Embed Size (px)
Citation preview

of June 16, 2018.This information is current as
InfectionsConcurrent Mycobacterial and Influenza Interactions between T Cells Responding to
and Matyas SandorHeninger, Shoua Vang, Krisna Wells, Yoshihiro Kawaoka Dominic O. Co, Laura H. Hogan, Jozsef Karman, Erika
http://www.jimmunol.org/content/177/12/8456doi: 10.4049/jimmunol.177.12.8456
2006; 177:8456-8465; ;J Immunol
Referenceshttp://www.jimmunol.org/content/177/12/8456.full#ref-list-1
, 31 of which you can access for free at: cites 53 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 2006 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on June 16, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on June 16, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

Interactions between T Cells Responding to ConcurrentMycobacterial and Influenza Infections1
Dominic O. Co,*† Laura H. Hogan,* Jozsef Karman,* Erika Heninger,* Shoua Vang,*Krisna Wells,‡ Yoshihiro Kawaoka,‡ and Matyas Sandor2*†
CD4� T cells are central in mediating granuloma formation and limiting growth and dissemination of mycobacterial infections.To determine whether T cells responding to influenza infection can interact with T cells responding to Mycobacterium bovis bacilleCalmette-Guerin (BCG) infection and disrupt granuloma formation, we infected mice containing two monoclonal T cell popula-tions specific for the model Ags pigeon cytochrome c (PCC) and hen egg lysozyme (HEL). These mice were chronically infectedwith PCC epitope-tagged BCG (PCC-BCG) and acutely infected with HEL epitope-tagged influenza virus (HEL-flu). In these mice,PCC-BCG infection is much more abundant in the liver than the lung, whereas HEL-flu infection is localized to the lung. Weobserve that both T cells have access to both inflammatory sites, but that PCC-specific T cells dominate the PCC-BCG inflam-matory site in the liver, whereas HEL-specific T cells dominate the HEL-flu inflammatory site in the lung. Influenza infection, inthe absence of an influenza-specific T cell response, is able to increase the activation state and IFN-� secretion of PCC-BCG-specific T cells in the granuloma. Activation of HEL-specific T cells allows them to secrete IFN-� and contribute to protection inthe granuloma. Ultimately, infection with influenza has little effect on bacterial load, and bacteria do not disseminate. In summary,these data illustrate complex interactions between T cell responses to infectious agents that can affect effector responses topathogens. The Journal of Immunology, 2006, 177: 8456–8465.
V arious CD4� T cells are critical in the long-term controlof mycobacterial infections by orchestrating the forma-tion of granulomas. The host-pathogen interface of the
granuloma is where mycobacteria live within infected macrophageand where the host immune response controls bacterial growth anddissemination. CD4� T cell-deficient mice are unable to formgranulomas and eventually succumb to infection (1–3). Adoptivetransfer of CD4� T cells to CD4� T cell-deficient mice is sufficientto restore granuloma formation and protection (4). The repertoireof granuloma-infiltrating CD4� T cells is broad, even at the levelof individual granulomas (5). However, infection of monoclonalpigeon cytochrome c (PCC)3-specific CD4� TCR transgenic (Tg)mice (5CC7 RAG�/�) with PCC epitope-tagged recombinantMycobacterium bovis bacille Calmette-Guerin (BCG), or PCC-BCG, demonstrates that a monoclonal T cell population is suffi-cient to mediate protective granuloma formation (5).
Although CD8� T cells are the primary T cells controlling in-fluenza infection, CD4� T cells also contribute to protection andthe establishment of T cell memory (6). Transfer of CD4� T cellclones can reduce viral titers, and CD4� T cells can in some cir-
cumstances exhibit direct cytolytic activity (7). CD4� T cells arealso important in providing B cell help for the production of pro-tective Ab against influenza infection (8).
To examine how CD4� T cell responses to BCG and influenzainteract, 5CC7 RAG�/� mice expressing a monoclonal T cellpopulation specific for PCC were infected with PCC-BCG.These mice were then adoptively transferred with spleen cellsfrom hen egg lysozyme (HEL)-specific CD4� 3A9 RAG�/�
TCR Tg mice and infected with recombinant influenza virusexpressing HEL (HEL-flu). This provided a system in whichPCC and HEL Ags are localized in anatomically distinct sites.
We tested how BCG-specific and influenza-specific CD4� Tcells distribute between the two inflammatory sites and how thesetwo T cells would interfere with each other. In addition, we ex-amined how these interactions affect granuloma formation, dis-semination, and control of BCG. We observe that activated T cellshave access to both the HEL-flu inflammatory site in the lung aswell as the PCC-BCG inflammatory site in the liver. However, Tcells specific for local Ag dominate each site. In addition, we ob-serve that activation of HEL-flu-specific T cells down-regulatesthe activation phenotype of recombinant BCG-targeted T cells inthe granuloma, but that activated HEL-flu-specific T cells maycompensate for this effect. Finally, we find that infection with in-fluenza does moderately increase bacterial load, but bacterial loadremains within the range of adequate control. These data demon-strate that T cells responding to an acute respiratory infection canmodulate host responses to an ongoing BCG infection.
Materials and MethodsMice
B10.BR RAG1�/� mice were a gift of D. Sant’Angelo (Memorial SloanKettering Cancer Center, New York, NY). 5CC7 RAG2�/� mice (9) spe-cific for PCC residues 88–104 in the context of I-Ek were purchased fromTaconic Farms Emerging Models Program and bred onto the B10.BRRAG1�/� background. 3A9 mice (10) specific for HEL residues 46–61 inthe context of I-Ak were purchased from The Jackson Laboratory and bred
*Department of Pathology and Laboratory Medicine, School of Medicine and PublicHealth, †Program in Cellular and Molecular Biology, and ‡Department of Pathobio-logical Sciences, School of Veterinary Medicine, University of Wisconsin, Madison,WI 53706
Received for publication June 22, 2006. Accepted for publication October 5, 2006.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported by National Institutes of Health Grants R01 AI48087, R01AI/HL46430, and R21 AI054893 (to M.S.).2 Address correspondence and reprint requests to Dr. Matyas Sandor, Department ofPathology and Laboratory Medicine, Medical Sciences Center, Room 5468, Univer-sity of Wisconsin, 1300 University Avenue, Madison, WI 53706. E-mail address:[email protected] Abbreviations used in this paper: PCC, pigeon cytochrome c; BCG, bacilleCalmette-Guerin; HEL, hen egg lysozyme; BAL, bronchoalveolar lavage; AFB, acid-fast bacilli; Tg, transgenic; wt, wild type.
The Journal of Immunology
Copyright © 2006 by The American Association of Immunologists, Inc. 0022-1767/06/$02.00
by guest on June 16, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

onto the B10.BR RAG1�/� background. Mice were housed at the Univer-sity of Wisconsin Animal Care Unit (Madison, WI) according to the guide-lines of the Institutional Animal Care and Use Committee.
Infections
Creation of kanamycin-resistant PCC-BCG from BCG Pasteur strain(Staten Serum Institut, Copenhagen, Denmark) was previously described(5). Growth and preparation of frozen stocks for infection were as de-scribed (5). For infection, 7 � 106 CFU of BCG were i.p. injected. WSN-HEL (HEL-flu) recombinant influenza virus was generated by reverse ge-netics from the A/WSN/33 parent strain wild-type influenza (wt flu) andproduced as previously described (11). For intranasal infection, mice werelightly anesthetized with isoflurane and 2.5 � 105 PFU of either HEL-fluor wt flu was pipetted into the nose of the sleeping mouse.
Cell isolation and flow cytometry
For bronchoalveolar lavage (BAL), the trachea was exposed and cannu-lated with a 20-gauge catheter. Cells were isolated by washing the lungsthree times with 1 ml of PBS, and the cell pellet was washed once withHBSS. Isolation of splenocytes and granuloma-infiltrating cells was per-formed as previously described (5) to produce single cell suspensions. Atotal of 106 cells were incubated for 30 min on ice with saturating con-centrations of labeled Abs and 40 �g/ml unlabeled 2.4G2 mAb to blockbinding to Fc receptors and washed three times. For intracellular staining,cells were first surface stained as outlined, then fixed for 20 min in Cyto-perm/Cytofix (BD Pharmingen). Cells were then washed in staining buffer(PBS plus 1% BSA) containing 0.1% saponin and stained with saturatingconcentrations of intracellular Abs for 30 min on ice. Cells were washed instaining buffer containing 0.1% saponin. Fixed samples were analyzed ona BD Biosciences FACSCalibur. Flow cytometric data was analyzed usingFlowJo software version 4.6.1 (Tree Star). For assessment of intracellularIFN-�, cells were cultured with complete RPMI 1640 plus 10% FBS orcomplete RPMI 1640 plus 10% FBS with 5 �g/ml anti-CD3 (145-2C11) inthe presence of GolgiStop (BD Pharmingen) for 6 h at 37°C in 5% CO2.Cells were harvested and washed twice with staining buffer and stained asoutlined above for intracellular IFN-�. Fluorochrome-labeled Abs againstCD4 (RM5-4), LFA-1 (7D4), TCR V�3 (KJ25), IFN-� (XMG1.2), andactivated caspase-3 (C92-605) were purchased from BD Pharmingen. Absagainst TCR V�3 (KJ25), I-Ak (10-2.16), Mac-1 (M1/70), and V�8(F23.1) were produced from hybridomas and labeled by standard methodswith biotin, FITC, or Cy5.
Immunofluorescence
All incubations were performed at room temperature unless otherwisestated. Thick cryosections (5 �m) were cut from OCT embedded tissuesand fixed for 30 min in 4% paraformaldehyde in PBS. Sections were thenwashed three times with PBS and outlined with a Pap pen. Sections wereblocked for 30 min with 1% BSA and 40 �g/ml 2.4G2 Ab and stained for30 min with Alexa Fluor 568-labeled KJ25 (anti-V�3) and FITC-labeled F23.1 (anti-V�8) in the presence of 1% BSA and 40 �g/ml 2.4G2.Unbound Ab was washed away by three washes in PBS and sections werecoverslipped with Gel/Mount (Biomeda). Confocal images were acquiredon a Bio-Rad MRC-1024 maintained by the W. M. Keck Laboratory forBiological Imaging (University of Wisconsin, Madison, WI).
TUNEL Staining
The 5-�m thick cryosections were cut from O.C.T. embedded liver tissuesamples and fixed for 30 min in 4% paraformaldehyde in PBS, then washedthree times with PBS and outlined with a Pap pen. Sections were blockedwith 40 �g/ml 2.4G2 Ab in 1% BSA for 30 min and then stained for 30 minusing Alexa Fluor 568-labeled KJ25 (anti-V�3) and Alexa Fluor 647-labeled F23.1 (anti-V�8). FITC-labeled TUNEL staining was per-formed following the manufacturer’s protocol (Roche Diagnostics).Confocal images were acquired on a Bio-Rad MRC-1024 maintained bythe W. M. Keck Laboratory for Biological Imaging (University of Wis-consin, Madison, WI).
Histopathology
Tissue was fixed in 10% neutral-buffered formalin and processed for par-affin embedding by standard methods. Thick sections (5 �m) were stainedwith H&E for tissue morphology and by the Ziehl-Neelsen method to iden-tify acid-fast bacilli (AFB). To quantitate granuloma lesion size, digitalimages of H&E stained sections were acquired at �400 total magnification,and the granuloma area was determined by outlining each lesion in theScion Image program version 1.62c (NIH Image software). Quantitation ofgranuloma burden was performed by counting the number of liver granu-
lomas per field at �100 total magnification. To quantitate bacterial load,the number of AFB per lesion were counted at �1000 total magnificationon Ziehl-Neelsen-stained slides.
Organ load
Bacterial colony formation was determined by plating serial dilutions of liverhomogenates on Middlebrook 7H10 agar plates (Difco) supplemented with10% oleic acid-dextrose-catalase, 50 �g/ml kanamycin, and 10 �g/ml cyclo-heximide. Colonies were counted after 3 wk of incubation at 37°C. Data arepresented as individual mouse values after a log 10 transformation.
RT-PCR
Homogenized liver and lung tissue was lysed in TRIzol (Invitrogen LifeTechnologies), and total RNA isolated according to manufacturer’s instruc-tions. cDNA was prepared using MMLV reverse transcriptase (InvitrogenLife Technologies) and PCR performed using �2-microglobulin primers(sense) 5�-TGACCGGCTTGTATGCTATC-3� and (antisense) 5�-CAGTGTGAGCCAGGATATAG-3� and influenza matrix protein primers (1) 5�-GGACTGCAGCGTAGACGCTT-3� and (2) 5�-CATCCTGTTGTATATGAGGCCCAT-3� (12) as previously described.
Databases for cross-reactivity
HEL and PCC sequences were searched against the mycobacterial se-quence (�www.sanger.ac.uk/Projects/M_bovis/�) and influenza sequence(�www.flu.lanl.gov�) databases using the BLAST (Basic Local AlignmentSearch Tool; National Center for Biotechnology Information).
ResultsInfluenza and BCG infections create two anatomically distinctinflammatory sites
The 5CC7 RAG�/� animals infected i.p. with PCC-BCG for 3 wkhave a high level of bacteria in liver granulomas, and a large por-tion of splenic, and granuloma-infiltrating T cells are activated asmeasured by LFA-1 surface expression. At 5 wk, bacterial load islow and liver granulomas induced by T cells prevent dissemination
FIGURE 1. BCG and influenza are localized to anatomically distinctinflammatory sites. A1, Liver and lung tissue homogenates from mice in-fected intranasally with WSN were titered on monolayers of MDCK cells.A2, RT-PCR was performed on RNA isolated from the same liver and lungtissue shown in A1 using primers specific for murine �2-microglobulin(�2-m) and influenza matrix protein gene (M gene). B, The 5-�m thicksections from paraffin-embedded liver (left) and lung (right) tissue of PCC-BCG-infected 5CC7 RAG�/� mice coinfected with HEL-flu were stainedby the Ziehl-Neelsen method for AFB. Arrowheads, Individual AFB. Orig-inal magnification was �1000.
8457The Journal of Immunology
by guest on June 16, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

of bacteria to the lung (Fig. 1B). At that stage, most T cells in bothspleen and granulomas express low levels of LFA-1, but also lowlevels of the naive T cell marker CD45RB, suggesting an Ag-experienced but resting phenotype (data not shown). This model isan example of chronically controlled mycobacterial infection. As amodel of concurrent acute infection, we chose intranasal infectionwith murine-adapted influenza A/WSN/33 to test whether granu-lomatous control of BCG will be affected. WSN replicates in thelungs, but not the liver of intranasally infected mice (13). Afterintranasal infection, viral titers on liver tissue demonstrate that noinfectious virus can be found in liver, at a time when virions areabundant in lungs (Fig. 1A1). No viral RNA is detectable in livertissues from the same mice (Fig. 1A2). In contrast, i.p. infectionwith BCG results in an infection that is largely localized to theliver, and spares the lung as judged by microscopic examination ofZiehl-Neelsen-stained liver and lung sections (Fig. 1B) and by�100-fold fewer CFU by quantitative microbiology (see Fig. 8C).Because in RAG�/� animals there is a strong dissemination ofBCG to the lung after initial infection, 5CC7 T cells are clearlyresponsible for the protective granuloma formation observed asreported previously (5). These data establish that the two infectiousagents, BCG and influenza, are restricted to two different anatom-ical sites in this model.
3A9 T cells do not cross-react with PCC-BCG
To test how CD4� 5CC7 T cells responding to two different in-fectious agents interact with each other, 3A9 T cells were adop-tively transferred into 5CC7 RAG�/� mice at a point when controlof BCG has been established. We have three lines of evidence that3A9 T cells do not cross-react with PCC-BCG. First, a BLASTsearch of the Mycobacterium tuberculosis genome for sequencesencoding amino acid sequences similar to the HEL 46–61 epitoperevealed no matches. Second, 3A9 T cells cocultured with PCC-
BCG do not up-regulate activation markers and do not expand,whereas 5CC7 T cells activated both responses (data not shownand (5). In addition, 3A9 T cells adoptively transferred into PCC-BCG infected 5CC7 RAG�/� mice do not up-regulate the activa-tion marker LFA-1 (data not shown), demonstrating that PCC-BCG does not activate 3A9 T cells in vivo. Also, 3A9 T cells donot expand in PCC-BCG-infected 5CC7 RAG�/� mice infectedwith wt flu, suggesting that they do not cross-react with influenzaAgs in vivo (Fig. 2B, upper left histogram). With regard to cross-reactivity of 5CC7 T cells for influenza Ags, a BLAST searchperformed on the Influenza Sequence Database did not identifyany significant matches. Also, although nearly all splenic 3A9 Tcells up-regulate LFA-1 in response to HEL-flu infection (Fig. 2B,lower left histogram), very little increase in activation is noted on5CC7 T cells when mice are infected with either wt flu or HEL-flu
FIGURE 2. LFA-1 expression of 5CC7 and 3A9 T cells in spleen, BAL,and liver granulomas in PCC-BCG coinfected with influenza 5CC7RAG�/� mice. Cell preparations from mice in Fig. 3 were stained forLFA-1 and analyzed by flow cytometry. A, Histograms represent LFA-1levels on 5CC7 T cells (CD4� V�3� lymphocytes). B, Histograms repre-sent LFA-1 levels on 3A9 T cells (CD4� V�8� lymphocytes). Numbershown for each histogram represents the percentage of Tg T cell populationexpressing high levels of LFA-1. Data are representative of three experi-ments each having three mice per group.
FIGURE 3. 3A9 T cell expansion in response to HEL-flu infection.PCC-BCG-infected 5CC7 RAG�/� mice were adoptively transferred with3A9 RAG�/� spleen cells and infected intranasally with wt flu or HEL-flu.Spleen cells were harvested at days 5, 6, and 7 and stained with Abs againstCD4 and V�8. Histograms represent V�8 expression on CD4� cells in thelymphocyte gate. Each number represents the percentage of CD4� T cellspositive for V�8. Data are representative of three experiments.
FIGURE 4. Inflammation in the lungs and liver in BCG coinfected withinfluenza 5CC7 RAG�/� mice. Sections from paraffin-embedded liver andlungs of 5CC7 RAG�/� mice infected with PCC-BCG alone or intranasallywith either wt flu or HEL-flu were stained with H&E. Total magnificationis �100.
8458 T CELL INTERACTIONS IN CONCURRENT INFECTION
by guest on June 16, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
(Fig. 2A, middle and lower left histograms), indicating that 5CC7T cells do not cross-react with wt flu or HEL-flu in vivo.
In vivo expansion of 3A9 T cells in response to HEL-fluinfection
PCC-BCG-infected 5CC7 RAG�/� mice, which control PCC-BCG infection, were adoptively transferred with 3A9 T cells, gen-erating a mouse with two monoclonal CD4� T cell populations. Incontrast to adoptive transfer of TCR Tg T cells into RAG�/� mice,there is little if any homeostatic expansion of 3A9 T cells in 5CC7RAG�/� mice (Fig. 3, top). Infection with HEL-flu results in arapid systemic expansion of 3A9 T cells in these mice (Fig. 3,bottom). At day 6, whereas 3A9 T cells only constitute �4% ofsplenic CD4� T cells in of PCC-BCG plus HEL-flu-infected 5CC7RAG�/� mice, �80% have an activated phenotype by LFA-1 ex-pression (Fig. 2B). Because only �10% of splenic 5CC7 T cells inPCC-BCG-infected mice are LFA-1high, the number of activated(LFA-1high) 3A9 and 5CC7 T cells is comparable. As this patternclosely repeats in various experiments, all additional experimentswere performed at day 6 postinfection with influenza.
Coinfection with influenza and BCG induces inflammation in theliver and lungs of 5CC7 RAG�/� mice
PCC-BCG infection of 5CC7 RAG�/� mice induces granuloma-tous inflammation in the liver, and the extent and character of theinflammation, is similar to that found in mice superinfected with
wt flu or HEL-flu (Fig. 4 left panels compare with Fig. 8A). Thelungs of PCC-BCG-infected 5CC7 RAG�/� mice have little if anyinflammation and in the sections that we examined, no granulomaswere found. However, influenza infection induced strong inflam-mation that is even more severe in HEL-flu-infected animals re-flected in the increased inflammatory infiltration and less air spacevisible in the sections (Fig. 4, right panels). As previous datashowed that influenza infection is present in the lungs and not theliver of infected mice, and that BCG infection is confined to theliver, these data show that both agents induce inflammation that ismostly restricted to the respective organ.
T cells have access to both BCG and influenza inflammatorysites but dominate where their Ag is present
Having established two separate inflammatory sites, one contain-ing HEL in the lung and the other PCC in the liver, we tested howthe two monoclonal T cell populations specific for these differentpeptides would distribute between these two inflammatory sites inthe context of these two infectious agents. To address this ques-tion, spleen cells, granuloma-infiltrating cells, and BAL cells wereisolated and stained to determine the relative proportions of thetwo Tg T cell populations in the doubly infected, 3A9 adoptivelytransferred 5CC7 RAG�/� mice. Fig. 5A shows that HEL-specificT cells undergo a 10-fold expansion in response to infection withHEL-flu vs wt flu (5.7 � 2.3% of CD4� T cells are HEL-specificin HEL-flu-infected mice vs 0.5 � 0.25 in wt flu-infected mice;
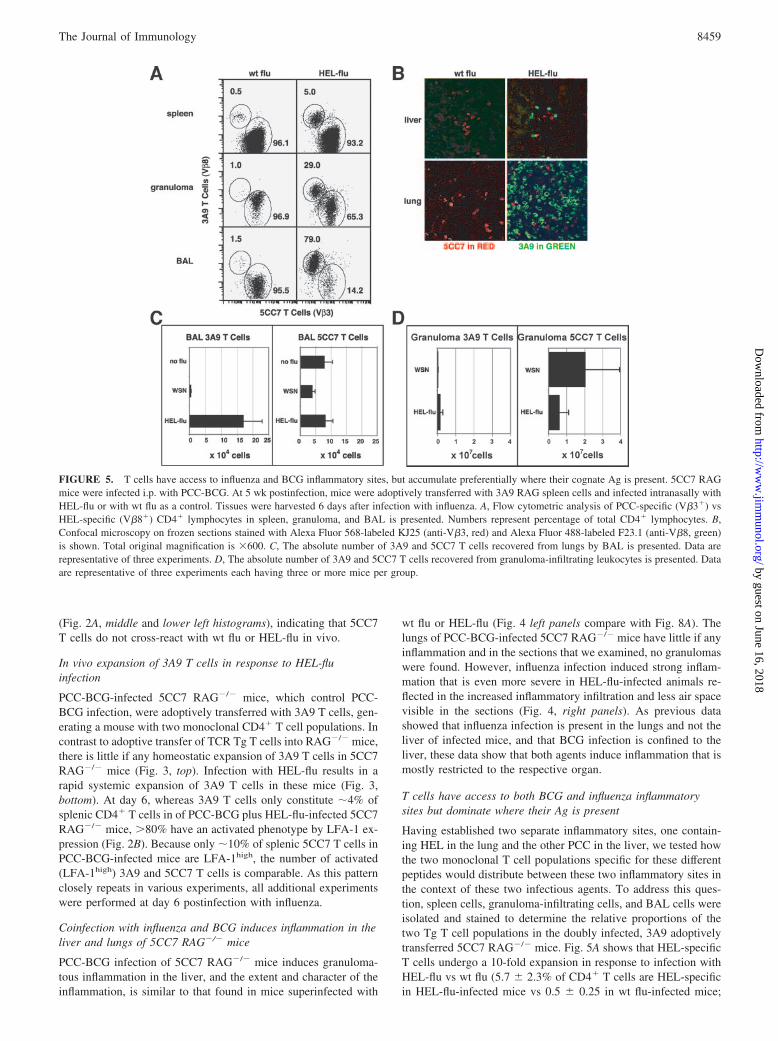
FIGURE 5. T cells have access to influenza and BCG inflammatory sites, but accumulate preferentially where their cognate Ag is present. 5CC7 RAGmice were infected i.p. with PCC-BCG. At 5 wk postinfection, mice were adoptively transferred with 3A9 RAG spleen cells and infected intranasally withHEL-flu or with wt flu as a control. Tissues were harvested 6 days after infection with influenza. A, Flow cytometric analysis of PCC-specific (V�3�) vsHEL-specific (V�8�) CD4� lymphocytes in spleen, granuloma, and BAL is presented. Numbers represent percentage of total CD4� lymphocytes. B,Confocal microscopy on frozen sections stained with Alexa Fluor 568-labeled KJ25 (anti-V�3, red) and Alexa Fluor 488-labeled F23.1 (anti-V�8, green)is shown. Total original magnification is �600. C, The absolute number of 3A9 and 5CC7 T cells recovered from lungs by BAL is presented. Data arerepresentative of three experiments. D, The absolute number of 3A9 and 5CC7 T cells recovered from granuloma-infiltrating leukocytes is presented. Dataare representative of three experiments each having three or more mice per group.
8459The Journal of Immunology
by guest on June 16, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

mean � SD of n � 3 experiments). HEL-specific T cells can befound both in the granuloma and in the lungs of HEL-flu-infectedmice. However, HEL-specific T cells significantly outnumberPCC-specific cells in the BAL of HEL-flu-infected mice (69.0 �14.0% of CD4� T cells are HEL-specific). Conversely, PCC-spe-cific cells outnumber HEL-specific cells in the granuloma by alower ratio (15.3 � 12.1% of CD4� T cells are HEL-specific).
Confocal microscopy on frozen sections of liver and lung tissuewas also used to determine the distribution of Tg T cells. Frozenliver and lung sections were stained with Abs against TCR V�3 toidentify PCC-specific T cells and against TCR V�8 to identifyHEL-specific T cells. As shown in Fig. 5B, HEL-specific cellspredominate in lungs of HEL-flu-infected mice, whereas PCC-spe-cific cells predominate in the liver granulomas. In addition, thesedata indicate that HEL-specific T cells are mostly localized ingranulomatous lesions and are rarely found in the liver paren-chyma. These data confirmed previous work that observed acti-vated T cells have access to inflammatory sites independent ofspecificity. However, the difference in the ratio of 5CC7 T cells to3A9 T cells between the BCG inflammatory site and the influenzainflammatory site suggests that other factors besides T cell acti-vation state play a role in determining what T cells accumulate atan inflammatory site.
In the BAL, reproducibility of the cell yield allowed us to de-termine the absolute number of 5CC7 and 3A9 T cells infiltratingthe lungs (Fig. 5C). There is clearly a 10-fold increase in the num-ber of 3A9 T cells infiltrating the lungs of mice infected withHEL-flu as compared with wt flu-infected mice. In contrast, theabsolute number of 5CC7 T cells in the lungs was similar in allgroups. Thus, 5CC7 T cells have access to the lung, but influenza-induced inflammation did not increase their recruitment to that site.In contrast, recovery of granuloma-infiltrating cells is much lessreproducible, restricting the interpretation of absolute numbers.Despite the fact that these numbers are questionable on technical
grounds, these data clearly support experiments (Fig. 5, A and B),indicating that there are more 5CC7 T cells in the granulomas ofHEL-flu-infected mice than there are 3A9 T cells.
In summary, activated T cells have access to both the influenzaand BCG inflammatory sites. However, T cells dominate the sitewhere their specific Ag is expressed.
Influenza infection increases expression of LFA-1 on 5CC7 Tcells in granulomas
Having determined the distribution of 5CC7 and 3A9 T cells dur-ing concurrent infection, we examined their activation phenotypeat the different inflammatory sites. 3A9 T cells in the spleen of wtflu-infected mice expressed low levels of LFA-1, whereas nearlyall 3A9 T cells in the spleen of HEL-flu-infected mice were LFA-1high (Fig. 2B, left column). Similarly, most of the granuloma- andlung-infiltrating 3A9 T cells in HEL-flu-infected mice were LFA-1high (Fig. 2B, bottom row, middle and right panels), demonstrat-ing that 3A9 T cells in HEL-flu-infected mice are acutely activatedat all sites. Interestingly, although few 3A9 T cells infiltrate gran-ulomas and BAL of wt flu-infected mice (Fig. 5A, left column),most 3A9 T cells also expressed high levels of LFA-1 (Fig. 2B, toprow), suggesting selection for activated T cells. However, the sig-nificance of this finding is restricted because these represent veryfew cells (see Fig. 5).
As mentioned earlier, at this stage in BCG infection, the numberof activated 5CC7 T cells in spleen and granuloma is decreasedrelative to earlier time points (Fig. 2A, upper row). The higherlevel of LFA-1 expression on 5CC7 T cells in the BAL probablyreflects the preference of higher activated T cells for nonspecificrecruitment to the lung. Interestingly, infection with wt flu morethan doubles the number of LFA-1high 5CC7 T cells in both thespleen and the granuloma, probably by an Ag-independent mech-anism. The mechanism by which LFA-1 is up-regulated on 5CC7T cells in these mice is not clear, but this response correlates with
FIGURE 6. Influenza-specific 3A9 T cells are pref-erentially eliminated from the granuloma by apoptosis.Frozen sections of liver (A) and lung (B) tissue fromPCC-BCG-infected mice coinfected with wt flu orHEL-flu were stained for 5CC7 T cells, 3A9 T cells, andTUNEL as a marker of apoptosis. Bars, 50 �m. Arrowpoints to a TUNEL positive.
8460 T CELL INTERACTIONS IN CONCURRENT INFECTION
by guest on June 16, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

higher levels of CD25 and CD69 expression as well as with anincreased ability to secrete IFN-� as assayed by intracellularcytokine staining (5%, data not shown, vs 10.8%, see Fig. 6). In-terestingly, in the presence of another activated T cell, this influ-enza-induced activation is much smaller. In the presence of a largenumber of acutely activated 3A9 T cells, the proportion of LFA-1high 5CC7 T cells is decreased in the granulomas and lungs ofHEL-flu-infected mice relative to wt flu-infected mice. Again, thisdecrease in LFA-1, CD69, and CD25 is also reflected in the pro-portion of 5CC7 T cells secreting IFN-� in the granuloma (see Fig.7A). These data suggest that recruitment of acutely activated non-specific T cells into the granuloma can interfere with the activationand function of local Ag-specific T cells in both the granuloma andthe lungs. These data suggest that the 5CC7 T cell compartmentsenses the activation state of the 3A9 T cells.
Activation of 3A9 T cells by HEL-flu infection results in similarlow levels of activated caspase-3 in granuloma-infiltrating 3A9and 5CC7 T cells
One possible mechanism for the differential accumulation of 5CC7vs 3A9 T cells in the granuloma is that local Ag may favor survivalof local Ag-specific T cells. To address this issue, the level ofactivated caspase-3 was determined as a marker for apoptosis. Weobserve that similar proportions (�10%) of both 3A9 (local Ag-nonspecific) and 5CC7 (local Ag-specific) T cells in HEL-flu-in-fected mice express high levels of activated caspase-3 in the gran-uloma at that time point.
These findings are confirmed by combined TUNEL and TCRstaining (Fig. 6). Note that the differential distribution of 3A9 and5CC7 T cells in the liver and lung shown in Fig. 5 is confirmed.HEL-Flu in the presence of 3A9 T cells results in more cell deathin the lungs and livers of infected mice that correlates with theheavier inflammation (Fig. 4). However, very few T cells colocal-ize with TUNEL staining, consistent with caspase staining de-scribed. Interestingly, although most 3A9 T cells are not apoptotic,there are more apoptotic 3A9 T cells than 5CC7 probably due toacute activation. Still, both caspase and TUNEL data suggest thatdifferential cell death is not the major mechanism of differentialdistribution of 3A9 and 5CC7 T cells at the inflammatory site.
HEL-flu-activated 3A9 T cells contribute IFN-� and increasemacrophage activation in BCG-induced granulomas
One of the major effector functions of CD4� T cells in thecontrol of mycobacterial infection is the secretion of IFN-� andsubsequent macrophage activation. To test the effect of HEL-fluinfection on the effector function of PCC-BCG-specific CD4� Tcells, staining for intracellular IFN-� was performed (Fig. 7A).It is clear that in HEL-flu-infected mice, a fraction of both5CC7 (V�3�) and 3A9 (V�3�) T cells in the spleen can pro-duce IFN-�. In the granuloma, infection with HEL-flu decreasesthe fraction of granuloma-infiltrating 5CC7 T cells secretingIFN-� relative to granuloma-infiltrating 5CC7 T cells frommice infected with wt flu. This finding is consistent with de-creased LFA-1 expression observed on granuloma-infiltrating5CC7 T cells of HEL-flu superinfected mice relative to wt flusuperinfected mice in Fig. 2A and might reflect a competition ofthese two T cells in the granuloma. Although HEL-flu infectiondecreased the proportion of granuloma-infiltrating 5CC7(V�3�) T cells secreting IFN-�, the proportion of 3A9 (V�3�)T cells secreting IFN-� increased. As a result, the total propor-tion of CD4� T cells secreting IFN-� in granulomas of HEL-flu-infected mice is similar to or greater than the proportion ingranulomas of wt flu-infected mice. These data suggest thatalthough HEL-flu infection partially suppresses IFN-� secretion
by 5CC7 T cells in the granuloma, HEL-flu activated 3A9 Tcells can compensate for this and potentially contribute to pro-tection through the secretion of IFN-�. In addition, these datasuggest that in mice coinfected with HEL-flu, 3A9 T cells arethe main source of IFN-� in the granuloma, despite the fact that 3A9T cells constitute a smaller proportion of granuloma-infiltrating Tcells.
Expression of MHC class II is dependent on secretion of IFN-�,and up-regulation of MHC class II is suggestive of IFN-� produc-tion in situ. As shown in Fig. 7B, HEL-flu infection did not appearto up-regulate MHC class II on splenic macrophages relative to wtflu infection. However, infection with HEL-flu moderately in-creased surface expression levels of MHC class II on granulomainfiltrating macrophages (Fig. 7B). Taken together, these data sug-gest that although IFN-� secretion by granuloma-infiltrating 5CC7T cells in HEL-flu-infected mice is decreased, activated 3A9 Tcells are able to provide enough IFN-� to up-regulate MHC classII expression on macrophages in granulomas. These finding sug-gest that T cells induced by different infectious agents can collab-orate in activation of macrophage to express anti-mycobacterialfunctions.
Effect of HEL-flu infection on control of bacteria and granulomaformation
Continuous activity of CD4� T cells is necessary for maintaininggranuloma function and preventing reactivation, expansion, anddissemination of bacteria (14). We tested whether infection withinfluenza and the induced CD4� T cell response would affect the
FIGURE 7. Influenza-specific 3A9 T cells secrete IFN-� and increasemacrophage activation in BCG-induced liver granulomas. A, Spleen andgranuloma cells were cultured for 6 h with anti-CD3 and stained for in-tracellular IFN-�. Dot plots represent V�3 surface staining and intracellu-lar IFN-� staining on CD4� gated lymphocytes. Number in each cornerrepresents the percentage of gated cells in the indicated quadrant. V�3-negative events shown in top left of each quadrant represent IFN-� pro-duction by 3A9 T cells as confirmed by V�8 staining (data not shown). B,Spleen and granuloma cells were stained with Mac-1 and anti-I-Ak Ab andanalyzed by flow cytometry. The number in each quadrant represents themean fluorescence intensity on Mac-1� cells. Data are representative ofthree experiments each having three mice per group.
8461The Journal of Immunology
by guest on June 16, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
effectiveness of granulomatous immune responses. It has been re-ported that in wt mice, infection with influenza promoted dissem-ination of mycobacterium from liver to lung (15) and suppressionof delayed-type hypersensitivity responses (16). Fig. 8A showsrepresentative micrographs of granulomas from 5CC7 RAG�/�
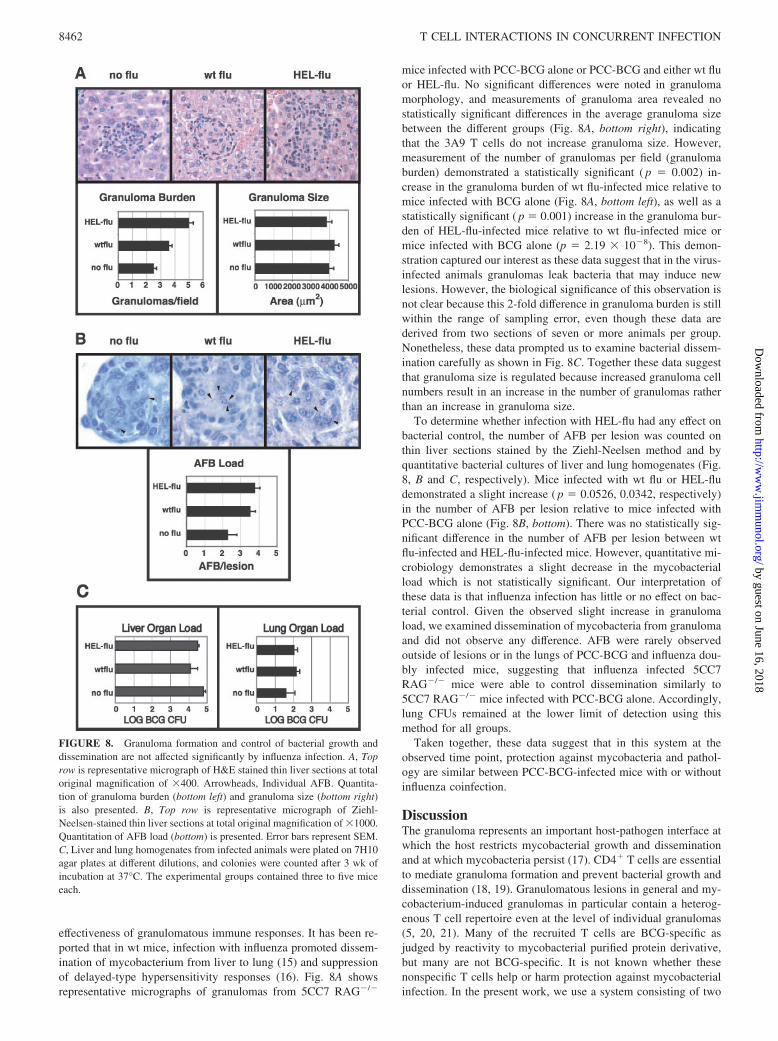
mice infected with PCC-BCG alone or PCC-BCG and either wt fluor HEL-flu. No significant differences were noted in granulomamorphology, and measurements of granuloma area revealed nostatistically significant differences in the average granuloma sizebetween the different groups (Fig. 8A, bottom right), indicatingthat the 3A9 T cells do not increase granuloma size. However,measurement of the number of granulomas per field (granulomaburden) demonstrated a statistically significant ( p � 0.002) in-crease in the granuloma burden of wt flu-infected mice relative tomice infected with BCG alone (Fig. 8A, bottom left), as well as astatistically significant ( p � 0.001) increase in the granuloma bur-den of HEL-flu-infected mice relative to wt flu-infected mice ormice infected with BCG alone (p � 2.19 � 10�8). This demon-stration captured our interest as these data suggest that in the virus-infected animals granulomas leak bacteria that may induce newlesions. However, the biological significance of this observation isnot clear because this 2-fold difference in granuloma burden is stillwithin the range of sampling error, even though these data arederived from two sections of seven or more animals per group.Nonetheless, these data prompted us to examine bacterial dissem-ination carefully as shown in Fig. 8C. Together these data suggestthat granuloma size is regulated because increased granuloma cellnumbers result in an increase in the number of granulomas ratherthan an increase in granuloma size.
To determine whether infection with HEL-flu had any effect onbacterial control, the number of AFB per lesion was counted onthin liver sections stained by the Ziehl-Neelsen method and byquantitative bacterial cultures of liver and lung homogenates (Fig.8, B and C, respectively). Mice infected with wt flu or HEL-fludemonstrated a slight increase ( p � 0.0526, 0.0342, respectively)in the number of AFB per lesion relative to mice infected withPCC-BCG alone (Fig. 8B, bottom). There was no statistically sig-nificant difference in the number of AFB per lesion between wtflu-infected and HEL-flu-infected mice. However, quantitative mi-crobiology demonstrates a slight decrease in the mycobacterialload which is not statistically significant. Our interpretation ofthese data is that influenza infection has little or no effect on bac-terial control. Given the observed slight increase in granulomaload, we examined dissemination of mycobacteria from granulomaand did not observe any difference. AFB were rarely observedoutside of lesions or in the lungs of PCC-BCG and influenza dou-bly infected mice, suggesting that influenza infected 5CC7RAG�/� mice were able to control dissemination similarly to5CC7 RAG�/� mice infected with PCC-BCG alone. Accordingly,lung CFUs remained at the lower limit of detection using thismethod for all groups.
Taken together, these data suggest that in this system at theobserved time point, protection against mycobacteria and pathol-ogy are similar between PCC-BCG-infected mice with or withoutinfluenza coinfection.
DiscussionThe granuloma represents an important host-pathogen interface atwhich the host restricts mycobacterial growth and disseminationand at which mycobacteria persist (17). CD4� T cells are essentialto mediate granuloma formation and prevent bacterial growth anddissemination (18, 19). Granulomatous lesions in general and my-cobacterium-induced granulomas in particular contain a heterog-enous T cell repertoire even at the level of individual granulomas(5, 20, 21). Many of the recruited T cells are BCG-specific asjudged by reactivity to mycobacterial purified protein derivative,but many are not BCG-specific. It is not known whether thesenonspecific T cells help or harm protection against mycobacterialinfection. In the present work, we use a system consisting of two
FIGURE 8. Granuloma formation and control of bacterial growth anddissemination are not affected significantly by influenza infection. A, Toprow is representative micrograph of H&E stained thin liver sections at totaloriginal magnification of �400. Arrowheads, Individual AFB. Quantita-tion of granuloma burden (bottom left) and granuloma size (bottom right)is also presented. B, Top row is representative micrograph of Ziehl-Neelsen-stained thin liver sections at total original magnification of �1000.Quantitation of AFB load (bottom) is presented. Error bars represent SEM.C, Liver and lung homogenates from infected animals were plated on 7H10agar plates at different dilutions, and colonies were counted after 3 wk ofincubation at 37°C. The experimental groups contained three to five miceeach.
8462 T CELL INTERACTIONS IN CONCURRENT INFECTION
by guest on June 16, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

monoclonal T cell populations specific for two different and ana-tomically separated infections, influenza in the lung, and BCG inthe liver, to ask three questions. First, how will the two T cellsdistribute between the two inflammatory sites when both T cellsare specific for a local Ag in one inflammatory site but not in theother? Second, will the T cell response to influenza alter the T cellresponse to BCG? In other words, can two concurrent T cell re-sponses induced at different anatomical sites interfere with eachother? Finally, will influenza infection diminish protective granu-loma formation and allow for reactivation of latent BCG infection?Although the dual monoclonal T cell system has inherent limita-tions, it allows easy identification of BCG-specific and BCG-non-specific T cells and may reveal properties of T cells interactionsthat might not be apparent in the context of the normal repertoire.
The major findings are as follows: T cells activated by HEL-fluinfection have access to both influenza- and BCG-induced inflam-matory sites, but T cells specific for local Ag dominate each site.This differential distribution does not appear to result from differ-ential cell death. Next, although most PCC-BCG-specific 5CC7 Tcells have a resting phenotype in chronic infection, infection withwt flu increases the proportion of LFA-1high and IFN-� secreting5CC7 T cells in the granuloma. Activation of 3A9 T cells by re-combinant influenza slightly decreases the proportion of LFA-1high
and IFN-�-secreting 5CC7 cells. Finally, despite infection withinfluenza, bacterial numbers and granuloma structure remain in therange of adequate control. These data demonstrate that secondaryinfections can modulate the function of recombinant BCG-targetedT cells and that activated virus-specific CD4� T cells can homeinto granulomatous lesions.
Access of T cells to inflammatory sites
Activated T cells have access to inflammatory sites regardlessof specificity for local Ag (22–24) and our results confirm thisfinding. This underlines the importance of understanding the in-teraction between specific and nonspecific T cells. Remarkably,although the number of activated PCC-BCG-specific and HEL-flu-specific T cells was comparable in the systemic compartment, theratio of the two T cells differed between the liver and lung inflam-matory sites with a preferential accumulation of local Ag-specificT cells. This dominance of local Ag-specific T cells can result fromdifferential entry of T cells into inflammatory sites or differentialsurvival, proliferation, and retention at these sites.
Differential entry of T cells to inflammatory sites is mediated byan array of chemokines and adhesion molecules. Lung homing Tcells express a unique pattern of adhesion molecules and chemo-kine receptors (25), but liver homing chemokines and adhesionmolecules have not yet been identified. In addition, T cells primedby dendritic cells in Peyer’s patches and mesenteric lymph nodescan imprint T cells to home to the intestine by expression of �4�7
integrin (26, 27). Thus, activation of HEL-specific T cells in me-diastinal lymph nodes may bias their migration back to the lung,whereas priming of PCC-specific T cells in liver draining lymphnodes may bias their return to liver granulomas. Alternatively, theinfluenza-induced inflammatory site may express a different set ofchemokines than the BCG-induced granuloma. Several systematicstudies have examined chemokines produced by purified proteinderivative bead-induced granulomas (28, 29), in vivo and in vitroM. tuberculosis infection (30–33), and influenza infection (34) andthose studies reveal some differences. In summary, it is possiblethat differential expression of T cell molecules required for traf-ficking, differential chemokine gradients or differential sites ofpriming may bias T cell accumulation in inflammatory sites in anAg-independent manner.
At the same time, the presence of Ag probably plays a centralrole in accumulation by its known capacity to enhance survival,proliferation, and retention of local Ag-specific T cells. Preferen-tial accumulation of OVA-specific T cells at an OVA-containinginflammatory site was not associated with proliferation at the siteas measured by short-term BrdU labeling (35). Instead, it was sug-gested that preferential accumulation is due to selective retentionat the site mediated by CD62E. Our data also demonstrate an in-crease in local Ag-specific T cells in both the influenza and BCGinflammatory sites. In the same vein, cycling cells are rarely ob-served in granuloma-infiltrating cells by analysis of forward scatterscans, propidium iodide staining, or staining for proliferating cellnuclear Ag, which is a marker of cells in cycle (data not shown).The macrophage dominant nature of granulomas makes detec-tion of apoptosis difficult because apoptotic bodies are rapidlyphagocytosed. Nevertheless, in this study, we show that few5CC7 or 3A9 T cells are activated caspase-3- or TUNEL-pos-itive. A somewhat higher proportion of granuloma-infiltrating3A9 T cells in HEL-flu-infected mice are TUNEL-positive, butthese represent a small fraction of the total granuloma-infiltrat-ing 3A9 T cells. This finding suggests that enhanced survival isnot a likely mechanism for preferential accumulation of 5CC7T cells in the granuloma. This simplified system gives us aunique model to analyze the relative involvement of Ag-depen-dent and Ag-independent mechanisms in accumulation of Tcells in inflammatory sites.
Interactions between immune responses to concurrent infection
Concurrent infections are likely to occur in chronic infections suchas tuberculosis. However, little is known about how immune re-sponses to different pathogens interact. We report that infectionwith influenza in the absence of a T cell response appears to in-crease LFA-1 expression of granuloma-infiltrating and BAL 5CC7T cells. This effect seems to correlate with an increase in theLFA-1 expression of systemic (splenic) 5CC7 T cells. This “by-stander activation” could occur through several mechanisms. Lessbacterial control would increase PCC Ag in granulomas and en-hance T cell activation. However, this option was not observed.Alternatively, influenza might possess or induce expression ofepitopes that are cross-reactive with 5CC7 T cells. This possibilityalso seems unlikely given that no sequences matching the PCCepitope were found using a BLAST search. In addition, the acti-vation observed in the spleen of 5CC7 mice in response to wt fluinfection is quite minimal when compared with activation of 3A9T cells in response to HEL-flu infection (compare Fig. 2A, leftmiddle histogram with Fig. 2B, left bottom histogram). Thus, pu-tative cross-reactivity is likely to be weak. A third possibility isthat influenza-induced cytokines may promote “true” bystanderactivation of influenza-nonspecific cells, which has been suggestedpreviously (36). Influenza infections are known to induce type IIFN, TNF, and IL-6 among other cytokines (37), which can induceproduction of IFN-� by human peripheral blood T cells in theabsence of Ag (38). Type I IFN induces IL-15 secretion and by-stander activation of CD8� as well as CD4� T cells (39, 40). Inaddition, a combination of IL-2, IL-6, and TNF has been shown toinduce T cell activation in vitro (41). Recent development of MHCtetramer technology has demonstrated that �40% of CD8� T cellsin primary influenza are specific for influenza, suggesting that by-stander activation is minimal in the context of a normal T cellrepertoire (42). It is clear that the effects of influenza infection onthe host immune response are complex.
8463The Journal of Immunology
by guest on June 16, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

We also report that the arrival of HEL-flu-activated 3A9 T cellsto both the influenza inflammatory site and the BCG-induced gran-uloma decreases the influenza-mediated activation of PCC-BCG-specific T cells in granuloma and lung, and decreased LFA-1 ex-pression correlates with decreased secretion of IFN-�. Thisdecreased activation may reflect competition between the two dif-ferent T cell populations for resources. T cells have been shown tocompete with each other for peptide-MHC complexes at the sur-face of APC (43–46). However, this competition is greater be-tween T cells specific for the same peptide-MHC complex than forT cells specific for different peptide-MHC complexes (47), and3A9 and 5CC7 T cells are restricted by different class II Ags. Somehave also suggested that T cells may compete for cytokines such asIL-2 (48). Recent work has also demonstrated that recently acti-vated T cells can inhibit the activation of naive T cells in vitro (49).Further work will be needed to determine the mechanism by whichthis response occurs, but it is clear that ongoing T cell responsesinterfere with each other, and that nonspecific activated T cells canalter specific T cell responses in the granuloma.
In previous work, using a similar two T cell network, we ob-served that recombinant BCG-targeted T cells were activated earlyduring infection, but this activation was down-regulated duringchronic infection. In this system, we found that activated nonspe-cific T cells competed with activated recombinant BCG-targeted Tcells during acute infection. In the present work, infection withinfluenza causes bystander activation of recombinant BCG-tar-geted T cells at the chronic stage of BCG infection. Activation ofHEL-specific T cells by HEL-flu infection competes with activatedrecombinant BCG-targeted T cells similar to acute infection in ourprevious work. Taken together, these results raise the possibilitythat activated nonspecific T cells compete with activated recom-binant BCG-targeted T cells.
Effect of secondary infection on control of BCG infection
Our results suggest complex effects of secondary infection on theT cell response to BCG infection. Influenza infection is known tocause immunosuppression (50) and in particular to suppress tuber-culin delayed-type hypersensitivity (16). Influenza infection hasalso been shown to interfere with immunity to Listeria, anotherintracellular pathogen of macrophages (51). Infection with influ-enza has been reported to allow dissemination of i.p. infected M.tuberculosis from liver granulomas to the lung (15). In the presentwork, we observed little effect on bacterial control or pathology.Granuloma formation appeared normal in these mice and no bac-teria could be found that had disseminated outside of lesions or tothe lungs of these mice. It should be noted that a prior work fol-lowed M. tuberculosis infection for 3 wk following influenza in-fection, whereas our study followed the mice for 1 wk. Effects ondissemination might be more apparent at later time points. Ourresults are consistent with the lack of any strong association re-ported in the clinical literature between influenza infection andreactivation of tuberculosis.
Interestingly, we report that 3A9 T cells present in granulomasof mice infected with HEL-flu are able to produce IFN-� and up-regulate expression of class II on macrophage. This finding sug-gests that activated T cells may participate in qualitatively similarimmune responses and cooperate with each other in providing pro-tection against infectious agents. Infection of Mycobacteriumavium-infected mice with the Th2-inducing pathogen Schistosomamansoni was able to skew the normally Th1 type M. avium gran-ulomas to Th2 behavior (52). However, infection with the Th2biasing helminth Nippostrongylus brasiliensis did not interferewith control of pulmonary BCG infection despite reduced Th1cytokine secretion by T cells in the lungs of these mice (53). This
is surprising given that IFN-� and other Th1 cytokines are knownto be critical in maintaining control of mycobacterial infections. Itwould be interesting to see whether we would observe similarresults in this two T cell system, coinfecting with BCG and aTh2-inducing infectious agent.
In summary, we show that T cells responding to two separateinfectious agents in anatomically distinct sites have access to bothinflammatory sites, but dominate where their Ag is present. Inaddition, nonspecific T cells can interfere with local Ag-specific Tcells in the granuloma. Finally, nonspecific T cells can contributeto protection. A two T cell system can focus attention on basicprinciples that may facilitate understanding of T cell interactions inmultiple infections.
AcknowledgmentsWe thank Stefan Hamm for assistance in production of WSN and WSN-HEL virus stocks, Khen Macvilay for assistance in granuloma prepara-tions, and Shin Il Kim for technical assistance. We also thank M. Suresh forcritical reading of the manuscript.
DisclosuresThe authors have no financial conflict of interest.
References1. Saunders, B. M., A. A. Frank, I. M. Orme, and A. M. Cooper. 2002. CD4 is
required for the development of a protective granulomatous response to pulmo-nary tuberculosis. Cell. Immunol. 216: 65–72.
2. Ladel, C. H., J. Hess, S. Daugelat, P. Mombaerts, S. Tonegawa, and S. H.Kaufmann. 1995. Contribution of �/� and �/� T lymphocytes to immunityagainst Mycobacterium bovis bacillus Calmette Guerin: studies with T cell re-ceptor-deficient mutant mice. Eur. J. Immunol. 25: 838–846.
3. Ladel, C. H., I. E. Flesch, J. Arnoldi, and S. H. Kaufmann. 1994. Studies withMHC-deficient knock-out mice reveal impact of both MHC I- and MHC II-dependent T cell responses on Listeria monocytogenes infection. J. Immunol.153: 3116–3122.
4. Feng, C. G., and W. J. Britton. 2000. CD4� and CD8� T cells mediate adoptiveimmunity to aerosol infection of Mycobacterium bovis bacillus Calmette-Guerin.J. Infect. Dis. 181: 1846–1849.
5. Hogan, L. H., K. MacVilay, B. Barger, D. Co, I. Malkovska, G. Fennelly, andM. Sandor. 2001. Mycobacterium bovis strain bacillus Calmette-Guerin-inducedliver granulomas contain a diverse TCR repertoire, but a monoclonal T cell pop-ulation is sufficient for protective granuloma formation. J. Immunol. 166:6367–6375.
6. Belz, G. T., D. Wodarz, G. Diaz, M. A. Nowak, and P. C. Doherty. 2002. Com-promised influenza virus-specific CD8�-T-cell memory in CD4�-T-cell-deficientmice. J. Virol. 76: 12388–12393.
7. Doherty, P. C., D. J. Topham, R. A. Tripp, R. D. Cardin, J. W. Brooks, andP. G. Stevenson. 1997. Effector CD4� and CD8� T-cell mechanisms in thecontrol of respiratory virus infections. Immunol. Rev. 159: 105–117.
8. Topham, D. J., and P. C. Doherty. 1998. Clearance of an influenza A virus byCD4� T cells is inefficient in the absence of B cells. J. Virol. 72: 882–885.
9. Seder, R. A., W. E. Paul, M. M. Davis, and B. F. de St. Groth. 1992. The presenceof interleukin-4 during in vitro priming determines the lymphokine-producingpotential of CD4� T cells from T cell receptor transgenic mice. J. Exp. Med. 176:1091–1098.
10. Ho, W. Y., M. P. Cooke, C. C. Goodnow, and M. M. Davis. 1994. Resting andanergic B cells are defective in CD28-dependent costimulation of naive CD4� Tcells. J. Exp. Med. 179: 1539–1549.
11. Walker, W. S., M. R. Castrucci, M. Y. Sangster, R. T. Carson, and Y. Kawaoka.1997. HEL-Flu: an influenza virus containing the hen egg lysozyme epitope rec-ognized by CD4� T cells from mice transgenic for an �� TCR. J. Immunol. 159:2563–2566.
12. van Elden, L. J., M. Nijhuis, P. Schipper, R. Schuurman, and A. M. van Loon.2001. Simultaneous detection of influenza viruses A and B using real-time quan-titative PCR. J. Clin. Microbiol. 39: 196–200.
13. Garcıa-Sastre, A., R. K. Durbin, H. Zheng, P. Palese, R. Gertner, D. E. Levy, andJ. E. Durbin. 1998. The role of interferon in influenza virus tissue tropism. J. Vi-rol. 72: 8550–8558.
14. Scanga, C. A., V. P. Mohan, K. Yu, H. Joseph, K. Tanaka, J. Chan, and J. L.Flynn. 2000. Depletion of CD4� T cells causes reactivation of murine persistenttuberculosis despite continued expression of interferon � and nitric oxide syn-thase 2. J. Exp. Med. 192: 347–358.
15. Volkert, M., C. Pierce, F. L. J. Horsfall, and R. J. Dubos. 1947. The enhancingeffect of concurrent infection with pneumotropic viruses on pulmonary tubercu-losis in mice. J. Exp. Med. 86: 202–213.
16. Massanari, R. M. 1979. Suppression of tuberculin hypersensitivity during influ-enza infection in mice. Infect. Immun. 24: 501–507.
17. Cosma, C. L., D. R. Sherman, and L. Ramakrishnan. 2003. The secret lives of thepathogenic mycobacteria. Annu. Rev. Microbiol. 57: 641–676.
8464 T CELL INTERACTIONS IN CONCURRENT INFECTION
by guest on June 16, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

18. Flynn, J. L., and J. Chan. 2001. Immunology of tuberculosis. Annu. Rev. Immu-nol. 19: 93–129.
19. Sandor, M., J. V. Weinstock, and T. A. Wynn. 2003. Granulomas in schistosomeand mycobacterial infections: a model of local immune responses. Trends Im-munol. 24: 44–52.
20. Hogan, L. H., M. Wang, M. Suresh, D. O. Co, J. V. Weinstock, and M. Sandor.2002. CD4� TCR repertoire heterogeneity in Schistosoma mansoni-inducedgranulomas. J. Immunol. 169: 6386–6393.
21. Mempel, M., P. Musette, B. Flageul, C. Schnopp, R. Remling, G. Gachelin,P. Kourilsky, J. Ring, and D. Abeck. 2002. T-cell receptor repertoire and cytokinepattern in granuloma annulare: defining a particular type of cutaneous granulo-matous inflammation. J. Invest. Dermatol. 118: 957–966.
22. Topham, D. J., M. R. Castrucci, F. S. Wingo, G. T. Belz, and P. C. Doherty. 2001.The role of antigen in the localization of naive, acutely activated, and memoryCD8� T cells to the lung during influenza pneumonia. J. Immunol. 167:6983–6990.
23. Stephens, R., D. A. Randolph, G. Huang, M. J. Holtzman, and D. D. Chaplin.2002. Antigen-nonspecific recruitment of Th2 cells to the lung as a mechanismfor viral infection-induced allergic asthma. J. Immunol. 169: 5458–5467.
24. Ely, K. H., L. S. Cauley, A. D. Roberts, J. W. Brennan, T. Cookenham, and D. L.Woodland. 2003. Nonspecific recruitment of memory CD8� T cells to the lungairways during respiratory virus infections. J. Immunol. 170: 1423–1429.
25. Campbell, J. J., C. E. Brightling, F. A. Symon, S. Qin, K. E. Murphy, M. Hodge,D. P. Andrew, L. Wu, E. C. Butcher, and A. J. Wardlaw. 2001. Expression ofchemokine receptors by lung T cells from normal and asthmatic subjects. J. Im-munol. 166: 2842–2848.
26. Mora, J. R., M. R. Bono, N. Manjunath, W. Weninger, L. L. Cavanagh, M.Rosemblatt, and U. H. Von Andrian. 2003. Selective imprinting of gut-homing Tcells by Peyer’s patch dendritic cells. Nature 424: 88–93.
27. Stagg, A. J., M. A. Kamm, and S. C. Knight. 2002. Intestinal dendritic cellsincrease T cell expression of �4�7 integrin. Eur. J. Immunol. 32: 1445–1454.
28. Chiu, B. C., C. M. Freeman, V. R. Stolberg, E. Komuniecki, P. M. Lincoln,S. L. Kunkel, and S. W. Chensue. 2003. Cytokine-chemokine networks in ex-perimental mycobacterial and schistosomal pulmonary granuloma formation.Am. J. Respir. Cell Mol. Biol. 29: 106–116.
29. Chiu, B. C., X. Z. Shang, V. R. Stolberg, E. Komuniecki, and S. W. Chensue.2002. Population analysis of CD4� T cell chemokine receptor transcript expres-sion during in vivo type-1 (mycobacterial) and type-2 (schistosomal) immuneresponses. J. Leukocyte Biol. 72: 363–372.
30. Rhoades, E. R., A. M. Cooper, and I. M. Orme. 1995. Chemokine response inmice infected with Mycobacterium tuberculosis. Infect. Immun. 63: 3871–3877.
31. Sadek, M. I., E. Sada, Z. Toossi, S. K. Schwander, and E. A. Rich. 1998. Che-mokines induced by infection of mononuclear phagocytes with mycobacteria andpresent in lung alveoli during active pulmonary tuberculosis. Am. J. Respir. CellMol. Biol. 19: 513–521.
32. Saukkonen, J. J., B. Bazydlo, M. Thomas, R. M. Strieter, J. Keane, andH. Kornfeld. 2002. Beta-chemokines are induced by Mycobacterium tuberculosisand inhibit its growth. Infect. Immun. 70: 1684–1693.
33. Algood, H. M., J. Chan, and J. L. Flynn. 2003. Chemokines and tuberculosis.Cytokine Growth Factor Rev. 14: 467–477.
34. Wareing, M. D., A. B. Lyon, B. Lu, C. Gerard, and S. R. Sarawar. 2004. Che-mokine expression during the development and resolution of a pulmonary leu-kocyte response to influenza A virus infection in mice. J. Leukocyte Biol. 76:886–895.
35. Reinhardt, R. L., D. C. Bullard, C. T. Weaver, and M. K. Jenkins. 2003. Pref-erential accumulation of antigen-specific effector CD4 T cells at an antigen in-
jection site involves CD62E-dependent migration but not local proliferation.J. Exp. Med. 197: 751–762.
36. Tripp, R. A., S. Hou, A. McMickle, J. Houston, and P. C. Doherty. 1995. Re-cruitment and proliferation of CD8� T cells in respiratory virus infections. J. Im-munol. 154: 6013–6021.
37. Van Reeth, K. 2000. Cytokines in the pathogenesis of influenza. Vet. Microbiol.74: 109–116.
38. Sareneva, T., S. Matikainen, M. Kurimoto, and I. Julkunen. 1998. Influenza Avirus-induced IFN-�/� and IL-18 synergistically enhance IFN-� gene expressionin human T cells. J. Immunol. 160: 6032–6038.
39. Tough, D. F., S. Sun, and J. Sprent. 1997. T cell stimulation in vivo by lipo-polysaccharide (LPS). J. Exp. Med. 185: 2089–2094.
40. Sun, S., X. Zhang, D. F. Tough, and J. Sprent. 1998. Type I interferon-mediatedstimulation of T cells by CpG DNA. J. Exp. Med. 188: 2335–2342.
41. Unutmaz, D., P. Pileri, and S. Abrignani. 1994. Antigen-independent activationof naive and memory resting T cells by a cytokine combination. J. Exp. Med. 180:1159–1164.
42. Belz, G. T., W. Xie, and P. C. Doherty. 2001. Diversity of epitope and cytokineprofiles for primary and secondary influenza A virus-specific CD8� T cell re-sponses. J. Immunol. 166: 4627–4633.
43. Kedl, R. M., W. A. Rees, D. A. Hildeman, B. Schaefer, T. Mitchell, J. Kappler,and P. Marrack. 2000. T cells compete for access to antigen-bearing antigen-presenting cells. J. Exp. Med. 192: 1105–1114.
44. Min, B., G. Foucras, M. Meier-Schellersheim, and W. E. Paul. 2004. Spontane-ous proliferation, a response of naive CD4 T cells determined by the diversity ofthe memory cell repertoire. Proc. Natl. Acad. Sci. USA 101: 3874–3879.
45. Ge, Q., A. Bai, B. Jones, H. N. Eisen, and J. Chen. 2004. Competition for self-peptide-MHC complexes and cytokines between naive and memory CD8� Tcells expressing the same or different T cell receptors. Proc. Natl. Acad. Sci. USA101: 3041–3046.
46. Hayball, J. D., B. W. Robinson, and R. A. Lake. 2004. CD4� T cells cross-compete for MHC class II-restricted peptide antigen complexes on the surface ofantigen presenting cells. Immunol. Cell Biol. 82: 103–111.
47. Kedl, R. M., B. C. Schaefer, J. W. Kappler, and P. Marrack. 2002. T cells down-modulate peptide-MHC complexes on APCs in vivo. Nat. Immunol. 3: 27–32.
48. Khetan, S., K. B. Sainis, S. Rath, and R. Kamat. 1993. Murine CD8� T sup-pressors against mycobacterial 65-kDa antigen compete for IL-2 and show lackof major histocompatibility complex-imposed restriction specificity in antigenrecognition. Eur. J. Immunol. 23: 2440–2447.
49. Duthoit, C. T., P. Nguyen, and T. L. Geiger. 2004. Antigen nonspecific suppres-sion of T cell responses by activated stimulation-refractory CD4� T cells. J. Im-munol. 172: 2238–2246.
50. Kavetsky, R. E., Z. D. Savtsova, V. I. Struk, L. V. Yakimenko, and Y. A.Umansky. 1977. Effect of influenza virus on the immune responsiveness of an-imals. Acta Virol. 21: 109–113.
51. Gardner, I. D. 1981. Suppression of antibacterial immunity by infection withinfluenza virus. J. Infect. Dis. 144: 225–231.
52. Sacco, R., M. Hagen, M. Sandor, J. V. Weinstock, and R. G. Lynch. 2002.Established TH1 granulomatous responses induced by active Mycobacteriumavium infection switch to TH2 following challenge with Schistosoma mansoni.Clin. Immunol. 104: 274–281.
53. Erb, K. J., C. Trujillo, M. Fugate, and H. Moll. 2002. Infection with the helminthNippostrongylus brasiliensis does not interfere with efficient elimination of My-cobacterium bovis BCG from the lungs of mice. Clin. Diagn. Lab. Immunol. 9:727–730.
8465The Journal of Immunology
by guest on June 16, 2018http://w
ww
.jimm
unol.org/D
ownloaded from