Embed Size (px)
Citation preview



Inês Filipa Rodrigues Vaz Influência dos Polimorfismos Genéticos TLR3 rs3775291e TLR7 rs179008 na sobrevivência de doentes com Linfomas Não-Hodgkin
Dissertação de Candidatura ao grau de Mestre em
Oncologia – Especialização em Oncologia
Molecular submetida ao Instituto de Ciências
Biomédicas de Abel Salazar da Universidade do
Porto
Orientador - Professor Doutor Rui Manuel de
Medeiros de Melo Silva
Categoria - Professor Associado Convidado com
Agregação
Afiliação - Instituto de Ciências Biomédicas Abel
Salazar da Universidade do Porto e Grupo de
Oncologia Molecular e Patologia Viral - Centro de
Investigação do Instituto Português de Oncologia
do Porto
Co-Orientadora - Doutora Ana Luísa Pereira
Teixeira
Afiliação - Grupo de Oncologia Molecular e
Patologia Viral - Centro de Investigação do
Instituto Português de Oncologia do Porto

Informação Técnica
TÍTULO:
Influência dos Polimorfismos Genéticos TLR3 rs3775291 e TLR7 rs179008 na
sobrevivência de doentes com Linfomas Não-Hodgkin
Tese de Candidatura ao Grau de Mestre em Oncologia - Especialização em Oncologia
Molecular submetida ao Instituto de Ciências Biomédicas Abel Salazar da Universidade
do Porto
AUTOR: Inês Filipa Rodrigues Vaz
DATA: Dezembro de 2015
EDITOR: Inês Filipa Rodrigues Vaz
MORADA: Rua Pedro Monjardino
LOCALIDADE: Lisboa
CÓDIGO POSTAL: 1600-892 Lisboa
CORREIO ELETRÓNICO: [email protected]
1ª EDIÇÃO: Setembro de 2015
2ª EDIÇÃO: Dezembro de 2015



Agradecimentos
Agradeço à comissão de coordenação do Mestrado em Oncologia, sob a pessoa da
Professora Doutora Berta Silva a oportunidade de ingressar a este mestrado, que me
enriqueceu em conhecimentos sobre Oncologia, uma área que sempre me despertou
curiosidade.
Ao Professor Doutor Rui Medeiros, meu orientador, um grande obrigado pela
oportunidade de realizar a minha Dissertação de Mestrado no grupo de Oncologia
Molecular e por todo apoio, compreensão e disponibilidade ao longo deste ano.
Do mesmo modo, quero agradecer à Doutora Ana Luísa, a minha co-orientadora e ao
grupo de Oncologia Molecular e Patologia Viral por toda a colaboração.
À Ana Bela companheira e amiga, por toda a compreensão e apoio que me deu ao
longo deste ano. Não esquecerei que esteve sempre comigo nos momentos mais difíceis.
À Ana Clara, uma das minhas melhoras amigas, sempre disponível a ajudar e a ouvir.
Agradeço-lhe do fundo do coração toda a ajuda que me deu ao longo desde percurso na
minha vida, mesmo estando longe.
À Inês Marques uma grande amiga de “A” maiúsculo.
Aos meus pais e ao meu irmão, o Nuno, hoje eu não estaria aqui se não fossem eles.
Em especial à minha mãe, o meu alicerce na vida, a quem eu quero dedicar este trabalho
como homenagem pela luta que ela também já travou contra o cancro.
Obrigado a todos, por tudo. Terei sempre presente em mim tudo o que vivi nesta longa
jornada, por contribuir para o que sou hoje esperando que cada vez mais mude para uma
pessoa melhor!


Abreviaturas
AP-1: Activator Protein-1
DNA: Deoxyribonucleic Acid
DAMPs: Damage-associated molecular pattern
dsRNA: Double-Stranded RNA
FADD: Fas-associated death domain protein
GVHD: Graft-versus-host disease
HSCT: Hematopoietic Stem Cell Transplantation
IL-1: Interleucina 1
IL-6: Interleucina 6
IL8: Interleucina 8
IL-12: Interleucina 12
IRAK: Interleukin-1 Receptor- Associated Kinase
IFNs: Interferões
IFN-β: Interferão β
IFN-α: Interferão α
IRF7: Interferon regulatory factor 7
IRF3: Interferon regulatory factor 3
IKK: I kappa B kinase
MyD88: Myeloid differentiation factor 88
MAPK: mitogen-activated protein kinases
NK: Natural Killer
NHL: Non-Hodgkin Lymphoma
NF-kB: Nuclear factor-kappa B
PAMPs: Pathogen- associated molecular patterns
Poly (I:C): Polyinosinic-polycytidylic acid
P: P-value
PCR: Polymerase Chain Reaction
RNA: Ribonucleic Acid
RE: Reticulo Endoplasmático
RIP1: Receptor interacting proteins 1
ssRNA: Single-stranded RNA
SNPs: Single Nucleotide Polymorphism
TLRs: Toll-like Receptors
TIR: Toll-IL1-receptor
TRIF: TIR-domain-containing adaptor inducing IFN-β
TRAF3: Tumor Necrose Factor Receptor-Associated Factor 3
TRAF6: Tumor Necrose Factor Receptor-Associated Factor 6

Abreviaturas
TNF- α: Tumor Necrose Factor - α
TBK1: TANK-binding kinase 1
TAK-1: Tumor Growth Factor- β- Associated- Kinase
TAB 1: TAK-1-Binding Protein 1
TAB 2: TAK-1-Binding Protein 2
UNC93B1: Uncoordinated 93B1
VIH: Vírus da Imunodeficiência Humana

Índice Geral
RESUMO .......................................................................................................................... 15
ABSTRACT ...................................................................................................................... 19
1. Introdução .................................................................................................................... 23
1.1 O Cancro ..................................................................................................................... 25
1.2 Linfomas ...................................................................................................................... 28
1.3 Sistema Imunitário: Toll-Like Receptors ..................................................................... 31
1.3.1 TLRs Endossomais: TLR3 e TLR7 .......................................................................... 34
1.4 Polimorfismos Genéticos nos genes TLR3 e TLR7 .................................................... 38
2. Objetivos ...................................................................................................................... 41
2.1 Objetivo Geral ............................................................................................................. 43
2.2 Objetivo Específico ..................................................................................................... 43
3. Materiais e Métodos .................................................................................................... 45
3.1 População em Estudo ................................................................................................. 47
3.2 Procedimento Laboratorial .......................................................................................... 47
3.2.1 Extração do DNA Genómico .................................................................................... 48
3.2.2 Genotipagem dos Polimorfismos TLR3 rs3775291 e TLR7 rs179008 ..................... 48
3.3 Análise Estatística ....................................................................................................... 50
4. Resultados ................................................................................................................... 51
4.1 Frequência dos polimorfismos genéticos TLR3 rs3775291 e TLR7 rs179008 em
doentes com NHL ............................................................................................................. 53
4.2 Influência do polimorfismo TLR3 rs3775291 na sobrevivência de doentes com NHL 54
4.3 Influência do polimorfismo TLR7 rs179008 na sobrevivência de doentes com NHL .. 55
5. Discussão .................................................................................................................... 57
6. Conclusão e Perspetivas Futuras ............................................................................. 63
7. Referências Bibliográficas ......................................................................................... 67

Índice das Figuras
Figura 1. Número de novos casos cancro estimados em 2012, em 21 áreas do mundo 25
Figura 2. Esquema das infeções oportunistas mais frequentes, ao longo das diferentes
fases, e do tempo (em dias) após HSCT .......................................................................... 31
Figura 3. Representação esquemática das vias de sinalização intracelular dependentes
de TRIF ............................................................................................................................. 35
Figura 4. Representação esquemática das vias de sinalização intracelular dependentes
de MyD88 .......................................................................................................................... 36
Figura 5. Representação esquemática da indução da apoptose, mediada pelo TLR3 .... 37
Figura 6. Representação de um Real-time PCR para o polimorfismo TLR3 rs3775291 . 49
Figura 7. Influência do polimorfismo TLR3 rs3775291 na sobrevivência A) a um ano e de
B) de longo termo nos doentes com NHL ......................................................................... 54
Figura 8. Influência do polimorfismo TLR7 rs179008 na sobrevivência A) a um ano e de
B) de longo termo em doentes com NHL do género feminino .......................................... 55
Figura 9. Influência do polimorfismo TLR7 rs179008 na sobrevivência A) a um ano e de
B) de longo termo em doentes com NHL do género masculino ....................................... 56
Figura 10. Influência da variante T do polimorfismo TLR3 rs3775291 nas funções
mediadas pelo recetor ....................................................................................................... 61

Índice das Tabelas
Tabela 1. Toll-like Receptors Humanos e os respetivos ligandos .................................... 32
Tabela 2. Características clínicas e patológicas gerais da população em estudo ........... 47
Tabela 3. Características clínicas dos doentes com NHL, relativamente à presença dos
polimorfismos genéticos TLR3 rs3775291 e TLR7 rs179008 .......................................... 53


Resumo


Resumo
17
Os linfomas são um grupo heterogéneo de doenças malignas com origem em
linfócitos B, linfócitos T e em menor frequência de células natural killer. Os linfomas Não-
Hodgkin (Non-Hodgkin Lymphoma: NHL) são o subgrupo de tumores malignos com
origem linfóide mais frequente. Os principais fatores de risco que condicionam o
desenvolvimento da doença relacionam-se essencialmente com alterações na função do
sistema imunitário. O sistema imunitário tem um papel central na manutenção da
homeostasia do organismo, reconhecendo agentes infeciosos e ao mesmo tempo pode
prevenir condições de inflamação crónica associada ao desenvolvimento de cancro. Os
Toll-like Receptors constituem parte do sistema imunitário inato, podendo ser ativados
por ácidos nucleicos provenientes do genoma de vírus assim como do próprio
hospedeiro, sendo assim fundamentais como uma primeira linha de defesa do
hospedeiro, e também como mediadores da resposta antitumoral.
Diversos estudos têm demonstrado que polimorfismos genéticos funcionais nos genes
TLR3 e TLR7 podem conferir não só maior suscetibilidade para determinadas infeções,
mas também modular o desenvolvimento/progressão tumoral. Deste modo, foi objetivo do
presente trabalho avaliar a influência dos polimorfismos genéticos TLR3 rs3775291 e
TLR7 rs179008 na sobrevivência a um ano e na sobrevivência de longo termo de
doentes diagnosticados com NHL.
Neste trabalho foram analisadas amostras de DNA de cento e vinte e nove (129)
indivíduos com diagnóstico de NHL. A análise dos polimorfismos genéticos TLR3
rs3775291 e TLR7 rs179008 foi efetuada por Real-Time PCR.
Relativamente ao polimorfismo TLR3 rs3775291, a análise das curvas de
sobrevivência de Kaplan-Meier evidenciou que os doentes homozigóticos CC
apresentaram uma sobrevivência de longo termo superior comparativamente aos doentes
portadores dos genótipos CT/TT (352,9 vesus 173,5 semanas, P=0,040). Quanto ao
polimorfismo TLR7 rs179008, observou-se que as doentes homozigóticas AA
apresentaram uma tendência para uma maior sobrevivência de longo termo
comparativamente a doentes portadoras dos genótipos AT/TT (231,4 versus 134,7
semanas, P=0,057). Contudo, não observamos diferenças estatisticamente significativas
na sobrevivência a um ano quanto aos dois polimorfismos estudados. De acordo com os
resultados obtidos podemos admitir que a ocorrência de polimorfismos genéticos
funcionais nos genes codificantes TLR3 e TLR7 podem modular um microambiente
celular podendo promover a progressão da doença após reconstituição total do sistema
imunológico dos doentes com NHL.
Futuramente, a utilização de agonistas destes recetores poderá ser uma estratégia
promissora no tratamento dos doentes com NHL, sendo que a definição de perfis

Resumo
18
genéticos segundo os polimorfismos estudados poderia ser útil na identificação de
subgrupos de doentes que apresentarão maior benefício na realização destas terapias.

Abstract


Abstract
21
Lymphomas are a heterogeneous group of malignancies arising from B and T
lymphocytes and less frequently from natural killer cells. The Non-Hodgkin Lymphoma
(NHL) is the most frequent subgroup of malignant tumors resulting from lymphoid origin.
The leading risk factors that predispose to development of diseases relate essentially to
changes in immune system function. The immune system has a central role not only in
response to external aggressions but in maintenance of cellular homeostasis, recognizing
infectious agents and at the same time it can prevent chronic inflammatory conditions
associated with cancer development. Toll-like Receptors (TLRs) are part of innate
immune system and can be activated by nucleic acids from virus genome and from host.
They represent a first line of defense and mediate the anti-tumoral immune response.
Several studies have been demonstrated that TLR3 and TLR7 genetic polymorphisms
might confer not only more susceptibility to infections but also can modulate tumoral
progression/development. Thus, the aim of the present study was to evaluate the
influence of TLR3 rs3775291 and TLR7 rs179008 genetic polymorphisms in one year
survival and long-term survival in patients diagnosed with NHL.
In this study we analyzed one hundred twenty nine (129) DNA samples from patients
with NHL. Both polymorphisms were analyzed using Real-Time PCR.
Relatively to TLR3 rs3775291 polymorphism, the Kaplan-Meier survival showed that
CC homozygous patients present a higher long-term survival comparing to CT/TT patients
(352.9 versus 173.5 weeks, P=0.040). Regarding the TLR7 rs179008 polymorphism, it
was observed that AA homozygous women presented a long-term survival tendency
comparing to AT/TT woman (231.4 versus 134.7 weeks, P=0.057). Moreover, we did not
observe any statistically significant differences in short-term survival regarding the two
polymorphisms studied.
According to the results, we can admit that occurrence of functional genetic
polymorphism in TLR3 and TLR7 genes can modulate a cellular microenvironmental can
promote disease progression after total immune reconstitution in NHL patients. In the
future, the use of TLRs agonists can be a promising strategy in the treatment of NHL
patients.


1. Introdução


1. Introdução
25
1.1 O Cancro
Atualmente o cancro é considerado um dos maiores problemas de saúde publica, no
mundo, por se revelar uma das principais causas de morte nos países economicamente
desenvolvidos e a terceira causa de morte em países subdesenvolvidos 1.
O crescimento e envelhecimento da população, refletem-se no aumento da esperança
média de vida, o que é considerado um bom indicador de saúde. Dado que, o
envelhecimento é uma característica funcional dos organismos biológicos,
consequentemente acarreta o desenvolvimento de doenças crónicas características, tais
como o cancro 2,3.
A figura 1 representa o número de novos casos de cancro ocorridos em 2012, na
população mundial. Nesse ano estimaram-se 14 090 100 de novos casos e a 8 201 600
de mortes provocadas por esta doença, sendo esperado que venha a aumentar no futuro 4.
Figura 1– Número de novos casos de cancro estimados em 2012, em 21 áreas do mundo.
(Fonte: Global Cancer Statistics 2012 4)
the all-sites cancer incidence rate for both sexes combinedin Western Europe is more than twice as high as that inEastern Africa (Table 2).
Although incidence rates for all cancers combined aretwice as high in more developed compared with less devel-oped countries, mortality rates are only 8% to 15% higherin more developed countries. This disparity primarilyreflects differences in cancer profiles and/or the availabilityof treatment. For example, liver cancer, a highly fatal can-cer, is much more common in less developed countries,thus contributing disproportionately to the overall cancermortality rate in these countries. Similarly, cancers aremore often detected at a later stage in less developed coun-tries (Fig. 3), which contributes to the disparity.
Selected Cancers
Female breast cancer
Breast cancer is the most frequently diagnosed cancer andthe leading cause of cancer death among females worldwide,with an estimated 1.7 million cases and 521,900 deaths in2012 (Fig. 2). Breast cancer alone accounts for 25% of allcancer cases and 15% of all cancer deaths among females.
More developed countries account for about one-half of allbreast cancer cases and 38% of deaths. Rates are generallyhigh in Northern America, Australia/New Zealand, andNorthern and Western Europe; intermediate in Central andEastern Europe, Latin America, and the Caribbean; and lowin most of Africa and Asia (Fig. 4). International variationin breast cancer incidence rates reflects differences in theavailability of early detection as well as risk factors. Risk fac-tors for breast cancer include reproductive and hormonal fac-tors such as a long menstrual history, recent use of oralcontraceptives, and never having children.12 Giving birth tochildren and breastfeeding decrease the risk of breast can-cer.12 Potentially modifiable risk factors include weight gainafter age 18 years, being overweight or obese (for postmeno-pausal breast cancer), use of menopausal hormone therapy(combined estrogen and progestin), physical inactivity, andalcohol consumption.12,13
Between 1980 and the late 1990s, breast cancer incidencerates rose approximately 30% in Western countries, likelybecause of changes in reproductive factors and the use ofmenopausal hormone therapy and more recently because ofincreased screening.14 Declining incidence rates in the early2000s have been attributed to the reduced use of menopausal
FIGURE 1. Estimated Number of New Cancer Cases in 21 World Areas, 2012.*Region estimates do not sum to the worldwide estimate due to calculation method.Source: GLOBOCAN 2012.
CA CANCER J CLIN 2015;00:00–00
VOLUME 00 _ NUMBER 00 _ MONTH 2015 3

1. Introdução
26
De entre os vários fatores de risco que condicionam o desenvolvimento de cancro, as
mutações hereditárias contribuem para o aparecimento de formas de cancro familiar,
correspondendo a 5-10% dos casos 5. A adoção de determinados estilos de vida, como
por exemplo os hábitos tabágicos, o consumo de álcool, uma dieta desequilibrada, o
sedentarismo, ou a exposição a fatores ambientais como a radiação, poluentes e ainda
microrganismos infeciosos, podem condicionar a ocorrência de formas esporádicas de
cancro 6.
Um cancro pode ser definido como um processo celular, onde as células adquirem
propriedades anormais podendo levar à transformação num fenótipo maligno, com
capacidade de proliferação descontrolada 7. Estas células sofrem múltiplas alterações no
ácido desoxirribonucleico (Deoxyribonucleic Acid: DNA), resultantes da interação entre a
genética do hospedeiro e a exposição prolongada a fatores ambientais, ocupacionais
entre outros que estão presentes no quotidiano. Deste modo, é possível inferir que uma
grande percentagem de casos de cancro possa ser reduzida, alterando os
comportamentos e estilos de vida de um individuo 5.
O cancro tem como principal origem alterações no genoma, que permitem a aquisição
de novas propriedades ao longo do processo de transformação. As competências
adquiridas pelas células caracterizam-se em dez hallmarks: auto-suficiência em fatores
de crescimento; evasão aos mecanismos supressores de crescimento; evasão ao
sistema imunitário; capacidade replicativa ilimitada; promoção da inflamação; mutações e
instabilidade genómica; resistência à morte celular programada; desregulação energética
das células; indução da angiogénese; capacidade de invasão e de metastização 8.
A carcinogénese é um processo complexo, multifactorial e pode ser dividido em três
fases a iniciação, a promoção e a progressão 9,10. A iniciação é o primeiro evento deste
processo proporcionada pela ação de agentes de diferente natureza, como por exemplo
agentes químicos genotóxicos, provocando alterações no DNA das células 9,11. Caso a
célula não tenha capacidade de reparar o dano sofrido, a exposição continua a agentes
carcinogénicos poderá permitir uma acumulação sucessiva de alterações no genoma,
que conferem vantagens de crescimento celular relativamente às células normais 12,13.
Posteriormente, a célula “iniciada” ao longo de todo processo da carcinogénese vai sofrer
um acumular de alterações genéticas em genes que regulam a proliferação celular (os
proto-oncogenes e os genes supressores tumorais), a morte celular programada e em
genes envolvidos na manutenção da integridade do genoma 14,15.
A segunda fase do processo da carcinogénese constitui a promoção, sendo mediada
por agentes promotores que estimulam a proliferação da célula “iniciada” 16. Estes podem
induzir alterações epigenéticas influenciando a expressão de genes que estimulam o

1. Introdução
27
crescimento celular, e assim, há um aumento da probabilidade de ocorrer erros na
replicação do DNA e de se gerarem novas mutações 17.
No fim da carcinogénese decorre a progressão, uma etapa irreversível, em que as
células do tumor continuam a proliferar progredindo para um fenótipo maligno 10,18. Ao
longo desta fase, as células pré-malignas vão acumulando novas alterações genéticas e,
também, epigenéticas, tornando-se autossuficientes na proliferação, independentemente
da presença ou ausência de estímulos 18. Subsequentemente, as células tornam-se
capazes de invadir os tecidos circundantes até alcançarem os vasos sanguíneos e
linfáticos de modo a invadir outros tecidos à distância – a metastização 17. Outro fator
importante para a progressão tumoral é a neoangiogénese, que consiste na formação de
novos vasos sanguíneos, aumentando o aporte de nutrientes e oxigénio para as células,
estando, também, implicado no processo de metastização, uma vez que pode fornecer
uma rota de entrada das células tumorais na corrente sanguínea11.
O microambiente tumoral engloba um tumor em desenvolvimento e um estroma
constituído por vasos sanguíneos, infiltrado de células inflamatórias e uma variedade de
células de outros tecidos 19. Associado a este microambiente encontra-se um clima
inflamatório que sustenta o desenvolvimento e progressão de um tumor, e ainda um
conjunto de moléculas produzidas pela interação entre o tumor e o estroma 20.

1. Introdução
28
1.2 Linfomas
Os linfomas são um grupo heterogéneo de doenças malignas, com origem em
linfócitos B, linfócitos T e em situações menos comuns provêm de células NK (natural
killer: NK) 21. O processo de carcinogénese ocorre devido à acumulação de alterações
genéticas e epigenéticas nas células percursoras linfóides, resultando na desregulação
dos mecanismos de diferenciação celular 22. A caracterização de um linfoma é baseada
na sua origem celular, nas suas características morfológicas e histológicas, no
imunofenótipo das células e ainda nas alterações genéticas características desta células
malignas 23.
Em 2008, a Organização Mundial de Saúde reformulou a classificação dos tumores
hematopoiéticos e dos tecidos linfóides, diferenciando-os em grupos: neoplasias de
células B maduras; neoplasias de células T e células NK maduras; linfomas de Hodgkin;
neoplasias histociticas e de células dendríticas; doenças linfoproliferativas pós-
transplante 24.
Uma vez que as características histológicas, moleculares e clínicas dos Linfomas de
Hodgkin são diferentes dos outros linfomas, os restantes são denominados de Linfomas
Não-Hodgkin (Non-Hodgkin Lymphoma: NHL) 25.
Os Linfomas de Hodgkin, também conhecidos como doença de Hodgkin, representam
10-12% de todos os linfomas 22,26. Os linfomas de Hodgkin clássicos são identificados
pela presença de células Reed-Sternberg, gigantes e multinucleadas, podendo subdividir-
se em grupos conforme a composição e morfologia das células. Os linfomas de Hodgkin
de predomínio linfocítico apresentam algumas semelhanças com os anteriores, embora
sejam considerados uma entidade distinta devido às diferenças nas suas características
clínicas 27.
Os NHL são o subgrupo de tumores malignos com origem linfóide mais frequente 28. A
nível mundial, em 2012 estimou-se a ocorrência de 385 700 novos casos e 199 700
mortes causadas por esta doença 4.
O desenvolvimento de NHL encontra-se associado a diversos fatores de risco os quais
podem condicionar a função do sistema imunitário. As doenças autoimunes, tais como o
lúpus eritematoso sistémico, doença celíaca ou tiroidite de Hashimoto e doenças
inflamatórias como por exemplo a artrite reumatóide podem contribuir para o
desenvolvimento de NHL 29. Os indivíduos submetidos a fármacos imunossupressores
também podem ter maior predisposição para a doença, uma vez que estes fármacos
afetam a função do sistema imunitário 22,30. Por exemplo a infeção pelo vírus da
imunodeficiência humana (VIH) provoca um descontrolo na imunidade celular, e assim a
infeção mantém-se levando à estimulação persistente dos linfócitos B, podendo estar na

1. Introdução
29
origem de NHL 31. Por outro lado, o vírus Epstein-Barr apresenta capacidade de infectar e
transformar os linfócitos B, sendo importante na etiologia de NHL com origem em células
B 32. O Helicobacter pylori parece estar associado ao aumento do risco de
desenvolvimento de NHL com origem em tecidos linfóides associados à mucosa
(mucosa-associated lymphoid tissue) do estômago. Estes são alguns dos principais
microrganismos patogénicos associados à etiologia de NHL 26.
Os linfócitos B são produzidos na medula óssea e migram até aos órgãos linfóides
secundários (gânglios linfáticos, mucosas e baço) 28,33. Estes tecidos contêm vários
compartimentos organizados em folículos, onde se encontra o centro germinativo, um
microambiente responsável pela proliferação e diferenciação de linfócitos B naive em
células B memória ou plasmócitos 34.
Os linfócitos B naive são estimulados por antigénios e estabelecem interações com
células T Helper, que controlam a proliferação e diferenciação 35,36. Posteriormente, os
linfócitos B migram para os folículos secundários (centro germinativo) sucedendo-se uma
série de eventos celulares com intuito de gerar linfócitos B com capacidade de
reconhecer e produzir anticorpos, com elevada afinidade para antigénios específicos 37.
Os NHL de células B são entidades distintas, derivam da transformação maligna das
células em determinadas fases da diferenciação celular 38. Deste modo, as células
comportam diversas alterações moleculares originadas durante o seu desenvolvimento
no centro germinativo. Por exemplo, alguns NHL apresentam a translocação t(14;18), o
que confere um aumento da expressão do gene BCL-2 o que pode proporcionar
resistência à morte celular programada 39.
Os NHL com origem em células T e NK são considerados um subgrupo raro, derivam
de diferentes subpopulações de células, com funções e perfis moleculares diferentes 40.
Para determinar o estadiamento dos linfomas é usado o sistema Ann Abor, o qual
descreve a extensão da doença no organismo dos indivíduos 41. As opções terapêuticas
dependem de vários fatores, principalmente o tipo de NHL e o seu comportamento clínico
(indolente ou agressivo) e o estadio da doença. De uma forma geral, o tratamento de
NHL consiste em protocolos de quimioterapia, sendo as doses ou a combinação de
fármacos dependente das características clínicas do linfoma. A radioterapia ou
imunoterapia, por exemplo com rituximab podem ser aplicados como monoterapia, em
conjunto ou em combinação com a quimioterapia 26,42.
No tratamento de doentes com NHL pode ser recomendado o transplante de células
estaminais hematopoiéticas (Hematopoietic Stem Cell Transplantation: HSCT), conforme
as características da doença 43. O HSCT é um procedimento realizado no âmbito do
tratamento de algumas neoplasias (tumores hematológicos e tumores sólidos), doenças
hematológicas, imunodeficiências e doenças genéticas 44,45. As fontes de células

1. Introdução
30
estaminais hematopoiéticas provêm da medula óssea, de células estaminais do sangue
periférico e ainda do sangue do cordão umbilical. A colheita das células pode ser feita
com recurso a um aspirado de medula óssea, num bloco operatório, ou pode ser feita por
aférese 46. Esta terapia tem como principal objectivo curar a doença em causa e
restabelecer a função da medula óssea 47.
O HSCT pode ser classificado em autólogo ou alogénico, tendo como base a relação
do dador e do doente 44. O HSCT autólogo consiste na colheita prévia de células
estaminais hematopoiéticas do doente, e posterior administração das mesmas 45. O
HSCT alogénico é um processo mais elaborado e complexo, onde os doentes recebem
um enxerto de células estaminais hematopoiéticas de um dador saudável 46.
Antes de se proceder ao HSCT, os doentes são submetidos a regimes de
quimioterapia ou a regimes de combinação de quimioterapia com radioterapia com o
intuito de erradicar o tumor residual e suprimir o sistema imunitário do doente, a fim de
prevenir a rejeição do enxerto 47,48. A aplicação de regimes mais intensivos nos doentes
relacionam-se com o desenvolvimento de toxicidade sendo a razão de elevadas taxas de
mortalidade, o que limita a sua aplicação em crianças ou doentes com mais de 50 anos
de idade 49. A doença do enxerto-contra-hospedeiro (Graft-versus-host disease: GVHD) é
uma complicação muito frequente após o HSCT alogénico, caracterizada pela reação de
células T dadoras contra antigénios presentes nas células do hospedeiro 50. Para a
profilaxia da doença são administrados fármacos imunossupressores, que causam uma
depressão no funcionamento do sistema imunitário dos doentes, tornando-os
imunocomprometidos 51. Assim, os doentes encontram-se mais suscetíveis a
determinadas infeções oportunistas, existindo uma ordem “cronológica” descrita em
guidelines, que pode ser observada consoante a fase do transplante 49. O esquema
representado na figura 2 representa a prevalência dos vários tipos de infeções
(bacterianas, virais e fúngicas) em cada uma das fases após HSCT.

1. Introdução
31
Figura 2- Esquema das infeções oportunistas mais frequentes, ao longo das diferentes fases, e do
tempo (em dias) após HSCT. (Adaptado de Tomblyn M et al 2009 49)
1.3 Sistema Imunitário: Toll-like Receptors
O sistema imunitário é constituído pela imunidade inata e adquirida, ambas integram a
defesa do hospedeiro contra microrganismos patogénicos 52.
A imunidade inata é a primeira linha de defesa do organismo, contra infeções
provocadas por agentes microbianos, onde os Toll-like-receptors (TLRs) têm um papel
fundamental na deteção destes agentes e posterior indução de uma resposta efetora 53. A
proteína Toll foi inicialmente descoberta na Drosophila melanogaster, sendo
posteriormente identificado em mamíferos recetores homólogos à proteína Toll, cuja
função estava envolvida na resposta inflamatória 54,55.
Os TLRs são uma família de dez recetores que pertencem aos pattern-recognition
receptors e são codificados por genes que se localizam em diferentes cromossomas 55.
Estes recetores têm capacidade de desencadear uma resposta imune contra agentes
infecciosos, uma vez que reconhecem padrões moleculares associados a patogénios
(Pathogen- associated molecular patterns: PAMPs), componentes estruturais de agentes
microbianos (bactérias, vírus, fungos), como por exemplo lipossacarideos, lipoproteínas,
peptidoglicanos, flagelos e oligossacarídeos (tabela 1) 56.
1. Introdução
32
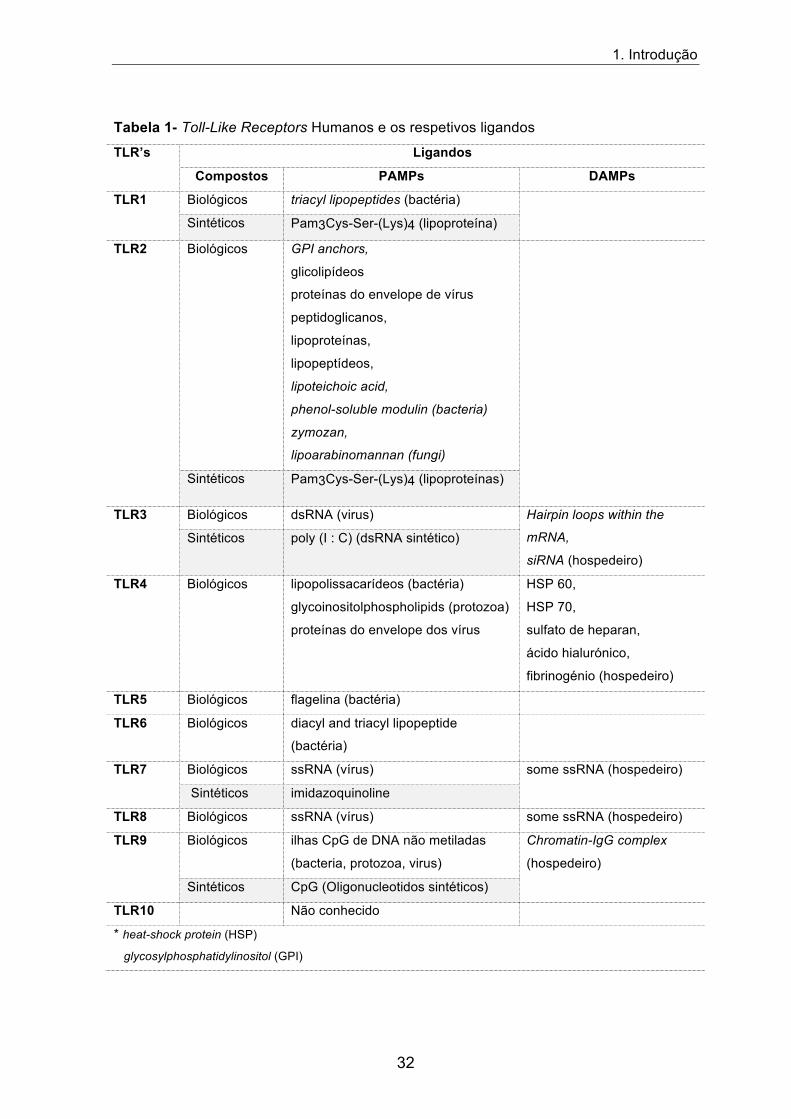
Tabela 1- Toll-Like Receptors Humanos e os respetivos ligandos
TLR’s Ligandos
Compostos PAMPs DAMPs
TLR1 Biológicos triacyl lipopeptides (bactéria)
Sintéticos Pam3Cys-Ser-(Lys)4 (lipoproteína)
TLR2 Biológicos GPI anchors,
glicolipídeos
proteínas do envelope de vírus
peptidoglicanos,
lipoproteínas,
lipopeptídeos,
lipoteichoic acid,
phenol-soluble modulin (bacteria)
zymozan,
lipoarabinomannan (fungi)
Sintéticos Pam3Cys-Ser-(Lys)4 (lipoproteínas)
TLR3 Biológicos dsRNA (virus) Hairpin loops within the
mRNA,
siRNA (hospedeiro)
Sintéticos poly (I : C) (dsRNA sintético)
TLR4 Biológicos lipopolissacarídeos (bactéria)
glycoinositolphospholipids (protozoa)
proteínas do envelope dos vírus
HSP 60,
HSP 70,
sulfato de heparan,
ácido hialurónico,
fibrinogénio (hospedeiro)
TLR5 Biológicos flagelina (bactéria)
TLR6 Biológicos diacyl and triacyl lipopeptide
(bactéria)
TLR7 Biológicos ssRNA (vírus) some ssRNA (hospedeiro)
Sintéticos imidazoquinoline
TLR8 Biológicos ssRNA (vírus) some ssRNA (hospedeiro)
TLR9 Biológicos ilhas CpG de DNA não metiladas
(bacteria, protozoa, virus)
Chromatin-IgG complex
(hospedeiro)
Sintéticos CpG (Oligonucleotidos sintéticos)
TLR10 Não conhecido
* heat-shock protein (HSP)
glycosylphosphatidylinositol (GPI)

1. Introdução
33
Os TLRs envolvidos no reconhecimento deste tipo de PAMPs são os TLR1, TLR2,
TLR4, TLR5 e TLR6 e encontram-se expressos à superfície da membrana celular 57. Os
TLRs também detetam sinais de “perigo” os padrões moleculares associados à lesão
(Damage-associated molecular pattern: DAMPs), ou seja, moléculas endógenas
libertadas por células em necrose ou por tecidos lesados na presença de estímulos
agressivos 58. Os TLRs também detetam ácidos nucleicos e desempenham um papel
fundamental no reconhecimento de vírus: o TLR3 reconhece moléculas de ácido
ribonucleico (Ribonucleic Acid: RNA) de cadeia dupla (doube-stranded RNA: dsRNA),
presentes na maioria dos vírus de RNA e de DNA; o TLR7 e o TLR8 reconhecem
moléculas de RNA de cadeia simples ssRNA (single-stranded RNA: ssRNA); e o TLR9
reconhece no DNA sequências CpG não metiladas 56.
Os TLRs são recetores transmembranares constituídos por um ectodominio rico em
múltiplos resíduos de leucina, um domínio transmembranar e um domínio citoplasmático
homólogo ao recetor da interleucina 1 (IL-1), o domínio TIR. Apesar da homologia
estrutural entre os TLRs, estes são capazes de distinguir diferentes PAMPs e DAMPs
(tabela 1) 59,60. Todos os TLRs reconhecem com especificidade os seus ligandos devido a
variações no número de resíduos de leucina presentes no ectodomínio de cada recetor 61.
Os TLRs podem ser expressos em muitos tipos de células como por exemplo as
células do sistema imunitário provenientes da linhagem mielóide e células provenientes
da linhagem linfóide (células T e células B) e ainda células epiteliais e fibroblastos, tendo
em consideração que a expressão destes recetores varia conforme o tipo de célula 62.
O reconhecimento dos agentes microbianos pelos TLRs promove a sua dimerização,
originando a formação de heterodímeros nos TLR1, TLR2 e TLR6. Por outro lado os
restantes TLRs depois de detetarem os respetivos ligandos formam homodímeros 63.
Deste modo, a interação do ligando-recetor conduz a uma alteração conformacional,
induzindo o domínio TIR e consequente ativação de uma cascata de sinalização
intracelular 64. O domínio citoplasmático TIR pode recrutar duas moléculas adaptadoras
independentes: a molécula adaptadora citoplasmática factor de diferenciação mielóide 88
(myeloid differentiation factor 88: MyD88) ou a molécula adaptadora citoplasmática
indutora de interferão β (IFN-β) (Domain-containing adaptor inducing IFN-β: TRIF), e
assim iniciam diferentes vias de sinalização, que vão levar à transcrição de genes de
citocinas pró-inflamatórias e de genes de interferões (IFNs), respectivamente 65.
Este passo é crucial para a maturação de células do sistema imunitário inato,
nomeadamente células apresentadoras de antigénios responsáveis por estabelecer uma
ligação com a imunidade adquirida 66.

1. Introdução
34
Apesar do papel crucial dos TLRs, na defesa do hospedeiro, a indução inadequada
destes recetores pode conduzir a um estado de inflamação crónica, que proporciona o
desenvolvimento de cancro 67.
1.3.1 TLRs Endossomais: TLR3 e TLR7
Os TLRs endossomais têm um papel de sentinela na entrada de microrganismos nas
células, particularmente vírus. A endocitose de agentes patogénicos ou de material
proveniente de células mortas permite que ácidos nucleicos de diferente origem (PAMPs
e DAMPs) sejam internalizados nos endossomas, o mesmo acontece durante a autofagia
de células infetadas 68,69.
Os TLRs intracelulares localizam-se no reticulo endoplasmático (RE) e sob
estimulação prévia deslocam-se para os endossomas. Estes recetores seguem o tráfego
intracelular comum à via de secreção, em que a partir do RE atravessam o aparelho de
Golgi e deste modo são integrados nos endossomas, onde sofrem processamento e
adquirem funcionalidade 68. Por exemplo, o TLR7 é processado por protéases da família
das furin-like propotein convertases e a expressão destas protéases aumenta perante
estímulos inflamatórios 70. A migração dos TLRs é garantida pela associação destes
recetores com muitas chaperones no RE. A proteína transmembranar UNC93B1
(Uncoordinated 93B1: UNC93B1) é essencial na migração do TLR3 e do TLR7, assim
como outras moléculas adaptadoras que são específicas no transporte de cada recetor 69.
Geralmente, o TLR3 é expresso no interior de muitos tipos de células, incluindo
células da microglia, mastócitos, eosinófilos, macrófagos, células NK, e células
dendríticas, contudo apresenta menor expressão em linfócitos B e T 71.
O gene TLR3 localiza-se no cromossoma 4 e codifica o recetor TLR3, apresentando
capacidade de se ligar a dsRNA e ao análogo sintético polyinosinic-polycytidylic acid
(Poly (I:C)) 55,62.
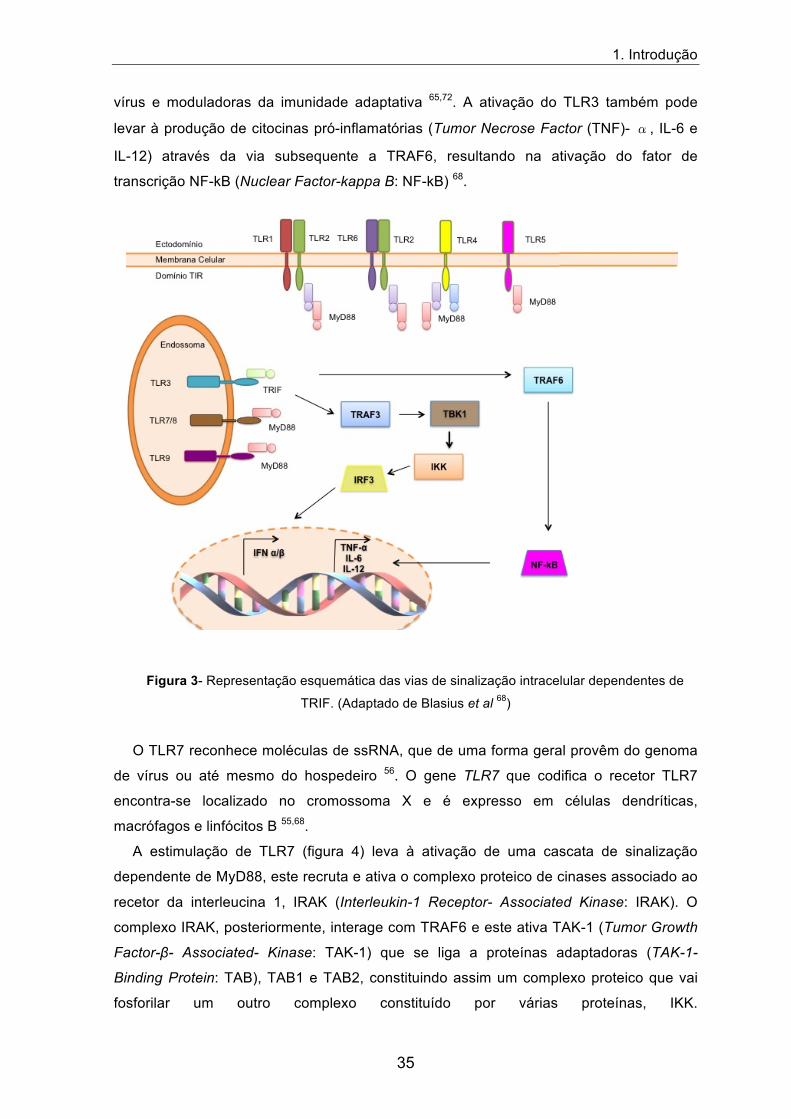
A estimulação de TLR3 inicia uma via de sinalização intracelular dependente da
molécula adaptadora TRIF. A molécula TRIF, quando ativada, pode recrutar TRAF3
(Tumor Necrose Factor Receptor-Associated Factor 3: TRAF3) ou TRAF6 (Tumor
Necrose Factor Receptor-Associated Factor 6: TRAF6). Ao recrutar TRAF3,
posteriormente este liga-se e ativa TBK1 (TANK-Binding Kinase 1: TBK1) e em seguida
ativa IKK (I kappa B Kinase: IKK), estas são proteínas cínases que em conjunto vão
fosforilar o fator regulador de interferão IRF3 (Interferon Regulatory Factor 3: IRF3), que é
translocado para o núcleo e induz a expressão de genes codificantes de IFNs do tipo I
(INF-�/�) (figura 3) 64. Os interferões tipo I são citocinas inibidoras da replicação dos
1. Introdução
35
vírus e moduladoras da imunidade adaptativa 65,72. A ativação do TLR3 também pode
levar à produção de citocinas pró-inflamatórias (Tumor Necrose Factor (TNF)- �, IL-6 e
IL-12) através da via subsequente a TRAF6, resultando na ativação do fator de
transcrição NF-kB (Nuclear Factor-kappa B: NF-kB) 68.
Figura 3- Representação esquemática das vias de sinalização intracelular dependentes de
TRIF. (Adaptado de Blasius et al 68)
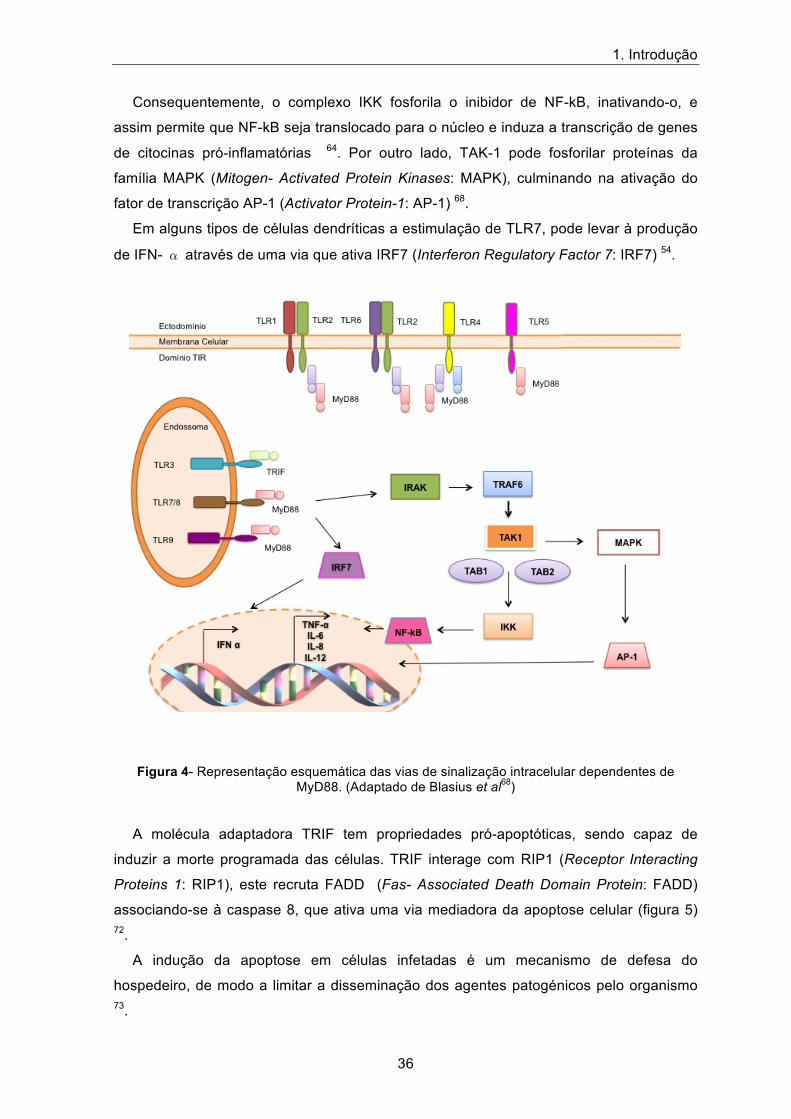
O TLR7 reconhece moléculas de ssRNA, que de uma forma geral provêm do genoma
de vírus ou até mesmo do hospedeiro 56. O gene TLR7 que codifica o recetor TLR7
encontra-se localizado no cromossoma X e é expresso em células dendríticas,
macrófagos e linfócitos B 55,68.
A estimulação de TLR7 (figura 4) leva à ativação de uma cascata de sinalização
dependente de MyD88, este recruta e ativa o complexo proteico de cinases associado ao
recetor da interleucina 1, IRAK (Interleukin-1 Receptor- Associated Kinase: IRAK). O
complexo IRAK, posteriormente, interage com TRAF6 e este ativa TAK-1 (Tumor Growth
Factor-β- Associated- Kinase: TAK-1) que se liga a proteínas adaptadoras (TAK-1-
Binding Protein: TAB), TAB1 e TAB2, constituindo assim um complexo proteico que vai
fosforilar um outro complexo constituído por várias proteínas, IKK.
1. Introdução
36
Consequentemente, o complexo IKK fosforila o inibidor de NF-kB, inativando-o, e
assim permite que NF-kB seja translocado para o núcleo e induza a transcrição de genes
de citocinas pró-inflamatórias 64. Por outro lado, TAK-1 pode fosforilar proteínas da
família MAPK (Mitogen- Activated Protein Kinases: MAPK), culminando na ativação do
fator de transcrição AP-1 (Activator Protein-1: AP-1) 68.
Em alguns tipos de células dendríticas a estimulação de TLR7, pode levar à produção
de IFN- � através de uma via que ativa IRF7 (Interferon Regulatory Factor 7: IRF7) 54.
Figura 4- Representação esquemática das vias de sinalização intracelular dependentes de
MyD88. (Adaptado de Blasius et al68)
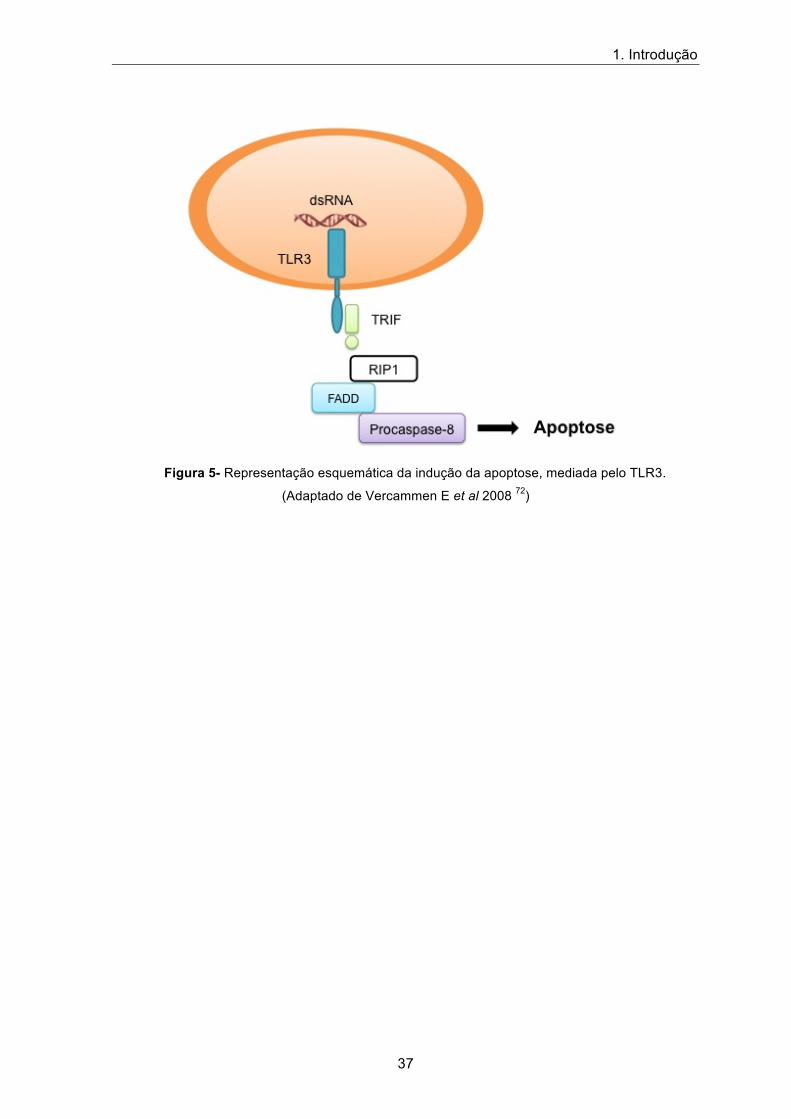
A molécula adaptadora TRIF tem propriedades pró-apoptóticas, sendo capaz de
induzir a morte programada das células. TRIF interage com RIP1 (Receptor Interacting
Proteins 1: RIP1), este recruta FADD (Fas- Associated Death Domain Protein: FADD)
associando-se à caspase 8, que ativa uma via mediadora da apoptose celular (figura 5) 72.
A indução da apoptose em células infetadas é um mecanismo de defesa do
hospedeiro, de modo a limitar a disseminação dos agentes patogénicos pelo organismo 73.
1. Introdução
37
Figura 5- Representação esquemática da indução da apoptose, mediada pelo TLR3.
(Adaptado de Vercammen E et al 2008 72)

1. Introdução
38
1.4 Polimorfismos Genéticos nos genes TLR3 e TLR7
Variações nos genes que codificam os Toll-like receptors podem conferir diferente
suscetibilidade para determinadas infeções, contribuindo assim para um risco aumentado
de desenvolvimento de linfomas 74. Assim ao modular as vias de sinalização ativadas por
estes retores promove-se um microambiente oncogénico indutor da progressão tumoral 75. Estas variações genéticas são consequência de polimorfismos genéticos que ocorrem
no genoma do individuo. Os polimorfismos podem ser definidos como variações no DNA
existentes nos indivíduos de uma população cuja variante menos frequente está presente
em pelo menos 1% dessa população 76. A maioria dos polimorfismos no genoma humano
são caracterizados pela alteração de apenas um nucleótido na sequência de DNA e são
denominados de SNPs (Single Nucleotide Polymorphism: SNPs). Os SNPs dependendo
da sua localização podem influenciar a função dos TLRs. Se a variação se encontrar na
região que codifica do ectodominio do recetor pode afetar a ligação deste com o ligando,
enquanto que a presença do SNP na região que codifica o domínio transmembranar ou
no domínio citoplasmático pode alterar a capacidade de transmitir o sinal para o interior
da célula e a sua interação com as moléculas adaptadoras intracelulares, respetivamente 77.
Estudos prévios demonstraram que a presença do polimorfismo genético TLR3
rs3775291 no gene TLR3 consiste na substituição de um C> T na posição 1234 na
sequência de cDNA. A ocorrência deste é responsável pela substituição aminoacídica
leucina à fenilanina na posição 412 na sequencia de aminoácidos 78. De acordo com
Ranjith-Kumar e colaboradores, a localização do polimorfismo TLR3 rs3775291 afeta o
ectodominio do recetor, reduzindo a afinidade da ligação com o agonista, e por sua vez
diminui a atividade da via de sinalização do TLR3 79.
O TLR3 encontra-se sobrexpresso em muitos tipos de cancro influenciando a atividade
das células neoplásicas 80. Um estudo realizado por Castro e os colaboradores
demonstrou que presença do polimorfismo TLR3 rs3775291 está associada a um pior
prognóstico em doentes com cancro coloretal. Os doentes portadores do genótipo TT têm
uma sobrevivência significativamente menor quando comparados com indivíduos
portadores dos genótipos CC e CT 81. A alteração da função mediada pelo recetor na
resposta imunitária e no processo de indução da morte celular podem explicar os
resultados obtidos por este autor e os colaboradores 73,81.
Estudos que caracterizaram o gene TLR7, identificaram o polimorfismo TLR7 rs179008
consiste numa substituição de A>T, na posição 32 de cDNA, sendo responsável por
glutamina à leucina na posição 11 na sequencia de aminoácidos 82. Esta substituição
provoca alterações na sequência de sinal da proteína, prejudicando as modificações pós-

1. Introdução
39
traducionais no recetor, que conferem menor capacidade de ativação ao TLR7 55,82. A
presença deste polimorfismo genético já foi associado à suscetibilidade para algumas
infeções, uma vez que a atividade do recetor encontra-se diminuída, levando à produção
de baixos níveis de INF-� 83. No estudo de Bordignon e colaboradores em que avaliaram
de que modo a presença do polimorfismo rs179008 no gene TLR7 pode influenciar a
suscetibilidade para o desenvolvimento de sarcoidose, uma doença caraterizada pelo
aumento de inflamação. Neste estudo, as mulheres portadoras do alelo T tinham um
risco significativamente menor para o desenvolvimento de sarcoidose, comparativamente
com mulheres portadoras do genótipo AA. Uma vez que o polimorfismo TLR7 rs179008
afeta a função do recetor levando a uma menor produção INF-� e de citocinas pró-
inflamatórias, as mulheres portadoras dos genótipo AT/TT apresentam menor
suscetibilidade relativamente ao da desenvolvimento da doença 84.
Dada a importância destes recetores na vigilância imunológica e na resposta
antitumoral, torna-se relevante analisar a influencia dos polimorfismos genéticos TLR3
rs3775291 e TLR7 rs179008 na sobrevivência de indivíduos com NHL.


2. Objetivos


2. Objetivos
43
2.1 Objetivo Geral:
Compreender a influência dos polimorfismos genéticos TLR3 rs3775291 e TLR7
rs179008 no desenvolvimento de NHL e a sua repercussão na sobrevivência de doentes
com NHL.
2.2 Objetivos Específicos:
1. Frequência dos polimorfismos genéticos TLR3 rs3775291 e TLR7 rs179008 em
indivíduos com NHL;
2. Analisar a influência do polimorfismo genético TLR3 rs3775291 na sobrevivência a
um ano e de longo termo de doentes com NHL;
3. Analisar a influência do polimorfismos genético TLR7 rs179008 na sobrevivência a
um ano e de longo termo de doentes com NHL;


3. Materiais e Métodos


3. Materiais e Métodos
47
3.1 População em Estudo
O estudo dos polimorfismos genéticos TLR3 rs3775291 e TLR7 rs179008 foi realizado
com base num estudo do tipo coorte retrospetivo, e a população em estudo foram
doentes com diagnóstico de NHL de células B, seguidos no Instituto Português de
Oncologia do Porto, Francisco Gentil. Todas as amostras biológicas são provenientes de
indivíduos caucasianos, com idade superior a 18 anos e foram utilizadas com o seu
conhecimento e consentimento prévio, de acordo com a declaração de Helsínquia.
Os dados demográficos (idade e género), clínicos e patológicos estão indicados na
tabela 2. Todas as informações foram recolhidas através da consulta dos processos
clínicos dos doentes.
Neste estudo participaram cento e vinte e nove (129) doentes com diagnóstico de NHL
de células B. Dos 129 indivíduos 48,1% são do género masculino e 51,9% são do género
feminino com uma média de idades de 58,3± 15,4.
Tabela 2- Características clínicas e patológicas gerais da população em estudo
Dados clínicos e patológicos Total %
Idade Média ± σ 58,3 ± 15,4
Género Masculino 48,1%
Feminino 51,9%
Estadio* Avançado 63,9%
Não avançado 36,1%
Alto grau Sim 47,2%
Não 52,8%
*Ann Arbor ≥ III
σ- Desvio Padrão
3.2 Procedimento laboratorial
A cada doente do presente estudo foi colhido uma amostra de sangue periférico de
aproximadamente 8 mL, através de uma técnica padronizada de colheita intravenosa,
para tubos contendo uma solução de EDTA.

3. Materiais e Métodos
48
3.2.1 Extração de DNA genómico
Em seguida, procedeu-se ao isolamento de DNA genómico a partir de células
nucleadas do sangue periférico, através do kit de extração GRS Genomic DNA Kit-
BroadRange (GRIPS®). A extração do DNA foi realizada mediante um sistema de colunas
de centrifugação, de acordo com as especificações do fornecedor.
3.2.2 Genotipagem dos polimorfismos TLR3 rs3775291 e TLR7 rs179008
Os polimorfismos genéticos TLR3 rs3775291 e TLR7 rs179008 foram analisados por
discriminação alélica através da técnica Real Time PCR (Polymerase Chain Reaction:
PCR). Os polimorfismos TLR3 rs3775291 e TLR7 rs179008 foram analisados recorrendo
à tecnologia TaqMan® (Applied Biosystems®) utilizando-se os seguintes assays:
C___1731425_10 e C___2259574_10 em que as sondas marcadas com fluorocromos
eram específicas para cada alelo: TLR3 rs3775291 VIC – alelo T
(ACTTGCTCATTCTCCCTTACACATATTCAACCTAACCAAGAATAAAATCTC), FAM –
alelo C (ACTTGCTCATTCTCCCTTACACATACTCAACCTAACCAAGAATAAAATCTC); e
TLR7 rs179008 VIC – alelo A
(TTTCCAATGTGGACACTGAAGAGACAAATTCTTATCCTTTTTAACATAATC),
FAM – alelo T
(TTTCCAATGTGGACACTGAAGAGACTAATTCTTATCCTTTTTAACATAATC).
A reação de amplificação efetuada para cada amostra, integrou um volume final de 6
µL por caso contendo 2.5 µL de 2x Taqman® Genotyping Master Mix, 0.125 µL de 40x
TaqMan® SNP Genotyping Assays e 2.375 µL de água à qual se adicionou 1 µL de DNA.
Os produtos amplificados foram detetados e analisados recorrendo ao aparelho Real-
Time 7300 ABI e através do software 7300 System Sequence Detection (versão 1.2.3
Applied Biosystems) (figura 6).
A reação de amplificação perfez as seguintes condições: ativação da Taq DNA
Polimerase a 95ºC durante 10 minutos, seguindo-se 45 ciclos de 92ºC por 15 segundos
para desnaturação e de 60ºC durante 1 minuto para emparelhamento dos primers e
extensão. A determinação do genótipo foi efectuada através do respetivo software, tendo-
se obtido uma razão entre a fluorescência dos fluorocromos VIC e FAM, sendo que cada
um deles corresponde a um único alelo.
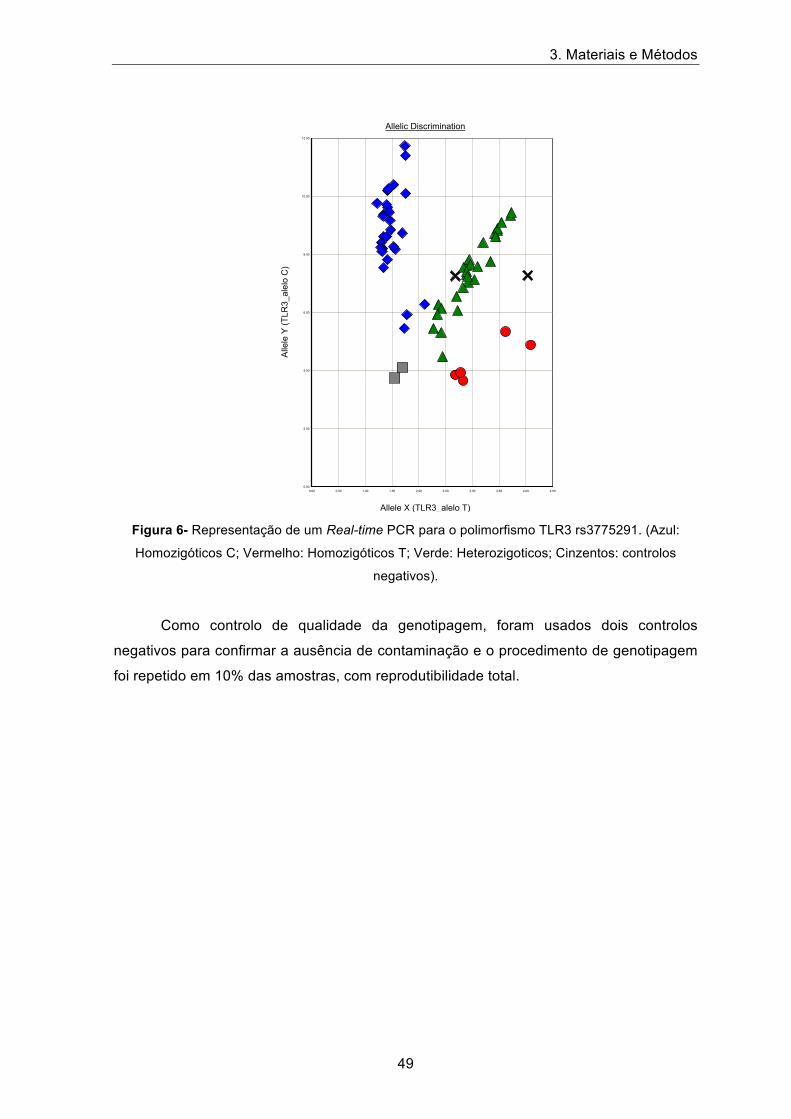
3. Materiais e Métodos
49
Figura 6- Representação de um Real-time PCR para o polimorfismo TLR3 rs3775291. (Azul:
Homozigóticos C; Vermelho: Homozigóticos T; Verde: Heterozigoticos; Cinzentos: controlos
negativos).
Como controlo de qualidade da genotipagem, foram usados dois controlos
negativos para confirmar a ausência de contaminação e o procedimento de genotipagem
foi repetido em 10% das amostras, com reprodutibilidade total.
Alle
le Y
(TLR
3_al
elo
C)
Allelic Discrimination
Allele X (TLR3_alelo T)
0.00
2.00
4.00
6.00
8.00
10.00
12.00
0.00 0.50 1.00 1.50 2.00 2.50 3.00 3.50 4.00 4.50
Allele X Allele Y Both NTC UndeterminedMarker: TLR3_rs3775291 Well(s): A1-H12
Page 5 of 5 (05/27/15 15:20:55) TLR3_15.04.2015_AD_2

3. Materiais e Métodos
50
3.3 Análise Estatística
A análise estatística dos resultados foi efetuada com recurso ao software estatístico
IBM® SPSS® Statistics para Windows (version 20.0).
O teste T-student foi usado para comparar as diferenças entre as médias de idade dos
doentes. O teste do χ2 foi aplicado para comparar as diferenças entre as variáveis
categóricas (dados clínicos e patológicos). Para estes dois testes estatísticos, o valor de
P obtido foi considerado estatisticamente significativo quando inferior a 0,05.
No presente estudo foram analisadas a sobrevivência a um ano, definida como o
tempo entre o diagnóstico e um ano após o mesmo, e a sobrevivência a longo termo
definida como o tempo após treze (13) meses até à ultima observação ou morte do
doente. As curvas de probabilidade de sobrevivência foram calculas pelo método
estatístico de Kaplan-Meier. A comparação entre as curvas referentes aos genótipos dos
polimorfismos estudados foram examinados pelo teste de Log-Rank, com um nível de
significância de 5%.

4. Resultados

4. Resultados
53
4.1 Frequência dos polimorfismos genéticos TLR3 rs3775291 e TLR7 rs179008 em
doentes com NHL
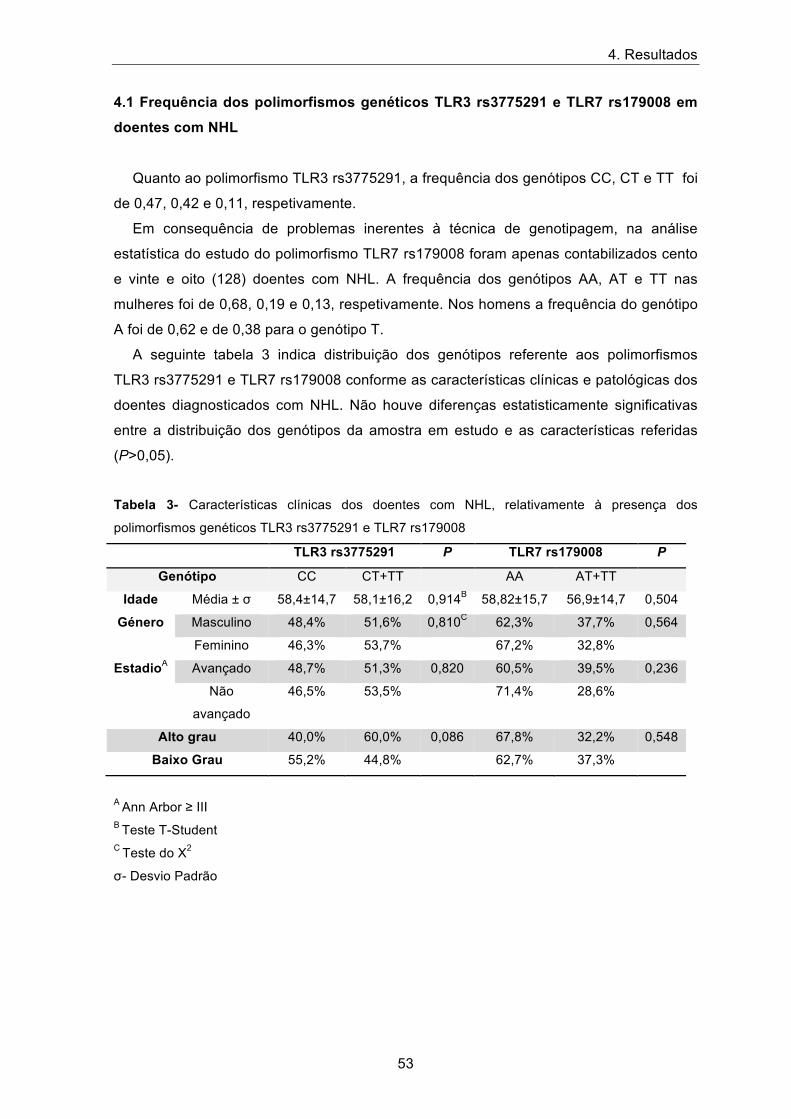
Quanto ao polimorfismo TLR3 rs3775291, a frequência dos genótipos CC, CT e TT foi
de 0,47, 0,42 e 0,11, respetivamente.
Em consequência de problemas inerentes à técnica de genotipagem, na análise
estatística do estudo do polimorfismo TLR7 rs179008 foram apenas contabilizados cento
e vinte e oito (128) doentes com NHL. A frequência dos genótipos AA, AT e TT nas
mulheres foi de 0,68, 0,19 e 0,13, respetivamente. Nos homens a frequência do genótipo
A foi de 0,62 e de 0,38 para o genótipo T.
A seguinte tabela 3 indica distribuição dos genótipos referente aos polimorfismos
TLR3 rs3775291 e TLR7 rs179008 conforme as características clínicas e patológicas dos
doentes diagnosticados com NHL. Não houve diferenças estatisticamente significativas
entre a distribuição dos genótipos da amostra em estudo e as características referidas
(P>0,05).
Tabela 3- Características clínicas dos doentes com NHL, relativamente à presença dos
polimorfismos genéticos TLR3 rs3775291 e TLR7 rs179008
TLR3 rs3775291 P TLR7 rs179008 P
Genótipo CC CT+TT AA AT+TT
Idade Média ± σ 58,4±14,7 58,1±16,2 0,914B 58,82±15,7 56,9±14,7 0,504
Género Masculino 48,4% 51,6% 0,810C 62,3% 37,7% 0,564
Feminino 46,3% 53,7% 67,2% 32,8%
EstadioA Avançado 48,7% 51,3% 0,820 60,5% 39,5% 0,236
Não
avançado
46,5% 53,5% 71,4% 28,6%
Alto grau 40,0% 60,0% 0,086 67,8% 32,2% 0,548
Baixo Grau 55,2% 44,8% 62,7% 37,3%
A Ann Arbor ≥ III B Teste T-Student C Teste do Χ2
σ- Desvio Padrão
4. Resultados
54
4.2 Influência do polimorfismo TLR3 rs3775291 na sobrevivência de doentes com
NHL
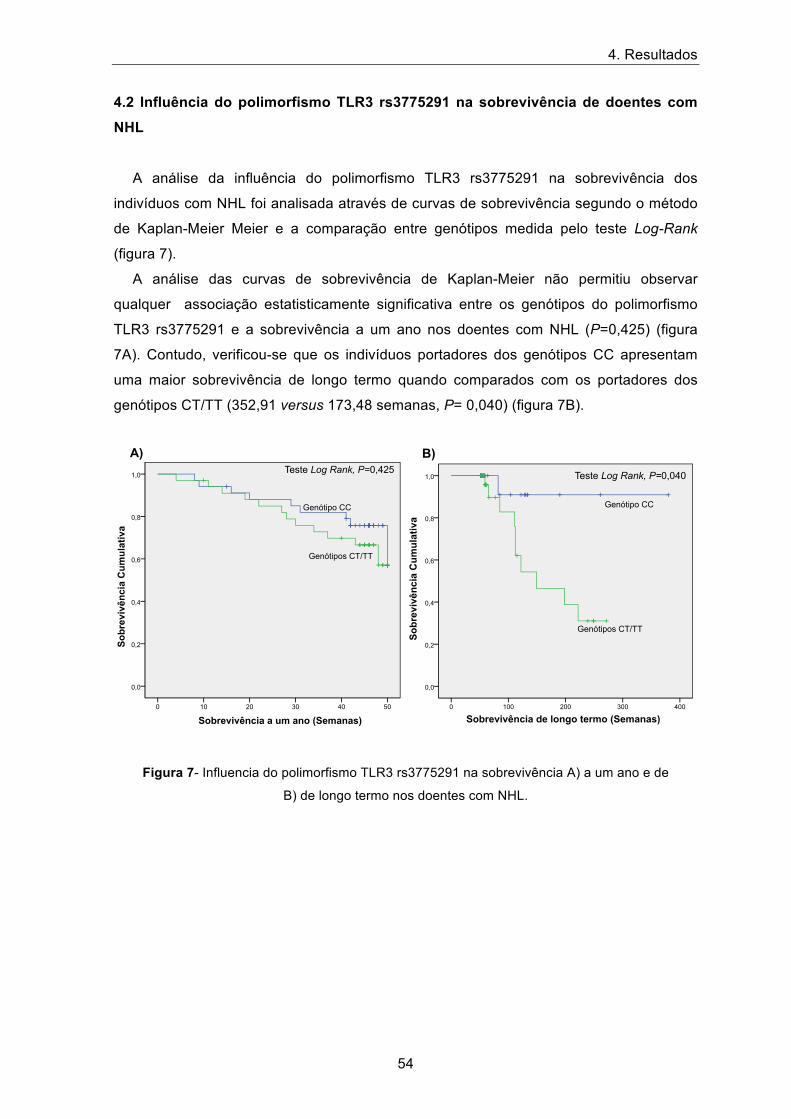
A análise da influência do polimorfismo TLR3 rs3775291 na sobrevivência dos
indivíduos com NHL foi analisada através de curvas de sobrevivência segundo o método
de Kaplan-Meier Meier e a comparação entre genótipos medida pelo teste Log-Rank
(figura 7).
A análise das curvas de sobrevivência de Kaplan-Meier não permitiu observar
qualquer associação estatisticamente significativa entre os genótipos do polimorfismo
TLR3 rs3775291 e a sobrevivência a um ano nos doentes com NHL (P=0,425) (figura
7A). Contudo, verificou-se que os indivíduos portadores dos genótipos CC apresentam
uma maior sobrevivência de longo termo quando comparados com os portadores dos
genótipos CT/TT (352,91 versus 173,48 semanas, P= 0,040) (figura 7B).
Figura 7- Influencia do polimorfismo TLR3 rs3775291 na sobrevivência A) a um ano e de
B) de longo termo nos doentes com NHL.
OS4003002001000
Cum
Sur
viva
l
1,0
0,8
0,6
0,4
0,2
0,0
Survival Functions
Followupminimun50weeks = 1
alelo T-censoredCC-censoredalelo TCC
TLR3_aleloT
KM OS BY TLR7_Tallele /STATUS=Obito(1) /PRINT NONE /PLOT SURVIVAL /TEST LOGRANK BRESLOW /TREND /COMPARE OVERALL STRATA.
Kaplan-Meier
[DataSet1] C:\Users\R\Desktop\InesVAz\TESTE_LNH_baseC_TLRmir101.sav
Page 6
Sobrevivência de longo termo (Semanas)
Sob
revi
vênc
ia C
umul
ativ
a
Teste Log Rank, P=0,040
Genótipo CC
Genótipos CT/TT
B)
OS50403020100
Cum
Sur
viva
l
1,0
0,8
0,6
0,4
0,2
0,0
Survival Functions
Followupminimun50weeks = 0
alelo T-censoredCC-censoredalelo TCC
TLR3_aleloT
Stratum: Followupminimun50weeks = 1
Page 5
Sobrevivência a um ano (Semanas)
Sob
revi
vênc
ia C
umul
ativ
a
Teste Log Rank, P=0,425
Genótipo CC
Genótipos CT/TT
A)
4. Resultados
55
4.3 Influência do polimorfismo TLR7 rs179008 na sobrevivência de doentes com
NHL
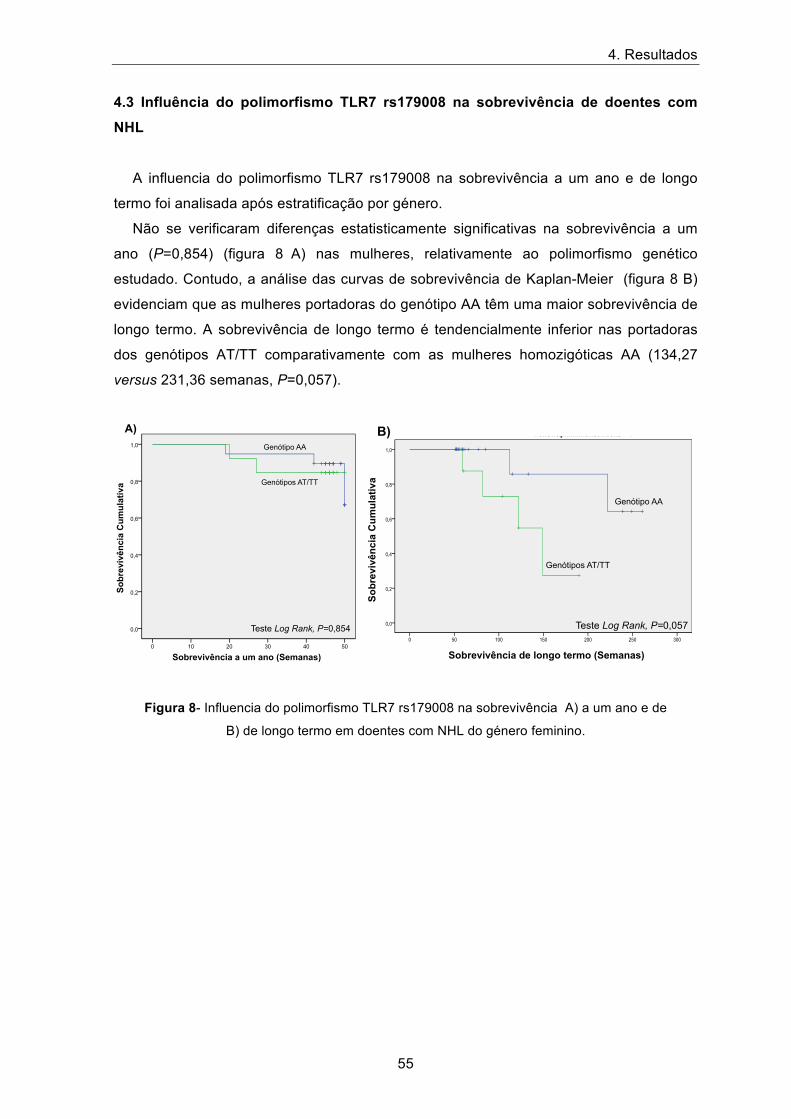
A influencia do polimorfismo TLR7 rs179008 na sobrevivência a um ano e de longo
termo foi analisada após estratificação por género.
Não se verificaram diferenças estatisticamente significativas na sobrevivência a um
ano (P=0,854) (figura 8 A) nas mulheres, relativamente ao polimorfismo genético
estudado. Contudo, a análise das curvas de sobrevivência de Kaplan-Meier (figura 8 B)
evidenciam que as mulheres portadoras do genótipo AA têm uma maior sobrevivência de
longo termo. A sobrevivência de longo termo é tendencialmente inferior nas portadoras
dos genótipos AT/TT comparativamente com as mulheres homozigóticas AA (134,27
versus 231,36 semanas, P=0,057).
Figura 8- Influencia do polimorfismo TLR7 rs179008 na sobrevivência A) a um ano e de
B) de longo termo em doentes com NHL do género feminino.
OS50403020100
Cu
m S
urv
ival
1,0
0,8
0,6
0,4
0,2
0,0
Survival Functions
Followupminimun50weeks = 0
TLR7_Tallele_lesssignallingtoIFNproduction
other-censored
TLR7_Tallele_lesssignallingtoIFNproduction
other
TLR7_Tallele
Stratum: Followupminimun50weeks = 1
OS300250200150100500
Cu
m S
urv
ival
1,0
0,8
0,6
0,4
0,2
0,0
Survival Functions
Followupminimun50weeks = 1
TLR7_Tallele_lesssignallingtoIFNproduction-censored
other-censoredTLR7_Tallele_lesssignallingtoIFNproductionother
TLR7_Tallele
FILTER OFF.USE ALL.EXECUTE.
Page 12
Teste Log Rank, P=0,057
Genótipo AA
Genótipos AT/TT
Sobrevivência de longo termo (Semanas)
Sob
revi
vênc
ia C
umul
ativ
a
B)
OS50403020100
Cum
Sur
viva
l
1,0
0,8
0,6
0,4
0,2
0,0
Survival Functions
Followupminimun50weeks = 0
TLR7_Tallele_lesssignallingtoIFNproduction
other-censored
TLR7_Tallele_lesssignallingtoIFNproduction
other
TLR7_Tallele
Stratum: Followupminimun50weeks = 1
OS300250200150100500
Cum
Sur
viva
l
1,0
0,8
0,6
0,4
0,2
0,0
Survival Functions
Followupminimun50weeks = 1
TLR7_Tallele_lesssignallingtoIFNproduction-censored
other-censoredTLR7_Tallele_lesssignallingtoIFNproductionother
TLR7_Tallele
FILTER OFF.USE ALL.EXECUTE.
Page 12
Teste Log Rank, P=0,854
Genótipo AA
Genótipos AT/TT
Sobrevivência a um ano (Semanas)
Sob
revi
vênc
ia C
umul
ativ
a
A)
4. Resultados
56
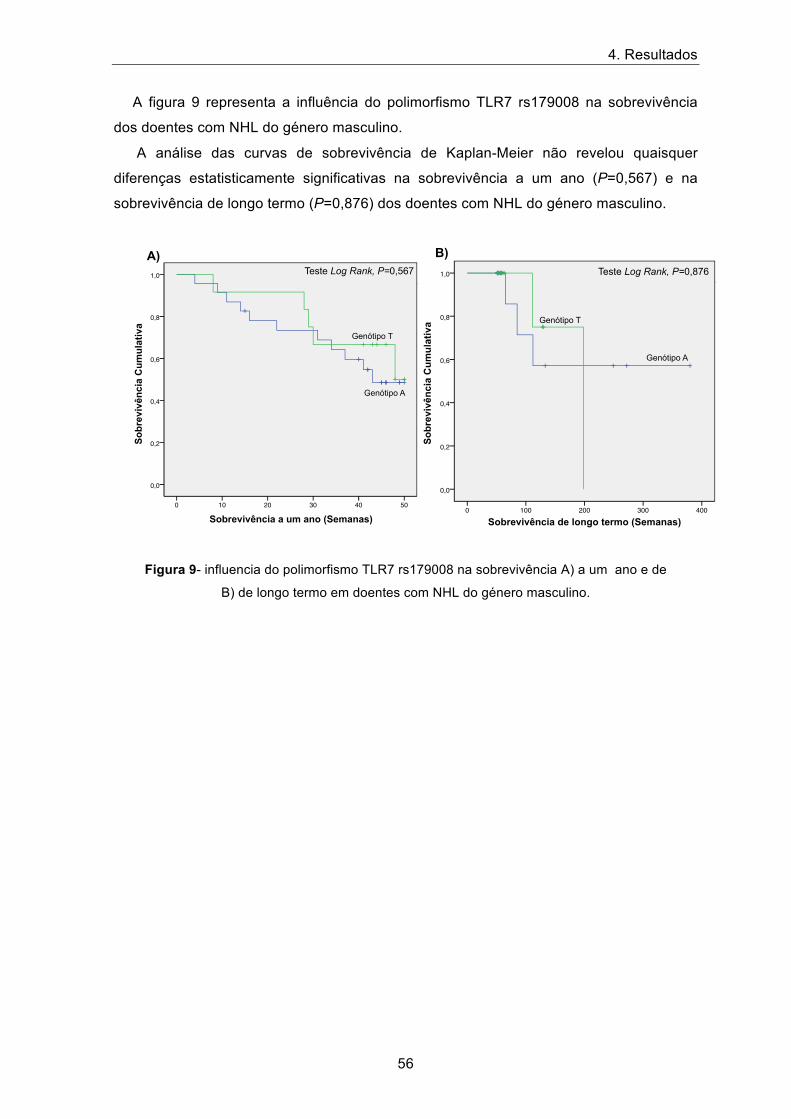
A figura 9 representa a influência do polimorfismo TLR7 rs179008 na sobrevivência
dos doentes com NHL do género masculino.
A análise das curvas de sobrevivência de Kaplan-Meier não revelou quaisquer
diferenças estatisticamente significativas na sobrevivência a um ano (P=0,567) e na
sobrevivência de longo termo (P=0,876) dos doentes com NHL do género masculino.
Figura 9- influencia do polimorfismo TLR7 rs179008 na sobrevivência A) a um ano e de
B) de longo termo em doentes com NHL do género masculino.
OS50403020100
Cum
Sur
viva
l
1,0
0,8
0,6
0,4
0,2
0,0
Survival Functions
Sexo = masculino
TLR7_Tallele_lesssignallingtoIFNproduction-
other-censored
TLR7_Tallele_lesssignallingtoIFNproduction
other
TLR7_Tallele
KM OSlong_after50weeks BY TLR3_aleloT /STRATA=Sexo /STATUS=Obito_longfollowafter50weeks(1) /PRINT MEAN /PLOT SURVIVAL /TEST LOGRANK BRESLOW /COMPARE OVERALL STRATA.
Kaplan-Meier[DataSet1] C:\Users\R\Desktop\R_2015jan-Jun\EmAnalise2015\InesVAz\TESTE_LNH_baseC_TLRmir101.sav
Page 12
Teste Log Rank, P=0,567
Genótipo A
Genótipo T
Sobrevivência a um ano (Semanas)
Sob
revi
vênc
ia C
umul
ativ
a
A)
OS4003002001000
Cum
Sur
viva
l
1,0
0,8
0,6
0,4
0,2
0,0
Survival Functions
Sexo = masculino
TLR7_Tallele_lesssignallingtoIFNproduction-
other-censored
TLR7_Tallele_lesssignallingtoIFNproduction
other
TLR7_Tallele
USE ALL.COMPUTE filter_$=(Followupminimun50weeks = 0).VARIABLE LABEL filter_$ 'Followupminimun50weeks = 0 (FILTER)'.VALUE LABELS filter_$ 0 'Not Selected' 1 'Selected'.FORMAT filter_$ (f1.0).FILTER BY filter_$.EXECUTE.KM OSlong_after50weeks BY TLR7_Tallele /STRATA=Sexo /STATUS=Obito_longfollowafter50weeks(1) /PRINT MEAN /PLOT SURVIVAL /TEST LOGRANK BRESLOW /COMPARE OVERALL STRATA.
Kaplan-Meier[DataSet1] C:\Users\R\Desktop\R_2015jan-Jun\EmAnalise2015\InesVAz\TESTE_LNH_baseC_TLRmir101.sav
Page 9
Sobrevivência de longo termo (Semanas)
Sob
revi
vênc
ia C
umul
ativ
a
Teste Log Rank, P=0,876
Genótipo A
Genótipo T
B)

5. Discussão


5. Discussão
59
Nos últimos anos observou-se um investimento significativo na área da epidemiologia
molecular com foco na definição de novos biomarcadores moleculares de risco de
desenvolvimento de cancro e preditivos do comportamento tumoral 85. A integração de
técnicas da biologia molecular em estudos epidemiológicos oferece a oportunidade de
compreender melhor a etiologia do cancro, identificar populações em risco de
desenvolver a doença, estabelecer medidas de quimioprevenção, e compreender os
mecanismo moleculares da doença 86. De facto, a identificação de polimorfismos
genéticos funcionais que afetam a dinâmica da célula, podem permitir um melhor
conhecimento dos processos moleculares envolvidos no desenvolvimento e progressão
tumoral, e no futuro poderão ser uma ferramenta útil no desenho de novas terapias
dirigidas 85.
O conhecimento sobre mecanismos moleculares envolvidos na carcinogénese e
progressão tumoral têm aumentado continuamente, tendo-se apostado na identificação
de genes responsáveis por manter a integridade celular e dos tecidos. O sistema
imunitário tem um papel central não só na resposta a agressões externas como também
na manutenção da homeostasia do organismo, tendo um papel fundamental na
prevenção do cancro. Desta forma, o sistema imunitário reconhece agentes infeciosos,
sendo responsável por eliminá-los ao mesmo tempo que pode prevenir condições de
inflamação crónica associadas ao desenvolvimento de cancro. Cumulativamente, este
sistema é capaz de detetar e eliminar células com fenótipo neoplásico 87. Os TLRs
constituem parte do sistema imunitário inato, constituindo não só a primeira linha de
defesa do hospedeiro contra agentes patogénicos como também têm a capacidade de
serem ativados por DAMPs libertados por células tumorais em necrose. Os TLRs
apresentam a capacidade de promover a carcinogénese e a progressão tumoral
condicionando a secreção de citocinas pro-inflamatórias, anti-apoptóticas, proliferativas,
podendo este efeito ser exercido por via autócrina ou parácrina 88. O TLR3 e o TLR7 são
recetores localizados no interior das células, ancorados à membrana dos endossomas. A
utilização de diferentes moléculas adaptadoras permite a indução de vias que levam à
produção de uma grande variedade de citocinas pró-inflamatórias 89. O TLR3 expresso
em células dendríticas e apresenta a capacidade de ser ativado por mRNA proveniente
de células em necrose, ocorrendo um aumento de sinalização desta via e
consequentemente maior secreção de TNF-α 90,91. Do mesmo modo se dá a ativação do
TLR7 promovendo a estimulação de células NK, que desempenha funções antitumorais e
antivirais 92. Os TLRs são também recetores cruciais na ativação e maturação de células
B, influenciando a sua diferenciação em células B de memória e plasmócitos 93.
De acordo com Pradere o TLR3 tem a capacidade de ativar múltiplas cascatas de
sinalização pro-inflamatórias incluindo o NF-kB, AP-1 e IRF3/7, sendo a produção de

5. Discussão
60
IFNs do tipo I os principais mediadores da resposta antitumoral 88. Polimorfismos
genéticos funcionais nos genes que codificam estes recetores podem contribuir para o
desequilíbrio de fenótipos celulares, uma vez que condicionam a ativação de vias de
sinalização intracelular 74. Em situações de NHL, estas alterações nos TLR3 e TLR7
podem condicionar o desenvolvimento tumoral, com impacto na sobrevivência dos
doentes.
Relativamente aos polimorfismos analisados no presente estudo, observou-se que os
doentes portadores dos genótipos CT/TT quanto ao polimorfismo TLR3 rs3775291
apresentam uma sobrevivência de longo termo inferior comparativamente aos portadores
CC (P=0,040). De acordo com Ranjith-Kumar, a ocorrência desta transição no exão 4
resulta numa alteração da função do TLR3 79. Um estudo in vitro realizado com células
HEK293 transfetadas com plasmídeos com a alteração +1234T, apresentam uma
atenuação da ativação da via do TLR3. De facto, estudos prévios têm associado a
variante T a um risco aumentado de desenvolvimento de carcinoma de células
escamosas da cavidade oral e de carcinoma hepatocelular 94. Cumulativamente, também
foi observado que os indivíduos diagnosticados com cancro colorectal e portadores do
genótipo TT apresentam maior risco de morte pela doença 81. De acordo com a literatura,
este polimorfismo está também associado a uma suscetibilidade para infeções pelo vírus
da hepatite B, aumentando também o risco de desenvolvimento de pneumonias
associadas à infeção pelo vírus Influenza A/H1N1 em crianças 95-97.
Num estudo realizado por Chen, observou-se que mulheres chinesas com cancro da
mama e portadoras do genótipo TT apresentavam uma sobrevivência livre de progressão
de doença inferior 98,99. Segundo o estudo de Castro e colaboradores, a presença do
polimorfismo TLR3 rs3775291 destabiliza a formação de dímeros uma vez que esta
variação altera a ligação recetor-ligando 81. Esta alteração está associada a uma redução
da capacidade de ligação ao dsRNA e inibindo a via de indução da apoptose e a via de
ativação de NF-KB e de IRF3 98. Deste modo, colocamos a hipótese que os indivíduos
portadores dos genótipos CT/TT apresentam menor capacidade/eficiência de ativação da
via de sinalização do TLR3, o que conduzirá a uma menor secreção de citocinas pro-
inflamatórias e IFNs. A produção de IFN do tipo I promove a estimulação de células NK e
células dendríticas, levando-as a exercer a sua ação efetora diretamente nas células
neoplásicas e o estabelecimento de uma resposta imune adquirida específica 88. Por
outro lado, a atenuação da ativação do TLR3 pode comprometer a ocorrência da
apoptose. O estudo realizado por Paone e os colaboradores permitiu verificar que a
ativação do TLR3 por poly I:C/dsRNA tem a capacidade de induzir a apoptose em linhas
celulares de cancro da próstata, revelando o seu potencial no controlo da progressão
tumoral 100,101. A ativação do TLR3 pelo agonista sintético por Poly(I:C) apresenta uma

5. Discussão
61
capacidade pró-apoptótica desencadeando um efeito antitumoral em células de
diferentes modelos tumorais, o que revela o seu elevado potencial no tratamento de
outros tipos de tumores 98,102,103. Deste modo, podemos concluir que o polimorfismo TLR3
rs3775291 poderá comprometer a resposta antitumoral e consequentemente a
sobrevivência de longo termo nos doentes com NHL (figura 10).
A imunossupressão a que estes doentes são submetidos é consequência das
abordagens terapêuticas, resultando na diminuição da imunovigilância, sendo necessário
até um período de 12 meses após a terapia para que ocorra a reconstituição do tecido
hematopoiético 49. De acordo com Worch e colaboradores, a reconstituição imunológica
começa usualmente 6 meses após o início da terapia ocorrendo a recuperação normal
entre os 9 e 12 meses 104. Esta evidência está de acordo com os resultados obtidos em
que apenas foi observado um efeito estatisticamente significativo do polimorfismo na
população em estudo após 12 meses do início do tratamento.
Figura 10 – Influência da variante T do polimorfismo TLR3 rs3775291 nas funções mediadas
pelo recetor
Relativamente ao TLR7, este também desempenha um papel essencial na resposta
anti-tumoral 105.
No presente estudo, de acordo com a análise do polimorfismo TLR7 rs179008
verificámos que as doentes portadoras do genótipo AT/TT apresentam uma tendência
para uma sobrevivência de longo termo inferior de 134,3 semanas comparativamente a
doentes portadoras do genótipo AA, em que se observou uma sobrevivência de longo
termo de 231,4 semanas (P=0,057). Segundo Hedge e Bernstein, a transição Gln>Leu
ocorre na sequência de sinal do TLR7 da região N-terminal, condicionando a localização

5. Discussão
62
e modificações pós-translacionais do recetor, alterando assim a sua funcionalidade 106. O
polimorfismo TLR7 rs179008 tem sido alvo de diversos estudos, estando associado a
resultados controversos, em consequência da falta de replicabilidade dos mesmos. De
acordo com o estudo realizado por Schott e colaboradores, as mulheres portadoras da
variante T apresentam maior suscetibilidade à infeção pelo vírus da hepatite C, não
respondendo à administração de IFN-α, o que sugere uma diminuição da resposta
antiviral associada à ativação do TLR7 107. Cumulativamente, esta variante tem sido
associada a diminuição da expressão de IFN-α, IL-29/INFλ, IL10R e IL28R 82. A produção
de INF do tipo I é crucial na resposta antitumoral, a estimulação do TLR7 e a
consequente produção de IFN-α é uma terapia eficaz no tratamento de melanoma, do
carcinoma de células renais e da leucemia de células pilosas 88,108. De facto, a
estimulação do TLR7 em células T CD4+ com o agonista sintético resiquimod aumentou a
produção de IFN-α, IL2 e IL10, assim como a proliferação destas células
independentemente da presença de células apresentadoras de antigénios 109. Por outro
lado, a literatura sugere que a estimulação do TLR7 é responsável pela modelação da
expansão policlonal das células B e ainda da sua diferenciação em células
apresentadoras de antigénios, podendo ativar células T citotóxicas e iniciar assim uma
resposta antitumoral específica 110. De facto, Dilillo e colaboradores verificaram que a
depleção das células B, num modelo de melanoma, promove o crescimento tumoral, e
está associado ao comprometimento da ativação de células T CD8+ e CD4+ 110. Deste
modo, podemos concluir que o polimorfismo genético TLR7 rs179008 poderá
comprometer a ativação do recetor e consequentemente a resposta antitumoral,
refletindo-se na diminuição da sobrevivência de longo termo dos doentes com NHL.
Aparentemente, ocorre um efeito mais significativo nas mulheres na presença do
polimorfismo estudado comparativamente aos homens. A incompleta inativação de um
dos cromossomas X poderá potenciar o efeito desta transição no TLR7 no género
feminino 111.
De acordo com os resultados, podemos admitir que a ocorrência de polimorfismos
genéticos funcionais nos genes codificantes TLR3 e TLR7 podem modular um
microambiente celular podendo promover a progressão da doença após reconstituição
total do sistema imunológico dos doentes com NHL. Futuramente, a utilização de
agonistas destes recetores poderá ser uma estratégia promissora no tratamento dos
doentes com NHL, sendo que a definição de perfis genéticos segundo os polimorfismos
estudados poderia ser útil na identificação de subgrupos de doentes que apresentarão
maior benefício na realização destas terapias.

6. Conclusão e Perspetivas Futuras


6. Conclusão e Perspetivas Futuras
65
Nos últimos anos, estudos de imunologia têm revelado que o sistema imunitário tem a
capacidade de detetar células tumorais, desencadeando uma resposta antitumoral.
De modo a compreender o papel do sistema imunitário no combate ao
desenvolvimento de cancro, a epidemiologia molecular tem realizado vários estudos com
intuito de compreender a influencia de determinados genes no controlo de vias
envolvidas na estimulação de células do sistema imunitário. Vários estudos mencionam
que os polimorfismos TLR3 rs3775291 e TLR7 rs179008 afetam o reconhecimento de
PAMPs e DAMPs e a posterior indução de uma via de sinalização intracelular que
culmina na produção de citocinas pró-inflamatórias e IFNs. A indução dos TLRs é crucial
na estimulação de células apresentadoras de antigénios, importantes no estabelecimento
da imunidade adquirida essencial no desenvolvimento de uma resposta anti-tumoral
específica. O presente estudo procurou determinar de que modo os polimorfismos TLR3
rs3775291 e TLR7 rs179008 influenciam a sobrevivência de doentes com NHL.
Na análise do polimorfismo TLR3 rs3775291, o presente estudo mostrou que os
doentes portadores dos genótipos CT/TT apresentam menor sobrevivência de longo
termo comparativamente com os portadores do genótipo CC. Quanto ao polimorfismo
TLR7 rs179008, a sobrevivência a longo termo das mulheres portadoras dos genótipos
AT/TT foi tendencialmente inferior comparativamente com as portadoras do genótipo AA.
Deste modo podemos concluir que a presença dos polimorfismos TLR3 rs3775291 e
TLR7 rs179008 diminui o tempo de sobrevivência de doentes com NHL após a
reconstituição do sistema imunológico.
No futuro, seria relevante a replicação deste trabalho, numa amostragem superior, a
fim de validar a influência dos polimorfismos estudados no prognóstico clínico dos
doentes com NHL. Seria importante que futuramente se pudesse compreender melhor o
papel dos TLR3 e TLR7 no desenvolvimento e diferenciação dos linfócitos B. Assim,
poder-se-á correlacionar de que forma os polimorfismos TLR3 rs3775291 e TLR7
rs179008 afetam o desenvolvimento das diferentes entidades de NHL de células B.
O recurso a agonistas dos TLRs tem permitido compreender melhor o seu modo de
funcionamento e o seu papel no combate a microrganismos patogénicos e ao cancro.
Consequentemente, tem proporcionado novos horizontes para a imunoterapia, podendo
vir a ser estudado se o uso de fármacos agonistas do TLR3 e TLR7 são benéficos no
tratamento de doentes com NHL.


7. Referências Bibliográficas


7. Referências Bibliográficas
69
1. Lindsey Torre M, Rebecca Siegel M, Ahmedin Jemal D, PhD. (2015) Global
Cancer Facts & Figures 3rd Edition. American Cancer Society.
2. Campisi J. (2013) Aging, cellular senescence, and cancer. Annual review of
physiology, 75:685-705.
3. Falandry C, Bonnefoy M, Freyer G, Gilson E. (2014) Biology of cancer and aging:
a complex association with cellular senescence. Journal of clinical oncology :
official journal of the American Society of Clinical Oncology, 32(24):2604-2610.
4. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. (2015) Global
cancer statistics, 2012. CA: a cancer journal for clinicians, 65(2):87-108.
5. Anand P, Kunnumakkara AB, Sundaram C, et al. (2008) Cancer is a preventable
disease that requires major lifestyle changes. Pharmaceutical research,
25(9):2097-2116.
6. Kolonel LN, Altshuler D, Henderson BE. (2004) The multiethnic cohort study:
exploring genes, lifestyle and cancer risk. Nature reviews. Cancer, 4(7):519-527.
7. Campisi J. (2003) Cancer and ageing: rival demons? Nature reviews. Cancer,
3(5):339-349.
8. Hanahan D, Weinberg RA. (2011) Hallmarks of cancer: the next generation. Cell,
144(5):646-674.
9. Martinez JD, Parker MT, Fultz KE, Ignatenko NA, Gerner EW. (2003) Molecular
Biology of Cancer. Burger's Medicinal Chemistry and Drug Discovery: John Wiley
& Sons, Inc.
10. Oliveira PA, Colaco A, Chaves R, Guedes-Pinto H, De-La-Cruz PL, Lopes C.
(2007) Chemical carcinogenesis. Anais da Academia Brasileira de Ciencias,
79(4):593-616.
11. Vincent TL, Gatenby RA. (2008) An evolutionary model for initiation, promotion,
and progression in carcinogenesis. International journal of oncology, 32(4):729-
737.
12. Dixon K, Kopras E. (2004) Genetic alterations and DNA repair in human
carcinogenesis. Seminars in cancer biology, 14(6):441-448.
13. Hursting SD, Slaga TJ, Fischer SM, DiGiovanni J, Phang JM. (1999) Mechanism-
Based Cancer Prevention Approaches: Targets, Examples, and the Use of
Transgenic Mice. Journal of the National Cancer Institute, 91(3):215-225.
14. Brennan P. (2002) Gene-environment interaction and aetiology of cancer: what
does it mean and how can we measure it? Carcinogenesis, 23(3):381-387.
15. Williams GM. (2001) Mechanisms of chemical carcinogenesis and application to
human cancer risk assessment. Toxicology, 166(1-2):3-10.

7. Referências Bibliográficas
70
16. Hennings H, Glick AB, Greenhalgh DA, et al. (1993) Critical aspects of initiation,
promotion, and progression in multistage epidermal carcinogenesis. Proceedings
of the Society for Experimental Biology and Medicine. Society for Experimental
Biology and Medicine (New York, N.Y.), 202(1):1-8.
17. Abel EL, DiGiovanni J. (2015) Environmental Carcinogenesis. In: Thompson
JMWGMHAIB, ed. The Molecular Basis of Cancer (Fourth Edition). Philadelphia:
Content Repository Only, 103-128.e102.
18. Tanaka T, Shimizu M, Kochi T, Moriwaki H. (2013) Chemical-induced
Carcinogenesis. Journal of Experimental & Clinical Medicine, 5(6):203-209.
19. Mohamed Adil A, Lavanya V, Jamal S, Ahmed N. (2014) Cancer immunotherapy:
Targeting immunosuppressive tumor microenvironment. Oncobiology and Targets,
1(1):16-22.
20. Whiteside TL. (2008) The tumor microenvironment and its role in promoting tumor
growth. Oncogene, 27(45):5904-5912.
21. Coupland SE. (2013) Molecular pathology of lymphoma. Eye (London, England),
27(2):180-189.
22. Fisher SG, Fisher RI. (2004) The epidemiology of non-Hodgkin's lymphoma.
Oncogene, 23(38):6524-6534.
23. Kuppers R, Duhrsen U, Hansmann ML. (2014) Pathogenesis, diagnosis, and
treatment of composite lymphomas. The Lancet. Oncology, 15(10):e435-446.
24. Campo E, Swerdlow SH, Harris NL, Pileri S, Stein H, Jaffe ES. (2011) The 2008
WHO classification of lymphoid neoplasms and beyond: evolving concepts and
practical applications. Blood, 117(19):5019-5032.
25. Janz M, Mathas S. (2008) The pathogenesis of classical Hodgkin’s lymphoma:
what can we learn from analyses of genomic alterations in Hodgkin and Reed-
Sternberg cells? Haematologica, 93(9):1292-1295.
26. Word ZH MM. (2012) Advances in the diagnosis and management of lymphoma.
Blood and Lymphatic Cancer: Targets and Therapy, 2:29—55.
27. Farrell K, Jarrett RF. (2011) The molecular pathogenesis of Hodgkin lymphoma.
Histopathology, 58(1):15-25.
28. Shankland KR, Armitage JO, Hancock BW. (2012) Non-Hodgkin lymphoma.
Lancet, 380(9844):848-857.
29. Baecklund E, Smedby KE, Sutton LA, Askling J, Rosenquist R. (2014) Lymphoma
development in patients with autoimmune and inflammatory disorders--what are
the driving forces? Seminars in cancer biology, 24:61-70.

7. Referências Bibliográficas
71
30. Bewtra M, Lewis JD. (2010) Update on the risk of lymphoma following
immunosuppressive therapy for inflammatory bowel disease. Expert review of
clinical immunology, 6(4):621-631.
31. Boffetta P. I. (2011) Epidemiology of adult non-Hodgkin lymphoma. Annals of
Oncology, 22(suppl 4):iv27-iv31.
32. Grulich AE, Vajdic CM. (2005) The epidemiology of non-Hodgkin lymphoma.
Pathology, 37(6):409-419.
33. Pieper K, Grimbacher B, Eibel H. (2013) B-cell biology and development. The
Journal of allergy and clinical immunology, 131(4):959-971.
34. Basso K, Dalla-Favera R. (2015) Germinal centres and B cell lymphomagenesis.
Nature reviews. Immunology, 15(3):172-184.
35. De Silva NS, Klein U. (2015) Dynamics of B cells in germinal centres. Nature
reviews. Immunology, 15(3):137-148.
36. Fernando A. Arosa EMC, Francisco C. Pacheco. (2007) Fundamentos de
Imunologia. LIDEL.
37. Kuppers R. (2005) Mechanisms of B-cell lymphoma pathogenesis. Nature reviews.
Cancer, 5(4):251-262.
38. Lenz G, Staudt LM. (2010) Aggressive lymphomas. The New England journal of
medicine, 362(15):1417-1429.
39. He B, Chadburn A, Jou E, Schattner EJ, Knowles DM, Cerutti A. (2004)
Lymphoma B Cells Evade Apoptosis through the TNF Family Members
BAFF/BLyS and APRIL. The Journal of Immunology, 172(5):3268-3279.
40. Pileri SA, Piccaluga PP. (2012) New molecular insights into peripheral T cell
lymphomas. The Journal of clinical investigation, 122(10):3448-3455.
41. Ansell SM. (2015) Non-Hodgkin Lymphoma: Diagnosis and Treatment. Mayo
Clinic Proceedings, 90(8):1152-1163.
42. Hennessy BT, Hanrahan EO, Daly PA. (2004) Non-Hodgkin lymphoma: an update.
The Lancet. Oncology, 5(6):341-353.
43. Goldstone AH, Kottaridis PD. (2003) The allogeneic effect in non-Hodgkin's
lymphoma. Leukemia & lymphoma, 44 Suppl 3:S91-97.
44. Saria MG, Gosselin-Acomb TK. (2007) Hematopoietic stem cell transplantation:
implications for critical care nurses. Clinical journal of oncology nursing, 11(1):53-
63.
45. Henig I, Zuckerman T. (2014) Hematopoietic stem cell transplantation-50 years of
evolution and future perspectives. Rambam Maimonides Med J, 5(4):e0028.

7. Referências Bibliográficas
72
46. Passweg JR, Halter J, Bucher C, et al. (2012) Hematopoietic stem cell
transplantation: a review and recommendations for follow-up care for the general
practitioner. Swiss medical weekly, 142:w13696.
47. Mohty B, Mohty M. (2011) Long-term complications and side effects after
allogeneic hematopoietic stem cell transplantation: an update. Blood Cancer
Journal,1:e16.
48. Bacigalupo A, Ballen K, Rizzo D, et al. (2009) DEFINING THE INTENSITY OF
CONDITIONING REGIMENS : working definitions. Biology of blood and marrow
transplantation : journal of the American Society for Blood and Marrow
Transplantation,15(12):1628-1633.
49. Tomblyn M, Chiller T, Einsele H, et al. (2009) Guidelines for preventing infectious
complications among hematopoietic cell transplantation recipients: a global
perspective. Biology of blood and marrow transplantation : journal of the American
Society for Blood and Marrow Transplantation, 15(10):1143-1238.
50. Ball LM, Egeler RM. (2008) Acute GvHD: pathogenesis and classification. Bone
marrow transplantation, 41(S2):S58-S64.
51. Passweg JR, Baldomero H, Peters C. (2014) Hematopoietic SCT in Europe: data
and trends in 2012 with special consideration of pediatric transplantation,
49(6):744-750.
52. Takeda K, Akira S. (2005) Toll-like receptors in innate immunity. International
immunology, 17(1):1-14.
53. Kollmann TR, Levy O, Montgomery RR, Goriely S. (2012) Innate immune function
by Toll-like receptors: distinct responses in newborns and the elderly. Immunity,
37(5):771-783.
54. Takeda K, Akira S. (2015) Toll-like receptors. Current protocols in immunology /
edited by John E. Coligan ... [et al.],109:14.12.11-14.12.10.
55. Georgel P, Macquin C, Bahram S. (2009) The heterogeneous allelic repertoire of
human toll-like receptor (TLR) genes, 4(11):e7803.
56. Nasu K, Narahara H. (2010) Pattern recognition via the toll-like receptor system in
the human female genital tract. Mediators of inflammation, 2010:976024.
57. Lester SN, Li K. (2014) Toll-like receptors in antiviral innate immunity. Journal of
molecular biology, 426(6):1246-1264.
58. Piccinini AM, Midwood KS. (2010) DAMPening Inflammation by Modulating TLR
Signalling. Mediators of inflammation, 2010:21.
59. Sen GC, Sarkar SN. (2005) Transcriptional signaling by double-stranded RNA:
role of TLR3. Cytokine & growth factor reviews, 16(1):1-14.

7. Referências Bibliográficas
73
60. Lee CC, Avalos AM, Ploegh HL. (2012) Accessory molecules for Toll-like
receptors and their function. Nature reviews. Immunology, 12(3):168-179.
61. Werling D, Jann OC, Offord V, Glass EJ, Coffey TJ. (2009) Variation matters: TLR
structure and species-specific pathogen recognition. Trends in immunology,
30(3):124-130.
62. Zhang SY, Herman M, Ciancanelli MJ, et al. (2013) TLR3 immunity to infection in
mice and humans. Current opinion in immunology, 25(1):19-33.
63. Newton K, Dixit VM. (2012) Signaling in innate immunity and inflammation. Cold
Spring Harbor perspectives in biology, 4(3): a006049.
64. Pandey S, Kawai T, Akira S. (2015) Microbial sensing by toll-like receptors and
intracellular nucleic acid sensors. Cold Spring Harbor perspectives in medicine,
5(1):a016246.
65. Xagorari A, Chlichlia K. (2008) Toll-like receptors and viruses: induction of innate
antiviral immune responses. The open microbiology journal, 2:49-59.
66. Yokota S-i, Okabayashi T, Fujii N. (2010) The Battle between Virus and Host:
Modulation of Toll-Like Receptor Signaling Pathways by Virus Infection. Mediators
of inflammation, 2010: 184328.
67. Yamashita M, Chattopadhyay S, Fensterl V, Zhang Y, Sen GC. (2012) A TRIF-
independent branch of TLR3 signaling. Journal of immunology, 188(6):2825-2833.
68. Blasius AL, Beutler B. (2010) Intracellular toll-like receptors. Immunity, 32(3):305-
315.
69. Brencicova E, Diebold SS. (2013) Nucleic acids and endosomal pattern
recognition: how to tell friend from foe? Frontiers in cellular and infection
microbiology, 3:37.
70. Hipp MM, Shepherd D, Gileadi U, et al. (2013) Processing of human toll-like
receptor 7 by furin-like proprotein convertases is required for its accumulation and
activity in endosomes. Immunity, 39(4):711-721.
71. Dunlevy F MNaGC. (2010) TLR3 Sensing of Viral Infection. The Open Infectious
Diseases Journal, 4:1-10.
72. Vercammen E, Staal J, Beyaert R. (2008) Sensing of viral infection and activation
of innate immunity by toll-like receptor 3. Clinical microbiology reviews, 21(1):13-
25.
73. Salaun B, Romero P, Lebecque S. (2007) Toll-like receptors' two-edged sword:
when immunity meets apoptosis. European journal of immunology, 37(12):3311-
3318.

7. Referências Bibliográficas
74
74. Nieters A, Beckmann L, Deeg E, Becker N. (2006) Gene polymorphisms in Toll-
like receptors, interleukin-10, and interleukin-10 receptor alpha and lymphoma risk.
Genes Immun, 7(8):615-624.
75. Huang B, Zhao J, Unkeless JC, Feng ZH, Xiong H. (2008) TLR signaling by tumor
and immune cells: a double-edged sword. Oncogene, 27(2):218-224.
76. Sanclemente G, Moreno A, Navasa M, Lozano F, Cervera C. (2014) Genetic
variants of innate immune receptors and infections after liver transplantation.
World journal of gastroenterology : WJG, 20(32):11116-11130.
77. GOMAZ A, PAVELI] J, GLAVAN TM. (2012) The polymorphisms in Toll-like
receptor genes and cancer risk. PERIODICUM BIOLOGORUM, 114(4):461–469.
78. Brown RA, Razonable RR. (2010) A Real-Time PCR Assay for the Simultaneous
Detection of Functional N284I and L412F Polymorphisms in the Human Toll-Like
Receptor 3 Gene. The Journal of Molecular Diagnostics, 12(4):493-497.
79. Ranjith-Kumar CT, Miller W, Sun J, et al. (2007) Effects of single nucleotide
polymorphisms on Toll-like receptor 3 activity and expression in cultured cells. The
Journal of biological chemistry, 282(24):17696-17705.
80. So EY, Ouchi T. (2010) The application of Toll like receptors for cancer therapy.
International journal of biological sciences, 6(7):675-681.
81. Castro FA, Forsti A, Buch S, et al. (2011) TLR-3 polymorphism is an independent
prognostic marker for stage II colorectal cancer. European journal of cancer,
47(8):1203-1210.
82. Askar E, Ramadori G, Mihm S. (2010) Toll-like receptor 7 rs179008/Gln11Leu
gene variants in chronic hepatitis C virus infection. Journal of medical virology,
82(11):1859-1868.
83. Oh DY, Baumann K, Hamouda O, et al. (2009) A frequent functional toll-like
receptor 7 polymorphism is associated with accelerated HIV-1 disease
progression. AIDS (London, England), 23(3):297-307.
84. Bordignon M, Bargagli E, Agostini C, et al. (2013) TLR7 Gln11Leu single
nucleotide polymorphism in patients with sarcoidosis. Sarcoidosis, vasculitis, and
diffuse lung diseases : official journal of WASOG / World Association of
Sarcoidosis and Other Granulomatous Disorders, 30(2):157-161.
85. Chen YC, Hunter DJ. (2005) Molecular epidemiology of cancer. CA: a cancer
journal for clinicians, 55(1):45-54; quiz 57.
86. Vineis P, Perera F. (2007) Molecular epidemiology and biomarkers in etiologic
cancer research: the new in light of the old. Cancer epidemiology, biomarkers &
prevention : a publication of the American Association for Cancer Research,
cosponsored by the American Society of Preventive Oncology, 16(10):1954-1965.

7. Referências Bibliográficas
75
87. Swann JB, Smyth MJ. (2007) Immune surveillance of tumors. Journal of Clinical
Investigation, 117(5):1137-1146.
88. Pradere JP, Dapito DH, Schwabe RF. (2014) The Yin and Yang of Toll-like
receptors in cancer. Oncogene, 33(27):3485-3495.
89. McGettrick AF, O'Neill LA. (2010) Localisation and trafficking of Toll-like receptors:
an important mode of regulation. Current opinion in immunology, 22(1):20-27.
90. Kariko K, Ni H, Capodici J, Lamphier M, Weissman D. (2004) mRNA is an
endogenous ligand for Toll-like receptor 3. The Journal of biological chemistry,
279(13):12542-12550.
91. Sato Y, Goto Y, Narita N, Hoon DS. (2009) Cancer Cells Expressing Toll-like
Receptors and the Tumor Microenvironment. Cancer microenvironment : official
journal of the International Cancer Microenvironment Society, 2 Suppl 1:205-214.
92. Guo Q, Zhang C. (2012) Critical role of Toll-like receptor signaling in NK cell
activation. Chin. Sci. Bull, 57(24):3192-3202.
93. Isaza-Correa JM, Liang Z, van den Berg A, Diepstra A, Visser L. (2014) Toll-like
receptors in the pathogenesis of human B cell malignancies. Journal of
hematology & oncology, 7:57-57.
94. Zeljic K, Supic G, Jovic N, et al. (2014) Association of TLR2, TLR3, TLR4 and
CD14 genes polymorphisms with oral cancer risk and survival. Oral diseases,
20(4):416-424.
95. Esposito S, Molteni CG, Giliani S, et al. (2012) Toll-like receptor 3 gene
polymorphisms and severity of pandemic A/H1N1/2009 influenza in otherwise
healthy children. Virology journal, 9:270.
96. Al-Qahtani A, Al-Ahdal M, Abdo A, et al. (2012) Toll-like receptor 3 polymorphism
and its association with hepatitis B virus infection in Saudi Arabian patients.
Journal of medical virology, 84(9):1353-1359.
97. Li G, Zheng Z. (2013) Toll-like receptor 3 genetic variants and susceptibility to
hepatocellular carcinoma and HBV-related hepatocellular carcinoma. Tumour
biology : the journal of the International Society for Oncodevelopmental Biology
and Medicine, 34(3):1589-1594.
98. Cheng YS, Xu F. (2010) Anticancer function of polyinosinic-polycytidylic acid.
Cancer biology & therapy, 10(12):1219-1223.
99. Chen DN, Song CG, Yu KD, Jiang YZ, Ye FG, Shao ZM. (2015) A Prospective
Evaluation of the Association between a Single Nucleotide Polymorphism
rs3775291 in Toll-Like Receptor 3 and Breast Cancer Relapse. PloS one,
10(7):e0133184.

7. Referências Bibliográficas
76
100. Salaun B, Zitvogel L, Asselin-Paturel C, et al. (2011) TLR3 as a biomarker for the
therapeutic efficacy of double-stranded RNA in breast cancer. Cancer research,
71(5):1607-1614.
101. Paone A, Starace D, Galli R, et al. (2008) Toll-like receptor 3 triggers apoptosis of
human prostate cancer cells through a PKC-alpha-dependent mechanism.
Carcinogenesis, 29(7):1334-1342.
102. Cheng M, Chen Y, Xiao W, Sun R, Tian Z. (2013) NK cell-based immunotherapy
for malignant diseases. Cell Mol Immunol, 10(3):230-252.
103. Lu H. (2014) TLR Agonists for Cancer Immunotherapy: Tipping the Balance
between the Immune Stimulatory and Inhibitory Effects. Frontiers in immunology,
5:83.
104. Worch J, Makarova O, Burkhardt B. (2015) Immunreconstitution and infectious
complications after rituximab treatment in children and adolescents: what do we
know and what can we learn from adults? Cancers, 7(1):305-328.105. Meyer T, Nindl I, Schmook T, Ulrich C, Sterry W, Stockfleth E. (2003) Induction of
apoptosis by Toll-like receptor-7 agonist in tissue cultures. The British journal of
dermatology, 149 Suppl 66:9-14.
106. Hegde RS, Bernstein HD. (2006) The surprising complexity of signal sequences.
Trends in biochemical sciences, 31(10):563-571.
107. Schott E, Witt H, Neumann K, et al. (2008) Association of TLR7 single nucleotide
polymorphisms with chronic HCV-infection and response to interferon-a-based
therapy. Journal of viral hepatitis, 15(1):71-78.
108. Kirkwood JM, Butterfield LH, Tarhini AA, Zarour H, Kalinski P, Ferrone S. (2012)
Immunotherapy of cancer in 2012. CA: a cancer journal for clinicians, 62(5):309-
335.
109. Caron G, Duluc D, Fremaux I, et al. (2005) Direct stimulation of human T cells via
TLR5 and TLR7/8: flagellin and R-848 up-regulate proliferation and IFN-gamma
production by memory CD4+ T cells. Journal of immunology (Baltimore, Md. :
1950),175(3):1551-1557.
110. DiLillo DJ, Yanaba K, Tedder TF. (2010) B cells are required for optimal CD4+ and
CD8+ T cell tumor immunity: therapeutic B cell depletion enhances B16 melanoma
growth in mice. Journal of immunology (Baltimore, Md. : 1950),184(7):4006-4016.
111. Libert C, Dejager L, Pinheiro I. (2010) The X chromosome in immune functions:
when a chromosome makes the difference. Nature reviews. Immunology,
10(8):594-604.

7. Referências Bibliográficas
77






























