Embed Size (px)
Citation preview

Biochimica et Biophysica Acta 1777 (2008) 826–833
Contents lists available at ScienceDirect
Biochimica et Biophysica Acta
j ourna l homepage: www.e lsev ie r.com/ locate /bbab io
Interrelated influence of superoxides and free fatty acids over mitochondrialuncoupling in skeletal muscle
Assunta Lombardi a, Paola Grasso a, Maria Moreno b, Pieter de Lange c,Elena Silvestri b, Antonia Lanni c, Fernando Goglia b,⁎a Dipartimento delle Scienze Biologiche, Sezione Fisiologia, Università degli Studi di Napoli “Federico II”, Via Mezzocannone 8, 80134 Napoli, Italyb Dipartimento di Scienze Biologiche ed Ambientali, Università degli Studi del Sannio, Via Port'Arsa 11, 82100 Benevento, Italyc Dipartimento di Scienze della Vita, Seconda Università di Napoli, Via Vivaldi 43, 81100 Caserta, Italy
⁎ Corresponding author. Tel.: +39 0824 305138; fax: +E-mail address: [email protected] (F. Goglia).
0005-2728/$ – see front matter © 2008 Elsevier B.V. Adoi:10.1016/j.bbabio.2008.04.019
A B S T R A C T
A R T I C L E I N F OArticle history:
Mitochondrial uncoupling p Received 15 November 2007Received in revised form 21 March 2008Accepted 14 April 2008Available online 18 April 2008Keywords:MitochondriaUncoupling protein 3Proton leak
rotein 3 (UCP3)-mediated uncoupling has been postulated to depend on severalfactors, including superoxides, free fatty acids (FFAs), and fatty acid hydroperoxides and/or their derivatives.We investigated whether there is an interrelation between endogenous mitochondrial superoxides and fattyacids in inducing skeletal muscle mitochondrial uncoupling, and we speculated on the possible involvementof UCP3 in this process. In the absence of FFAs, no differences in proton-leak kinetic were detected betweensuccinate-energized mitochondria respiring in the absence or presence of rotenone, despite a large differencein complex I superoxide production. The addition of either arachidic acid or arachidonic acid induced anincrease in proton-leak kinetic, with arachidonic acid having the more marked effect. The uncoupling effectof arachidic acid was independent of the presence of GDP, rotenone and vitamin E, while that of arachidonicacid was dependent on these factors. These data demonstrate that FFA and O2− play interrelated roles ininducing mitochondrial uncoupling, and we hypothesize that a likely formation of mitochondrial fatty acidhydroperoxides is a key event in the arachidonic acid-induced GDP-dependent inhibition of mitochondrialuncoupling.
© 2008 Elsevier B.V. All rights reserved.
1. Introduction
The flux of substrate through the tricarboxylic acid cycle and theflux of electrons through the respiratory chain are coupled to ATPsynthesis via the formation of a proton gradient across the mito-chondrial inner membrane. ATP is generated from ADP and Pi as pro-tons pass down their electrochemical gradient through the action ofATPsynthase. However, in some conditions a dissipation of theelectrochemical gradient may take place [in the absence of ADP orin the presence of inhibitors of the proton flux through the Fo portionof ATPsynthase, for instance, the protons pumped out of the matrixpass back into the mitochondria through other proton-conductanceroutes (proton leak)].The proportion of respiration that is used to drivethe energy-dissipating futile proton cycle across the mitochondrialinnermembrane is high [1,2], and up to 20% of the basal metabolic ratemay be used to drive this process [3].
Although the proton leak has been related to the phospholipidcomposition of the inner membrane [4,5], studies on liposomes con-stituted by inner membrane phospholipids have revealed that theyare several-fold less proton-permeable than native mitochondria [6].Moreover, the proton conductance is the same in such liposomes
39 0824 23013.
ll rights reserved.
prepared from mitochondria with very different endogenous protonconductances [7]. Thus, simple diffusion through bulk regions of themembrane bilayer does not explain the proton conductance, and pro-tein components present in the mitochondrial inner membrane mustbe involved. Besides the basal proton conductance, mitochondria alsohave an inducible proton conductance that requires the presence ofcofactors for its activation.
Among the proteins involved in this inducible proton conductanceare adenine nucleotide translocase (ANT) [8] and the uncoupling pro-teins (UCPs) [9,10].
In brown adipose tissue, uncoupling protein 1 (UCP1) catalyzes theinducible proton conductance, generating physiologically importantheat production in response to physiological stimuli such as cold andthe animal's diet. Within the last decade, several genes have beendiscovered that encode proteins closely related to UCP1, includinguncoupling protein 2 (UCP2) and uncoupling protein 3 (UCP3). Despitethe ubiquitous presence of UCP2 mRNA, UCP2 protein has been de-tected in only a few tissues [10], and a causal role in pancreatic betacell dysfunction and type 2 diabetes has been attributed to it [10].UCP3 – because of its preferential expression in skeletal muscle (ametabolically very active tissue endowed with a high mitochondrialactivity) – has attracted the interest of several researchers. Manystudies have been carried out to try to clarify the role played by thisprotein, but its physiological function is still in doubt. It seems likely to

827A. Lombardi et al. / Biochimica et Biophysica Acta 1777 (2008) 826–833
contribute to inducible proton leak [9] as it does not uncouple unless itis first activated. Indeed, growing evidence suggests that the presenceof cofactors is crucial for UCP3-mediated uncoupling [11–13], and thatthe absence of one of them can adversely affect UCP3 uncouplingactivity [13,14]. While studying the changes in UCP3 expression/ac-tivity occurring during the transition hypo-hyperthyroidism, we firstdemonstrated that UCP3-mediated uncoupling can be observed onlyin the presence of free fatty acids (FFAs) [14]. Moreover, it could beargued that an absence of mitochondrial uncoupling, despite anupregulation of UCP3 protein, does not necessarily mean that UCP3 isdevoid of the capacity to uncouple. The above situations might be theresult of changes in the levels/production of one or more putativecofactors. Indeed, a requirement for additional activators has beenindicated by studies on reconstituted systems in isolated mitochon-dria and on animal tissues [11–13], and it seems likely that UCP3 is notfunctional in the absence of activators. However, it is not clear if and inwhat way these cofactors are related to each other.
The identities of the activators for UCP3 remain unknown, butpossible activators include free fatty acids [9], CoQ [11,15,16], super-oxides [12,17,18], AAPH (a carbon-centered radical generator) [19],reactive alkenals such as 4-hydroxy-2-nonenal (HNE) [20], and alkenalanalogs such as cinnamate and retinal [20].
It has been shown that UCP3 is a highly active H+ transporter whenactivated by CoQ [16], and that CoQ, superoxide, and AAPH workindirectly, by generating carbon-centered radicals on the polyunsatu-rated fatty acid chains of the phospholipids located in the innermitochondrial membrane. The idea that the action of CoQ on UCP3-mediated uncoupling occurs via O2− seems to be confirmed by studies inwhich the activation of UCP3 induced by adding CoQ to mitochondriadisappeared when superoxide dismutase was also added [15]. Super-oxides, therefore, seem toplay a pivotal role in activatingUCPs [12,17,18],even though compounds formed downstream of superoxide formationhave also been proposed to be involved in the activation of UCPs. Thesedownstream compounds include hydroperoxy fatty acids [21,22] andlipid peroxidationproducts such asHNE [20,23] and/or 4-hydroxinoneicacid [24].
Since: i) the proton leak contributes significantly to the setting ofthe level of the metabolic rate in animals [2,25] (ii) among all tissuesskeletal muscle mitochondria exhibit the highest proton leak [2], iii) achange in the skeletalmusclemitochondrial proton conductance couldsignificantly affect the energy expenditure of the animal, and iv) acti-vation of proton-leak processes is a potential target for the treatment ofpathologies such as obesity via a modulation of metabolic rate, wethought it both interesting and important to try to gain a deeper insightinto the mechanism by which the skeletal muscle proton leak may beinduced.
In the present study, we therefore investigated the interrelatedinfluences of superoxides and fatty acids over the mitochondriaproton-leak kinetic in skeletal muscle (and we discuss the putativeinvolvement of UCP3 in this process). To this end, we evaluated proton-leak kinetics while modulating both mitochondrial endogenous su-peroxide production and levels of FFAs. In addition, we tested theeffects of rotenone, vitamin E, and GDP on FFA-induced uncoupling.
2. Material and methods
2.1. Animals
Male Wistar rats (250–300 g) (Charles River) were used throughout. They werekept one per cage in a temperature-controlled room at a thermoneutral temperature of28 °C under a 12-h light, 12-h dark cycle. A commercial mash and water were availablead libitum. Rats were anesthetized by means of an i.p. injection of chloral hydrate(40 mg/100 g BW), then killed by decapitation.
2.2. Isolation of mitochondria and evaluation of respiratory parameters
Subsarcolemmal skeletal muscle mitochondria were isolated by differential cen-trifugation at 8000 ×g, as previously described [14]. They were then immediately used
for measurement either of respiratory rate or membrane potential. We chose toperform our experiments on subsarcolemmal mitochondria because the mitochondrialH2O2 release per unit of O2 consumed is reportedly 2-fold higher in subsarcolemmalmitochondria than in intermyofibrillar ones [26].
Mitochondrial oxygen consumptionwas measured polarographically using a Clark-type electrode, themeasurements being carried out in duplicate using succinate (6mM)as substrate. The analyses were performed at 37 °C in a final volume of 0.5 ml ofstandard incubation medium [consisting of 80 mM KCl, 50 mM HEPES (pH 7.2–7.4),1 mM EGTA, 5 mM K2HPO4, 5 mM MgCl2, 4 μM rotenone, and 0.3% bovine serumalbumin (BSA) (w/v)], either in the absence or presence of ADP (300 μM).
The respiratory control ratio (RCR) was calculated as the ratio of respiration mea-sured in the presence of ADP to that measured in its absence.
2.3. Proton-leak kinetic assay
To evaluate proton conductancewe detected the kinetic response of proton leak toa change in membrane potential. Respiration rate andmembrane potential (ΔΨ) weremeasured simultaneously, the latter being detected using a triphenylmethylpho-sphonium (TPMP+)-sensitive electrode. For these measurements, 0.5 mg mitochon-drial proteins was incubated in 1 ml standard incubation medium from whichrotenone was omitted (unless otherwise specified) and to which 1 μg/ml oligomycin,80 ng/ml nigericin, and 0.3% BSAwere added. At the concentration of BSA used, BSA isthe predominant protein, and endogenous FFAs can therefore be considered to bebuffered by the albumin.
First, mitochondria were incubated in the respiratory medium for 2 min to calibratetheTPMP+-sensitive electrode, calibrationbeing achievedbymeansof sequential additionsof up to 2 μM TPMP+. Then, mitochondria were energized using 6 mM succinate, andrespiration was titrated with increasing amounts of malonate (up to 2 mM).
Next, we tested the effect of FFAs on mitochondrial proton-leak kinetics. Since:i) among FFAs, polyunsaturated fatty acids (PUFAs) display the highest sensitivity tooxidative damage, and ii) many studies have shown that free-radical damage and lipidperoxidation each increase as a function of the degree of unsaturation of the fatty acids[27], we decided to evaluate the effect of two different FFAs with the same chain length,but a different saturation index, on mitochondrial uncoupling. These FFAs were ar-achidonic acid and arachidic acid. Arachidonic acid, being a PUFA, easily enters per-oxidative processes. Moreover, Beck et al. [28] reported that among the free fatty acidstested, arachidonic acid had the highest capacity to induce proton conductance inplanar lipid membranes reconstituted with purified recombinant human uncouplingproteins.
To test the effect of the above FFAs on mitochondrial uncoupling, malonate titra-tions of respiration rate were performed in the presence of either arachidonic acid(30 μM) or arachidic acid (30 μM). In the case of these two FFAs, the binding affinityconstant of BSA is known only for arachidonic acid [29], and so we will refer in thisstudy to the nominal amount of each fatty acid added.
To test the involvement of O2− production via electron reverse from complex II tocomplex I on both the mitochondrial basal and FFA-inducible proton leaks, in someexperiments rotenone was present in the respiration medium (at a concentration of4 μM). To test the effect of vitamin E on both the basal and FFA-inducible proton leaks, insome experiments it was present in the respirationmedium at a concentration of 1mM.The effect of GDP on both the basal and FFA-induced proton leaks was tested by addingit to the incubationmedium at a concentration of 0.5mM. Each of the above compoundswas added to the incubation medium before the mitochondria were added.
In some experiments, to exclude the involvement of adenine nucleotide translocase(ANT) in fatty acid-induced uncoupling, carboxyatractiloside (CAT; 10 μM)was added tothe incubation medium. When CAT was present, to test the effect of GDP on fatty acid-induced uncoupling, malonate titrations of respiration rate were performed in thepresence of either arachidonic acid (60 μM) or arachidic acid (60 μM), in each case withor without GDP (0.5 mM).
The concentrations of arachidonic acid used in the basal condition and in thepresence of CAT were chosen after carrying out arachidonic-acid titration of theoligomycin-inhibited respiration rate. From the arachidonic-acid titration data, wechose the concentrations of arachidonic acid that were able to induce the same milduncoupling in the two different experimental conditions.
Baseline correction was obtained by the addition of 0.2 μM FCCP, which inducescomplete release of TPMP+. A TPMP+-binding correction of 0.4/(μl/mg protein) wasapplied, as reported by Rolfe et al. [2].
In all the experimental conditions, EGTA was present in the incubation medium toprevent the opening of transition pores [30].
2.4. Mitochondrial H2O2 release
The rate of mitochondrial H2O2 release was measured at 37 °C the following, using afluorimeter, the linear increase in fluorescence (ex 312 nm and em 420 nm) that occurreddue to the oxidation of homovanillic acid (HVA) by H2O2 in the presence of horseradishperoxidase (HRP) [31]. Reaction conditions were 0.2 mg of mitochondrial protein/ml, 6 U/ml HRP, 0.1mMHVA, 5mMsuccinate, and 75U/ml superoxide dismutase, dissolved in thesame incubation buffer as that used for the oxygen-consumption measurements.
The effects of arachidonic acid, arachidic acid,and other compounds (rotenone,vitamin E, GDP, CAT) on mitochondrial H2O2 release was detected by adding them(individually or in combination) to the incubation medium. Mitochondria were
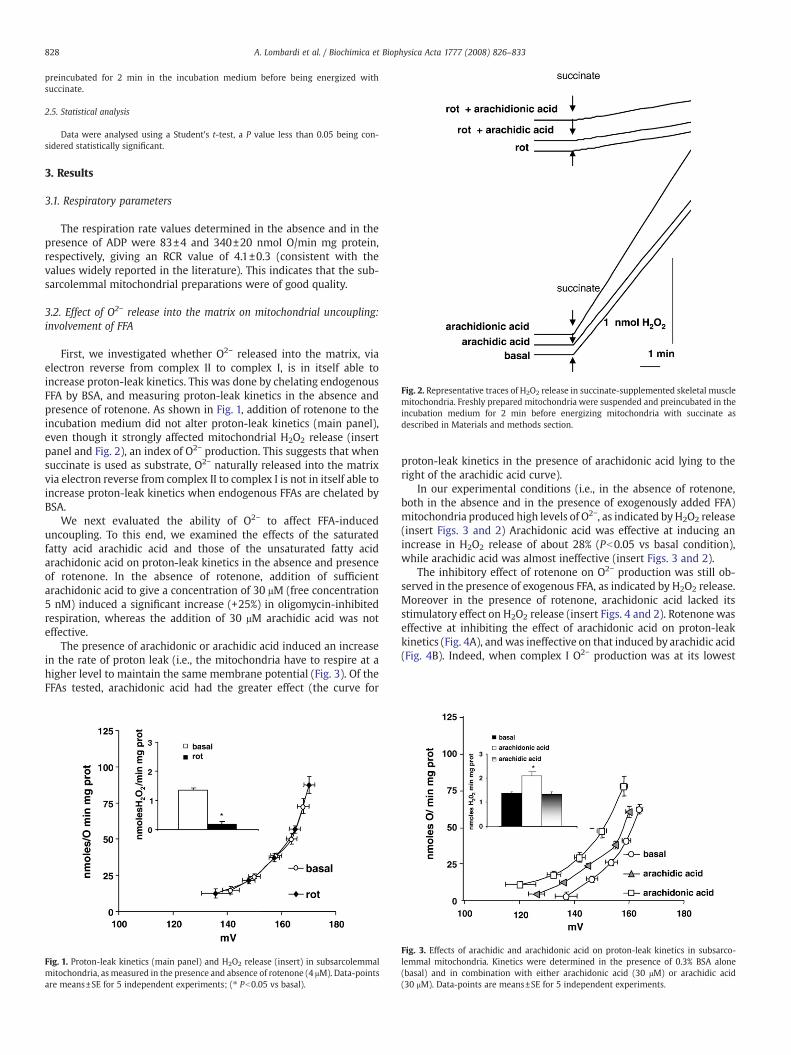
Fig. 2. Representative traces of H2O2 release in succinate-supplemented skeletal musclemitochondria. Freshly prepared mitochondria were suspended and preincubated in theincubation medium for 2 min before energizing mitochondria with succinate asdescribed in Materials and methods section.
828 A. Lombardi et al. / Biochimica et Biophysica Acta 1777 (2008) 826–833
preincubated for 2 min in the incubation medium before being energized withsuccinate.
2.5. Statistical analysis
Data were analysed using a Student's t-test, a P value less than 0.05 being con-sidered statistically significant.
3. Results
3.1. Respiratory parameters
The respiration rate values determined in the absence and in thepresence of ADP were 83±4 and 340±20 nmol O/min mg protein,respectively, giving an RCR value of 4.1±0.3 (consistent with thevalues widely reported in the literature). This indicates that the sub-sarcolemmal mitochondrial preparations were of good quality.
3.2. Effect of O2− release into the matrix on mitochondrial uncoupling:involvement of FFA
First, we investigated whether O2− released into the matrix, viaelectron reverse from complex II to complex I, is in itself able toincrease proton-leak kinetics. This was done by chelating endogenousFFA by BSA, and measuring proton-leak kinetics in the absence andpresence of rotenone. As shown in Fig. 1, addition of rotenone to theincubation medium did not alter proton-leak kinetics (main panel),even though it strongly affected mitochondrial H2O2 release (insertpanel and Fig. 2), an index of O2− production. This suggests that whensuccinate is used as substrate, O2− naturally released into the matrixvia electron reverse from complex II to complex I is not in itself able toincrease proton-leak kinetics when endogenous FFAs are chelated byBSA.
We next evaluated the ability of O2− to affect FFA-induceduncoupling. To this end, we examined the effects of the saturatedfatty acid arachidic acid and those of the unsaturated fatty acidarachidonic acid on proton-leak kinetics in the absence and presenceof rotenone. In the absence of rotenone, addition of sufficientarachidonic acid to give a concentration of 30 μM (free concentration5 nM) induced a significant increase (+25%) in oligomycin-inhibitedrespiration, whereas the addition of 30 μM arachidic acid was noteffective.
The presence of arachidonic or arachidic acid induced an increasein the rate of proton leak (i.e., the mitochondria have to respire at ahigher level to maintain the same membrane potential (Fig. 3). Of theFFAs tested, arachidonic acid had the greater effect (the curve for
Fig. 1. Proton-leak kinetics (main panel) and H2O2 release (insert) in subsarcolemmalmitochondria, as measured in the presence and absence of rotenone (4 μM). Data-pointsare means±SE for 5 independent experiments; (⁎ Pb0.05 vs basal).
proton-leak kinetics in the presence of arachidonic acid lying to theright of the arachidic acid curve).
In our experimental conditions (i.e., in the absence of rotenone,both in the absence and in the presence of exogenously added FFA)mitochondria produced high levels of O2−, as indicated byH2O2 release(insert Figs. 3 and 2) Arachidonic acid was effective at inducing anincrease in H2O2 release of about 28% (Pb0.05 vs basal condition),while arachidic acid was almost ineffective (insert Figs. 3 and 2).
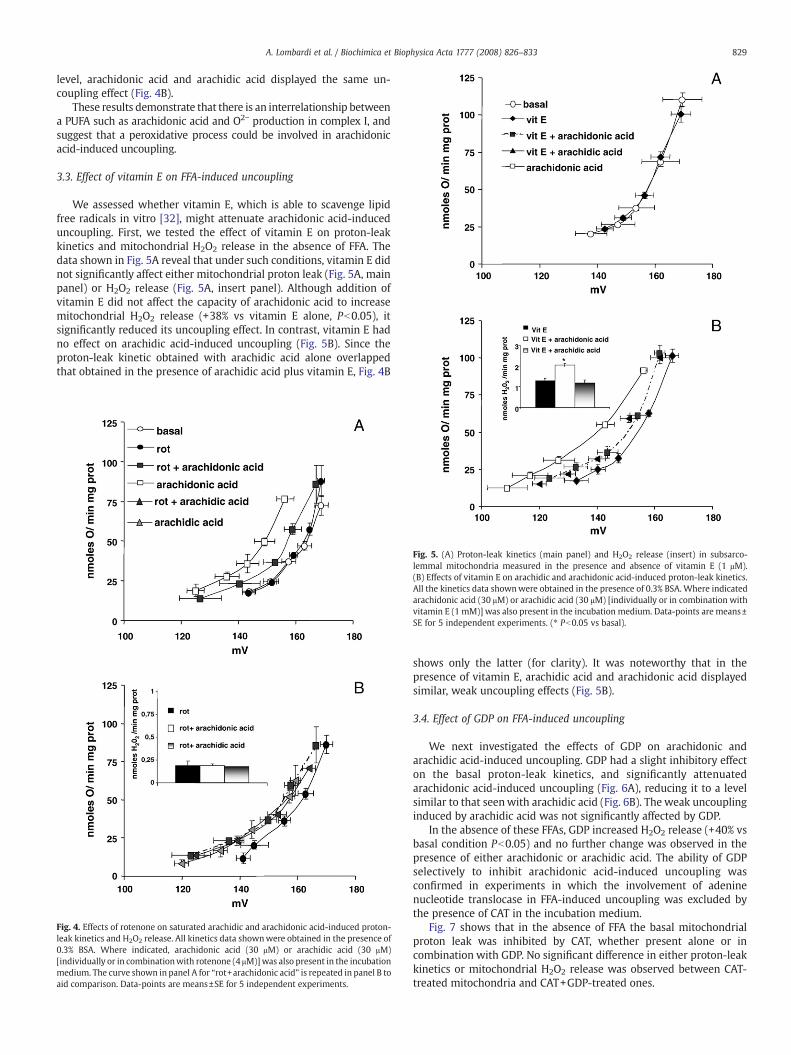
The inhibitory effect of rotenone on O2− production was still ob-served in the presence of exogenous FFA, as indicated by H2O2 release.Moreover in the presence of rotenone, arachidonic acid lacked itsstimulatory effect on H2O2 release (insert Figs. 4 and 2). Rotenone waseffective at inhibiting the effect of arachidonic acid on proton-leakkinetics (Fig. 4A), andwas ineffective on that induced by arachidic acid(Fig. 4B). Indeed, when complex I O2− production was at its lowest
Fig. 3. Effects of arachidic and arachidonic acid on proton-leak kinetics in subsarco-lemmal mitochondria. Kinetics were determined in the presence of 0.3% BSA alone(basal) and in combination with either arachidonic acid (30 μM) or arachidic acid(30 μM). Data-points are means±SE for 5 independent experiments.
829A. Lombardi et al. / Biochimica et Biophysica Acta 1777 (2008) 826–833
level, arachidonic acid and arachidic acid displayed the same un-coupling effect (Fig. 4B).
These results demonstrate that there is an interrelationship betweena PUFA such as arachidonic acid and O2− production in complex I, andsuggest that a peroxidative process could be involved in arachidonicacid-induced uncoupling.
3.3. Effect of vitamin E on FFA-induced uncoupling
We assessed whether vitamin E, which is able to scavenge lipidfree radicals in vitro [32], might attenuate arachidonic acid-induceduncoupling. First, we tested the effect of vitamin E on proton-leakkinetics and mitochondrial H2O2 release in the absence of FFA. Thedata shown in Fig. 5A reveal that under such conditions, vitamin E didnot significantly affect either mitochondrial proton leak (Fig. 5A, mainpanel) or H2O2 release (Fig. 5A, insert panel). Although addition ofvitamin E did not affect the capacity of arachidonic acid to increasemitochondrial H2O2 release (+38% vs vitamin E alone, Pb0.05), itsignificantly reduced its uncoupling effect. In contrast, vitamin E hadno effect on arachidic acid-induced uncoupling (Fig. 5B). Since theproton-leak kinetic obtained with arachidic acid alone overlappedthat obtained in the presence of arachidic acid plus vitamin E, Fig. 4B
Fig. 5. (A) Proton-leak kinetics (main panel) and H2O2 release (insert) in subsarco-lemmal mitochondria measured in the presence and absence of vitamin E (1 μM).(B) Effects of vitamin E on arachidic and arachidonic acid-induced proton-leak kinetics.All the kinetics data shownwere obtained in the presence of 0.3% BSA. Where indicatedarachidonic acid (30 μM) or arachidic acid (30 μM) [individually or in combination withvitamin E (1 mM)] was also present in the incubation medium. Data-points are means±SE for 5 independent experiments. (⁎ Pb0.05 vs basal).
Fig. 4. Effects of rotenone on saturated arachidic and arachidonic acid-induced proton-leak kinetics and H2O2 release. All kinetics data shownwere obtained in the presence of0.3% BSA. Where indicated, arachidonic acid (30 μM) or arachidic acid (30 μM)[individually or in combinationwith rotenone (4 μM)]was also present in the incubationmedium. The curve shown in panel A for “rot+arachidonic acid” is repeated in panel B toaid comparison. Data-points are means±SE for 5 independent experiments.
shows only the latter (for clarity). It was noteworthy that in thepresence of vitamin E, arachidic acid and arachidonic acid displayedsimilar, weak uncoupling effects (Fig. 5B).
3.4. Effect of GDP on FFA-induced uncoupling
We next investigated the effects of GDP on arachidonic andarachidic acid-induced uncoupling. GDP had a slight inhibitory effecton the basal proton-leak kinetics, and significantly attenuatedarachidonic acid-induced uncoupling (Fig. 6A), reducing it to a levelsimilar to that seenwith arachidic acid (Fig. 6B). The weak uncouplinginduced by arachidic acid was not significantly affected by GDP.
In the absence of these FFAs, GDP increased H2O2 release (+40% vsbasal condition Pb0.05) and no further change was observed in thepresence of either arachidonic or arachidic acid. The ability of GDPselectively to inhibit arachidonic acid-induced uncoupling wasconfirmed in experiments in which the involvement of adeninenucleotide translocase in FFA-induced uncoupling was excluded bythe presence of CAT in the incubation medium.
Fig. 7 shows that in the absence of FFA the basal mitochondrialproton leak was inhibited by CAT, whether present alone or incombination with GDP. No significant difference in either proton-leakkinetics or mitochondrial H2O2 release was observed between CAT-treated mitochondria and CAT+GDP-treated ones.
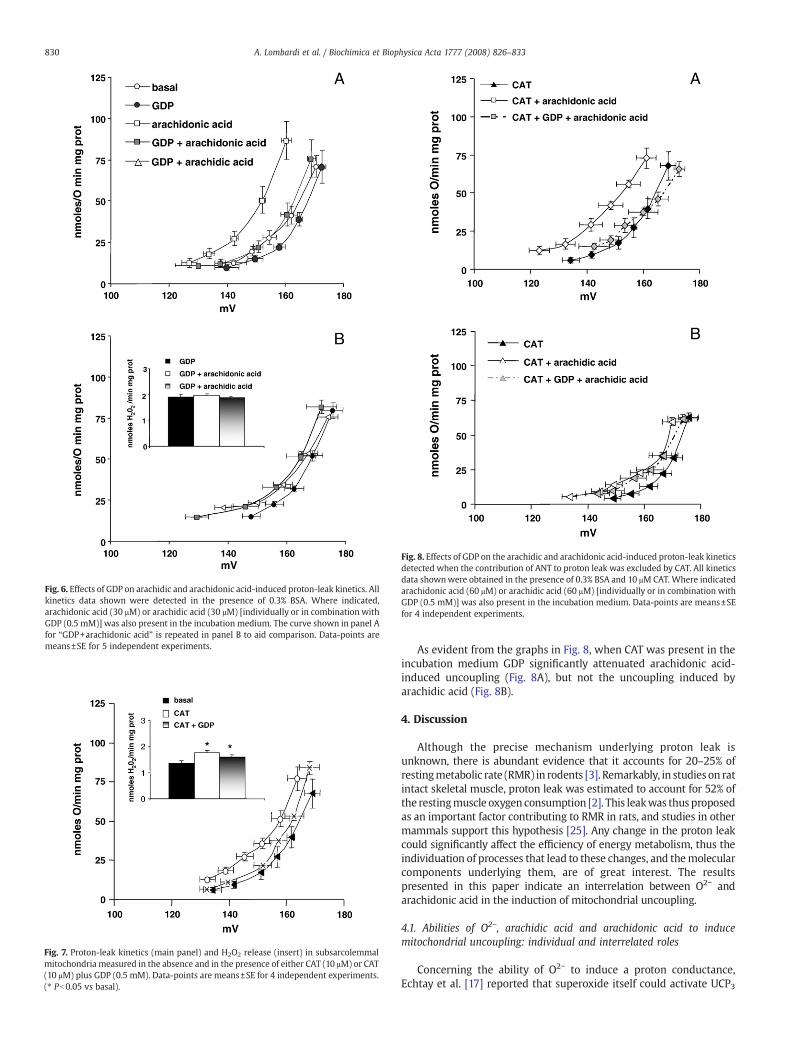
Fig. 8. Effects of GDP on the arachidic and arachidonic acid-induced proton-leak kineticsdetected when the contribution of ANT to proton leak was excluded by CAT. All kineticsdata shownwere obtained in the presence of 0.3% BSA and 10 μM CAT. Where indicatedarachidonic acid (60 μM) or arachidic acid (60 μM) [individually or in combination withGDP (0.5 mM)] was also present in the incubation medium. Data-points are means±SEfor 4 independent experiments.
Fig. 7. Proton-leak kinetics (main panel) and H2O2 release (insert) in subsarcolemmalmitochondria measured in the absence and in the presence of either CAT (10 μM) or CAT(10 μM) plus GDP (0.5 mM). Data-points are means±SE for 4 independent experiments.(⁎ Pb0.05 vs basal).
Fig. 6. Effects of GDP on arachidic and arachidonic acid-induced proton-leak kinetics. Allkinetics data shown were detected in the presence of 0.3% BSA. Where indicated,arachidonic acid (30 μM) or arachidic acid (30 μM) [individually or in combination withGDP (0.5 mM)] was also present in the incubation medium. The curve shown in panel Afor “GDP+arachidonic acid” is repeated in panel B to aid comparison. Data-points aremeans±SE for 5 independent experiments.
830 A. Lombardi et al. / Biochimica et Biophysica Acta 1777 (2008) 826–833
As evident from the graphs in Fig. 8, when CAT was present in theincubation medium GDP significantly attenuated arachidonic acid-induced uncoupling (Fig. 8A), but not the uncoupling induced byarachidic acid (Fig. 8B).
4. Discussion
Although the precise mechanism underlying proton leak isunknown, there is abundant evidence that it accounts for 20–25% ofrestingmetabolic rate (RMR) in rodents [3]. Remarkably, in studies on ratintact skeletal muscle, proton leak was estimated to account for 52% ofthe restingmuscle oxygen consumption [2]. This leakwas thus proposedas an important factor contributing to RMR in rats, and studies in othermammals support this hypothesis [25]. Any change in the proton leakcould significantly affect the efficiency of energy metabolism, thus theindividuation of processes that lead to these changes, and themolecularcomponents underlying them, are of great interest. The resultspresented in this paper indicate an interrelation between O2− andarachidonic acid in the induction of mitochondrial uncoupling.
4.1. Abilities of O2−, arachidic acid and arachidonic acid to inducemitochondrial uncoupling: individual and interrelated roles
Concerning the ability of O2− to induce a proton conductance,Echtay et al. [17] reported that superoxide itself could activate UCP3

831A. Lombardi et al. / Biochimica et Biophysica Acta 1777 (2008) 826–833
uncoupling activity from the matrix side. Subsequently, they ascribedthis effect of O2− to the end-products of the lipoperoxidation cascade,such as HNE [18–20], which would activate UCP3. The same group hadpreviously suggested that i) the role played by FFAs, in inducing UCP3-mediated uncoupling (when they are added to a mitochondria-containing incubation mixture) was restricted to the import of O2−
into the mitochondrial matrix once it had been exogenously producedby a xanthine plus xanthine oxidase system [18], and ii) when O2− wasendogenously produced by mitochondria and directly released intothe matrix (due to electron reverse from complex II to complex I), thisprocess was independent of the presence of FFAs and sufficient toactivate UCP3-mediated proton conductance indirectly through mem-brane lipid peroxidation products. Our results, however, show thatwhen mitochondrial endogenous FFAs were chelated by BSA, nodifferences in proton-leak kinetics were detected between the ab-sence and presence of rotenone, indicating that O2− itself was not ableto increase proton-leak kinetic (see Fig. 1).
Concerning FFA-induceduncoupling,we found thatwhen succinate-energized mitochondria were unable to produce high levels of O2−
in complex I because of rotenone-induced inhibition, FFAs induced aslight uncoupling that did not depend on the degree of unsaturationof their lateral chain [since both arachidic and arachidonic acid, whichhave the same chain length, showed the same uncoupling capacity(Fig. 4B)].
From the present data, it is also evident that changes in O2−
production did not affect arachidic acid-induced uncoupling (Fig. 4B),although the same changes significantly affected arachidonic acid-induced uncoupling (Fig. 4A). This suggests the involvement of aperoxidative process in arachidonic acid-induced uncoupling. More-over, the presence of the lipid antioxidant vitamin E [32] selectivelyinhibited arachidonic acid-induced uncoupling.
Under our experimental conditions, vitamin E did not affect mito-chondrial H2O2 release, in the absence of BSA, but increased its releasein the presence of arachidonic acid. These results indicated that theability of vitamin E to reduce arachidonic acid-induced uncouplingcould be attributable to its capacity to block the chain reaction in-volving the peroxyl-radical [32] rather than to an effect on mito-chondrial O2− levels. However, as we did not measure the levels oflipid hydroperoxides this conclusion is speculative.
The above results suggest that arachidonic acid and O2− exert acombined effect on mitochondrial uncoupling. This combined effectmay be relevant in certain conditions in which both an increase inmitochondrial FFA levels and O2− release into the matrix take place.
4.2. Capacity of arachidonic acid to induce mitochondrial H2O2 release
In the present study we observed that of the two FFA acid tested,arachidonic acid was the more effective at inducing both mitochon-drial H2O2 release and mitochondrial uncoupling. These effects areopposite to what would be expected from the hypothesis of “milduncoupling”; indeed, according such a hypothesis, high mitochondrialROS can be generated only under high membrane potential/lowelectron flux conditions [33]. However, we cannot exclude the pos-sibility that the uncoupling effect of arachidonic acid may limit aneven higher ROS production induced by arachidonic acid itself. Theidea is supported by the finding that when arachidonic acid-induceduncoupling was limited by vitamin E, the induction of H2O2 release byarachidonic acid was increased by about one-third (38% in absence ofvitamin E vs 28% in its absence).
Our finding that arachidonic acid induced an increase in H2O2
release, confirms the reports byother authors [34–36].Wealsoobservedthat rotenone, a known inhibitor of the electron reverse from complex IIto complex I, completely blocked arachidonic acid-stimulated H2O2
release. These results are consistent with the involvement of complex Iin arachidonic acid-induced mitochondrial H2O2 release and are inagreementwith the finding of Nethery et al. [35]who demonstrated the
ability of arachidonic acid to induce the formation of reactive oxygenspecies in succinate-energized diaphragm mitochondria, an effectinhibited by rotenone.
It has also been shown that arachidonic acid has the ability to affectboth mitochondrial transition pores [37,38] and cell death [39] even ifthe molecular mechanisms underlying these effects are not wellestablished. The relationship between apoptosis and ROS production iswidely documented (for reviews see [40,41]) and ROS could mediatethe ability of arachidonic acid to induce apoptosis. However, it is notclear if activation of transition pores is involved in arachidonic acid-induced apoptosis. Indeed, Dymkowska et al. [36] reported thatarachidonic acid induced apoptosis mainly by stimulating ROS pro-duction via a mechanism independent of activation of the mitochon-drial permeability transition, whereas Catisti and Vercesi [42]hypothesized an involvement of the pyridine nucleotide redox stateand of ROS in the arachidonic acid-induced permeability transition inrat liver mitochondria.
In our study, the involvement of the transition pore in arachidonicacid-induced uncoupling is unlikely because of the presence of EGTAin the medium. In addition, we did not observe any mitochondrialswelling in our experimental conditions (data not shown).
4.3. Possible involvement of UCP3 in the combined arachidonic acid/O2−
induced uncoupling effect
In vivo, UCP3 levels are upregulated principally when mitochondriallipid utilization – a process leading to high FFA levels [43–45] and a largerelease of O2− into the matrix [46] – is enhanced. In this condition, lipidperoxidation, a process that initiates chain reactions producing veryaggressive oxidants, take places [47]. The upregulation of UCP3 in theabove situation suggests that it could be involved in protecting againstlipid-induced oxidative damage at the mitochondrial level [48,49], andthat lipid hydroperoxides [21,22], or their metabolite HNE[20], mightactivate UCP3-mediated uncoupling. Concerning this lipid hydroper-oxide metabolite, Echtay et al. [20] showed that HNE stimulated theuncoupling activity of UCPs, and proposed a covalent modification ofUCPs by HNE as a molecular mechanism for the activation of UCPs [20].However, in that studyHNEwasexogenouslyadded tomitochondria in aconcentration that, as admitted by the authors [20], is much higher thanthat eventually induced by O2−. The Echtay hypothesis has recently beenquestioned by a study that failed to uncover any HNE-induced cova-lent modification of UCP1 protein, and indeed the later authorssuggested that the uncoupling effect of HNE could depend upon itsconversion to 4-hydroxynonenoic acid [24].
One of us first suggested that UCPs might be implicated in theexport of hydroperoxide fatty acid anions from the matrix [21], ahypothesis later extended by Jaburek et al. [22]. The latter group, usingEscherichia coli-expressed UCP2 reconstituted into liposomes, reportedthat although linoleic acid induces purine nucleotide-sensitive H+
uniport in liposomes containing UCP2, it does so with a lower affinitythan that observed for linoleic acid hydroperoxide. Indeed, the Km
value of linoleic acid hydroperoxide was almost 3-times that oflinoleic acid. Moreover, they showed that synthesized linoleic acidhydroperoxides caused a fast flip-flop-dependent acidification ofliposomes, thus suggesting that UCP2 may transport peroxidised fattyacids and mediate a fatty acid peroxide-cycling mechanism. Jabureket al. [22], on the other hand, added lipid hydroperoxide directly toUCP2-reconsituted proteoliposomes. This system could be useful fordemonstrating the effectiveness of UCP2 in the transport of lipidhydroperoxides, but it lacks physiological components that might in-activate lipid hydroperoxides, such as glutathione peroxidase [50,51].It therefore leaves without a definitive answer the question of theexistence of a possible flip-flop mechanism.
In the present paper, we induced endogenous O2− productionand its release inside the mitochondrial matrix, and we added only asmall amount of arachidic acid or arachidonic acid to BSA-treated

832 A. Lombardi et al. / Biochimica et Biophysica Acta 1777 (2008) 826–833
mitochondria. This should allow FFA and O2− to interact directly witheach other within the mitochondria, with the putative consequentformation in situ of lipid hydroperoxides and their metabolite.
In the present study, we observed that GDP, a compound oftenused to inhibit UCP3 uncoupling activity, significantly reduced ar-achidonic acid-induced uncoupling, although it was ineffective atinhibiting arachidic acid-induced uncoupling (Fig. 6). This led us tospeculate that UCP3 might be involved in arachidonic acid-induceduncoupling. Recently, however, the specificity of GDP for UCPs hasbeen questioned on the grounds that GDP could also inhibit ANT andother mitochondrial carriers [52–54], although, Parker et al. [52] re-ported that the interactions of GDP with carriers other than UCPappeared to be blocked by fatty acids. In our study, the above effect ofGDP was also observed when the contribution of ANT to fatty acid-induced uncoupling was inhibited by CAT, a finding that supports thepossible role of UCP3 in arachidonic acid-induced uncoupling.However, as we have no clear indications as to the capacity of GDPto inhibit the arachidonic acid-induced proton leak through carriersother than UCP3 and ANT, we can only postulate a possible in-volvement of UCP3, and we cannot exclude possible contributions byof other carriers.
A question that arises from the present data concerns the possiblefunctional relevance of the arachidonic acid/O2− combined effect (andthe consequent putative lipid hydroperoxide formation) in theactivation of proton leak. In the light of the hypothesis that UCP3catalyzes fatty acid hydroperoxide export, the above processes mayrepresent a protective mechanism for mitochondrial activity. Indeed,peroxidation of lipids initiates chain reactions producing very ag-gressive oxidants [47], and if lipid peroxides are translocated outsidethe matrix by UCP3, then in this way the matrix targets (mtDNA,aconitase, and other matrix-localized components) may be defended.The importance of such a defense is also demonstrated by the fact thatthe longevity of mammals shows a reverse correlationwith the degreeof oxidation of mtDNA, while mtDNA oxidation is in turn proportionalto the degree of mitochondrial fatty acid unsaturation [55]. Theimportance of an antioxidant defense of the mitochondrial matrixspace is also highlighted by the observation that knockouts of genesfor cytosolic superoxide dismutase result in mild, non-lethal pheno-types [56], whereas knockout of the gene for the matrix superoxidedismutase gives rise to a lethal neonatal phenotype characterized bymtDNA oxidative damage as well as by abnormalities of the respi-ratory chain and Krebs cycle [57]. The hypothesis that UCP3 mayexport fatty acid hydroperoxides, however, does not exclude thepossibility that lipid peroxidation metabolites such as 4-HNE couldactivate UCP3. Indeed, the formation of 4-HNE could signal to UCP3 thepresence of high levels of lipid hydroperoxides that need to be re-duced by their transport outside the matrix.
Acknowledgement
This work was supported by Grant MIUR-COFIN 2006 Prot2006051517; REGIONE CAMPANIA LR 28/5/02 N.5-2003 Prot 20050166044.
References
[1] M.D. Brand, The proton leak across the mitochondrial inner membrane, Biochim.Biophys. Acta 1018 (1990) 128–133.
[2] D.F.S. Rolfe, A.J. Hulbert, M.D. Brand, Characteristics of mitochondrial proton leakand control of oxidative phosphorylation in the major oxygen-consuming tissuesof the rat, Biochem. Biophys. Acta 1118 (1994) 405–416.
[3] D.F.S. Rolfe, M.D. Brand, The physiological significance of mitochondrial protonleak in animal cells and tissues, Biosci. Rep. 17 (1997) 9–16.
[4] R.K. Porter, A.J. Hulbert, M.D. Brand, Allometry of mitochondrial proton leak:influence of membrane surface and fatty acid composition, Am. J. Physiol 271(1996) R1550–R1560.
[5] M.D. Brand, N. Turner, A. Ocloo, P.L. Else, A.J. Hulbert, Proton conductance and fattyacyl composition of liver mitochondria correlates with body mass in birds,Biochem. J. 376 (2003) 741–748.
[6] P.S. Brookes, D.S.F. Rolfe, M.D. Brand, The proton permeability of liposomes madefrom mitochondrial inner membrane phospholipids: comparison with isolatedmitochondria, J. Membr. Biol. 155 (1997) 167–174.
[7] P.S. Brookes, A.J. Hulbert, M.D. Brand, The permeability of liposomes madefrommitochondrial inner membrane phospholipids: no effect of fatty acidcomposition, Biochim. Biophys. Acta 1330 (1997) 157–170.
[8] V.P. Skulachev, Fatty acid circuit as a physiological mechanism of uncouplingoxidative phosphorylation, FEBS Lett. 294 (1991) 158–162.
[9] D. Ricquier, F. Bouillaud, The uncoupling protein homologues: UCP1, UCP2, UCP3,StUCP and AtUCP, Biochem. J. 345 (2000) 161–179.
[10] S. Krauss, C. Zhang, B.B. Lowell, The uncoupling protein homologues, Nature 6(2005) 248–261.
[11] K.S. Echtay, E. Winkler, M. Klingenberg, Coenzyme Q is an obligatory cofactor foruncoupling protein function, Nature 408 (2000) 609–613.
[12] K.S. Echtay, D. Roussel, J. St-Pierre,M.B. Jekabsons, S. Cadenas, J.A. Stuart, J.A. Harper,S.J. Roebuck, A. Morrison, S. Pickering, J.C. Clapham, M.D. Brand, Superoxideactivates mitochondrial uncoupling proteins, Nature 415 (2002) 96–99.
[13] M. Moreno, A. Lombardi, P. de Lange, E. Silvestri, M. Ragni, A. Lanni, F. Goglia,Fasting, lipid metabolism, and triiodothyronine in rat gastrocnemius muscle:interrelated roles of uncoupling protein 3, mitochondrial thioesterase, andcoenzyme Q, FASEB J. 17 (2003) 1112–1114.
[14] A. Lanni, L. Beneduce, A. Lombardi, M. Moreno, O. Boss, P. Muzzin, J.P. Giacobino, F.Goglia, Expression of uncoupling protein-3 and mitochondrial activity in thetransition from hypothyroid to hyperthyroid state in rat skeletal muscle, FEBS Lett.444 (1999) 250–254.
[15] K.S. Echtay, M.D. Brand, Coenzyme Q induces GDP-sensitive proton conductance inkidney mitochondria, Biochem. Soc. Trans. 29 (2001) 763–768.
[16] K.S. Echtay, E.Winkler, K. Frischmuth, M. Klingenberg, Uncoupling proteins 2 and 3are highly active H(+) transporters and highly nucleotide sensitive when activatedby coenzyme Q (ubiquinone), Proc. Natl. Acad. Sci. U. S. A. 98 (2001) 1416–1421.
[17] K.S. Echtay, M.P. Murphy, R.A. Smith, D.A. Talbot, M.D. Brand, Superoxide activatesmitochondrial uncoupling protein 2 from the matrix side. Studies using targetedantioxidants, J. Biol. Chem. 277 (2002) 47129–47135.
[18] D.A. Talbot, A.J. Lambert, M.D. Brand, Production of endogenous matrix superoxidefrom mitochondrial complex I leads to activation of uncoupling protein 3, FEBSLett. 556 (2004) 111–115.
[19] M.P. Murphy, K.S. Echtay, F.H. Blaikie, J. Asin-Cayuela, H.M. Cocheme, K. Green, J.A.Buckingham, E.R. Taylor, F. Hurrell, G. Hughes, S. Miwa, C.E. Cooper, D.A.Svistunenko, R.A. Smith, M.D. Brand, Superoxide activates uncoupling proteinsby generating carbon-centered radicals and initiating lipid peroxidation: studiesusing a mitochondria-targeted spin trap derived from alpha-phenyl-N-tert-butylnitrone, J. Biol. Chem. 278 (2003) 48534–48545.
[20] K.S. Echtay, T.C. Esteves, J.L. Pakay, M.B. Jekabsons, A.J. Lambert, M. Portero-Otin,R. Pamplona, A.J. Vidal-Puig, S.J. Wang, M.D. Roebuck Brand, A signalling role for4-hydroxy-2-nonenal in regulation of mitochondrial uncoupling, EMBO J. 22(2003) 4103–4110.
[21] F. Goglia, V.P. Skulachev, A function for novel uncoupling proteins: antioxidantdefense of mitochondrial matrix by translocating fatty acid peroxides from theinner to the outer membrane leaflet, FASEB J. 12 (2003) 1585–1591.
[22] M. Jaburek, S. Miyamoto, P. Di Mascio, K.D. Garlid, P. Jezek, Hydroperoxy fatty acidcycling mediated by mitochondrial uncoupling protein UCP2, J. Biol. Chem. 279(2004) 53097–53102.
[23] T.C. Esteves, M.D. Brand, The reactions catalysed by the mitochondrial uncouplingproteins UCP2 and UCP3, Biochim. Biophys. Acta 1709 (2005) 35–44.
[24] I.G. Shabalina, N. Petrovic, T.V. Kramarova, J. Hoeks, B. Cannon, J. Nedergaard, UCP1and defense against oxidative stress. 4-Hydroxy-2-nonenal effects on brown fatmitochondria are uncoupling protein 1-independent, J. Biol. Chem. 281 (2006)13882–13893.
[25] R.K. Porter, M.D. Brand, Body mass dependence of H+ leak in mitochondria and itsrelevance to metabolic rate, Nature 362 (1993) 628–630.
[26] S. Servais, K. Couturier, H. Koubi, J.L. Rouanet, D. Desplanches, M.H. Sornay-Mayet, B.Sempore, J.M. Lavoi, R. Favier, Effect of voluntary exercise on H2O2 release bysubsarcolemmal and intermyofibrillarmitochondria, Free Radic. Biol. Med. 35 (2003)24–32.
[27] R. Pamplona, M. Bring-Otin, D. Riba, J.R. Requena, S.R. Thorpe, M. Lopez-Torres, G.Barja, Low fatty acid unsaturation: a mechanism for lowered lipoperoxidativemodification of tissue proteins in mammalian species with long life spans,J. Gerontol. A Biol. Sci. Med. Sci. 55 (2000) B286–B291.
[28] V. Beck, M. Jaburek, T. Demina, A. Rupprecht, R.K. Porter, P. Jezek, E.E. Pohl,Polyunsaturated fatty acids activate human uncoupling proteins 1 and 2 in planarlipid bilayers, FASEB J. 21 (2007) 1137–1144.
[29] V.G. Richieri, A. Anel, A.M. Kleinfeld, Interactions of long-chain fatty acids andalbumin: determination of free fatty acid levels using the fluorescent probeADIFAB, Biochemistry 32 (1993) 7574–7580.
[30] M. Crompton, The mitochondrial permeability transition pore and its role in celldeath, Biochem. J. 341 (1999) 233–249.
[31] G. Barja, Mitochondrial free radical production and aging in mammals and birds,Ann. N. Y. Acad. Sci. 854 (1998) 224–238.
[32] M.B. Gavazza, A. Catala, The effect of alpha-tocopherol on lipid peroxidation ofmicrosomes and mitochondria from rat testis, Prostaglandins Leukot. Essent. FattyAcids 74 (2006) 247–254.
[33] S. Papa, V.P. Skulachev, Reactive oxygen species, mitochondria apoptosis andaging, Mol. Cell. Biochem, 174 (1997) 305–319.
[34] T. Cocco, M. Di Paola, S. Papa, M. Lo Russo, Arachidonic acid interaction with themitocondrial electron transport chain promotes reactive oxygen species genera-tion, Free Rad. Biol. Med. 27 (1999) 51–59.

833A. Lombardi et al. / Biochimica et Biophysica Acta 1777 (2008) 826–833
[35] D. Nethery, L.A. Callahan, D. Stofan, R. Mattera, A. Di Marco, G. Supinski, PLA(2)dependence of diaphragm mitochondrial formation of reactive oxygen speciesPLA2, J. Appl Physiol. 89 (2000) 72–80.
[36] D. Dymkowska, J. Szczepanowska, M.R. Wieckowski, L. Wojtczak, Short-term andlong-term effects of fatty acids in rat hepatoma AS-30d cells: the way to apoptosis,BBA 1763 (2006) 152–163.
[37] M. Di Paola, M. Lorusso, Interaction of free fatty acids with mitochondria: coupling,uncoupling and permeability transition, BBA 1757 (2006) 1330–1337.
[38] M. Di Paola, P. Zaccagnino, C. Oliveros-Celis, M. Lorusso, Arachidonic acid inducesspecific membrane permeabilità in heart mitochondria, FEBS Lett. 580 (2006)775–781.
[39] D. Penzo, C. Tagliapietra, R. Colonna, V. Petronilli, P. Bernardi, Effects of fatty acidson mitochondria: implications for cell death, BBA 1555 (2002) 160–165.
[40] H.U. Simon, A. Haj-Yehia, A. Levi-Shaffer, Role of reactive oxygen species (ROS) inapoptosis induction, Apoptosis 5 (2000) 415–418.
[41] C. Fleury, B. Mignotte, J.L. Vayssiere, Mitochondrial reactive oxygen species in celldeath signalling, Biochimie 84 (2002) 131–141.
[42] R. Catisti, A.E. Vercesi, The participation of pyridine nucleotides redox state andreactive oxygen in the fatty acid-induced permeability transition in rat liver, FEBSLett. 464 (1999) 97–101.
[43] J. Himms-Hagen, M.E. Harper, Physiological role of UCP3 may be export of fattyacids from mitochondria when fatty acid oxidation predominates: an hypothesis,Exp. Biol. Med. 226 (2001) 78–84.
[44] E. Silvestri, M. Moreno, A. Lombardi, M. Ragni, P. de Lange, S.E. Alexson, A. Lanni, F.Goglia, Thyroid-hormone effects on putative biochemical pathways involved inUCP3 activation in rat skeletal muscle mitochondria, FEBS Lett. 579 (2005)1639–1645.
[45] E. Silvestri, P. de Lange, M. Moreno, A. Lombardi, M. Ragni, A. Feola, L. Schiavo, F.Goglia, A. Lanni, Fenofibrate activates the biochemical pathways and the de novoexpression of genes related to lipid handling and uncoupling protein-3 functionsin liver of normal rats, Biochim. Biophys. Acta 1757 (2006) 486–495.
[46] J. St-Pierre, J.A. Buckingham, S.J. Roebuck, M.D. Brand, Topology of superoxideproduction from different sites in the mitochondrial electron transport chain,J. Biol. Chem. 277 (2002) 44784–44790.
[47] K. Yagi, Lipid peroxides and human diseases, Chem. Phys. Lipids 45 (1987)337–351.
[48] J. Hoekes, M.K. Hesselink, P. Schrauwen, Involvement of UCP3 in mild uncouplingand lipotoxicity, Exp Gerontol. 41 (2006) 658–662.
[49] J. Hoeks, M.K. Hesserlin, W. Sluiter, G. Schaart, J. Willems, A. Morrisson, J.C.Clapham, W.H. Saris, P. Schrauwen, The effect of high-fat feeding on intramuscularlipid and lipid peroxidation levels in UCP3-ablated mice, FEBS Lett. 580 (2006)1371–1375.
[50] H. Imai, Y. Nakagawa, Biological significance of phospholipid hydroperoxideglutathione peroxidase (PHGPX, GPXA) in mammalian cells, Free Radic. Biol. Med.34 (2003) 145–169.
[51] K. Nomura, H. Imai, T. Koumura, T. Kobayashi, Y. Nakagawa, Mitochondrialphospholipid hydroperoxide glutathione peroxidase inhibits the release ofcytochrome c from mitochondria by suppressing the peroxidation of cardiolipinin hypoglycaemia-induced apoptosis, Biochem. J. 351 (2000) 183–193.
[52] N. Parker, D. Humphry, K. Green, M.D. Brand, Abstract 14th European BioenergeticConference, Moscow, 22–27 July, Biochim. Biophys. Acta Bioenergetics 14 (2006)385.
[53] L.S. Khailova, E.A. Prikhodko, V.I. Dedukhova, E.N. Mokhova, V.N. Popov, V.P.Skulachev, Participation of ATP/ADP antiporter in oleate- and oleate hydroper-oxide-induced uncoupling suppressed by GDP and carboxyatractylate, Biochim.Biophys. Acta 1757 (2006) 1324–1329.
[54] N. Parker, C. Affourtit, A.J. Vidal-Puig, M.D. Brand, Energisation- dependentendogenous activation of proton conductance in skeletal muscle mitochondria,Biochem. J. 412 (1) (2008) 131–139.
[55] G. Barja, Rate of generation of oxidative stress-related damage and animallongevity, Free Radic. Biol. Med. 33 (2002) 1167–1172.
[56] L.M. Carlsson, J. Jonsson, T. Edlund, S.L. Marklund, Mice lacking extracellularsuperoxide dismutase are more sensitive to hyperoxia, Proc. Natl. Acad. Sci. U. S. A.92 (1995) 6264–6268.
[57] A.G. Reaume, J.I. Elliott, E.K. Hoffman, N.W. Kowal, R.J. Ferrante, D.F. Siwek, H.M.Wilcox, D.C. Flood, M.F. Beal, R.H. Brown, et al., Motor neurons in Cu/Zn superoxidedismutase-deficient mice develop normally but exhibit enhanced cell death afteraxonal injury, Nat. Genet. 13 (1996) 43–47.





























