin R L, Fox D J, B inkley J S, Defrees D J, Baker J, Stewart J P, Head-Gordon M, Gonzalez C & Pople J A, Gaussian, Inc., Pittsburgh PA, 1995.
1 8 Scott A P & Radom L, J phys Chern, 100 ( 1 996) 16502. 1 9 Hazel A & Mukhopadhyay A, Acta Cryst, B36 ( 1 980)
747.





























